Submitted:
08 December 2025
Posted:
09 December 2025
You are already at the latest version
Abstract
Immunotherapy (IT) and especially immune checkpoint blockade (ICB) changed the therapeutic approaches in non-small cell lung cancer (NSCLC). Still, primary or second-ary resistance and a percentage of long responders and survivors have been observed. The objective is to have a deeper understanding of the complex mechanisms of primary and secondary resistance to IT involving tumor cells, TME and host, to find strategies to over-come it. In this aim a search of the key words has been performed to identify the relevant evidence in the literature. The most available approach thus far is the combination of IT with chemotherapy (CT) and/or radiotherapy (RT), relying on the synergistic effect in enhancement of immunogenic cell death. Even so, a lot of questions are to be answered, as a dual role has been observed, considering the complex effect, especially on the TME. Preclinical and clinical studies investigate the best sequencing and timing of chemoradiation with IT, the optimal RT volumes, sites, dose/fractionation regimens, to favor immunostimulation over suppression on the TME. Moving forward, multiple agents addressing coinhibitory or costimulatory receptors, on immune or tumor cells, are under evalua-tion. In perspective, a huge potential of combination therapies is arising, but to which tar-get, to what patient, at what time and sequence to be administered, considering the complex mechanisms of resistance, needs dynamic biomarkers to guide the decisions, to-wards more personalized treatments.
Keywords:
chemo-radio-immunotherapy
; immune resistance
; NSCLC
1. Introduction
In the last decade immunotherapy and especially immune checkpoint inhibitors (ICIs) revolutionized cancer treatment and changed the therapeutic strategies in many cancers as well as in lung cancer, the most frequently diagnosed cancer, responsible for almost 2.5 million of new cases and the leading cause of cancer death, with an estimated 1.8 million deaths [1].
Cancer immunotherapy has a history of more than 130 years, going back to William B. Coley who is considered it’s promoter, as he observed in 1891 that a patient with sarcoma presenting post-surgery erysipelas infection with streptococcus pyogenes experienced a complete regression of his cancer. He explained this remission as immune reaction and started to treat cancer patients with bacterial products [2]. A century of controversies followed, until the interest of the role of immune system in cancer patients reemerged and drove to the birth of immuno-oncology [3].
1.1. Cancer Immune Surveillance and Immunoediting
In the 1950s a Hypothesis of “cancer immune surveillance” was elaborated independently by M.F. Burnet and L. Thomas, sustaining that the immune system can recognize tumor antigens and can eliminate tumor cells. This theory raised a lot of debates, but only in 2001, R.D. Schreiber and colleagues evidenced that chemically induced sarcoma grew better in immune-incompetent mice than in wild animals. Furthermore, they showed that 40% of chemically induced tumors from immune-incompetent mice were rejected when transplanted to immune-competent hosts, due to T-cell mediated immune responses and followed by immunologic memory [3].
Finally, a recognition was obtained that immune surveillance represents only one dimension of the complex interaction between the immune system and cancer, inducing a new concept of cancer immune editing and “the three Es” theory, which assumes three phases: elimination, equilibrium and escape, [3,4]. In the first phase of elimination or immune surveillance, the immune system is capable to eradicate the developing tumor cells. A cancer immune cycle was described in seven steps by Daniel S. Chen and Ira Mellman in 2013 [5]. Antigens released by tumors are taken over by dendritic cells. In the process of priming which takes place in the lymph node, these antigens are presented to the T lymphocytes, which undergo activation and clonal expansion. Trafficking through blood vessels, and the penetration between endothelial cells, drives the infiltration of T cells into the tumor where they are becoming able to recognize and to destroy tumor cells and produce more antigens to argument the process, Figure 1. This elimination phase is considered immunogenic cell death.
The process is more complex with several interactions between the tumor cells, their immunogenic capacity, mutational load, sensitivity to immune effector and the general immune status of the host, characterized for example by lymphocyte count, infiltration with T cells, the presence or absence of immune checkpoints [6].
Although these interactions, spontaneous tumor regressions are seldom because of progressive developments of inhibition mechanisms of the immune system. For a long time, which can take several years an equilibrium phase is developing between immune surveillance and the inhibition mechanisms of the immune system, but still with the capability of controlling the disease. Following genetic instability and immune selection, new variants of tumor cells are immunologically “sculpted by immune editors”. Finally, these variants of tumor cells manage to escape from immune surveillance and proliferate by becoming clinically detectable.
1.2. Immune Checkpoint Blockade (ICB)
In the aim of better understanding of the process of immune evasion, one of the first mechanisms evidenced was the direct inhibition of T cells. James P Allison and Tasuku Honjo, discovered the immune checkpoint function of receptors like Cytotoxic T Lymphocyte Associated Antigen 4 (CTLA-4) and Programmed Death 1 (PD-1) on the surface of T-cells, acting like “brakes” of the immune system, to prevent autoimmune reactions. For their vision regarding cancer therapy by inhibiting the negative immune regulation by antibody blockade of these receptors, they received the Nobel Prize in Physiology and Medicine in 2018. Based on the clinical benefits, the US Food and Drug Administration (FDA) approved several monoclonal antibodies (mAb) like ipilimumab, targeting CTLA-4, first in melanoma, then in lung cancer, nivolumab, pembrolizumab targeting the receptor PD-1 on T cells, atezolizumab and durvalumab targeting the ligand PDL-1 on lung tumor cells, preventing thus the binding to the PD-1 receptor on the T-cell. The first PD-1 inhibitor, nivolumab, was approved in 2014, for previously treated advanced squamous and non-squamous NSCLC, according to the results of CheckMate 017 and CheckMate 057 trials [7,8,9].
In the meantime, the combined long-term results of the two trials showed a significant benefit for nivolumab with 5-year survival of 13.4% compared to 2.6% for the standard second-line treatment with docetaxel, Figure 2, [10]. The results proved to be even better for patients with PDL1 expression ≥ 1%, obtaining 18,3% vs. 3,4%.
2. Primary and Secondary Resistance
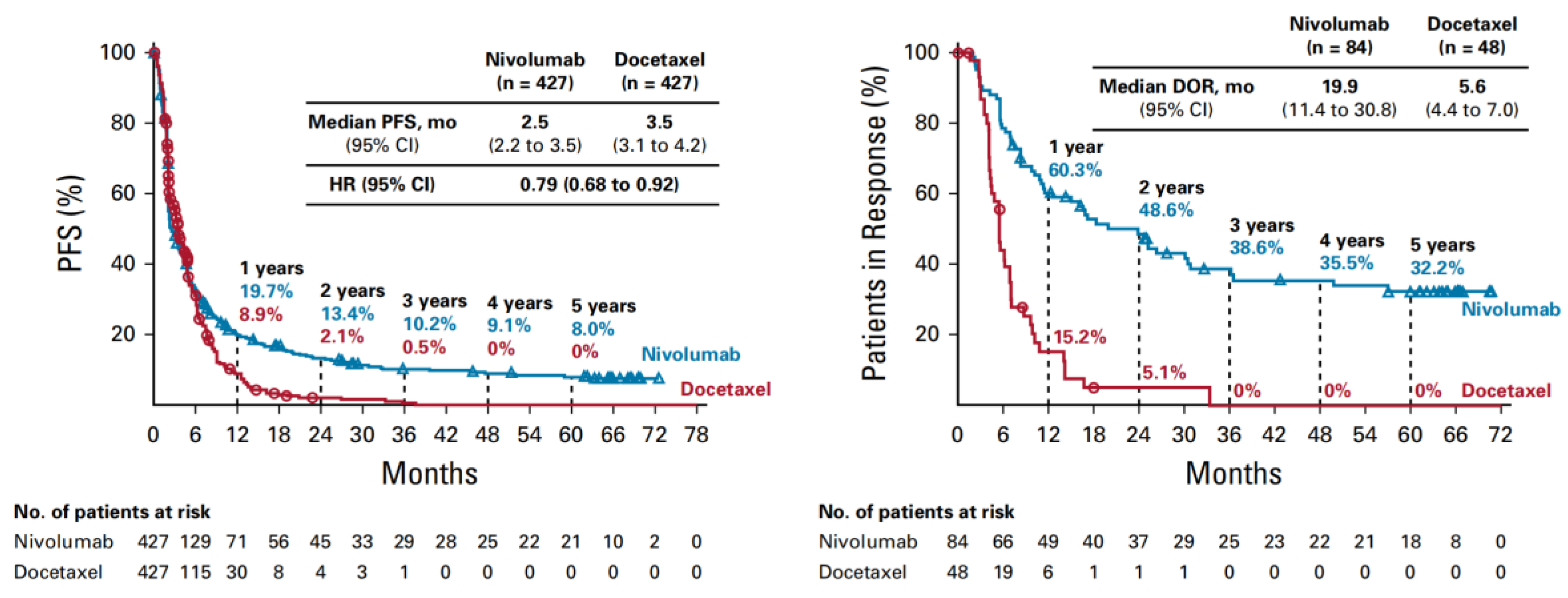
Despite these unprecedented results, the survival curves revealed in the first 6 months an overlapping, steep part of the curves, and afterwards a progressive falling apart of the curves, with leveling after 3-years of the nivolumab arm, obtaining a percentage of long-term survivors. PFS curves also overlap in the first 6 months attaining at about 40% of patients without disease progression. The initial steep part of the nivolumab curve can be interpreted as a an intrinsic, primary resistance, meaning a lack of initial response, whereas the second part, an acquired, secondary resistance, translated into disease progression after an initial response to IT. Patients in response were 60% at 1 year and at about 40% of patients with progressive disease (Figure 3) [10]. The main question is how to identify these categories of patients, including the long survivors.
In the OAK trial patients who received one to two previous platinum-based chemotherapy regimens for stage IIIB or IV NSCLC with atezolizumab in second- or third-line treatment versus docetaxel, had progressive disease in about 44% [11].
In the Keynote 024 trial, in first line treatment with pembrolizumab versus platinum doublet, in advanced NSCLC, PDL1≥50% and no sensitizing mutation of the epidermal growth factor receptor gene (EGFR) or translocation of the anaplastic lymphoma kinase gene (ALK), after overlapping curves in the first 3 months, PFS at 6 months was 62% vs. 50.3%, meaning that 38% of patients were in progression or dead [12].
In the Keynote 042 trial patients who had previously untreated advanced NSCLC with PD-L1 expression at least 50% of tumor cells and EGFR, ALK negative, were randomized to receive in first line either pembrolizumab, or the investigator’s choice of platinum-based chemotherapy. The response rate was higher in the pembrolizumab group than in the chemotherapy group, 44.8% vs. 27.8% and 21% of patients progressed [13].
Zhou S. et al [14] emphasized that primary resistance to IT in NSCLC patients is around 21-27% for nivolumab alone or combined with another ICI like ipilimumab as first-line treatment [15,16] and 40-44% in second line. When combining IT with chemotherapy as first line treatment the incidence of primary resistance dropped around 9-11%. [17,18,19]. Secondary resistance to IT in first line treatment, was 52% in the Keynote 042 study, and in second line treatment was 55% in the OAK study and 64% in the pooled analysis of the CheckMate 017 and CheckMate 057 trials, at four years of follow-up.
In april 2019, the Society for Immunotherapy of Cancer (SITC) developed expert consensus definitions for the PD-L1 inhibitor resistance, including clinical definitions of primary resistance, secondary resistance, and resistance that develops after discontinuation of therapy [20].
Despite no uniform agreement on every issue, primary resistant disease was considered in a patient who has disease progression (DP), or stable disease (SD) less than 6 months, after receiving at least 6 weeks of exposure to PD-L1 checkpoint inhibitors, but no more than 6 months. RECIST1.1 was used as response criteria and additionally immune RECIST (iRECIST) for mixed responses and pseudo progression. Although evaluating diameters of tumors might not be accurate in many circumstances, as an excavating tumor is witnessed often after IT. On the other hand, caveats on primary resistance are late responders after initial progression, with a frequency varying between drugs and tumor types, or treatments beyond progression, especially in oligo-progression, where local therapy to the progressing lesion can be associated. Patients who experience early toxicities, needing steroids and cessation of PD-L1 inhibitors are difficult to assess for primary resistance.
Secondary resistance can be considered, when a patient treated with PD-L1 checkpoint inhibitors, has a documented, confirmed objective response or prolonged SD more than 6 months, and then has DP. Reconfirmation by imaging procedures is needed within 4-12 weeks after the first evidence of disease progression, in the setting of ongoing treatment, although the timeframe may need to be rethought in indolent tumor types. All definitions are to be based on patients being treated with systemic anti- PD-L1 monotherapy, and combinations with other ICIs and other types of systemic or local therapies were not addressed [20].
3. Mechanisms of Resistance
The resistance mechanisms can be awarded to tumor cells, immune cells and host [21]. Primary resistance may result mainly of low tumor immunogenicity, impaired antigen presentation, immunosuppressive TME, whereas the complex mechanisms of secondary resistance involve tumor cells and TME. Tumor cells are characterized by heterogeneity and may gain clonal neoantigen depletion. The impact of ICB might be a sub-clonal selection of tumor cells with genomic and epigenetic transformation, responsible for immune inhibitory signals. Immune cells can undergo exhaustion or dysfunctions, responsible for defective priming, immune suppressive signals, with the polarization of the TME cells towards immune-suppressive cells like myeloid derived suppressive cells (MDSCs), regulatory T cells, (T regs), type 2 macrophages (TAM2) converted from type 1 macrophages (TAM1), or type 2 neutrophils (TAN2) from type1 neutrophils (TAN1) respectively and cancer associated fibroblasts (CAFs). Host factors can consist of altered antigen recognition according to HLA loss, defective metabolic adaptation or cytokine production and the micro-biome, diet, and medication like antibiotics and steroids can be encountered.
Mechanisms of resistance can be also classified according to each phase of the cancer immunity cycle. Firstly, a low tumor mutation burden (TMB) and a reduction of neoantigens can take place, affecting tumor immunogenicity. Next-generation sequencing applications found that gene mutations affecting TMB and PD-L1 expression may be involved in primary and secondary, acquired resistance to IT. For example, in oncogene driver mutated subsets of NSCLC cells like EGFR, HER2 or ALK, ROS, RET and MET fusions, limited benefit was obtained from ICB, despite enhanced PD-L1 expression, but down-regulation of TMB and tumor infiltrated lymphocytes (TILs), leading to resistance to IT [14,22]. Thus, the most applied biomarkers TMB and PDL1 expression seem to be independent and influenced by the complex mechanisms of resistance involved.
Secondary, impaired recruitment and dysfunction of DCs and antigen processing and presentation have been encountered. Human Leukocyte Antigen (HLA) gene loss affects neo-antigen presentation [14,23], conducting to an impaired priming or activation of the T cells. In 40% of cases, NSCLC cells are characterized by allele-specific HLA Loss, related to high TMB and positive PDL1, but poor response to ICIs [24,25].
Recent studies evidenced that mutations in KRAS, SKT11, KEAP1, JAK1/2, B2M, APC, MTOR and TP53 and co-mutations of these genes are influencing the metabolic changes in cancer cells, cytotoxic T cells, being thus the main determinants of mechanisms of resistance to ICI, in NSCLC patients [26,27].
The inhibition of T cell trafficking by down regulation of chemokines such as CXCL9 and CXCL10, followed by prevention of T cell infiltration in the tumor, by up-regulation of VEGF and TGF β conducting to immune desert or exclusion and finally to the inability of eliminating tumor cells also by up-regulation of immune-suppressive cells, cytokines and co-expression of multiple immune checkpoints [21].
The main objective consists of overcoming resistance mechanisms to IT. Regarding the complexity of this mechanism, combination treatments might be the rational answer. But it is unlikely to target all these mechanisms simultaneously. The most available approach, thus far, is the combination of CT and RT with IT.
4. Preclinical Studies Combining Immunotherapy with Chemotherapy and Radiotherapy
Evidence from preclinical studies underlined a synergistic anti-tumor effect, by combining immunotherapy with chemotherapy and radiotherapy, assuring increased cytotoxicity, enhancement of immunogenic cell death, tumor necrosis and neoantigen creation, thus challenging the multi-modality treatment strategy. But also, a double edge effect has been described [2,6,7].
4.1. Chemotherapy
CT demonstrates great value in combination with IT. Despite immunosuppressive effect on the bone marrow, is responsible of induction of immunogenic cell death by direct cytotoxicity producing DNA lethal and sub-lethal damage, assuring neoantigen production and has also immunoregulatory functions by enhancing APC and effector T cell response, and disrupting immune suppressive pathways. The most frequently used drugs in NSCLC are platinum compounds, cisplatin or carboplatin, etoposide, and third generation drugs like vinorelbine, paclitaxel and pemetrexed, the latter used in non-squamous NSCLC [7,28,29,30].
Platinum compounds promote up-regulation of MHC class I expression, release of tumor antigens, emission of danger-associated molecular patterns (DAMPs), recruitment and proliferation of effector cells, up regulation of cytotoxic effectors and down regulation of the immune-suppressive microenvironment.
Whereas the immunomodulatory role of etoposide is not fully understood with suppression of inflammatory cytokine levels and apoptosis of peripheral activated lymphocytes [17], Gameiro and al. evidenced that vinorelbine a semi-synthetic vinca alkaloid which interferes with the polymerization of tubulin, contained in microtubules, blocking thus cell division, in combination with cisplatin, enhances MHC class I expression, increases sensitivity to perforin/granzyme-mediated cytotoxic T Lymphocyte killing by modulation of tumor phenotype, cytokine/chemokine expression and the proapoptoic/antiapoptoic gene ratio, such as tumor necrosis factor-α (TNFα), IL8, CXCL5, and B cell lymphoma-2 gene (BCL-2). The data are complementary to that of immunogenic cell death, suggesting the benefit of combining chemotherapy with immunotherapy [32].
Paclitaxel, naturally produced in the bark and needles of Taxus brevifolia, increases the rate of apoptosis in tumor cells, releasing tumor antigens, enhancing thus the antigen presentation and phagocytosis of the APCs. Furthermore, paclitaxel increases the proinflammatory cytokines as IL-10 and decreases the number and activity of T regs [33].
Pemetrexed used in non-squamous NSCLC, inhibits folate metabolism, stimulates the activity and infiltration of T cells, making it a candidate for combination with radiotherapy and immunotherapy [34].
4.2. Radiotherapy
Preclinical studies evidenced also an immune activation by RT, which produces DNA damage, directly or by generation of free radicals, inducing the release of DAMPs, producing a series of biological events like neoantigen generation, APC recruitment and antigen presentation, priming and T cell proliferation, trafficking and infiltration of the tumor and finally enabling to recognize and destroy tumor cells in the irradiated volume or even far from it, named abscopal effect. Thus, local RT can trigger systemic antitumor response, at distant non-irradiated sites through infiltrating immune cells and reprogrammed TME, alteration of cytokines and chemokines, inducing the abscopal effect. This is more frequently observed when irradiation is combined with IT [2].
Frey et al [35] demonstrated that hypo fractionated RT of 2 fractions of 5 Gy induced a significant infiltration of APCs on day 5-8 in colorectal cancer. CD8+ T cells recruited by activated APCs, were enhanced on day 8. As the immune cell infiltration takes place in a narrow window time, there can be assumed that RT effect can be boosted by IT. Furthermore, infiltration of immune suppressive cells such as T regs and MDSC are less influenced by hypo fractionated RT. There is a slight increase in T regs on day 8-10, following infiltration of cytotoxic T cells, suggesting that reirradiation of the tumor on days 9-10 would be optimal. RT also impacts recruitment and activation of APCs and T cells according to dose, at an administration of 2Gy/fraction but even more at 5-8 Gy/fraction. Vanpouille et al. showed that 3 doses of 8Gy produces double strand DNA accumulation in the cytosol and activation of INF-I pathway via cyclic GMP-AMP (cGAMP) synthase (cGAS) and its downstream stimulation of interferon genes (STING), producing INFβ, activating DCs, and priming tumor specific CD8 T cells. Furthermore, in the presence of ICIs, mediated regression of irradiated tumors takes place demonstrating the augmentation of abscopal effect. Whereas, single fraction, high dose, with a threshold between 12-18Gy induces activation of exonuclease Trex1 at levels that degrade the accumulated DNA in the cytosol of irradiated tumor cells, making unable the INF-I pathway. These data have an important implication for the choice of radiation dose and fractionation in the clinic, to convert unresponsive patients into responders to IT [36].
Furthermore Dovedi et al. [37,38] evidenced better survival curves when RT 5 × 2Gy was administered concurrently with an PD-L1 antibody on day 1 or 5, than sequentially on day 7. And finally, Demaria et al. [39], demonstrated the abscopal effect, the reduction of tumor growth outside the field of radiation in mice who received RT and growth factor Flt3-Ligand (Flt3-L), but not by Flt3-L or RT alone, or in mice with T cell deficiency, emphasizing their role in this process. Moreover, her group demonstrated that fractionated but no single dose radiotherapy induces an immune mediated abscopal effect when combined with anti-CTLA-4 antibodies [40].
Also studies in murine lymphoma models demonstrated that the combination of RT with stimulatory CD40 mAb produce an enhancement of DC functions, through increased expression of major histocompatibility complex (MHC) molecules and proinflammatory cytokines finally stimulating T cell trafficking and M1 polarized macrophages [41,42].
RT produces also an activation of mTOR signaling, as part of the DNA damage response, leading to an increase of peptide presentation by tumors and T-cell activation [43,44].
On the other hand, preclinical studies also demonstrated immune suppression by RT on circulating lymphocytes B or T and lymphoid tissue which are highly sensitive to irradiation, whereas CD4+ T-cells or T-regs are more resistant and a slight increase was obtained on days 8 to 10 of the letters [35]. L Deng et al. [45] showed growth of PD-L1 expression after RT. Derer, Spiljar et al. [46] suggested a tumor cell mediated up-regulation of PD-L1 expression following chemoradiation and dependent on fractionation of RT, offering a good ration for combination with ICIs. Furthermore, the dual role of RT on TME has been emphasized. Z Zang et al. described in detail the immunostimulatory effect, produced by damaged DNA on the cGAS-STING and Type I interferon signaling, activating the DC and priming the T cells. On the contrary, immunoinhibitory effects on tumor cells, stromal and immune cells, are leading to reprogramming the TME. To influence this balance various complex interactions, modulations of the secretion of chemokines and cytokines or growth factors take place [2]. For example, DCs migration is favored by chemokines CCL7, CCL21. T-cell priming, CD8+Tcell cytotoxicity is mediated by interleukin IL-12, NK enhancement by IL-18, TAM1 polarization by TNFɑ, TAN1 polarization by INFβ. On the other hand, immunoinhibitory effects like TAM2 infiltration and differentiation are favored by CXCL2, CXCL12, CCL2,3,5, MDSC and Tregs recruitment by CCL2 and CCL22, 28 respectively. IL-10 promotes APC inhibition, Treg action and IL-1 induction of MDCs. Growth factors like colony stimulating factor 1 (CSF1) favors TAM mobilization and proliferation and MDCs recruitment, whereas G-CSF neutrophiles mobilization and VEGFA Treg proliferation. TGF-β favors TAM2 and TAN2 polarization, CD4+ T-cell transformation in Tregs and NK suppression.
The main question is regarding the optimal RT schedule to elicit an immune response, to transform a tumor from “cold to hot”, and stimulate the TME, or to prevent a polarization of TME cells into immunosuppressive cells. Regarding the best dose per fraction preclinical studies suggested the classical 2Gy/fraction. Higher doses like 5-8 Gy/fraction proved to be even better, but unlikely single fraction, high dose of 12-18Gy. Concerning the number of fractions the clinical abscopal effect was observed following 3-5 fractions whereas protracted RT may induce more lymphopenia. Also, questions arose about the total duration of RT and the timing with the immunotherapy, concurrently or sequentially?
Other questions are linked to the optimal RT target, should be single or multiple targets, the primary tumor or metastases, one or all metastases should be irradiated, taking in account the sub clonal neoantigens, and the abscopal effect. Should we irradiate the whole lesion and how many lymph nodes, considering the sensitivity of lymph nodes or circulating lymphocytes? With the advent of the association of radio-immunotherapy the philosophy of irradiation towards deescalation of the doses and definition of target volumes would be changed. More likely the clinical target volume would be reduced as much as possible as large volumes would cover more lymphoid tissue. Besides these questions of modulation of dose, time, volume, the sequencing of therapies, type of chemotherapy and immunotherapy are parameters that require further investigations in clinical studies [47].
5. Clinical Studies
After the good results of Pembrolizumab versus chemotherapy in the Keynote 024 and 042 studies for patients with PD-L1 expression ≥50% [12,13], subsequently the combination of pembrolizumab with different chemotherapy regimens, in the Keynote 189 and 407 trials demonstrated a further significant improvement of patients survival, compared to chemotherapy alone, regardless of PD-L1 status, thus becoming standard of care in the guidelines for advanced non-squamous and squamous lung carcinoma with good performance status and without oncogenic driver mutations. The Keynote 189 trial for no squamous NSCLC [48], compared pembrolizumab versus placebo up to 35 cycles associated with pemetrexed and a platinum compound for four cycles, followed by maintenance pemetrexed, obtaining 5y-OS of 19.4% versus 11.3%. The Keynote 407 trial for squamous NSCLC [49] compared pembrolizumab versus placebo, for up to 35 cycles with carboplatin and paclitaxel, or nab-paclitaxel, for four cycles. The median overall survival was 15.9 months in the pembrolizumab combination arm and 11.3 months in the placebo combination arm, hazard ratio for death, 0.64, p<0.001.
Further combinations of tri-modality strategies of chemoradiotherapy with immunotherapy in clinical trials followed two approaches: immunotherapy to boost chemoradiotherapy in locally advanced lung cancer, or radiotherapy to boost immunotherapy or immuno-chemotherapy in advanced lung cancer.
The first approach occurred in the Pacific Trial [50] for patients with stage III locally advanced NSCLC treated with concurrent chemoradiotherapy followed by immunotherapy with durvalumab for 12 months for the non-progressive patients, with unexpected good long term results at five years of 33.1% vs. 19% PFS, 16.9 vs. 5.6 months in median value and 42.9 vs. 33.4% OS, 47.5 vs. 29.1months respectively. For patients with PD-L1 expression ≥ 1%, results proved to be even better at 5 years OS with 50% vs. 36,9%, thus becoming standard of care. Due to these unprecedented results, a lot of trials followed, for less favorable categories of patients including patients with sequential chemoradiotherapy, the Pacific-R [51] or Pacific 6 [52], or even for those with only radiotherapy, to be followed by consolidation durvalumab, in the DUART trial [53], with promising results.
However, up to one third of patients are not eligible for consolidation IT, due to disease progression, during or after concurrent chemoradiotherapy, radiation pneumonitis, or other adverse events.
A lot of randomized phases II and III trials with chemoradiotherapy and IT followed. A further step was to bring IT in concurrent setting with chemoradiotherapy. The Keynote 997 phase II trial presented at ASCO meeting 2021 evidenced that pembrolizumab and concurrent chemoradiotherapy and 14 cycles of consolidation Pembrolizumab, present promising antitumor activity, with an ORR of 70%, regardless PDL1 and histology, and manageable safety profile in stage III NSCLC [54]. But the final reports of the Pacific 2 trial presented at ELCC 2024 in Prague concluded that starting IT with durvalumab concurrently with chemoradiotherapy, followed by consolidation durvalumab in patients with unresectable stage III NSCLC does not improve outcomes compared to chemoradiotherapy alone. At a median follow-up of 30.5 months, no significant difference was observed either in PFS (13.8 months compared to 9.4 months respectively, HR 0.85; 95%CI: 0.65-1.12; p=0.247), nor in OS (36.4 vs. 29.5 months HR 1.03; 95%CI:0.78-1.39; p=0.823). Regarding the safety profile in the first months of concurrent treatment, a higher number of adverse events leading to death (13.7% versus 10.2%), or discontinuation (25.6% versus 12.0%) occurred in the durvalumab arm [55]. Also, in the recently reported results of the Checkmate -73L phase III trial, simultaneous nivolumab with concurrent chemoradiotherapy followed by consolidation IT with nivolumab and ipilimumab, did not improve, PFS versus the Pacific regimen [56].
As the intrathoracic recurrence in 80.6% of patients was the main pattern of disease progression in the Pacific study, hypo fractionated regimens started to be evaluated. A split course of hypo fractionated RT concurrently with weekly Docetaxel and Cisplatin and 1 year consolidation immunotherapy with PD-1/PD-L1 inhibitors was presented at ASTRO 2024, obtaining a better PFS, 25.7 months vs. 16.7 months in the control arm, p=0.044, and absolute lymphocyte count (ALC) proved to be an independent factor in correlation with PFS [57].
IT was evaluated even in neoadjuvant settings in combination therapies. Altkori et al [58] conducted a phase II trial for early stage, operable NSCLC with neoadjuvant Durvalumab, alone or combined with immunomodulatory doses of stereotactic radiation of 3x8Gy, obtaining major pathological response of 6.7% versus 53.3% and furthermore 3-year DFS of 63% compared to 67%, in the dual therapy arm.
For metastatic NSCLC, two trials of pembrolizumab, with or without RT, were negative regarding overall response rate, but became positive in the pooled analysis [59]. In the Pembro-RT trial the first dose of pembrolizumab was given sequentially, less than one week after the last dose of radiotherapy, 24Gy in 3 fractions. In the MDACC trial, two regimens of RT were administrated, 50 Gy in 4 fractions, or 45Gy in 15 fractions and pembrolizumab was given concurrently with the first dose of RT. The pooled analysis of the two trials encountered 148 patients, 76 received pembrolizumab with RT, 72 pembrolizumab alone. Of the 148 patients, 124 (84%) had non-squamous carcinoma and 111 (75%) received previous chemotherapy. Best abscopal response rate was 19.7% with pembrolizumab versus 41.7% with pembrolizumab plus RT (p=0.0039) and best abscopal control rate was 43.4% with pembrolizumab versus 65.3% with pembrolizumab plus RT (p=0.0071). The median PFS was 4.4 months with pembrolizumab alone versus 9 months with pembrolizumab and RT. The median OS was 8.7 months in the pembrolizumab arm versus 19.2 months for the pembrolizumab combined with RT arm.
6. Biomarkers
The most applied biomarkers PD-L1 expression, TMB, tumor immune cell infiltrates and any intrinsic or extrinsic factor affecting them may predict response to IT, as it resulted from the complex mechanisms of resistance described [14].
Tumor immune cell infiltrates evidently predict better response in patients receiving IT, than in cases with tumors characterized by immune desert. Even more available, the absolute lymphocyte count (ALC) has been reported to predict RT outcomes and abscopal effect [44,60,61,62]. In an analysis of 165 patients from three prospective trials evaluating combination of RT and IT found that pre-RT ALC was correlated with significantly better PFS regardless stereotactic or traditional RT. The data from three institutional phase 1/2 trials reported that for post-RT ALC higher than the median value, the abscopal response rate was 34.2% vs. 3.9% in patients with lower than the median value. In the experience of “Ion Chiricuta Oncology Institute”, using different anti-PD-1 checkpoint combination strategies, for first -line advanced NSCLC treatment, the 4-year survival was significantly higher, 32.3% vs. 8.2%, in patients with neutrophils to lymphocytes ratio (NLR) ≤3.81 than in those with NLR>3.81, p<0.01. Older age, impaired PS, corticotherapy in the first month of IT and NLR>3.81 were independent unfavorable prognostic factors in the multivariate analysis of survival [63].
High PD-L1 expression in tumor cells is generally associated with better outcomes to PD-1/PD-L1 inhibitors in NSCLC patients. PD-L1 expression ≥ 1% was correlated with improved survival in the combined long-term results of the CheckMate 017 and CheckMate 057 trials [7,8,9,10], or after chemoradiotherapy in the Pacific trial [50]. But in the Keynote 189 and 407 trials, IT combined with CT, gave better outcomes, regardless of PD-L1 status [48,49].
High TMB assumes tumor specific neoantigens that contribute to the immune recognition of tumor cells and is associated with better outcomes to ICIs, however tumors with HLA loss behave like tumors with low TMB. However, obtaining information by whole exome sequencing using tumor biopsy samples is hardly achievable in everyday clinical practice. Moreover, non-invasive strategies like blood-based assays using circulating tumor DNA (ctDNA), are of great potential but under investigation [64].
As shown above, driver mutations in NSCLC patients proved limited benefit from ICB. However high PD-L1 expression was found in 19-20% of cases with classic EGFR, EGFR exon 20 and HER2 mutant tumors and 34-55% in tumors with ALK, BRAF, ROS, RET or MET alterations [22].
Despite works that have shown an increase of PD-L1 expression via the IL-6/JAK/STAT3 [65] or p-ERK1/2/p-c-Jun signaling but not through p-AKT/p-S6 pathway [66], Biton et al found that EGFR mutated NSCLC cells, were found to have a lower PD-L1 expression, and weaker immunogenic TME [67].
KRAS alterations are the most frequent oncogenic driver mutations in NSCLC, but characterized by a phenotypic heterogeneity, and a higher TMB has been observed [67,68].
BRAF mutations were associated with greater clinical benefit from ICB, that may be attributed to higher TMB and PD-L1 expression in tumor cells [22].
RET and HER2 mutations attenuated PD-L1 expression, while ALK, ROS, and MET enhanced PD-L1 expression but down-regulates TMB and TILs, leading to resistance to ICIs, [14,22]. However, EGFR, HER2 mutations and ALK, ROS, RET and MET fusions, define NSCLC subsets with minimal benefit from ICB, despite high PDL1 expression indicating that PD-L1 expression and TMB seem to have an independent impact on sensitivity to IT and influenced by the complex mechanisms of resistance involved [22].
Chen et al found that the APOBEC (apolipoprotein B mRNA editing enzyme catalytic polypeptide-like) signature mutational activity, is correlated with the high TMB and PD-L1, LAG3 immune checkpoint associated gene markers and CD8+, CD4+ T immune cell infiltration markers. Furthermore, the APOBEC gene family is correlated with the IL2-STAT5 gene involvement in the INF gamma signaling pathway. Individual gene mutations IFNGR1 or VTCN1 were also found associated with response whereas PTEN with resistance to IT [69].
Also, Xu et al. revealed that patients with NFE2L2/KEAP1 gene mutation are correlated with higher TMB and PD-L1 expression, improving clinical outcome to IT [70].
Recent studies evidenced that mutations in KRAS, SKT11, KEAP1, JAK1/2, B2M, APC, MTOR and TP53 and co-mutations of these genes are the main determinants of ICI response in non-small-cell lung cancer (NSCLC) patients, influencing the metabolic changes in cancer cells, cytotoxic T cells and the efficacy of ICIs [26,27].
TP53 mutation with STK11/EGFR WT identified a tumor immune profile by enriched expression of PD-L1 and CD8+ T cells in the TME, by the up-regulation of chemokines CXCL9, 10, 11, and a stronger expression of genes involved in antigen processing and MHC-1 presentation indicating a higher immunogenicity.
On the contrary, combinations of TP53 and STK11 alterations were associated with lower PD-L1 expression and immune cell infiltration, indicating that the effect of STK11 mutation dominates over the TP53 mutation. [67]. Dong et al using gene set enrichment analysis, to determine potentially relevant gene expression, to predict response to ICB found that co-mutant TP53 and KRAS are responsible for enhanced PD-L1 expression, TMB and CD8+Tcell infiltration [68].
Resistance mutations were identified in 27.8% of patients who received IT and included acquired loss-of-function mutations in STK11, B2M, APC, MTOR, KEAP1, and JAK1/2. These acquired alterations were not observed in the control groups. Immunophenotyping of matched pre- and post-ICI samples demonstrated significant decreases in intratumoral lymphocytes, CD3e+ and CD8a+ T cells, and PD-L1/PD-1 engagement, as well as increased distance between tumor cells and CD8+PD-1+ T cells. There was a significant decrease in HLA class I expression in the immunotherapy cohort at the time of acquired resistance compared with the control groups receiving chemotherapy (p =0 .005) and targeted therapy (p =0 .01) cohorts [27]. Furthermore Ricciuti et al [71] analyzed clinical outcomes to PD-1/PD-L1 inhibition according to KRAS, STK11, and KEAP1 mutation status in two independent cohorts, at the Dana-Farber Cancer Institute/Massachusetts General Hospital cohort and the Memorial Sloan Kettering Cancer Center/MD Anderson Cancer Center cohort, and found that STK11 and KEAP1 mutations confer worse outcomes to IT among patients with KRAS mutant but not among KRAS wild type lung adenocarcinoma. Tumors harboring co-occurring genomic alterations in STK11 or KEAP1 genes to KRAS mutations define a distinct immune profile in terms of gene expression and immune cell infiltration.
The STK11 gene regulates diverse cellular metabolic functions. STK11 loss occurs in approximately 15% of lung adenocarcinomas and is associated with a lack of PD-L1 expression, reduced tumor-infiltrating cytotoxic CD8+ T lymphocytes, and resistance to ICI in patients with KRAS mutant NSCLC. KEAP1 is a negative regulator of Nrf2, which is responsible for oxidative damage response. KEAP1 loss occurs in approximately 20% of NSCLC and is associated with an immunosuppressive microenvironment characterized by low infiltration of CD8+ T cells [71].
Zielinski et al. correlated also resistance mechanisms with genetic mutation like STK11, KEAP1, JAK1/2. Moreover, STK11 mutations, besides M2 macrophage polarization and T cell dysfunction produce epigenetic changes responsible for metabolic alterations like methylation, histone acetylation, and increased lactate production. KEAP1 is responsible for increased consumption of glucose and glutamine by tumor cells, producing deprivation of nutrients of the T-cells. JAK1/2 mutation disrupts interferon-gamma signaling, responsible for DC activation [72], suggesting therapeutic strategies to correct this metabolic alteration.
7. Future Directions
7.1. New Actionable Checkpoints
As only a percentage of cases respond to CTLA4, PD-1-PD-L1 axis blocking, multiple agents are in development to address to new actionable checkpoints, like TIGIT, LAG3, TIM3, NKG2A, CD-73 [44,72,73,74,75].
The T cell Immune-receptor with immune-globulin and ITIM domain (TIGIT) is a co-inhibitory immune-modulatory checkpoint receptor, present on T cells CD8+, CD4+, T regs and NK cells, when binds to ligands CD112 and CD115, on tumor cells and APCs.
The co-stimulatory receptor CD226 competes with TIGIT for binding ligands CD112 and CD115, restoring immune antitumor response. In the Cityscape trial, the combination of tiragolumab (an anti TIGIT Ab) + atezolizumab vs. atezolizumab in PD-L1 positive, recurrent, or metastatic NSCLC, in first line treatment, obtained a response rate of 31% vs. 16%, mPFS of 5.4 months vs. 3.6 months, HR 0.57, p=0.015 [73]. Serious treatment related adverse events were 21% vs. 18%.
Lymphocyte activation gene-3 (LAG-3) is an inhibitory molecule on CD8+, CD4+ T-cells and other cells. Increased expression and co-expression of PD-1 results in a greater T cell dysfunction and is associated with resistance to PD-1 blockade. The Relativity-104 phase II trial, presented at ESMO 2024, evaluated relatlimab a human LAG-3 blocking Ab with nivolumab and platinum doublet chemotherapy vs. nivolumab and platinum doublet chemotherapy obtaining benefit in median duration of response and PFS, HR 0.88, especially in prespecified groups like PD-L1>1% (mPFS 9.8 vs. 6.1 months, HR 0.63) with manageable safety profile [74]. Trials with relatlimab and even bispecific LAG-3 and PD-1 antibodies are ongoing.
Other receptors expressed on NK cells, DCs, Monocytes, Macrophages, responsible with immune tolerance are T cell immunoglobulin and mucin domain containing protein 3 (TIM-3), Natural killer group protein 2A (NKG-2A) which triggers immune suppression and CD-73 overexpressed on TAMs, T regs, exhausted T cells, responsible of TME suppression [75].
Also, there are additional inhibitory checkpoints on T cells APCs, tumor cells, under investigation.
7.2. Novel Types of Immunotherapies
Moving beyond checkpoint inhibitors, novel types of immunotherapy emerge like STING agonists, which stimulates downstream production of the Type I interferon, responsible of activation of DCs, amongst other immunostimulatory events, CD40 agonists, which are enhancing DC function, stimulates T cell trafficking and activates M1 polarized macrophages and Toll-like Receptor agonists that activate T cells and convert MDSC into immunostimulatory antigen presenting cells [44]. These agonists might be therapeutic strategies in JAK1/2 mutations which disrupt interferon-gamma signaling. Whenever in STK11 and KEAP1 mutations, corrections of the metabolic changes, like DNA methyltransferase inhibitors or histone deacetylase inhibitors respectively glutaminase inhibitors can be associated with IT.
8. Conclusions and Perspectives
Many combination therapies like chemo-radio-immunotherapy studies are ongoing and results are awaited. Taking in account the clinical considerations affecting tumor immunogenicity, further investigations are required to study RT dose/fractionations and the effects on TME, RT volume and sites, as primary tumor, lymph nodes or metastases, sequencing and timing the multimodality treatments.
The genomic and immunophenotypic heterogeneity of resistance to IT in NSCLC, will need to be considered when developing novel therapeutic strategies aimed to overcome resistance.
Concerning the multiple agents addressing coinhibitory receptors, multiple additional inhibitory targets on T-cells, APCs, and tumor cells, co-stimulatory receptors need to be confirmed by further studies.
A huge potential of combination therapies is arising, but to which target, to what patient, at what time and sequence to be administered, considering the complex mechanisms of resistance, needs dynamic biomarkers to guide the decisions, towards more personalized treatments.
Conflicts of Interest
R.-A.R.-P. reports no conflict of interest, P.R. reports personal fees from Roche, Astra Zeneca, BMS, Magna Pharm, Accord, outside the submitted work, T.-E.C. reports personal fees from Astellas Pharma, Janssen, MSD, Merck Serono, Amgen, Roche, Pfizer, Sanofi Genzyme, Servier, Ipsen, Astra Zeneca, Lilly, Novartis, Boehringer Ingelheim, BMS, Magna Pharm, Accord, Angelini, and Takeda outside the submitted work.
References
- Bray, F; Laversanne, M; Sung, H; Ferlay, J; Siegel, RL; Soerjomataram, I; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin.;PubMed 2024, 74(3), 229–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z; Liu, X; Chen, D; Yu, J. Radiotherapy combined with immunotherapy: the dawn of cancer treatment. Signal Transduct Target Ther;PubMed 2022, 7(1), 258. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fridman, WH. From Cancer Immune Surveillance to Cancer Immunoediting: Birth of Modern Immuno-Oncology. J Immunol;PubMed 2018, 201(3), 825–826. [Google Scholar] [CrossRef] [PubMed]
- Dunn, GP; Old, LJ; Schreiber, RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity;PubMed 2004, 21(2), 137–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, DS; Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity;PubMed 2013, 39(1), 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chajon, E; Castelli, J; Marsiglia, H; De Crevoisier, R. The synergistic effect of radiotherapy and immunotherapy: A promising but not simple partnership. Crit Rev Oncol Hematol;PubMed 2017, 111, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Käsmann, L; Eze, C; Taugner, J; Roengvoraphoj, O; Dantes, M; Schmidt-Hegemann, NS; Schiopu, S; Belka, C; Manapov, F. Chemoradioimmunotherapy of inoperable stage III non-small cell lung cancer: immunological rationale and current clinical trials establishing a novel multimodal strategy. Radiat Oncol;PubMed 2020, 15(1), 167. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brahmer, J; Reckamp, KL; Baas, P; Crinò, L; Eberhardt, WE; Poddubskaya, E; Antonia, S; Pluzanski, A; Vokes, EE; Holgado, E; Waterhouse, D; Ready, N; Gainor, J; Arén Frontera, O; Havel, L; Steins, M; Garassino, MC; Aerts, JG; Domine, M; Paz-Ares, L; Reck, M; Baudelet, C; Harbison, CT; Lestini, B; Spigel, DR. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med;PubMed Epub. 2015, 373(2), 123–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Borghaei, H; Paz-Ares, L; Horn, L; Spigel, DR; Steins, M; Ready, NE; Chow, LQ; Vokes, EE; Felip, E; Holgado, E; Barlesi, F; Kohlhäufl, M; Arrieta, O; Burgio, MA; Fayette, J; Lena, H; Poddubskaya, E; Gerber, DE; Gettinger, SN; Rudin, CM; Rizvi, N; Crinò, L; Blumenschein, GR, Jr.; Antonia, SJ; Dorange, C; Harbison, CT; Graf Finckenstein, F; Brahmer, JR. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med;PubMed 2015, 373(17), 1627–39. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Borghaei, H.; Gettinger, S.; Vokes, E. E.; Chow, L. Q. M.; Burgio, M. A.; de Castro Carpeno, J.; Pluzanski, A.; Arrietac, O.; Frontera, O. A.; Chiari, R.; Butts, C.; Wójcik-Tomaszewska, J.; Coudert, B.; Garassino, M. C.; Ready, N.; Felip, E.; García, M. A.; Waterhouse, D.; Domine, M.; Brahmerc, J. Five-year outcomes from the randomized, phase iii trials checkmate 017 and 057: nivolumab versus docetaxel in previously treated non-small-cell lung cancer. Journal of Clinical Oncology 2021, 39(7), 723–733. [Google Scholar] [CrossRef]
- Rittmeyer, A; Barlesi, F; Waterkamp, D; Park, K; Ciardiello, F; von Pawel, J; Gadgeel, SM; Hida, T; Kowalski, DM; Dols, MC; Cortinovis, DL; Leach, J; Polikoff, J; Barrios, C; Kabbinavar, F; Frontera, OA; De Marinis, F; Turna, H; Lee, JS; Ballinger, M; Kowanetz, M; He, P; Chen, DS; Sandler, A; Gandara, DR; OAK Study Group. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet Erratum in: Lancet. 2017 Apr 8;389(10077):e5. doi: 10.1016/S0140-6736(17)30904-2. PMID: 27979383; PMCID: PMC6886121. [PubMed]. 2017, 389(10066), 255–265. [Google Scholar] [CrossRef] [PubMed]
- Martin Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N Engl J Med 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K. K.; Bondarenko, I.; Kubota, K.; Lubiniecki, G. M.; Zhang, J.; Kush, D.; Lopes, G. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomized, open-label, controlled, phase 3 trial. The Lancet 2019, 393(Issue 10183), Pages 1819–1830. [Google Scholar] [CrossRef]
- Zhou, S; Yang, H. Immunotherapy resistance in non-small-cell lung cancer: From mechanism to clinical strategies. Front Immunol;PubMed 2023, 14, 1129465. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carbone, DP; Reck, M; Paz-Ares, L; Creelan, B; Horn, L; Steins, M; Felip, E; van den Heuvel, MM; Ciuleanu, TE; Badin, F; Ready, N; Hiltermann, TJN; Nair, S; Juergens, R; Peters, S; Minenza, E; Wrangle, JM; Rodriguez-Abreu, D; Borghaei, H; Blumenschein, GR, Jr.; Villaruz, LC; Havel, L; Krejci, J; Corral Jaime, J; Chang, H; Geese, WJ; Bhagavatheeswaran, P; Chen, AC; Socinski, MA. CheckMate 026 Investigators. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N Engl J Med.;PubMed 2017, 376(25), 2415–2426. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hellmann, MD; Paz-Ares, L; Bernabe Caro, R; Zurawski, B; Kim, SW; Carcereny Costa, E; Park, K; Alexandru, A; Lupinacci, L; de la Mora Jimenez, E; Sakai, H; Albert, I; Vergnenegre, A; Peters, S; Syrigos, K; Barlesi, F; Reck, M; Borghaei, H; Brahmer, JR; O’Byrne, KJ; Geese, WJ; Bhagavatheeswaran, P; Rabindran, SK; Kasinathan, RS; Nathan, FE; Ramalingam, SS. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. Epub N Engl J Med;PubMed 2019, 381(21), 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- West, H; McCleod, M; Hussein, M; Morabito, A; Rittmeyer, A; Conter, HJ; Kopp, HG; Daniel, D; McCune, S; Mekhail, T; Zer, A; Reinmuth, N; Sadiq, A; Sandler, A; Lin, W; Ochi Lohmann, T; Archer, V; Wang, L; Kowanetz, M; Cappuzzo, F. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol;Epub;PubMed 2019, 20(7), 924–937. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L; Rodríguez-Abreu, D; Gadgeel, S; Esteban, E; Felip, E; De Angelis, F; Domine, M; Clingan, P; Hochmair, MJ; Powell, SF; Cheng, SY; Bischoff, HG; Peled, N; Grossi, F; Jennens, RR; Reck, M; Hui, R; Garon, EB; Boyer, M; Rubio-Viqueira, B; Novello, S; Kurata, T; Gray, JE; Vida, J; Wei, Z; Yang, J; Raftopoulos, H; Pietanza, MC; Garassino, MC. KEYNOTE-189 Investigators. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med;PubMed 2018, 378(22), 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L; Luft, A; Vicente, D; Tafreshi, A; Gümüş, M; Mazières, J; Hermes, B; Çay Şenler, F; Csőszi, T; Fülöp, A; Rodríguez-Cid, J; Wilson, J; Sugawara, S; Kato, T; Lee, KH; Cheng, Y; Novello, S; Halmos, B; Li, X; Lubiniecki, GM; Piperdi, B; Kowalski, DM. KEYNOTE-407 Investigators. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med;PubMed 2018, 379(21), 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Kluger, HM; Tawbi, HA; Ascierto, ML; Bowden, M; Callahan, MK; Cha, E; Chen, HX; Drake, CG; Feltquate, DM; Ferris, RL; Gulley, JL; Gupta, S; Humphrey, RW; LaVallee, TM; Le, DT; Hubbard-Lucey, VM; Papadimitrakopoulou, VA; Postow, MA; Rubin, EH; Sharon, E; Taube, JM; Topalian, SL; Zappasodi, R; Sznol, M; Sullivan, RJ. Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J Immunother Cancer;PubMed 2020, 8(1), e000398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Said, SS; Ibrahim, WN. Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives. Pharmaceutics;PubMed 2023, 15(4), 1143. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Negrao, MV; Skoulidis, F; Montesion, M; Schulze, K; Bara, I; Shen, V; Xu, H; Hu, S; Sui, D; Elamin, YY; Le, X; Goldberg, ME; Murugesan, K; Wu, CJ; Zhang, J; Barreto, DS; Robichaux, JP; Reuben, A; Cascone, T; Gay, CM; Mitchell, KG; Hong, L; Rinsurongkawong, W; Roth, JA; Swisher, SG; Lee, J; Tsao, A; Papadimitrakopoulou, V; Gibbons, DL; Glisson, BS; Singal, G; Miller, VA; Alexander, B; Frampton, G; Albacker, LA; Shames, D; Zhang, J; Heymach, JV. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J Immunother Cancer;PubMed 2021, 9(8), e002891. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Campoli, M; Ferrone, S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene;PubMed 2008, 27(45), 5869–85. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McGranahan, N; Rosenthal, R; Hiley, CT; Rowan, AJ; Watkins, TBK; Wilson, GA; Birkbak, NJ; Veeriah, S; Van Loo, P; Herrero, J; Swanton, C; TRACERx Consortium. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell;PubMed 2017, 171(6), 1259–1271.e11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Montesion, M; Murugesan, K; Jin, DX; Sharaf, R; Sanchez, N; Guria, A; Minker, M; Li, G; Fisher, V; Sokol, ES; Pavlick, DC; Moore, JA; Braly, A; Singal, G; Fabrizio, D; Comment, LA; Rizvi, NA; Alexander, BM; Frampton, GM; Hegde, PS; Albacker, LA. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov.;PubMed 2021, 11(2), 282–292. [Google Scholar] [CrossRef] [PubMed]
- Otegui, N; Houry, M; Arozarena, I; Serrano, D; Redin, E; Exposito, F; Leon, S; Valencia, K; Montuenga, L; Calvo, A. Cancer Cell-Intrinsic Alterations Associated with an Immunosuppressive Tumor Microenvironment and Resistance to Immunotherapy in Lung Cancer. Cancers (Basel);PubMed 2023, 15(12), 3076. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ricciuti, B; Lamberti, G; Puchala, SR; Mahadevan, NR; Lin, JR; Alessi, JV; Chowdhury, A; Li, YY; Wang, X; Spurr, L; Pecci, F; Di Federico, A; Venkatraman, D; Barrichello, AP; Gandhi, M; Vaz, VR; Pangilinan, AJ; Haradon, D; Lee, E; Gupta, H; Pfaff, KL; Welsh, EL; Nishino, M; Cherniack, AD; Johnson, BE; Weirather, JL; Dryg, ID; Rodig, SJ; Sholl, LM; Sorger, P; Santagata, S; Umeton, R; Awad, MM. Genomic and Immunophenotypic Landscape of Acquired Resistance to PD-(L)1 Blockade in Non-Small-Cell Lung Cancer. J Clin Oncol;PubMed 2024, 42(11), 1311–1321. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chiang, A.; Detterbeck, F. C.; Stewart, T.; Decker, R. H.; Tanoue, L. Non–small-cell Lung Cancer, Chapter 48 in DeVita, Hellman, and Rosenberg’s Cancer Principles & Practice of Oncology 11th edition; Wolters Kluwer, 2019; pp. 1135–1223. [Google Scholar]
- Ciuleanu, T. E. Tumorile toracice. Cap în Compendiu de Oncologie Medicala: principii şi practica. In Ed. Casa Cartii de Stiinta; 2021; ISBN 978-606-17-1798-9. [Google Scholar]
- Zheng, H; Zeltsman, M; Zauderer, MG; Eguchi, T; Vaghjiani, RG; Adusumilli, PS. Chemotherapy-induced immunomodulation in non-small-cell lung cancer: a rationale for combination chemoimmunotherapy. Immunotherapy;PubMed 2017, 9(11), 913–927. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ferraro, C; Quemeneur, L; Fournel, S; Prigent, AF; Revillard, JP; Bonnefoy-Berard, N. The topoisomerase inhibitors camptothecin and etoposide induce a CD95-independent apoptosis of activated peripheral lymphocytes. Cell Death Differ;PubMed 2000, 7(2), 197–206. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, SR; Caballero, JA; Hodge, JW. Defining the molecular signature of chemotherapy-mediated lung tumor phenotype modulation and increased susceptibility to T-cell killing. Cancer Biother Radiopharm;Epub;PubMed 2012, 27(1), 23–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhu, L; Chen, L. Progress in research on paclitaxel and tumor immunotherapy. Cell Mol Biol Lett;PubMed 2019, 24, 40. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schaer, DA; Geeganage, S; Amaladas, N; Lu, ZH; Rasmussen, ER; Sonyi, A; Chin, D; Capen, A; Li, Y; Meyer, CM; Jones, BD; Huang, X; Luo, S; Carpenito, C; Roth, KD; Nikolayev, A; Tan, B; Brahmachary, M; Chodavarapu, K; Dorsey, FC; Manro, JR; Doman, TN; Donoho, GP; Surguladze, D; Hall, GE; Kalos, M; Novosiadly, RD. The Folate Pathway Inhibitor Pemetrexed Pleiotropically Enhances Effects of Cancer Immunotherapy. Clin Cancer Res;PubMed 2019, 25(23), 7175–7188. [Google Scholar] [CrossRef] [PubMed]
- Frey, B; Rückert, M; Weber, J; Mayr, X; Derer, A; Lotter, M; Bert, C; Rödel, F; Fietkau, R; Gaipl, US. Hypofractionated Irradiation Has Immune Stimulatory Potential and Induces a Timely Restricted Infiltration of Immune Cells in Colon Cancer Tumors. Front Immunol;PubMed 2017, 8, 231. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vanpouille-Box, C; Alard, A; Aryankalayil, MJ; Sarfraz, Y; Diamond, JM; Schneider, RJ; Inghirami, G; Coleman, CN; Formenti, SC; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumor immunogenicity. Nat Commun;PubMed 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dovedi, SJ; Adlard, AL; Lipowska-Bhalla, G; McKenna, C; Jones, S; Cheadle, EJ; Stratford, IJ; Poon, E; Morrow, M; Stewart, R; Jones, H; Wilkinson, RW; Honeychurch, J; Illidge, TM. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res;PubMed 2014, 74(19), 5458–68. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, SJ; Cheadle, EJ; Popple, AL; Poon, E; Morrow, M; Stewart, R; Yusko, EC; Sanders, CM; Vignali, M; Emerson, RO; Robins, HS; Wilkinson, RW; Honeychurch, J; Illidge, TM. Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin Cancer Res.;PubMed 2017, 23(18), 5514–5526. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S; Ng, B; Devitt, ML; Babb, JS; Kawashima, N; Liebes, L; Formenti, SC. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys;PubMed 2004, 58(3), 862–70. [Google Scholar] [CrossRef] [PubMed]
- Dewan, MZ; Galloway, AE; Kawashima, N; Dewyngaert, JK; Babb, JS; Formenti, SC; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res;PubMed 2009, 15(17), 5379–88. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vonderheide, RH; Glennie, MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res;PubMed 2013, 19(5), 1035–43. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dovedi, SJ; Lipowska-Bhalla, G; Beers, SA; Cheadle, EJ; Mu, L; Glennie, MJ; Illidge, TM; Honeychurch, J. Antitumor Efficacy of Radiation plus Immunotherapy Depends upon Dendritic Cell Activation of Effector CD8+ T Cells. Cancer Immunol Res;Epub;PubMed 2016, 4(7), 621–630. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reits, EA; Hodge, JW; Herberts, CA; Groothuis, TA; Chakraborty, M; Wansley, EK; Camphausen, K; Luiten, RM; de Ru, AH; Neijssen, J; Griekspoor, A; Mesman, E; Verreck, FA; Spits, H; Schlom, J; van Veelen, P; Neefjes, JJ. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med;PubMed 2006, 203(5), 1259–71. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Colton, M.; Cheadle, E.J.; Honeychurch, J.; et al. Reprogramming the tumor microenvironment by radiotherapy: implications for radiotherapy and immunotherapy combinations. Radiat Oncol Published. 2020). https://doi.org/10.1186/s13014-020-01678-1. Published: 04 November 2020, Volume 15, article number 254, (2020, 15, 254 15, article number 254. [Google Scholar] [CrossRef]
- Deng, L; Liang, H; Burnette, B; Beckett, M; Darga, T; Weichselbaum, RR; Fu, YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest;PubMed 2014, 124(2), 687–95. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Derer, A; Spiljar, M; Bäumler, M; Hecht, M; Fietkau, R; Frey, B; Gaipl, US. Chemoradiation Increases PD-L1 Expression in Certain Melanoma and Glioblastoma Cells. Front Immunol;PubMed 2016, 7, 610. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pointer, KB; Pitroda, SP; Weichselbaum, RR. Radiotherapy and immunotherapy: open questions and future strategies. Trends Cancer;PubMed 2022, 8(1), 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S; Gray, JE; Rizzo, MT; Peterson, P; Kim, JS; Rodríguez-Abreu, D. Pemetrexed and Platinum Plus Pembrolizumab in Patients With Metastatic Nonsquamous NSCLC by Tumor Burden at Baseline: A Post Hoc Efficacy Analysis of KEYNOTE-189. JTO Clin Res Rep.;PubMed 2022, 3(11), 100389. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Paz-Ares, L; Luft, A; Vicente, D; Tafreshi, A; Gümüş, M; Mazières, J; Hermes, B; Çay Şenler, F; Csőszi, T; Fülöp, A; Rodríguez-Cid, J; Wilson, J; Sugawara, S; Kato, T; Lee, KH; Cheng, Y; Novello, S; Halmos, B; Li, X; Lubiniecki, GM; Piperdi, B; Kowalski, DM. KEYNOTE-407 Investigators. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med;PubMed 2018, 379(21), 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S. J.; Villegas, A.; Daniel, D. Durvalumab after Chemoradiotherapy in stage III Non-small-Cell Lung Cancer. N Engl J Med 2017, 2017;377, 1919–1929. [Google Scholar] [CrossRef]
- Girard, N.; Bar, J.; Garrido, B.; et al. Treatment Characteristics and Real-World Progression-Free Survival in Patients With Unresectable Stage III NSCLC Who Received Durvalumab After Chemoradiotherapy: Findings From the PACIFIC-R Study. J Thorac Onc 2023, (18) 2, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Garassino, MC; Mazieres, J; Reck, M; Chouaid, C; Bischoff, H; Reinmuth, N; Cove-Smith, L; Mansy, T; Cortinovis, D; Migliorino, MR; Delmonte, A; Sánchez, JG; Chara Velarde, LE; Bernabe, R; Paz-Ares, L; Perez, ID; Trunova, N; Foroutanpour, K; Faivre-Finn, C. Durvalumab After Sequential Chemoradiotherapy in Stage III, Unresectable NSCLC: The Phase 2 PACIFIC-6 Trial. J Thorac Oncol;PubMed Epub. 2022, 17(12), 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Filippi, AR; Dziadziuszko, R; García Campelo, MR; Paoli, JB; Sawyer, W; Díaz Pérez, IE. DUART: durvalumab after radiotherapy in patients with unresectable, stage III NSCLC who are ineligible for chemotherapy. Future Oncol.;PubMed 2021, 17(34), 4657–4663. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, SK; Lee, KH; Frost, N; Breder, V; Kowalski, DM; Pollock, T; Levchenko, E; Reguart, N; Martinez-Marti, A; Houghton, B; Paoli, JB; Safina, S; Park, K; Komiya, T; Sanford, A; Boolell, V; Liu, H; Samkari, A; Keller, SM; Reck, M. Pembrolizumab Plus Concurrent Chemoradiation Therapy in Patients With Unresectable, Locally Advanced, Stage III Non-Small Cell Lung Cancer: The Phase 2 KEYNOTE-799 Nonrandomized Trial. JAMA Oncol;PubMed Epub ahead of print. 2021, 7(9), 1–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bradley, JD; Sugawara, S; Lee, KH; Ostoros, G; Demirkazik, A; Zemanova, M; Sriuranpong, V; Gelatti, ACZ; de Menezes, JJ; Zurawski, B; Newton, M; Chander, P; Jia, N; Bielecka, ZF; Özgüroğlu, M. PACIFIC-2 Investigators. Simultaneous Durvalumab and Platinum-Based Chemoradiotherapy in Unresectable Stage III Non-Small Cell Lung Cancer: The Phase III PACIFIC-2 Study. J Clin Oncol;PubMed 2025, 43(33), 3610–3621. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Ruysscher, D; Ramalingam, S; Urbanic, J; Gerber, DE; Tan, DSW; Cai, J; Li, A; Peters, S. CheckMate 73L: A Phase 3 Study Comparing Nivolumab Plus Concurrent Chemoradiotherapy Followed by Nivolumab With or Without Ipilimumab Versus Concurrent Chemoradiotherapy Followed by Durvalumab for Previously Untreated, Locally Advanced Stage III Non-Small-Cell Lung Cancer. Clin Lung Cancer;PubMed 2022, 23(3), e264–e268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R; Liu, F; Zhang, H; Wang, D; Zhang, P; Zheng, S; Liu, Y; Chen, L; Guo, J; Zou, Y; Rong, YM; Liu, H; Qiu, B. Fraction Dose Escalation of Hypofractionated Radiotherapy with Concurrent Chemotherapy and Subsequent Consolidation Immunotherapy in Locally Advanced Non-Small Cell Lung Cancer: A Phase I Study. Clin Cancer Res.;PubMed 2024, 30(13), 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Altorki, NK; Walsh, ZH; Melms, JC; Port, JL; Lee, BE; Nasar, A; Spinelli, C; Caprio, L; Rogava, M; Ho, P; Christos, PJ; Saxena, A; Elemento, O; Bhinder, B; Ager, C; Amin, AD; Sanfilippo, NJ; Mittal, V; Borczuk, AC; Formenti, SC; Izar, B; McGraw, TE. Neoadjuvant durvalumab plus radiation versus durvalumab alone in stages I-III non-small cell lung cancer: survival outcomes and molecular correlates of a randomized phase II trial. Nat Commun Erratum in: Nat Commun. 2024 Jan 3;15(1):225. doi: 10.1038/s41467-023-44575-3. PMID: 38114518; PMCID: PMC10730562. [PubMed]. 2023, 14(1), 8435. [Google Scholar] [CrossRef]
- Theelen, WSME; Chen, D; Verma, V; Hobbs, BP; Peulen, HMU; Aerts, JGJV; Bahce, I; Niemeijer, ALN; Chang, JY; de Groot, PM; Nguyen, QN; Comeaux, NI; Simon, GR; Skoulidis, F; Lin, SH; He, K; Patel, R; Heymach, J; Baas, P; Welsh, JW. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: a pooled analysis of two randomised trials. Lancet Respir Med Erratum in: Lancet Respir Med. 2021 Mar;9(3):e29. doi: 10.1016/S2213-2600(21)00012-6. PMID: 33096027. [PubMed]. 2021, 9(5), 467–475. [Google Scholar] [CrossRef]
- Angell, H; Galon, J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Curr Opin Immunol;PubMed 2013, 25(2), 261–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, D; Patel, RR; Verma, V; Ramapriyan, R; Barsoumian, HB; Cortez, MA; Welsh, JW. Interaction between lymphopenia, radiotherapy technique, dosimetry, and survival outcomes in lung cancer patients receiving combined immunotherapy and radiotherapy https://doi.org/10.1016/j.radonc.2020.05.051. Epub 2020 Jun 7. Radiother Oncol Erratum in: Radiother Oncol. 2021 May;158:332-334. doi: 10.1016/j.radonc.2021.01.017. PMID: 32525003. [PubMed]. 2020, 150, 114–120. [Google Scholar] [CrossRef]
- Chen, D; Verma, V; Patel, RR; Barsoumian, HB; Cortez, MA; Welsh, JW. Absolute Lymphocyte Count Predicts Abscopal Responses and Outcomes in Patients Receiving Combined Immunotherapy and Radiation Therapy: Analysis of 3 Phase 1/2 Trials https://doi.org/10.1016/j.ijrobp.2020.01.032. Int J Radiat Oncol Biol Phys Erratum in: Int J Radiat Oncol Biol Phys. 2021 Jun 1;110(2):623. doi: 10.1016/j.ijrobp.2021.01.012. PMID: 32036004. [PubMed]. 2020, 108(1), 196–203. [Google Scholar] [CrossRef] [PubMed]
- Preda, AC; Ciuleanu, TE; Todor, N; Vlad, C; Iancu, DI; Mocan, C; Bandi-Vasilica, M; Albu, F; Todor-Bondei, IM; Hapca, MC; Kubelac, MP; Kubelac-Varro, AD. Use of Different Anti-PD-1 Checkpoint Combination Strategies for First-Line Advanced NSCLC Treatment-The Experience of Ion Chiricuță Oncology Institute. Cancers (Basel);PubMed 2024, 16(11), 2022. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pilard, C; Ancion, M; Delvenne, P; Jerusalem, G; Hubert, P; Herfs, M. Cancer immunotherapy: it’s time to better predict patients’ response. Br J Cancer;PubMed 2021, 125(7), 927–938. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, N; Zeng, Y; Du, W; Zhu, J; Shen, D; Liu, Z; Huang, JA. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol;Epub;PubMed 2016, 49(4), 1360–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, N; Fang, W; Zhan, J; Hong, S; Tang, Y; Kang, S; Zhang, Y; He, X; Zhou, T; Qin, T; Huang, Y; Yi, X; Zhang, L. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol;PubMed 2015, 10(6), 910–23. [Google Scholar] [CrossRef] [PubMed]
- Biton, J; Mansuet-Lupo, A; Pécuchet, N; Alifano, M; Ouakrim, H; Arrondeau, J; Boudou-Rouquette, P; Goldwasser, F; Leroy, K; Goc, J; Wislez, M; Germain, C; Laurent-Puig, P; Dieu-Nosjean, MC; Cremer, I; Herbst, R; Blons, H; Damotte, D. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin Cancer Res;PubMed 2018, 24(22), 5710–5723. [Google Scholar] [CrossRef] [PubMed]
- Dong, ZY; Zhong, WZ; Zhang, XC; Su, J; Xie, Z; Liu, SY; Tu, HY; Chen, HJ; Sun, YL; Zhou, Q; Yang, JJ; Yang, XN; Lin, JX; Yan, HH; Zhai, HR; Yan, LX; Liao, RQ; Wu, SP; Wu, YL. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res;PubMed 2017, 23(12), 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Chen, H; Chong, W; Teng, C; Yao, Y; Wang, X; Li, X. The immune response-related mutational signatures and driver genes in non-small-cell lung cancer. Cancer Sci;PubMed 2019, 110(8), 2348–2356. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, X; Yang, Y; Liu, X; Cao, N; Zhang, P; Zhao, S; Chen, D; Li, L; He, Y; Dong, X; Wang, K; Lin, H; Mao, N; Liu, L. NFE2L2/KEAP1 Mutations Correlate with Higher Tumor Mutational Burden Value/PD-L1 Expression and Potentiate Improved Clinical Outcome with Immunotherapy. Oncologist;PubMed 2020, 25(6), e955–e963. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ricciuti, B; Arbour, KC; Lin, JJ; Vajdi, A; Vokes, N; Hong, L; Zhang, J; Tolstorukov, MY; Li, YY; Spurr, LF; Cherniack, AD; Recondo, G; Lamberti, G; Wang, X; Venkatraman, D; Alessi, JV; Vaz, VR; Rizvi, H; Egger, J; Plodkowski, AJ; Khosrowjerdi, S; Digumarthy, S; Park, H; Vaz, N; Nishino, M; Sholl, LM; Barbie, D; Altan, M; Heymach, JV; Skoulidis, F; Gainor, JF; Hellmann, MD; Awad, MM. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J Thorac Oncol;PubMed 2022, 17(3), 399–410. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zieliński, P; Stępień, M; Chowaniec, H; Kalyta, K; Czerniak, J; Borowczyk, M; Dwojak, E; Mroczek, M; Dworacki, G; Ślubowska, A; Markiewicz, H; Ałtyn, R; Dobosz, P. Resistance in Lung Cancer Immunotherapy and How to Overcome It: Insights from the Genetics Perspective and Combination Therapies Approach. Cells;PubMed 2025, 14(8), 587. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cho, BC; Abreu, DR; Hussein, M; Cobo, M; Patel, AJ; Secen, N; Lee, KH; Massuti, B; Hiret, S; Yang, JCH; Barlesi, F; Lee, DH; Ares, LP; Hsieh, RW; Patil, NS; Twomey, P; Yang, X; Meng, R; Johnson, ML. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol;PubMed 2022, 23(6), 781–792. [Google Scholar] [CrossRef] [PubMed]
- Girard, N.; Burotto, M.; Paz-Ares, L.G.; Reck, M.; Schenker, M.; Lingua, A.; Orlandi, F.J.; Naidoo, J.; Beardlsey, E.K.; Velcheti, V.; Martinengo, G.L.; Felip, E.; Zhang, Y.; Kasbekar, P.; Chowdhury, M.; Spires, T.; Tendolkar, A.; Cobo Dols, M. LBA53 Nivolumab (NIVO) plus relatlimab with platinum-doublet chemotherapy (PDCT) vs. NIVO + PDCT as first-line (1L) treatment (tx) for stage IV or recurrent NSCLC: Results from the randomized phase II RELATIVITY-104 study. Ann onc 2024, 35, s1243–1244. [Google Scholar] [CrossRef]
- He, X; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res;PubMed Epub. 2020, 30(8), 660–669. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
Figure 1.
The Cancer Immunity Cycle. D.S. Chen et al. [5].
Figure 1.
The Cancer Immunity Cycle. D.S. Chen et al. [5].
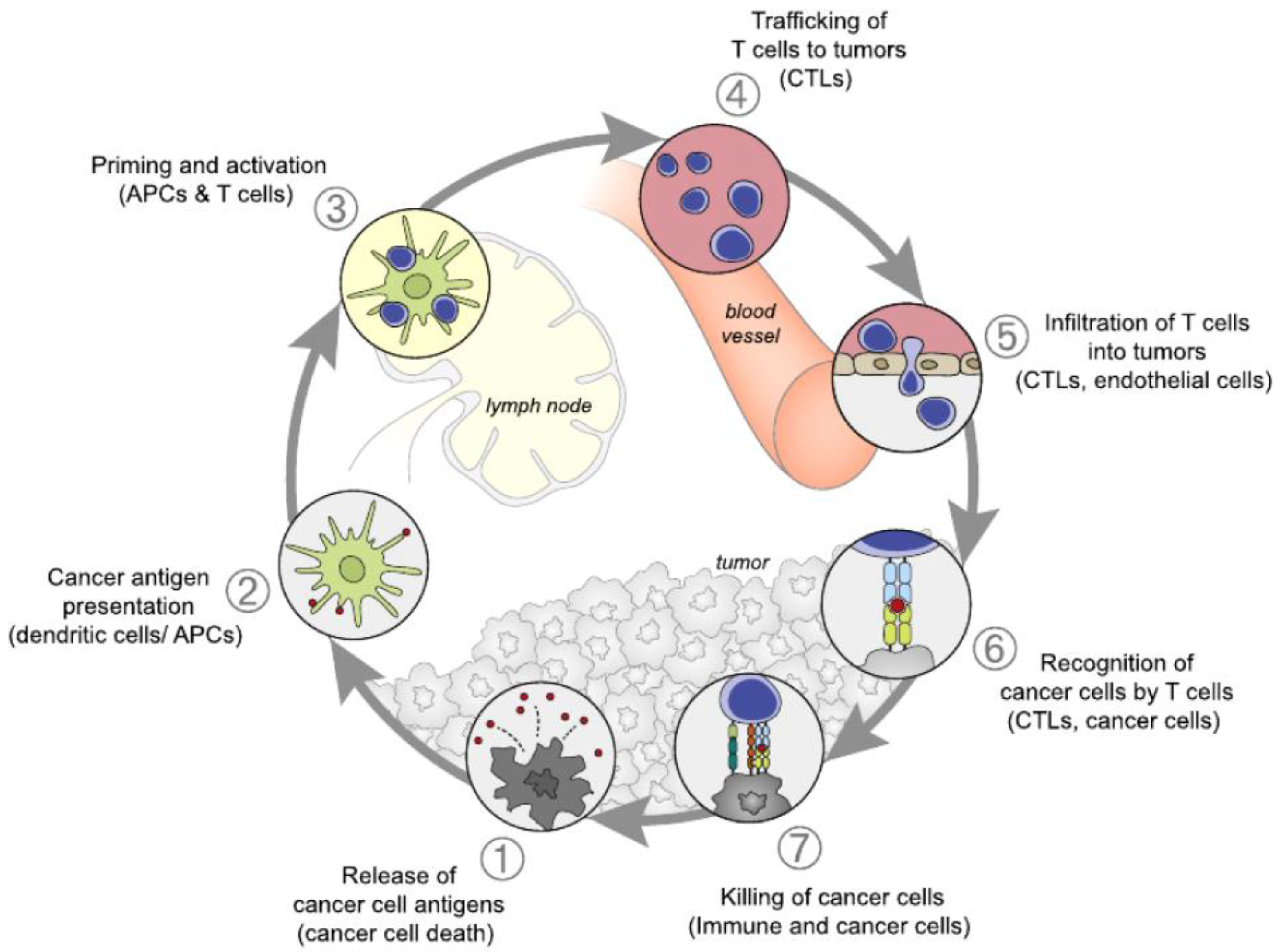
Figure 2.
Overall survival curves of nivolumab versus docetaxel in Previously Treated Non-Small-Cell Lung Cancer patients [10].
Figure 2.
Overall survival curves of nivolumab versus docetaxel in Previously Treated Non-Small-Cell Lung Cancer patients [10].
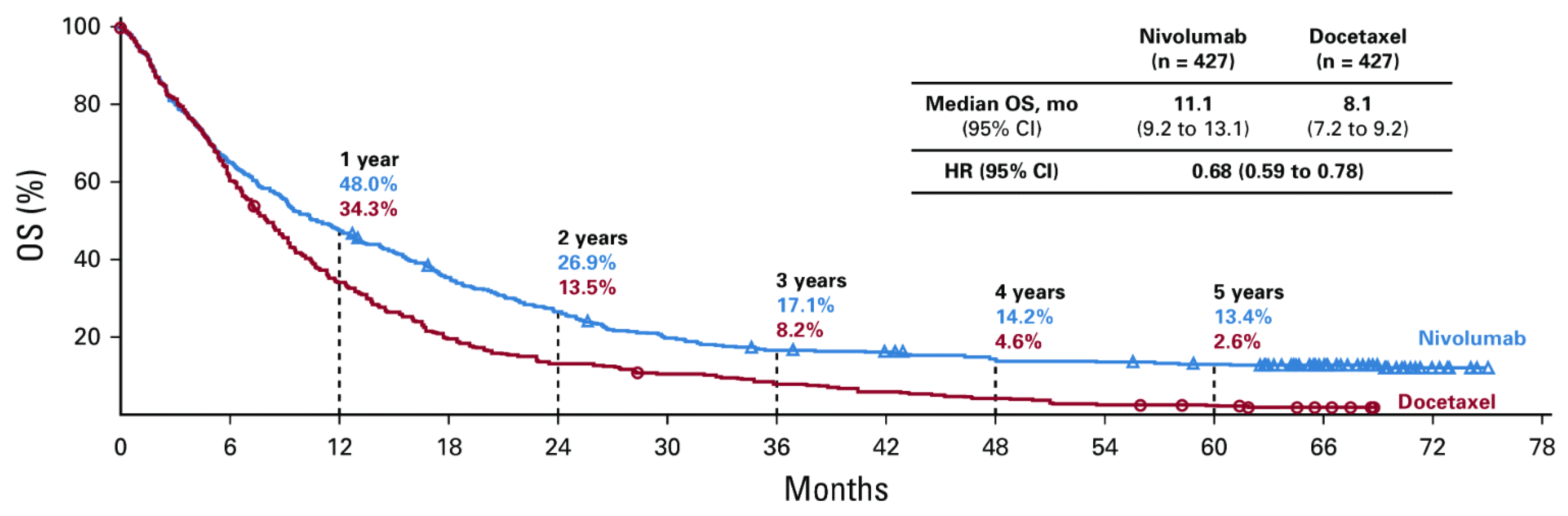
Figure 3.
PFS and patients in response with nivolumab vs. Docetaxel [10].
Figure 3.
PFS and patients in response with nivolumab vs. Docetaxel [10].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



