
Submitted:
03 December 2025
Posted:
04 December 2025
You are already at the latest version
Abstract
The development of Multi-Target-Directed Ligands (MTDLs) offers a compelling therapeutic strategy for multifactorial diseases like cancer and Alzheimer's disease (AD), which share pathological pathways, notably microtubule abnormalities. This study introduces and validates a state-of-the-art computational pipeline, the QSAR-MD-DCCM workflow, designed to accelerate the discovery of dual-acting agents targeting tubulin polymerization and acetylcholinesterase (AChE). Two highly predictive QSAR models (R2 > 0.83), built upon the trimethoxyphenyl scaffold, guided the rational design of 16 novel compounds. Subsequent ADMET screening identified compounds 15 and 16 as optimal leads, demonstrating excellent physicochemical properties and CNS penetrability. Molecular docking and rigorous 100 ns Molecular Dynamics (MD) simulations confirmed strong, persistent binding to both targets (PDB ID: 4O2B for tubulin; 1EVE for AChE), with the compounds showing complementary, target-differentiated potency. Subsequent MM-GBSA/MM-PBSA binding free energy calculations provided the essential energetic validation, confirming highly favorable binding for both leads. Crucially, Dynamic Cross-Correlation Map (DCCM) analysis provided novel mechanistic insights into the functional allosteric coupling of residues upon ligand binding, reinforcing the stability and distinct dynamic modes of action for both compounds. This integrated methodological approach successfully delivered two highly validated virtual MTDL candidates, establishing a robust and predictive platform for accelerating dual-target drug discovery.
Keywords:
Machine Learning (ML)
; Quantitative Structure-Activity Relationship (QSAR)
; Molecular Dynamics (MD)
; Dynamic Cross-Correlation Map (DCCM)
; Multi-Target-Directed Ligand (MTDL)
; tubulin
; Acetylcholinesterase (AChE)
1. Introduction
The pharmaceutical landscape is continually shifting from the “one disease, one target, one drug” paradigm toward sophisticated therapeutic strategies capable of addressing the complex, multifactorial nature of prevalent age-related disorders. This transition has been mandated by the clinical limitations of single-target agents, which often succumb to pathway redundancy, compensatory biological mechanisms, and the high failure rate associated with modulating a single node within a dense biological network. Consequently, the concept of Multi-Target-Directed Ligands (MTDLs) has emerged as a cornerstone of modern drug discovery. MTDLs, also known as polypharmacology agents, are single chemical entities designed to simultaneously and beneficially modulate two or more validated targets implicated in a disease cascade. This approach is particularly advantageous for neurodegenerative and oncological diseases, where complex, chronic pathology necessitates intervention at multiple points to achieve clinical efficacy, improve patient adherence, and potentially mitigate drug resistance [1,2,3,4,5].
The strategic importance of MTDLs is perhaps most evident in the context of global health challenges posed by cancer and Alzheimer’s disease (AD). Together, these two pathologies represent a massive public health burden, characterized by high morbidity, soaring socioeconomic costs, and a global patient population that rises exponentially with age [6,7]. While seemingly disparate, accumulating epidemiological evidence points to an intriguing and often paradoxical inverse relationship between cancer and AD incidence, suggesting shared underlying molecular foundations [8,9,10,11,12,13]. At the cellular level, both diseases converge on dysregulated processes, including chronic inflammation, oxidative stress, mitochondrial dysfunction, and, critically, abnormalities in microtubule (MT) dynamics [14,15,16,17]. This shared pathology establishes a robust rationale for designing dual-acting agents, which can exploit these common mechanisms to provide synergistic therapeutic benefits and address the complications arising from co-morbidity in elderly patients.
One of the most compelling common denominators is the microtubule system. Microtubules, dynamic polymers of α- and β-tubulin heterodimers, are essential cellular components performing distinct but vital roles in both cancer and neurodegeneration. In cancer cells, microtubules form the mitotic spindle, and inhibitors targeting tubulin polymerization are staples of chemotherapy (vinca alkaloids and taxanes). These agents destabilize or hyper-stabilize MTs, leading to mitotic arrest and apoptosis in rapidly dividing cells [18,19]. In Alzheimer’s disease, however, MTs are central to neuronal architecture, axonal transport, and synaptic integrity. Hyperphosphorylation of the microtubule-associated protein tau causes MT destabilization, leading to the collapse of the axonal network and the formation of neurofibrillary tangles, which is a hallmark of AD pathophysiology [20,21,22]. Therefore, a single MTDL capable of binding to the tubulin interface, specifically the colchicine binding site on the β-tubulin subunit offers a promising mechanism: preventing uncontrolled proliferation in cancer and protecting against cytoskeletal collapse and neural degeneration in AD [23,24]
Complementing this structural target, the acetylcholinesterase (AChE) enzyme remains the primary symptomatic target for AD. AChE inhibitors (Donepezil, Rivastigmine, etc.) function by blocking the hydrolysis of the neurotransmitter acetylcholine (ACh) in the synaptic cleft, thereby enhancing cholinergic transmission and temporarily improving cognitive function [25,26] However, these single-target agents fail to address the underlying disease progression or the MT pathology. The inclusion of an AChE inhibitory domain in a tubulin-targeting scaffold creates a synergistic MTDL: the tubulin-modulating component targets the neurodegenerative etiology, while the AChE-inhibitory component provides necessary symptomatic relief. Crucially, the structural requirements for compounds targeting both the β-tubulin colchicine site and the AChE catalytic/peripheral anionic sites often involve common aromatic and trimethoxyphenyl scaffolds [27,28]. This chemical compatibility reduces the synthetic complexity of creating a potent MTDL, making the dual-target approach both biologically and chemically sound.
The complexity inherent in designing a compound to fit two distinct binding sites and navigate the necessary pharmacokinetic barriers (such as the Blood-Brain Barrier (BBB), crucial for central nervous system targets like AChE) makes traditional high-throughput screening prohibitively costly and time-consuming. This challenge necessitates the application of advanced Computer-Aided Drug Design (CADD) methodologies, an approach central to the field of Computational Chemistry and Chemical Informatics [29,30]. Quantitative Structure-Activity Relationship (QSAR) modeling stands out as an indispensable tool, enabling the rapid and cost-effective prediction of biological activity based on key molecular descriptors. QSAR transforms empirical activity data into mathematical models that can rationally guide chemical modifications and filter large virtual libraries before synthesis [31,32]. Following QSAR-guided optimization, molecular docking provides an essential structural validation, predicting the preferred binding pose and affinity (ΔGbinding) of the MTDL for each target [33,34].
However, static docking predictions often fail to account for the essential dynamic nature of protein-ligand interactions within a physiological environment. Proteins, particularly flexible targets like tubulin and multi-domain enzymes like AChE, undergo significant conformational changes upon ligand binding. This methodological gap is bridged by Molecular Dynamics (MD) simulation, which provides a high-resolution, time-dependent view of complex stability, pose persistence, and the nature of continuous residue contacts (hydrophobic, hydrogen bonding) over hundreds of nanoseconds [35,36]. Furthermore, advanced post-MD analyses, such as the Dynamic Cross-Correlation Map (DCCM), move beyond simple stability checks (Root Mean Square Deviation, RMSD) to uncover essential allosteric or dynamic coupling effects between distant protein domains [37]. Applying this integrated QSAR-MD-DCCM pipeline ensures that predicted MTDL candidates are not only computationally potent but also dynamically stable and structurally valid under simulated physiological conditions, a level of rigor demanded by contemporary chemical informatics research.
Driven by the need for structurally validated MTDLs against the converging pathologies of cancer and AD, and leveraging the immense power of integrated computational methodologies, the present study introduces a robust QSAR-MD-DCCM workflow for the rational design and virtual validation of novel dual-acting inhibitors. Starting from a focused library of trimethoxyphenyl-containing analogues, we established two predictive QSAR models to guide the rational design of sixteen novel compounds. These compounds were subsequently subjected to extensive in silico ADMET and toxicity profiling to prioritize candidates with optimal drug-like properties and BBB permeability. The most promising candidates were then evaluated against the colchicine binding site of β-tubulin (PDB ID: 4O2B) and the active site of AChE (PDB ID: 1EVE) via molecular docking and rigorous 100 ns MD simulations, and binding free energy calculations (MM-GBSA/MM-PBSA). We conclude by presenting a detailed analysis of ligand stability, binding differentiation, and complex dynamics, using DCCM to provide unprecedented mechanistic insights into the functional allosteric consequences of MTDL binding (Figure 1).
2. Results and Discussion
2.1. Predictive Validation: QSAR Model Performance
2.1.1. Statistical Validation and Predictive Power
The systematic application of the GA coupled with Multiple Linear Regression (MLR) yielded two statistically robust QSAR models, Model 1 and Model 2, aimed at predicting the inhibitory activity (pIC50) of trimethoxyphenyl analogs against tubulin polymerization. The performance metrics (Table 1) confirmed that both models adhered strictly to the stringent OECD validation criteria, demonstrating exceptional internal consistency and external predictive capability.
The high R2 values (Model 1: 0.8319; Model 2: 0.8384) demonstrated that over 83% of the variance in the experimental pIC50 was explained by the selected descriptors. Critically, the strong cross-validation metrics, including Q2 (LOO) (>0.79) and (>0.85), confirmed the excellent internal predictability and external generalization of both models, respectively, ensuring they are not overfitted to the training data. Furthermore, the Y-randomization test results (Model 1: = 0.0866; Model 2: = 0.0861) demonstrated a clear absence of chance correlation, firmly validating the mathematical significance of the derived equations and underpinning the reliability of the predictive models.
Model 1 Equation (Topological/Electrostatic Fit):
Model 2 Equation (Polarizability/Flexibility):
2.1.2. Mechanistic Interpretation of Key Descriptors
The efficacy of the QSAR approach, a pillar of this integrated methodology, is strongly validated by the chemical relevance of the selected descriptors, which collectively characterize the optimal molecular features for inhibiting tubulin polymerization at the colchicine binding site (Table S3, SI).
of0ug (Molecular Polarizability) emerged as the most dominant determinant, featuring the highest coefficient in both models (Model 1: 34.0115; Model 2: 33.5921). This descriptor quantifies a molecule’s ability to redistribute its electron density, indicating that high electronic polarizability is essential for strong activity. This property facilitates strong van der Waals and induced dipole interactions, which are characteristic of non-covalent binding within the highly aromatic environment of the tubulin colchicine site.
map4_26 (Topological Complexity), present in both models, represents the topological complexity and molecular architecture. Its positive contribution suggests that a precise degree of molecular complexity and specific branching pattern is required for optimal recognition and fit within the constricted colchicine binding pocket.
ETA_shape_p (3D Conformational Flexibility), also highly weighted in both models (coefficient around 6.5), quantifies the responsiveness of the molecule’s three-dimensional shape to conformational changes. The strong positive correlation confirms that a certain level of molecular flexibility and shape adaptability is advantageous, enabling the ligand to optimally adjust its conformation to the dynamic structural changes that occur in β-tubulin during polymerization inhibition.
The two models are further differentiated by unique descriptors: R3s++ (Steric Accessibility) in Model 1 emphasizes that reduced steric hindrance is a prerequisite for effective binding within the spatially confined pocket. Conversely, Mor15i (Electronic Density) in Model 2, a 3D-MoRSE descriptor, reinforces the need for electronic complementarity and specific mass distribution to fine-tune the inhibitory potential.
Given its slightly superior R2 and the inclusion of descriptors emphasizing both electronic density (Mor15i) and flexibility, Model 2 was prioritized as the primary design tool. Model 1 served as an essential cross-validation tool focused on steric fit, ensuring that the subsequent designed analogs were simultaneously robust across key electronic and spatial parameters, a dual requirement that critically informed the rational structural modifications detailed next.
2.2. Design and Preclinical Filtering: Novel Analogs and ADMET Analysis
2.2.1. QSAR-Guided Rational Design
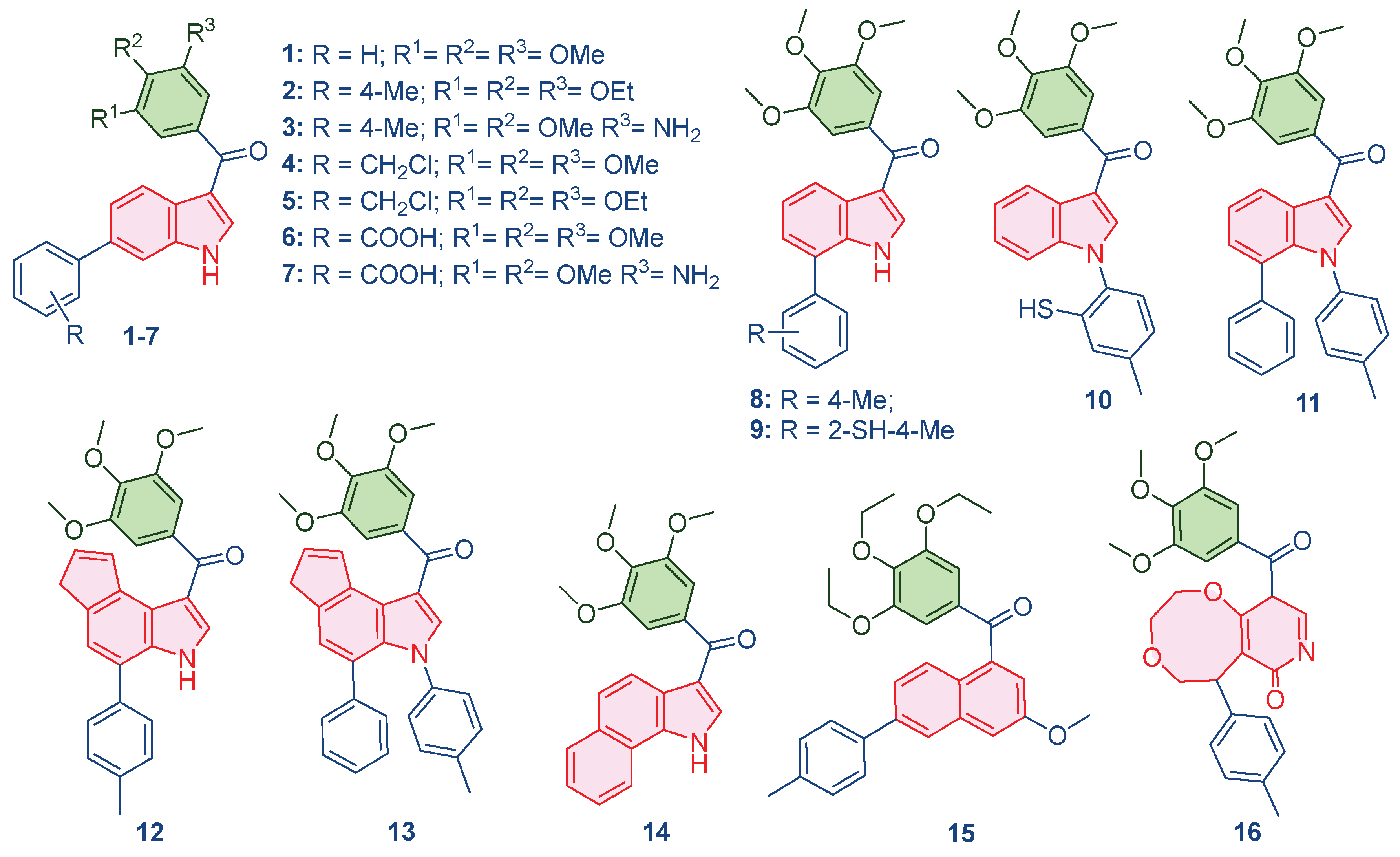
Leveraging the mechanistic insights generated by the predictive QSAR models, particularly the high importance placed on molecular polarizability (of0ug), topological complexity (map4_26), and conformational flexibility (ETA_shape_p), a panel of 16 novel compounds was rationally designed around the trimethoxyphenyl core. The primary objective was to systematically perturb these key descriptors to optimize the predicted pIC50 and enhance the compound’s dual-targeting profile. Structural modifications included the addition of polar groups (amino, benzoic acid), substitution of heteroatoms (chlorine, sulfur), and the incorporation of fused aromatic rings (naphthalene, pyridone-fused systems). These changes were targeted to refine lipophilicity, rigidity, and the capacity for π-stacking interactions essential for both tubulin and AChE binding sites. The specific design rationale for each compound (Table 2), systematically links the chemical modification to the targeted descriptor perturbation. All newly conceived structures were optimized using the MMFF94 force field in MarvinSketch to obtain energetically favorable 3D coordinates, which served as the consistent input for all subsequent in silico validation steps.
Figure 2.
Designed compounds using generated QSAR model.
2.2.2. ADMET Profile and Lead Candidate Selection
Following the rational design, all 16 novel compounds underwent comprehensive ADMET and toxicity screening using ADMETlab 3.0 and ProTox-II platforms to ensure their preclinical viability (Table 3). This pre-screening filter was critical for identifying candidates with favorable pharmacokinetic profiles before proceeding to resource-intensive structural simulations. Given the dual requirement for systemic efficacy (cancer) and CNS penetration (AChE targeting), selection criteria prioritized candidates with high BBB permeability, favorable lipophilicity (logP ideally 2-5) for good oral bioavailability, and minimal predicted safety liabilities.
This systematic, multi-parameter evaluation led to the identification of Compounds 15 and 16 as the optimal lead candidates. Compound 15 displayed favorable lipophilicity (log P of 2.417, log D of 2.718) and a predicted BBB value of 0.564, indicating high bioavailability and the potential to cross the BBB for efficient AChE targeting. Its ProTox analysis suggested a low probability of key toxicities. Compound 16 showed optimized lipophilicity (log P of 3.229, log D of 3.073) and an exceptionally high BBB penetration value (0.998), suggesting highly effective CNS targeting. While it exhibited a safe profile for neurotoxicity and cardiotoxicity, its elevated hepatotoxicity score and CYP2E1 interaction were noted as areas requiring future experimental investigation. The strong, yet complementary, dual-target pharmacokinetic profiles of Compounds 15 and 16 justified their advancement for the next rigorous stage: molecular docking and dynamic simulations to validate their structural binding hypotheses.
2.3. Structural Validation: Dual-Target Molecular Docking and Affinity Assessment
The initial structural validation of the rigorous computational workflow was performed by assessing the static binding affinity (ΔG) and detailed interaction profiles of the QSAR-filtered lead candidates, compounds 15 and 16, against their respective targets. For the anticancer target, the colchicine-binding site of Tubulin (PDB ID: 4O2B) was used, and for the anti-Alzheimer’s target, the active site of AChE (PDB ID: 1EVE) was employed. The accuracy of the docking protocol was rigorously validated by successfully reproducing the crystallographic poses of the reference ligands, Colchicine (RMSD: 0.172 Å) and Donepezil (RMSD: 0.532 Å), confirming the geometric accuracy of the method (Figure 3).
The calculated binding free energies (Table 4) revealed a critical target-differentiated potency, strongly supporting the feasibility of the dual-targeting strategy. Compound 16 showed a marginally superior predicted affinity for the Tubulin binding site (ΔG: -10.0 kcal/mol), making it the prime anticancer candidate, while compound 15 exhibited superior predicted affinity for the AChE active site (ΔG: -10.6 kcal/mol), confirming its strong potential as an anti-Alzheimer’s agent.
2.3.1. Tubulin Binding Profile (Anticancer Target)
The affinity of compound 16 (ΔG: -10.0 kcal/mol) was nearly identical to the positive control, Colchicine (ΔG: -10.1 kcal/mol), suggesting highly effective molecular recognition at the interface of the α- and β-tubulin subunits. Compound 16 achieved this potent binding via extensive interaction profile: Hydrogen Bonding with key residues Gln11, Asn101, and Ala180 (Chain A) and Asn258 (Chain B); and π-Alkyl Contacts with hydrophobic residues Leu248, Ala250, Lys254, and Leu255 (Chain B). This widespread engagement, spanning both chains, explains its superior affinity and strong propensity to destabilize the microtubule structure. Compound 15 (ΔG: -9.7 kcal/mol) displayed a similar but less extensive profile, relying primarily on π-Alkyl interactions (Chain B) and a single hydrogen bond to Asn258.
2.3.2. Acetylcholinesterase Binding Profile (Anti-Alzheimer’s Target).
Compound 15 demonstrated a strong affinity (ΔG of -10.6 kcal/mol), approaching the clinical standard Donepezil (ΔG: -11.8 kcal/mol) and confirming its potential for cholinesterase inhibition. Its interaction profile was diverse and indicative of a dual-binding site mechanism: Hydrophobic Interactions, including π-Alkyl contacts at Phe290 and Phe331; π-π Stacking interactions with aromatic residues, notably Tyr121, Trp279, Phe330, and Tyr334; and Hydrogen Bonding with Asp72 and Gly118. The trimethoxyphenyl scaffold effectively engaged the peripheral anionic site (PAS) residues, while the rest of the molecule penetrated deeper into the active site Gorge. Compound 16 (ΔG: -9.7 kcal/mol) also formed favorable contacts, but lacked the extensive π-π stacking seen in compound 15, resulting in a diminished affinity. The complementary target affinity profiles observed in the docking study were used to guide the subsequent rigorous dynamic validation step.
2.4. Dynamic Validation: 100 ns MD Simulation
2.4.1. MD Simulation and Stability Analysis
To move beyond static binding predictions and perform a rigorous validation of the integrated workflow, 100 ns all-atom MD simulations were conducted for the four final complexes, demonstrating the dynamic behavior of the lead compounds under simulated physiological conditions. This process served as a high-resolution filter, confirming the stability and permanence of the strong binding poses observed in the docking study.
The RMSD relative to the protein served as the primary metric for tracking conformational stability (Figure 4). The compound 16-Tubulin complex exhibited superior stability, maintaining RMSD values largely within the 1.0 - 4.0 Å range, with the main body of the compound remaining fixed inside the colchicine binding site for the entire simulation. The Compound 15-Tubulin complex also proved highly rigid, staying well below 2.0 Å RMSD for most of the trajectory. Conversely, the AChE complexes showed slightly higher, yet constrained, fluctuations: Compound 15-AChE fluctuated between 1.8 - 4.8 Å, settling into a stable secondary conformation after an initial ring rotation, while compound 16-AChE remained rigid, oscillating between 1.6 - 3.2 Å.
The RMSF of the ligands further differentiated the binding dynamics, indicating that the compounds utilize complementary stabilization mechanisms. Compound 15 displayed high flexibility in the AChE site (RMSF: 3.0 Å), suggesting adaptation to engage multiple hydrophobic residues and continuous H-bonding, but low flexibility in the Tubulin site (RMSF: 1.5 Å). Compound 16 showed the reverse trend: low flexibility in the AChE pocket (RMSF: 1.5 Å) but higher flexibility in the Tubulin pocket (RMSF: 3.0 Å), indicating the motion required to react to the fluctuating tubulin structure and interact with multiple residues.
2.5. Energetic Validation: MM-GBSA/MM-PBSA Analysis
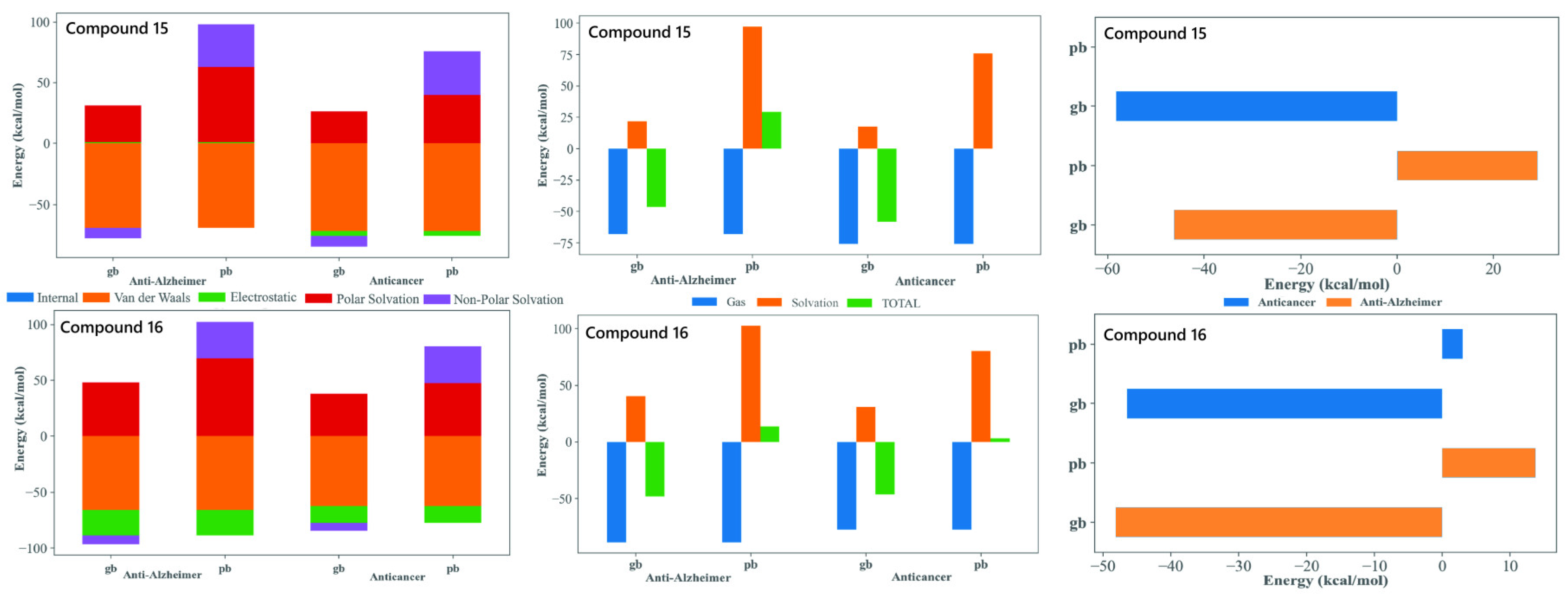
The MM-GBSA/MM-PBSA decomposition of the MD trajectories provided valuable insights into the energetic basis of binding for compounds 15 and 16 with both tubulin and AChE, supporting their potential as dual-target anticancer and anti-Alzheimer’s agents. In all four complex systems, the gas-phase contributions were markedly favorable and were dominated by van der Waals and electrostatic factors. This highlighted the importance of tight packing and aromatic stacking with residues embedded within both the colchicine site of tubulin and the aromatic active site region of AChE. The consistently stabilizing van der Waals profiles reflected the contribution of the tri-methoxy substituted phenyl core enabling the extensive hydrophobic complementarity. The polar solvation penalties observed are typical for the positioning of ligand within deep hydrophobic binding sites. Both compounds display comparably favorable total binding energies across tubulin and AChE, supporting their suitability as dual-acting candidates. Compound 15, with an extended biphenyl substituent, achieved stable binding through enhanced π-stacking and hydrophobic enclosure. This is particularly advantageous for tubulin interactions but also remains beneficial in AChE. Compound 16, with a heterocyclic imide-linked fused ring system, showed a good balance between electrostatic interactions and solvation effects. This behavior works together with the hydrophobic contribution of its trimethoxyphenyl group, allowing it to interact favorably within both binding pockets. This difference in structural features between the two ligands helps explain why both maintained favorable binding energies across the two targets despite of their varying energetic contributions. Overall, the MM-GBSA/MM-PBSA results supported the selection of 15 and 16 as strong candidates for dual inhibition. Both compounds demonstrated the ability to bind effectively to tubulin as well as AChE. They used slightly different but complementary interaction patterns. Their binding energies remained stable over the full 100 ns MD simulations. Their structural flexibility allowed them to adapt well within two very different binding environments. Together, these findings highlighted their promise as multifunctional therapeutic leads with potential relevance in both cancer treatment and neurodegenerative disease management.
Figure 5.
MM-GBSA/MM-PBSA analysis of binding free energy (ΔGbind) for compounds 15 and 16. The analysis compares the energetic contributions (van der Waals, Electrostatic, Solvation) to the stable binding of the lead candidates against the Anti-Alzheimer (AChE) and Anticancer (Tubulin) targets. Favorable ΔGbind values across both MM-GBSA (gb) and MM-PBSA (pb) methods support their dual-inhibitory potential.
Figure 5.
MM-GBSA/MM-PBSA analysis of binding free energy (ΔGbind) for compounds 15 and 16. The analysis compares the energetic contributions (van der Waals, Electrostatic, Solvation) to the stable binding of the lead candidates against the Anti-Alzheimer (AChE) and Anticancer (Tubulin) targets. Favorable ΔGbind values across both MM-GBSA (gb) and MM-PBSA (pb) methods support their dual-inhibitory potential.
2.6. Functional Validation: Post-MD Mechanistic Insights
The final methodological steps involved performing functional analysis on the 100 ns trajectories, serving as the ultimate verification point of the predictive workflow.
2.6.1. The Ramachandran Plot Analysis
This analysis was performed on the final (100 ns) snapshot of each molecular dynamics trajectory using the MolProbity server to rigorously validate the stereochemical quality and conformational integrity of the target proteins post-ligand binding (Figure 6). The results consistently demonstrated that the ligand-induced conformations for all four complexes were structurally sound and highly reliable. Specifically, the complexes showed high percentages of residues in the favored regions (ranging from 86.5% to 91.2%) and allowed regions (ranging from 96.8% to 99.1%). A quality threshold of >85% of residues in the favored regions is considered excellent for post-MD analyses, and the low number of outliers, confined primarily to flexible loop regions, unequivocally confirmed the stereochemical validity of the models.
2.6.2. Dynamic Cross-Correlation Matrix (DCCM) Analysis
The final, high-value component of the integrated pipeline was the DCCM Analysis. This analysis quantifies the concerted motions between residue pairs, providing a functional, allosteric context for the observed stability. The DCCM maps (Figure 7) confirmed the compounds’ complementary dynamic signatures.
For Tubulin, compound 15 exhibited a complex pattern of both positive and negative correlations, suggesting it engages in broader, more widespread structural adjustments across the α/β interface (greater structural flexibility). Conversely, compound 16 showed a more simplified and localized correlation profile, aligning with its rigid, focused binding mode.
For AChE, both complexes maintained positive correlations in critical pairs (Tyr70-Trp84, Tyr121-Glu199, and Tyr334-His440). Crucially, compound 15 showed a distinct negative correlation between Gly441-Tyr70, a key pair regulating the gorge entrance. The presence of this anti-correlation in 15 suggests it is more effective at modulating the dynamic opening/closing of the AChE gorge, reinforcing its superior predicted inhibitory affinity and deeper mechanistic engagement observed in the docking study.
The DCCM analysis serves as the functional validation of the entire predictive workflow, reinforcing that compound 15 employs a broader, more flexible engagement, while compound 16 uses a more focused, rigid strategy, confirming the versatility and therapeutic promise of the leads.
3. Materials and Methods
3.1. Dataset Preparation and QSAR Modeling Rationale
The development of robust QSAR models commenced with an exhaustive literature search utilizing the ChEMBL and BindingDB databases to create a high-quality dataset of 57 molecules (Table S3, SI). The key inclusion criterion was the presence of the trimethoxyphenyl scaffold, a pharmacophore known for its affinity to the colchicine binding site on tubulin and documented tubulin polymerization inhibitory activity (IC50). Biological activity values (IC50) were uniformly converted to the negative logarithmic scale (pIC50) to ensure a normal distribution and reduce data skewness, which is essential for stable Multiple Linear Regression (MLR) modeling. The final dataset of 57 compounds was systematically divided to ensure rigorous validation: 10 compounds (≈ 17.5%) were designated as the external validation set to assess predictive capability on unseen data, while the remaining 47 compounds formed the training set for model generation and internal cross-validation. This division strategy maximized the training set size, ensuring model stability and adequate descriptor space coverage while maintaining substantial external validation power, adhering strictly to OECD principles for QSAR development [31,38].
3.2. Descriptor Generation & Pre-Filtering
Molecular structures for all 57 compounds were rigorously optimized using the MMFF94 force field within MarvinSketch to ensure consistent, low-energy three-dimensional conformations. Subsequently, a massive initial pool of 11,829 molecular descriptors was generated using specialized software packages, including PaDEL-Descriptor, alvaDesc, MORDRED, and MERA via the OCHEM server [39]. This extensive descriptor space, encompassing constitutional, topological, geometrical, electronic, and hybrid properties, was intentionally broad to provide the Genetic Algorithm (GA) with the widest possible feature set for robust structure-activity relationship identification. To mitigate the inherent risks of data overfitting and multicollinearity associated with such large descriptor pools, a systematic two-stage pre-filtering procedure was implemented. First, descriptors exhibiting the same value across more than 80% of the dataset were removed; second, a threshold of 95% correlation was applied to eliminate redundant descriptor pairs. This methodical pre-filtering eliminated 7,943 descriptors, resulting in a refined and highly informative set of 3,886 descriptors for the subsequent QSAR model construction phase.
3.3. Molecular Sketching and Geometry Optimization
Leveraging the mechanistic insights derived from the QSAR models, a series of 16 novel compounds were rationally designed based on the original trimethoxyphenyl scaffold. The design strategy was guided by the positive contribution of the identified key descriptors. For instance, structural modifications such as the addition of chlorine or ethoxy groups were introduced to modulate Mor15i (electronic density) and MAP4_26 (topological complexity), while the introduction of fused rings or changes in substituent positions were aimed at optimizing ETA_shape_p (conformational flexibility) and steric fit (R3s++). Specific modifications included altering the aromaticity, introducing flexible linkers, and changing the polarity of key functional groups to maximize the predicted pIC50 for tubulin inhibition. All newly sketched structures were subsequently subjected to geometry optimization using the MMFF94 force field in MarvinSketch to obtain energetically favorable 3D coordinates, which served as the input for all subsequent ADMET and docking calculations [40,41,42,43].
3.4. ADMET Prediction and Selection of Lead Candidates
Following the rational QSAR-guided design, all 16 novel compounds were subjected to comprehensive ADMET profiling to rigorously filter candidates before proceeding to resource-intensive structural simulations. This critical in silico screen employed the ADMETlab 3.0 and ProTox-II platforms to predict essential pharmacokinetic parameters, including Caco-2 permeability, BBB penetration, aqueous solubility (logS), lipophilicity (logP and logD), metabolism via CYP isoforms, and general toxicity risks such as hepatotoxicity and mutagenicity. Given the dual-target nature of the study, requiring systemic efficacy for cancer and CNS penetration for Alzheimer’s Disease, the selection criteria were stringent, prioritizing candidates with high BBB permeability (essential for AChE targeting), favorable logP (ideally 2-5) for oral bioavailability, and minimal predicted safety liabilities. This systematic, multi-parameter evaluation led to the identification of Compounds 15 and 16 as the optimal lead candidates. Compound 15 was selected for its balanced physicochemical profile and predicted BBB permeability, while Compound 16 was advanced due to its exceptional BBB penetration prediction, justifying their prioritization for the subsequent molecular docking and dynamics simulations to validate their structural binding hypotheses [44].
3.5. Molecular Docking & Validation
The dual-target potential of the two selected leads, Compounds 15 and 16, was structurally evaluated using molecular docking simulations against their respective targets. For the anticancer target, the crystal structure of β-tubulin in complex with colchicine (PDB ID: 4O2B) was used. For the anti-Alzheimer’s target, the crystal structure of Acetylcholinesterase (AChE) (PDB ID: 1EVE) was utilized. The proteins were pre-processed (removing water molecules, standardizing protonation states) using the Discovery Studio Visualizer and SwissPDB Viewer. Docking calculations were performed using AutoDock Vina integrated within the CB-Dock2 server, which automatically defines the optimal binding site cavity, ensuring an unbiased “blind docking” approach [45,46]. Validation of the docking methodology was performed by redocking the co-crystallized ligands (Colchicine for 4O2B and Donepezil for 1EVE). The method was accepted only after the Root Mean Square Deviation (RMSD) between the docked and crystal poses was less than 2.0 Å [47]. The final predicted binding affinities (ΔG in kcal/mol) and the nature of the specific protein-ligand interactions (H-bonds, π-stacking, hydrophobic contacts) were meticulously analyzed using Discovery Studio Visualizer and UCSF Chimera for both compounds against both targets [48].
3.6. Molecular Dynamics (MD) Simulation and Post-MD Analysis
To comprehensively assess the stability, flexibility, and permanence of the protein-ligand interactions observed in the static docking poses, 100 ns all-atom MD simulations were conducted for the four final complexes: 15-Tubulin, 16-Tubulin, 15-AChE, and 16-AChE. The simulations were performed using the Desmond module within the Schrödinger Suite. Each system was prepared by placing the docked complex in a periodic boundary condition box solvated with TIP3P water molecules and neutralized with Na+ and Cl- ions. The OPLS3e force field was used to describe all atomic interactions. After a thorough minimization and pre-equilibration phase, the production run was executed for 100 ns under constant temperature (300 K) and pressure (1 bar) conditions, with trajectories saved every 50 ps [49].
Post-MD Analysis was critical for interpreting the dynamic stability and functional integrity: trajectory analysis (Protein/Ligand RMSD and residue RMSF) monitored stability and flexibility; interaction timelines tracked the persistence of specific residue contacts; the Ramachandran Plot validation ensured the stereochemical quality and conformational suitability of the final complex structures using the MolProbity server [50]; and finally, the advanced Dynamic Cross-Correlation Matrix (DCCM) analysis, performed with the MD-TASK suite, was used to quantify the functional connectivity and allosteric communication within the protein induced by ligand binding [51].
To provide the energetic basis for the observed dynamic stability, the binding free energy (ΔGbind) for the four protein-ligand complexes (15-Tubulin, 16-Tubulin, 15-AChE, and 16-AChE) was calculated using the MM-GBSA and MM-PBSA methodologies. This was performed on the molecular dynamics trajectories using the Prime module of the Schrödinger Suite. The analysis was conducted on the final 50 ns (500 frames) of the 100 ns production run to ensure the inclusion of only equilibrated conformations. The free energy of binding was calculated according to the general equation:
where ΔEMM represents the change in molecular mechanics energy (sum of ΔEvdw and ΔEelec), ΔGsolv represents the change in solvation energy (polar and non-polar components), and TΔS is the change in conformational entropy. The non-polar contribution to solvation energy was calculated using the Solvent Accessible Surface Area (SASA) model. The entropy term (TΔS) was neglected due to its computational intensity, which is common practice for comparative studies. The focus was on the most favorable non-covalent terms: van der Waals and electrostatic energies, as they dominate the gas-phase energy in these systems.
4. Conclusions
This study successfully established an integrated QSAR-MD-DCCM pipeline for the rational discovery and dynamic validation of multi-target-directed ligands (MTDLs), satisfying all stringent OECD criteria. Two highly accurate, statistically robust QSAR models (R2 > 0.83) were developed from a focused trimethoxyphenyl scaffold library, where mechanistic interpretation demonstrated that molecular polarizability (of0ug), topological complexity (map4_26), and conformational flexibility (ETA_shape_p) are the indispensable key determinants for effective tubulin polymerization inhibition. These quantitative insights directly guided the rational design of 16 novel analogues, demonstrating the pipeline’s utility in early-stage Machine Learning application. The leads identified through this initial filtering, compounds 15 and 16, were selected based on optimized drug-likeness and their high predicted capacity for BBB penetration, an essential feature for effective AChE targeting. Molecular docking and rigorous 100 ns MD simulations confirmed dynamically stable and persistent binding for both leads against both Tubulin (PDB ID: 4O2B) and AChE (PDB ID: 1EVE). Furthermore, MM-GBSA/MM-PBSA binding free energy calculations provided essential energetic validation, showing highly favorable total binding energies (ΔGbind) that primarily stemmed from stabilizing van der Waals interactions. This analysis revealed a complementary, target-differentiated potency: Compound 16 showed marginally superior predicted affinity for the Tubulin binding site (ΔG: -10.0 kcal/mol), while compound 15 exhibited superior predicted affinity for the AChE active site (ΔG: -10.6 kcal/mol). Post-MD Ramachandran analysis confirmed the high stereochemical integrity of all four protein-ligand complexes. Furthermore, the DCCM analysis provided the final and highest level of functional validation, revealing complementary dynamic signatures: Compound 15 was found to induce a broader range of dynamic interactions and allosteric coupling, suggesting a more flexible engagement, while compound 16 exhibited a more focused and rigid inhibition strategy. In conclusion, this rigorous, integrated QSAR-MD-DCCM pipeline represents a highly predictive and robust platform for the de novo design and dynamic validation of multi-target therapeutics. The successful virtual validation of two distinct dual-acting candidates (15 and 16) serves as compelling proof-of-concept for this advanced computational workflow, ready for immediate experimental synthesis and in vitro validation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, T.J.P., G.M. and S.S.N.; methodology, T.J.P., S.S.N., and D.F.C.; software, P.D.; validation, S.C., P.K.K., D.F.C., G.M. and S.S.N.; formal analysis, T.J.P., P.D., M.F., G.M. and S.S.N.; investigation, S.C., P.D., P.K.K., S.L., A.D.R. and S.S.N.; resources, S.S.N.; data curation, S.C., S.L., A.D.R. and S.S.N.; writing—original draft preparation, S.S.N. and A.D.R.; writing—review and editing, T.J.P., S.S.N., D.F.C., P.K.K., M.F., and G.M.; visualization, S.L.; supervision, T.J.P., G.M. and S.S.N.; project administration, T.J.P. G.M. and S.S.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia [Grant No. xxxx].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting the findings of this study, including all molecular structures, computational models, raw and processed trajectory data, and scripts required for the full replication of the integrated QSAR-MD-DCCM pipeline, are openly available in a public GitHub repository: https://github.com/tusharpawar49/QSAR-MD-DCCM_Dual-Target_Tubulin-AChE
Acknowledgments
We gratefully acknowledge the creators and developers of the non-commercial software and platforms that were critical to this integrated workflow. Specifically, we thank the teams responsible for QSARINS and the Online Chemical Modeling Environment (OCHEM) for predictive modeling; AutoDock Vina and ADMETlab 3.0 for efficient structural filtering; D. E. Shaw Research and Schrödinger, LLC for the fundamental methods and tools used in the all-atom Molecular Dynamics simulations and MM-GBSA/MM-PBSA calculations; and the developers of the MD-TASK suite and MolProbity for enabling the rigorous dynamic and stereochemical validation. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem 2014, 57, 7874–7887. [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem 2005, 48, 6523–6543. [CrossRef]
- Sang, Z.; Wang, K.; Bai, P.; Wu, A.; Shi, J.; Liu, W.; Zhu, G.; Wang, Y.; Lan, Y.; Chen, Z.; Zhao, Y.; Qiao, Z.; Wang, C.; Tan Z. Design, synthesis and biological evaluation of novel O-carbamoyl ferulamide derivatives as multi-target-directed ligands for the treatment of Alzheimer’s disease. Eur J Med Chem 2020, 194, 112265. [CrossRef]
- Ramsay, R. R.; Popovic-Nikolic, M. R.; Nikolic, K.; Uliassi, E.; Bolognesi, M. L. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med 2018, 7, 3. [CrossRef]
- Makhoba, X. H.; Viegas, C. J.; Mosa, R. A.; Viegas, F. P. D.; Pooe, O. J. Potential Impact of the Multi-Target Drug Approach in the Treatment of Some Complex Diseases. Drug Des Devel Ther 2020, 14, 3235–3249. [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R. L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [CrossRef]
- 2025 Alzheimer’s disease facts and figures. Alzheimers Dement 2025, 21, e70235. [CrossRef]
- Musicco, M.; Adorni, F.; Di Santo, S.; Prinelli, F.; Pettenati, C.; Caltagirone, C.; Palmer, K.; Russo, A. Inverse occurrence of cancer and Alzheimer disease: a population-based incidence study. Neurology 2013, 81, 322–328. [CrossRef]
- Friedman, J. R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2012, 2, a006346. [CrossRef]
- Roe, C. M.; Behrens, M. I.; Xiong, C.; Miller, J. P.; Morris, J. C. Alzheimer disease and cancer. Neurology 2005, 64, 895–898. [CrossRef]
- Freedman, D. M.; Wu, J.; Chen, H.; Kuncl, R. W.; Enewold, L. R.; Engels, E. A.; Freedman, N. D.; Pfeiffer, R. M. Associations between cancer and Alzheimer’s disease in a U.S. Medicare population. Cancer Med 2016, 5, 2965–2976. [CrossRef]
- Sasco, A. J.; Secretan, M. B.; Straif, K. Tobacco smoking and cancer: a brief review of recent epidemiological evidence. Lung Cancer Amst Neth 2004, 45 Suppl 2, S3–S9. [CrossRef]
- Swerdlow, R. H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J Alzheimers Dis JAD 2018, 62, 1403–1416. [CrossRef]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S. P.; Wu, J. F.; Carney, J. M.; Lovell, M.; Markesbery, W. R.; Butterfield, D. A. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem 1995, 65, 2146–2156. [CrossRef]
- Jordan, M. A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004, 4, 253–265. [CrossRef]
- Baas, P. W. Microtubules and Neuronal Polarity: Lessons from Mitosis. Neuron 1999, 22, 23–31. [CrossRef]
- Kavalallaris, M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer 2010, 10, 194–204. [CrossRef]
- Dumontet, C.; Jordan, M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 2010, 9, 790–803. [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat Rev Neurosci 2016, 17, 5–21.
- Ballatore, C.; Lee, V. M.-Y.; Trojanowski, J. Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 2007, 8, 663–672. [CrossRef]
- Brunden, K. R.; Trojanowski, J. Q.; Smith 3rd, A. B.; Lee, V. M.-Y.; Ballatore, C. Microtubule-stabilizing agents as potential therapeutics for neurodegenerative disease. Bioorg Med Chem 2014, 22, 5040–5049. [CrossRef]
- Brunden, K. R.; Yao, Y.; Potuzak, J. S.; Ferrer, N. I.; Ballatore, C.; James, M. J.; Hogan, A. M.; Trojanowski, J. Q.; Smith, A. B. 3rd; Lee, V. M. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol Res 2011, 63, 341–351. [CrossRef]
- Zhang, B.; Carroll, J.; Trojanowski, J. Q.; Yao, Y.; Iba, M.; Potuzak, J. S.; Hogan, A. M.; Xie, S. X.; Ballatore, C.; Smith 3rd, A. B.; Lee, V. M.; Brunden, K. R. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci Off J Soc Neurosci 2012, 32, 3601–3611. [CrossRef]
- Francis, P. T.; Palmer, A. M.; Snape, M.; Wilcock, G. K. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry 1999, 66, 137–147. [CrossRef]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 2006, 2006, CD005593. [CrossRef]
- Kos, J.; Strharsky, T.; Stepankova, S.; Svrckova, K.; Oravec, M.; Hosek, J.; Imramovsky, A.; Jampilek, J. Trimethoxycinnamates and Their Cholinesterase Inhibitory Activity. Appl Sci 2021, 11, 4691. [CrossRef]
- La Regina, G.; Edler, M. C.; Brancale, A.; Kandil, S.; Coluccia, A.; Piscitelli, F.; Hamel, E.; De Martino, G.; Matesanz, R.; Díaz, J. F.; Scovassi, A. I.; Prosperi, E.; Lavecchia, A.; Novellino, E.; Artico, M.; Silvestri, R. Arylthioindole Inhibitors of Tubulin Polymerization. 3. Biological Evaluation, Structure-Activity Relationships and Molecular Modeling Studies. J Med Chem 2007, 50, 2865–2874. [CrossRef]
- Cherkasov, A.; Muratov, E. N.; Fourches, D.; Varnek, A.; Baskin, I. I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y. C.; Todeschini, R.; Consonni, V.; Kuz’min, V. E.; Cramer, R.; Benigni, R.; Yang, C.; Rathman, J.; Terfloth, L.; Gasteiger, J.; Richard, A.; Tropsha, A. QSAR modeling: where have you been? Where are you going to? J Med Chem 2014, 57, 4977–5010. [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E. W. J. Computational methods in drug discovery. Pharmacol Rev 2014, 66, 334–395. [CrossRef]
- Gramatica, P. Principles of QSAR Modeling: Comments and Suggestions From Personal Experience. Int J Quant Struct-Prop Relatsh IJQSPR 2020, 5, 61–97.
- OECD. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models. 2007.
- Trott, O.; Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31, 455–461. [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 2011, 7, 146–157. [CrossRef]
- Hollingsworth, S. A.; Dror, R. O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [CrossRef]
- Karplus, M.; McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat Struct Biol 2002, 9, 646–652. [CrossRef]
- Brown, D. K.; Penkler, D. L.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A. R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: a software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017, 33, 2768–2771. [CrossRef]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J Comput Chem 2014, 35, 1036–1044. [CrossRef]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A. K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V. V.; Tanchuk, V. Y.; Todeschini, R.; Varnek, A.; Marcou, G.; Ertl, P.; Potemkin, V.; Grishina, M.; Gasteiger, J.; Schwab, C.; Baskin, I. I.; Palyulin, V. A.; Radchenko, E. V.; Welsh, W. J.; Kholodovych, V.; Chekmarev, D.; Cherkasov, A.; Aires-de-Sousa, J.; Zhang, Q.-Y.; Bender, A.; Nigsch, F.; Patiny, L.; Williams, A.; Tkachenko, V.; Tetko, I. V. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J Comput Aided Mol Des 2011, 25, 533–554. [CrossRef]
- Karatoprak, G. Ş.; Küpeli Akkol, E.; Genç, Y.; Bardakcı, H.; Yücel, Ç.; Sobarzo-Sánchez, E. Combretastatins: An Overview of Structure, Probable Mechanisms of Action and Potential Applications. Molecules 2020, 25, 2560. [CrossRef]
- Fantacuzzi, M.; Carradori, S.; Giampietro, L.; Maccallini, C.; De Filippis, B.; Amoroso, R.; Ammazzalorso, A. A novel life for antitumor combretastatins: Recent developments of hybrids, prodrugs, combination therapies, and antibody-drug conjugates. Eur J Med Chem 2025, 281, 117021. [CrossRef]
- Majchrzak, M. W.; Kotełko, A.; Guryn, R.; Lambert, J. B.; Szadowska, A.; Kowalczyk, K. Synthesis and action on the central nervous system of mescaline analogues containing piperazine or homopiperazine rings. J Pharm Sci 1983, 72, 304–306. [CrossRef]
- Kos, J.; Strharsky, T.; Stepankova, S.; Svrckova, K.; Oravec, M.; Hosek, J.; Imramovsky, A; Jampilek, J. Trimethoxycinnamates and Their Cholinesterase Inhibitory Activity. Appl Sci 2021, 11, 4691. [CrossRef]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; Lyu, A.; Zeng, X.; Zhao, W.; Hou, T.; Cao, D. ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res 2024, 52, W422–W431. [CrossRef]
- Trott, O.; Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31, 455–461. [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A. F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J Chem Inf Model 2021, 61, 3891–3898. [CrossRef]
- Bell, E. W.; Zhang, Y. DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J Cheminformatics 2019, 11, 40. [CrossRef]
- Meng, E. C.; Goddard, T. D.; Pettersen, E. F.; Couch, G. S.; Pearson, Z. J.; Morris, J. H.; Ferrin, T. E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci Publ Protein Soc 2023, 32, e4792. [CrossRef]
- Bowers, K. J.; Chow, D. E.; Xu, H.; Dror, R. O.; Eastwood, M. P.; Gregersen, B. A.; Klepeis, J. L.; Kolossvary, I.; Moraes, M. A.; Sacerdoti, F. D.; Salmon, J. K.; Shan, Y.; Shaw, D. E. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proc. 2006 ACM/IEEE Conf. Supercomput., New York, NY, USA, November 11–17, 2006; Association for Computing Machinery: New York, NY, USA, 2006, p. 84-es. [CrossRef]
- Williams, C. J.; Headd, J. J.; Moriarty, N. W.; Prisant, M. G.; Videau, L. L.; Deis, L. N.; Verma, V.; Keedy, D. A.; Hintze, B. J.; Chen, V. B.; Jain, S.; Lewis, S. M.; Arendall 3rd, W. B.; Snoeyink, J.; Adams, P. D.; Lovell, S. C.; Richardson, J. S.; Richardson, D. C. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci Publ Protein Soc 2018, 27, 293–315. [CrossRef]
- Ichiye, T.; Karplus, M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 1991, 11, 205–217. [CrossRef]
Figure 1.
Overlapping Pathophysiology of Alzheimer’s Disease (AD) and Cancer: The Rationale for and Implementation of the Integrated Computational Pipeline.
Figure 1.
Overlapping Pathophysiology of Alzheimer’s Disease (AD) and Cancer: The Rationale for and Implementation of the Integrated Computational Pipeline.
Figure 3.
Visualization of the molecular docking results for compounds 15 and 16 against the dual targets, providing the initial structural validation of the MTDL strategy.
Figure 3.
Visualization of the molecular docking results for compounds 15 and 16 against the dual targets, providing the initial structural validation of the MTDL strategy.
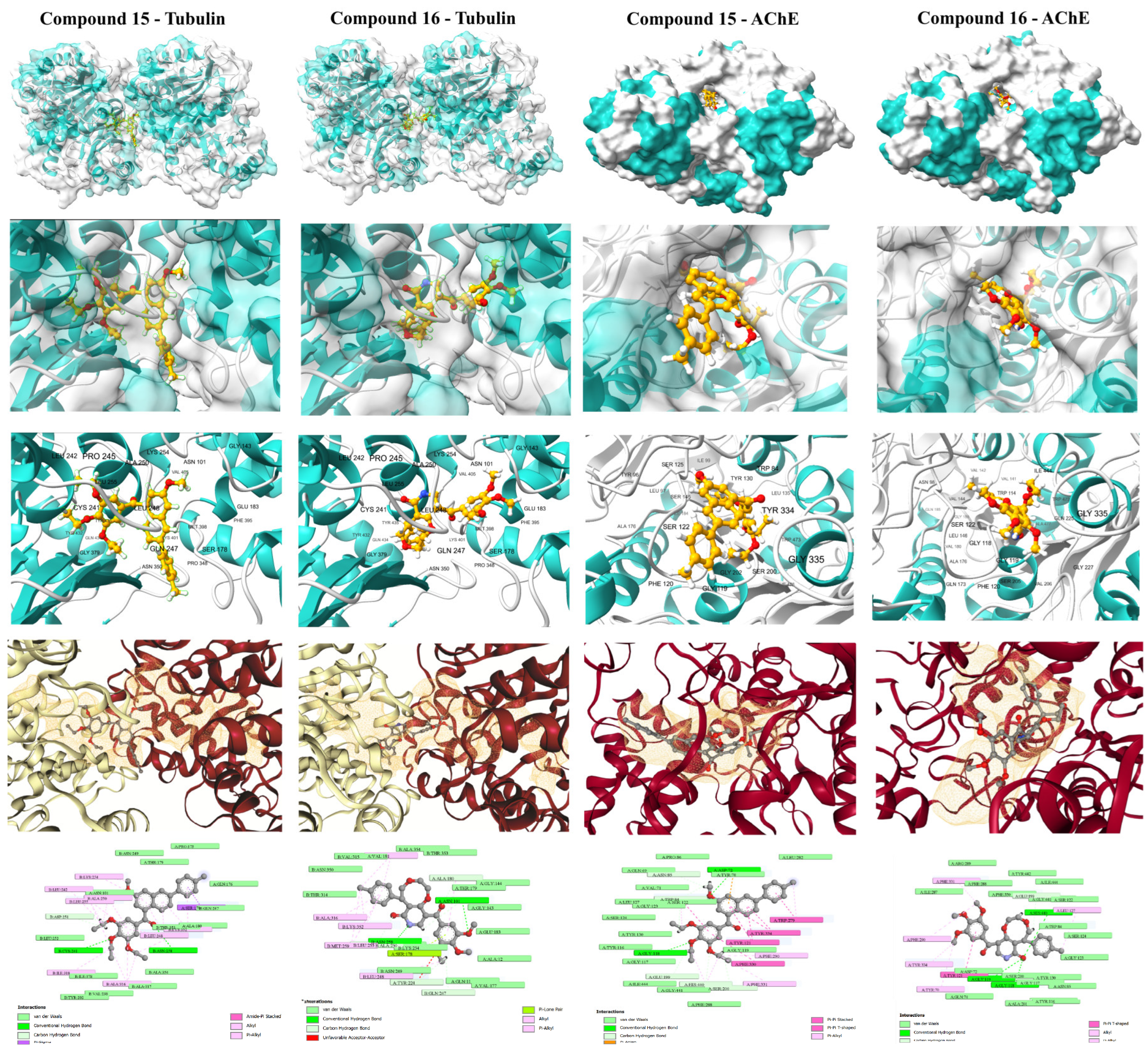
Figure 4.
Molecular dynamic simulation results for compound 15 (A: Anticancer activity/Tubulin; C: Anti-Alzheimer’s activity/AChE) and 16 (B: Anticancer activity/Tubulin; D: Anti-Alzheimer’s activity/AChE). Each panel typically shows (Top Left) Protein and Ligand RMSD over 100 ns; (Bottom Left) Interaction fractions (H-bonds, Hydrophobic, Ionic, Water bridges) with key residues; and (Right) Interaction timelines tracking the persistence of specific residue contacts over the 100 ns trajectory.
Figure 4.
Molecular dynamic simulation results for compound 15 (A: Anticancer activity/Tubulin; C: Anti-Alzheimer’s activity/AChE) and 16 (B: Anticancer activity/Tubulin; D: Anti-Alzheimer’s activity/AChE). Each panel typically shows (Top Left) Protein and Ligand RMSD over 100 ns; (Bottom Left) Interaction fractions (H-bonds, Hydrophobic, Ionic, Water bridges) with key residues; and (Right) Interaction timelines tracking the persistence of specific residue contacts over the 100 ns trajectory.
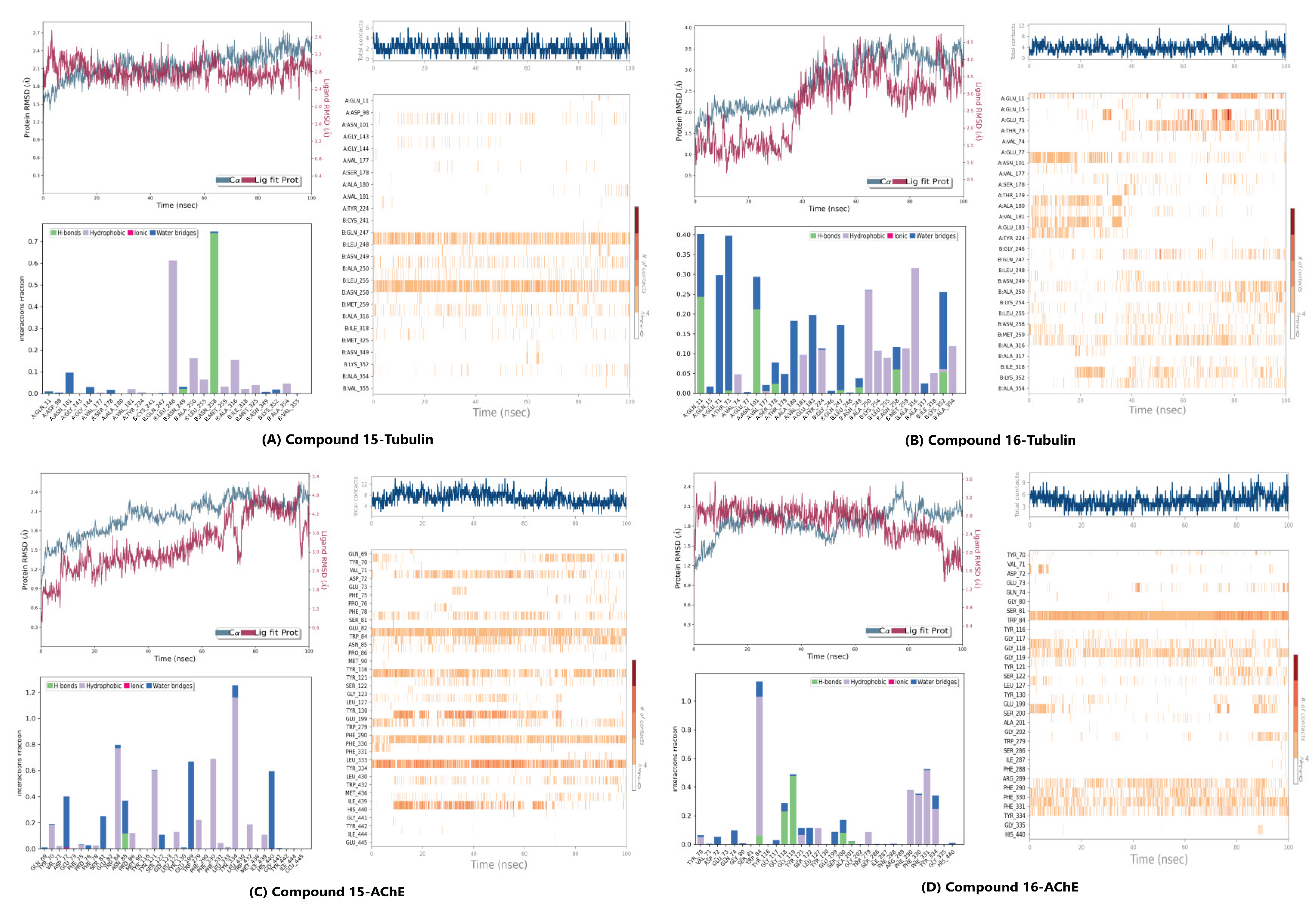
Figure 6.
Ramachandran Plot analysis of the modeled protein structures after the 100 ns Molecular Dynamics simulations. The plots show the backbone dihedral angle distribution of residues, confirming the stereochemical quality of the final ligand-bound complexes: (A) Compound 15-Tubulin, (B) Compound 16-Tubulin, (C) Compound 15-AChE, and (D) Compound 16-AChE. The high percentage of residues in the favored and allowed regions validates the conformational suitability of all four systems for further analysis.
Figure 6.
Ramachandran Plot analysis of the modeled protein structures after the 100 ns Molecular Dynamics simulations. The plots show the backbone dihedral angle distribution of residues, confirming the stereochemical quality of the final ligand-bound complexes: (A) Compound 15-Tubulin, (B) Compound 16-Tubulin, (C) Compound 15-AChE, and (D) Compound 16-AChE. The high percentage of residues in the favored and allowed regions validates the conformational suitability of all four systems for further analysis.
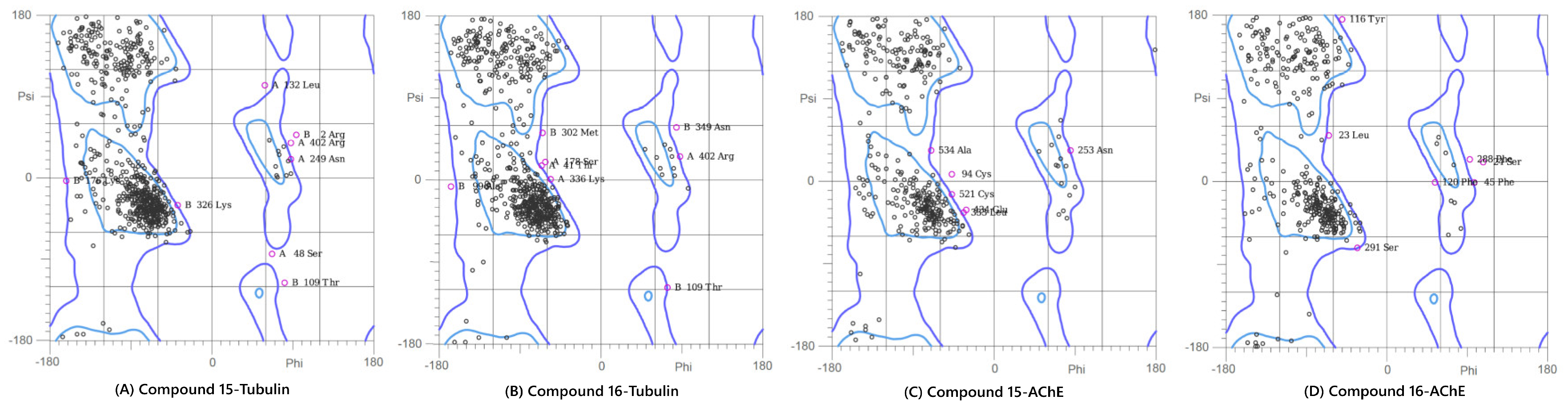
Figure 7.
Dynamic cross correlation matrix of compounds 15 and 16.
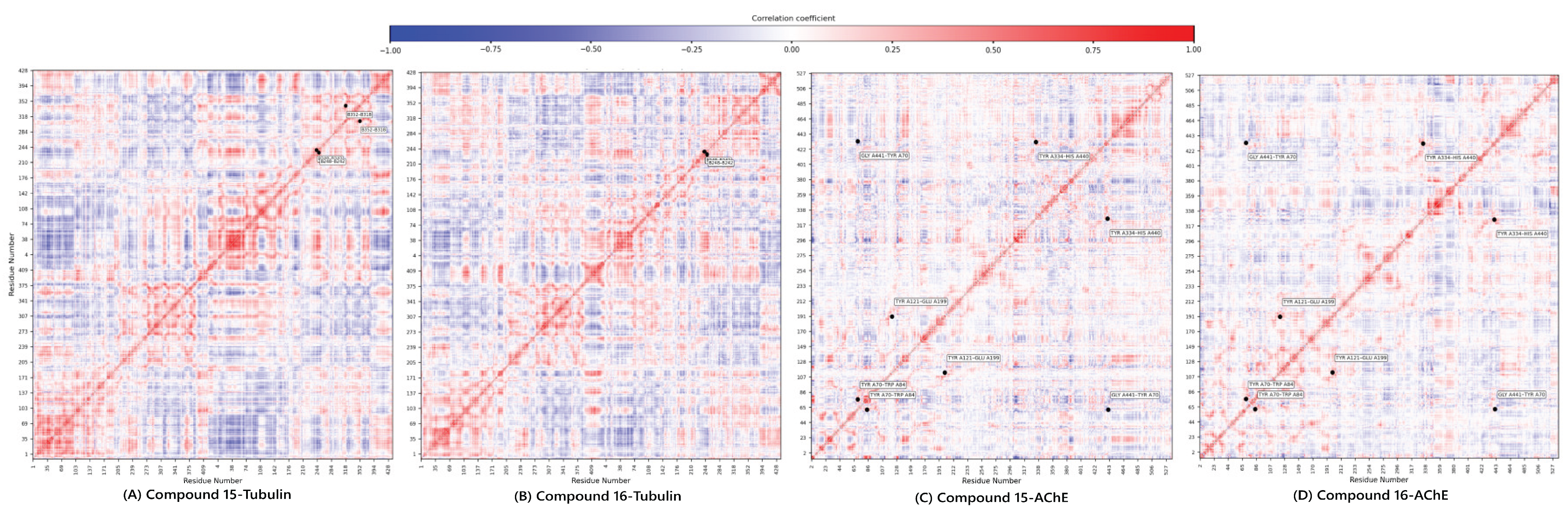

Table 1.
Model validation parameters.
| Metric | Model 1 | Model 2 | OECD Threshold |
| 0.0866 | 0.0861 | <0.20 | |
| -0.1499 | -0.1512 | <0.20 | |
| R2 (Training) | 0.8319 | 0.8384 | >0.60 |
| Adjusted R2 | 0.8159 | 0.8230 | Close to R2 |
| Q2 (LOO Cross-validation) | 0.7940 | 0.7976 | >0.50 |
| Q2 (LMO Cross-validation) | 0.7804 | 0.7976 | >0.60 |
| (External Prediction Set) | 0.8565 | 0.8633 | >0.60 |
| CCC (External) | 0.9197 | 0.9252 | >0.85 |
| RMSE (Training) | 0.1360 | 0.1333 | Lower is better |
| MAE (training) | 0.1067 | 0.1082 | Lower is better |
| r2m (average) | 0.7926 | 0.8022 | >0.50 |
| Δr2m | 0.0340 | 0.0097 | <0.20 |
Table 2.
Drug design using QSAR model.
| Comp No | Targeted Descriptors | Design Rationale |
|---|---|---|
| 1 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Phenyl addition to 6th position enhances shape and aromaticity for optimized binding. |
| 2 | Mor15i, MAP4_26, of0ug, ETA_shape_p, R3s+ | Ethoxy substitution modifies lipophilicity and molecular geometry for better receptor interaction. |
| 3 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Methoxy-to-amino replacement alters polarity and shape for improved receptor targeting. |
| 4 | Mor15i, MAP4_26, of0ug, ETA_shape_p, R3s+ | Chlorine addition alters electron density and lipophilicity, impacting binding and molecular shape. |
| 5 | Mor15i, MAP4_26, of0ug, ETA_shape_p, R3s+ | Chlorine and ethoxy substitution affect hydrophobicity, shape, and receptor fit. |
| 6 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Benzoic acid replaces Tolyl, increasing aromaticity and modifying shape for better binding. |
| 7 | Mor15i, MAP4_26, of0ug, ETA_shape_p, R3s+ | Amino substitution and benzoic acid replaces Tolyl, enhancing hydrophilicity, shape, and binding. |
| 8 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Tolyl group affects topological and spatial properties for optimized binding. |
| 9 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Tolyl and SH substitution increases aromaticity, modifying binding interactions. |
| 10 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Tolyl at 1st position with SH alters shape and polarity for improved receptor fit. |
| 11 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Tolyl at 1st position and phenyl at 7th position modifies geometry and receptor interactions. |
| 12 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Tolyl at 7th position and fused 5-membered ring enhances aromaticity and binding. |
| 13 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Tolyl at 1st position, phenyl at 7th, and fused 5-membered ring improves aromaticity and binding. |
| 14 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Fused 6-membered ring increases aromaticity and rigidity for better receptor interaction. |
| 15 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Indole replaced with naphthalene alters shape and flexibility for improved interaction. |
| 16 | Mor15i, MAP4_26, ETA_shape_p, R3s+ | Pyridone replacement with fused eight-membered ring adjusts shape and electronic characteristics. |
Table 3.
ADMET profiles of the designed compounds.
| Parameters | 14 | 3 | 5 | 1 | 2 | 6 | 7 | 4 | 8 | 9 | 10 | 11 | 12 | 13 | 15 | 16 |
| MW | 484.220 | 443.210 | 386.160 | 435.120 | 477.170 | 431.140 | 416.140 | 401.160 | 433.130 | 477.190 | 439.180 | 515.210 | 361.130 | 387.150 | 465.180 | 437.180 |
| Vol | 522.820 | 475.775 | 408.797 | 439.098 | 490.986 | 438.831 | 423.741 | 423.887 | 442.396 | 511.197 | 464.582 | 551.892 | 374.635 | 406.591 | 470.190 | 446.740 |
| QED | 0.222 | 0.297 | 0.375 | 0.293 | 0.197 | 0.408 | 0.318 | 0.440 | 0.303 | 0.240 | 0.376 | 0.212 | 0.529 | 0.468 | 0.603 | 0.585 |
| Synth | 2.343 | 2.302 | 2.279 | 2.277 | 2.434 | 2.202 | 2.339 | 2.202 | 2.552 | 2.341 | 2.765 | 2.819 | 2.194 | 2.079 | 3.800 | 3.204 |
| Fsp3 | 0.258 | 0.250 | 0.125 | 0.160 | 0.250 | 0.120 | 0.083 | 0.160 | 0.160 | 0.129 | 0.179 | 0.147 | 0.136 | 0.125 | 0.346 | 0.320 |
| logS | -8.469 | -7.311 | -6.296 | -6.537 | -7.145 | -5.230 | -5.158 | -6.338 | -6.070 | -7.309 | -6.837 | -7.719 | -5.832 | -5.759 | -4.386 | -4.965 |
| logD | 5.152 | 4.436 | 3.206 | 3.530 | 4.179 | 2.645 | 2.459 | 3.749 | 3.642 | 4.552 | 3.738 | 4.548 | 3.494 | 3.569 | 2.718 | 3.073 |
| logP | 6.644 | 5.656 | 4.173 | 4.193 | 5.185 | 3.806 | 3.691 | 4.527 | 4.301 | 5.407 | 4.710 | 5.765 | 3.948 | 4.167 | 2.417 | 3.229 |
| DILI | 0.994 | 0.977 | 0.958 | 0.892 | 0.951 | 0.992 | 0.993 | 0.941 | 0.999 | 0.989 | 0.962 | 0.996 | 0.952 | 0.950 | 0.835 | 0.926 |
| Ames | 0.621 | 0.476 | 0.788 | 0.826 | 0.717 | 0.629 | 0.787 | 0.611 | 0.759 | 0.713 | 0.742 | 0.874 | 0.746 | 0.638 | 0.438 | 0.433 |
| FDAMDD | 0.866 | 0.837 | 0.859 | 0.837 | 0.864 | 0.685 | 0.761 | 0.792 | 0.271 | 0.818 | 0.852 | 0.913 | 0.795 | 0.793 | 0.686 | 0.615 |
| caco2 | -4.647 | -4.736 | -5.016 | -4.962 | -4.714 | -5.034 | -5.166 | -4.990 | -5.140 | -4.899 | -4.853 | -4.804 | -4.909 | -4.958 | -4.658 | -4.906 |
| PAMPA | 0.099 | 0.191 | 0.237 | 0.013 | 0.033 | 0.800 | 0.932 | 0.010 | 0.002 | 0.006 | 0.012 | 0.010 | 0.008 | 0.040 | 0.090 | 0.016 |
| hia | 0.000 | 0.000 | 0.000 | 0.001 | 0.000 | 0.002 | 0.006 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.001 | 0.000 | 0.000 | 0.000 |
| BBB | 0.110 | 0.283 | 0.358 | 0.722 | 0.283 | 0.026 | 0.006 | 0.923 | 0.257 | 0.973 | 0.941 | 0.981 | 0.913 | 0.590 | 0.564 | 0.998 |
Table 4.
Docking results of designed compounds for anticancer and anti-Alzheimer activity.
| Ligand | Target | ΔG (kcal/mol | Benchmark (ΔG) | Target affinity profile |
| 15 | AChE | -10.6 | Donepezil (-11.8) | Strong Inhibitor |
| 15 | Tubulin | -9.7 | Colchicine (-10.1) | Moderate Inhibitor |
| 16 | AChE | -9.7 | Donepezil (-11.8) | Moderate Inhibitor |
| 16 | Tubulin | -10.0 | Colchicine (-10.1) | Strong Inhibitor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



