
Submitted:
08 November 2025
Posted:
10 November 2025
You are already at the latest version
Abstract
Heart failure (HF) is often accompanied by cognitive and mood disturbances, yet the underlying neurobiological mechanisms remain unclear. Oxytocin (OT) signaling in the hypothalamus–amygdala axis, particularly within the central amygdala (CeA), is known to regulate mood and anxiety-like behaviors. Using an ischemic rat model of HF, we investigated whether altered OTergic signaling contributes to mood dysfunction. We found that CeA OTergic innervation primarily originates from the supraoptic nucleus (SON). Although SON→CeA OT neuron numbers were unchanged, OT content and release at CeA terminals were reduced, accompanied by downregulation of neuronal and astrocytic OT receptors and impaired OT receptor–driven GABAergic transmission. These findings reveal that HF disrupts hypothalamus-to-amygdala OTergic circuitry, suggesting that targeting OT signaling or amygdalar GABAergic function may help mitigate mood impairments in HF.
Keywords:
heart failure
; congnitive impairment
; oxytocin
Chronic Heart Failure attenuates Oxytocin Signaling in the Central Amygdala.
Chronic heart failure (CHF) is one of the leading public health concerns throughout the world. Globally, around 64 million people are suffering from CHF [1]. In the United States, approximately 6.7 million people over the age of 20 years, are suffering from heart failure, and the number is expected to increase to 8.7 million by 2030 [2]. Classical manifestations of CHF include neurohormonal imbalance, in particular; increased sympathetic activity and elevated circulating levels of vasopressin (VP) and oxytocin (OT), atrial natriuretic peptide (ANP), angiotensin II, aldosterone, and catecholamines [3,4,5,6]. Hence, most of the previous studies related to CHF are focused on the mechanisms of these neurohormonal activations.
In recent years, accumulating evidence in the clinical literature has demonstrated that the late stage of CHF is closely associated with anxiety [7], depression [8], and cognitive impairments [9,10]. Similarly, rat model of CHF has been reported to be associated with cognitive disorders, including elevated anxiety and anhedonia [11,12]. However, the precise mechanism contributing to this anxiety, depression, and cognitive impairment in the late stage of CHF remains unknown.
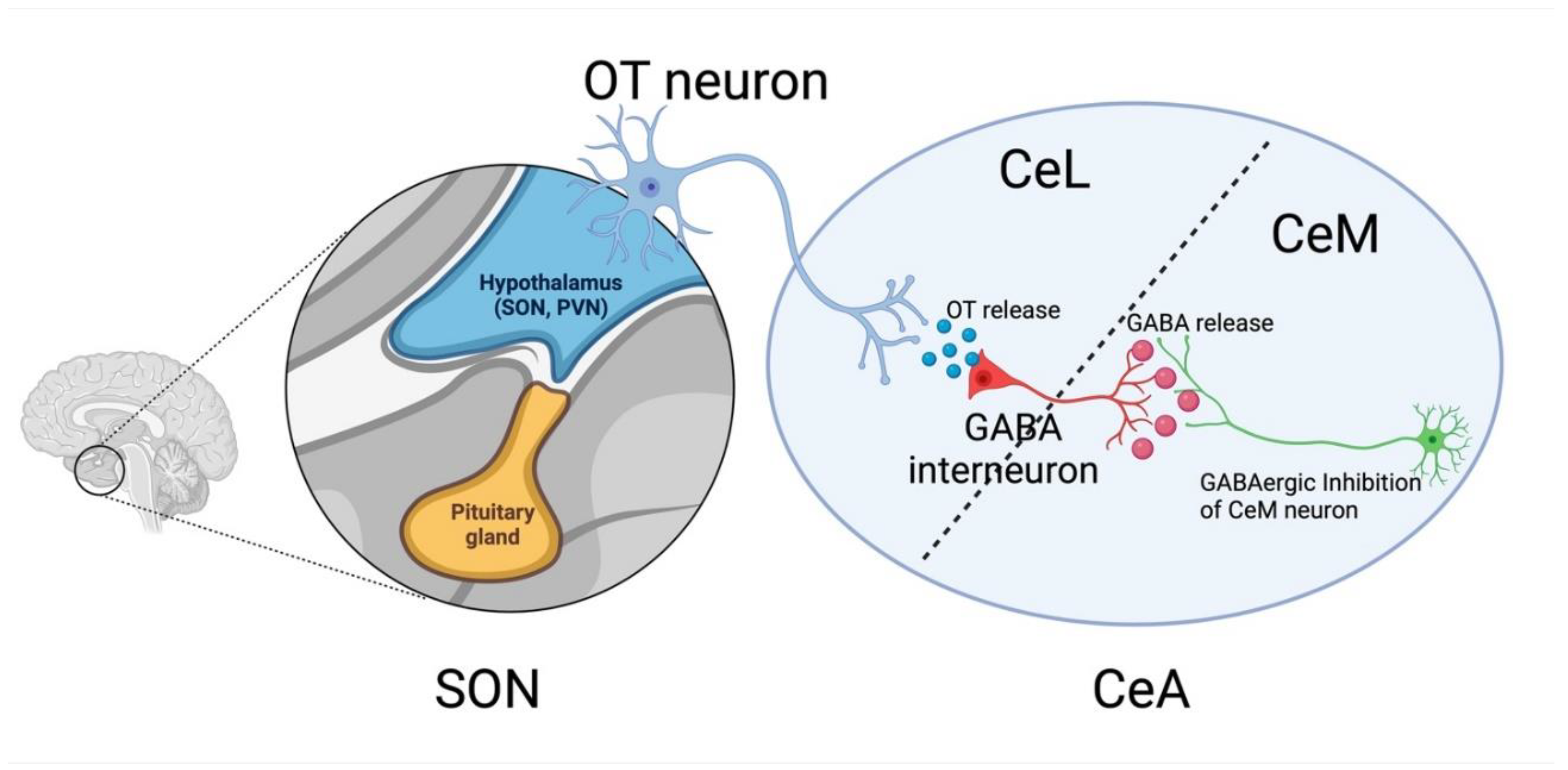
OT plays a critical role in the context of social and emotional cues and has been shown to have anxiolytic and antidepressant effects [13,14]. The action of OT related to this behavioral effect is predominantly reported in the central nucleus of the amygdala (CeA) [15], where OT regulates potent GABAergic circuits [16]. OT neurons in PVN and SON project to the CeA. Upon activation of this CeA-projecting OT neurons, it releases OT to GABAergic interneurons located in the lateral portion of the central nucleus of the amygdala (CeL). GABAergic interneurons in CeL project to the medial portion of the central nucleus of the amygdala (CeM). And hence, the OT release to the CeL results in robust GABAergic inhibition of the CeM [17,18] (Figure 1), which produces a robust anxiolytic effect [16] and eliminates previous fearful memory [17]. To date, it remains to be elucidated whether the OT signaling mechanism in the CeA is altered in CHF, and thus contributes to the development of cognitive decline and mood disorder in CHF. Taken together, the authors in this paper [19] hypothesized that CHF is associated with depleted OT release and OT receptor expression in the CeA, thereby altering the GABAergic circuitry in the CeA.
To test the hypothesis, the authors first validated whether hypothalamic OT neurons project to the CeL in a rat model. To do so, they injected OT cell type-specific recombinant adeno-associated virus (AAV-OTp-Venus) into the SON to determine OT fiber density in the CeA. Results demonstrated that SON provides a significantly denser OT neuronal projection to the CeL.
Next, they investigated whether this denser SON OT neuronal projection to the CeL is altered in CHF rats compared to sham. To do so, they first generated CHF rat models by ligating the left anterior descending coronary artery followed by echocardiography (ejection fraction <40% was considered CHF).
At the level of SON:
Confocal microscopy revealed that there is no difference in the number of OT cells between sham and CHF, indicating CHF does not alter the OT neuronal expression in SON. Next, they investigated whether the number of CeL-projecting SON OT neurons is altered in CHF.
To investigate this, they performed dual viral tracer injections AAV_OTp-DIO-GFP into the SON, to label SON OT neurons; and CAV2-Cre in the CeL, to retrogradely label SON neurons. In both CHF and sham, a similar proportion (15%) of SON OT neurons project to the CeL, indicating that there is no difference in the number of SON OT neurons that project to the CeL between sham and CHF rat.
Furthermore, they investigated whether CHF altered the electrophysiological property of the CeL-projecting SON OT neurons. In this end, they performed patch clamp electrophysiology from the CeL-projecting SON OT neurons. Results demonstrated that the number of evoked action potentials per current pulse and the input resistance were not different between sham and CHF rats. Taken together, these results suggest that the total number and electrophysiological properties of the CeL-projecting SON OT neurons were not altered in CHF.
At the level of CeL:
Next, they investigated whether the innervation of the CeL-projecting SON OT neurons in CeL was altered in the CHF. Immunohistochemistry in the CeL brain sections revealed that OT immunoreactivity was significantly lowered in the CHF rats, indicating a reduction of the OT content at the axonal terminal in the CeL. To further strengthen these findings, they used complementary sniffer-OT biosensor cells to determine the activity-dependent OT release from CeL-projecting SON OT neuron fibers in CeL. Results demonstrated that in the CeL, Ca2+ responsiveness of the sniffer-OT cell was significantly lowered in the CHF rats compared to sham. Taken together, these results suggest that in the CeL, not only the OT content of the CeL-projecting SON OT terminals were reduced, but also the activity-dependent OT release from the CeL-projecting SON OT terminals was also reduced in the CHF compared to sham.
In addition to the decreased OT content and activity-dependent OT release, they next investigated whether CHF is associated with reduced OT receptors (OTRs) expression in the CeL. By measuring the mRNA expression using qPCR, it was revealed that CHF significantly reduced OTR mRNA expression in the CeL neurons. They also investigated whether this decreased OTR mRNA expression is reflected to the decreased (OTR) protein level. Results demonstrated that consistent with the decreased mRNA expression, CHF reduced the OTR protein in CeL. Taken together, these results indicate that CHF causes a blunted OT signaling mechanism in the CeL division of CeA.
At the level of CeM:
An activated CeL-projecting SON OT neurons release the OT to the CeL, which binds to the GABAergic interneurons and subsequently inhibits the neurons projecting to the CeM[16]. Here, they investigated whether CHF alters this local GABAergic circuitry dynamics. To do so, they implemented optogenetic stimulation coupled with electrophysiology. As expected, optogenetic stimulation of the SON OT neurons causes a robust hyperpolarization (inhibition) of the CeM neurons in sham rats. However, in CHF rats, similar optogenetic stimulations cause a blunted inhibition. In addition to the optogenetic stimulation, when CeL-projecting SON OT neurons were activated by OTR agonist (TGOT), they found a similar blunted inhibitory response in CHF. Taken together, these results indicate that CHF blunts the effect of OT in the CeM, most likely due to the reduced presynaptic OT release and/or reduced post-synaptic OTR expression.
Next, they investigated whether this blunted OT effect in the CeM was mediated by the activity of the GABAergic interneuron. To do so, they measured the GABAergic inhibitory post-synaptic current (IPSC) in CeM neurons. As expected, TGOT causes a robust increase in the amplitude and frequency of the GABAergic IPSCs in CeM of sham rats. However, this robust increase in the amplitude and frequency of the GABAergic IPSCs was significantly blunted in CHF rats. These results indicate that CHF blunts the OT-mediated CeL to CeM GABAergic transmission.
At the level of Astrocyte:
It was previously shown that astrocytic OTR contributes to the activation of CeL and inhibition of the CeM neurons[18]. This effect is mediated by the NMDA receptors in the CeL neurons[18]. The authors wanted to investigate whether OTR-astrocyte-CeL pathway is altered in CHF. To do so, they blocked NMDA receptors by amino-5-phosphonovaleric acid (APV) and then stimulated the CeL-projecting SON OT neurons by TGOT. TGOT induces a robust increase in GABA IPSCs in the CeM of sham rats. However, the robust increase in GABA IPSCs was severely blunted in CHF. APV did not prevent the blunted effect of TGOT, which indicates that the OT signaling mechanism in the astrocyte did not contribute to the OT-mediated blunted CeL to CeM GABAergic transmission observed in CHF.
A previous study demonstrated that altered GABAergic signaling in CeA contributes to cognitive decline [20]. Results presented in this study suggest that CHF causes disruption of OT-mediated CeL to CeM GABAergic signaling pathway in the CeA. These findings are significant because the OT-mediated GABAergic signaling pathway could emerge as the novel pathway for the development of cognitive decline in CHF. In the future, it can also serve as a therapeutic target for treating cognitive decline in CHF patients. However, one limitation is that the authors did not perform any behavioral experiments in this study. Therefore, it needs further behavioral studies to validate the behavioral consequences of this OT-mediated GABAergic signaling pathway. Moreover, CHF is associated with hypoxia [11], neuroinflammation[11], and elevated circulating peptides (VP[21], Angiotensin II [22]) all of these factors might contribute to the altered OT-mediated pathway presented in this study. The previous study demonstrated the presence of OT-mediated GABAergic signaling in CeA in both sexes [18]. However, only the male sex was studied in this paper. Therefore, further studies are needed to include both males and females to validate any possible sex differences.
Acknowledgments
I would like to thank Dr. Angela Mabb for her constructive feedback and guidance in preparation for this Journal Club Preview.
References
- Khan, M.S., et al. Global epidemiology of heart failure. Nature Reviews Cardiology, 1-18 (2024).
- Bozkurt, B., et al. HF STATS 2024: Heart Failure Epidemiology and Outcomes Statistics An Updated 2024 Report from the Heart Failure Society of America. J. Card. Fail., S1071-9164 (1024) 00232-X (2024).
- Sigurdsson, A., Held, P. & Swedberg, K. Short-and long-term neurohormonal activation following acute myocardial infarction. Am. Heart J. 126, 1068-1075 (1993).
- Persson, H., et al. Neurohormonal activation in heart failure after acute myocardial infarction treated with beta-receptor antagonists. Eur. J. Heart Fail. 4, 73-82 (2002).
- McAlpine, H., et al. Neuroendocrine activation after acute myocardial infarction. Heart 60, 117-124 (1988).
- Rouleau, J.L., et al. Activation of neurohumoral systems following acute myocardial infarction. The American journal of cardiology 68, 80-86 (1991).
- Konstam, V., Moser, D.K. & De Jong, M.J. Depression and anxiety in heart failure. J. Card. Fail. 11, 455-463 (2005).
- Rutledge, T., Reis, V.A., Linke, S.E., Greenberg, B.H. & Mills, P.J. Depression in heart failure: a meta-analytic review of prevalence, intervention effects, and associations with clinical outcomes. J. Am. Coll. Cardiol. 48, 1527-1537 (2006).
- Hammond, C.A., et al. Long-term cognitive decline after newly diagnosed heart failure: longitudinal analysis in the CHS (Cardiovascular Health Study). Circ. Heart Fail. 11, e004476 (2018).
- Leto, L. & Feola, M. Cognitive impairment in heart failure patients. Journal of geriatric cardiology: JGC 11, 316 (2014).
- Althammer, F., et al. Angiotensin II–mediated neuroinflammation in the hippocampus contributes to neuronal deficits and cognitive impairment in heart failure rats. Hypertension 80, 1258-1273 (2023).
- Parent, M.B., et al. Heart failure impairs mood and memory in male rats and down-regulates the expression of numerous genes important for synaptic plasticity in related brain regions. Behav. Brain Res. 414, 113452 (2021).
- Meyer-Lindenberg, A., Domes, G., Kirsch, P. & Heinrichs, M. Oxytocin and vasopressin in the human brain: social neuropeptides for translational medicine. Nature Reviews Neuroscience 12, 524-538 (2011).
- Neumann, I.D. & Slattery, D.A. Oxytocin in general anxiety and social fear: a translational approach. Biol. Psychiatry 79, 213-221 (2016).
- Huber, D., Veinante, P. & Stoop, R. Vasopressin and oxytocin excite distinct neuronal populations in the central amygdala. Science 308, 245-248 (2005).
- Knobloch, H.S., et al. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron 73, 553-566 (2012).
- Hasan, M.T., et al. A fear memory engram and its plasticity in the hypothalamic oxytocin system. Neuron 103, 133-146. e138 (2019).
- Wahis, J., et al. Astrocytes mediate the effect of oxytocin in the central amygdala on neuronal activity and affective states in rodents. Nat. Neurosci. 24, 529-541 (2021).
- Althammer, F., et al. Impaired oxytocin signalling in the central amygdala in rats with chronic heart failure. J. Physiol. 602, 6259-6280 (2024).
- Jie, F., et al. Stress in regulation of GABA amygdala system and relevance to neuropsychiatric diseases. Front. Neurosci. 12, 562 (2018).
- Iovino, M., et al. Vasopressin in Heart Failure. Endocr. Metab. Immune Disord. Drug Targets 18, 458-465 (2018).
- Opie, L.H. & Sack, M.N. Enhanced Angiotensin II Activity in Heart Failure. Circ. Res. 88, 654-658 (2001).
Figure 1.
Hypothalamic OT neuron-mediated GABAergic inhibition of the medial CeA neuron. (Figure is generated using Biorender).
Figure 1.
Hypothalamic OT neuron-mediated GABAergic inhibition of the medial CeA neuron. (Figure is generated using Biorender).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



