
Submitted:
12 November 2025
Posted:
13 November 2025
You are already at the latest version
Abstract
Background/Objectives: Sickle cells disease (SCD) and -thalassemia are autosomal recessive disorders of erythroid cells due to gene mutations occurring at the level of the -globin gene. The severe forms of these hemoglobinopathies observed in individuals homozygous for these defective genes need intensive treatments, are associated with a poor quality of life and allogeneic hematopoietic stem cell represents the only curative treatment option that can be offered only to a limited proportion of patients. Methods: This work is a narrative review supported by a systematic literature search and analysis. Results: To bypass this limitation, autologous hematopoietic stem cell transplantation has been developed in these patients in which patients’ HSCs are harvested and genetically modified ex vivo, then transplanted back into patients after conditioning for stem cell transplantation. There are two different approaches for gene therapy of hemoglobinopathies’, one based on gene addition or gene silencing using lentiviruses as vectors and the other based on gene editing strategies using CRISPR-Caspase 9 technology or base editing. Several gene therapy products have been successfully evaluated in these patients achieving transfusion independence and correction of hematological abnormalities durable in the time. Conclusions Several gene therapy products have been approved for the treatment of SCD and -thalassemic patients and offer a potentially curative treatment for these patients.
Keywords:
hemoglobin
; gene therapy
; sickle cell disease
; beta thalassemia
; stem cell transplantation
; gene editing
1. Introduction
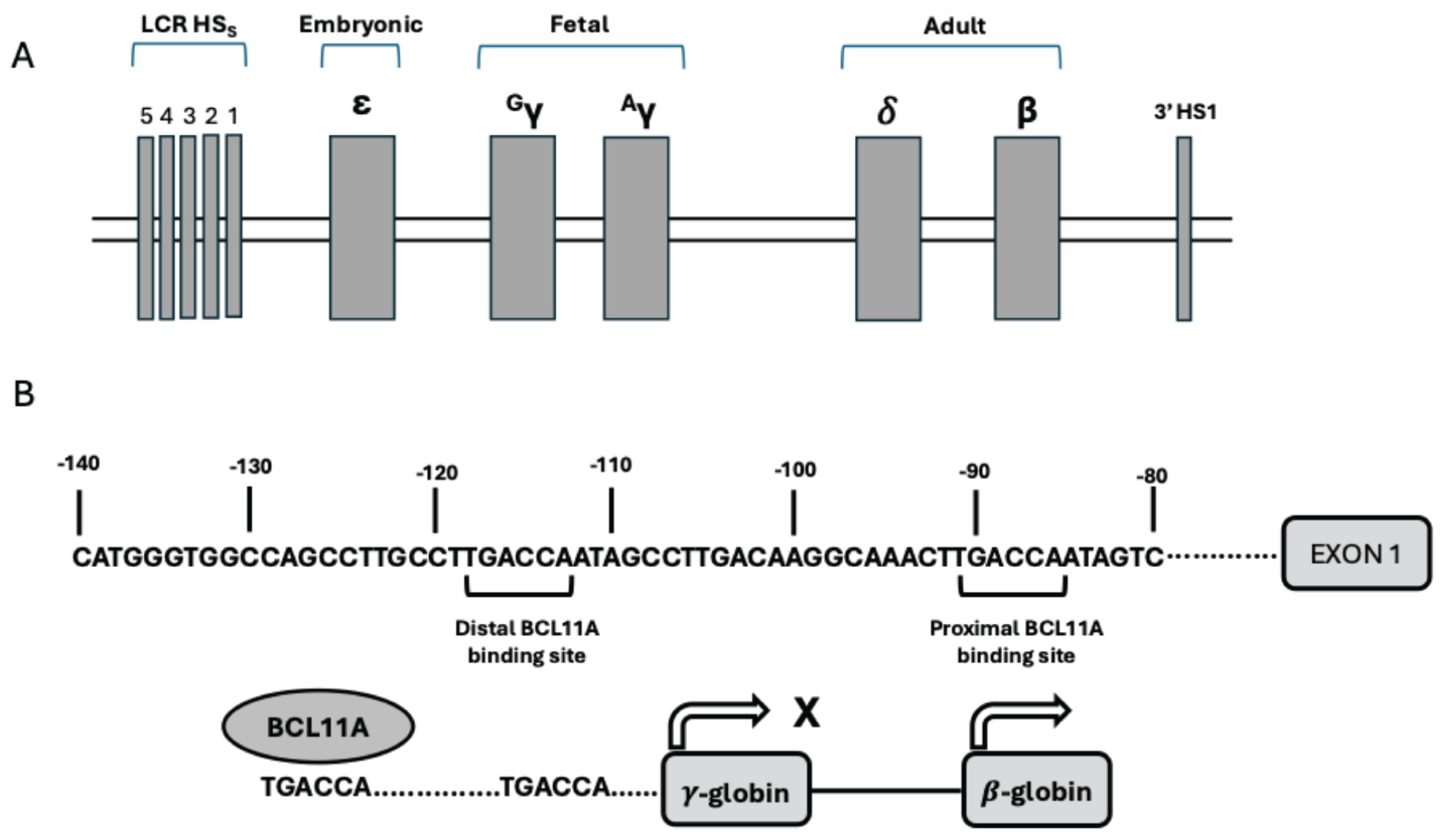
Hemoglobin is a key functional protein of red blood cells (RBCs), constituted by a tetramer α2β2 bound to a heme group; this protein plays a unique and fundamental role as O2 transporter in the body. The level of globin synthesis markedly increases during erythroid differentiation/maturation and is finely controlled through the modulation of the transcriptional activity of α- and β-globin genes. α- and β-globin genes are organized in gene clusters located at the level of chromosomes 16 and 11, respectively.
The HBA (α-globin) locus contains an embryonic gene (ξ-globin) and two adult genes HBA1 (α1-globin) and HBA2 (α2-globin); the expression of these genes is controlled by distal enhancers that are active in erythroid cells at various stages of development [1]. The HBB locus (β-globin gene cluster) is composed by 5 β-like globin genes [ε (HBE), γ2 (HBG2), γ1 (HBG1), δ (HBGD) and β (HBB)]. The physical arrangement of these genes corresponds to their developmental expression: during the embryonic life, the yolk sac primarily synthesizes embryonic hemoglobin (HBE, α2ε2); around the second month of development, transcription of β-like genes shifts to the γ-globin genes (HBG1, α2γ12 and HBG2, α2γ22) and the main site of erythroid cell production becomes the liver; at later stages of development, γ-globin chain syn thesis is progressively replaced by β-globin chain synthesis (HbF to HbA hemoglobin switching) and this process is fully completed only after birth at 9-12 months of age [1]. The activation of the expression of the various β-globin-like genes is controlled by the upstream locus control region (LCR), an enhancer element active at all stages of erythroid development. LCR controls globin genes through physical interaction with the promoters of individual globin genes by looping out the intervening DNA; this looping is mediated by DNA-binding proteins and transcription factors that bridge the LCR and promoters, bringing them in contact, despite their consistent physical distance. These interactions between LCR and specific promoters play a key role in activating gene transcription at a given developmental stage [1]. Thus, the LCR loops to the γ-globin genes in fetal erythroid and not to the β-globin gene in adult erythroid cells, resulting in their reciprocal expression. Enforced looping that generates contact between the LCR and the γ-globin gene promoters determines high levels of γ-globin expression and reduced β-globin expression [1].
Silencing of globin gene is controlled by the promoter regions of developmentally expressed globin genes. An important example of these silencing mechanisms is given by the repression of the HBG gene expression in adult erythroid cells mediated recruitment of repressor proteins to the HBG promoters with BCL11A binding 115 bp upstream of the transcription start site [1].
BCL11A is a zinc-finger protein predominantly expressed in brain and in erythroid cells. The BCL11A expression is transcriptionally controlled during development: in fetal erythroid cells, BCL11A expression is inhibited by the repressor HIC2, whose expression is high in fetal cells but low in adult adult erythroid cells [2]. The expression of BCL11A in erythroid cells is controlled by an erythroid-specific enhancer located at position +58 in intron 2 of the BCL11A gene [3]. The +58 BCL11A intronic enhancer contains a binding site for GATA1, which is required to sustain erythroid-specific expression of BCL11A [3].
In addition to BCL11A, another repressor, the LRF/ZBTB7A transcription factor inhibits γ-globin gene transcription in adult erythroid cells [4]. This factor binds to a specific binding element present in the γ-globin promoter at -197 [4]. The mutation -198T>C is responsible for a form of hereditary persistence of fetal hemoglobin (HPFH), generating a binding site for the HbF activator KLF1. BCL11A and ZBTB7A independently repress expression of HbF.
Mutations in the β-globin gene cause two autosomal recessive diseases, sickle cell disease (SCD) and β-thalassemia. In SCD, a point mutation of the β-globin gene which determines the substitution of glutamic acid at position 6 with a valine residue results in the production of an abnormal hemoglobin (HbS) that in its deoxygenated form had a tendency to polymerization; this polymerization of HbS alters the architecture and flexibility of the sickle RBCs with increased hemolysis and tendency of veno-occlusive events with consequent tissue damage. [5] β-thalassemias are characterized by reduced/absent β-globin chain synthesis due to more than 400 different mutations at the level of the β-globin gene cluster, including point mutations, minor deletions or insertions, or gross deletions in either the β-globin gene or in its flanking, noncoding regions. The reduced/absent β-globin chain synthesis results in a markedly decreased/absent synthesis of HbA, with an excess of free α-globin chains that are unstable, precipitate, and induce oxidative damage with alterations of the membrane of RBCs and consequent premature death (dyserythropoiesis). [6] The only possible curative effect for both SCD and β-thalassemia patients is represented by allogeneic HSCT. Allo-HSCT allows for a one-time cure of SCD or β-thalassemia; however, only a minority (about 25%) of these patients could find a suitable donor due to immunoincompatibility, limiting accessibility.
Figure 1.
Figure 1. A- Genomic organization of the β-globin gene cluister on chromosome 11. The genes are physically arranged in their order of expression during development, including ε-globin (embryonic) Gγ- and Aγ-globins (fetal) and δ and β-globins (adult). LCR corresponds to the locus control region (LCR), with its 5 hypersensitivity sites. HS1 denotes the hypersensitivity site 1. B- Control of γ-globin gene gene expression mediated by BCL11A. Top panel: nucleotide sequence of the gene promoter of HBG1 and HBG2 genes: in these promoters, two BCL11A TGACCA binding sites are present (a distal -118 to -113 and a proximal -91 to -86). Bottom panel: the distal site actively binds BCL11A in adult erythroid cells and represses γ-globin gene expression.
Figure 1.
Figure 1. A- Genomic organization of the β-globin gene cluister on chromosome 11. The genes are physically arranged in their order of expression during development, including ε-globin (embryonic) Gγ- and Aγ-globins (fetal) and δ and β-globins (adult). LCR corresponds to the locus control region (LCR), with its 5 hypersensitivity sites. HS1 denotes the hypersensitivity site 1. B- Control of γ-globin gene gene expression mediated by BCL11A. Top panel: nucleotide sequence of the gene promoter of HBG1 and HBG2 genes: in these promoters, two BCL11A TGACCA binding sites are present (a distal -118 to -113 and a proximal -91 to -86). Bottom panel: the distal site actively binds BCL11A in adult erythroid cells and represses γ-globin gene expression.
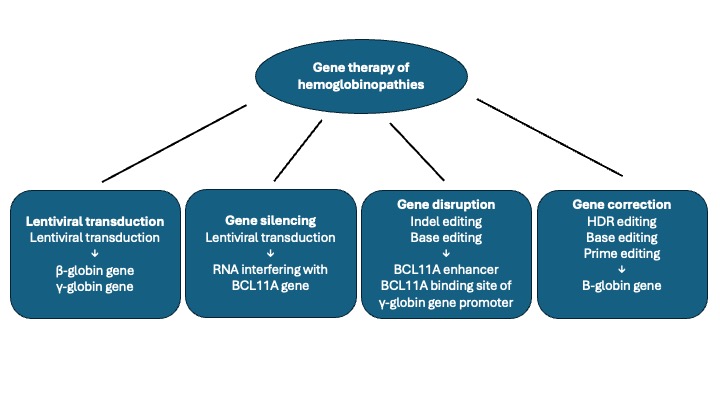
To bypass these limitations, autologous HSC gene therapy was developed, in which patients’ HSCs are harvested and genetically modified ex vivo, then transplanted back into patient after conditioning with busulfan or other cytotoxic agents. Basically, there are two distinct approaches for gene therapy of hemoglobinopathies: one approach is based on gene addition strategies based on lentiviral vectors to add functional copies of the gene encoding β-globin in defective HSCs or to silence faulty genes such as BCL11A to induce HbF synthesis to replace or to counteract the defective β-globin; the second approach is based on gene editing strategies involving the use of CRISPR-Cas9, transcription activator-like effector protein nuclease and zinc finger nuclease techniques either for directly repair the underlying genetic cause of disease or to induce HbF production by gene disruption [7,8].
2. Gene Therapy Strategies
Basically, there are two main approaches for gene therapy for the treatment of hereditary hemoglobinopathies. One approach is based on gene insertion, a procedure in which a therapeutic globin gene is introduced in hematopoietic stem cells. In the approach, patients continue to express the pathologic β-thalassemic or βS globin gene and the therapeutic gene inserted determines the synthesis of a globin gene competing with the pathologic β-gobin gene, inducing the generation of a scenario similar to that observed in patients with β-thalassemic trait or with the sickle cell trait. Alternatively, the gene inserted in HSCs encodes the synthesis of a gene such as an RNA interfering with the BCL11A gene, thus reactivating HbF synthesis that counteracts HbS or replaces defective HbA synthesis in β-thalassemic patients. The second approach is based on gene editing through genetic procedures causing disruption of a HbF repressor or correction of the disease-causing mutation.
In the gene therapy approaches by gene insertion it is of fundamental importance to evaluate the minimal percentage of the transgene that needs to be expressed In vivo in comparison with the pathologic β-globin; furthermore, an additional important determinant is also the cellular distribution of the therapeutic transgene at the level of RBCs. Considering the clinical experience with hemoglobinopathy patients treated with hematopoietic stem cell transplantation, it was evaluated that a minimal expression of 20% donor myeloid chimerism is required to reverse the sickle phenotype; in β-thalassemic patients a lower level around 15% seems to be necessary to correct ineffective erythropoiesis and reverse the anemic condition [9].
It is important to note that the same principle can be applied to gene therapy studies aiming to reactivate HbF synthesis, where a minimum level of HbF synthesis is required to efficiently correct hematopoietic defects observed in SCD or in β-thalassemic patients.
All the procedures of treatment of SCD and β-thalassemic patients with ex vivo genetically manipulated HSCs(HPCs are complex (Table 1). [10] The initial steps, preceding the transplantation of engineered HSCs/HPCs, including initial evaluation with patient selection and preparation, mobilization of HSCs/HPCs, isolation of HSCs/HPCs and ex vivo manufacturing requires considerable scientific expertise and a GMP laboratory authorized for the manipulation and culture of human HSCs for clinical use. Importantly, during the initial evaluation, patients are assessed for the availability of a full sibling who would be HLA-matched; in this eventuality, allo-HSCT is evaluated as a valuable therapeutic option [10]. The subsequent clinical steps of administration of conditioning chemotherapy, transfusion of engineered autologous HSCs/HPCs and monitoring of patients post-transplantation require at least 4-6 weeks of hospitalization in a specialized clinical unit [10].
This section analyzes the various clinical studies carried out in patients with hemoglobinopathies and based on a gene addition strategy using lentiviral vectors.
3.1. Different Lentiviral Vectors Used in Clinical Trials for Hemoglobinopathies
Gene therapy approaches of gene addition were based on the use of lentiviral vectors. These vectors offer the advantage of delivering high levels of transgene in populations of quiescent cells such as hematopoietic stem cells and of conferring long-term, stable expression of the transduced transgene.
The construction of lentiviral vectors is based on some elements required to ensure a consistent level of safety and a high level of expression in erythroid cells. (Table 2) Concerning the safety, to reduce the risk of inappropriate oncogene activation, current lentiviral vectors utilize self-inactivating mechanisms of viral promoter present in the long-terminal repeats (LTR), acting upon vector integration; these inactivation mechanisms limit the risk of inappropriate activation of nearby genes. Particularly, lentiviruses are produced as replication-defective viral particles in which the RNA genome contains a self-inactivating (SIN) 3′LTR, which upon reverse transcription into a linear, double stranded DNA genes gives rise to an U3-deleted enhancer and promoter-less 5′ LTR [11].
The induction of high expression in erythroid cells requires the presence of internal lineage-specific promoters that drive the hyperexpression of the integrated vectors in erythroid cells; these enhancers are present in the β-LCR and in the intron-2 and 3′ untranslated region of the β-globin gene [12]. The key elements in the β-LCR are represented by five hypersensitive sites, named HS1 to 5. For the construction of a genomically stable vector suitable for globin gene therapy the presence of DNA sequences encompassing HS2, HS3 and HS4 of the LCR together with β-globin gene proximal regulatory elements provide therapeutic levels of β-globin expression [13]. The presence of intron 2 of the β-globin gene in the retroviral vector is fundamental because this region contains an essential enhancer element [14].
Interestingly, through the study of a library of short sequences derived from developmentally active elements in erythropoiesis, a clinically relevant enhancer was identified, replacing the canonical LCR and ensuring therapeutic levels of β-globin transgene expression in a β-thalassemia lentiviral vector [15].
A fundamental element of the lentiviral vector is represented by the transgene. Four different vectors have been used in clinical trials for SCD and β-thalassemia. The choice of the transgene is related to its function and its capacity to replace the pathologic β-globin gene. Concerning the gene therapy, there are multiple possible transgenes related to their capacity to efficiently counteract the sickling effect of βS; in this context, γ-globin chains possess a strong anti-sickling activity related to their higher oxygen affinity and to structural properties related to the glutamine at position 87 of the γ-globin chains. The analysis of the properties of γ-chains in their interaction with βS and with α chains allowed the development a β chain mutant incorporating 3 aminoacidic changes T87Q, E22A and G16D, allowing the generation of mutated β-chains with anti-sickling properties similar to those of γ-chains and with an affinity for α chains higher than that of WT β-chains; these three mutations incorporated into a β-chain, allowed the generation of βAS3 globin, exhibiting enhanced anti-sickling capacities [16,17]. The introduction of a G16D mutation in the γ-globin allowed the generation of a γ-globin chain with enhanced anti-sickling activity [18].
The four different vectors used in gene therapy trials were: (i) a vector expressing a WT β-globin gene (Vebeglogene Autotemcel); vector expressing a modified β-globin, βT87Q (Levotibiglogene); a vector expressing βAS3; a vector expressing a modified γ-globin, γG16D.
Some lentiviral vectors used for gene therapy of hemoglobinopathies contain insulator elements, corresponding to DNA sequences that control the activity of enhancer elements present in these vectors. Thus, Cabriolu and coworkers showed that HS1 and HS2 LCR elements present in the lentiviral vectors can be activated in early hematopoietic cells, including HSCs; this activity may result in inappropriate transcriptional activation of nearby genes [19]. The introduction of an insulator element, such as A1 insulator, reduced non-erythroid expression, while expression in erythroid cells was unaffected; importantly, incorporation of the A1 insulator in a therapeutic globin vector was not detrimental to vector titer and hemoglobin production in globin vector-treated thalassemic mice [19].
The most relevant clinical trials of gene therapy involving the use of lentiviral vectors are reported in Table 3
3.2. Lovo-Cel Therapy for Sickle Cell Disease
Lovotibeglogene (Lovo-Cel, BB111; Lenti-Globin for SCD) gene therapy is administered by autologous stem cell transplantation with cells transduced with the BB05 lentiviral vector encoding the modified β-globin gene (βT87Q), resulting in the production of HbAT87Q protein with anti-sickling properties. The HbAT87Q protein was specifically designed to sterically inhibit HbS polymerization and preclinical studies have shown the capacity of Lovo-Cel-mediated endogenous expression of βT87Q to inhibit HbS polymerization [20].
Interestingly, studies in an erythroid cell line expressing βS showed that expression of βT87Q in these cells induces a significant reduction of βS synthesis [21]. This observation suggests that βT87Q exerts an anti-sickling effect through two different mechanisms, one related to a direct competition of βT87Q with βS and the other related to an inhibitory effect of βT87Q on βS synthesis through an undefined mechanism [21].
The Lenti-Globin was evaluated in patients with SCD in the context of the clinical trial HB-205. Initially one SCD patient was treated with Lenti-Globin BB305 resulting in persistent expression of the transgene in vivo, with a level of βT87Qcorresponding to about 50% of total hemoglobin, with no recurrence of sickle crises [22]. An initial report of HGB-205 trial included 3 SCD patients: 2/3 patients displayed a long-term clinical response and 1/3 displayed a reduced transfusional need [23]. After the initial proof of the efficacy and safety of Lenti-Globin-based gene therapy of SCD patients, the phase I/II HGB-206 trial was started using Lenti-Globin BB305 in 50 adults and adolescent patients with severe SCD. In the group A, seven patients were treated and achieved only a partial therapeutic effect due to inappropriately low levels of HbAT87Q (0.46 g/dL) due to low copy number per cell observed 6 months after gene therapy [24]. To bypass these limitations, the procedure of gene transfer was improved and Plerixafor mobilization of HPC progenitors was used in patients to improve the recovery of CD34+ cells. Using this strategy, higher levels of VCN and of HbAT87Q were obtained in the group B of two patients [24]. Using these manufacturing modifications and optimized HPC mobilization, in the phase C, 36 SCD patients were treated with a single autologous infusion of CD34+ cells transduced with Lenti-Globin leading to an increase in total hemoglobin levels from 8.5 g per deciliter at baseline to 11g or more from 6 months through 36 months after infusion; HbAT87Q mean levels were 5.2 g/dL and contributed at least 40% of total hemoglobin and was distributed across over a mean of 85±8% of RBC; VCN was 1.5 copy/cell [25]. All evaluable patients had resolution of severe vaso-occlusive events and reduction of hemolysis [25]. It is important to note that in the HGB-206 trial the enrollment criteria included only SCD patients who had at least four vaso-occlusive events in the 24 months before enrollment [25]. In 2020, the phase III clinical study HGB-210 involving the enrollment of pediatric patients aged 12-18 years, was initiated. Recent reports provided updated analyses of the clinical data observed in 36 SCD patients enrolled in the context of HGB-206 study and 19 SCD patients enrolled in the context of HGB-210 study [26,27]. The results of this combined analysis showed that HbAT87Q levels remained stable during follow-up and were similar in adult and pediatric patients, with a mean percentage of HbT87Q of 49% (range 26%-63%). The large majority of patients had complete resolution of the vascular occlusion episodes, including severe occlusion episodes, in the two years following gene therapy [26,27]. The analysis of pharmacodynamic parameters, such as transduction efficiency, the total Hb level, the HbAT87Q levels are sensitive and early predictors of the clinical response of SCD patients to Lovo-Cel-based gene therapy [28].
Several studies have evaluated the socio-economic impact of Lovo-Cell gene therapy in SCD patients. An analysis of cost effectiveness suggested remarkable improvements in survival quality of life and other outcomes for Lovo-Cel compared to common care [29]. The costs associated with Lovo-Cel were in part offset by predicted reductions in other heath system and societal costs [29].
Importantly, in December 2023 the FDA approved the use of Lovo-Cel for the treatment of SCD patients who have a history of vaso-occlusive crises.
An ongoing follow-up study (LTF-307) will provide data on the long-term safety and efficacy of Lovo-Cel therapy in SCD patients.
3.3. Gene Therapy of SCD Using Lenti/G-βAS3-FB Vector
In 2003, an anti-sickling β-globin variant was developed by Townes laboratory. βAS3 involves three β-chain substitutions, including the T/Q change present in Lenti-Globin and the substitutions of glutamic acid in codon 22 with alanine (E22A) and of glycine at codon 16 with aspartic acid (G16D). A lentivirus encoding the βAS3 and containing also the FB insulator (Lenti/G-βAS3-FB) was shown to be active in preclinical models to correct hematological parameters in a SCD mouse model and was used for clinical studies [30]. A recent study reported the clinical results obtained in the first five treated patients reporting a significant improvement in only two of these five SCD patients [31]. Given these non-optimal results the study was closed to future enrollment.
A second study confirmed the results observed in this study. Thus, Sabrino and coworkers in a phase I/II clinical study (NCT 03964472) evaluated the safety and the efficacy of gene therapy of SCD by transplantation of autologous HSCs/HPCs transduced ex vivo with the DREPANOGLOBE lentivirus vector expressing the βAS3 globin gene [30]. The study of the first four SCD patients treated showed variable results from one patient to another, with only 50% of patients achieving correction of the clinical phenotype with absence of severe vaso-occlusive events [30]. Single-cell transcriptomic studies showed that reduced engraftment of DREPANIGLOBE-modified CD34+ cells was associated with an inflammatory signature [32].
3.4. Gene Therapy of SCD Using Lentiviral Vectors Encoding HbFG16D
A recent study reported the clinical results observed in SCD patients treated with gene addition therapy based on autologous stem cells transduced with a vector expressing a modified γ-globin gene [33]. This gene therapy was based on the use of γ-globin lentiviral vector, GbG, encoding human Aγ globin gene exons and β-globin non-coding (untranslated regions, introns and promoter) to ensure high expression of the γ-globin gene in adult erythroid cells [33]. This vector was modified introducing a change at the 16th codon in γ-globin exon 1 to change glycine (G) to aspartic acid (D), generating a vector expressing γG16D globin [34]. Experimental studies in SCD mice supported a more potent anti-sickling activity of HbFG16D compared to HbAT87Q [34] Autologous CD34+ cells from 7 SCD patients with severe disease were transduced and at least 4x106 transduced CD34+ cells were reinfused to the patients [32]. Before infusion of autologous stem cells, the patients were submitted to a reduced intensity conditioning regimen using a reduced-intensity dose of melphalan [34]. One year after infusion, an average vector copy number of >0.01 copies per cell was observed. All seven treated patients displayed sustained HbFG16Dexpression and >80% reduction in severe vaso-occlusive events [34]. The use fo reduced-intensity conditioning instead of myeloablative busulfan decreased the duration of thrombocytopenia and neutropenia and length of hospital stay [34]. The most common adverse events were grade 4 thrombocytopenia and grade 4 neutropenia [34]. The results of this trial need to be confirmed in a larger cohort of patients.
3.5. Lovo-Cel Gene Therapy for β-Thalassemia
Several studies have evaluated the safety and the efficacy of Lenti-Globin BB305 in patients with transfusion-dependent β-thalassemia. The HGB-204 and HGB-205 clinical trials evaluated the safety and the efficacy of Lenti-Globin 3305 in 22 patients with transfusion-dependent β-thalassemia: autologous CD34+ cells were transduced ex vivo with Lenti-Globin BB305 vector and reinfused to patients after myeloablative busulfan conditioning [35]. Efficacy evaluation involved levels of total Hb, of HbAT87Q, transfusion requirements and average vector copy number [35]. All but 1 of the 13 β-thal patients with non-β° genotype stopped to receive red cell transfusions; in 9 patients with β°/β° genotype or two copies of IVS1-110 mutation, the median transfusion request was decreased by 73% and transfusions were discontinued in 3 patients [35]. Adverse events were those typically associated with autologous SCT [35].
Given the good outcomes of this study, two phase III trials, HGB-207 (Northstar-2) and HGB-212 (Northstar-3) studies were carried out to more carefully evaluate the safety and efficacy of Lenti-Globin in transfusion-dependent β-thalassemic patients [36]. In the HGB-207 study β-thal patients with non-β°/β° genotype and in the HGB-212 study β-thal patients with β°/β°, β°/β+IVS-I110 genotypes were enrolled. In this study, a hypertransfusion regimen for at least two months prior HSC/HPC mobilization to maintain Hb levels >11g/dL was adopted; furthermore, an optimized manufacturing process with improved transduction efficiencies was developed, achieving a mean copy vector number after 24 months of 1.99 copies/cell, compared to a VCN of 0.7 in the HGB-204 and 1.3 in the HGB-205 study [36]. In the HGB-207 study, 20 out of 22 patients reached a condition of transfusion independence with a median HbAT87Q level of 8.7 g/dL (ranging from 5.2 to 10.6 g/dL) at 12 months post-infusion and an average total Hb level of 11.7 g/dL [36]. The HGB-212 study enrolled 18 β-thal patients (12 with β°/β° genotype, 3 with β°/β+IVS-I110 genotype and 3 with β+IVS-I110/β+IVS-I110 genotype); 89% of these patients reached and maintained a condition of transfusion independence [37]. The two patients who did not achieve transfusion independence had genotypes β°/β° and βIVS-110/βIVS-110 and had significantly lower VCN (0.199 and 0.320, respectively) compared to those who achieved transfusion independence [37]. Since these two patients received a cell product displaying an adequate vector copy number, the low VCN observed 24 months after infusion suggests a reduced in vivo engraftment of transduced cells. There were no serious adverse events directly related to gene therapy and no deaths [37].
Gibson et al. reported post-approval, real-world experience in 9 β-thal patients with Beta-Cel gene therapy [38]. All patients reached transfusion-independence. Patients experienced prolonged platelet engraftment time and high platelet transfusion requirements, which were associated with severe bleeding in patients with veno-occlusive disease or HLA Class I alloimmunization [38]. This observation suggests the need for additional studies aiming to mitigate bleeding complications in these patients. In a real-world setting in Germany, Betabeglogene gene therapy resulted to be safe and effective in 8 transfusion-dependent non-β°/β° thalassemic patients [39]. All these patients reached and maintained transfusion independence. The safety profile was acceptable and most of patients displayed pituitary-endocrine dysfunction; one patient developed a depression and anxiety syndrome; one patient developed fatigue [39].
3.6. LV-GLOBE Gene-Therapy of β-Thalassemia
In a phase I/II clinical study (TIGET-BTHAL), a modified vector lacking HS4 LCR element (LV-GLOBE) encoding modified β-globin βAS3 was used for the transduction of autologous CD34+ cells derived from 3 adults and 6 children with β° or β+ thalassemia [40]. 3 out of 4 pediatric patients discontinued RBC transfusions, while transfusion requirement was reduced in the adults [40]. Younger age and persistence of higher number in the repopulating stem cells were associated with better outcome [40].
3.7. Modified LV Globin Gene Therapy for Pediatric β°/β°
Li et al. have reported an interim analysis of a single-arm pilot trial evaluating a β-globin expression-optimized and insulator-engineered lentivirus-modified autologous HSCs in β°/β° pediatric β-thalassemic patients [41]. This study was based on the construction of a vector (BD211) containing the WT-β-globin gene as transgene, various elements of the LCR, the enhancers of BCL11A and glycophorin A promoter to ensure erythroid-specific expression of the HBB cDNA and a short insulator derived from foamy viruses, which reduced the genotoxicity potential of the U3 region of the LTR [42]. The distinctive properties of this vector consisted in: (i) an insulator design integrated into the lentiviral vector to minimize the risk deriving from potential insertional mutagenesis; (ii) an optimized β-globin gene expression cassette engineered to achieve physiological hemoglobin levels compared to those observed in healthy subjects [42]. Preclinical studies supported the safety and the efficacy of BD211 CD34+ cells in NCG-X mice (triple-immunodeficient mouse with a point mutation in c-kit); particularly, these cells efficiently engrafted and differentiated into human erythroid cells within the mouse bone marrow and blood [41].
A clinical study reported the efficacy of autologous BD211 CD34+ autologous cells in two female pediatric β°/β° thal patients [42]. Engraftment of genetically modified HSCs and HPCs was successful and sustained in both patients; both patients reached a condition of durable transfusion independence; both patients showed a stable vector copy number of 2-4 copies per cell [42]. Single-cell analysis showed vector inserts in 33-65% of cells and no dominant clones were observed.
3.8. Lentiviral BCL11A Short Hairpin mRNA
The Boston Children’s Hospital developed a gene therapy based on the lentiviral vector LVV BCH-BB694 encoding a micro RNA-adapted short hairpin (shRNAmiR) targeting BCL11A [43]. In preclinical studies this vector efficiently transduced CD34+ cells with a high gene marking and inducing HbF synthesis [40]. Thus, a phase I clinical study (NCT 03282656) evaluated the safety and efficacy of HbF synthesis induction by lentiviral gene addition with a short hairpin RNA embedded in a micro RNA (shmiR) against BCL11A driven by an erythroid-specific promoter [44]. A first report on this study showed the results on the first six SCD patients enrolled, showing a significant gene marking in whole blood (VCN 0.49-1.49) 6 months after infusion, with a significant and stable induction of HbF synthesis (20-41%, median level 31%) [44]. Three severe adverse events were reported, including influenza infection, leg pain and recurrent priapism [44]. No events of vascular occlusion, acute chest syndrome or stroke after infusion were reported [41]. An updated analysis of this study was extended to 10 SCD patients, showing that post-transcriptional gene knockdown of BCL11A was safe, associated with stable HbF induction and significant mitigation of vascular occlusive events: in 6 SCD patients with frequent vascular occlusive events before treatment, reported between 0 and 1 events of vascular occlusion after gene therapy; in 3 SCD patients treated with chronic transfusion regimens before treatment, transfusion independence was reached in 2/3 cases [45]. A recent long-term follow-up of these 10 patients treated with LVV BCH-BB694 gene therapy confirmed the results previously reported [46]. Among the 10 treated patients, the patient with the lowest in vivo VCN had the lowest HbF post-infusion level of 14.1% that remained stable during the whole follow-up; a robust Hb F induction was observed in the other 9 treated patients, ranging from 20% to 38.2% and F-cells ranging from 55% to 89% and HbF content/F-cell ranging from 9.4 pg to 13.6 pg [46]. The analysis of patient and parent-reported outcomes post-treatment with LVV-BCH-BB694 supported a significant improvement of quality of life related to the decrease of vascular occlusive events and of transfusion rates [47].
A single-cell analysis on RBCs of SCD patients treated with LVV-BB694 gene therapy, compared to RBCs derived from SCD patients treated with hydroxyurea showed in the first group of patients fewer RBCs with high content of HbS that could be susceptible to sickling compared to the second group of patients; conversely, RBCs from patients treated with LVV-BB694 are more resistant to sickling at physiologic O2 tension compared to those from SCD patients treated with hydroxyurea [48].
Finally, whole-genome sequencing of individual HSCs/HPCs of six SCD patients undergoing BCH-BB694 gene therapy was evaluated, showing that no clonal expansions of gene-modified or un modified cells were observed; however, an increased frequency of potential driver mutations associated with clonal hematopoiesis or with myeloid neoplasms (DNMT3A and EZH2 mutated clones) was observed both in genetically modified and unmodified cells, thus suggesting a positive selection of mutant clones during gene therapy [49].
4. Gene Editing Therapy of Hemoglobinopathies
Gene editing therapy in hemoglobinopathies is a recently developed technology to modify genetic information in patients with hemoglobinopathies. This technology offers the advantage with respect to gene addition studies using lentiviral vectors to enable gene modification without the need of generating an expression lentiviral vector and of introducing exogenous DNA. Two different versions of gene-editing were used for therapy of hemoglobinopathies: (i) gene editing for correction of single nucleotide mutations such as SCD mutation at the level of β-globin gene; (ii) gene editing of a regulatory DNA sequence to induce HbF synthesis (SCD and β-thalassemia). Gene editing does not require the use of lentiviral vectors and is based into the introduction in patient’s cells of engineered nucleases for site-specific editing.
Gene editing techniques can be subdivided into two different groups: gene editing based on double-strand break; gene editing without double-strand DNA break.
4.1. Gene Editing Based on Double-Strand Break
Several engineered nucleases for site-specific editing are available, including the CRISPR/CRISPR-associated protein 9 (Cas9) system, zinc finger nucleases and transcription activator-like effector nucleases. (Table 4) Among these various systems, the CRISPR/Cas9 system is the most frequently used in gene therapy studies for its high efficiency of gene he most primitive and quiescent compartments of HSCs and HPCs editing.
The CRISPR/Cas9 is a two-component system based on the utilization of nucleases allowing the generation of DNA double-strand breaks into specific DNA sequences of the genome; this system is composed of a caspase 9 (Cas9) that is driven on a specific DNA target sequence by a single-guide RNA (gRNA); the Cas9 recognizes this sequence and cleaves DNA. The cell attempts to repair double-stand breaks by using two different repair mechanisms. Non-homologous end joining repair (NHEJ) and homology-directed repair (HDR). NHEJ creates insertions and deletions (indel) at the cut site which can inactivate the gene by disrupting the coding sequence or can alternatively increase or decrease gene expression by modifying the binding site of a transcriptional repressor or activator, respectively. However, it is difficult to drive exactly the nature of the editing process in terms of insertion or deletion and the extent of these deletions or insertion in terms of the number of nucleotides inserted or deleted. Alternatively, the HDR system utilizes a donor DNA template sequence which directs the host genome to repair the cut site, matching the template [50]. These two repair processes differ not only in their mechanisms, but also in their occurrence in different cell cycle phases: HDR-based editing is confined to the S/G2 cell cycle phases, thus limiting it to the most primitive and quiescent compartment of HSCs/HPC, while NHEJ editing system operates in quiescent cells. (Table IV)
CRISPR/Cas 9 was used as a gene editing tool in clinical studies aiming to disrupt regulatory elements, such as the erythroid enhance of BCL11A gene or the BCL11A-binding elements present in the promoter of HBG1 and HBG2 genes or to correct the point mutation observed in SCD patients.
4.2. Gene Editing Without Double-Strand DNA Break
Base editing and prime editing represent two strategies to modify DNA sequences without inducing a double-strand break.
Base editing is a genome editing approach that uses components of the CRISPR systems (catalytically inactive Cas9 nickase or dead Cas9) with other enzymes (deaminases) to directly install point mutations into cellular DNA or RNA without making double-strand DNA breaks. Base editors directly convert one base or base pair into another, enabling the efficient installation of point mutations in non-dividing cells [51]. PAM (protospacer adjacent motif) is a short DNA sequence required for CRISPR-Cas editing to occur mediating Cas enzyme binding, unwinding of DNA and cutting of the target. PAM sequence is located immediately downstream of the target DNA sequence recognized by the guide RNA.
A base editing system was developed to correct the SCD genetic defect using a PAM nickase specifically recognizing the SCD mutation site, fused to a adenine deaminase [49]. Electroporation of this base editing product in CD34+ cells resulted in 80% conversion of the SCD mutation into the non-pathogenic HbGMakassar variant (β6 Glu⟶Ala) [52]. Base edited CD34+ cells maintained their gene-editing in their blood progeny (44% gene-edited cells 16 weeks after transplantation in mice) [52]. This gene editing significantly inhibited RBC sickling [52]. HbG-Makassar is a naturally occurring variant that is clinically asymptomatic. Studies in transgenic mice model expressing homozygous HbG-Makassar showed a normal RBC physiology and RBC physiology comparable to WT HbA; RBCs HbGS displayed a hematological phenotype intermediate between HbAS and HbSS, supporting the use of base editing of HbS to HbG-Makassar in SCD patients [52]. Importantly, base-edited CD34+ βS cells converted to βMakassar, displayed an efficient engraftment capacity in nonhuman primates, rapidly regenerating all hematopoietic lineages [53].
Several studies have shown that base editing of DNA sequences involved in the control of HbF synthesis represents an important strategy for potential therapeutic implications. Thus, the mutation of the -175 γ-globin nucleotide A to G determines a strong induction of HbF synthesis at levels higher than those elicited by Cas9 strategies targeting a BCL11A motif in the γ-globin promoter or a BCL11A erythroid enhancer [54]. Comparison with strategies using Cas9 showed that disruption of the BCL11A-binding motif at the HBG1/HBG2 promoters elicited sustained HbF synthesis in healthy and β-thal patient HSCs/HPCs [55]. Importantly, base editing of the γ-globin gene promoter induces potent HbF synthesis without detectable off-target mutations in HSCs/HPCs [55].
The base editing may represent an efficient strategy to disrupt a DNA regulatory, with the aim of blocking its function. In fact, a recent study by Fontana and coworkers provided evidence that multiplex base editing represents an efficient strategy to disrupt the +58K and +55 enhancer of BCL11A gene, resulting in a reactivation of HbF to levels exceeding those achieve with CRISP-Cas9-induced editing, minimizing double-strand breaks and genomic rearrangements [56].
An alternative approach was proposed by Rajendiran and coworkers who have base-edited the zinc finger domain (Znf4, ZnF5 and ZnF6) used by BCL11A to interact and repress the HBG1 and HBG2 promoters [54]. Base editing of ZnF4 and ZnF6 induced elevated HbF synthesis without affecting normal hematopoiesis [57].
Base editing strategy was under evaluation in clinical studies assessing the efficacy of autologous HSCs/HPCs base-edited at the level of HBG1 and HBG2 promoter sequences involved in repression of HbF synthesis in adult erythroid cells.
Prime editing is a technology of gene editing allowing the direct writing of new genetic information into a targeted DNA sequence. This technology uses a prime editing guide RNA (peg RNA) capable of identifying the target site and providing the new genetic information to replace the target DNA nucleotides and a fusion protein, formed by a catalytically impaired Cas9 endonuclease fused to an engineered reverse transcriptase enzyme. Prime editing is capable of mediating targeted insertions, deletions, and base-to-base conversions without the need for double strand breaks or donor DNA templates [58]. The prime editing methodology was recently improved through the development of prime editors exhibiting a uniquely high level of editing precision and are highly error prone [59].
Everette et al. showed that prime editing can correct the SCD allele βS to WT βA at frequencies of 14-41% in HSCs/HPCs derived from patients with SCD [60]. Several weeks after transplantation into immunodeficient mice, prime-edited SCD maintained βA levels and displayed engraftment rates and lineage differentiation capacities comparable to those of WT HSCs [60].
Furthermore, a recent study provided evidence that multiple editing of γ globin gene promoters by prime editing in erythroid cells induced an enhanced capacity to reactivate HbF synthesis compared to cells with individual mutations [61].
Fiumara and coworkers have compared base-editing and Cas9 in human HSCs/HPCs concerning editing efficiency, cytotoxicity, transcriptomic changes and on-target and genome-wide genotoxicity [62]. Base editing and prime editing induced detrimental transcriptional responses that reduced editing efficiency and hematopoietic repopulation in xenotransplants and also generated double-strand breaks and genotoxic products at lower frequency than Cas9 [62]. These findings raised concerns about the genotoxicity potential of base editing and prime editing and suggested careful additional evaluations in view of clinical applications [62]. Cas9-mediated double-strand breaks induce large deletions at a frequency approximately 20-fold higher than base editing and prime editing [63].
4.3. Specific Transcriptional and Epigenetic Modulation Using Dead Caspase 9 (dCas9)
The considerable progresses made in gene editing technologies have provided the rationale for the development of a technology aiming to specifically and stably modulate gene expression though epigenetic mechanisms, thus providing a tool for therapeutic interventions without altering the target DNA sequence. This approach involves CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) and requires the use of catalytically dead Cas9 (dCas9) fused to different transcriptional effectors [64,65]. The CRISPRa systems were based on transcriptional activators, while CRISPRi systems involve transcriptional repressors. The CRISPi represents a programmable and reversible strategy of gene silencing; in this technique, specific single guide RNAs drive dCas9 at the level of promoter regions or in proximity of start transcription sites inhibiting RNA polymerase binding, thus blocking gene expression [66].
dCas9 may also be fused to epigenetic effectors to modulate gene expression by epigenetic mechanisms. Interestingly, a recent study showed that modulation of methylation status of the promoter of HBG by epigenome editing may represent a potential therapeutic for β-hemoglobinopathies [[67].
4.4. CRISPR-Cas9 Editing of BCL11A Enhancer: Studies in β-Thalassemia
A pivotal study by Ye and coworkers showed that the CRISPR-Cas9 genome-editing technology can be used to modify the β-gene locus using NHEJ repair to generate modifications suitable for therapy of patients with SCD and β-thalassemia [68]. In 2019, Wu et al. showed that Cas9/sg RNA-mediated cleavage within a GATA1 binding site at the +58 BCL11A enhancer results in disruption of this motif, reduction of BCL11A expression, and induction of fetal γ-globin [69]. Gene edition of BCL11A erythroid enhancer appeared to represent an important strategy to induce therapeutically-relevant high levels of HbF synthesis in SCD and β-thalassemia [69].
A pilot clinical trial provided the first evidence in favor of the safety and efficacy of CRISPR/Cas9 gene editing of the BCL11A erythroid promoter in SCD and β-thalassemia patients. The report on the first two patients enrolled in the CLIMB-THAL 111 and CLIMB-SCD 122 studies, one β°/β+ thal and one βS/βSSCD patient, showed a rapid rise of HbF and Hb levels after infusion od autologous CD34+ cells gene-edited, a pancellular distribution of HbF at the level of RBCs and the permanence of gene-edited cells in peripheral blood [70]. Both thalassemic and SCD patient become transfusion-independent and in the patient with SCD no vaso-occlusive events occurred [70].
The results observed in the phase III CLIMB-THAL 111 study were recently published; in this study, 52 β°/β°, β°/β+, β°/β°like with transfusion-dependent disease received autologous CD34+ cells. Gene-edited ith CRISPR-Cas9 at the level of BCL-11A erythroid enhancer (with the commercial name of Exaglamblogene, Exa-Cel) [71]. 91% of these patients become transfusion-independent, with a mean Hb level of 13.1 g/dL and mean HbF level of 11.9 g/dL, distributed in 94% of RBCs; only 9% of patients did not reach transfusion-independence but decreased their transfusion rates [71]. Allelic editing remained stable during the first 6 months after gene therapy. The safety profile of Exa-Cel-treated patients was consistent with busulfan conditioning [71]. An updated analysis extended to 54 β-thal patients confirmed transfusion-independence in 94% of patients receiving Exa-Cel, with sustained increases in HbF and total Hb for up to 5 years of follow-up [72]. An analysis of these patients with a follow-up extended up to 6 years showed that Exa-Cel-based gene therapy not only induced transfusion independence but also led to to a normalization of iron metabolism: in fact, after Exa-Cel treatment, iron was successfully removed by iron-replating therapy [73]. This observation further supports a curative effect of Exa-Cel in β-thal patients [73].
A similar approach was used by Fu et al. who initially reported the results on two β-thal β°/β° pediatric patients transfusion-dependent treated with autologous HSCs/HPCs BCL11A enhancer-edited in the context of the phase I/II trial NCT 04211480 [74]. Both patients achieved transfusion-independence and their Hb increased from 8.0 to 10.8 g/dL at screening to 15.0 and 14.0 g/dL, with >85% editing persistence in bone marrow cells [74]. An updated report on this study displayed the results observed in 10 β-thal patients (5 β°/β°, 4 β+/β°, 1 β+/β+) showing a sustained rise of HbF and F cells, reaching values of 98-99% [75]. Adverse events were related to busulfan conditioning. A more recent update to 15 β-thal patients 8 β°/β°, 4 β+/β°, 3 β+/β+ confirmed and extended the results observed in previous studies [76].
4.5. CRISPR-Cas9 Editing of BCL11A Enhancer: Studies in Sickle Cell Disease
Exa-Cel gene therapy was investigated also in SCD patients. The phase III CLIMB SCD-121 clinical trial explored the safety and the therapeutic efficacy of CD34+ autologous cells gene-edited using Exa-Cel in 44 transfusion-dependent SCD patients [77]. The large majority of treated patients were free from hospitalizations for severe vaso-occlusive crises; only 6 events of severe vaso-occlusive events were observed in patients whose increase in total Hb and HbF levels was similar to that observed in patients without vaso-occlusive crises [77]. Total Hb levels increased from <9 g/dL to 12.5 g/dL six months after the start of therapy, with 44.5% of HbF; all patients displayed a decrease of hemolysis and became transfusion-independent. Adverse events were those expected for patients receiving a conditioning with busulfan and autologous HSC transplantation [77]. An updated analysis of the results of this trial with a more extended follow-up englobing 46 SCD patients showed that elimination of vascular occlusive events was observed in 90% of patients receiving Exa-Cel, with a marked increase of total Hb (>12 g/dL) and of HbF (>40%) [78]. In CLIMB SCD-121 and CLIMB-131 combined, 100% of patients were free of severe vascular occlusive crises for ≥12 consecutive months and 100% were free from in patient hospitalization for severe occlusive crises [79]. These observations support a curative effect in SCD patients undergoing Exa-Cel-based gene therapy [79].
The long-ter effects of Exa-Cel-based gene therapy observed in the CLIMB SCD-121 study are under evaluation in the context of CLIMB 131 study. The analysis of quality of life of these patients showed a remarkable improvement of quality of life both for adolescent and adult patients [80].
In the CLIMB THAL-141 and CLIMB SCD-151 stuides Exa-Cel was infused following busulfan myeloablation to patients aged 2-11 years with a history of transfusion-dependent β-thalassemia or SCD with ≥2 severe occlusive events per year for 2 years before screening [81]. Efficacy and safety data for the first 26 patients (13 SCD and 13 β-thal) are consistent with the data observed in patients aged ≥12 years, with clear signs of clinical benefit and with a safety profile consistent with busulfan conditioning and autologous HSCT [81].
4.6. Gene Editing of γ-Globin Gene Promoters Using CRISPR-Cas9 Technology
The expression of human γ-globin genes is finely regulated during development by multiple mechanisms. The expression of HBG1 and HBG2 genes in human erythroid cells is almost completely repressed through efficient transcriptional and post-transcriptional mechanisms [82]. Some gene therapy studies aimed to reactivate HbF synthesis in adult erythroid cells by interfering with the genetic mechanisms that mediate the transcriptional repression of HBG1 and HBG2. In this context, the two main targets are represented by the binding sites of the transcriptional repressors BCL11A and LRF present in the promoter region of HBG1 and HBG2 genes. In a recent study, Wongbrisuth et al. have comparatively evaluated the effect of the disruption of LRF or BCL11A binding sites of the γ-globin gene promoter by CRISP/Cas9 gene editing in CD34+ cells isolated from healthy or β°/HbE individuals: both disruptions similarly increased HbF synthesis, without affecting erythroid differentiation and with minimal off-target effects [83].
Some studies used CRISPR-Cas9 to edit sequences of HBG1 and HBG2 genes involved in gene repression. In this context, an initial study by Traxler and coworkers showed that the introduction of a mutation in the -102 to -114 HBG1 promoter by CRISPR-Cas9 enabled the production of erythroid cells with an increased HbF synthesis capacity [84]. Disruption of the HBG1/HBG2 gene promoter motif that is bound by BCL11A using CRIOSPR/Cas9-mediated gene editing in CD34+ cells resulted in the generation of erythroid cells exhibiting a consistently increased HbF synthesis [85].
These studies have provided the preclinical basis for a clinical trial involving gene editing of HBG1 and HBG2 gene promoters using CRISPR-Cas9 technology. To this end, Sharm aet al. in a first set of experiments have first defined the guide RNA for optimal targeting of HBG1 and HBG2 promoters; then, using gRNA 68 that was selected for clinical studies, have edited CD34+ cells isolated from SCD patients [80]. The editing product was defined as OTQ923. Three SCD patients were treated with OTQ923 and displayed a significant increase of HbF levels, ranging from 19% to 25%; all three patients had a significant improvement of their hematological levels but still displayed signs of mild hemolysis; all the three treated patients had at least one episode of vaso-occlusive crises [80]. These findings suggest that the levels of HbF synthesis observed after infusion of gen-edited autologous HSCs/HPCs were not sufficient to inhibit HbS polymerization completely [86].
4.7. Base Editing of BCL11A Binding Site in γ-Globin Gene Promoter
BEAM-101 is a gene therapy for hemoglobinopathies that uses adenine base editors to introduce single base changes (A to G substitutions) in the γ-globin genes HBG1 and HBG2 in patients’ HSCs/HPCs ex vivo. These changes disrupt the binding of BCL11A repressor, increasing the expression of fetal hemoglobin production. Preclinical studies showed that base editing potently induced HbF synthesis (>60%) and proportionately reduced HbS (<40%). Two recent studies reported the preliminary results observed in the first 6 SCD patients enrolled in the phase I/II clinical study BEACON involving autologous stem cells gene-edited with BEAM-101 [87]. These results supported the efficacy of this treatment in terms of increase of HbF levels and inhibition of vascular occlusive events [87,88]. One patient died during treatment of respiratory insufficiency related to busulfan conditioning [87,88].
Beam Therapeutics recently updated the results of BEACON study reporting the results on the first 17 enrolled patients [89]. The presented data showed that all 17 patients achieved HbF levels over 60% and HbS levels below 40%, with these effects lasting for up to 15 months; lack of vaso-occlusive crises post-treatment was observed in all patients; markers of hemolysis normalized or improved, and erythropoietin levels decreased, indicating better oxygen delivery; the safety profile remained consistent with the expected results of busulfan conditioning [89]. A very recent update extended to the first 26 patients confirmed the results reported in previous reports and showed that in all treated patients a robust HbF synthesis was observed with total Hb levels reaching 15.6 g/dL by 6 months after infusion of engineered HSCs; peripheral blood ceil editing was high, with >72% of cells gene-edited 6 months after infusion of gene-edited HSCs [90]. It is important to note that the BEAM-101treatment process is associated with high editing efficiency and rapid neutrophil and platelet engraftment, showing the potential to minimize hospitalization [90].
Wang and coworkers reported the development of a transformer base editor (tBE) system involving a cleavable deoxycytidine inhibitor (dCI) that induces efficient editing with only background levels of genome-wide and transcriptome-wide off-target mutations [91]. After being produced, the tBE remains inactive at off-target sites with the fusion of a cleavable dCI, therefore eliminating unintended mutations [91]. Correspondence Sequence Therapeutics (CST) used the tBE for the editing of the γ-globin HBG1 and HBG2 promoters (CS-101 product). Preclinical studies showed that CS-101-mediated editing of HBG1 and HBG2 promoters inducing robust HbF synthesis, without causing adverse events on the engraftment or differentiation of HSCs in mice after transplantation [92]. The preliminary results obtained in the first 11 β-thal patients transplanted with autologous HSCs gene-edited with CS-101 showed clinically significant increases in both total Hb and HbF, prompt and durable engraftment and therapeutic benefits (transfusion independence) [93,94]. The treatment elicited a rapid increase of HbF levels with pancellular distribution and stable editing efficiency [93,94]. Very recently, Chen et al. reported at the ASH Meeting 2025 the results on the first 14 β-thalassemic patients treated with CS-101 showing transfusion-independence in 12/14 patients, with HbF levels above 12 g/dL from month 4 onwards; the allele base editing efficiency in bone marrow and peripheral blood nucleated cells remained stable over time [95].
4.8. Cas12 Editing of BCL11A Binding Site in γ-Globin Genes Promoter
Another study of gene editing of the HGB1 and HBG2 promoters utilized the CRSIPR-Cas12 technology. There are several remarkable differences between Cas-12 and Cas-9; the most remarkable difference is that Cas-12a possesses a single nuclease domain and intrinsic RNA processing activity, allowing multigene editing of RNA transcripts and results in stagged DNA end, promoting HDR repair instead of NHEJ [96]. The CRISPR-Cas12a system was used to edit the distal CCAAT-box region of the HBG1 and HBG2 promoters, resulting in a high editing rate of these DNA sites, associated with the induction of elevated HbF synthesis in the erythroid progeny (about 40%) [97]. Importantly, gene edited HSCs efficiently repopulate the hematopoietic system and no off-target editing was observed [89]. Using this approach it was developed the EDIT-301 gene editing product. Using this product, two ongoing clinical trials were initiated. The RUBY trial is evaluating the safety and efficacy of EDIT-301 in adult and adolescent participants with severe SCD, while the EdiThal trial evaluated the safety and efficacy of EDIT-301 in transfusion-dependent β-thalassemia. The initial results of the first 7 and 2 patients enrolled in the RUBY and EdiThal studies were reported [98]. Successful engraftment was observed in all patients: in SCD a rapid and sustained normalization of Hb after 4 months after infusion was observed, associated with a marked increase of HbF synthesis, associated with resolution of vaso-occlusive events and normalization of the markers of hemolysis; in the two β-thalassemic patients rapid improvements of hematological parameters were observed, associated with development of transfusion-independence [98]. The NCT 03663760 study will evaluate the long-term safety and efficacy of EDIT-301 in SCD and β-thalassemia patients.
An updated report of the RUBY trial included the results on 21 SCD patients supporting the efficacy of EDIT-301 (renamed as Reni-Cel): total Hb was 14.2±2 g/dL at month 6 and was maintained up to last follow-up; mean percentage of HbF was 48.2% at month 6 and was maintained at >40% through last follow-up; markers of hemolysis improved or normalized; all patients were vascular event-free [99]. The safety profile was consistent with myeloablative conditioning with busulfan [99]. A very recent report updated the results the RUBY trial on 32 SCD patients, with 31/32 patients reaching a condition of absence of of vascular occlusive events after Reni-Cel infusion and with all patients reaching a normalization of Hb levels (13.8±1.8 g/dL), with >40% of HbF [100].
An updated analysis on 7 β-thalassemic patients treated in the control of Edit-Thal trial was recently reported [101]. In all treated patients, HbF concentration increased early and was 11.3±1.7 g/dL by month 6; the percentage of F cells was >99%; all 7 patients reached a transfusion-independence status [92]. A high level of gene editing (>75%) was observed in PB cells and in CD34+ cells [101]. The safety profile was consistent myeloablative conditioning with busulfan.
4.9. Gene Correction Studies Using CRISPR-Cas9 Gene Editing
Uchida et al. reported an efficient gene correction strategy for the SCD mutation in the β-globin gene with electroporation-mediated delivery of editing tools achieving therapeutic-level correction at the DNA level (about 30%) and the protein level (about 80%) [102]. This virus-free gene correction system, used to correct βS mutation, utilizes SCD mutation-Targeting guide RNA, Cas9 mRNA/protein and donor ssDNA encoding the normal β-globin sequence [102]. The gene-edited CD34+ cells with correction to normal β-globin gene sequence were engraftable in mouse and primate models [102].
The Cedar phase I/II clinical study evaluated the efficacy of autologous HSCs with correction of a single nucleotide mutation (GPH 101) to convert HbS to HbA for treating severe SCD [103]. Thus, GPH 101 is an investigational, autologous, HSC drug product designed to correct the SCD mutation in the β-globin gene ex vivo using a high-fidelity Cas9 paired with AAV6 (adenovirus-associated virus type 6), efficiently harnessing the natural HDR repair pathway [103]. In preclinical studies, β-globin gene in SCD donor HSCs resulted in ≥60% gene-corrected alleles in vitro with minimal off-target effects [103].
In the phase I/II Cedar trial for HSC gene-correction therapy in SCD with GPH 101, poor engraftment of HDR-edited CD34+ cells was observed. In the first treated patients, 2.3% editing in BM cells was detected 26 weeks post-gene therapy; however, blood cell recovery was delayed since platelet and RBC transfusions were required until 26 weeks and 38 weeks post-gene therapy, respectively [104]. This finding implies the absolute need of an improvement in the engraftment capacity of HDR-edited CD34+ cells for HSC gene-correction therapy for SCD. Clinically, the patient showed improvement in the quality of life with absent vascular occlusive events [104].
The reduced engraftment capacity of HDR-edited HSCs was supported by an experimental study in rhesus macaques [105]. Thus, Lee et al. have coinfused HSCs transduced with a barcoded GFP-expressing lentiviral vector and HDR-edited at the CD33 locus. CRISPR/HDR-edited cells showed a two-log decrease by 2 months following transplantation in comparison to minimal loss of lentivirus-transduced cells long term [105]. Furthermore, HDR long-term clonality was oligoclonal in contrast to highly polyclonal lentivirus-transduced HSCs [105]. These observations suggest marked differences in the impact of genetic modification approaches on HSCs.
4.10. Zinc Finger Nuclease-Mediated Gene Editing of BCL11A Erythroid Enhancer in HSCs
BIVV003 is a gene-edited autologous cell therapy in which HScs are genetically modified with mRNA encoding zinc finger nuclease (ZNF) that target and disrupt a specific regulatory GATAA motif present in the BCL11A erythroid-specific enhancer [106]. Zinc finger proteins combined with the nuclease domain of the restriction endonuclease Fok1 create double-strand breaks at precisely defined genomic locations. Repair of the double strand breaks via NHEJ or MMEJ results in target sequence disruption [106]. ZNF-mediated gene editing of the BCL11A erythroid-specific enhancer markedly reactivates HbF synthesis in erythroid cells without affecting in vivo engraftment of gene-edited HSCs [106]. Preliminary results obtained in the context of the PRECIZN-1 phase I-II study in 7 SCD patients showed that BIVV003 was well-tolerated and elicited an increased total Hb and HbF levels, associated with absent vaso-occlusive events [106].
5. Gene Therapy of β-Hemoglobinopathies Through In Vivo Gene Editing
As above discussed, the various approaches used for ex vivo gene therapy are very complex and expensive. The in vivo gene editing and gene therapy through intravenous or intraosseous delivery of gene therapy vectors could considerably simplify the manufacturing process, to overcome the numerous challenges associated with manufacturing of gene therapy products required for ex vivo HSCs/HPCs engineering and to reduce the costs of the whole procedure.
Various preclinical studies have supported the therapeutic efficacy of in vivo gene editing. Li et al. reported a gene therapy approach for SCD based on gene editing of the βS globin gene in HSCs in vivo by infusion of a prime editing vector in mouse bone marrow [98]. Using this approach, Le et al. reported the achievement of therapeutic levels of β-globin gene correction in mouse HSCs in vivo using a prime editing vector administered intravenously in mice [98]. In this study, prime vector was represented by an adenoviral vector with high affinity for CD46, a membrane receptor expressed on HSCs [107]. The prime vector was infused into mice with HSCs/HPCs mobilized using G-CSF/Plerixafor. With a single infusion of adenovirus carrying prime editors about 40% of βS alleles were corrected and replaced by β-WT [98]. Importantly, this level of gene correction was maintained in secondary transplants, thus supporting the efficacious gene editing of repopulating HSCs [107]. The procedure of in vivo gene editing using this delivery machinery may be simplified replacing G-CSF as mobilizing agent with WU-106, an inhibitor of integrin α4β1, obtaining a rapid and efficient mobilization of HSCs [108].
A similar approach using an adenoviral vector expressing all-in-one adenine base editor to convert the βS mutation into the benign HbGMakassar variant was used for efficient ex vivo and in vivo correction of SCD mutation [109]. The in vivo treated animals demonstrated correction of disease features without significant side effects [109].
Other studies have adopted lipid nanoparticles as in vivo delivery system alternative to viruses to vehiculate gene-editing machinery. Thus, Breda et al. have developed a CD117/LNP-messenger RNA, a lipid nanoparticle (LPN) that encapsulates mRNA and is targeted to stem cell receptor (CD117) on HSCs [101]. Delivery of the anti-human CD117/LNP-based editing system elicited a nearly complete correction of SCD mutation in hematopoietic cells [110]. Using the same anti-CD117/LNP delivery system, ex vivo delivery of adenine base editors to SCD HSCs/HPCs resulted in an efficient (88%) conversion to HbGMakassar with up to 91.7% increase in HBGMakassar protein and a nearly complete absence of sickled RBCs in mature erythroid elements [110].
Lian et al. have explored several bone marrow-homing nanoparticles and showed that they deliver mRNA to a broad group of hematopoietic cell types, including HSCs and HPCs [102]. Using these LPR-nanoparticles, CRISPR/Bas9 and base editing was achieved in a mouse model expressing human sickle cell disease phenotypes for potential HbF reactivation and conversion from sickle to non-sickle alleles [111].
Xu and coworkers reported the development of an efficient LPN-based delivery system for in vivo gene editing of BCL11A enhancer and of HBG promoter [112]. This system uses antibody-free targeted nanoparticles (LPNs) for mRNA delivery to HSCs in vivo, allowing efficient base editing of the HBG1 and HBG2 promoters in human HSCs [112]. The system uses ABE8e, an adenine base editor with an active efficient adenine conversion and showed high efficient on-target adenine base edits at the level of regulatory regions of BCL11A and HBG genes [113]. Delivery of ABE8e/sg RNA with optimized LPNs (containing lipid-168 possessing enhanced delivery efficiency in vivo) achieved efficient in vivo base editing of the HBG1 and HBG2 promoters in β-thal HSCs engrafted into immunodeficient mice, showing restored globin chain balance in erythroid cells [113].
Milani et al. recently reported an interesting observation related to the high trafficking of HSCs/HPCs observed in newborn mice as well as in humans after birth, thus showing an ontogenic window suitable for in vivo gene therapy studies [114]. The system of in vivo gene editing of HSCs is optimized also through the use of lentiviruses engineered for in vivo administration and tissue targeted expression [115]. Using this system, it was reported the efficient in vivo gene transfer into long-term HSCs in newborn mice [114]. The efficacy of this in vivo strategy of HSC gene therapy was tested in the context of mouse models of various hereditary HSC defects [114].
Other studies showed the highly efficient transduction in vivo of HSCs using adenovirus-associated virus serotype 6 (AAV6) vectors as in vivo delivery system [116]. In situ gene editing of HSCs can be achieved via AAV6-delivered CRISPR guide RNA [117]. The efficacy of this in vivo gene editing system was shown in a SCD mouse model treated with AAV6 delivering a Cas9 editing system enabling induction of therapeutic levels of HbF [117].
A recent study explored the use of extracellular vesicles (EVs, membrane limited particles secreted by the cells) as a delivery system for in vivo gene therapy [118]. Bhoorasingh et al. have used EVs engineered by loading Cas9 RNP complexes and by expressing kit ligand for efficient targeting of HSCs [118]. These EVs have an efficient targeting of c-kit expressing cells and may vehiculate CRISPR-Cas9 RNP complexes to HSCs/HPCs and may be suitable for in vivo gene therapy of SCD [118].
Other studies used a new technology of gene editing developed by Tessera Therapeutics, based on RNA gene writers that use an RNA template and a protein to make chenges to DNA without making double strand breaks. This system involves an RNA template and an RNA encoding for the writer protein packaged into LNPs for delivery into cells; into the cells, the mRNA is translated into the writer protein which then binds to the RNA template. This complex travels to the genome, nicks one strand of the DNA and uses the RNA template to write new DNA into the genome [119]. Using LPR-nanoparticles targeting HSCs and vehiculating RNA Gene Writers designed to make the wild-type correction or the HBMakassar correction of βS it was shown an efficient in vivo editing of HbS to HbA or to HbMakassar (in primate models, an in vivo editing efficiency of 20-24% was observed) [120].
In conclusion, in vivo delivery of gene therapy through viral vectors or nanoparticles offers some consistent advantages compared to ex vivo gene therapy consisting in the opportunity to avoid all the complex ex vivo procedures required for the collection and isolation of HSCs/HPCs, their ex vivo culture and genetic engineering and infusion to patients, thus simplifying the clinical gene therapy protocol. Furthermore, in vivo gene therapy approaches offer the opportunity to broaden access of patients to this therapy. However, the introduction of in vivo delivery of gene editing agents has to face many challenges, such as achieving therapeutically efficient amounts of editing agents to the target cells; avoiding base-editing at the level of nontarget cells and tissues; reducing the immunogenicity of the vector (particularly for adenovirus-based vectors) [121].
6. Affordability of Gene Therapies for Hemoglobinopathies
The approval of Exa-Cel, Beta-Cel and Lov-Cel as a potentially curative agent for SCD and β-thalassemia represented a milestone in the field and one of the most remarkable successes of modern medicine. However, these treatments based on ex vivo gene therapy are resource-intensive and not suited for the treatment of large number of patients. Various factors represent major challenges to the diffusion of these therapies. First, among them, the high cost of these therapies, estimated to amount 2.2 $ per patients for Exa-Cel and 3.1 $ million for Lov-Cel. A second limiting factor is that these treatments are feasible only in highly specialized medical centers and skilled personnel and substantial infrastructures are required during the whole treatment. The obvious consequence is that market-driven pricing and the need of adequate health care infrastructures required for delivering these therapies, will completely limit gene therapy treatments in the developing world.
These considerations have animated a consistent debate to critically evaluate affordability and accessibility of gene therapy of hemoglobinopathies. In this context, an important element of discussion is the comparison of transplantation based-studies and gene therapy studies in terms of costs and patients’ eligibility.
Allogeneic hematopoietic cell transplantation (allo-HSCT) is a curative therapy in SCD but, until recently, it was constrained by limited donor availability, by the risk of graft versus host disease (GVHD), graft rejection and death [122]. However, recent important advances have considerably mitigated these barriers. In fact, recent studies showed that haploidentical bone marrow transplant in SCD patients after non-myeloablative conditioning regimens was associated with a high rate of engraftment (88% to 95%), 2-year overall survival ranging from 94% to 95%, acute GVHD ranging from 4% to 10%, chronic GVHD ranging from 10% to 22% and a transplantation-related death rate of 5%-7% [123,124]. The use of allo-HSCT in β-thalassemia is more consolidated and retrospective analysis of long-term survival and of late effects showed a non-relapse mortality of about 10% (mostly observed during the first year after allo-HSCT) and an overall survival of 81.4% after 39 years and a cumulative incidence of secondary solid cancers of 16.4% [125,126]. The incidence of secondary solid cancers was four-fold to six-fold higher than for hematopoietic cell donors and patients non-transplanted [125,126].
At the moment, several considerations related to the cost (the cost of allo-HSCT is about five- six-fold lower than the cost of approved gene therapies of hemoglobinopathies) and to the long-term effects (that are well known for allo-HSCT, but only in part established for gene therapy treatments) favor allo-HSCT over gene therapy treatments [122].
These recommendations are important for an optimal selection of patients with hemoglobinopathies for gene therapy studies. A decision-making algorithm was developed by a group of experts in hemoglobinopathies and/or transplantation of the EHA and EBMT who discussed the selection of SCD patients for gene therapy/editing studies [127]. The team of experts has presented a document proposing to select the access of gene therapy to SCD patients in good clinical condition with no HLA-matched donor available, without irreversible severe organ impairment and able to tolerate myeloablative conditioning, since these patients are likely to obtain most benefit with the lowest risk [127]. This approach is fully justified by both the limited clinical experience so far accumulated during clinical studies and the limited availability of gene therapy [127].
There is no demonstration of feasibility or safety of HSC-based gene therapy after failed allo-HSCT. Therefore, the decision whether to pursue an autologous gene therapy after previous unsuccessful allo-HSCT may need to be made on a case-by-case basis [128].
Given the high cost and the elevated technological and infrastructural requirement, it is not surprising that multiple barriers compound challenges in gene therapy access, exacerbating existing health iniquities. These include the existence of some major barriers at financial, geographic and sociocultural levels, all of which will contribute to determine who receives these therapies first or not all [129]. Kelkar et al. considering all these factors have proposed eligibility criteria for SCD and β-thalassemia patients for gene therapy treatments. These criteria are broader than eligibility criteria of clinical trials but narrower than FDA approval criteria. Thus, fear prioritization criteria are proposed for SCD and transfusion-dependent β-thalassemia patients for gene therapy studies: i) proportional equality: patients are prioritized in a 3:1 of SCD to β-thalassemia; ii) modified sickest first: patients with impeding organ failure; iii) lack of alternative therapy: patients without a matched donor for allo-HSCT; iv) lottery: random sequencing of multiple patients within the same priority category [129].
7. Conclusions
Three approaches have been adopted for gene therapy of β-hemoglobinopathies: (i) lentiviral vector-based approaches supporting a gene addition strategy using integrating lentiviruses to introduce ex vivo into patient’s HSCs a copy of a β-globin coding sequence together with the DNA machinery required to support its high expression in erythroid cells; (ii) nuclease-based approaches supporting a gene editing strategy aiming to directly target the β-globin gene regulatory elements and consequently to reactivate HbF synthesis at therapeutic levels; (iii) base-editing approaches allowing the insertion of point mutations at level of a specific sequence in a locus, without the need for double-strand breaks and for an exogenous DNA template. Currently, three products of clinical HSC-based gene therapy for β-hemoglobinopathies have been approved by federal regulatory agencies. Thus, Betibeglogene autotemcel (with the commercial name of ZYNTEGLB), based on βA-T87Q globin gene addition via lentivirus was approved by EMA in 2019 and by FDA in 2022 for transfusion-dependent β-thalassemia; Lovotibeglogene autotemcel (with the commercial name of LYFGENIA), based on βA-T87Q globin gene addition via lentivirus was approved by FDA in 2023 for SCD; Exagamglogene autotemcel, based on CRISPR-Cas9 gene editing of BCL11A enhancer via electroporation (with the commercial name of CASGEVY) was approved by EMA in 2023 and FDA in 2023/2024 for both SCD and therapy-dependent β-thalassemia [130]. The three gene therapy products approved for SCD and β-thal treatment support a possible curative effect and represent some of the most remarkable successes of modern medicine.
Although these treatments may offer a curative approach for the treatment of SCD and β-thalassemia, face numerous challenges due to their high costs, the complexity of manufacturing the therapeutic agents and some safety concerns.
Concerning the safety concerns it has to be emphasized that the safety profile of lentivirus and genome editing based therapies is consistent with myeloablative conditioning and autologous HSCT. In this context, myeloablative conditioning represents an important limitation of current gene therapies since it can result in both severe acute and chronic toxicities. Several gene therapies studies of β-hemoglobinopathies have suggested that myeloablative conditioning is required to maximize engraftment of genetically-engineered CD34+ cells. In the three approved gene therapy studies, fully myeloablative conditioning with busulfan was used. In the study HGB-206 of lentiviral-mediated βT87Q-globin transduction, the first seven patients received a busulfan conditioning regimen with a busulfan exposure target area under the curve (AUC) 65-74 mg*h/L and a cell dose of ≥ 1.5x106 CD34+ cells/kg; a durable expression of the transgene was observed but suboptimal HBAT87Q concentration of 0.51 to 1,17 g/dL were observed, requiring an adjustment of busulfan conditioning, with increased target AUC to 82 mg*h/L in group B and C [36]. In the HGB-204, -205, -207 and -212 trials, transfusion dependent β-thalassemia patients received a myeloablative conditioning with a busulfan dose of HGB-207 and -212 with a busulfan AUC of 59-82 mg*h/L [36]. In the studies of Examglogene-based gene therapy of SCD and β-thal patients a myeloablative busulfan-base regimen was used with a busulfan AUC dose of AUC of 74-90 mg*h/L [71,76]. Also, other studies of gene editing of HBG1 and HBG2 promoters have incorporated myeloablative busulfan conditioning into their designs [83].
In the TIGET-Bthal trial a reduced toxicity myeloablative regimen with treosulfan and thiotepa was used and transfusion independence was reached only in a part of treated patients [40]. Furthermore, in a small phase I trial carried out with a lentiglobin vector (TNS9.3.55) the patients received a reduced-intensity busulfan conditioning regimen; low but stable hematopoietic gene marking was observed and patients not reached transfusion independence [131] Non-myeloablative conditioning regimens may significantly reduce the risk of these acute and chronic toxicities, may allow a wider patient eligibility and may lower resource use reducing the need for extensive hospital care. In this context, reduced-intensity conditioning was adopted in few gene therapy studies of β-hemoglobinopathies, supporting that non-myeloablative conditioning can achieve durable stem cell engraftment [34]. The study of Grimley et al. showed that the use of reduced-intensity melphalan conditioning instead of myeloablative busulfan in 7 SCD patients undergoing autologous stem cell transplantation with CD34+ cells transduced with a lentivirus vector inducing enforced expression of γ-globin G16D, decreased the duration of thrombocytopenia and neutropenia and accelerated neutrophil and platelet engraftment and reduced the time of hospitalization after transplantation compared to what observed in the studies with currently approved gene therapy products [32]. In this study, after the treatment of the first two patients, an improved strategy of HSCs/HPCs mobilization and collection was adopted with multiple collections; furthermore, melphalan dose was adjusted according to glomerular filtration [34]. The success of reduced-intensity conditioning in this study is seemingly related to a more potent anti-sickling efficacy of γG16D than βT87Q since patients with βT87Q levels in the HGB-206 study similar to those observed for γG16D in the study of Grimley et al. had suboptimal clinical benefit [24]. Although the results are encouraging, observations on large number of patients and on different gene therapy vectors are required to assess the effect of reduced-intensity conditioning compared to myeloablative conditioning.
Another important issue of safety is related to the risk of secondary hematological malignancies that is complex in its multifactorial origin, not related only to the risk of insertional mutagenesis. The risk of insertional mutagenesis was related to the studies involving the use of integrating lentiviral vectors. Third-generation lentiviral-based vectors, as well as those used in Beta-Cel and Lov-Cel therapies, have been engineered to be replication incompetent and self-inactivating, thus reducing their risk of insertional mutagenesis. No events of insertional oncogenesis have been observed in β-thal patients treated with Beta-Cel; however, two SCD patients who received Lov-Cel developed AML at 3 and 5.5 years after infusion: both patients did not show insertional oncogenesis; in one patient, leukemic cells contained the BB305 lentiviral vector [132,133]. The emergence of myeloid malignancy in gene therapy recipients with SCD is likely related to regeneration stress induced by the gene therapy procedure, triggering rapid proliferation of clones with driver mutations. These clones are related to clonal hematopoiesis, characterized by the presence of somatic mutations in the peripheral blood with a variant allele frequency ≥2% in genes associated with hematologic malignancies. Both patients with SCD [134,135] and β-thalassemia [136] display a significantly increased rate of clonal hematopoiesis compared to age-matched healthy individuals. Clonal hematopoiesis is increasingly recognized in SCD patients at younger ages than in the general population, potentially due to chronic inflammation, erythropoietic stress and exposure to cytotoxic therapies [137,138]. Particularly patients with CH-related mutations TP53 and PPM1D may have an increased risk to develop leukemia post-transplantation [137,138].
An elevated mutation rate observed in some patients with SCD and some pressure on HSCs containing pre-existing driver mutations represent mechanisms that could increase leukemia risk in patients undergoing gene therapy for SCD and β-thalassemia [46]. A non-myeloablative conditioning regimen offers also the advantage of reducing the risk of positive selection of pre-existing clonal hematopoiesis-associated driver mutations.
Another limiting factor emerging from gene therapy studies is that a fully curative effect is not frequently observed in patients most severely affected by β-thalassemia. This suggests that the efficacy of gene therapy treatments could be obtained by achieving higher levels of therapeutic Hb levels. In this context, recent studies have provided evidence that gene editing of both the BCL11A +58 and +55 enhancers elicited a more efficacious stimulation of HbF synthesis than a single gene editing [53,125,126]. In this setting of double editing strategies, several strategies reduced the risk of genome aberrations caused by CRISPR-Cas9-mediated double strand breaks, such as the use of base-editing and not CRISPR-Cas9 editing, the omission of ex vivo culture of gene-edited HSCs/HPCs avoiding the deleterious effects of cellular proliferation stimulated by ex vivo culture [139] and the conditioning procedure (nonprogrammed long deletions were disfavored in engrafting cells in animals conditioned by a CD45 antibody drug conjugate compared to animals conditioned using busulfan) [140].
CRISPR-Cas9 editing and lentiviral transduction have shown consistent clinical benefits, but it remains unclear which approach is superior. Alternatively base editing showed also promising results and could reduce risks of genotoxicity. A recent study compared three different gene therapy approaches, such as CRISPR-Cas9 editing of the BCL11A enhancer to reactivate HbF, lentiviral anti-sickling βT87Q gene addition, and base editing converting sickle β-globin to βMakassar globin in an immunodeficient mouse model [141]. All three gene-editing methods showed therapeutic potential; however, base editing and lentiviral transduction showed superior outcomes over CRISPR-Cas9-mediated editing in a competitive murine transplantation model [141].
Author Contributions
G.C. and E.P. were involved in researching, writing, and editing the manuscript. U.T. was involved in conceptualization, organization, research, and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Orkin, S.H. The fetal-to-adult hemoglobin switch—mechanisms and therapy. N Engl J Med 2025, 392, 2135–2149. [Google Scholar] [CrossRef]
- Huang, P.; Pesiak, C.A.; Ren, R.; Khandros, E.; Qin, K.; Keller, C.A.; Giardine, B.; Bell, H.W.; Lan, X.; Sharma, M.; et al. HIC2 controls developmental hemoglobin switching by repressing BCL11A transcription. Nat Genet 2022, 54, 1417–1426. [Google Scholar] [CrossRef]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013, 342, 2453–2457. [Google Scholar] [CrossRef]
- Masuda, T.; Wang, X.; Maedfa, M.; Canver, M.C.; Sher, F.; Funnel, A.; Fisher, C.; Suclu, M.; Martyn, G.; Norton, L.; et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef]
- Piel, F.; Steinberg, M.; Rees, D.C. Sickle cell disease. N Engl J Med 2017, 376, 1561–1573. [Google Scholar] [CrossRef]
- Taher, A.; Musallam, K.; Cappellini, D. β-thalassemias. N Engl J Med 2021, 384, 727–743. [Google Scholar] [CrossRef]
- Jones-Wonni, B. A review of gene therapies for hemoglobinopathies. Hemoglobin 2024, 48, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Butt, H.; Sathish, S.; London, E.; Johnson, T.L.; Essaewi, K.; Leonard, A.; Tisdale, J.F.; Demirici, S. Genome editing strategies for targeted correction of β-globin mutation in sickle cell disease: from bench to bedside. Mol Ther 2025, 33, 2154–2171. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.D.; Cordes, S.; Taylor, T.; Cotes, W.; Roskom, K.; Link, M.; Hsieh, M.M.; Tisdale, J.F. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood 2017, 130, 1946–1948. [Google Scholar] [CrossRef]
- Frangoul, H.; Stuits, A.; Bruce, K.; Domm, J.; Carroll, C.; Aide, S.; Duckworth, M.; Evans, M.; McManus, M. Best practices in gene therapy for sickle cell disease and transfusion-dependent β-thalassemia. Transpl Cell Therapy.
- Poletti, V.; Mavilio, F. Designing lentiviral vectors for gene therapy of genetic diseases. Viruses 2021, 13, 1526. [Google Scholar] [CrossRef] [PubMed]
- Ballantine, J.; Tisdale, J.F. Gene therapy for sickle cell disease: recent advances, clinical trials and future directions. Cytotherapy 2025, 27, 826–834. [Google Scholar] [CrossRef]
- Sadelain, M.; Wang, C.H.; Antoniou, M.; Grosveld, F.; Mulligan, R.C. Generation of a high-titer retroviral vector capable of expressing high levels of the human beta-globin gene.
- Miller, A.D.; Bender, M.A.; Harris, E.; Kalako, M.; Gelinas, R.E. Design of retrovirus vectors for transfer and expression of the human β-globin gene. J Virol 1988, 62, 4337–4345. [Google Scholar] [CrossRef]
- Psatha, N.; Sava, P.; Georgopoulos, G.; Psehaudi, K.; Iwata, M.; Bloom, J.; Ulyanova, T.; Wang, H.; Kiztsau, A.; Vasiloudis, N.I.; et al. Large-scale discovery of potent, compact and erythroid specific enhancers for gene therapy vectors. Nat Commun 2025, 16, 4325. [Google Scholar] [CrossRef]
- Pawliuk, R.; Westerman, K.A.; Fabry, M.E.; Payen, E.; Bouhassira, E.E.; Acharya, S.A.; Ellis, J.; London, I.M.; Eaves, C.J.; Humphries, R.K.; et al. Correction of sickle-cell disease in transgenic mouse models by gene therapy. Science 2001, 294, 2368–2371. [Google Scholar] [CrossRef]
- McCune, S.L.; Reilly, M.P.; Chomo, M.J.; Asakura, T.; Townes, T.M. Recombinant human hemoglobins designed for gene therapy of sickle cell disease. Proc Natl Acad Sci USA 1994, 91, 9852–9856. [Google Scholar] [CrossRef]
- Grimley, M.; Sherestha, A.; Felker, S.; Lutzko, C.; Arumugam, P.I.; Wititng, S.; et al. Safety and efficacy of Aru-1801 in patients with sickle cell disease: early results from the phase 1-2 momentum study of a modified gamma globin gene therapy and reduced intensity conditioning. Blood 2021, 138 (suppl.1), 3970–3972. [Google Scholar] [CrossRef]
- Cabriolu, A.; Odek, A.; Zamparo, L.; Yuan, H.; Leslie, C.D.; Sadelain, M. Globin vector regulatory elements are active in early hematopoietic progenitor cells. Mol Ther 2022, 30, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E. Gene editing. Gene genich cures sickle cell in mice. Science 2011, 294, 2268. [Google Scholar]
- Demirci, S.; Gudmundsdottir, B.; Li, Q.; Haro-Mora, J.J.; Nassehi, T.; Drysdale, C.; Yapundich, M.; Gamer, J.; Seifuddin, F.; Tisdale, J.F.; et al. βT87Q-globin gene therapy reduces sickle hemoglobin production, allowing for ex vivo anti-sickling activity in human erythroid cells. Methods Clin Dev 2020, 17, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene therapy in a patient with sickle cell disease. N Engl J Med 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Magrin, E.; Semeraro, M.; Hebert, N.; Joseph, L.; Magnani, A.; Chalumeau, A.; Gabrion, A.; Roudaut, C.; Marouene, J.; Lfrere, F.; et al. Long-term outcomes of lentiviral therapy for the β-hemoglobinopathies: the HGB-205 trial. Nat Med 2022, 28, 81–88. [Google Scholar] [CrossRef]
- Kanter, J. Thompson, A.A.; Piercey, F.J.; Hsieh, M.; Uchida, N.; Leboulch, P.; Schmidt, M.; Bonner, M.; Guo, R.; Miller, A.; et al. Loco-cel gene therapy for sickle cell disease: treatment process evolution and outcomes in the initial groups of the HGB-206 study. Am J Hematol 2023, 98, 11–22. [Google Scholar] [CrossRef]
- Kanter, J. Walters, M.C.; Krishnamurti, L.; Mapara, M.Y.; Kwiatkowski, J.L.; Rifkin-Zenenberg, S.; Aygun, B.; Kasow, K.A.; Pierciey, F.J.; Bonner, M.; et al. Biologic and clinical efficacy of LentiGloibin for sickle cell disease. N Engl J Med. 2022, 386, 617‐628.
- Rifkin-Zenenberg, S.; Kanter, J.; Kinney, M.A.; Kwiatkowski, J.L.; Nickel, R.S.; Walters, M.C.; Parikh, S.; Thompson, A.; George, A.P.; Mapara, M.; et al. An update on Lovotibeglogene autotemcel (Lovo-cel) for sickle cell disease (SCD) and analysis of early predictors of response to lovo-cel. Blood 2024, 144 (suppl.1), 511–512. [Google Scholar] [CrossRef]
- Kanter, J.; Chawla, A.; Thompson, A.A.; Kwiatkowski, J.L.; Parikh, S.; Mapara, M.Y.; Rifkin-Zenenberg, S.; Aygun, B.; Kasow, K.A.; Gupta, A.O.; et al. Lovotibeglogene autotemcel gene therapy for sickle cell disease: 60 months follow-up. J Sickle Dis 2024, 1, 3–4. [Google Scholar] [CrossRef]
- Kinney, M.A.; Shestopalov, I.; Christiansen, L.; Jiang, H.; Foos, M.; Elliot, H.; Chawla, A.; Piercey, F.J. Predictors of biologic efficacy with Lovotibeglogene autotemcel (Lovo-cel) gene therapy in patients with sickle cell disease. Transpl Cell Therapy 2024, 30 (supplement 2), 302. [Google Scholar] [CrossRef]
- Heering, W.L.; Gallagher, M.E.; Shah, N.; Morse, K.C.; Mladsi, D.; Dong, O.M.; Chawrla, A.; Leiding, J.; Zhang, L.; Paramore, C.; et al. Cost-effectiveness for Lovotibeglogene autotemcel (Lovo-Cel) gene therapy for patients with sickle cell disease and recurrent vaso-occlusive events in the United States. PharmacoEconomics 2024, 42, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Urbinati, F.; Fernandez, B.C.; Masiuk, K.E.; Poletti, V.; Hollis, R.P.; Koziol, C.; Kaufman, M.L.; Brown, D.; Mavilio, F.; Kohn, D.B. Gene therapy for sickle cell disease: a lentiviral vector comparison study. Human Gene Therapy 2018, 29, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Prueksapraopong, C.; Fernandes, A.; Fernandez, B.C.; Roy, S.; Habtemariam, B.; Romero, Z.; Moore, T.M.; Schiller, G.J.; Kohn, D.B. Clinical outcomes of Lenti/G-βAS3-FB lentiviral vector gene therapy for sickle cell disease. Transpl Cell Ther 2025, 31, S255–S256. [Google Scholar] [CrossRef]
- Sobrino, S.; Joseph, L.; Magrin, E.; Chalumeau, A.; Hebert, N.; Corsia, A.; Denis, A.; Roudaut, C.; Aussel, C.; : Leblanc, O.; et al. Severe inflammation and lineage skewing are associated with poor engraftment of engineered hematopoietic stem cells in patients with sickle cell disease. Nat Commun 2025, 16, 3137. [Google Scholar] [CrossRef]
- Perumbeti, A.; Higashimoto, T.; Urbinati, F.; Franco, R.; Meiselman, H.J.; Witte, D.; Lalik, P. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood 2009, 114, 1174–1185. [Google Scholar] [CrossRef]
- Grimley, M.; Davies, S.M.; Shrestha, A.; Shova, A.; Asnani, M.; Kent, M.; Sayani, F.; Quin, C.; Niss, O.; Lutzko, C.; et al. Lentiviral gene therapy with reduced-intensity conditioning for sickle cell disease: a phase 1-2 trial. Nat Med 2025, 31, 2204–2212. [Google Scholar]
- Thompson,A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.; Ribeil,J.A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2022, 386, 617‐628.
- Locatelli, F.; Thompson, A.A.; Kwiatkowski, J.; Porter, J.B.; Thrasher, A.J.; Hongerng, S.; Sauer, M.G.; Thuret, I.; Lal, A.; Algeri, M.; et al. Betibeglogene autotemcel gene therapy for non-β°/β° genotype β-thalassemia. N Engl J Med 2022, 386, 415–427. [Google Scholar]
- Kwiatkowski, J.; Walters, M.C.; Hongeng, S.; Yannaki, E.; Kulozik, A.E.; Kunz,J.B.; Sauer, M.G.; Tharasher, A.J.; Thuret, I.; Lal, A.; et al. Betibeglogene autotemcel gene therapy in patients with thalassemiadependent, severe genotype β‐thalassemia (HGB‐212): a non‐randomised, multicentre, single‐arm, openlabel, single‐dose, phase 3 trial. Lancet 2024, 404, 2175‐2186 .
- Gibson, N.M.; Friedman, D.F.; Elgarten, C.W.; Haimed, A.; Khandros, E.; Worster, E.; Bardahl, J.; Wray, L.; Wang, Y.; Thompson, A.A.; et al. Post-approval, real-world experience with Betibeglogene Autotemcel for transfusion-dependent beta thalassemia. Transpl Cell Therapy 2025, 31 (suppl. 1). S254.. [Google Scholar] [CrossRef]
- Mizza, A.; Ritsert, M.L.; Tao, G.; Thakar, H.; Labitz, S.; Heine, S.; Kosher, L.; Durken, M.; Schmitt, A.; Pavel, P.; et al. Gene therapy in transfusion-dependent non-β°/β° genotype β-thalassemia: first real-world experience of beti-cel. Blood Adv 2025, 9, 29–38. [Google Scholar]
- Marktel, S.; Scaramuzza, S.; Cicalese, M.P.; Giglio, F.; Galimberti, S.; Lidonnici, M.R.; Calbi, V.; Assanelli, A.; Bernardo, M.E.; Rossi, C.; et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent β-thalassemia. Nat Med 2019, 25, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, Z.; Chen, S.; Huang, Y.; Ling, S.; Wang, Q.; Wang, Q. Preclinical efficacy and safety evaluation of BD211 autologous CD34+ hematopoietic stem cell injection for transfusion-dependent β-thalassemia in NCG-X mice. Front Cell Dev Biology 2025, 13, 1607707. [Google Scholar]
- Li, S.; Ling, S.; Wang, D.; Wang, X.; Hao, F.; Yin, L.; Yuan, Z.; Liu, L.; Zhang, L.; Li, Y.; et al. Modified lentiviral globin gene therapy for pediatric β°/β° transfusion-dependent β-thalassemia: a single-center, single-arm pilot trial. Cell Stem Cell 2024, 31, 961–973. [Google Scholar]
- Brendel, C.; Negre, O.; Rothe, M.; Guda, S.; Parson, G.; Harris, C.; McGiuness, M.; Abriss, D.; Tsytsykova, A.; Klatt, D.; Bentler, M.; et al. Preclinical evaluation of a novel lentiviral vector driving lineage-specific BCL11A knockdown fro sickle cell gene therapy. Mol Therapy Methods Clin Dev 2020, 17, 589–595. [Google Scholar] [CrossRef]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med 2021, 384, 205–215. [Google Scholar]
- Esrick, E.B.; Federico, A.; Abriss, D.; Armant, M.; Boardman, K.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; Dansereau, C.; Fernandes, A.; et al. Induction of fetal hemoglobin and reduction of clinical manifestations in patients with severe Ahmir-base lentiviral gene therapy for post-transcriptional gene editing of BCL11A: updated results from pilot and feasibility trial. Blood 2022, 140 (suppl. 1), 10665–10667. [Google Scholar]
- Esrick, E.B.; Lehmann, L.E.; Federico, A.; Daley, H.; Dansereau, C.; DeOliveira, S.; Everett, J.; Kao, P.C.; Liu, B.; Moore, T.; et al. Long-term follow-up of the firsty in human post-transcriptional genetic silencing of BCL11A in sickle cell disease in a phase 1 pilot and feasibility study. Blood 2025, 146 (suppl.1), abs25–2853. [Google Scholar]
- Jimenez-Kurlander, L.; Kao, P.C.; Morris, E.; Federico, A.; Moore, T.; DeOliveira, S.; Fernandes, A.; Kohn, D.B.; London, W.B.; Williams, D.A.; et al. Patient and parent-reported outcomes post-treatment with Shmir-based lentiviral gene therapy for sickle cell disease. Blood 2023, 142 (suppl.1), 3750. [Google Scholar] [CrossRef]
- De Souza, D.C.; Hebert, N.; Esrick, E.B.; Ciulescu, M.F.; Archer, N.A.; Armant, M.; Audereau, E.; Brendel, C.; Di Caprio, G.; et al. Genetic reversal of the globin switch concurrently modulates both fetal and sickle hemoglobin and reduces red cell sickling. Nat Commun 2023, 14, 5850. [Google Scholar] [CrossRef] [PubMed]
- Spencer Chapman, M.S.; Cull, A.H.; Ciuculescu, M.F.; Esrick, E.B.; Mitchell, E.; Jung, H.; O’Neill, L.; Roberts, K.; Fabre, M.A.; Williams, N.; et al. Clonal selection of hematopoietic stem cells after gene therapy for sickle cell disease. Nature 2023, 29, 3175–3183. [Google Scholar] [CrossRef]
- Xue, C.; Greene, E.C. DNA repair pathway choices in CRISPR-Cas9-mediated genome editing. Trends Genet 2021, 37, 639–656. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Kostamo, Z.; Ortega, M. A: Xu, C.; Feliciano, P.R.; Budak, E.; Lam, D.; Winton, V.; Jenkins, R.; Venugopal, A.; Zhang, M.; et al. Base editing HbS to HbG-Makassar improves hemoglobin function supporting its use in sicke cell disease. Nat Commun. 2025, 16, 1441.
- Radtke, S.; Fields, E.; Swing, K.; Kanestrom, G.; Yen, J.S.; Pande, D.; Enstrom, M.R.; Humbert, O.; Weiss, M.J.; Liu, D.R.; et al. Engraftment and persistence of HBB base-edited hematopoietic stem cells in nonhuman primates. Sci Transl Med 2025, 17, eadn2601. [Google Scholar] [CrossRef]
- Mayuranathan, T.; Newby, G.A.; Feno, R.; Yao, Y.; Mayberry, K.D.; Lazzarotto, C.R.; LI, Y.; Levine, R.M.; Nimmagadda, N.; Dempsey, E.; Kang, G.; et al. Potent and uniform fetal hemoglobin induction via base editing. Nat Genet 2023, 55, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Qiu, H.Y.; Sun, S.; Fu, Z.C.; Wang, Q.Q.; Qian, X.; Wang, L.; Zhai, X.; Wei, J.; Wang, Y.; et al. Base editing of the HBG promoter induces potent fetal hemoglobin expression with no detectable off-target mutations in human HSCs. Cell Stem Cell 2023, 30, 1624–1639. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Martinucci, P.; Amistadi, S.; Felix, T.; Mombled, M.; Tachtsidi, A.; Corre, G.; Chalumeau, A.; Hardouin, G.; Martin, J.; et al. Multiplex base editing of BCL11A regulatory elements to treat sickle cell disease. Cell Rep Med 2025, 26, 102376. [Google Scholar] [CrossRef]
- Rajendiran, V.; Devaraju, N.; Haddad, M.; Ravi, N.S.; Panigrahi, L.; Paul, J.; Gopalakrishnan, C.; Wyman, S.; Ariudainambi, K.; Mahalingam, G.; et al. Base editing of key residues in the BCL11A-XL-specific zinc finger domains derepresses fetal globin expression. Mol Ther 2024, 32, 663–677. [Google Scholar] [CrossRef]
- Chen, P.J.; Liu, D.R. Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet 2023, 24, 161–177. [Google Scholar] [CrossRef]
- Chauhan, V.; Sharp, P.A.; Langer, R. Engineered prime editors with minimal genomic errors. Nature 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Everette, K.A. ;Newby GA, Levine, R.M.; Mayberry, K.; Jang, Y.; Mayuranathan, T.; Nimmagadda, N.; Dempsey, E.; Li, Y.; Bhoopala, N.; et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat Biomed Eng. 2023, 7, 616‐626.
- Chalumeau, A.; Dames, M.B.; Fontana, L.; Amistadi, S.; Anatoniou, P.; Loganathan, P.; Mombled, M.; Corre, G.; Peterka, M.; Amendola, M.; et al. A prime editing strategy to rewrite the γ-globin promoters and reactivate fetal hemoglobin for sickle cell disease. Blood 2025, in press. [Google Scholar] [CrossRef]
- Fiumara, M.; Ferrari, S.; Omer-Javed, A.; Beretta, S.; Albano, L.; Canarutto, D.; Varesi, A.; Gaddoni, C.; Brombin, C.; Cuganta, F.; et al. Genotoxic effects of base and prime editing in human haematopoietic stem cells. Nature Biotechnol 2024, 42, 877–891. [Google Scholar] [CrossRef]
- Hwang, G.H.; Lee, S.H.; Oh, M.; Kim, S.; Habib, O.; Jang, H.K.; Kim, H.S.; Kim, Y.; Kim, C.H.; Kim, S.; et al. Large DNA deletions occur during DNA repair at 20-fold lower frequency for base editors and prime editors than for Cas9 nucleases. Nature Biomed Eng 2025, 9, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2021, 184, 844. [Google Scholar] [CrossRef]
- Bell, H.W.; Feng, R.; Shah, M.; Yao, Y.; Douglas, J.; Doerfler, P.A.; Mayuranathan, T.; O’Dea, M.F.; Li, Y.; Wang, Y.D.; et al. Removal of promoter CpG methylation by epigenome editing reverses HBG silencing. Nat Commun 2025, 16, 6919. [Google Scholar] [CrossRef]
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Zie, F.; Muench, M.O.; Kan, Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia. Proc Natl Acad Sci USA 2016, 113, 10661–10665. [Google Scholar] [CrossRef]
- We,Y. ; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 2019, 25, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Althshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foeli, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Locatelli, F.; Lang, P.; Wall, D.; Meisel, R.; Carbacioglu, S.; Li, A.M.; d e la Fuente, J.; shah, A.J.; Carpentier, B.; Kwiatkowski, J.L.; et al. Examglogene autotemcel for transfusion-dependent β-thalassemia. N Engl J Med 2024, 390, 1663–1676. [Google Scholar] [CrossRef]
- Frangoul, H.; Locatelli, F.; Lang, P.; Meisel, R.; Wall, D.A.; Carbacioglu, S.; Li, A.; de la Fuente, J.; Shah, A.J.; Carpenter, B.; et al. Durable clinical benefits in transfusion-dependent β-thalassemia with Examglogene Autotemcel. Transpl Cel Ther. 2023, 31 (suppl.1) S252.
- Locatelli, F.; Meisel, R.; Carbacioglu, S.; de la Fuente, J.; Algeri, M.; Ruppechjt, J.; Kuo, K.; Shah, A.; Lang, P.; Merkeley, H.; et al. Correction of ineffective erythropoiesis and durable clinical benefit with exagamglogene autotemcel for transfusion-dependent β-thalassemia. Blood 2025, 146 (suppl.1), abs25–8312. [Google Scholar]
- Fu, B.; Liao, J.; Chen, S.; Li, W.; Wang, Q.; Hu, J.; Yang, F.; Hsiao, S.; Jiang, Y.; Wang, L.; et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric β°/β° transfusion-dependent β-thalassemia. Nat Med 2022, 28, 1573–1580. [Google Scholar] [CrossRef]
- Zheng, B.; Liu, R.; Zhang, X.; Fu, B.; Xu, Y.; Shi, J.; Feng, X.; Wang, L.; Wang, C.; Liang, R.; Tan, L.; et al. Efficacy and safety of BRL-101, CRISPR-Cas9-mediated gene editing of the Bcl11A enhancer in transfusion-dependent β-thalassemia. Blood 2023, 142, 4995–4996. [Google Scholar] [CrossRef]
- Zheng, B.; Liu, R.; Zhang, X.; Fu, B.; Xu, Y.; Shi, J.; Feng, X.; Li, D.; Wu, Y.; Liu, M.; et al. Efficacy and safety of BRL-101, CRISPR-Cas9-mediated gene editing of the Bcl11A enhancer in transfusion-dependent beta-thalassemia. EHA Library. 2024, p1514.
- Frangoul,H. ; Locatelli, F.; Sharma, A.; Bhatia, M.; Mapara, M.; Liem, R.I.; Telfer, P.; Shah, A.J.; Cavazzana, M.; Corbacioglu, S.; et al. Examglogene autotemcel for severe sickle cell disease. N Engl J Med 2024, 390, 16749–1662. [Google Scholar]
- Grupp, S.A.; Locatelli, F.; Sharma, A.; Bhatia, M.; Mapara, M.; Liem, R.I.; Wall, D.A.; Molinari, L.; Dedeken, L.; Kuo, K.; et al. Durable clinical benefits in severe sickle cell disease with exagamglogene autotemcel. Transl Cell Therapy. 2025, 31 (suppl. 1) S20‐S21.
- Grupp, S.; Locatelli, F.; Sharma, A.; Bhatia, M.; Mapara, M.; Liem, R.; Dedeken, L.; Molinari, L.; Eckrich, M.; Kuo, K.; et al. Long-term follow-up demonstrates durable clinical benefits of examglogene autotemcel for sickle cell disease with recurrent vaso-occlusive crises: final results of Climb SCD-121. Blood 2025, 146 (suppl.1), abs25–801. [Google Scholar]
- Sharma, S.A.; Locatelli, F.; Bhatia, M.; Molinari, L.; Mapara, M.; Liem, R.; Wall, D.A.; Molinari, L.; Dedekan, L.; Wall, D.; et al. Improvements in health-reklated quality of life in patients with severe sickle cell disease after examglogene autotemcel. Blood Adv 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; de la Fuente, J.; Algeri, M.; Chopra, Y.; Amrolia, P.; Sharma, A.; Meisel, R.; Cappellini, M.D.; Carbacioglu, S.; Kattamis, A.; et al. First results of examglogene autotemcel in pediatric patients aged 5-11 years with transfusion-dependent β-thalassemia or sickle cell disease with recurrent severe vaso-occlusive crises. Blood 2025, 146 (suppl.1), abs25–8416. [Google Scholar]
- Ye, D.; Chen, M.; Zhu, Y.; Feng, X.; Xu, L.; Huang, H. Comprehensive regulation of γ-globin expression by epigenetic modification s and protein post-translational modifications. Clin Epigenetics 2025, 17, 173. [Google Scholar] [CrossRef]
- Wongabirisuth, C.; Innachai, P.; Saisawang, C.; Tabsuwan, A.; Jearawirslyaparisan, N.; Kaewprommal, P.; Piryapoagsa, J.; Chiangyong, W.; Anurathapani, U.; Sandeis, D.; et al. Disrupting ZBTB7A or BCL11A binding sites reactivates fetal hemoglobin in erythroblasts from healthy and β°/HbE individuals. Scient Rep 2025, 15, 25580. [Google Scholar] [CrossRef] [PubMed]
- Traxler, E.A.; Yao, Y.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A genome editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 2016, 22, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Métais, J.Y.; Doerfler, P.A.; Mayuranathan, T.; Bauer, D.E.; Fowler, S.C.; Hsieh, M.M.; Katta, V.; Kerwala, S.; Lazzarotto, C.R.; Luk, K.; et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv 2019, 3, 3379–3392. [Google Scholar] [CrossRef]
- Sharma, A.; Boelens, J.J.; Cancio, M.; Hanking, J.S.; Bhad, P.; Azizy, M.; Lewandoski, A.; Zhao, X.; Chitnis, S.; Peddinti, R.; et al. CRISPR-Cas9 editing of the HBBG1 and HBBG2 promoters to treat sickle cell disease. N Engl J Mded 2023, 389, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.O.; Sharma, A.; Frangoul, H.; Dalai, J.; Kanter, J.; Alavi, A.; DiPersio, J.; Eapen, M.; Jaroscak, J.J.; Ayala, E.; et al. Initial results from the BEACON clinical study: a phase 1-2 study evaluating the safety and efficacy of a single dose of autologous CD34+ base edited hematopoietic stem cells (BEAM-101) in patients with sickle cell disease with severe vaso-occlusive crises. Blood 2024, 144 (suppl.1), 513. [Google Scholar] [CrossRef]
- Chockalingram, P.S.; Chen, G.; Minella, A.C.; Chen, Y.; Shehan, V.; Zhang, N.; Armant, M.; Zaidi, A.U.; Goodrich, R.; Hines, P.C.; et al. Impact of BEAM-101 treatemnt on red blood cell hemoglobin expression, rheology and sickling properties: initial data from the BEACON phase 1-2 study of autologous CD34+ base edited hematopoietic stem cells in sickle cell disease. Blood 2024, 144 (suppl.1), 4957–4958. [Google Scholar] [CrossRef]
- Gupta, A.O.; Sharma, A.; Frangoul, H.; Dalai, J.; Kanter, J.; Alavi, A.; DiPersio, J.; Eapen, M.; Jaroscak, J.J.; Ayala, E.; et al. Base editing for sickle cell disease: ongoing results from the Beacon study evaluating the safety and efficacy of BEAM-101, the first base-edited autologous CD34+ HSPC one-time cell therapy. EHA Meeting 2025. abst. PF 1151.
- Gupta, A.; Sharma, A.; Frangoul, H.; Kanter, J.; Mapara, M.; Dalai, J.; Alavi, A.; Jaroscak, J.; Ayala, E.; DiPersio, J.; et al. Robust HbF induction and improvement of anemia and hemolysis with base editing in sickle cell disease: safety and efficacy findings from the ongoing BEACON study. Blood 2025, 146 (suppl.1), abs25–2531. [Google Scholar]
- Wang, L.; Xue, W.; Zhang, H.; Gao, R.; Qiu, H.; Wei, J.; Zhou, L.; Lei, Y.N.; Wu, X.; Li, X.; et al. Eliminating base-editor-induced genome-wide and transcriptome-wide off-target mutations. Nat Cell Biol 2021, 23, 552–563. [Google Scholar] [CrossRef]
- Wang, L.; Li, P.; Wang, Y.; Lan, K.; Zheng, H.; Zhu, D.; Zhang, Y.; Guo, R.; Ma, H.; He, J.; et al. Development of best-in-class gene editing therapy for β-hemoglobinopathies using innovative transformer base editor (tBE). Blood 2023, 142 (suppl.1), 5243. [Google Scholar] [CrossRef]
- Lai, Y.; Liu, R.; Wang, L.; Ma, X.K.; Li, Y.; Yang, G.; Shi, L.; Wei, J.; Wei, Z.; Zhou, X.; et al. Rapid, efficient and durable fetal hemoglobin production following CS-101 treatment in transfusion-dependent β-thalassemia participants: an autologous, ex vivo edited CD34+ stem cell product using the innovative transformer base editor (tBE). Blood 2024, 144 (suppl.1), 4962–4963. [Google Scholar] [CrossRef]
- Lai, Y.; Liu, R.; Wang, L.; Ma, X.K.; Li, Y.; Yang, G.; Shi, L.; Wei, J.; Wei, Z.; Zhou, X.; et al. Treatment of patients with severe transfusion-dependent β-thalassemia with CS-101, an autologous, ex vivo edited, CD34+ hematopoietic stem cell product using innovative transformer base editor (TBE). 2025, EHA Meeting abst. S291.
- Chen, J.; Lai, Y.; Zhai, X.; Zhao, W.L.; Liu, R.; Qian, X.; Tang, W.; Wang, L.; Ma, X.K.; LI, Y.; et al. Rapid, efficient and durable fetal hemoglobin production following CS-101 treatment in transfusion-dependent β-thalassemia participants; an autologous, ex vivo edited CD34+ stem cell product using the innovative transformer base editor (tBE). Blood 2025, 146 (suppl.1), abs25–8614. [Google Scholar]
- Paul, B.; Montoya, G. CRISPR-Cas12a: functional overview and applications. Biochem J 2020, 43, 8–17. [Google Scholar] [CrossRef]
- De Dreuzy, E.; Haeth, G.; Zuris, J.A.; Sousa, P.; Viswanathan, R.; Scott, S.; Da Silva, J.; Ta, T.; Copehart, S.; Wang, T.; et al. EDIT-301: an experimental autologous cell therapy comprising Cas12a-RNP modified mPB-CD34+ cells for the potential treatment of SCD. Blood. 2019;134(suppl 1):4636.
- Hanna, R.; Frangoul, H.; McKinney, C.; Pineiro, L.; Mapara, M.; Chang, K.H.; Jaskolka, M.; Kim, K.; Farrington, D.L.; Wally. M.; et al. EDIT-301: an experimental autologous cell therapy comprising Cas12a-RNP-modified mPB-CD34+ cells for the potential treatment of SCD. Blood 2019, 134 (suppl.1), 4636. [Google Scholar]
- Hanna, R.; Frangoul, H.; McKinney, C.; Pineiro, L.; Mapara, M.; Dalal, J.; Rangarajan, H.; Atkins, H.; Chang, K.H.; Mei, B.; et al. As Cas12a gene editing of HBG1/2 promoters with EDIT-301 results in rapid and sustained normalization of hemoglobin with increased fetal hemoglobin in patients with severe sickle cell disease and transfusion-dependent beta-thalassemia. Blood 2023, 142 (suppl.1), 4996. [Google Scholar] [CrossRef]
- Hanna, R.; Frangoul, H.; Pineiro, L.; McKinney, C.; Mapara, M.; Dalai, J.; Rangarajan, H.; Atkins, H. ; Bhatia, Chellapandian, D.; et al. CRISPR-Cas12a gene editing of the HBG 1-2 promoters leads to sustained normalization of total hemoglobin and increased fetal hemoglobin in patients with severe sickle cell disease: updated results from the RUBY study. Blood 2025, 146 (suppl.1), abs25-8372. [Google Scholar]
- Frangoul, H.; Hanna, R.; Walters, >M. C.; Chang, K.H.; Jaskolka, M.; Kim, K.; Mei, B.; Afonja, O.; Thompson, A. Reni-Cel, an investigational As Cas12a gene-edited cell medicine, led to substantial engraftment, increased hemoglobin, and reduced transfusion dependence in patients with transfusion-dependent beta-thalassemia treated in the Edithal trial. Blood 2024, 144 (suppl.1), 7456–7457. [Google Scholar]
- Uchida, N.; Li, L.; Nassehi, T.; Drysdale, C.M.; Yapundich, M.; Gasner, J.; Haro-Mora, J.; Demirci, S.; Leonard, A.; Bonifacino, A.C.; et al. Preclinical evaluation for engraftment of CD34+ cells gene-edited at the sickle disease locus in xenograft mouse and non-human primate. Cell Rep Med 2021, 2, 100247. [Google Scholar] [CrossRef] [PubMed]
- Kanter, J.; DiPersio, J.F.; Leavey, P.; Shyr, D.C.; Thompson, A.A.; Porteus, M.H.; Intondi, A.; Lahiri, P.; Dever, D.P.; Petrusich, A.; et al. Cedar trial in progress: a first in human, phase 1-2 study of the correction of a single nucleotide mutation in autologous HSCs (GPH 101) to convert HbS to HbA for treating severe SCD. Blood 2021, 138 (suppl.1), 1864–1865. [Google Scholar] [CrossRef]
- Shyr, D.C.; Lowsky, R.; Miller, W.; Schroeder, M.A‐; Bucholz, T.; Dougall, K.; Intondi, A.; Charles, A.; Leher, J.; Lehrer, J.; et al. One year follow‐up on the first patient treated with Nula‐Cel: an autologous CRISPR/Cas9 gene corrected CD34+ cell product to treat sickle cell disease. Blood 2023, 142 (suppl.1), 5000.
- Lee, B.; Gin, A.; Wu, C.; Singh, K.; Grice, M.; Mortlock, R.; Abraham, D.; Fan, X.; Zhou, Y.; Aljanahi, A.; et al. Impact of CRISPR/HDR editing versus lentiviral transduction on long-term engraftment and clonal dynamics of HSPCs in rhesus macaques. Cell Stem Cell 2024, 31, 455–466. [Google Scholar] [CrossRef]
- Lessard, S.; Rimmelé, P.; Ling, H.; Moran, K.; Vieira, B.; Lin, Y.D.; Rajani, G.M.; Hong, V.; Reik, A.; Boismenu, R.; et al. Zinc finger nuclease-mediated gene editing in hematopoietic stem cells results in reactivation of fetal hemoglobin in sickle cell disease. Scient Rep 2024, 14, 24298. [Google Scholar] [CrossRef]
- Li, C.; Georgakopoulou, A.; Newly, G.A.; Chen, P.J.; Everette, K.A.; Paschoudi, K.; Vlachaki, E.; Gil, S.; Anderson, A.K.; Koob, T.; et al. In vivo HSC prime editing rescues sickle cell disease in a mouse model. Blood 2023, 141, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- LiC.; C.; Anderson, A.K.; Ruminski, P.; Kapova, D.; Kidem, H.P.; DiPersio, J.F.; Lieber, A. A simplified GCSF‐ free procedure allows for in vivo HSC gene therapy of sickle cell disease in a mouse model. Blood Adv 2024, 8, 4089‐4095.
- Li, C.; Georgakopoulou, A.; Paschoudi, K.; Anderson, A.K.; Huang, L.; Gil, S.; Giannaki, M.; Vlachaki, E.; Newby, G.A.; Liu, D.R.; et al. Introducing a hemoglobin G-Makassar variant in HSCs by in vivo base editing treats sickle cell disease in mice. Mol Ther 2024, 32, 4353–4371. [Google Scholar] [CrossRef] [PubMed]
- Breda, L.; Papp, T.E.; Triebwasser, M.P.; Yadegari, A.; Fedorky, M.T.; Tanaka, N.; Abdulmalik, O.; Pavani, G.; Wang, Y.; et al. In vivo hematopoietic stem cell modification by mRNA delivery. Science 2023, 381, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Chatterjee, S.; Dilliard, S.A.; Moore, S.; Xiao, Y.; Bian, X.; Yamada, K.; Sung, Y.C.; Levine, R.M.; Mayberry, K.; et al. Bone-marrow-homing lipid nanoparticles for genome editing in diseased and malignant hematopoietic stem cells. Nat Biotechnol 2024, 19, 1409–1417. [Google Scholar]
- Xu, S.; Liang, D.; Wang, Q.; Cheng, Y.; Xie, D.; Gui, Y.; Zhang, H.; Feng, C.; Zhao, F.; Ren, W.; et al. In vivo genome editing of human hematopoietic stem cells for treatment of blood disorders using mRNA delivery. Nat Biomed Eng 2025, in press. [Google Scholar] [CrossRef]
- Liao, J.; Chen, S.; Hsiao, S.; Jiang, Y.; Yang, Y.; Zhang, Y.; Wang, X.; Lai, Y.; Bauer, D.E.; Wu, Y. Therapeutic base adenine editing of human hematopoietic stem cells. Nat Commun 2023, 14, 207. [Google Scholar] [CrossRef]
- Milani, M.; Fabiano, A.; Perez-Rodriguez, M.; Hernandez, R.J.; Zecchillo, A.; Zonari, E.; Ottonello, S.; Basso-Ricci, L.; Canepari, C.; Volpin, M.; et al. In vivo hematopoietic stem cell gene therapy enabled by postnatal trafficking. Nature 2025, 643, 1097–1106. [Google Scholar] [CrossRef]
- Milani, M.; Annoni, A.; Moalli, F.; Liu, T.; Cesana, D.; Calabria, A.; Bartolaccini, S.; Biffi, M.; Russo, F.; Visigalli, I.; et al. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Soc Transl Med 2019, 11, eaay7325. [Google Scholar] [CrossRef]
- Charlesworth, C.T.; Homma, S.; Amaya, A.K.; Dib, C.; Vaidyanathan, S.; Tan, T.K.; Miyauchi, M.; Nakauchi, Y.; Suchy, F.P.; Wang, S.; et al. Highly efficient in vivo hematopoietic stem cell transduction using an optimized self-complementary adeno-associated virus. Mol Ther Methods Clin Dev 2025, 33, 1–10. [Google Scholar] [CrossRef]
- Karimzadeh, A.; Kim, R.; Garcia, V.; Fiorea, M.; Peacker, B.L.; Kobayashi, S.; Watkins, D.; Messemer, K.; Zheng, J.; Bauer, D.E.; et al. In situ gene editing of hematopoietic stem cells via AAV-delivered CRISPR guide RNAs. Blodd Adv 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Bhoorasingh, M.; Vail, E.; Yoon, Y.; Black, R.; Reddy, M.; Jeong, S. Engineered extracellular vesicles for in vivo therapy in sickle cell disease. Blood 2025, 146 (suppl.1), abs25–2915. [Google Scholar]
- Tozzi, L.; Schiroli, G.; Heshmati, Y.; Cao, Y.; Monte, T.; Wangweerawong, A.; Rottman, J.B.; Palchaudhuri, R.; Ribeil, J.A.; Manis, J.; et al. In vivo HSC gene editing for correction of the sickle cell mutation using RNA gene writers. Blood 2024, 144 (suppl.1), 515. [Google Scholar] [CrossRef]
- Heshmati, Y.; Schiroli, G.; Tozzi, L.; Monte, M.; Beytour, N.; Wangweerawong, A.; Rottman, J.; Salomon, W.E.; Palchaudhuri, R.; Wang, J.; et al. In vivo correction of the sickle cell disease mutation in hematopoietic stem cells using RNA gene writers. Blood 2025, 146 (suppl.1), abs25–7314. [Google Scholar]
- Raguram, A.; Banskota, S.; Liu, D.R. Therapeutic in vivo delivery of gene editing agents. Cell 2022, 185, 2806–2827. [Google Scholar] [CrossRef]
- Jones, R.J.; Kassim,A:; Brodsky, R. A.; De Baun, M. Is allogeneic transplantation for sickle cell disease still relevant in the era of gene therapy? Blood Adv 2025, 9, 877–883. [Google Scholar] [CrossRef]
- Kassim,A. A.; de la Funte, J.; Wilkerson, K.L.; Alahmari, A.; Seber, A.; Bonfim, C.; Simoes, B.P.; Alzahrani, M.; Eckrich, M.J.; Horn, B.; et al. An international learning collaborative phase 2 trial for haploidentical bone marrow transplant in sickle cell disease. Blood 2024, 143, 2654–2663. [Google Scholar] [CrossRef]
- Kassim, A.A.; Walters, M.C.; Eapen, M.; Smith, M.; Logan, B.R.; Solh, M.; McKinney, C.; Nieder, M.; Ross, M.; Kent, M.; et al. Haploidentical bone marrow transplantation for sickle cell disease. NEJM Evidence 2025, 4, EVIDoa2400192. [Google Scholar] [CrossRef] [PubMed]
- Santorone, S.; Angelini, S.; Natale, A.; Vaddinelli, D.; Spadano, R.; Casciani, P.; Papola, F.; DiLembo, E.; Iannetti, G.; Di Bartolomeo, P. survival and late effects of hematopoietic cell transplantation in patients with thalassemia major. Bone Marrow Trasplant 2022, 57, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Santorone, S.; Pepe, A.; Meloni, A.; Natale, A.; Pistoia, L.; Olioso, P.; Papalinetti, G.; Cuccia, L.; Spasiano, A.; Lisi, R.; et al. Secondary solid cancer following hematopoietic transplantation in patients with thalassemia major. Bone Marrow Transpl 2018, 53, 39–43. [Google Scholar] [CrossRef]
- De Franceschi, L.; Locatelli, F.; Rees, D.; Chabannon, C.; Dalle, J.H.; Rivella, S.; Iolascon, A.; Lobitz, S.; Abboud, M.R.; de la Fuente, J.; et al. Selecting patients with sickle cell disease for gene addition or gene editing-based therapeutic approaches: report on behalf of a joint EHA specialized working group and EBMT hemoglobinopathies working party consensus conference. HemaSphere 2025, 9, e70089. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; John, T. To pursue gene therapy or not? Is it feasible after graft failure in allogeneic hematopoietic cell transplant recipients? Blood Adv. 2025, 9, 3845‐3849.
- Kelkar, A.H.; Achebe, M.O.; Hantel, A. An ethical allocation scheme for scarce gene therapies in sickle cell disease and transfusion-dependent β-thalassemia. Blood Adv 2025, 9, 4502–4510. [Google Scholar] [CrossRef]
- John, T.; Czechowiez, A. Clinical hematopoietic stem cell-based gene therapy. Mol Therapy 2025 33, 2663–2683. [CrossRef] [PubMed]
- Boulad, F.; Maggio, A.; Wang, X.; Moi, P.; Acuto, S.; Kogel, F.; Takpradit, C.; Mansilla-Soto, J.; Cabriolu, A.; Odak, A.; et al. Lentiviral globin gene therapy with reduced-intensity conditioning in adults with β-thalassemia: a phase 1 trial. Nat Med 2022, 28, 63–70. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Bonner, M.; Pierciey, F.J.; Uchida, N.; Rottman, J.; Demopoulos, L.; et al. Myelodysplastic syndrome unrelated to lentiviral vector in patient treated with gene therapy for sickle cell disease. Blood Adv 2020, 4, 2058–2063. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.; Tisdale, J.; Schmidt, M. , Kanter, J.; Jaroscak, J.; Whitney, D.; et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med 2022, 386, 138–147. [Google Scholar] [CrossRef]
- Liggett, L.A.; Cato, L.D.; Weinstock, J.S.; Zhang, Y.; Nourale, S.M.; Gladwin, M.T.; Garrett, M.E.; Ashley-Koch, A.; Telen, M.J.; Custer, S.; et al. Clonal hematopoiesis in sickle cell disease. J Clin Invest 2022, 132, e156060. [Google Scholar] [CrossRef]
- Pincez, T.; Lee, S.; Ilboudo, Y.; Preuss, M.; Hung d’Alexandry d’Orengiani, A.L.; Bartolucci, P.; Galacteros, F.; Loly, P.; Bauer, D.E.; Loos, R.; et al. Clonal hematopoiesis in sickle cell disease. Blood 2021, 138, 2148–2152. [Google Scholar] [CrossRef]
- Solomou, E.E.; Delaporta, P.; Stamatia, L.; Katsika, E.V.; Catzieleftheriou, M.; Toutoudaki, K.; Glentis, S.; Stamatopoulos, K.; Catzidimitriou, A.; Kattamis, A. High incidence of clonal hematopoiesis in transfusion-dependent thalassemia patients. Blood 2022, 140 (suppl.1), 8213–8214. [Google Scholar] [CrossRef]
- De Luna, G.; Cretin, J.; Redjoul, R.; Beckerich, F.; Habiobi, A.; Menouche, D.; Hebert, N.; Giovannangeli, C.; Maury, S.; Bartolucci, P.; Sloma, I. Screening for clonal hematopoiesis in patients with β-hemoglobinopathies who are candidates to transplant approaches. Blood 2025, 146 (suppl.1), abs25–2407. [Google Scholar]
- Weeks, L.; Fitzhugh, C.; Pollock, S.; Osei, M.; Rickles-Young, M.; Murdock, M.; Townsend, M.; Reilly, C.; Dinardo, C.; Sabino, E.; et al. Sickle cell disease is associated with early onset clonal hematopoiesis involving DNA damage response pathway mutations. Blood 2025, 146 (suppl.1), abs25–7115. [Google Scholar]
- Zeng, J.; Nguyen, M.A.; Liu, P.; Ferreira da Silva, L.; Levesque, S.; Lin, L.Y.; Justus, D.G.; Petri, K.; Clement, K.; Porter, S.N. Gene editing without ex vivo culture evades genetoxicity in human hematopoietic cells. Cell Stem Cell 2025, 32, 191–208. [Google Scholar] [CrossRef]
- Demirci, S.; Zeng, J.; Pelchaudhuri, R.; Wu, C.; Abraham, D.M.; Hayal, T.B.; Essawi, K.; Nguyen, M.A.; Stasula, U.; Chu, R.; et al. BCL11A +58/+55 enhancer-editing facilitates HSPC engraftment and HbF induction in rhesus macaques conditioned with a CD45 antibody-drug conjugate. Cell Stem Cell 2025, 32, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Butt, H.; Sathish, S.; London, E.; Le, A.; Li, Q.; Gudmundsdottir, B.; Lee, D.Y.; Burke, E.V.; Yates, B.P.; Liu, D.R.; et al. Comparative analysis of CRISPR-Cas9, lentiviral transduction, and base editing for sickle cell disease in a murine model. Blood Adv 2025, in press. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Main steps required for gene therapy of SCD and β-thalassemic patients with ex vivo engineered autologous HSCs/HPCs.3. Gene therapy studies based on gene addition using lentiviral vectors.
Table 1.
Main steps required for gene therapy of SCD and β-thalassemic patients with ex vivo engineered autologous HSCs/HPCs.3. Gene therapy studies based on gene addition using lentiviral vectors.
| Initial evaluation: patient selection and preparation |
| HSC mobilization with G-CSF and Plexifor, apheresis and HSCs/HPCs collection |
| CD34+ HSC/HPC purification and genetic manipulation (lentiviral transfection or gene editor electroporation) |
| Patient hospitalization and myeloablative conditioning |
| Infusion of engineered HSCs/HPCs; post-transplantation hospitalization and supportive care |
| Clinical support, monitoring and follow-up (including post-transplantation support, evaluation of hematological outcomes of gene-modified hematopoiesis, evaluation of late effects) |
Table 2.
Main genetic components of lentiviral vectors used for gene therapy of β-hemoglobinopathies.
Table 2.
Main genetic components of lentiviral vectors used for gene therapy of β-hemoglobinopathies.
| Main genetic components of lentiviral vectors used for gene therapy of hemoglobinopathies |
|
|
|
|
Table 3.
Main gene therapy studies in beta-hemoglobinopathies.
| Therapy and Clinical trial-gov. No. |
Disease | Gene therapy approach | Status and number of patients |
|---|---|---|---|
| Lov-Cel (Blue Bird Bio) NCT 02140554 HGB-205 and HGB-206 |
Sickle Cell Disease | Lentiviral globin gene addition (modified β-globin βT87Q) |
Approved (FDA 2023) 55 patients |
| Beti-Cel (Blue Bird Bio) NCT 01745120 HGB-204, HGB-205, HGB-207, HGB-212 |
β°/β°, β+/β° thalassemia |
Lentiviral globin gene addition (modified β-globin βT87Q) |
Approved (EMA 2019, FDA 2022) 40 patients |
| BCH-BB694 (Boston Children Hospital) NCT 03282656 |
Sickle Cell Disease | Lentiviral BCL11A-shmiR addition | Trial in progress 10 patients |
| LVV GbGM NCT 02186418 |
Sickle Cell Disease | Lentiviral globin gene addition (modified γ-globin γG16D) |
Trial in progress 7 patients |
| Exa-Cel (CRISPR Therapeutics/Vertex Pharmaceuticals) NCT 03745287 CLIMB SCD-121/131 |
Sickle Cell Disease | CRISPR-Cas9 editing of BCL11A enhancer | Approved (EMA 2023, FDA 2023/2024) 44 patients |
| Exa-Cel (CRISPR Therapeutics/Vertex Pharmaceuticals) NCT 03655678 CLIMB THAL-111 |
β°/β°, β+/β° thalassemia |
CRISPR-Cas9 editing of BCL11A enhancer | Approved 52 patients |
| Reni-Cel (Editas Medicine) NCT 05444894 EdiThal |
β°/β°, β+/β°like thalassemia |
Cas 12 editing of BCL11A binding sited in γ-globin promoter | Trial in progress 7 patients |
| Reni-Cel (Editas Medicine) NCT 04853576 Ruby |
Sickle Cell Disease | Cas 12 editing of BCL11A binding sited in γ-globin promoter | Trial in progress 21 patients |
| BEAM-101 (Beam Therapeutics) NCT 05456880 |
Sickle Cell Disease | Base editing of BCL11A binding site in -globin promoter | Trial in progress 5 patients |
| BRL-101 (BRL Medicine Inc) NCT 04211480 NCT 04205435 |
β°/β°, β+/β°, β+/β+ thalassemia |
CRISPR Cas 9 editing of BCL11A enhancer | Trial in progress (15 patients) |
Table 4.
Different CRISPR-based genome editing strategies used for gene therapy studies of β-hemoglobinopathies.
Table 4.
Different CRISPR-based genome editing strategies used for gene therapy studies of β-hemoglobinopathies.
| Gene editing strategy Editing efficacy |
Specifity | Main advantage | Main limitations | Translational applications |
|---|---|---|---|---|
| CRISPR/Cas9 High |
Moderate | Cost effectiveness, speed, efficiency, ease of use, flexibility and versatility, multiplexing | Off-target effects, double-strand breaks induced genotoxicity Active only in proliferating cells (HDR repair) |
Disruption of regulators elements regulating HbF synthesis (NHEJ repair) Correction of point mutations (HDR repair) Approved for clinical use. |
| CRISPR/Cas12 High |
High | Efficiency in HDR repair, multiplexing, fewer off-target effects, more precision in gene targeting than CRISPR/Cas9 | Double-strand breaks induced genotoxicity, possible off-target effects | Disruption of regulators elements regulating HbF synthesis Clinical trials in progress |
| Base editing Very high |
High | Efficient editing No donor template needed, versatile applications, no double-strand breaks, precise correction of single-base changes |
Bystander effects, PAM dependency, limited conversion for some base pairs | Base-editing of regulatory elements in HBG1 and HBG2 gene promoters Clinical trials in progress |
| Prime editing Moderate |
Very high | High versatility (base substitutions, small insertions and deletions), precision, reduced off-target effects | Low efficiency (particularly for some DNA sequences), some possible off-target effects Difficult delivery for in vivo studies |
Pre-clinical studies |
| CRISPR a/i | Very high | Efficient activation or inhibition of specific genes | Effect on gene expression insufficient for therapeutic purposes | Pre-clinical studies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



