
Submitted:
19 November 2025
Posted:
20 November 2025
You are already at the latest version
Abstract
Immunothrombosis can affect substantially the course and outcomes of severe infections and immune-mediated diseases. While inflammatory thrombi are neutrophil-rich, impact of neutrophils on clot contraction, a key modulator of thrombus stability and obstructiveness, was unknown. This study investigated how neutrophils and neutrophil extracellular traps (NETs) affect the rate and extent of platelet-driven clot contraction. Isolated human neutrophils were activated with phorbol-12-myristate-13-acetate (PMA) to induce NETosis, confirmed by fluorescence microscopy and scanning electron microscopy. Thrombin-induced clots, formed from whole blood or platelet-rich plasma, were supplemented with non-activated or PMA-activated neutrophils. Clot contraction kinetics and viscoelasticity were analyzed. PMA-activated neutrophils significantly enhanced the rate and final extent of clot contraction compared to controls. This promoting effect was abolished by DNAse I, confirming that it was mediated by NETs embedded in the fibrin network. The factor Xa inhibitor rivaroxaban also abrogated this effect, indicating a role for NET-induced endogenous thrombin generation and platelet hyperactivation. Thromboelastography revealed that NETs made clots softer and more deformable. We conclude that activated neutrophils promote clot contraction via NETs embedded into the fibrin network, which enhance platelet contractility via endogenous thrombin production and increase clot deformability, suggesting that inflammatory thrombosis may require treatments addressing this enhanced contractility.
Keywords:
neutrophils
; neutrophil extracellular traps
; clot retraction
; immunothrombosis
1. Introduction
Inflammatory thrombosis, or immunothrombosis, represents a pathophysiological intersection between inflammation and blood clotting, playing an important role in various infectious and immune-mediated inflammatory diseases with a relatively high prevalence globally and remaining a significant cause of illness and death [1,2,3]. Thrombosis is a key driver of morbidity and mortality in sepsis and severe viral infections such as COVID-19 [4,5], as well as in many autoimmune conditions like antiphospholipid syndrome, systemic lupus erythematosus, and rheumatoid arthritis, with thromboembolic events occurring 2- to 10-fold more often than in the general population [6,7,8,9]. Immunothrombosis is associated with the activation of immune cells, particularly neutrophils and monocytes, which contribute to thrombus formation through the release of procoagulant agents and formation of neutrophil extracellular traps (NETs) [10] that amplify both blood clotting and local inflammatory responses [11,12].
An important yet understudied aspect of thrombosis is the process of platelet-driven clot contraction (retraction), which has long been recognized in vitro as a manifestation and measure of platelet functionality [13,14,15], but it also occurs in vivo as a part of hemostasis and thrombosis [16,17,18]. Platelet-driven contraction modulates key intravascular thrombus properties, including obstructiveness, susceptibility to lysis, stiffness, porosity, and resistance to rupture, all of which determine clinical course and outcomes of thrombosis [19]. However, the interrelation between blood clot contraction and immunothrombosis remains poorly understood.
It is known that clot contraction is highly dependent on thrombus cellular composition, in particular the number of platelets and volume fraction of red blood cells (RBCs) [20]. Although RBCs passively resist clot shrinkage, it has been recently shown that aggregation of RBCs within a clot may reinforce platelet-driven contraction and promote clot compaction when there are few and/or dysfunctional platelets [21]. Given that inflammatory thrombi are enriched with neutrophils, their presence likely alters contraction dynamics compared to conventional non-inflammatory thrombi. Activation of neutrophils is accompanied by NETosis, the extrusion of nuclear chromatin components, in particular DNA and histones (NETs), under the influence of inflammatory stimuli [10,22]. NETs formation is known to enhance blood clotting and promote platelet activation, but the real effects of NETs on blood clot contraction remain unknown.
This study fills a gap in understanding the interplay between immunothrombosis and blood clot contraction. The specific aim of the current work was to study the effects of the activated neutrophils on the rate and extent of platelet-driven contraction of blood clots. We have shown that activated neutrophils promote blood clot contraction and this effect is associated with formation of extracellular traps embedded into the fibrin network. NETs stimulated clot contraction by enhancing the production of endogenous thrombin as well as affecting the mechanical properties of blood clots.
2. Materials and Methods
2.1. Blood Collection and Fractionation
All procedures involving human subjects received approval from the Ethical Committee of Kazan Federal University (December 28, 2020, protocol #27; renewed February 26, 2025, protocol #53). Written informed consent was obtained from healthy subjects enrolled in the study. For in vitro model experiments, blood was collected from 30 donors, comprising 20 (67%) men and 10 (33%) women, aged 18 to 36 years (average 26±4 years). Only subjects who had not taken anticoagulants or non-steroidal anti-inflammatory drugs within 2 weeks prior to blood drawing were included in the study. Blood collection and handling adhered to approved guidelines and followed standard pre-analytical requirements. Venous blood was collected by venipuncture into vacutainers containing 3.2% trisodium citrate (Vacuette, Greiner Bio-one, USA) and mixed 9:1 by volume. One portion of citrated blood samples was used for neutrophil isolation (see below). The second portion was centrifuged (200g, 10 min, room temperature) to obtain platelet-rich plasma (PRP). A portion of PRP was centrifuged at 2,000g for 10 min to obtain platelet-poor plasma (PPP) and a part of PPP was centrifuged at 10,000g for 5 min to obtain platelet-free plasma (PFP). Whole blood and its fractions were used no later than 4 h after blood collection.
2.2. Isolation and Activation of Neutrophils to Produce Neutrophil Extracellular Traps (NETs)
Neutrophils were isolated from 5 ml of fresh citrated blood layered on 5 ml of the Lympholyte-Poly Cell Separation Media (Cedarlane, Canada) by centrifugation at 500g for 35 min at room temperature. Following centrifugation, the opaque layer containing neutrophils was collected, and the cells were washed three times with Ca2+- and Mg2+-free Hank’s balanced salt solution (HBSS) by resuspension and centrifugation for 10 min at 300g. After the last wash, the cells were resuspended in 250 μl of HBSS. The number, purity and morphology of neutrophil preparations were assessed using light microscopy in a hemocytometer, flow cytometry, and scanning electron microscopy, respectively (see below). A portion of the isolated neutrophils was activated by adding 100 nM (final concentration) phorbol-12-myristate-13-acetate (PMA) and incubating 3.5 hours at 37ºC in a CO2 incubator (New Brunswick Galaxy 170R, Marshall Scientific, USA) to induce the release of NETs. In further experiments, two sources of NETs were used: i) suspension of PMA-activated neutrophils and ii) supernatant of PMA-activated neutrophils formed after gravitational sedimentation of the cells.
2.3. Flow Cytometry of Isolated Neutrophils
To assess purity and functional state of neutrophils, flow cytometry with fluorescently labeled monoclonal antibodies was used. Isolated neutrophils, either non-activated or PMA-activated, were incubated for 10 min in the dark in the presence of two types of labeled antibodies: i) anti-human CD16 (BD Biosciences, USA) used as a neutrophil-specific marker; ii) anti-human-activated CD11b (Biolegend, USA) used as a marker of neutrophil activation since CD11b is the αM subunit of the αMβ2 (Mac-1) integrin. Anti-CD16 antibodies were labeled with phycoerythrin (PE), while anti-CD11b antibodies were labeled with allophycocyanin (APC). Flow cytometry was conducted using either a FACSCalibur (Beckman Dickinson, USA) or CytoFlex (Beckman Coulter, USA) instrument, and data were analyzed using FlowJo software (Beckman Dickinson, USA). For each measurement, 5000 neutrophils were analyzed. Neutrophils were initially gated based on their forward scatter (FSC) and side scatter (SSC) characteristics and quantified further as CD16+ signals. Within a CD16+ gate, activated neutrophils were identified as CD11b+ events and presented as a fraction of CD16+ signals taken as 100%.
2.4. (Immuno)Histochemical Examination of PMA-Activated Neutrophils and NETs
For histological studies, PRP clots containing non-activated or PMA-activated neutrophils were formed in a tube pre-lubricated with 1% Pluronic F-127 (MilliporeSigma, USA) to prevent the clot sticking to the walls. A PRP sample containing ~48×106 platelets was added to ~0.8×106 neutrophils in HBSS (~60:1 physiological cell type ratio) to a final volume of 500 μl, and clot formation was initiated by the addition of 2 mM CaCl2 and 1 U/ml thrombin (final concentrations). After incubating for 30 min, the clots were fixed in 10% neutral buffered formalin for 1 hour, then rinsed with water, treated with ascending concentrations of isopropanol and xylene using a tissue processor (STP420ES, Thermo Scientific, USA), and embedded in paraffin. Four micrometer-thick sections were deparaffinized and stained with a hematoxylin and eosin histochemical kit (Thermo Scientific, USA). Another set of 4-μm-thick slices was used for immunohistochemical staining. For live confocal microscopy of hydrated unfixed samples, PRP clots containing PMA-activated neutrophils were formed in confocal dishes and subjected to immunofluorescent analysis. The same staining techniques were used to visualize cells and NETs in clots and isolated PMA-activated neutrophils.
For the peroxidase method of antigen visualization, clot sections were first deparaffinized and rehydrated using standard techniques. For antigen retrieval, the slices were boiled for 30 minutes in 0.01M citrate buffer (pH 6.0), left in the same buffer for another 15-20 minutes, and then transferred to Dulbecco’s phosphate buffered saline (DPBS). To block endogenous peroxidase activity, the samples were incubated for 20 minutes in a 3% hydrogen peroxide solution in DPBS. To reduce non-specific binding, all preparations were incubated for 2 hours in 200 μl of a blocking solution containing 10% donkey serum (d9663, Sigma-Aldrich, USA) and 1% bovine serum albumin (Dia-M, USA) in DPBS. The next step was overnight incubation with primary rabbit antibodies against human citrullinated histones H3 (R2+8+17, H3Cit; cat. # ab5103; Abcam, UK) diluted 1:400 in DPBS containing 1% BSA. For control, clots were formed in the absence of neutrophils, without primary antibodies (DPBS instead of antibodies) or by adding mouse IgG1 for isotype control. After washing with DPBS, the sections were incubated with secondary biotinylated antirabbit antibodies (VECSTATIN ABC-HRP Kit, Vector Laboratories, USA) for 30 minutes, washed and incubated with the peroxidase-containing solution of the kit for 30 minutes. The washed slices were processed using 3,3’-diaminobenzidine (DAB) solution (K3467, Dako, USA), a chromogenic peroxidase substrate. Alternatively, fluorescently labeled secondary antibodies (donkey anti-rabbit IgG (H+L), Alexa Fluor™ 488, A21206, Invitrogen, USA) were used at a 1:400 dilution and incubated for 1 hour in the dark. To visualize DNA, the clot was washed in DPBS, fixed for one hour in 10% buffered paraformaldehyde, and then subjected to DNA-specific 4’,6-diamidino-2-phenylindole (DAPI, BioLegend, USA) staining in the dark. Histological imaging was performed using a Hamamatsu NanoZoomer 560 scanner (Hamamatsu Photonics, Japan); confocal fluorescent light microscopy was performed using a laser scanning confocal microscope LSM 780 (Carl Zeiss, Germany).
2.5. Scanning Electron Microscopy of Neutrophils and NETs
For high-resolution scanning electron microscopy, the following samples were prepared i) isolated non-activated and PMA-activated neutrophils; ii) supernatant of PMA-activated neutrophils; iii) thrombin-induced PFP clots formed in the presence of PMA-activated neutrophils. Fixed isolated neutrophils and the supernatant were layered on coverslips pre-treated with 0.01% poly-L-lysine (EMD Millipore, USA) overnight and incubated for 30 min for attachment of cells and NETs on a cover glass followed by fixation in 2% glutaraldehyde for 90 min at room temperature. Thrombin-induced PFP clots in the presence of activated neutrophils (see above) were allowed to form on coverslips for 20 min and then fixed in 2% glutaraldehyde for 90 min at room temperature. All the fixed samples were rinsed three times with a 50 mM cacodylate buffer, containing 150 mM NaCl, pH 7.4, for 10 min, dehydrated in ascending concentrations of ethanol, immersed in hexamethyldisilazane, and air-dried overnight. A thin film of gold palladium was layered on the samples using a sputter coater Quorum Q 150T ES (Quorum, Lewes, UK). Micrographs were taken with a scanning electron microscope (Merlin, Carl Zeiss, Germany).
2.6. Blood Clot Contraction Assay
Contraction of thrombin-induced clots in whole citrated human blood or PRP was tracked optically as a reduction of clot size in the absence or presence of non-activated or PMA-activated neutrophils. In the control samples, the corresponding volume of HBSS buffer was added. The kinetics of clot contraction was followed using the Thrombodynamics Analyzer System (HemaCore Ltd., Russia). To study effects of isolated non-activated or PMA-activated neutrophils, 60-80 μl HBSS containing ~8×105 neutrophils were added to 120-140 μl of blood or PRP to the final volume of 200 μl. The samples were activated with 2 mM CaCl2 and 1 U/ml human thrombin (Sigma-Aldrich, USA) (final concentrations). Before clot formation, the activated blood samples (80 µl) were quickly transferred to 12 mm × 7 mm × 1 mm transparent plastic cuvettes, which were pre-lubricated with a layer of 4% v/v Triton X-100 in 150 mM NaCl for blood or 1% Pluronic for PRP to prevent the clot sticking to the walls of the chamber without affecting the clot structure and platelet functionality. The cuvette was placed at 37ºC into the temperature-controlled chamber of the optical analyzer. Light scattering-based images of the contracting clots were taken every 15 sec for 20 min to track changes in the clot size. The serial images were analyzed computationally to plot a kinetic curve of clot contraction and extract the following parameters: i) the extent of contraction (%) calculated from the initial and final clot size at the 20-min end point; ii) lag time (sec) determined as the time from the addition of thrombin until the clot reaches 95% of its initial size; iii) the average contraction rate, which is a final extent of contraction divided by the time; and iv) the area under the contraction kinetic curve (a.u.), an integral characteristic of the intensity of clot contraction.
2.7. Thromboelastography (TEG)
Thromboelastograms were recorded using the TEG 5000 instrument (Haemoscope, USA). Citrated blood or PRP samples were mixed with either a physiological buffer (control) or isolated neutrophils, non-activated or PMA-activated. Next, CaCl2 (final concentration 2 mM) and human thrombin (Sigma-Aldrich, USA) (final concentration 1 U/ml) were added, after which 360 μl of the mixture was quickly transferred into a TEG cuvette preheated to 37 °C, and the measurement was started. After recording a thromboelastogram, the following TEG parameters were determined: R — reaction time, i.e., the time from the initiation of clotting to the onset of fibrin formation; G’ – the storage (elastic) modulus of a clot calculated from the maximal amplitude (MA in mm) according to the manufacturer’s formula: G’ (dyn/cm2)=[5000*MA/(100-MA)].
2.8. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, USA). Normality of data distribution was assessed with the Shapiro–Wilk and D’Agostino–Pearson criteria. Pairwise statistical differences were estimated using the Student’s paired t-test (parametric analysis) and Mann-Whitney U test or Wilcoxon test (nonparametric analysis). Statistical differences for multiple comparisons were estimated using one-way ANOVA with the post-hoc Sidak’s test. Multi-group column Repeated Measures-analysis for data with non-normal distribution was performed using the nonparametric Friedman test. The level of significance was 95% (p < 0.05).
3. Results
3.1. Effects of Activated Neutrophils on Clot Contraction
To mimic major aspects of thromboinflammation, we studied effects of PMA-activated neutrophils on clot contraction in comparison with non-activated neutrophils added to whole blood or PRP at the amount corresponding to a physiological neutrophil count (8,000/μl). The averaged kinetic curves of clot contraction in whole blood (Figure 1A) and PRP (Figure 1B) in the absence (control) and presence of non-activated or PMA-activated neutrophils showed that the final extent of contraction was higher in the presence of PMA-activated neutrophils. Numerical data analysis confirmed that there was a significant difference in the final extent of clot contraction in the presence of PMA-activated neutrophils versus non-activated neutrophils and control samples without addition of exogenous neutrophils both in whole blood (Figure 1C) and PRP (Figure 1D).
Although the kinetics of clot shrinkage in the curves look somewhat similar, the overall dynamics reflecting the rate of contraction was significantly higher in the presence of the PMA-activated neutrophils (p<0.0001, Friedman test). The numerical values of the parameters studied and statistical analysis are presented in Table S1. Notably, there was a significant decrease in the average lag time of contraction in whole blood clots and an increase in the average rate of contraction in PRP in the presence of PMA-activated neutrophils compared to non-activated neutrophils and neutrophil-free control samples (Figure S1, Table S1). Other kinetic parameters of whole blood clot contraction also showed a trend toward acceleration of clot contraction by the PMA-treated neutrophils but they were statistically insignificant. Taken together, these differences suggest accelerated platelet activation [23] and increased contractility in the presence of activated neutrophils.
To exclude the possibility that the observed stimulating effects of PMA-treated neutrophils on clot contraction were due to direct effects of the residual PMA on platelets or other blood cells, we performed a control experiment in which PMA alone (without neutrophils) was added to PRP in the same final concentration (2.2 nmol) as with the PMA-treated neutrophil preparations. The results showed that PMA at this low concentration affected neither the rate nor the final extent of clot contraction, remaining at the level of 80-83% (Figure S2, Table S2). Therefore, a potential artifact associated with the contamination of neutrophils with PMA has been excluded.
3.2. Kinetic Phase Analysis of Clot Contraction in the Presence of Activated Versus Non-Activated Neutrophils
It has been previously shown that blood clot contraction occurs in three kinetic phases: initiation of contraction (phase 1), linear contraction (phase 2), and mechanical stabilization (phase 3) [20]. Here, we performed the same type of phase analysis for individual kinetic curves of clot contraction in whole blood and PRP averaged in Figure S3.
Both in whole blood and PRP clots, in the presence of PMA-activated neutrophils, the duration of the initiation phase (phase 1) increased (Figure S3; Table S3). In combination with the higher final extents of clot contraction in the presence of PMA-activated neutrophils compared to non-activated neutrophils (Figure 1), this finding suggests a key and dominant role of phase 1 reactions in the overall enhancement of clot contraction caused by the PMA-activated neutrophils. Considering that phase 1 reflects platelet activation caused by exogenous and endogenous thrombin, the observed extension of the first phase in the presence of PMA-activated neutrophils suggests gradual generation of the endogenous thrombin and prolonged but enhanced platelet activation.
Unlike the durations, the rates of each kinetic phase were less consistent and had a different dependency on the presence of activated neutrophils. Regression analyses conducted on the kinetic curves of contraction obtained in whole blood clots (Figure S3A) revealed that in the presence of PMA-activated neutrophils the average rate constants of phases 2 and 3 were reduced compared to non-activated neutrophils (Table S3). The same trend was revealed in the kinetics of contraction in PRP clots (Table S3), altogether indicating facilitated mechanical compaction of the clots in the presence of activated neutrophils. Since phases 2 and 3 are largely mechanical, i.e., reflecting progressive clot compression and mechanical stabilization [20], the observed differences in the phase rates suggest changes of the mechanical properties of fibrin in the presence of activated neutrophils.
3.3. Visualization of NETs Produced by PMA-Activated Neutrophils
Treating isolated neutrophils with 100 nM PMA at 37ºC for 3.5 hours caused their activation as assessed by the surface expression of active integrin Mac-1 (CD11b) measured by flow cytometry (Figure S4). Within the neutrophil gate of CD16+ signals taken as 100%, the average fraction of neutrophils expressing active CD11b in the control untreated samples was only 0.9±1.3% (Figure S4A), while in the PMA-treated samples, the average fraction of neutrophils expressing the active integrin Mac-1 reached 49±16% (p=0.03) (Figure S4B) indicating strong activation of neutrophils by PMA.
Most importantly, under the conditions applied, the PMA-activated neutrophils formed NETs, which could be visualized directly either (immuno)histochemically after staining for histones and DNA (Figure S5A-C) or with scanning electron microscopy (Figure S5D-G). As a valid sign of NETosis in the PMA-activated neutrophil preparations, NET-specific citrullinated histones H3 and DNA were clearly detected as extracellular structures (Figure S5A-C). The scanning electron micrographs captured an early stage of NETosis with a nascent NET (Figure S5D) and the end of NETosis, where a free NET was no longer connected to a neutrophil (Figure S5E). NETs could be visualized not only amid activated neutrophils, but also in the supernatant of a PMA-treated neutrophil preparation (Figure S5F,G). Remarkably, the high-resolution scanning electron microscopy showed that the plasma membrane of PMA-treated neutrophils was highly porous (Figure S5D), confirming that uncondensed chromatin, the structural basis of NETs, could be extruded through these pores as shown earlier [24,25].
3.4. The Effects of Activated Neutrophils on Clot Contraction Are Associated with NETs Embedded Into a Clot
To test if the observed effects of PMA-activated neutrophils on clot contraction are due to the NETs revealed in the neutrophil preparations, we first tried to see if NETs are existent in PRP clots formed in the presence of the activated neutrophils. Figure 2 provides direct evidence that NETs are incorporated into the structure of the clot, since they could be visualized histologically with and without fluorescence (Figure 2A-F) as well as with scanning electron microscopy (Figure 2G,H), although sometimes it is hard to clearly distinguish the fibrillar structures of NETs from fibrin [26]. These findings show that blood clotting in the presence of neutrophils pre-activated for a few hours with PMA results in the embedding of NETs into the clot, strongly suggesting that the observed effects of PMA-activated neutrophils on clot contraction are associated with NETs.
This hypothesis of the key role of NETs in clot contraction has been confirmed by experiments in which NETs in a clot were destroyed by enzymatic cleavage with DNAse I. The effects of DNAse I on clot contraction in the absence or presence of PMA-activated neutrophils are shown for whole blood (Figure 3, Table S4) and PRP (Figure S6, Table S5). DNAse I did not induce any changes by itself, but it completely abolished the stimulating effect of PMA-activated neutrophils on the final extent and average rates of clot contraction in whole blood (Figure 3A,D; Table S4). A similar inhibitory effect of DNAse I on clot contraction was observed in PRP-clots in which contraction was initially enhanced with activated neutrophils but overturned by DNase I, as assessed by the final extent of clot contraction and area under the kinetic curve (Figure S6A,C; Table S5). Taken together, these findings indicate that the stimulating effects of PMA-activated neutrophils on clot contraction are associated with NETs incorporated into the whole blood or plasma clots.
3.5. The Stimulating Effect of NETs on Clot Contraction Is Mediated by Enhanced Generation of Endogenous Thrombin
Based on the stimulating effect of PMA-activated neutrophils on the initiation of clot contraction during phase 1 (Figure S3), a conceivable mechanism underlying the amplification of blood clot contraction by NETs is the enhancement of endogenous thrombin production, causing amplified secondary platelet activation and enhanced contractility. To test this assumption, we used rivaroxaban, an inhibitor of factor Xa capable of preventing the formation of endogenous thrombin. Rivaroxaban was added to whole blood (Figure 4, Table S6) or PRP (Figure S7, Table S7) containing non-activated or PMA-activated neutrophils before thrombin-induced clot formation and contraction. Unlike with non-activated neutrophils, in the presence of rivaroxaban the final extent of clot contraction, area under the kinetic curve, and average rate were reduced significantly in whole blood with the PMA-activated neutrophils (Figure 4, Table S6). Similar effects were observed in PRP samples (Figure S7, Table S7), such that in the presence of rivaroxaban the final extent of clot contraction and average rate were substantially lowered with PMA-activated neutrophils, while there were no visible effects of rivaroxaban in the presence of non-activated neutrophils (Figure S7A,D; Table S7).
Therefore, suppression of endogenous thrombin generation via inhibition of factor Xa abrogates the stimulating effect of NETs on clot contraction. The results obtained suggest that in the presence of PMA-activated neutrophils that form NETs, the enhanced clot contraction is due to increased production of endogenous thrombin that hyperactivates platelets, making them more contractile.
3.6. NETs Make the Fibrin Clot Softer
As suggested by the variations in the kinetics of the mechanical stages of clot contraction during phases 2 and 3 (Figure S3, Table S3), another conceivable mechanism underlying the promoting effect of PMA-activated neutrophils on clot contraction is the effect of NETs on the clot mechanical properties or clot deformability. To test this assumption, we utilized a common technique and apparatus to study clot elasticity, namely thromboelastography (TEG), which records changes in clot stiffness over time in response to shear. In addition to the blood clotting time (R in seconds), TEG provides information about a clot mechanical feature known as the maximal clot firmness (MA, maximal amplitude in millimeters), which can be converted to absolute clot stiffness or elasticity (storage modulus, G’, in Pascal). First, we looked at the effects of PMA-activated and non-activated neutrophils on the clotting time and clot stiffness in whole blood and PRP. In whole blood clots, there was no noticeable influence of PMA-activated neutrophils on the parameters of TEG (Figure 5A,B; Table S8), while in PRP clots, NETs slightly prolonged the clotting time and, most importantly, reduced the final stiffness of the clot (Figure 5C,D; Table S8), indicating that NETs make the clot softer.
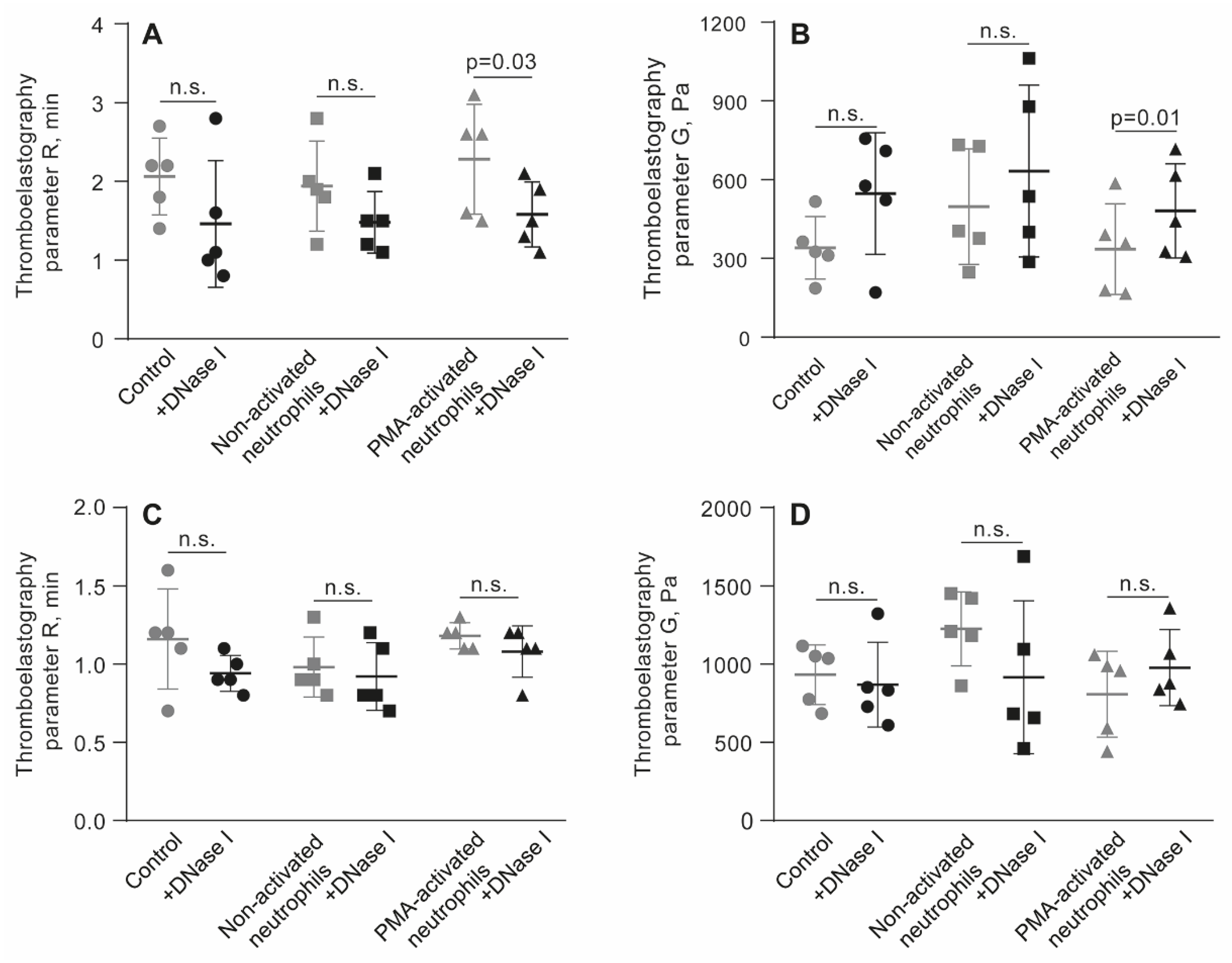
The link between clot stiffness and NETs was additionally tested by applying DNAse I and assessing the effects of NETs cleavage on TEG parameters in whole blood and PRP in the presence of PMA-activated neutrophils. Before thrombin-induced initiation of clotting and registration of TEG, blood or PRP samples were mixed with untreated or PMA-activated neutrophils followed by addition of DNAse I and incubation for 30 min in 37 °C (Figure 6, Table S9). Treatment with DNAse I increased an average storage modulus (G’) in whole blood clots with PMA-activated neutrophils, suggesting that NETs were responsible for the reduced stiffness of the clot. Accordingly, there was no effect of DNAse I in the blood samples containing non-activated neutrophils (Figure 6B, Table S9). Surprisingly, unlike in whole blood, in PRP addition of DNAse I did not affect the TEG parameters (Figure 6C,D; Table S9), perhaps due to the much stiffer plasma clots with a higher volume fraction of fibrin compared to whole blood, as seen from the maximal values on the Y-axes in Figure 6B,D.
4. Discussion
Inflammatory thrombosis or immunothrombosis is a life-threatening complication in which dysregulated co-activation of the immune and blood clotting systems leads to the formation of obstructive venous or arterial thrombi with a strong inflammatory component [27,28]. This dysregulation is a pathogenic mechanism developing in a wide range of severe inflammatory conditions [29,30]. The clinical course and outcomes of inflammatory thrombosis, including severe organ damage and thromboembolic events, as well as efficacy of treatments, are largely dictated by the composition and dynamic properties of the resultant thrombus, such as mechanical stability, permeability, and susceptibility to lysis [31]. One of the major determinants of these properties is the maturation and remodeling of the thrombus, known as platelet-driven clot contraction (aka retraction) [16,19]. Although the fundamental role of platelets, RBCs and plasma proteins in modulating clot contraction is well-established [20], the impact of leukocytes, especially activated neutrophils embedded into the blood clot, remains unclear. To address this question directly, we have employed a model system of neutrophil-enriched human blood or plasma clots, enabling us to analyze how neutrophil activation and NETosis affect the clot contraction process.
The main finding of this study is that activated neutrophils promote the contraction of blood clots, and this effect is directly related to the formation of NETs embedded within the fibrin network. Clot contraction is enhanced substantially in the presence of PMA-stimulated neutrophils (Figure 1) producing extracellular traps, which comprise uncondensed chromatin built of DNA and histones (Figure S5, Figure 2). We have provided direct evidence that NETs are incorporated into the structure of a fibrin clot, since they have been visualized using histochemical techniques (Figure 2A-F), as well as with scanning electron microscopy (Figure 2G,H). Notably, the stimulating effect of PMA-activated neutrophils on clot contraction in both whole blood and PRP (Figure 3A,D; Figure S6A,C) has been shown to be abolished by treatment with DNase I, which catalyzes cleavage and elimination of NETs. Taken together, these results indicate that NETs are essential for the promoting effect of the activated neutrophils on clot contraction. These new data are in line with the growing evidence identifying NETs not merely as ubiquitous components of thrombi but as active, strong (pro)thrombotic structures that promote formation of stable thrombi in various conditions, including deep vein thrombosis, acute ischemic stroke, and myocardial infarction [10,12,29,32,33,34,35].
Trying to decipher the underlying mechanisms, we have identified at least two distinct pathways through which NETs facilitate clot contraction: i) by enhancing the local production of endogenous thrombin and ii) by affecting the mechanical properties of the fibrin network within inflammatory clots.
It has been shown that the histone-DNA complexes within NETs potentiate thrombin generation via platelet-dependent and platelet-independent mechanisms [36,37]. Since thrombin is a potent platelet activator known to enhance their contractility [14,20,38], we assumed that prevention of the NET-induced local generation of thrombin within the clot would reduce or abrogate the promoting effect of the activated neutrophils on clot contraction. Indeed, in the presence of rivaroxaban (an inhibitor of factor Xa), the rate and extent of clot contraction in the presence of PMA-activated neutrophils have been significantly reduced to the level of control non-activated neutrophils (Figure 4A,C,D; Figure S7A,D), strongly suggesting the critical role of NET-mediated endogenous thrombin generation as a mechanisms promoting contraction of inflammatory thrombi. This observation is in line with the findings in which activated monocytes promote clot contraction via expression of tissue factor and local thrombin generation, the most powerful physiological platelet agonist promoting platelet contractility [39]. This observation is also in line with publications showing that NETs promote blood clotting [10,40,41,42].
As an alternative or additional mechanism for boosting clot contraction by the activated neutrophils, we have found that NETs have altered the mechanical properties of clots, decreasing their stiffness, as determined by rheological measurements using TEG (Figure 5D; Figure 6B). Accordingly, the enzymatic cleavage of NETs with DNase I has restored the clot firmness, although this effect was more pronounced in whole blood compared to PRP clots (Figure 6C,D). Altogether, these data suggest that NETs soften blood clots, making the fibrin scaffold less resilient to the platelet-generated contractile forces, thus facilitating clot shrinkage and compaction. A similar conclusion was drawn from the effects of DNA and histones on the mechanical stability of fibrin clots formed by staphylocoagulase [43]. It is noteworthy that the observed softening of plasma clots in the presence of NETs seemingly contradicts the results obtained by Longstaff et al. [44], showing that fibrin clots formed in the presence of the mixture of histones and DNA were more resistant to shear forces. However, when added separately, DNA and histones had opposing effects on fibrin stiffness, such that in the presence of DNA alone, fibrin was more sensitive to mechanical shear. This contradiction suggests complex relations between the components of NETs and fibrin mechanics depending on the concentrations, applied shear stresses, and perhaps other conditions. It is conceivable that clot formation kinetics affected by NETs contribute to the diversity of clot structure which is a major determinant of blood clot mechanics [43,44,45].
The enhanced clot contractility under the influence of activated neutrophils and NETs has important pathophysiological implications, affecting at least three key pathogenic features of in vivo thrombi: obstructiveness, susceptibility to fibrinolysis, and embologenicity. Firstly, the increased contraction of NET-enriched thrombi is likely to reduce the overall clot volume, thereby decreasing the degree of vascular occlusion. This is relevant to inflammatory venous thrombosis (e.g., sepsis-associated or cancer-associated deep vein thrombosis), where highly compacted thrombi may cause less severe flow obstruction compared to voluminous thrombi [46]. Secondly, densification of fibrin during clot contraction is known to increase clot’s susceptibility to endogenous fibrinolysis due to the proximity of fibrin fibers to each other and an increase the t-PA to fibrin ratio [47]. Presumably, NET-enhanced contraction of inflammatory thrombi accelerates natural fibrinolysis, promoting faster resolution and reducing the thrombus durability [48,49,50]. The latter idea is supported by the data showing that following the initial lytic stability, NET-rich fibrin network is ultimately cleaved by plasmin, suggesting self-restraint of inflammatory thrombosis [44,51]. Thirdly, the embologenicity of inflammatory thrombi is potentially diminished because a well-contracted blood clot is more stable mechanically and less prone to rupture and fragmentation [17,52]. In summary, although inflammation creates a hypercoagulable state that promotes thrombosis, the resulting NET-enhanced contraction may paradoxically lower the risk of thrombotic embolization and support gradual dissolution of a thrombus rather than its accidental breakdown [10,36].
The severity of inflammation, and, consequently, the degree of activated neutrophils and NETs involvement, is of great clinical importance for treatment efficacy. Our data suggest that inflammatory NET-rich thrombi are prone to enhanced contraction, resulting in denser occlusive thrombi that may be less susceptible to thrombolytic therapy and mechanical thrombectomy. The reinforced compaction and low porosity of NET-rich thrombi may reduce the efficacy of therapeutic thrombolysis, since contracted clots are less sensitive to the external fibrinolysis [44,47,53,54,55]. It has been shown that embedding histone-DNA complexes into fibrin results in the higher resistance of fibrin clots to external lysis [43,44]. Since the enhanced clot contraction results in formation of mechanically strong and stiff thrombi [32,45,56,57,58], it may be more difficult to remove an inflammatory NET-containing thrombus during mechanical thrombectomy.
Based on the peculiarities of mechanical and structural rearrangement of inflammatory thrombi, assessing the level of soluble NET biomarkers in the blood [29,35,59] and/or developing imaging techniques to detect NET-rich thrombi [60,61] could improve diagnostics and help with the risk prognosis in immunothrombosis [62]. Potentially, adjuvant therapies aimed at cleaving NETs with DNase I or suppressing NETosis with inhibitors of peptidylarginine deiminase type 4 (PAD4), an enzyme that catalyzes citrullination of H3 histones essential for NETosis [63,64], could improve the outcomes of immunothrombosis by rendering inflammatory thrombi more susceptible to therapeutic thrombolysis and mechanical thrombectomy.
5. Conclusions
Activated neutrophils promote blood clot contraction via the formation of NETs. Clinically, the enhancement of clot contraction under the influence of inflammatory cells has both positive and negative aspects. The positive consequences include those related to the pathophysiology of immunothrombosis, namely, reduced thrombus obstructiveness, increased susceptibility to intrinsic lysis, and decreased embologenic potential. Conversely, the negative aspects of enhanced contractility in an inflammatory thrombus include difficulties in its removal, specifically, reduced susceptibility to exogenous thrombolysis and more challenging mechanical thrombectomy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Kinetic parameters of clot contraction in whole blood and PRP clotted by thrombin in the absence and presence of non-activated or PMA-activated neutrophils; Figure S2. Parameters of clot contraction in PRP clotted in the absence (control) and presence of 2.2 nM PMA, corresponding to the final concentration of PMA added with PMA-activated neutrophils; Figure S3. Comparative phase analysis of the averaged kinetic curves of contraction of clots formed in whole blood (A) and PRP (B) in the presence of non-activated and PMA-activated neutrophils; Figure S4. Representative flow cytometry of non-activated and PMA-activated isolated neutrophil preparations; Figure S5. Visualization of NETs formed by isolated PMA-activated neutrophils with immunohistochemistry and scanning electron microscopy; Figure S6. Effects of DNase I on the parameters of clot contraction in PRP clotted in the absence (control) and presence of non-activated or PMA-activated neutrophils; Figure S7. Effects of rivaroxaban on the parameters of clot contraction in PRP clotted in the presence of non-activated or PMA-activated neutrophils; Table S1. Parameters of clot contraction in whole blood and PRP clotted in the absence and presence of non-activated or PMA-activated neutrophils; Table S2. Parameters of clot contraction in PRP in the absence (control) and presence of 2.2 nM PMA, corresponding to the final concentration of PMA added with PMA-activated neutrophil preparations; Table S3. Parameters of comparative phase analysis (phase duration and rate constant) of the averaged kinetic curves of contraction of clots formed in the whole blood and PRP; Table S4. Effects of DNase I on the parameters of clot contraction in whole blood clotted in the absence (control) and presence of non-activated or PMA-activated neutrophils; Table S5. Effects of DNase I on the parameters of clot contraction in PRP clotted in the absence (control) and presence of non-activated or PMA-activated neutrophils; Table S6. Effects of rivaroxaban on the parameters of clot contraction in whole blood clotted in the absence and presence of non-activated or PMA-activated neutrophils; Table S7. Effects of rivaroxaban on the parameters of clot contraction in PRP clotted in the presence of non-activated or PMA-activated neutrophils; Table S8. Parameters of TEG - the reaction time (R) and clot elastic modulus (G’) - in the absence (control) presence of non-activated and PMA-activated neutrophils; Table S9. Effect of DNAse I on the parameters of thromboelastogram (TEG) in the absence (control) and presence of non-activated or PMA-activated neutrophils in whole blood and in PRP.
Author Contributions
Conceptualization, R.I.L. and J.W.W.; methodology, R.I.L.; software, A.I.K.; validation, R.R.K., R.I.L. and J.W.W.; formal analysis, R.R.K.; investigation, S.M.S.; resources, S.M.S.; data curation, A.I.K.; writing—original draft preparation, R.R.K.; writing—review and editing, R.I.L.; visualization, R.R.K.; supervision, R.I.L.; project administration, J.W.W.; funding acquisition, J.W.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health grant PO1-HL146373.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. All procedures involving human subjects received approval from the Ethical Committee of Kazan Federal University (December 28, 2020, protocol #27; renewed February 26, 2025, protocol #53).
Informed Consent Statement
Written informed consent was obtained from healthy subjects enrolled in the study.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.:
Acknowledgments
The authors thank HemaCore Ltd. (Russia) for providing the Thrombodynamics Analyzer System used in this study.
Conflicts of Interest
The authors declare no conflicts of interest. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| NETs | Neutrophil extracellular traps |
| PMA | Phorbol-12-myristate-13-acetate |
| PRP | Platelet-rich plasma |
| DNAse I | Deoxyribonuclease I |
| COVID-19 | Coronavirus disease 2019 |
| RBCs | Red blood cells |
| PPP | Platelet-poor plasma |
| PFP | Platelet-free plasma |
| DNA | Deoxyribonucleic acid |
| HBSS | Hanks’ balanced salt solution |
| DPBS | Dulbecco’s phosphate-buffered saline |
| TEG | Thromboelastography |
| ANOVA | Analysis of variance |
| R | Reaction time |
| G’ | Storage (elastic) modulus |
| MA | Maximal amplitude |
References
- Engelmann, B.; Massberg, S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [CrossRef]
- Ma, L.; Willey, J. The Interplay Between Inflammation and Thrombosis in COVID-19: Mechanisms, Therapeutic Strategies, and Challenges. Thromb. Update 2022, 8, 100117. [CrossRef]
- Schrottmaier, W.C.; Assinger, A. The Concept of Thromboinflammation. Hamostaseologie 2024, 44, 21–30. [CrossRef]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [CrossRef]
- Zhang, J.J.; Dong, X.; Liu, G.H.; Gao, Y.D. Risk and Protective Factors for COVID-19 Morbidity, Severity, and Mortality. Clin. Rev. Allergy Immunol. 2023, 64, 90–107. [CrossRef]
- Duarte-García, A.; Pham, M.M.; Crowson, C.S.; Amin, S.; Moder, K.G.; Pruthi, R.K.; Warrington, K.J.; Matteson, E.L.; et al. The Epidemiology of Antiphospholipid Syndrome: A Population-Based Study. Arthritis Rheumatol. 2019, 71, 1545–1552. [CrossRef]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; DE Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International Consensus Statement on an Update of the Classification Criteria for Definite Antiphospholipid Syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [CrossRef]
- Jorge, A.; Wallace, Z.S.; Lu, N.; Zhang, Y.; Choi, H.K. Renal Transplantation and Survival Among Patients With Lupus Nephritis: A Cohort Study. Ann. Intern. Med. 2019, 170, 240–247. [CrossRef]
- Holmqvist, M.E.; Neovius, M.; Eriksson, J.; Mantel, Ä.; Wållberg-Jonsson, S.; Jacobsson, L.T.; Askling, J. Risk of Venous Thromboembolism in Patients With Rheumatoid Arthritis and Association With Disease Duration and Hospitalization. JAMA 2012, 308, 1350–1356. [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA Traps Promote Thrombosis. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15880–15885. [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled Up in NETs. Blood 2014, 123, 2768–2776. [CrossRef]
- Sun, Y.; Myers, D.R.; Nikolov, S.V.; Oshinowo, O.; Baek, J.; Bowie, S.M.; Lambert, T.P.; Woods, E.; Sakurai, Y.; Lam, W.A.; et al. Platelet Heterogeneity Enhances Blood Clot Volumetric Contraction: An Example of Asynchrono-Mechanical Amplification. Biomaterials 2021, 274, 120828. [CrossRef]
- Carr, M.E., Jr. Development of Platelet Contractile Force as a Research and Clinical Measure of Platelet Function. Cell Biochem. Biophys. 2003, 38, 55–78. [CrossRef]
- Myers, D.R.; Qiu, Y.; Fay, M.E.; Tennenbaum, M.; Chester, D.; Cuadrado, J.; Sakurai, Y.; Baek, J.; Tran, R.; Ciciliano, J.C.; et al. Single-Platelet Nanomechanics Measured by High-Throughput Cytometry. Nat. Mater. 2017, 16, 230–235. [CrossRef]
- Cines, D.B.; Lebedeva, T.; Nagaswami, C.; Hayes, V.; Massefski, W.; Litvinov, R.I.; Rauova, L.; Lowery, T.J.; Weisel, J.W. Clot Contraction: Compression of Erythrocytes Into Tightly Packed Polyhedra and Redistribution of Platelets and Fibrin. Blood 2014, 123, 1596–1603. [CrossRef]
- Khismatullin, R.R.; Abdullayeva, S.; Peshkova, A.D.; Sounbuli, K.; Evtugina, N.G.; Litvinov, R.I.; Weisel, J.W. Extent of Intravital Contraction of Arterial and Venous Thrombi and Pulmonary Emboli. Blood Adv. 2022, 6, 1708–1718. [CrossRef]
- Stalker, T.J.; Welsh, J.D.; Tomaiuolo, M.; Wu, J.; Colace, T.V.; Diamond, S.L.; Brass, L.F. A Systems Approach to Hemostasis: 3. Thrombus Consolidation Regulates Intrathrombus Solute Transport and Local Thrombin Activity. Blood 2014, 124, 1824–1831. [CrossRef]
- Litvinov, R.I.; Weisel, J.W. Blood Clot Contraction: Mechanisms, Pathophysiology, and Disease. Res. Pract. Thromb. Haemost. 2022, 7, 100023. [CrossRef]
- Tutwiler, V.; Litvinov, R.I.; Lozhkin, A.P.; Peshkova, A.D.; Lebedeva, T.; Ataullakhanov, F.I.; Spiller, K.L.; Cines, D.B.; Weisel, J.W. Kinetics and Mechanics of Clot Contraction Are Governed by the Molecular and Cellular Composition of the Blood. Blood 2016, 127, 149–159. [CrossRef]
- Peshkova, A.D.; Rednikova, E.K.; Khismatullin, R.R.; Kim, O.V.; Muzykantov, V.R.; Purohit, P.K.; Litvinov, R.I.; Weisel, J.W. Red Blood Cell Aggregation Within a Blood Clot Causes Platelet-Independent Clot Shrinkage. Blood Adv. 2025, 9, 3418–3428. [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J. Exp. Med. 2020, 217, e20200652. [CrossRef]
- Mitchell, J.L.; Dunster, J.L.; Kriek, N.; Unsworth, A.J.; Sage, T.; Mohammed, Y.M.M.; De Simone, I.; Taylor, K.A.; Bye, A.P.; Ólafsson, G.; et al. The Rate of Platelet Activation Determines Thrombus Size and Structure at Arterial Shear. J. Thromb. Haemost. 2023, 21, 2248–2259. [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J. Cell Biol. 2007, 176, 231–241. [CrossRef]
- Muraro, S.P.; De Souza, G.F.; Gallo, S.W.; Da Silva, B.K.; De Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory Syncytial Virus Induces the Classical ROS-Dependent NETosis Through PAD-4 and Necroptosis Pathways Activation. Sci. Rep. 2018, 8, 14166. [CrossRef]
- Onouchi, T.; Shiogama, K.; Matsui, T.; Mizutani, Y.; Sakurai, K.; Inada, K.; Tsutsumi, Y. Visualization of Neutrophil Extracellular Traps and Fibrin Meshwork in Human Fibrinopurulent Inflammatory Lesions: II. Ultrastructural Study. Acta Histochem. Cytochem. 2016, 49, 117–123. [CrossRef]
- Stark, K.; Massberg, S. Interplay Between Inflammation and Thrombosis in Cardiovascular Pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [CrossRef]
- Noubouossie, D.F.; Reeves, B.N.; Strahl, B.D.; Key, N.S. Neutrophils: Back in the Thrombosis Spotlight. Blood 2019, 133, 2186–2197. [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight 2020, 5, e138999. [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Exploring the Thrombus Niche: Lessons Learned and Potential Therapeutic Opportunities. Blood 2025, 146, 1389–1399. [CrossRef]
- Ducroux, C.; Di Meglio, L.; Loyau, S.; Delbosc, S.; Boisseau, W.; Deschildre, C.; Ben Maacha, M.; Blanc, R.; Redjem, H.; Ciccio, G.; et al. Thrombus Neutrophil Extracellular Traps Content Impair tPA-Induced Thrombolysis in Acute Ischemic Stroke. Stroke 2018, 49, 754–757. [CrossRef]
- Savchenko, A.S.; Martinod, K.; Seidman, M.A.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil Extracellular Traps Form Predominantly During the Organizing Stage of Human Venous Thromboembolism Development. J. Thromb. Haemost. 2014, 12, 860–870. [CrossRef]
- Laridan, E.; Denorme, F.; Desender, L.; François, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Ischemic Stroke Thrombi. Ann. Neurol. 2017, 82, 223–232. [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, M.; Jakowitsch, J.; Panzenböck, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary Neutrophil Extracellular Trap Burden and Deoxyribonuclease Activity in ST-Elevation Acute Coronary Syndrome Are Predictors of ST-Segment Resolution and Infarct Size. Circ. Res. 2015, 116, 1182–1192. [CrossRef]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.; Weitz, J.I.; Liaw, P.C. Neutrophil Extracellular Traps Promote Thrombin Generation Through Platelet-Dependent and Platelet-Independent Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [CrossRef]
- von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, Neutrophils, and Platelets Cooperate to Initiate and Propagate Venous Thrombosis in Mice In Vivo. J. Exp. Med. 2012, 209, 819–835. [CrossRef]
- Carr, M.E., Jr.; Martin, E.J.; Carr, S.L. Delayed, Reduced or Inhibited Thrombin Production Reduces Platelet Contractile Force and Results in Weaker Clot Formation. Blood Coagul. Fibrinolysis 2002, 13, 193–197. [CrossRef]
- Peshkova, A.D.; Le Minh, G.; Tutwiler, V.; Andrianova, I.A.; Weisel, J.W.; Litvinov, R.I. Activated Monocytes Enhance Platelet-Driven Contraction of Blood Clots via Tissue Factor Expression. Sci. Rep. 2017, 7, 5149. [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular Histones Promote Thrombin Generation Through Platelet-Dependent Mechanisms: Involvement of Platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal Coupling of Coagulation and Innate Immunity via Neutrophil Serine Proteases. Nat. Med. 2010, 16, 887–896. [CrossRef]
- Ruf, W.; Ruggeri, Z.M. Neutrophils Release Brakes of Coagulation. Nat. Med. 2010, 16, 851–852. [CrossRef]
- Komorowicz, E.; Farkas, V.J.; Szabó, L.; Cherrington, S.; Thelwell, C.; Kolev, K. DNA and Histones Impair the Mechanical Stability and Lytic Susceptibility of Fibrin Formed by Staphylocoagulase. Front. Immunol. 2023, 14, 1233128. [CrossRef]
- Longstaff, C.; Varjú, I.; Sótonyi, P.; Szabó, L.; Krumrey, M.; Hoell, A.; Bóta, A.; Varga, Z.; Komorowicz, E.; Kolev, K. Mechanical Stability and Fibrinolytic Resistance of Clots Containing Fibrin, DNA, and Histones. J. Biol. Chem. 2013, 288, 6946–6956. [CrossRef]
- Litvinov, R.I.; Weisel, J.W. Fibrin Mechanical Properties and Their Structural Origins. Matrix Biol. 2017, 60-61, 110–123. [CrossRef]
- Wakefield, T.W.; Myers, D.D.; Henke, P.K. Mechanisms of Venous Thrombosis and Resolution. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 387–391. [CrossRef]
- Tutwiler, V.; Peshkova, A.D.; Le Minh, G.; Zaitsev, S.; Litvinov, R.I.; Cines, D.B.; Weisel, J.W. Blood Clot Contraction Differentially Modulates Internal and External Fibrinolysis. J. Thromb. Haemost. 2019, 17, 361–370. [CrossRef]
- Henke, P.K.; Wakefield, T. Thrombus Resolution and Vein Wall Injury: Dependence on Chemokines and Leukocytes. Thromb. Res. 2009, 123, S72–S78. [CrossRef]
- Saha, P.; Humphries, J.; Modarai, B.; Mattock, K.; Waltham, M.; Evans, C.E.; Ahmad, A.; Patel, A.S.; Premaratne, S.; Lyons, O.T.; et al. Leukocytes and the Natural History of Deep Vein Thrombosis: Current Concepts and Future Directions. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 506–512. [CrossRef]
- Kolev, K.; Machovich, R. Molecular and Cellular Modulation of Fibrinolysis. Thromb. Haemost. 2003, 89, 610–621. [CrossRef]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases Prevent Vascular Occlusion by Neutrophil Extracellular Traps. Science 2017, 358, 1202–1206. [CrossRef]
- Peshkova, A.D.; Malyasyov, D.V.; Bredikhin R.A.; Le Minh, G.; Andrianova, I.A.; Tutwiler, V.; Nagaswami, C.; Weisel, J.W.; Litvinov, R.I. Reduced Contraction of Blood Clots in Venous Thromboembolism Is a Potential Thrombogenic and Embologenic Mechanism. TH Open 2018, 2, e104–e115. [CrossRef]
- Varjú, I.; Longstaff, C.; Szabó, L.; Farkas, Á.Z.; Varga-Szabó, V.J.; Tanka-Salamon, A.; Machovich, R.; Kolev, K. DNA, Histones and Neutrophil Extracellular Traps Exert Anti-Fibrinolytic Effects in a Plasma Environment. Thromb. Haemost. 2015, 113, 1289–1298. [CrossRef]
- Tan, Q.; Guo, P.; Zhou, J.; Zhang, J.; Zhang, B.; Lan, C.; Xian, J.; Ge, M.; Feng, H.; Chen, Z. Targeting Neutrophil Extracellular Traps Enhanced tPA Fibrinolysis for Experimental Intracerebral Hemorrhage. Transl. Res. 2019, 211, 139–146. [CrossRef]
- Kumar, R.; Patil, G.; Dayal, S. NLRP3-Induced NETosis: A Potential Therapeutic Target for Ischemic Thrombotic Diseases?. Cells 2023, 12, 2709. [CrossRef]
- Litvinov, R.I.; Weisel, J.W. What Is the Biological and Clinical Relevance of Fibrin?. Semin. Thromb. Hemost. 2016, 42, 333–343. [CrossRef]
- Collet, J.P.; Allali, Y.; Lesty, C.; Tanguy, M.L.; Silvain, J.; Ankri, A.; Blanchet, B.; Dumaine, R.; Gianetti, J.; Payot, L.; et al. Altered Fibrin Architecture Is Associated With Hypofibrinolysis and Premature Coronary Atherothrombosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2567–2573. [CrossRef]
- Wufsus, A.R.; Macera, N.E.; Neeves, K.B. The Hydraulic Permeability of Blood Clots as a Function of Fibrin and Platelet Density. Biophys. J. 2013, 104, 1812–1823. [CrossRef]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Ten Cate, H.; Hofstra, L.; et al. Elevated Levels of Circulating DNA and Chromatin Are Independently Associated With Severe Coronary Atherosclerosis and a Prothrombotic State. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [CrossRef]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ. Res. 2014, 114, 947–956. [CrossRef]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [CrossRef]
- de Maat, M.P.; van Schie, M.; Kluft, C.; Leebeek, F.W.; Meijer, P. Biological Variation of Hemostasis Variables in Thrombosis and Bleeding: Consequences for Performance Specifications. Clin. Chem. 2016, 62, 1639–1646. [CrossRef]
- Sabnis, R.W. Novel Peptidylarginine Deiminase Type 4 (PAD4) Inhibitors. ACS Med. Chem. Lett. 2022, 13, 1537–1538. [CrossRef]
- Aiken, S.G.; Grimes, T.; Munro, S.; Zarganes-Tzitzikas, T.; La Thangue, N.B.; Brennan, P.E. A Patent Review of Peptidylarginine Deiminase 4 (PAD4) Inhibitors (2014–Present). Expert Opin. Ther. Pat. 2025, 35, 611–621. [CrossRef]
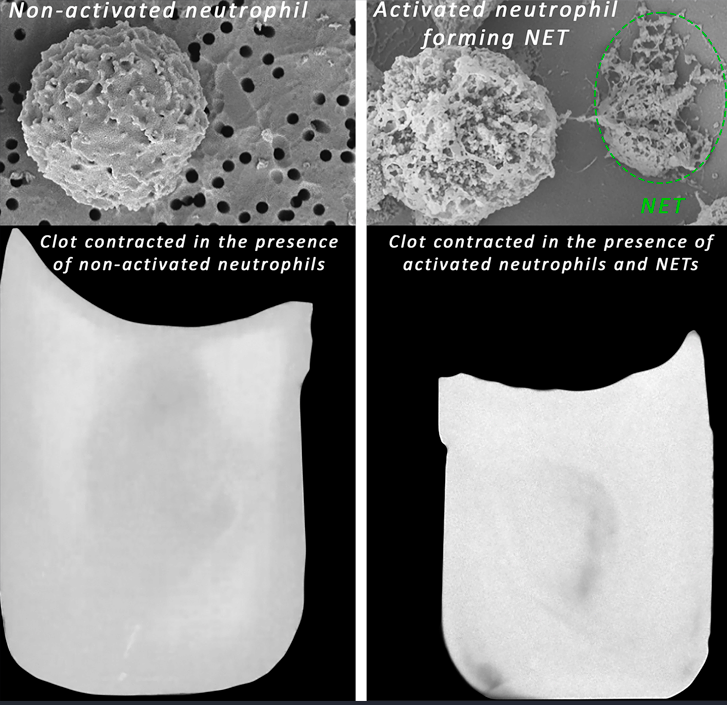
Figure 1.
Effects of PMA-activated vs. non-activated neutrophils on the kinetics of clot contraction. Thrombin-induced clots were formed in whole blood (A, C) or PRP (B, D) in the absence (control) and presence of non-activated or PMA-activated neutrophils followed by optical tracking of the clot size. The results are presented as the averaged kinetic curves (A, B) and the final extents (C, D) of clot contraction. Other kinetic parameters of clot contraction are shown in Figure S1. Data points in the curves and the values of the extent of clot contraction represent the mean ± SD (n=5 for blood, n=4 for PRP, individual donors). One-way ANOVA was used for comparisons. Abbreviation: n.s. - not significant.
Figure 1.
Effects of PMA-activated vs. non-activated neutrophils on the kinetics of clot contraction. Thrombin-induced clots were formed in whole blood (A, C) or PRP (B, D) in the absence (control) and presence of non-activated or PMA-activated neutrophils followed by optical tracking of the clot size. The results are presented as the averaged kinetic curves (A, B) and the final extents (C, D) of clot contraction. Other kinetic parameters of clot contraction are shown in Figure S1. Data points in the curves and the values of the extent of clot contraction represent the mean ± SD (n=5 for blood, n=4 for PRP, individual donors). One-way ANOVA was used for comparisons. Abbreviation: n.s. - not significant.
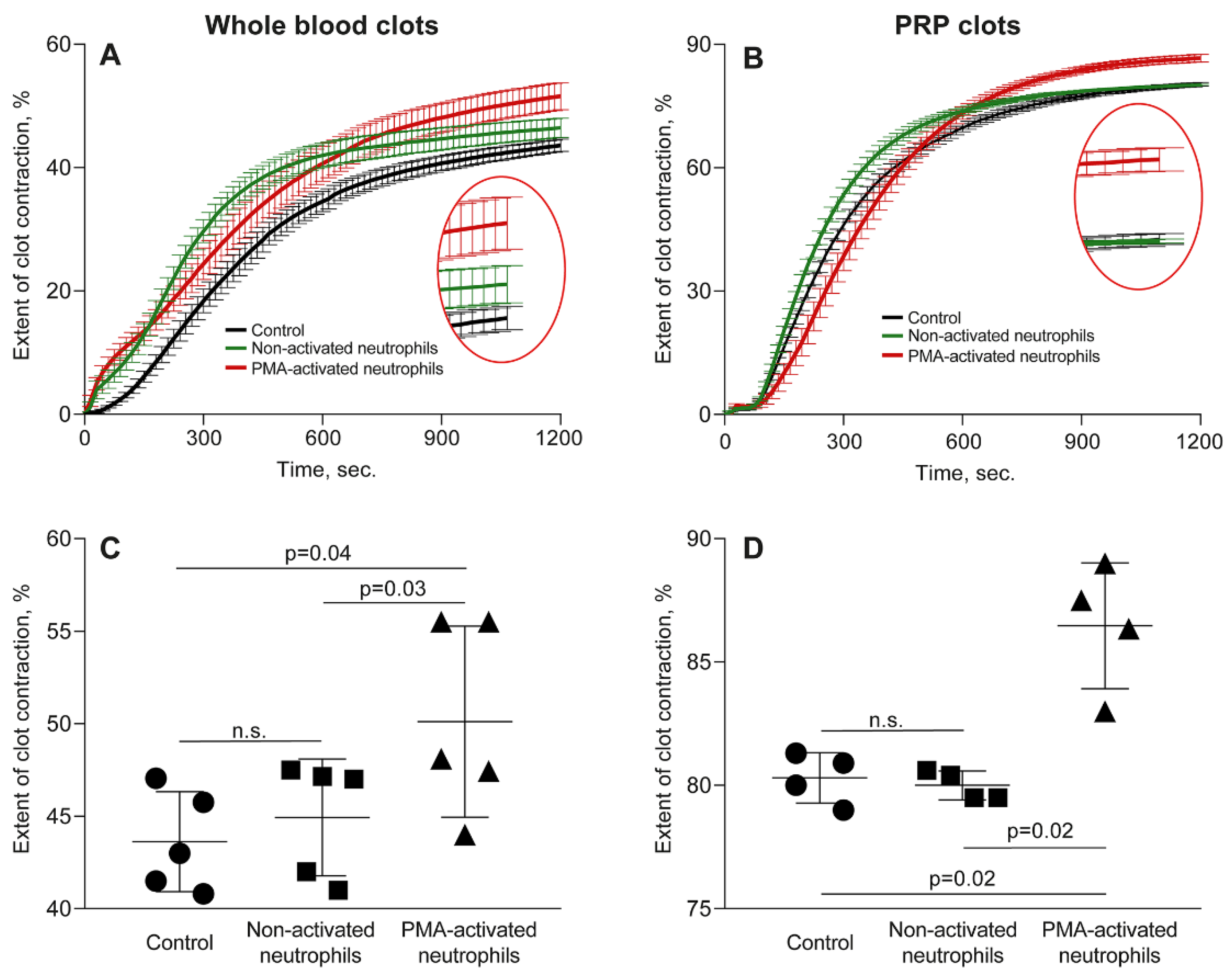
Figure 2.
Representative light microscopy and scanning electron microscopy images of NETs released from PMA-activated neutrophils and embedded within PRP clots. (A-C) Fluorescent microscopy of PMA-activated neutrophils within a PRP clot stained for citrullinated histones CitH3 (A), DNA (B), and overlay (C). (D, E) Non-activated, mainly segmented nuclear neutrophils (green arrows) and PMA-activated predominantly enucleated neutrophils of irregular shapes with indistinct cell membranes and/or pericellular structures (red arrows) within a PRP clot. (F) A PMA-activated neutrophil within a clot stained for citrullinated histones CitH3 (dashed circle) on top of hematoxylin &eosin stain to visualize the cell nucleus and pericellular uncondensed chromatin (NETs). (G, H) Representative scanning electron micrographs of a PRP clots showing the incorporation of PMA-activated neutrophils and surrounding fibrillar structures that comprise NETs and fibrin. Magnification bars: 20 µm (A-C), 50 µm (D, F), 25 µm (E), and 1 µm (G, H).
Figure 2.
Representative light microscopy and scanning electron microscopy images of NETs released from PMA-activated neutrophils and embedded within PRP clots. (A-C) Fluorescent microscopy of PMA-activated neutrophils within a PRP clot stained for citrullinated histones CitH3 (A), DNA (B), and overlay (C). (D, E) Non-activated, mainly segmented nuclear neutrophils (green arrows) and PMA-activated predominantly enucleated neutrophils of irregular shapes with indistinct cell membranes and/or pericellular structures (red arrows) within a PRP clot. (F) A PMA-activated neutrophil within a clot stained for citrullinated histones CitH3 (dashed circle) on top of hematoxylin &eosin stain to visualize the cell nucleus and pericellular uncondensed chromatin (NETs). (G, H) Representative scanning electron micrographs of a PRP clots showing the incorporation of PMA-activated neutrophils and surrounding fibrillar structures that comprise NETs and fibrin. Magnification bars: 20 µm (A-C), 50 µm (D, F), 25 µm (E), and 1 µm (G, H).
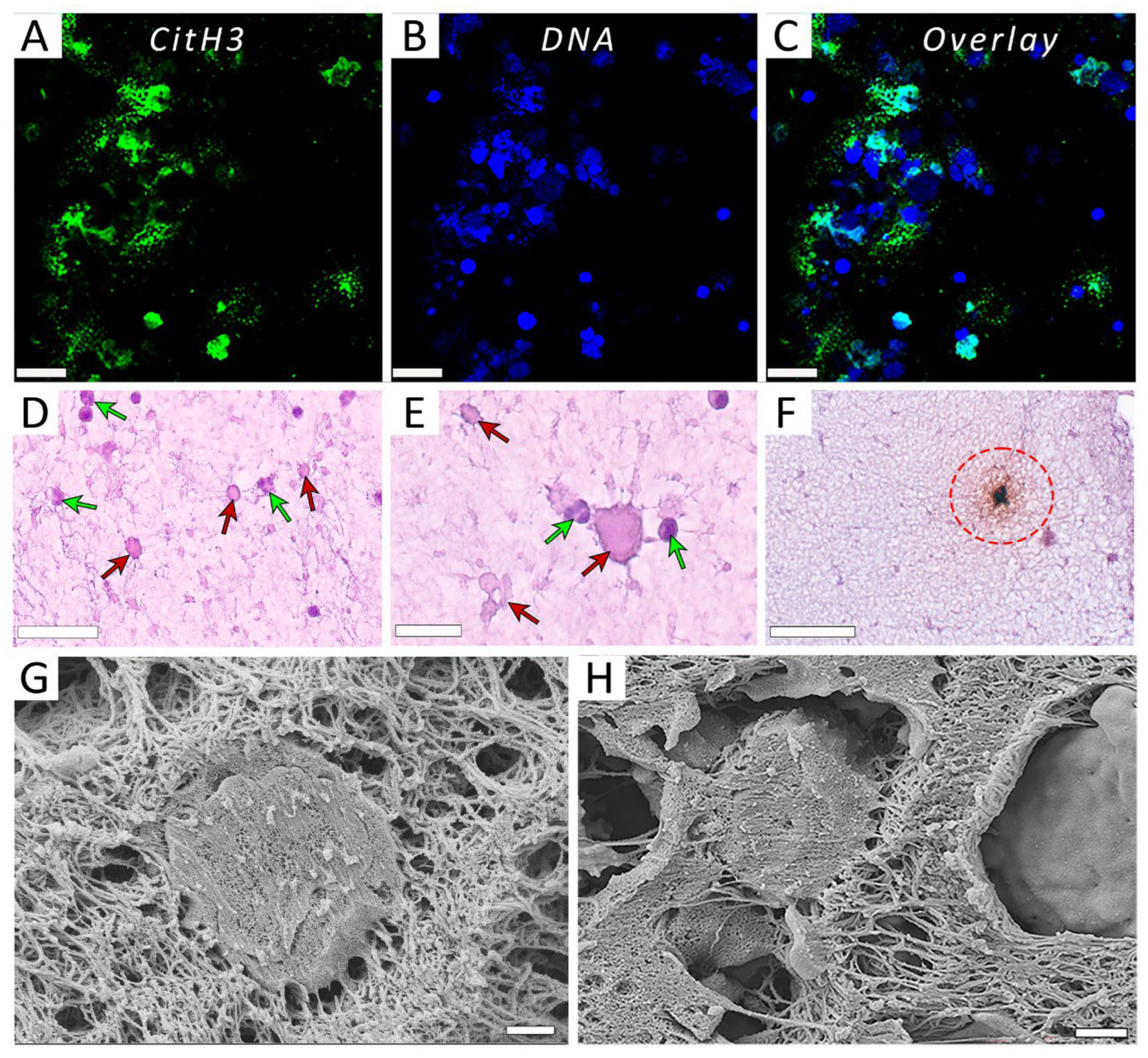
Figure 3.
Effect of DNase I on the parameters of clot contraction in whole blood clotted in the absence (control) and presence of non-activated or PMA-activated neutrophils. Clot formation and contraction were induced by 1 U/ml thrombin followed by the optical tracking of a clot size. The final extent of clot contraction (A), lag time (B), area under the curve (C), and average rate (D) were measured in clots formed in blood samples obtained from independent donors. Results are presented as the mean ± SD (n=5). The Student’s paired t-test was used for comparisons. Abbreviation: n.s. - not significant. For numerical data and detailed statistical analysis see Table S4.
Figure 3.
Effect of DNase I on the parameters of clot contraction in whole blood clotted in the absence (control) and presence of non-activated or PMA-activated neutrophils. Clot formation and contraction were induced by 1 U/ml thrombin followed by the optical tracking of a clot size. The final extent of clot contraction (A), lag time (B), area under the curve (C), and average rate (D) were measured in clots formed in blood samples obtained from independent donors. Results are presented as the mean ± SD (n=5). The Student’s paired t-test was used for comparisons. Abbreviation: n.s. - not significant. For numerical data and detailed statistical analysis see Table S4.
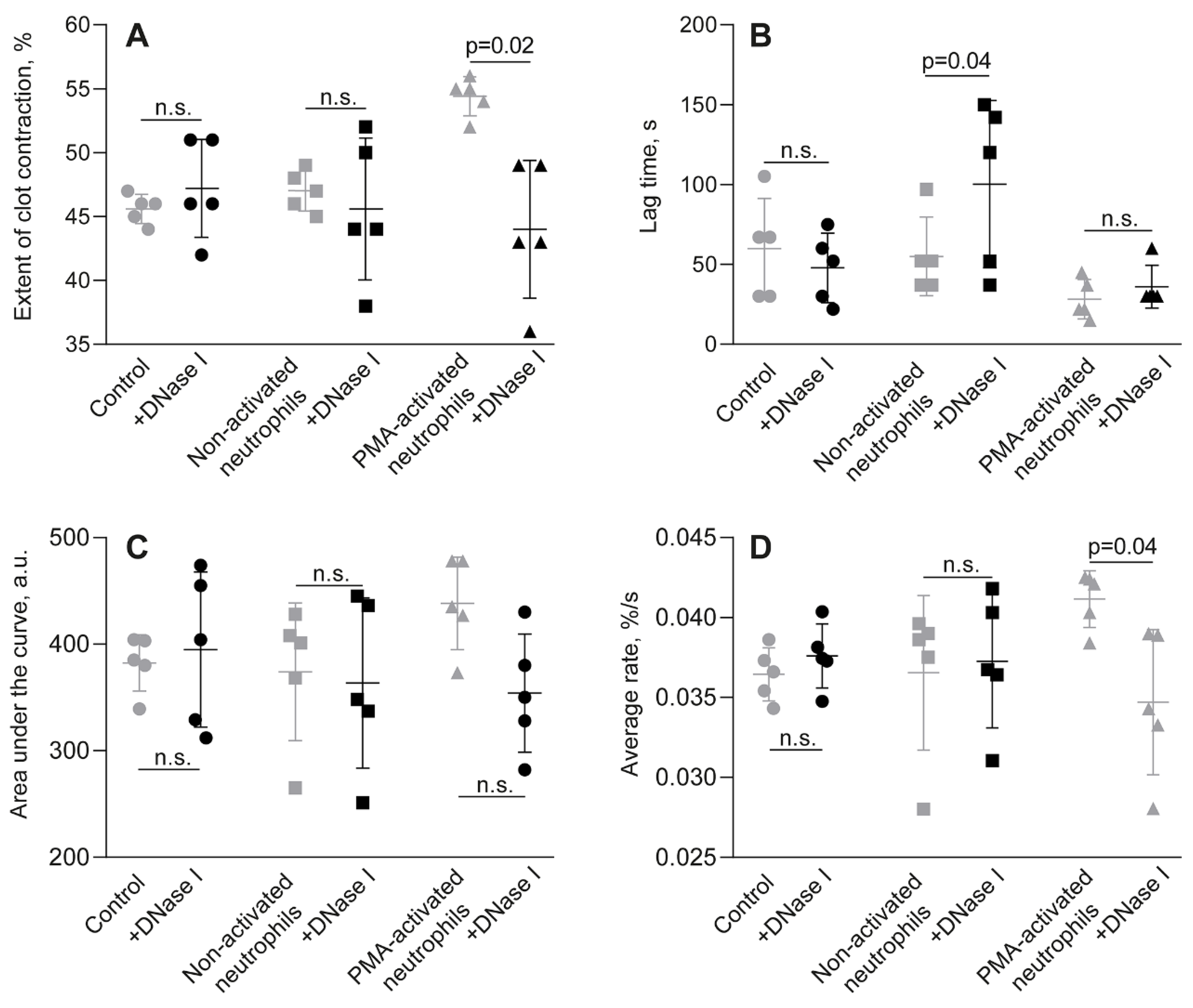
Figure 4.
Effect of rivaroxaban on the parameters of clot contraction in whole blood clotted in the presence of non-activated or PMA-activated neutrophils. Clot formation and contraction were induced by 1 U/ml thrombin followed by the optical tracking of the clot size. The final extent of clot contraction (A), lag time (B), area under the curve (C), and average rate (D) were measured in clots formed in blood samples obtained from independent donors. Results are presented as the mean ± SD (n=5). The Student’s paired t-test was used for comparisons. Abbreviation: n.s. - not significant. For numerical data and detailed statistical analysis see Table S6.
Figure 4.
Effect of rivaroxaban on the parameters of clot contraction in whole blood clotted in the presence of non-activated or PMA-activated neutrophils. Clot formation and contraction were induced by 1 U/ml thrombin followed by the optical tracking of the clot size. The final extent of clot contraction (A), lag time (B), area under the curve (C), and average rate (D) were measured in clots formed in blood samples obtained from independent donors. Results are presented as the mean ± SD (n=5). The Student’s paired t-test was used for comparisons. Abbreviation: n.s. - not significant. For numerical data and detailed statistical analysis see Table S6.
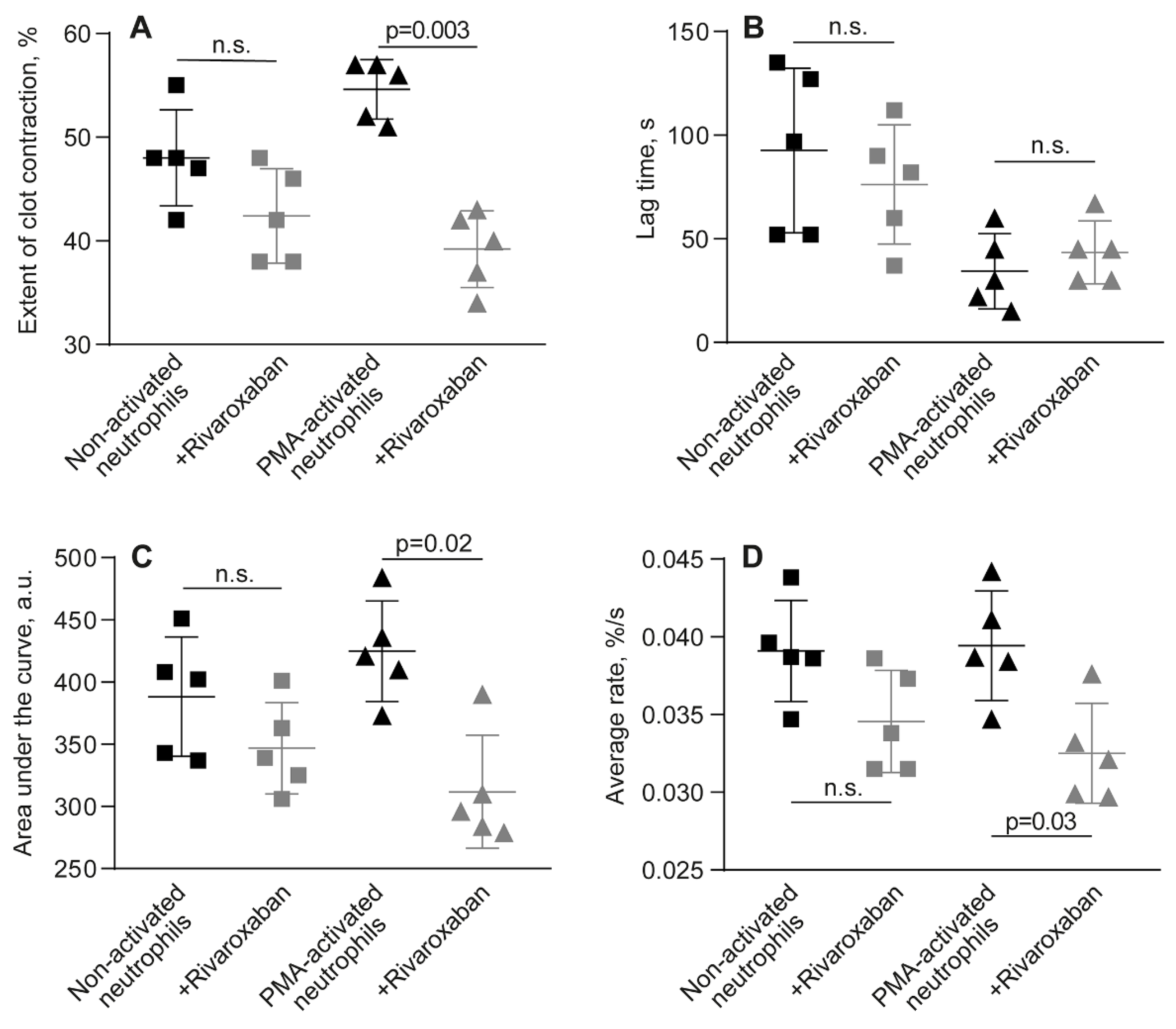
Figure 5.
Parameters of TEG - the reaction time (R) and clot elastic modulus (G’) – measured in the absence (control) and presence of non-activated or PMA-activated neutrophils. Clot formation and contraction were induced by 1 U/ml thrombin in whole blood (A, B) or PRP (C, D) obtained from independent donors. Results are presented as the mean ± SD (n=5). One-way ANOVA. Abbreviation: n.s. - not significant. For numerical data and detailed statistical analysis see Table S8.
Figure 5.
Parameters of TEG - the reaction time (R) and clot elastic modulus (G’) – measured in the absence (control) and presence of non-activated or PMA-activated neutrophils. Clot formation and contraction were induced by 1 U/ml thrombin in whole blood (A, B) or PRP (C, D) obtained from independent donors. Results are presented as the mean ± SD (n=5). One-way ANOVA. Abbreviation: n.s. - not significant. For numerical data and detailed statistical analysis see Table S8.
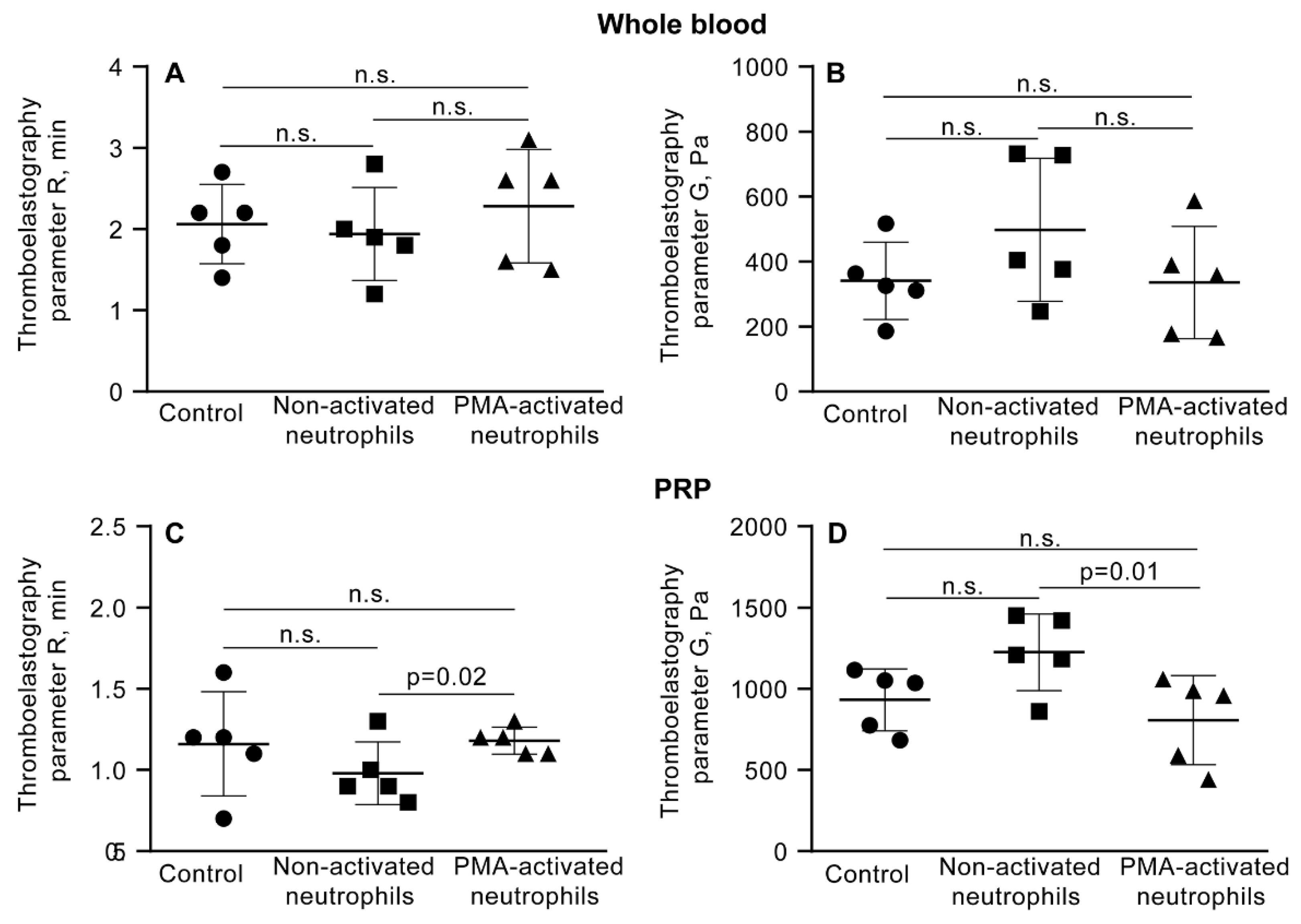
Figure 6.
Effects of DNase I on the parameters of TEG, the reaction time (R) and clot elastic modulus (G’), in the absence (control) and presence of non-activated or PMA-activated neutrophils. Clot formation and contraction were induced by 1 U/ml thrombin in whole blood (A, B) or PRP (C, D) obtained from independent donors. Results are presented as the mean ± SD (n=5). The Student’s paired t-test was used for comparisons. Abbreviation: n.s. - not significant. For numerical data and detailed statistical analysis see Table S9.
Figure 6.
Effects of DNase I on the parameters of TEG, the reaction time (R) and clot elastic modulus (G’), in the absence (control) and presence of non-activated or PMA-activated neutrophils. Clot formation and contraction were induced by 1 U/ml thrombin in whole blood (A, B) or PRP (C, D) obtained from independent donors. Results are presented as the mean ± SD (n=5). The Student’s paired t-test was used for comparisons. Abbreviation: n.s. - not significant. For numerical data and detailed statistical analysis see Table S9.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



