Submitted:
21 October 2025
Posted:
22 October 2025
You are already at the latest version
Abstract
TARDBP (TDP-43), a multifunctional RNA-binding protein, has emerged as a critical host factor controlling HIV-1 replication by destabilizing the viral capsid protein (CA) 55 kDa Gag precursor (Pr55Gag). TARDBP promotes HDAC6-mediated autophagic degradation of HIV-1 Pr55Gag and Vif, impairing nascent virion assembly and infectivity. Simultaneously, TARDBP disrupts viral entry by modulating HDAC6-dependent microtubule (MT) deacetylation, blocking the viral core at the pore fusion step in target cells. These dual mechanisms position TARDBP as a central antiviral defender, paralleling the CA (viral core)-targeting activity of TRIM5α and novel therapeutic inhibitors such as lenacapavir. This review synthesizes evidence for TARDBP’s roles in HIV-1 restriction, highlighting its potential to destabilize the CA-formed viral core during both viral assembly and entry. We propose that enhancing TARDBP activity, combined with destabilizing CA-binding drugs, could offer a synergistic strategy to combat drug-resistant HIV-1 strains and target viral reservoirs, providing hope for functional cure approaches.
Keywords:
TARDBP (TDP-43)
; HIV
; Pr55Gag degradation
; capsid (CA) and viral core
; virus replication and infection
; potential antiviral target
1. Introduction
Human immunodeficiency virus type 1 (HIV-1) 55 kDa Gag precursor protein (Pr55Gag) mediates viral core formation (i.e., by assembling the capsid protein (CA) [1,2,3]) and budding which makes it essential for viral replication ([2,4,5], reviewed in [6]). While antiretrovirals mainly target later stages of the viral life cycle (e.g., integrase, protease) [7,8], vulnerabilities in CA-viral core formation had remained underexplored until the use of the next-gen CA inhibitor lenacapavir [8,9,10]. This new drug selectively binds to the hexamer subunits of the HIV-1 CA protein, disrupting the viral core lattice and impairing both the nuclear import of viral DNA and viral CA generation [11,12,13,14,15,16], thereby acting at the early and late stages of the HIV life cycle.
Host proteins such as tripartite motif-containing protein 5 alpha (TRIM5α) exemplify the innate CA-targeted restriction of retroviruses. Briefly, the mechanism of TRIM5α-mediated retroviral restriction involves a multistep process initiated by the multivalent binding of TRIM5α to the incoming viral capsid lattice, which can induce premature disassembly, or uncoating, and directly inactivate HIV-1 and other retroviral cores [17]. This binding event, while potentially disruptive on its own [18,19,20,21,22], is significantly enhanced by the E3 ubiquitin ligase activity of the Really Interesting New Gene (RING) finger domain, which promotes self-ubiquitination and recruits the ubiquitin‒proteasome system to accelerate nonproductive uncoating and degrade core components [20,23,24,25,26,27,28,29]. In fact, HIV-1 can evade the host’s innate immune response by using its viral core to conceal its nucleic acids from detection. Thus, when the HIV-1 CA carries mutations that disrupt its interactions with host proteins, this evasion fails [30]. These viral cores bearing CA mutants may also alter viral trafficking within the cell, leading to premature reverse transcription; this exposes the viral DNA to host-cell sensors, which then triggers a signaling pathway involving the transcription factors nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and interferon regulatory factor 3 (IRF3) to induce type 1 interferon (IFN) production [30]. The role of the proteasome in accelerating TRIM5α-mediated restriction appears to be secondary, as TRIM5α binding alone can divert the core into an off-pathway trajectory that disables infectivity even when reverse transcription proceeds [27,31,32,33]. On the other hand, TRIM5α could activate autophagy effectors (called virophagy), although this pathway seems redundant for nuclear inactivation. [34,35,36,37,38].
Recent work identified transactive response DNA-binding protein (TARDBP), also known as transactive response DNA binding protein 43 kDa (TDP-43), as a novel regulator of Pr55Gag stability [39,40]. TARDBP, best known for its role in neurodegenerative diseases (reviewed in [41]), unexpectedly suppresses HIV-1 by degrading Pr55Gag and limiting its incorporation into nascent virions thus hampering viral production, [39] and possibly disrupting subsequent capsid maturation. Moreover, TARDBP establishes the permissive status of cells infected by HIV-1 through the regulation of the histone deacetylase enzyme 6 (HDAC6) mRNA and protein expression levels, which restricts HIV-1 entry by its microtubule (MT) deacetylation activity [40]. In fact, TARDBP is a nuclear RNA-binding protein able to process RNA, acting on transcription splicing, mRNA transport, stability and translation, and pri-miRNA processing [42,43,44,45,46,47,48,49,50]. TARDBP recognizes UG-rich mRNA sequences [45,46,51] and can therefore influence the processing of hundreds to thousands of transcripts, including the one coding for the cytoplasmic enzyme HDAC6 (i.e., one of the transcripts that is mainly regulated by TARDP) [40,52]. Notably, HDAC6 promotes the deacetylation of the α-tubulin subunit in MTs, modulating cytoskeleton dynamics [40,53,54,55,56,57,58,59,60,61] and pore fusion formation during the first steps of the HIV-1 infection process [55,57,58], thereby affecting the infection activity of the HIV-1 envelope glycoprotein complex (Env) [40,55,56,62]. Therefore, TARDBP regulates infection by acting at the early and late stages of the HIV-1 life cycle.
This review explores TARDBP’s dual antiviral mechanisms, highlights its potential alongside lenacapavir which was approved by the Food and Drug Administration (FDA) in 2022 for clinical use in combination with other antiretroviral(s) in heavily treatment-experienced HIV-1 patients with multidrug-resistance (reviewed in [63]), and proposes strategies to harness TARDBP for clinical intervention.
2. TARDBP Regulating Virion Formation and Infectiveness
A sentinel Against Pr55Gag Correct Processing and Virion Incompetence
TARDBP is a nuclear RNA-binding protein that performs crucial biological tasks in cells, such as splicing, transcription and translation; mRNA transport; mRNA stability; and pri-miRNA processing [40,42,43,44,45,46,47,48,49,51,64]. TARDBP is able to interact with hundreds to thousands of transcripts, including the proautophagic and anti-HIV-1 factor HDAC6-tubulin deacetylase enzyme, which is highly regulated by TARDBP [39,40,52]. The level of expression of the TARDBP protein directly affects the mRNA and protein levels of HDAC6, which has been previously reported to control Pr55Gag stability and the expression of the auxiliary viral infectivity factor (Vif) protein of HIV-1 [65]. It is thought that HIV-1 uses Vif in combination with the protein core binding factor β (CBF-β) to promote the degradation of the host restriction factor apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G or A3G) [66,67,68], thereby increasing viral infectivity. Notably, cellular HDAC6 counteracts this viral strategy by forming a complex with A3G, which stabilizes A3G by competing with Vif for binding [65]. HDAC6 directly interacts with Vif itself via its C-terminal binder of ubiquitin zinc finger (BUZ) domain [65] (also known as the zinc-finger ubiquitin-binding (ZnF-UBP) or the polyubiquitin-associated zinc finger (PAZ) domain) [69,70,71] and, in a process requiring its deacetylase activity, promotes the autophagic degradation of Vif without affecting its CBF-β partner [65]. This dual action of HDAC6, which stabilizes A3G and degrades Vif, antagonizes the proviral Vif/CBF-β complex, reduces the amount of Vif incorporated into new virions, and consequently diminishes HIV-1 infectivity. Furthermore, HDAC6 acts against HIV-1 infection in a later step of the viral cycle. HDAC6 limits viral production and infection by promoting the aggresome/autophagic degradation of the viral polyprotein Pr55Gag [65,72], an effect independent of the presence of A3G in virions, thus identifying a new HDAC6/A3G cellular complex involved in antiviral defense [65] by targeting Vif and the CA precursor, the polyprotein Pr55Gag, for autophagy degradation [65,72].
As for other restriction factors, HIV-1 counteracts the antiviral activity of HDAC6 using the negative regulatory factor (Nef) of HIV-1 [72]. Nef induces HDAC6 degradation through an acidic/endosomal-lysosomal process, which is mediated by its polyproline-rich region (P72xxP75, aa 69-77) and a di-leucine motif (ExxxLL160-165). By degrading HDAC6, Nef stabilizes the Pr55Gag and Vif viral proteins, ensures the proper localization and aggregation of Pr55Gag at the plasma membrane for viral egress, and ultimately enhances the infectivity of viral particles [72]. This interplay between HIV-1 Nef and cellular HDAC6 could be a key determinant of viral infection and pathogenesis, indicating that both molecules are critical therapeutic targets for combating HIV-1.
Similarly, TARDBP and the TARDBP/HDAC6 axis have been reported to control HIV-1 infection [39]. We suggest that TARDBP functions as a key cellular regulator of the late stages of the HIV-1 viral cycle by stabilizing the anti-HIV-1 factor HDAC6. The overexpression of TARDBP in virus-producing cells increases HDAC6 levels, which in turn triggers the autophagic degradation of the CA precursor, the viral protein Pr55Gag, together with HIV-1 Vif, thereby inhibiting viral particle production and reducing virion infectivity by limiting the incorporation of these proteins into new particles [39].
Conversely, TARDBP knockdown reduces HDAC6 expression, increases the stability of HIV-1 Vif and Pr55Gag, and enhances both virion production and infectious capacity (Figure 1, summary of the TARDBP presented data).
In turn, TARDBP knockdown that diminishes the level of expression of the HDAC6 enzyme or HDAC6-tubulin deacetylase chemical inhibition promotes stabilization of acetylated MTs [39,40,57], which favors Pr55Gag association with MTs and its transport to plasma membrane where Pr55Gag orchestrates virus budding and egress of infectious viral particles (reviewed in [73]).
The inability of a nuclear localization signal (NLS)-TARDBP mutant to control HIV-1 confirmed the functional importance of this pathway. These findings establish the TARDBP /HDAC6 axis as a critical cellular mechanism for controlling HIV-1 production and infectiveness, with a direct relationship observed between the virion content of Vif and the CA precursor Pr55Gag and the resulting infection capacity [39].
3. TARDBP at the Portal
Blocking Viral Core Entry and Early Infection via HDAC6 Regulation
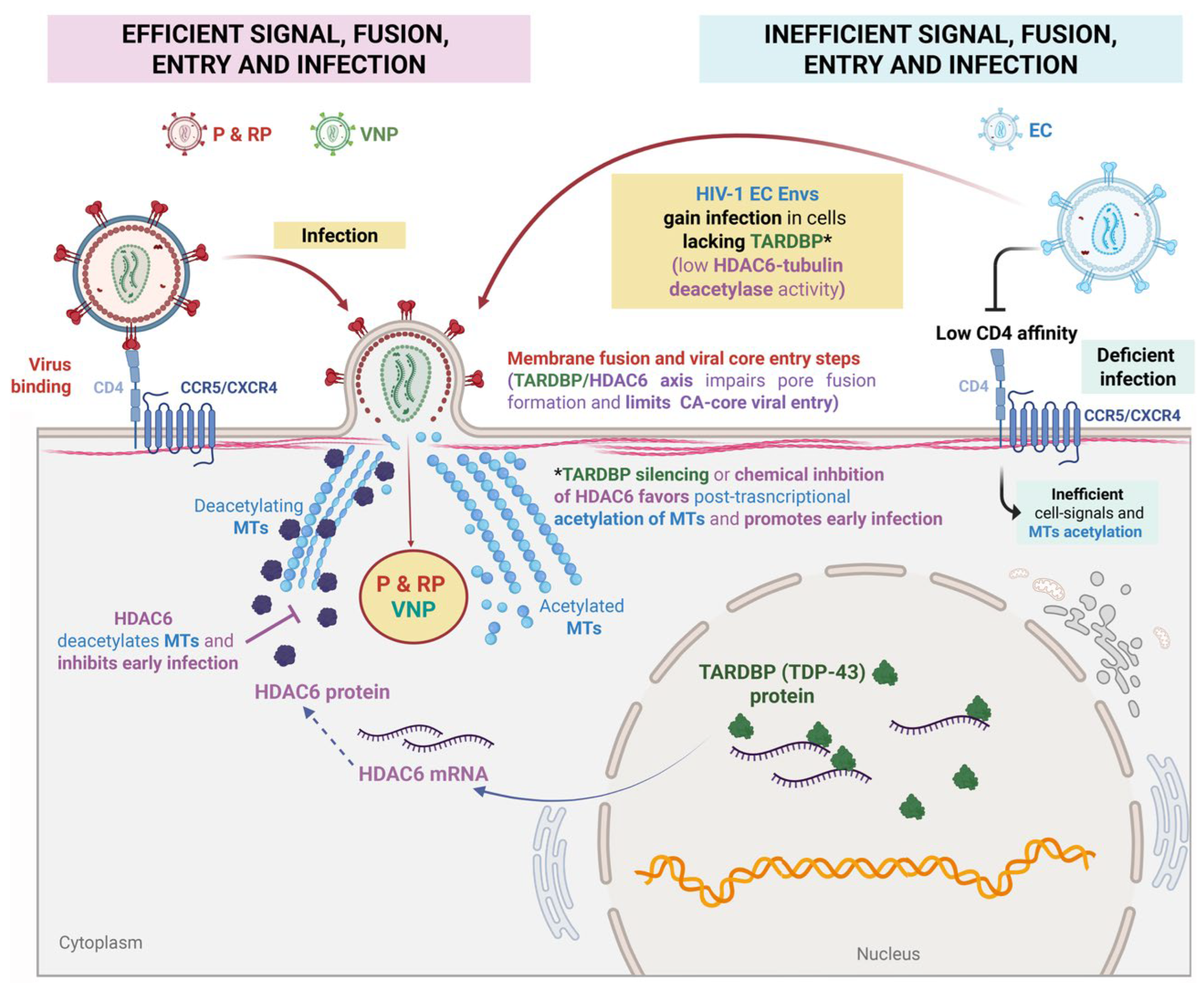
The second arm of TARDBP’s antiviral activity involves disrupting viral entry and early steps of infection [40]. TARDBP shapes CD4+ T-cell permissivity to HIV-1 infection by regulating HDAC6-tubulin deacetylase levels. HDAC6 deacetylates MTs at the Lys60 residue of the α-tubulin subunit [53,57], destabilizing MT networks and impairing pore fusion formation and HIV-1 infection [40,57]. In fact, the acetylation of MTs is a cytoskeleton event required to facilitate HIV-1 envelope (Env)-mediated membrane fusion, subsequent viral core entry and infection of target cells [57]. Therefore, HDAC6-tubulin deacetylase activity prevents CA-associated viral core entry and infection. In this context, overexpression of TARDBP stabilizes the mRNA of HDAC6 and increases the level of expression of the enzyme, which impairs HIV-1 infection by reducing the fusion capacity of the viral Env glycoprotein complex (i.e., formed by trimers of the noncovalently associated gp41/gp120 (glycoprotein 120) viral Env proteins (reviewed in [74,75])). Likewise, this effect is independent of HIV-1 Env tropism [40]. Conversely, silencing endogenous TARDBP decreases HDAC6 and increases HIV-1 Env-mediated fusion and infection, a process that is directly dependent on Env/CD4-mediated stabilization of acetylated MTs [40,55,56,57,58,62]. This regulatory mechanism of TARDBP and the TARDBP/HDAC6 axis was confirmed by analyzing the function of HIV-1 Envs from viruses isolated from patients with different disease progression and clinical outcomes.
Thus, activation of the TARDBP/HDAC6 axis reduces infection of virus-bearing functional Envs from virus of viremic nonprogressor (VNP) and rapid progressor (RP) patients [40] to low levels observed with virions bearing non-functional Envs from virus of long-term nonprogressor elite controller (LTNP-EC) individuals [40]. In contrast, silencing of the TARDBP/HDAC6 axis enhances the infectivity of all primary HIV-1 Envs, including those deficient in function isolated from viruses of LTNP-ECs [40].
Thus, TARDBP controls HDAC6-tubulin deacetylase activity, regulating virus-cell membrane fusion and subsequent CA-associated viral core entry, thereby conditioning cell permissivity to HIV-1 infection (Figure 2, summarizes these TARDBP presented data).
Taken together, these findings suggest that TARDBP could be relevant for the pathogenesis of HIV-1 infection, as recent works indicate that the infective properties of the HIV-1 Env complex of viruses from infected individuals are directly related to different infection progression rates in vivo [40,55,56,62]. Thus, CD4 binding, fusion and cell-to-cell viral transfer, and the cell-signal capacities of the HIV-1 Env complex affect the infectivity of HIV-1 [40,55,56,57,62], which is under the control of the endogenous TARDBP/HDAC6 axis. Likewise, nonfunctional LTNP-EC Envs are unable to overcome this barrier and stabilize acetylated MTs to efficiently trigger pore fusion, and only functional viral Env complexes from patients with detectable viremia (i.e., VNPs, Ps and RPs) are efficient by stabilizing acetylated MTs against the endogenous TARDBP/HDAC6 axis [40].
4. Perspective
TARDBP as a Potential Clinical Target
Besides the well-defined role of TARDBP in the HIV life cycle, an imbalance in the expression and function of TARDBP and HDAC6 has been associated with neurodegeneration and disease in the central nervous system (CNS) [76,77,78,79,80,81,82,83]. In addition, HDAC6 has been shown to protect against neurotoxicity induced by the HIV-1 Env subunit gp120 in cortical neurons [84]. These findings suggest that the activation of TARDBP (i.e., gene/protein expression and/or protein stability) and/or the TARDBP/HDAC6 axis may play a protective role against HIV-1-mediated toxicity in the CNS. Conversely, dysregulation of their expression and function could exacerbate HIV-1-induced damages, a hypothesis that requires further investigation. Nevertheless, HDAC6 inhibitors (e.g., the potent and selective inhibitor tubastatin A and the nonspecific trichostatin A (TSA) inhibitor [57,58,85]) enhance cell permissivity to HIV-1 infection and trigger HIV-1 transcription [57,58,86], suggesting that TARDBP acts as a natural HDAC6 agonist, as indicated by the anti-HIV-1 activities of the TARDBP/HDAC6 axis [39,40] (Figure 1 and Figure 2). Among the mechanisms involved in this effect, autophagy promotion is a key antiviral function of the TARDBP/HDAC6 axis. Considering the relevance of autophagy in HIV-1 infection (reviewed in [87]), it could be of interest to develop new drugs and anti-HIV-1 strategies at that level.
In particular, TARDBP could be considered a potential target against HIV-1 [39,40], considering the potential side effects of drugs and/or treatments on the CNS and immune system (reviewed in [88]). Thus, the clinical translation of screening-based compounds that could enhance TARDBP-mediated targeting and clearance of HIV-1 Pr55Gag could be considered to avoid CA-associated viral core formation and, therefore, impairing virus production and HIV-1 spread in reservoirs and the organisms. It is conceivable that these TARDBP-boosting agents (i.e., activating selective drugs for gene expression or stabilizing the protein) could act in a similar fashion as lenacapavir (Sunlenca®), a new anti-HIV drug that targets the CA lattice that constitutes the viral core of HIV [8,9,10,11,12,13,14,15,16]. As reported for TARDBP, lenacapavir inhibits viral replication at both the early and late stages of the HIV-1 life cycle. On 22 August 2022, lenacapavir was approved in the EU for use in combination with other antiretroviral(s) in adults with multidrug-resistant HIV-1 infection, for whom it is otherwise not possible to construct a suppressive antiviral regimen ([8,9,10,11,12,13,14,15,16], reviewed in [63]).
Therefore, recent advances in viral core/CA inhibitors, such as lenacapavir, which stabilizes capsid intermediates to block nuclear import, may synergize with TARDBP-enhancing therapies. For example, lenacapavir’s “hyperstable capsid” phenotype might render HIV-1 more susceptible to TARDBP-mediated HIV-1 Pr55Gag degradation in infected, virus-producing cells, creating a potential two-pronged attack in combination trials.
5. Conclusions
TARDBP has dual roles in HIV-1 restriction, limiting i) virus production and infectivity and ii) viral entry and infection by targeting Pr55Gag, positioning it as a master regulator of CA-induced vulnerability of the viral core. By integrating its mechanisms with emerging CA inhibitors such as lenacapavir, researchers could pioneer therapies that overcome drug resistance. However, considering TARDBP’s neuroprotective-antiviral duality, any anti-HIV-1 strategy will require innovative delivery systems (e.g., targeted nanocarriers) to selectively act on anatomical, cellular and molecular HIV-1 reservoirs (reviewed in [89]) to avoid undesirable side effects on the CNS. As the field revisits CA (viral core)-centric strategies, TARDBP has emerged as a potential target for new HIV-1 therapeutics for functional cure approaches.
Abbreviations
APOBEC3G (or A3G), cytidine deaminase restriction factor apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G
BUZ domain, binder of ubiquitin zinc finger
CA, capsid protein
CBF-β, core binding factor β
Env, envelope (HIV envelope glycoproteins complex)
FDA, Food and Drug Administration
gp41, glycoprotein 41 (of the HIV-1 Env complex)
gp120, glycoprotein 120 (of the HIV-1 Env complex)
HDAC6, histone deacetylase enzyme 6
HIV-1, human immunodeficiency virus type 1
IFN, interferon
IRF3, interferon regulatory factor 3
LTNP-EC, long-term nonprogressor elite controller (HIV individuals)
mRNA, messenger ribonucleic acid
MT, microtubule
Nef, negative regulatory factor (of HIV)
NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells
NLS, nuclear localization signal
PAZ, polyubiquitin-associated zinc finger
Pr55Gag, 55 kDa Gag precursor protein (of HIV)
Ps, progresors (HIV patients)
RING, Really Interesting New Gene (it is a finger domain)
RPs; rapid progressors (HIV patients)
TARDBP, transactive response DNA-binding protein
TDP-43, transactive response DNA binding protein 43 kDa
TRIM5α, tripartite motif-containing protein 5 alpha
TSA, trichostatin A
Vif, viral infectivity factor
VNPs, viremic nonprogressors (HIV patients)
ZnF-UBP, the zinc-finger ubiquitin-binding domain
Author Contributions
AV-F, conceptualization, funding acquisition, project administration, writing-original draft, drew the figures, and writing-review and editing. RC-R, conceptualization, drew the figures, writing-original draft, writing-review and editing. JB, BT, and RT-G, conceptualization, writing-original draft, writing-review and editing. CC, MP, AP-G, IL-S and IR-C, writing-review and editing. All authors read and approved the final manuscript.
Funding
This research and A.V.-F.’s laboratory are supported by Grant PID2021-123031OB-I00 funded by MICIU/AEI/10.13039/501100011033 and by “ERDF A way of making Europe” (MICIU (“Ministerio de Ciencia, Innovación y Universidades”)/AEI (“Agencia Española de Investigación”), Spain), Grant PID2024-155444OB-I00 funded by MICIU/AEI and by “ERDF A way of making Europe”, UNLL10-3E-783 (The European Regional Development Fund (ERDF) and “Fundación CajaCanarias”) and “SEGAI-ULL”. I.L-S. was founded by Grant PID2021-123031OB-I00 (MICIU/AEI/10.13039/501100011033, Spain, and “ERDF A way of making Europe”) and is now founded by “Ayuda formación de investigadores del Gobierno de Canarias/Fondo Social Europeo Plus (Exp.: FPI2024010073)”). RC-R is funded by Grant PID2024-155444OB-I00 (“MICIU/AEI and by “ERDF A way of making Europe”). J.B. is a researcher from “Fundació Institut de Recerca en Ciències de la Salut Germans Trias i Pujol” and IrsiCaixa, supported by the Health Department of the Catalonian Government/Generalitat de Catalunya Through the CERCA program. Work in J.B. lab is cofunded by ISCIII and Europe and funds through the Grant number PI23/01269. C.C. and M.P. are researchers from “Centro Nacional de Microbiología” supported by Science and Technology Department of Spanish Government and ISCIII Grant number PI20/00093, ISCIII.
Acknowledgments
Designs and templates were created with BioRender. The English grammar has been edited and corrected by AJE through our institutional cooperation.
Conflicts of Interest
The authors declare that they have no competing interest.
References
- Ganser-Pornillos BK, Cheng A, Yeager M: Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 2007, 131, 70–79. [CrossRef]
- Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI: Assembly and analysis of conical models for the HIV-1 core. Science 1999, 283, 80–83. [CrossRef]
- Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P: Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [CrossRef]
- Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC: The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol 2004, 11, 672–675. [CrossRef]
- Li S, Hill CP, Sundquist WI, Finch JT: Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 2000, 407, 409–413. [CrossRef]
- Campbell EM, Hope TJ: HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol 2015, 13, 471–483. [CrossRef] [PubMed]
- Department of Health and Human Services (Washington D: Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents with HIV. HHS Panel on Antiretroviral Guidelines for Adults and Adolescents—A Working Group of the NIH Office of AIDS Research Advisory Council (OARAC) 2024. https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/guidelines-adult-adolescent-arv.pdf.
- Sever B, Otsuka M, Fujita M, Ciftci H: A Review of FDA-Approved Anti-HIV-1 Drugs, Anti-Gag Compounds, and Potential Strategies for HIV-1 Eradication. Int J Mol Sci 2024, 25.
- Segal-Maurer S, DeJesus E, Stellbrink HJ, Castagna A, Richmond GJ, Sinclair GI, Siripassorn K, Ruane PJ, Berhe M, Wang H, et al. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. N Engl J Med 2022, 386, 1793–1803. [Google Scholar] [CrossRef]
- Bekker LG, Das M, Abdool Karim Q, Ahmed K, Batting J, Brumskine W, Gill K, Harkoo I, Jaggernath M, Kigozi G, et al. Twice-Yearly Lenacapavir or Daily F/TAF for HIV Prevention in Cisgender Women. N Engl J Med 2024, 391, 1179–1192. [Google Scholar] [CrossRef]
- Link JO, Rhee MS, Tse WC, Zheng J, Somoza JR, Rowe W, Begley R, Chiu A, Mulato A, Hansen D, et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Bekerman E, Yant SR, VanderVeen L, Hansen D, Lu B, Rowe W, Wang K, Callebaut C: Long-acting lenacapavir acts as an effective preexposure prophylaxis in a rectal SHIV challenge macaque model. J Clin Invest 2023, 133.
- Marcelin AG, Charpentier C, Jary A, Perrier M, Margot N, Callebaut C, Calvez V, Descamps D: Frequency of capsid substitutions associated with GS-6207 in vitro resistance in HIV-1 from antiretroviral-naive and -experienced patients. J Antimicrob Chemother 2020, 75, 1588–1590. [CrossRef] [PubMed]
- Dzinamarira T, Almehmadi M, Alsaiari AA, Allahyani M, Aljuaid A, Alsharif A, Khan A, Kamal M, Rabaan AA, Alfaraj AH, et al. Highlights on the Development, Related Patents, and Prospects of Lenacapavir: The First-in-Class HIV-1 Capsid Inhibitor for the Treatment of Multi-Drug-Resistant HIV-1 Infection. Medicina (Kaunas) 2023, 59. [Google Scholar]
- Hitchcock AM, Kufel WD, Dwyer KAM, Sidman EF: Lenacapavir: A novel injectable HIV-1 capsid inhibitor. Int J Antimicrob Agents 2024, 63, 107009. [CrossRef] [PubMed]
- Subramanian R, Tang J, Zheng J, Lu B, Wang K, Yant SR, Stepan GJ, Mulato A, Yu H, Schroeder S, et al. Lenacapavir: A Novel, Potent, and Selective First-in-Class Inhibitor of HIV-1 Capsid Function Exhibits Optimal Pharmacokinetic Properties for a Long-Acting Injectable Antiretroviral Agent. Mol Pharm 2023, 20, 6213–6225. [Google Scholar] [CrossRef] [PubMed]
- Perron MJ, Stremlau M, Lee M, Javanbakht H, Song B, Sodroski J: The human TRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J Virol 2007, 81, 2138–2148. [CrossRef]
- Wagner JM, Roganowicz MD, Skorupka K, Alam SL, Christensen D, Doss G, Wan Y, Frank GA, Ganser-Pornillos BK, Sundquist WI, Pornillos O: Mechanism of B-box 2 domain-mediated higher-order assembly of the retroviral restriction factor TRIM5alpha. Elife 2016, 5.
- Wagner JM, Christensen DE, Bhattacharya A, Dawidziak DM, Roganowicz MD, Wan Y, Pumroy RA, Demeler B, Ivanov DN, Ganser-Pornillos BK, et al. General Model for Retroviral Capsid Pattern Recognition by TRIM5 Proteins. J Virol 2018, 92. [Google Scholar]
- Diaz-Griffero F, Vandegraaff N, Li Y, McGee-Estrada K, Stremlau M, Welikala S, Si Z, Engelman A, Sodroski J: Requirements for capsid-binding and an effector function in TRIMCyp-mediated restriction of HIV-1. Virology 2006, 351, 404–419. [CrossRef]
- Li YL, Chandrasekaran V, Carter SD, Woodward CL, Christensen DE, Dryden KA, Pornillos O, Yeager M, Ganser-Pornillos BK, Jensen GJ, Sundquist WI: Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. Elife 2016, 5.
- Javanbakht H, Diaz-Griffero F, Yuan W, Yeung DF, Li X, Song B, Sodroski J: The ability of multimerized cyclophilin A to restrict retrovirus infection. Virology 2007, 367, 19–29. [CrossRef]
- Kutluay SB, Perez-Caballero D, Bieniasz PD: Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog 2013, 9, e1003214.
- Lukic Z, Hausmann S, Sebastian S, Rucci J, Sastri J, Robia SL, Luban J, Campbell EM: TRIM5alpha associates with proteasomal subunits in cells while in complex with HIV-1 virions. Retrovirology 2011, 8, 93. [CrossRef] [PubMed]
- Danielson CM, Cianci GC, Hope TJ: Recruitment and dynamics of proteasome association with rhTRIM5alpha cytoplasmic complexes during HIV-1 infection. Traffic 2012, 13, 1206–1217. [CrossRef]
- Danielson CM, Hope TJ: Using antiubiquitin antibodies to probe the ubiquitination state within rhTRIM5α cytoplasmic bodies.
- Roa A, Hayashi F, Yang Y, Lienlaf M, Zhou J, Shi J, Watanabe S, Kigawa T, Yokoyama S, Aiken C, Diaz-Griffero F: RING domain mutations uncouple TRIM5alpha restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J Virol 2012, 86, 1717–1727. [CrossRef] [PubMed]
- Fletcher AJ, Christensen DE, Nelson C, Tan CP, Schaller T, Lehner PJ, Sundquist WI, Towers GJ: TRIM5alpha requires Ube2W to anchor Lys63-linked ubiquitin chains and restrict reverse transcription. EMBO J 2015, 34, 2078–2095. [CrossRef]
- Campbell EM, Weingart J, Sette P, Opp S, Sastri J, O’Connor SK, Talley S, Diaz-Griffero F, Hirsch V, Bouamr F: TRIM5α-Mediated Ubiquitin Chain Conjugation Is Required for Inhibition of HIV-1 Reverse Transcription and Capsid Destabilization.
- Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ: HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [CrossRef]
- Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD: Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J Virol 2005, 79, 15567–15572. [CrossRef]
- Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ: Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci U S A 2006, 103, 7465–7470. [CrossRef] [PubMed]
- Anderson JL, Campbell Em Fau - Wu X, Wu X Fau - Vandegraaff N, Vandegraaff N Fau - Engelman A, Engelman A Fau - Hope TJ, Hope TJ: Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. 2006.
- O’Connor C, Pertel T, Gray S, Robia SL, Bakowska JC, Luban J, Campbell EM: p62/sequestosome-1 associates with and sustains the expression of retroviral restriction factor TRIM5alpha. J Virol 2010, 84, 5997–6006. [CrossRef]
- Mandell MA, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Münch J, Kirchhoff F, Simonsen A, et al.: TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition.
- Keown JR, Black MM, Ferron A, Yap M, Barnett MJ, Pearce FG, Stoye JP, Goldstone DC: A helical LC3-interacting region mediates the interaction between the retroviral restriction factor Trim5α and mammalian autophagy-related ATG8 proteins.
- Tanida I, Ueno T Fau - Kominami E, Kominami E: LC3 and Autophagy.
- Imam S, Talley S, Nelson RS, Dharan A, O’Connor C, Hope TJ, Campbell EM: TRIM5α Degradation via Autophagy Is Not Required for Retroviral Restriction.
- Cabrera-Rodriguez R, Perez-Yanes S, Lorenzo-Sanchez I, Estevez-Herrera J, Garcia-Luis J, Trujillo-Gonzalez R, Valenzuela-Fernandez A: TDP-43 Controls HIV-1 Viral Production and Virus Infectiveness. Int J Mol Sci 2023, 24.
- Cabrera-Rodriguez R, Perez-Yanes S, Montelongo R, Lorenzo-Salazar JM, Estevez-Herrera J, Garcia-Luis J, Inigo-Campos A, Rubio-Rodriguez LA, Munoz-Barrera A, Trujillo-Gonzalez R, et al.: Transactive Response DNA-Binding Protein (TARDBP/TDP-43) Regulates Cell Permissivity to HIV-1 Infection by Acting on HDAC6. Int J Mol Sci 2022, 23.
- Zeng J, Luo C, Jiang Y, Hu T, Lin B, Xie Y, Lan J, Miao J: Decoding TDP-43, the molecular chameleon of neurodegenerative diseases. Acta Neuropathol Commun 2024, 12, 205. [CrossRef]
- Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, Bampton A, Lee FCY, Masino L, Qi YA, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 2022, 603, 131–137. [Google Scholar] [CrossRef]
- Buratti E, Baralle FE: The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol 2010, 7, 420–429. [CrossRef] [PubMed]
- Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A: TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem 2012, 287, 15635–15647. [CrossRef] [PubMed]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 2011, 14, 452–458. [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Kawahara Y, Mieda-Sato A: TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci U S A 2012, 109, 3347–3352. [CrossRef]
- Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR, Higginbottom A, Raman R, Ferraiuolo L, Cooper-Knock J, et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol 2014, 40, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Ma X, Ying Y, Xie H, Liu X, Wang X, Li J: The Regulatory Role of RNA Metabolism Regulator TDP-43 in Human Cancer. Front Oncol 2021, 11, 755096. [CrossRef]
- Ma XA-O, Prudencio MA-O, Koike YA-O, Vatsavayai SC, Kim GA-O, Harbinski F, Briner AA-O, Rodriguez CM, Guo C, Akiyama TA-O, et al.: TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A.
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Fiesel FC, Voigt A, Weber SS, Van den Haute C, Waldenmaier A, Gorner K, Walter M, Anderson ML, Kern JV, Rasse TM, et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J 2010, 29, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP: HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [CrossRef]
- Zilberman Y, Ballestrem C, Carramusa L, Mazitschek R, Khochbin S, Bershadsky A: Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J Cell Sci 2009, 122, 3531–3541. [CrossRef] [PubMed]
- Casado C, Marrero-Hernandez S, Marquez-Arce D, Pernas M, Marfil S, Borras-Granana F, Olivares I, Cabrera-Rodriguez R, Valera MS, de Armas-Rillo L, et al. Viral Characteristics Associated with the Clinical Nonprogressor Phenotype Are Inherited by Viruses from a Cluster of HIV-1 Elite Controllers. mBio 2018, 9. [Google Scholar]
- Pérez-Yanes S, Pernas M, Marfil S, Cabrera-Rodríguez R, Ortiz R, Urrea V, Rovirosa C, Estévez-Herrera J, Olivares I, Casado C, et al. The Characteristics of the HIV-1 Env Glycoprotein Are Linked With Viral Pathogenesis. Frontiers in Microbiology 2022, 13. [Google Scholar]
- Valenzuela-Fernandez A, Alvarez S, Gordon-Alonso M, Barrero M, Ursa A, Cabrero JR, Fernandez G, Naranjo-Suarez S, Yanez-Mo M, Serrador JM, et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol Biol Cell 2005, 16, 5445–5454. [Google Scholar] [CrossRef]
- Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F: HDAC6, a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008, 18, 291–297. [CrossRef]
- Ling L, Hu F, Ying X, Ge J, Wang Q: HDAC6 inhibition disrupts maturational progression and meiotic apparatus assembly in mouse oocytes. Cell Cycle 2018, 17, 550–556. [CrossRef]
- Osseni A, Ravel-Chapuis A, Thomas JL, Gache V, Schaeffer L, Jasmin BJ: HDAC6 regulates microtubule stability and clustering of AChRs at neuromuscular junctions. J Cell Biol 2020, 219.
- Bershadsky AD, Ballestrem C, Carramusa L, Zilberman Y, Gilquin B, Khochbin S, Alexandrova AY, Verkhovsky AB, Shemesh T, Kozlov MM: Assembly and mechanosensory function of focal adhesions: experiments and models. Eur J Cell Biol 2006, 85, 165–173. [CrossRef] [PubMed]
- Cabrera-Rodriguez R, Hebmann V, Marfil S, Pernas M, Marrero-Hernandez S, Cabrera C, Urrea V, Casado C, Olivares I, Marquez-Arce D, et al. HIV-1 envelope glycoproteins isolated from Viremic Non-Progressor individuals are fully functional and cytopathic. Sci Rep 2019, 9, 5544. [Google Scholar] [CrossRef]
- Paik J: Lenacapavir: First Approval. Drugs 2022, 82, 1499–1504. [CrossRef]
- Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 2022, 603, 124–130. [Google Scholar] [CrossRef]
- Valera MS, de Armas-Rillo L, Barroso-Gonzalez J, Ziglio S, Batisse J, Dubois N, Marrero-Hernandez S, Borel S, Garcia-Exposito L, Biard-Piechaczyk M, et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar]
- Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, Gross JD: CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol Cell 2013, 49, 632–644. [CrossRef]
- Zhou X, Han X, Zhao K, Du J, Evans SL, Wang H, Li P, Zheng W, Rui Y, Kang J, Yu XF: Dispersed and conserved hydrophobic residues of HIV-1 Vif are essential for CBFbeta recruitment and A3G suppression. J Virol 2014, 88, 2555–2563. [CrossRef]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP: The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [CrossRef]
- Seigneurin-Berny D, Verdel A Fau - Curtet S, Curtet S Fau - Lemercier C, Lemercier C Fau - Garin J, Garin J Fau - Rousseaux S, Rousseaux S Fau - Khochbin S, Khochbin S: Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways.
- Hook SS, Orian A Fau - Cowley SM, Cowley Sm Fau - Eisenman RN, Eisenman RN: Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes.
- Marrero-Hernandez S, Marquez-Arce D, Cabrera-Rodriguez R, Estevez-Herrera J, Perez-Yanes S, Barroso-Gonzalez J, Madrid R, Machado JD, Blanco J, Valenzuela-Fernandez A: HIV-1 Nef Targets HDAC6 to Assure Viral Production and Virus Infection. Front Microbiol 2019, 10, 2437.
- Cabrera-Rodriguez R, Perez-Yanes S, Lorenzo-Sanchez I, Trujillo-Gonzalez R, Estevez-Herrera J, Garcia-Luis J, Valenzuela-Fernandez A: HIV Infection: Shaping the Complex, Dynamic, and Interconnected Network of the Cytoskeleton. Int J Mol Sci 2023, 24.
- Ward AB, Wilson IA: Insights into the trimeric HIV-1 envelope glycoprotein structure.
- Ward AB, Wilson IA: The HIV-1 envelope glycoprotein structure: nailing down a moving target.
- Simoes-Pires C, Zwick V, Nurisso A, Schenker E, Carrupt PA, Cuendet M: HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs? Mol Neurodegener 2013, 8, 7. [CrossRef] [PubMed]
- Gao J, Wang L, Huntley ML, Perry G, Wang X: Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem 2018.
- Guo W, Van Den Bosch L: Therapeutic potential of HDAC6 in amyotrophic lateral sclerosis. Cell Stress 2017, 2, 14–16.
- Odagiri S, Tanji K, Mori F, Miki Y, Kakita A, Takahashi H, Wakabayashi K: Brain expression level and activity of HDAC6 protein in neurodegenerative dementia. Biochem Biophys Res Commun 2013, 430, 394–399. [CrossRef]
- Lemos M, Stefanova N: Histone Deacetylase 6 and the Disease Mechanisms of alpha-Synucleinopathies. Front Synaptic Neurosci 2020, 12, 586453. [CrossRef]
- Trzeciakiewicz H, Ajit D, Tseng JH, Chen Y, Ajit A, Tabassum Z, Lobrovich R, Peterson C, Riddick NV, Itano MS, et al. An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat Commun 2020, 11, 5522. [Google Scholar] [CrossRef]
- Cykowski MD, Powell SZ, Peterson LE, Appel JW, Rivera AL, Takei H, Chang E, Appel SH: Clinical Significance of TDP-43 Neuropathology in Amyotrophic Lateral Sclerosis. J Neuropathol Exp Neurol 2017, 76, 402–413. [CrossRef] [PubMed]
- Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ: TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol 2007, 114, 63–70. [CrossRef]
- Wenzel ED, Speidell A, Flowers SA, Wu C, Avdoshina V, Mocchetti I: Histone deacetylase 6 inhibition rescues axonal transport impairments and prevents the neurotoxicity of HIV-1 envelope protein gp120. Cell Death Dis 2019, 10, 674. [CrossRef]
- Pérez-Yanes S, Lorenzo-Sánchez I, Cabrera-Rodríguez RA-O, García-Luis JA-O, Trujillo-González RA-O, Estévez-Herrera J, Valenzuela-Fernández AA-O: The ZIKV NS5 Protein Aberrantly Alters the Tubulin Cytoskeleton, Induces the Accumulation of Autophagic p62 and Affects IFN Production: HDAC6 Has Emerged as an Anti-NS5/ZIKV Factor. LID - 10.3390/cells13070598 [doi] LID - 598. [CrossRef]
- Bartholomeeusen K, Fujinaga K, Xiang Y, Peterlin BM: Histone deacetylase inhibitors (HDACis) that release the positive transcription elongation factor b (P-TEFb) from its inhibitory complex also activate HIV transcription.
- Cabrera-Rodriguez R, Perez-Yanes S, Estevez-Herrera J, Marquez-Arce D, Cabrera C, Espert L, Blanco J, Valenzuela-Fernandez A: The Interplay of HIV and Autophagy in Early Infection. Front Microbiol 2021, 12, 661446.
- Lanman T, Letendre S, Ma Q, Bang A, Ellis R: CNS Neurotoxicity of Antiretrovirals. J Neuroimmune Pharmacol 2021, 16, 130–143. [CrossRef] [PubMed]
- Henderson LJ, Reoma LB, Kovacs JA, Nath A: Advances toward Curing HIV-1 Infection in Tissue Reservoirs. J Virol 2020, 94.
Figure 1.
Scheme illustrating the functional role of TARDBP in late stages of the HIV-1 life cycle. Left, the increase in TARDBP activity enhances the action of the autophagic HDAC6 enzyme (i.e., by first stabilizing the HDAC6 mRNA levels; not shown on the illustration), thereby targeting HIV-1 Pr55Gag for autophagic degradation together with the viral Vif protein. The scheme shows that the transcription and translation of the integrated HIV-1 genome generate viral RNA+ and proteins, with the structural Pr55Gag polyprotein being relevant for recognizing viral RNA+ and recruiting a complex to stable MTs to travel to the plasma membrane; however, under these conditions, Pr55Gag is degraded, limiting virus assembly, budding and egress. Similarly, the MT-associated HDAC6 enzyme deacetylates stable MTs, impeding Pr55Gag cell-surface localization. Right, in contrast, TARDBP silencing (low levels of the cellular protein) of chemical inhibition of HDAC6 favors the stabilization of the Pr55Gag and Vif proteins, together with the acetylation of MTs, thereby promoting the trafficking of the HIV-1 Pr55Gag MT-dependent cellular machinery to the cell surface, where it assembles and buds to form viral particles that incorporate Vif (red dots; which are not shown in virions on the left panel-virus scheme)) to ensure the infectivity of nascent HIV-1. Designs and templates were created with BioRender and licensed are obtained for publication.
Figure 1.
Scheme illustrating the functional role of TARDBP in late stages of the HIV-1 life cycle. Left, the increase in TARDBP activity enhances the action of the autophagic HDAC6 enzyme (i.e., by first stabilizing the HDAC6 mRNA levels; not shown on the illustration), thereby targeting HIV-1 Pr55Gag for autophagic degradation together with the viral Vif protein. The scheme shows that the transcription and translation of the integrated HIV-1 genome generate viral RNA+ and proteins, with the structural Pr55Gag polyprotein being relevant for recognizing viral RNA+ and recruiting a complex to stable MTs to travel to the plasma membrane; however, under these conditions, Pr55Gag is degraded, limiting virus assembly, budding and egress. Similarly, the MT-associated HDAC6 enzyme deacetylates stable MTs, impeding Pr55Gag cell-surface localization. Right, in contrast, TARDBP silencing (low levels of the cellular protein) of chemical inhibition of HDAC6 favors the stabilization of the Pr55Gag and Vif proteins, together with the acetylation of MTs, thereby promoting the trafficking of the HIV-1 Pr55Gag MT-dependent cellular machinery to the cell surface, where it assembles and buds to form viral particles that incorporate Vif (red dots; which are not shown in virions on the left panel-virus scheme)) to ensure the infectivity of nascent HIV-1. Designs and templates were created with BioRender and licensed are obtained for publication.
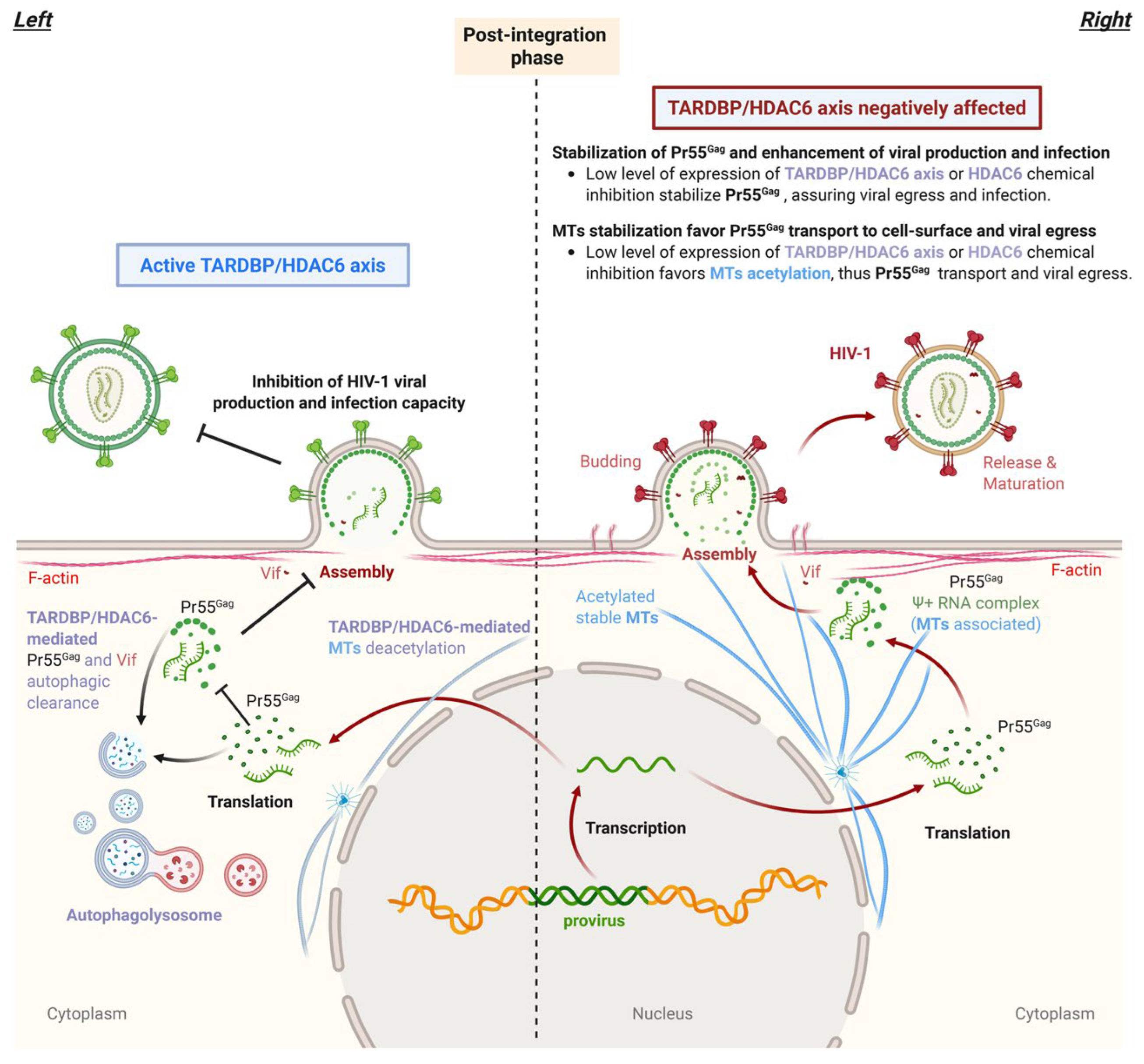
Figure 2.
Scheme illustrating the functional role of TARDBP in the early stages of HIV-1 infection. Functional HIV-1 Envs from progressors, (P) rapid progressors (RP) and viremic nonprogressors (VNP) patients trigger MT acetylation in a CD4-dependent manner to promote pore fusion and evade the antiviral action of the endogenous tubulin-deacetylase HDAC6, which is under the control of TARDBP (TDP-43). Thus, activation of the TARDBP/HDAC6 axis by overexpressing TARDBP, stabilizes HDAC6 mRNA and increases HDAC6-tubulin deacetylase enzyme activity, inhibiting HIV-1-mediated pore fusion formation, viral core entry and early infection. Functional HIV-1 Env complexes, such as those from viruses of P, RP and VNP patients, are able to stabilize acetylated MTs, overcoming the endogenous activity of TARDBP and the antiviral TARDBP/HDAC6 axis. In this sense, deficient HIV-1 Envs from viruses of long-term nonprogressor Elite Controller (LTNP-EC) individuals are insufficient to form the fusion pore and infect because of their inability to escape the antiviral TARDBP/HDAC6-tubulin-deacetylase barrier. Notably, viral particles bearing EC-Envs gain infection function when HDAC6 or the HDAC6/TARDBP axis is inhibited by either HDAC6 inhibitors or their nonactive mutants or interference with HDAC6/TARDBP mRNA, thereby stabilizing MTs to generate a permissive state for infection. Designs and templates were created with BioRender and licensed are obtained for publication.
Figure 2.
Scheme illustrating the functional role of TARDBP in the early stages of HIV-1 infection. Functional HIV-1 Envs from progressors, (P) rapid progressors (RP) and viremic nonprogressors (VNP) patients trigger MT acetylation in a CD4-dependent manner to promote pore fusion and evade the antiviral action of the endogenous tubulin-deacetylase HDAC6, which is under the control of TARDBP (TDP-43). Thus, activation of the TARDBP/HDAC6 axis by overexpressing TARDBP, stabilizes HDAC6 mRNA and increases HDAC6-tubulin deacetylase enzyme activity, inhibiting HIV-1-mediated pore fusion formation, viral core entry and early infection. Functional HIV-1 Env complexes, such as those from viruses of P, RP and VNP patients, are able to stabilize acetylated MTs, overcoming the endogenous activity of TARDBP and the antiviral TARDBP/HDAC6 axis. In this sense, deficient HIV-1 Envs from viruses of long-term nonprogressor Elite Controller (LTNP-EC) individuals are insufficient to form the fusion pore and infect because of their inability to escape the antiviral TARDBP/HDAC6-tubulin-deacetylase barrier. Notably, viral particles bearing EC-Envs gain infection function when HDAC6 or the HDAC6/TARDBP axis is inhibited by either HDAC6 inhibitors or their nonactive mutants or interference with HDAC6/TARDBP mRNA, thereby stabilizing MTs to generate a permissive state for infection. Designs and templates were created with BioRender and licensed are obtained for publication.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



