
Submitted:
04 December 2025
Posted:
05 December 2025
You are already at the latest version
Abstract
Fraser Syndrome (FS) is an extremely rare genetic disorder with a strong pattern of inheritability that follows the autosomal recessive fashion; the fundamental clinical features include congenital anomalies such as cryptophthalmos, syndactyly, as well as, renal damage. The FRAS1 gene is one of the principal genes implicated in FS. The role of genetic mutations in the development of FS has not been comprehensively elucidated for the present, and novel mutations are still being identified. Hence, the current article addresses two patients who were clinically diagnosed with Fraser Syndrome and included two unique mutations (c.7777C˃T; (p. Q2593X) and c.9821G˃C; (p. R3274P) that are found in the FRAS1 gene. Both patients had clinical features concerning Fraser Syndrome including renal abnormalities and cryptophthalmos, which addresses the severe phenotype associated with FRAS1 mutations. These two novel mutations discovered have expanded the genetic heterogeneity of Fraser Syndrome and have extended the mutational list of the FRAS1 gene. These outcomes might have a profound impact on the further approaches to the therapy of FS and methods of genetic counseling and diagnosis.
Keywords:
fraser syndrome
; FRAS1 gene
; novel mutations
; cryptophthalmos
; syndactyly
1. Introduction
The Fraser condition (FS) is a rare congenital autosomal recessive condition that is marked by syndactyly, cryptophthalmos, and problems with the respiratory and urogenital tracts [1,2]. It occurs in 0.43 out of every 100,000 live births and 11.06 out of every 100,000 stillbirths [1,3]. George Fraser first identified the diagnosis in 1962 after observing two siblings who had renal agenesis, laryngeal stenosis, ambiguous genitalia, renal abnormalities, and optic deformities [4]. About 250 cases have been reported as of the present time. Usually, renal abnormalities, laryngeal deformities, or both cause newborns to die at delivery [1]. Fraser syndrome has genetic variation, so the people affected can have different expressions of the disorder and an even more stressful level of intensity [5]. Since the specific features of Fraser syndrome can differ in each patient and throughout his or her development, diagnosis and treatment of the disease may be complicated [6]. The extracellular matrix proteins required for adhesion between the basement membrane of the epidermis and the connective tissues of the dermic layer during embryonic development are encoded by the genes FRAS1, FREM1, FREM2, and GRIP1 [7].
1.1. Aims and Objectives
The present case reports aim at raising awareness of further cases of Fraser syndrome and adding to the data on the identification of new mutations in the FRAS1 gene. To shed more light on people’s genetics, thus offering a better understanding of the phenotypic heterogeneity associated with FRAS1 mutations, we report two cases here. These reports are useful for clinicians and genetic counselors as they educate them on the significance of genetic testing as an approach to making an accurate diagnosis of FS, which is usually complex due to its heterogeneity. In addition, the study offers an understanding of the pathways involved with this disorder and, therefore, informs potential future studies, as well as intervention approaches. Finally, increasing awareness about these new mutations can contribute to the diagnosis and better management of patients with those findings, thus increasing the quality of care and prognosis.
1.1.1. Importance of FRAS1 Gene in Fraser Syndrome
Studies have explored the impact of Fraser syndrome a rare, congenital disease associated with a definite pattern of dysmorphia on the FRAS1 gene [1]. This gene is involved in the function of a protein that is critical in the development of the epithelial layers and maintenance of the tissue architecture, especially in embryonic development [8]. FRAS1 mutations interfere with the normal function of adhesion-related tissues and signal transduction between cells, which results in several symptoms of Fraser syndrome-like ocular dermoid, finger and toe fusions, and urogenital abnormalities [9]. A basic role of the FRAS1 protein is to provide structural support to molecules that make up the extracellular matrix of tissues through binding with other components [10]. Mutation in this gene leads to crucial developmental problems, and this means that the gene is crucial in regulating cell interactions vital for the formation of an organ [11]. Furthermore, clinical findings in individuals with Fraser syndrome would also indicate that mutations in FRAS1 can present with a wide range of phenotypic expression, thereby complicating clinical classification as well as diagnosis and management.
Moreover, the role of the FRAS1 gene in diagnosis also means acknowledging the role of genetic counseling and explaining to families the patterns of inheritance and recurrence risks [12]. The Fraser syndrome is a rare genetic disorder, and a genetic test for the FRAS1 mutation is vital for early diagnosis of the various complications since management can be immediate [1]. However, continued preclinical work to further understand the molecular function of FRAS1 could offer suggestive therapeutic targets, improving hope for improved clinical intervention for affected people with this complicated syndrome [13]. To sum up, the FRAS1 gene not only plays a crucial role in the genetic basis of Fraser syndrome but also offers hope in the argument for broadening the knowledge of congenital diseases and promoting the betterment of the patient’s condition.
2. Materials and Methods
2.1. Case.01
2.1.1. History of Patient
A 32-year-old patient (G:6 P:5 A:0 L:1) in the 31st week of pregnancy, colliding with the date of her last menstruation, was referred to our hospital from an external center with a diagnosis of severe oligohydramnios and intrauterine growth retardation. Her ultrasound revealed fetal measurements consistent with 26 weeks of gestation, along with monitoring of fetal cardiac activity and observation of severe oligohydramnios. The bilateral kidneys could not be monitored (bilateral renal agenesis), and the lungs were observed to be congested. A detailed fetal examination could not be performed due to oligohydramnios. The patient was admitted for a Caesarean section with evidence of a previous Caesarean section and a breech presentation after the onset of labor. A male newborn weighing 920 grams and measuring 31 cm with an Apgar score of ½ was delivered by breech cesarean section. The newborn suffered from various abnormalities and spontaneous breathing and was subjected to positive-pressure ventilation. When his breathing could not be started, an attempt was made to intubate the newborn, but the intubation was not possible due to his larynx being too narrow. The newborn died of respiratory failure in the 30th postnatal minute.
2.1.2. Diagnosis
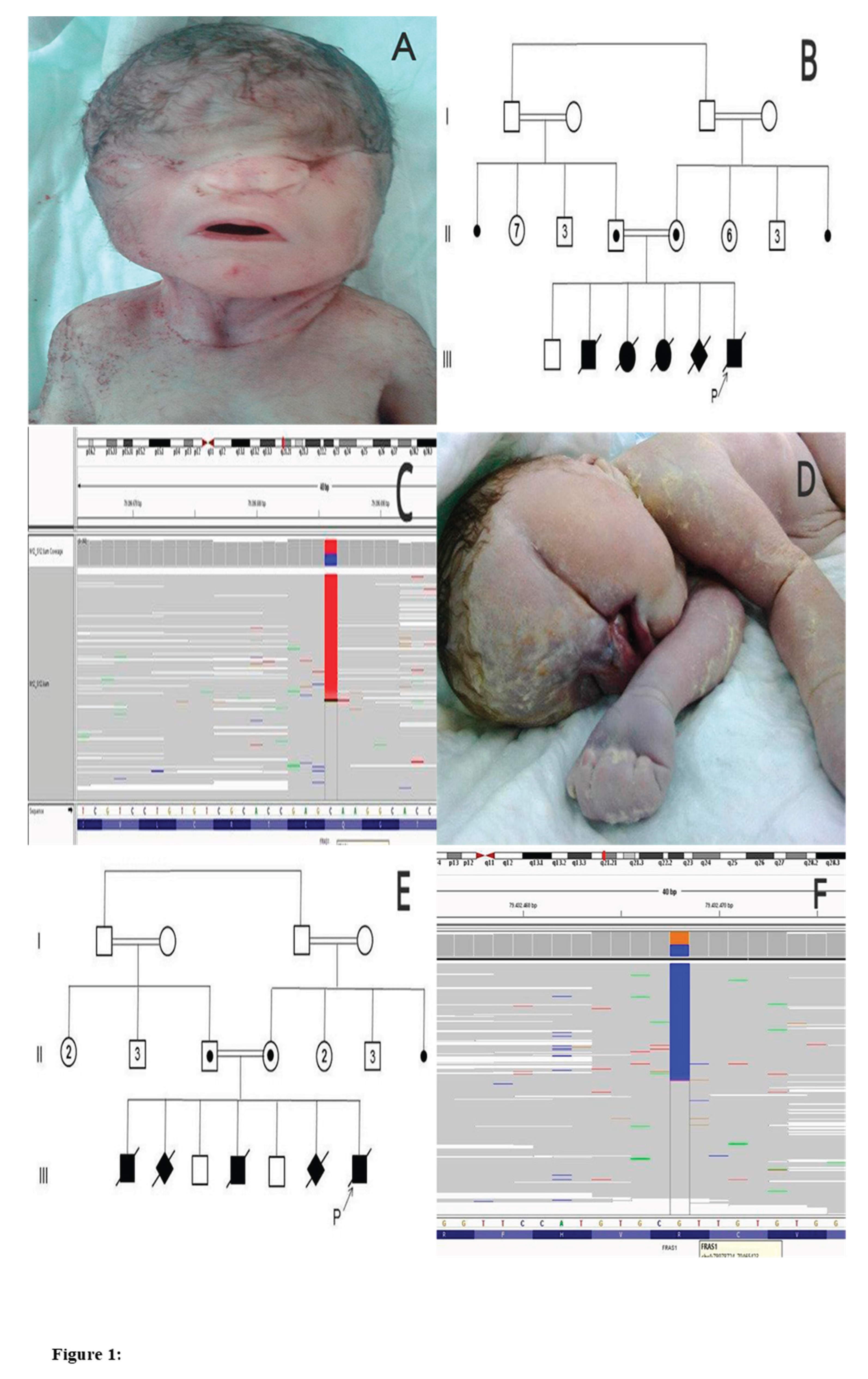
Postmortem examination led to the observation of bilateral cryptophthalmos, low hairline, depressed nasal root, nasal hypoplasia, low ears, turbinate malformation, microstomia, narrow chest, cutaneous syndactyly in the left hand, and umbilical hernia, as shown in Figure 1A.
Karyotype analysis revealed 46XY. The case was diagnosed with Fraser syndrome based on clinical findings. A blood sample was taken from the sample for further investigations into gene mutations, which again could not be carried out due to technical deficiencies. Information about the parents suggested that they were relatives (first cousins), and the mother’s birth history indicated that she had given birth to a healthy male child in her first pregnancy. In her four consecutive pregnancies, one year apart from the first pregnancy, there has been a history of infant deaths with similar abnormalities, as shown in Figure 1B.
The family had not received genetic counseling during or after their previous pregnancies. After birth, blood samples were collected from parents for prospective gene mutation studies. DNA was obtained from blood samples by standard procedures. Therefore, all 75 protein-coding exons and splice sites of the FRAS1 gene were sequenced using the next-generation sequencing method on the Illumina Miseq platform. Both results reported a heterozygous premature stop codon mutation c.7777C˃T (p. Q2593X) in exon 54 of the FRAS-1 gene. This mutation has been classified as pathogenic by various online prediction tools (Mutation-Taster). The mutation identified in the gene mutation study thus conducted was reported to be a novel mutation that had not been previously described, as shown in Figure 1C. HGMD was used to describe the mutations as novel. The family was transferred to the genetics department of our hospital and received genetic counseling for Fraser syndrome.
2.2. Case. 02
2.2.1. History of Patient
A 28-year-old patient (G:7 P:6 A:0 L:2) in the 40th week of pregnancy, matching the date of her last menstrual period, presented to our clinic with the onset of labor pains. The patient had not undergone previous prenatal follow-up, and her ultrasound failed to observe the fetal heartbeat and had severe oligohydramnios. The bilateral kidneys had a hypoechoic appearance (renal cystic dysplasia), and the lungs were hyperdense. The patient gave birth vaginally to a male newborn weighing 3700 g and 51 cm with an Apgar score of 0. Postnatal autopsy of the newborn revealed unilaterally incomplete cryptophthalmos, low and dysmorphic ears, cleft lip and palate, skin tip syndactyly in fingers and toes, bilateral cryptorchidism, and imperforate anus, as shown in Figure 1D. The case was diagnosed with Fraser syndrome based on clinical findings. The couple (first cousins) were in a blood marriage. The obstetric history showed that the couple had two healthy and live male children and experienced the birth and postnatal deaths of infants with similar anomalies in four additional pregnancies, as shown in Figure 1E.
2.2.2. Diagnosis
The family, which had not previously received genetic counseling, provided blood samples for further research into gene mutations. All 75 coding exons and splice sites of the FRAS1 gene were sequenced using the next-generation sequencing method on the Illumina Miseq platform. The results of the gene mutation study were reported as a heterozygous missense mutation c.9821G˃C (p. R3274P) in exon 64 of the FRAS1 gene for both parents. This mutation has been classified as pathogenic by various online prediction tools (Mutation-Taster and PolyPhen-2). In addition, the report said that the mutation so identified was a novel mutation that had not been previously described. HGMD was used to describe the mutations as novel. The family received genetic counseling for Fraser syndrome in the genetics department of our hospital, as shown in Figure 1F.
3. Discussion
In this report two unusual cases of Fraser syndrome, an autosomal recessive genetic disorder characterized by birth defects such as cryptophthalmos, syndactyly and multiple craniofacial anomalies are presented. In both patients, the FRAS1 gene that encodes the main protein associated with Fraser syndrome was sequenced and novel pathogenic mutations previously absent in literature were identified. As incidents between 0.043 and 0.11 per 100,000 live births have been estimated for Fraser syndrome yearly, the disorder is considered to be exceptionally rare among people [3]. This condition arises from biallelic mutations at genes crucial for the development of extracellular matrix proteins and is mostly connected with FRAS1 [14]. So far as over a hundred distinct pathogenic variants have been described in literature affecting FRAS1 gene and causing Frasier syndrome [1]. The identification of new mutations further supports the genetic heterogeneity of this uncommon disease highlighting the importance of extensive familial genetic screening.
In the first case, Fraser syndrome was identified in the patient who had many congenital abnormalities including craniofacial abnormalities, syndactylism and bilateral cryptophthalmos. Sanger sequencing analysis showed homozygous mutation c.7777C>T (p.Q2593X) in the exon 54 of the FRAS1 gene. A change of codon causes the premature stop mutation that leads to the formation of non- functional fras1 protein hence disturbing ECM cell deposition and organ growth [15]. The parents were first-degree cousins, thus giving the autosomal recessive pattern of Fraser syndrome. The second case was of a fetus that was postnatally suspected of having Fraser syndrome and was found to have many characteristics, such as unilateral cryptophthalmos, cleft lip/palate, and syndactyly. The molecular study was performed on this patient and a new heterozygous missense mutation c. 9821G>C (p. R3274P) in exon 64 of FRAS1 was identified.
According to various in silico pathogenicity prediction tools, this amino acid substitution is expected to reduce protein function [16]. Notably, both parents were also reported to be heterozygous for this variant, which is also in line with the recessive nature of Fraser syndrome. Thus, the reported identification of new, until now unknown FRAS1 mutations can be considered for expanding the known list of Fraser syndrome mutations . Awareness of all these pathogenic variants is necessary for several reasons, as will be discussed below. First, it leads to a better definition of this disease on a genetic level, which is crucial for the clinical approach to the patient as well as for successful prenatal screening and further family planning [17]. Second, as mentioned above, detailed characterization of newly revealed variants may help in understanding the molecular nature of the FRAS1 protein and the pathways that regulate normal growth and development [18]. The results of such research may one day contribute to the creation of individualized treatment for Fraser syndrome.
Both families in the present report had a history of recurrent pregnancy losses, and still-born infants with the presented congenital anomalies. This makes it imperative that couples, especially in consanguineous marriages, undergo genetic counseling and tests to reduce the effects of autosomal recessive disorders such as Fraser syndrome [19]. However, the families had never had any previous genetic counseling, making it evident that there is a dire desire for people to seek genetic services, especially in areas where most people still practice consanguinity [20]. Therefore, Fraser syndrome can present with a wide clinical spectrum, from simple isolated cryptophthalmos to severe multiorgan involvement and potentially lethal outcomes [21]. Herein, the newborns displayed a range of dysmorphic features, including craniofacial abnormalities of different severity, hand malformations such as syndactyly, and multiple urogenital system anomalies [22]. These findings are under the phenotypic genotype mapping of Fraser syndrome, which could affect eyes, ears, nose, mouth, limbs, kidneys, and genitalia [8]. The variability of the symptoms manifesting in Fraser syndrome may be complex and sometimes even misleading, especially when there are only certain vague symptoms [5]. In such circumstances, it is very helpful in management when a confirmatory test, such as genetic testing, has been done [23]. In the present cases, the identification of pathogenic variants in the FRAS1 gene offers a distinct molecular diagnosis, direct genetic counseling, and an estimation of risk during the next pregnancies.
Furthermore, as highlighted in the present report, both families in this study had a history of repeated abortions and stillbirths with similar congenital problems [24]. It therefore stresses the need for counseling and testing, especially in consanguineous marriages that are hereditarily endowed with autosomal recessive conditions like the Fraser syndrome [25]. However, these families reported no prior genetic counseling, pointing to the dearth of genetic awareness and the availability of such consulting in those areas where consanguinity is more prevalent [26]. Similarly, in our present work, we have identified new FRAS1 mutations and therefore added new data on the genetics of Fraser syndrome. The FRAS1 gene encodes a large extracellular matrix protein that is involved in embryonic development as well as in the formation of the basement membrane [27]. Loss of function of FRAS1, as exemplified by the present two heterozygous variants, can cause the spectrum of congenital anomalies associated with Fraser syndrome [28]. It can be assumed that the identification of new FRAS1 mutations might have implications for future therapeutic management, not only for refining diagnostic capacities [29]. Given the current study of the underlying molecular mechanisms that define Fraser syndrome, there could be the development of molecular therapies or gene therapies [4]. Yet, the existence of such a disease and the availability of affected people are quite rare, which is a major problem for the development of such approaches.
Furthermore, when the causative genes for Fraser syndrome become much clearer, disease-targeting drugs or gene therapies might be produced [30]. It is never easy to design proper therapy because of the disease’s dynamics and effects on many systems; epidemiology requirements also enter [2]. At present, studies are necessary on potential management approaches in Fraser syndrome with the purpose of enhancing the outcome and existence of affected individuals with this uncommon disease [2]. Joint work of geneticists, developmental biologists, and clinicians is necessary for enhancing the understanding of the illness and for searching for the cure [1]. However, Fraser syndrome is not among the many genetic syndromes that can be managed today or which development of future genetics can assist in managing the syndrome.
4. Conclusion
The discovery of new mutations of FRAS1 is expanding the understanding of Fraser syndrome, an autosomal recessive disorder characterized by complex malformations at birth [28]. Amongst the known genes, FRAS1 is crucial for consistent embryonic development particularly the formation of the basement membrane and a major extracellular matrix protein [31]. These anomalies can be produced due to disruption of FRAS1 function and it has been seen in the two new variations described in the following study. Such mutations might improve diagnostics alongside providing potential therapeutic gene or gene-targeted spots [32]. However, the development of such treatments is severely frustrating due to the rarity of Fraser syndrome and the corresponding number of affected patients [1]. Frequent genetic testing along with proper counseling constitute the focus of the paper especially on the populations at higher risk for autosomal recessive diseases.
Funding
This research received no external funding.
Informed Consent Statement
Written informed consent has been obtained from the patients to publish this paper.
Acknowledgments
We thank Dr. Salih Coşkun for his contributions to our study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bouaoud J, Olivetto M, Testelin S, Dakpé S, Bettoni J, Devauchelle B. Fraser syndrome: review of the literature illustrated by a historical adult case. International Journal of Oral and Maxillofacial Surgery. 2020, 49(10), 1245-1253.
- Das D, Modaboyina S, Raj S, Agrawal S, Bajaj MS. Clinical features and orbital anomalies in Fraser syndrome and a review of management options. Indian Journal of Ophthalmology. 2022, 70(7), 2559-2563.
- Neri ID, Vela APF, Mendoza RLA, Roa RG, Vizcaino GCR. Prenatal hydrometrocolpos as an unusual finding in Fraser syndrome. Case report. Case Reports in Perinatal Medicine. 2023, 12(1). [CrossRef]
- Laminou L, Habou O, Amadou M, Hadjia A K Y, Abdou A. Fraser syndrome: About A Case and Review of the Literature. Journal of Surgery and Research. 2022, 5(4), 585-587.
- Sajoura C, Ech-Chebab M, Ayyad A, Messaoudi S, Amrani R. Fraser Syndrome: A Case Report. Open Journal of Pediatrics. 2024, 14(3), 476-481.
- Felton A. What Is Fraser Syndrome? 2022, WebMD. https://www.webmd.com/ children/what-is-fraser-syndrome.
- Esho T, Kobbe B, Tufa SF, Keene DR, Paulsson M, Wagener R. The Fraser Complex Proteins (Frem1, Frem2, and Fras1) Can Form Anchoring Cords in the Absence of AMACO at the Dermal–Epidermal Junction of Mouse Skin. International Journal of Molecular Sciences. 2023, 24(7), 6782.
- Ikeda S, Akamatsu C, Ijuin A, Nagashima A, Sasaki M, Mochizuki A, Nagase H, Enomoto Y, Kuroda Y, Kurosawa K. Prenatal diagnosis of Fraser syndrome caused by novel variants of FREM2. Human genome variation. 2020, 7(1), 32.
- Arcot Sadagopan K. Genetics in Oculoplastics. Smith and Nesi’s Ophthalmic Plastic and Reconstructive Surgery. 2021, 1115-1143.
- Wang G, Wang Z, Lu H, Zhao Z, Guo L, Kong F, Wang A, Zhao S. Comprehensive analysis of FRAS1/FREM family as potential biomarkers and therapeutic targets in renal clear cell carcinoma. Frontiers in Pharmacology. 2022, 13, 972934.
- Ou T-Y, Tsai M-C, Kuo P-L, Lee N-C, Chou Y-Y. Whole exome sequencing identifies a novel FRAS1 mutation and aids in vitro fertilization with preimplantation genetic diagnosis in Fraser syndrome. Taiwanese Journal of Obstetrics and Gynecology. 2022, 61(3), 521-524.
- Tsuji Y, Yamamura T, Horinouchi T, Sakakibara N, Ishiko S, Aoto Y, Rossanti R, Okada E, Tanaka E, Tsugawa K. Systematic review of genotype- phenotype correlations in Frasier syndrome. Kidney International Reports. 2021, 6(10), 2585-2593.
- Chańska W. The principle of nondirectiveness in genetic counseling. Different meanings and various postulates of normative nature. Medicine, Health Care and Philosophy. 2022, 25(3), 383-393.
- Landau-Prat D, Kim DH, Bautista S, Strong A, Revere KE, Katowitz WR, Katowitz JA. Cryptophthalmos: associated syndromes and genetic disorders. Ophthalmic Genetics. 2023, 44(6), 547-552.
- Murali S, Almahmoudi FH, Traboulsi EI. SECTION IV Systematic Pediatric Ophthalmology. Taylor and Hoyt’s Pediatric Ophthalmology and Strabismus. 2022, 179.
- Skalniak A, Trofimiuk-Müldner M, Surmiak M, Totoń-Żurańska J, Jabrocka-Hybel A, Hubalewska-Dydejczyk A. Whole-exome screening and analysis of signaling pathways in multiple endocrine neoplasia type 1 patients with different outcomes: insights into cellular mechanisms and possible functional implications. International Journal of Molecular Sciences. 2024, 25(2), 1065.
- Kumaran K, Abirami S, Ajeesh A, Hemarangan J, Vasanth Kanth T, Shriya P, Aruljothi K. Prenatal Screening and Counseling for Rare Genetic Disorders. In Rare Genetic Disorders: Advancements in Diagnosis and Treatment. 2024, (pp.61-76). Springer.
- Lamandé S R, Bateman JF. Genetic disorders of the extracellular matrix. The anatomical record. 2020, 303(6), 1527-1542.
- Sahin E, İnciser Paşalak Ş, Seven M. Consanguineous marriage and its effect on reproductive behavior and uptake of prenatal screening. Journal of Genetic Counseling. 2020, 29(5), 849-856.
- Camats N, Flück CE, Audí L. Oligogenic origin of differences of sex development in humans. International Journal of Molecular Sciences. 2020, 21(5), 1809.
- Koprulu M, Kumare A, Bibi A, Malik S, Tolun A. The first adolescent case of Fraser syndrome 3, with a novel nonsense variant in GRIP1. American Journal of Medical Genetics Part A. 2021,185(6), 1858-1863.
- Shrestha S, Thani KP, Keshari M, Shrestha A. Fraser Syndrome: A Rare Case Report. Journal of Karnali Academy of Health Sciences. 2022, 5(3).
- Fortin O, Mulkey SB, Fraser JL. Advancing fetal diagnosis and prognostication using comprehensive prenatal phenotyping and genetic testing. Pediatric Research. 2024, 1-11.
- Ryan CE, Schust DJ. Recurrent pregnancy loss. Clinical Maternal-Fetal Medicine, 2021, 4.1-4.13.
- Elmugadam FM, Ahmed H, Karamelghani M, Ali A, Ali I, Ahmed A, Salman M, Mohamed W, Ahmed EA, Ahmed KAHM. Awareness of consanguineous marriage burden and willingness towards premarital genetic testing in Sudan: a national cross-sectional study. Annals of Medicine and Surgery. 2024, 86(7), 3959-3971.
- Provenzano A, Palazzo V, Reho P, Pagliazzi A, Marozza A, Farina A, Zuffardi O, Giglio S. Noninvasive prenatal diagnosis in a family at risk for Fraser syndrome. Prenat Diagn. 2020, 40(7): 905-908.
- Zhu L, Shen S, Pan C, Lan X, Li J. Bovine FRAS1: mRNA Expression Profile, Genetic Variations, and Significant Correlations with Ovarian Morphological Traits, Mature Mature Follicle, and Corpus Luteum. Animals (Basel). 2024, 14(4):597. [CrossRef]
- Maksiutenko EM, Barbitoff YA, Nasykhova YA, Pachuliia OV, Lazareva TE, Bespalova ON, Glotov AS. The Landscape of Point Mutations in Human Protein Coding Genes Leading to Pregnancy Loss. International Journal of Molecular Sciences. 2023, 24(24), 17572.
- Al-Hamed MH, Sayer JA, Alsahan N, Tulbah M, Kurdi W, Ambusaidi Q, Ali W, Imtiaz F. Novel loss of function variants in FRAS1 AND FREM2 underlie renal agenesis in consanguineous families. Journal of nephrology. 2021, 34, 893-900.
- Kunz F, Kayserili H, Midro A, de Silva D, Basnayake S, Güven Y, Borys J, Schanze D, Stellzig-Eisenhauer A, Bloch-Zupan A. Characteristic dental pattern with hypodontia and short roots in Fraser syndrome. American Journal of Medical Genetics Part A. 2020, 182(7), 1681-1689.
- Borgio JF. Heterogeneity in biomarkers, mitogenome and genetic disorders of the Arab population with special emphasis on large-scale whole-exome sequencing. Archives of Medical Science: AMS. 2023, 19(3), 765.
- Garrison Jr LP, Lo AW, Finkel RS, Deverka PA. A review of economic issues for gene-targeted therapies: Value, affordability, and access. Am J Med Genet C Semin Med Genet. 2023, 193(1):64-76.
Figure 1.
A: Postnatal appearance of the first case. B: Pedigree of the family. FRAS1 gene was fist sequenced in parents. Unaffected couple were heterozygous for c.7777C>T mutation (p.Q2593X). C: Result of the FRAS1 gene sequencing encompassing the c.7777C>T (exon 54). The single nucleotide substitution in the FRAS1 gene is observed in the middle column in sample. At the top of the figure, the location of the sequenced region was indicated according to the genome reference consortium. The original sequence, codons and corresponding amino acids are shown at the bottom of the figure. D: Postnatal appearance of the second case. E: Pedigree of the family. FRAS1 gene was fist sequenced in parents. Unaffected couple were heterozygous forc.9821G>C mutation (p.R3274P). F: Result of the FRAS1 gene sequencing encompassing the c.9821G>C (exon 64). The single nucleotide substitution in the FRAS1gene is observed in the middle column in sample. At the top of the figure, the location of the sequenced region was indicated according to the genome reference consortium. The original sequence, codons and corresponding aminoacids are shown at the bottom of the figure.
Figure 1.
A: Postnatal appearance of the first case. B: Pedigree of the family. FRAS1 gene was fist sequenced in parents. Unaffected couple were heterozygous for c.7777C>T mutation (p.Q2593X). C: Result of the FRAS1 gene sequencing encompassing the c.7777C>T (exon 54). The single nucleotide substitution in the FRAS1 gene is observed in the middle column in sample. At the top of the figure, the location of the sequenced region was indicated according to the genome reference consortium. The original sequence, codons and corresponding amino acids are shown at the bottom of the figure. D: Postnatal appearance of the second case. E: Pedigree of the family. FRAS1 gene was fist sequenced in parents. Unaffected couple were heterozygous forc.9821G>C mutation (p.R3274P). F: Result of the FRAS1 gene sequencing encompassing the c.9821G>C (exon 64). The single nucleotide substitution in the FRAS1gene is observed in the middle column in sample. At the top of the figure, the location of the sequenced region was indicated according to the genome reference consortium. The original sequence, codons and corresponding aminoacids are shown at the bottom of the figure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



