Submitted:
06 December 2025
Posted:
09 December 2025
You are already at the latest version
Abstract
Protein aggregation is a defining pathological hallmark of major neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and tauopathies. These disorders share a convergent failure of proteostasis—the integrated network that regulates protein synthesis, folding, trafficking, and clearance—rather than isolated, disease-specific anomalies. Neurons, as long-lived post-mitotic cells, are particularly vulnerable to proteostasis collapse, which promotes accumulation of misfolded proteins such as amyloid-β, α-synuclein, tau, and TDP-43. Aggregates exhibit structural polymorphism, prion-like propagation, and strain-specific conformations that dictate regional vulnerability and clinical heterogeneity. Mechanistically, clearance pathways—including the ubiquitin–proteasome system, autophagy–lysosomal axis, endosomal–lysosomal trafficking, and molecular chaperones—operate in a highly coordinated manner; their dysfunction accelerates aggregation and neurotoxicity. Emerging evidence implicates liquid-liquid phase separation as a precursor to amyloid formation, while post-translational modifications such as phosphorylation, ubiquitination, and truncation modulate aggregation kinetics and pathogenicity. Aging further exacerbates proteostasis failure through impaired autophagic flux, lysosomal dysfunction, and glymphatic clearance decline, amplifying aggregate burden and prion-like dissemination. Despite extensive development of aggregation inhibitors, immunotherapies, and proteostasis enhancers, clinical translation remains limited due to poor blood–brain barrier penetration, off-target effects, and incomplete understanding of intermediate toxic species. Future strategies should integrate structural biology, seed amplification assays, and systems-level approaches to target early aggregation events, reinforce clearance pathways, and modulate neuroinflammation. This review synthesizes mechanistic insights, structural data, and therapeutic advances, emphasizing the complexity of protein aggregation as a dynamic, multifactorial process in neurodegeneration.
Keywords:
protein aggregation
; proteostasis
; prion‐like propagation
; autophagy
; mixed proteinopathies
; neurodegeneration
; α‐synuclein
; tauopathies
; amyloid‐β
; TDP‐43
1. Introduction
Neurodegenerative diseases are defined by the characteristic neuropathological findings, particularly the presence of specific misfolded protein aggregates in distinct areas of the central nervous system (CNS). These aggregates vary by disease: Lewy bodies (LBs) in Parkinson’s disease (PD), amyloid-β (Aβ) plaques and neurofibrillary tangles in Alzheimer’s disease (AD), mutant huntingtin inclusions in Huntington’s disease (HD), glial cytoplasmic inclusions in multiple system atrophy (MSA), prion-derived Aβ plaques in prion diseases, and ubiquitinated protein aggregates in amyotrophic lateral sclerosis (ALS) (Figure 1). Despite differences in protein identity and anatomical distribution—nuclear and cytoplasmic inclusions in HD, cytoplasmic aggregates in PD and ALS, and extracellular plaques in AD—the convergence on protein aggregation suggests a shared mechanism of proteostasis failure rather than isolated, disease-specific anomalies [1].
Although the term proteinopathy is often used interchangeably with abnormal protein aggregation, a more precise definition considers proteinopathy as a broader impairment of proteostasis. Proteostasis refers to the dynamic maintenance of a functional proteome through coordinated regulation of protein synthesis, folding, trafficking, and clearance [2]. Disruption of this network can lead to misfolded protein accumulation, overwhelming cellular quality-control systems and triggering neurodegeneration. Importantly, proteostasis failure and protein aggregation are not exclusive to classical neurodegenerative disorders; they also occur in certain non-neurodegenerative movement disorders, underscoring the relevance of protein homeostasis across a wider spectrum of neurological conditions.
Liquid-liquid phase separation (LLPS) has emerged as a critical intermediate in the transition from soluble proteins to pathogenic aggregates in neurodegenerative diseases. LLPS enables intrinsically disordered proteins such as α-synuclein (αSyn), tau, and TDP-43 to form dynamic condensates that concentrate aggregation-prone species within membranelles compartments. These condensates exhibit liquid-like properties initially but can mature into gel-like or solid amyloid states under stress conditions, post-translational modifications (PTMs), or altered ionic environments. LLPS is modulated by protein isoforms, charge distribution, and cofactors such as RNA and lipids, which influence condensate stability and aggregation kinetics. Recent studies demonstrate that αSyn condensates recruit synaptic vesicles and SNARE proteins, linking phase separation to synaptic dysfunction [3], while tau LLPS accelerates nucleation of paired helical filaments in the presence of polyanions [4]. Importantly, LLPS provides a mechanistic framework for understanding how cellular stress and crowding bias aggregation pathways, suggesting that targeting condensate formation or maturation could represent a novel therapeutic strategy [5,6].
Building on these emerging concepts, this review integrates mechanistic, structural, and translational perspectives to dissect the complexity of protein aggregation in neurodegenerative diseases. We first examine the architecture and dynamics of proteostasis networks—including the ubiquitin–proteasome system (UPS), autophagy–lysosomal pathways, endosomal trafficking, and molecular chaperones—that collectively safeguard neuronal protein homeostasis. Next, we explore how their failure converges with aggregation-prone proteins such as Aβ, αSyn, tau, and TDP-43, emphasizing structural polymorphism, prion-like propagation, and modulatory factors such as PTMs, lipid interactions, and aging. Finally, we synthesize current and emerging therapeutic strategies, ranging from aggregation inhibitors and immunotherapies to network-level interventions targeting epichaperomes and proteostasis enhancers. By framing aggregation as a dynamic, multifactorial process rather than a static endpoint, this review underscores the need for integrated approaches that combine structural biology, systems-level analysis, and precision therapeutics to effectively mitigate neurodegeneration.
2. Protein Clearance Mechanisms – Intraneuronal
Neurons are particularly vulnerable to proteostasis disruption because they are long-lived, post-mitotic cells that cannot dilute misfolded proteins through cell division. This intrinsic limitation means that even minor perturbations in protein homeostasis can have profound consequences for neuronal function and survival. To counteract this risk, neurons rely on an intricate proteostasis network comprising molecular chaperones, the UPS, and autophagy-lysosomal pathways, which collectively regulate protein folding, trafficking, and clearance (Figure 2) [7].
2.1. UPS
UPS is the primary pathway for selective intracellular protein degradation, ensuring proteostasis by removing misfolded, damaged, or short-lived proteins. Ubiquitin acts as a molecular tag, covalently attached to substrate proteins through an enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ubiquitin ligases. Substrates can be modified with a single ubiquitin or polyubiquitin chains, which dictate their fate—typically proteasomal degradation for K48-linked chains or signaling for other linkages [8,9]. The 26S proteasome recognizes ubiquitinated substrates and catalyzes their degradation into peptides, maintaining neuronal homeostasis. Given neurons’ post-mitotic nature and high metabolic demand, UPS dysfunction leads to accumulation of toxic aggregates, a hallmark of neurodegenerative diseases such as PD, AD, and HD [10].
UPS activity is tightly regulated by ubiquitin ligases and deubiquitinating enzymes (DUBs), which control the addition and removal of ubiquitin chains. Genetic mutations affecting these enzymes have been implicated in movement disorders. For example, pathogenic variants in ataxin-3 (ATXN3), a DUB, cause spinocerebellar ataxia type 3 (SCA3) [11], while mutations in E3 ligases such as STUB1/CHIP lead to autosomal recessive cerebellar ataxia (SCAR16) and spinocerebellar ataxia type 48 (SCA48) [12,13]. Similarly, RNF216 mutations underlie Gordon Holmes syndrome [14], and AMFR variants cause hereditary spastic paraplegia (SPG89) [15,16].
Beyond genetic causes, UPS impairment is exacerbated by aging and cellular stress, reducing proteasomal efficiency and promoting aggregation of proteins. Recent studies emphasize that UPS dysfunction often coexists with autophagy deficits, forming a dual failure of proteostasis in neurodegeneration [17]. Therapeutic strategies targeting UPS components—such as small-molecule activators of proteasome function, modulators of E3 ligases, and DUB inhibitors—are under investigation to restore protein clearance and mitigate disease progression [12].
2.2. Endosomal-Lysosomal System
The endosomal–lysosomal system is a critical arm of the cellular proteostasis network, responsible for degrading membrane proteins, extracellular cargo, and aggregate-prone species that escape proteasomal clearance. Proteins destined for disposal are sorted into endosomes and subsequently trafficked to lysosomes for degradation. This process relies on the coordinated action of Rab GTPases, which regulate vesicle budding, trafficking, and fusion events, and the endosomal sorting complexes required for transport (ESCRT) machinery, which mediates cargo sequestration into intraluminal vesicles (ILVs) within multivesicular bodies (MVBs) [18,19]. These ILVs are typically degraded upon lysosomal fusion; however, they can also be secreted as exosomes, contributing to intercellular communication and, in pathological contexts, the spread of misfolded proteins such as tau and αSyn [20].
Genetic disruptions in endosomal–lysosomal components underscore their importance in movement disorders. Mutations in ESCRT-I subunit UBAP1 cause hereditary spastic paraplegia type 80 (SPG80), characterized by axonal degeneration linked to defective endosomal cargo sorting [21]. Similarly, variants in Rab GTPases, such as RAB32, have been associated with atypical parkinsonism, implicating impaired vesicular trafficking and mitochondrial-endolysosomal crosstalk in disease pathogenesis [22].
Beyond genetic causes, emerging evidence suggests that endosomal–lysosomal dysfunction synergizes with autophagy impairment and proteasomal overload in neurodegenerative conditions. Defective ESCRT-mediated membrane repair and lysosomal degradation promote accumulation of toxic aggregates and exacerbate prion-like propagation of pathogenic proteins via exosomes [23]. Therapeutic strategies aimed at enhancing lysosomal biogenesis, modulating Rab GTPase activity, or targeting ESCRT components are being explored to restore vesicular homeostasis and mitigate disease progression [24].
2.3. Autophagy-Lysosomal System
The autophagy–lysosomal pathway (ALP) is a major degradative system that maintains neuronal homeostasis by eliminating misfolded proteins, aggregates, and damaged organelles. During macroautophagy, cytoplasmic cargo is sequestered into double-membraned autophagosomes, which then fuse with lysosomes for degradation by acidic hydrolases [25]. This process is highly dependent on vesicular trafficking regulators such as Rab GTPases and the HOPS complex, which mediate autophagosome–lysosome fusion. Genetic evidence underscores the importance of these components: mutations in VPS16 and VPS41 [26], encoding HOPS subunits, cause early-onset dystonia and spastic ataxia (SCAR29, DYT30), highlighting the link between autophagy failure and movement disorders [27].
Beyond canonical PI3P-dependent autophagy, recent studies have identified a noncanonical pathway mediated by PI5P, which can substitute for PI3P in recruiting autophagy machinery [28]. PI5P synthesis by PIKfyve and its regulation by PI5P4 kinases modulate autophagosome biogenesis independently of VPS34. Inhibition of PI5P4Ks enhances clearance of aggregate-prone proteins, including mutant huntingtin, positioning these kinases as promising therapeutic targets for neurodegenerative diseases [29].
Autophagy dysfunction is increasingly recognized as a convergent mechanism in AD and PD, where impaired autophagosome formation or lysosomal fusion promotes accumulation of tau and αSyn aggregates [30]. Factors such as excess αSyn, nitric oxide signaling, and mutations in trafficking proteins like VPS35 exacerbate these defects [31], while GWAS implicates loci such as CALM [32], which regulate SNARE-mediated vesicle fusion, in tau pathology. Therapeutic strategies aimed at enhancing lysosomal function, modulating PI5P signaling, or restoring HOPS-mediated fusion hold promise for mitigating neurodegeneration by reinforcing the autophagy–lysosomal axis [33].
2.4. Chaperone System
Molecular chaperones, primarily heat shock proteins (HSPs), are essential components of the proteostasis network, ensuring proper protein folding and preventing aggregation under physiological and stress conditions. HSP families such as HSP70 and HSP90 bind nascent or misfolded polypeptides, facilitating correct folding or refolding, and directing terminally misfolded proteins toward degradation pathways like the UPS or autophagy [34,35]. These chaperones act in concert with co-chaperones and ATP-dependent cycles to maintain neuronal protein homeostasis, which is particularly critical in post-mitotic neurons vulnerable to proteotoxic stress. Dysregulation of chaperone activity contributes to the accumulation of pathogenic aggregates driving neurodegenerative processes [36].
The expression of HSPs is transcriptionally regulated by heat shock factor 1 (HSF1), a master stress-response regulator that activates heat shock genes during proteotoxic stress. HSF1 dysfunction has been implicated in movement disorders; notably, recent genetic studies identified intronic VNTR expansions in HSF1 that reduce its expression and confer risk for essential tremor, linking impaired chaperone induction to disease pathogenesis [37]. Beyond genetic factors, aging and chronic stress diminish HSF1 activity, weakening the cellular defense against misfolded proteins. Pharmacological strategies aimed at modulating HSF1 or enhancing HSP expression—such as small molecules that disrupt HSP90–HSF1 interaction to activate HSF1—are emerging as promising therapeutic approaches for neurodegenerative and movement disorders [38].
Recent advances highlight the therapeutic potential of targeting chaperone systems directly. Approaches include HSP90 inhibitors, which reduce tau and Aβ toxicity in AD models, and HSP70 activators, which promote disaggregation and clearance of misfolded proteins [39]. Novel concepts such as epichaperomes—stable, disease-specific chaperone assemblies—offer network-level intervention strategies to restore proteostasis in complex disorders [40]. Unlike transient chaperone complexes, epichaperomes act as scaffolding hubs that rewire protein–protein interaction networks, amplifying synaptic dysfunction and proteotoxic stress. Their persistence under chronic stress creates a maladaptive connectivity state that cannot be corrected by conventional chaperone modulators. Recent studies highlight small molecules such as PU-H71 and PU-AD (icapamespib) that selectively dismantle epichaperomes, restoring dynamic chaperone function and network integrity without broadly inhibiting heat shock proteins [40,41]. Chemical biology approaches have advanced clickable probes (PU-TCO) for single-cell mapping of epichaperomes in AD and PD, enabling precision diagnostics and therapeutic stratification [42]. Systems-level interactome analyses confirm that epichaperome disruption reverses synaptic and cognitive deficits in preclinical models, positioning these assemblies as actionable targets for network-centric interventions [43]. This strategy contrasts with single-protein aggregation inhibitors by addressing upstream proteostasis failure and restoring connectivity across signaling and trafficking pathways.
2.5. Additional Selective Clearance Pathways
Beyond canonical UPS and macroautophagy, selective clearance of protein aggregates leverages ESCRT-driven microautophagy, proteaphagy of proteasomes, receptor-guided aggrephagy (NBR1, OPTN, TOLLIP), and LLPS chaperone control (HSP70/RNA), together forming an integrated network that funnels diverse substrates to lysosomes or stabilizes pre-aggregate condensates.
Autophagy-linked FYVE domain-containing protein (ALFY, encoded by WDFY3) is a large (400 kDa), evolutionarily conserved scaffold essential for selective macroautophagy (aggrephagy). ALFY mediates clearance of ubiquitinated aggregates by bridging cargo and autophagic machinery. Its FYVE domain binds PI3P on autophagic membranes, WD40 repeats interact with Atg5 and GABARAP, and its PH-BEACH domain binds p62/SQSTM1, enabling recruitment of autophagosomes to ubiquitinated inclusions [44,45]. Under basal conditions, ALFY localizes to the nucleus but translocates to the cytoplasm upon stress or aggregation, colocalizing with pathogenic proteins such as mutant huntingtin, TDP-43, and SOD1 [46,47]. Loss of ALFY impairs selective autophagic degradation of detergent-insoluble aggregates, accelerating neurodegenerative phenotypes in models of HD [44,48]. Importantly, ALFY is dispensable for starvation-induced autophagy, underscoring its specificity for aggregate clearance [49].
Proteomic analysis of insoluble aggregates identified CRAM-1, a worm protein with a ubiquitin-binding domain, as a key modulator. Knockdown of CRAM-1 reduced aggregate burden by up to 86%, despite its minor representation in aggregate mass, indicating a leveraged effect [50]. Mechanistically, CRAM-1 binds ubiquitin and oligoubiquitin with high affinity, interfering with p62/sequestosome-mediated delivery of misfolded proteins to proteasomes and autophagosomes. Functional assays confirmed that CRAM-1 knockdown rescues motility and chemotaxis in Aβ-expressing worms, but only when proteasome activity is intact, implicating CRAM-1 as a negative regulator of proteasomal clearance [51].
CRAM-1 knockdown extended worm lifespan by 11%, consistent with prior findings for its mammalian orthologs (SURF1/2, HYPC). However, worms lacking CRAM-1 exhibited reduced early fecundity, suggesting evolutionary retention of aggregation-promoting proteins due to reproductive advantage despite late-life proteotoxicity [50]. This supports the antagonistic pleiotropy hypothesis, where genes beneficial for early fitness persist despite deleterious aging effects.
3. Protein Clearance Mechanisms - Extraneuronal
3.1. Glymphatic System
The glymphatic system facilitates bulk clearance of interstitial solutes and proteins through a perivascular network. Cerebrospinal fluid (CSF) enters the brain parenchyma from periarterial spaces, propelled by arterial pulsatility and regulated by aquaporin-4 (AQP4) channels concentrated on astrocytic endfeet [52]. This arrangement enables efficient CSF–interstitial fluid exchange and drives convective flow through the neuropil [53]. After solute exchange, CSF exits along perivenous spaces toward the subarachnoid space and cervical lymphatics, forming a continuous clearance route for extracellular metabolites and proteins.
Glymphatic activity is state-dependent: tracer studies demonstrate markedly increased clearance during non-REM sleep and under anesthesia, while wakefulness significantly reduces flow [54]. Human imaging studies using intrathecal gadolinium-enhanced MRI confirmed this pathway in vivo, validating its role in solute transport [55]. Initially, Aβ was identified as a glymphatic substrate, linking this system to AD pathophysiology [52]. More recent evidence shows αSyn clearance via glymphatic flow, implicating it in PD and related synucleinopathies [56].
3.2. Glial Cells
Glial cells are essential for maintaining extracellular proteostasis through phagocytosis of protein aggregates and cellular debris. Microglia, the brain’s resident immune cells, exhibit the highest phagocytic capacity among glial populations. They internalize αSyn aggregates via receptor-mediated endocytosis involving Toll-like receptors (TLR2, TLR4) and Fcγ receptors, followed by degradation through lysosomal pathways. In vivo studies using human αSyn transgenic mice (hαSyn-Tg) confirmed microglial uptake and clearance of fibrillar αSyn, which is accompanied by activation of inflammatory signaling cascades [57,58]. Microglia also phagocytose tau and Aβ aggregates, with efficiency influenced by complement receptor pathways (CR3) and triggering receptor expressed on myeloid cells 2 (TREM2) [59].
Astrocytes contribute to extracellular clearance by engulfing αSyn and tau aggregates through endocytosis and macropinocytosis, followed by lysosomal degradation. Recent studies show astrocytes accumulate αSyn inclusions in PD models, suggesting a secondary clearance role when microglial capacity is exceeded [60]. Astrocytic uptake is mediated by LRP1 and other scavenger receptors, and astrocytes can form glial scars that sequester aggregates, limiting their spread. Oligodendrocytes and oligodendrocyte precursor cells (OPCs) also exhibit phagocytic activity, particularly relevant in MSA, where they internalize αSyn and contribute to glial cytoplasmic inclusion formation [61].
4. Protein Aggregation
Neurons bearing protein aggregates exhibit widespread cellular damage (Figure 3). Aggregated species such as αSyn and tau disrupt nuclear integrity, causing DNA double-strand breaks and transcriptional stress, while RNA metabolism is impaired through sequestration of RNA-binding proteins [62]. These aggregates activate innate immune pathways, including TLR signaling and inflammasome components, amplifying neuroinflammation and accelerating degeneration [63]. Cytoskeletal collapse is another hallmark: aggregates destabilize microtubules and actin networks, impairing axonal transport and synaptic vesicle trafficking, which compromises neuronal connectivity and cortical circuit function [64].
Mitochondrial impairment emerges as a critical consequence of aggregation rather than a primary trigger. Experimental models using optogenetic control of αSyn aggregation demonstrate that mitochondrial depolarization, ATP depletion, and fragmentation occur after aggregate formation, mediated by cardiolipin externalization and mitophagy activation [65,66]. These changes coincide with reduced oxidative phosphorylation and metabolic collapse, ultimately engaging apoptotic and necroptotic pathways. Importantly, overexpression of αSyn without aggregation does not reproduce these mitochondrial defects, reinforcing the causative role of aggregate formation in bioenergetic failure [65].
Protein aggregates propagate through prion-like mechanisms, spreading along neuronal networks and amplifying pathology in connected regions [67]. Computational models integrating connectome architecture and prion-like kinetics replicate observed progression patterns, identifying hubs that accelerate dissemination [68]. Despite widespread exposure, not all neurons succumb to aggregate toxicity, suggesting cell-intrinsic resilience mechanisms, such as enhanced lysosomal degradation or tissue-specific anti-aggregation pathways like SAPA [69]. These protective programs highlight heterogeneity in neuronal vulnerability and represent key determinants of disease trajectory.
Although Aβ, αSyn, tau, and TDP-43 share common principles such as β-sheet-driven assembly and prion-like propagation, their aggregation pathways exhibit distinct molecular signatures. Aβ forms extracellular cross-β fibrils with high polymorphism and secondary nucleation dominance [70], whereas αSyn aggregates in the cytoplasm via NAC-driven fibrillization strongly influenced by lipid interactions [71]. Tau aggregation requires polyanionic cofactors and displays isoform-dependent filament folds (3R vs 4R) [72], while TDP-43 undergoes phase separation before solidification, producing non-classical amyloid-like structures modulated by diverse PTMs [73]. These differences in architecture, nucleation dynamics, cellular localization, and regulatory mechanisms are summarized in Table 1.
4.1. Aβ Aggregation
Amyloid fibrils display a cross-β arrangement in which β-strands are perpendicular to the fibril axis and β-sheets stack parallel, yielding 4.8 Å inter-strand and 10 Å inter-sheet spacings reproducibly resolved by X-ray and cryo-EM across sequences [74]. Representative cryo-EM structures of Aβ filaments associated with AD, cerebral amyloid angiopathy, and Down syndrome are shown in Figure 4 [75,76,77,78]. In human AD brain, Aβ42 filaments adopt two S-shaped protofilament folds (Type I/II) with Type I enriched in sporadic AD and Type II in familial AD, and these in vivo folds differ from in vitro assemblies; AppNL-F knock-in mice recapitulate Type II deposits [79]. Brain-seeded samples expand this repertoire beyond S-shapes to ν- and υ-shaped cores with opposite helical handedness and intersubunit salt bridges (e.g., K16–A42; partial K28–A42) that define distinct packing topologies and solvent accessibilities [80]. Aβ40 filaments from leptomeningeal vessels (CAA) show anti-parallel packing of H14–G37 within protofilament pairs and ordered cores spanning D1–G38, establishing architecture that is proteinopathy-specific and distinct from parenchymal Aβ42 [76].
Aβ aggregation is governed by primary nucleation → elongation → secondary nucleation, with surface-catalyzed secondary nucleation on existing fibrils emerging as the dominant microscopic step that amplifies oligomer production under physiological conditions [81]. Denaturant-specific effects dissect these steps: urea reduces overall aggregation by decreasing fibril surface coverage and suppressing secondary/primary nucleation more than elongation, whereas low GuHCl accelerates aggregation via electrostatic screening before higher concentrations restore denaturation-dominated slowing [82]. pH provides step selectivity; titration near the histidine pKa (7.0) enhances primary nucleation while secondary nucleation and elongation remain pH-independent, linking acidic endo-lysosomes to accelerated nucleus formation without changing elongation [83]. These observations align with the backbone-driven nature of cross-β assembly (hydrogen-bond ladders), with side-chain and solvent constraints selecting polymorph-specific interfaces and tissues [84].
Aβ oligomers are the principal cytotoxins, producing ion-conducting pores that dysregulate Ca²⁺ homeostasis and trigger apoptotic/inflammatory signaling in neurons [85]. Single-channel electrophysiology in model bilayers shows Aβ1-42 pores < 5 Å in diameter with short-lived openings and conductances comparable to gramicidin, supporting an Aβ channelopathy [86]. High-resolution oligomer structures in membrane mimetics reveal tetramers/octamers with hydrophilic edge residues forming lipid-stabilized “edge-conductivity” pores that allow water permeation and leakage [87]. Lipid composition is decisive: outer-leaflet phosphatidylserine (≥ 10 mol %) accelerates Aβ1-42 oligomerization and pore formation within 30 min, whereas neutral DOPC resists adsorption/leakage [88]. Free POPC in extracellular microdomains co-aggregates with Aβ42, increasing compactness/sphericity of octamer/decamer oligomers via hydrophobic tail interactions and exposed hydrophobic patches that may nucleate further oligomerization [89].
Aβ–tau and Aβ–αSyn interactions reshape aggregation pathways. Aβ oligomers increase tau oligomer binding and internalization at human synapses via membrane/endocytic proteins, mechanistically linking Aβ burden to tau spread [90]. Tau microtubule-binding repeats form hetero-adducts with Aβ, altering Aβ aggregation kinetics and toxicity in intra- and extracellular milieus [91]. For αSyn, secondary nucleation on fibril surfaces is the dominant source of oligomers under neutral pH/ionic strength [92], mirroring Aβ’s surface catalysis [93]. Direct Aβ–α-syn co-assembly shows Aβ42 heterogeneous nucleation of α-syn condensation, whereas Aβ40 is sequestered into α-syn condensates that accelerate the liquid-to-solid transition to amyloid, accounting for mixed deposits in AD/Lewy body spectra [94].
Not all cross-β assemblies are pathological. The Drosophila Orb2 (CPEB) functional amyloid forms three-fold symmetric filaments with a hydrophilic glutamine-packed core that switches Orb2 from translation repressor to activator, providing a stable yet regulatable substrate for memory persistence [95]. This hydrophilic-core architecture contrasts with the hydrophobic cores typical of pathogenic fibrils, illustrating how sequence-context and packing encode biological function versus toxicity [95].
4.2. αSyn Aggregation
αSyn is a 140-amino-acid, intrinsically disordered presynaptic protein whose misfolding and self-assembly into oligomers and fibrils underlie the hallmark Lewy body and Lewy neurite pathology of PD and related synucleinopathies [96]. Over the last five years, converging structural, biophysical, and cellular studies have refined our understanding of how α-syn transitions from dynamic monomers to conformationally diverse assemblies, how membranes/lipids and post-translational modifications (PTMs) modulate this process, and how strain polymorphism and prion-like mechanisms shape propagation and disease heterogeneity [97,98].
High-resolution cryo-EM has mapped multiple α-syn fibril polymorphs, highlighting distinct protofilament interfaces, buried cores, and asymmetric architectures (Figure 5) [78,99,100,101,102]. Notably, brain-derived fibrils from MSA exhibit protofilaments with extensive interfacial contacts and asymmetric pairing, distinct from recombinant fibrils [103]. Familial mutations reconfigure fibril structure; for example, A53T alters the protofilament interface and increases neuronal seeding/toxicity [104]. Methodological advances now routinely achieve ≤2.5 Å maps and robust workflows for helical reconstruction, extending resolution of residues 36–97 in the fibril core and newly resolving N-terminal density islands [103,105].
Beyond classical nucleation–elongation, recent work shows α-syn undergoes LLPS to form protein condensates that can mature via liquid-to-solid phase transitions into amyloid [5,106]. Alternative splice isoforms modulate LLPS and aggregation kinetics: even minor fractions of α-syn-112 significantly accelerate phase separation and co-aggregation of full-length α-syn-140 [107]. SNARE interactions provide a cellular handle on LLPS. VAMP2 drives α-syn phase separation via electrostatic contacts between its juxtamembrane domain and the α-syn C-terminus, with condensates that sequester vesicles and recruit complexins, linking LLPS to synaptic organization [3]. Importantly, solution conditions decouple LLPS and amyloid formation. Salt promotes LLPS monotonically, but aggregation shows non-monotonic salt dependence, reflecting different intra- vs. intermolecular interaction landscapes [6]. These observations place condensate biology at the gateway of α-syn aggregation and suggest that cellular context (protein isoforms, binding partners, ionic milieu) chooses between metastable condensates and irreversible fibrils [106,107].
α-syn’s amphipathic N-terminus binds negatively charged phospholipids, and lipids are now recognized as active co-reactants in aggregation, not merely passive surfaces [108,109]. Kinetic and scattering studies show that binding to anionic vesicles remodels membranes, producing small lipid–protein particles that subsequently co-assemble into fibrils with lipid incorporation in the fibril material [109,110]. At organelle contact sites, α-syn localizes to mitochondria-associated ER membranes (MAMs), where it perturbs phosphatidylserine metabolism; lipidomics of human PD brain reveals region-specific phosphatidylserine alterations aligned with vulnerability [111]. These findings support a reciprocal coupling: lipid composition and curvature bias α-syn assembly pathways, while aggregates feedback to alter membrane structure and lipid homeostasis [108,111].
PTMs on soluble α-syn gate amplification of pathological seeds. Systematic mapping of human α-syn uncovered new phosphorylation and acetylation sites whose presence modulates seeded transmission in site- and conformation-specific ways [112]. Non-enzymatic PTMs linked to metabolism (e.g., glycation by methylglyoxal) enhance aggregation, increase pSer129-positive deposits, and activate microglia, providing molecular traction for the PD–diabetes comorbidity [113,114]. Conversely, O-GlcNAcylation has context-dependent effects on monomer dynamics and early oligomer formation: glycosylation at T72 slows aggregation whereas S87 may favor small oligomer formation without accelerating late fibrillation, underscoring residue-specific outcomes [115]. Collectively, PTMs serve as rheostats that redirect α-syn across condensed, oligomeric, and fibrillar states [112,115].
A consensus continues to crystallize that soluble oligomers are the primary cytotoxic species, with polymorphic oligomers showing distinct membrane disruption, mitochondrial impairment, and proteostasis stress [116,117]. Structural work distinguishes off-pathway, highly toxic oligomers—stable, often α-helical-rich assemblies—from on-pathway species that mature into less dynamic fibrils [116,118]. In vivo, neuronal fibrils penetrating the nucleus have been observed in MSA, where early nuclear envelope damage and loss of lamin integrity tie aggregation state to rapid neurodegeneration [119]. These data support a damage continuum in which oligomer–membrane interactions, organelle stress, and nuclear invasion are mechanistically linked [116,119].
Recent studies refine the prion-like propagation framework. Soluble oligomers/protofibrils display seeding capacity that depends on assembly size/shape and exposed epitopes [120]. Strain propagation occurs independently of cellular prion protein (PrPᴄ), pointing to alternate receptors/pathways for uptake and neuroinvasion [121]. In vivo imaging and computational modeling reveal network-like atrophy after PFF inoculation that depends on epicenter (striatal vs. hippocampal) and host genotype, emphasizing connectivity and regional vulnerability over a simplistic domino spread [120,122]. Complementarily, stochastic misfolding under identical conditions yields multiple strains in vitro and in vivo, offering a parsimonious explanation for clinical heterogeneity across synucleinopathies [123].
Mounting clinical, neuropathological, and experimental data implicate the gut–brain axis in initiation and modulation of α-syn pathology. Reviews and models support ENS-to-brainstem propagation via the vagus pathway in a “gut-first” PD subtype, consistent with early gastrointestinal prodromes [124,125,126]. Microbiome dysbiosis and metabolites shape aggregation and inflammation: short-chain fatty acids aggravate α-syn deposition and NLRP3-mediated neuroinflammation via GPR43, linking peripheral metabolism to central pathology [127,128]. These lines of evidence situate aggregation biology within a systems framework, where peripheral immune–metabolic states and barrier integrity bias α-syn assembly and spread [124,127].
4.3. Tau Aggregation
Tau aggregation is strongly driven by its primary structure, particularly the microtubule-binding repeats (R1–R4) located between residues 244–369. Within these repeats, two hexapeptide motifs—VQIINK (R2, residues 275–280) and VQIVYK (R3, residues 306–311)—are critical nucleation sites for β-sheet formation and filament assembly [129]. These motifs exhibit high β-propensity and form steric zippers in crystallographic studies, providing the structural basis for paired helical filament (PHF) formation [130]. Mutations within these motifs, such as ΔK280 or P301L, enhance aggregation by stabilizing β-structure, whereas proline insertions disrupt fibrillization [131]. Additionally, cysteine residues at positions 291 and 322 can form intermolecular disulfide bonds, further promoting filament elongation [132].
Tau is intrinsically disordered in its monomeric state, exhibiting minimal α-helix or β-sheet content [133]. However, aggregation involves a coil-to-β-sheet transition, particularly within R2 and R3, which nucleates filament growth [134,135]. Cryo-EM studies reveal that β-sheet formation in these repeats underpins the amyloid-like cross-β architecture of PHFs and straight filaments [136]. Tertiary structural rearrangements, such as the opening of the “paper-clip” conformation—where N- and C-terminal domains interact—are prerequisites for aggregation [137]. Phosphorylation at Thr231 within the proline-rich region induces cis-trans isomerization via Pin1 [138], destabilizing local α-helices and favoring aggregation-prone conformations [139].
Aggregated tau exhibits remarkable structural polymorphism, with disease-specific filament folds revealed by cryo-EM (Figure 6) [78,136,140,141,142]. AD filaments incorporate residues 273–380, forming paired helical and straight filaments, whereas Pick’s disease filaments (3R isoforms) span residues 254–378 and adopt distinct conformations [143]. Corticobasal degeneration filaments (4R isoforms) display unique folds involving residues 274–380. Heparin-induced filaments in vitro recapitulate some, but not all, disease-specific architectures, emphasizing the influence of cofactors and isoform composition on structural outcomes [144]. These polymorphic assemblies likely represent distinct “strains” of tau, analogous to prions, and may underlie phenotypic diversity across tauopathies [145].
Tau aggregation follows a nucleation-elongation mechanism, where oligomeric intermediates precede fibril formation [146]. In vitro, polyanions such as heparin or RNA accelerate nucleation by neutralizing electrostatic repulsion between positively charged repeat domains [147]. Seeding experiments demonstrate that short fibrils are the most potent species for templating aggregation, consistent with prion-like propagation observed in transgenic mouse models [145]. Hexapeptide deletion mutants lacking VQIINK or VQIVYK fail to seed aggregation, confirming their essential role in templated assembly [148]. Moreover, aggregation is concentration-dependent and modulated by PTMs, including phosphorylation and acetylation, which alter charge distribution and steric accessibility of nucleating motifs [149].
PTMs of misfolded proteins are increasingly recognized as critical determinants of aggregation dynamics, pathogenicity, and clinical outcomes in neurodegenerative diseases. In AD, phosphorylation of tau at threonine 217 (pT217) is strongly associated with progressive cognitive decline and serves as a predictive biomarker for conversion from mild cognitive impairment (MCI) to AD [150]. Furthermore, tau seeding activity and specific PTMs exhibit domain-specific correlations with cognitive impairment and can differentiate AD-related tau aggregation from primary age-related tauopathies, highlighting their diagnostic relevance [151].
Direct inhibition of tau aggregation has been achieved using small molecules that interfere with β-sheet stacking or hexapeptide interactions. Rhodanine derivatives exhibit submicromolar IC₅₀ values for both aggregation inhibition and fibril disassembly, mediated by hydrogen bonding and hydrophobic interactions with repeat domains [152]. Phenylthiazolyl-hydrazides and benzothiazoles similarly disrupt filament elongation, with structure-activity relationships emphasizing planarity, aromaticity, and hydrogen bond donors as key pharmacophores [153]. Methylene blue (methylthioninium chloride), a phenothiazine derivative, demonstrated clinical efficacy in reducing cognitive decline in patients with AD, supporting the therapeutic potential of aggregation inhibitors [154]. However, challenges remain regarding blood-brain barrier permeability, off-target effects, and the risk of generating toxic oligomeric intermediates during disassembly. Rational optimization of these scaffolds requires integration of pharmacokinetic and pharmacodynamic parameters early in development.
4.4. TDP-43 Aggregation
TDP-43 is a ubiquitously expressed RNA/DNA-binding protein whose pathological aggregation is a defining feature of ALS and frontotemporal lobar degeneration (FTLD), occurring in >97% of ALS and 45% of FTLD cases [155,156]. Cryo-EM studies of patient-derived inclusions reveal that TDP-43 filaments adopt an amyloid-like architecture comprising a single protofilament with a double-spiral fold spanning residues 282–360 in the low-complexity domain (LCD) [157], distinct from in vitro assemblies (Figure 7) [78,157,158,159]. This fold is stabilized by glycine-rich turns and polar residues, resulting in short β-strands and an absence of classical cross-β stacking, producing chemically heterogeneous surfaces that may mediate ligand interactions [160,161]. Solid-state NMR further demonstrates structural polymorphism among LCD fragments (e.g., 300–414 vs. 314–414), where minor sequence truncations generate fibrils with comparable morphology but distinct local packing, highlighting the role of aggregation-prone segments in polymorphic assembly and neurotoxicity [160].
Pathological TDP-43 aggregation is initiated by nuclear clearance and cytoplasmic mislocalization, followed by LLPS and transition to solid fibrils [162]. Disease-associated mutations and stressors (oxidative, nitrosative) accelerate this process. S-nitrosylation of cysteine residues promotes disulfide-linked oligomerization and prion-like propagation in neurons and in vivo models [163]. Recent work shows that amyloidogenic oligomers derived from the LCD act as potent nucleators, inducing phase separation of full-length TDP-43 and enhancing phosphorylation of cytosolic condensates, particularly in ALS-linked mutants such as A315T [164]. These oligomers exhibit high toxicity and recruit stress granule components, while chaperones like HSP70 modulate their material state, delaying irreversible gelation [164]. PTMs critically regulate aggregation. Hyperphosphorylation at C-terminal serines, long considered a pathological hallmark, may paradoxically antagonize LLPS and aggregation under certain contexts [165,166]. Conversely, SUMO2/3 conjugation during oxidative stress stabilizes RNA-free TDP-43 in stress granules, preventing irreversible aggregation, whereas inhibition of SUMOylation accelerates fibrillization [167]. Novel PTMs such as citrullination have been identified as irreversible modifications that alter TDP-43 structure and promote pathology progression [168].
TDP-43 aggregates propagate via prion-like seeding, templating misfolding of native TDP-43 and spreading along corticospinal and frontotemporal networks, consistent with neuropathological staging of ALS and FTLD [156,169]. Seeding experiments in neurons demonstrate that aggregate uptake triggers loss of nuclear TDP-43, cryptic exon activation (e.g., STMN2, UNC13A), and disruption of autoregulatory feedback, establishing a toxic feed-forward loop [155,170]. Aggregates also sequester RNA and splicing factors, impairing transcriptome integrity, while mitochondrial dysfunction emerges as a downstream effect. Mislocalized TDP-43 interacts with mitochondrial membranes, activating the mitochondrial unfolded protein response (UPRmt) and perturbing oxidative phosphorylation [171,172]. Heterotypic interactions further exacerbate pathology— αSyn co-assembly with TDP-43 LCD generates distinct fibril polymorphs with enhanced protease resistance and neurotoxicity, explaining mixed protein deposits in LATE and Lewy body dementias [173].
5. Protein-Based Self-Propagation Mechanism
The concept of templated conversion, first established in prion biology, describes how misfolded proteins act as seeds that recruit native monomers, inducing them to adopt the same pathogenic conformation. This mechanism drives fibril elongation through iterative cycles of monomer incorporation and structural corruption [174]. In Lewy body disorders, a similar process occurs with αSyn. Soluble monomeric αSyn undergoes conformational refolding into β-sheet-rich fibrils, forming the characteristic Lewy fold [175].
Recent studies confirm that αSyn aggregates propagate via prion-like mechanisms, meaning they self-template and spread across interconnected neural circuits [176]. Misfolded αSyn seeds can be internalized by neurons, escape endosomal compartments, and induce conformational conversion of endogenous αSyn, amplifying pathology [177,178]. This process underlies the spatial and temporal progression of Lewy pathology described by Braak, which begins in the olfactory bulb and dorsal motor nucleus of the vagus and ascends to limbic and cortical regions [179].
Cryo-EM studies reveal that αSyn fibrils exhibit polymorphic structures, with distinct protofilament interfaces and strain-specific conformations that dictate neurotoxicity and regional targeting [103,180]. These strains behave analogously to prions: they maintain biochemical identity upon serial propagation and produce disease-specific phenotypes in experimental models [121,181]. Strain adaptation during cross-species transmission has also been documented, suggesting selective pressures shape αSyn prion variants [182].
αSyn seeds initiate aggregation through nucleation-dependent polymerization, recruiting monomers into growing fibrils. Advanced assays such as real-time quaking-induced conversion (RT-QuIC) and seed amplification assays (SAA) exploit this property to detect pathogenic αSyn in cerebrospinal fluid and blood, demonstrating that amplified seeds retain strain-specific features [183,184,185].
Emerging evidence shows that αSyn interacts with other amyloidogenic proteins, notably tau, enabling cross-seeding and synergistic aggregation. This interplay contributes to overlapping pathologies in PD, dementia with LBs, and MSA, challenging traditional disease classifications [72].
6. Impact of Aging on Protein Aggregation
Aging drives a global erosion of proteostasis in the nervous system, so neurons accumulate misfolded/aggregated proteins and become more vulnerable to prion-like spread within networks [186]. For αSyn, aging acts as a rate-limiting cofactor that promotes both aggregation and long-range propagation from enteric/peripheral sites to brainstem and cortex according to recent syntheses of age-dependent α-syn biology [187,188]. Also, Morley et al. demonstrated that huntingtin aggregation increases with aging in in vivo models using C. elegans [189]. Parallel analyses of protein degradation and cellular senescence show age-driven declines in proteome turnover and intracellular proteolysis, further linking aging to aggregate accumulation across tissues [190].
The autophagy–lysosome axis exhibits region- and cell-type specific aging phenotypes. Certain brain regions (e.g., dentate gyrus, prefrontal cortex) show reduced autolysosome abundance/flux with age, while other tissues display compensatory or even increased autophagy—underscoring that “autophagy decline” is not uniform and must be assessed as flux rather than expression alone [191]. Integrated reviews detail how aging perturbs autophagosome formation, trafficking, fusion, and lysosomal pH/ion homeostasis, tipping neurons toward aggregate retention and organelle stress, with these failures emerging as primary drivers of late-age neurodegeneration [25]. At the organelle level, lysosome-repair capacity itself appears to fail with aging, fueling dys-proteostasis and sterile inflammation in aged and AD-derived neurons—providing a mechanistic bridge between normal aging and AD pathology [192]. Complementary syntheses position lysosomes as dynamic regulators of metabolic signaling in aging and neurodegeneration, tying lysosomal homeostasis to systemic hallmarks such as inflammation and mitochondrial dysfunction [193].
Beyond intracellular mechanisms, brain-wide clearance pathways deteriorate with age. Human diffusion-MRI demonstrates a mismatch in glymphatic influx/efflux in older adults—higher arterial-driven influx yet lower paravenous efflux—implicating age-related changes in aquaporin-4 polarization and vascular pulsatility in waste retention (Aβ, tau, α-syn) [194]. Large MRI cohorts further show an age-related fall in the ALPS index (diffusion along perivascular spaces), corroborating glymphatic slowing across adulthood [195]. Multimodal MRI now links declining BBB water exchange to lower ALPS and higher white-matter free water—indicating coupled BBB–glymphatic failure in healthy aging [196]. In vivo, aging enhances α-syn gut-to-brain propagation and aggregate maturation/resistance, with autonomic dysfunction reminiscent of clinical PD—positioning impaired clearance and aging-biased aggregate dynamics as convergent drivers of late-life synucleinopathy risk [197].
7. Strategies to Limit Protein Aggregation and Spread
Strategies to mitigate protein aggregation encompass diverse mechanisms, including RNA interference to reduce expression, stabilization of non-toxic conformers, inhibition of oligomerization and fibril elongation, and enhancement of proteostasis through autophagy or proteasome activation [198]. Immunotherapeutic approaches—both passive and active—aim to neutralize extracellular seeds and limit prion-like propagation, while emerging modalities such as PROTACs and AUTOTACs enable targeted intracellular degradation. Natural compounds and rationally designed peptides further expand the therapeutic landscape, although challenges such as blood–brain barrier penetration and off-target effects persist. These approaches, along with their mechanisms, representative agents, and translational limitations, are summarized in Table 2.
7.1. Immunization
The mechanisms of passive immunization are hypothesized to inhibit αSyn aggregation; decrease αSyn release; inhibit cell-to-cell transmission. Other possible mechanisms are related to the active immunization with immune generated antibodies [199].
Pagano et al. [200] and Lang et al. [201] evaluated two anti-αSyn monoclonal antibodies—prasinezumab and cinpanemab—in early PD through randomized, double-blind, placebo-controlled trials. Prasinezumab, which targets the C-terminal region of aggregated αSyn (aa 118–122), achieved target engagement but did not significantly slow clinical progression over 52 weeks, although imaging biomarkers suggested biological activity [200]. Cinpanemab, directed against the N-terminal epitope (aa 1–10), similarly failed to demonstrate meaningful benefit in motor or non-motor outcomes compared to placebo [201].
Beyond these two agents, multiple antibodies are under investigation to disrupt αSyn propagation in PD and MSA. MEDI1341/TAK-341 (AstraZeneca-Takeda) binds both monomeric and aggregated αSyn at aa 102–130 [202] and is in Phase II for MSA [203], while LuAF82422 (Lundbeck) [204] and Exidavemab (BioArctic) [205] target aggregated species with distinct C-terminal epitopes [206]. Despite strong mechanistic rationale, major limitations persist, including poor brain penetration of large immunoglobulins and uncertain reduction of αSyn in cerebrospinal fluid. Future strategies may involve smaller constructs such as single-chain antibodies or nanobodies, which offer improved blood-brain barrier permeability but shorter half-life [207].
Active immunization strategies targeting αSyn aim to elicit endogenous antibody production to reduce pathogenic aggregates in synucleinopathies. UB-312 (Vaxxinity) is a synthetic peptide vaccine that induces antibodies against insoluble αSyn at the C-terminal region, successfully completing Phase I by generating anti- αSyn antibodies and lowering aggregated species in cerebrospinal fluid (CSF) of PD patients, though without affecting monomeric forms [208]. Similarly, AFFiRiS-developed peptides PDO1A and PDO3A target the same C-terminal epitope, with PDO1A demonstrating a more robust and sustained antibody response and advancing to Phase II trials [209]. Despite promising immunogenicity, these approaches face challenges including off-target binding, neuroinflammation, and potential autoimmune reactions. Future directions may involve adeno-associated virus (AAV)-mediated delivery of optimized αSyn sequences to enhance specificity and durability of immune responses [210].
Beyond kinetics, modifying oligomer physicochemical properties—size and hydrophobicity—affects toxicity. Antibodies that enlarge oligomers may reduce membrane penetration, though increased surface hydrophobicity can offset benefits, yielding negligible net toxicity change.
7.2. Natural Compounds
Natural polyphenolic compounds such as epigallocatechin gallate (EGCG), gallic acid, and fisetin exhibit antioxidant and anti-inflammatory properties and can interact with hydrophobic regions of αSyn, thereby inhibiting oligomerization and aggregation in preclinical models [211]. Curcumin, which binds both monomeric and aggregated αSyn, has been evaluated in several clinical trials but failed to demonstrate significant disease-modifying effects, likely due to poor bioavailability and low target specificity [212]. In contrast, squalamine and its synthetic derivative ENT-01 (Enterin) prevent αSyn aggregation and have shown additional benefits in alleviating constipation in PD patients, with Phase II trials completed successfully [213]. Despite these promising mechanisms, major limitations—including low CNS penetration and rapid metabolism—continue to hinder clinical translation, emphasizing the need for improved formulations or delivery systems to enhance therapeutic efficacy.
7.3. Small Molecules
Small-molecule inhibitors targeting αSyn aggregation represent an alternative to immunotherapy for disease modification in synucleinopathies. Anle138b (Emrusolmin), developed by MODAG-Teva, prevents αSyn oligomerization and aggregation and also inhibits tau and prion protein aggregation, currently in Phase Ib for PD and Phase II for MSA [214]. PBT434 (ATH434), an iron chelator from Alterity, reduces iron-mediated aggregation of αSyn and is in Phase II trials, leveraging the link between metal dysregulation and synuclein pathology [215]. NPT200-11 (UCB-0599/minzasolmin) binds αSyn to prevent misfolding and aggregation, with Phase II completed for MSA [216].
Despite promising mechanisms, challenges remain. Off-target effects and limited CNS penetration pose significant safety and efficacy concerns. SynuClean-D, identified as a potential therapeutic for PD due to its ability to inhibit aggregation and degrade existing fibrils, did not progress to clinical trials, reflecting translational hurdles [217]. Future directions include optimizing pharmacokinetics and specificity to minimize adverse effects while enhancing target engagement.
7.4. Peptides
Peptide-based inhibitors of αSyn aggregation span rationally selected sequences, macrocyclic scaffolds, and cationic arginine-rich constructs, each acting at distinct steps of the Aβ pathway. High-throughput yeast screening identified K84s and K102s, two short peptides that suppress αSyn toxicity and oligomerization at sub-stoichiometric ratios [218]; K84s additionally reduced aggregation in human cells, and in silico analyses mapped favorable binding interfaces on αSyn consistent with direct anti-aggregation mechanisms. Designed derivatives TO2 (monomer-binding) and TO5 (oligomer-binding) follow the same logic—targeting regions proximal to the NAC domain to lower fibril formation in vitro—paralleling other NAC/N-terminal–focused inhibitory motifs that decelerate nucleation and elongation in ThT assays [219].
Macrocyclic peptides provide kinetic blockade at fibril ends or induce off-pathway remodeling. A RaPID-selected macrocycle (BD1) binds αSyn fibril termini to halt extension without altering fibril morphology, demonstrating a tractable “end-capping” mode of action; solubility optimization further enhanced inhibitory potency [220]. Complementarily, self-assembled cyclic d,l-α-peptides (e.g., CP-2) interact with the N-terminal/NAC regions to redirect αSyn toward amorphous, non-toxic species and disaggregate preformed fibrils in vitro and in cells overexpressing αSyn [221]. These macrocyclic strategies expand the peptide toolbox beyond linear sequence mimetics by exploiting conformational preorganization and multivalent display to engage dynamic aggregation intermediates [222].
Cationic poly-arginine peptides such as R18D (an 18-mer of D-arginine) reduce both intracellular αSyn aggregation and neuronal uptake of αSyn seeds—by 38% and 78%, respectively—in primary cortical neurons, and also diminish monomer aggregation in cell-free assays, highlighting combined anti-seeding and anti-aggregation effects [223]. Notwithstanding these preclinical signals, translation is constrained by short in vivo half-life, blood–brain barrier permeability, and potential off-target interactions; current solutions include sequence/structure optimization (e.g., β-sheet breakers [224], D-residue incorporation [225], and macrocyclization [226]), computational epitope scaffolding to improve conformational selectivity, and peptidomimetic design to enhance pharmacokinetics and CNS delivery [227].
TAT-βSyn-Degron is a rationally engineered, cell-penetrating peptide that selectively binds endogenous αSyn and directs it to the proteasome for targeted degradation, thereby reducing intracellular aggregates and ameliorating motor deficits in PD mouse models [228,229]. This construct incorporates three functional domains: a TAT sequence for efficient cellular uptake, a β-synuclein–derived binding motif for specific recognition of αSyn, and a degron tag to recruit proteasomal machinery, enabling clearance of pathogenic species. Preclinical studies demonstrated significant reductions in αSyn burden and associated neuropathology, supporting its potential as a disease-modifying strategy. However, translational challenges remain, including short systemic half-life and limited blood–brain barrier penetration, underscoring the need for advanced delivery platforms or chemical modifications to enhance CNS bioavailability [228,229].
7.5. PTMs
PTMs exert a pivotal influence on αSyn homeostasis and aggregation dynamics. Monoubiquitination of αSyn by the E3 ligase seven in absentia homolog (SIAH) facilitates its proteasomal degradation but paradoxically also promotes aggregation in dopaminergic cells, indicating a context-dependent role [230]. Conversely, USP9X-mediated deubiquitination reverses this modification, stabilizing αSyn and thereby increasing its aggregation propensity [231]. In addition, SUMOylation catalyzed by PIAS2 enhances αSyn stability and fosters pathological fibril formation, while reciprocal regulation between SUMOylation and ubiquitination determines degradation efficiency [232]. Engelender et al. are developing a peptide-based therapeutic designed to inhibit αSyn SUMOylation, a modification that promotes aggregation and neurotoxicity. In vivo studies using nigral injection of AAV2/1 encoding αSyn A53T demonstrated that treatment with this peptide significantly improved motor performance and reduced both nigral neurodegeneration and levels of S129-phosphorylated αSyn [233].
7.6. Proteolysis-Targeting Chimeras
Proteolysis-targeting chimeras (PROTACs) represent a rational approach to eliminate pathogenic αSyn by leveraging the ubiquitin-proteasome system. Arginine-based PROTACs utilize a bifunctional design in which one moiety binds the E3 ubiquitin ligase UBR1, while a benzothiazole-aniline group engages αSyn, forming a ternary complex that facilitates polyubiquitination and subsequent proteasomal degradation [234]. This catalytic mechanism enables repeated cycles of target elimination, offering a potentially efficient strategy for reducing intracellular αSyn burden. However, limitations include restricted proteasomal capacity to degrade large aggregates, incomplete specificity for αSyn, and poor blood–brain barrier (BBB) penetration, necessitating structural optimization and advanced delivery systems [235].
7.7. Autophagy-Targeting Chimeras
Autophagy-targeting chimeras (AUTOTACs) provide an alternative degradation pathway by redirecting αSyn aggregates to autophagosomes [236]. ATC161, a fusion of the aggregation inhibitor Anle138b and the ZZ domain of p62, selectively targets aggregated αSyn for autophagic clearance without affecting monomeric species [237]. In αSynPFF mouse models, ATC161 reduced cell-to-cell transmission of αSyn and improved striatal pathology, supporting its therapeutic potential. Currently in Phase I trials in South Korea, AUTOTACs address some limitations of PROTACs by exploiting lysosomal degradation; however, challenges such as BBB permeability and aggregate size remain critical barriers to clinical translation [238].
7.8. Activators of Autophagy
Autophagy-enhancing agents have emerged as potential disease-modifying strategies in PD by promoting clearance of misfolded αSyn. Trehalose, a disaccharide, activates autophagy through both mTOR-dependent and mTOR-independent pathways and has completed Phase I trials for sporadic and LRRK2-associated PD [239]. Metformin induces autophagy via AMPK activation and is currently in Phase II trials for PD, with epidemiological studies linking its use to reduced dementia risk [240,241]. Rapamycin, an mTOR inhibitor, enhances autophagy and has shown neuroprotective effects in mild cognitive impairment and AD, though PD-specific trials remain lacking [242]. Lithium, acting through an mTOR-independent mechanism, has been associated with a 77% reduction in PD risk in observational studies [243], but clinical trials for PD have not yet been initiated [244].
Despite promising mechanisms, these compounds present significant translational challenges. Lithium is related to renal and cardiac toxicity [245]. Rapamycin’s immunosuppressive profile limits long-term use, while metformin’s gastrointestinal side effects and variable CNS bioavailability complicate dosing strategies [246,247]. Furthermore, indiscriminate activation of autophagy may lead to degradation of essential proteins, underscoring the need for selective modulators and biomarker-driven trials to optimize therapeutic benefit while minimizing adverse effects.
7.9. Activators of the Proteasome
Currently, no compound has demonstrated robust activation of the proteasome for therapeutic purposes in PD. Preclinical efforts have explored agents that indirectly enhance proteasomal activity, such as selective phosphodiesterase inhibitors (e.g., roflumilast), which increase intracellular cAMP and upregulate proteasome function [248]. Other candidates include modified tetramethylpyrazine [249] and fluspirilene [250], identified through high-throughput screening for proteasome stimulators [251]. While these molecules show modest effects on proteasomal activity in vitro, their efficacy in reducing αSyn aggregates remains limited, and none have advanced to clinical trials. Major challenges include insufficient potency, off-target interactions, and poor blood–brain barrier penetration, highlighting the need for rational design of proteasome activators with improved specificity and pharmacokinetics.
8. Contradictory Points
8.1. Neurodegeneration Without Misfolded Protein Aggregates
Neurodegeneration can occur independently of protein misfolding, as demonstrated by hippocampal sclerosis (HS), which is characterized by severe gliosis and neuronal loss in the CA1 region of the hippocampus. HS may present as an isolated pathology or co-occur with other neurodegenerative conditions, including AD, frontotemporal dementia, temporal lobe epilepsy, multiple sclerosis, ALS, and TDP-43 proteinopathy [252]. Clinically, HS is associated with progressive memory impairment and cognitive decline, although at a slower rate compared to AD. These observations underscore that neuronal loss and gliosis alone can drive cognitive deterioration, highlighting that misfolded protein aggregates are not an obligatory feature of all neurodegenerative processes.
8.2. LBs: Beyond αSyn Aggregates
Although αSyn is the predominant component of LBs, these inclusions represent highly complex pathological structures. Proteomic analyses have identified more than 300 proteins within LBs, including molecular chaperones, components of the ubiquitin-proteasome system, and markers of axonal injury, indicating that Lewy pathology involves widespread disruption of proteostasis and neuronal integrity [253,254,255]. Advanced correlative light and electron microscopy studies have revealed that LBs are not homogeneous fibrillar aggregates but rather crowded membranous environments enriched with fragmented organelles, lipid membranes, autophagosomes, and lysosomes, reflecting severe impairment of intracellular trafficking and degradation pathways [256]. Experimental models further demonstrate that αSyn seeding initiates a cascade of secondary pathological events, culminating in the recruitment of diverse cellular components and the formation of complex LB-like inclusions. Critically, Mahul-Mellier et al. showed that the process of LB maturation—rather than αSyn fibrillization alone—is a major driver of neurodegeneration, underscoring that Lewy pathology is a multifactorial phenomenon involving protein misfolding, organellar dysfunction, and failure of cellular clearance mechanisms [257].
8.3. Mixed Pathologies
Autopsy studies reveal that αSyn pathology co-occurs in more than 50% of AD cases, often alongside tau aggregates, which are also frequently detected in Lewy body disease [253,258]. These concomitant pathologies complicate diagnostic accuracy and therapeutic development, as overlapping molecular signatures may obscure disease-specific biomarkers and treatment targets [259]. Clinically, co-pathologies are associated with accelerated cognitive decline and a more aggressive disease course compared to cases with isolated pathology [260,261].
Large-scale histopathological and proteomic studies confirm that mixed proteinopathies are frequent in aged brains; however, the prevalence and distribution of co-occurring protein seeds remain incompletely characterized across different neurodegenerative conditions [262]. Importantly, even low-level protein aggregation appears to be nearly ubiquitous in advanced age, suggesting that cases entirely devoid of misfolded proteins are rare [259].
Fein et al. demonstrated that approximately 24% of Aβ-positive synaptic terminals also contain phosphorylated tau, with the strongest co-localization observed in hippocampal regions, suggesting a synaptic interface for dual pathology [263]. Complementing this, Aksman et al. used a data-driven approach to map progression patterns and confirmed the existence of amyloid-first and tau-first subtypes, while highlighting their interaction within hippocampal circuits [264].
Using ultrasensitive seeding assays and large-scale neuropathological analyses, Manca et al. demonstrated that tau seeds are present in prodromal stages and occur across multiple neurodegenerative disorders, including Lewy body disease and other tauopathies [265]. This early appearance of tau pathology suggests that misfolded tau may act as an initiating or accelerating factor in disease progression, particularly when combined with αSyn or other protein aggregates. The frequent co-occurrence of tau and αSyn inclusions complicates diagnostic interpretation and may contribute to more severe cognitive impairment and faster clinical decline compared to cases with isolated pathology.
8.4. Rethinking Protein Aggregation
The persistent failure of anti-aggregation therapies in clinical trials for PD and AD underscores the need to reassess the amyloid hypothesis [266]. Despite thousands of reported aggregation inhibitors, no disease-modifying therapy has succeeded clinically. This gap may reflect oversimplified models and reductionist approaches that fail to capture the complexity of aggregation in vivo, where protein misfolding occurs within a dynamic cellular environment rich in lipids, organelles, and PTMs [267,268].
Advances in cryo-EM reveal that fibrils generated in vitro differ significantly from those isolated from diseased brains, both structurally and biochemically [175,269]. LBs and amyloid plaques are not homogeneous fibrillar deposits but complex assemblies of truncated proteins, lipids, and organelles. Moreover, polymorphism in αSyn and tau aggregates correlates with distinct seeding activities and possibly clinical heterogeneity, challenging the notion of a single toxic species [270].
PTMs such as phosphorylation, ubiquitination, and truncation are abundant in pathological aggregates and act as regulators rather than mere markers. Evidence suggests that phosphorylation often delays aggregation, while C-terminal truncation of αSyn promotes fibril compaction and Lewy body maturation [257,271]. These findings imply that PTMs orchestrate aggregation dynamics and interactions with cellular degradation systems, influencing neurotoxicity.
Data from neuronal models indicate that fibril formation alone is insufficient to induce neurodegeneration; toxicity emerges during the transition to Lewy body-like inclusions, which disrupt mitochondrial function, synaptic integrity, and protein homeostasis [257]. This process-centric view suggests that therapeutic strategies should target early aggregation and inclusion formation rather than solely removing mature deposits, as damage likely occurs during intermediate stages.
Given the heterogeneity of aggregates and co-pathologies, a single-target approach is unlikely to succeed. Future strategies should combine aggregation inhibitors, PTM modulators, and enhancers of proteostasis while addressing neuroinflammation and metabolic dysfunction [272]. Embracing complexity—rather than avoiding it—will be key to developing effective disease-modifying therapies.
9. Future Studies
Several critical questions remain unresolved. When do self-propagating assemblies acquire clinical relevance? Defining the temporal threshold between silent seeding and symptomatic disease requires longitudinal studies integrating seed amplification kinetics (e.g., RT-QuIC lag phases) with neuroimaging markers of network atrophy and clinical conversion rates. Another priority is clarifying whether a single structural strain can account for clinicopathological heterogeneity, or if mixed conformers drive divergent phenotypes. This is particularly relevant given the frequent co-occurrence of proteoforms—such as 3R/4R tau and αSyn—in both primary synucleinopathies and tauopathies, where dual seeding activity reaches near-universal prevalence in Lewy body disease cases examined [72,180].
Future research should also dissect the biochemical complexity of seeds, including PTMs and their impact on amplification efficiency and regional vulnerability. PTM-resolved cryo-EM and proteomics could reveal structural fingerprints that distinguish disease subtypes and predict propagation patterns [273]. Additionally, the role of LLPS and hydrogel-like transitions in vivo remains unexplored. While in vitro studies show that condensates mature into highly resistant amyloid states, direct evidence for hydrogel-to-Lewy body conversion in brain tissue is lacking [4]. Advanced imaging and biorthogonal labeling in animal models should address whether these transitions occur physiologically and influence aggregate resilience.
The failure of single-target approaches—such as aggregation inhibitors or immunotherapies—underscores the complexity of neurodegenerative diseases. These disorders are driven not only by misfolded protein accumulation but also by systemic proteostasis collapse, neuroinflammation, and metabolic dysfunction. Targeting aggregation alone often fails because toxic intermediates, impaired clearance pathways, and inflammatory cascades remain unchecked, allowing pathology to progress despite partial reduction of fibrils. Future strategies should adopt a multi-target paradigm that combines aggregation inhibitors with proteostasis enhancers (e.g., autophagy activators, proteasome modulators) and neuroinflammation modulators (e.g., microglial phenotype regulators, inflammasome inhibitors). Such combinatorial interventions address upstream and downstream drivers of neurodegeneration, restoring protein homeostasis while mitigating secondary damage. Integrating these approaches with biomarker-guided patient stratification and systems-level modeling may overcome translational barriers and deliver durable disease modification
Finally, the discovery of w-tau, a human-specific isoform generated by intron 12 retention, introduces a natural anti-aggregation mechanism. Its polycationic segment antagonizes polyanion-driven assembly, and reduced levels in AD brains suggest a protective role [135,274]. Structural characterization of w-tau and development of peptide mimetics could yield biomimetic strategies to competitively inhibit nucleation sites within the microtubule-binding region. These directions, combined with strain-aware modeling in iPSCs [275] and yeast systems [246], will enable mechanistic dissection of aggregation pathways and accelerate precision-targeted interventions.
10. Conclusions
Proteostasis is a fundamental requirement for neuronal integrity across both neurodegenerative and select non-neurodegenerative movement disorders. Protein clearance systems operate in a highly integrated manner to maintain proteome stability. Aging progressively impairs these mechanisms, while aggregation-prone proteins exploit this vulnerability, initiating a self-amplifying cycle of dysfunction. Misfolded proteins not only accumulate but also propagate through prion-like mechanisms, achieving disease-level burden and driving network-wide pathology. Their ability to seed, amplify, and maintain structural fidelity underlies distinct clinical phenotypes and experimental transmissibility. These features, combined with the interdependence of clearance pathways, underscore the complexity of targeting aggregation and proteostasis therapeutically, demanding multifactorial strategies rather than single-target interventions.
Author Contributions
Conceptualization, J.P.R. and A.L.F.C.; methodology, A.L.F.C.; software, A.L.F.C.; validation, A.L.F.C. and J.P.R.; formal analysis, A.L.F.C.; investigation, A.L.F.C.; resources, A.L.F.C.; data cu-ration, J.P.R.; writing—original draft preparation, J.P.R.; writing—review and editing, J.P.R.; vis-ualization, A.L.F.C.; supervision, A.L.F.C.; project administration, A.L.F.C.; funding acquisition, J.P.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data are presented in the manuscript.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| Aβ | Amyloid-beta |
| CNS | Central nervous system |
| HD | Huntington’s disease |
| LB | Lewy bodies |
| LLPS | Liquid-liquid phase separation |
| MSA | Multiple system atrophy |
| PD | Parkinson’s disease |
| PTMs | Post-translational modifications |
| UPS | Ubiquitin-proteasome system |
| αSyn | α-Synuclein |
References
- Soto, C. Unfolding the Role of Protein Misfolding in Neurodegenerative Diseases. Nat Rev Neurosci 2003, 4, 49–60. [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [CrossRef]
- Agarwal, A.; Chandran, A.; Raza, F.; Ungureanu, I.-M.; Hilcenko, C.; Stott, K.; Bright, N.A.; Morone, N.; Warren, A.J.; Lautenschläger, J. VAMP2 Regulates Phase Separation of α-Synuclein. Nat Cell Biol 2024, 26, 1296–1308. [CrossRef]
- Mukherjee, S.; Sakunthala, A.; Gadhe, L.; Poudyal, M.; Sawner, A.S.; Kadu, P.; Maji, S.K. Liquid-Liquid Phase Separation of α-Synuclein: A New Mechanistic Insight for α-Synuclein Aggregation Associated with Parkinson’s Disease Pathogenesis. J Mol Biol 2023, 435, 167713. [CrossRef]
- Ruiz-Ortega, E.D.; Wilkaniec, A.; Adamczyk, A. Liquid-Liquid Phase Separation and Conformational Strains of α-Synuclein: Implications for Parkinson’s Disease Pathogenesis. Front Mol Neurosci 2024, 17, 1494218. [CrossRef]
- Sternke-Hoffmann, R.; Sun, X.; Menzel, A.; Pinto, M.D.S.; Venclovaitė, U.; Wördehoff, M.; Hoyer, W.; Zheng, W.; Luo, J. Phase Separation and Aggregation of α-Synuclein Diverge at Different Salt Conditions. 2024. [CrossRef]
- Tseng, C.-S.; Chao, Y.-W.; Liu, Y.-H.; Huang, Y.-S.; Chao, H.-W. Dysregulated Proteostasis Network in Neuronal Diseases. Front Cell Dev Biol 2023, 11, 1075215. [CrossRef]
- Xue, Y.; Liu, X.; Chen, X.; Zhang, S.; Liu, W. Ubiquitination in the Nervous System: From Molecular Mechanisms to Disease Implications. Mol Neurobiol 2025, 62, 15365–15389. [CrossRef]
- Church, T.R.; Margolis, S.S. Mechanisms of Ubiquitin-Independent Proteasomal Degradation and Their Roles in Age-Related Neurodegenerative Disease. Front Cell Dev Biol 2024, 12, 1531797. [CrossRef]
- Santos, L.S.; Moreira-de-Carvalho, G.O.A.; Fortes, F.S.A.; de Souza, A.L.F. Autophagy Pathways, Ubiquitin-Proteasome System and Neurodegenerative Diseases: A Scopus Review. Brazilian Journal of Biological Sciences 2025, 12, e149–e149. [CrossRef]
- Hernández-Carralero, E.; Quinet, G.; Freire, R. ATXN3: A Multifunctional Protein Involved in the Polyglutamine Disease Spinocerebellar Ataxia Type 3. Expert Rev Mol Med 2024, 26, e19. [CrossRef]
- Liu, N.; Lin, M.-M.; Wang, Y. The Emerging Roles of E3 Ligases and DUBs in Neurodegenerative Diseases. Mol Neurobiol 2023, 60, 247–263. [CrossRef]
- Ronnebaum, S.M.; Patterson, C.; Schisler, J.C. Emerging Evidence of Coding Mutations in the Ubiquitin-Proteasome System Associated with Cerebellar Ataxias. Hum Genome Var 2014, 1, 14018. [CrossRef]
- Husain, N.; Yuan, Q.; Yen, Y.-C.; Pletnikova, O.; Sally, D.Q.; Worley, P.; Bichler, Z.; Shawn Je, H. TRIAD3/RNF216 Mutations Associated with Gordon Holmes Syndrome Lead to Synaptic and Cognitive Impairments via Arc Misregulation. Aging Cell 2017, 16, 281–292. [CrossRef]
- Deng, R.; Medico-Salsench, E.; Nikoncuk, A.; Ramakrishnan, R.; Lanko, K.; Kühn, N.A.; van der Linde, H.C.; Lor-Zade, S.; Albuainain, F.; Shi, Y.; et al. AMFR Dysfunction Causes Autosomal Recessive Spastic Paraplegia in Human That Is Amenable to Statin Treatment in a Preclinical Model. Acta Neuropathol 2023, 146, 353–368. [CrossRef]
- Gupta, R.; Begum, M.Y.; Kumar, R.; Gupta, J.; Nagraik, R.; Panda, S.P.; Abomughaid, M.M.; Lakhanpal, S.; D, A.; Osmani, R.A.M.; et al. The Ubiquitin-Proteasome System in Brain Disorders: Pathogenic Pathways, Post-Translational Tweaks, and Therapeutic Frontiers. J Neuroimmune Pharmacol 2025, 20, 105. [CrossRef]
- Le Guerroué, F.; Youle, R.J. Ubiquitin Signaling in Neurodegenerative Diseases: An Autophagy and Proteasome Perspective. Cell Death Differ 2021, 28, 439–454. [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu Rev Cell Dev Biol 2014, 30, 255–289. [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The Many Functions of ESCRTs. Nat Rev Mol Cell Biol 2020, 21, 25–42. [CrossRef]
- Udayar, V.; Chen, Y.; Sidransky, E.; Jagasia, R. Lysosomal Dysfunction in Neurodegeneration: Emerging Concepts and Methods. Trends Neurosci 2022, 45, 184–199. [CrossRef]
- Nan, H.; Ichinose, Y.; Tanaka, M.; Koh, K.; Ishiura, H.; Mitsui, J.; Mizukami, H.; Morimoto, M.; Hamada, S.; Ohtsuka, T.; et al. UBAP1 Mutations Cause Juvenile-Onset Hereditary Spastic Paraplegias (SPG80) and Impair UBAP1 Targeting to Endosomes. J Hum Genet 2019, 64, 1055–1065. [CrossRef]
- Gustavsson, E.K.; Follett, J.; Trinh, J.; Barodia, S.K.; Real, R.; Liu, Z.; Grant-Peters, M.; Fox, J.D.; Appel-Cresswell, S.; Stoessl, A.J.; et al. RAB32 Ser71Arg in Autosomal Dominant Parkinson’s Disease: Linkage, Association, and Functional Analyses. Lancet Neurol 2024, 23, 603–614. [CrossRef]
- Hu, C.; Yan, Y.; Jin, Y.; Yang, J.; Xi, Y.; Zhong, Z. Decoding the Cellular Trafficking of Prion-like Proteins in Neurodegenerative Diseases. Neurosci Bull 2024, 40, 241–254. [CrossRef]
- Lu, T.-W.; Frost, A.; Moss, F.R. 3rd Organelle Homeostasis Requires ESCRTs. Curr Opin Cell Biol 2025, 93, 102481. [CrossRef]
- Nixon, R.A.; Rubinsztein, D.C. Mechanisms of Autophagy-Lysosome Dysfunction in Neurodegenerative Diseases. Nat Rev Mol Cell Biol 2024, 25, 926–946. [CrossRef]
- Steel, D.; Zech, M.; Zhao, C.; Barwick, K.E.S.; Burke, D.; Demailly, D.; Kumar, K.R.; Zorzi, G.; Nardocci, N.; Kaiyrzhanov, R.; et al. Loss-of-Function Variants in HOPS Complex Genes VPS16 and VPS41 Cause Early Onset Dystonia Associated with Lysosomal Abnormalities. Ann Neurol 2020, 88, 867–877. [CrossRef]
- Monfrini, E.; Zech, M.; Steel, D.; Kurian, M.A.; Winkelmann, J.; Di Fonzo, A. HOPS-Associated Neurological Disorders (HOPSANDs): Linking Endolysosomal Dysfunction to the Pathogenesis of Dystonia. Brain 2021, 144, 2610–2615. [CrossRef]
- Vicinanza, M.; Korolchuk, V.I.; Ashkenazi, A.; Puri, C.; Menzies, F.M.; Clarke, J.H.; Rubinsztein, D.C. PI(5)P Regulates Autophagosome Biogenesis. Mol Cell 2015, 57, 219–234. [CrossRef]
- Boffey, H.K.; Rooney, T.P.C.; Willems, H.M.G.; Edwards, S.; Green, C.; Howard, T.; Ogg, D.; Romero, T.; Scott, D.E.; Winpenny, D.; et al. Development of Selective Phosphatidylinositol 5-Phosphate 4-Kinase γ Inhibitors with a Non-ATP-Competitive, Allosteric Binding Mode. J Med Chem 2022, 65, 3359–3370. [CrossRef]
- Xiaojie Zhang; Huan Zhang; Jie Dong; Huaibin Cai; Weidong Le Advances in Autophagy for Parkinson’s Disease Pathogenesis and Treatment. Ageing and Neurodegenerative Diseases 2025, 5, 6. [CrossRef]
- Nixon, R.A. The Role of Autophagy in Neurodegenerative Disease. Nat Med 2013, 19, 983–997. [CrossRef]
- Moreau, K.; Fleming, A.; Imarisio, S.; Lopez Ramirez, A.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. PICALM Modulates Autophagy Activity and Tau Accumulation. Nat Commun 2014, 5, 4998. [CrossRef]
- van der Beek, J.; Klumperman, J. Trafficking to the Lysosome: HOPS Paves the Way. Curr Opin Cell Biol 2025, 94, 102515. [CrossRef]
- Singh, M.K.; Han, S.; Ju, S.; Ranbhise, J.S.; Ha, J.; Yeo, S.G.; Kim, S.S.; Kang, I. Hsp70: A Multifunctional Chaperone in Maintaining Proteostasis and Its Implications in Human Disease. Cells 2025, 14. [CrossRef]
- Wei, H.; Zhang, Y.; Jia, Y.; Chen, X.; Niu, T.; Chatterjee, A.; He, P.; Hou, G. Heat Shock Protein 90: Biological Functions, Diseases, and Therapeutic Targets. MedComm (2020) 2024, 5, e470. [CrossRef]
- Sharma, A.; Shah, O.P.; Sharma, L.; Gulati, M.; Behl, T.; Khalid, A.; Mohan, S.; Najmi, A.; Zoghebi, K. Molecular Chaperones as Therapeutic Target: Hallmark of Neurodegenerative Disorders. Mol Neurobiol 2024, 61, 4750–4767. [CrossRef]
- Bi, H.; Wan, Y.; Zhao, R.; Pan, S.; Luan, M.; Wu, W.; Qiu, Y.; Yu, J.; Sun, Y.; Wei, L.; et al. Intronic VNTRs Downregulate Expression of HSF1 and Confer Genetic Risk of Essential Tremor. Brain 2025, 148, 4098–4111. [CrossRef]
- Ho, A.K.; Jeganathan, F.; Bictash, M.; Chen, H.-J. Identification of Novel Small Molecule Chaperone Activators for Neurodegenerative Disease Treatment. Biomed Pharmacother 2025, 187, 118049. [CrossRef]
- Rai, P. Conformational Dynamics of Hsp90 and Hsp70 Chaperones in Treating Neurodegenerative Diseases: Insights from the Drosophila Model. Proceedings of the Indian National Science Academy 2024, 90, 628–637. [CrossRef]
- Pasala, C.; Digwal, C.S.; Sharma, S.; Wang, S.; Bubula, A.; Chiosis, G. Epichaperomes: Redefining Chaperone Biology and Therapeutic Strategies in Complex Diseases. RSC Chem Biol 2025, 6, 678–698. [CrossRef]
- Sharma, S.; Joshi, S.; Kalidindi, T.; Digwal, C.S.; Panchal, P.; Lee, S.-G.; Zanzonico, P.; Pillarsetty, N.; Chiosis, G. Unraveling the Mechanism of Epichaperome Modulation by Zelavespib: Biochemical Insights on Target Occupancy and Extended Residence Time at the Site of Action. Biomedicines 2023, 11. [CrossRef]
- Bay, S.; Digwal, C.S.; Rodilla Martín, A.M.; Sharma, S.; Stanisavljevic, A.; Rodina, A.; Attaran, A.; Roychowdhury, T.; Parikh, K.; Toth, E.; et al. Synthesis and Characterization of Click Chemical Probes for Single-Cell Resolution Detection of Epichaperomes in Neurodegenerative Disorders. Biomedicines 2024, 12. [CrossRef]
- Inda, M.C.; Joshi, S.; Wang, T.; Bolaender, A.; Gandu, S.; Koren Iii, J.; Che, A.Y.; Taldone, T.; Yan, P.; Sun, W.; et al. The Epichaperome Is a Mediator of Toxic Hippocampal Stress and Leads to Protein Connectivity-Based Dysfunction. Nat Commun 2020, 11, 319. [CrossRef] [PubMed]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.G.; Lamark, T.; et al. The Selective Macroautophagic Degradation of Aggregated Proteins Requires the PI3P-Binding Protein Alfy. Mol Cell 2010, 38, 265–279. [CrossRef]
- Lystad, A.H.; Ichimura, Y.; Takagi, K.; Yang, Y.; Pankiv, S.; Kanegae, Y.; Kageyama, S.; Suzuki, M.; Saito, I.; Mizushima, T.; et al. Structural Determinants in GABARAP Required for the Selective Binding and Recruitment of ALFY to LC3B-Positive Structures. EMBO Rep 2014, 15, 557–565. [CrossRef]
- Han, H.; Wei, W.; Duan, W.; Guo, Y.; Li, Y.; Wang, J.; Bi, Y.; Li, C. Autophagy-Linked FYVE Protein (Alfy) Promotes Autophagic Removal of Misfolded Proteins Involved in Amyotrophic Lateral Sclerosis (ALS). In Vitro Cell Dev Biol Anim 2015, 51, 249–263. [CrossRef]
- Dragich, J.M.; Kuwajima, T.; Hirose-Ikeda, M.; Yoon, M.S.; Eenjes, E.; Bosco, J.R.; Fox, L.M.; Lystad, A.H.; Oo, T.F.; Yarygina, O.; et al. Autophagy Linked FYVE (Alfy/WDFY3) Is Required for Establishing Neuronal Connectivity in the Mammalian Brain. Elife 2016, 5, e14810. [CrossRef] [PubMed]
- Isakson, P.; Holland, P.; Simonsen, A. The Role of ALFY in Selective Autophagy. Cell Death Differ 2013, 20, 12–20. [CrossRef]
- Simonsen, A.; Birkeland, H.C.G.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a Novel FYVE-Domain-Containing Protein Associated with Protein Granules and Autophagic Membranes. J Cell Sci 2004, 117, 4239–4251. [CrossRef] [PubMed]
- Ayyadevara, S.; Balasubramaniam, M.; Gao, Y.; Yu, L.-R.; Alla, R.; Shmookler Reis, R. Proteins in Aggregates Functionally Impact Multiple Neurodegenerative Disease Models by Forming Proteasome-Blocking Complexes. Aging Cell 2015, 14, 35–48. [CrossRef]
- Balasubramaniam, M.; Ayyadevara, S.; Shmookler Reis, R.J. Structural Insights into Pro-Aggregation Effects of C. Elegans CRAM-1 and Its Human Ortholog SERF2. Sci Rep 2018, 8, 14891. [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci Transl Med 2012, 4, 147ra111. [CrossRef]
- Ciurea, A.V.; Mohan, A.G.; Covache-Busuioc, R.-A.; Costin, H.P.; Saceleanu, V.M. The Brain’s Glymphatic System: Drawing New Perspectives in Neuroscience. Brain Sci 2023, 13. [CrossRef]
- Hablitz, L.M.; Plá, V.; Giannetto, M.; Vinitsky, H.S.; Stæger, F.F.; Metcalfe, T.; Nguyen, R.; Benrais, A.; Nedergaard, M. Circadian Control of Brain Glymphatic and Lymphatic Fluid Flow. Nat Commun 2020, 11, 4411. [CrossRef] [PubMed]
- Ringstad, G.; Valnes, L.M.; Dale, A.M.; Pripp, A.H.; Vatnehol, S.-A.S.; Emblem, K.E.; Mardal, K.-A.; Eide, P.K. Brain-Wide Glymphatic Enhancement and Clearance in Humans Assessed with MRI. JCI Insight 2018, 3, 121537. [CrossRef]
- Lopes, D.M.; Llewellyn, S.K.; Bury, S.E.; Wang, J.; Wells, J.A.; Gegg, M.E.; Verona, G.; Lythgoe, M.F.; Harrison, I.F. The Influence of the Glymphatic System on α-Synuclein Propagation: The Role of Aquaporin-4. Brain 2025, awaf255. [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia Clear Neuron-Released α-Synuclein via Selective Autophagy and Prevent Neurodegeneration. Nat Commun 2020, 11, 1386. [CrossRef]
- Lv, Q.-K.; Tao, K.-X.; Wang, X.-B.; Yao, X.-Y.; Pang, M.-Z.; Liu, J.-Y.; Wang, F.; Liu, C.-F. Role of α-Synuclein in Microglia: Autophagy and Phagocytosis Balance Neuroinflammation in Parkinson’s Disease. Inflamm Res 2023, 72, 443–462. [CrossRef] [PubMed]
- Qin, Q.; Wang, M.; Yin, Y.; Tang, Y. The Specific Mechanism of TREM2 Regulation of Synaptic Clearance in Alzheimer’s Disease. Front Immunol 2022, 13, 845897. [CrossRef]
- Brash-Arias, D.; García, L.I.; Pérez-Estudillo, C.A.; Rojas-Durán, F.; Aranda-Abreu, G.E.; Herrera-Covarrubias, D.; Chi-Castañeda, D. The Role of Astrocytes and Alpha-Synuclein in Parkinson’s Disease: A Review. NeuroSci 2024, 5, 71–86. [CrossRef]
- Yu, Z.; Shi, M.; Stewart, T.; Fernagut, P.-O.; Huang, Y.; Tian, C.; Dehay, B.; Atik, A.; Yang, D.; De Giorgi, F.; et al. Reduced Oligodendrocyte Exosome Secretion in Multiple System Atrophy Involves SNARE Dysfunction. Brain 2020, 143, 1780–1797. [CrossRef] [PubMed]
- Welch, G.; Tsai, L.-H. Mechanisms of DNA Damage-Mediated Neurotoxicity in Neurodegenerative Disease. EMBO Rep 2022, 23, e54217. [CrossRef]
- Saramowicz, K.; Siwecka, N.; Galita, G.; Kucharska-Lusina, A.; Rozpędek-Kamińska, W.; Majsterek, I. Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease. Int J Mol Sci 2023, 25. [CrossRef]
- Louros, N.; Schymkowitz, J.; Rousseau, F. Mechanisms and Pathology of Protein Misfolding and Aggregation. Nat Rev Mol Cell Biol 2023, 24, 912–933. [CrossRef]
- Lurette, O.; Martín-Jiménez, R.; Khan, M.; Sheta, R.; Jean, S.; Schofield, M.; Teixeira, M.; Rodriguez-Aller, R.; Perron, I.; Oueslati, A.; et al. Aggregation of Alpha-Synuclein Disrupts Mitochondrial Metabolism and Induce Mitophagy via Cardiolipin Externalization. Cell Death Dis 2023, 14, 729. [CrossRef]
- Chand, J.; Fanai, H.L.; Antony, A.; Subramanian, G. Neurotoxicity by Hypoxia an Intermediate Between Alpha-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease. Journal of Pharmacology and Pharmacotherapeutics 2024, 15, 253–263. [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.-Y. Protein Transmission in Neurodegenerative Disease. Nat Rev Neurol 2020, 16, 199–212. [CrossRef]
- Alexandersen, C.G.; Brennan, G.S.; Brynildsen, J.K.; Henderson, M.X.; Iturria-Medina, Y.; Bassett, D.S. Network Models of Neurodegeneration: Bridging Neuronal Dynamics and Disease Progression. arXiv preprint 2025. arXiv:2509.05151.
- Jung, R.; Lechler, M.C.; Fernandez-Villegas, A.; Chung, C.W.; Jones, H.C.; Choi, Y.H.; Thompson, M.A.; Rödelsperger, C.; Röseler, W.; Kaminski Schierle, G.S.; et al. A Safety Mechanism Enables Tissue-Specific Resistance to Protein Aggregation during Aging in C. Elegans. PLoS Biol 2023, 21, e3002284. [CrossRef]
- Niu, Z.; Gui, X.; Feng, S.; Reif, B. Aggregation Mechanisms and Molecular Structures of Amyloid-β in Alzheimer’s Disease. Chemistry 2024, 30, e202400277. [CrossRef] [PubMed]
- Xu, L.; Nussinov, R.; Ma, B. Coupling of the Non-Amyloid-Component (NAC) Domain and the KTK(E/Q)GV Repeats Stabilize the α-Synuclein Fibrils. Eur J Med Chem 2016, 121, 841–850. [CrossRef]
- Padilla-Godínez, F.J.; Vázquez-García, E.R.; Trujillo-Villagrán, M.I.; Soto-Rojas, L.O.; Palomero-Rivero, M.; Hernández-González, O.; Pérez-Eugenio, F.; Collazo-Navarrete, O.; Arias-Carrión, O.; Guerra-Crespo, M. α-Synuclein and Tau: Interactions, Cross-Seeding, and the Redefinition of Synucleinopathies as Complex Proteinopathies. Front Neurosci 2025, 19, 1570553. [CrossRef]
- Koszła, O.; Sołek, P. Misfolding and Aggregation in Neurodegenerative Diseases: Protein Quality Control Machinery as Potential Therapeutic Clearance Pathways. Cell Commun Signal 2024, 22, 421. [CrossRef] [PubMed]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril Structure of Amyloid-β(1-42) by Cryo-Electron Microscopy. Science 2017, 358, 116–119. [CrossRef] [PubMed]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer’s Brain Tissue. Nat Commun 2019, 10, 4760. [CrossRef]
- Yang, Y.; Murzin, A.G.; Peak-Chew, S.; Franco, C.; Garringer, H.J.; Newell, K.L.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM Structures of Aβ40 Filaments from the Leptomeninges of Individuals with Alzheimer’s Disease and Cerebral Amyloid Angiopathy. Acta Neuropathol Commun 2023, 11, 191. [CrossRef] [PubMed]
- Fernandez, A.; Hoq, M.R.; Hallinan, G.I.; Li, D.; Bharath, S.R.; Vago, F.S.; Zhang, X.; Ozcan, K.A.; Newell, K.L.; Garringer, H.J.; et al. Cryo-EM Structures of Amyloid-β and Tau Filaments in Down Syndrome. Nat Struct Mol Biol 2024, 31, 903–909. [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res 2021, 49, W431–W437. [CrossRef]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM Structures of Amyloid-β 42 Filaments from Human Brains. Science 2022, 375, 167–172. [CrossRef]
- Lee, M.; Yau, W.-M.; Louis, J.M.; Tycko, R. Structures of Brain-Derived 42-Residue Amyloid-β Fibril Polymorphs with Unusual Molecular Conformations and Intermolecular Interactions. Proc Natl Acad Sci U S A 2023, 120, e2218831120. [CrossRef]
- Meisl, G.; Yang, X.; Frohm, B.; Knowles, T.P.J.; Linse, S. Quantitative Analysis of Intrinsic and Extrinsic Factors in the Aggregation Mechanism of Alzheimer-Associated Aβ-Peptide. Sci Rep 2016, 6, 18728. [CrossRef]
- Weiffert, T.; Meisl, G.; Curk, S.; Cukalevski, R.; Šarić, A.; Knowles, T.P.; Linse, S. Influence of Denaturants on Amyloid Β42 Aggregation Kinetics. Frontiers in Neuroscience 2022, 16, 943355. [CrossRef]
- Tian, Y.; Viles, J.H. pH Dependence of Amyloid-β Fibril Assembly Kinetics: Unravelling the Microscopic Molecular Processes. Angew Chem Int Ed Engl 2022, 61, e202210675. [CrossRef]
- Pal, S.; Goswami, S.; Das, D. Cross β Amyloid Assemblies as Complex Catalytic Machinery. Chem Commun (Camb) 2021, 57, 7597–7609. [CrossRef]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J Biol Chem 2005, 280, 17294–17300. [CrossRef]
- Xu, Y.; Bukhteeva, I.; Potsiluienko, Y.; Leonenko, Z. Amyloid β 1-42 Can Form Ion Channels as Small as Gramicidin in Model Lipid Membranes. Membranes (Basel) 2025, 15. [CrossRef]
- Ciudad, S.; Puig, E.; Botzanowski, T.; Meigooni, M.; Arango, A.S.; Do, J.; Mayzel, M.; Bayoumi, M.; Chaignepain, S.; Maglia, G.; et al. Aβ(1-42) Tetramer and Octamer Structures Reveal Edge Conductivity Pores as a Mechanism for Membrane Damage. Nat Commun 2020, 11, 3014. [CrossRef]
- Robinson, J.; Sarangi, N.K.; Keyes, T.E. Role of Phosphatidylserine in Amyloid-Beta Oligomerization at Asymmetric Phospholipid Bilayers. Phys Chem Chem Phys 2023, 25, 7648–7661. [CrossRef] [PubMed]
- King, K.M.; Cleveland, E.M.; Pennington, A.; Fuccello, S.; Brown, A.M. The Influence of POPC as a Coaggregate in Amyloid-β Oligomer Formation. ACS Chem Neurosci 2025, 16, 3886–3898. [CrossRef] [PubMed]
- Kadamangudi, S.; Marcatti, M.; Zhang, W.-R.; Fracassi, A.; Kayed, R.; Limon, A.; Taglialatela, G. Amyloid-β Oligomers Increase the Binding and Internalization of Tau Oligomers in Human Synapses. Acta Neuropathol 2024, 149, 2. [CrossRef] [PubMed]
- Kim, M.; Lin, Y.; Nam, E.; Kang, D.M.; Lim, S.; Kim, Y.K.; Lee, Y.-H.; Lim, M.H. Interactions with Tau’s Microtubule-Binding Repeats Modulate Amyloid-β Aggregation and Toxicity. Nat Chem Biol 2025, 21, 1709–1718. [CrossRef]
- Hoyt, F.; Standke, H.G.; Artikis, E.; Schwartz, C.L.; Hansen, B.; Li, K.; Hughson, A.G.; Manca, M.; Thomas, O.R.; Raymond, G.J.; et al. Cryo-EM Structure of Anchorless RML Prion Reveals Variations in Shared Motifs between Distinct Strains. Nat Commun 2022, 13, 4005. [CrossRef]
- Xu, C.K.; Meisl, G.; Andrzejewska, E.A.; Krainer, G.; Dear, A.J.; Castellana-Cruz, M.; Turi, S.; Edu, I.A.; Vivacqua, G.; Jacquat, R.P.B.; et al. α-Synuclein Oligomers Form by Secondary Nucleation. Nat Commun 2024, 15, 7083. [CrossRef]
- Röntgen, A.; Toprakcioglu, Z.; Morris, O.M.; Vendruscolo, M. Amyloid-β Modulates the Phase Separation and Aggregation of α-Synuclein. Proc Natl Acad Sci U S A 2025, 122, e2501987122. [CrossRef]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM Structure of a Neuronal Functional Amyloid Implicated in Memory Persistence in Drosophila. Science 2020, 367, 1230–1234. [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [CrossRef]
- Arias-Carrión, O.; Guerra-Crespo, M.; Padilla-Godínez, F.J.; Soto-Rojas, L.O.; Manjarrez, E. α-Synuclein Pathology in Synucleinopathies: Mechanisms, Biomarkers, and Therapeutic Challenges. Int J Mol Sci 2025, 26. [CrossRef] [PubMed]
- Mehra, S.; Gadhe, L.; Bera, R.; Sawner, A.S.; Maji, S.K. Structural and Functional Insights into α-Synuclein Fibril Polymorphism. Biomolecules 2021, 11. [CrossRef]
- Fan, Y.; Sun, Y.; Yu, W.; Tao, Y.; Xia, W.; Liu, Y.; Zhao, Q.; Tang, Y.; Sun, Y.; Liu, F.; et al. Conformational Change of α-Synuclein Fibrils in Cerebrospinal Fluid from Different Clinical Phases of Parkinson’s Disease. Structure 2023, 31, 78-87.e5. [CrossRef]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of Full-Length α-Synuclein Reveals Fibril Polymorphs with a Common Structural Kernel. Nat Commun 2018, 9, 3609. [CrossRef] [PubMed]
- Yan, N.L.; Candido, F.; Tse, E.; Melo, A.A.; Prusiner, S.B.; Mordes, D.A.; Southworth, D.R.; Paras, N.A.; Merz, G.E. Cryo-EM Structure of a Novel α-Synuclein Filament Subtype from Multiple System Atrophy. FEBS Lett 2025, 599, 33–40. [CrossRef]
- Sokratian, A.; Zhou, Y.; Xu, E.; Viverette, E.; Dillard, L.; Yuan, Y.; Li, J.Y.; Matarangas, A.; Bouvette, J.; Borgnia, M. Structural and Functional Landscape of α-Synuclein Fibril Conformations Amplified from Cerebrospinal Fluid. BioRxiv 2022, 2022–07.
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM Structure of Alpha-Synuclein Fibrils. Elife 2018, 7. [CrossRef]
- Sun, Y.; Hou, S.; Zhao, K.; Long, H.; Liu, Z.; Gao, J.; Zhang, Y.; Su, X.-D.; Li, D.; Liu, C. Cryo-EM Structure of Full-Length α-Synuclein Amyloid Fibril with Parkinson’s Disease Familial A53T Mutation. Cell Res 2020, 30, 360–362. [CrossRef]
- Sanchez, J.C.; Pierson, J.A.; Borcik, C.G.; Rienstra, C.M.; Wright, E.R. High-Resolution Cryo-EM Structure Determination of a-Synuclein-A Prototypical Amyloid Fibril. Bio Protoc 2025, 15, e5171. [CrossRef]
- Han, W.; Wei, M.; Xu, F.; Niu, Z. Aggregation and Phase Separation of α-Synuclein in Parkinson’s Disease. Chem Commun (Camb) 2024, 60, 6581–6590. [CrossRef]
- Röntgen, A.; Toprakcioglu, Z.; Tomkins, J.E.; Vendruscolo, M. Modulation of α-Synuclein in Vitro Aggregation Kinetics by Its Alternative Splice Isoforms. Proc Natl Acad Sci U S A 2024, 121, e2313465121. [CrossRef] [PubMed]
- Makasewicz, K.; Linse, S.; Sparr, E. Interplay of α-Synuclein with Lipid Membranes: Cooperative Adsorption, Membrane Remodeling and Coaggregation. JACS Au 2024, 4, 1250–1262. [CrossRef] [PubMed]
- Dear, A.J.; Teng, X.; Ball, S.R.; Lewin, J.; Horne, R.I.; Clow, D.; Stevenson, A.; Harper, N.; Yahya, K.; Yang, X.; et al. Molecular Mechanism of α-Synuclein Aggregation on Lipid Membranes Revealed. Chem Sci 2024, 15, 7229–7242. [CrossRef] [PubMed]
- Galvagnion, C.; Barclay, A.; Makasewicz, K.; Marlet, F.R.; Moulin, M.; Devos, J.M.; Linse, S.; Martel, A.; Porcar, L.; Sparr, E.; et al. Structural Characterisation of α-Synuclein-Membrane Interactions and the Resulting Aggregation Using Small Angle Scattering. Phys Chem Chem Phys 2024, 26, 10998–11013. [CrossRef] [PubMed]
- Barbuti, P.A.; Guardia-Laguarta, C.; Yun, T.; Chatila, Z.K.; Flowers, X.; Wong, C.; Santos, B.F.R.; Larsen, S.B.; Lotti, J.S.; Hattori, N.; et al. The Role of Alpha-Synuclein in Synucleinopathy: Impact on Lipid Regulation at Mitochondria-ER Membranes. NPJ Parkinsons Dis 2025, 11, 103. [CrossRef]
- Zhang, S.; Zhu, R.; Pan, B.; Xu, H.; Olufemi, M.F.; Gathagan, R.J.; Li, Y.; Zhang, L.; Zhang, J.; Xiang, W.; et al. Post-Translational Modifications of Soluble α-Synuclein Regulate the Amplification of Pathological α-Synuclein. Nat Neurosci 2023, 26, 213–225. [CrossRef] [PubMed]
- Vasili, E.; König, A.; Al-Azzani, M.; Bosbach, C.; Gatzemeier, L.M.; Thom, S.; Chegão, A.; Miranda, H.V.; Steinem, C.; Erskine, D.; et al. Glycation of Alpha-Synuclein Enhances Aggregation and Neuroinflammatory Responses. NPJ Parkinsons Dis 2025, 11, 307. [CrossRef] [PubMed]
- Baldensperger, T.; Preissler, M.; Becker, C.F.W. Non-Enzymatic Posttranslational Protein Modifications in Protein Aggregation and Neurodegenerative Diseases. RSC Chem Biol 2025, 6, 129–149. [CrossRef] [PubMed]
- Gamage, K.; Wang, B.; Hard, E.R.; Van, T.; Galesic, A.; Phillips, G.R.; Pratt, M.; Lapidus, L.J. Post-Translational Modification of α-Synuclein Modifies Monomer Dynamics and Aggregation Kinetics. bioRxiv 2024, 2024–05.
- Yoo, J.M.; Lin, Y.; Heo, Y.; Lee, Y.-H. Polymorphism in Alpha-Synuclein Oligomers and Its Implications in Toxicity under Disease Conditions. Front Mol Biosci 2022, 9, 959425. [CrossRef]
- Du, X.-Y.; Xie, X.-X.; Liu, R.-T. The Role of α-Synuclein Oligomers in Parkinson’s Disease. Int J Mol Sci 2020, 21. [CrossRef]
- Mondal, A.; Dolui, S.; Dhabal, S.; Kundu, S.; Das, L.; Bhattacharjee, A.; Maiti, N.C. Structure Specific Neuro-Toxicity of α-Synuclein Oligomer. Int J Biol Macromol 2023, 253, 126683. [CrossRef]
- Wiseman, J.A.; Halliday, G.M.; Dieriks, B.V. Neuronal α-Synuclein Toxicity Is the Key Driver of Neurodegeneration in Multiple System Atrophy. Brain 2025, 148, 2306–2319. [CrossRef]
- Zampar, S.; Di Gregorio, S.E.; Grimmer, G.; Watts, J.C.; Ingelsson, M. “Prion-like” Seeding and Propagation of Oligomeric Protein Assemblies in Neurodegenerative Disorders. Front Neurosci 2024, 18, 1436262. [CrossRef]
- Khedmatgozar, C.R.; Holec, S.A.M.; Woerman, A.L. The Role of α-Synuclein Prion Strains in Parkinson’s Disease and Multiple System Atrophy. PLoS Pathog 2024, 20, e1011920. [CrossRef]
- Tullo, S.; Park, J.S.; Rahayel, S.; Gallino, D.; Park, M.; Mar, K.; Zheng, Y.-Q.; del Cid-Pellitero, E.; Fon, E.A.; Luo, W. In Vivo and in Silico Alpha-Synuclein Propagation Dynamics: The Role of Genotype, Epicentre, and Connectivity. bioRxiv 2025, 2025–07. [CrossRef]
- Wojewska, M.J.; Otero-Jimenez, M.; Guijarro-Nuez, J.; Alegre-Abarrategui, J. Beyond Strains: Molecular Diversity in Alpha-Synuclein at the Center of Disease Heterogeneity. Int J Mol Sci 2023, 24. [CrossRef]
- Takahashi, R.; Yamakado, H.; Uemura, N.; Taguchi, T.; Ueda, J. The Gut-Brain Axis Based on α-Synuclein Propagation-Clinical, Neuropathological, and Experimental Evidence. Int J Mol Sci 2025, 26. [CrossRef]
- Oliver, P.J.; Civitelli, L.; Hu, M.T. The Gut-Brain Axis in Early Parkinson’s Disease: From Prodrome to Prevention. J Neurol 2025, 272, 413. [CrossRef]
- Rissardo, J.P.; Caprara, A.L. A Narrative Review on Biochemical Markers and Emerging Treatments in Prodromal Synucleinopathies. Clinics and Practice 2025, 15. [CrossRef]
- Qu, Y.; An, K.; Wang, D.; Yu, H.; Li, J.; Min, Z.; Xiong, Y.; Xue, Z.; Mao, Z. Short-Chain Fatty Acid Aggregates Alpha-Synuclein Accumulation and Neuroinflammation via GPR43-NLRP3 Signaling Pathway in a Model Parkinson’s Disease. Mol Neurobiol 2025, 62, 6612–6625. [CrossRef] [PubMed]
- Mahbub, N.U.; Islam, M.M.; Hong, S.-T.; Chung, H.-J. Dysbiosis of the Gut Microbiota and Its Effect on α-Synuclein and Prion Protein Misfolding: Consequences for Neurodegeneration. Front Cell Infect Microbiol 2024, 14, 1348279. [CrossRef]
- von Bergen, M.; Barghorn, S.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Tau Aggregation Is Driven by a Transition from Random Coil to Beta Sheet Structure. Biochim Biophys Acta 2005, 1739, 158–166. [CrossRef] [PubMed]
- Jeganathan, S.; von Bergen, M.; Mandelkow, E.-M.; Mandelkow, E. The Natively Unfolded Character of Tau and Its Aggregation to Alzheimer-like Paired Helical Filaments. Biochemistry 2008, 47, 10526–10539. [CrossRef]
- Barghorn, S.; Zheng-Fischhöfer, Q.; Ackmann, M.; Biernat, J.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. Structure, Microtubule Interactions, and Paired Helical Filament Aggregation by Tau Mutants of Frontotemporal Dementias. Biochemistry 2000, 39, 11714–11721. [CrossRef]
- Schweers, O.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Oxidation of Cysteine-322 in the Repeat Domain of Microtubule-Associated Protein Tau Controls the in Vitro Assembly of Paired Helical Filaments. Proc Natl Acad Sci U S A 1995, 92, 8463–8467. [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Physical and Chemical Properties of Purified Tau Factor and the Role of Tau in Microtubule Assembly. J Mol Biol 1977, 116, 227–247. [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural Polymorphism of 441-Residue Tau at Single Residue Resolution. PLoS Biol 2009, 7, e34. [CrossRef]
- Hernández Pérez, F.; Ferrer, I.; Pérez Martínez, M.M.; Zabala, J.C.; del Rio, J.A.; Ávila, J. Tau Aggregation. Neuroscience 2022. [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [CrossRef]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.-J.; Mandelkow, E. Global Hairpin Folding of Tau in Solution. Biochemistry 2006, 45, 2283–2293. [CrossRef]
- Lin, Y.-T.; Cheng, J.-T.; Liang, L.-C.; Ko, C.-Y.; Lo, Y.-K.; Lu, P.-J. The Binding and Phosphorylation of Thr231 Is Critical for Tau’s Hyperphosphorylation and Functional Regulation by Glycogen Synthase Kinase 3beta. J Neurochem 2007, 103, 802–813. [CrossRef] [PubMed]
- Sibille, N.; Huvent, I.; Fauquant, C.; Verdegem, D.; Amniai, L.; Leroy, A.; Wieruszeski, J.-M.; Lippens, G.; Landrieu, I. Structural Characterization by Nuclear Magnetic Resonance of the Impact of Phosphorylation in the Proline-Rich Region of the Disordered Tau Protein. Proteins 2012, 80, 454–462. [CrossRef] [PubMed]
- Shi, Y.; Murzin, A.G.; Falcon, B.; Epstein, A.; Machin, J.; Tempest, P.; Newell, K.L.; Vidal, R.; Garringer, H.J.; Sahara, N.; et al. Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease with PET Ligand APN-1607. Acta Neuropathol 2021, 141, 697–708. [CrossRef]
- Hallinan, G.I.; Hoq, M.R.; Ghosh, M.; Vago, F.S.; Fernandez, A.; Garringer, H.J.; Vidal, R.; Jiang, W.; Ghetti, B. Structure of Tau Filaments in Prion Protein Amyloidoses. Acta Neuropathol 2021, 142, 227–241. [CrossRef]
- Qi, C.; Lövestam, S.; Murzin, A.; Peak-Chew, S.; Franco, C.; Bogdani, M.; Latimer, C.; Murrell, J.; Cullinane, P.; Jaunmuktane, Z. Tau Filaments with the Alzheimer Fold in Cases withMAPTmutations V337M and R406W. 2024.
- Scheres, S.H.; Zhang, W.; Falcon, B.; Goedert, M. Cryo-EM Structures of Tau Filaments. Current Opinion in Structural Biology 2020, 64, 17–25. [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of Microtubule-Associated Protein Tau into Alzheimer-like Filaments Induced by Sulphated Glycosaminoglycans. Nature 1996, 383, 550–553. [CrossRef]
- Goedert, M.; Spillantini, M.G. Propagation of Tau Aggregates. Mol Brain 2017, 10, 18. [CrossRef] [PubMed]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased Levels of Granular Tau Oligomers: An Early Sign of Brain Aging and Alzheimer’s Disease. Neurosci Res 2006, 54, 197–201. [CrossRef]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA Stimulates Aggregation of Microtubule-Associated Protein Tau into Alzheimer-like Paired Helical Filaments. FEBS Lett 1996, 399, 344–349. [CrossRef]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of Tau Protein into Alzheimer Paired Helical Filaments Depends on a Local Sequence Motif ((306)VQIVYK(311)) Forming Beta Structure. Proc Natl Acad Sci U S A 2000, 97, 5129–5134. [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The Acetylation of Tau Inhibits Its Function and Promotes Pathological Tau Aggregation. Nat Commun 2011, 2, 252. [CrossRef] [PubMed]
- Schöll, M.; Vrillon, A.; Ikeuchi, T.; Quevenco, F.C.; Iaccarino, L.; Vasileva-Metodiev, S.Z.; Burnham, S.C.; Hendrix, J.; Epelbaum, S.; Zetterberg, H.; et al. Cutting through the Noise: A Narrative Review of Alzheimer’s Disease Plasma Biomarkers for Routine Clinical Use. J Prev Alzheimers Dis 2025, 12, 100056. [CrossRef] [PubMed]
- Browne, D.F.; Smirnov, D.S.; Coughlin, D.G.; Peng, I.; Standke, H.G.; Kim, Y.; Pizzo, D.P.; Unapanta, A.; Andreasson, T.; Hiniker, A.; et al. Early Alzheimer’s Disease with Frequent Neuritic Plaques Harbors Neocortical Tau Seeds Distinct from Primary Age-Related Tauopathy. Nat Commun 2025, 16, 1851. [CrossRef] [PubMed]
- Bulic, B.; Pickhardt, M.; Mandelkow, E.-M.; Mandelkow, E. Tau Protein and Tau Aggregation Inhibitors. Neuropharmacology 2010, 59, 276–289. [CrossRef]
- Pickhardt, M.; Larbig, G.; Khlistunova, I.; Coksezen, A.; Meyer, B.; Mandelkow, E.-M.; Schmidt, B.; Mandelkow, E. Phenylthiazolyl-Hydrazide and Its Derivatives Are Potent Inhibitors of Tau Aggregation and Toxicity in Vitro and in Cells. Biochemistry 2007, 46, 10016–10023. [CrossRef]
- Berrocal, M.; Caballero-Bermejo, M.; Gutierrez-Merino, C.; Mata, A.M. Methylene Blue Blocks and Reverses the Inhibitory Effect of Tau on PMCA Function. Int J Mol Sci 2019, 20. [CrossRef]
- Zeng, Y.; Lovchykova, A.; Akiyama, T.; Rayner, S.L.; Maheswari Jawahar, V.; Liu, C.; Sianto, O.; Guo, C.; Calliari, A.; Prudencio, M. TDP-43 Nuclear Loss in FTD/ALS Causes Widespread Alternative Polyadenylation Changes. Nature Neuroscience 2025, 1–10. [CrossRef] [PubMed]
- Pongrácová, E.; Buratti, E.; Romano, M. Prion-like Spreading of Disease in TDP-43 Proteinopathies. Brain Sciences 2024, 14, 1132. [CrossRef]
- Arseni, D.; Hasegawa, M.; Murzin, A.G.; Kametani, F.; Arai, M.; Yoshida, M.; Ryskeldi-Falcon, B. Structure of Pathological TDP-43 Filaments from ALS with FTLD. Nature 2022, 601, 139–143. [CrossRef]
- Guenther, E.L.; Ge, P.; Trinh, H.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Gonen, T.; Zhou, Z.H.; Eisenberg, D.S. Atomic-Level Evidence for Packing and Positional Amyloid Polymorphism by Segment from TDP-43 RRM2. Nat Struct Mol Biol 2018, 25, 311–319. [CrossRef] [PubMed]
- Arseni, D.; Chen, R.; Murzin, A.G.; Peak-Chew, S.Y.; Garringer, H.J.; Newell, K.L.; Kametani, F.; Robinson, A.C.; Vidal, R.; Ghetti, B.; et al. TDP-43 Forms Amyloid Filaments with a Distinct Fold in Type A FTLD-TDP. Nature 2023, 620, 898–903. [CrossRef]
- Shenoy, J.; Lends, A.; Berbon, M.; Bilal, M.; El Mammeri, N.; Bertoni, M.; Saad, A.; Morvan, E.; Grélard, A.; Lecomte, S.; et al. Structural Polymorphism of the Low-Complexity C-Terminal Domain of TDP-43 Amyloid Aggregates Revealed by Solid-State NMR. Front Mol Biosci 2023, 10, 1148302. [CrossRef]
- Harrison, R.S.; Sharpe, P.C.; Singh, Y.; Fairlie, D.P. Amyloid Peptides and Proteins in Review. Rev Physiol Biochem Pharmacol 2007, 159, 1–77. [CrossRef]
- Liu, Y.; Xiang, J.; Gong, H.; Yu, T.; Gao, M.; Huang, Y. The Regulation of TDP-43 Structure and Phase Transitions: A Review. Protein J 2025, 44, 113–132. [CrossRef]
- Pirie, E.; Oh, C.-K.; Zhang, X.; Han, X.; Cieplak, P.; Scott, H.R.; Deal, A.K.; Ghatak, S.; Martinez, F.J.; Yeo, G.W.; et al. S-Nitrosylated TDP-43 Triggers Aggregation, Cell-to-Cell Spread, and Neurotoxicity in hiPSCs and in Vivo Models of ALS/FTD. Proc Natl Acad Sci U S A 2021, 118. [CrossRef]
- Chen, B.P.-W.; Lee, C.-C.; He, R.-Y.; Huang, A.-C.; Huang, J.-R.; Chan, J.C.C.; Huang, J.J.-T. Amyloidogenic Oligomers Derived from TDP-43 LCD Promote the Condensation and Phosphorylation of TDP-43. Chem Sci 2025. [CrossRef]
- Mosna, S.; Dormann, D. TDP-43 Phosphorylation: Pathological Modification or Protective Factor Antagonizing TDP-43 Aggregation in Neurodegenerative Diseases? Bioessays 2025, e70084. [CrossRef]
- Kellett, E.A.; Bademosi, A.T.; Walker, A.K. Molecular Mechanisms and Consequences of TDP-43 Phosphorylation in Neurodegeneration. Mol Neurodegener 2025, 20, 53. [CrossRef]
- Verde, E.M.; Antoniani, F.; Mediani, L.; Secco, V.; Crotti, S.; Ferrara, M.C.; Vinet, J.; Sergeeva, A.; Yan, X.; Hoege, C.; et al. SUMO2/3 Conjugation of TDP-43 Protects against Aggregation. Sci Adv 2025, 11, eadq2475. [CrossRef]
- Saunders, C.; Rocha-Rangel, P.; Desai, R.; Quadri, Z.; Lui, H.; Hunt Jr, J.B.; Liang, H.; Rogers, C.; Nash, K.; Tsoi, P.S. Citrullination of TDP-43 Is a Key Post-Translation Modification Associated with Structural and Functional Changes and Progressive Pathology in TDP-43 Mouse Models and Human Proteinopathies. bioRxiv 2025, 2025–02. [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The Role of TDP-43 Propagation in Neurodegenerative Diseases: Integrating Insights from Clinical and Experimental Studies. Experimental & molecular medicine 2020, 52, 1652–1662.
- Mamede, L.D.; Hu, M.; Titus, A.R.; Vaquer-Alicea, J.; French, R.L.; Diamond, M.I.; Miller, T.M.; Ayala, Y.M. TDP-43 Aggregate Seeding Impairs Autoregulation and Causes TDP-43 Dysfunction. 2025. [CrossRef] [PubMed]
- Lépine, S.; Nauleau-Javaudin, A.; Deneault, E.; Chen, C.X.-Q.; Abdian, N.; Franco-Flores, A.K.; Haghi, G.; Castellanos-Montiel, M.J.; Maussion, G.; Chaineau, M.; et al. Homozygous ALS-Linked Mutations in TARDBP/TDP-43 Lead to Hypoactivity and Synaptic Abnormalities in Human iPSC-Derived Motor Neurons. iScience 2024, 27, 109166. [CrossRef]
- Liu, Z.; Qiang, Y.; Shan, S.; Wang, S.; Liu, Z.; Yang, Y.; Huang, Z.; Song, M.; Zhao, X.; Song, F. Aberrant Mitochondrial Aggregation of TDP-43 Activated Mitochondrial Unfolded Protein Response and Contributed to Recovery of Acetaminophen Induced Acute Liver Injury. Toxicol Res (Camb) 2024, 13, tfae008. [CrossRef]
- Dhakal, S.; Wyant, C.E.; George, H.E.; Morgan, S.E.; Rangachari, V. Prion-like C-Terminal Domain of TDP-43 and α-Synuclein Interact Synergistically to Generate Neurotoxic Hybrid Fibrils. J Mol Biol 2021, 433, 166953. [CrossRef] [PubMed]
- Prusiner, S.B.; Scott, M.R.; DeArmond, S.J.; Cohen, F.E. Prion Protein Biology. Cell 1998, 93, 337–348. [CrossRef]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-Synuclein Filaments from Multiple System Atrophy. Nature 2020, 585, 464–469. [CrossRef] [PubMed]
- Pitton Rissardo, J.; McGarry, A.; Shi, Y.; Fornari Caprara, A.L.; Kannarkat, G.T. Alpha-Synuclein Neurobiology in Parkinson’s Disease: A Comprehensive Review of Its Role, Mechanisms, and Therapeutic Perspectives. Brain Sciences 2025, 15, 1260. [CrossRef]
- Wu, S.; Schekman, R.W. Intercellular Transmission of Alpha-Synuclein. Front Mol Neurosci 2024, 17, 1470171. [CrossRef]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int J Mol Sci 2021, 22. [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol Aging 2003, 24, 197–211. [CrossRef]
- Schmitz, M.; Candelise, N.; Canaslan, S.; Altmeppen, H.C.; Matschke, J.; Glatzel, M.; Younas, N.; Zafar, S.; Hermann, P.; Zerr, I. α-Synuclein Conformers Reveal Link to Clinical Heterogeneity of α-Synucleinopathies. Transl Neurodegener 2023, 12, 12. [CrossRef]
- Lau, A.; So, R.W.L.; Lau, H.H.C.; Sang, J.C.; Ruiz-Riquelme, A.; Fleck, S.C.; Stuart, E.; Menon, S.; Visanji, N.P.; Meisl, G.; et al. α-Synuclein Strains Target Distinct Brain Regions and Cell Types. Nat Neurosci 2020, 23, 21–31. [CrossRef]
- Holec, S.A.M.; Khedmatgozar, C.R.; Schure, S.J.; Pham, T.; Woerman, A.L. A-Synuclein Prion Strains Differentially Adapt after Passage in Mice. PLoS Pathog 2024, 20, e1012746. [CrossRef]
- Okuzumi, A.; Hatano, T.; Matsumoto, G.; Nojiri, S.; Ueno, S.-I.; Imamichi-Tatano, Y.; Kimura, H.; Kakuta, S.; Kondo, A.; Fukuhara, T.; et al. Propagative α-Synuclein Seeds as Serum Biomarkers for Synucleinopathies. Nat Med 2023, 29, 1448–1455. [CrossRef]
- Bsoul, R.; Simonsen, A.H.; Frederiksen, K.S.; Svenstrup, K.; Bech, S.; Salvesen, L.; Hejl, A.-M.; Rossi, M.; Parchi, P.; Lund, E.L.; et al. Seeding Amplification Assay with Universal Control Fluid: Standardized Detection of α-Synucleinopathies. PLoS One 2025, 20, e0326568. [CrossRef] [PubMed]
- Rissardo, J.P.; Fornari Caprara, A.L. Alpha-Synuclein Seed Amplification Assays in Parkinson’s Disease: A Systematic Review and Network Meta-Analysis. Clin Pract 2025, 15. [CrossRef]
- Voicu, V.; Toader, C.; Șerban, M.; Covache-Busuioc, R.-A.; Ciurea, A.V. Systemic Neurodegeneration and Brain Aging: Multi-Omics Disintegration, Proteostatic Collapse, and Network Failure Across the CNS. Biomedicines 2025, 13. [CrossRef]
- Song, D.-Y.; Li, J.-Y. Revisiting the Advance of Age-Dependent α-Synuclein Propagation and Aggregation. Ageing and Neurodegenerative Diseases 2025, 5, N-A. [CrossRef]
- Yaribash, S.; Mohammadi, K.; Sani, M.A. Alpha-Synuclein Pathophysiology in Neurodegenerative Disorders: A Review Focusing on Molecular Mechanisms and Treatment Advances in Parkinson’s Disease. Cell Mol Neurobiol 2025, 45, 30. [CrossRef] [PubMed]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The Threshold for Polyglutamine-Expansion Protein Aggregation and Cellular Toxicity Is Dynamic and Influenced by Aging in Caenorhabditis Elegans. Proc Natl Acad Sci U S A 2002, 99, 10417–10422. [CrossRef] [PubMed]
- Hamazaki, J.; Murata, S. Relationships between Protein Degradation, Cellular Senescence, and Organismal Aging. J Biochem 2024, 175, 473–480. [CrossRef]
- Singh, S.; Carosi, J.M.; Dang, L.; Sargeant, T.J. Autophagy Does Not Always Decline with Ageing. Nat Cell Biol 2025, 27, 712–715. [CrossRef] [PubMed]
- Vrancx, C.; Annaert, W. Lysosome Repair Fails in Ageing and Alzheimer’s Disease. Nat Cell Biol 2025, 27, 553–555. [CrossRef]
- He, Y.; Fan, Y.; Ahmadpoor, X.; Wang, Y.; Li, Z.A.; Zhu, W.; Lin, H. Targeting Lysosomal Quality Control as a Therapeutic Strategy against Aging and Diseases. Med Res Rev 2024, 44, 2472–2509. [CrossRef]
- Han, G.; Zhou, Y.; Zhang, K.; Jiao, B.; Hu, J.; Zhang, Y.; Wang, Z.; Lou, M.; Bai, R. Age- and Time-of-Day Dependence of Glymphatic Function in the Human Brain Measured via Two Diffusion MRI Methods. Front Aging Neurosci 2023, 15, 1173221. [CrossRef]
- Dai, Z.; Yang, Z.; Chen, X.; Zheng, W.; Zhuang, Z.; Liao, Y.; Li, M.; Chen, S.; Lin, D.; Wu, X.; et al. The Aging of Glymphatic System in Human Brain and Its Correlation with Brain Charts and Neuropsychological Functioning. Cereb Cortex 2023, 33, 7896–7903. [CrossRef] [PubMed]
- Bhattarai, A.; Zhu, Y.D.; Albuhwailah, B.; Maillard, P.; DeCarli, C.; Fan, A.P. Multimodal MRI Insights into Glymphatic Clearance and Blood-Brain Barrier Integrity in Healthy Aging. bioRxiv 2025, 2025–11. [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamgüney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing Promotes Pathological Alpha-Synuclein Propagation and Autonomic Dysfunction in Wild-Type Rats. Brain 2021, 144, 1853–1868. [CrossRef]
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging Treatment Approaches for Parkinson’s Disease. Front Neurosci 2018, 12, 693. [CrossRef]
- Henriquez, G.; Narayan, M. Targeting α-Synuclein Aggregation with Immunotherapy: A Promising Therapeutic Approach for Parkinson’s Disease. Exploration of Neuroprotective Therapy 2023, 3, 207–234. [CrossRef]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N Engl J Med 2022, 387, 421–432. [CrossRef]
- Lang Anthony E.; Siderowf Andrew D.; Macklin Eric A.; Poewe Werner; Brooks David J.; Fernandez Hubert H.; Rascol Olivier; Giladi Nir; Stocchi Fabrizio; Tanner Caroline M.; et al. Trial of Cinpanemab in Early Parkinson’s Disease. New England Journal of Medicine 2022, 387, 408–420. [CrossRef]
- Shering, C.; Pomfret, M.; kubiak, R.; Pouliquen, I.; Yin, W.; Simen, A.; Ratti, E.; Perkinton, M.; Tan, K.; Chessell, I. Reducing α-Synuclein in Human CSF; an Evaluation of Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of MEDI1341, an α-Synuclein-Specific Antibody, in Healthy Volunteers and Parkinson’s Disease Patients (P1-11.007). Neurology 2023, 100, 2469. [CrossRef]
- Padmanabhan, J.; Ratti, E.; Khudyakov, P.; Yin, W.; Zicha, S.; Golonzhka, O.; Kangarloo, T.; Harel, B.; Goodman, N.; Magueur, E.; et al. Study Design of TAK-341-2001: A Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of TAK-341 in Subjects With Multiple System Atrophy (P9-9.003). Neurology 2023, 100, 0247. [CrossRef]
- Buur, L.; Wiedemann, J.; Larsen, F.; Ben Alaya-Fourati, F.; Kallunki, P.; Ditlevsen, D.K.; Sørensen, M.H.; Meulien, D. Randomized Phase I Trial of the α-Synuclein Antibody Lu AF82422. Mov Disord 2024, 39, 936–944. [CrossRef]
- Boström, E.; Bachhav, S.S.; Xiong, H.; Zadikoff, C.; Li, Q.; Cohen, E.; Dreher, I.; Torrång, A.; Osswald, G.; Moge, M.; et al. Safety, Tolerability, and Pharmacokinetics of Single Doses of Exidavnemab (BAN0805), an Anti-α-Synuclein Antibody, in Healthy Western, Caucasian, Japanese, and Han Chinese Adults. J Clin Pharmacol 2024, 64, 1432–1442. [CrossRef] [PubMed]
- Tran, H.T.; Chung, C.H.-Y.; Iba, M.; Zhang, B.; Trojanowski, J.Q.; Luk, K.C.; Lee, V.M.Y. A-Synuclein Immunotherapy Blocks Uptake and Templated Propagation of Misfolded α-Synuclein and Neurodegeneration. Cell Rep 2014, 7, 2054–2065. [CrossRef]
- Butler, Y.R.; Liu, Y.; Kumbhar, R.; Zhao, P.; Gadhave, K.; Wang, N.; Li, Y.; Mao, X.; Wang, W. α-Synuclein Fibril-Specific Nanobody Reduces Prion-like α-Synuclein Spreading in Mice. Nat Commun 2022, 13, 4060. [CrossRef]
- Eijsvogel, P.; Misra, P.; Concha-Marambio, L.; Boyd, J.D.; Ding, S.; Fedor, L.; Hsieh, Y.-T.; Sun, Y.S.; Vroom, M.M.; Farris, C.M.; et al. Target Engagement and Immunogenicity of an Active Immunotherapeutic Targeting Pathological α-Synuclein: A Phase 1 Placebo-Controlled Trial. Nature Medicine 2024, 30, 2631–2640. [CrossRef] [PubMed]
- Meissner, W.G.; Traon, A.P.-L.; Foubert-Samier, A.; Galabova, G.; Galitzky, M.; Kutzelnigg, A.; Laurens, B.; Lührs, P.; Medori, R.; Péran, P.; et al. A Phase 1 Randomized Trial of Specific Active α-Synuclein Immunotherapies PD01A and PD03A in Multiple System Atrophy. Mov Disord 2020, 35, 1957–1965. [CrossRef]
- Björklund, A.; Mattsson, B. The AAV-α-Synuclein Model of Parkinson’s Disease: An Update. J Parkinsons Dis 2024, 14, 1077–1094. [CrossRef]
- Xiang, L.; Wang, Y.; Liu, S.; Liu, B.; Jin, X.; Cao, X. Targeting Protein Aggregates with Natural Products: An Optional Strategy for Neurodegenerative Diseases. Int J Mol Sci 2023, 24. [CrossRef]
- Emani, L.S.; Rao, J.K.; Kumar Dasappa, J.; Pecchio, M.; Lakey-Beitia, J.; Rodriguez, H.; Cruz-Mora, J.; Narayan, P.; Anand, N.; Mullur, G.; et al. Studies on Curcumin-Glucoside in the Prevention of Alpha-Synuclein Aggregation. J Alzheimers Dis Rep 2025, 9, 25424823251347260. [CrossRef]
- Camilleri, M.; Subramanian, T.; Pagan, F.; Isaacson, S.; Gil, R.; Hauser, R.A.; Feldman, M.; Goldstein, M.; Kumar, R.; Truong, D.; et al. Oral ENT-01 Targets Enteric Neurons to Treat Constipation in Parkinson Disease : A Randomized Controlled Trial. Ann Intern Med 2022, 175, 1666–1674. [CrossRef]
- Levin, J.; Sing, N.; Melbourne, S.; Morgan, A.; Mariner, C.; Spillantini, M.G.; Wegrzynowicz, M.; Dalley, J.W.; Langer, S.; Ryazanov, S.; et al. Safety, Tolerability and Pharmacokinetics of the Oligomer Modulator Anle138b with Exposure Levels Sufficient for Therapeutic Efficacy in a Murine Parkinson Model: A Randomised, Double-Blind, Placebo-Controlled Phase 1a Trial. EBioMedicine 2022, 80, 104021. [CrossRef]
- Bailey, D.K.; Clark, W.; Kosman, D.J. The Iron Chelator, PBT434, Modulates Transcellular Iron Trafficking in Brain Microvascular Endothelial Cells. PLoS One 2021, 16, e0254794. [CrossRef]
- Mercier, J.; Bani, M.; Colson, A.-O.; Germani, M.; Lalla, M.; Plisson, C.; Huiban, M.; Searle, G.; Mathy, F.-X.; Nicholl, R.; et al. Evaluation and Application of a PET Tracer in Preclinical and Phase 1 Studies to Determine the Brain Biodistribution of Minzasolmin (UCB0599). Mol Imaging Biol 2024, 26, 310–321. [CrossRef]
- Peña-Díaz, S.; Pujols, J.; Vasili, E.; Pinheiro, F.; Santos, J.; Manglano-Artuñedo, Z.; Outeiro, T.F.; Ventura, S. The Small Aromatic Compound SynuClean-D Inhibits the Aggregation and Seeded Polymerization of Multiple α-Synuclein Strains. J Biol Chem 2022, 298, 101902. [CrossRef]
- Borah, P.; Mattaparthi, V.S. Investigation into the Interaction Sites of the K84s and K102s Peptides with α-Synuclein for Understanding the Anti-Aggregation Mechanism: An In Silico Study. Current Biotechnology 2023, 12, 103–117.
- Popova, B.; Wang, D.; Rajavel, A.; Dhamotharan, K.; Lázaro, D.F.; Gerke, J.; Uhrig, J.F.; Hoppert, M.; Outeiro, T.F.; Braus, G.H. Identification of Two Novel Peptides That Inhibit α-Synuclein Toxicity and Aggregation. Front Mol Neurosci 2021, 14, 659926. [CrossRef] [PubMed]
- Ikenoue, T.; Oono, M.; So, M.; Yamakado, H.; Arata, T.; Takahashi, R.; Kawata, Y.; Suga, H. A RaPID Macrocyclic Peptide That Inhibits the Formation of α-Synuclein Amyloid Fibrils. Chembiochem 2023, 24, e202300320. [CrossRef] [PubMed]
- Chemerovski-Glikman, M.; Rozentur-Shkop, E.; Richman, M.; Grupi, A.; Getler, A.; Cohen, H.Y.; Shaked, H.; Wallin, C.; Wärmländer, S.K.T.S.; Haas, E.; et al. Self-Assembled Cyclic d,l-α-Peptides as Generic Conformational Inhibitors of the α-Synuclein Aggregation and Toxicity: In Vitro and Mechanistic Studies. Chemistry 2016, 22, 14236–14246. [CrossRef] [PubMed]
- Hornung, S.; Vogl, D.P.; Naltsas, D.; Volta, B.D.; Ballmann, M.; Marcon, B.; Syed, M.M.K.; Wu, Y.; Spanopoulou, A.; Feederle, R.; et al. Multi-Targeting Macrocyclic Peptides as Nanomolar Inhibitors of Self- and Cross-Seeded Amyloid Self-Assembly of α-Synuclein. Angew Chem Int Ed Engl 2025, 64, e202422834. [CrossRef] [PubMed]
- Robinson, E.C.; Gorecki, A.M.; Pesce, S.R.; Bagda, V.; Anderton, R.S.; Meloni, B.P. Novel Poly-Arginine Peptide R18D Reduces α-Synuclein Aggregation and Uptake of α-Synuclein Seeds in Cortical Neurons. Biomedicines 2025, 13. [CrossRef] [PubMed]
- Kim, Y.S.; Lim, D.; Kim, J.Y.; Kang, S.J.; Kim, Y.-H.; Im, H. Beta-Sheet-Breaking Peptides Inhibit the Fibrillation of Human Alpha-Synuclein. Biochem Biophys Res Commun 2009, 387, 682–687. [CrossRef]
- Horsley, J.R.; Jovcevski, B.; Pukala, T.L.; Abell, A.D. Designer D-Peptides Targeting the N-Terminal Region of α-Synuclein to Prevent Parkinsonian-Associated Fibrilization and Cytotoxicity. Biochim Biophys Acta Proteins Proteom 2022, 1870, 140826. [CrossRef]
- Hsueh, S.C.C.; Aina, A.; Roman, A.Y.; Cashman, N.R.; Peng, X.; Plotkin, S.S. Optimizing Epitope Conformational Ensembles Using α-Synuclein Cyclic Peptide “Glycindel” Scaffolds: A Customized Immunogen Method for Generating Oligomer-Selective Antibodies for Parkinson’s Disease. ACS Chem Neurosci 2022, 13, 2261–2280. [CrossRef]
- Allen, S.G.; Meade, R.M.; White Stenner, L.L.; Mason, J.M. Peptide-Based Approaches to Directly Target Alpha-Synuclein in Parkinson’s Disease. Mol Neurodegener 2023, 18, 80. [CrossRef]
- Qu, J.; Ren, X.; Xue, F.; He, Y.; Zhang, R.; Zheng, Y.; Huang, H.; Wang, W.; Zhang, J. Specific Knockdown of α-Synuclein by Peptide-Directed Proteasome Degradation Rescued Its Associated Neurotoxicity. Cell Chem Biol 2020, 27, 751-762.e4. [CrossRef]
- Jin, J.W.; Fan, X.; Del Cid-Pellitero, E.; Liu, X.-X.; Zhou, L.; Dai, C.; Gibbs, E.; He, W.; Li, H.; Wu, X.; et al. Development of an α-Synuclein Knockdown Peptide and Evaluation of Its Efficacy in Parkinson’s Disease Models. Commun Biol 2021, 4, 232. [CrossRef]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of Alpha-Synuclein by Seven in Absentia Homolog (SIAH) Promotes Its Aggregation in Dopaminergic Cells. J Biol Chem 2008, 283, 3316–3328. [CrossRef]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. α-Synuclein Fate Is Determined by USP9X-Regulated Monoubiquitination. Proc Natl Acad Sci U S A 2011, 108, 18666–18671. [CrossRef]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Abd Elghani, F.; Bandopadhyay, R.; Engelender, S. SUMOylation and Ubiquitination Reciprocally Regulate α-Synuclein Degradation and Pathological Aggregation. Proc Natl Acad Sci U S A 2017, 114, 13176–13181. [CrossRef] [PubMed]
- Engelender, S.; Stefanis, L.; Oddo, S.; Bellucci, A. Can We Treat Neurodegenerative Proteinopathies by Enhancing Protein Degradation? Mov Disord 2022, 37, 1346–1359. [CrossRef] [PubMed]
- Shen, L.; Zhang, J.; Wang, Z.; Liu, Y.; Cui, S.; Rao, H. Targeted Degradation of α-Synuclein by Arginine-Based PROTACs. J Biol Chem 2025, 301, 110449. [CrossRef] [PubMed]
- Kargbo, R.B. PROTAC Compounds Targeting α-Synuclein Protein for Treating Neurogenerative Disorders: Alzheimer’s and Parkinson’s Diseases. ACS Med Chem Lett 2020, 11, 1086–1087. [CrossRef]
- Kumar, G.; Thakur, V.; Sardana, S.; Tiwari, V.; Sharma, D.; Kumar, A. Contemporary Trends in Targeted Protein Degradation for Neurodegenerative Diseases. Eur J Med Chem 2025, 300, 118110. [CrossRef]
- Lee, J.; Sung, K.W.; Bae, E.-J.; Yoon, D.; Kim, D.; Lee, J.S.; Park, D.-H.; Park, D.Y.; Mun, S.R.; Kwon, S.C.; et al. Targeted Degradation of ⍺-Synuclein Aggregates in Parkinson’s Disease Using the AUTOTAC Technology. Mol Neurodegener 2023, 18, 41. [CrossRef]
- Lee, J.; Yoon, D.; Sung, K.W.; Bae, E.-J.; Park, D.-H.; Suh, Y.H.; Kwon, Y.T. Targeted Degradation of SNCA/α-Synuclein Aggregates in Neurodegeneration Using the AUTOTAC Chemical Platform. Autophagy 2024, 20, 463–465. [CrossRef]
- Moon, S.H.; Kwon, Y.; Huh, Y.E.; Choi, H.J. Trehalose Ameliorates Prodromal Non-Motor Deficits and Aberrant Protein Accumulation in a Rotenone-Induced Mouse Model of Parkinson’s Disease. Arch Pharm Res 2022, 45, 417–432. [CrossRef]
- AlRasheed, H.A.; Bahaa, M.M.; Elmasry, T.A.; Elberri, E.I.; Kotkata, F.A.; El Sabaa, R.M.; Elmorsi, Y.M.; Kamel, M.M.; Negm, W.A.; Hamouda, A.O.; et al. Randomized, Double-Blind, Placebo-Controlled Pilot Study of Metformin as an Adjunctive Therapy in Parkinson’s Disease. Front Pharmacol 2025, 16, 1497261. [CrossRef]
- Ping, F.; Jiang, N.; Li, Y. Association between Metformin and Neurodegenerative Diseases of Observational Studies: Systematic Review and Meta-Analysis. BMJ Open Diabetes Res Care 2020, 8. [CrossRef]
- Zhang, G.; Yin, L.; Luo, Z.; Chen, X.; He, Y.; Yu, X.; Wang, M.; Tian, F.; Luo, H. Effects and Potential Mechanisms of Rapamycin on MPTP-Induced Acute Parkinson’s Disease in Mice. Ann Palliat Med 2021, 10, 2889–2897. [CrossRef] [PubMed]
- Guttuso, T.J.; Russak, E.; De Blanco, M.T.; Ramanathan, M. Could High Lithium Levels in Tobacco Contribute to Reduced Risk of Parkinson’s Disease in Smokers? J Neurol Sci 2019, 397, 179–180. [CrossRef]
- Guttuso, T.J.; Shepherd, R.; Frick, L.; Feltri, M.L.; Frerichs, V.; Ramanathan, M.; Zivadinov, R.; Bergsland, N. Lithium’s Effects on Therapeutic Targets and MRI Biomarkers in Parkinson’s Disease: A Pilot Clinical Trial. IBRO Neurosci Rep 2023, 14, 429–434. [CrossRef] [PubMed]
- Truedson, P.; Ott, M.; Lindmark, K.; Ström, M.; Maripuu, M.; Lundqvist, R.; Werneke, U. Effects of Toxic Lithium Levels on ECG-Findings from the LiSIE Retrospective Cohort Study. J Clin Med 2022, 11. [CrossRef]
- Khurana, V.; Lindquist, S. Modelling Neurodegeneration in Saccharomyces Cerevisiae: Why Cook with Baker’s Yeast? Nat Rev Neurosci 2010, 11, 436–449. [CrossRef]
- Sorrenti, V.; Benedetti, F.; Buriani, A.; Fortinguerra, S.; Caudullo, G.; Davinelli, S.; Zella, D.; Scapagnini, G. Immunomodulatory and Antiaging Mechanisms of Resveratrol, Rapamycin, and Metformin: Focus on mTOR and AMPK Signaling Networks. Pharmaceuticals (Basel) 2022, 15. [CrossRef] [PubMed]
- Desouky, M.A.; George, M.Y.; Michel, H.E.; Elsherbiny, D.A. Roflumilast Escalates α-Synuclein Aggregate Degradation in Rotenone-Induced Parkinson’s Disease in Rats: Modulation of the Ubiquitin-Proteasome System and Endoplasmic Reticulum Stress. Chem Biol Interact 2023, 379, 110491. [CrossRef]
- Zhou, H.; Shao, M.; Guo, B.; Li, C.; Lu, Y.; Yang, X.; ShengnanLi; Li, H.; Zhu, Q.; Zhong, H.; et al. Tetramethylpyrazine Analogue T-006 Promotes the Clearance of Alpha-Synuclein by Enhancing Proteasome Activity in Parkinson’s Disease Models. Neurotherapeutics 2019, 16, 1225–1236. [CrossRef]
- Fiolek, T.J.; Keel, K.L.; Tepe, J.J. Fluspirilene Analogs Activate the 20S Proteasome and Overcome Proteasome Impairment by Intrinsically Disordered Protein Oligomers. ACS Chemical Neuroscience 2021, 12, 1438–1448. [CrossRef]
- Staerz, S.D. Discovery and Design of Novel 20S Proteasome Activators for Targeted Protein Degradation and Their Evaluation in Neurodegeneration Models; Michigan State University, 2024; ISBN 979-8-3822-4002-2.
- Smirnov, D.S.; Galasko, D.; Hansen, L.A.; Edland, S.D.; Brewer, J.B.; Salmon, D.P. Trajectories of Cognitive Decline Differ in Hippocampal Sclerosis and Alzheimer’s Disease. Neurobiol Aging 2019, 75, 169–177. [CrossRef]
- Leverenz, J.B.; Umar, I.; Wang, Q.; Montine, T.J.; McMillan, P.J.; Tsuang, D.W.; Jin, J.; Pan, C.; Shin, J.; Zhu, D.; et al. Proteomic Identification of Novel Proteins in Cortical Lewy Bodies. Brain Pathol 2007, 17, 139–145. [CrossRef]
- Xia, Q.; Liao, L.; Cheng, D.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic Identification of Novel Proteins Associated with Lewy Bodies. Front Biosci 2008, 13, 3850–3856. [CrossRef]
- Licker, V.; Turck, N.; Kövari, E.; Burkhardt, K.; Côte, M.; Surini-Demiri, M.; Lobrinus, J.A.; Sanchez, J.-C.; Burkhard, P.R. Proteomic Analysis of Human Substantia Nigra Identifies Novel Candidates Involved in Parkinson’s Disease Pathogenesis. Proteomics 2014, 14, 784–794. [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy Pathology in Parkinson’s Disease Consists of Crowded Organelles and Lipid Membranes. Nat Neurosci 2019, 22, 1099–1109. [CrossRef]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The Process of Lewy Body Formation, Rather than Simply α-Synuclein Fibrillization, Is One of the Major Drivers of Neurodegeneration. Proc Natl Acad Sci U S A 2020, 117, 4971–4982. [CrossRef]
- Lau, V.Z.; Awogbindin, I.O.; Frenkel, D.; Whitehead, S.N.; Tremblay, M.-È. A Hypothesis Explaining Alzheimer’s Disease, Parkinson’s Disease, and Dementia with Lewy Bodies Overlap. Alzheimers Dement 2025, 21, e70363. [CrossRef]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative Disease Concomitant Proteinopathies Are Prevalent, Age-Related and APOE4-Associated. Brain 2018, 141, 2181–2193. [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.-Y.; Trojanowski, J.Q. Parkinson’s Disease Dementia: Convergence of α-Synuclein, Tau and Amyloid-β Pathologies. Nat Rev Neurosci 2013, 14, 626–636. [CrossRef] [PubMed]
- Toledo, J.B.; Gopal, P.; Raible, K.; Irwin, D.J.; Brettschneider, J.; Sedor, S.; Waits, K.; Boluda, S.; Grossman, M.; Van Deerlin, V.M.; et al. Pathological α-Synuclein Distribution in Subjects with Coincident Alzheimer’s and Lewy Body Pathology. Acta Neuropathol 2016, 131, 393–409. [CrossRef]
- Walker, L.; Stefanis, L.; Attems, J. Clinical and Neuropathological Differences between Parkinson’s Disease, Parkinson’s Disease Dementia and Dementia with Lewy Bodies - Current Issues and Future Directions. J Neurochem 2019, 150, 467–474. [CrossRef] [PubMed]
- Fein, J.A.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Yang, F.; Cole, G.M.; Gylys, K.H. Co-Localization of Amyloid Beta and Tau Pathology in Alzheimer’s Disease Synaptosomes. Am J Pathol 2008, 172, 1683–1692. [CrossRef]
- Aksman, L.M.; Oxtoby, N.P.; Scelsi, M.A.; Wijeratne, P.A.; Young, A.L.; Alves, I.L.; Collij, L.E.; Vogel, J.W.; Barkhof, F.; Alexander, D.C.; et al. A Data-Driven Study of Alzheimer’s Disease Related Amyloid and Tau Pathology Progression. Brain 2023, 146, 4935–4948. [CrossRef] [PubMed]
- Manca, M.; Standke, H.G.; Browne, D.F.; Huntley, M.L.; Thomas, O.R.; Orrú, C.D.; Hughson, A.G.; Kim, Y.; Zhang, J.; Tatsuoka, C.; et al. Tau Seeds Occur before Earliest Alzheimer’s Changes and Are Prevalent across Neurodegenerative Diseases. Acta Neuropathol 2023, 146, 31–50. [CrossRef]
- Lashuel, H.A. Rethinking Protein Aggregation and Drug Discovery in Neurodegenerative Diseases: Why We Need to Embrace Complexity? Curr Opin Chem Biol 2021, 64, 67–75. [CrossRef]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.G.; Falcon, B.; Kotecha, A.; van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-Based Classification of Tauopathies. Nature 2021, 598, 359–363. [CrossRef]
- Limorenko, G.; Lashuel, H.A. Revisiting the Grammar of Tau Aggregation and Pathology Formation: How New Insights from Brain Pathology Are Shaping How We Study and Target Tauopathies. Chem Soc Rev 2022, 51, 513–565. [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau Filaments from Multiple Cases of Sporadic and Inherited Alzheimer’s Disease Adopt a Common Fold. Acta Neuropathol 2018, 136, 699–708. [CrossRef]
- So, R.W.L.; Watts, J.C. α-Synuclein Conformational Strains as Drivers of Phenotypic Heterogeneity in Neurodegenerative Diseases. J Mol Biol 2023, 435, 168011. [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The Many Faces of α-Synuclein: From Structure and Toxicity to Therapeutic Target. Nat Rev Neurosci 2013, 14, 38–48. [CrossRef]
- Kuzu, O.F.; Granerud, L.J.T.; Saatcioglu, F. Navigating the Landscape of Protein Folding and Proteostasis: From Molecular Chaperones to Therapeutic Innovations. Signal Transduct Target Ther 2025, 10, 358. [CrossRef] [PubMed]
- Kametani, F.; Tahira, M.; Takao, M.; Matsubara, T.; Hasegawa, K.; Yoshida, M.; Saito, Y.; Murayama, S.; Hasegawa, M. Analysis and Comparison of Post-Translational Modifications of α-Synuclein Filaments in Multiple System Atrophy and Dementia with Lewy Bodies. Sci Rep 2024, 14, 22892. [CrossRef] [PubMed]
- García-Escudero, V.; Ruiz-Gabarre, D.; Gargini, R.; Pérez, M.; García, E.; Cuadros, R.; Hernández, I.H.; Cabrera, J.R.; García-Escudero, R.; Lucas, J.J. A New Non-Aggregative Splicing Isoform of Human Tau Is Decreased in Alzheimer’s Disease. Acta Neuropathologica 2021, 142, 159–177. [CrossRef]
- Yefroyev, D.A.; Jin, S. Induced Pluripotent Stem Cells for Treatment of Alzheimer’s and Parkinson’s Diseases. Biomedicines 2022, 10. [CrossRef] [PubMed]
Figure 1.
Protein aggregates in neurodegenerative diseases.
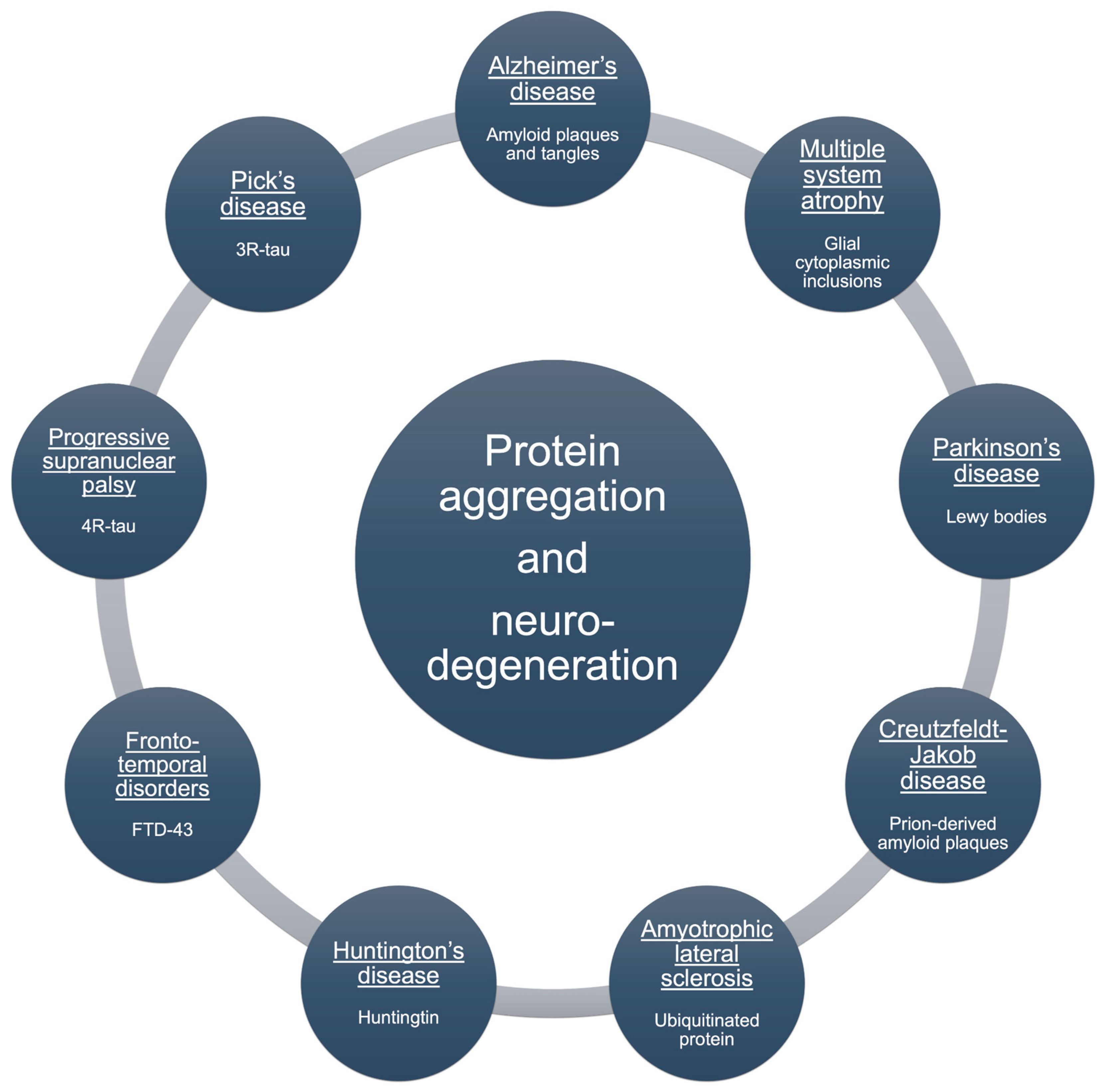
Figure 2.
Intraneuronal Protein Clearance Mechanisms. Schematic representation of major path-ways involved in protein quality control within neurons, including the UPS, molecular chaperones, the autophagy–lysosomal pathway, and the endosomal–lysosomal system.
Figure 2.
Intraneuronal Protein Clearance Mechanisms. Schematic representation of major path-ways involved in protein quality control within neurons, including the UPS, molecular chaperones, the autophagy–lysosomal pathway, and the endosomal–lysosomal system.
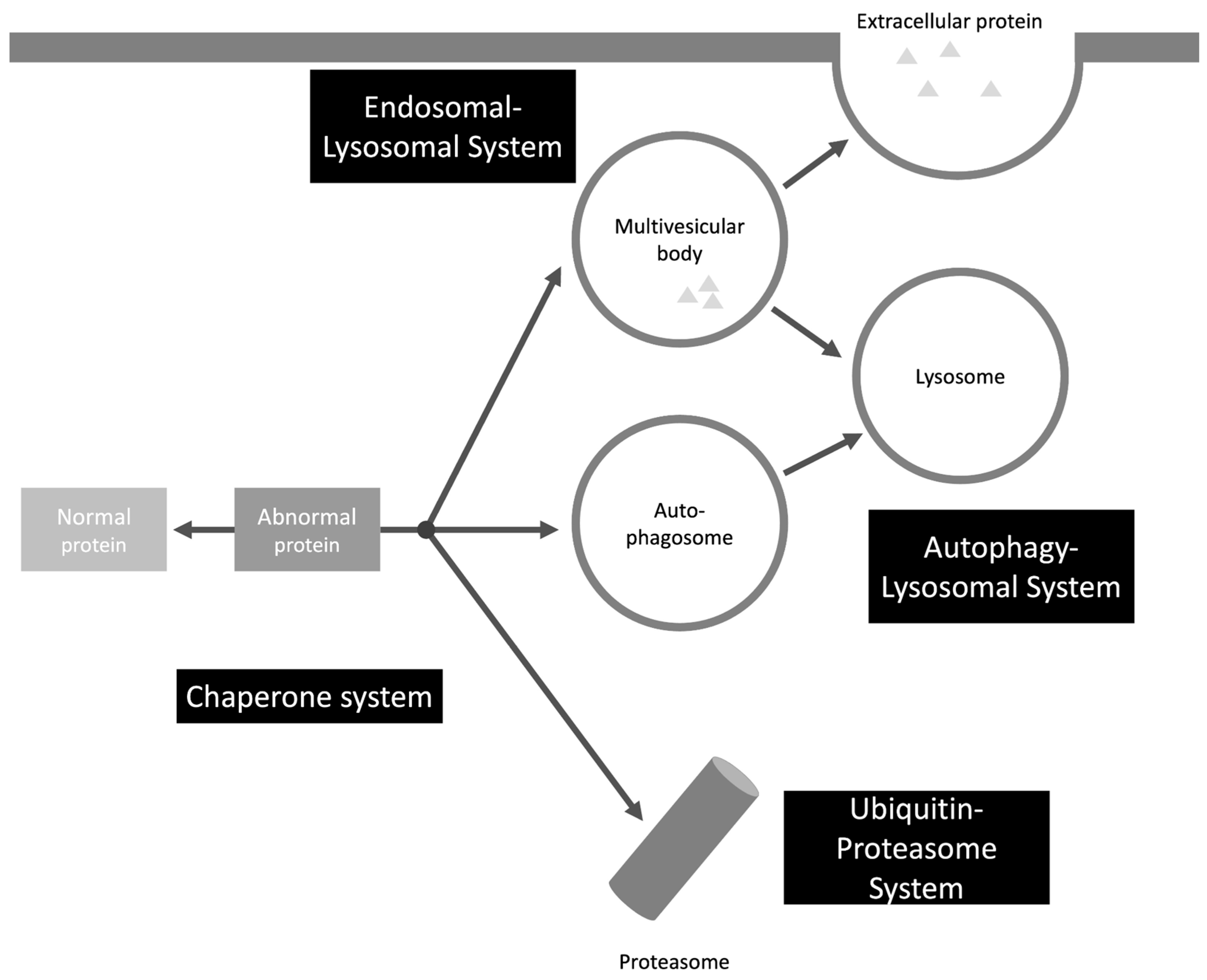
Figure 3.
Schematic representation of protein aggregation pathways. Proteins in their native state can undergo misfolding, forming oligomers that serve as intermediates for different aggregation routes. These include fibril elongation, leading to ordered Aβ fibrils, or the formation of amorphous, disorganized aggregates. Native proteins may also assemble into quaternary structures or crystalline forms under specific conditions.
Figure 3.
Schematic representation of protein aggregation pathways. Proteins in their native state can undergo misfolding, forming oligomers that serve as intermediates for different aggregation routes. These include fibril elongation, leading to ordered Aβ fibrils, or the formation of amorphous, disorganized aggregates. Native proteins may also assemble into quaternary structures or crystalline forms under specific conditions.
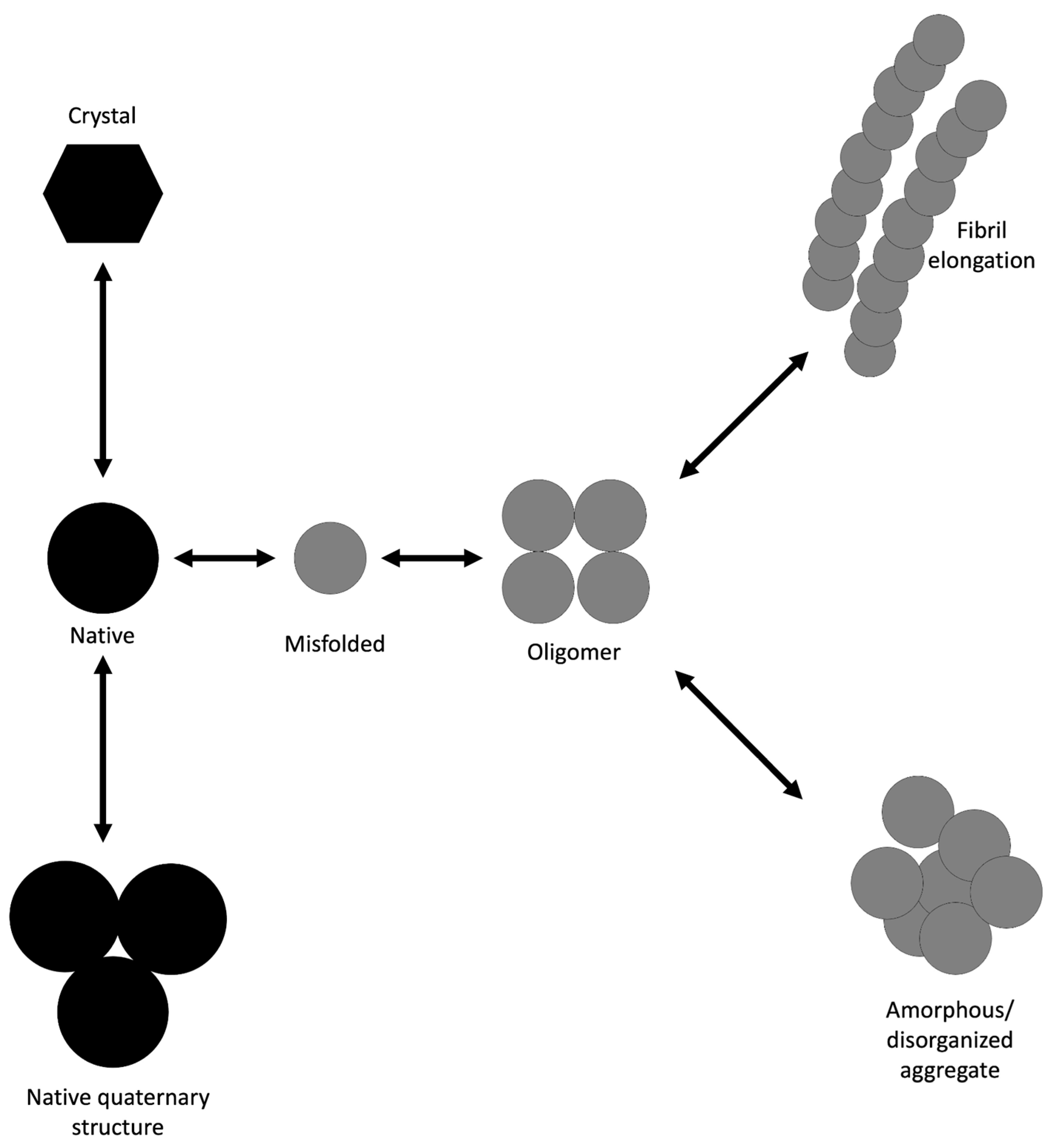
Figure 4.
Structural diversity of human Aβ filaments in different pathological conditions. The panel shows representative 3D structures of Aβ extracted from human brain tissue in AD, cerebral amyloid angiopathy, and Down syndrome with AD. These cryo-EM structures reveal distinct filament conformations and polymorphs associated with disease-specific pathology. Data from Kollmer et al. [75], Yang et al. [76], and Fernandez et al. [77]. Image created using RCSB PDB Mol 3D Viewer by Sehnal et al. [78].
Figure 4.
Structural diversity of human Aβ filaments in different pathological conditions. The panel shows representative 3D structures of Aβ extracted from human brain tissue in AD, cerebral amyloid angiopathy, and Down syndrome with AD. These cryo-EM structures reveal distinct filament conformations and polymorphs associated with disease-specific pathology. Data from Kollmer et al. [75], Yang et al. [76], and Fernandez et al. [77]. Image created using RCSB PDB Mol 3D Viewer by Sehnal et al. [78].
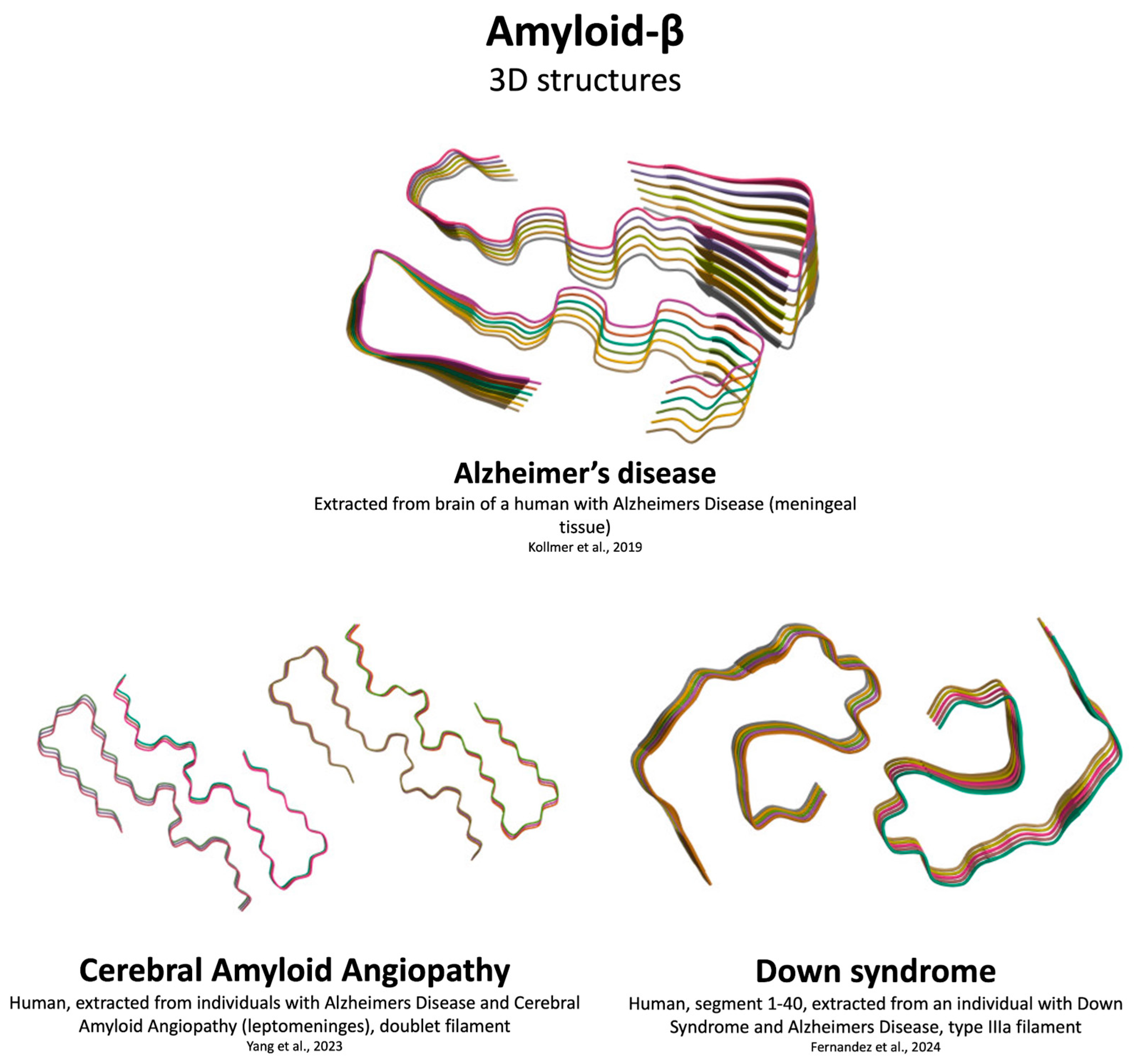
Figure 5.
Structural diversity of human αSyn in different states and disease conditions. The panel shows representative 3D structures of αSyn in its recombinant form and aggregated states associated with neurodegenerative disorders. Structures were obtained from cryo-EM studies and illustrate distinct fibril conformations and polymorphs linked to disease-specific pathology. Data from Li et al. [100], Sokratian et al. [102], Fan et al. [99], and Yan et al. [101]. Image created using RCSB PDB Mol 3D Viewer by Sehnal et al. [78].
Figure 5.
Structural diversity of human αSyn in different states and disease conditions. The panel shows representative 3D structures of αSyn in its recombinant form and aggregated states associated with neurodegenerative disorders. Structures were obtained from cryo-EM studies and illustrate distinct fibril conformations and polymorphs linked to disease-specific pathology. Data from Li et al. [100], Sokratian et al. [102], Fan et al. [99], and Yan et al. [101]. Image created using RCSB PDB Mol 3D Viewer by Sehnal et al. [78].
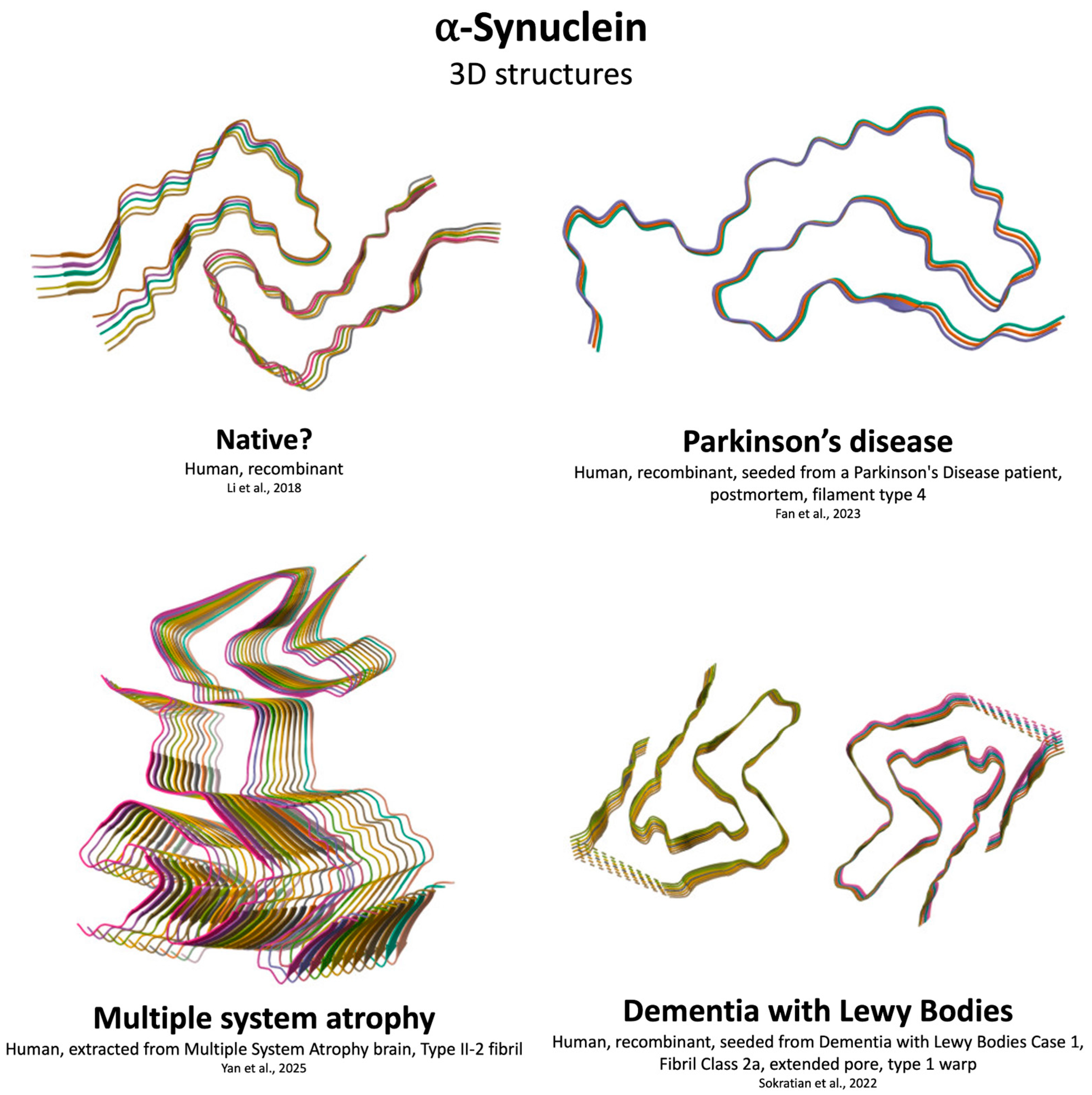
Figure 6.
Structural diversity of human tau protein in different pathological conditions. The panel shows representative 3D structures of tau filaments extracted from human brain tissue. These cryo-EM structures illustrate distinct filament conformations and polymorphs as-sociated with disease-specific pathology. Data from Shi et al. [140], Fitzpatrick et al. [136], Hallinan et al. [141], and Qi et al.[142]. Image created using RCSB PDB Mol 3D Viewer by Sehnal et al. [78].
Figure 6.
Structural diversity of human tau protein in different pathological conditions. The panel shows representative 3D structures of tau filaments extracted from human brain tissue. These cryo-EM structures illustrate distinct filament conformations and polymorphs as-sociated with disease-specific pathology. Data from Shi et al. [140], Fitzpatrick et al. [136], Hallinan et al. [141], and Qi et al.[142]. Image created using RCSB PDB Mol 3D Viewer by Sehnal et al. [78].
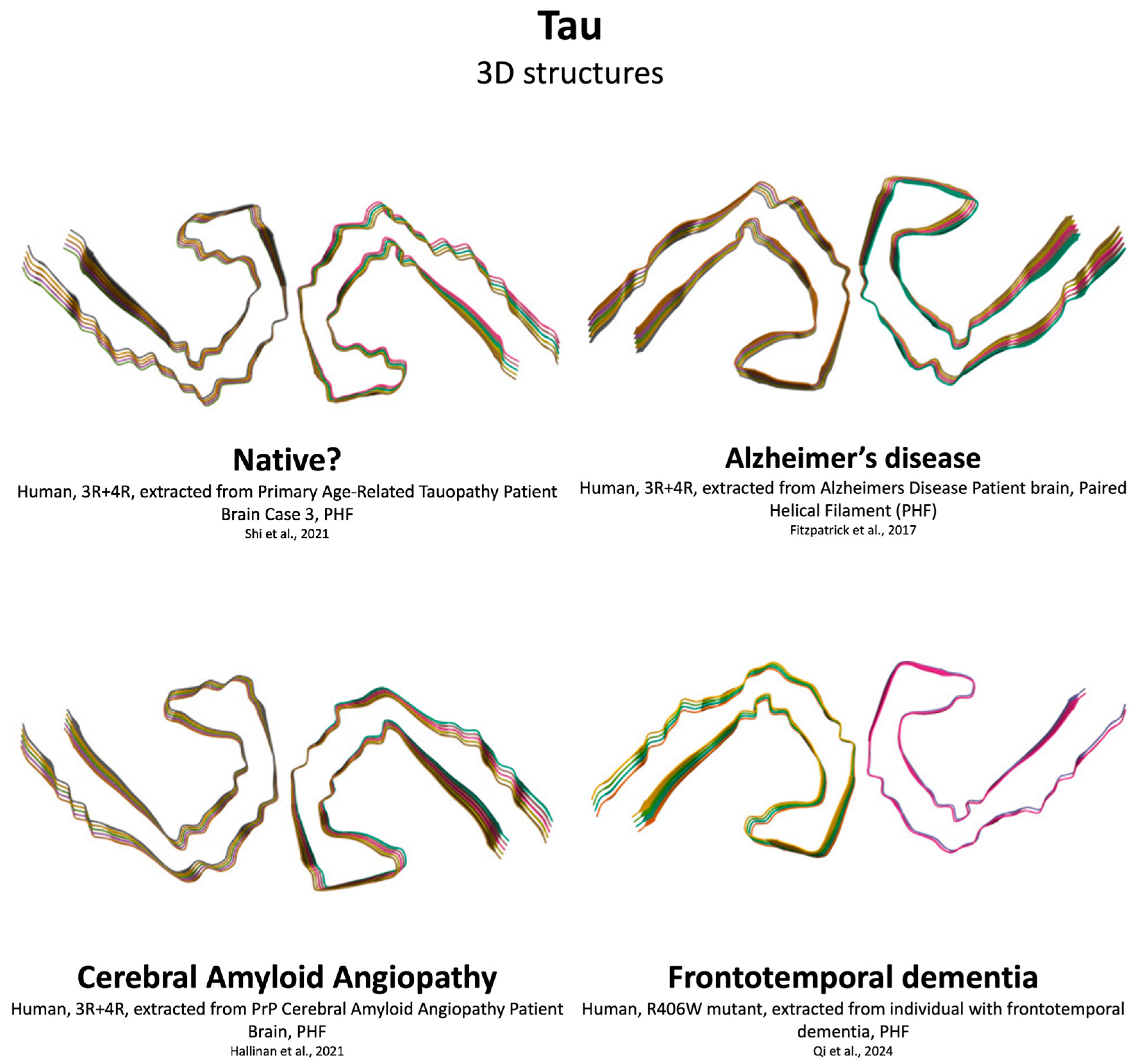
Figure 7.
Structural diversity of human TDP-43 filaments in different conditions. The panel shows representative 3D structures of TDP-43 in a synthetic fragment and aggregated states associated with neurodegenerative disorders. These cryo-EM structures reveal distinct filament conformations and polymorphs linked to disease-specific pathology. Data from Guenther et al. [158], Arseni et al. [157], and Arseni et al. [159]. Image created using RCSB PDB Mol 3D Viewer by Sehnal et al. [78].
Figure 7.
Structural diversity of human TDP-43 filaments in different conditions. The panel shows representative 3D structures of TDP-43 in a synthetic fragment and aggregated states associated with neurodegenerative disorders. These cryo-EM structures reveal distinct filament conformations and polymorphs linked to disease-specific pathology. Data from Guenther et al. [158], Arseni et al. [157], and Arseni et al. [159]. Image created using RCSB PDB Mol 3D Viewer by Sehnal et al. [78].
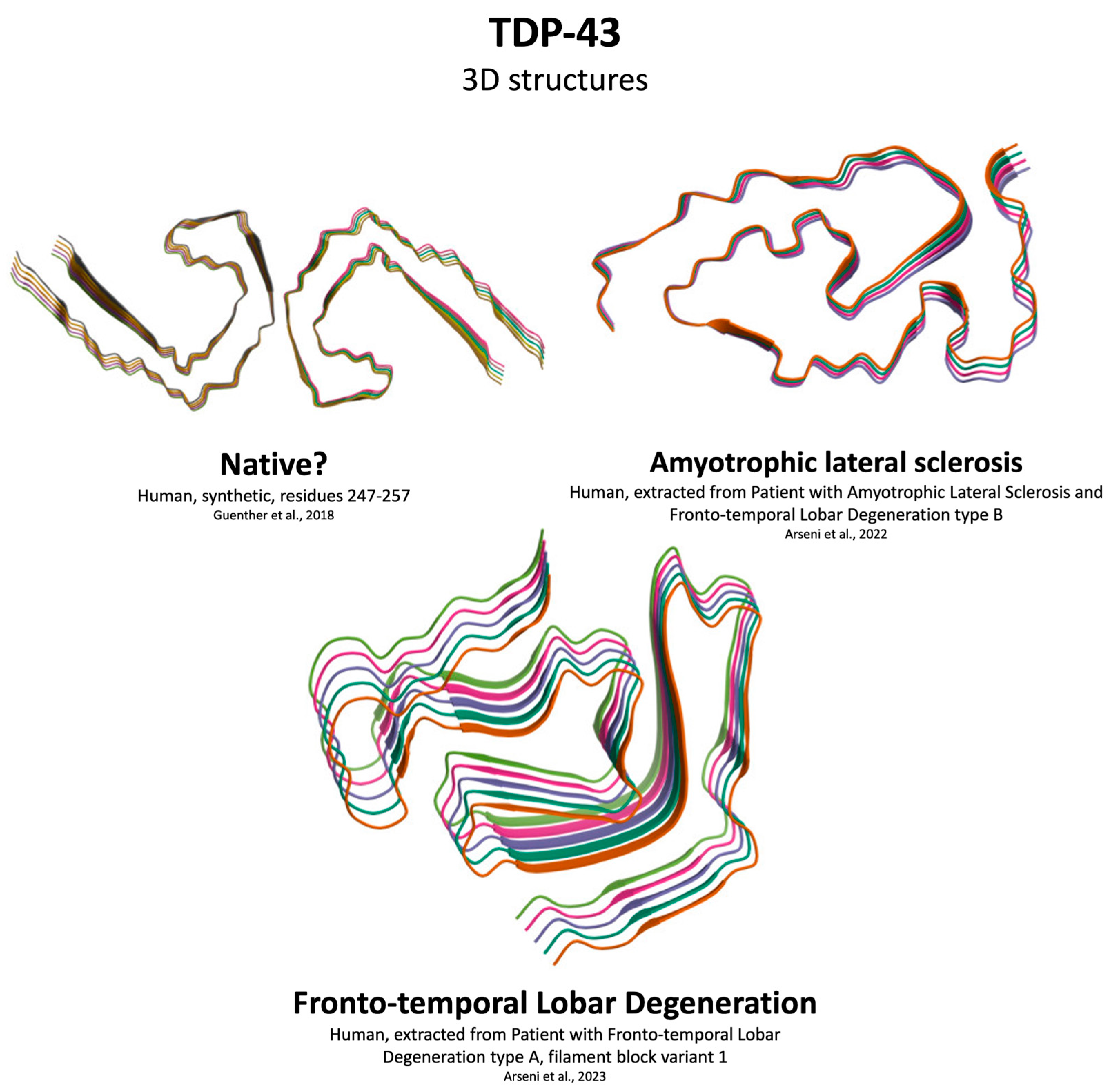

Table 1.
Comparative Structural and Mechanistic Features of Aβ, αSyn, Tau, and TDP-43 Aggregation.
| Feature | Aβ | αSyn | Tau | TDP-43 |
| Primary aggregation domain | Full-length (Aβ40/42), hydrophobic core (residues 17–21, 30–42) | NAC region (61–95) critical for fibrillization | Microtubule-binding repeats (R2, R3; VQIINK, VQIVYK motifs) | Low-complexity domain (LCD; residues 282–360) |
| Architecture | Canonical cross-β fibrils; cryo-EM shows S-shaped, ν-shaped polymorphs | Cross-β fibrils; multiple polymorphs (MSA vs PD) | Paired helical filaments (PHFs) and straight filaments; disease-specific folds | Amyloid-like but non-classical cross-β; short β-strands, spiral folds |
| Nucleation mechanism | Primary nucleation + secondary nucleation dominates oligomer formation | Primary nucleation; secondary nucleation accelerates oligomer growth | Heparin-induced nucleation in vitro; secondary nucleation less defined | LLPS precedes solidification; oligomeric seeds nucleate fibrils |
| Polymorphism | High; Aβ40 vs Aβ42 differ; multiple brain-derived folds | High; strain-specific folds linked to disease phenotype | High; AD vs PiD vs CBD folds differ by isoform composition (3R vs 4R) | Present; LCD truncations yield distinct fibril cores |
| Cellular localization | Extracellular plaques (parenchyma, vasculature) | Cytoplasmic inclusions (LBs, neurites) | Cytoplasmic neurofibrillary tangles; axonal threads | Cytoplasmic inclusions; nuclear clearance |
| PTMs influencing aggregation | N-terminal truncation, oxidation | Phosphorylation, nitration | Hyperphosphorylation, acetylation | Phosphorylation, ubiquitination, SUMOylation, citrullination |
| Propagation | Prion-like seeding; extracellular spread | Prion-like; vesicle-mediated transmission | Prion-like; templated misfolding | Prion-like; LLPS-driven seeding and spread |
| Toxic species | Soluble oligomers (membrane pore formation) | Oligomers; disrupt vesicle trafficking | Oligomers; impair microtubule stability | Oligomers; RNA sequestration, splicing defects |
| Abbreviations: Aβ, amyloid-beta; AD, Alzheimer’s disease; CAA, cerebral amyloid angiopathy; EM, electron microscopy; FTLD, frontotemporal lobar degeneration; LCD, low-complexity domain; LLPS, liquid–liquid phase separation; MSA, multiple system atrophy; PHF, paired helical filament; PTM, post-translational modification; SSNMR, solid-state nuclear magnetic resonance. | ||||
Table 2.
Therapeutic Strategies Targeting Protein Aggregation.
| Strategy | Mechanism of action | Representative agents | Current Status / Limitations |
| Passive Immunotherapy | Monoclonal antibodies bind aggregated αSyn to block propagation and promote clearance | Prasinezumab (C-terminal epitope), Cinpanemab (N-terminal epitope), MEDI1341/TAK-341, LuAF82422, Exidavemab | Phase II trials; limited efficacy, poor BBB penetration |
| Active Immunotherapy | Vaccines induce endogenous antibodies against aggregated αSyn | UB-312, PD01A, PD03A | Phase I–II; risk of neuroinflammation, variable immunogenicity |
| Small Molecule Aggregation Inhibitors | Block oligomerization or fibril elongation | Anle138b, NPT200-11, PBT434 | Phase I–II; off-target effects, CNS penetration issues |
| Natural Compounds | Polyphenols disrupt hydrophobic interactions and reduce oxidative stress | EGCG, Curcumin, Squalamine/ENT-01 | Preclinical/Phase II; poor bioavailability |
| Peptide-Based Inhibitors | Target NAC domain or fibril ends to prevent nucleation and elongation | K84s, TO2, TO5, macrocyclic peptides (BD1), poly-arginine peptides (R18D) | Preclinical; short half-life, BBB limitations |
| Proteolysis-Targeting Chimeras (PROTACs) | Recruit E3 ligases to ubiquitinate αSyn for proteasomal degradation | Arginine-based PROTACs | Preclinical; limited aggregate clearance, BBB penetration |
| Autophagy-Targeting Chimeras (AUTOTACs) | Redirect aggregates to autophagosomes for lysosomal degradation | ATC161 | Phase I; BBB penetration remains a challenge |
| Autophagy Activators | Enhance autophagic flux to clear aggregates | Trehalose, Metformin, Rapamycin, Lithium | Phase I–II; systemic toxicity, non-selective degradation |
| Proteasome Activators | Stimulate proteasomal activity to degrade misfolded proteins | Roflumilast, modified tetramethylpyrazine | Preclinical; insufficient potency, poor CNS penetration |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



