
Submitted:
10 November 2025
Posted:
11 November 2025
You are already at the latest version
Abstract
Acute injuries to the central nervous system (CNS) converge on a shared pathophysiology of energetic failure, vascular disruption, and neuroimmune activation. These events destabilize the same metabolic and arousal networks governed by the orexin (hypocretin) system, a hypothalamic hub that synchronizes energy balance, wakefulness, and motivation. Experimental evidence across ischemic, hemorrhagic, traumatic, and systemic models shows that orexin signaling falls sharply during metabolic crisis and gradually reactivates with recovery of vigilance and homeostasis. Controlled enhancement of this system through peptide, pharmacological, or physiological means can limit secondary injury, stabilize autonomic function, and foster motor and cognitive rehabilitation. This review integrates preclinical and clinical evidence linking orexin modulation to neuroprotection and functional restoration, and discusses opportunities and challenges for translating mechanistic insight into therapy. It also outlines practical strategies to enhance orexinergic tone, highlighting the recent development of selective small-molecule orexin agonists and non-invasive delivery routes that make translational exploration increasingly feasible. Orexin emerges as a unifying neuromodulatory target that bridges acute metabolic rescue with long-term behavioral recovery.
Keywords:
orexin/hypocretin
; acute CNS injury
; neuroprotection
; arousal
; neurorehabilitation
; spinal cord injury
; stroke
; traumatic brain injury
; orexin receptor agonists
1. Introduction
Acute central nervous system (CNS) damage, whether caused by stroke, trauma, cardiac arrest, or systemic inflammation, remain among the most devastating medical emergencies, with few therapies capable of altering long-term outcomes. The initial mechanical or vascular insult is only the beginning: what follows is a cascade of metabolic collapse, neurovascular failure, and maladaptive inflammation that often extends far beyond the primary lesion (Dirnagl et al., 1999; Lo et al., 2003; Sofroniew, 2015). Despite advances in reperfusion, neurocritical care, and rehabilitation, survivors often face persistent cognitive, motor, and motivational deficits. These limitations reflect not only structural loss but a sustained failure of neuromodulatory systems that coordinate arousal, metabolism, and adaptive behavior (Maas et al., 2022).
Among these systems, the orexin (hypocretin) network of the lateral hypothalamus has emerged as a surprisingly powerful regulator of recovery potential. Originally characterized for its role in maintaining wakefulness, orexin signaling also links metabolic status to autonomic control, emotion, and goal-directed action (Burdakov, 2021; J. Li et al., 2014; Sakurai, 2014). Through widespread projections to monoaminergic, cholinergic, and motor circuits, orexin neurons sustain not just vigilance, but the capacity to mobilize effort and purposeful movement. These integrative properties place the orexin system at the intersection of nearly every process disrupted after acute CNS injury: energy collapse, disordered arousal, and impaired motivation.
This perspective recasts orexin not as a sleep peptide, but as a homeostatic integrator of survival, one that synchronizes energy availability with behavioral demand. Its dysfunction may therefore explain a wide spectrum of post-injury states, from coma and hypoarousal to fatigue and apathy. Conversely, its controlled activation might help the injured brain transition from metabolic shutdown to coordinated recovery. Recent progress on both peptide-based and small-molecule orexin receptor agonists (Seigneur & de Lecea, 2020) has turned a once theoretical idea into a tangible pharmacological strategy, renewing interest in its translational potential across acute and chronic neurorepair.
Here, we review current evidence linking orexin signaling to recovery from acute CNS damage, from pathophysiological foundations to translational strategies. We outline the shared biological cascades that underlie these conditions, summarize how orexin integrates cellular and systemic responses to injury, and examine experimental and clinical interventions that enhance orexinergic tone. Finally, we discuss the practical and conceptual challenges that must be resolved for orexin-based therapies to progress from laboratory insight to clinical reality.
2. Pathophysiology of Acute CNS Damage: Common Cascades and Points of Divergence
2.1. Common Temporal Sequence Across Etiologies
Acute insults to the central nervous system arise from distinct causes, including vascular occlusion or rupture in stroke, mechanical impact in traumatic brain or spinal injury, or global ischemia and systemic inflammatory crises. Despite etiological differences, they all converge on a remarkably similar biological logic. Each begins with a primary event that abruptly disrupts perfusion or tissue integrity, followed by secondary cascades that propagate damage and shape recovery potential. Immediately, loss of perfusion or mechanical disruption precipitates energy failure, ionic disequilibrium, and excitotoxic glutamatergic drive, triggering necrotic and apoptotic cell death (Hilkens et al., 2024; Maas et al., 2022). Within hours to days, blood–brain and blood–spinal-cord barrier dysfunction, microvascular failure, neuroinflammation, and evolving edema amplify the lesion. Over subsequent weeks, glial and stromal remodeling forms a compartmentalized injury border and fibrotic core (Dorrier et al., 2022; O’Shea et al., 2024). Circuit-level adaptations and plasticity then determine the balance between persistent deficits and compensation. This shared backbone underlies heterogeneous clinical entities and sets the stage for targeted, timing-sensitive therapies.
2.2. Cardiovascular Etiologies: Focal Arterial Occlusion, Parenchymal Bleeding, and Whole-Brain Ischemia
2.2.1. Ischemic Stroke
In ischemic stroke, abrupt arterial occlusion produces a core–penumbra architecture: an irreversibly injured core surrounded by hypoperfused, electrically silent but potentially salvageable tissue whose fate depends on reperfusion timing and collateral flow (Hilkens et al., 2024). The biological substrate is rapid ATP depletion, membrane depolarization, Na⁺/Ca²⁺ influx, and excitotoxicity, followed by oxidative stress, mitochondrial injury, and microvascular failure. Reperfusion can rescue penumbra but also introduces reperfusion injury and inflammatory amplification. Clinically, workflow aims to restore perfusion while limiting edema and hemorrhagic transformation (Campbell & Khatri, 2020). BBB dysfunction is an early and dynamic feature, driven by tight-junction alterations, increased transcytosis, pericyte changes, and endothelial inflammatory signaling. It not only contributes to edema formation but also critically influences drug delivery strategies. (O’Shea et al., 2024).
2.2.2. Intracerebral Hemorrhage (ICH)
In ICH, primary parenchymal bleeding abruptly elevates local pressure and shears tissue. Early outcomes are dominated by hematoma expansion, which is influenced by hypertension and anticoagulation status. Secondary injury reflects perihematomal edema, iron toxicity, and sterile inflammation around the clot (Sun et al., 2022). Care bundles that couple rapid blood pressure reduction, urgent anticoagulant reversal when indicated, and specialist stroke-unit care improve survival; minimally invasive evacuation strategies are being refined, though the impact on functional outcomes remains under active evaluation (Magid-Bernstein et al., 2022). The underlying pathophysiology remains a blend of mass effect, BBB injury, and innate immune activation, with timing critical for each countermeasure.
2.2.3. Subarachnoid Hemorrhage (SAH) and Global Ischemia After Cardiac Arrest
Although subarachnoid hemorrhage originates from a focal vascular rupture, extravasated blood rapidly disseminates through the cerebrospinal fluid, triggering diffuse vasospasm, metabolic failure, and impaired arousal. Consequently, its secondary cascades resemble those of global hypoxic–ischemic injury rather than purely focal stroke (Campbell & Khatri, 2020). This hybrid nature justifies grouping SAH alongside post–cardiac arrest brain injury. Both share a prominent microvascular and inflammatory signature superimposed on diffuse ischemic stress (Perkins et al., 2024). Following return of spontaneous circulation, the brain experiences a post-resuscitation syndrome marked by diffuse edema, excitotoxicity, and impaired autoregulation; targeted temperature and perfusion management are designed around these mechanisms (Yenari & Han, 2012). In SAH, delayed cerebral ischemia from vasospasm and BBB failure drives secondary deterioration on top of the hemorrhagic insult. The unifying thread is global ischemic and inflammatory stress in a brain with fragile hemodynamics.
2.3. Traumatic CNS Injury: Mechanical Initiation, Biochemical Propagation
2.3.1. Traumatic Brain Injury (TBI)
TBI begins with mechanical forces (linear/angular acceleration, impact, blast) producing focal contusions and diffuse axonal injury. The primary physical insult is rapidly followed by ionic flux, excitotoxic neurotransmission, mitochondrial dysfunction, and metabolic crisis, all of which broaden tissue loss (Maas et al., 2022). Cerebrovascular dysfunction and BBB failure promote vasogenic edema, while sterile inflammation recruits microglia, infiltrating leukocytes, and cytokine networks that can be both reparative and damaging (Theus, 2024). The field now recognizes TBI as a multisystem disorder of the neurovascular unit and immune network, not only of neurons and axons, which explains the variability in trajectories and the potential for targeted interventions beyond pure neuroprotection. During the subacute phase, astrocyte-rich borders and a fibrotic stromal core form to contain damage. These structures, once termed a “glial scar,” are now viewed as coordinated compartments that regulate axon guidance and immune traffic (Dorrier et al., 2022; O’Shea et al., 2024). Understanding when to modulate versus harness these barriers is crucial for designing interventions.
2.3.2. Spinal Cord Injury (SCI)
SCI mirrors TBI’s two-stage logic: a primary mechanical insult (contusion, compression, laceration) followed by secondary injury comprising ischemia, ionic dysregulation, excitotoxicity, free radical stress, edema, and robust inflammation (Courtine & Sofroniew, 2019). Vascular compromise at and around the lesion and barrier breakdown extend tissue loss beyond the impact site. Over time, the cord assembles border astrocyte territories and a fibrotic core with perivascular/meningeal fibroblasts and extracellular matrix, forming a lesion compartment similar in principle to brain scars yet anatomically constrained by spinal architecture (O’Shea et al., 2024). These processes intersect with systemic responses, neuropathic pain pathways, and autonomic dysregulation.
2.4. Systemic and Metabolic Causes of Acute CNS Dysfunction
Beyond focal or mechanical lesions, systemic crises such as sepsis, hepatic failure, or metabolic collapse can trigger acute brain dysfunction. These events produce a secondary encephalopathy dominated by endothelial dysfunction, barrier leakage, and cytokine-driven neuroinflammation rather than direct necrosis (Perkins et al., 2024; Sonneville et al., 2013). Experimental sepsis models reproduce the clinical phenotype of fluctuating arousal and delayed cognition through BBB leakage, astrocytic swelling, and microglial activation (Barichello et al., 2022). Similar cascades appear in hypoxia, hypoglycemia, and hepatic or toxic encephalopathies (Bémeur & Butterworth, 2013). Despite diverse triggers, these conditions converge with focal injuries on energy failure, excitotoxicity, inflammation, and maladaptive plasticity, explaining why survivors of sepsis or metabolic coma often experience long-term cognitive and affective deficits comparable to those after stroke or TBI. Current management remains largely supportive, focused on hemodynamic stabilization, infection control, and mitigation of metabolic derangements.
2.5. Cross-Cutting Mechanisms and Therapeutic Objectives
Across vascular, traumatic, spinal, and systemic injuries, several intertwined mechanisms determine outcome. Excitotoxic ionic failure is almost universal: as ATP stores collapse, glutamate accumulates and NMDA/AMPA receptors flood neurons with Ca²⁺, initiating protease activation and mitochondrial dysfunction. Parallel neurovascular-unit breakdown exposes the brain to edema and leukocyte infiltration; endothelial tight-junction loss and increased transcytosis compromise the barrier, while pericyte stress and astrocyte detachment further weaken microvascular stability (O’Shea et al., 2024). The resulting leakage amplifies inflammation, which in turn exacerbates oxidative and metabolic stress (Maas et al., 2022).
Inflammatory amplification follows within hours: activated microglia and infiltrating monocytes release cytokines, chemokines, and reactive oxygen species that can extend cell loss yet are indispensable for debris clearance and repair (Theus, 2024). In ICH and systemic inflammation, heme and cytokine storms add further toxicity. Over subsequent days, astrocytes and fibroblasts remodel the lesion into spatially distinct domains: a glial border that isolates viable tissue and a fibrotic core that provides structural stability but restricts axonal regrowth (Dorrier et al., 2022). Paralleling glial responses take place in diffuse systemic encephalopathies.
Together, these processes form a continuum that spans etiologies: excitotoxic and inflammatory stresses initiate damage, barrier failure amplifies it, and glial–stromal remodeling seals it off. Understanding this shared logic provides the conceptual foundation for targeting mechanisms, such as neuromodulatory systems of arousal and metabolism, that operate across injury types and time scales.
2.6. Therapeutic Objectives Across Time: What Current Care Tries to Achieve
Hyperacute (minutes–hours). The goal is to limit primary and imminent secondary injury. In ischemic stroke, this means rapid reperfusion of the penumbra by thrombolysis or thrombectomy; in intracerebral hemorrhage, preventing hematoma expansion through prompt blood-pressure control and reversal of anticoagulation. After cardiac arrest, physiology-directed post-resuscitation care (i.e., stabilizing oxygenation, perfusion, and temperature) aims to reduce global ischemic damage (Perkins et al., 2024). In systemic crises such as sepsis or metabolic failure, early hemodynamic and metabolic stabilization and infection control are crucial to prevent further hypoperfusion and inflammatory injury (Sonneville et al., 2013). Across etiologies, the intent is to rescue vulnerable tissue, stabilize systemic physiology, and avoid iatrogenic harm during this labile phase.
Acute–subacute (hours–days). Priorities shift toward containing secondary cascades: managing edema, preventing hemorrhagic transformation, and controlling seizures or delirium. Barrier and microvascular stabilization become central, alongside calibrated modulation of inflammation, suppressing excess cytokine activity without blocking necessary repair. In systemic insults, maintaining perfusion, oxygenation, and glycemic balance prevents additional neural injury. Early specialist-unit protocols and rehabilitation principles already begin to influence trajectory. Approaches such as minimally invasive hematoma evacuation, targeted blood pressure management, or structured post-sepsis bundles exemplify the effort to translate mechanistic insight into functional gains.
Chronic (weeks–months and beyond). Long-term care extends from survival to functional independence, cognition, mood, fatigue, and pain control. Persistent deficits after stroke, trauma, or sepsis often reflect incomplete resolution of secondary cascades rather than residual lesion size. Despite progress in acute interventions, effective strategies to restore arousal stability, motivation, and adaptive plasticity remain scarce. These gaps motivate exploration of neuromodulatory approaches capable of enhancing wakefulness, drive, and circuit reorganization during rehabilitation.
Why this scaffold matters? This time-ordered framework defines the windows of biological opportunity where therapies targeting arousal, inflammation, and barrier integrity might act, while clarifying the constraints of each condition: reperfusion biology in ischemia, mass effect in hemorrhage, diffuse metabolic-vascular instability after sepsis or cardiac arrest, and compartmentalized scarring in TBI or SCI. The next sections build upon this scaffold to (i) outline orexin biology relevant to these processes, (ii) describe practical means to boost orexinergic tone, (iii) review experimental and clinical evidence, and (iv) discuss a translational outlook that is timing-aware and mechanism-linked.
3. The Orexin/Hypocretin System in Brief
3.1. Cellular Mechanisms and Receptor Signaling
Orexin-A (hypocretin-1) and orexin-B (hypocretin-2) are neuropeptides encoded by the HCRT gene and produced by a small but widely projecting population of neurons in the lateral, perifornical, and dorsomedial hypothalamus (de Lecea et al., 1998; Sakurai et al., 1998). Despite numbering only thousands of cells, their axons innervate virtually every brain region, including cortex, thalamus, brainstem, and spinal cord, forming an integrative hub that links metabolic state and external conditions to arousal and behavior.
Two G-protein–coupled receptors mediate orexin signaling. OX₁R binds preferentially to orexin-A and couples mainly to Gq/11, activating phospholipase C, inositol-trisphosphate (IP₃) production, and intracellular Ca²⁺ mobilization. OX₂R binds both peptides with comparable affinity and can signal through Gq/11 and Gi/o, enabling mixed excitatory and modulatory actions (Saitoh & Sakurai, 2023; Sakurai, 2014). Receptor activation closes K⁺ leak channels and opens non-selective cation channels, producing slow depolarizations and sustained firing in target neurons (Kukkonen et al., 2024).
Downstream cascades couple to CaMKII and ERK/MAPK pathways that support long-lasting potentiation and gene transcription, whereas AMPK–mTOR signaling adjusts firing rate to cellular energy status (Kukkonen et al., 2024). Through these interactions, the orexin system functions as a metabolic tuner, maintaining neuronal excitability under fluctuating energy conditions. By integrating energy-balance signals with arousal circuitry, orexins thereby support sustained network activation and protects against synaptic fatigue during prolonged wakefulness (Milbank & López, 2019). This combination of excitatory and metabolic effects explains why orexin activity stabilizes behavioral states over minutes to hours rather than milliseconds.
3.2. State Control and Homeostatic Integration
Orexin neurons operate as sentinels of vigilance and internal balance. They fire tonically during wakefulness, particularly during active exploration or stress, and fall silent in sleep (J. Li et al., 2014). Their projections innervate every component of the ascending arousal system (e.g., the locus coeruleus (LC), dorsal raphe nucleus (DRN), tuberomammillary nucleus (TMN), laterodorsal/pedunculopontine tegmentum (LDT/PPT), and basal forebrain (BF)) where they excite monoaminergic and cholinergic neurons to maintain cortical activation (Peyron et al., 1998). Degeneration of this network produces the pathognomonic loss of wake stability seen in narcolepsy type 1 (Thannickal et al., 2000). Conversely, selective OX₂R agonists such as danavorexon and ALKS-2680 sustain alertness without the tolerance typical of psychostimulants (Saitoh & Sakurai, 2023).
Orexin neurons also integrate autonomic and metabolic cues. They are activated by fasting, hypoglycemia, and hypercapnia, and inhibited by glucose and leptin (Peleg-Raibstein et al., 2023; Yamanaka et al., 2003). Projections to the paraventricular nucleus and rostral ventrolateral medulla drive sympathetic outflow, increasing blood pressure, thermogenesis, and respiratory rate (Kuwaki, 2011). Through AMPK–mTOR coupling, their excitability reflects intracellular ATP levels, ensuring that behavioral activation occurs when energy availability permits (Martins et al., 2016).
This orchestration makes the orexin network a physiological bridge between homeostasis and action: it estimates energetic need, mobilizes autonomic effectors, and maintains wakefulness long enough for adaptive behavior to occur. In the context of CNS injury, when energy failure, disordered sleep, and autonomic instability are common, this capacity to synchronize metabolism and arousal becomes especially relevant.
3.3. From Drive to Movement: Orexins and the Neural Implementation of Action
The transition from internal drive to noticeable movement depends on linking motivational state to motor command, a process in which orexins play a central role. Orexin neurons project to motor cortex, striatum, red nucleus, superior colliculus, cerebellum, and spinal cord (Peyron et al., 1998; van den Pol, 1999). Their activation precedes and facilitates voluntary movements: optogenetic or chemogenetic stimulation elicits locomotion and posture adjustments even in resting animals (Karnani et al., 2020). Recordings show that orexin cells discharge vigorously just before self-initiated actions, encoding not mere arousal but the intention to move (Burdakov, 2019).
At the supraspinal level, orexin inputs to the nucleus accumbens and ventral tegmental area potentiate dopaminergic signaling, biasing behavior toward high-effort rewards (Burdakov, 2021; Korotkova et al., 2006). Within motor cortex and pontine reticular formation, orexins enhance neuronal excitability and synchrony, promoting readiness for complex actions (Mileykovskiy et al., 2005). These influences suggest that orexins provide a motivational “gain” on motor networks, ensuring that energy investment aligns with behavioral goals.
Descending orexinergic projections reach spinal motoneurons and interneurons, where OX₁R and OX₂R activation depolarizes cells, augments persistent inward currents, and amplifies reflex output (Biswabharati et al., 2018). Functionally, this translates into increased muscle tone and motor responsiveness. Such descending facilitation may be critical for maintaining posture and motor engagement during recovery after injury, when descending monoaminergic tone is blunted. By coupling cortical intention, limbic motivation, and spinal excitability, orexins effectively transform internal drive into coordinated movement, a principle directly applicable to post-lesional motor rehabilitation.
3.4. System-Wide Neuromodulatory Integration
Orexin neurons exist within a dense, reciprocal web of neuromodulatory circuits that collectively shape global brain states. Through these interactions, orexins act more as the conductor of a multi-system orchestra than as an isolated transmitter.
In the locus coeruleus, orexins excite noradrenergic neurons via OX₁R-dependent depolarization, promoting sustained firing and enhancing cortical norepinephrine levels (Horvath et al., 1999). The LC, in turn, provides excitatory input back to the hypothalamus, generating a positive feedback loop that sustains alertness (J. Li et al., 2014). In the dorsal raphe nucleus, orexin-A increases the activity of serotonergic neurons while simultaneously modulating GABAergic inhibition, producing state-dependent shifts in serotonin tone (R.-J. Liu et al., 2002). During stress or novelty, this coupling promotes resilience and behavioral engagement, but chronic over-activation can yield anxiety-like hyperarousal. In the ventral tegmental area, orexin-A potentiates glutamatergic drive onto dopamine neurons through NMDA-receptor phosphorylation, linking arousal and reward (Korotkova et al., 2006). Consequently, orexinergic release scales with motivational salience and anticipated effort, ensuring that dopaminergic reinforcement matches energetic readiness (Burdakov, 2020). Additional projections to the tuberomammillary nucleus and basal forebrain excite histaminergic and cholinergic neurons, further amplifying cortical desynchronization and attention (Eriksson et al., 2001).
These multiple reciprocal connections enable orexins to synchronize the brain’s arousal, affective, and motor systems into coherent states suited for the organism’s goals. Noradrenergic and dopaminergic excitation heightens vigilance and initiative, serotonergic and cholinergic pathways refine mood and cognition, and inhibitory feedback from GABAergic and serotonergic neurons provides the damping force that prevents metabolic overload. The result is a flexible yet stable framework for adaptive behavior, precisely the sort of global coordination disrupted after CNS injury and that orexinergic modulation could help restore.
4. Strategies for Enhancing Orexinergic Tone
Efforts to enhance orexinergic tone have grown substantially in the last two decades. However, despite this expanding interest, progress has been slower than for antagonists. While dual orexin receptor antagonists rapidly reached clinical approval for insomnia (Herring et al., 2019), strategies to boost orexin signaling have faced pharmacokinetic, safety, and delivery challenges. Yet, a diverse toolbox now exists (summarized in Table 1), spanning from small-molecule agonists in late-stage trials to experimental gene and cell therapies, each offering distinct translational opportunities.
4.1. Synthetic Small-Molecule Agonists
The most advanced pharmacological tools are small-molecule agonists, mostly OX2R-selective given its central role in promoting stable wakefulness (Saitoh & Sakurai, 2023). Leading compounds include danavorexton (TAK-925), oveporexton (TAK-861), E2086, and alixorexton (ALKS-2680), developed by Takeda, Eisai, and Alkermes, respectively (Cheng, Jocelyn, 2025; Dauvilliers et al., 2025; Evans et al., 2022; Plazzi, Giuseppe, 2025). These agents consistently reproduce the wake-promoting actions of endogenous orexins with acceptable tolerability. Danavorexton, an intravenous short-acting agonist, and oveporexton, an oral longer-lasting analogue, have shown the strongest clinical efficacy and are nearing regulatory approval for narcolepsy type 1 (Dauvilliers, 2025). Their success has established proof of concept for pharmacological orexin replacement.
A newer dual OX1R/OX2R agonist, RTOXA-43, increases wakefulness and sleep–wake consolidation in aged mice (D. Zhang et al., 2021). Although still preclinical, dual compounds may more closely mimic native orexin signalling across motivational and arousal domains. Overall, OX2R-selective agonists currently represent the most translationally ready option, while OX1R activation remains an unexplored therapeutic frontier.
4.2. Peptide-Based Orexin Replacement
Native orexin-A, with high affinity for both receptors, remains the reference ligand for restoring dual orexinergic tone despite its short half-life and limited brain penetration. Central administration demonstrated early feasibility: intracerebroventricular or intrathecal delivery robustly increased wakefulness and locomotion in rodents (España et al., 2001; Piper et al., 2000), but invasiveness, instability, and potential desensitisation restrict clinical translation (Gao & Horvath, 2014; Sakurai, 2007). Intranasal delivery provides a less invasive route, reaching the CNS via olfactory and trigeminal pathways (Illum, 2000; Lochhead & Thorne, 2012). Studies in humans and animal models confirmed central activity, including modulation of vigilance, sympathetic tone, and even neuroprotection after acute injury (Baier et al., 2011; Meusel et al., 2022). Yet rapid clearance (Dhuria et al., 2010) and variable deposition (Djupesland, 2013) limit reproducibility. Optimised sprays and mucoadhesive compounds, already successful for intranasal insulin and oxytocin (Born et al., 2002, p. 20; Quintana et al., 2021), could make orexin delivery clinically viable.
Carrier systems, including nanoparticles, liposomes, exosomes, and BBB-shuttle conjugates, offer additional ways to protect peptides and improve brain access. Transferrin-targeted liposomes increased orexin-A delivery in rats (Lai et al., 2018), and similar designs with oxytocin or PACAP38 have shown enhanced stability and CNS penetration (T. Wang et al., 2021; Wong et al., 2021, p. 202). Although complex and still experimental, these technologies outline a promising route toward long-acting peptide formulations.
4.3. Physiological and Behavioral Modulation
Because orexin neurons integrate metabolic, circadian, and motivational cues, physiological interventions that influence these domains can enhance orexinergic signaling endogenously.
Orexin neurons’ firing follows the daily rhythm of sleep and activity, rising with arousal and energy demand. Stable light–dark schedules and consolidated sleep maintain this oscillation, while fragmentation or circadian misalignment suppresses orexin expression and dampens its amplitude (Islam et al., 2022; J. Li et al., 2014). In contrast, acute sleep deprivation transiently increases prepro-orexin mRNA and orexin-A levels in rodents and humans, suggesting a compensatory mechanism that supports wakefulness under high arousal drive (Yoshida et al., 2001; Zeitzer et al., 2003). Interestingly, when sleep deprivation has been applied before CNS injury, it appears to engage orexin neurons in a way that mitigates subsequent damage (Pace et al., 2017). Still, this effect is preventive rather than curative, making its clinical translation challenging.
Physical activity is another powerful physiological driver of the orexin system. Exercise increases hypothalamic c-Fos and circulating orexin-A concentrations (Messina et al., 2014), and orexin population activity scales closely with locomotion and energy turnover (Tesmer et al., 2025). Conversely, orexin signaling itself promotes voluntary movement and endurance, maintaining locomotor drive even when competing with rewarding alternatives (Tesmer et al., 2024). This reciprocal relationship positions regular exercise as a natural enhancer of orexin tone and a behavioral co-therapy capable of improving both energy balance and motivation.
Nutritional and thermogenic influences further modulate orexinergic excitability. These neurons are activated by fasting, ghrelin, and cold exposure, and inhibited by glucose and leptin (Peleg-Raibstein et al., 2023; Yamanaka et al., 2003). Diets that stabilize glycemia or enhance thermogenesis through higher protein intake or moderate ketogenic composition help preserve basal orexin activity. In turn, orexins promote brown-fat thermogenesis and sympathetic outflow, closing a feedback loop between metabolic demand and arousal (Kuwaki, 2011). Notably, nutritional interventions engaging orexin pathways, such as branched-chain amino acid supplementation, have restored sustained wakefulness in experimental models of brain injury (Lim et al., 2013).
Beyond metabolic regulation, orexin neurons are also responsive to novelty, reward, and social complexity. Enriched or stimulating environments increase orexin gene expression and neuronal activation (Burdakov, 2021), reflecting their role as integrators of arousal and motivation. Such experiences likely enhance orexinergic tone through dopaminergic and limbic circuits, providing a behavioral route to maintain engagement without pharmacological intervention. By linking energy balance, exploration, and reward, orexin neurons thus emerge as a central hub through which physiological state and behavioral drive remain aligned.
4.4. Neuromodulation and Electrical Stimulation
Neuromodulation has expanded beyond its original therapeutic indications to encompass a wide range of neurological and metabolic disorders (Gouveia et al., 2024; Lozano et al., 2019), offering potential strategies to enhance orexinergic tone. At the cortical level, transcranial stimulation enhances alertness and cortical excitability, and accumulating experimental evidence indicates that such interventions can also activate hypothalamic orexin pathways. Recent studies have shown that both transcranial pulsed and direct-current stimulation increase orexin-A levels and OX1R expression, engaging downstream signaling cascades associated with neural recovery and homeostatic regulation (Ren et al., 2024; Yao et al., 2025). These findings support a potential top-down route by which cortical activation can recruit orexinergic circuits, thereby linking brain stimulation to arousal and metabolic control.
Peripheral stimulation provides another pathway to influence orexin-related networks. Both invasive and transcutaneous VNS activate ascending noradrenergic and serotonergic nuclei targeted by orexin projections, and increases hypothalamic orexin mRNA in rodents (X.-Y. Dong & Feng, 2018). VNS also improves vigilance and autonomic regulation in humans, supporting indirect orexinergic recruitment (Y. Chen et al., 2023). Other peripheral approaches, including taVNS and median nerve stimulation (MNS), further broaden clinical feasibility. Daily taVNS in obese mice reduced appetite and weight gain via orexin-dependent hypothalamic circuits (Y. Zhang et al., 2025), while MNS evoked orexin-A release contributing to analgesia through endocannabinoid mechanisms in the periaqueductal grey (Y.-H. Chen et al., 2018). Orexinergic involvement has also been described in VNS and MNS paradigms after CNS injury (see Section 5).
Although invasive, direct deep-brain stimulation (DBS) of the hypothalamus has been explored in humans with refractory conditions. For example, bilateral DBS of the lateral hypothalamic area (LHA) in patients with severe obesity increased resting metabolic rate and was performed safely in a pilot study (Whiting et al., 2013). Systematic reviews confirm that hypothalamic DBS is technically feasible and has been applied in disorders ranging from chronic cluster headache to aggressive behavior and metabolic disease (Mofatteh et al., 2025). In these settings, such stimulation may engage orexinergic and other arousal-related circuits, thereby offering a potential adjunctive strategy to boost arousal, autonomic regulation and recovery after central-nervous-system damage.
Compared with pharmacological or peptide-based strategies, neuromodulation bypasses pharmacokinetic barriers and acts at the circuit level, though it demands precise control, individualized calibration, and sustained application to achieve reproducible effects. While the specific contribution of orexins relative to other neuromodulators remains to be clarified, converging evidence across modalities supports its involvement, positioning neuromodulation as a promising complement to orexin-targeted pharmacotherapy.
4.5. Cell and Gene Therapy
Strategies aimed at biologically restoring orexin signaling have advanced considerably in recent years, driven largely by efforts to cure narcolepsy type 1, where selective orexin cell loss is the primary pathology. Transplantation studies show that grafted orexin-producing cells can survive, integrate, and alleviate symptoms in mouse models. Fluorescent-activated cell sorting enabled transplantation of purified orexin neurons into orexin/ataxin-3 mice, improving sleep–wake regulation and reducing cataplexy (Equihua-Benítez et al., 2020). More recently, immortalised orexin cell lines engineered for stable expression were grafted into the dorsal raphe, restoring motor–arousal synchrony and suppressing cataplexy, effects enhanced by chemogenetic activation (Pintwala et al., 2023). Gene therapy approaches have similarly re-expressed prepro-orexin in targeted brain regions, partially restoring wakefulness and reducing cataplexy (Blanco-Centurion et al., 2013; M. Liu et al., 2016). These findings confirm that durable orexin re-expression can rescue behavioral phenotypes in relevant models.
Such strategies, however, are best suited to diseases caused by primary orexin cell loss. In acquired CNS injuries, orexinergic dysfunction is typically secondary, making full replacement less applicable. Moreover, immune rejection, long-term survival, tumourigenicity, and precise integration remain major challenges (Christiansen & Kirkeby, 2024; Rahimi Darehbagh et al., 2024). Still, recent progress demonstrates that orexin replacement is biologically feasible and increasingly reliable, with immortalised lines and stem-cell–derived hypothalamic neurons offering scalable sources. While direct use in stroke, trauma, or sepsis is improbable, adapted approaches (e.g., viral tools to boost residual orexin output or circuit-specific interventions) may eventually emerge from this line of work.
Taken together, available approaches span a continuum from molecular precision to systemic physiological modulation. Direct agonists offer target specificity but face delivery challenges; behavioral and neuromodulatory strategies harness the system’s endogenous plasticity with broader but gentler effects. In practice, combinations of pharmacological and physiological modulation may prove most effective, for instance, using lifestyle or stimulation paradigms to sustain network responsiveness while pharmacological agents provide targeted boosts. The following section examines how these strategies have performed in experimental and clinical contexts of acute CNS damage.
5. Experimental and Clinical Evidence of Orexinergic Modulation in Acute CNS Damage
Acute injury to the central nervous system disrupts the same homeostatic variables that normally drive orexin neurons, including energy balance, vigilance, and autonomic control. The resulting disruption of arousal stability and metabolic regulation creates a context in which the orexin system becomes both highly vulnerable and potentially reparative. Experimental and clinical observations converge on two consistent findings: orexin signaling is altered soon after brain or spinal insults, and interventions that restore or mimic its activity can modulate multiple secondary processes relevant to recovery.
Thus, reductions in cerebrospinal orexin-A levels and changes in hypothalamic or brainstem expression have been reported across ischemic, traumatic, and systemic conditions, often correlating with impaired alertness, motivation, and autonomic function. Conversely, exogenous orexin-A or pharmacological agonists reinstate wakefulness, stabilize metabolic and cardiovascular parameters, and influence neuroinflammatory tone. Together, these data suggest that the orexin system acts as a sensor and regulator of neural integrity, linking cellular stress to organism-level arousal.
In the following sections, experimental and clinical evidence are examined exhaustively by pathological origin, emphasizing mechanistic insights and the temporal windows in which orexin modulation exerts its effects. Relevant literature with this regard is compiled in Table 2.
5.1. Cerebrovascular and Global Ischemic Injuries
5.1.1. Orexin Alterations After Ischemic or Hemorrhagic Events
Cerebrovascular insults rapidly disturb hypothalamic and brainstem circuits that sustain orexinergic tone. Across ischemic, hemorrhagic, and global ischemic models, early reductions in orexin-A levels mirror the combined impact of energy failure, oxidative stress, and autonomic dysregulation.
In ischemic stroke, both clinical and experimental data consistently demonstrate acute depression of orexin signaling. Patients with acute ischemic stroke show decreased CSF orexin-A within the first 48–72 h, inversely correlating with infarct volume and neurological severity (Fu et al., 2025; Kotan et al., 2013; Scammell et al., 2001). Other cohorts reveal that higher serum orexin-A during the subacute period predicts faster neurological improvement and better sleep–wake regulation (Hu et al., 2023). Rodent models of middle-cerebral-artery occlusion (MCAO) confirm this temporal pattern: sharp declines in hypothalamic prepro-orexin mRNA and peptide content occur within the first 6–12 h, with partial normalization over the next 3–7 days (Kitamura et al., 2010; C.-M. Wang et al., 2017; Wu et al., 2022; Xiong et al., 2013; Xu Y. et al., 2011; Zhu et al., 2024). Up-regulation of OX₁R expression within the lesion core but not OX₂R (Irving et al., 2002; Nakamachi et al., 2005) suggests a stress-induced receptor shift. These data indicate that orexin depletion is an early, transient hallmark of ischemic injury, paralleling loss of arousal and global metabolic suppression.
After intracerebral hemorrhage (ICH), hypothalamic and cortical orexin-A immunoreactivity decline sharply in the acute phase, consistent with acute stress inhibition (Dohi et al., 2008; T. Li et al., 2020; Rejdak et al., 2005; Wu et al., 2022). Subsequent rebound of circulating orexin-A accompanies sympathetic activation and cortisol rise during recovery. In subarachnoid hemorrhage (SAH), CSF orexin-A levels correlate with consciousness state and display biphasic shifts: initial depression during coma followed by transient elevation associated with vasospasm and autonomic instability (Ang et al., 2005; Dohi et al., 2005; Rejdak et al., 2005). Collectively, clinical and experimental evidence establish that orexinergic signaling is profoundly sensitive to perfusion and metabolic status, with depletion marking acute neuronal silencing and delayed normalization accompanying systemic recovery.
Global ischemic and post-cardiac-arrest models show analogous dynamics. In rats subjected to asphyxial cardiac arrest, CSF orexin-A decreases during coma and gradually normalizes as EEG activity and neurological scores recover (Akbari et al., 2012; Dohi et al., 2006; Y. Guo et al., 2023, 2024; Y.-J. Kang et al., 2017; Koenig et al., 2009; Sherman et al., 2021). These studies indicate that, in experimental models of cardiac arrest, restoration of orexin tone parallels the recovery of arousal and autonomic stability.
5.1.2. Experimental Modulation of Orexin Signaling
Acute-phase interventions. Prompt orexin replacement during the hyperacute window mitigates ischemic and hemorrhagic damage in multiple models. In MCAO, ICV orexin-A administered within 1–3 h reduces infarct volume, improves neurological scores, and dampens apoptosis and inflammatory signaling through ERK, Akt, NF-κB, and MAPK pathways (C.-M. Wang et al., 2017; Xiong et al., 2013; D. Xu, Kong, Shao, et al., 2021; Yuan et al., 2011; Zhu et al., 2024). These effects depend on OX₁R activation, as receptor antagonism abolishes neuroprotection (T. Li et al., 2020; Matsuura et al., 2020).
Indirect upregulation of the orexin system also confers protection. In a sleep-deprivation paradigm, endogenous orexin elevation prior to stroke reduced infarct size and pro-inflammatory gene expression, suggesting a pre-conditioning benefit (Pace et al., 2015, 2017). Similarly, focal neck stimulation (FNS) preconditioning in MCAO rats increased hypothalamic prepro-orexin and orexin-A levels while decreasing OX₁R expression, leading to attenuated brain injury when applied 1–24 hours post-insult (Xu Y. et al., 2011). Pharmacological strategies not directly targeting orexin have also been shown to upregulate this system: intravenous parecoxib (a COX-2 inhibitor) increased orexin-positive cells and hypothalamic orexin-A concentration at 72 hours post-MCAO, coinciding with reduced damage and improved behavioral scores (F.-T. Li et al., 2018). Comparable neuroprotective outcomes have been reported in chemical and hypoxia–ischemia models, where both orexin-A and orexin-B enhance neuronal viability via Akt and MEK signaling cascades (Palomba et al., 2020; Sokołowska et al., 2014; Zawilska, 2015).
In ICH, early orexin-A administration via ICV or intranasal routes reduces perihematomal edema, limits BBB disruption, and suppresses pro-inflammatory cytokines such as IL-1β and TNF-α (Dohi et al., 2008; T. Li et al., 2020; D. Zhang et al., 2022). In SAH, delivery during the vasospasm-prone period improves microvascular reactivity and correlates with better arousal scores (Ang et al., 2005; Dohi et al., 2005). In global ischemia and post-cardiac-arrest models, ICV or intranasal orexin-A accelerates EEG recovery, stabilizes autonomic output, and shortens coma duration (Akbari et al., 2012; Koenig et al., 2009; Modi et al., 2017; Sherman et al., 2021). Notably, interventions that enhance endogenous orexinergic tone may offer parallel benefits. Caloric restriction applied overnight prior to cardiac arrest reduced neurological deficits and neurodegeneration while modulating systemic metabolism, including lower stress-induced hyperglycemia and elevated ketone bodies, despite no changes in BDNF or SIRT1 expression (Azadian et al., 2021). Together these studies show that timely orexinergic stimulation can stabilize metabolism and arousal, while inappropriate dosing or timing may amplify sympathetic drive and cardiovascular load (Y. Guo et al., 2024; Matsuura et al., 2020; Zhu et al., 2024).
Subacute-phase interventions. In MCAO and related models, sleep-deprivation preconditioning that increases endogenous orexin-A reduces infarct size and modifies inflammatory and cell-cycle gene programs at 3–7 days post-insult, with concurrent changes in REM architecture (Pace et al., 2015, 2017). Experimental enhancement of orexin signaling also supports subacute functional recovery: orexin overexpression improves neurological scores and sleep structure from 1 to 10 days after MCAO (Wu et al., 2022). In hemorrhagic models, exogenous orexin-A reduces edema and neuronal damage and improves behavioral outcomes across subacute time points, with effects extending to 28 days in some protocols (T. Li et al., 2020; D. Zhang et al., 2022). In global ischemia/post–cardiac arrest, improvements in neurological indices persist beyond the immediate hours into several days when orexin-A is delivered early, indicating carry-over benefits into the subacute window (Akbari et al., 2012). Taken together, these data support the view that moderating orexin signaling beyond the hyperacute phase can aid network re-engagement and sleep–wake organization, particularly in settings where interventions (genetic or pharmacologic) were sustained or where endogenous tone was boosted (Akbari et al., 2012; T. Li et al., 2020; Pace et al., 2015, 2017; Wu et al., 2022; D. Zhang et al., 2022).
Chronic and delayed interventions. Evidence into the chronic period remains limited but suggestive in hemorrhagic injury. In ICH models, benefits of orexin-A on neurological function, brain water content, and tissue preservation are reported through 2–4 weeks post-injury (T. Li et al., 2020; D. Zhang et al., 2022). For ischemic stroke and global ischemia, most datasets concentrate on acute and early subacute endpoints (hours to ~10 days), so claims beyond that window should be considered provisional. Mechanistically, sustained normalization of sleep–wake structure and autonomic balance provides a plausible route for longer-term gains, yet controlled chronic-phase studies and clinical trials are still lacking. Indeed, behavioral or pharmacological strategies that indirectly raise orexin tone, including exercise or structured wake therapy, ameliorate daytime somnolence and improve drive (Burdakov, 2021). Although direct orexin agonists have not yet been trialed clinically after stroke or hypoxic encephalopathy, their pharmacology suggests potential utility for restoring vigilance, metabolic regulation, and motor engagement during rehabilitation (Saitoh & Sakurai, 2023). Overall, current animal data indicate a shift in therapeutic emphasis from cytoprotection early on to circuit and behavioral support in later phases, with the strongest chronic-interval evidence to date coming from ICH models (T. Li et al., 2020; D. Zhang et al., 2022).
5.2. Traumatic Brain and Spinal Cord Injury
5.2.1. Orexin Alterations After Mechanical Trauma
Mechanical trauma to the brain or spinal cord perturbs hypothalamic–brainstem networks that couple arousal, autonomic output, and metabolic control. In traumatic brain injury (TBI), human studies report reduced CSF orexin-A during the acute phase that is still present at 6 months post-injury and correlates to post-traumatic hypersomnia (Baumann et al., 2005, 2007). Post-mortem analysis shows reduced orexin immunoreactivity, consistent with functional suppression or injury of lateral-hypothalamic neurons (Baumann et al., 2009). Prospective data further support the prognostic value of orexin: low CSF orexin-A concentrations in coma patients with TBI or stroke predict poor neurological recovery and higher mortality at 7–14 days, independently of initial GCS score (Cangiano-Heath et al., 2020).
Experimental fluid-percussion and controlled-cortical-impact models confirm this phenotype: mice and rats show decreased Hcrt mRNA and peptide content within 24 h, which partially recovers over one to two weeks as metabolic stability returns (Elliott et al., 2018; Lim et al., 2013; Skopin et al., 2015; Somach et al., 2023; Thomasy et al., 2017; Thomasy & Opp, 2019; Willie et al., 2012). This suppression is accompanied by dynamic changes in orexin receptor expression: in a CCI model, OX1R immunoreactivity emerges around the injury penumbra within 6 h, peaks at 24 h, and gradually decreases over the subsequent days. Initially associated with activated microglia, OX1R expression later becomes neuronal, indicating a potential shift from early immune modulation to longer-term synaptic plasticity (Mihara et al., 2011). In the subchronic interval, persistent alterations emerge. TBI mice develop chronic hypersomnolence and fragmented wakefulness associated with reduced hypothalamic activation (Lim et al., 2013), implying sustained orexinergic impairment. Similar sleep–wake instability and attentional deficits are reported during weeks 2–4 (Skopin et al., 2015; Thomasy et al., 2017; Thomasy & Opp, 2019), as well as abnormalities in sleep homeostasis and spectral power distribution during recovery (Somach et al., 2023). Notably, hypothalamic–brainstem coupling was impaired under the same temporal window, underlying behavioral hypoarousal (Craig et al., 2024). Collectively, these studies show that incomplete recovery of orexin activity in the subacute and subchronic stages contributes to fatigue, poor vigilance, and reduced engagement with the environment. These deficits may be driven not only by reduced peptide availability but also by structural disruption of downstream orexinergic circuits. In repeat mTBI models, orexin-producing neurons in the lateral hypothalamus are diminished, and projections to the periaqueductal gray, critical for defensive responses and nociception, are weakened (Christensen et al., 2023). These anatomical changes coincide with altered nociceptive thresholds and reduced anxiety-like behaviors, indicating dysregulation of the arousal–pain interface.
To date, no study has directly examined orexin alterations after spinal cord injury (SCI). However, SCI induces several hypothalamic changes, including metabolic, neuropeptidergic, and autonomic, that could plausibly affect orexinergic function. Reported adaptations include disrupted leptin and adipokine signaling (Bigford et al., 2012), altered hypothalamic POMC and NPY expression (Bigford et al., 2013), and plasticity within lateral-hypothalamic circuits that modulate locomotor recovery (Bonizzato et al., 2021). These indirect findings highlight a critical gap and support investigating whether hypothalamic orexin signaling contributes to post-injury dysautonomia and recovery.
5.2.2. Experimental Modulation of Orexin Signaling
Acute-phase interventions. In TBI, multiple studies demonstrate that enhancing orexin signaling or stimulating orexin-related circuits within the first 24–72 h after trauma mitigates secondary injury and accelerates early neurological recovery. Direct administration of orexin-A or activation of its downstream networks improves consciousness, reduces inflammation, and limits cell death in several rodent models. Exogenous orexin-A administration following cortical injury attenuates ferroptosis and oxidative damage, leading to preserved tissue structure and improved neurological scores (J. Kang, Ren, et al., 2024). In parallel, several neuromodulatory strategies have been shown to activate endogenous orexinergic circuits and promote functional recovery. These include vagus nerve stimulation (VNS), trigeminal nerve stimulation (TNS), and median nerve stimulation (MNS), all of which increase hypothalamic orexin activity and ameliorate early neurological deficits via OX₁R-dependent pathways (X. Dong et al., 2018, 2021; X.-Y. Dong & Feng, 2018; Du et al., 2022; Zhong et al., 2015). Similarly, deep brain stimulation of the lateral hypothalamus (LH-DBS) delivered during the first 12 hours post-injury enhances consciousness and increases both orexin-A and OX1R expression in the hypothalamus. These effects are abolished by OX1R antagonism, confirming their dependence on orexinergic signaling (X. Dong et al., 2021). Another promising approach is low-intensity focused ultrasound stimulation (LIFUS), applied at 3 days post-TBI. LIFUS produced parallel protective effects, reducing edema, inflammasome activation, and oxidative stress while improving behavioral outcomes (Huang et al., 2022). Collectively, these neuromodulation-based interventions converge on a shared mechanism: they upregulate hypothalamic orexin output, stabilizing autonomic tone, reducing neuroinflammation, and promoting arousal recovery.
Subacute-phase administration. At later time points, orexin-related manipulations continue to influence recovery. In a fluid-percussion injury model, branched-chain amino acid (BCAA) supplementation begun 2 days post-injury and maintained through days 4–30 improved wakefulness and partially restored orexin-neuron activation, reversing the TBI-induced increases in NREM sleep and wake fragmentation (Lim et al., 2013).
Conversely, chronic down-biasing of orexin signaling with dual orexin receptor antagonists (DORA-22) produced different outcomes. In mice exposed to controlled cortical impact, daily oral DORA-22 for 30 days decreased post-traumatic seizures and enhanced GABAergic inhibition but increased overall sleep pressure and homeostatic drive (Konduru et al., 2022). Thus, while orexin suppression may have anticonvulsant benefits, it compromises arousal maintenance. Complementary circuit analyses indicate that impaired hypothalamic–brainstem coupling during this period contributes to behavioral hypoarousal and delayed cortical reactivation (Craig et al., 2024). Together, these studies reveal that subacute to subchronic interventions can either sustain or blunt orexin-linked arousal depending on whether signaling is enhanced or inhibited.
Chronic-phase modulation. At longer intervals, persistent orexin deficits are evident across species. In patients with moderate-to-severe TBI and excessive daytime sleepiness, CSF orexin-A concentrations remained low six months post-injury (Baumann et al., 2007). In parallel animal studies, a single DORA administration after controlled cortical impact increased sleep fragmentation and sleepiness measured at 24 h, 2 weeks, 1 month, and 2 months, confirming that acute orexin blockade can exacerbate hypersomnolence throughout recovery (Craig et al., 2024). Recent evidence highlights, however, that chronic orexin-A supplementation is not necessarily beneficial and may worsen outcomes. In a rat model of repeated mild TBI, intracerebroventricular administration of orexin-A beginning five days post-injury and continued chronically over four weeks exacerbated anxiety-like behaviour, impaired motor performance, and altered CSF metabolomic and proteomic profiles, in stark contrast to modafinil, which improved these parameters despite acting on arousal systems (Christensen et al., 2024). These findings suggest that orexinergic interventions during the chronic phase must be approached with caution, as prolonged stimulation of the system may disrupt homeostatic recovery mechanisms and precipitate maladaptive arousal states. Collectively, human and animal data indicate that long-term orexinergic dysfunction contributes to chronic hypersomnolence and reduced vigilance after TBI, but also that untimely or excessive replacement may be counterproductive.
Evidence in SCI remains limited but points toward both direct and indirect hypothalamic involvement. In rats with complete spinal transection, it has been showed that intrathecal orexin-A administered once daily for three days enhanced locomotor recovery and normalized glutamatergic transmission within the first post-injury week, indicating that orexinergic stimulation can acutely modulate spinal network excitability and metabolic balance (He et al., 2025). Importantly, a groundbreaking recent study showed that deep brain stimulation of the lateral hypothalamus markedly improved walking ability and motor coordination in individuals with chronic SCI (Cho et al., 2024). Although LH-DBS activates diverse neuronal populations, the recovery of volitional movement and arousal likely involves the re-engagement of descending hypothalamic circuits that include orexin neurons (X. Dong et al., 2021). Together, these findings suggest that reactivating hypothalamic output, either through targeted orexinergic stimulation or broader neuromodulatory approaches, may promote spinal motor recovery after SCI. However, controlled studies addressing orexin-specific contributions and long-term outcomes remain to be conducted.
5.3. Systemic Biological and Toxic Insults
5.3.1. Orexin Alterations During Sepsis and Systemic Metabolic Failure
Systemic inflammatory and toxic insults profoundly disturb the neural systems that sustain arousal and homeostatic balance. The orexin network, situated at the interface of metabolic, immune, and autonomic regulation, appears particularly vulnerable to this disruption.
In clinical sepsis, cerebrospinal fluid (CSF) orexin-A concentrations are consistently reduced during the acute phase, recovering gradually with resolution of systemic inflammation (Ogawa et al., 2021, 2022). This dynamic normalization parallels improvements in consciousness and autonomic stability, suggesting that orexin suppression is state-dependent rather than degenerative. A similar pattern is seen in isolated case reports where severe sepsis was accompanied by low CSF orexin-A and blood–brain barrier leakage, which reversed upon clinical recovery (Ogawa et al., 2021).
In rodent models, peripheral inflammation rapidly silences hypothalamic orexin neurons and alters their transcriptional activity. Lipopolysaccharide (LPS) exposure causes dose-dependent reductions in orexin immunoreactivity within hours, often followed by partial rebound at later time points (Perekrest et al., 2009, 2011, 2013). These changes coincide with reduced wakefulness and increased NREM sleep, mimicking human sepsis-associated hypoarousal (Tanaka et al., 2015, 2016). Transgenic loss-of-orexin lines exhibit exaggerated sickness behavior, prolonged inactivity, and higher mortality after LPS, indicating that endogenous orexins normally exert a counter-inflammatory and arousal-supporting role (Hirota et al., 2018; Takekawa et al., 2018). In vivo mapping studies confirm that LPS differentially modulates orexin subpopulations depending on behavioral context and circadian phase, suppressing Fos expression more strongly in lateral than medial orexin neurons during exploratory behavior, and blunting activation across both orexin and histaminergic neurons in the dark period (Gaykema & Goehler, 2009). Additional studies show that LPS reduces prepro-orexin expression in the hypothalamus and lowers orexin-A levels in cortex, hippocampus and pons, especially in orexin-deficient mice, further confirming region-wide suppression and its functional consequences (Deutschman et al., 2013).
Interestingly, in a non-infectious model of systemic inflammation, rats subjected to acute pancreatitis displayed elevated brain levels of orexin, β-endorphin, and oxytocin as early as 12–24 h post-injury, in the absence of increased cytokine expression or microglial activation (Hamasaki et al., 2016a). This suggests that orexin upregulation may serve as an early alarm signal prior to immune cell recruitment, possibly initiating neuroendocrine responses to distant tissue damage. Similarly, alcohol-induced coma suppresses hypothalamic orexin activity, while exogenous orexin-A or -B accelerates awakening and reduces delta power in cortical EEG, an effect partially dependent on histaminergic, noradrenergic and serotonergic transmission (Jia et al., 2012). Overall, the available evidence supports a view in which systemic inflammation and metabolic stress transiently suppress (or in some cases dysregulate) orexinergic tone, contributing to hypoarousal and altered autonomic balance during acute illness.
5.3.2. Experimental Modulation of Orexin Signaling
Acute-phase interventions. During the acute phase of systemic inflammatory or metabolic insults, multiple studies support a time-sensitive, OX₁R- or OXR2-mediated protective role for orexin-A. In rodent models of endotoxemia induced by LPS, centrally or peripherally administered orexin-A (as it crosses the compromised BBB) improves survival, restores cardiovascular tone and thermoregulation, and reduces both plasma and brain cytokine expression, effects that are dependent on OX₁R activation and vagal cholinergic signaling (Igarashi et al., 2020; Ogawa et al., 2016). In a polymicrobial sepsis model (cecal ligation and puncture, CLP), intranasal orexin-A delivered within 30 minutes post-insult reduces mortality, cerebral edema, BBB disruption, microglial activation, and cognitive impairment through OXR2-mediated suppression of the RAS/MAPK cascade (J. Guo et al., 2024). Conversely, delayed central administration at 24–48 hours post-CLP fails to confer benefit and may exacerbate lethargy and neuroendocrine suppression (Deutschman et al., 2013), highlighting the critical importance of early intervention. Chemogenetic activation of orexinergic neurons in CLP mice, as well as pharmacological stimulation with xanomeline, also improved physiological arousal and suppressed cytokine production (Nedeljkovic-Kurepa et al., 2025). These effects were reversed by dual orexin receptor blockade, supporting a causal link between orexin activity and host resilience. Beyond infectious or inflammatory challenges, orexin also modulates arousal in toxic-metabolic states as seen in a model of alcohol-induced coma, where icv orexin-A or orexin-B shortened the duration of unconsciousness and normalized righting reflexes (Jia et al., 2012). These peptides enhanced cortical excitability and reduced EEG delta-power through coordinated recruitment of histaminergic, noradrenergic, and serotonergic pathways. Collectively, these findings identify orexin-A as a fast-acting neuromodulator that bridges metabolic, immune, and arousal systems: when administered early, it stabilizes autonomic and neuroimmune function, enhances survival, and restores consciousness across diverse acute systemic and metabolic insults.
Subacute-phase interventions. In the subacute post-septic phase (24–72 h), the focus shifts from survival to restoration of arousal and cognitive function. Data extending into the subacute phase suggest that orexinergic activity may guide recovery of wakefulness and neurovascular function once systemic inflammation subsides. In humans, CSF orexin-A progressively normalizes over the first 1–3 weeks following sepsis, coinciding with improved consciousness and autonomic tone (Ogawa et al., 2021, 2022). In rodents, the benefits of early orexin-A dosing persist at 7 days post-CLP, with sustained improvements in cognition and structural integrity (J. Guo et al., 2024). Although no interventions were initiated exclusively during the subacute period, the sustained benefits of early orexin exposure into this window support its potential to accelerate functional restoration and counteract sickness behavior.
Chronic and delayed interventions. Persistent orexin deficiency after sepsis or critical illness may contribute to post-intensive-care syndrome, characterized by fatigue, sleep disturbance, and cognitive impairment (Sonneville et al., 2013). However, chronic-phase data after sepsis are not yet available. Whether long-lasting alterations in orexin tone contribute to post-ICU fatigue, cognitive slowing, or metabolic inertia remains speculative. However, the recovery pattern observed clinically implies that orexin neurons retain plasticity and can rebound once systemic homeostasis is re-established, providing a possible therapeutic window for longer-term neuromodulation strategies.
5.4. Mechanistic Convergence Across Injury Types
5.4.1. Acute Phase: Metabolic Crisis, Excitotoxicity, and Orexin Silencing
Across ischemic, traumatic, and systemic insults, the earliest hours are dominated by abrupt energy collapse and ionic disequilibrium. ATP depletion, glutamate overflow, and calcium overload trigger neuronal depolarization and mitochondrial failure (Dirnagl et al., 1999; Lo et al., 2003). Within this landscape, orexin neurons rapidly down-regulate firing and transcription. Hypothalamic Hcrt mRNA and peptide levels fall within 2–6 h after stroke, TBI, SCI, or sepsis (C.-M. Wang et al., 2017; Yuan et al., 2011). Reduced orexinergic activity in the lateral hypothalamus parallels EEG suppression and hypoarousal (Baumann et al., 2007; Koenig et al., 2009).
This silencing likely serves a protective, energy-saving function: orexin neurons integrate metabolic cues such as glucose, lactate, and pH via ATP-sensitive K⁺ channels (Burdakov et al., 2006; Yamanaka et al., 2003). When energetic failure and hypoxia dominate, inhibitory feedback curtails orexin drive to minimize metabolic demand. However, prolonged suppression propagates instability in the arousal and autonomic axes, contributing to coma, hypotension, and respiratory irregularity. Experimental orexin replacement during this window reverses these features, reducing infarct size, edema, and mortality across ischemic, hemorrhagic, and septic models (Akbari et al., 2012; J. Guo et al., 2024; D. Zhang et al., 2022). Mechanistically, OX₁R activation engages ERK/Akt signaling, inhibits caspase-3, and limits NF-κB-dependent cytokine cascades (Palomba et al., 2020; Sokołowska et al., 2014; D. Xu, Kong, Shao, et al., 2021). The pattern is consistent: early, moderate orexin restoration stabilizes cellular energetics and systemic homeostasis, whereas excessive stimulation risks hyperexcitability or sympathetic overshoot.
5.4.2. Subacute Phase: Neurovascular Stabilization, Inflammation Tuning, and Circuit re-engagement
During the ensuing days, secondary injury cascades (i.e., barrier breakdown, edema, and inflammation) become dominant determinants of outcome (Lo et al., 2003; Sofroniew, 2015). Across etiologies, orexinergic reactivation coincides with microvascular and glial remodeling. Partial recovery of orexin tone restores BBB integrity by tightening junction proteins and reducing endothelial transcytosis (Y. Guo et al., 2024; Zhao et al., 2021). Orexin-treated animals show decreased microglial activation and lower IL-1β and TNF-α levels after ischemia, trauma, or sepsis (Modi et al., 2017; Xiong et al., 2013; D. Xu, Kong, Shao, et al., 2021). These effects may reflect both central and peripheral modulation: OX₁R engagement might restrain NF-κB signaling within microglia, whereas OX₂R activation possibly attenuates sympathetic overdrive and systemic cytokine release.
At the circuit level, orexin neurons begin re-firing in response to metabolic normalization and sensory input. Their ascending projections to the locus coeruleus, raphe nuclei, and basal forebrain resynchronize cortical rhythms, facilitating sleep–wake consolidation and behavioral engagement (Burdakov, 2021). Subacute orexin supplementation during this window enhances locomotion, cognition, and thermoregulation in stroke, TBI, and sepsis models (T. Li et al., 2020; D. Zhang et al., 2022). This phase thus marks a shift from neuroprotection to neuromodulation, where orexin’s influence on attention, motivation, and autonomic balance reinforces recovery trajectories rather than simply preventing cell death.
5.4.3. Chronic Phase: Arousal, Plasticity, and Long-Term Maladaptations
Weeks after injury, residual orexin dysregulation manifests as fatigue, sleep fragmentation, chronic pain, or dysautonomia, common across stroke, TBI, SCI, and post-sepsis survivors (Baumann et al., 2007; Ogawa et al., 2021; Thomasy & Opp, 2019). Persistent hypo-orexinergic function contributes to apathy and cognitive slowing, while hyper-orexinergic states favor anxiety or sleep disturbances (Ten-Blanco et al., 2023). The balance between OX₁R- and OX₂R-mediated signaling becomes crucial: the former modulates nociception and sympathetic tone; the latter governs sleep–wake stability (Saitoh & Sakurai, 2023). Chronic therapeutic strategies therefore diverge from acute neuroprotection toward fine-tuning network excitability and supporting behavioral rehabilitation.
Preclinical evidence shows that sustained, mild orexinergic stimulation enhances BDNF expression, hippocampal neurogenesis, and mitochondrial function (Chieffi et al., 2017). Lifestyle factors that raise endogenous orexin, including regular physical activity, enriched environments, consistent light cycles, improve motivation and motor learning in recovery paradigms (Burdakov, 2021). Pharmacological partial agonists currently in development could target chronic hypoarousal and cognitive fatigue without provoking sympathetic excess (Saitoh & Sakurai, 2023). Across etiologies, these approaches transform orexins from emergency neuromodulators into rehabilitation co-factors linking energy state to goal-directed behavior.
5.4.4. Mechanistic Convergence Across Injury Types
As summarized in Figure 1, orexinergic modulation exerts a coordinated sequence of effects that unfold from immediate stabilization to long-term functional recovery.
Short-term actions include metabolic preservation and systemic homeostasis. By supporting glucose utilization and mitochondrial efficiency through AMPK and Akt pathways, orexinergic signaling stabilizes neuronal energetics during hypoperfusion or inflammation. This underlies the rapid restoration of arousal and autonomic control observed across models, improving vigilance and therapeutic readiness while limiting coma or respiratory irregularity. In parallel, orexins dampen glial and cytokine activation, curbing NF-κB-dependent cascades and oxidative stress that drive secondary injury. Reinforcement of endothelial tight-junction integrity help preserve blood–brain and blood–spinal barriers, thereby limiting edema and perivascular infiltration. Collectively, these processes prevent the amplification of tissue damage and support early neuroprotection, though excessive stimulation can transiently heighten sympathetic tone.
Long-term effects emerge as the system transitions from protection to repair. Sustained orexin signaling promotes synaptic plasticity, circuit rebalancing, and behavioral engagement. Through its influence on monoaminergic networks, it recalibrates arousal and motivational tone, enabling goal-directed movement, cognitive performance, and emotional stability. Balanced activity across OX₁R- and OX₂R-mediated pathways thus supports motivation and recovery without inducing maladaptive hyperarousal.
Together, these layers illustrate how orexins act as a unifying homeostatic regulator: metabolic and anti-apoptotic in the acute phase, anti-inflammatory and neurovascular in the subacute phase, and behavioral and motivational in the chronic phase. The same molecular system thereby bridges cellular, circuit, and behavioral recovery, linking short-term survival mechanisms to long-term rehabilitation potential.
6. Translational Opportunities, Challenges and Perspectives
6.1. Temporal Precision: From Injury Phase to Circadian Alignment
Translating orexin biology into therapy requires precision not only in when after injury treatment is initiated, but also in when within the day it is delivered. The first dimension (phase-specific timing) reflects the evolving biology of injury, from metabolic crisis to recovery, as detailed previously. The second dimension (the circadian phase) acknowledges that orexin neurons are integral components of the sleep–wake oscillator and that their activation must respect intrinsic rhythmicity to remain beneficial.
In the hyperacute phase, when perfusion and metabolic collapse dominate, orexinergic stimulation must remain modest and continuous to prevent excitotoxicity or sympathetic overload. In contrast, later phases present a unique opportunity for chronotherapeutic alignment. During the subacute and chronic periods, circadian rhythmicity gradually re-emerges, and orexin activity again peaks in the light–active period. Interventions that synchronize with this rhythm, administered in the morning or early active phase, can amplify arousal and motor engagement while preserving sleep–dependent consolidation at night (Burdakov, 2021; Saitoh & Sakurai, 2023).
This principle opens a translational avenue for time-tailored pharmacology. Short-acting orexin agonists such as danavorexton or TAK-925, with 1–3 h half-lives, could be delivered once daily to reinforce morning wakefulness and rehabilitation sessions (Seigneur & de Lecea, 2020). By contrast, longer-acting analogues or controlled-release peptides may sustain daytime motivation in patients with post-injury fatigue or apathy (Saitoh & Sakurai, 2023). Aligning drug pharmacokinetics with endogenous circadian and activity rhythms could reduce side effects and enhance behavioral gains, a strategy already successful in other neuromodulatory fields such as dopaminergic and corticosteroid therapies (Smolensky et al., 2016).
Ultimately, orexin therapy should be understood as chronobiological modulation rather than static replacement. Its effectiveness will depend on delivering the right magnitude of stimulation, at the right injury stage, and at the right time of day to cooperate with endogenous arousal cycles and rehabilitation schedules. This dual precision (i.e., phase-sensitive and circadian-sensitive) represents the conceptual cornerstone of orexin translation.
6.2. Delivery Routes and Formulation Strategies
Therapeutic translation depends on reaching central targets efficiently and safely, which in turn reflects whether the compound is a peptide or a small molecule.
Peptide-based orexin analogues are rapidly degraded and cross the blood–brain barrier poorly, so systemic delivery is ineffective. Intranasal administration is the most practical route, enabling direct olfactory and trigeminal transport and measurable hypothalamic activation within minutes (Meusel et al., 2022). Formulations using nanoparticles or lipid carriers further improve uptake while limiting peripheral exposure (Lochhead & Thorne, 2012; Wong et al., 2021). These approaches are best suited to experimental or short-term use during the acute and subacute phases, when transient modulation of hypothalamic tone is beneficial.
Small-molecule orexin receptor agonists overcome these limitations. Drugs such as danavorexton (TAK-925) and its oral derivatives (<600 Da) display high receptor selectivity, central penetration, and favorable pharmacokinetics (Seigneur & de Lecea, 2020). Intravenous danavorexton produces rapid, controllable arousal in humans, while oral analogues like TAK-994 offer convenient dosing for longer-term therapy, though safety optimization continues. Their flexibility enables parenteral use for short-term hypoarousal in intensive care and oral use during rehabilitation to sustain motivation and energy.
Future formulations may combine fast intranasal peptides with long-acting oral or subcutaneous small molecules, or incorporate circadian-timed release to align orexin exposure with natural wakefulness peaks. Collectively, these delivery options turn orexin modulation into a realistic pharmacological strategy adaptable to both acute stabilization and chronic recovery.
6.3. Receptor Selectivity and Subtype Balance
Balancing activity across orexin receptor subtypes is crucial for both efficacy and safety. OX₁R and OX₂R share structural homology but mediate distinct physiological domains: OX₁R drives rapid excitatory and analgesic responses, whereas OX₂R stabilizes wakefulness and autonomic tone (Sakurai, 2014; Ten-Blanco et al., 2023). Preclinical evidence suggests that OX₁R activation underlies much of orexin’s cytoprotective and anti-apoptotic signaling after ischemia or trauma, while OX₂R contributes to metabolic and arousal regulation.
Pharmacological development, however, remains asymmetric. Most clinical progress has focused on OX₂R-selective agonists, notably danavorexton and TAK-994, which reliably enhance vigilance with minimal sympathetic load (Seigneur & de Lecea, 2020). In contrast, no selective OX₁R agonists and very few true dual agonists are currently available, leaving a therapeutic gap for conditions where OX₁R signaling predominates, such as neuroprotection or pain modulation. Developing safe small-molecule OX₁R agonists and balanced dual agonists therefore represents a priority for translational neuroscience. Such compounds could reproduce the full spectrum of orexin’s benefits while allowing phase-specific receptor targeting to minimize adverse autonomic effects.
6.4. Combination and Multimodal Approaches
Because orexins regulate metabolism, arousal, and motivation, its therapeutic effects will likely be maximized through complementary interventions rather than monotherapy. Orexin neurons closely interact with serotonin, noradrenaline, dopamine, and histamine systems (Burdakov, 2021; Sakurai, 2014), suggesting that modest co-activation of these circuits could enhance recovery while avoiding excessive sympathetic drive. For instance, serotonergic or noradrenergic agents may stabilize mood and cortical excitability in patients treated with OX₂R agonists, partially compensating for the current lack of selective OX₁R agonists.
Behavioral and neuromodulatory combinations hold equal promise. Physical training, environmental enrichment, and cognitive engagement naturally stimulate orexinergic firing and promote plasticity; short-acting orexin agonists given before rehabilitation sessions could therefore prime motivation and learning (Karnani et al., 2020). Likewise, vagus-nerve or transcranial stimulation can boost endogenous orexin release and autonomic stability, potentially amplifying pharmacological effects (X. Dong et al., 2018).
These integrative approaches position orexins not as stand-alone drug targets but as a hub within a therapeutic network, aligning pharmacological, behavioral, and physiological interventions toward shared recovery goals. Systematic testing of such multimodal paradigms will be essential to define optimal combinations and maximize functional outcomes.
6.5. Safety Considerations and Potential Risks
Because orexins modulate multiple physiological systems, safety evaluation must extend beyond the CNS. The most consistent preclinical concern is sympathetic overactivation, leading to hypertension, tachycardia, or metabolic strain (Saitoh & Sakurai, 2023; Seigneur & de Lecea, 2020). Excessive OX₁R stimulation can precipitate anxiety-like behavior and, at very high doses, seizures (H.-T. Li et al., 2024). Conversely, OX₂R-biased compounds exhibit a wider safety margin, producing smoother arousal with minimal cardiovascular load (Saitoh & Sakurai, 2023). Chronic overstimulation could also disrupt sleep architecture (Tsuneki et al., 2018).
In systemic illness, metabolic demands must be carefully matched: in sepsis, early orexin enhancement improves survival, but if applied before hemodynamic stabilization, it may increase oxygen consumption (Deutschman et al., 2013). Thus, dose titration and patient monitoring will be indispensable for safe translation.
7. Conclusions and Future Perspectives
Acute injuries to the central nervous system, whether vascular, traumatic, or systemic, share a fundamental biological logic: a rapid energetic collapse, vascular failure, and neuroimmune activation, followed by slow attempts at network repair. Across this continuum, the orexin system operates as a dynamic regulator of arousal, metabolism, and stress resilience. Evidence from experimental models shows that early orexin suppression parallels metabolic depression, while its later recovery accompanies restoration of barrier integrity, motivation, and motor engagement. Controlled enhancement of this signaling can therefore influence not only survival but the quality and pace of functional recovery.
Translational progress, however, is still uneven. The biological rationale for orexinergic intervention is strongest where arousal and motivational deficits dominate: post-stroke or post-traumatic fatigue, chronic hypoarousal after sepsis or cardiac arrest, and disorders of vigilance emerging in the subacute or chronic stages. These indications align with the current toolset of OX₂R-selective small-molecule agonists, already in human trials for sleep-wake disorders and adaptable to circadian-timed dosing. By contrast, hyperacute cytoprotection in focal ischemia, hemorrhage, or spinal trauma still requires additional preclinical mapping of dose, timing, and receptor-specific safety to define whether early orexin stimulation is protective or excessive. Gaps also remain in the development of selective OX₁R agonists and balanced dual agonists, as well as in understanding how sex, age, and systemic factors shape responsiveness.
Future work should therefore follow a two-track strategy: cautious clinical exploration in arousal-related syndromes using available OX₂R agents, alongside deeper mechanistic and pharmacological research in acute injury models. Integrating orexinergic modulation with rehabilitation, circadian entrainment, and metabolic support could convert diffuse biological promise into measurable functional benefit. This requires multidisciplinary collaboration, bridging neuropharmacology, chronobiology, and clinical neuroscience to transform conceptual plausibility into therapeutic reliability.
In summary, orexinergic modulation offers a biologically grounded strategy to bridge acute neuroprotection with long-term functional recovery. Orexin’s broad integrative role invites a new therapeutic philosophy: to treat recovery as a coordinated energetic process rather than a purely structural one. By aligning orexinergic modulation with the body’s intrinsic healing chronology, it may be possible to shorten the trajectory from survival to functional independence, redefining how post-injury repair is understood and pursued.
Author Contributions
Conceptualization, Á. Flores; investigation, P. Otero-López, X. Madrid-González, and Á. Flores; data curation, P. Otero-López and X. Madrid-González; visualization, P. Otero-López and X. Madrid-González; writing - original draft preparation, P. Otero-López, X. Madrid-González and Á. Flores; writing - review and editing, V. Fernández-Dueñas and Á. Flores; supervision, V. Fernández-Dueñas and Á. Flores; funding acquisition, Á. Flores. All authors have read and agreed to the published version of the manuscript. (P. Otero-López and X. Madrid-González contributed equally to this work; Á. Flores is the corresponding author.).
Funding
This work was supported by the Ministerio de Ciencia e Innovación (grant PID2022-139396OA-I00). P. Otero-López was supported by a Formación del Profesorado Universitario (FPU) predoctoral fellowship from the Ministerio de Universidades (Government of Spain). X. Madrid-González was supported by an internal research grant from the University of Barcelona, Bellvitge Campus.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Akbari: Y., Maybhate, A., Chen, C., Greenwald, E., Liang, L., Buitrago-Blanco, M., Jia, X., Thakor, N., & Geocadin, R. (2012). Abstract 2: Orexin-A Improves Arousal from Post--Cardiac Arrest Coma in a Rodent Model. Circulation, 126(suppl_21), A2-A2. [CrossRef]
- Ang, B. T., Tan, W. L., Lim, J., & Ng, I. (2005). Cerebrospinal fluid orexin in aneurysmal subarachnoid haemorrhage – a pilot study. Journal of Clinical Neuroscience, 12(7), 758-762. [CrossRef]
- Azadian, M., Tian, G., Bazrafkan, A., Maki, N., Rafi, M., Chetty, N., Desai, M., Otarola, I., Aguirre, F., Zaher, S. M., Khan, A., Suri, Y., Wang, M., Lopour, B. A., Steward, O., & Akbari, Y. (2021). Overnight Caloric Restriction Prior to Cardiac Arrest and Resuscitation Leads to Improved Survival and Neurological Outcome in a Rodent Model. Frontiers in Neuroscience, 14. [CrossRef]
- Baier, P. C., Hallschmid, M., Seeck-Hirschner, M., Weinhold, S. L., Burkert, S., Diessner, N., Göder, R., Aldenhoff, J. B., & Hinze-Selch, D. (2011). Effects of intranasal hypocretin-1 (orexin A) on sleep in narcolepsy with cataplexy. Sleep Medicine, 12(10), 941-946. [CrossRef]
- Barichello, T., Generoso, J. S., Singer, M., & Dal-Pizzol, F. (2022). Biomarkers for sepsis: More than just fever and leukocytosis-a narrative review. Critical Care (London, England), 26(1), 14. [CrossRef]
- Baumann, C. R., Bassetti, C. L., Valko, P. O., Haybaeck, J., Keller, M., Clark, E., Stocker, R., Tolnay, M., & Scammell, T. E. (2009). Loss of hypocretin (orexin) neurons with traumatic brain injury. Annals of Neurology, 66(4), 555-559. [CrossRef]
- Baumann, C. R., Stocker, R., Imhof, H.-G., Trentz, O., Hersberger, M., Mignot, E., & Bassetti, C. L. (2005). Hypocretin-1 (orexin A) deficiency in acute traumatic brain injury. Neurology, 65(1), 147-149. [CrossRef]
- Baumann, C. R., Werth, E., Stocker, R., Ludwig, S., & Bassetti, C. L. (2007). Sleep–wake disturbances 6 months after traumatic brain injury: A prospective study. Brain, 130(7), 1873-1883. [CrossRef]
- Bémeur, C., & Butterworth, R. F. (2013). Liver-brain proinflammatory signalling in acute liver failure: Role in the pathogenesis of hepatic encephalopathy and brain edema. Metabolic Brain Disease, 28(2), 145-150. [CrossRef]
- Bigford, G. E., Bracchi-Ricard, V. C., Keane, R. W., Nash, M. S., & Bethea, J. R. (2013). Neuroendocrine and cardiac metabolic dysfunction and NLRP3 inflammasome activation in adipose tissue and pancreas following chronic spinal cord injury in the mouse. ASN Neuro, 5(4), 243-255. [CrossRef]
- Bigford, G. E., Bracchi-Ricard, V. C., Nash, M. S., & Bethea, J. R. (2012). Alterations in mouse hypothalamic adipokine gene expression and leptin signaling following chronic spinal cord injury and with advanced age. PloS One, 7(7), e41073. [CrossRef]
- Biswabharati, S., Jean-Xavier, C., Eaton, S. E. A., Lognon, A. P., Brett, R., Hardjasa, L., & Whelan, P. J. (2018). Orexinergic Modulation of Spinal Motor Activity in the Neonatal Mouse Spinal Cord. eNeuro, 5(5), ENEURO.0226-18.2018. [CrossRef]
- Blanco-Centurion, C., Liu, M., Konadhode, R., Pelluru, D., & Shiromani, P. J. (2013). Effects of orexin gene transfer in the dorsolateral pons in orexin knockout mice. Sleep, 36(1), 31-40. [CrossRef]
- Bonizzato, M., James, N. D., Pidpruzhnykova, G., Pavlova, N., Shkorbatova, P., Baud, L., Martinez-Gonzalez, C., Squair, J. W., DiGiovanna, J., Barraud, Q., Micera, S., & Courtine, G. (2021). Multi-pronged neuromodulation intervention engages the residual motor circuitry to facilitate walking in a rat model of spinal cord injury. Nature Communications, 12(1), 1925. [CrossRef]
- Born, J., Lange, T., Kern, W., McGregor, G. P., Bickel, U., & Fehm, H. L. (2002). Sniffing neuropeptides: A transnasal approach to the human brain. Nature Neuroscience, 5(6), 514-516. [CrossRef]
- Burdakov, D. (2019). Reactive and predictive homeostasis: Roles of orexin/hypocretin neurons. Neuropharmacology, 154, 61-67. [CrossRef]
- Burdakov, D. (2020). How orexin signals bias action: Hypothalamic and accumbal circuits. Brain Research, 1731, 145943. [CrossRef]
- Burdakov, D. (2021). Subsecond Ensemble Dynamics of Orexin Neurons Link Sensation and Action. Frontiers of Neurology and Neuroscience, 45, 52-60. [CrossRef]
- Burdakov, D., Jensen, L. T., Alexopoulos, H., Williams, R. H., Fearon, I. M., O’Kelly, I., Gerasimenko, O., Fugger, L., & Verkhratsky, A. (2006). Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron, 50(5), 711-722. [CrossRef]
- Campbell, B. C. V., & Khatri, P. (2020). Stroke. Lancet (London, England), 396(10244), 129-142. [CrossRef]
- Cangiano-Heath, A., Wirkowski, E., Gu, Y., Adachi, J., Ashcroft, B., Fiore, S., Mofakham, S., & Wright, B. (2020). Pilot Study of Orexin as an Indicator of Coma Recovery (820). Neurology, 94(15_supplement), 820. [CrossRef]
- Chen, Y., Lu, X., & Hu, L. (2023). Transcutaneous Auricular Vagus Nerve Stimulation Facilitates Cortical Arousal and Alertness. International Journal of Environmental Research and Public Health, 20(2), 1402. [CrossRef]
- Chen, Y.-H., Lee, H.-J., Lee, M. T., Wu, Y.-T., Lee, Y.-H., Hwang, L.-L., Hung, M.-S., Zimmer, A., Mackie, K., & Chiou, L.-C. (2018). Median nerve stimulation induces analgesia via orexin-initiated endocannabinoid disinhibition in the periaqueductal gray. Proceedings of the National Academy of Sciences of the United States of America, 115(45), E10720-E10729. [CrossRef]
- Cheng, Jocelyn. (2025, septiembre 8). E2086, a Selective Orexin Receptor-2 Agonist, Study for Promoting Wakefulness in Patients With Narcolepsy Type-1. World Sleep Congress, Singapore. https://ws2025.abstractserver.com/program/#/details/presentations/1510.
- Chieffi, S., Messina, G., Villano, I., Messina, A., Esposito, M., Monda, V., Valenzano, A., Moscatelli, F., Esposito, T., Carotenuto, M., Viggiano, A., Cibelli, G., & Monda, M. (2017). Exercise Influence on Hippocampal Function: Possible Involvement of Orexin-A. Frontiers in Physiology, 8, 85. [CrossRef]
- Cho, N., Squair, J. W., Aureli, V., James, N. D., Bole-Feysot, L., Dewany, I., Hankov, N., Baud, L., Leonhartsberger, A., Sveistyte, K., Skinnider, M. A., Gautier, M., Laskaratos, A., Galan, K., Goubran, M., Ravier, J., Merlos, F., Batti, L., Pages, S., … Courtine, G. (2024). Hypothalamic deep brain stimulation augments walking after spinal cord injury. Nature Medicine, 30(12), 3676-3686. [CrossRef]
- Christensen, J., MacPherson, N., Li, C., Yamakawa, G. R., & Mychasiuk, R. (2023). Repeat mild traumatic brain injuries (RmTBI) modify nociception and disrupt orexinergic connectivity within the descending pain pathway. The Journal of Headache and Pain, 24(1), 72. [CrossRef]
- Christensen, J., Vlassopoulos, E., Barlow, C. K., Schittenhelm, R. B., Li, C. N., Sgro, M., Warren, S., Semple, B. D., Yamakawa, G. R., Shultz, S. R., & Mychasiuk, R. (2024). The beneficial effects of modafinil administration on repeat mild traumatic brain injury (RmTBI) pathology in adolescent male rats are not dependent upon the orexinergic system. Experimental Neurology, 382, 114969. [CrossRef]
- Christiansen, J. R., & Kirkeby, A. (2024). Clinical translation of pluripotent stem cell-based therapies: Successes and challenges. Development (Cambridge, England), 151(7), dev202067. [CrossRef]
- Courtine, G., & Sofroniew, M. V. (2019). Spinal cord repair: Advances in biology and technology. Nature Medicine, 25(6), 898-908. [CrossRef]
- Craig, J., Carey, S., Gibbons, A., Kaplan, G., & Zielinski, M. (2024). 0049 Sleep and Electroencephalogram Effects from a Dual Orexin Receptor Antagonist in Mice with Traumatic Brain Injury. Sleep, 47(Supplement_1), A22. [CrossRef]
- Dauvilliers, Y. (2025, septiembre 8). The Radiant Light: Efficacy and Safety of Oveporexton (TAK-861), an Oral Orexin Receptor 2 Agonist for the Treatment of Narcolepsy Type 1. World Sleep Congress, Singapore. https://ws2025.abstractserver.com/program/#/details/presentations/3429.
- Dauvilliers, Y., Plazzi, G., Mignot, E., Lammers, G. J., Del Río Villegas, R., Khatami, R., Taniguchi, M., Abraham, A., Hang, Y., Kadali, H., Lamberton, M., Sheikh, S., Stukalin, E., Neuwirth, R., Swick, T. J., Tanaka, S., von Hehn, C., von Rosenstiel, P., Wang, H., … Olsson, T. (2025). Oveporexton, an Oral Orexin Receptor 2-Selective Agonist, in Narcolepsy Type 1. The New England Journal of Medicine, 392(19), 1905-1916. [CrossRef]
- de Lecea, L., Kilduff, T. S., Peyron, C., Gao, X., Foye, P. E., Danielson, P. E., Fukuhara, C., Battenberg, E. L., Gautvik, V. T., Bartlett, F. S., Frankel, W. N., van den Pol, A. N., Bloom, F. E., Gautvik, K. M., & Sutcliffe, J. G. (1998). The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proceedings of the National Academy of Sciences of the United States of America, 95(1), 322-327. [CrossRef]
- Deutschman, C. S., Raj, N. R., McGuire, E. O., & Kelz, M. B. (2013). Orexinergic Activity Modulates Altered Vital Signs and Pituitary Hormone Secretion in Experimental Sepsis. Critical Care Medicine, 41(11), e368. [CrossRef]
- Dhuria, S. V., Hanson, L. R., & Frey, W. H. (2010). Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. Journal of Pharmaceutical Sciences, 99(4), 1654-1673. [CrossRef]
- Dirnagl, U., Iadecola, C., & Moskowitz, M. A. (1999). Pathobiology of ischaemic stroke: An integrated view. Trends in Neurosciences, 22(9), 391-397. [CrossRef]
- Djupesland, P. G. (2013). Nasal drug delivery devices: Characteristics and performance in a clinical perspective-a review. Drug Delivery and Translational Research, 3(1), 42-62. [CrossRef]
- Dohi, K., Nishino, S., Nakamachi, T., Ohtaki, H., Morikawa, K., Takeda, T., Shioda, S., & Aruga, T. (2006). CSF orexin A concentrations and expressions of the orexin-1 receptor in rat hippocampus after cardiac arrest. Neuropeptides, 40(4), 245-250. [CrossRef]
- Dohi, K., Ripley, B., Fujiki, N., Ohtaki, H., Shioda, S., Aruga, T., & Nishino, S. (2005). CSF hypocretin-1/orexin-A concentrations in patients with subarachnoid hemorrhage (SAH). Peptides, 26(11), 2339-2343. [CrossRef]
- Dohi, K., Ripley, B., Fujiki, N., Ohtaki, H., Yamamoto, T., Goto, Y., Nakamachi, T., Shioda, S., Aruga, T., & Nishino, S. (2008). CSF orexin-A/hypocretin-1 concentrations in patients with intracerebral hemorrhage (ICH). Regulatory Peptides, 145(1), 60-64. [CrossRef]
- Dong, X., Papa, E. V., Liu, H., Feng, Z., Huang, F., & Liao, C. (2018). Vagus nerve stimulation causes wake-promotion by affecting neurotransmitters via orexins pathway in traumatic brain injury induced comatose rats.
- Dong, X., Ye, W., Tang, Y., Wang, J., Zhong, L., Xiong, J., Liu, H., Lu, G., & Feng, Z. (2021). Wakefulness-Promoting Effects of Lateral Hypothalamic Area–Deep Brain Stimulation in Traumatic Brain Injury-Induced Comatose Rats: Upregulation of α1-Adrenoceptor Subtypes and Downregulation of Gamma-Aminobutyric Acid β Receptor Expression Via the Orexins Pathway. World Neurosurgery, 152, e321-e331. [CrossRef]
- Dong, X.-Y., & Feng, Z. (2018). Wake-promoting effects of vagus nerve stimulation after traumatic brain injury: Upregulation of orexin-A and orexin receptor type 1 expression in the prefrontal cortex. Neural Regeneration Research, 13(2), 244-251. [CrossRef]
- Dorrier, C. E., Jones, H. E., Pintarić, L., Siegenthaler, J. A., & Daneman, R. (2022). Emerging roles for CNS fibroblasts in health, injury and disease. Nature Reviews. Neuroscience, 23(1), 23-34. [CrossRef]
- Du, Q., Huang, L., Tang, Y., Kang, J., Ye, W., & Feng, Z. (2022). Median Nerve Stimulation Attenuates Traumatic Brain Injury–Induced Comatose State by Regulating the Orexin-A/RasGRF1 Signaling Pathway. World Neurosurgery, 168, e19-e27. [CrossRef]
- Elliott, J. E., De Luche, S. E., Churchill, M. J., Moore, C., Cohen, A. S., Meshul, C. K., & Lim, M. M. (2018). Dietary therapy restores glutamatergic input to orexin/hypocretin neurons after traumatic brain injury in mice. Sleep, 41(3), zsx212. [CrossRef]
- Equihua-Benítez, A. C., Equihua-Benítez, J. A., Guzmán-Vásquez, K., Prospero-García, O., & Drucker-Colín, R. (2020). Orexin cell transplant reduces behavioral arrest severity in narcoleptic mice. Brain Research, 1745, 146951. [CrossRef]
- Eriksson, K. S., Sergeeva, O., Brown, R. E., & Haas, H. L. (2001). Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 21(23), 9273-9279. [CrossRef]
- España, R. A., Baldo, B. A., Kelley, A. E., & Berridge, C. W. (2001). Wake-promoting and sleep-suppressing actions of hypocretin (orexin): Basal forebrain sites of action. Neuroscience, 106(4), 699-715. [CrossRef]
- Evans, R., Kimura, H., Alexander, R., Davies, C. H., Faessel, H., Hartman, D. S., Ishikawa, T., Ratti, E., Shimizu, K., Suzuki, M., Tanaka, S., Yukitake, H., Dauvilliers, Y., & Mignot, E. (2022). Orexin 2 receptor-selective agonist danavorexton improves narcolepsy phenotype in a mouse model and in human patients. Proceedings of the National Academy of Sciences of the United States of America, 119(35), e2207531119. [CrossRef]
- Fu, T., Zhang, W., Guo, R., He, S., Yu, S., Wang, H., Zhang, Y., & Wu, Y. (2025). Inclusion of hypocretin-1 improved performance of poor sleep quality prediction for elderly patients with acute ischemic stroke: A prospective cohort study. Frontiers in Aging Neuroscience, 16. [CrossRef]
- Gao, X.-B., & Horvath, T. (2014). Function and dysfunction of hypocretin/orexin: An energetics point of view. Annual Review of Neuroscience, 37, 101-116. [CrossRef]
- Gaykema, R. P. A., & Goehler, L. E. (2009). Lipopolysaccharide challenge-induced suppression of Fos in hypothalamic orexin neurons: Their potential role in sickness behavior. Brain, Behavior, and Immunity, 23(7), 926-930. [CrossRef]
- Gouveia, F. V., Warsi, N. M., Suresh, H., Matin, R., & Ibrahim, G. M. (2024). Neurostimulation treatments for epilepsy: Deep brain stimulation, responsive neurostimulation and vagus nerve stimulation. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics, 21(3), e00308. [CrossRef]
- Guo, J., Kong, Z., Yang, S., Da, J., Chu, L., Han, G., Liu, J., Tan, Y., & Zhang, J. (2024). Therapeutic effects of orexin-A in sepsis-associated encephalopathy in mice. Journal of Neuroinflammation, 21(1), 131. [CrossRef]
- Guo, Y., Gharibani, P., Agarwal, P., Cho, S.-M., Thakor, N. V., & Geocadin, R. G. (2023). Hyperacute autonomic and cortical function recovery following cardiac arrest resuscitation in a rodent model. Annals of Clinical and Translational Neurology, 10(12), 2223-2237. [CrossRef]
- Guo, Y., Gharibani, P., Agarwal, P., Modi, H., Cho, S.-M., Thakor, N. V., & Geocadin, R. G. (2024). Endogenous orexin and hyperacute autonomic responses after resuscitation in a preclinical model of cardiac arrest. Frontiers in Neuroscience, 18. [CrossRef]
- Hamasaki, M. Y., Barbeiro, H. V., Barbeiro, D. F., Cunha, D. M. G., Koike, M. K., Machado, M. C. C., & Silva, F. P. da. (2016a). “Neuropeptides in the brain defense against distant organ damage”. Journal of Neuroimmunology, 290, 33-35. [CrossRef]
- Hamasaki, M. Y., Barbeiro, H. V., Barbeiro, D. F., Cunha, D. M. G., Koike, M. K., Machado, M. C. C., & Silva, F. P. da. (2016b). “Neuropeptides in the brain defense against distant organ damage”. Journal of Neuroimmunology, 290, 33-35. [CrossRef]
- He, G., Chu, W., Li, Y., Sheng, X., Luo, H., Xu, A., Bian, M., Zhang, H., Wang, M., & Zheng, C. (2025). [Orexin-A promotes motor function recovery of rats with spinal cord injury by regulating ionotropic glutamate receptors]. Nan Fang Yi Ke Da Xue Xue Bao = Journal of Southern Medical University, 45(5), 1023-1030. [CrossRef]
- Herring, W. J., Connor, K. M., Snyder, E., Snavely, D. B., Morin, C. M., Lines, C., & Michelson, D. (2019). Effects of suvorexant on the Insomnia Severity Index in patients with insomnia: Analysis of pooled phase 3 data. Sleep Medicine, 56, 219-223. [CrossRef]
- Hilkens, N. A., Casolla, B., Leung, T. W., & de Leeuw, F.-E. (2024). Stroke. Lancet (London, England), 403(10446), 2820-2836. [CrossRef]
- Hirota, K., Takekawa, D., Kushikata, T., & Kudo, M. (2018). Orexinergic tone affects survival in sepsis in rat. British Journal of Anaesthesia, 120(5), e19-e20. [CrossRef]
- Horvath, T. L., Peyron, C., Diano, S., Ivanov, A., Aston-Jones, G., Kilduff, T. S., & van Den Pol, A. N. (1999). Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. The Journal of Comparative Neurology, 415(2), 145-159.
- Hu, S., Ren, L., Wang, Y., Lei, Z., Cai, J., & Pan, S. (2023). The association between serum orexin A and short-term neurological improvement in patients with mild to moderate acute ischemic stroke. Brain and Behavior, 13(1), e2845. [CrossRef]
- Huang, L., Kang, J., Chen, G., Ye, W., Meng, X., Du, Q., & Feng, Z. (2022). Low-intensity focused ultrasound attenuates early traumatic brain injury by OX-A/NF-κB/NLRP3 signaling pathway. Aging, 14(18), 7455-7469. [CrossRef]
- Igarashi, S., Nozu, T., Ishioh, M., Kumei, S., Saito, T., Toki, Y., Hatayama, M., Yamamoto, M., Shindo, M., Tanabe, H., & Okumura, T. (2020). Centrally administered orexin prevents lipopolysaccharide and colchicine induced lethality via the vagal cholinergic pathway in a sepsis model in rats. Biochemical Pharmacology, 182, 114262. [CrossRef]
- Illum, L. (2000). Transport of drugs from the nasal cavity to the central nervous system. European Journal of Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical Sciences, 11(1), 1-18. [CrossRef]
- Irving, E. A., Harrison, D. C., Babbs, A. J., Mayes, A. C., Campbell, C. A., Hunter, A. J., Upton, N., & Parsons, A. A. (2002). Increased cortical expression of the orexin-1 receptor following permanent middle cerebral artery occlusion in the rat. Neuroscience Letters, 324(1), 53-56. [CrossRef]
- Islam, M. T., Rumpf, F., Tsuno, Y., Kodani, S., Sakurai, T., Matsui, A., Maejima, T., & Mieda, M. (2022). Vasopressin neurons in the paraventricular hypothalamus promote wakefulness via lateral hypothalamic orexin neurons. Current Biology: CB, 32(18), 3871-3885.e4. [CrossRef]
- Jia, X., Yan, J., Xia, J., Xiong, J., Wang, T., Chen, Y., Qi, A., Yang, N., Fan, S., Ye, J., & Hu, Z. (2012). Arousal effects of orexin A on acute alcohol intoxication-induced coma in rats. Neuropharmacology, 62(2), 775-783. [CrossRef]
- Kang, J., Ren, B., Huang, L., Dong, X., Xiong, Q., & Feng, Z. (2024). Orexin-A alleviates ferroptosis by activating the Nrf2/HO-1 signaling pathway in traumatic brain injury. Aging, 16(4), 3404-3419. [CrossRef]
- Kang, J., Ren, B., Wang, J., Tang, Y., & Dong, X. (2025). Vagus nerve stimulation: A promising strategy to combat pyroptosis and inflammation in traumatic brain injury through the OX-A/NLRP3/caspase-1/GSDMD signaling pathway. European Journal of Medical Research, 30(1), 586. [CrossRef]
- Kang, J., Zhou, Y., Xiong, Q., & Dong, X. (2024). Trigeminal nerve electrical stimulation attenuates early traumatic brain injury through the TLR4/NF-κB/NLRP3 signaling pathway mediated by orexin-A/OX1R system. Aging, 16(9), 7946-7960. [CrossRef]
- Kang, Y.-J., Tian, G., Bazrafkan, A., Farahabadi, M. H., Azadian, M., Abbasi, H., Shamaoun, B. E., Steward, O., & Akbari, Y. (2017). Recovery from Coma Post-Cardiac Arrest Is Dependent on the Orexin Pathway. Journal of Neurotrauma, 34(19), 2823-2832. [CrossRef]
- Karnani, M. M., Schöne, C., Bracey, E. F., González, J. A., Viskaitis, P., Li, H.-T., Adamantidis, A., & Burdakov, D. (2020). Role of spontaneous and sensory orexin network dynamics in rapid locomotion initiation. Progress in Neurobiology, 187, 101771. [CrossRef]
- Kitamura, E., Hamada, J., Kanazawa, N., Yonekura, J., Masuda, R., Sakai, F., & Mochizuki, H. (2010). The effect of orexin-A on the pathological mechanism in the rat focal cerebral ischemia. Neuroscience Research, 68(2), 154-157. [CrossRef]
- Koenig, M. A., Jia, X., Kang, X., Velasquez, A., Thakor, N. V., & Geocadin, R. G. (2009). Intraventricular orexin-A improves arousal and early EEG entropy in rats after cardiac arrest. Brain Research, 1255, 153-161. [CrossRef]
- Konduru, S. R., Isaacson, J. R., Lasky, D. J., Zhou, Z., Rao, R. K., Vattem, S. S., Rewey, S. J., Jones, M. V., & Maganti, R. K. (2022). Dual orexin antagonist normalized sleep homeostatic drive, enhanced GABAergic inhibition, and suppressed seizures after traumatic brain injury. Sleep, 45(12), zsac238. [CrossRef]
- Korotkova, T. M., Brown, R. E., Sergeeva, O. A., Ponomarenko, A. A., & Haas, H. L. (2006). Effects of arousal- and feeding-related neuropeptides on dopaminergic and GABAergic neurons in the ventral tegmental area of the rat. The European Journal of Neuroscience, 23(10), 2677-2685. [CrossRef]
- Kotan, D., Deniz, O., Aygul, R., & Yıldırım, A. (2013). Acute cerebral ischaemia: Relationship between serum and cerebrospinal fluid orexin-A concentration and infarct volume. Journal of International Medical Research, 41(2), 404-409. [CrossRef]
- Kukkonen, J. P., Jacobson, L. H., Hoyer, D., Rinne, M. K., & Borgland, S. L. (2024). International Union of Basic and Clinical Pharmacology CXIV: Orexin Receptor Function, Nomenclature and Pharmacology. Pharmacological Reviews, 76(5), 625-688. [CrossRef]
- Kuwaki, T. (2011). Orexin links emotional stress to autonomic functions. Autonomic Neuroscience: Basic & Clinical, 161(1-2), 20-27. [CrossRef]
- Lai, F., Cucca, F., Frau, R., Corrias, F., Schlich, M., Caboni, P., Fadda, A. M., & Bassareo, V. (2018). Systemic Administration of Orexin a Loaded Liposomes Potentiates Nucleus Accumbens Shell Dopamine Release by Sucrose Feeding. Frontiers in Psychiatry, 9, 640. [CrossRef]
- Li, F.-T., Yao, C.-H., Yao, L., Huo, Z.-F., & Liu, J. (2018). Study on the effects of parecoxib on hypothalamus orexin neuron of cerebral infarction rats. European Review for Medical and Pharmacological Sciences, 22(5), 1499-1505. [CrossRef]
- Li, H.-T., Viskaitis, P., Bracey, E., Peleg-Raibstein, D., & Burdakov, D. (2024). Transient targeting of hypothalamic orexin neurons alleviates seizures in a mouse model of epilepsy. Nature Communications, 15(1), 1249. [CrossRef]
- Li, J., Hu, Z., & de Lecea, L. (2014). The hypocretins/orexins: Integrators of multiple physiological functions. British Journal of Pharmacology, 171(2), 332-350. [CrossRef]
- Li, T., Xu, W., Ouyang, J., Lu, X., Sherchan, P., Lenahan, C., Irio, G., Zhang, J. H., Zhao, J., Zhang, Y., & Tang, J. (2020). Orexin A alleviates neuroinflammation via OXR2/CaMKKβ/AMPK signaling pathway after ICH in mice. Journal of Neuroinflammation, 17(1), 187. [CrossRef]
- Lim, M. M., Elkind, J., Xiong, G., Galante, R., Zhu, J., Zhang, L., Lian, J., Rodin, J., Kuzma, N. N., Pack, A. I., & Cohen, A. S. (2013). Dietary Therapy Mitigates Persistent Wake Deficits Caused by Mild Traumatic Brain Injury. Science Translational Medicine, 5(215), 215ra173-215ra173. [CrossRef]
- Liu, M., Blanco-Centurion, C., Konadhode, R. R., Luan, L., & Shiromani, P. J. (2016). Orexin gene transfer into the amygdala suppresses both spontaneous and emotion-induced cataplexy in orexin-knockout mice. The European Journal of Neuroscience, 43(5), 681-688. [CrossRef]
- Liu, R.-J., van den Pol, A. N., & Aghajanian, G. K. (2002). Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 22(21), 9453-9464. [CrossRef]
- Lo, E. H., Dalkara, T., & Moskowitz, M. A. (2003). Mechanisms, challenges and opportunities in stroke. Nature Reviews. Neuroscience, 4(5), 399-415. [CrossRef]
- Lochhead, J. J., & Thorne, R. G. (2012). Intranasal delivery of biologics to the central nervous system. Advanced Drug Delivery Reviews, 64(7), 614-628. [CrossRef]
- Lozano, A. M., Lipsman, N., Bergman, H., Brown, P., Chabardes, S., Chang, J. W., Matthews, K., McIntyre, C. C., Schlaepfer, T. E., Schulder, M., Temel, Y., Volkmann, J., & Krauss, J. K. (2019). Deep brain stimulation: Current challenges and future directions. Nature Reviews. Neurology, 15(3), 148-160. [CrossRef]
- Maas, A. I. R., Menon, D. K., Manley, G. T., Abrams, M., Åkerlund, C., Andelic, N., Aries, M., Bashford, T., Bell, M. J., Bodien, Y. G., Brett, B. L., Büki, A., Chesnut, R. M., Citerio, G., Clark, D., Clasby, B., Cooper, D. J., Czeiter, E., Czosnyka, M., … InTBIR Participants and Investigators. (2022). Traumatic brain injury: Progress and challenges in prevention, clinical care, and research. The Lancet. Neurology, 21(11), 1004-1060. [CrossRef]
- Magid-Bernstein, J., Girard, R., Polster, S., Srinath, A., Romanos, S., Awad, I. A., & Sansing, L. H. (2022). Cerebral Hemorrhage: Pathophysiology, Treatment, and Future Directions. Circulation Research, 130(8), 1204-1229. [CrossRef]
- Martins, L., Seoane-Collazo, P., Contreras, C., González-García, I., Martínez-Sánchez, N., González, F., Zalvide, J., Gallego, R., Diéguez, C., Nogueiras, R., Tena-Sempere, M., & López, M. (2016). A Functional Link between AMPK and Orexin Mediates the Effect of BMP8B on Energy Balance. Cell Reports, 16(8), 2231-2242. [CrossRef]
- Matsuura, W., Nakamoto, K., & Tokuyama, S. (2020). Involvement of descending pain control system regulated by orexin receptor signaling in the induction of central post-stroke pain in mice. European Journal of Pharmacology, 874, 173029. [CrossRef]
- Messina, G., Dalia, C., Tafuri, D., Monda, V., Palmieri, F., Dato, A., Russo, A., De Blasio, S., Messina, A., De Luca, V., Chieffi, S., & Monda, M. (2014). Orexin-A controls sympathetic activity and eating behavior. Frontiers in Psychology, 5, 997. [CrossRef]
- Meusel, M., Voß, J., Krapalis, A., Machleidt, F., Vonthein, R., Hallschmid, M., & Sayk, F. (2022). Intranasal orexin A modulates sympathetic vascular tone: A pilot study in healthy male humans. Journal of Neurophysiology, 127(2), 548-558. [CrossRef]
- Mihara, Y., Dohi, K., Yofu, S., Nakamachi, T., Ohtaki, H., Shioda, S., & Aruga, T. (2011). Expression and Localization of the Orexin-1 Receptor (OX1R) After Traumatic Brain Injury in Mice. Journal of Molecular Neuroscience, 43(2), 162-168. [CrossRef]
- Milbank, E., & López, M. (2019). Orexins/Hypocretins: Key Regulators of Energy Homeostasis. Frontiers in Endocrinology, 10, 830. [CrossRef]
- Mileykovskiy, B. Y., Kiyashchenko, L. I., & Siegel, J. M. (2005). Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron, 46(5), 787-798. [CrossRef]
- Modi, H. R., Wang, Q., Gd, S., Sherman, D., Greenwald, E., Savonenko, A. V., Geocadin, R. G., & Thakor, N. V. (2017). Intranasal post-cardiac arrest treatment with orexin-A facilitates arousal from coma and ameliorates neuroinflammation. PLOS ONE, 12(9), e0182707. [CrossRef]
- Mofatteh, M., Mohamed, A., Mashayekhi, M. S., Skandalakis, G. P., Neudorfer, C., Arfaie, S., MohanaSundaram, A., Sabahi, M., Anand, A., Aboulhosn, R., Liao, X., Horn, A., & Ashkan, K. (2025). Deep brain stimulation of the hypothalamic region: A systematic review. Acta Neurochirurgica, 167(1), 33. [CrossRef]
- Nakamachi, T., Endo, S., Ohtaki, H., Yin, L., Kenji, D., Kudo, Y., Funahashi, H., Matsuda, K., & Shioda, S. (2005). Orexin-1 receptor expression after global ischemia in mice. Regulatory Peptides, 126(1), 49-54. [CrossRef]
- Nedeljkovic-Kurepa, A., Abraham, M. N., Fernandes, T. D., Yaipen, O., Brewer, M. R., Taylor, M. D., Pavlov, V. A., & Deutschman, C. S. (2025). Loss of M1 Acetylcholine Receptor-mediated Orexinergic Activity Contributes to Immune Dysfunction in Experimental Sepsis. Research Square. [CrossRef]
- Ogawa, Y., Ezaki, S., Shimojo, N., & Kawano, S. (2021). Case Report: Reduced CSF Orexin Levels in a Patient With Sepsis. Frontiers in Neuroscience, 15. [CrossRef]
- Ogawa, Y., Irukayama-Tomobe, Y., Murakoshi, N., Kiyama, M., Ishikawa, Y., Hosokawa, N., Tominaga, H., Uchida, S., Kimura, S., Kanuka, M., Morita, M., Hamada, M., Takahashi, S., Hayashi, Y., & Yanagisawa, M. (2016). Peripherally administered orexin improves survival of mice with endotoxin shock. eLife, 5, e21055. [CrossRef]
- Ogawa, Y., Shimojo, N., Ishii, A., Tamaoka, A., Kawano, S., & Inoue, Y. (2022). Reduced CSF orexin levels in rats and patients with systemic inflammation: A preliminary study. BMC Research Notes, 15(1), 221. [CrossRef]
- O’Shea, T. M., Ao, Y., Wang, S., Ren, Y., Cheng, A. L., Kawaguchi, R., Shi, Z., Swarup, V., & Sofroniew, M. V. (2024). Derivation and transcriptional reprogramming of border-forming wound repair astrocytes after spinal cord injury or stroke in mice. Nature Neuroscience, 27(8), 1505-1521. [CrossRef]
- Pace, M., Adamantidis, A., Facchin, L., & Bassetti, C. (2017). Role of REM Sleep, Melanin Concentrating Hormone and Orexin/Hypocretin Systems in the Sleep Deprivation Pre-Ischemia. PLOS ONE, 12(1), e0168430. [CrossRef]
- Pace, M., Baracchi, F., Gao, B., & Bassetti, C. (2015). Identification of Sleep-Modulated Pathways Involved in Neuroprotection from Stroke. Sleep, 38(11), 1707-1718. [CrossRef]
- Palomba, L., Motta, A., Imperatore, R., Piscitelli, F., Capasso, R., Mastroiacovo, F., Battaglia, G., Bruno, V., Cristino, L., & Di Marzo, V. (2020). Role of 2-Arachidonoyl-Glycerol and CB1 Receptors in Orexin-A-Mediated Prevention of Oxygen–Glucose Deprivation-Induced Neuronal Injury. Cells, 9(6), 1507. [CrossRef]
- Peleg-Raibstein, D., Viskaitis, P., & Burdakov, D. (2023). Eat, seek, rest? An orexin/hypocretin perspective. Journal of Neuroendocrinology, 35(9), e13259. [CrossRef]
- Perekrest, S. V., Abramova, T. V., & Novikova, N. S. (2009). [Comparative analysis of orexin-containing neurons reactions in the rat hypothalamus after lipopolysaccharide injection in different doses]. Rossiiskii Fiziologicheskii Zhurnal Imeni I.M. Sechenova, 95(12), 1336-1345.
- Perekrest, S. V., Shainidze, K. Z., Loskutov, I. V., Abramova, T. V., Novikova, N. S., & Korneva, E. A. (2011). [Immunoreactivity of hypothalamic orexin neurons and expression level of preproorexin gene in them after lipopolysaccharide injection]. Rossiiskii fiziologicheskii zhurnal imeni I.M Sechenova, 97(6), 573-579.
- Perekrest, S. V., Shainidze, K. Z., Loskutov, Yu. V., Abramova, T. V., Novikova, N. S., & Korneva, E. A. (2013). Immunoreactivity of Orexin-Containing Business in the Hypothalamus and the Level of Expression of the Preproorexin Gene in These Cells after Administration of Lipopolysaccharide. Neuroscience and Behavioral Physiology, 43(2), 256-260. [CrossRef]
- Perkins, G. D., Neumar, R., Hsu, C. H., Hirsch, K. G., Aneman, A., Becker, L. B., Couper, K., Callaway, C. W., Hoedemaekers, C. W. E., Lim, S. L., Meurer, W., Olasveengen, T., Sekhon, M. S., Skrifvars, M., Soar, J., Tsai, M.-S., Vengamma, B., Nolan, J. P., & International Liaison Committee on Resuscitation. (2024). Improving Outcomes After Post-Cardiac Arrest Brain Injury: A Scientific Statement From the International Liaison Committee on Resuscitation. Resuscitation, 201, 110196. [CrossRef]
- Peyron, C., Tighe, D. K., van den Pol, A. N., de Lecea, L., Heller, H. C., Sutcliffe, J. G., & Kilduff, T. S. (1998). Neurons containing hypocretin (orexin) project to multiple neuronal systems. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 18(23), 9996-10015. [CrossRef]
- Pintwala, S. K., Fraigne, J. J., Belsham, D. D., & Peever, J. H. (2023). Immortal orexin cell transplants restore motor-arousal synchrony during cataplexy. Current Biology: CB, 33(8), 1550-1564.e5. [CrossRef]
- Piper, D. C., Upton, N., Smith, M. I., & Hunter, A. J. (2000). The novel brain neuropeptide, orexin-A, modulates the sleep-wake cycle of rats. The European Journal of Neuroscience, 12(2), 726-730. [CrossRef]
- Plazzi, Giuseppe. (2025, septiembre 8). Vibrance-1: A Randomized Phase 2 Study Evaluating Safety and Efficacy of the Orexin 2 Receptor Agonist Alixorexton (ALKS 2680) in Patients with Narcolepsy Type 1. World Sleep Congress, Singapore. https://ws2025.abstractserver.com/program/#/details/presentations/1499.
- Quintana, D. S., Lischke, A., Grace, S., Scheele, D., Ma, Y., & Becker, B. (2021). Advances in the field of intranasal oxytocin research: Lessons learned and future directions for clinical research. Molecular Psychiatry, 26(1), 80-91. [CrossRef]
- Rahimi Darehbagh, R., Seyedoshohadaei, S. A., Ramezani, R., & Rezaei, N. (2024). Stem cell therapies for neurological disorders: Current progress, challenges, and future perspectives. European Journal of Medical Research, 29(1), 386. [CrossRef]
- Rejdak, K., Petzold, A., Lin, L., Smith, M., Kitchen, N., & Thompson, E. J. (2005). Decreased CSF hypocretin-1 (orexin-A) after acute haemorrhagic brain injury. Journal of Neurology, Neurosurgery & Psychiatry, 76(4), 597-598. [CrossRef]
- Ren, B., Kang, J., Wang, Y., Meng, X., Huang, Y., Bai, Y., & Feng, Z. (2024). Transcranial direct current stimulation promotes angiogenesis and improves neurological function via the OXA-TF-AKT/ERK signaling pathway in traumatic brain injury. Aging, 16(7), 6566-6587. [CrossRef]
- Saitoh, T., & Sakurai, T. (2023). The present and future of synthetic orexin receptor agonists. Peptides, 167, 171051. [CrossRef]
- Sakurai, T. (2007). The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nature Reviews. Neuroscience, 8(3), 171-181. [CrossRef]
- Sakurai, T. (2014). The role of orexin in motivated behaviours. Nature Reviews. Neuroscience, 15(11), 719-731. [CrossRef]
- Sakurai, T., Amemiya, A., Ishii, M., Matsuzaki, I., Chemelli, R. M., Tanaka, H., Williams, S. C., Richardson, J. A., Kozlowski, G. P., Wilson, S., Arch, J. R., Buckingham, R. E., Haynes, A. C., Carr, S. A., Annan, R. S., McNulty, D. E., Liu, W. S., Terrett, J. A., Elshourbagy, N. A., … Yanagisawa, M. (1998). Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell, 92(4), 573-585. [CrossRef]
- Scammell, T. E., Nishino, S., Mignot, E., & Saper, C. B. (2001). Narcolepsy and low CSF orexin (hypocretin) concentration after a diencephalic stroke. Neurology, 56(12), 1751-1753. [CrossRef]
- Seigneur, E., & de Lecea, L. (2020). Hypocretin (Orexin) Replacement Therapies. Medicine in Drug Discovery, 8, 100070. [CrossRef]
- Sherman, D. L., Williams, A., Gd, S., Modi, H. R., Wang, Q., Thakor, N. V., & Geocadin, R. G. (2021). Intranasal Orexin After Cardiac Arrest Leads to Increased Electroencephalographic Gamma Activity and Enhanced Neurologic Recovery in Rats. Critical Care Explorations, 3(2), e0349. [CrossRef]
- Skopin, M. D., Kabadi, S. V., Viechweg, S. S., Mong, J. A., & Faden, A. I. (2015). Chronic Decrease in Wakefulness and Disruption of Sleep-Wake Behavior after Experimental Traumatic Brain Injury. Journal of Neurotrauma, 32(5), 289-296. [CrossRef]
- Smolensky, M. H., Hermida, R. C., Reinberg, A., Sackett-Lundeen, L., & Portaluppi, F. (2016). Circadian disruption: New clinical perspective of disease pathology and basis for chronotherapeutic intervention. Chronobiology International, 33(8), 1101-1119. [CrossRef]
- Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nature Reviews. Neuroscience, 16(5), 249-263. [CrossRef]
- Sokołowska, P., Urbańska, A., Biegańska, K., Wagner, W., Ciszewski, W., Namiecińska, M., & Zawilska, J. B. (2014). Orexins Protect Neuronal Cell Cultures Against Hypoxic Stress: An Involvement of Akt Signaling. Journal of Molecular Neuroscience, 52(1), 48-55. [CrossRef]
- Somach, R. T., Jean, I. D., Farrugia, A. M., & Cohen, A. S. (2023). Mild Traumatic Brain Injury Affects Orexin/Hypocretin Physiology Differently in Male and Female Mice. Journal of Neurotrauma, 40(19-20), 2146-2163. [CrossRef]
- Sonneville, R., Verdonk, F., Rauturier, C., Klein, I. F., Wolff, M., Annane, D., Chretien, F., & Sharshar, T. (2013). Understanding brain dysfunction in sepsis. Annals of Intensive Care, 3(1), 15. [CrossRef]
- Sun, Y., Li, Q., Guo, H., & He, Q. (2022). Ferroptosis and Iron Metabolism after Intracerebral Hemorrhage. Cells, 12(1), 90. [CrossRef]
- Takekawa, D., Kushikata, T., Akaishi, M., Nikaido, Y., & Hirota, K. (2018). Influence of Orexinergic System on Survival in Septic Rats. Neuropsychobiology, 77(1), 45-48. [CrossRef]
- Tanaka, S., Takizawa, N., Honda, Y., Koike, T., Oe, S., Toyoda, H., Kodama, T., & Yamada, H. (2016). Hypocretin/orexin loss changes the hypothalamic immune response. Brain, Behavior, and Immunity, 57, 58-67. [CrossRef]
- Tanaka, S., Toyoda, H., Honda, Y., Seki, Y., Sakurai, T., Honda, K., & Kodama, T. (2015). Hypocretin/orexin prevents recovery from sickness. Biomedical Reports, 3(5), 648-650. [CrossRef]
- Ten-Blanco, M., Flores, Á., Cristino, L., Pereda-Pérez, I., & Berrendero, F. (2023). Targeting the orexin/hypocretin system for the treatment of neuropsychiatric and neurodegenerative diseases: From animal to clinical studies. Frontiers in Neuroendocrinology, 69, 101066. [CrossRef]
- Tesmer, A. L., Li, X., Bracey, E., Schmandt, C., Polania, R., Peleg-Raibstein, D., & Burdakov, D. (2024). Orexin neurons mediate temptation-resistant voluntary exercise. Nature Neuroscience, 27(9), 1774-1782. [CrossRef]
- Tesmer, A. L., Viskaitis, P., Donegan, D., Bracey, E. F., Grujic, N., Patriarchi, T., Peleg-Raibstein, D., & Burdakov, D. (2025). Orexin population activity precisely reflects net body movement across behavioral and metabolic states. eLife, 14, RP103738. [CrossRef]
- Thannickal, T. C., Moore, R. Y., Nienhuis, R., Ramanathan, L., Gulyani, S., Aldrich, M., Cornford, M., & Siegel, J. M. (2000). Reduced number of hypocretin neurons in human narcolepsy. Neuron, 27(3), 469-474. [CrossRef]
- Theus, M. H. (2024). Neuroinflammation and acquired traumatic CNS injury: A mini review. Frontiers in Neurology, 15, 1334847. [CrossRef]
- Thomasy, H. E., Febinger, H. Y., Ringgold, K. M., Gemma, C., & Opp, M. R. (2017). Hypocretinergic and cholinergic contributions to sleep-wake disturbances in a mouse model of traumatic brain injury. Neurobiology of Sleep and Circadian Rhythms, 2, 71-84. [CrossRef]
- Thomasy, H. E., & Opp, M. R. (2019). Hypocretin Mediates Sleep and Wake Disturbances in a Mouse Model of Traumatic Brain Injury. Journal of Neurotrauma, 36(5), 802-814. [CrossRef]
- Tsuneki, H., Wada, T., & Sasaoka, T. (2018). Chronopathophysiological implications of orexin in sleep disturbances and lifestyle-related disorders. Pharmacology & Therapeutics, 186, 25-44. [CrossRef]
- van den Pol, A. N. (1999). Hypothalamic hypocretin (orexin): Robust innervation of the spinal cord. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 19(8), 3171-3182. [CrossRef]
- Wang, C.-M., Pan, Y.-Y., Liu, M.-H., Cheng, B.-H., Bai, B., & Chen, J. (2017). RNA-seq expression profiling of rat MCAO model following reperfusion Orexin-A. Oncotarget, 8(68), 113066-113081. [CrossRef]
- Wang, T., Li, Y., Guo, M., Dong, X., Liao, M., Du, M., Wang, X., Yin, H., & Yan, H. (2021). Exosome-Mediated Delivery of the Neuroprotective Peptide PACAP38 Promotes Retinal Ganglion Cell Survival and Axon Regeneration in Rats With Traumatic Optic Neuropathy. Frontiers in Cell and Developmental Biology, 9, 659783. [CrossRef]
- Whiting, D. M., Tomycz, N. D., Bailes, J., de Jonge, L., Lecoultre, V., Wilent, B., Alcindor, D., Prostko, E. R., Cheng, B. C., Angle, C., Cantella, D., Whiting, B. B., Mizes, J. S., Finnis, K. W., Ravussin, E., & Oh, M. Y. (2013). Lateral hypothalamic area deep brain stimulation for refractory obesity: A pilot study with preliminary data on safety, body weight, and energy metabolism. Journal of Neurosurgery, 119(1), 56-63. [CrossRef]
- Willie, J. T., Lim, M. M., Bennett, R. E., Azarion, A. A., Schwetye, K. E., & Brody, D. L. (2012). Controlled Cortical Impact Traumatic Brain Injury Acutely Disrupts Wakefulness and Extracellular Orexin Dynamics as Determined by Intracerebral Microdialysis in Mice. Journal of Neurotrauma, 29(10), 1908-1921. [CrossRef]
- Wong, J. C., Shapiro, L., Thelin, J. T., Heaton, E. C., Zaman, R. U., D’Souza, M. J., Murnane, K. S., & Escayg, A. (2021). Nanoparticle encapsulated oxytocin increases resistance to induced seizures and restores social behavior in Scn1a-derived epilepsy. Neurobiology of Disease, 147, 105147. [CrossRef]
- Wu, G., Zhang, X., Li, S., Zhou, D., Bai, J., Wang, H., & Shu, Q. (2022). Overexpression of ORX or MCH Protects Neurological Function Against Ischemic Stroke. Neurotoxicity Research, 40(1), 44-55. [CrossRef]
- Xiong, X., White, R. E., Xu, L., Yang, L., Sun, X., Zou, B., Pascual, C., Sakurai, T., Giffard, R. G., & Xie, X. (Simon). (2013). Mitigation of Murine Focal Cerebral Ischemia by the Hypocretin/Orexin System is Associated With Reduced Inflammation. Stroke, 44(3), 764-770. [CrossRef]
- Xu, D., Kong, T., Cheng, B., Zhang, R., Yang, C., Chen, J., & Wang, C. (2021). Orexin-A alleviates cerebral ischemia-reperfusion injury by inhibiting endoplasmatic reticulum stress-mediated apoptosis. Molecular Medicine Reports, 23(4), 266. [CrossRef]
- Xu, D., Kong, T., Shao, Z., Liu, M., Zhang, R., Zhang, S., Kong, Q., Chen, J., Cheng, B., & Wang, C. (2021). Orexin-A alleviates astrocytic apoptosis and inflammation via inhibiting OX1R-mediated NF-κB and MAPK signaling pathways in cerebral ischemia/reperfusion injury. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1867(11), 166230. [CrossRef]
- Xu, D., Kong, T., Zhang, S., Cheng, B., Chen, J., & Wang, C. (2021). Orexin-A protects against cerebral ischemia-reperfusion injury by inhibiting excessive autophagy through OX1R-mediated MAPK/ERK/mTOR pathway. Cellular Signalling, 79, 109839. [CrossRef]
- Xu Y., Miao J., Jia Y., Dong W., & Xie P. (2011). Expression of the orexinergic system in ischemic cerebral injury and the modulation of the cerebellar fastigial nucleus through electrical stimulation. Chinese Journal of Physical Medicine and Rehabilitation, 100-105.
- Yamanaka, A., Beuckmann, C. T., Willie, J. T., Hara, J., Tsujino, N., Mieda, M., Tominaga, M., Yagami, K. ichi, Sugiyama, F., Goto, K., Yanagisawa, M., & Sakurai, T. (2003). Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron, 38(5), 701-713. [CrossRef]
- Yao, P., Zhou, Q., Ren, B., Yang, L., Bai, Y., & Feng, Z. (2025). Transcranial pulsed current stimulation alleviates neuronal pyroptosis and neurological dysfunction following traumatic brain injury via the orexin-A/NLRP3 pathway. Neuropeptides, 110, 102501. [CrossRef]
- Yenari, M. A., & Han, H. S. (2012). Neuroprotective mechanisms of hypothermia in brain ischaemia. Nature Reviews. Neuroscience, 13(4), 267-278. [CrossRef]
- Yoshida, Y., Fujiki, N., Nakajima, T., Ripley, B., Matsumura, H., Yoneda, H., Mignot, E., & Nishino, S. (2001). Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. The European Journal of Neuroscience, 14(7), 1075-1081. [CrossRef]
- Yuan, L., Dong, H., Zhang, H.-P., Zhao, R., Gong, G., Chen, X., Zhang, L., & Xiong, L. (2011). Neuroprotective Effect of Orexin-A Is Mediated by an Increase of Hypoxia-inducible Factor-1 Activity in Rat. Anesthesiology, 114(2), 340. [CrossRef]
- Zawilska, J. (2015). A new face of orexins action—Neuroprotection. SpringerPlus, 4(1), L59. [CrossRef]
- Zeitzer, J. M., Buckmaster, C. L., Parker, K. J., Hauck, C. M., Lyons, D. M., & Mignot, E. (2003). Circadian and homeostatic regulation of hypocretin in a primate model: Implications for the consolidation of wakefulness. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 23(8), 3555-3560. [CrossRef]
- Zhang, D., Cui, Y., Zhao, M., Zheng, X., Li, C., Wei, J., Wang, K., & Cui, J. (2022). Orexin-A exerts neuroprotective effect in experimental intracerebral hemorrhage by suppressing autophagy via OXR1-mediated ERK/mTOR signaling pathway. Frontiers in Cellular Neuroscience, 16. [CrossRef]
- Zhang, D., Perrey, D. A., Decker, A. M., Langston, T. L., Mavanji, V., Harris, D. L., Kotz, C. M., & Zhang, Y. (2021). Discovery of Arylsulfonamides as Dual Orexin Receptor Agonists. Journal of Medicinal Chemistry, 64(12), 8806-8825. [CrossRef]
- Zhang, Y., Song, D., Liu, R., Gong, H., Wang, C., Zhao, M.-G., & Zhang, K. (2025). Transcutaneous auricular vagus nerve stimulation inhibits food intake and body weight gain through the orexin dependent pathway in high fat diet mice. Scientific Reports, 15(1), 19286. [CrossRef]
- Zhao, D., Zeng, Z., Mo, H., Hu, W., Tian, S., Hu, D., Gong, L., & Hu, K. (2021). Orexin-A inhibits cerebral ischaemic inflammatory injury mediated by the nuclear factor-κB signalling pathway and alleviates stroke-induced immunodepression in mice. Brain Research Bulletin, 174, 296-304. [CrossRef]
- Zhong, Y.-J., Feng, Z., Wang, L., & Wei, T.-Q. (2015). Wake-promoting actions of median nerve stimulation in TBI-induced coma: An investigation of orexin-A and orexin receptor 1 in the hypothalamic region. Molecular Medicine Reports, 12(3), 4441-4447. [CrossRef]
- Zhu, M., Li, X., Guo, J., Zhang, Z., Guo, X., Li, Z., Lin, J., Li, P., Jiang, Z., & Zhu, Y. (2024). Orexin A protects against cerebral ischemia-reperfusion injury by enhancing reperfusion in ischemic cortex via HIF-1α-ET-1/eNOS pathway. Brain Research Bulletin, 218, 111105. [CrossRef]
Figure 1.
Proposed mechanistic framework for orexin-mediated recovery after acute CNS injury. Integrative model summarizing convergent findings across ischemic, traumatic, and systemic insults. Orexinergic activation supports early metabolic preservation (↑ glucose use, ↑ mitochondrial efficiency), barrier stabilization (↑ tight-junction integrity, ↓ MMP-9), and neuroimmune modulation (↓ NF-κB/ROS, ↓ cytokines), thereby limiting secondary injury and autonomic collapse. In later phases, balanced OX₁R/OX₂R signaling promotes synaptic plasticity, arousal and motivational recalibration, and behavioral recovery. The framework is hypothetical, integrating experimental evidence rather than depicting a confirmed pathway. Abbreviations: LH, lateral hypothalamus; LC, locus coeruleus; DR, dorsal raphe; TMN, tuberomammillary nucleus; PFC, prefrontal cortex; NAc, nucleus accumbens; BBB, blood–brain barrier; BSCB, blood–spinal cord barrier; ROS, reactive oxygen species; NF-κB, nuclear factor κB; MMP-9, matrix metalloproteinase-9; OX₁R/₂R, orexin receptor 1/2. Created in BioRender.
Figure 1.
Proposed mechanistic framework for orexin-mediated recovery after acute CNS injury. Integrative model summarizing convergent findings across ischemic, traumatic, and systemic insults. Orexinergic activation supports early metabolic preservation (↑ glucose use, ↑ mitochondrial efficiency), barrier stabilization (↑ tight-junction integrity, ↓ MMP-9), and neuroimmune modulation (↓ NF-κB/ROS, ↓ cytokines), thereby limiting secondary injury and autonomic collapse. In later phases, balanced OX₁R/OX₂R signaling promotes synaptic plasticity, arousal and motivational recalibration, and behavioral recovery. The framework is hypothetical, integrating experimental evidence rather than depicting a confirmed pathway. Abbreviations: LH, lateral hypothalamus; LC, locus coeruleus; DR, dorsal raphe; TMN, tuberomammillary nucleus; PFC, prefrontal cortex; NAc, nucleus accumbens; BBB, blood–brain barrier; BSCB, blood–spinal cord barrier; ROS, reactive oxygen species; NF-κB, nuclear factor κB; MMP-9, matrix metalloproteinase-9; OX₁R/₂R, orexin receptor 1/2. Created in BioRender.
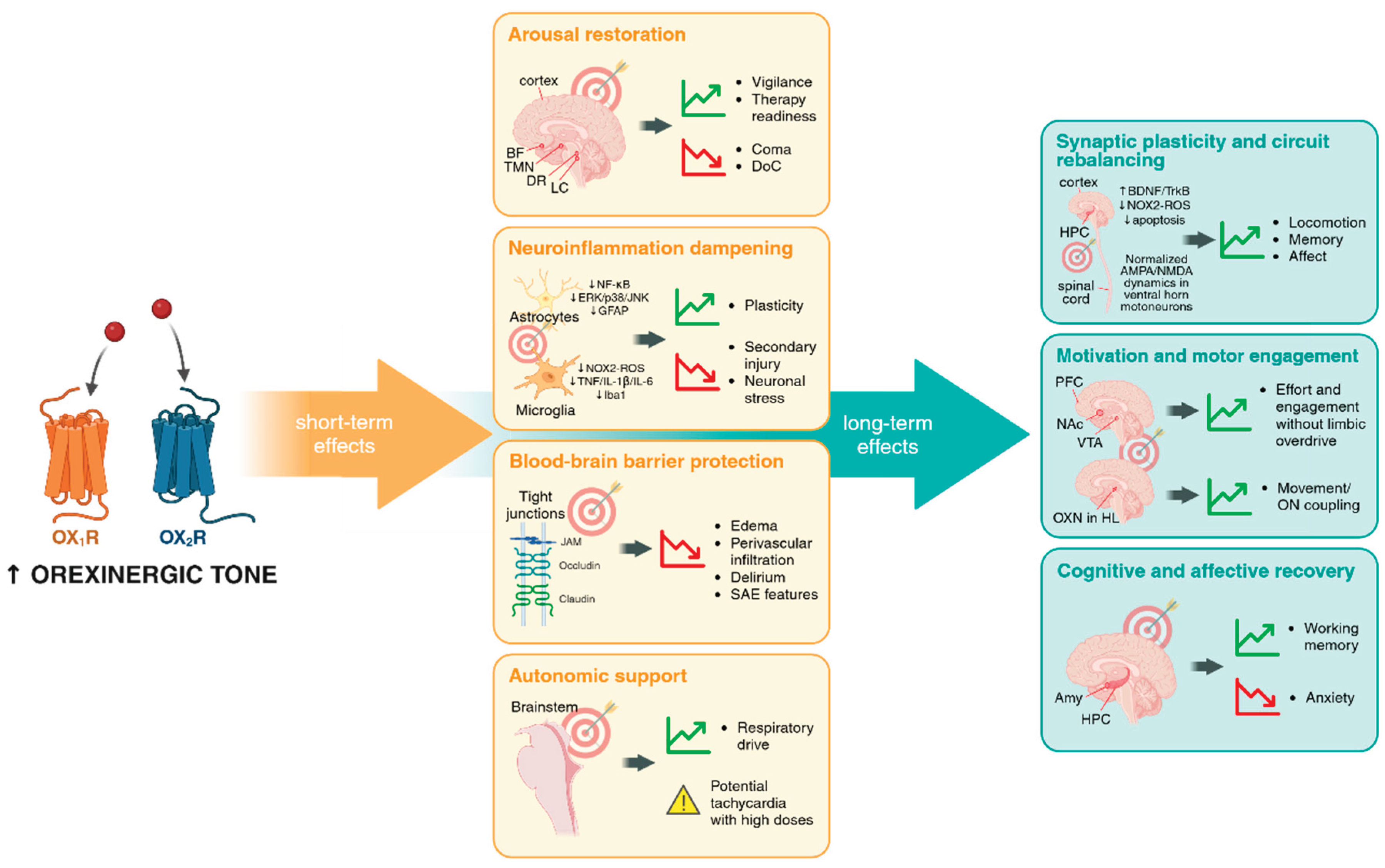

Table 1.
Strategies to enhance orexinergic tone for therapeutic application after acute CNS injury. Summary of current and emerging approaches aimed at increasing central orexin signaling. Each strategy is described by its main pharmacological or physiological features, stage of development, advantages, limitations, and potential utility in acquired CNS injury. Abbreviations: BBB, blood–brain barrier; OXA, orexin-A; OX1R/2R, orexin receptor 1/2; IN, intranasal; SCI, spinal cord injury; MNS, median-nerve stimulation; VNS, vagus-nerve stimulation; DBS, deep-brain stimulation; AAV, adeno-associated virus.
Table 1.
Strategies to enhance orexinergic tone for therapeutic application after acute CNS injury. Summary of current and emerging approaches aimed at increasing central orexin signaling. Each strategy is described by its main pharmacological or physiological features, stage of development, advantages, limitations, and potential utility in acquired CNS injury. Abbreviations: BBB, blood–brain barrier; OXA, orexin-A; OX1R/2R, orexin receptor 1/2; IN, intranasal; SCI, spinal cord injury; MNS, median-nerve stimulation; VNS, vagus-nerve stimulation; DBS, deep-brain stimulation; AAV, adeno-associated virus.
| Strategy | Main features | Development stage | Advantages | Limitations | Potential in acute CNS injury |
|---|---|---|---|---|---|
| Synthetic small-molecule agonists (OX2R selective) | Oral/IV, BBB-penetrant; phase 2–3 efficacy in narcolepsy | Late-stage clinical | Drug-like; scalable; robust wake-promoting effects; clear PK; feasible in rodents & humans. | Only OX2R (no OX1R agonists yet); risk of insomnia, CV load; timing critical. | IV: arousal rescue in coma/ICU. Oral: time-locked to rehab to sustain vigilance and engagement. |
| Peptide replacement (orexin-A/B) | Non-invasive, tested in humans (pilot narcolepsy studies, healthy volunteers); robust preclinical benefit in injury models | Early translational | Engages both receptors; strong biological effect; non-invasive (IN). | Short half-life; repeated dosing; variable nasal deposition. | Acute rescue of arousal; peri-rehab adjunct to enhance plasticity, cognition, and BBB protection. |
| Advanced delivery systems (nano, liposomes, exosomes, BBB-shuttles) | Liposomes, nanoparticles, exosomes tested with other peptides (oxytocin, TRH, PACAP) | Preclinical (conceptual for orexin) | Stabilise peptides; prolong CNS action; targeting possible. | No orexin-specific primaries (post-2018); regulatory and manufacturing barriers. | Could enable sustained intranasal OXA or regional spinal depots in SCI. Proof-of-concept only. |
| Endogenous modulation (dietary/behavioral) | Exercise, diet (BCAA), metabolic cues | Preclinical/observational | Accessible, low-cost, scalable; aligns with rehab (mobilisation, exercise, diet). | Nonspecific; state-dependent; effects variable. | Exercise/diet as orexin primers; potential adjunct to pharmacology in rehab protocols. |
| Neuromodulation (MNS, VNS, DBS) | MNS/VNS tested in animals & humans (non-orexin fields), orexin mechanism shown in models | Experimental/Clinical (for other indications) | Provides causal proof; circuit-specific; inspires hybrid strategies. | Invasive (DBS/VNS); protocols not standardised; mostly animal models. | Proof-of-principle; could combine with low-dose agonists or IN-OXA to maximise benefit. |
| Cell/gene therapy replacement | Orexin grafts restore cataplexy in narcoleptic mice; AAV orexin reduces cataplexy | Preclinical/Translational (for narcolepsy) | Potential durable restoration of orexin tone. | Invasive; integration/tumorigenesis risks; distant from clinical use. | Unlikely near-term for CNS injury; future use may involve enhancing residual orexin neurons via vectors. |
Table 2.
Summary of clinical and preclinical studies reporting orexinergic alterations or interventions in models of acute CNS injury and systemic inflammatory or metabolic insults.
Table 2.
Summary of clinical and preclinical studies reporting orexinergic alterations or interventions in models of acute CNS injury and systemic inflammatory or metabolic insults.
| Disease context | Setting | Model / Condition | Intervention | Assessment phase | Primary outcomes | References |
|---|---|---|---|---|---|---|
| Ischemic stroke | Clinical | Acute ischemic stroke | N/A | Acute phase (1 - 30 dpi) |
• ↓ CSF and serum Orexin-A, correlated with poor sleep quality and neurological deficit. Excess of Orexin-A associated with post-stroke insomnia | (Fu et al., 2025; Hu et al., 2023; Kotan et al., 2013; Scammell et al., 2001) |
| Preclinical | CCAO MCAO |
N/A | Acute phase (1- 7 dpi) |
• ↑ cell death • ↑ OX1R expressed in neurons, astroglia and oligodendroglia within lesion core |
Irving et al., 2002; Nakamachi et al., 2005 | |
| BCAO MCAO 4-Vo |
icv Orexin-A pre- or post-injury | Acute phase (1 - 9 dpi) |
Orexin-A ameliorates stroke consequences: • ↓ infarct volume, ↓ neurological deficit, ↓ pain, ↑ blood pressure, flow and heart rate • Neuroprotection: ↓ apoptosis, ↓ autophagy in vitro and in vivo (via ↓ p-ERK1/2 and ↑ p-mTOR), ↑ OXR1, ↑ HIF-1α, eNOS, NO and BDNF expression, ↓ ET-1 expression • ↓ neuroinflammation: ↓ excessive activation of astrocytes, ↓ pro-inflammatory cytokines, NF-κB p65 pathway and MAPK pathway |
Kitamura et al., 2010; Matsuura et al., 2020; C.-M. Wang et al., 2017; Xiong et al., 2013; D. Xu, Kong, Cheng, et al., 2021; D. Xu, Kong, Shao, et al., 2021; D. Xu, Kong, Zhang, et al., 2021; Yuan et al., 2011; Zhao et al., 2021; Zhu et al., 2024 | ||
| MCAO CCAO |
Sleep deprivation pre-injury | Acute phase (3 - 7 dpi) |
Sleep deprivation ameliorates stroke consequences: • ↓ infarct volume, ↑ REM • ↓ upregulation of genes related to immune response and cell division |
Pace et al., 2015, 2017 | ||
| MCAO | FNS | Acute phase (1 - 24 hpi) |
FNS preconditioning ameliorates stroke consequences: • ↑ prepro-orexin Orexin-A in hypothalamus, ↓ OX1R |
(Xu Y. et al., 2011) | ||
| MCAO | Orexin overexpression plasmid 3 days before MCAO | Acute/Subacute phase (1 - 10 dpi) | Plasmid ameliorates stroke consequences: • ↓ infarct volume, ↑ sleep structure and neurological function (via Glu uptake and GABA levels) • Neuroprotection: ↓ neuronal apoptosis |
Wu et al., 2022 | ||
| MCAO | iv parecoxib (COX2 inhibitor) | Acute phase (72 hpi) |
Parecoxib ameliorates stroke consequences: • ↑ orexin positive cells and orexin levels in hypothalamus |
(F.-T. Li et al., 2018) | ||
| Hemorrhagic stroke | Clinical | ICH SAH |
N/A | Acute/Subacute phase (1 - 36 dpi) | • ↓ CSF orexin-A, correlated to ↓ consciousness and ↑ neurological deficit | (Ang et al., 2005; Dohi et al., 2005, 2008; Rejdak et al., 2005) |
| Preclinical | Injection of autologous blood into basal ganglion (ICH) | Intranasal or icv Orexin-A | Acute/Subacute/ Chronic phase (1 - 28 dpi) |
Orexin-A ameliorates ICH consequences: • ↓ neurological deficit, ↓ brain water content • Neuroprotection: ↓ neuronal damage, ↓ autophagy (via ERK/mTOR pathway), ↑ p-CaMKKβ, p-AMPK, and anti-inflammatory cytokines, ↓ p-NF-κB and pro-inflammatory cytokines |
(T. Li et al., 2020; D. Zhang et al., 2022) | |
| Cardiac arrest (CA) | Preclinical | ACA Major cardiac vessels compression (transient global ischaemia) |
N/A | Acute phase (0 - 7 dpi) |
• ↑ cell death. Better neurological function correlated with ↑ HR post-resuscitation, ↑ LF/HF ratio and ↑ gamma band power • ↑ CFS Orexin-A at 24 hpi but ↓ CSF Orexin-A at 2 and 4 dpi |
(Dohi et al., 2006; Y. Guo et al., 2023) |
| ACA | icv or intranasal Orexin-A 20-30 min after resucitation | Acute (0 - 72 hpi) and Subacute phase (12 dpi) | Orexin-A meliorates CA consequences: • ↓ neurological deficit, ↑ EEG entropy (IQ values), ↑ arousal (EEG gamma fraction) • ↑ OX1R expression • ↓ neuroinflammation: ↓ TNF-a, iNOS, CD11b |
Akbari et al., 2012; Koenig et al., 2009; Modi et al., 2017; Sherman et al., 2021 | ||
| ACA | ip suvorexant (OX1/OX2 antagonist) | Acute phase (0 - 72 hpi) |
Suvorexant has detrimental effects: • No neurological recovery, ↓ HR, ↓ LF/HF ratio |
(Y. Guo et al., 2024; Y.-J. Kang et al., 2017 | ||
| ACA | Caloric restriction O/N pre-injury | Acute phase (0 - 72 hpi) |
Caloric restriction ameliorates CA consequences: • ↓ neurological deficit • ↓ stress-induced hyperglycemia, ↑ blood ketone levels, no change in SIRT-1 • Neuroprotection: ↓ neurodegeneration, no change in BDNF |
(Azadian et al., 2021) | ||
| Hypoxia-Ischemia | Preclinical | Transient focal ischaemia in vivo | Orexin-A | Acute phase | Orexin-A ameliorates hypoxia-ischaemia consequences: • ↓ infarct volume • Neuroprotection: ↓ ROS accumulation and neuronal death, ↑ PI3K/Akt survival pathway |
(Palomba et al., 2020) |
| Cobalt chloride on primary cortical neuronal cell culture | Orexin-A/B | Acute phase (24 - 48 hpi) |
Orexin-A/B ameliorate hypoxia-ischaemia consequences: • Neuroprotection: ↑ neuronal viability (via Akt activation) |
(Sokołowska et al., 2014) | ||
| Chemical hypoxia | Orexin-A/B | Acute phase | Orexin-A/B ameliorate hypoxia-ischaemia consequences: • Neuroprotection: ↑ neuronal viability, ↓ oxidative stress (via MEKK and Akt activation) |
(Zawilska, 2015) | ||
| TBI | Clinical | Moderate to severe TBI | N/A | Acute (0 - 14 dpi), chronic (6 mpi) or post-mortem phase | • ↓ CSF orexin-A, correlated to ↑ daytime sleepiness and predictive of poor outcome | (Baumann et al., 2005, 2007, 2009; Cangiano-Heath et al., 2020) |
| Preclinical | CCI Fluid perfusion injury |
N/A | Acute/Subacute phase (1 - 30 dpi) |
• ↑ cognitive deficit, ↓ wakefulness, ↑ REM and NREM sleep, ↑ depressive-like symptoms, ↓ motor activity • ↓ Orexin-A, orexinergic neurons and orexinergic activity, • ↑ astrogliosis |
(Mihara et al., 2011; Skopin et al., 2015; Somach et al., 2023; Thomasy et al., 2017; Thomasy & Opp, 2019; Willie et al., 2012) | |
| Modified Feeney's method | icv orexin-A post-injury | Acute phase (12 - 72 hpi) |
Orexin-A ameliorates TBI consequences: • ↓ neurological deficit, ↓ lesion volume • Neuroprotection: ↓ neuronal damage, ↓ ferroptosis (via Nrf2/HO-1 pathway) • ↓ pro-inflammatory citokines |
(J. Kang, Ren, et al., 2024) | ||
| CCI | DORA (dual orexinergic antagonist) | Acute/Subacute/ Chronic (1 week - 3 mpi) |
Orexinergic inhibition contributed to TBI consequences: • ↑ sleep fragmentation, ↑ sleepines |
(Craig et al., 2024) | ||
| CCI | tPCS post-injury | Acute phase (1 - 7 dpi) |
tPCS ameliorates TBI consequences: • ↓ neurological, motor and cognitive deficit • ↑ orexin-A • Neuroprotection: ↓ tissue damage All effects are OX1R-dependent |
(Yao et al., 2025) | ||
| Free fall drop / Modified Feeney's method |
VNS post-injury MNS post-injury TNS post-injury |
Acute phase (1 - 24 hpi) |
VNS, MNS and TNS ameliorate TBI consequences: • ↑ consciousness (via ↑ RasGRF1 pathway), ↓ neurological deficit, ↓ brain edema • ↑ orexin-A and OX1R expression, • Neuroprotection: ↓ brain cellular and tissue damage, ↓ neuronal pyroptosis (via ↓ NLRP3/Caspase-1/GSDMD) • ↓ pro-inflammatory cytokines, ↓ TLR4/NF-κB/NLRP3 inflammasome All effects are OX1R-dependent |
(X. Dong et al., 2018; X.-Y. Dong & Feng, 2018; Du et al., 2022; J. Kang, Zhou, et al., 2024; J. Kang et al., 2025; Zhong et al., 2015) | ||
| Free fall drop | LH-DBS post-injury | Acute phase (1 - 12 hpi) |
LH-DBS ameliorates TBI consequences: • ↑ consciousness • ↑ orexin-A/OX1R expression All effects are OX1-dependent |
(X. Dong et al., 2021) | ||
| Free fall drop | LIFUS post-injury | Acute phase (3 dpi) |
LIFUS ameliorates TBI consequences: • ↓ neurological deficits, ↓ brain edema • ↑ Orexin-A and OX1R expression • Neuroprotection: ↓ tissue damage, ↓ necrotic neuronal degeneration • ↓ pro-inflammatory cytokines, ↓ NF-κB/NLRP3 inflammasome |
(Huang et al., 2022) | ||
| Fluid percussion injury | BCAA dietary supplementation | Acute/Subacute/ Chronic (4 - 30 dpi) |
BCAA ameliorates TBI consequences: • ↑ wakefulness, ↓ fragmented wake bouts ↑ Orexin neurons activation (via ↑ glutamatergic presynaptic density) |
(Elliott et al., 2018; Lim et al., 2013) | ||
| Repeated mild TBI | Chronic ICV orexin-A (daily, 5–33 dpi) | Chronic phase (2–4 wpi) | • ↓ orexin+ neurons, ↓ orexinergic projections to PAG • ↑ nociceptive sensitivity, ↓ anxiety-like behavior Orexin-A worsened outcomes: • ↑ anxiety and motor deficits • ↑ CSF metabolomic disruption |
(Christensen et al., 2023, 2024) | ||
| SCI | Preclinical | Complete spinal cord transection at T9 | intrathecal orexin-A post-Injury | Acute phase (1 - 7 dpi) |
Orexin-A ameliorates SCI consequences: • ↑ motor recovery • ↓ overactive/dysregulated Glu dynamics |
(He et al., 2025) |
| Systemic biological and toxic insults | Clinical | Meningitis, Encephalitis |
N/A | Acute phase (1 - 20 dpi) |
• ↓ CSF orexin-A levels • ↓ BBB integrity (Orexin-A leaked into the blood) |
(Ogawa et al., 2021, 2022) |
| Preclinical | ip LPS in WT and/or orexin-ablated (OX/AT3 TG) mice | N/A | Acute phase (1 - 3 dpi) |
• ↓ gait speed and exploration, ↑ nREM sleep, ↓ wakefulness • ↓ CSF orexin-A levels, ↓ orexin neurons, ↓ prepro-orexin expression, ↓ Fos expression in orexin neurons • ↑ cytokines In orexin-ablated mice: • ↓ survival |
(Gaykema & Goehler, 2009; Hirota et al., 2018; Ogawa et al., 2022; Perekrest et al., 2009, 2011, 2013; Takekawa et al., 2018; Tanaka et al., 2015, 2016) | |
| Acute pancreatitis (non-infectious) | N/A | Acute phase (12-24 hpi) |
• ↑ brain levels of β-endorphin, orexin, and oxytocin Neuropeptide upregulation occurred before local cytokine increase or microglial activation |
(Hamasaki et al., 2016b) | ||
| Cecal ligation & puncture | Icv or intranasal Orexin-A post-injury | Acute phase (1 - 7 dpi) |
Orexin-A ameliorates sepsis consequences: • ↑ survival, ↓ cognitive and emotional deficit, ↓ brain edema, ↑ responsiveness, ↑ pituitary function • ↓ tissue damage, ↓ BBB disruption, • ↓ neuroinflammation: ↓ pro-inflammatory citokines, ↓ microglia activation Effects mediated via OX2R and RAS/MAPK pathway |
(Deutschman et al., 2013; J. Guo et al., 2024) | ||
| Cecal ligation & puncture | ip xanomeline (mAChR agonist) post-injury | Acute phase (48 hpi) |
Xanomeline ameliorates sepsis consequences: • ↑ orexinergic activity, ↑ temp, HR and RR • ↑ pituitary function • ↓ pro-inflammatory cytokines Effects reversed by OX1/2R antagonist (almorexant) |
(Nedeljkovic-Kurepa et al., 2025) | ||
| LPS-induced endotoxemia | ict, ip or sc orexin-A post-injury | Acute phase (1-3 dpi) |
Central, peripheral orexin-A (crosses BBB in endotoxemia): • ↑ survival • restores body temperature, cardiovascular tone • ↓ cytokine levels • ↑ catecholamines, ↑ corticosterone Effects blocked by OX1R antagonist, vagotomy, or atropine |
(Igarashi et al., 2020; Ogawa et al., 2016) | ||
| Alcohol-induced coma | icv orexin-A or orexin-B | Acute phase (hpi) | Orexin-A/B ameliorates intoxication consequences: • ↓ coma duration, restores righting reflex • ↓ delta power in EEG, ↑ prefrontal cortex activity |
(Jia et al., 2012) |
Abbreviations: ACA = Asphyxial cardiac arrest; BCAA = Branched-chain amino acids; BBB = Blood–brain barrier; BDNF = Brain-derived neurotrophic factor; BCAO = Bilateral common carotid artery occlusion; CCAO = Common carotid artery occlusion; CCI = Controlled cortical impact; CLP = Cecal ligation and puncture; CNS = Central nervous system; dpi = days post-injury; EEG = Electroencephalogram; ERK = Extracellular signal-regulated kinase; FNS = Facial nerve stimulation; HR = Heart rate; hpi = hours post-injury; icv = Intracerebroventricular; ict = Intracisternal; ICH = Intracerebral hemorrhage; i.p. = Intraperitoneal; IV = Intravenous; LF/HF = Low frequency/high frequency heart rate variability ratio; LIFUS = Low-intensity focused ultrasound; LPS = Lipopolysaccharide; MCAO = Middle cerebral artery occlusion; MNS = Median nerve stimulation; mPI = months post-injury; mTBI = mild traumatic brain injury; NF-κB = Nuclear factor kappa B; NLRP3 = NOD-, LRR- and pyrin domain-containing protein 3; NO = Nitric oxide; NREM = Non-rapid eye movement sleep; O/N = Overnight; OX1R/OX2R = Orexin receptor 1/2; PAG = Periaqueductal gray; RAS = Renin–angiotensin system; REM = Rapid eye movement sleep; SAH = Subarachnoid hemorrhage; SCI = Spinal cord injury; sc = Subcutaneous; subPI = subacute post-injury; suv = Suvorexant; tPCS = Transcranial pulsed current stimulation; TBI = Traumatic brain injury; TLR4 = Toll-like receptor 4; TNS = Trigeminal nerve stimulation; VNS = Vagus nerve stimulation; wpi = weeks post-injury; WT = Wild type.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



