
Submitted:
27 October 2025
Posted:
28 October 2025
You are already at the latest version
Abstract
Background. Genetic studies justified that germline BRCA1 gene mutation is the origin of highly increased cancer risk. Clinical studies suggested that increased cancer risk in type-2 diabetes maybe attributed to unhealthy lifestyle factors and bad habits. Purpose. Patients with either BRCA1 gene mutation or type-2 diabetes similarly exhibit increased cancer risk, insulin resistance and fertility disorders. It was suggested that these three alterations derive from a common genomic failure and its recognition may shed light on the unsolved secret of cancer. Results. 1. Germline mutations on ESR1, BRCA1, and CYP19A genes encoding estrogen receptor alpha (ERα), genome safeguarding BRCA1 protein and CYP19 aromatase enzyme, cause genomic instability. BRCA1 and ESR1 gene mutations cause particularly breast cancer, while the error of CYP19A gene leads to cancers in the endometrium, ovaries and thyroid. 2. ERα, BRCA1 and CYP19 aromatase proteins are transcription factors creating the crucial DNA stabilizer circuit driven by estrogen regulation. Liganded ERα drives a second regulatory circuit for controlling cell proliferation as well, in partnership with various growth factors. In a third regulatory circuit, liganded ERα drives cellular glucose supply in close interplay with insulin, IGF-1 and glucose transporters. 3. Weakening expression or activation of each transcription factor of the triad, leads to defective estrogen signaling and endangers regular cell proliferation, insulin sensitivity and fertility. 4. Weakening estrogen signaling caused by either genetic or lifestyle factor, is alarming for the hypothalamus and it sends neural and hormonal commands throughout the body so as to restoring estrogen signaling. 5. When the compensatory actions cannot restore estrogen guideline, the breakdown of genomic regulation leads to cancer initiation. 6. Lifestyle factors upregulating estrogen signaling, decrease, while downregulating estrogen signaling increases the risk for cancer. Conclusion. Increased cancer risk, insulin resistance and infertility all are originating from defective estrogen signaling.
Keywords:
alcohol consumption
; DNA damage
; DNA repair
; endocrine disruptor
; estrogen
; estrogen receptor
; growth factor receptor
; immune reaction
; lifestyle factors
; mutation
; smoking
1. Introduction
Cancer is a major challenge globally, attributed to its rapidly increasing morbidity and mortality rates worldwide [1]. The mechanism of cancer initiation and progression is not exactly clarified until now and the results of enormous therapeutic efforts are fairly questionable.
According to the reigning opinion, cancer initiation derives from the incidence of oncogenic mutations that maybe either inherited, increasing the susceptibility, or acquired arising under harmful environmental exposure [2]. An accumulation of mutations in crucial genes presumably disrupt normal genomic regulation leading to unrestrained cell proliferation. The majority of cancers may develop through an unfortunate interaction between genetic predispositions and environmental influences [3]. Since only a small percentage of cancers exhibit solely inherited origin, small genetic differences among individuals may define the response to environmental exposures leading to increased or decreased risk for cancer.
Analysis of a huge number of cancer genomes resulted in around 300 genes that were mutated in at least one type of cancers [4]. Thoroughly examining the features of oncogenic mutations, their accurate roles in cancer development were not clarified. Increased prevalence of certain somatic mutations in the genes of various cancers cannot justify their oncogenic impact, but rather a genome repairing effort maybe supposed [5].
Historically, the first causal factor for cancer affecting the female breast in particular was introduced at the end of the 19th century. The clinically observed pulsation of tumors parallel with the menstrual cycle mistakenly suggested that breast cancer growth maybe associated with the ovulatory peak of serum estrogen level. High concentration of estrogen hormone emerged as being a unique causal factor for premenopausal breast cancer. Surgical oophorectomy was applied as a causal therapy of breast cancer targeting ovarian estrogen synthesis [6]. Nevertheless, the regression of breast tumors via surgical estrogen withdrawal was modest and transient [7].
The absurdity of estrogen induced breast cancer and the therapeutic blockade of estrogen signaling has remained a gold standard for breast cancer care through centuries until today. The concept of estrogen induced breast cancer yielded further mistakes in cancer research and fairly held back the understanding of the key to human health and the development of cancer therapy [8].
In 1988, a causal factor for human diseases (and later for cancer) was introduced into the scientific world. Gerald Reaven established and popularized his concept in his Banting Lecture that a defect of insulin assisted glucose uptake, nominated as insulin resistance is the source of human diseases [9]. Insulin resistance was linked with a cluster of metabolic disorders and they were summarized as Metabolic Syndrome or Syndrome X. Metabolic syndrome is characterized by impaired glucose metabolism, high blood pressure, central obesity, and low HDL-cholesterol coupled with increased triglyceride levels [10].
In the early stage of insulin resistance, compensatory hyperinsulinemia maintains the blood glucose levels within the physiological range via stimulating the insulin secretion of pancreatic β-cells [11]. Advanced insulin resistance leads to metabolic syndrome, obesity and nonalcoholic fatty liver disease [12]. Type-2 diabetes develops, when the compensatory insulin synthesis cannot keep up with the insulin demand of insulin resistant target cells leading to hyperglycemia [13]. Thorough investigations equivocally revealed that insulin resistance is the underlying disorder for developing cardiovascular diseases and cancers, which are the leading causes of human mortality [14,15]. Since insulin resistance leads to various, quite different chronic human conditions, clarifying its origin seemed to be crucial for the recognition of the principal regulator of human genome.
Insulin resistant status shows gender related differences as men exhibit a fairly higher prevalence of metabolic diseases as compared to women. In addition, among women, premenopausal cases are protected from the defect of glucose uptake via their active hormonal cycles, whilst postmenopausal cases exhibit a quite similar incidence rate of insulin resistance like age matched man [16]. These gender and menopause related differences in the prevalence of insulin resistant cases led to putting forward that estrogen hormone has crucial role in glucose homeostasis and energy balance [17]. Moreover, estrogen deficiency or impaired estrogen receptor activation seemed to be a causal factor for insulin resistance and the development of metabolic diseases [18,19].
Gender related differences are conspicuous in the prevalence of cardiovascular diseases as well [20]. Healthy premenopausal women are typically protected from cardiovascular diseases and hypertension compared to age matched men suggesting protective effects of cycling estrogen hormone levels [21]. Estrogen plays a crucial regulatory role in the health of cardiovascular system [22]. After menopause, decreasing estrogen levels and the associated insulin resistance are important causal factors in the pathogenesis of atherosclerosis, myocardial dysfunction, cardiac hypertrophy, heart failure, and myocardial ischemia.
Pan-cancer analyses support that men in general show a higher cancer incidence and worse survival than women across most cancer types [23]. These sex-based differences in cancer incidence are likely due to a combination of biological, environmental, and behavioral factors; however, a robust estrogen signaling in women seems to be protective against cancer. Oral cancer incidence shows conspicuous gender related differences [24]. Premenopausal women are highly protected from this tumor, while after menopause, estrogen loss is associated with a steeply increasing prevalence of the disease even in non smoker, non drinker cases.
Breast cancer incidence is significantly higher in postmenopausal women as compared with premenopausal cases showing increasing risk with age after menopause [25]. In postmenopausal women, the loss of estrogen increases the risk for type-2 diabetes and obesity and these chronic conditions are high risks for breast cancer.
During the fight against insulin resistance, clinical investigations revealed that certain lifestyle factors and bad habits may induce and worsen insulin resistance [26]. At the same time, positive life-style changes of patients and abandoning their bad habits are associated with surprising improvement in metabolic diseases and decreasing risk for cancer development [27].
Parallel with thorough investigations on the origin and progression of insulin resistance, there was a continuous fight against excessive estrogen signaling in breast cancer cases. Antiestrogens including estrogen receptor blockers and aromatase inhibitors were introduced against estrogen receptor positive (ER+) breast cancers [28]. Breast cancer therapy has remained a challenge until now as the half of targeted ER-positive breast cancers proved to be primarily non responsive to antiestrogen therapy. In addition, the earlier responsive tumors lost their responsiveness during the therapy [29]. This behavior of breast cancers was evaluated as primary and secondary therapy resistance. In reality, ER-positive tumors that are not responsive to the inhibitory therapy are not antiestrogen resistant. They are incapable of counteraction or exhausted in the fight against the genomic damage caused by the inadequate therapy [30].
The mechanism of insulin resistance induced oncogenesis has remained highly debated. Investigators follow the old erroneous theory suggesting that an increased concentration of certain molecular players and somatic mutations of their genes are causal factors for cancer initiation and progression. During the past decades, hyperinsulinemia [15], hyperglycemia [31], overexpression of proinflammatory cytokines [32] and increased growth factor signaling [33] were supposed as cancer initiators in insulin resistant cases. Although, the presence of these alterations is characteristic for insulin resistance, they may be regarded as compensatory efforts for restoring genomic functions, rather than facilitating cancer development [8].
This work introduces the defective estrogen signaling as the causal factor of cancer initiation and presents the wonderful genome-repairing efforts of healthy cells, tumor cells and the whole body in humans. These facts recommend that natural biotherapies are highly superior over the inhibition of genomic pathways and the blockade of immune responses.
2. Hyperinsulinemia Is Not the Predecessor of Insulin Resistance
There has been a continuous debate on the cause-effect relationship between insulin resistance and hyperinsulinemia; however, it can equivocally be established that their coexistence leads to serious chronic conditions and increasing mortality [34].
Until the beginning of the 21th century, insulin resistance itself was regarded as the origin of disorders leading to metabolic syndrome and type-2 diabetes, while the associated excessive insulin synthesis seemed to be a compensatory action helping cellular glucose uptake [35]. The origin of insulin resistance seemed to be variable, and the relative importance of each presumed causal factor has been under thorough investigation [36]. Searching for the causal factors of insulin resistance, authors focused on the role of inflammation, altered lipid metabolism, and the dysbiosis of gastrointestinal microbiota. Nevertheless, these factors are counteractions to insulin resistance, rather than being its initiators.
Recent studies established that hyperinsulinemia equivocally precedes insulin resistance becoming a causal factor for the defective glucose uptake, obesity, type-2 diabetes, cardiovascular diseases and cancer [37,38,39]. In PCOS (polycystic ovary syndrome) cases, hyperinsulinemia seemed to be the predecessor of insulin resistance creating a self-generating circuit for worsening the reproductive and metabolic defects [40]. Nevertheless, the initiator of either insulin resistance or hyperinsulinemia remained quite obscure.
Recently, genetic background together with over-nutrition seems to be responsible for inducing hyperinsulinemia that shows primacy over insulin resistance. Hyperinsulinemia is regarded as a major driver for obesity, type-2 diabetes and the associated chronic conditions [41,42]. Presumably, energy rich food intake drives the increased insulin synthesis of pancreatic β-cells. Over time, β-cells become exhausted and high insulin levels cannot keep up with the insulin demand of resistant cells resulting in elevated blood sugar level and type-2 diabetes. It was supposed that hyperinsulinemia deregulates the balance of insulin-GH-IGF axis promoting lipid deposition coupled with low energy expenditure.
Development of type-2 diabetes via hyperinsulinemia is explained by a supposed interplay among insulin, GH and IGF-I [43]. Type-2 diabetes develops attributed to increased insulin levels of portal vein that increases liver sensitivity for growth hormone (GH). Liver increases insulin-like growth factor-1 (IGF-1) levels, which means a negative feedback on the hypothalamus decreasing significantly the GH levels. The low GH/insulin ratio stimulates lipogenesis and increases the body weight. Nevertheless, the limited regulatory axis of three molecular players is erroneously picked out from the whole regulatory network.
In clinical practice, diabetes treatment using insulin with high affinity for IGF-I receptors, improves the lipid profile, increases insulin sensitivity and helps glucose metabolism. [44]. IGF-I shows cardioprotective effects and improvement of insulin sensitivity in patients being after a myocardial infarct [45]. These results suggest that insulin and GH-IGF-I axis improve endocrine regulation and metabolic balance instead of disrupting them.
3. The Origin of Insulin Resistance Is a Defect of Estrogen Signaling, While Hyperinsulinemia Is a Compensatory Effort for Improving Estrogen Regulation
Since glucose supply is the prerequisite of cellular health being the fuel for all cellular functions, glucose deprival seriously affects the continuous work of the main regulatory circuits of ER alpha. These circuits control genome stabilization, cell growth/proliferation as well as glucose supply [46]. Insulin and IGF-I together are important players in the regulatory circuit of ERs controlling glucose supply and metabolic balance. A weakening ER expression/activation or low estrogen level upregulates compensatory insulin secretion leading to hyperinsulinemia that is an effort for the restoration of estrogen signaling, even when it is not successful.
Weakening estrogen signaling poses an emergency situation for mammalian cells by disrupting glucose and lipid metabolism and at the same time endangering DNA stability. The hypothalamus-hypophysis unit then sends neuro-endocrine signalings to the adipose tissue as commands for the upregulation of estrogen synthesis and ER activation. Restoration of estrogen signaling improves glucose uptake, metabolic homeostasis and energy expenditure in insulin resistant patients [47,48].
Insulin and estrogen (specifically 17β-estradiol) signalings work in synergism on the activation of glucose transporter 4 (GLUT4) by promoting its translocation to the plasma membrane [49]. Incorporation of GLUT4 into the plasma membrane allows the entrance of glucose into the cell. In estrogen deficiency, increased insulin level may exert a complementary effect on GLUT4 translocation facilitating cellular glucose uptake.
Insulin-like growth factor-1 (IGF-1) has a direct role on glucose uptake, working in partnership with insulin, particularly in peripheral sites like adipose tissue and muscles [50]. IGF-I promotes glucose uptake by activating its own IGF-I receptors and hybrid receptors reacting to both IGF-I and insulin receptors (IRs). Amplified insulin levels facilitate IGF-I signaling and at the same time IGF-I activation increases the unliganded activation of membrane associated ERs closing the regulatory circuit [30].
Low grade inflammation is a characteristic finding in insulin resistant adipose tissue. Insulin and IGF-1 signaling have complex regulatory effects on the development of inflammation. In healthy adipose tissue, they are acting as anti-inflammatory agents. Conversely, in insulin resistance, including obesity and ageing, paralelly increased insulin and IGF-1 signalings promote inflammation [51]. In insulin resistance, macrophages and immune competent cells recruit for increasing pro-inflammatory cytokine expression, facilitating aromatase enzyme concentration and estrogen synthesis [52]. When estrogen concentration can reach an appropriate level, estrogen signaling improves cellular glucose uptake. Restored glucose uptake silences the inflammation and lowers estrogen concentration [8]. In insulin resistant adipose tissue, the inflammation continuously remains low grade as estrogen level cannot achieve suitably high concentration for the improvement of glucose uptake.
Estrogen and insulin show a close interplay in regulating the balance of lipolysis/lipogenesis. In insulin resistance, the defect of estrogen signaling liberates free fatty acids (FFAs), while hyperinsulinemia cannot exert its powerful anti-lipolytic effect [53]. In obesity, abundant FFAs in the circulation are pathologically deposited in non adipose organs and tissues; such as liver, pancreas, skeletal muscles and heart [54].
In postmenopausal women, estrogen deficiency decreases the body's ability to oxidize (burn) lipids, leading to insulin resistance and fat accumulation. In obesity, weak estrogen signaling in the background leads to a dysregulation of fatty acid metabolism and visceral lipid deposition [55]. Phosphodiesterase 3B (PDE3B) is an enzymatic hydrolyser of cAMP and cGMP pathways in partnership with estrogen. They are co-regulating the key metabolic actions of estrogen: lipolysis, energy homeostasis and insulin secretion. Using a pharmacological blockade of AKT phosphorylation and PDE3B expression promoted lipolysis even in the presence of insulin [56]. These findings support that in insulin resistance, an estrogen deficiency induced damage of AKT and PDE3B pathways inhibit the antilipolytic action of insulin in spite of its high concentrations.
Takeuchi et al. published in Nature that in humans, increased carbohydrate metabolism by the gut microbiota contributes to the development of insulin resistance [57]. In insulin resistant patients, increased fecal carbohydrate content associated with microbial metabolism and the inflammatory cytokines of patients showed strong correlations. Recently, it was established that gut bacteria associated with insulin sensitivity and insulin resistance, exhibit different patterns of carbohydrate metabolism [58]. Bacteria associated with insulin sensitivity, improved the insulin resistance in a mouse model.
Unique bacterial components of the gut microbiome nominated as estrobolome, play a crucial role in the reactivation of inactive, bound estrogens helping the maintenance of insulin synthesis in the pancreatic β cells [59]. Inactive, conjugated estrogens are arriving from the liver via bile secretion and bacteria of the estrobolome showing high β-glucuronidase activity, may reactivate them in the gut. In conclusion, the accumulation of gut bacteria with high β-glucuronidase activity improves insulin sensitivity via increasing the levels of bioactive estrogens. In postmenopausal women, a partnership was observed between estrogen deficiency and dysbiosis of gut bacteria leading to the development of type-2 diabetes [60].
Recently, gut bacterial sequences were found in healthy brain samples suggesting that there is a microbiome in the brain [61]. Considering the importance of high β-glucuronidase activity in the gut, it can be presumed that intestinal bacteria reactivate conjugated estrogens in the brain as well, increasing the levels of bioactive estrogen.
Mitochondrial dysfunction is important finding in insulin resistance. Dysfunction of mitochondria leads to decreased β-oxidation, low ATP creation and increased ROS production [62]. Estrogen maintains mitochondrial function by affecting mitochondrial respiration, mitochondrial biogenesis, and mitochondrial quality control [63]. Insulin and estrogen signaling come together on Sirt1, mTOR, and PI3K signaling pathways in the joint regulation of autophagy and mitochondrial metabolism [64]. In insulin resistance, hyperinsulinemia is an effort for compensating the lack of estrogen regulation on mitochondria.
In conclusion, estrogen signaling as the chief regulator of metabolic processes shows strong interplay with insulin and IGF-I and with other molecular players participating in the control of glucose uptake. Weakening estrogen signaling leads to a failure of glucose uptake, while recruits insulin and IGF-1 for restoring the regulation of glucose and lipid metabolism. In crisis situations, growth factor receptors including IGF-1 receptor are capable of the unliganded activation of ERs.
4. The Defect of Estrogen Signaling Is the Origin of Genomic Instability and Insulin Resistance in BRCA Gene Mutation Carriers
The identifications of BRCA1 and BRCA2 genes were enormous steps in cancer research [65,66]. The products of these genes, BRCA1 and BRCA2 proteins proved to be safeguards of genomic integrity controlling transcriptional processes, DNA replication and recombination as well as the repair of DNA damages [67].
Inheritable mutations of BRCA1 and BRCA2 genes led to an increased risk of hereditary cancers via genomic instability particularly in the female breasts and ovaries [68,69]. Well functioning BRCA proteins exhibit pivotal role in the genomic stability of all cell types in men and women. Nevertheless, the specific risk for breast cancer development in BRCA1 mutation carriers, mistakenly suggested an excessive estrogen signaling in the background.
The receptor landscape of BRCA1 mutant breast cancers show typically ER-alpha negativity and histologically they are poorly differentiated, while clinically they exhibit a rapid growth and poor prognosis [70]. The vast majority of BRCA1 mutant cancers are ER-alpha negative and ER-PR-HER2 negative (triple negative), being apparently independent of estrogen regulation [71,72]. In non familial, sporadically occurring ER-alpha negative breast cancers, a decreased expression of BRCA1 protein and reduced ER-alpha mRNA level reflect a defective interplay between the two regulatory proteins [73]. These findings reveal that BRCA1 gene mutation inhibits the transcriptional activity of ER-alpha via weakening its liganded activation. In BRCA1 mutation carriers, weakening estrogen signaling causes increased risk for cancer development, metabolic diseases and sex hormone imbalance with infertility [46].
In BRCA1 mutation carriers, decreased estrogen signaling may be the origin of ER-negative and TNBC type tumors, instead of excessive estrogen activation [74]. The conspicuous prevalence of breast cancers among BRCA1 mutation carrier women may be explained by the unique demand of female breast for the balanced liganded and unliganded ER activation. In postmenopausal women with type-2 diabetes and obesity, estrogen loss increases the metabolic and hormonal imbalances leading to increased risk for TNBC development.
Molecular investigations tried to reveal the key to interactions between ER alpha and BRCA1 proteins. Wild type BRCA1 protein could suppress the transcriptional activity of ER alpha [75] inhibiting near all genes that are regulated by estrogen [76]. BRCA1 could inhibit ER acetylation conferred by p300 that is crucial for ER transactivation [77]. Conversely, BRCA1 protein could induce an upregulation of p300, which is a coactivator of ER alpha [78]. Similarly, BRCA1 was capable of facilitating Cyclin D binding to ER alpha that upregulated the transcriptional activity of ERs [79]. These ambiguous findings suggest a complex interplay between BRCA1 and ER alpha signalings.
BRCA and ER proteins are also capable of direct binding for regulation of each other’s activation [80]. In reality, a decreased expression or inactivation of either ER alpha or BRCA1 protein deregulates their interaction, jeopardizing both estrogen signaling and genomic stability [46]. Healthy BRCA1 upregulates ER alpha activation via amino-terminus binding; however, its carboxyl-terminus binding confers repressive effect on ER alpha.
In healthy breast cells with BRCA1 gene mutation, BRCA1 protein synthesis is low together with decreased expression of ER alpha mRNA and ER alpha protein [81]. In BRCA1 gene mutation carrier breast cancer cells, the liganded activation of ERs was decreased [75] and ER alpha also showed a decreased expression [82].
Molecular investigations revealed that both healthy and tumor cells carrying BRCA mutation, are intelligent and respond to the danger of estrogen defect via driving the regulatory circuits of ERs through different pathways [5]. BRCA1 mutation promotes an increased expression of epidermal growth factor receptors (IGFs) in healthy epithelial breast cells [83]. In BRCA mutation carrier cells, increased growth factor receptor signaling and P13K/Akt cascade activated ERs via an unliganded manner [84]. In fibrous adipocytes of the breast, BRCA protein defect facilitates increased estrogen synthesis choosing the PII promoter region on aromatase gene [85]. In mammary luminal progenitor cells with BRCA protein deficiency, a further coactivator of ER alpha, nuclear factor kappaB (NF-κB) showed persistent activation [86] as a compensatory effort for the improvement of estrogen signaling.
In ovarian tumor cells with BRCA protein deficiency, ER alpha showed an unusual, conspicuously high unliganded transcriptional activity [87]. In a BRCA mutation carrier tumor cell line, increased activation of p300 (an ER coactivator) facilitated the transcriptional activity of ERs [78]. In breast cancer cells having BRCA gene mutation, Cyclin D1, another activator of ER transcription was highly expressed [88].
Mutant variants of p53 (genome safeguarding protein) were observed in the luminal cells of healthy breasts in BRCA mutation carriers [89]. Mutant TP53 gene was found in both familial and sporadic TNBC type tumors. These findings support that in germline BRCA mutation carriers, somatic mutation of TP53 maybe an effort for replacing the lost genome safeguarding function of BRCA protein.
In BRCA gene mutation carrier women, anovulatory infertility frequently occurs [90] attributed to the weakness of liganded estrogen signaling. Early menopause caused by ovarian failure is a feature finding in BRCA mutation carriers [91]. In the majority of women with BRCA1 mutation, the lack of functional BRCA1 protein was associated with compensatory increased aromatase levels and abundant estrogen synthesis [92]. In BRCA gene mutation carriers, defective ER signaling promotes insulin resistance; however, compensatory high levels of insulin and IGF-1 are not successful in the improvement of glucose uptake [74].
Development of central obesity and metabolic syndrome are markers of insulin resistance that correlate with a higher penetrance of BRCA mutation [93]. BRCA mutation associated damage of estrogen signaling disturbs not only genomic stability but also further regulatory circuits of ERs including those for glucose and lipid metabolism [46].
In conclusion, genomic instability is originated from the defect of liganded ER activation, rather than from an excessive estrogen concentration. In addition, both healthy and tumor cells have protective efforts against genomic damages through rising estrogen signaling via gene amplification and activating mutations. In BRCA mutation carrier women, the defect of liganded ER activation explains metabolic and fertility disorders and the increased risk of breast cancer.
5. Polycystic Ovary Syndrome Originates from the Disruption of Estrogen Signaling via CYP19A Gene Mutation
In premenopausal women, having functioning ovaries, polycystic ovary syndrome (PCOS) is the manifestation of insulin resistance syndrome. PCOS presents in adolescent girls with multiple ovarian cysts, menstrual disorders, anovulatory infertility and hirsutism besides metabolic syndrome and obesity [94]. These findings suggest a failure of genomic estrogen regulation. In young men with insulin resistance, a damage of semen quality and infertility may be found in the background of the predominant metabolic alterations attributed to the defect of estrogen signaling [95].
In PCOS, the defect of neuro-hormonal signaling along hypothalamic-pituitary-gonadal axis deregulates the whole hormonal system [96]. In PCOS cases, the glucose uptake of cells is defective, and insulin resistance leads to compensatory hyperinsulinemia as an effort for improving the glucose supply of cells [97]. In vitro studies revealed that high insulin level may augment both basal and LH stimulated androgen synthesis in ovarian theca cells leading to hyperandrogenism in women with PCOS [98].
Development of cystic ovaries is a characteristic symptom in women with genetically defined serious defects of estrogen signaling, caused by a mutation of CYP19 aromatase or ESR1 gene [99,100]. In aromatase deficient girls with CYP19 aromatase gene mutation, cystic ovaries and delayed bone maturation develop during childhood. At puberty, primary amenorrhea, failure of breast development, virilization, and hypergonadotrophic hypogonadism may be experienced [99]. In women with PCOS, the increased androgen synthesis of ovaries and adrenal glands is a compensatory answer to low estrogen levels; however, there is no conversion from androgen to estrogen attributed to aromatase deficiency [100].
The germline mutation of ESR1 gene, encoding ER alpha, resulted in deep estrogen resistance, delayed puberty and polycystic ovaries in a young girl [101]. Despite the compensatory sky-high serum levels of estrogen, the absence of breast development, small uterus and enlarged polycystic ovaries revealed missing estrogen signaling. ER resistance repressed the insulin assisted uptake of glucose and hyperinsulinemia was observed as a counteraction to deepening insulin resistance.
Women with PCOS clearly exhibit a higher risk for early cardiovascular diseases, attributed to their metabolic dysfunction and abnormal hormonal pattern [102]. In PCOS cases, increased risk for clotting disorders and low fibrinolytic activity is a characteristic feature promoting the development of thrombotic complications [103]. In women with PCOS, immune responses are dysregulated. Insulin resistance and hyperandrogenism are associated with immune cell dysfunction and cytokine imbalance leading to long term inflammatory environment [104]. In PCOS cases, all these complications and co-morbidities are associated with the defect of aromatase synthesis and decreased estrogen signaling.
In women with PCOS, an increased risk maybe observed for gynecological tumors. Increased prevalence of endometrial, ovarian and thyroid cancers was found in PCOS cases [105]. Endocrine disorders with anovulatory infertility including PCOS are associated with highly increased risk for endometrial cancer [106].
Recently, PCOS studies in humans and rodents revealed that a disruption of estrogen signaling is highly associated with the development of the syndrome [107]. These experiences led paradoxically to the therapeutic targeting of estrogen by using antiestrogen clomiphene or aromatase inhibitor letrozole therapy so as to induce ovulation [108]. Fortunately, the inhibition of an earlier defective estrogen signaling provokes extreme counteractions abruptly upregulating both estrogen synthesis and ER activation, which explains the achievement of good pregnancy outcomes.
6. Estrogen Is the Principal Regulator of All Cellular Functions in Mammalians
Estrogens (estrone, estriol, and estradiol) are unique hormones, as they have no harmfully high concentrations like other hormones [46]. Activated estrogen receptors (ERs) can choose from several genes for expression and activation and at the same time they may facilitate or silence the regulatory processes using their coactivators or corepressors. Enormous upregulation of estrogen signaling is necessary for ovulation and during pregnancy for the growth of the uterus and for embryonic development. Conversely, the low level of estrogen or the defect of ER activation is an emergency situation as it leads to genomic deregulation throughout the body in men and women.
ERs have possibilities for liganded (estrogen bound) activation through their activation function-2 (AF2) domain and for unliganded activation by transduction molecules, such as growth factor receptors (GFRs) through the ancient activation function-1 (AF1) domain [109] (Figure 1). In estrogen deficient periods, increased growth factor receptor (GFR) expression and activation may transiently maintain the appropriate ER activation via unliganded pathway [110,111]. Balanced liganded and unliganded activation of ERs stimulates estrogen synthesis and ER expression ensuring DNA stabilization and upregulation of the whole genomic machinery [112]. Artificial inhibition of either liganded or unliganded activation of ERs induces a strong compensatory upregulation of the unaffected domain, while the failure of restorative efforts may lead to a breakdown of the whole genomic regulation.
The genomic machinery is driven by estradiol (E2) activated ERs via stimulating and silencing all physiological pathways through regulatory circuits [113]. The main regulatory circuits of ERs control DNA stabilization, cell proliferation and cellular glucose supply. Activated ERs harmonize all physiological actions via balanced upregulation or downregulation of its regulatory circuits. The upregulation of estrogen signaling is always reparative effort aiming the restoration of defective genomic functions
Figure 1.
Liganded and unliganded activation possibilities for estrogen receptors. 1. Liganded estradiol (E2) activated estrogen receptors (ERs) bind directly to the gene promoter region. 2. E2 activated ERs bind indirectly to the gene promoter region, through transcription factor (TF). 3. Growth factor (GF) activated growth factor receptors (GF-Rs) activate membrane associated ERs that can further activate nuclear ERs, bound to the gene promoter region. 4. GF activated GF-Rs activate nuclear ERs through TF, bound to the gene promoter region. P: phosphorylation, RNA: ribonucleic acid, N: nucleus.
Figure 1.
Liganded and unliganded activation possibilities for estrogen receptors. 1. Liganded estradiol (E2) activated estrogen receptors (ERs) bind directly to the gene promoter region. 2. E2 activated ERs bind indirectly to the gene promoter region, through transcription factor (TF). 3. Growth factor (GF) activated growth factor receptors (GF-Rs) activate membrane associated ERs that can further activate nuclear ERs, bound to the gene promoter region. 4. GF activated GF-Rs activate nuclear ERs through TF, bound to the gene promoter region. P: phosphorylation, RNA: ribonucleic acid, N: nucleus.
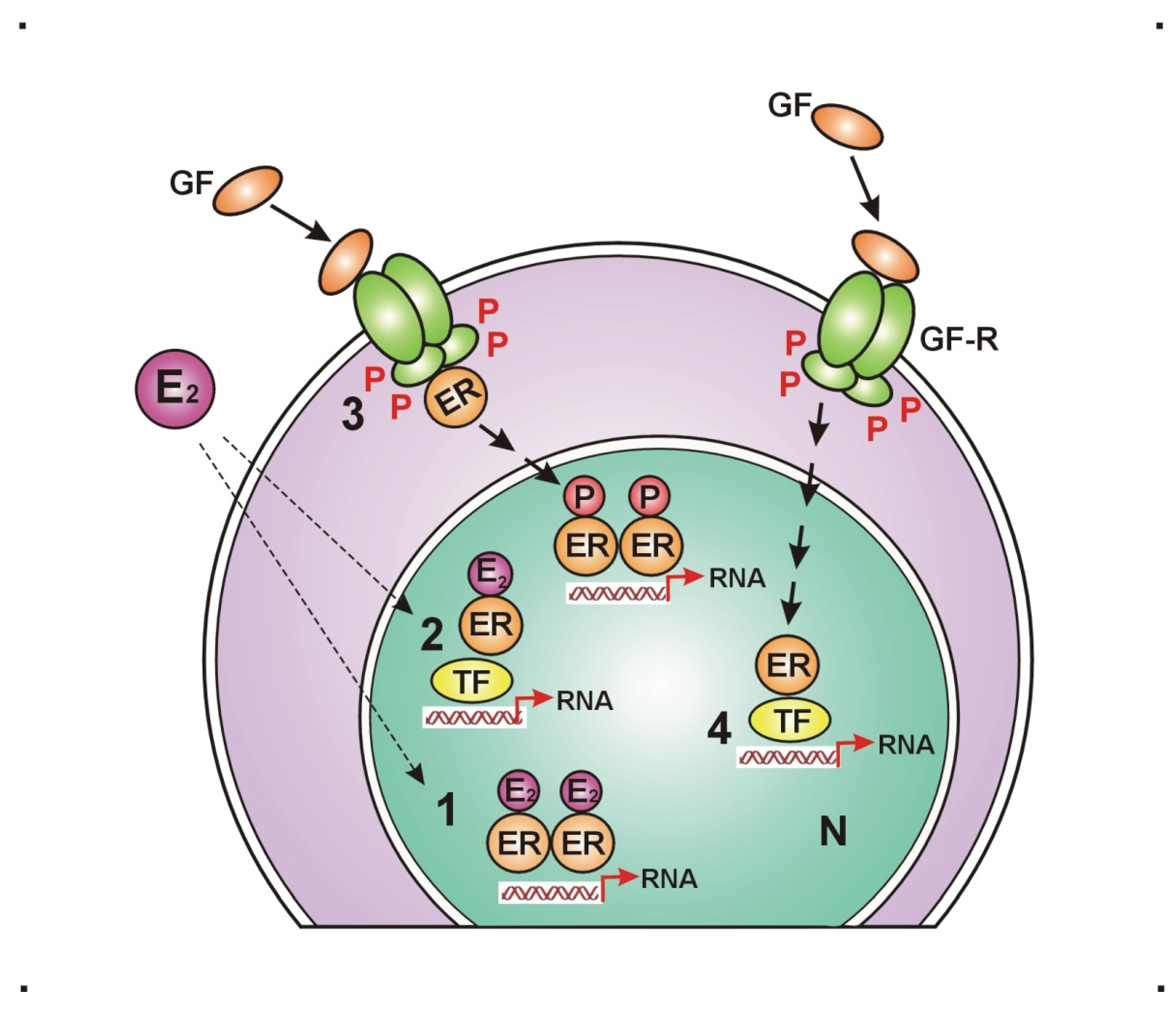
The principal regulatory round of ERs is the circuit of DNA stabilization [46]. It initiates with the estrogen activation of ER alpha and then inducing upregulation of further ER expression. Abundant activated ERs work via upregulation of genome safeguarding protein, such as BRCA1. The transcription proteins, ER alpha and BRCA1 are in close interplay via direct binding and they are capable of either rising or silencing each other’s activation. BRCA1 is responsible for the balance between ER alpha expression and aromatase enzyme activation. When ER alpha expression or its activation is weakening, BRCA protein facilitates aromatase enzyme expression and activation leading to compensatory increased estrogen synthesis [114]. In conclusion, the E2ER – BRCA – aromatase – E2 – E2ER circuit ensures the continuous maintenance of both genome stabilization and estrogen signaling.
In the regulatory circuit of cell proliferation estrogen activated ERs drive and control the cell growth and proliferation at all sites of the body in strong interplay with growth factor receptors: epidermal growth factor receptors (EGFRs) and insulin-like growth factor-1 receptors (IGF-1Rs) [111]. Estrogen activated ERs control the expression and activation of growth factors (GFs) and growth factor receptors (GFRs). Transduction of growth factor signal (GFS) induces kinase cascade pathways sending further unliganded activation to nuclear ERs [30]. Low estrogen level is an emergency state leading to increased expression/activation of GFRs and compensatory unliganded activation of ERs. In tumors, a therapeutic inhibition of estrogen signaling activates growth factor kinase cascades providing a compensatory stimulation of ER activation through unliganded pathway.
In the regulatory circuits of glucose supply, estrogen drives and controls all steps of cellular glucose uptake and glucose homeostasis [115]. Estrogen safeguards the vitality of pancreatic islet cells protecting them from lipid deposition [116]. Estrogen controls insulin expression, activation and secretion [117]. Estrogen controls insulin assisted glucose uptake through helping the expression and translocation of intracellular glucose transporters (GLUTs) [118]. Defects of glucose uptake always reflect the failure of estrogen signaling in the background [100].
7. Adipose Tissue Ensures Metabolic Balance and Energy Homeostasis via Estrogen Regulation
Circulating estrogen regulates body fat distribution, remodeling, and the maintenance of adipose tissue health [119]. Males tend to accumulate adipose tissue in visceral location promoting metabolic disorders and increasing the risk for chronic conditions like cardiovascular diseases and cancers. In contrast, females show inclination to the deposition of subcutaneous fatty tissue in the gluteofemoral region that is not associated with metabolic disorders. After menopause, estrogen loss is associated with male-like abdominal deposition of fatty tissue in women, becoming similarly endangered by metabolic disorders like men [120].
Adipose tissue mass is an estrogen regulated endocrine organ that exhibits the highest activity of estrogen synthesis among extragonadal sites [121]. Circulating and locally produced estrogens exert their regulatory effects locally on growth, metabolism and estrogen signal of adipocytes in an intracrine manner. In addition the hormonal regulation and energy supply is extended to the adjacent organs and tissues in a paracrine manner [122]. Adipose tissue health maintenance is regulated chiefly by estrogen in partnership with other hormonal and neural signalings [123].
Estrogen exerts its regulatory effects on estrogen responsive adipocytes depending on the intensity of their receptor expression [124]. Estrogen signaling regulates glucose metabolism, lipolysis/lipogenesis [48] and energy balance [125,126] in the whole body. In adipose tissue, defects of estrogen signaling result in deregulation in all regulatory functions of adipocytes leading to metabolic disorders, obesity and chronic conditions in the fat regulated organs and tissues [18].
Subcutaneously positioned fatty tissue supplies energy and estrogen regulation for the skeletal muscles and skin. The abdominally located visceral adipose tissue surrounds and nurses the visceral organs, gonads and cardiovascular structures [127]. Female breasts and the bone marrow enjoy extremely good energy supply as breast lobules and bone marrow cell populations are closely intermingled with abundant adipose tissue. Female breasts require strict hormonal regulation and high energy supply parallel with the hormonal changes of menstrual cycles [128]. In the bone marrow, adipocytes are actively influencing the proliferation and functional activity of hemopoietic and immune competent cells via their estrogen signaling and hormonal secretome [129]. Bone marrow fat supplies energy and regulatory command for the bones as well.
Abdominal adipose tissue has crucial physiological secretory functions [130]. The secretory activities of adipose tissue are defined by its estrogen supply and estrogen sensitivity [119]. Suitable estrogen signaling ensures the health and functional activity of adipocytes. Visceral fat as a central endocrine organ regulates metabolic balance, vascular health, appetite, body weight, and immune system among many others [131].
Sexual hormones, adipokines, cytokines and growth factors are important signaling molecules in the adipose tissue and their estrogen regulated activation ensures the health of the whole body. In postmenopausal women and men, the vast majority of estrogens are produced from circulating adrenal androgens via conversion by aromatase enzyme [132]. In addition, adipose tissue is capable of synthesizing sexual steroids locally, de novo from cholesterol. In adipose tissue, estrogen synthesis and appropriate estrogen signaling regulates the expression of numerous genes and the harmonized synthesis of various signaling molecules [123].
Adipokines are regulatory hormones secreted by the adipose tissue in health and disease. Circulating leptin ensures the energy balance in the hypothalamus through anorexinogenic and lipolytic effects. Estrogen increases the expression and activation of leptin receptors increasing the leptin-sensitivity of different cells [133]. Adiponectin improves insulin sensitivity silencing inflammatory reactions, and restoring endothelial functions. In adult mice, oophorectomy facilitates adiponectin synthesis, while estradiol substitution restores the normal levels [134]. Resistin is an anti-obesity hormone showing paralelly increased levels with weight gain. Estradiol benzoate treatment decreased resistin levels in subcutaneous adipose tissue [135].
Ghrelin is a multifaceted hormone secreted by gastric and intestinal cells showing endocrine function. Ghrelin is known as the hunger hormone, increasing growth hormone release, food intake and fat storage, when the stomach is empty [136]. Estrogen regulates ghrelin secretion and ghrelin receptor activation controlling ghrelin effect on appetite, energy balance, and metabolic changes [137].
In conclusion, estrogen signaling controls all hormones regulating appetite, hunger, food intake and fat deposition. Weakening ER activation or estrogen deficiency obviously disturbs the balance of food intake and energy expenditure promoting weight gain and the development of obesity. .
Estrogen deficiency and/or ER resistance deregulates all signaling functions of adipocytes. Adipocytes, losing their ER expression and estrogen synthesizing capacity, exhibit an excessive lipid deposition, parallel with becoming insulin resistant [138]. Considering that weakening estrogen signaling endangers DNA stability as well, abdominal obesity is not the origin of cancer development in visceral organs, but rather defects of estrogen function is the common initiator of both obesity and cancer.
In obese adipocytes, lipogenesis become predominant over lipolysis and deepening estrogen deficiency strengthens this imbalance. In thin stromal adipose cells, increased insulin levels stimulate aromatase expression and estrogen synthesis [139]. This observation justifies that in obesity, increased insulin and IGF-1 levels exert anti-obesity effects via facilitation of estrogen synthesis. Regulatory defect of obese adipose tissue promotes further disorders in the adjacent insulin resistant organs, resulting in serious co-morbidities, such as metabolic disorders, fatty degeneration and malignancies [140,141].
In health and disease, adipose tissue produces inflammation regulating cytokines that are regulatory proteins having great role in either anti-inflammatory or pro-inflammatory activities. Cytokines are secreted by adipocytes, fibroblasts and immune competent cells within the adipose tissue influencing systemic metabolic and immune functions [142].
In obesity, adipose tissue mass exhibits low grade inflammation with abundant macrophages and T-cells [143] targeting the improvement of glucose uptake. Macrophages produce proinflammatory cytokines, including tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6). Proinflammatory cytokines drive increased expression and activation of aromatase enzyme leading to augmented estrogen synthesis [144]. In obese adipose tissue, overexpression of ER alpha increases estrogen sensitivity of fat adipocytes, silences inflammation, rapidly promotes lipolysis and improves the signaling function [145].
Estrogens have pivotal roles in the improvement of metabolic dysfunctions via orchestrating inflammatory changes and immune responses [131]. Low grade inflammation in obese, insulin resistant fatty tissue is a compensatory process for improving the regulatory capacity of adipocytes via increasing aromatase expression and estrogen concentration [5].
Dense inflammatory reaction in the vicinity of HER2-rich and TNBC type breast tumors is an effort for facilitating aromatase expression and estrogen synthesis helping the regulatory improvement of these poorly differentiated cancers [8]. In case of ER positive, highly differentiated breast cancers, immune competent cells are not recruited as the adjacent adipocytes increase their estrogen synthesis, helping the genomic repair of tumors. Conversely, the tamoxifen blockade of ER positive tumors activates a neighboring inflammation, which is a compensatory response to the sudden inhibition of estrogen signaling.
Adipocytes synthesize insulin like growth factor 1 (IGF-1) as well and they exhibit IGF-1 receptor expression (IGF-1R). IGF-1 signaling works within the growth hormone (GH)–IGF-I axis regulating growth, development, and metabolism in mammals, particularly during childhood [146]. GH, produced by the pituitary gland, stimulates the adipose tissue, liver and skeletal muscles for IGF-1 secretion. IGF-1 then acts directly on various tissues, including bone, muscle, and cartilage, to regulate protein synthesis, cellular growth and cell division.
IGF-1 shows regulatory functions on glucose and lipid metabolism in partnership with insulin and estrogen. IGF-1 regulates glucose and fatty acid uptake, improving insulin sensitivity and reducing body weight as it shares similar signaling pathways with insulin [147]. Increased IGF-1 levels paralelly augment insulin synthesis leading to compensatory hyperinsulinemia, in the early phase of insulin resistance [148]. IGF-1 is important player in the regulatory circuits of estrogen for controlling cell proliferation and glucose uptake [113].
In adipose tissue, estrogens regulate both IGF-1 synthesis and IGF-1R expression of adipocytes. Estrogen stimulated IGF-1 synthesis and IGF-1 receptor activation upregulate the AKT and MAPK pathways facilitating an increased unliganded activation of ERs [149]. In the absence of estrogen, IGF-1 receptor signaling maintains the unliganded activation of ERs ensuring a transient, genome wide expression of estrogen regulated genes [109]. In conclusion, in insulin resistance and obesity, an upregulation of IGF-1 signaling improves glucose uptake and helps the loss of body weight via increasing the unliganded activation of ERs.
Adipose tissue is a crucial organ in the regulation of immune reactions and immune defense [150]. It actively participates in host defense against pathogens and regulates the quality and intensity of inflammatory processes. In the adipose tissue, inflammation is a means for the improvement of genomic stability in metabolic diseases as well and provides help for tumors in their fight to achieve a self-directed apoptotic death [8].
Estrogen acts as a major regulator of the immune system, influencing the development, function, and activity of various immune cells, including lymphocytes, monocytes, macrophages, as well as T and B lymphocytes. Estrogen exerts these effects by binding to its specific receptors, which activate gene expression and signaling pathways modulating both innate and adaptive immunity [151].
There is a thorough interaction between adipocytes and immune competent cells, such as macrophages, T cells, and B cells in both healthy and obese adipose tissue [152]. In lean adipose tissue, eosinophil granulocytes show IL-4 cytokine secretion and regulatory T cells activate M2 type macrophages promoting the expression of arginase and anti-inflammatory cytokines including IL-10. Conversely, in obese adipose tissue, increased number of M1 type macrophages and abundant secretion of pro-inflammatory cytokines including TNFα and IL-6 may be observed [143]. In obese female mice, estrogen treatment improved insulin sensitivity, silenced the inflammation and reduced obesity [153].
In conclusion, insulin resistance and obesity equally originate from the defect of estrogen signaling. Estrogen loss inhibits glucose uptake and increases the lipid deposition of adipocytes weakening their regulatory functions. Heavily lipid laden deregulated adipocytes cannot control DNA surveillance, increasing the risk for cancer development in visceral organs.
8. Skeletal Muscle Contraction Improves Insulin Sensitivity via Rapid Unliganded Activation of ERs by IGF-1 Receptor
Physical activity is in strong correlation with human health. The skeletal muscle mass accounts for about 40% of the total body, and the regulation of its glucose metabolism has a crucial role in the glucose homeostasis of the whole body [154]. Regular exercise improves mental health, enhances the quality of life, and it is capable of improving existing chronic conditions including obesity and type 2 diabetes [155].
In skeletal muscles, glucose uptake is mainly insulin dependent at rest, while during exercise, glucose uptake is stimulated by muscle contraction and increased blood flow via insulin-independent pathways [156]. In resting muscle, insulin binding to its membrane receptor, triggers a cascade of intracellular signalings culminating in the activation of GLUT4 that migrates to the plasma membrane and allows the entry of glucose. By contrast, muscle contraction induces changes in levels of intracellular molecules such as ATP and calcium, regulating GLUT4 translocation and glucose uptake, even in the absence of insulin.
Estrogen is the principal regulator of cellular glucose uptake and metabolism. Estrogens regulate pancreatic β-cell survival [116] and activate insulin biosynthesis and release [157]. Estradiol treatment facilitates GLUT4 expression and intracellular translocation to the plasma membrane [118]. Weakening estrogen signaling is an emergency state and requires compensatory efforts for the improvement of glucose supply.
Under estrogen regulation, IGF-1 signaling pathway is strongly linked to insulin signaling, working synergistically on muscle insulin sensitivity, growth, metabolism and overall health [158]. Insulin and IGF-1 signaling initiate a cascade activating IRS-1 and IRS-2 signaling proteins initiating downstream pathways of PI3-kinase and MAPK.
Estrogen treatment may stimulate a rapid cellular response within seconds to minutes through the activation of cell membrane associated ERs [159]. This rapid response avoids the time consuming transcription and translation processes that can take several hours or even one day. These membrane-located rapid ER responses work through the activation of kinases, calcium mobilization, and G protein cascades, promptly realizing different cellular functions.
The technique of rapid ER response to estrogen was studied on muscle cells and adipocytes [49,118]. Following estradiol treatment, a prompt translocation of ER alpha was observed from the nucleus to the cell membrane so as to interacting with membrane associated growth factors and fulfilling a rapid, non genomic signaling. The rapid response to estradiol facilitated GLUT4 expression and translocation to the plasma membrane improving glucose uptake in both muscle cells and adipocytes.
Facilitating the glucose uptake through a rapid response to estrogen may have a great importance in the physiology of mammalians as glucose shortage highly endangers the integrity and function of cells. In addition, a rapid activation of non-nuclear estrogen receptor signaling was observed conferring protection against vascular injury [160].
Contraction in muscles requires high energy utilization evoking a rapid translocation of ER alpha from the nucleus to the cell membrane, similarly, like under estrogen treatment. Along the cell membrane, IGF-1 receptors are accumulated waiting for unliganded estrogen receptors in an estrogen deficient milieu. ER alpha, arriving to the cell membrane gains rapid, non genomic activation from the IGF-1 receptor. Activated ERs induce increased expression and activation of GLUT4 vesicles and promote their migration to and incorporation into the cell membrane of muscle cells. Increased glucose entrance into the muscle cells explains the improvement of insulin resistance attributed to contraction. Membrane associated ERs activated by IGF-1 receptor exhibit further possibilities for the activation of nuclear ERs as well [Figure 2].
The activation of the ER network in the huge mass of skeletal muscles explains the improvement of glucose uptake in the whole body in correlation with physical exercise.
9. Estrogen Regulation of Multitude Functions of the Liver
Liver development, integrity and functional activity are regulated by the neuro-hormonal signalings of hypothalamus-hypophysis system. GH/IGF-1 axis is one of the major regulators of liver as liver has a primacy in IGF-1 synthesis [161]. Moreover, estrogen hormones also are acting on the liver centrally, controlling the pituitary GH secretion and peripherally, by modulating GHR-JAK2-STAT5 signaling pathway [162]. Estrogens and GHs show thorough interplay in liver physiology, while a disruption of either GH or estrogen signaling leads to dramatic changes in the function of liver both during development and in adulthood.
Estrogen regulates directly the functions of liver as it expresses abundant ERα. Estrogen controls liver development, growth and regeneration [163] and regulates hepatic lipid and glucose metabolism [164].
In addition to the maintenance of hepatic health, estrogen signaling controls multitude functions of the liver influencing the regulation of the whole body. The liver is the major secretor of IGF-1, producing approximately 75% of circulating IGF-1, highly surpassing the IGF-1 synthesis of other tissues like adipose tissue and skeletal muscle [165]. This liver-derived IGF-1 behaves as an endocrine hormone, participating in the regulation of further tissues via distribution in the circulation. Conversely, in other tissues, locally secreted IGF-1 may function via paracrine and autocrine mechanisms.
Estrogen controls IGF-1 signaling pathways interacting in a complex manner in the regulation of glucose supply [166]. In turn, IGF-1 receptor is capable of unliganded activation of ERs via growth factor kinase cascades. This interplay between ER and IGF-1 receptor involves multiple levels, including their direct binding. Moreover, ER may modulate IGF-1 binding protein levels leading to increasing or decreasing bioavailability of IGF-1.
ER and IGF-1 receptor signaling interact in the protection of cardiovascular health. IGF-1 regulates cardiac development and improves the output, stroke volume, contractility, and ejection fraction of heart [45]. In human studies, IGF-1 stimulates contractility and tissue remodeling improving heart function after myocardial infarction. Low serum levels of free or total IGF-1 lead to an increased risk of cardiovascular and cerebro-vascular diseases.
Endogenous estrogen is the chief regulator of the liver in balanced production of clotting and fibrinolytic factors. The liver synthesizes coagulation factors, anticoagulants, proteins involved in fibrinolysis and the platelet production regulator, thrombopoeitin, deriving from megakaryocytes [167]. In cirrhotic patients, hepatic dysfunction perturbs the clotting process [168]. The risk for thrombosis in patients treated with synthetic estrogen and/or progestin derives from pharmaceutical mistakes [112]. The production of chemically modified hormones, such as ethinylestradiol, and medroxyprogesterone results in a blockade of unliganded ER activation on AF1 domain and compensatory activation on AF2 domain. This regulatory imbalance leads to unforeseeable toxic effects of synthetic hormones in human practice.
Liver also have crucial roles in immune responses [169]. Hepatocytes exhibit immune functions via expressing pattern-recognition receptors (PPRs), secreting complement components and cytokines. Hepatocytes also show immunoglobulin secretory capacity.
In the liver, estrogen controls insulin sensitivity and glucose uptake via a balanced activation of glycogen synthesis and glycolysis. In animal experiment, ER alpha knockout mice showed insulin resistance paralelly in the liver and skeletal muscles [170]. The hepatic lipid metabolism is regulated by estrogens. Estradiol treatment showed antidiabetic and antiobesity effects in mice kept on high fat diet. Estrogen decreased the expression of lipogenic genes in adipose tissue and liver, and suppressed the expression of hepatic G-6-Pase. Estradiol treatment in aged rats decreased the lipid peroxidation and improved the functional parameters of the liver [171].
In humans, estrogen has pivotal role in the hepatic regulation of serum lipid levels. In postmenopausal women, estrogen loss resulted in higher total and LDL cholesterol levels, and increased triglyceride levels compared to premenopausal women [172]. Menopausal estrogen therapy decreases the risk of cardiovascular diseases and this result maybe conferred by the advantageous changes of plasma lipid profile [173].
Insulin resistance in hepatocytes deregulates glucose metabolism, particularly the control of glucose output into the circulation. In insulin resistance, hepatic fatty acid synthesis becomes stimulated leading to hepatic steatosis. Type-2 diabetes is a significant risk factor for progressive hepatic steatosis, from non alcoholic fatty liver to hepatic cirrhosis [174]. Recent data show that type-2 diabetes and obesity are risk factors for hepatocellular carcinoma [175].
10. Hypothalamic Estrogen Signaling Is the Central Regulator of Somatic, Reproductive and Mental Health
The hypothalamus is situated in the ventral brain above the pituitary gland creating a central regulatory entity with it [176]. The hypothalamus exhibits integrative role in linking mental and somatic functions for well-being and adaptability. The hypothalamus continuously receives outer, environmental and internal stimuli from the body and answers by regulatory commands so as to maintain the internal balance.
Light signals arriving through the eye play a crucial role in visual perception but also evoke a non-image-forming vision by stimulating hypothalamic nuclei. The light-eye-body axis is a neuro-hormonal signaling network thoroughly influencing the genomic regulation of the entire body [177]. The hypothalamus regulates the hormone production of hypophysis and sends neural impulses through the autonomic nervous system. In the hypothalamus, estrogen is the chief coordinator between circadian system and gene expression/activation adapting all mental, somatic and reproductive functions to the changes of light and darkness in mammals [178].
Estrogen is the master regulator of bio-energetic systems in the brain and the body via hypothalamic activation [179]. Hypothalamus-pituitary unit exhibits interplay with sex steroids, modulating all neuro-hormonal signalings deriving from the central nervous system. Estrogen regulates cognitive functions by acting on the hypothalamus and other brain regions, promoting synaptic plasticity and neuron survival through both genomic and non-genomic ER activation [180]. HPG axis driven regulation shows various links with stress signaling and their somatic impacts [181]. HPG axis regulates the immune system showing strong correlations between sex steroid activation and immune responses [182].
Hypothalamic estrogen signaling regulates adipose tissue metabolism and energy homeostasis via hypothalamus-pituitary adipose tissue (HPA) axis [183]. There is a gender-specific protection for females against metabolic diseases and increased disease susceptibility for males. Adipose tissue regulation is driven by the activation of estrogen receptor alpha (ERα) through neuro-hormonal interactions together with other hormones, particularly insulin and adipokines [119].
Hormonal and neural signalings arrive from all regions of the adipose tissue to the hypothalamus conferring their own energy homeostasis and those of various adjacent organs and tissues [184]. In certain hypothalamic regions, the loss of ER alpha positive cells results in obesity and fertility disorders. The hypothalamic arcuate nucleus (ARC) is widely recognized as a central regulator of appetite [185]. The complex hormonal network regulating appetite, hunger and food intake is under estrogen control [186].
Disrupted estrogen regulation of HPA axis contributes to the development of insulin resistance, metabolic syndrome, obesity and type-2 diabetes, as estrogen plays pivotal role in the regulation of metabolism and energy homeostasis of fatty tissue.
11. The Origin of Cancer Development by Unhealthy Lifestyle Factors and Bad Habits: A Defect of Estrogen Signaling and the Associated Insulin Resistance
Much of the burden of cancer may be associated with modifiable lifestyle factors that increase one's risk for the disease [187]. There are epidemiological evidences on the contributions of major risk factors to cancer incidence and mortality: tobacco use, alcohol consumption, unhealthy dietary patterns and physical inactivity.
Smoking habit. Tobacco use is the major cause of cancer among harmful lifestyle factors affecting several regions, such as oral cavity, digestive tract, upper respiratory tract, lung, urinary tract, cervix uteri and ovaries [188].
Smoking is linked with insulin resistance in a dose-dependent manner [189]. Smoking increases the risk for insulin resistance, mainly via deregulation of hormones participating in glucose uptake, and causing a shift to lipogenesis and lipid deposition promoting abdominal obesity [190]. Studies have demonstrated that in animal models and in human tissues, nicotine and its metabolites inhibit aromatase enzyme in a dose-dependent manner decreasing androstenedione conversion to estrogen [191] In smokers, nicotine inhibits aromatase enzyme expression and activity in the brain, negatively influencing some basic psychological activities, such as cognition, libido and appetite [192].
Summarizing the smoking associated insulin resistance and the aromatase inhibition of nicotine, the defect of estrogen signaling and the associated failure of glucose uptake may explain the strong cancer inducing capacity of tobacco use at several sites.
Alcohol consumption. Chronic, heavy alcohol consumption deregulates the glucose homeostasis and is associated with the development of insulin resistance [193]. Excessive alcohol consumption is an independent risk factor for type-2 diabetes [194]. Alcohol drinking is in close correlation with cancer development of oral cavity, pharynx, larynx, esophagus, [squamous cell carcinoma only], colorectum, liver [hepatocellular carcinoma only], and female breast [188].
Multiple molecular mechanisms contribute to cancer development mediated by drinking [195]. Acetaldehyde, deriving from ethanol metabolism is a potent carcinogen capable of inducing DNA damage and mutations. During alcohol metabolism, oxidative stress can damage cellular components, including DNA, proteins, and lipids, contributing to genetic mutations and genomic instability [196].
Alcohol abuse disrupts the hypothalamic regulation of endocrine system, affecting various hormones that regulate growth, metabolism, stress and reproduction leading to metabolic disorders, cognitive decline and infertility [197]. Alcohol upregulates aromatase activity and increases estrogen levels in heavy drinker women that are presumably causal factors for their increased risk for breast and ovarian cancers [198]. In reality, increased aromatase activity and estrogen synthesis are compensatory actions in drinkers, against the inhibition of ERs and the developing insulin resistance.
The toxic, antiestrogenic effects of ethanol were studied on osteoblasts in female rats and on cultured osteoblasts in vitro [199]. Ethanol inhibited estrogen receptor signaling via the blockade of both ER alpha and ER beta activation, while estradiol treatment restored the health of osteoblasts. These results explain that in highly alcohol-exposed females, bone loss develops, while estrogen treatment exhibits protective effects.
The alcohol induced blockade of ER activation deregulates all genomic functions and increases the risk for several cancers in both men and women, in spite of the increased compensatory estrogen concentration, The aromatase inhibition of smoking and the ER blockade of drinking clearly explains the amplification of cancer risk in patients having smoking and drinking habits together
Unhealthy Dietary Patterns. Overeating and excess body weights are associated with increased risk for cancer in esophagus (adenocarcinoma only), gastric cardia, colorectum, liver, pancreas, endometrium, ovary, kidney, thyroid and female breast (postmenopausal cancers only) [200]. Interestingly, among premenopausal women, consistent inverse associations have been observed between obesity and breast cancer risk [201]. Overeating in premenopausal women with healthy hormonal balance, leads to gluteofemoral adiposity that is independent of metabolic disorders. It is not obesity but rather cycling estrogen levels and insulin sensitivity of abdominal adipocytes that protects obese young women from metabolic diseases and breast cancer [202].
The quality of diet is a significant factor in the development of insulin resistance. Studies show that a high-energy, high-fat, high-carbohydrate, and low-fiber diet as well as taking ultra-processed foods can increase the risk for type-2 diabetes [203]. Diet and nutrition seem to be modifiable risk factors for the development of several cancers, but the associations may be faulty due to inherent bias of investigations. [204]. It was established that only few single food or nutrient proved to be strongly or suggestively associated with cancer risk as tumor development is influenced by multitude genetic and environmental factors beyond the quality of diet.
Sedentary lifestyle. Sedentary behaviors have wide-ranging adverse impacts on the human body including high risks of metabolic disorders such as diabetes mellitus, and obesity, musculoskeletal disorders such as sarcopenia and osteoporosis, as well as depression and cognitive impairment [205]. Lack of physical activity increases the risk for cardiovascular diseases and cancers and raises the all-cause mortality in populations.
Physical activity is strongly linked to a lower risk of developing several types of cancer, including bladder, breast, colon, and lung cancer [206]. Regular exercise decreases cancer risk by maintaining a healthy weight, regulating hormonal balance particularly that of estrogen and insulin, and strengthening the immune system. Experimental studies show that physical activity improves insulin sensitivity through skeletal muscle contraction both immediately during exercise and for up to 48 hours afterward [207]. Muscle contraction is capable of increasing glucose uptake by a coordinated increase in microvascular perfusion and at the level of glycogen synthase.
Considering the crucial role of estrogen signaling in glucose uptake, muscle contraction may be associated with a rapid, non genomic ER activation facilitating glucose uptake. Muscle contraction improves insulin sensitivity via rapid, unliganded ER activation and regular exercise decreases the risk for cancer.
12. Conclusion
Oncogenic mutations are regarded as initiators of cancer development. Cancers with a strong family history may develop by inherited germline mutations, while bad habits and unhealthy lifestyle factors may presumably induce cancers conferred by somatic mutations.
Patients exhibiting either germline mutation or clinically diagnosed type-2 diabetes, similarly exhibit insulin resistance, fertility disorder and increased cancer risk. According to a new idea, it was suggested that these strongly associated three alterations derive from a common genomic failure and its recognition may shed light on the unsolved secret of cancer.
Germline mutations on ESR1, BRCA1, and CYP19A genes encoding estrogen receptor alpha (ERα), genome safeguarding BRCA1 protein and CYP19 aromatase enzyme, similarly cause genomic instability and increased cancer risk. BRCA1 and ESR1 gene mutations cause particularly breast cancer, while the error of CYP19A gene leads to cancers in the endometrium, ovaries and thyroid.
ERα, BRCA1 and CYP19 aromatase proteins are transcription factors creating the crucial DNA stabilizer circuit driven by the estrogen activation of ERα. Estrogen bound ERα drives a second regulatory circuit as well, for controlling cell proliferation in partnership with various growth factors and kinase cascades. In a third regulatory circuit, liganded ERα drives cellular glucose supply in close interplay with insulin, IGF-1 and glucose transporters. In the DNA stabilizer circuit, aromatase produced estrogen completes the round via a liganded activation of ERα. In the second and third circuit, growth factor signalings complete the round via an unliganded activation of ERα.
Weakening expression or activation of each transcription factor of the triad; ERα, BRCA1 or CYP19 aromatase, leads to defective estrogen synthesis or estrogen refractory receptors. Any defect of estrogen signaling endangers together the regular cell proliferation, cellular glucose supply and reproduction. Estrogen regulated hypothalamus gathers the neuro-endocrine signals of the body and the environmental influences so as to ensure adaptive answers to the internal and external changes. Weakening estrogen signaling caused by either endogenous or environmental factor is alarming for the hypothalamus and it sends neural and hormonal commands for the whole body aiming the restoration of estrogen signaling.
In the first, compensated phase of defective estrogen signaling, patients are apparently healthy. Hyperinsulinemia is a whisper of the genome notifying about the successful restoration of estrogen signaling and glucose uptake at the expense of excessive insulin synthesis. Later, the strengthening defect of estrogen signaling and impaired glucose uptake result in metabolic syndrome with various symptoms: hypertension, obesity and laboratory findings showing defects in lipid metabolism. The compensatory low grade inflammation with pro-inflammatory cytokine expression and high levels of insulin and IGF-1 are warnings from the endangered genome having difficulties in the restoration of estrogen signaling and DNA stability. The deepening defect of estrogen signaling leads to type-2 diabetes. Serum glucose levels are increasing in spite of high insulin and IGF-1 levels and other compensatory actions. At the same time organs are suffering of the lack of glucose and excessive lipid deposition. The complete breakdown of estrogen signaling results in DNA damage and uncontrolled cell proliferation leading to cancer initiation in the affected organ. Cancer development is a cry of the genome asking for help that supports its combat against deregulation.
Human body maintains its integrity and functional activity via appropriate estrogen signaling. Advantageous lifestyle factors drive genomic machinery via the upregulation of estrogen signaling ensuring the maintenance of human health. Conversely, bad habits and unhealthy lifestyle slow down estrogen synthesis or inhibit ER activation endangering all genomic functions and leading to metabolic diseases, reproductive failures and increased cancer risk.
References
- Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, Jemal A. Global cancer statistics 2022: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Phys 2024; 74(3): 229-63. Epub 2024 Apr 4. PMID: 38572751. [CrossRef]
- Kontomanolis EN, Koutras A, Syllaios A, Schizas D, Mastoraki A, Garmpis N, Diakosavvas M, Angelou K, Tsatsaris G, Pagkalos A, Ntounis T, Fasoulakis Z. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020 Nov;40(11):6009-6015. PMID: 33109539. [CrossRef]
- Mbemi A, Khanna S, Njiki S, Yedjou CG, Tchounwou PB. Impact of Gene-Environment Interactions on Cancer Development. Int J Environ Res Public Health. 2020 Nov 3;17(21):8089. PMID: 33153024; PMCID: PMC7662361. [CrossRef]
- Sinkala M. Mutational landscape of cancer-driver genes across human cancers. Sci Rep. 2023 Aug 7;13(1):12742. PMID: 37550388; PMCID: PMC10406856. [CrossRef]
- Suba Z. DNA Damage Responses in Tumors Are Not Proliferative Stimuli, but Rather They Are DNA Repair Actions Requiring Supportive Medical Care. Cancers (Basel). 2024 Apr 19;16(8):1573. PMID: 38672654; PMCID: PMC11049279. [CrossRef]
- Beatson G.T. On the treatment of inoperable cases of carcinoma of the mamma: Suggestions for a new method of treatment, with illustrative cases. Lancet. 1896;2:104–107. [CrossRef]
- Boyd S. OOPHORECTOMY IN CANCER OF THE BREAST. BMJ. 1902;1:110–111. [CrossRef]
- Suba Z. Estrogen Regulated Genes Compel Apoptosis in Breast Cancer Cells, Whilst Stimulate Antitumor Activity in Peritumoral Immune Cells in a Janus-Faced Manner. Curr Oncol. 2024 Aug 24;31(9):4885-4907. PMID: 39329990; PMCID: PMC11431267. [CrossRef]
- Reaven GM. Banting Lecture 1988. Role of insulin resistance in human disease. 1988. Nutrition. 1997 Jan;13(1):65; discussion 64, 66. PMID: 9058458. [CrossRef]
- Prasad H, Ryan DA, Celzo MF, Stapleton D. Metabolic syndrome: definition and therapeutic implications. Postgrad. Med. 2012 Jan;124(1):21-30. PMID: 22314111. [CrossRef]
- Thomas DD, Corkey BE, Istfan NW, Apovian CM. Hyperinsulinemia: An Early Indicator of Metabolic Dysfunction. J Endocr Soc. 2019 Jul 24;3(9):1727-1747. PMID: 31528832; PMCID: PMC6735759. [CrossRef]
- Nolan CJ, Prentki M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diab Vasc Dis Res. 2019 Mar;16(2):118-127. Epub 2019 Feb 15. PMID: 30770030. [CrossRef]
- Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, Ostolaza H, Martín C. Pathophysiology of Type 2 Diabetes Mellitus. Int J Mol Sci. 2020 Aug 30;21(17):6275. PMID: 32872570; PMCID: PMC7503727. [CrossRef]
- Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuñiga FA. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol. 2018 Aug 31;17(1):122. PMID: 30170598; PMCID: PMC6119242. [CrossRef]
- Szablewski L. Insulin Resistance: The Increased Risk of Cancers. Curr Oncol. 2024 Feb 13;31(2):998-1027. PMID: 38392069; PMCID: PMC10888119. [CrossRef]
- Ciarambino T, Crispino P, Guarisco G, Giordano M. Gender Differences in Insulin Resistance: New Knowledge and Perspectives. Curr Issues Mol Biol. 2023 Sep 27;45(10):7845-7861. PMID: 37886939; PMCID: PMC10605445. [CrossRef]
- Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013 Jun;34(3):309-38. Epub 2013 Mar 4. PMID: 23460719; PMCID: PMC3660717. [CrossRef]
- Suba Z. Low Estrogen Exposure and/or Defective Estrogen Signaling Induces Disturbances in Glucose Uptake and Energy Expenditure. J Diabetes Metab 2013, 4:5. [CrossRef]
- Hevener AL, Clegg DJ, Mauvais-Jarvis F. Impaired estrogen receptor action in the pathogenesis of the metabolic syndrome. Mol Cell Endocrinol. 2015 Dec 15;418 Pt 3(Pt 3):306-21. Epub 2015 May 29. PMID: 26033249; PMCID: PMC5965692. [CrossRef]
- Betai D, Ahmed AS, Saxena P, Rashid H, Patel H, Shahzadi A, Mowo-Wale AG, Nazir Z. Gender Disparities in Cardiovascular Disease and Their Management: A Review. Cureus. 2024 May 5;16(5):e59663. PMID: 38836150; PMCID: PMC11148660. [CrossRef]
- Reckelhoff JF. Sex steroids, cardiovascular disease, and hypertension: unanswered questions and some speculations. Hypertension. 2005 Feb;45(2):170-4. Epub 2004 Dec 6. PMID: 15583070. [CrossRef]
- Xiang D, Liu Y, Zhou S, Zhou E, Wang Y. Protective Effects of Estrogen on Cardiovascular Disease Mediated by Oxidative Stress. Oxid Med Cell Longev. 2021 Jun 28;2021:5523516. PMID: 34257804; PMCID: PMC8260319. [CrossRef]
- Selvaraj RC, Cioffi G, Waite KA, Jackson SS, Barnholtz-Sloan JS. A Pan-Cancer Analysis of Age and Sex Differences in Cancer Incidence and Survival in the United States, 2001-2020. Cancers (Basel). 2025 Jan 24;17(3):378. PMID: 39941747; PMCID: PMC11815994. [CrossRef]
- Suba Z. Gender-related hormonal risk factors for oral cancer. Pathol Oncol Res. 2007;13(3):195-202. Epub 2007 Oct 7. PMID: 17922048. [CrossRef]
- Heer E, Harper A, Escandor N, Sung H, McCormack V, Fidler-Benaoudia MM. Global burden and trends in premenopausal and postmenopausal breast cancer: a population-based study. Lancet Glob Health. 2020 Aug;8(8):e1027-e1037. PMID: 32710860. [CrossRef]
- Ohkuma T, Iwase M, Fujii H, Ide H, Kaizu S, Jodai T, Kikuchi Y, Idewaki Y, Sumi A, Nakamura U, Kitazono T. Joint impact of modifiable lifestyle behaviors on glycemic control and insulin resistance in patients with type 2 diabetes: the Fukuoka Diabetes Registry. Diabetol Int. 2017 Feb 17;8(3):296-305. PMID: 30603335; PMCID: PMC6224889. [CrossRef]
- Bruckner F, Gruber JR, Ruf A, Edwin Thanarajah S, Reif A, Matura S. Exploring the Link between Lifestyle, Inflammation, and Insulin Resistance through an Improved Healthy Living Index. Nutrients. 2024 Jan 29;16(3):388. PMID: 38337673; PMCID: PMC10857191. [CrossRef]
- Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006;6(3):181-202. PMID: 16515478.
- Hayes DF. Tamoxifen: Dr. Jekyll and Mr. Hyde? J Natl Cancer Inst. 2004 Jun 16;96(12):895-7. PMID: 15199102. [CrossRef]
- Suba Z. Compensatory Estrogen Signal Is Capable of DNA Repair in Antiestrogen-Responsive Cancer Cells via Activating Mutations. J Oncol. 2020 Jul 28;2020:5418365. PMID: 32774370; PMCID: PMC7407016. [CrossRef]
- Ramteke P, Deb A, Shepal V, Bhat MK. Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality. Cancers (Basel). 2019 Sep 19;11(9):1402. PMID: 31546918; PMCID: PMC6770430. [CrossRef]
- Lee HM, Lee HJ, Chang JE. Inflammatory Cytokine: An Attractive Target for Cancer Treatment. Biomedicines. 2022 Aug 29;10(9):2116. PMID: 36140220; PMCID: PMC9495935. [CrossRef]
- Rho O, Kim DJ, Kiguchi K, Digiovanni J. Growth factor signaling pathways as targets for prevention of epithelial carcinogenesis. Mol Carcinog. 2011 Apr;50(4):264-79. Epub 2010 Jul 20. PMID: 20648549; PMCID: PMC3005141. [CrossRef]
- Abdul-Ghani M, DeFronzo RA. Insulin Resistance and Hyperinsulinemia: the Egg and the Chicken. J Clin Endocrinol Metab. 2021 Mar 25;106(4):e1897-e1899. PMID: 33522574; PMCID: PMC7993580. [CrossRef]
- Wilcox G. Insulin and insulin resistance. Clin Biochem Rev. 2005 May;26(2):19-39. PMID: 16278749; PMCID: PMC1204764.
- Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell. 2013 Feb 14;152(4):673-84. PMID: 23415219. [CrossRef]
- Freeman AM, Acevedo LA, Pennings N. Insulin Resistance. 2023 Aug 17. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 29939616.
- Janssen JAMJL. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int J Mol Sci. 2021 Jul 21;22(15):7797. PMID: 34360563; PMCID: PMC8345990. [CrossRef]
- Zhang AMY, Wellberg EA, Kopp JL, Johnson JD. Hyperinsulinemia in Obesity, Inflammation, and Cancer. Diabetes Metab J. 2021 May;45(3):285-311. Epub 2021 Mar 29. Erratum in: Diabetes Metab J. 2021 Jul;45(4):622. PMID: 33775061; PMCID: PMC8164941. https://doi.org/10.4093/dmj.2021.0131. [CrossRef]
- Houston EJ, Templeman NM. Reappraising the relationship between hyperinsulinemia and insulin resistance in PCOS. J Endocrinol. 2025 Mar 12;265(2):e240269. PMID: 40013621; PMCID: PMC11906131. [CrossRef]
- Rangraze IR, El-Tanani M, Arman Rabbani S, Babiker R, Matalka II, Rizzo M. Diabetes and its Silent Partner: A Critical Review of Hyperinsulinemia and its Complications. Curr Diabetes Rev. 2025;21(9):e15733998311738. PMID: 39192649. [CrossRef]
- Janssen JAMJL. Overnutrition, Hyperinsulinemia and Ectopic Fat: It Is Time for A Paradigm Shift in the Management of Type 2 Diabetes. Int J Mol Sci. 2024 May 17;25(10):5488. PMID: 38791525; PMCID: PMC11121669. [CrossRef]
- Nijenhuis-Noort EC, Berk KA, Neggers SJCMM, Lely AJV. The Fascinating Interplay between Growth Hormone, Insulin-Like Growth Factor-1, and Insulin. Endocrinol Metab (Seoul). 2024 Feb;39(1):83-89. Epub 2024 Jan 9. PMID: 38192102; PMCID: PMC10901670. [CrossRef]
- Giustina A, Berardelli R, Gazzaruso C, Mazziotti G. Insulin and GH-IGF-I axis: endocrine pacer or endocrine disruptor? Acta Diabetol. 2015 Jun;52(3):433-43. Epub 2014 Aug 14. PMID: 25118998. [CrossRef]
- Macvanin M, Gluvic Z, Radovanovic J, Essack M, Gao X, Isenovic ER. New insights on the cardiovascular effects of IGF-1. Front Endocrinol (Lausanne). 2023 Feb 9;14:1142644.. PMID: 36843588; PMCID: PMC9947133. [CrossRef]
- Suba Z. DNA stabilization by the upregulation of estrogen signaling in BRCA gene mutation carriers. Drug Des Devel Ther. 2015 May 15;9:2663-75. PMID: 26028963; PMCID: PMC4440422. [CrossRef]
- Yan H, Yang W, Zhou F, Li X, Pan Q, Shen Z, Han G, Newell-Fugate A, Tian Y, Majeti R, Liu W, Xu Y, Wu C, Allred K, Allred C, Sun Y, Guo S. Estrogen Improves Insulin Sensitivity and Suppresses Gluconeogenesis via the Transcription Factor Foxo1. Diabetes. 2019 Feb;68(2):291-304. Epub 2018 Nov 28. PMID: 30487265; PMCID: PMC6341301. [CrossRef]
- Kuryłowicz A. Estrogens in Adipose Tissue Physiology and Obesity-Related Dysfunction. Biomedicines. 2023 Feb 24;11(3):690. PMID: 36979669; PMCID: PMC10045924. [CrossRef]
- Gregorio KCR, Laurindo CP, Machado UF. Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation. Cells. 2021 Jan 7;10(1):99. PMID: 33430527; PMCID: PMC7827878. [CrossRef]
- Rajpathak SN, Gunter MJ, Wylie-Rosett J, Ho GY, Kaplan RC, Muzumdar R, Rohan TE, Strickler HD. The role of insulin-like growth factor-I and its binding proteins in glucose homeostasis and type 2 diabetes. Diabetes Metab Res Rev. 2009 Jan;25(1):3-12. PMID: 19145587; PMCID: PMC4153414. [CrossRef]
- Salminen A, Kaarniranta K, Kauppinen A. Insulin/IGF-1 signaling promotes immunosuppression via the STAT3 pathway: impact on the aging process and age-related diseases. Inflamm Res. 2021 Dec;70(10-12):1043-1061. Epub 2021 Sep 2. PMID: 34476533; PMCID: PMC8572812. [CrossRef]
- Ohlsson C, Hammarstedt A, Vandenput L, Saarinen N, Ryberg H, Windahl SH, Farman HH, Jansson JO, Movérare-Skrtic S, Smith U, Zhang FP, Poutanen M, Hedjazifar S, Sjögren K. Increased adipose tissue aromatase activity improves insulin sensitivity and reduces adipose tissue inflammation in male mice. Am J Physiol Endocrinol Metab. 2017 Oct 1;313(4):E450-E462. Epub 2017 Jun 27. PMID: 28655716; PMCID: PMC5668598. [CrossRef]
- Saponaro C, Gaggini M, Carli F, Gastaldelli A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients. 2015 Nov 13;7(11):9453-74. PMID: 26580649; PMCID: PMC4663603. [CrossRef]
- Chakrabarti P, Kim JY, Singh M, Shin YK, Kim J, Kumbrink J, Wu Y, Lee MJ, Kirsch KH, Fried SK, Kandror KV. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol Cell Biol. 2013 Sep;33(18):3659-66. Epub 2013 Jul 15. PMID: 23858058; PMCID: PMC3753874. [CrossRef]
- Ko SH, Kim HS. Menopause-Associated Lipid Metabolic Disorders and Foods Beneficial for Postmenopausal Women. Nutrients. 2020 Jan 13;12(1):202. PMID: 31941004; PMCID: PMC7019719. [CrossRef]
- Yin C, Liu WH, Liu Y, Wang L, Xiao Y. PID1 alters the antilipolytic action of insulin and increases lipolysis via inhibition of AKT/PKA pathway activation. PLoS One. 2019 Apr 16;14(4):e0214606. Erratum in: PLoS One. 2019 Jun 17;14(6):e0218721. doi: 10.1371/journal.pone.0218721. PMID: 30990811; PMCID: PMC6467375. [CrossRef]
- Takeuchi T, Kubota T, Nakanishi Y, Tsugawa H, Suda W, Kwon AT, Yazaki J, Ikeda K, Nemoto S, Mochizuki Y, Kitami T, Yugi K, Mizuno Y, Yamamichi N, Yamazaki T, Takamoto I, Kubota N, Kadowaki T, Arner E, Carninci P, Ohara O, Arita M, Hattori M, Koyasu S, Ohno H. Gut microbial carbohydrate metabolism contributes to insulin resistance. Nature. 2023 Sep;621(7978):389-395. Epub 2023 Aug 30. PMID: 37648852; PMCID: PMC10499599. [CrossRef]
- Semo D, Reinecke H, Godfrey R. Gut microbiome regulates inflammation and insulin resistance: a novel therapeutic target to improve insulin sensitivity. Signal Transduct Target Ther. 2024 Feb 21;9(1):35. PMID: 38378663; PMCID: PMC10879501. [CrossRef]
- Wang H, Shi F, Zheng L, Zhou W, Mi B, Wu S, Feng X. Gut microbiota has the potential to improve health of menopausal women by regulating estrogen. Front Endocrinol (Lausanne). 2025 Jun 9;16:1562332. PMID: 40551890; PMCID: PMC12183514. [CrossRef]
- Rishabh, Bansal S, Goel A, Gupta S, Malik D, Bansal N. Unravelling the Crosstalk between Estrogen Deficiency and Gut-biotaDysbiosis in the Development of Diabetes Mellitus. Curr Diabetes Rev. 2024;20(10):e240124226067. PMID: 38275037. [CrossRef]
- Link CD. Is There a Brain Microbiome? Neurosci Insights. 2021 May 27;16:26331055211018709. PMID: 34104888; PMCID: PMC8165828. [CrossRef]
- Kim J-a, Wei Y, Showers JR. Role of Mitochondrial Dysfunction in Insulin Resistance. Circulation Research 2008; 102(4) 401-14. [CrossRef]
- Huang, X., Pan, CH., Yin, F. et al. The Role of Estrogen in Mitochondrial Disease. Cell Mol Neurobiol 2025 45, 68. [CrossRef]
- Tao Z, Cheng Z. Hormonal regulation of metabolism-recent lessons learned from insulin and estrogen. Clin Sci (Lond). 2023 Mar 31;137(6):415-434. PMID: 36942499; PMCID: PMC10031253. [CrossRef]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266(5182): 66-71.
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378(6559): 789-92.
- Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002; 108: 171–182.
- Gorski JJ, Kennedy RD, Hosey AM, Harkin DP. The complex relationship between BRCA1 and ER alpha in hereditary breast cancer. Clin Cancer Res 2009; 15(5): 1514-8. [CrossRef]
- Wang L, Di LJ. BRCA1 and estrogen/estrogen receptor in breast cancer: where they interact? Int J Biol Sci 2014; 10(5): 566-75.
- Lakhani SR, Van De Vijver MJ, Jacquemier J, et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol. 2002; 20: 2310–8. [CrossRef]
- Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001; 98: 10869–74.
- Santana dos Santos, E.; Lallemand, F.; Petitalot, A.; Caputo, S.M.; Rouleau, E. HRness in Breast and Ovarian Cancers. Int. J. Mol. Sci. 2020, 21, 3850. [CrossRef]
- Spillman MA, Bowcock AM. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene. 1996; 13: 1639–45.
- Suba Z. Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: preventive and therapeutic implications. OncoTargets Ther 2014; 7: 147-64.
- Fan S, Wang J, Yuan R, et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 1999; 284: 1354–6. [CrossRef]
- Xu J, Fan S, Rosen EM. Regulation of the estrogen-inducible gene expression profile by the breast cancer susceptibility gene BRCA1. Endocrinology 2005; 146: 2031–47.
- Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG. et al. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Molecular Endocrinology 2010; 24: 76–90. [CrossRef]
- Fan S, Ma YX, Wang C, et al. p300 Modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Res 2002; 62: 141–51.
- Wang C, Fan S, Li Z, Fu M, Rao M, Ma Y. et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor alpha activity. Cancer research 2005; 65: 6557–67.
- Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, Erdos M, Goldberg ID, Webb P, Kushner PJ, Pestell RG, Rosen EM. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001; 20(1): 77-87. [CrossRef]
- Russo J., Russo I. H. Toward a unified concept of mammary carcinogenesis Aldaz M. C. Gould M. N. McLachlan J. Slaga T. J. eds. Progress in Clinical and Biological Research: 1-16, Wiley-Liss New York 1997.
- Hosey AM, Gorski JJ, Murray MM, Quinn JE, Chung WY, Stewart GE. et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J Natl Cancer Inst 2007; 99: 1683–94. [CrossRef]
- Burga, L.N., Hu, H., Juvekar, A. et al. Loss of BRCA1 leads to an increase in epidermal growth factor receptor expression in mammary epithelial cells, and epidermal growth factor receptor inhibition prevents estrogen receptor-negative cancers in BRCA1-mutant mice. Breast Cancer Res 2011; 13: R30.
- Ma Y, Hu Ch, Riegel AT, Fan S, Rosen EM. Growth factor signaling pathways modulate BRCA1 repression of estrogen receptor-alpha activity. Mol Endocrinol 2007; 21(8): 1905-23.
- Ghosh S, Lu Y, Katz A, Hu Y, Li R. Tumor suppressor BRCA1 inhibits a breast cancer-associated promoter of the aromatase gene (CYP19) in human adipose stromal cells. Am J Physiol Endocrinol Metab 2007; 292: 246–252. [CrossRef]
- Sau A, Lau R, Cabrita MA, Nolan E, Crooks PA et al. Persistent Activation of NF-κB in BRCA1-Deficient Mammary Progenitors Drives Aberrant Proliferation and Accumulation of DNA Damage Cell Stem Cell 2016; 19(1): 52-65.
- Zheng L, Annab LA, Afshari CA, Lee WH, Boyer TG. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci USA. 2001; 98:9587–92. [CrossRef]
- Arnold A , Papanikolaou A. Cyclin D1 in Breast Cancer Pathogenesis. J Clin Oncol 2005; 23(18): 4215-24.
- Wang X, El-Halaby AA, Zhang H, Yang Q, Laughlin TS, Rothberg PG, Skinner K, Hicks DG. p53 alteration in morphologically normal/benign breast luminal cells in BRCA carriers with or without history of breast cancer. Hum Pathol. 2017 Oct;68:22-25. Epub 2017 Apr 21. PMID: 28438622. [CrossRef]
- Oktay K, Kim JY, Barad D, Babayev SN. Association of BRCA1 mutations with occult primary ovarian insufficiency: a possible explanation for the link between infertility and breast/ovarian cancer risks. J Clin Oncol 2010; 28(2): 240-4.
- Lin WT, Beattie M, Chen LM, Oktay K, Crawford SL, Gold EB, Cedars M, Rosen M. Comparison of age at natural menopause in BRCA1/2 mutation carriers with a non-clinic-based sample of women in northern California. Cancer 2013; 119(9): 1652-9.
- Chand AL, Simpson ER, Clyne CD. Aromatase expression is increased in BRCA1 mutation carriers. BMC Cancer. 2009; 9: 148. [CrossRef]
- Bruno E, Manoukian S, Venturelli E, Oliverio A, Rovera F, Iula G, Morelli D, Peissel B, Azzolini J, Roveda E, Pasanisi P. Adherence to Mediterranean Diet and Metabolic Syndrome in BRCA Mutation Carriers. Integr Cancer Ther. 2018 Mar;17(1):153-160. Epub 2017 Jul 25. PMID: 28741383; PMCID: PMC5950953. [CrossRef]
- Lentscher JA, Decherney AH. Clinical Presentation and Diagnosis of Polycystic Ovarian Syndrome. Clin Obstet Gynecol. 2021 Mar 1;64(1):3-11. PMID: 32701517; PMCID: PMC10683967. [CrossRef]
- Zańko A, Siewko K, Krętowski AJ, Milewski R. Lifestyle, Insulin Resistance and Semen Quality as Co-Dependent Factors of Male Infertility. Int J Environ Res Public Health. 2022 Dec 30;20(1):732. PMID: 36613051; PMCID: PMC9819053. [CrossRef]
- Dong J, Rees DA. Polycystic ovary syndrome: pathophysiology and therapeutic opportunities. BMJ Med. 2023 Oct 12;2(1):e000548. PMID: 37859784; PMCID: PMC10583117. [CrossRef]
- Blackwood SJ, Tischer D, Pontén M, Moberg M, Katz A. Relationship between insulin sensitivity and hyperinsulinemia in early insulin resistance is sex-dependent. J Clin Endocrinol Metab. 2025 May 13:dgaf282. Epub ahead of print. PMID: 40356550. [CrossRef]
- Hernández-Jiménez JL, Barrera D, Espinoza-Simón E, González J, Ortíz-Hernández R, Escobar L, Echeverría O, Torres-Ramírez N. Polycystic ovarian syndrome: signs and feedback effects of hyperandrogenism and insulin resistance. Gynecological Endocrinology. 2022 Jan 2;38(1):2-9.
- Bulun SE. Aromatase and estrogen receptor α deficiency. Fertil Steril. 2014 Feb;101(2):323-9. PMID: 24485503; PMCID: PMC3939057. [CrossRef]
- Suba Z. Diverse pathomechanisms leading to the breakdown of cellular estrogen surveillance and breast cancer development: new therapeutic strategies. Drug Des Devel Ther. 2014 Sep 11;8:1381-90. PMID: 25246776; PMCID: PMC4166254. [CrossRef]
- Quaynor SD, Stradtman EW Jr, Kim HG, Shen Y, Chorich LP, Schreihofer DA, Layman LC. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med. 2013 Jul 11;369(2):164-71. PMID: 23841731; PMCID: PMC3823379. [CrossRef]
- Scicchitano P, Dentamaro I, Carbonara R, Bulzis G, Dachille A, Caputo P, Riccardi R, Locorotondo M, Mandurino C, Matteo Ciccone M. Cardiovascular Risk in Women With PCOS. Int J Endocrinol Metab. 2012; 10(4):611-8. Epub 2012 Sep 30. PMID: 23843832; PMCID: PMC3693634. [CrossRef]
- Bird ST, Hartzema AG, Brophy JM, Etminan M, Delaney JA. Risk of venous thromboembolism in women with polycystic ovary syndrome: a population-based matched cohort analysis. CMAJ. 2013 Feb 5;185(2):E115-20. Epub 2012 Dec 3. PMID: 23209115; PMCID: PMC3563911. [CrossRef]
- Wang J, Yin T, Liu S. Dysregulation of immune response in PCOS organ system. Front Immunol. 2023 May 5;14:1169232. PMID: 37215125; PMCID: PMC10196194. [CrossRef]
- Yuk JS, Noh JH, Han GH, Yoon SH, Kim M. Risk of cancers in women with polycystic ovary syndrome: Cohort study based on health insurance database in South Korea. Int J Gynaecol Obstet. 2025 Sep 27. Epub ahead of print. PMID: 41014020. [CrossRef]
- Ignatov A, Ortmann O. Endocrine Risk Factors of Endometrial Cancer: Polycystic Ovary Syndrome, Oral Contraceptives, Infertility, Tamoxifen. Cancers (Basel). 2020 Jul 2;12(7):1766. PMID: 32630728; PMCID: PMC7408229. [CrossRef]
- Xu XL, Deng SL, Lian ZX, Yu K. Estrogen Receptors in Polycystic Ovary Syndrome. Cells. 2021 Feb 21;10(2):459. PMID: 33669960; PMCID: PMC7924872. [CrossRef]
- Casper RF, Mitwally MF. Use of the aromatase inhibitor letrozole for ovulation induction in women with polycystic ovarian syndrome. Clin Obstet Gynecol. 2011 Dec;54(4):685-95. PMID: 22031258. [CrossRef]
- Maggi A. Liganded and unliganded activation of estrogen receptor and hormone replacement therapies. Biochim Biophys Acta 2011; 1812(8): 1054–1060. [CrossRef]
- Curtis SW, Washburn T, Sewall C, DiAugustine R, Lindzey J, Couse JF et al. Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci USA 1996; 93(22): 12626–30. [CrossRef]
- Levin ER. Bidirectional Signaling between the Estrogen Receptor and the Epidermal Growth Factor Receptor. Mol Endocrinol 2003; 17(3): 309–317. [CrossRef]
- Suba, Z. Amplified crosstalk between estrogen binding and GFR signaling mediated pathways of ER activation drives responses in tumors treated with endocrine disruptors. Recent Pat Anticancer Drug Discov (2018) 13(4): 428-444. [CrossRef]
- Suba Z. Rosetta Stone for Cancer Cure: Comparison of the Anticancer Capacity of Endogenous Estrogens, Synthetic Estrogens and Antiestrogens. Oncol Rev. 2023 Apr 19;17:10708. PMID: 37152665; PMCID: PMC10154579. [CrossRef]
- Suba Z. Activating Mutations of ESR1, BRCA1 and CYP19 Aromatase Genes Confer Tumor Response in Breast Cancers Treated with Antiestrogens. Recent Pat Anticancer Drug Discov. 2017;12(2):136-147. PMID: 28245776. [CrossRef]
- Barros RPA, Gustafsson JÅ. Estrogen Receptors and the Metabolic Network. Cell Metabolism (2011) 14(3): 289-299. [CrossRef]
- Tiano JP, Mauvais-Jarvis F. Importance of oestrogen receptors to preserve functional β-cell mass in diabetes. Nat Rev Endocrinol (2012) 8(6): 342-51. [CrossRef]
- Choi SB, Jang JS, Park S. Estrogen and exercise may enhance beta-cell function and mass via insulin receptor substrate 2 induction in ovariectomized diabetic rats. Endocrinology (2005) 146(11): 4786-94. [CrossRef]
- Campello RS, Fátima LA, Barreto-Andrade JN, Lucas TF, Mori RC, Porto CS, Machado UF. Estradiol-induced regulation of GLUT4 in 3T3-L1 cells: involvement of ESR1 and AKT activation. J Mol Endocrinol (2017) 59(3): 257-268. [CrossRef]
- Steiner BM, Berry DC. The Regulation of Adipose Tissue Health by Estrogens. Front Endocrinol (Lausanne). 2022 May 26;13:889923. PMID: 35721736; PMCID: PMC9204494. [CrossRef]
- Lizcano F, Guzmán G. Estrogen Deficiency and the Origin of Obesity during Menopause. Biomed Res Int. 2014;2014:757461. Epub 2014 Mar 6. PMID: 24734243; PMCID: PMC3964739. [CrossRef]
- Barakat R, Oakley O, Kim H, Jin J, Ko CJ. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016; 49(9): 488-96.
- Labrie F, Bélanger A, Luu-The V, et al. DHEA and the intracrine formation of androgens and estrogens in peripheral target tissues: its role during aging. Steroids. 1998; 63: 322–328.
- Bjune J-I, Strømland PP, Jersin RÅ, Mellgren G, Dankel SN. Metabolic and Epigenetic Regulation by Estrogen in Adipocytes. Front Endocrinol (Lausanne) 2022; 13: 828780. [CrossRef]
- Dieudonné MN, Leneveu MC, Giudicelli Y, Pecquery R. Evidence for functional estrogen receptors α and β in human adipose cells: regional specificities and regulation by estrogens. Am J Physiol-Cell Physiol 2004; 286(3): C655-C661.
- Kim JH, Cho HT, Kim YJ. The role of estrogen in adipose tissue metabolism: insights into glucose homeostasis regulation. Endocr J. 2014;61(11):1055-67. Epub 2014 Aug 9. PMID: 25109846. [CrossRef]
- Ahmed F, Kamble PG, Hetty S, Fanni G, Vranic M, Sarsenbayeva A, Kristófi R, Almby K, Svensson MK, Pereira MJ, Eriksson JW, Role of Estrogen and Its Receptors in Adipose Tissue Glucose Metabolism in Pre- and Postmenopausal Women. The Journal of Clinical Endocrinology & Metabolism, 2022; 107(5), e1879–e1889. [CrossRef]
- Donohoe CL, Doyle SL, Reynolds JV. Visceral adiposity, insulin resistance and cancer risk. Diabetol Metab Syndr 2011; 3: 12.
- Pelekanou V, Leclercq G. Recent insights into the effect of natural and environmental estrogens on mammary development and carcinogenesis The Int J of Develop Biol 2011; 55(7-9): 869-78.
- Wang H, Leng Y, Gong Y. Bone Marrow Fat and Hematopoiesis. Front. Endocrinol 2018; 9: 694. [CrossRef]
- Wang P, Mariman E, Renes J, Keijer J. The Secretory Function of Adipocytes in the Physiology of White Adipose Tissue. J Cell Physiol 2008; 216: 3–13.
- Monteiro R, Teixeira D, Calhau C. Estrogen signaling in metabolic inflammation. Mediators Inflamm. 2014; 2014: 615917. Epub 2014 Oct 23. PMID: 25400333; PMCID: PMC4226184. [CrossRef]
- Boon WC, Jenny D.Y. Chow JDY, Simpson ER. The Multiple Roles of Estrogens and the Enzyme Aromatase. 2010; 181: 209-32. [CrossRef]
- Clegg DJ, Brown LM, Woods SC, Benoit SC. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes. 2006; 55: 978–987.
- Combs TP, Pajvani UB, Berg AH, et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology. 2004; 145(1): 367–383. [CrossRef]
- Steppan CM, Bailey ST, Bhat S, et al. The hormone resistin links obesity to diabetes. Nature. 2001; 409(6818): 307–12.
- Pradhan G, Samson SL, Sun Y. Ghrelin: much more than a hunger hormone. Curr Opin Clin Nutr Metab Care. 2013 Nov;16(6):619-24. PMID: 24100676; PMCID: PMC4049314. [CrossRef]
- Smith A, Woodside B, Abizaid A. Ghrelin and the Control of Energy Balance in Females. Front Endocrinol (Lausanne). 2022 Jul 15;13:904754. PMID: 35909536; PMCID: PMC9334675. [CrossRef]
- Suba Z. Crossroad between obesity and cancer: a defective signaling function of heavily lipid laden adipocytes (Online First). In: Crosstalk in Biological Processes. Ed: El-Esawi MA. InTechOpen, London, May 3rd 2019. [CrossRef]
- Lueprasitsakul P, Latour D, Longcope C. Aromatase activity in human adipose tissue stromal cells: effect of growth factors. Steroids. 1990 Dec;55(12):540-4. PMID: 1965238. [CrossRef]
- Armani A, Berry A, Cirulli F, Caprio M. Molecular mechanisms underlying metabolic syndrome: the expanding role of the adipocyte. FASEB J. 2017; 31(10): 4240-4255.
- De Pergola G, Silvestris F. Obesity as a major risk factor for cancer. J Obes. 2013; 2013: 291546.
- Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol. 2021 Mar 1;320(3):C375-C391. Epub 2020 Dec 23. PMID: 33356944; PMCID: PMC8294624. [CrossRef]
- Huh JY, Park YJ, Ham M, and Kim JB. Crosstalk between Adipocytes and Immune Cells in Adipose Tissue Inflammation and Metabolic Dysregulation in Obesity. Mol Cells 2014; 37(5): 365–371.
- Purohit A, Newman SP, Reed MJ. The role of cytokines in regulating estrogen synthesis: implications for the etiology of breast cancer. Breast Cancer Research 2002; 4: 65.
- Hope MC, Unger CA, Kettering MC, Socia CE, Aladhami AK, Rice BC, Niamira DS, Wiznitzer BP, Altomare D, Cotham WE, Enos RT. Adipose Tissue Estrogen Receptor-Alpha Overexpression Ameliorates High-Fat Diet-Induced Adipose Tissue Inflammation. J Endocr Soc. 2025 Aug 19;9(10):bvaf134. PMID: 40980540; PMCID: PMC12448864. [CrossRef]
- Blum WF, Alherbish A, Alsagheir A, El Awwa A, Kaplan W, Koledova E, Savage MO. The growth hormone-insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocr Connect. 2018 Jun;7(6):R212-R222. Epub 2018 May 3. PMID: 29724795; PMCID: PMC5987361. [CrossRef]
- Yakar S, Adamo ML. Insulin-like growth factor 1 physiology: lessons from mouse models. Endocrinol Metab Clin North Am. 2012 Jun;41(2):231-47, v. Epub 2012 May 15. PMID: 22682628; PMCID: PMC5488279. [CrossRef]
- Boucher J, Tseng J-H, Kahn CR. Insulin and insulin-like growth factor receptors act as ligand-specific amplitude modulators of a common pathway regulating gene. J Biol Chem 2010; 285(22): 17235-17245.
- Caizzi L, Ferrero G, Cutrupi S, Cordero F, Ballaré C, Miano V, et al. Genome-wide activity of unliganded estrogen receptor-α in breast cancer cells. Proc Natl Acad Sci USA 2014; 111(13): 4892-7.
- Barthelemy J, Bogard G, Wolowczuk I. Beyond energy balance regulation: The underestimated role of adipose tissues in host defense against pathogens. Front Immunol. 2023 Mar 2;14:1083191. PMID: 36936928; PMCID: PMC10019896. [CrossRef]
- Millas I, Duarte Barros M. Estrogen receptors and their roles in the immune and respiratory systems. Anat Rec (Hoboken). 2021 Jun; 304(6): 1185-1193. Epub 2021 Apr 15. PMID: 33856123. [CrossRef]
- Bradley D, Deng T, Shantaram D, Hsueh WA. Orchestration of the Adipose Tissue Immune Landscape by Adipocytes. Annu Rev Physiol. 2024 Feb 12;86:199-223. PMID: 38345903. [CrossRef]
- Stubbins RE, Najjar K, Holcomb VB, Hong J, Núñez NP. Oestrogen alters adipocyte biology and protects female mice from adipocyte inflammation and insulin resistance. Diabetes, Obesity and Metabolism. 2012; 14(1): 58–66.
- Wang T, Wang J, Hu X, Huang XJ, Chen GX. Current understanding of glucose transporter 4 expression and functional mechanisms. World J Biol Chem. 2020 Nov 27;11(3):76-98. PMID: 33274014; PMCID: PMC7672939. [CrossRef]
- Warburton DE, Nicol CW, Bredin SS. Health benefits of physical activity: the evidence. CMAJ. 2006 Mar 14;174(6):801-9. PMID: 16534088; PMCID: PMC1402378. [CrossRef]
- Pereira RM, Pereira de Moura L, Muñoz VR et als. Molecular mechanisms of glucose uptake in skeletal muscle at rest and in response to exercise. 2017, Motriz Revista de Educação Física 23(spe). [CrossRef]
- Merino B, García-Arévalo M. Sexual hormones and diabetes: The impact of estradiol in pancreatic β cell. Int Rev Cell Mol Biol. 2021; 359: 81-138. Epub 2021 Mar 16. PMID: 33832654. [CrossRef]
- Khan MZ, Zugaza JL, Torres Aleman I. The signaling landscape of insulin-like growth factor 1. J Biol Chem. 2025 Jan;301(1):108047. Epub 2024 Dec 3. PMID: 39638246; PMCID: PMC11748690. [CrossRef]
- Schwartz N, Verma A, Bivens CB, Schwartz Z, Boyan BD. Rapid steroid hormone actions via membrane receptors. Biochim Biophys Acta. 2016 Sep;1863(9):2289-98. Epub 2016 Jun 22. PMID: 27288742. [CrossRef]
- Mendelsohn ME, Karas RH. Rapid progress for non-nuclear estrogen receptor signaling. J Clin Invest. 2010 Jul;120(7):2277-9. Epub 2010 Jun 23. PMID: 20577045; PMCID: PMC2898619. [CrossRef]
- Dichtel LE, Cordoba-Chacon J, Kineman RD. Growth Hormone and Insulin-Like Growth Factor 1 Regulation of Nonalcoholic Fatty Liver Disease. J Clin Endocrinol Metab. 2022 Jun 16;107(7):1812-1824. PMID: 35172328; PMCID: PMC9202731. [CrossRef]
- Fernández-Pérez L, Guerra B, Díaz-Chico JC, Flores-Morales A. Estrogens regulate the hepatic effects of growth hormone, a hormonal interplay with multiple fates. Front Endocrinol (Lausanne). 2013 Jun 3;4:66. PMID: 23761784; PMCID: PMC3670000. [CrossRef]
- Chaturantabut S, Shwartz A, Evason KJ, Cox AG, Labella K, Schepers AG, Yang S, Acuña M, Houvras Y, Mancio-Silva L, Romano S, Gorelick DA, Cohen DE, Zon LI, Bhatia SN, North TE, Goessling W. Estrogen Activation of G-Protein-Coupled Estrogen Receptor 1 Regulates Phosphoinositide 3-Kinase and mTOR Signaling to Promote Liver Growth in Zebrafish and Proliferation of Human Hepatocytes. Gastroenterology. 2019 May;156(6):1788-1804.e13. Epub 2019 Jan 12. PMID: 30641053; PMCID: PMC6532055. [CrossRef]
- Faulds MH, Zhao C, Dahlman-Wright K, Gustafsson JÅ. The diversity of sex steroid action: regulation of metabolism by estrogen signaling. J Endocrinol. 2012 Jan;212(1):3-12. Epub 2011 Apr 21. PMID: 21511884. [CrossRef]
- Yakar S, Adamo ML. Insulin-like growth factor 1 physiology: lessons from mouse models. Endocrinol Metab Clin North Am. 2012 Jun;41(2):231-47, v. Epub 2012 May 15. PMID: 22682628; PMCID: PMC5488279. [CrossRef]
- Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000 Jun 16;275(24):18447-53. PMID: 10749889. [CrossRef]
- Flores B, Trivedi HD, Robson SC, Bonder A. Hemostasis, bleeding and thrombosis in liver disease. J Transl Sci. 2017 May;3(3):10.15761/JTS.1000182. Epub 2017 Mar 4. PMID: 30221012; PMCID: PMC6136435. [CrossRef]
- Muciño-Bermejo J, Carrillo-Esper R, Méndez-Sánchez N, Uribe M. Thrombosis and hemorrhage in the critically ill cirrhotic patients: five years retrospective prevalence study. Ann Hepatol. 2015 Jan-Feb;14(1):93-8. PMID: 25536646.
- Kostallari E, Schwabe RF, Guillot A. Inflammation and immunity in liver homeostasis and disease: a nexus of hepatocytes, nonparenchymal cells and immune cells. Cell Mol Immunol. 2025 Oct;22(10):1205-1225. Epub 2025 Jul 1. PMID: 40595432; PMCID: PMC12480768. [CrossRef]
- Bryzgalova G, Lundholm L, Portwood N, Gustafsson JA, Khan A, Efendic S, Dahlman-Wright K. Mechanisms of antidiabetogenic and body weight-lowering effects of estrogen in high-fat diet-fed mice. Am J Physiol Endocrinol Metab. 2008 Oct;295(4):E904-12. Epub 2008 Aug 12. PMID: 18697913; PMCID: PMC2575902. [CrossRef]
- Hamden, K., Carreau, S., Ellouz, F. et al. Protective effect of 17β-estradiol on oxidative stress and liver dysfunction in aged male rats. J. Physiol. Biochem. 63, 195–201 (2007). [CrossRef]
- Anagnostis P, Stevenson JC, Crook D, Johnston DG, Godsland IF. Effects of menopause, gender and age on lipids and high-density lipoprotein cholesterol subfractions. Maturitas. 2015 May;81(1):62-8. Epub 2015 Mar 6. PMID: 25804951. [CrossRef]
- Anagnostis P, Lambrinoudaki I, Stevenson JC, Goulis DG. Menopause-associated risk of cardiovascular disease. Endocr Connect. 2022 Apr 22;11(4):e210537. PMID: 35258483; PMCID: PMC9066596. [CrossRef]
- Cetin EG, Demir N, Sen I. The Relationship between Insulin Resistance and Liver Damage in non-alcoholic Fatty Liver Patients. Sisli Etfal Hastan Tip Bul. 2020 Dec 11;54(4):411-415. PMID: 33364879; PMCID: PMC7751232. [CrossRef]
- Talamantes S, Lisjak M, Gilglioni EH, Llamoza-Torres CJ, Ramos-Molina B, Gurzov EN. Non-alcoholic fatty liver disease and diabetes mellitus as growing aetiologies of hepatocellular carcinoma. JHEP Rep. 2023 Jun 9;5(9):100811. PMID: 37575883; PMCID: PMC10413159. [CrossRef]
- Goel M, Mittal A, Jain VR, Bharadwaj A, Modi S, Ahuja G, Jain A, Kumar K. Integrative Functions of the Hypothalamus: Linking Cognition, Emotion and Physiology for Well-being and Adaptability. Ann Neurosci. 2025 Apr;32(2):128-142. Epub 2024 Jun 12. PMID: 39544638; PMCID: PMC11559822. [CrossRef]
- Zeng Y, Rong R, You M, Zhu P, Zhang J, Xia X. Light-eye-body axis: exploring the network from retinal illumination to systemic regulation. Theranostics. 2025 Jan 2;15(4):1496-1523. PMID: 39816683; PMCID: PMC11729557. [CrossRef]
- Alvord VM, Kantra EJ, Pendergast JS. Estrogens and the circadian system. Semin Cell Dev Biol. 2022 Jun;126:56-65. Epub 2021 May 9. PMID: 33975754; PMCID: PMC8573061. [CrossRef]
- Rettberg JR, Yao J, Brinton RD. Estrogen: a master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol. 2014 Jan;35(1):8-30. Epub 2013 Aug 29. PMID: 23994581; PMCID: PMC4024050. [CrossRef]
- Hara Y, Waters EM, McEwen BS, Morrison JH. Estrogen Effects on Cognitive and Synaptic Health Over the Lifecourse. Physiol Rev. 2015 Jul;95(3):785-807. PMID: 26109339; PMCID: PMC4491541. [CrossRef]
- Acevedo-Rodriguez A, Kauffman AS, Cherrington BD, Borges CS, Roepke TA, Laconi M. Emerging insights into hypothalamic-pituitary-gonadal axis regulation and interaction with stress signaling. J Neuroendocrinol. 2018 Oct;30(10):e12590. Epub 2018 Aug 7. PMID: 29524268; PMCID: PMC6129417. [CrossRef]
- Taneja V. Sex Hormones Determine Immune Response. Front Immunol. 2018 Aug 27;9:1931. PMID: 30210492; PMCID: PMC6119719. [CrossRef]
- Torres Irizarry VC, Jiang Y, He Y, Xu P. Hypothalamic Estrogen Signaling and Adipose Tissue Metabolism in Energy Homeostasis. Front Endocrinol (Lausanne). 2022 Jun 9;13:898139. PMID: 35757435; PMCID: PMC9218066. [CrossRef]
- Rossi MA. Control of energy homeostasis by the lateral hypothalamic area. Trends Neurosci. 2023 Sep;46(9):738-749. Epub 2023 Jun 22. PMID: 37353461; PMCID: PMC10524917. [CrossRef]
- Sun X, Liu B, Yuan Y, Rong Y, Pang R, Li Q. Neural and hormonal mechanisms of appetite regulation during eating. Front Nutr. 2025 Mar 24;12:1484827. PMID: 40201582; PMCID: PMC11977392. [CrossRef]
- Butera PC. Estradiol and the control of food intake. Physiol Behav. 2010 Feb 9;99(2):175-80. Epub 2009 Jun 23. PMID: 19555704; PMCID: PMC2813989. [CrossRef]
- Islami F, Marlow EC, Thomson B, McCullough ML, Rumgay H, Gapstur SM, Patel AV, Soerjomataram I, Jemal A. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States, 2019. CA Cancer J Clin. 2024 Sep-Oct;74(5):405-432. Epub 2024 Jul 11. PMID: 38990124. [CrossRef]
- IARC Working Group. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Volume 100E: Personal Habits and Indoor Combustions. IARC Press; 2012.
- Cena H, Fonte ML, Turconi G. Relationship between smoking and metabolic syndrome. Nutr Rev. 2011 Dec;69(12):745-53. Erratum in: Nutr Rev. 2013 Apr;71(4):255. PMID: 22133198. [CrossRef]
- Artese A, Stamford BA, Moffatt RJ. Cigarette Smoking: An Accessory to the Development of Insulin Resistance. Am J Lifestyle Med. 2017 Aug 23;13(6):602-605. PMID: 31662726; PMCID: PMC6796230. [CrossRef]
- Barbieri RL, Gochberg J, Ryan KJ. Nicotine, cotinine, and anabasine inhibit aromatase in human trophoblast in vitro. J Clin Invest. 1986 Jun;77(6):1727-33. PMID: 3711333; PMCID: PMC370526. [CrossRef]
- Biegon A, Alia-Klein N, Fowler JS. Potential contribution of aromatase inhibition to the effects of nicotine and related compounds on the brain. Front Pharmacol. 2012 Nov 6;3:185. PMID: 23133418; PMCID: PMC3490106. [CrossRef]
- Wan Q, Liu Y, Guan Q, Gao L, Lee KO, Zhao J. Ethanol feeding impairs insulin-stimulated glucose uptake in isolated rat skeletal muscle: role of Gs alpha and cAMP. Alcohol Clin Exp Res. 2005 Aug;29(8):1450-6. PMID: 16131853. [CrossRef]
- Wannamethee SG, Shaper AG, Perry IJ, Alberti KG. Alcohol consumption and the incidence of type II diabetes. J Epidemiol Community Health. 2002 Jul;56(7):542-8. PMID: 12080164; PMCID: PMC1732195. [CrossRef]
- Johnson C.H., Golla J.P., Dioletis E., Singh S., Ishii M., Charkoftaki G., Thompson D.C., Vasiliou V. Molecular Mechanisms of Alcohol-Induced Colorectal Carcinogenesis. Cancers. 2021;13:4404. [CrossRef]
- Orywal K., Szmitkowski M. Alcohol Dehydrogenase and Aldehyde Dehydrogenase in Malignant Neoplasms. Clin. Exp. Med. 2017;17:131–139. [CrossRef]
- Rachdaoui N, Sarkar DK. Effects of alcohol on the endocrine system. Endocrinol Metab Clin North Am. 2013 Sep;42(3):593-615. PMID: 24011889; PMCID: PMC3767933. [CrossRef]
- Fanfarillo F, Caronti B, Lucarelli M, Francati S, Tarani L, Ceccanti M, Piccioni MG, Verdone L, Caserta M, Venditti S, Ferraguti G, Fiore M. Alcohol Consumption and Breast and Ovarian Cancer Development: Molecular Pathways and Mechanisms. Curr Issues Mol Biol. 2024 Dec 20;46(12):14438-14452. PMID: 39727994; PMCID: PMC11674816. [CrossRef]
- Chen JR, Lazarenko OP, Haley RL, Blackburn ML, Badger TM, Ronis MJ. Ethanol impairs estrogen receptor signaling resulting in accelerated activation of senescence pathways, whereas estradiol attenuates the effects of ethanol in osteoblasts. J Bone Miner Res. 2009 Feb;24(2):221-30. PMID: 18847333; PMCID: PMC3276356. [CrossRef]
- Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K; International Agency for Research on Cancer Handbook Working Group. Body Fatness and Cancer--Viewpoint of the IARC Working Group. N Engl J Med. 2016 Aug 25;375(8):794-8. PMID: 27557308; PMCID: PMC6754861. [CrossRef]
- Anderson GL, Neuhouser ML. Obesity and the risk for premenopausal and postmenopausal breast cancer. Cancer Prev Res (Phila). 2012 Apr;5(4):515-21. Epub 2012 Mar 5. PMID: 22392012. [CrossRef]
- Suba Z. Circulatory estrogen level protects against breast cancer in obese women. Recent Pat Anticancer Drug Discov. 2013 May;8(2):154-67. PMID: 23061769; PMCID: PMC3636519. [CrossRef]
- Gong D, Lai WF. Dietary patterns and type 2 diabetes: A narrative review. Nutrition. 2025 Dec;140:112905. Epub 2025 Jul 8. PMID: 40749645. [CrossRef]
- Papadimitriou N, Markozannes G, Kanellopoulou A, Critselis E, Alhardan S, Karafousia V, Kasimis JC, Katsaraki C, Papadopoulou A, Zografou M, Lopez DS, Chan DSM, Kyrgiou M, Ntzani E, Cross AJ, Marrone MT, Platz EA, Gunter MJ, Tsilidis KK. An umbrella review of the evidence associating diet and cancer risk at 11 anatomical sites. Nat Commun. 2021 Jul 28;12(1):4579. PMID: 34321471; PMCID: PMC8319326. [CrossRef]
- Park JH, Moon JH, Kim HJ, Kong MH, Oh YH. Sedentary Lifestyle: Overview of Updated Evidence of Potential Health Risks. Korean J Fam Med. 2020 Nov;41(6):365-373. Epub 2020 Nov 19. PMID: 33242381; PMCID: PMC7700832. [CrossRef]
- McTiernan A, Friedenreich CM, Katzmarzyk PT, Powell KE, Macko R, Buchner D, Pescatello LS, Bloodgood B, Tennant B, Vaux-Bjerke A, George SM, Troiano RP, Piercy KL; 2018 PHYSICAL ACTIVITY GUIDELINES ADVISORY COMMITTEE*. Physical Activity in Cancer Prevention and Survival: A Systematic Review. Med Sci Sports Exerc. 2019 Jun;51(6):1252-1261. PMID: 31095082; PMCID: PMC6527123. [CrossRef]
- Sjøberg KA, Frøsig C, Kjøbsted R, Sylow L, Kleinert M, Betik AC, Shaw CS, Kiens B, Wojtaszewski JFP, Rattigan S, Richter EA, McConell GK. Exercise Increases Human Skeletal Muscle Insulin Sensitivity via Coordinated Increases in Microvascular Perfusion and Molecular Signaling. Diabetes. 2017 Jun;66(6):1501-1510. Epub 2017 Mar 14. PMID: 28292969. [CrossRef]
Figure 2.
Skeletal muscle cell in contraction. 1. Estrogen receptors (ERs) are migrating from the nucleus to the cell membrane. 2. Insulin like growth factor 1 (IGF-1) activated receptors (IGF-1Rs) carry out rapid, unliganded activation on the arriving ERs. 3. Activated ERs induce GLUT4 expression and rapid translocation of GLUT4 vesicles (GVs) to the cell membrane. 4. The incorporation of GLUT4s into the cell membrane allows glucose entry into the cytoplasm. 5. Membrane associated, IGF-1R activated ERs may transfer their activation for the nuclear ERs. IR: insulin receptor, P: phosphorylation, E2. estradiol, TF: transcription factor, N: nucleus.
Figure 2.
Skeletal muscle cell in contraction. 1. Estrogen receptors (ERs) are migrating from the nucleus to the cell membrane. 2. Insulin like growth factor 1 (IGF-1) activated receptors (IGF-1Rs) carry out rapid, unliganded activation on the arriving ERs. 3. Activated ERs induce GLUT4 expression and rapid translocation of GLUT4 vesicles (GVs) to the cell membrane. 4. The incorporation of GLUT4s into the cell membrane allows glucose entry into the cytoplasm. 5. Membrane associated, IGF-1R activated ERs may transfer their activation for the nuclear ERs. IR: insulin receptor, P: phosphorylation, E2. estradiol, TF: transcription factor, N: nucleus.
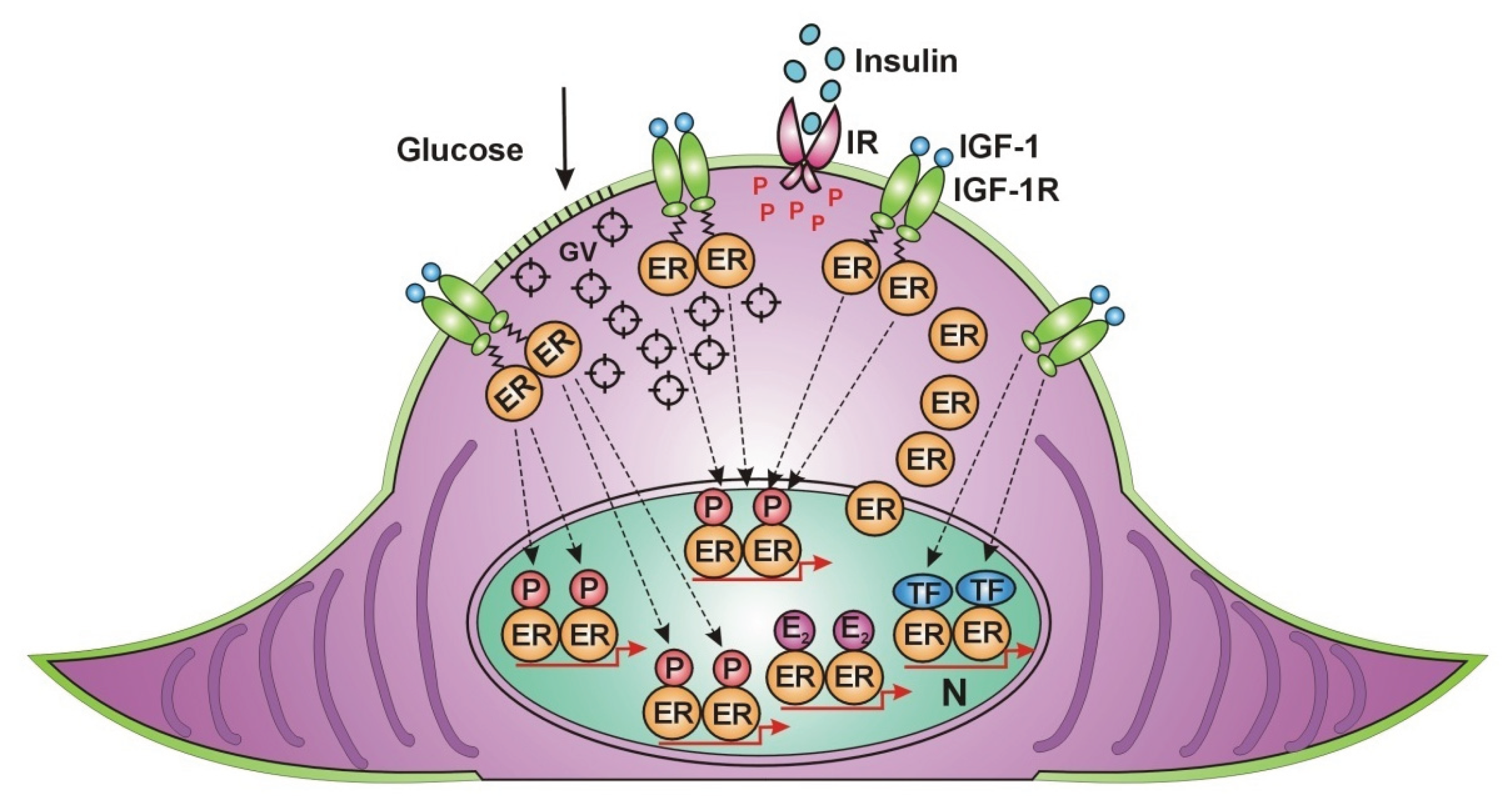
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



