Submitted:
17 October 2025
Posted:
17 October 2025
You are already at the latest version
Abstract
Cutting-edge Artificial Intelligence (AI) techniques keep reshaping our view of the world. For example, Large Language Models (LLMs) based applications such as ChatGPT have shown the capability of generating human-like conversation on extensive topics. Due to the impressive performance on a variety of language-related tasks (e.g., open-domain question answering, translation, and document summarization), one can envision the far-reaching impacts that can be brought by the LLMs with broader real-world applications (e.g., customer service, education and accessibility, and scientific discovery). Inspired by their success, this paper will offer an overview of state-of-the-art LLMs and their integration into a wide range of academic disciplines, including: (1) arts, letters, and law (e.g., history, philosophy, political science, arts and architecture, law), (2) economics and business (e.g., finance, economics, accounting, marketing), and (3) science and engineering (e.g., mathematics, physics and mechanical engineering, chemistry and chemical engineering, life sciences and bioengineering, earth sciences and civil engineering, computer science and electrical engineering). Integrating humanity and technology, in this paper, we will explore how LLMs are shaping research and practice in these fields, while also discussing key limitations, open challenges, and future directions in the era of generative AI. The review of how LLMs are engaged across disciplines—along with key observations and insights—can help researchers and practitioners interested in exploiting LLMs to advance their works in diverse real-world applications.
Keywords:
large language models
; interdisciplinary research
; scientific discovery
1. Introduction
Nowadays, cutting-edge technologies in Artificial Intelligence (AI) keep reshaping our view of the world. For example, as a foundation language model based on the Generative Pre-trained Transformer (GPT) architecture, ChatGPT [1] has shown its capability of generating human-like conversation on extensive topics, which makes it the fastest-growing application (i.e., with more than 100 million users within the first two months of its launch) [2]. Although limitations such as robustness and truthfulness it still remains, due to the impressive performance on a variety of language-related tasks (e.g., open-domain question answering, translation, and document summarization), ChatGPT could have a wide range of potential applications (e.g., customer service, personal assistants, and medical diagnosis). Besides the models like ChatGPT in Natural Language Procession (NLP), the pre-trained foundation models in Computer Vision (CV) such as Florence/Florence-2 [3] and Qwen2.5-VL can achieve state-of-the-art performance on various vision-tasks (e.g., object detection, image segmentation, and video reasoning), which make them particularly useful for applications such as facial recognition, medical image analysis, and self-driving cars. This cross-domain convergence underscores the pivotal role of large language models (LLMs), which provide the representational and reasoning framework for embedding other modalities, positioning them as central components in the evolving ecosystem of AI-powered research and applications.
Motivated by recent advances, this paper surveys cutting-edge LLMs and their integration into a wide range of academic disciplines, including: (1) arts, letters, and law (history, philosophy, political science, arts and architecture, law), (2) economics and business (finance, economics, accounting, marketing), and (3) science and engineering (mathematics, physics and mechanical engineering, chemistry and chemical engineering, life sciences and bioengineering, earth sciences and civil engineering, computer science and electrical engineering). At the intersection of humanistic inquiry and technology, we examine how LLMs may reshape research workflows and professional practice in each area, while also outlining major limitations, unresolved challenges, and promising directions in the era of generative AI. By synthesizing cross-disciplinary uses and distilling key takeaways, this review is intended to guide researchers and practitioners seeking to harness LLMs to advance their work in real-world applications. In the following, we outline the organization of the paper.
Building on recent breakthroughs, in Chapter 2, we ground the reader in what LLMs are and how to assess them. We begin with precise definitions and a concise history of LLMs. We then map the state of the art with an overview and focused profiles of major model families—GPT-series, OpenAI reasoning models, Claude 3, Gemini 2, Gork, Llama 3, Qwen 2, and DeepSeek—highlighting design choices and capabilities. We conclude with evaluation: the core task types, representative benchmarks, and commonly used methods, followed by a performance-at-a-glance synthesis. Together, these sections aim to provide a background, a comparative map of current models, and practical guidance for reading results and making methodologically sound choices.
For each academic discipline within the three clusters—arts, letters, and law; economics and business; and science and engineering—we begin by introducing the discipline through an overview of its major research tasks and traditional methodologies, with the highlight of its key contributions and significant impacts. After identifying common research challenges that could be assisted by AI—particularly LLMs, we integrate disciplinary research with LLMs by providing a taxonomy that aligns with established disciplinary tasks while mapping them onto a unified computational input–output framework. This ensures both disciplinary relevance and algorithmic consistency for model development, benchmarking, and comparative analysis. Within each category, we review existing works on LLM-powered research and applications, examine current limitations, and explore future research directions. Finally, we conclude with representative benchmarks and critical discussions.
In Chapter 3, we survey how LLMs are transforming the humanities and law, moving from evidence to practice. In history, we cover narrative and interpretive uses (e.g., narrative generation and analysis), quantitative and scientific approaches (e.g., simulating historical psychological responses), and comparative and cross-disciplinary work, with benchmarks and a brief discussion. In philosophy, we review normative and interpretive applications (e.g., debate/dialogue generation), analytical and logical ones (e.g., symbol grounding diagnostics), and comparative and cross-disciplinary studies with benchmarks. In political science, we examine text analysis for policy insights, opinion simulation and forecasting, and the generation and framing of political messaging, integrating these with benchmark summaries and reflections. In arts and architecture, we outline model-assisted creation in visual, literary, and performing arts, as well as LLM-aided architectural design, creation, and analysis, followed by evaluations and takeaways. Finally, in law, we cover legal consultant question answering, contract and brief drafting, legal document understanding and case analysis, and judgment prediction, concluding with representative benchmarks and discussion.
In Chapter 4, we review how LLMs are being used across economics and business. In finance, we survey LLMs for trading and investment research, corporate finance, market analysis, financial intermediation and risk management, sustainable finance, and fintech, as well as how these systems are benchmarked. In economics, we cover behavioral and experimental studies, macroeconomic simulation and agent-based modeling, strategic and game-theoretic interactions, and economic reasoning/knowledge representation, with dedicated evaluations. In accounting section, we examine auditing, financial and managerial accounting, and taxation, alongside benchmarking. In marketing section, we review consumer insight and behavior analysis, content creation and campaign design, and market-intelligence and trend analysis, again with performance benchmarks.
In Chapter 5, we chart how LLMs are used across science and engineering. We start with mathematics, including proof assistance, theoretical exploration and pattern recognition, math education, and targeted benchmarks. In physics and mechanical engineering, we cover documentation-centric tasks, design ideation and parametric drafting, simulation-support and modeling interfaces, multimodal lab and experiment interpretation, as well as interactive reasoning, followed by domain-specific evaluations and a discussion of opportunities and limits. In chemistry and chemical engineering, we examine molecular structure and reaction reasoning, property prediction, materials optimization, test/assay mapping, property-oriented molecular design, and reaction-data knowledge organization, followed by a comparison of benchmark suites. In life sciences and bioengineering section, we include genomic sequence analysis, clinical structured-data integration, biomedical reasoning and understanding, and hybrid outcome prediction, with attention to validation standards. In earth sciences and civil engineering section, we review geospatial and environmental data tasks, simulation and physical modeling, document workflows, monitoring and predictive maintenance, plus design/planning tasks, again with benchmarks. Finally, we close the chapter with computer science and electrical engineering: code generation and debugging, large-codebase analysis, hardware description language code generation, functional verification, and high-level synthesis, followed by purpose-built benchmarks and a concluding discussion of impacts and open challenges.
In Chapter 6, we conclude—“Navigating the Present, Shaping the Future”—by synthesizing what we learn across domains. We first outline emerging frontiers, then synthesize evidence across three arenas: (i) arts, letters, and law—shared opportunities, limitations, and use paradigms from historical analysis to legal reasoning; (ii) economics and business—signals from finance, accounting, economics, and marketing translated into strategy with concrete paradigms; and (iii) science and engineering—models as instruments, with discipline probes yielding cross-cutting opportunities, constraints, and workflow-ready paradigms. We conclude with a path forward that integrates schema-aligned multimodality and grounded attribution; tool-augmented computation under formal constraints; rule-governed, reproducible agent simulation; temporal-causal adaptation; decision support with calibrated uncertainty and domain controls; human-in-the-loop oversight and transparent governance; and education-led capacity building with embedded safety—providing a practical, auditable, and scalable blueprint for cross-disciplinary adoption.
Altogether, in this paper, we chart the LLM landscape end-to-end—foundations and evaluation, then concrete uses across arts, letters, and law, economics and business, and science and engineering—showing what works today, where capabilities remain fragile, and how to measure progress. Readers can take away a common vocabulary and task taxonomy; guidance for selecting models and tools; recipes for building rigorous evaluations and benchmarks; and practical patterns for deployment that balance utility with safety, compliance, and human oversight. The content of this paper may not be exhaustive, and certain perspectives presented herein may be open to debate; furthermore, with the rapid advancement and continuous evolution of technology especially in the field of AI, the disciplines reviewed in this study are expected to witness ongoing developments. However, as an initial effort, this paper may help readers identify promising problem formulations, design defensible evaluations, estimate potential impact, and anticipate failure modes in their respective disciplines. We hope this synthesis equips researchers, practitioners, and policymakers to navigate the present responsibly—and to shape a future where LLMs deliver reliable, auditable, and genuinely useful capabilities—across a wide spectrum of academic disciplines.
2. Background
In this chapter, we orient the reader within the rapidly advancing field of LLMs by clarifying both what these models are and how their performance can be meaningfully assessed. To set the stage, we begin with precise definitions and a concise historical overview, tracing the key developments that have shaped the present landscape. Building on this foundation, the chapter then turns to the current state of the art (SOTA), presenting comparative portraits of major model families—including the GPT lineage, OpenAI’s reasoning-focused systems, Claude 3, Gemini 2, Gork, Llama 3, Qwen 2, and DeepSeek—highlighting the architectural choices and distinctive capabilities that define each. From this survey, we move naturally to the question of evaluation, examining how these models are tested in practice: the principal categories of tasks, the benchmark suites most widely referenced, and the methodological approaches commonly employed. We conclude with an integrated synthesis of comparative performance, offering readers both a high-level view and practical guidance. Together, these sections establish the necessary background, provide a structured map of the model landscape, and equip readers to interpret results and make sound methodological choices in the chapters that follow.
2.1. What are Large Language Models (LLMs)?
2.1.1. Definition of LLMs
Large Language Models (LLMs) are artificial intelligence systems designed to comprehend and generate human language by modeling the probability distribution of word sequences, typically using neural network architectures like Transformers [4,5,6,7]. At their core, LLMs rely on deep learning techniques, especially self-attention mechanisms, allowing parallel processing of text [8]. The fundamental logic behind LLMs involves two main phases: pre-training and fine-tuning [8]. In the pre-training phase, LLMs are trained on extensive text corpora using self-supervised learning tasks, such as predicting masked tokens (words) or the next word in a sequence. This process allows them to implicitly learn grammar, syntax, semantics, and factual knowledge directly from raw text data, without explicit human labeling [7,9]. Subsequently, in the fine-tuning phase, these models are further trained on task-specific datasets or prompted with task examples, enabling them to perform diverse language understanding and generation tasks efficiently and effectively [10,11,12]. From an application perspective, this pretrain–finetune paradigm allows non-experts, including professionals outside computer science, to benefit from powerful language understanding capabilities without requiring deep technical expertise or extensive labeled data. Users can adapt LLMs to their specific needs through minimal customization, making advanced AI tools more accessible across domains such as healthcare, law, and biology.
- How to Define “Large”? LLMs are characterized as “large” by their exceptionally high number of parameters—typically ranging from billions to hundreds of billions—paired with extensive training data. For instance, OpenAI’s GPT series contains hundreds of billions of parameters, allowing it to capture a wide range of linguistic patterns and knowledge. Beyond sheer size, the term “large” also signifies a critical scale at which emergent capabilities arise—abilities that are not present in smaller models and often cannot be predicted simply by extrapolating from smaller-scale performance [13]. Driven by advances in computing power and guided by scaling laws, the size of language models has increased rapidly over recent years. Models once regarded as state-of-the-art have been quickly surpassed and are now considered relatively small by current standards. For example, GPT-2, released in 2019, contained 1.5 billion parameters [5], whereas the smallest variants of contemporary LLMs typically begin at 7 billion parameters. This shift highlights the field’s rapid progression and the evolving definition of what constitutes a “large” model.
How to Categorize LLMs? While there are many ways to categorize LLMs, the choice often depends on the intended application and user needs. Based on the current development of LLMs, we highlight two complementary perspectives, functionality-based [14] and reasoning-based [15], that offer practical guidance for both researchers and domain professionals aiming to exploit LLMs effectively.
From the functionality-based perspective, LLMs can be broadly divided into general-purpose and domain-specific models. General-purpose LLMs, such as GPT-3 and GPT-4 [1,7], are trained on large, diverse corpora and perform well across a wide array of tasks. In contrast, domain-specific LLMs are fine-tuned on specialized data to enhance performance in focused areas, as general-purpose models may often under-perform in specialized domains due to their lack of domain-specific knowledge [16]. For example, BioBERT [17] targets biomedical texts, while SciBERT [18] is optimized for scientific literature. This distinction is especially important for professionals in domains like healthcare, legal services, and scientific research, who require models that understand domain-specific terminology and context, yet retain general linguistic competence.
From the reasoning-based perspective, LLMs can be distinguished by their capacity for complex inference. Reasoning-capable models, such as GPT-4 with chain-of-thought prompting [19], GPT o1 [20], and Deepseek-R1 [21], are designed for multi-step reasoning tasks like mathematical problem solving or logical deduction. These models are well suited for analytical or decision-support tasks where explainability and intermediate steps matter. In contrast, non-reasoning models—though less adept at complex inference—excel in tasks where surface-level language understanding is sufficient, such as summarization, classification, or named entity recognition [8]. These models are often more efficient and robust, making them preferable in real-time applications or resource-constrained settings.
2.1.2. History of LLMs
Figure 1 illustrates the key milestones in the development of LLMs, which will be introduced in detail below.
Rule-based Systems: Symbolic Foundation. The emergence of LLMs is the culmination of several decades of progress in natural language processing (NLP), moving from manual rules to statistical models to modern deep learning approaches [8,22]. Early attempts at machine language understanding were rule-based and symbolic – researchers hand-crafted grammatical rules or expolited expert systems to parse and generate text [23,24,25]. While insightful, these systems were brittle and could not easily scale to the diversity of real-world language. By the 1990s, the field had shifted toward statistical methods: instead of manual rules, systems learned from data. For example, n-gram language models were used to predict text by learning probabilities of word sequences from large text corpora. Notably, IBM’s alignment models in the 1990s applied statistical methods to tasks like translation [26,27], and by the 2000s researchers were building ever larger text datasets (“web-scale” corpor [28]) to train statistical language models [29,30]. These data-driven models outperformed earlier symbolic approaches, as evidenced by the fact that by 2009, statistical language models had largely overtaken rule-based ones on many tasks. However, classical statistical models still had limitations — they typically considered only limited context (e.g. a fixed window of previous words) and could not capture long-range dependencies or deeper meanings effectively [8].
Recurrent Neural Networks: Learning Sequences. A major paradigm shift came in the 2010s with the rise of neural network approaches to NLP [31,32]. Inspired by successes in computer vision, researchers began applying deep learning to language. Recurrent neural networks (RNNs) [32,33], especially ones with gating mechanisms like LSTM [34,35], were used to model sequences of text. RNN-based language models could maintain a hidden state that in principle captured information about prior words in a sequence, allowing them to handle longer contexts than n-gram models [36,37]. By the mid-2010s, RNNs became state-of-the-art for tasks like translation – for instance, in 2016 Google Translate switched from a phrase-based system to a neural sequence-to-sequence model (an encoder-decoder network using LSTMs) for improved accuracy [38,39]. This neural takeover marked an improvement in handling context and generating more fluent outputs. Nonetheless, standard RNNs had drawbacks: they processed words sequentially and had difficulty with very long sequences or capturing long-term dependencies due to issues like vanishing gradients [8].
Early progress was driven by advances in word embeddings. The introduction of Word2Vec (Mikolov et al., 2013) enabled the learning of dense vector representations of words, capturing rich semantic relationships and laying the groundwork for neural NLP. Building on this, the application of sequence-to-sequence (Seq2Seq) models with attention mechanisms (Bahdanau et al., 2015) enabled substantial improvements in tasks such as machine translation and text summarization.
Transformers: a Parallel Paradigm Shift. The next major breakthrough came with the introduction of the Transformer architecture in 2017, which profoundly transformed the development of language models [4]. Transformer introduced the mechanism of self-attention to handle sequences, allowing the model to consider all words in a sentence in parallel rather than sequentially [4]. Transformers could thus capture long-range relationships more effectively and be trained in parallel batches, dramatically improving scalability. This architecture fundamentally shifted the field, enabling the training of significantly larger models on vastly greater datasets than was previously feasible.
As shown in Figure 2, Transformer model consists of two primary components: an encoder and a decoder, both built by stacking multiple identical layers. Each encoder layer contains two main sublayers: a multi-head self-attention mechanism and a feedforward neural network. The multi-head self-attention mechanism allows the model to attend to different positions of the input sequence across multiple subspaces, thereby capturing diverse semantic relationships. The self-attention mechanism computes attention weights based on the dot-product between linearly projected queries (Q) and keys (K), which are then used to aggregate values (V) through a weighted sum. By deploying multiple attention heads in parallel, the model learns rich, context-aware representations.
The decoder shares a similar structure with the encoder but includes an additional encoder-decoder attention sublayer between the self-attention and feedforward layers. This component enables the decoder to focus on relevant parts of the input sequence during generation, which is critical for producing accurate and coherent outputs.
Within a year of its introduction, large Transformer-based models began to emerge. Notably, BERT (2018) employed the Transformer encoder to achieve unprecedented performance on natural language understanding tasks [9]. The success of BERT and subsequent models like T5 [40] popularized the pretraining-finetuning paradigm, where models are first pretrained on large corpora using objectives like masked language modeling and then fine-tuned for specific downstream tasks. Around the same time, OpenAI introduced the first Generative Pre-trained Transformer (GPT-1)[5], which leveraged a Transformer decoder and an autoregressive training objective (predicting the next token) to specialize in text generation. GPT-1 is often regarded as the first modern LLM, despite its relatively modest scale of 117 million parameters by today’s standards. Unlike encoder-based models such as BERT, which are primarily designed for understanding tasks and lack autoregressive generation capability[9,41,42], GPT-1 marked the beginning of a new paradigm: decoder-only architectures optimized for text generation.

Table 1.
Several important models in different time nodes.
| Time Nodes | Model | Core Tech | Contribution/Feature | Major Lab/Company | Release Time |
|---|---|---|---|---|---|
|
Rule-based Pre 2010 |
ELIZA [43] | Rule-based communication | First NLP communication system | MIT | 1966 |
| n-grams [44] | Markov language model | Based on statistical frequency modeling | IBM | 1992 | |
| PCFG [45] | Context-independent probabilistic | Syntactic analysis & structural prediction | Brown Uni. | 1998 | |
|
RNN 2010s |
RNNLM [46] | Based on RNN | First RNN-based language model | Microsoft | 2010 |
| Seq2Seq [47] | Based on GRU / LSTM | Pioneered encoder-decoder for NLP | 2014 | ||
|
Transformer 2017-2019 |
Transformer [4] | Self-attention | Breaks sequential constraint, enables parallelism | 2017 | |
| BERT [9] | Masked LM + fine-tuning | Contextualized language representation | 2018 | ||
| T5 [40] | Text-to-text transfer learning | Reformulates all NLP tasks as text generation | 2019 | ||
|
GPT & ChatGPT 2020-2023 |
GPT-1/2 [5,6] | Autoregressive transformer | Unified architecture for generation | OpenAI | 2018/2019 |
| GPT-3 [7] | Large-scale autoregressive model | Kickstarted large model era | OpenAI | 2020 | |
| ChatGPT [48] | Fine-tuned GPT-3 with RLHF | Interactive and aligned chatbot | OpenAI | 2022 | |
| GPT-4 [1] | Multimodal, tool-use capabilities | Generalized across wide range of tasks | OpenAI | 2023 | |
|
Other LLMs 2020-2023 |
Codex [49] | Code-focused GPT fine-tuning | Natural language to code translation | OpenAI | 2021 |
| FLAN-T5 [50] | Instruction-tuned T5 | Strong zero-shot generalization | 2022 | ||
| QWen [51] | Tool-augmented transformer | Open-source model with strong instruction-following | Alibaba | 2023 | |
| LLaMA [52] | Efficient transformer variants | High performance with fewer resources | Meta | 2023 | |
|
Emerging Frontier Till Now |
Mixtral [53] | Sparse Mixture-of-Experts | Efficient inference with high performance | MixtralAI | 2023 |
| DeepSeek-R1 [21] | Neural-symbolic reasoning | Multi-step logic | DeepSeek | 2024 | |
| o1 [20] | Experimental AGI prototype | Focus on generalized reasoning skills | OpenAI | 2024 |
GPT and ChatGPT: From Language Modeling to General Intelligence. The GPT series by OpenAI, based on the Transformer architecture, incorporated key design innovations that positioned it as a landmark in the advancement of LLMs. Unlike BERT’s masked token prediction, GPT models use causal (autoregressive) language modeling, predicting the next token given all previous tokens [5,6,7]. This seemingly simple change allows GPT models to generate long, coherent passages of text. The architecture is decoder-only, stacking multiple Transformer blocks to model the distribution over sequences. The evolution from GPT-1 [5] through GPT-2 [6] to GPT-3 [7] (2018–2020) was marked by exponential growth in both model size and training data [8]. GPT-3 [7] with 175 billion parameters, was trained on a massive and diverse dataset covering web text, books, Wikipedia, and more. It demonstrated few-shot and even zero-shot learning: the ability to perform unseen tasks when given just a prompt or a few examples, without additional training. This behavior, previously unseen, signaled an emergent form of general-purpose linguistic intelligence.
In fact, it was the release of ChatGPT in late 2022 that truly rendered LLMs accessible and practically useful at scale. While based on GPT-3.5 [7] and later GPT-4 [1], ChatGPT introduced instruction tuning and reinforcement learning from human feedback (RLHF) to align the model with human values, dialogue etiquette, and safety constraints [1,54]. Unlike vanilla GPT-3, which was often unpredictable, ChatGPT could follow instructions, engage in multi-turn conversations, and avoid unsafe outputs—making it viable for deployment in education, research, programming, and creative tasks.
However, it was the release of ChatGPT (OpenAI, late 2022) that truly democratized access to LLMs, introducing a conversational interface that brought LLM capabilities to millions of users and triggered a global wave of LLM applications.
The difference between GPT and vanilla Transformer is thus not just in architecture (both use the Transformer as a backbone), but in how GPT leverages generative pretraining, scaling laws [55,56], few-shot prompting [57,58], and instruction alignment [59] to transition from a language model to a general-purpose AI assistant. ChatGPT represents a milestone because it closes the loop between model, user, and feedback, making LLMs interactive, helpful, and widely usable.
Reasoning: The Emerging Frontier. With the widespread adoption of LLMs (LLMs), researchers have observed that these models often struggle with multi-step problems and abstract logical tasks. To further enhance their capabilities and move closer to the goal of artificial general intelligence (AGI), substantial efforts have been made to improve their reasoning abilities. The introduction of chain-of-thought prompting [60] has emerged as a promising approach to address this limitation. By enabling models to decompose problems into intermediate reasoning steps, this method mirrors the way humans tackle complex tasks [61,62]. This paradigm shift has empowered LLMs to handle tasks requiring sequential logical reasoning, ranging from mathematical problem solving to complex decision-making processes [62].
The year 2023 saw continued innovation with the release of GPT-4, a multimodal model capable of processing both text and images, along with Claude (Anthropic), which emphasized alignment and safety through Constitutional AI, and LLaMA (Meta), which spurred a vibrant open-source LLM community. Most recently, GPT-4o (2024) introduced real-time multimodal capabilities—integrating text, vision, and audio—with improved conversational alignment and latency, signaling a new phase in the evolution of interactive and multimodal AI systems.
Complementing this approach, self-consistency techniques [63] have been proposed to further refine reasoning performance [62]. These methods sample multiple reasoning paths and select the most coherent outcome, significantly reducing error rates and enhancing the reliability of model outputs. Additionally, a variety of reinforcement learning-based strategies have been employed to improve reasoning capabilities. One notable example is the DeepSeek-R1 [21] model, which contains 671 billion parameters and is specifically optimized for tasks involving mathematics, programming, and logical reasoning. It leverages reinforcement learning with a rule-based reward mechanism (rule base reward + RL) to enhance its reasoning proficiency [21]. As models continue to improve in planning and reasoning over complex problems, humanity appears to be gradually approaching AGI.
2.2. State-of-the-art LLMs
Figure 3.
Chronological display of current SOTA LLMs.
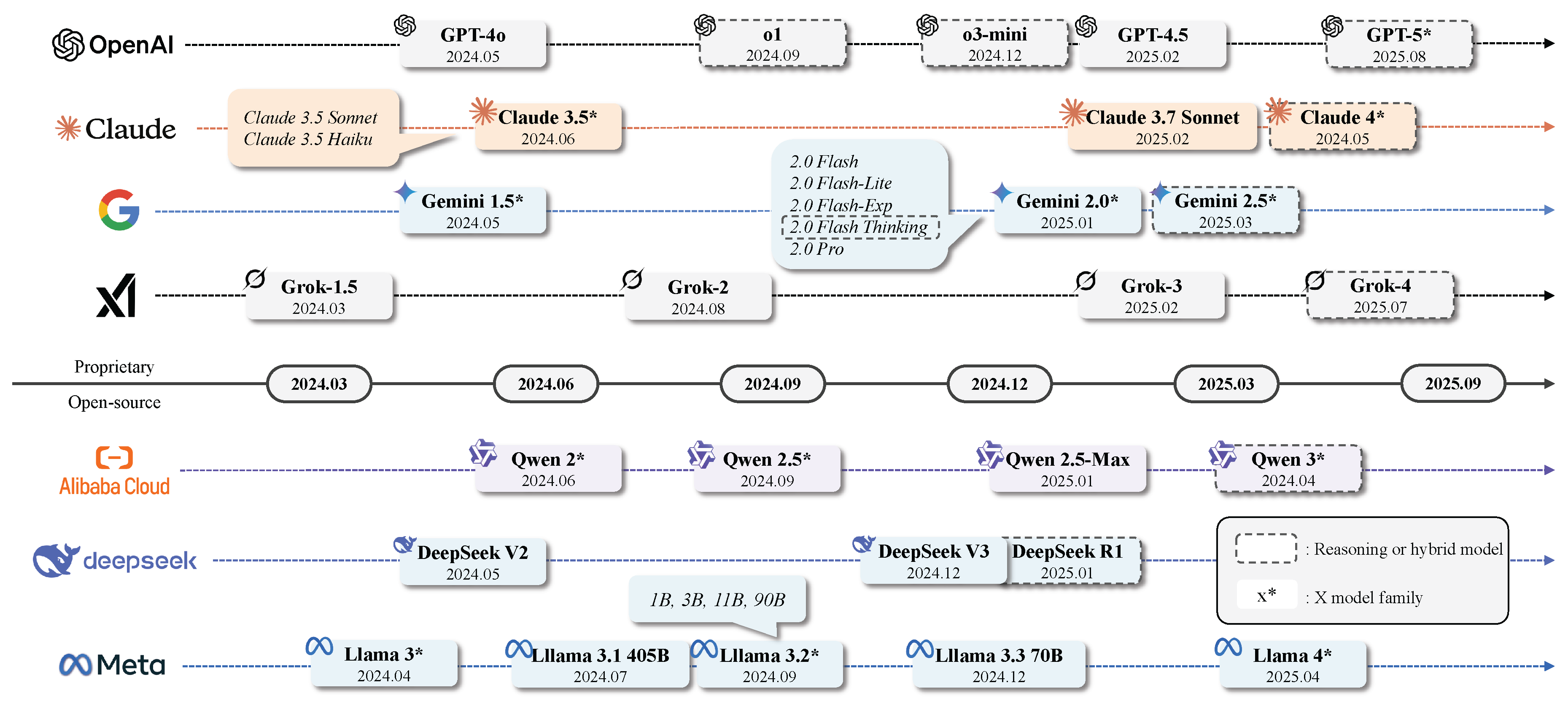
2.2.1. Overview
Nowadays, a variety of LLMs are available to address different downstream tasks. These models can be broadly categorized as closed-source (accessed through APIs) or open-source (deployed locally). Given varied requirements such as latency, performance, and input/output modalities, no single LLM can simultaneously satisfy all use cases. In general, closed-source LLMs outperform open-source LLMs due to large model sizes, extensive training corpora, and training recipes. They also tend to support larger context windows and higher token throughput, enabling more complex and longer interactions. However, they often require API usage, resulting in high time to first token (TFT), also called latency, and may incur substantial costs, especially for reasoning models, such as OpenAI o1-pro. Additionally, off-site inference can raise privacy concerns when handling sensitive data. By contrast, open-source LLMs typically feature smaller model sizes, allowing cost-effective deployment on local hardware. Open-source LLMs often yield faster response times (depending on the user’s infrastructure) and stricter data confidentiality. Aside from the aforementioned basic factors, LLM capabilities should also be considered. We summarize the key features of SOTA LLMs in Table 2. In the subsequent sections, we undertake a systematic analysis of each SOTA LLM model family in detail. Note that with the rapid development of AI technology, LLMs are also evolving quickly. This section only covers the mainstream SOTA models up to the time of publication. As GPT plays a central and milestone role in the development of LLMs—by establishing the methodological paradigm, advancing large-scale modeling, and shaping the surrounding application ecosystem—we begin by introducing and discussing the GPT-series models in the following section (Section 2.2.2).
Table 3.
Guidelines for LLMs selection based on tasks and corresponding constraints.
| Task | Modality | Context Window | Latency | Privacy | Budget | Hardware | Perf. | LLMs |
| Conversational Task | T, (I, A) | ≥ 200K | Yes | No | Low | – | Med | Gemini 2 |
| T, (I) | < 200K | Yes | No | Med | – | High | ChatGPT-4o | |
| T | < 200K | No | Yes | – | Med | – | Qwen 2.5 | |
| T, (I) | < 200K | No | Yes | – | High | – | Llama 3.2 MM | |
| Reasoning Task | T, (I, A) | ≥ 200K | Yes | No | – | – | High | Gemini 2.5 Pro |
| T | < 200K | No | No | Med | – | Med | OpenAI Reasoning | |
| T | < 200K | Yes | Yes | – | High | – | DeepSeek-R1 | |
| Coding Task | T | < 200K | Yes | No | Low | – | Med | Claude 3.5 |
| T | < 200K | No | No | High | – | High | Claude 3.7 | |
| T | ≥ 200K | No | Yes | – | Med | – | Qwen 2.5-1M | |
| Open-Domain QA | T, (I, A) | ≥ 200K | Yes | No | Low | – | Med | Gemini 2 |
| T | < 200K | Yes | No | Med | – | High | GPT-4o | |
| T | < 200K | Yes | Yes | – | Med | – | QwQ 32B | |
| Focused Tasks (Fine-tune) | T | < 200K | Yes | No | Low | – | – | GPT-4o mini |
| T, (I) | < 200K | No | Yes | – | Med | – | LLama 3.2 MM | |
| T | < 200K | Yes | Yes | – | Low | – | DeepSeek-R1 Distill |
Notes: T, I, and A denote Text, Image, and Audio, respectively. Brackets surround optional input modality. In latency, YES indicates the task has a latency requirement. For privacy, YES represents that the input data may be confidential. For budget, hardware, and performance(perf.), we report in low, med (median), and high, based on the relative criteria. For example, LOW BUDGET prefers costs less than $1 per 1M tokens, HIGH HARDWARE indicates a requirement to run LLMs with model size greater than or equal to 90B. The recommended LLMs are based on the performance, requirements, and strengths provided in the provider official websites, Artificial Analysis, and Vellum LLM Leaderboard. These provided LLMs in each scenario are merely for referenes, and are not guaranteed to be the optimal solutions. Users should consider their application scenarios when choosing the LLMs.
2.2.2. GPT-series Models
The Generative Pre-training Transformer (GPT) series represents a family of autoregressive language models based on Transformer architecture [4], introduced by OpenAI 1, These models employ a self-supervised learning paradigm that generates contextually coherent text through sequential token prediction. Due to the excellent performance in generative language tasks, GPT models have introduced multiple breakthroughs in the NLP community [8,75]. In this section, we will discuss each GPT model and its contributions in detail.
GPT-1 [5]. Prior to the emergence of GPT, conventional NLP models are trained over large amounts of task-specific annotated datasets, leading to limited generalization capabilities across tasks beyond trained datasets. To address this challenge, OpenAI developed GPT-1 in 2018, a decoder-only transformer architecture with 117 million parameters, and adopts a two-stage training recipe: (i) unsupervised pre-training on large text corpus; and (ii) supervised fine-tuning. One of the successes of GPT-1 is the excellent zero-shot performance across multiple downstream tasks, including sentiment analysis, question answering, etc. Besides the success in model performance, the underlying principle to model natural language text, i.e., next token (word) prediction, also has a profound influence on the development of subsequent LLMs [76].
GPT-2 [6]. In late 2019, OpenAI released GPT-2 which employed similar architecture of GPT-1, with 10 times the size of GPT-1 (117M to 1.5B parameters). The model is trained over a newly collected webpage dataset, called WebText, which contains slightly over 8 million documents [6]. GPT-2 sought to perform multi-task via unsupervised learning, without explicit fine-tuning over labeled datasets. Motivated by existing studies for the probabilistic framework with task condition [77,78,79], GPT-2 introduces a probabilistic framework for multi-task solving, formulated as
which generates output conditioned on the input and task information. Here, the task information can be regarded as the pioneer of the concept In-Context Learning (ICL) in the current LLMs community. Besides the proposed probabilistic model, the success of GPT-2 under the unsupervised multi-task learning settings is rooted in their training philosophy: the global minimum of the unsupervised objective is also the global minimum of the supervised objective [6].
GPT-3 and GPT-3.5 [7] . Although GPT-2 has provided significant insights into the LLM community, the overall performance of GPT-2 is lower than supervised SOTA models. OpenAI extended GPT-2 to GPT-3 in 2020, which demonstrated incremental performance compared to GPT-2 and supervised fine-tuning models, by scaling up the model architectures to 175B parameters, i.e., the largest language model ever at that time. Although not explicitly stated, the performance gain of GPT-3 over GPT-2 validates the scaling law [56] that large models, in terms of model parameters, have stronger capabilities. Another contribution of GPT-3 is the introduction of ICL, which instructs LLMs with a few demonstrations of the task at inference time. These demonstrations can be viewed as task conditioning in Equation 1. To enhance the capability over complex tasks, such as code completion and math problems, OpenAI develops a stronger capability model than GPT-3 in complex problem solving, by training over code dataset, called GPT-3.5 [49]. In addition to capability improvement, GPT-3.5 is also trained with a three-stage reinforcement learning algorithm from human feedback (RLHF) (first introduced in InstructGPT [80]), which helps enhance the ability to follow instructions and ease concerns regarding LLMs producing toxic responses or violation of local policies. The research contribution of GPT-3.5 and RLHF can be summarized in three directions: training LLMs (i) using human feedback, (ii) to assist human evaluation, and (iii) to do alignment research [8,81].
ChatGPT. OpenAI launched a conversation model called ChatGPT in November 2022, which achieves a pivotal milestone in the AI research community. ChatGPT is a sibling model to InstructGPT, while specially optimized on a human-generated conversation dataset [48]. ChatGPT demonstrates superior ability in communications with humans, and the additional support of the plugin mechanisms enables ChatGPT to obtain external knowledge by interacting with plugins, such as a calculator and web search etc. The plugin support can be viewed as the prototype of MCP [82]. The success of ChatGPT marks the exploration of LLMs and has a significant impact on future research in the LLM domain.
GPT-4* [1,64]. The aforementioned GPT series models only support text input. To extend the model input from single text to multimodal signals (text and image), OpenAI published GPT-4 in March 2023. GPT-4 outperforms earlier GPT models, including ChatGPT, in various benchmark datasets. Moreover, as reported in GPT-4 technical report [1], OpenAI spent six months on human alignment training in RLHF to alleviate the safety concerns of GPT-4. Besides the remarkable performance gain and alleviation of safety concerns, GPT-4 is trained over a new principle called predictable scaling, which refers to the development of infrastructure that allows for reliable extrapolation of the performance across scales of compute and model sizes. This approach is adopted by most later LLMs to minimize the need for extensive model-specific tuning, making the training process more efficient and systematic. Several GPT-4* models are released in late 2024 or early 2025, i.e., GPT-4o [83], GPT-4o mini [84], and GPT-4.5 [64], which step further than GPT-4. GPT-4o and GPT-4o mini are multilingual, multimodal (text, image, audio, and video) models that generate any combination of text, audio, and image as output. GPT-4.5 emphasizes improved writing capabilities, enhanced world knowledge, and a refined interaction experience.
GPT-5* [65]. GPT-5 is introduced as a model family with multiple sizes, including GPT-5, GPT-5-mini, and GPT-5-nano, and two principal configurations: a standard model and a thinking variant optimized for extended, deliberate reasoning. The release highlights step-change improvements in core capabilities: stronger multi-step reasoning, more capable multimodality, and more reliable tool use and orchestration. On the training and alignment side, GPT-5 adopts an expanded RLHF pipeline and upgraded data curation, which together aim to improve instruction following, factuality, and controllability. Safety systems are deepened with tighter gating and continuous monitoring, particularly for sensitive domains such as biosecurity, cybersecurity, and model autonomy. Moreover, according to their report [65], GPT-5 has improved calibration and robustness, enabling more dependable model behavior under varied prompts and contexts. Operationally, GPT-5 delivers lower latency and better cost efficiency than prior GPT-4-class models, supported by serving-side and architectural optimizations. Taken together, these changes position GPT-5 as a more capable, controllable, and practical foundation model for both general and high-stakes reasoning use cases, with a specialized thinking configuration for complex problem solving.
2.2.3. OpenAI Reasoning Models
OpenAI Reasoning Models, such as OpenAI o1 and o3-mini, represent a significant evolution in LLM design, specifically engineered to address complex, multi-step reasoning tasks [85]. These models, collectively known as the "o" series, introduce innovations including reasoning tokens and advanced reinforcement learning techniques. Such features facilitate a structured "chain-of-thought" reasoning process, wherein the model generates internal reasoning steps prior to producing a final response. This approach proves particularly effective in domains demanding sophisticated problem-solving, including advanced programming, scientific inquiry, and strategic planning. Moreover, the models are available in configurations with varying computational demands, allowing users to balance speed and accuracy based on application-specific requirements. This emphasis on explicit, structured reasoning represents a departure from traditional LLM architectures, aiming to generate outputs that are not only accurate but also traceable through logical inference. OpenAI’s release of its reasoning models marks the onset of heightened competition among leading technology firms in the realm of reasoning-oriented artificial intelligence.
2.2.4. Claude 3 Model Family
Claude 3 [66,67] is a family of LLMs developed by Anthropic (https://www.anthropic.com/), a company founded in 2021 by former OpenAI employees. Claude utilizes Transformer architecture and has gone through versions Claude 3 Opus, Claude 3.5 Sonnet, Claude 3.5 Haiku, and Claude 3.7 Sonnet from 2024 to 2025. At the heart of Claude, the task is training LLMs to be helpful, honest and harmless [66]. Claude achieves this by incorporating a Constitution, which contains predefined ethical and behavioral guidelines that shape the outputs of LLMs. Most of the principles in the Constitution are introduced in their earlier post [86], with an additional principle based on feedback from the public input process that directs Claude to be empathetic and accessible to people with disabilities, thereby reducing model stereotype bias.
The Claude 3 family offers various models with capabilities to meet specific needs. Claude 3.5 Haiku, the fastest model in the Claude 3 family, is optimized for near-instant responses, which is suitable for real-time tasks to mimic human interactions, such as customer support, content moderation, etc. Claude 3.5 Sonnet balances performance and speed, excelling in enterprise workloads like data processing and code generation at a low cost. Claude 3.7 Sonnet, the latest Claude model, is a hybrid reasoning model that provides the thinking process in the output. It includes a toggleable "extended thinking" model in which Claude produces a sequence of tokens as a "thinking process" to work through complex problems before delivering the final response. This mode, trained through reinforcement learning, allows for detailed step-by-step reasoning, which can be adjusted by the user to specify a token limit. Experiments reported in Claude 3.7 Sonnet System Card [67] demonstrate that the "extend thinking" model is particularly valuable for challenging tasks, such as mathematical problems, complex analysis, and multi-step reasoning tasks. Overall, the Claude 3 models are distinguished by their multi-modal capabilities, allowing them to process visual inputs alongside text. They demonstrate SOTA performance on vision-related benchmarks and quantitative reasoning tasks [87]. Additionally, Claude 3.5 Sonnet stands as the SOTA model in coding benchmarks, and maintains strong performance for routine programming tasks in practical applications.
2.2.5. Gemini 2 Model Family
Gemini 2.0 represents Google’s latest advancement in multimodal LLMs, encompassing a comprehensive suite of models tailored to diverse computational needs [68]. Central to this suite is Gemini 2.0 Flash, a high-performance model optimized for rapid response and efficient handling of general-purpose tasks. Accompanying this is Gemini 2.0 Flash-Lite, a more cost-effective variant designed to maintain substantial performance while reducing computational overhead. Additionally, Gemini 2.0 Pro demonstrates particular strengths in code generation, tool use, and the processing of complex prompts with its 2 million context window. Key innovations in the Gemini 2.0 series include native tool usage capabilities, image generation, and speech synthesis, all within an expanded multimodal framework [88]. Enhanced context window size and improved integration of multiple input-output modalities position Gemini 2.0 as a pivotal tool in the evolution toward agentic AI systems.
2.2.6. Gork Model Family
Grok 3, developed by xAI, integrates extensive pretraining with enhanced reasoning capabilities enabled by the Colossus supercluster, which offers an order-of-magnitude increase in computational power over previous state-of-the-art models [89]. Grok 3 exhibits marked improvements in areas such as logical reasoning, mathematics, coding, factual recall, and adherence to complex instructions. Its reasoning proficiency is reinforced through large-scale reinforcement learning, enabling the model to engage in extended problem-solving, correct internal inconsistencies, and explore alternative solution pathways. This iterative training process yields more accurate and dependable outputs. Benchmark evaluations reveal that Grok 3 achieves a leading Elo score of 1402 in the Chatbot Arena [89] (the score may change over time). In addition, xAI has released Grok 3 mini, a more computationally efficient variant that retains strong reasoning capabilities. However, at the time of writing, APIs for the Grok 3 series remain unavailable to the public.
Next, we provide reviews for open-source LLMs, i.e., GPT-OSS, Llama 3, Qwen, and DeepSeek Model Family, that the model parameters are available on public platforms, such as Huggingface (https://huggingface.co/) and Github (https://github.com/). While open-source, the licensing terms of each model may vary significantly, necessitating careful consideration when deploying them in research or production environments.
2.2.7. GPT-OSS
GPT Open-Source Series (GPT-OSS) [69] is a family of open-weight language models released by OpenAI under the Apache 2.0 license in August 2025, marking a notable shift in the company’s stance toward open-source AI. The models employ autoregressive MoE transformer architectures and come in two sizes: gpt-oss-120b and gpt-oss-20b. Specifically, gpt-oss-120b consists of 36 layers, with 116.8B total parameters and 5.1B “active” parameters per token per forward pass, while gpt-oss-20b has 24 layers with 20.9B total and 3.6B active parameters. Training combines RL with methods informed by OpenAI’s most advanced internal system, e.g., o3 and related frontier models. The models standardize on an extended o200k_harmony tokenizer and a harmony chat schema that encodes role hierarchy and channels for CoT, tool calls, and final outputs, which support reliable multi-turn, agentic behavior and seamless interleaving of reasoning with function execution. In evaluation, gpt-oss-120b achieves near-parity with o4-mini on core reasoning benchmarks, while running efficiently on a single 80 GB GPU. The gpt-oss-20b model delivers performance comparable to o3-mini on common benchmarks and can run on consumer devices with just 16 GB of memory, making it ideal for on-device use cases, local inference, or rapid iteration without costly infrastructure. Both models also perform strongly on tool use, few-shot function calling, and CoT reasoning. Together, these elements yield an open, deployable blueprint for long-context, tool-using, and compute-adaptive reasoning systems.
2.2.8. Llama 3 Model Family
Large Language Model Meta AI (Llama) is a family of LLMs introduced by Meta AI (https://www.meta.ai/), first released in February 2023. It serves as Meta’s response to OpenAI’s GPT models and is designed as a foundational model for various NLP tasks [90,91]. Llama 3 Model Family [74] has gone through versions Llama 3, to 3.3, with model parameters ranging from 1B to 405B. All of these models are auto-regressive decoder-only models based on the Transformer with slight modifications for efficiency purposes. Specifically, Llama 3 leverages grouped query attention [92] with eight heads to improve inference speed and to reduce the size of key-value caches during decoding. The performance gain of Llama 3 compared with previous versions, i.e., Llama 1 & 2, is primarily driven by improvement in data quality and diversity, as well as by increased training scale. Unlike other LLM model families that employ RLHF for human preference alignment, Llama 3 adopts Direct Preference Optimization (DPO), which directly optimizes for the policy best satisfying the preferences with a simple classification object. Meta AI also explores Proximal Policy Optimization (PPO) [93], but found that DPO requires less computing for large-scale models and performs better. The success of Llama is rooted in the efficient training recipe and the huge high-quality training data.
2.2.9. Qwen 2 Model Family
Qwen 2 model family is developed by Alibaba Cloud (https://www.alibabacloud.com), also known as Tongyi Qianwen. These models are designed for a variety of downstream tasks, including NLP, multimodal understanding, and coding assistance. The first version of the Qwen 2 model family is Qwen 2 [70] with model parameters ranging from 0.5B to 72B. In September 2024, an extended version, called Qwen 2.5, was released with more model size options compared to Qwen 2, such as 3B, 14B, and 32B. In earlier 2025, Alibaba Cloude further released an advanced version, Qwen 2.5 Max, and a reasoning model called QWQ-32B. The architecture of Qwen 2 model family is similar to Llama 3, which adopt Transformer-based decoder architecture with GQA [92] for efficient KV cache, SwiGLU activation [94] for non-linear activation, and RoPE [95] for encoding position information [71]. All of the aforementioned Qwen 2 models are available at Huggingface. One success of the Qwen 2 model family is relevant to their innovative Mixture of Expert (Moe) [96], which efficiently allocates computational resources, improving scalability and performance. Moreover, Qwen 2 models support multilingualism across 29+ languages for global applications.
2.2.10. DeepSeek Model Family
DeepSeek-V3. DeepSeek-V3, released by DeepSeek in December 2024, employs a Mixture-of-Experts (MoE) architecture comprising 671 billion parameters, with 37 billion parameters activated per token [73]. This dynamic routing mechanism allows the model to selectively activate relevant subsets of parameters based on input characteristics, enhancing both computational efficiency and model performance. Trained on a vast multilingual dataset totaling 14.8 trillion tokens—primarily in English and Chinese—over a 55-day period, the training process utilized 2,048 NVIDIA H800 GPUs at an estimated cost of $5.6 million. This is significantly more cost-efficient than comparable models such as GPT-4, whose training expenditures are estimated to range between $50–100 million. Benchmark results indicate that DeepSeek-V3 surpasses models such as LLaMA 3.1 and Qwen 2.5, and achieves parity with leading models like GPT-4o and Claude 3.5 Sonnet.
DeepSeek-R1. DeepSeek-R1 [21], introduced in January 2025 by DeepSeek, advances the reasoning capabilities of its predecessor through an enhanced MoE architecture and a multi-stage training regimen [21]. Like DeepSeek-V3, R1 consists of 671 billion parameters with 37 billion activated per token, optimizing the balance between scale and computational efficiency. A defining feature of DeepSeek-R1 is its emphasis on reinforcement learning (RL) to cultivate advanced reasoning behaviors. The model was initially subjected to supervised fine-tuning using a curated dataset of chain-of-thought exemplars—a phase referred to as the "cold start." This was followed by large-scale RL using the Group Relative Policy Optimization (GRPO) algorithm, which incentivizes autonomous development of reasoning strategies, including self-verification and error correction. This robust training strategy enables DeepSeek-R1 to attain reasoning performance on par with OpenAI’s o1 model, while maintaining significantly lower training costs. Crucially, DeepSeek-R1 has been released under the permissive MIT License, granting the research community unrestricted access to its model weights and outputs, thereby fostering transparency and collaborative innovation in AI development.
2.3. Evaluation on LLMs
Understanding the landscape of state-of-the-art (SOTA) LLMs requires not only a grasp of their architectural innovations, capabilities, and training paradigms, but also a clear evaluation of how these models perform across real-world tasks. While the previous section outlines the defining characteristics of leading LLMs—ranging from GPT-4.5 and Claude 3.7 Sonnet to open-source models like Llama 3 and DeepSeek-R1—this alone does not provide a full picture of their practical effectiveness. In this section, we shift focus to rigorous evaluation methods and benchmark results that quantify these models’ strengths and trade-offs across diverse tasks such as reasoning, coding, multilingual understanding, and tool use. By linking architectural design with empirical performance, we aim to guide practitioners and researchers in making informed decisions when selecting LLMs for specific applications.
2.3.1. Tasks
The core function of LLMs is language modeling—predicting the next token based on the current input. This inherently requires both understanding and generating human language. Leveraging this foundational capability, LLMs can perform a wide variety of downstream tasks. We broadly categorize four key types of tasks that LLMs are capable of performing:
Text Understanding [9,42,97]. Text understanding is a fundamental NLP task focused on identifying the intent, topic, or semantics of a given input, often within a long-context. It answers the question: "What is this text about?" This category includes several sub-tasks: (1) Sentiment detection determines the writer’s emotional tone—positive, negative, or neutral. For example, “I like this apple” expresses positivity, while “This is a total waste of time” conveys a negative sentiment. More nuanced categories such as anger, joy, or frustration can also be captured. (2) Information extraction involves identifying specific entities or facts from text, such as names, dates, locations, or actions. From “Apple announced a new iPhone on March 15,” a model could extract “Apple” (organization), “iPhone” (product), and “March 15” (date). (3) Relationship understanding tracks how entities are related across sentences. For instance, in “Sarah gave the book to Mike. He thanked her,” a model must infer that “he” refers to Mike and “her” refers to Sarah. (4) Summarization reduces lengthy text to a concise version that preserves the main ideas. It may also simplify complex language or adjust the tone or style for different audiences.
Text Generation [7,40,98]. Text generation refers to the ability of an LLM to produce coherent, fluent, and contextually appropriate text. This extends beyond stringing words together—it requires logic, relevance, and creativity. Key sub-tasks include: (1) Question answering, which involves generating natural, complete answers based on a question and its context. This is essential in open-domain systems like digital assistants and educational tools. (2) Style transfer rewrites text in a different tone or style while preserving its original meaning. For example, the formal sentence “I regret to inform you” might be rendered more casually as “Just a heads-up.” (3) Text completion involves filling in or finishing partially written content. It powers autocomplete tools in emails, messaging apps, and writing assistants. (4) Machine translation converts text from one language to another, not just literally, but with attention to grammar, idioms, and cultural nuance to preserve meaning and tone.
Complex Reasoning [49,99,100,101]. Complex reasoning involves deeper cognitive abilities such as logical inference, problem-solving, and structured thinking that go beyond simple pattern matching. Sub-tasks include: (1) Code generation, where natural language instructions are translated into executable code. This allows users to generate scripts or programs using plain English descriptions. (2) Multi-step inference requires synthesizing information across several logical steps. For example, answering “Which country hosted the Olympics after China in 2008?” requires knowing that China hosted in 2008 and the UK hosted in 2012. (3) Logical reasoning tests the ability to apply deductive logic and identify valid conclusions or contradictions. A classic example: “If all cats are animals and some animals are black, can some cats be black?” (4) Commonsense reasoning leverages everyday knowledge. For instance, given “He put the ice cream on the table in the sun,” the model should infer that the ice cream will melt.
Knowledge Utilization [102,103,104,105]. Knowledge utilization refers to an LLM’s ability to access, retrieve, and apply factual or procedural knowledge—either from internal memory or external sources—to solve tasks accurately. This includes: (1) Open-domain question answering, where models retrieve and use up-to-date information to answer questions. For example, responding to “What are the current COVID-19 travel guidelines for Japan?” may require accessing recent data. (2) Tool-augmented reasoning enhances LLM capabilities by integrating external tools such as calculators, databases, or code interpreters. For instance, to compute the square root of 41,324, a model may call a calculator tool. (3) Conversational search and retrieval allows models to engage in interactive, multi-turn queries while dynamically retrieving and integrating relevant information. For example, answering “What are the side effects of this medication?” followed by “How does it compare to ibuprofen?” involves iterative search and context maintenance.
2.3.2. Benchmarks
As LLMs continue to advance, it becomes increasingly important to assess their capabilities across various domains, tasks, and reasoning skills. To evaluate how well LLMs perform in real-world scenarios, numerous benchmarks have been proposed across tasks such as mathematical reasoning, long-context understanding, and tool usage. We collect a selection of these benchmarks and categorize them by task type in Table 4. Below, we highlight several key benchmarks that are widely used to evaluate LLMs’ abilities.
- MMLU [135]: The Massive Multitask Language Understanding (MMLU) benchmark evaluates multitask accuracy across 57 diverse subjects, including humanities, social sciences, STEM fields, and professional domains like law and medicine. Each question is multiple-choice with four options, covering difficulty levels from elementary to professional. Questions are sourced from standardized test prep materials (e.g., GRE, USMLE) and university-level courses. The dataset comprises 15,908 questions split into training, validation, and test sets. MMLU assesses models in both zero-shot and few-shot settings, reflecting real-world conditions where no task-specific fine-tuning is applied. Human performance baselines are also provided, ranging from average crowdworkers to expert-level participants.
- BIG-Bench [136]: The Beyond the Imitation Game Benchmark (BIG-Bench) is a large-scale suite of 204 tasks designed to test LLMs on capabilities not captured by conventional benchmarks. Tasks span areas such as linguistics, mathematics, biology, social bias, and software engineering, and were contributed by researchers and institutions worldwide. Human experts also completed the tasks to establish reference baselines. BIG-Bench includes JSON tasks (with structured inputs/outputs) and programmatic tasks (which allow custom metrics and interaction). Evaluation metrics include accuracy, exact match, and calibration. A smaller curated subset, BIG-Bench Lite, contains 24 JSON tasks for lightweight and efficient evaluation.
- HumanEval [49]: HumanEval is a benchmark for evaluating the functional correctness of code generation. It consists of 164 original Python programming problems, each with a function signature, descriptive docstring, and empty function body. A solution is deemed correct if it passes predefined unit tests, aligning with how developers assess code quality. The benchmark targets abilities such as comprehension, algorithmic reasoning, and basic mathematics. For safety, all code is executed in a secure sandbox to mitigate risks posed by untrusted or potentially harmful code.
- TruthfulQA [151]: This benchmark is designed to assess whether LLMs generate truthful answers and avoid perpetuating misconceptions or factual inaccuracies. It includes 817 questions across 38 domains, such as health, finance, and law. The questions—typically concise, with a median length of 9 words—are crafted to exploit known weaknesses in LLMs, particularly their tendency to imitate common yet incorrect human text. The benchmark imposes rigorous truthfulness criteria, evaluating answers based on factual accuracy as supported by public sources like Wikipedia. Each question includes both true and false reference answers.
- GSM8K [140]: The Grade School Math 8K (GSM8K) dataset comprises 8.5K human-written arithmetic word problems suitable for gradeschool-level mathematics. Of these, 7.5K are training problems and 1K are test problems. Each problem typically requires 2 to 8 reasoning steps and involves basic arithmetic. The dataset emphasizes: (1) high quality, with a reported error rate below 2%; (2) high diversity, avoiding repetitive templates and encouraging varied linguistic expression; (3) moderate difficulty, solvable using early algebra without advanced math concepts; and (4) natural language solutions, favoring everyday phrasing over formal math notation.
2.3.3. Evaluation Methods
Evaluating LLMs typically involves a combination of automatic and human-centered metrics, depending on the specific task at hand. We categorize existing evaluation methods into the following four classes:
Basic Automatic Evaluation Metrics. Quantitative metrics are essential for assessing the performance of LLMs. These metrics vary based on the task type, such as classification, generation, or translation. For instance, in classification tasks, metrics like accuracy and F1-score are commonly used to compare predicted outputs with ground-truth labels. In contrast, for generative tasks such as question answering, metrics like BLEU (Bilingual Evaluation Understudy) [159], ROUGE (Recall-Oriented Understudy for Gisting Evaluation) [160], and BERTScore [161] are employed to assess the semantic similarity between the generated and reference texts. Additionally, domain-specific evaluations exist. For example, in code generation, metrics like pass@k and functional correctness [49] are utilized to measure the quality of the generated code.
Advanced Automatic Evaluation Metrics Beyond Correctness. Beyond correctness, comprehensive evaluation of LLMs must account for broader behavioral attributes such as trustworthiness, toxicity, fairness, robustness, and reasoning quality [150,162,163,164,165]. As LLMs are increasingly deployed in real-world applications—including education, healthcare, legal advising, and customer interaction—accuracy alone becomes an inadequate measure of performance. For instance, trustworthiness metrics assess whether models avoid hallucinations, misinformation, or unsupported claims [163], while toxicity evaluation captures the frequency of harmful, biased, or offensive content [164]. Reasoning quality, on the other hand, examines whether a model can follow logically consistent, step-by-step problem-solving processes [165]. Fairness metrics [166] further ensure that model behavior remains equitable across diverse user groups, preventing systematic biases that could reinforce societal inequities. Additionally, robustness measures [167] how stable model outputs are under adversarial or distributional shifts, and calibration evaluates whether a model’s confidence levels meaningfully reflect its likelihood of being correct. In safety-critical or socially impactful scenarios, transparency and explainability become essential [168], helping users understand, validate, or contest model outputs. These advanced metrics—often requiring dedicated datasets, probing techniques, or alignment with human values—offer a more holistic view of LLM capabilities and limitations. Moving beyond narrow accuracy-based benchmarks, they are essential for building LLMs that are not only powerful but also reliable, fair, and aligned with human expectations.
Human Evaluation. Although automatic metrics offer scalability and efficiency, they often fail to capture nuanced qualities such as reasoning depth, factual accuracy, and practical utility [169]. Human evaluation addresses these gaps by involving human raters who assess LLM outputs for clarity, engagement, structure, and alignment with user intent [169]. This ensures higher reliability, safety, and real-world applicability. However, human evaluation is not without drawbacks: it is costly, time-consuming, and prone to variability due to subjective interpretations and potential rater bias. Moreover, it lacks scalability, making it infeasible for evaluating responses at large scale.
LLM-as-a-Judge. To overcome the limitations of human evaluation, the concept of using LLMs themselves as evaluators, LLM-as-a-judge [170], emerged. In this paradigm, human-aligned LLMs are employed to replace human raters [171,172]. A typical approach involves prompting the LLM to compare and rank two candidate answers. This method is more scalable, faster, and generally more cost-effective than relying on human reviewers, despite the computational costs of querying LLMs. Additionally, it allows for real-time feedback. However, this approach still faces several challenges, such as susceptibility to prompt sensitivity, potential biases in judgment, and the risk of hallucinations in comparative reasoning.
2.3.4. Performance at a Glance
In this section, we provide a performance-at-a-glance synthesis of selected SOTA LLM models. Table 5 and Table 6 report the performance of selected LLMs across a range of benchmarks. Table 5 presents results on widely adopted basic benchmarks, while Table 6 highlights performance on more challenging reasoning tasks. The results are aggregated from both Chatbot Arena2 and the Vellum LLM Leaderboard3. We apply different colors to indicate the top-3 performances per benchmark. The selected evaluations cover diverse capabilities, including commonsense reasoning, code generation, tool use, multilingual understanding, and mathematical problem-solving, providing a holistic view of current LLM competence.
From Table 5, models such as GPT-4.5, GPT-4o, and DeepSeek R1 exhibit strong and consistent performance across a broad array of tasks. Notably, GPT-4.5 sets the state of the art on HumanEval (92.40%) and MATH (96.40%), while also remaining competitive on MMLU. DeepSeek R1 places in the top three on MMLU (90.80%), GPQA (71.50%), and MATH (97.30%), underscoring its strength in both reasoning and quantitative domains. Additionally, GPT-o1 and GPT-o3-mini deliver compelling results in mathematical reasoning (e.g., MATH: 97.90%) and multilingual understanding (MGSM: up to 92.00%). Table 6 further differentiates models based on their performance on advanced reasoning tasks. OpenAI o1 and Grok 3 Beta attain competitive scores on GPQA Diamond, MATH 500, and AIME 2024, reflecting solid reasoning abilities. Notably, Claude 3.7 Sonnet (reasoner) achieves leading results in tool use (Airline: 86.10%), IF-Eval (93.20%), and MATH 500 (96.20%), highlighting its proficiency in multi-step reasoning and task completion. DeepSeek R1 continues to demonstrate top-tier performance, maintaining high scores on MATH 500 (97.30%) and AIME 2024 (79.80%).
From the foregoing analysis of LLM performance across a range of benchmarks, we draw the following observations:
- Insight 1: No single LLM dominates across all tasks. GPT-4.5 ranks highest on Chatbot Arena (1398), suggesting strong general chatbot capabilities. However, it does not lead in benchmarks like GPQA (reasoning) or math. Conversely, DeepSeek R1 achieves the best scores on MMLU (90.8%) and Math (97.3%) but lacks results in HumanEval (coding) and multilingual tasks, indicating that top performance in one area does not translate to all domains.
- Insight 2: Reasoning models outperform others in logical and structured tasks. Claude 3.7 Sonnet (reasoner) achieves the highest GPQA Diamond score (84.8%), the best Tool Use (Retail) result (81.2%), and leads in MMMLU (86.1%). These benchmarks emphasize reasoning and complex task execution, showcasing the value of models fine-tuned for reasoning.
- Insight 3: Specialized models often come with trade-offs. Claude 3.5 Haiku performs well in general chatbot interaction but has one of the lowest GPQA scores (41.6%) and math scores (69.4%). Gemini 1.5 Pro and Gemini 2.0 Flash perform reasonably well in multilingual (MGSM) and tool-use (BFCL) tasks, but underperform in reasoning and coding, highlighting performance sacrifices in general-purpose versus specialized capabilities.
- Insight 4: Different models excel at different tasks, and should be selected accordingly. For chatbot dialogue, GPT-4.5 and GPT-4o are top performers. Claude 3.7 Sonnet (reasoner) excels in reasoning, tool use, and multilingual benchmarks. Claude 3.5 Sonnet leads in coding with the best HumanEval score (93.7%). For math-heavy benchmarks, DeepSeek R1 and GPT-03-mini both surpass 97% on the Math benchmark. Thus, model selection should be guided by the specific task requirements.
3. LLMs for Arts, Letters, and Law
In this chapter, we explore how LLMs are reshaping the humanities and law, shifting emphasis from evidence to application. Specifically, we review five disciplines—history, philosophy, political science, arts and architecture, and law. In history, we address narrative and interpretive practices (e.g., story generation and analysis), quantitative and scientific methods (e.g., modeling historical psychological responses), as well as interdisciplinary and comparative approaches, supported by benchmarks and brief commentary. In philosophy, we consider normative and interpretive applications (e.g., generating debate or dialogue), analytical and logical domains (e.g., symbol grounding diagnostics), along with comparative and cross-disciplinary analyses, again connected to benchmark studies. In political science, we investigate text-based methods for extracting policy insights, simulating and forecasting opinions, and shaping and framing political messaging, while linking these to benchmark assessments and reflective discussion. In arts and architecture, we present model-enabled creation across the visual, literary, and performing arts, alongside LLM-supported architectural design, production, and analysis, followed by evaluations and key lessons. Finally, in law, we examine LLM use in consultant-style question answering, drafting contracts and briefs, parsing and analyzing legal documents and cases, and predicting judgments, concluding in representative benchmarks and discussion.
3.1. History
3.1.1. Overview
Introduction. History is a continuous process of interaction between the historian and his facts, an unending dialogue between the present and the past [173]. It helps us understand how the world has changed over time and provides valuable context for interpreting the present and imagining the future [174,175]. At its core, history is about reconstructing narratives, analyzing cause and effect, and interpreting the motivations and consequences of human actions across time [176,177].
Figure 4.
The history research in the era of LLMs.
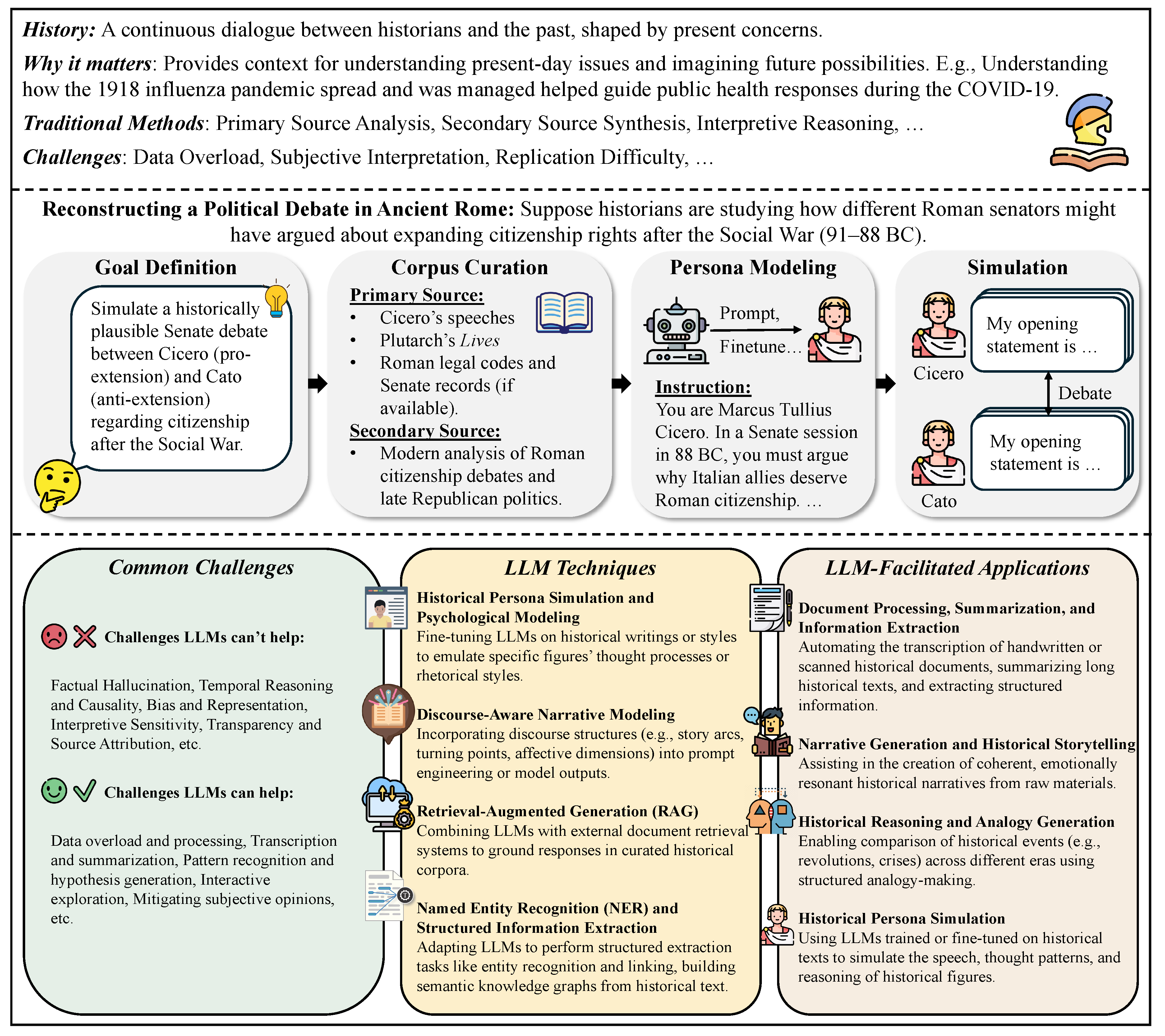
Traditionally, history research involves careful examination of primary sources—such as letters, legal documents, newspaper articles, and government records—as well as secondary analyses written by other historians [178]. Scholars use these materials to craft explanations of past events, build timelines, uncover social patterns, and offer interpretations grounded in context [179,180]. This process relies heavily on human reading, note-taking, and interpretive reasoning [181].
However, traditional methods face growing limitations. First, the volume of historical data—digitized archives, scanned manuscripts, oral histories, and digital media—has grown far beyond what any individual or team can process manually [182,183,184]. Second, interpreting history often requires synthesizing multiple perspectives, which can be slow and subjective [185]. Third, historical research is time- and labor-intensive, making it difficult to scale or replicate. These challenges have prompted interest in computational tools that can support, accelerate, or expand historical inquiry.
The role of LLMs. LLMs offer a powerful new toolkit for historical research. Trained on vast corpora of text, LLMs can read, summarize, translate, and generate human-like text at scale. They can identify patterns across large document collections, extract names and dates, simulate alternative narratives, or respond to questions using knowledge from multiple sources. These capabilities make LLMs especially suited for analyzing unstructured historical text, processing archival documents, and enabling interactive exploration of the past.
LLMs can assist historians in a variety of ways: by automating the transcription of handwritten documents, summarizing long articles or books, clustering related texts, or generating hypotheses about social or political dynamics. They can also support the creation of historical simulations or dialogue systems, enabling new forms of engagement with historical knowledge. Researchers have begun to explore LLM-based systems for historical thinking, comparative analysis, and even the modeling of historical psychology.
Limitations of LLMs. Despite their promise, LLMs face important limitations when applied to historical research: Factual Hallucination: LLMs may generate plausible-sounding but false or unverifiable historical claims, which undermines academic rigor and trust. Temporal Reasoning: LLMs often struggle with chronology, causality, and contextual nuance—essential features of historical reasoning. Bias and Representation: Because LLMs reflect the biases in their training data, they may reproduce skewed or incomplete views of history, overlooking marginalized voices or reinforcing dominant narratives. Interpretive Sensitivity: History is not just about facts but about interpretation. LLMs may oversimplify or flatten complex debates by offering overly confident or decontextualized summaries. Transparency and Source Attribution: LLMs generally do not cite specific sources, making it difficult for historians to verify information or trace interpretive lineage.
As such, LLMs should not be seen as replacements for human historians, but rather as tools that can extend their reach, suggest new questions, and assist in data exploration—especially when paired with domain expertise and critical oversight.
Taxonomy. To better understand the potential of LLMs in history research, we organize the field into three broad categories, based on methodological approaches and research goals:
Table 7.
Applications and insights of LLMs in historical research
| Historical Category | LLM Application Areas | Use Case-Inspired Research Question | Key Insights and Contributions | References |
|---|---|---|---|---|
| Narrative and Interpretive History | Narrative Generation and Analysis | Can LLMs generate historically coherent and emotionally resonant narratives from primary source texts? | LLMs struggle with coherence and diversity; discourse-aware prompts improve storytelling; useful for story arcs and emotional analysis. | [186,187] |
| Historical Research Assistance | How can LLMs simulate historical personas or provide conversational access to archives for education and research? | Used in chat-based exploration, simulated historical figures, and layered tools (e.g., KleioGPT); helpful for both scholars and students. | [188,189,190] | |
| Historical Interpretation | Can LLMs improve consistency and reduce bias in interpretive historical analysis? | LLMs show more consistent judgments than humans; useful for reducing subjectivity in analysis. | [191] | |
| Quantitative and Scientific History | Historical Thinking | How do LLMs enhance access to and reflection on historical content through transcription and summarization? | Help transcribe handwriting and summarize texts; make primary sources easier to explore and reflect on. | [192] |
| Historical Data Processing | Can LLMs scale up entity extraction and temporal reasoning across vast historical corpora? | Enable name/date extraction, timeline generation, and semantic linking at scale. | [193,194] | |
| Simulating Historical Psychological Responses | Is it possible to model the psychological tendencies of past societies using LLMs trained on historical texts? | Can mimic cultural mindsets from historical texts, but results may reflect biases of elite sources. | [188] | |
| Comparative and Cross-Disciplinary History | Historical Analogy Generation | How can LLMs retrieve or generate meaningful analogies between past and present events? | Generate cross-era comparisons; reduce hallucinations with structured frameworks and similarity checks. | [195] |
| Interdisciplinary Information Seeking | Can LLMs facilitate the discovery of relevant insights across disciplines for historical inquiry? | Tools like DiscipLink help explore diverse sources and connect ideas across fields. | [196] |
- Narrative and Interpretive History: This area emphasizes descriptive, subjective, and human-centered accounts of the past. It uses storytelling and meaning-making to explain events in context. LLMs can assist in narrative generation, reconstruct voices from historical texts, and interpret language use in personal accounts or memoirs.
- Quantitative and Scientific History: This approach applies statistical, computational, and formal methods to study historical data and trends. LLMs can process large datasets, simulate historical psychological responses, aid in historical reasoning tasks, and evaluate knowledge systems.
- Comparative and Cross-Disciplinary History: This domain integrates methods from sociology, economics, political science, and other disciplines to study similarities and differences across historical contexts. LLMs can Support comparative analysis, generate historical analogies, link concepts across eras or cultures.
Our taxonomy aligns with the framework outlined in reference [197], though it uses different terminology. It is conceptually consistent with how scholars commonly categorize historical research: narrative and interpretive history corresponds to traditional, humanistic approaches; quantitative and scientific history reflects data-driven, computational, and social-scientific methods; and comparative and cross-disciplinary history encompasses global, integrative, and hybrid perspectives.
3.1.2. Narrative and Interpretive History
Narrative and interpretive history refers to the study of the past through storytelling and critical interpretation, focusing on the meanings, motives, and lived experiences behind historical events. This method is often what people first imagine when they think of history: a crafted narrative that explains what happened, why it happened, and how it was experienced. Rather than just listing facts or dates, this approach aims to understand how individuals and societies made sense of their world. Historians working in this tradition analyze sources like letters, speeches, and cultural artifacts to reconstruct events with nuance, paying close attention to human intentions, emotions, and consequences. It is about weaving the past into coherent stories that connect with readers and shed light on the complexity of human life.
Narrative Generation and Analysis. Recent work has critically examined the narrative generation and comprehension capacities of LLMs. [186] introduce a comprehensive computational framework that evaluates LLM storytelling through three discourse-level aspects: story arcs (macro), turning points (meso), and affective dimensions (micro). Their analysis reveals substantial discrepancies between human- and machine-generated narratives: LLM outputs tend to be homogeneously positive, poorly paced, and lacking in suspense and diversity. Benchmarking on tasks such as story arc and turning point identification further confirms that models like GPT-4 and Claude underperform relative to humans in discourse-level narrative reasoning. However, the authors show that explicit integration of discourse structures into prompting strategies significantly improves storytelling outcomes, including greater emotional engagement and narrative diversity. Complementing this, the WNU 1.4 benchmark [187] offers a cross-lingual, multi-task diagnostic tool to assess narrative understanding in LLMs. It reveals deficiencies in narrative coherence and character goal modeling, especially in low-resource languages. Together, these studies advocate for discourse-aware approaches in both evaluation and generation, marking a crucial step toward more human-like narrative competence in LLMs.
Historical Research Assistance. Recent research also integrates LLMs into historical inquiry, proposing new methodologies that enhance both analysis and pedagogy. Varnum et al. [188] advocate for using LLMs trained on historical texts to simulate responses of historical figures and generate contextualized insights, revealing both the interpretive potential and epistemological tensions inherent in such applications. Zeng [189] introduces HistoLens, an LLM-powered, multi-layered framework for historical text analysis, demonstrated via the Confucian-Legalist debates in the Western Han dynasty’s Yantie Lun. This framework combines thematic word frequency analysis, named entity recognition, knowledge graph construction, GIS-based spatial mapping, and machine teaching to construct interpretative pipelines that blend traditional scholarship with computational scalability. Complementing these innovations, Gonzalez Garcia and Weilbach [190] present KleioGPT, a retrieval-augmented conversational interface that enables historians to interact with curated corpora through natural language prompts. Their evaluation shows LLMs can assist with complex historiographic tasks, such as question answering and data extraction, while also underscoring challenges like hallucination and source attribution. Together, these works signal a paradigm shift in historical methodology, foregrounding LLMs as both analytical tools and collaborators in the humanities.
Historical Interpretation. In addition, LLMs have extended their application to the domain of historical reasoning, focusing on interpretative consistency. Celli and Spathoulas [191] empirically examine interpretative agreement between humans and LLMs over historical annotations using two cyclical theories—Structural Demographic Theory and the Big Cycle model. They find that LLMs, particularly large-scale models like GPT-4 and Claude 3.5, achieve significantly higher inter-annotator agreement than humans, suggesting a promising role for LLMs in minimizing subjective bias in historical analysis. Together, these studies reveal that LLMs not only possess the capacity to generate meaningful historical analogies but also exhibit interpretive consistency that may surpass human annotators, raising both opportunities and concerns for digital humanities and historical scholarship.
3.1.3. Quantitative and Scientific History
Quantitative and scientific history applies statistical, mathematical, and computational techniques to historical data in order to uncover patterns, test hypotheses, and make systematic comparisons. This method treats history a bit like a science experiment. Instead of focusing solely on individuals or events, it looks at big-picture trends—such as population changes, economic growth, or voting behavior—often using data like census records, trade statistics, or social surveys. It helps answer questions like “How did literacy rates change over a century?” or “What economic factors were linked to revolutions?” Thanks to digital tools and vast data archives, historians can now analyze much more information than before, revealing trends that would be impossible to spot just by reading documents one by one.
Historical Thinking. The article [192] by Cameron Blevins explores the use of LLMs like ChatGPT in historical research, particularly in transcribing and interpreting primary sources. LLMs can accurately transcribe handwritten historical documents, which were traditionally considered “machine-unreadable.” The article provides an example of an 1886 letter transcribed by ChatGPT with impressive accuracy. These tools can generate concise and accurate summaries of historical texts, as demonstrated with Benjamin Curtis’s letter. However, errors can occur, especially with complex documents or non-English sources. A minor transcription mistake in the Curtis letter highlights how a lack of historical context can lead to missed nuances. Thus, LLMs can serve as research assistants, helping with tedious tasks like transcription while refining historians’ own thoughts and interpretations.
Historical Data Processing. Recent advancements in the processing of historical data through LLMs have demonstrated significant potential in enhancing the accuracy and efficiency of information retrieval and analysis. The work [193] introduces a framework for the intelligent extraction and visualization of Indonesian historical narratives using transformer-based LLMs. By integrating Named Entity Recognition (NER) and text classification models, the system efficiently identifies and links key entities and events within historical texts, offering an enriched semantic representation of historical knowledge. Complementarily, the comprehensive survey [194] categorizes and evaluates the capabilities of LLMs across diverse tasks, highlighting their utility in temporal reasoning, fact-checking, and structured data understanding, which are pivotal in historical research. Together, these studies underscore the transformative impact of LLMs on historical data processing, enabling more nuanced analysis, enhanced data curation, and scalable knowledge extraction from complex archival sources.
Simulating Historical Psychological Responses. The integration of historical LLMs into behavioral science marks a novel methodological advance, enabling the simulation of psychological responses from temporally distant populations. Varnum et al. [188] argue that by training generative language models on corpora of historical texts—ranging from letters and fiction to scholarly works—it is possible to approximate the mental frameworks of historical societies. This approach allows researchers to transcend the temporal constraints of traditional psychological methods, which are inherently limited to living participants, and to infer the psychology of past cultures with greater fidelity than indirect proxies such as word frequency analyses. HLLMs could facilitate empirical investigations into cultural change, test the generalizability of psychological theories across time, and enrich historical scholarship with quantifiable insights. Despite promising early applications, such as MonadGPT and XunziALLM, the authors note significant challenges, including limited and elite-skewed training data, difficulties in benchmarking historical accuracy, and the risk of anachronistic biases. Nonetheless, with careful curation and interdisciplinary validation, HLLMs hold the potential to illuminate the cognitive landscapes of bygone eras and serve as valuable tools in both psychology and the historical sciences.
3.1.4. Comparative and Cross-Disciplinary History
Comparative and cross-disciplinary history involves analyzing historical phenomena across different cultures, regions, or time periods, often incorporating insights from other disciplines like sociology, anthropology, or political science. This method steps back to take a wide-angle view. Instead of looking at a single nation or event, comparative history puts multiple cases side by side to ask questions like, “Why did one country experience a revolution while another did not?” or “How did different empires manage trade or power?” Cross-disciplinary approaches further enrich this analysis by borrowing theories and methods from other fields, allowing historians to explore issues like class, race, environment, or technology with more depth. It’s a powerful way to understand not just what happened, but why it happened in some places and not others.
Historical Analogy Generation. Li et al. [195] introduce the novel task of historical analogy acquisition, which seeks to identify past events that are analogous to contemporary ones across multiple cognitive dimensions—topic, background, process, and result. They propose both retrieval-based and generative approaches, and notably develop a self-reflection framework to mitigate hallucinations and biases in free-form generation. Their automatic multi-dimensional similarity metric demonstrates high alignment with human evaluations, affirming the potential of LLMs in historical analogy tasks.
Interdisciplinary Information Seeking. Interdisciplinary historical research necessitates traversing diverse disciplinary landscapes, often hindered by scattered knowledge, domain-specific terminologies, and cognitive load in assimilating unfamiliar perspectives. The DiscipLink [196] system exemplifies how LLMs can be harnessed to support such complex information-seeking endeavors through a human-AI co-exploration paradigm. Integrating mixed-initiative workflows, DiscipLink scaffolds the nonlinear process of interdisciplinary information seeking (IIS)—comprising orientation, opening, and consolidation—by eliciting exploratory questions (EQs) tailored to users’ research goals, expanding search queries with domain-specific vocabulary, and synthesizing retrieved literature into contextualized themes. The system’s design foregrounds researcher agency, allowing iterative engagement with LLM-generated suggestions while mitigating the risks of hallucination and bias. Empirical evaluations, including usability studies and case analyses, underscore its utility in helping scholars—especially those in fields like history—to navigate and integrate knowledge from adjacent disciplines such as sociology, education, and psychology. This work demonstrates the potential of LLMs not as autonomous agents but as collaborative partners in exploratory, interdisciplinary historical research.
3.1.5. Benchmarks
TimeTravel [198] is a benchmark designed to evaluate large multimodal models (LMMs) on historical and cultural artifacts. It consists of 10,250 expert-verified samples spanning 266 cultures across 10 major historical regions. Unlike existing benchmarks that focus on modern objects and landmarks, TimeTravel prioritizes historical knowledge, contextual reasoning, and cultural preservation, providing structured datasets for AI-driven analysis of manuscripts, artworks, inscriptions, and archaeological findings. The benchmark enables the assessment of AI models in classification, interpretation, and historical comprehension, helping researchers identify strengths and limitations. TimeTravel sets a new standard for evaluating AI in cultural heritage preservation and historical discovery, with results demonstrating gaps in current AI capabilities while providing a foundation for future improvements.
Table 8.
Overview of Benchmarks for Evaluating LLMs on Historical Data
| Benchmark | Scope and Focus | Data Composition | Evaluation Tasks | Key Insights |
|---|---|---|---|---|
| TimeTravel [198] | Multimodal evaluation of historical and cultural artifacts | 10,250 expert-verified samples across 266 cultures and 10 regions; includes manuscripts, artworks, inscriptions, and archaeological findings | Classification, interpretation, and historical reasoning | Highlights LMMs’ limitations in cultural/historical context understanding; sets new standards for AI in cultural heritage preservation |
| AC-EVAL [199] | Ancient Chinese language understanding by LLMs | 3,245 multiple-choice questions on historical facts, geography, customs, classical poetry, and philosophy, categorized into three difficulty levels | General knowledge, short text understanding, long text comprehension | Chinese-trained models outperform English-trained ones; ancient Chinese remains a low-resource challenge; few-shot often introduces noise |
| Hist-LLM [193] | Global historical knowledge evaluation using structured data | Subset of Seshat Global History Databank: 600 societies, 36,000 data points | Multiple-choice on historical facts across global regions and eras | Models show moderate accuracy; better on early periods and the Americas; weaker in Sub-Saharan Africa and Oceania |
AC-EVAL [199] is a benchmark designed to evaluate LLMs’ understanding of ancient Chinese, addressing gaps in existing benchmarks that primarily focus on modern Chinese. It consists of 3,245 multiple-choice questions spanning historical facts, geography, social customs, philosophy, classical poetry, and prose, structured into three difficulty levels: general historical knowledge, short text understanding, and long text comprehension. The evaluation reveals that Chinese-trained LLMs outperform English-trained ones, highlighting ancient Chinese as a low-resource challenge for models like GPT-4. While chain-of-thought reasoning enhances performance in larger models, few-shot learning often introduces noise rather than improving accuracy. AC-EVAL provides a structured and comprehensive framework for assessing LLMs’ proficiency in ancient Chinese, aiming to advance their application in language education and scholarly research.
HiST-LLM [193] is a benchmark for evaluating LLMs’ historical knowledge using a subset of the Seshat Global History Databank. This dataset, covering 600 historical societies and 36,000 data points, enables the assessment of LLMs across various world regions and time periods. Seven models from OpenAI, Llama, and Gemini families were tested using multiple-choice questions on historical facts, showing accuracy ranging from 33.6% to 46%, outperforming random guessing but falling short of expert comprehension. The results indicate that models perform better on earlier historical periods and exhibit regional discrepancies, with stronger performance for the Americas and weaker results for Sub-Saharan Africa and Oceania. While LLMs demonstrate some expert-level historical knowledge, the benchmark reveals significant gaps and opportunities for improvement.
3.1.6. Discussion
Opportunities and Impact. The integration of LLMs into historical research marks a profound shift in how the past can be explored, analyzed, and understood. As demonstrated across narrative and interpretive history [186,188,191], quantitative and scientific history [188,192,193], and comparative and cross-disciplinary history [195,196], LLMs offer unprecedented capabilities for processing vast corpora of historical texts, assisting with complex interpretive tasks, and supporting interdisciplinary inquiries.
By automating labor-intensive processes such as transcription [192], entity recognition [193], and analogical reasoning [195], LLMs can significantly augment the productivity of historians and open new avenues for exploring historical narratives and mentalities. They also enable novel methodologies, such as historical psychology simulation [188] and interdisciplinary information retrieval [196], bridging gaps between traditional humanities approaches and computational scalability. In doing so, LLMs have the potential to democratize access to historical research, enhance interpretive diversity, and foster richer engagements with the past.
Challenges and Limitations. Despite their transformative potential, LLMs present serious limitations when applied to historical inquiry. Factual hallucination remains a critical risk [188,190], undermining trust and academic rigor, especially when models generate plausible but unverified historical claims. Temporal reasoning deficiencies [186] hinder models’ ability to understand chronology, causality, and contextual nuance—core competencies in historical scholarship.
Bias amplification is another major concern [191]. LLMs inherit the systemic biases present in their training data, which can lead to the overrepresentation of dominant historical narratives while marginalizing underrepresented perspectives. Moreover, the interpretive nature of history poses challenges: while LLMs can simulate interpretative consistency [191], they may oversimplify complex historiographic debates or falsely present contested interpretations as settled facts.
Transparency and source attribution issues also persist [190]. Without clear references to underlying sources, it becomes difficult for historians to verify claims, trace intellectual lineage, or engage in critical evaluation. Furthermore, the limited historical coverage and elite bias of available training corpora [188] constrain the representativeness of LLM-assisted insights, particularly for studies focused on marginalized or non-Western histories.
Research Directions. Building on current advances, several promising research directions emerge:
- Temporal and Causal Reasoning Enhancement. Advances in temporal modeling, causal inference, and discourse-level narrative comprehension [186] are critical for enabling LLMs to reason more accurately about historical sequences and cause-effect relationships.
- Explainable and Source-Grounded Historical AI. Building models that produce verifiable outputs, grounded in cited historical documents or structured datasets, can strengthen academic trust and facilitate critical engagement [190].
- Collaborative Human-AI Historical Research. Systems like DiscipLink [196] suggest a promising future where LLMs act as exploratory partners rather than authoritative experts, supporting iterative, mixed-initiative workflows that preserve human scholarly agency.
- Ethics and Epistemology of Digital History. Further interdisciplinary studies are needed to critically examine how LLMs reshape historical knowledge production, interpretive authority, and educational practices, ensuring that technological augmentation remains aligned with historical rigor and ethical standards [191].
Conclusion. LLMs offer powerful new tools for historical research, enabling the scaling of traditional methods and the exploration of novel forms of inquiry. However, realizing their potential responsibly demands rigorous attention to their limitations: factual integrity, interpretive nuance, fairness, and transparency. Future advances must emphasize domain-specific fine-tuning, temporal reasoning, source attribution, and collaborative workflows that maintain critical human oversight. As the discipline of history adapts to the possibilities of the LLMs era, the most promising path lies in a balanced integration—where machine assistance amplifies, but does not replace, the historian’s craft.
3.2. Philosophy
3.2.1. Overview
Introduction. Philosophy is the discipline that investigates the most fundamental questions concerning existence, knowledge, value, reason, consciousness, and language. Unlike the natural sciences, which rely on empirical observation and experimentation, philosophy employs logical reasoning, conceptual analysis, and critical reflection to examine the foundational assumptions that underlie all domains of thought [200,201]. It seeks to clarify the principles that govern how we understand the world and our place within it.
In simpler terms, philosophy is the pursuit of wisdom about life and the universe. It asks questions such as: What is truly real? Can we know anything with certainty? What makes an action right or wrong? Do we have free will? What constitutes a meaningful life? These questions may not yield definite answers, but the process of exploring them shapes how we think, live, and build societies [202,203].
As Aristotle observed, “All men by nature desire to know” [203]; Kant formulated four core philosophical questions—“What can I know? What ought I to do? What may I hope? What is man?”—to map the field of inquiry [202]; and Russell noted that philosophy’s value lies not in the certainty of its answers, but in the breadth of its questions [201].
Philosophy has not only shaped abstract systems of thought but has also directly influenced real-world political institutions and historical events. A prominent example is the impact of John Locke’s social contract theory on the development of modern liberal democracy. Locke argued that all individuals possess natural rights to “life, liberty, and property,” and that governments are legitimate only insofar as they derive their authority from the consent of the governed [204]. This idea became foundational for Enlightenment political philosophy and deeply influenced the American and French revolutions.
In particular, Locke’s philosophy was central to the drafting of the Declaration of Independence by Thomas Jefferson in 1776. Jefferson echoed Locke’s ideas by asserting that “all men are created equal” and endowed with “unalienable rights,” including “life, liberty, and the pursuit of happiness” [205]. These principles were later institutionalized through the United States Constitution and the Bill of Rights, embedding philosophical notions of individual rights, limited government, and rule of law into the very fabric of a modern state [206].
Thus, what began as a philosophical inquiry into the legitimacy of authority and the moral basis of government became a blueprint for real-world governance, illustrating philosophy’s enduring capacity to shape societal norms and legal systems.
Core Domains of Philosophical Inquiry. Although philosophy spans a wide intellectual terrain, its major questions can be organized into four interrelated domains, each addressing a distinct class of foundational issues: metaphysics and rpistemology explore the nature of reality and the conditions of knowledge. Metaphysics asks what kinds of things exist—such as time, space, causality, and identity—while epistemology investigates how we come to know what we know, and what counts as justified belief [202,207]. These domains ground the philosophical enterprise itself and provide the basis for reflection in all other fields. ethics and political philosophy concern the principles of right action and just social arrangements. Ethics examines what is good, right, or virtuous in human behavior, while political philosophy analyzes the legitimacy of authority, the nature of justice, and the structure of political institutions [208,209]. These areas provide normative frameworks that influence law, governance, and interpersonal conduct. philosophy of mind and human existence addresses consciousness, selfhood, perception, and the existential conditions of human life. This includes questions about the mind–body relationship, free will, intentionality, and the meaning of life [210,211]. In the context of neuroscience and artificial intelligence, these issues have gained renewed urgency and interdisciplinary relevance. logic, language, and philosophy of science analyze the structure of reasoning, the nature of meaning, and the epistemic foundations of scientific inquiry. This domain encompasses symbolic logic, theories of reference, and debates about realism, explanation, and the limits of scientific knowledge [212,213]. It bridges the gap between abstract thought and empirical investigation. These domains not only define the contours of philosophy but also serve as its point of contact with science, politics, art, and human experience.
Figure 5.
The philosophy research in the era of LLMs.
The role of LLMs. LLMs offer a transformative new toolkit for philosophical research. Trained on enormous corpora of texts—including classical treatises, contemporary papers, and interdisciplinary works—LLMs can read, summarize, translate, and generate nuanced arguments at scale. They are capable of identifying patterns in philosophical discourse, drawing connections between disparate ideas, and even simulating dialectical debates. Such capabilities make LLMs particularly well-suited for tasks like literature synthesis, hypothesis generation, and preliminary conceptual analysis.
LLMs can assist philosophers in several ways: by automating the review of extensive philosophical literature, summarizing dense arguments, clustering related ideas, and even generating novel perspectives that bridge established schools of thought. They also enable interactive exploration of complex theoretical landscapes, providing new insights and sparking innovative research questions. Early applications include simulating ethical debates, modeling epistemological frameworks, and mapping out ontological structures across diverse traditions.
Limitations of LLMs. Despite their promise, LLMs have significant limitations when applied to philosophical research: Factual and Logical Hallucination: LLMs may produce coherent yet unfounded or erroneous arguments, which can compromise academic rigor and the reliability of conclusions [214]. Abstract Conceptualization: Deeply abstract or nuanced philosophical concepts may be oversimplified or misunderstood by LLMs, limiting their effectiveness in capturing the full complexity of theoretical ideas [215]. Bias and Partial Perspectives: Because LLMs are trained on existing literature, they may inadvertently reproduce dominant paradigms while overlooking marginalized or alternative viewpoints [216]. Transparency and Source Attribution: LLM-generated content often lacks explicit citations, making it difficult for researchers to verify arguments or trace the lineage of ideas [217].
Taxonomy. To better understand the potential of LLMs in philosophical research, we organize the field into three broad categories based on methodological approaches and research goals:
- Normative and Interpretive Philosophy: This area emphasizes the analysis of moral values, ethical dilemmas, and interpretive narratives within philosophical traditions. LLMs can assist in generating novel normative arguments, reconstructing historical ethical discourses, and offering fresh interpretations of canonical texts.
- Analytical and Logical Philosophy: This approach applies formal logic, precise argumentation, and rigorous conceptual analysis to explore philosophical problems. LLMs can support the systematic breakdown of complex arguments, facilitate comparative analyses of theoretical positions, and enhance clarity in logical reasoning.
- Comparative and Cross-Disciplinary Philosophy: This domain integrates insights from diverse fields—such as political science, sociology, and linguistics—to study philosophical questions from multiple angles. LLMs can aid in cross-cultural comparisons, generate analogies between disparate theories, and help synthesize interdisciplinary perspectives.
Our taxonomy aligns with frameworks found in contemporary philosophical research, reflecting the balance between traditional humanistic inquiry and data-driven, computational methods.
3.2.2. Normative and Interpretive Philosophy
Normative and interpretive philosophy addresses the ethical, cultural, and historical dimensions of human life. It centers on the values that govern actions, the moral frameworks that shape decision-making, and the narratives that inform how individuals and societies understand right and wrong. This domain does not prioritize formal rigor or abstract universals alone, but instead embraces the complexity of lived experience, emphasizing context, positionality, and meaning. Philosophers working in this tradition engage deeply with literature, history, and socio-linguistic patterns, interpreting moral claims not merely as logical propositions but as situated expressions of ethical life.
LLMs have recently been deployed to explore normative reasoning, with striking results. Dillion et al.[218] demonstrated that GPT-4 was often perceived by laypeople as offering moral judgments that were more convincing than those of trained ethicists. This suggests a shifting public trust in algorithmic agents as sources of ethical insight. In related work, Kempt and Lavie[219] emphasized the role of socio-linguistic appropriateness over formal consistency in ethical dialogues, proposing a new conversational norm that better reflects human ethical discourse. Wang et al.[220] extended this discussion by drawing on Deweyan models of moral growth to argue that iterative learning processes in LLMs—particularly Reinforcement Learning from Human Feedback (RLHF)—mirror developmental trajectories of ethical learning, albeit without achieving full conceptual maturity. Colombatto and Fleming[216] added another layer by analyzing how people attribute consciousness and moral agency to LLMs based on their behavioral cues, raising important questions about anthropomorphism, folk psychology, and ethical projection.
Taken together, these studies suggest that LLMs are not merely tools for generating ethical content—they are reshaping our expectations of what it means to reason morally in an era of artificial intelligence. Yet this promise is tempered by deep concerns about context-awareness, the reproduction of cultural biases, and the limits of machine understanding in normative domains.
3.2.3. Analytical and Logical Philosophy
Analytical philosophy prioritizes clarity, precision, and argumentative rigor. It involves the careful dissection of philosophical problems, attention to formal structure, and an emphasis on logical coherence. In this domain, philosophical inquiry often proceeds through systematic critique, deductive reasoning, and conceptual analysis, frequently borrowing from tools in mathematics, linguistics, and computer science.
LLMs have become increasingly relevant in this tradition. Myung et al.[221] argued that LLMs challenge traditional epistemological categories by producing compressed representations of knowledge that function differently from standard propositional structures. Heersmink et al.[217] examined the epistemic status of LLM outputs, emphasizing the tension between the black-box nature of the models and the need for transparency and traceability in epistemic justification. Coeckelbergh[214] addressed the implications of hallucination and misinformation generated by LLMs, highlighting their potential to disrupt the epistemic infrastructure of democratic discourse. Overgaard and Kirkeby-Hinrup[215] further investigated the conditions under which LLMs could be attributed with a form of consciousness, proposing conceptual criteria grounded in contemporary philosophy of mind. Finally, Harnad[222] revisited the symbol grounding problem, arguing that LLMs, despite their linguistic fluency, lack true semantic understanding, thereby illuminating the limits of machine "comprehension."
What makes analytical philosophy particularly significant in the context of LLMs is its focus on testability, validity, and formal consistency. With the rise of AI-generated reasoning, long-standing debates in philosophy of language, such as the distinction between syntax and semantics or the nature of reference, have acquired new urgency. LLMs serve not only as subjects for testing classical problems like the Chinese Room argument or Turing Test variations but also as experimental platforms for rethinking those challenges.
Moreover, LLMs may assist philosophers in creating formal argument maps, identifying unstated premises, and evaluating deductive soundness. These capabilities have pedagogical value in teaching formal logic and informal argumentation alike. However, their effectiveness depends heavily on prompt design and internal alignment mechanisms, which themselves are subject to philosophical scrutiny.
Importantly, the proliferation of LLMs is prompting philosophers to revisit foundational questions: Can a non-conscious system produce true knowledge? What counts as understanding in a computational model? Are the outputs of LLMs assertions, simulations, or performative acts? These inquiries do not just engage epistemology and logic—they reframe the very nature of philosophical methodology in the 21st century.
3.2.4. Comparative and Cross-Disciplinary Philosophy
Comparative and cross-disciplinary philosophy bridges traditions and methodologies across cultures and academic domains. It addresses how different societies formulate responses to universal philosophical questions and seeks to integrate perspectives from political theory, sociology, cognitive science, and psychoanalysis, among others. Rather than pursuing a single analytic style or normative framework, this approach values synthesis, pluralism, and dialogue.
LLMs play a unique role in this area by enabling the integration of heterogeneous data sources and philosophical paradigms. Colombatto and Fleming[216] demonstrated how folk attributions of consciousness differ across demographic groups, revealing cultural variability in how AI is morally framed. Overgaard and Kirkeby-Hinrup[215] provided a cross-cultural critique of consciousness attribution by examining competing theories from Western and non-Western traditions. Heimann and Hübener[223] employed a Lacanian psychoanalytic framework to analyze metaphorical disruptions in LLM outputs, drawing parallels between psychotic structures and the disjointed metaphor use in machine-generated language. Harnad[222] contributed to this dialogue by situating LLMs in the broader context of human symbolic evolution, suggesting that computational models both reflect and distort the underlying architecture of meaning.
Through these diverse engagements, LLMs serve as tools for comparative ontology, cultural critique, and interdisciplinary synthesis. They not only illuminate philosophical differences but also generate new frameworks for exploring global patterns of thought.
In summary, LLMs are reshaping philosophical inquiry across multiple domains. In normative and interpretive philosophy, they offer fresh ethical narratives and challenge traditional modes of value judgment. In analytical and logical philosophy, they test the boundaries of meaning, truth, and justification. In comparative and cross-disciplinary philosophy, they foster pluralistic dialogue and conceptual innovation. While LLMs do not replace the depth of human reflection, they open up transformative avenues for reimagining what philosophy can be in an age of machine intelligence.
3.2.5. Benchmarks
To evaluate the ability of LLMs to understand abstract concepts, perform long-form reasoning, and support scholarly exploration, several curated datasets from the philosophy and humanities domains have been adopted for model training and assessment. Table 9 highlights two foundational resources that have played a central role in philosophy-aware AI development: Project Gutenberg and PhilPapers.
Project Gutenberg provides public domain access to thousands of classical texts authored by major philosophical and literary figures from antiquity to the modern era. These digitized volumes cover a wide range of traditions, including ancient Greek philosophy, Enlightenment rationalism, and 20th-century existentialism. Although originally designed for public access, the corpus has become a rich source for language modeling, long-form reasoning, and document understanding tasks. It is widely used in philosophical question answering, argument structure analysis, and stylistic adaptation, enabling LLMs to learn from centuries of philosophical prose and debate.
PhilPapers is a comprehensive bibliographic database that indexes over 2.5 million philosophy-related academic works, including journal articles, preprints, books, and conference papers. Each entry includes metadata, subfield categorization, and often abstracts or keywords, allowing for precise topic modeling, concept clustering, and academic stance detection. The resource supports document retrieval and citation graph analysis, and it has been used in tasks such as philosophical influence modeling and automated literature reviews. Its taxonomy of philosophical subfields makes it ideal for evaluating an LLM’s ability to reason within structured conceptual frameworks.
Together, these benchmarks provide broad coverage of philosophy as both a historical corpus and a living academic discipline. They are instrumental in assessing how well LLMs can engage with complex, ambiguous, and abstract content—traits that are central to human philosophical inquiry.
3.2.6. Discussion
Opportunities and Impact. The adoption of LLMs in philosophical research introduces powerful new methods for engaging with age-old questions of ethics, meaning, and knowledge. As demonstrated across normative reasoning [218,219], analytical logic [217,221], and comparative traditions [216], LLMs assist in the interpretation of dense arguments, simulation of ethical debate, and synthesis of diverse traditions. These models support the analysis of canonical texts, the generation of novel conceptual variations, and the mapping of ontological structures across schools of thought.
LLMs reduce barriers of scale and access by automating tasks such as argument classification, stylistic translation, and comparative summarization. They allow philosophers to simulate dialogues between traditions (e.g., utilitarian vs. deontological), emulate historical voices, and frame philosophical problems through alternative lenses. In doing so, LLMs may broaden access to philosophical education and enable new forms of inquiry that complement traditional methods.
Challenges and Limitations. Despite these advantages, the use of LLMs in philosophy remains constrained by serious risks. Hallucination and fabrication pose threats to intellectual rigor, especially when models generate arguments without grounding in sourceable literature [214]. LLMs may oversimplify abstract ideas or misrepresent debates, particularly where nuance and interpretive depth are essential. Issues of bias and dominance arise when models reproduce Western-centric canons while marginalizing global or alternative voices [216].
Another challenge is the lack of transparency and epistemic accountability: models often fail to attribute claims or cite sources, limiting their usefulness in rigorous philosophical debate [217]. Finally, the absence of true intentionality, phenomenological understanding, or cultural embodiment limits the kinds of questions LLMs can meaningfully engage with. While they simulate reasoning, they do not possess the self-awareness or lived context that underlies philosophical reflection.
Research Directions. Drawing on recent progress, several promising avenues for future research can be identified:
- Philosophical Fine-Tuning and Corpus Design. Curating balanced and inclusive corpora that represent diverse traditions can mitigate bias and expand philosophical reach [226].
- Logical Structure and Argument Mining. Developing tools to extract, visualize, and compare philosophical arguments enhances interpretive transparency and pedagogical value [221].
- Ontology Mapping and Comparative Frameworks. Leveraging LLMs to compare metaphysical and ethical schemas across cultures can support pluralistic theorizing and cross-cultural ethics [215].
- Interactive Dialogue Systems for Teaching. Deploying LLMs as Socratic partners or role-played historical thinkers can deepen student engagement and simulate philosophical exchange [218].
- Epistemology and AI Ethics. Interdisciplinary work is needed to assess the epistemic status of LLM outputs, their limits of understanding, and their role in reshaping human inquiry [217].
Conclusion. LLMs offer compelling tools for augmenting philosophical research and education. From dialogical simulations to conceptual experimentation, they enhance scalability and diversify access to philosophical content. Yet realizing this promise responsibly requires deeper attention to epistemic integrity, bias, and philosophical coherence. Future developments must focus on transparency, inclusivity, and alignment with human critical faculties. Rather than replacing human reflection, LLMs should be regarded as tools for extending and enriching the practice of philosophy in contemporary contexts.
3.3. Political Science
3.3.1. Overview
Introduction. Political science is the systematic study of power, governance, institutions, behavior, and the distribution of authority in societies. It analyzes how decisions are made, how policies are formulated and implemented, and how political actors compete, cooperate, and justify their legitimacy within local and global systems [227,228,229].
In simpler terms, political science explores how people organize themselves to make collective decisions and resolve conflicts. It studies elections, governments, protests, ideologies, and laws—how they work, why they fail, and what values they serve.
For example, understanding why democracies thrive or collapse, how authoritarian regimes maintain control, or how public opinion shapes policy are central political questions. Political science informs not only academics, but journalists, activists, diplomats, and ordinary citizens navigating civic life.
As Robert Dahl famously noted, “The key characteristic of democracy is the continued responsiveness of the government to the preferences of its citizens” [227]. Max Weber described politics as the “striving to share power or influence the distribution of power” [229], while David Easton defined it as “the authoritative allocation of values for a society” [228]—capturing how politics structures both resources and meaning in human life.
Figure 6.
The political research in the era of LLMs.
Traditional Political Research. Political science has long employed a diverse toolkit of methodologies to investigate questions of power, governance, and behavior. These approaches include: Textual and Historical Analysis. The close reading of constitutions, political speeches, and canonical works to uncover ideological foundations and institutional change [230]. Comparative Case Studies. Cross-national or historical comparisons to identify causal patterns in political systems and transitions [231]. Survey Research and Public Opinion Analysis. Empirical measurement of citizens’ beliefs, voting behavior, and attitude structures through questionnaires and polls [232]. Formal Modeling and Game Theory. Use of abstract models and strategic reasoning to represent decision-making among rational actors [233]. Statistical and Computational Approaches. Application of regression, network analysis, and text-as-data methods to detect patterns in political discourse and social dynamics [234]. Together, these methods span philosophical, empirical, and quantitative traditions. However, they also face challenges in scalability, integration, and the interpretation of complex social meaning.
The role of LLMs. LLMs could potentially reshape how political scientists analyze texts, simulate behaviors, and generate hypotheses. Their ability to process vast volumes of unstructured political data—such as speeches, manifestos, news articles, and social media posts—has opened new avenues for studying ideology, public opinion, and discourse dynamics [235]. LLMs can assist in automatically classifying sentiment and ideological alignment [234], generating realistic responses for survey simulation, and constructing policy or campaign narratives tailored to specific frames. In archival work, they can support interactive exploration of political history by summarizing, translating, or rephrasing historical texts. These capabilities expand the scope and speed of political analysis, reducing the burden of manual coding while enabling rapid experimentation.
Limitations of LLMs. Despite their promise, LLMs face significant limitations when applied to political research: Hallucination: LLMs often generate plausible but factually incorrect content, posing risks for misinformation and faulty inference [236]. Temporal and Institutional Fragility: They may struggle to track historical timelines, legislative sequences, or institutional nuances across contexts. Bias Propagation: Because they inherit biases from training data, LLMs may reinforce stereotypes or marginalize dissenting perspectives, raising concerns about fairness and representation [237]. Shallow Interpretation: LLMs often lack the ability to perform deep causal or normative reasoning, limiting their utility in theory-building or critical analysis. Opacity and Attribution: Their outputs typically lack transparent source citations, complicating verification and undermining reproducibility.
For these reasons, LLMs are best viewed not as replacements for interpretive or empirical political methods, but as augmentative tools that can enhance—but not substitute—core disciplinary judgment.
Taxonomy. To organize the expanding applications of LLMs in political science, we propose a task-based taxonomy that reflects how these models are reshaping core research practices. This framework consists of three domains:
- Text Analysis for Political Insight: LLMs assist in analyzing political texts such as speeches, party platforms, policy documents, and social media discourse. They are widely used for tasks like sentiment analysis, policy position classification, ideological scaling, and automated topic modeling, enabling scalable and consistent textual interpretation at unprecedented scale.
- Opinion Simulation and Forecasting: LLMs can simulate public opinion, generate synthetic survey respondents (“silicon samples”), and forecast electoral outcomes through multi-step reasoning. These capabilities are particularly valuable for behavioral modeling, comparative analysis, and data augmentation in contexts where real-world survey data is limited.
- Generation and Framing of Political Messaging: LLMs are increasingly used to craft persuasive political language, adapt messages to different audiences, and analyze framing strategies. These applications include generating campaign slogans and policy narratives, auditing ideological bias in generated outputs, and studying the persuasive dynamics of AI-authored political content.
3.3.2. Text Analysis for Political Insight
A core area where LLMs have shown significant promise is in the automated analysis of political text. Political scientists routinely rely on textual data—speeches, party platforms, social media posts, legislative records—to extract insights about ideology, sentiment, issue salience, and elite communication. LLMs offer scalable tools that augment or replace traditional methods of content analysis, often with comparable or superior performance.
Ornstein et al. [238] demonstrate that few-shot prompting with GPT-3.5 and GPT-4 can perform complex classification and topic modeling tasks on political documents without requiring extensive labeled data. Le Mens and Gallego [239] propose an “ask-and-average” framework that enables LLMs to position political texts within ideological space with remarkable correlation to expert surveys and roll-call–based measures. Similarly, O’Hagan and Schein [240] show that LLM-based measurement of ideological positions can yield consistent and interpretable estimates that rival traditional text scaling methods, while requiring significantly fewer assumptions.
Beyond ideological scaling, LLMs also excel in classification tasks central to empirical political research. Liu and Shi [241] introduce PoliPrompt, a cost-effective framework for political text classification that achieves high accuracy in stance detection and sentiment labeling by combining prompt engineering with consensus-based inference. Törnberg [242] further finds that ChatGPT-4 surpasses both expert coders and crowd workers in classifying political tweets, even in zero-shot settings.
Together, these studies show that LLMs are not only capable of interpreting political language at scale, but also of generating high-fidelity annotations and latent constructs that expand the methodological toolkit of computational political science. As LLMs continue to evolve, their integration into political text analysis promises to accelerate empirical research while also raising important questions about transparency, replicability, and conceptual rigor.
3.3.3. Opinion Simulation and Forecasting
LLMs are increasingly used to simulate public attitudes, generate synthetic survey data, and forecast electoral outcomes—tasks that lie at the core of political behavior research. These models can act as proxies for survey respondents, offering cost-effective tools for hypothesis testing in contexts where traditional data collection is limited or infeasible.
Qu and Wang [243] demonstrate that ChatGPT can simulate public opinion across diverse issue domains with surprising fidelity, particularly on moral and political questions. However, they also note that the model’s representational capacity varies across regions and demographics, raising concerns about generalizability. Yu et al. [244] propose a multi-step simulation framework for modeling political decision-making using LLMs, incorporating voter profiles and ideological cues to emulate realistic behavioral patterns at scale.
Role-conditioning has emerged as a key strategy to enhance realism. Karanjai et al. [245] show that embedding demographic and personality attributes into prompts enables LLMs to generate diverse and policy-relevant opinion distributions—what they call “synthetic public spheres.” Meanwhile, Lee et al. [246] empirically test whether LLMs can estimate population-level opinion distributions on global warming, finding promising accuracy when covariates are incorporated, but persistent bias in underrepresented subgroups.
At the predictive frontier, Bradshaw et al. [247] introduce a novel distribution-based method for forecasting U.S. electoral outcomes using token probability distributions from LLMs. Their work highlights how generative uncertainty can be turned into a probabilistic prediction tool, offering new metrics for electoral inference.
Collectively, these studies suggest that LLMs are more than textual tools—they are evolving into simulators of political behavior, capable of reflecting, and at times amplifying, the complexities of democratic publics. Yet, these advances also necessitate careful scrutiny of bias, alignment, and methodological transparency.
3.3.4. Generation and Framing of Political Messaging
LLMs are increasingly being used not only to analyze but to generate persuasive political content. This includes tasks such as crafting campaign messages, personalizing voter appeals, reframing controversial issues, and, in darker contexts, synthesizing disinformation. These capabilities place LLMs at the center of contemporary political communication, raising both practical opportunities and urgent ethical questions.
Hackenburg et al. [248] show that the persuasive effectiveness of political messages generated by LLMs increases logarithmically with model size. Using 24 different models and 720 U.S.-focused prompts, their study finds that while larger models are more persuasive, returns diminish rapidly beyond a certain scale. This implies that performance can often be optimized through prompt engineering rather than model expansion alone. In related work, Aldahoul et al. [249] evaluate the ideological positioning of LLMs and show that many models exhibit politically extreme and inconsistent behaviors across prompts—yet still persuade users effectively, even when merely conveying neutral information.
One emerging trend is the use of LLMs for personalized persuasion at scale. Matz et al. [250] demonstrate that ChatGPT, when tailored to users’ psychological profiles, can generate messages that are significantly more effective than generic ones. These findings point toward a future where AI-driven campaign strategies are hyper-individualized, adapting tone, framing, and issue salience to individual voters.
On the other hand, the same generative capacities pose risks. Williams et al. [251] assess how easily LLMs can be prompted to generate false but persuasive election-related narratives. Their DisElect dataset and experimental findings show that several popular LLMs consistently produce high-quality disinformation that is difficult for humans to distinguish from truth. These results underscore the importance of guardrails, transparency, and disinformation detection frameworks.
Finally, Šola et al. [252] combine AI-driven eye-tracking and LLM-based message generation to redesign political campaign materials. Their study highlights that human-centered design principles—when merged with generative models—can enhance attention and emotional engagement in political communication.
In sum, LLMs are not passive describers of political reality; they are active agents in shaping it. Whether optimizing legitimate political messaging or amplifying manipulative content, their generative potential reconfigures the dynamics of persuasion, participation, and propaganda in the digital public sphere.
3.3.5. Benchmarks
To evaluate the capabilities of LLMs in political analysis and discourse understanding, several specialized benchmarks have been introduced across media, legislative, and rhetorical domains. Table 10 summarizes five representative datasets that capture different layers of political language processing—from ideology classification and rhetorical structure to factual consistency and bias detection.
POLITICS Dataset is a large-scale corpus of 3.6 million news articles collected from left-, center-, and right-leaning media outlets. It supports tasks such as political stance classification, ideological alignment detection, and media bias recognition. By modeling political signals embedded in content and hyperlinks, the dataset helps identify framing strategies and polarization in news discourse. It has been widely used for training and evaluating fairness-aware and ideology-sensitive language models.
U.S. Congressional Records provide a structured textual record of congressional debates and proceedings, enabling fine-grained modeling of formal political speech. Tasks supported by this dataset include legislative summarization, policy intent classification, and speaker-style modeling. These records are particularly valuable for studying deliberative argumentation, procedural discourse, and political framing across party lines.
The American Presidency Project compiles over two centuries of presidential communications, including speeches, executive orders, and press statements. This resource supports rhetorical analysis and temporal comparison of executive discourse. It enables longitudinal studies of leadership language, agenda-setting patterns, and institutional tone shifts across administrations.
The Media Bias and Fact-check Dataset combines factuality ratings and political bias labels from platforms like MediaBiasFactCheck and AllSides. It supports tasks such as source bias prediction, factual reliability assessment, and misinformation flagging. As LLMs are increasingly used in content moderation and source attribution, this dataset is essential for stress-testing their alignment with factual standards and political neutrality.
TruthfulQA is designed to test whether language models can resist producing factually incorrect or misleading responses to questions based on false premises or human misconceptions. With 817 curated QA items across 38 categories—including politics, law, and health—it evaluates truthfulness and informativeness under adversarial conditions. The benchmark reveals that larger models may generate more fluent yet less truthful answers, underscoring the need for truth-aligned training in politically sensitive domains.
Collectively, these political NLP benchmarks offer a multi-faceted framework for evaluating LLMs’ ability to understand, generate, and fact-check politically charged content. They are essential for ensuring the safety, fairness, and integrity of AI systems deployed in democratic discourse and governance contexts.
3.3.6. Discussion
Opportunities and Impact. LLMs are transforming the scope and method of political science by automating textual analysis, simulating voter behavior, and generating persuasive political content. As demonstrated across core tasks like text classification [238,239,240], synthetic opinion generation [243,245], and campaign message design [248,250], LLMs enable political scientists to explore ideological dynamics, electoral predictions, and persuasive framing at unprecedented scale and speed. These models open new avenues for behavioral experimentation, hypothesis testing, and the synthesis of fragmented discourse.
LLMs also contribute to political pedagogy and public engagement by simulating survey responses, summarizing legislative debates, and facilitating multilingual access to policy documents. In low-resource environments or emerging democracies, they can support faster information processing and broader access to civic discourse. Moreover, LLMs enhance reproducibility and consistency in textual coding, long a bottleneck in political communication and media studies.
Challenges and Limitations. Despite these gains, LLM-based political analysis faces serious risks. Hallucination and misinformation generation remain persistent challenges [236], particularly when models are prompted with politically charged or adversarial inputs. Issues of fairness, bias amplification, and ideological skew [237] raise concerns about LLMs reinforcing dominant narratives while marginalizing dissent.
Interpretive shallowness further limits LLM utility in theory-building: while models can simulate preference distributions or generate policy arguments, they rarely reflect causal depth, institutional nuance, or normative coherence. Their black-box nature and lack of source attribution complicate scholarly transparency and verification. Finally, the political misuse of LLMs—for generating disinformation [251] or manipulating electoral behavior [249] —raises urgent ethical questions that require governance and auditing frameworks.
Research Directions. Building on recent developments, we can outline several promising directions for future research:
- Fine-Tuned Political Models. Domain-specific fine-tuning on legislative records, public opinion corpora, and multilingual political texts can enhance accuracy and reduce ideological bias.
- Causal and Temporal Modeling. Combining LLMs with structured models or reasoning frameworks may improve their ability to infer causality, detect agenda dynamics, and simulate policy feedback.
- Bias Detection and Auditing. Systematic tools to audit, mitigate, and document political biases in LLM outputs are essential for responsible deployment.
- Interactive Political Simulation. LLMs can be embedded in deliberative platforms to support role-playing, voter education, or negotiation training in civic and educational settings.
- Disinformation Defense. Techniques such as adversarial prompting, fact-checking augmentation, and truth-conditioned training can be used to defend against political misuse.
Conclusion. LLMs offer transformative potential for political science, enabling scalable behavioral modeling, discourse analysis, and personalized messaging. Yet, their integration into democratic contexts must be critically governed to preserve transparency, fairness, and civic trust. Future efforts should combine computational innovation with domain-sensitive safeguards, ensuring that political AI supports deliberation and accountability rather than distortion and manipulation.
3.4. Arts and Architecture
3.4.1. Overview
Introduction. Art is the conscious use of imagination and creative skill to produce works that express aesthetic, emotional, or conceptual ideas, often through mediums such as visual images, sound, performance, or language [258]. Architecture is the art of designing and constructing buildings and physical structures, combining functionality, aesthetics, and environmental consideration to shape human environments [259,260].
Figure 7.
The art and architecture research in the era of LLMs.
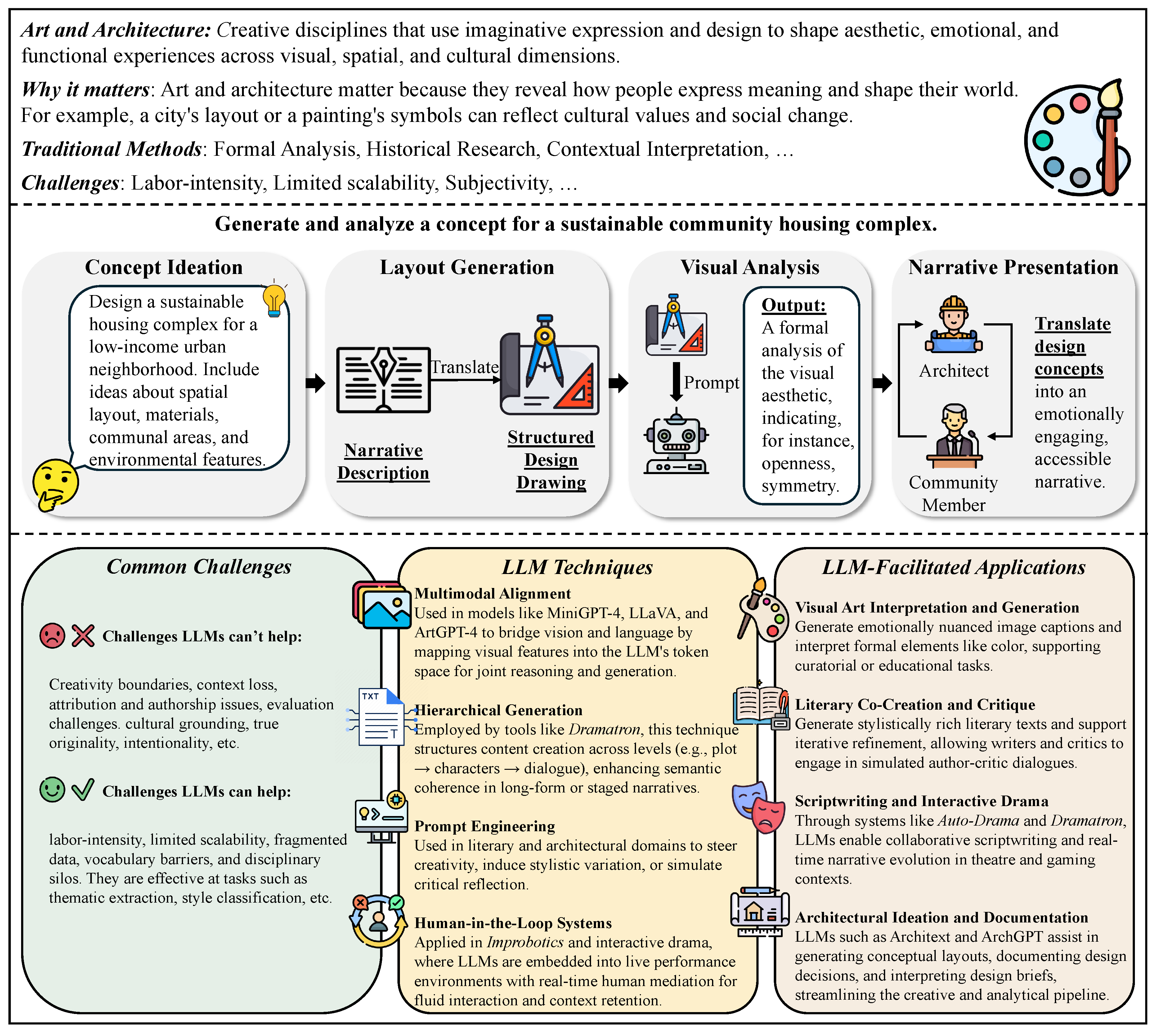
Research in art and architecture traditionally combines theoretical inquiry, historical investigation, and practice-based exploration [261]. In the arts, scholars engage with artworks through methods such as formal analysis (examining visual elements like color, line, and composition) [262], iconography (interpreting symbols and motifs), and contextual analysis (situating works within their social, political, or historical setting) [263]. Literary and performance studies may also involve close reading, narrative analysis, and ethnographic observation, as well as archival research to uncover original scripts, notes, or recordings [264].
In architecture, research spans multiple domains—from studying the evolution of design movements and analyzing urban development to conducting material testing or building performance simulations. Scholars may investigate the sociocultural impact of built environments, model spatial behavior, or explore the use of sustainable technologies [265]. Methodologies often blend technical modeling (e.g., CAD, BIM), environmental simulation, historical documentation, and conceptual critique [266,267]. In both art and architecture, qualitative approaches (interviews, fieldwork, visual diaries) and quantitative methods (e.g., surveys, pattern recognition in design databases) are increasingly used in combination [268].
Despite this methodological richness, traditional research in these fields faces several limitations. It is often labor-intensive, time-consuming, and limited by the scale of what a researcher can manually process or observe. Analyzing thousands of artworks, texts, or architectural plans for patterns or cross-cultural comparisons is rarely feasible without computational tools. Furthermore, insights are often locked behind domain-specific expertise or specialized vocabularies, making it challenging to connect findings across artistic disciplines or integrate them with computational models. These challenges highlight the need for scalable, cross-modal, and linguistically aware systems—such as LLMs—to support the next generation of art and architectural research.
Table 11.
LLM Applications and Insights in Art and Architecture
| Type of Art | Subtasks | Use Case-Inspired Research Question | Insights and Contributions | Citations |
|---|---|---|---|---|
| Visual Art | Creation (Prompting, Image Generation) | How can LLM-vision models generate visually compelling and stylistically diverse art through prompt-based interaction? | Multimodal models like MiniGPT-4 and LLaVA support creative generation; ArtGPT-4 enhances aesthetics using image adapters. | [269,270,271] |
| Analysis (Symbolism, Style Classification) | Can LLMs classify artistic styles and interpret visual symbolism without dedicated visual encoders? | GalleryGPT reduces hallucinations via structured prompts; CognArtive uses SVGs to enable symbolic reasoning without visual models. | [272,273] | |
| Literary Art | Creation (Storytelling, Stylistic Writing) | How can LLMs be guided to produce stylistically rich and emotionally resonant literary texts? | Prompt tuning and temperature settings support diverse styles; enables multi-voice storytelling and creative expression. | [274,275] |
| Analysis (Critique, Interpretation) | Can LLMs simulate literary critics or authors to support textual analysis and interpretation? | Interactive and self-reflective prompting enables critical reading and style-aware interpretation. | [276,277] | |
| Performing Art | Creation (Scriptwriting, Interactive Drama) | How can LLMs co-write scripts and structure dramatic arcs in interactive performances? | Tools like Dramatron build layered plots; Auto-Drama uses classical structure (e.g., Aristotelian elements) to shape narrative flow. | [278,279] |
| Performance (Live Interaction, Improvisation) | Can LLMs participate in live improvisational performance alongside human actors? | Improbotics blends live dialogue with LLM input for co-creative scenes; handles multi-party flow and timing. | [280] | |
| Architecture | Design and Creation (Concept Ideation, Layout Generation, Decision Records) | How can LLMs support early-stage design, spatial layout, and architectural decision-making processes? | Tools like Architext generate floorplans; GPT-based pipelines assist with ideation and accurate design documentation. | [281,282,283] |
| Analysis (Heritage Assessment, Design Comparison) | Can LLMs assist in heritage assessment and design analysis by integrating semantic knowledge with user intent? | ArchGPT retrieves context-aware insights; supports restoration, regulation checks, and expert collaboration. | [284] |
The Role of LLMs. LLMs offer new possibilities for supporting, augmenting, and democratizing research in the arts and architecture. These models can analyze and generate text, describe images, interpret creative language, and assist in ideation. Their ability to understand and produce language-rich content makes them particularly useful in domains that rely on creative description, interpretation, or storytelling.
In the arts, LLMs are being used to co-create visual and literary works, simulate historical or fictional voices, summarize critical interpretations, and generate poetic or narrative content. In architecture, LLMs assist in conceptual design, help interpret building codes, summarize design briefs, and even simulate conversations with historical architects. Multimodal extensions of LLMs (e.g., combining text with image or spatial data) further enhance their role in creative and analytical workflows.
Limitations of LLMs. Despite their growing adoption, LLMs introduce challenges when applied to creative disciplines: Creativity Boundaries: LLMs can generate stylistic content but lack true originality, intentionality, or cultural grounding. Their outputs may mimic but not innovate meaningfully without human curation. Context Loss: Art and architecture are deeply tied to historical, social, and cultural contexts. LLMs often fail to retain or interpret this embedded context, leading to surface-level or anachronistic outputs. Attribution and Authorship: The use of LLMs in co-creation raises ethical questions about authorship, originality, and credit—especially in arts communities where individual expression is core. Evaluation Challenges: Unlike scientific domains, there are no objective benchmarks for creativity or artistic value, making it hard to evaluate LLM-generated contributions meaningfully.
Thus, LLMs should be viewed as collaborators or assistants in the creative process—not as substitutes for human imagination, expertise, or cultural insight.
Taxonomy. Based on the current literature, we outline a taxonomy of how LLMs are being applied across different artistic and design domains:
- Visual Arts. Visual arts include creative practices such as painting, photography, illustration, and video. LLMs assist artists by generating image prompts, describing visual scenes, and helping with conceptual development. They also support analysis by interpreting symbolism, classifying art styles, and summarizing critical commentary.
- Literary Arts. Literary arts encompass creative writing forms such as poetry, drama, fiction, and essays. LLMs can generate literary content, mimic specific authorial styles, and assist in drafting narratives. They also enable textual analysis, such as thematic extraction, stylistic comparison, and literary critique.
- Performing Arts. Performing arts include music, dance, theater, and opera—forms that rely on embodied performance. LLMs can generate scripts, lyrics, or librettos, and simulate performative dialogue for interactive settings. They also help tag and analyze archival materials, enabling exploration of movement, expression, and performance history.
- Architecture. Architecture focuses on designing functional and aesthetic spaces, integrating art, engineering, and environmental factors. LLMs support architects by generating design narratives, proposing spatial ideas, and interpreting project briefs. They also assist in summarizing regulations, comparing architectural styles, and evaluating design decisions.
3.4.2. Visual Art
Visual art is formally defined as any art form that is primarily visual in nature, encompassing disciplines such as painting, drawing, photography, sculpture, printmaking, and video art. It emphasizes the use of imagery, color, composition, and form to convey meaning, emotion, or aesthetic value. In simpler terms, visual art is what we usually think of when we visit an art gallery or museum—works that are meant to be seen. These can include a painting on canvas, a photograph on a wall, or even a digital video installation. Visual art can be realistic or abstract, decorative or political, and it often reflects how artists interpret the world around them.
Visual Art Creation. Recent advancements in LLMs have significantly enhanced the capabilities of AI in visual art creation. Notably, models such as MiniGPT-4 [269], LLaVA [270], and ArtGPT-4 [271] demonstrate varied architectural innovations to improve artistic image understanding and generation. MiniGPT-4 employs a frozen vision encoder and LLM backbone, utilizing a Q-former for feature mapping, which facilitates efficient training but limits alignment flexibility. LLaVA overcomes this by enabling partial fine-tuning of the LLM, achieving better multimodal integration at the cost of computational efficiency. ArtGPT-4 innovates further by introducing trainable image adapters within the LLM architecture, allowing for parameter-efficient fine-tuning that preserves the interpretive depth of artistic content. These adapters, implemented with bottleneck structures and placed after attention layers, help the model capture subtle artistic features and emotional undertones with human-like sensitivity. Moreover, ArtGPT-4’s superior performance on benchmarks such as ArtEmis and ArtMM demonstrates its proficiency in conveying nuanced emotional and aesthetic qualities of visual art.
Visual Art Analysis. Also, the advancements in LLMs have significantly enhanced the capacity for automated visual art analysis by integrating visual perception with linguistic reasoning. GalleryGPT [272], a specialized LMM fine-tuned on a high-quality dataset called PaintingForm, exemplifies this trend by addressing the limitations of traditional image-text models which often exhibit “LLM-biased visual hallucination”—an overreliance on memorized textual knowledge rather than genuine visual understanding. By focusing on formal analysis—encompassing visual features like composition, color, and light—GalleryGPT leverages structured supervision to interpret and describe artworks based purely on their visual elements. Meanwhile, CognArtive [273] explore an alternative strategy by translating image content into SVG-based textual formats, enabling off-the-shelf language models to perform visual reasoning, classification, and even generative tasks without dedicated visual encoders [285]. These methods highlight a paradigm shift from recognition-centric models to visually-grounded analytical systems, empowering LMMs to articulate nuanced, expert-level interpretations of art.
3.4.3. Literary Art
Literary art refers to creative works expressed through written or spoken language, including genres such as poetry, fiction, drama, and essays. It is characterized by the imaginative use of words to convey ideas, emotions, and stories, often with attention to aesthetics, structure, and cultural significance. Put simply, literary art is the art of storytelling, whether through novels, plays, or poems. It helps people share experiences, express feelings, and explore ideas using language. We encounter literary art not only in classic literature but also in everyday forms like spoken word poetry or online short stories.
Literary Art Creation. The interdisciplinary methods presented in recent studies highlight how LLMs can significantly enhance literary art creation by offering a dynamic interplay between creative generation and critical interpretation. The work [274] demonstrates that techniques such as creative dialogue, temperature modulation, and multi-voice prompting can elicit complex, stylistically nuanced texts from LLMs, which in turn become fertile material for literary critical analysis. These methods invite iterative engagement, mirroring traditional author-critic interactions and foregrounding the process of refinement and self-critique. Literary critical frameworks—concerned not with appraisal but with interpretive depth—allow researchers to investigate elements like diction, narrative coherence, and stylistic innovation in AI-generated outputs, thereby moving beyond reductive measures of quality or creativity. Similarly, the evaluation protocols outlined in the comparative study of LLMs on creative writing tasks emphasize human-centered assessment across dimensions of craft, originality, and stylistic fidelity, reaffirming the necessity of qualitative, rubric-based evaluation to capture the performative and affective aspects of literary creation [275].
Literary Art Analysis. Recent advancements in LLMs have catalyzed innovative methodologies for facilitating literary art analysis. [274] demonstrate that techniques such as interactive prompting, temperature modulation, and multi-voice generation not only enable the generation of literarily rich texts but also support critical examination through a literary lens. Their qualitative evaluation [276] reveals that creative dialogue between a user and an LLM, characterized by iterative feedback and stylistic refinement, mirrors traditional pedagogical practices in creative writing. Modulating the temperature parameter allows for varying degrees of stylistic experimentation, producing texts ranging from conventional to lexically and syntactically avant-garde, thus expanding the interpretive canvas for literary critics. Moreover, the self-reflective, multi-voice generation method [277]—where the model simulates both authorial and critical roles—exemplifies a novel approach to self-critique and textual development, further blurring the lines between analysis and creation. These methods not only challenge conventional notions of authorship and creativity but also provide new tools for exploring intertextuality, narrative voice, and stylistic nuance within computational frameworks.
3.4.4. Performing Art
Performing art is defined as artistic expression conveyed through live performance, typically involving the human body, voice, or presence, and includes disciplines such as music, dance, theatre, and opera. These art forms unfold over time and are often intended for an audience, emphasizing temporality, embodiment, and interaction. In everyday terms, performing art includes concerts, plays, dance shows, or operas—any art form you watch and experience as it happens. It can be scripted or improvised, classical or experimental, and often brings together multiple art forms like movement, music, and storytelling.
Performing Art Creation. LLMs significantly influenced performing art creation, particularly in scriptwriting and dramaturgy. One prominent example is Dramatron [278], a hierarchical story generation tool that leverages LLMs to co-author theatre scripts and screenplays with human writers. By structuring content generation across multiple layers—from log lines and character descriptions to plot outlines and dialogues—Dramatron addresses the challenge of long-term semantic coherence, which is often a limitation in traditional flat LLM generations. Auto-Drama [279] significantly advances the field of interactive drama by enabling dynamic, immersive storytelling experiences. The authors introduce a comprehensive framework for LLM-based interactive drama, grounded in six redefined Aristotelian dramatic elements: plot, character, thought, diction, spectacle, and interaction. This model facilitates rich, player-driven narrative construction where users can engage directly with characters and environments. Key methodological innovations, such as the Narrative Chain, offer granular control over narrative development by segmenting the storyline into interconnected sub-narratives, allowing seamless plot progression while preserving player autonomy.
Interaction Performance. The methods [280] employed in the Improbotics study demonstrate significant advancements in facilitating interactive performance between AI and human actors, particularly within the dynamic context of improvisational theatre. By integrating LLMs into live performances through a human-in-the-loop system, the researchers enabled real-time, multi-party dialogue that simulates authentic co-creative interaction. The system employed speech recognition, manual metadata input, and curated line selection, allowing the AI (presented as a “Cyborg”) to contribute meaningfully to spontaneous scene development. This architecture addresses core challenges of multi-party conversational AI, such as speaker identification and context retention, by relying on both automated and human-mediated input. Iterative design with actor feedback ensured that the AI’s role evolved from a comedic object to a viable ensemble member, encouraging collaboration rather than mere novelty. The curated stream of LLM-generated responses enabled performers to incorporate AI contributions fluidly, supporting narrative coherence and audience engagement. Furthermore, game formats such as “Speed Dating” and “Wedding Speech” were strategically developed to evaluate the AI’s responsiveness to social cues and narrative arcs. While limitations in speech recognition and delayed response selection occasionally hindered fluidity, the hybrid human-AI interaction model fostered audience curiosity and performer creativity. Overall, this approach reframes AI from a static content generator to a dynamic performance partner, illustrating the potential of LLMs in embodied, context-sensitive artistic collaboration.
3.4.5. Architecture
Architecture is formally defined as the art and science of designing and constructing buildings and physical spaces that fulfill functional, structural, and aesthetic requirements. It involves the integration of form, environment, culture, and technology to shape the built environment. More simply, architecture is what surrounds us every day—from houses and schools to skyscrapers and public plazas. It is not just about making buildings stand up, but about how they make us feel, how they function, and how they reflect the identity of communities. Good architecture blends beauty, usefulness, and meaning.
Architecture Design and Creation. Architectural design is facilitated by methods like Architext [281], which leverage LLMs to generate valid and diverse floorplans from natural language prompts. By fine-tuning PLMs on synthetic semantic representations of layouts, Architext enables intuitive and scalable design workflows, reducing dependency on expert knowledge or specialized software. Its models consistently achieve high validity and correctness, demonstrating that language-based generation can serve as an effective tool for structured design tasks, transforming conceptual architectural descriptions directly into usable layout configurations. The work [282] facilitate architecture design and creation by enabling the generation of large, diverse sets of conceptual solutions through LLMs. By manipulating generative parameters and employing various prompt engineering strategies—including critique prompting and few-shot learning—the research demonstrates how LLMs like GPT-4 can support early-stage design ideation. These approaches provide designers with rapid access to broad solution spaces, enhancing creativity and overcoming fixation during concept development stages. In addition, [283] facilitate architectural design by enabling the generation of Architecture Decision Records (ADRs) from contextual inputs. Through zero-shot, few-shot, and fine-tuning approaches, LLMs can assist architects in documenting architectural decisions efficiently. Notably, fine-tuned smaller models like Flan-T5 demonstrate performance comparable to large models, offering scalable, privacy-conscious solutions. These methods enhance knowledge management and streamline decision-making, though human oversight remains essential for quality assurance.
Architecture Analysis. ArchGPT [284] facilitates architecture analysis by leveraging LLMs to parse user intents, retrieve domain-specific knowledge, and coordinate external tools. Through task-specific modules—such as image classification, semantic retrieval, and visual rendering—ArchGPT streamlines complex renovation tasks. Its structured workflow enhances interdisciplinary communication, automates heritage assessment, and ensures compliance with architectural guidelines. This methodology bridges expert knowledge with AI reasoning, offering efficient, adaptive solutions for heritage conservation and restoration.
3.4.6. Benchmarks
ArtBench-10 [286] is introduced as the first standardized, class-balanced, and high-quality benchmark dataset for artwork generation. It consists of 60,000 images spanning 10 distinct artistic styles and addresses common issues in previous datasets, such as class imbalance, noisy labels, and near-duplicates. The authors provide extensive benchmarking experiments using various generative models, including GANs, VAEs, and diffusion-based methods, and analyze their performance using metrics like FID, IS, precision, recall, and KID. The results highlight StyleGAN2 + ADA as the leading approach, while Projected GAN struggles with diversity. ArtBench-10 establishes a rigorous framework for evaluating generative models in the context of artwork synthesis
Table 12.
Overview of Benchmarks for Evaluating LLMs and Generative Models in Art and Architecture Domains
Table 12.
Overview of Benchmarks for Evaluating LLMs and Generative Models in Art and Architecture Domains
| Benchmark | Scope and Focus | Data Composition | Evaluation Tasks | Key Insights |
|---|---|---|---|---|
| ArtBench-10 [286] | Artwork generation and style classification | 60,000 curated images spanning 10 artistic styles with balanced classes and cleaned labels | Generative modeling using GANs, VAEs, diffusion; FID, IS, KID, precision/recall scores | StyleGAN2+ADA achieves best results; highlights diversity and fidelity gaps in GAN variants |
| AKM [283] | Architectural design decisions from text prompts | Context-rich design scenarios and model inputs for GPT/T5-family models | Zero-shot, few-shot, and fine-tuned generation of architectural decisions | GPT-4 performs best in zero-shot; GPT-3.5 effective with few-shot; Flan-T5 benefits from fine-tuning |
| ADD (DRAFT) [287] | Domain-specific architectural decision generation | 4,911 real-world Architectural Decision Records (ADRs) with labeled rationale and context | Few-shot vs. RAG vs. fine-tuning using DRAFT for architectural reasoning | DRAFT outperforms baselines in accuracy and efficiency; avoids reliance on large proprietary LLMs |
| AGI & Arch [288] | Generative AI’s knowledge of architectural history | 101M+ Midjourney prompts + qualitative and factual analysis of style descriptions | Historical style recognition, hallucination detection, generative image-text alignment | ChatGPT shows inconsistencies in confidence vs. accuracy; Midjourney trends analyzed via reverse prompts |
| WenMind [289] | Chinese Classical Literature and Language Arts (CCLLA) | 42 fine-grained tasks across Ancient Prose, Poetry, and Literary Culture; tested on 31 LLMs | Question answering, translation, rewriting, interpretation across genres | ERNIE-4.0 best performer with 64.3 score; major gap in LLM proficiency for classical Chinese content |
| AIST++ [290] | Music-conditioned 3D dance motion generation | 5.2 hours of 3D motion data across 10 dance genres + musical accompaniment | Motion synthesis with FACT transformer; evaluation via FID, diversity, beat alignment | FACT model generates realistic, synchronized, long-sequence dances better than prior baselines |
AKM [283] is a benchmark the effectiveness of LLMs in generating architectural design decisions based on a given context. The study evaluates GPT and T5-based models using zero-shot, few-shot, and fine-tuning approaches, measuring their performance against human-level decision-making. Results indicate that GPT-4 performs best in zero-shot settings, producing relevant and accurate decisions, while cost-effective models like GPT-3.5 achieve comparable results with few-shot learning. Additionally, smaller models such as Flan-T5 perform competitively after fine-tuning, suggesting that LLMs can assist in generating architectural decisions but require further research to reach human-level performance and standardization.
ADD [287] The paper evaluates the effectiveness of DRAFT—Domain-specific Retrieval Augmented Few-shot Tuning—for generating Architectural Design Decisions (ADDs). The benchmark includes comparisons against few-shot learning, retrieval-augmented generation (RAG), and fine-tuning across a dataset of 4,911 Architectural Decision Records (ADRs). DRAFT consistently outperforms other approaches in accuracy and relevance while maintaining efficiency. Automated metrics and human evaluations indicate that DRAFT generates high-quality ADDs without requiring proprietary or resource-intensive LLMs, making it a viable solution for organizations facing privacy and infrastructure constraints.
AGI & Arch [288] benchmarks the architectural knowledge of generative AI models—specifically ChatGPT for text generation and Midjourney for image generation—by systematically analyzing their ability to recognize and accurately describe historical architectural styles. Through quantitative assessments, the authors evaluate ChatGPT’s reliability in identifying styles, naming architects, and avoiding hallucinations, finding inconsistencies in its confidence versus factual accuracy. Meanwhile, Midjourney’s capability to generate images based on architectural style prompts is assessed by reversing the generation process to see if the AI can correctly describe its own outputs. The benchmark includes a large-scale analysis of over 101 million Midjourney queries to determine popular architectural styles and trends. Ultimately, the study exposes the strengths and limitations of generative AI models in preserving and understanding architectural history, offering a structured methodology for evaluating their knowledge fidelity.
WenMind [289] is a newly proposed benchmark designed to evaluate LLMs in the domain of Chinese Classical Literature and Language Arts (CCLLA). It encompasses three sub-domains—Ancient Prose, Ancient Poetry, and Ancient Literary Culture—spanning 42 fine-grained tasks across multiple question formats and evaluation scenarios. Through rigorous testing of 31 representative LLMs, the study finds that even the best-performing model, ERNIE-4.0, scores only 64.3, highlighting a significant gap in LLM proficiency in CCLLA. The benchmark provides insights into the strengths and weaknesses of various models and underscores the importance of pre-training data in achieving better results. WenMind sets a new standardized baseline for CCLLA research, offering a valuable resource for future advancements in this domain.
AIST++ [290] is the largest multi-modal dataset of 3D dance motion and music, along with the Full-Attention Cross-modal Transformer (FACT) network for generating 3D dance motion conditioned on music. AIST++ serves as a benchmark for music-conditioned dance generation, providing 5.2 hours of 3D motion across 10 dance genres. The FACT model outperforms prior state-of-the-art methods by employing full-attention transformers with future-N supervision, generating realistic and diverse long-sequence motions. The benchmark evaluations include motion quality (FID scores), diversity, and motion-music correlation (Beat Alignment Score), showing that the FACT model produces more natural and synchronized dance movements compared to existing approaches.
3.4.7. Discussion
Opportunities and Impact. LLMs are catalyzing a profound evolution in how creative disciplines like art and architecture are practiced, analyzed, and reimagined. As demonstrated across visual arts [271,272], literary arts [274,275], performing arts [278,280], and architecture [281,284], LLMs offer unprecedented capabilities for augmenting creative processes, automating interpretive analysis, and expanding access to specialized knowledge.
By facilitating narrative generation, multimodal interpretation, and conceptual ideation, LLMs can assist artists, writers, performers, and architects in overcoming traditional bottlenecks related to scale, documentation, and cross-disciplinary synthesis. They democratize access to creative exploration, allowing a broader range of individuals—including those without specialized training—to engage with and contribute to artistic and architectural production. Furthermore, LLMs open up new methodological possibilities: structured formal analysis of artworks [272], hybrid human-AI dramaturgy [278], iterative literary critique [274], and language-driven spatial design [281] exemplify how machine collaboration can enhance and diversify creative inquiry.
Challenges and Limitations. Nonetheless, the application of LLMs in artistic and architectural domains raises significant challenges. First, creativity remains bounded: while LLMs can generate stylistic approximations, they lack genuine intentionality, emotional grounding, and cultural specificity, often producing outputs that are derivative or decontextualized [271,274]. Second, contextual sensitivity is a critical limitation. Art and architecture are deeply embedded in social, historical, and material contexts that LLMs may fail to adequately capture or respect, leading to superficial interpretations or anachronistic outputs [272].
Authorship and originality also present complex ethical questions. In disciplines where individual vision and innovation are central, the blending of human and machine contributions challenges traditional notions of creative ownership, citation, and value attribution [278,280]. Moreover, evaluation remains difficult: unlike objective domains, assessing artistic quality or creative utility is inherently subjective, requiring nuanced, context-sensitive frameworks that go beyond automated metrics [275].
Finally, multimodal integration—especially in architecture and performing arts—remains in its early stages. Current LLM systems still struggle to fully align spatial, temporal, and embodied aspects of creativity within coherent generative frameworks [280,284].
Research Directions. Several promising research directions emerge to address these challenges:
- Ethical Authorship and Attribution Frameworks. Establishing clear guidelines for authorship attribution, ethical usage, and creative credit in LLM-augmented works will be critical to ensuring fair recognition and responsible innovation [278].
- Qualitative Evaluation Metrics. Developing domain-specific, human-centered evaluation rubrics—focused on creativity, authenticity, and emotional resonance—will be necessary to meaningfully assess LLM contributions in artistic fields [275].
Conclusion. LLMs are expanding the boundaries of creative exploration, offering powerful tools for augmenting artistic production, critical analysis, and interdisciplinary synthesis. Yet their application demands careful attention to contextual sensitivity, creative agency, and ethical responsibility. As art, literature, performance, and architecture increasingly intersect with AI, the future lies not in replacing human creativity, but in forging synergistic partnerships—where machine intelligence extends human imagination, enriches critical engagement, and fosters new forms of cultural expression.
3.5. Law
3.5.1. Overview
Introduction. Law shapes nearly every aspect of our lives—from signing a lease, starting a business, or getting married, to protecting free speech, settling disputes, and ensuring fairness in society. It is a system of rules, created and enforced by governments and institutions, that helps organize how people and organizations interact. These rules are found in many forms: laws passed by legislatures, decisions made by courts, regulations from agencies, and long-standing customs that have legal force [291,292,293].
In short, law acts as society’s rulebook. It lays out our rights and responsibilities, provides tools to resolve conflicts, and guides behavior in everything from everyday transactions to high-stakes corporate or constitutional decisions. Understanding and applying the law, however, is not always straightforward. Legal documents are often long, complex, and written in specialized language. Figuring out what a regulation means, how a court case applies, or what terms go into a contract often requires time, training, and expertise.
Legal work is deeply text-based. Lawyers and legal professionals spend much of their time reading and writing—interpreting laws, analyzing past court decisions (precedents), drafting agreements, responding to client questions, and preparing arguments. This work traditionally relies on years of professional training, manual research, and reasoning skills developed through legal education and practice [294,295,296]. Tools like Westlaw and LexisNexis have helped by making legal materials easier to search, but they still depend on users knowing exactly what to look for and how to navigate technical legal databases.
More broadly, legal research refers to the process of discovering, understanding, and analyzing legal information to answer specific questions or develop arguments. It covers a wide range of activities: finding relevant statutes and regulations, identifying controlling precedents from court rulings, comparing legal principles across jurisdictions, and understanding how laws apply in different factual situations. To solve these research problems, legal professionals rely on doctrinal analysis [295,297,298], case law reasoning [296,299,300], analogical reasoning [301,302], and manual document review—skills refined through rigorous training and experience [294].
Despite these advances, legal research still faces uniquely challenges. A single word in a statute can carry enormous weight, but its meaning often depends on how courts have interpreted it over time. Arguments must be built on precedent, logic, and interpretation—while also accounting for evolving social, political, and institutional contexts. Such biases, the blend of rigorous logic, textual analysis, and human judgment, are the most important issues in legal research. At the same time, the legal world is facing new challenges. The amount of legal text—court rulings, statutes, filings, contracts, and more—is growing faster than ever. Legal professionals must now sift through massive amounts of information, stay up to date on changing rules, and make sense of complex relationships across documents. Doing this manually is time-consuming and expensive, and it creates barriers for individuals or smaller organizations that cannot afford high-end legal support.
Figure 8.
Overview of LLM Applications in Legal Research. This figure illustrates the core challenges in legal work, key tasks enabled by LLMs, and the dominant techniques used to deploy them. It highlights how LLMs address traditional limitations—such as bias and information overload—by supporting tasks like legal Q&A, document drafting, case analysis, and prediction. On the methods side, it showcases powerful techniques including Retrieval-Augmented Generation (RAG), Domain-Specific Fine-Tuning, Prompt Engineering, and Hybrid Modeling.
Figure 8.
Overview of LLM Applications in Legal Research. This figure illustrates the core challenges in legal work, key tasks enabled by LLMs, and the dominant techniques used to deploy them. It highlights how LLMs address traditional limitations—such as bias and information overload—by supporting tasks like legal Q&A, document drafting, case analysis, and prediction. On the methods side, it showcases powerful techniques including Retrieval-Augmented Generation (RAG), Domain-Specific Fine-Tuning, Prompt Engineering, and Hybrid Modeling.
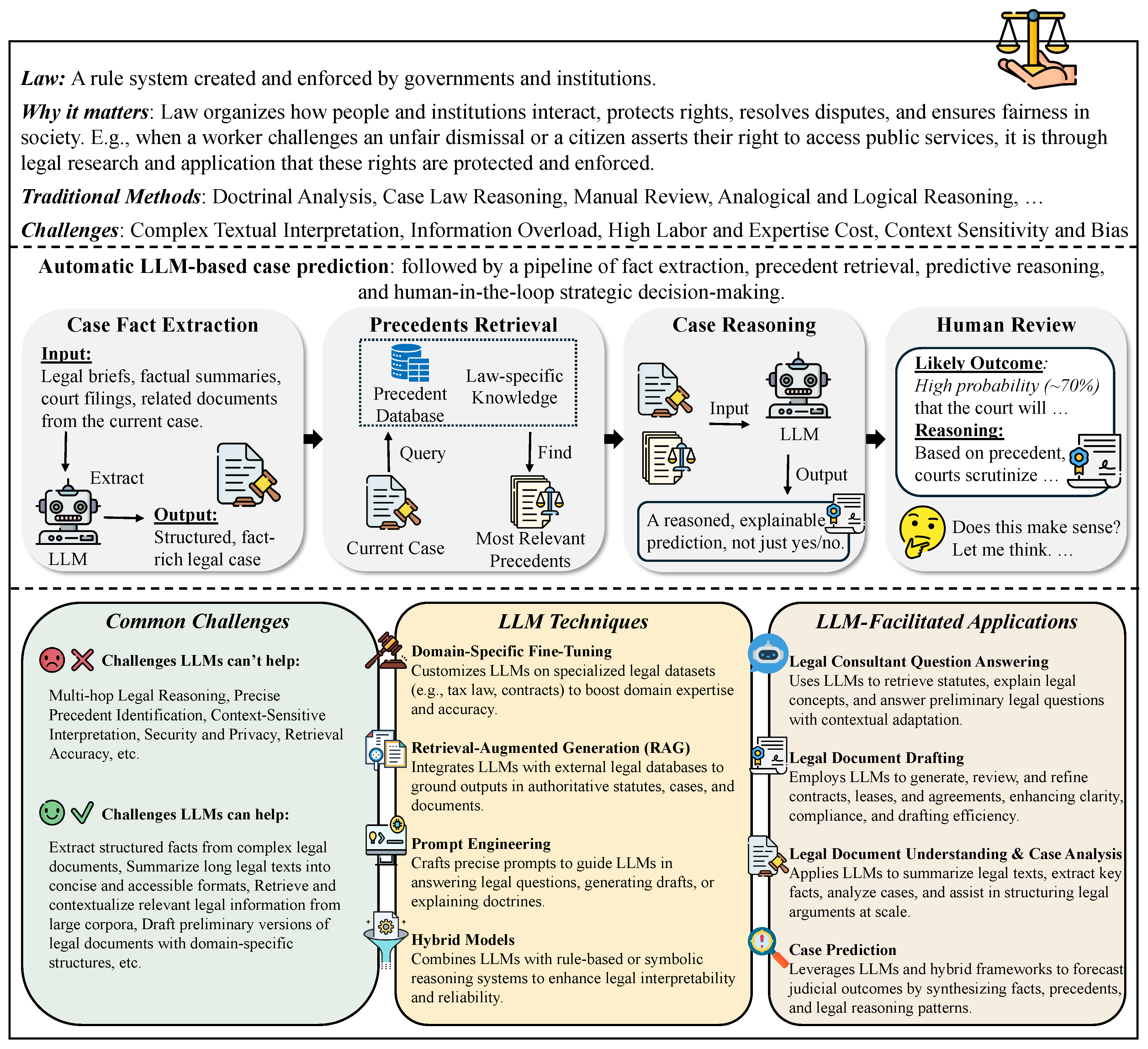
The role of LLMs. LLMs are advanced AI systems trained to understand and generate human language in an unbiased way. Their strengths in natural language understanding, summarization, retrieval, and question answering position them well to support legal tasks. Recent advances show that LLMs can extract structured facts from legal documents, identify key issues, generate coherent legal drafts, and assist users in navigating legal systems [303,304]. Unlike keyword-based tools, LLMs can engage with legal text at the level of meaning, making them more adaptable and user-friendly.
Figure 9.
LLM-Driven Workflow in Legal Tasks. This figure illustrates the end-to-end paradigm of applying LLMs in various legal processes. For each task—Consultant QA, Document Drafting, Case Analysis, and Case Prediction—the figure outlines typical inputs (e.g., legal queries, cases, statutes), how the LLM processes these inputs, and the corresponding outputs such as query answers, contract drafts, key legal facts, and judicial predictions.
Figure 9.
LLM-Driven Workflow in Legal Tasks. This figure illustrates the end-to-end paradigm of applying LLMs in various legal processes. For each task—Consultant QA, Document Drafting, Case Analysis, and Case Prediction—the figure outlines typical inputs (e.g., legal queries, cases, statutes), how the LLM processes these inputs, and the corresponding outputs such as query answers, contract drafts, key legal facts, and judicial predictions.
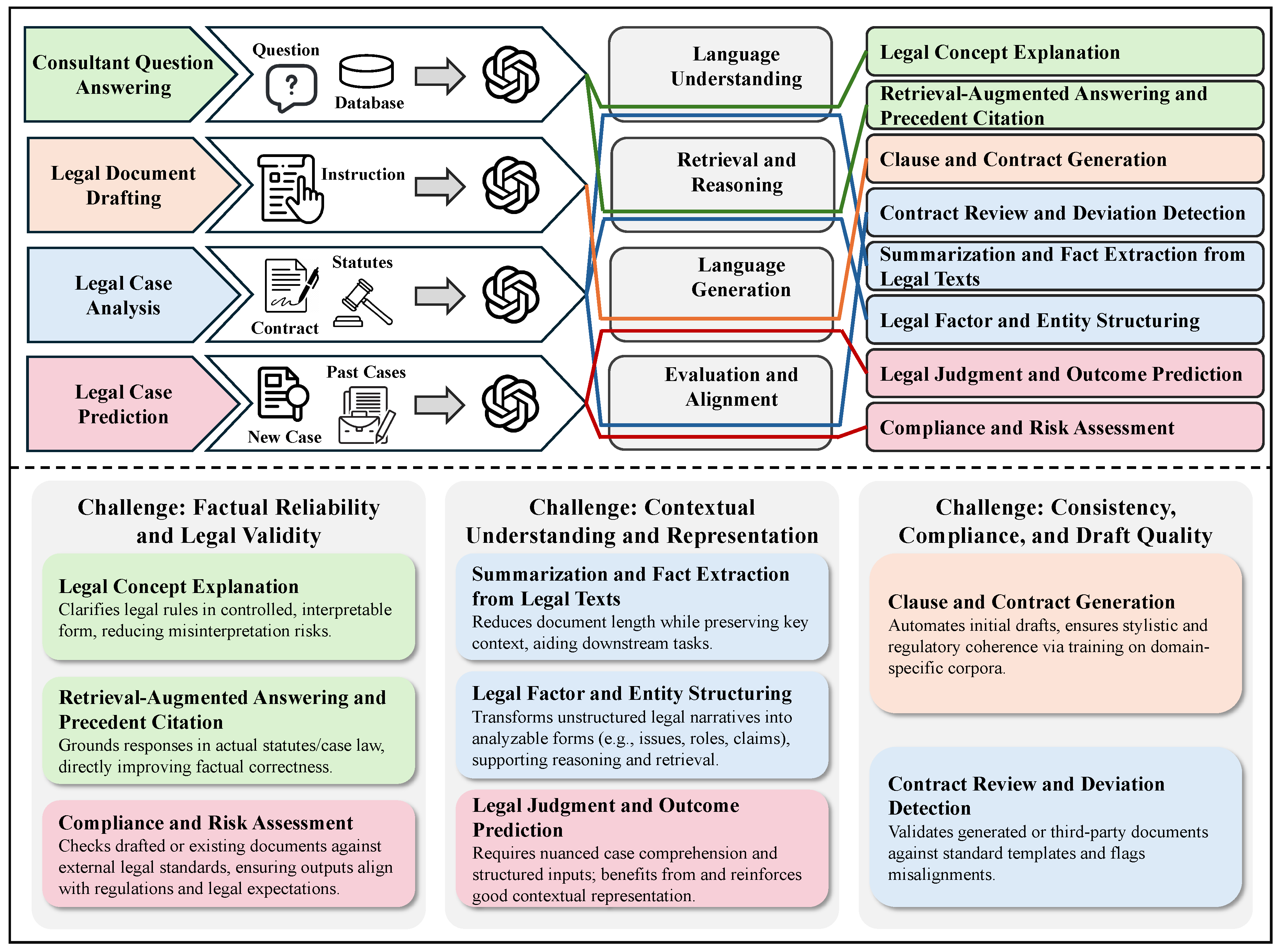
Limitations of LLMs. Despite their promise, LLMs also face important limitations in legal applications. Security and Privacy: Legal documents often contain sensitive or privileged information. Using cloud-hosted LLMs risks inadvertent disclosure of confidential data, raising ethical and regulatory concerns—especially in jurisdictions with strict data protection laws. Multi-hop Reasoning: Many legal questions require synthesizing information across multiple sources—e.g., linking a statute to case interpretations and factual contexts. While LLMs are fluent in language generation, they often struggle with the structured, multi-step reasoning needed in legal contexts [304]. Retrieval Accuracy: Legal outcomes frequently hinge on identifying the most relevant precedents. Yet LLMs may retrieve plausible but non-binding cases, miss key controlling authorities, or fail to distinguish nuances in legal reasoning.
Taxonomy. To better understand how LLMs contribute to legal research and practice, we propose a taxonomy that captures core categories of legal tasks where language plays a central role:
- Legal Consultant Question Answering. Many legal queries—such as “What are the elements of negligence?” or “Does the GDPR apply to this situation?”—involve retrieving statutory definitions, summarizing doctrines, or explaining precedent. LLMs can function as legal assistants, offering plain-language explanations, surfacing relevant laws, and contextualizing rules. This enables broader access to legal knowledge and supports both laypersons and professionals during early-stage legal reasoning.
- Legal Document Drafting. Drafting contracts, policies, and filings involves significant repetition and domain knowledge. LLMs can generate initial templates, propose clauses, and adapt documents to specific scenarios or jurisdictions. This accelerates document production, reduces drafting overhead, and promotes standardization—especially useful for small firms or high-volume legal operations.
- Legal Document Understanding & Case Analysis. Interpreting statutes, summarizing opinions, or identifying relevant facts is core to legal analysis. LLMs can extract key information, highlight legal entities or issues, and support case comparison. This improves comprehension, reduces time spent on manual review, and helps structure arguments and decisions based on large textual corpora.
- Case Prediction. Predicting legal outcomes—based on case facts, prior rulings, and jurisdictional context—is valuable for risk assessment and litigation strategy. While final outcomes are shaped by human judgment and evolving law, LLMs can surface patterns, suggest likely outcomes, and support probabilistic reasoning based on precedent, helping users plan and prioritize cases.
Table 13.
Applications and insights of LLMs in legal research and practice
| Legal Domain | LLM Application Areas | Use Case-Inspired Research Question | Key Insights and Contributions | References |
|---|---|---|---|---|
| Legal Consultant Question Answering | Interactive Legal Q&A Systems | Can LLMs answer basic legal questions with accurate references to statutes and case law? | Emergent legal reasoning observed; retrieval-augmented prompting reduces hallucination; GPT-4 can approximate legal explanations with improved accuracy. | [305,306,307] |
| Domain-Specific Legal Models | How can LLMs be fine-tuned to better address legal reasoning tasks? | Models like LawLLM improve U.S. law reasoning through fine-tuning and task adaptation (retrieval, precedent matching, judgment prediction). | [308] | |
| Legal Factor Extraction | Can LLMs extract and define core legal factors from court opinions? | Supports building expert systems; improves structure and consistency of legal analysis. | [309] | |
| Legal Document Drafting | Clause Generation | Can LLMs autonomously generate domain-compliant legal clauses? | LLMs generate grammatically and legally sound clauses; useful in reducing drafting effort. | [310,311] |
| Draft Comparison via NLI | How can LLMs verify consistency between generated and template contracts? | NLI tasks help identify deviations and inconsistencies, enabling automated review. | [312] | |
| Legal Validity of Prompt-Based Contracts | What legal risks arise when contracts are generated using prompts? | Raises issues with doctrines like the parol evidence rule; prompt provenance matters. | [313] | |
| Legal Document Understanding and Case Analysis | Document Summarization & Entity Extraction | How well can LLMs extract facts and citations from unstructured legal texts? | Enhanced summarization, fact extraction, and legal entity recognition using retrieval-based and fine-tuned models. | [306,308,314] |
| Large-Scale Legal Analysis | Can LLMs support empirical research over large legal corpora? | Enables scalable judgment pattern extraction; useful for comparative legal studies. | [307] | |
| E-Discovery and Compliance | Can LLMs assist in regulatory compliance and legal review at scale? | RAG-based systems improve due diligence and compliance decisions; multi-agent LLMs aid in document relevance prediction. | [315,316] | |
| Legal Case Prediction | Judgment Forecasting | Can LLMs accurately predict legal case outcomes? | LLMs outperform traditional models; retrieval-augmented LLMs improve consistency and generalization. | [317,318] |
| Hybrid Legal Reasoning | How can LLMs be integrated with expert systems to improve prediction accuracy? | Hybrid systems improve interpretability and performance by aligning LLM outputs with legal logic. | [319] |
3.5.2. Legal Consultant Question Answering
Legal Consultant Question Answering is the task of addressing fundamental legal inquiries using LLMs [314]. For example, questions such as “What are the elements of negligence?” or “Does the GDPR apply to this case?” typically require swift reference to statutes, case law, and legal principles [320,321]. Serving as the initial layer of legal support, these systems offer immediate, scalable, and context-aware legal assistance, thereby democratizing access to legal knowledge and alleviating the routine workload of human professionals [321].
Traditional legal question-answering systems have historically relied on rule-based processes or keyword searches within carefully curated legal databases, as described by Ashley [320]. However, these conventional approaches often suffer from limitations in terms of coverage, sensitivity to linguistic variations, and difficulties in adapting across different jurisdictions [320]. In contrast, recent advances in LLMs have opened up new possibilities for addressing these challenges.
For instance, Nay et al. [305] presented a case study where LLMs were adapted to function as tax attorneys. Their work demonstrated that by providing relevant legal texts and a few examples, the model’s performance in handling complex tax law queries could be substantially enhanced, thereby showcasing the emergent legal capabilities of LLMs in specialized domains. Similarly, Savelka et al. [306] investigated the use of GPT-4 to explain legal concepts. They found that augmenting the model with retrieval mechanisms not only improved the clarity of the generated legal explanations but also significantly reduced factual inaccuracies, making the outputs more reliable for legal practitioners.
Shu et al. [308] introduced LawLLM, a LLM tailored specifically for the U.S. legal system. LawLLM demonstrated strong performance in tasks such as similar case retrieval, precedent recommendation, and legal judgment prediction, benefiting greatly from domain-specific fine-tuning and retrieval augmentation. This work highlights how targeted adaptations can enable LLMs to effectively support legal decision-making in practice.
In another direction, Gray et al. [309] proposed a novel method that leverages LLMs to automatically extract legal factors from court opinions. By processing raw judicial texts and generating a set of legal factors with associated definitions, their approach assists legal analysis and supports the development of expert systems that can summarize complex legal information efficiently.
Finally, Choi [307] explored the practical application of LLMs in empirical legal research. This study examined how LLMs can be employed to analyze legal documents, providing a detailed evaluation of best practices and highlighting both the potential benefits and limitations of using LLMs in data-driven legal analysis.
In summary, these studies underscore the growing capabilities of LLMs to democratize access to legal information. They also highlight the importance of contextual adaptation, continuous updates, and robust safety mechanisms when deploying such systems in practical legal settings.
3.5.3. Legal Document Drafting
Legal Document Drafting refers to the task of generating, reviewing, and ensuring the compliance of standardized legal documents—such as contracts, leases, and other legal agreements—by integrating legal principles into clear and consistent texts [320,322]. This task requires the accurate formulation of legal clauses and the adherence to regulatory standards to minimize ambiguity and reduce potential disputes [322]. Its objective is not only to produce legally sound documents but also to achieve consistency and clarity, thereby streamlining legal processes and reducing manual effort [320].
Traditional methods for legal document drafting have predominantly relied on manual drafting by experienced legal professionals or on rule-based systems that utilize pre-defined templates and keyword searches within carefully curated legal databases [320,322]. However, these approaches often suffer from several limitations: they are labor-intensive, inflexible, and prone to human error, which can lead to inconsistent language and difficulties in adapting to evolving legal standards [322]. In contrast, recent advances in LLMs offer significant advantages; LLM-based systems can process unstructured legal texts, leverage semantic understanding to retrieve pertinent information, and generate coherent, legally grounded drafts with minimal human intervention [306,314]. Moreover, when further fine-tuned on domain-specific legal corpora or enhanced with retrieval-augmented generation techniques, these models can produce more accurate, consistent, and compliant legal documents, ultimately increasing the efficiency and reliability of the drafting process [308].
Recent advancements in LLMs have catalyzed innovative approaches in legal document drafting. Zhang et al. [310] investigate the feasibility of generating contract clauses automatically by leveraging pre-trained language models on large-scale legal corpora. Their study demonstrates that, with sufficient training data, LLMs can generate contract clauses that are both grammatically coherent and legally compliant, thereby reducing manual drafting efforts and increasing drafting efficiency.
Building on this, Wang et al. [312] propose a novel approach using Natural Language Inference (NLI) tasks to compare generated contracts with standard templates. Their method is designed to identify deviations and inconsistencies between the drafted document and established templates, thereby providing an automated mechanism for quality control during contract negotiations and template refinement.
In another vein, Sato and Nakamura [313] analyze the legal status of prompt-based generation in contract drafting. Their work offers an in-depth discussion of how the use of prompts in generating contract clauses interacts with traditional legal doctrines—such as the parol evidence rule—and examines both the advantages and potential legal risks associated with prompt-based systems.
Furthermore, Liu et al. [311] explore the adaptation of open-source LLMs for domain-specific contract drafting. They demonstrate that by fine-tuning open-source models on specialized legal datasets, it is possible to significantly enhance text clarity, incorporate domain-specific terminology, and tailor the drafting process to meet the unique requirements of fields like construction contracts.
In summary, these studies illustrate that LLM-based approaches offer transformative potential for legal document drafting by automating the generation of precise legal clauses, enabling systematic quality checks via NLI, addressing the legal implications of prompt usage, and adapting general models for specialized domains. Collectively, these innovations not only streamline the drafting process but also enhance the consistency, accuracy, and compliance of legal documents.
3.5.4. Legal Document Understanding and Case Analysis
Understanding legal texts—such as statutes, court opinions, and filings—is essential in the legal process, especially for judges, clerks, and legal researchers [307]. Traditionally, legal document analysis has involved manual review, annotation, and summarization by legal experts, often supplemented by rule-based systems or keyword-driven information retrieval to extract key facts and legal citations [320]. However, these conventional methods are time-consuming, prone to inconsistencies, and struggle to cope with the ever-growing volume and complexity of legal materials [320].
In contrast, recent advancements in LLMs offer significant advantages. Modern LLM-based systems are capable of automatically summarizing lengthy legal documents, extracting crucial facts and citations, tagging legal entities, and highlighting pertinent issues with remarkable speed and consistency [306,314]. By leveraging deep semantic understanding and contextual learning, these models can process unstructured legal texts to generate coherent, legally grounded analyses, thereby facilitating more efficient legal decision-making in the judicial process [308]. Moreover, the integration of retrieval-augmented techniques and domain-specific fine-tuning further enhances their performance, enabling LLMs to provide more accurate and contextually relevant insights in legal research and case analysis [307,309].
Understanding and analyzing legal documents—such as statutes, court opinions, and filings—is fundamental to the legal process, especially for judges, clerks, and legal researchers [307]. Traditionally, legal document analysis relied on manual review and rule-based methods, where experts would meticulously annotate texts or employ keyword searches within structured legal databases [320,323]. However, these approaches are often labor-intensive and struggle to scale in the face of ever-increasing volumes of complex legal materials.
Recent advancements in LLMs have led to significant breakthroughs in automating legal document understanding and case analysis. For instance, Shu et al. [308] introduced LawLLM, a multi-task model specifically designed for the U.S. legal system. Beyond its capabilities in legal judgment prediction, LawLLM is adept at retrieving relevant statutes and cases, thus facilitating comprehensive document analysis. In a similar vein, Nay et al. [305] explored the application of LLMs in the tax law domain, demonstrating that with the provision of pertinent legal texts and a few examples, the models can leverage logical reasoning to parse complex tax regulations and case details effectively.
Further expanding the scope of automated legal analysis, Choi [307] illustrated how LLMs can be employed for empirical legal research by analyzing large-scale legal corpora to extract patterns and summarize judicial opinions. In another study, Savelka et al. [306] evaluated GPT-4’s performance in generating legislative explanations and found that incorporating retrieval-augmented methods significantly enhances the clarity and factual accuracy of the generated legal interpretations. Gray et al. [309] proposed a novel approach to automatically extract key legal factors and definitions from raw court opinions, thereby providing structured insights that can assist legal analysts and support the development of expert systems.
Beyond these foundational studies, additional research has extended LLM applications to practical legal workflows. For instance, Lahiri et al. [315] proposed DISCOvery Graph, a hybrid framework that combines graph neural networks with LLMs to enhance retrieval and reasoning in eDiscovery. By structuring litigation data into graphs and enabling LLM-based reasoning over them, their system improves document relevance prediction and supports large-scale legal review.
Similarly, Chang et al. [316] introduced MAIN-RAG, a multi-agent filtering framework for retrieval-augmented generation. Their approach leverages multiple LLM agents to collaboratively score and filter retrieved documents before generation, thereby increasing the accuracy, consistency, and interpretability of generated outputs—particularly useful in high-stakes domains such as regulatory compliance and legal decision support.
Recent research has demonstrated the transformative potential of LLMs in the domain of e-discovery. Lahiri et al. [324] proposed DISCOvery Graph, a hybrid system that combines graph-based document modeling with LLM reasoning to enhance relevance prediction in legal document review. Their system achieved substantial improvements in precision, recall, and F1 score while drastically reducing the cost of document screening. Wickramasekara et al. [325] conducted a comprehensive review of the applicability of LLMs to digital forensic investigations, highlighting both the opportunities for improved traceability and the limitations regarding auditability and legal compliance. Yin et al. [326] further explored how LLMs can reshape the digital forensic process, emphasizing challenges such as hallucination and bias, and proposing best practices to integrate LLMs responsibly. Additionally, a comparative evaluation by Pai et al. [327] examined multiple open-source LLMs in practical e-discovery tasks, offering insights into their strengths in summarization, key evidence detection, and adaptability across diverse legal corpora.
Collectively, these studies underscore the growing capabilities of LLM-based methods in automating tasks such as document summarization, relevance filtering, and legal fact extraction. They pave the way for more efficient, scalable, and consistent legal discovery processes, while calling for robust safeguards and domain-specific customization in real-world deployment.
3.5.5. Legal Judgment Prediction
Legal Judgment Prediction is a core task in the intersection of legal informatics and artificial intelligence, aiming to forecast the outcome of judicial decisions based on structured or unstructured descriptions of legal cases. It is of paramount importance in practice, as it supports litigation strategy, risk assessment, and policy analysis. Traditional approaches to Legal Judgment Prediction have relied heavily on statistical models trained on manually curated features and structured legal records [320], which, while useful, often lack the capacity to scale across jurisdictions or adapt to the subtleties of legal reasoning embedded in natural language narratives.
With the emergence of LLMs, the field has witnessed a paradigm shift. Unlike earlier methods, LLMs are capable of understanding and generating complex legal texts, making it possible to model legal reasoning as a language generation or classification task. These models bring strong capabilities in semantic representation, contextual reasoning, and few-shot learning, making them well-suited to judicial prediction, especially when combined with retrieval or reasoning modules.
A series of recent studies have validated the potential of LLMs in enhancing the accuracy, robustness, and interpretability of Legal Judgment Predictio systems. Cao et al. [328] introduced the PILOT framework, which combines precedent retrieval with time-aware modeling to better reflect the evolving nature of legal principles. Evaluated on the ECHR2023 dataset, PILOT significantly outperformed baseline models, demonstrating the importance of temporal and precedent-aware reasoning in outcome prediction.
Building on this notion, Wu et al. [329] proposed the PLJP framework, integrating LLMs with domain-specific modules to enhance both interpretability and predictive performance. By aligning the outputs of LLMs with key legal factors extracted from precedents, PLJP enables models to ground their predictions in structured legal reasoning rather than purely statistical associations.
Shui et al. [330] contributed a thorough empirical study that evaluates the performance of several LLMs on legal judgment tasks. Their findings show that prompting strategies, such as including in-context examples and using multiple-choice formulations, substantially affect prediction accuracy. These insights are crucial for practitioners seeking to optimize LLM usage without fine-tuning.
To push beyond static prediction, Deng et al. [331] proposed the ADAPT framework, which decomposes a legal case into fact segments, possible charges, and corresponding outcomes. This discriminative reasoning structure enables more transparent and structured inference, particularly in complex multi-label judgment scenarios such as criminal law.
Lastly, Nigam et al. [332] explored the challenges of deploying LLMs in real-world legal environments, using the Indian Supreme Court corpus. Their work emphasized the importance of retrieval-augmented generation (RAG) and jurisdiction-specific adaptation, especially in legal systems with different precedent norms or linguistic styles. The study demonstrated that models like GPT-3.5 Turbo can, with proper domain integration, match or exceed traditional benchmarks in outcome prediction.
3.5.6. Benchmarks
The evaluation of LLMs in the legal domain has become an increasingly active research area, propelled by the urgent need for AI systems that can understand, retrieve, and reason over complex legal texts. Compared to general NLP tasks, legal applications require precise reasoning under rigid formal logic, interpretive nuance, and jurisdiction-specific language. Thus, benchmark datasets are essential not only for comparing model performance but also for formalizing what constitutes legal competence in machine systems.
Before the rise of LLMs, legal NLP primarily relied on task-specific benchmarks such as CUAD, CaseHOLD, EUR-Lex, and COLIEE. These benchmarks typically focused on narrow sub-tasks like contract clause extraction, case conclusion classification, or statute retrieval. Models in this era were mainly based on SVMs, rule-based systems, or fine-tuned BERT variants. Evaluation was conducted using task-specific metrics like accuracy, precision, or recall, often without generalization testing or complex inference.
With the emergence of foundation models such as GPT-3, GPT-4, and their multilingual counterparts, the legal community began designing new benchmarks that match the scale and depth of LLM capabilities. These new benchmarks reflect a shift from task-isolated pipelines toward comprehensive, multi-task and multi-language evaluations emphasizing zero-shot, few-shot, and instruction-following reasoning.
Table 14.
Pre-LLM Era Benchmarks in Legal NLP
| Benchmark | Scope and Focus | Data Composition | Evaluation Tasks | Key Insights |
|---|---|---|---|---|
| CUAD [333] | Contract clause extraction and risk detection in legal documents | 13,000+ expert-annotated examples across 41 clause types, sourced from real commercial contracts | Clause identification, named entity recognition (NER), binary classification | Focused on practical contract review tasks; emphasized precision in extraction under legal ambiguity; widely used in contract AI |
| CaseHOLD [334] | Case law judgment understanding via conclusion prediction | 53,000+ U.S. appellate court case summaries with multiple-choice legal holdings | Multiple-choice question answering; outcome selection from candidates | Tests nuanced legal entailment and fact-to-holding inference; served as early benchmark for transformer-based legal models |
| EUR-Lex [335] | Multilabel legal topic classification for EU directives and regulations | 55,000+ European legal documents tagged with 3,956 EuroVoc labels | Multilabel text classification | One of the earliest and most cited legal NLP datasets; highly imbalanced and hierarchical label space inspired development of label-aware classifiers |
| SCOTUS [336] | Supreme Court decision classification and ideological alignment analysis | U.S. Supreme Court opinions, annotated with justice ideology, vote splits, and case topics | Binary and multiclass classification, ideological trend analysis | Used in political science and legal prediction; supported early quantitative legal studies using machine learning |
| COLIEE [337] | Legal information retrieval and entailment challenge (competition format) | Multiple years of formal tasks including Japanese Bar Exam questions and Canadian legal cases/statutes | IR, entailment classification, statute retrieval, legal QA | Serves as international benchmark challenge; evaluated both retrieval and inference under strict logic constraints |
Table 15 summarizes six representative legal benchmarks developed in the post-LLM era: LegalBench, LawBench, LexGLUE, LAiW, Swiss-Judgment-Prediction, and LJP-IV. These benchmarks collectively span English and Chinese legal systems, multiple legal traditions (common law, civil law), and a variety of task formats (classification, retrieval, generation, and reasoning). Each is designed to capture a particular aspect of legal intelligence, including factual recall, statutory alignment, interpretive inference, and multilingual adaptability.
LegalBench. LegalBench is one of the most fine-grained and widely used benchmarks for legal reasoning, introduced by the Hazy Research group at Stanford. It contains 162 subtasks across six categories of legal reasoning (rule recall, rule application, contextual interpretation, rhetorical analysis, etc.), with prompts written by legal experts. LegalBench emphasizes few-shot and zero-shot settings, testing whether models can generalize to complex legal logic without task-specific fine-tuning. Evaluation is primarily based on accuracy and exact match. Leaderboard results are presented in Table 16.
LawBench. LawBench evaluates Chinese legal models through 20 distinct tasks structured across three tiers: memory, comprehension, and application. It features over 90,000 QA pairs built from legal statutes and court rulings. It is designed to examine how well Chinese LLMs can perform legal retrieval, clause classification, and open-domain legal analysis. It serves as a counterpart to LegalBench in the Chinese legal environment.
LexGLUE. LexGLUE unifies seven English-language legal datasets—including ECtHR, EUR-Lex, and SCOTUS—into a benchmark suite for legal classification and judgment prediction. It supports both general-purpose and domain-pretrained models, providing a standardized platform for evaluating legal NLP systems. LexGLUE helped establish best practices for legal model development and evaluation in European and U.S. contexts.
LAiW. The LAiW benchmark targets typical Chinese legal tasks such as statute retrieval, judgment prediction, and article matching. Based on real-world Chinese judicial data, it simulates courtroom decision flows and legal reasoning chains. LAiW exposes domain adaptation gaps in general Chinese LLMs and highlights the need for legal domain alignment in foundation models.
Swiss-Judgment-Prediction. This benchmark addresses multilingual judgment prediction in Switzerland’s trilingual legal system (German, French, Italian). It consists of over 85,000 annotated cases and emphasizes robustness to temporal drift and cross-lingual inference. It enables the evaluation of legal AI systems in multi-jurisdictional settings where language and law evolve concurrently.
LJP-IV. LJP-IV introduces a third label—“innocent”—to the widely used Chinese legal judgment prediction datasets, enabling trichotomous classification (guilty, partially guilty, innocent). It emphasizes fine-grained reasoning and helps investigate fairness and model bias in criminal judgment prediction.
Evaluation tasks and metrics. Across both pre-LLM and post-LLM benchmarks, common legal NLP tasks include legal question answering (QA), statutory article retrieval, case outcome prediction, legal summarization, and precedent matching. Evaluation metrics vary by task: classification tasks use accuracy, F1, and exact match; generation tasks use BLEU, ROUGE, or GPTScore; retrieval tasks use recall@K or mean average precision (MAP). Notably, post-LLM benchmarks increasingly incorporate zero/few-shot reasoning settings and human evaluation of legal correctness and explainability.
LegalBench leaderboard. Among all benchmarks, LegalBench is the most prominent for leaderboard-based performance comparison. Table 16 reports the results of leading models such as Gemini 2.5 Pro Exp, GPT-4.1, and Grok 3. While Gemini Pro leads in accuracy, cost-effective models like Gemini Flash Preview deliver comparable results with lower latency and API cost. This suggests that highly competitive legal reasoning models can now be deployed efficiently at scale.
Summary. Compared to the pre-LLM era, where legal benchmarks targeted narrow tasks with limited scope and manually tuned models, the post-LLM era features broad, multi-faceted, and more cognitively demanding benchmarks. These new benchmarks reflect the increasing ambition to model real legal reasoning rather than isolated subtasks. Still, current benchmarks remain limited in areas such as procedural simulation, adversarial robustness, and cross-jurisdiction alignment. Bridging this gap will require new benchmarks that reflect practical legal workflows, account for legal change over time, and support real-time AI-human collaboration in high-stakes legal environments.
3.5.7. Discussion
Opportunities and Impact. LLMs are reshaping the landscape of legal practice by enhancing efficiency, accessibility, and interpretability across core tasks such as legal question answering, document drafting, case analysis, and judgment prediction. These models enable the automation of traditionally manual and expert-dependent processes, thereby democratizing access to legal knowledge and reducing costs associated with legal services. For instance, in legal consultant question answering, LLMs offer context-aware and scalable assistance, improving upon the limitations of rule-based systems [305,306]. In legal document drafting, LLMs support the generation of compliant, precise clauses through semantic understanding and template alignment [310,312]. Document understanding and case analysis are similarly enhanced, with models like LawLLM and GPT-4 enabling structured extraction of legal factors and empirical insights from complex legal texts [308,309]. Finally, in legal judgment prediction, models are beginning to capture the nuances of precedent, legal reasoning, and jurisdictional variation through frameworks such as PILOT and PLJP [328,329], offering significant utility in litigation planning and risk assessment.
Challenges and Limitations. Despite these promising developments, significant challenges remain. One key limitation is factual reliability: LLMs may hallucinate statutes or precedents if not coupled with robust retrieval mechanisms [306]. Moreover, the interpretability of model outputs—especially in high-stakes legal decisions—can hinder trust and accountability, particularly when models act as opaque black boxes [332]. Domain specificity also poses a critical barrier; general-purpose models often fail to capture jurisdictional nuances or evolving regulatory standards, requiring continual fine-tuning and dataset curation [311]. In drafting tasks, questions of legal liability arise when AI-generated text is ambiguous or misaligned with established legal doctrines, as highlighted by concerns surrounding prompt-based generation and doctrines like the parol evidence rule [313]. Furthermore, in judgment prediction, risks of reinforcing historical biases, legal inequities, or overfitting to precedent without understanding intent must be carefully managed [331,332].
Research Directions. To address these challenges and realize the full potential of LLMs in legal domains, several research directions merit emphasis:
- Retrieval-Augmented and Fact-Verified Generation. Integrating retrieval-augmented generation (RAG) systems that anchor responses in verifiable legal texts can reduce hallucination and improve accuracy [309].
- Jurisdiction-Aware and Temporal Modeling. Developing models sensitive to jurisdictional differences and evolving case law—such as time-aware frameworks like PILOT—can enhance the contextual reliability of legal predictions [328].
- Ethical and Regulatory Frameworks. Establishing governance standards for the deployment of LLMs in legal contexts—including audit trails, liability attribution, and responsible AI usage—will be essential to mitigate misuse and legal uncertainty [325].
Conclusion. LLMs are emerging as transformative tools in legal informatics, streamlining workflows, enhancing access to justice, and supporting complex legal reasoning. However, their successful deployment demands careful alignment with legal standards, domain expertise, and interpretability requirements. Rather than replacing legal professionals, LLMs function best as assistive technologies—amplifying legal insight, accelerating analysis, and fostering more equitable, data-informed legal systems.
4. LLMs for Economics and Business
In this chapter, we examine how LLMs are applied in economics and business domains. In particular, we review four disciplines—finance, economics, accounting, and marketing. In finance, we cover trading and investment research, corporate finance, market analytics, financial intermediation and risk management, sustainable finance, and fintech, and explain how these tools are evaluated. In economics, we consider behavioral and experimental studies, macroeconomic and agent-based simulation, strategic and game-theoretic interactions, and systems for economic reasoning and knowledge representation, with targeted assessments. In accounting section, we examine auditing, financial and managerial accounting, and taxation, together with benchmarking. In marketing section, we review consumer insight and behavior analysis, content creation and campaign design, and market-intelligence and trend analysis, with associated performance benchmarks.
4.1. Finance
4.1.1. Overview
Introduction. Finance is the study of how individuals, institutions, and governments acquire, spend and manage money and other financial resources over time under conditions of uncertainty [344,345]. It encompasses the mechanisms of asset valuation, the behavior of financial markets, and the strategic decision-making of economic agents under risk. In simpler terms, finance is about managing money—how people and organizations save, invest, borrow, and plan for the future while dealing with risks. Whether it’s a family budgeting for a home, a company raising capital, or a trader investing in the stock market, finance provides the principles and tools to make informed decisions.
Finance research covers a broad range of tasks, including asset pricing, portfolio optimization, risk management, financial forecasting, and corporate decision analysis. Traditionally, these tasks are approached using mathematical modeling, econometric techniques, statistical inference, and more recently, machine learning algorithms [346,347,348]. For example, regression models and time-series analysis are widely used for forecasting [349,350,351], while stochastic calculus and optimization methods are central to asset pricing and portfolio theory [352,353].
Traditional quantitative methods have been instrumental in advancing finance as a rigorous and impactful discipline. Groundbreaking models like the Capital Asset Pricing Model (CAPM) [354], Black-Scholes option pricing [355], and the Fama-French factor models [356,357]have not only deepened our understanding of market dynamics but also shaped financial regulation, investment strategies, and risk management practices. The integration of economics, statistics, and computation in finance has driven scientific innovation and enabled practical tools used daily by investors, policymakers, and financial institutions.
Despite recent advancements, finance research continues to face persistent and multifaceted challenges. One major hurdle is the explosion of unstructured data, including news articles, earnings call transcripts, regulatory filings, and social media posts [358]. These sources are rich in information but difficult to analyze systematically due to their variability in format, tone, and relevance. For example, assessing the market impact of a CEO’s offhand comment during an earnings call requires deep contextual understanding beyond traditional models.
Another key challenge is the manual workload involved in processing and analyzing financial data. Tasks like data labeling, report summarization, or sentiment annotation often rely on significant human effort, making them time-consuming, resource-intensive, and prone to error. This bottleneck can slow down decision-making and hinder scalability in fast-paced financial environments.
Finance also operates within a highly dynamic environment, where market conditions, policy landscapes, and investor behaviors change rapidly. Static models trained on historical data frequently fail to adapt to new patterns, such as those observed during financial crises or global events like the COVID-19 pandemic. This volatility complicates the development of robust, long-lasting analytical models.
Finally, interpretability remains a critical concern. Many high-performing models, especially those based on deep learning, function as “black boxes,” offering little transparency into how predictions are made. This lack of clarity can undermine trust in model outputs, particularly in high-stakes decisions such as credit approval, fraud detection, or investment recommendations, where explainability is essential for compliance and stakeholder confidence.
In this context, LLMs represent a significant technological shift. Their ability to process and reason over vast amounts of both structured and unstructured text data positions them as valuable tools for supporting and enhancing financial research [359].
Figure 10.
Overview of LLMs’ Applications in Finance Research.
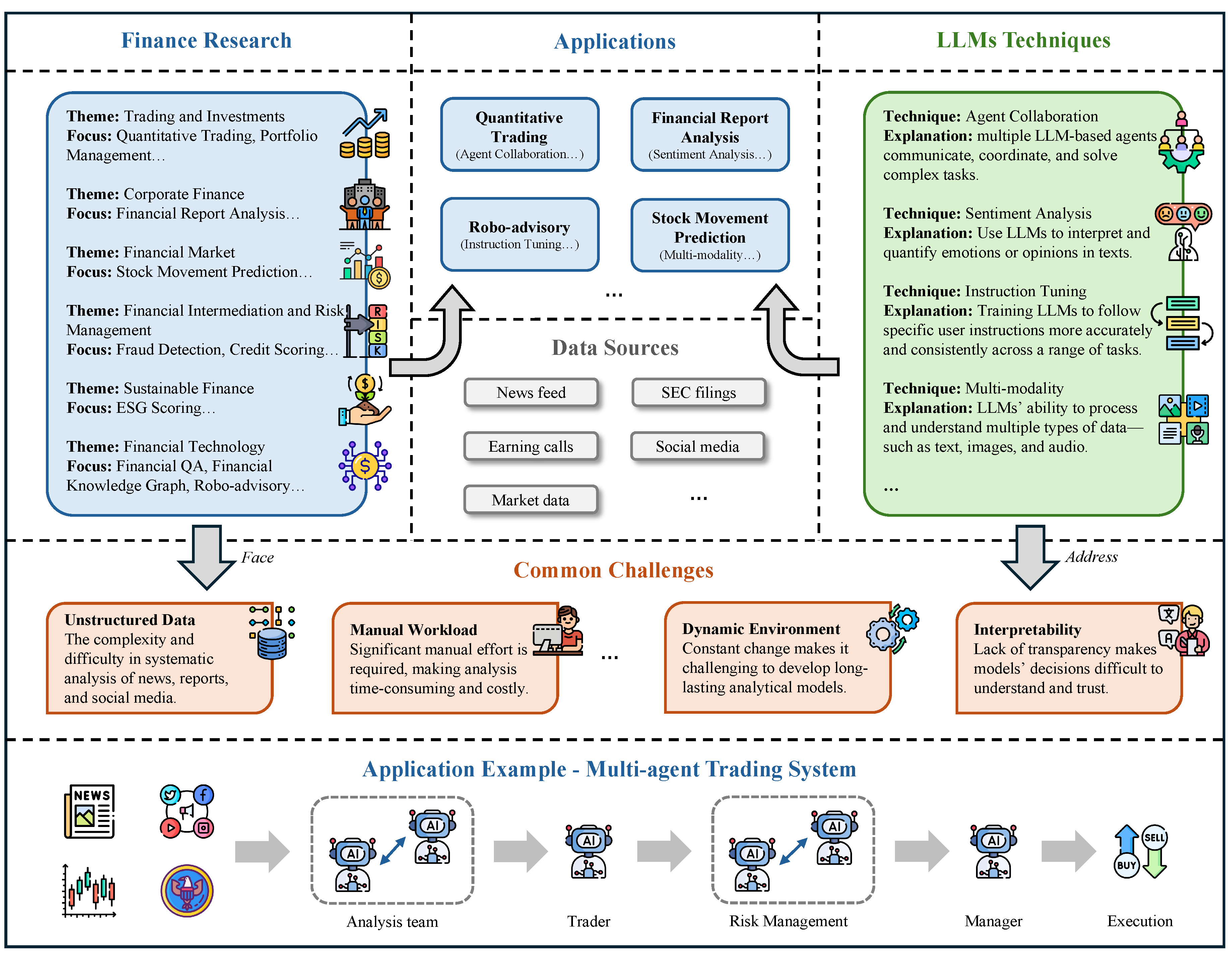
The Role of LLMs. However, integrating LLMs into financial research must be approached with caution. Certain problem areas remain beyond their current capabilities. Tasks that require precise numerical computation, high-frequency decision-making, or real-time financial modeling demand low-latency inference, quantitative rigor, and often, regulatory robustness—areas where traditional models still hold a clear advantage. That said, there are specific categories of research problems where LLMs are especially well-positioned to contribute. These include extracting structured information from unstructured financial documents [360], interpreting and generating textual financial reports [361], and answering complex domain-specific questions [362]. The strength of LLMs lies in their ability to process and synthesize large volumes of text, adapt to domain-specific jargon through fine-tuning or prompt engineering, and facilitate interactive exploration of financial knowledge [363]. As such, LLMs should be seen not as replacements for traditional tools, but as powerful complements—particularly in domains where language and knowledge representation play a central role.
Table 17.
Applications and insights of LLMs in finance research
| Field | Subfield | Key Insights and Contributions | Examples | Citations |
|---|---|---|---|---|
|
Trading and Investments |
Quantitative Trading | LLM-based trading agents demonstrate enhanced interpretability, adaptability, and profitability in simulating and executing financial strategies across diverse market conditions | FinCon [364]: Multi-agent LLM system improves trading via verbal risk reinforcement. FinMem [365]: Layered memory and character design enhance LLM-based trading decisions. | [364,365,366,367,368,369,370,371,372,373,374,375] |
| Portfolio Management | LLMs enhance financial decision-making by enabling adaptive, explainable, and multi-modal portfolio strategies through agent collaboration, sentiment reasoning, and dynamic alpha mining | Ko & Lee [376]: ChatGPT enhances portfolio diversification and asset selection across classes. Kou et al. [377]: LLM-driven agents mine multimodal alphas, dynamically adapt trading strategies. | [376,377,378,379,380,381,382,383] | |
| Corporate Finance | Financial Report Analysis | LLMs enhance financial report analysis by enabling accurate, explainable, and scalable extraction and generation through multimodal processing, domain-specific fine-tuning, and tool-augmented reasoning | XBRL-Agent [384]: LLM agent analyzes XBRL reports using retriever and calculator tools. | [384,385,386] |
| Financial Markets | Stock Movement Prediction | LLMs can effectively predict and explain stock movements by extracting sentiment, factors, and insights from financial text through self-reflection, instruction tuning, and domain-specific prompting | Koa et al. [387]: Self-reflective LLM explains stock predictions via reinforcement learning framework. Ni et al. [388]: QLoRA-fine-tuned LLM predicts stocks using rich earnings data. | [387,388,389,390,391,392] |
|
Financial Intermediation and Risk Management |
Fraud Detection | LLMs significantly enhance financial fraud detection by enabling accurate, scalable, and robust identification of anomalies and manipulations through prompt engineering, hybrid modeling, and adversarial benchmarking | Fraud-R1 [393]: Benchmark tests LLM defenses against multi-round fraud and phishing. RiskLabs [394]: LLM fuses multi-source data to forecast volatility and financial risk. | [393,394,395,396,397] |
| Credit Scoring | LLMs enhance credit risk assessment by improving prediction accuracy, generalization, and explainability through hybrid modeling, text integration, and domain-specific fine-tuning | CALM [398]: LLM scores credit risk across tasks with fairness checks. LGP [399]: Prompted LLMs use Bayesian logic to generate insightful risk reports. | [398,399,400,401] | |
| Sustainable Finance | ESG Scoring | Leverage LLMs for classification, rule learning, data extraction, greenwashing detection, readability assessment, and multi-lingual understanding, thereby enhancing transparency and decision-making in sustainable finance. | ESGReveal [402]: Leverage LLMs and RAG to systematically extract and analyze ESG data from corporate reports. | [170,402,403,404,405,406,407,408,409,410] |
|
Finance Technology |
Financial Question Answering | LLMs enhance financial QA in accurate, context-aware reasoning over complex, multi-source financial data. | TAT-QA [411]: a Financial QA benchmark that contains over 16,000 questions built from real-world financial reports that combines tabular and textual data. | [411,412,413,414,415,416,417,417] |
| Knowledge Graph Construction | LLMs enable automated KG construction from financial data, retrieval from KG and support multi-document financial QA. | FinKG [418]: A curated core financial knowledge graph built from authoritative sources like corporate reports and stock data, structured to enable systematic analysis and applications in financial forecasting, risk assessment, and decision-making through semantically rich relationships between entities. | [417,418,419,420,421,422] | |
| Robo-advisory | LLMs enhance robo-advisors for novice investors, but still lag behind humans in performance and trust. | Jung et al [423]: A earlier work that propose the concept "Robo-Advisory" that leverage AI to provide automatic financial advisory servies for a broader range of investors. | [404,423,424,425,426,427,426] |
Taxonomy. To understand the potential application of LLMs in finance research, we propose a taxonomy that reflects the diversity of tasks across the field:
- Trading and Investments. Trading pursues short-term gains while investment emphasizes long-term value through diversification and analysis. Traditional methods struggle with large-scale, complex data, whereas LLMs offer new capabilities for processing unstructured information, enhancing forecasting, and supporting strategies in quantitative trading and portfolio management.
- Corporate Finance. Corporate finance manages funding, capital structure, and investment to drive growth. Conventional approaches like financial modeling and discounted cash flow analysis are labor-intensive and limited under fast-changing conditions. LLMs streamline tasks such as financial report analysis, improving efficiency and accuracy in strategic decision-making.
- Financial Markets. Financial markets allocate resources and manage risk through the trading of assets. While econometric models and machine learning aid analysis, they face challenges with today’s data scale and complexity. LLMs advance this field by processing unstructured information and enabling applications such as stock movement prediction.
- Financial Intermediation and Risk Management. Banks and insurers channel capital while managing risks, but traditional statistical models and manual processes lag in dynamic environments. LLMs improve performance by analyzing diverse datasets, with emerging applications in fraud detection and credit scoring.
- Sustainable Finance. Sustainable finance incorporates ESG factors into investment decisions. Standard scoring systems often overlook rich unstructured data from reports and media. LLMs can extract and synthesize such information, offering more context-aware and adaptive ESG insights.
- Financial Technology. FinTech reshapes financial services through innovations like digital banking, blockchain, and robo-advisory. Traditional solutions emphasize automation but lack flexibility. LLMs expand FinTech by powering financial question answering, knowledge graph construction, and conversational advisory, enhancing personalization and accessibility.
Each of these areas presents distinct opportunities for LLMs to enhance or extend traditional approaches. For instance, In Financial Report Analysis, LLMs can interpret complex narratives in earnings reports, extract key metrics or risk factors, and even flag inconsistencies or anomalies that may be missed by rule-based systems [384]. In ESG Scoring, LLMs can analyze qualitative disclosures across environmental, social, and governance dimensions, enabling more comprehensive and up-to-date assessments that incorporate nuanced language cues and public sentiment [428].
Across these use cases, LLMs offer new forms of insight, not by replacing models that price assets or manage risk, but by bridging the gap between language and data, enabling better contextual awareness, more transparent decision-making, and broader access to financial knowledge.
4.1.2. Trading and Investment
Trading and investment constitute the bedrock of capital markets, encompassing the strategic allocation of financial resources to generate returns and manage risk. From a financial perspective, trading often focuses on short-term opportunities, driven by price volatility, liquidity dynamics, and market microstructure [429], while investment emphasizes long-term value creation through fundamental analysis, asset diversification, and portfolio optimization [430]. These activities, though distinct in horizon and methodology, share a common goal: the maximization of expected utility under uncertainty.
Historically, trading and investment decisions have relied heavily on traditional methodologies, such as fundamental analysis—assessing the intrinsic value of assets based on economic indicators, company financials, and industry conditions—and technical analysis, which examines historical price patterns and market trends [431,432]. However, these conventional methods face notable limitations, including the challenge of systematically processing vast amounts of information, susceptibility to subjective bias, and difficulty in accurately interpreting complex, unstructured data.
Recent advancements in LLMs have provided promising tools to overcome these limitations. By capturing and analyzing nuanced sentiments and patterns hidden within structured and unstructured massive textual data, these advanced models can offer improved insights, enhanced forecasting capabilities, and more informed decision-making processes for trading and investment professionals. Two notable applications in this evolving landscape include Quantitative Trading and Portfolio Management, which are further discussed below:
Quantitative Trading. Quantitative trading involves the use of mathematical models and algorithms to identify and exploit trading opportunities [433]. LLMs can be used to incorporate textual data (e.g., news sentiment, social media, analyst reports) into predictive models, enhancing signal generation for algorithmic trading strategies.
Recent research in LLM-driven financial trading agents has advanced across several interconnected fronts, notably in memory architecture, agent individuality, multimodal intelligence, and simulation environments. A foundational trend is the emergence of LLM agents equipped with layered memory and character design, as seen in TradingGPT [374], FinMem [365], and FinCon [364]. These systems introduce cognitive structures mimicking human memory stratification (short, medium, and long-term) while embedding distinctive agent personalities. This not only improves interpretability and decision diversity but also enhances adaptability in volatile markets.
Building on the concept of self-improvement, QuantAgent [373] proposes a dual-loop learning system that evolves its domain-specific financial knowledge through iterative simulation and real-world feedback. Similarly, FS-ReasoningAgent [366] innovatively segments reasoning into factual and subjective channels to optimize cryptocurrency trades, revealing nuanced insights into LLM reasoning preferences under different market conditions.
A parallel direction emphasizes realistic multi-agent financial ecosystems. TradingAgents [375] introduces a hierarchical LLM-agent framework that mirrors the structure of real-world trading firms, organizing agents into specialized roles (e.g., fundamental analysts, sentiment analysts, traders, risk managers). Through structured inter-agent communication and decision protocols, it fosters collaborative, debate-driven decision-making, demonstrating superior cumulative returns and Sharpe ratios in simulation. Complementarily, FinCon [364] proposes a synthesized manager-analyst hierarchy with dual-level risk control—one handling within-episode volatility via CVaR alerts, and another guiding strategic belief updates across episodes via conceptual verbal reinforcement—achieving strong performance in both single-stock and portfolio trading tasks.
Meanwhile, simulation-based evaluation of trader behavior remains an active area. ASFM [371] and StockAgent [368] construct rich multi-agent environments with LLMs as trader proxies, allowing researchers to analyze policy impacts and behavioral biases in realistic, yet controlled, financial ecosystems. These studies highlight the use of LLMs not only as traders but as tools to deepen our understanding of market dynamics and agent interactions.
In parallel, LLMs’ capacity for sentiment-driven trading has also been substantiated [372], where models like GPT-3 significantly outperform traditional sentiment dictionaries in predicting returns, achieving strong Sharpe ratios and cumulative profits. MarketSenseAI [367] and FinAgent [369] further extend this line with multimodal inputs and tool-augmented architectures, synthesizing text, charts, and fundamentals to simulate generalist financial agents with impressive profitability and reasoning transparency.
Collectively, these works mark a shift toward intelligent, explainable, and increasingly autonomous AI trading systems grounded in human-aligned cognition, hierarchical coordination, and complex environmental simulations.
Portfolio Management. Portfolio management involves strategically selecting and overseeing a set of financial assets, such as stocks, bonds, commodities, currencies, and cryptocurrencies, to meet specified investment objectives [434]. LLMs can analyze large volumes of qualitative and quantitative data, interpret market news and sentiments, forecast trends, and assist in strategic decision-making.
The application of LLMs in portfolio and trading strategy research has evolved rapidly, branching into several thematic directions. A foundational area centers on strategy generation and alpha mining. Alpha-GPT [383] introduces a human-AI interactive paradigm for alpha discovery, empowering quants to translate intuitive trading ideas into formulaic signals via natural language interfaces. Similarly, Kou et al. [377] extends this concept through a multi-agent and multimodal framework that dynamically evaluates market conditions and adapts trading strategies accordingly.
A second important line of work investigates portfolio construction and management via LLMs. Studies like Ko and Lee [376] and Abe et al. [379] explore how ChatGPT and persona-based ensembles enhance asset selection and diversification. These systems often outperform random or traditional strategies, particularly during specific market conditions such as rising inflation or high volatility. Gu et al. [381] takes this further by introducing a margin trading model that adaptively reallocates between long and short positions using an LLM-RL hybrid system, combining real-time reasoning and transparency for better risk management.
Parallel to these efforts, researchers have focused on news-driven sentiment analysis and reinforcement learning (RL). Wu [382] and Unnikrishnan [380] demonstrate how LLMs can extract actionable sentiment from financial news, significantly improving the performance of RL agents when managing portfolios or single-stock trading. These works confirm that LLM-enhanced strategies outperform baseline RL models and even historical benchmarks under various market scenarios.
Lastly, in the domain of crypto portfolio management, Luo et al. [378] proposes an LLM-powered multi-agent system that integrates multimodal data and collaborative reasoning across expert agents. This approach captures the complex nature of digital assets, showing strong performance in both asset selection and portfolio return across leading cryptocurrencies.
Together, these studies illustrate a shift from traditional, rule-based approaches to more adaptive, interpretable, and human-aligned systems powered by LLMs. They mark a significant leap in integrating advanced language technologies into financial decision-making and portfolio optimization.
4.1.3. Corporate Finance
Corporate finance is formally defined as the area of finance that focuses on how corporations handle their sources of funding, capital structuring, and investment decisions [345,435]. In simpler terms, corporate finance is about how companies manage their money, deciding where to get funds, how to invest them effectively, and how to ensure the company grows profitably and sustainably over time.
Traditionally, corporate finance relies heavily on methods such as financial modeling, discounted cash flow analysis [436], capital budgeting techniques [437], and sensitivity analysis. These conventional approaches, though proven effective, often face critical limitations—they tend to be labor-intensive, heavily reliant on manual computations, susceptible to human error [438], and sometimes inflexible when dealing with rapidly changing market conditions or complex financial scenarios.
In recent years, LLMs have emerged as powerful tools capable of addressing these limitations. LLMs have the potential to automate and enhance many corporate finance tasks, such as investment analysis, merge and acquisition (M&A) forecasting and insolvency forecasting, enabling more accurate, insightful, and efficient financial management [439]. Among these applications, one particularly promising area is financial report analysis, which plays a critical role in informing corporate financial strategies. Specifically, we discuss the recent progress in financial report analysis in the following section.
Financial Report Analysis. Financial report analysis is crucial for understanding corporate performance and guiding investment decisions. Traditionally involving labor-intensive methods like ratio and trend analysis, it has increasingly benefited from automation via LLMs, which excel at extracting, summarizing, and interpreting complex financial information.
Recent studies highlight various applications of LLMs in financial analysis. Han et al. [384] demonstrated improvements in eXtensible Business Reporting Language (XBRL) analysis by integrating retrieval-augmented generation and specialized calculation tools, while Le [385] illustrated the effectiveness of financially fine-tuned LLMs for earnings report generation. Ziegler [386] underscored the superior accuracy of multimodal approaches for ESG data extraction. Moving forward, key research opportunities include advancing multimodal integration, refining domain-specific fine-tuning strategies, enhancing interpretability for robust decision-making, and developing comprehensive platforms that seamlessly integrate LLM-based analyses into financial workflows.
4.1.4. Financial Market Analysis
A financial market can be formally defined as a structured marketplace where individuals, institutions, and governments engage in the buying and selling of financial instruments such as stocks, bonds, commodities, derivatives, and currencies [440]. It serves as a pivotal platform that facilitates efficient allocation of resources, liquidity management, and risk distribution, underpinning economic growth and financial stability on a global scale [441,442]. In simpler terms, financial markets function similarly to any marketplace, like a grocery store or an auction, but instead of groceries or antiques, they deal with assets like shares of companies, government bonds, or commodities such as gold and oil. Buyers and sellers come together to trade these financial assets, each seeking profit, stability, or a way to mitigate their risks.
Historically, traditional methods employed within financial markets have revolved primarily around statistical and econometric models [443], fundamental analysis [444], and machine learning algorithms [445]. While these approaches have served as the foundation for investment decisions for decades, they possess inherent limitations, particularly concerning their predictive accuracy, adaptability, and capacity to handle vast, complex, and unstructured datasets prevalent in today’s fast-moving digital financial landscape [446,447].
In recent years, advances in LLMs present promising solutions to address many limitations encountered by traditional methodologies. By leveraging their remarkable abilities to process extensive amounts of textual and numerical data, understand contextual nuances, and discern patterns, LLMs offer powerful tools to interpret financial data more accurately, effectively enhancing decision-making processes in the financial industry [448]. One notable application of these models, Stock Movement Prediction, are attracting considerable interest, and will be discussed in subsequent section.
Stock Movement Prediction. Stock movement prediction involves forecasting the direction or magnitude of stock price changes, which is a pivotal task for investors and financial analysts [449]. With recent advancements, LLMs have shown substantial promise in this domain by leveraging vast amounts of unstructured textual data, such as financial news, earnings reports, and investor sentiment, which traditional numerical models might overlook or fail to adequately analyze.
Recent research has seen a surge of interest in leveraging LLMs for stock movement prediction, evolving from simple sentiment analysis to sophisticated, explainable, and data-integrated forecasting frameworks. A foundational line of work demonstrates that LLMs can extract economically meaningful sentiment signals from financial headlines. For example, Lopez-Lira and Tang [389] show that ChatGPT can predict short-term stock returns from news headlines without explicit financial fine-tuning, outperforming traditional methods and revealing that model size correlates with economic efficacy. Extending this, Zhang et al. [392] explore LLMs in the context of Chinese financial texts, comparing different LLM architectures for sentiment factor extraction and establishing a standardized pipeline for backtesting trading strategies derived from Chinese news sentiment. Similarly, Bhat and Jain [390] focus on distilled LLMs to perform emotion analysis on headlines, showing that emotion-labeled news can predict stock trends as reliably as traditional financial indicators.
Moving beyond raw sentiment, several works emphasize explainability and structured prediction. Koa et al. [387] introduce the Summarize-Explain-Predict (SEP) framework, where a self-reflective LLM iteratively trains itself to generate human-readable justifications for stock predictions, eliminating the need for expert-annotated data and offering robust performance in both classification and portfolio tasks. Wang et al. [391] propose LLMFactor, which extracts interpretable economic “factors” from news via prompt engineering (Sequential Knowledge-Guided Prompting), significantly improving explainability and aligning predictions with market-relevant insights. Meanwhile, Ni et al. [388] tailor LLMs for earnings report analysis using a QLoRA-enhanced model that integrates both firm-specific financials and external macro factors, outperforming GPT-4 in predictive accuracy.
Together, these studies reflect a broader transition in the field: from sentiment extraction as a proxy for return signals to multimodal, interpretable, and fine-tuned approaches that push the boundaries of AI-enabled financial forecasting. They underscore the growing ability of LLMs to not only predict stock movement but also to articulate why and how such predictions are made, thereby enhancing transparency and trust in AI-driven investment decisions.
4.1.5. Financial Intermediation and Risk Management
Financial Intermediation and Risk Management refers to the systematic process through which financial institutions, such as banks, insurance companies, and investment firms, facilitate the efficient allocation of capital by channeling funds from savers to borrowers, while simultaneously managing the inherent financial risks associated with such transactions [450]. In simpler terms, financial intermediation involves institutions acting as middlemen who pool money from individuals and businesses with surplus funds, and then lend or invest those funds in activities that need financing. Risk management involves strategies that these institutions use to identify, assess, and control risks, ensuring the stability and health of the financial system.
Traditionally, financial intermediation and risk management have relied heavily on statistical methods, manual reviews, and rule-based systems [451,452]. Institutions typically used historical data analysis, regulatory frameworks, and expert judgment to make lending and investment decisions, as well as to evaluate and mitigate risks [453]. However, these traditional methods have notable limitations, including their reliance on historical data, which may not effectively capture rapidly changing market dynamics or unexpected events. Additionally, manual processes can be time-consuming, error-prone, and inefficient when handling large volumes of data, thus limiting the ability of financial institutions to respond quickly and effectively to new financial risks.
Advances in LLMs have the potential to significantly enhance financial intermediation and risk management practices. LLMs can process vast amounts of structured and unstructured data efficiently, while offer novel capabilities in analyzing textual data, interpreting complex patterns, and providing actionable insights, thus enabling institutions to manage risk more proactively and dynamically. Two important applications where LLMs show promise are Fraud Detection and Credit Scoring, which will be introduced in the following sections.
Fraud Detection. Fraud detection is essential for safeguarding financial institutions from illicit activities that result in significant financial losses and damage stakeholder trust [454]. LLMs offer powerful capabilities for enhancing fraud detection, as they can efficiently process vast volumes of textual and numerical data, detect subtle anomalies, and recognize complex patterns indicative of fraudulent behavior. Leveraging LLMs enables financial institutions to improve accuracy, reduce detection time, and swiftly adapt to emerging threats.
Recent advancements illustrate the potential of LLMs across diverse financial fraud scenarios. Notably, Bakumenko et al. [396] present a compelling methodology that leverages sentence-transformer embeddings to encode non-semantic categorical features in financial journal entries. This approach effectively mitigates feature sparsity and heterogeneity, yielding substantial performance gains across multiple classification models. Their results suggest that LLM-based embeddings capture latent structures traditional encodings overlook, marking a pivotal step in audit-grade anomaly detection.
Complementing this, Korkanti [395] integrates customized LLMs with state-of-the-art predictive analytics and anomaly detection techniques, producing a robust framework that significantly boosts both precision and recall. The model demonstrates heightened sensitivity to subtle, high-risk indicators within real-time transactional and communication datasets, addressing key gaps in adaptability and responsiveness prevalent in existing systems.
Meanwhile, studies like Boskou et al. [397] and Cao et al. [394] affirm the broader versatility of LLMs. The former employs prompt-engineered interactions with ChatGPT-4 to identify deception in corporate disclosures, yielding moderately successful classification scores without fine-tuning. The latter, through a multi-modal fusion of earnings calls, time-series data, and news content, shows promise for general financial risk prediction, though its direct fraud-detection utility remains underexplored.
On the benchmarking front, Yang et al. [393] introduce the Fraud-R1 dataset—an extensive, multilingual, multi-round benchmark designed to rigorously assess LLM robustness against realistic phishing and fraud scenarios. Their results expose persistent weaknesses in role-play and cross-lingual settings, emphasizing the need for multilingual, adaptive, and context-aware models.
In sum, LLMs are reshaping the fraud detection landscape by enabling deeper contextual comprehension and cross-modal reasoning. Future efforts should focus on real-time multilingual capabilities, explainability, and adversarial robustness to ensure deployment-ready solutions in dynamic financial environments.
Credit Scoring. Credit scoring is a critical tool in financial risk management used by financial institutions to assess the likelihood that individuals or entities will fulfill their credit obligations. Traditionally, it relies on statistical methods such as logistic regression and decision trees [455]. However, recent advancements of LLMs offer the potential to enhance predictive accuracy and provide deeper contextual understanding through textual data analysis. LLMs can complement traditional numeric risk indicators, improving both predictive performance and interpretability.
The integration of LLMs into credit risk assessment has opened new avenues for enhancing both predictive performance and interpretability across diverse financial settings. One line of research explores LLMs as generalist credit scoring tools, exemplified by Feng et al.’s CALM model [398], which leverages instruction tuning across nine datasets to create a versatile and benchmarked LLM for credit and risk assessment. This approach reveals LLMs’ promise in democratizing access to sophisticated scoring tools while also highlighting concerns around fairness and potential bias.
Another core stream investigates LLMs for hybrid modeling and interpretability enhancement. Teixeira et al. [399] introduce Labeled Guide Prompting (LGP), which augments GPT-4 outputs with Bayesian network reasoning and labeled examples to generate credit reports preferred by human analysts, blending human-like judgment with machine consistency.
Several studies address LLMs in specialized or constrained financial environments. Sanz-Guerrero and Arroyo [400] apply a fine-tuned BERT model to peer-to-peer (P2P) lending platforms, where traditional financial variables are sparse. By extracting risk signals from loan narratives, they show that LLM-derived scores can improve prediction and reshape how credit models weigh traditional features, especially across different loan purposes. Similarly, Drinkall et al. [401] conduct a critical evaluation of generative LLMs in corporate credit rating prediction, finding that despite strong textual encoding, models like GPT underperform compared to multimodal baselines like XGBoost when numerical reasoning is essential.
Collectively, these works illustrate a maturing field where LLMs are not merely augmenting legacy models, but increasingly redefining how creditworthiness is evaluated—particularly in terms of generalization, explainability, and ethical oversight. They point to a future in which credit assessment becomes more adaptive, inclusive, and transparent through the responsible deployment of AI.
4.1.6. Sustainable Finance
The integration of Environmental, Social, and Governance (ESG) factors into investment decisions has gained significant traction in recent years. Traditional ESG scoring methodologies often rely on structured data and predefined metrics. However, the increasing availability of unstructured textual data related to ESG, such as news articles and company reports, offers a rich source of information that LLMs are well-suited to process. Recent research [404] indicates that LLMs can potentially contribute to ESG scoring by learning implicit evaluation criteria directly from text. Moreover, several studies [405,406,407] have explored the use of LLMs to extract structured information from sustainability reports, which can then be employed for ESG-relevant classification tasks [408,409]. Beyond information extraction and classification, the concept of using LLMs as evaluators in ESG scoring is also gaining attention [402]. A recent survey [170] mentions the promising applications of the "LLM-as-a-judge" paradigm in various financial domains, including ESG scoring. This suggests a future where LLMs could synthesize information from diverse textual sources and make an overall judgment on a company’s ESG performance, potentially mimicking the role of human ESG analysts. This could lead to more dynamic and context-aware scoring mechanisms that go beyond traditional data aggregation methods. However, it is important to note that research has also highlighted the potential brittleness of LLMs when processing long contexts, such as large sustainability reports, which could impact their reliability in ESG scoring tasks [410].
4.1.7. Financial Technology
Financial Technology (Fintech) refers to the integration of technology into the offerings of financial services companies to improve their use and delivery to consumers [456]. FinTech has implications not merely for the financial industry, and includes virtually all forms of e-commerce and spreads to other industry fields, especially in blockchain technology, such as insurance, banking services, trading on capital markets, and risk management [457].
Financial QA. The capability to quickly and accurately access financial information is crucial in the fast-paced world of finance. LLMs are emerging as powerful tools to build sophisticated financial question answering systems. Studies in this topic aim to explore the capabilities of LLMs to understand and respond to a wide range of financial queries. For instance, FinCausal 2025 shared task [412] proposes a task specifically focused on the ability of LLMs to detect causal relationships within financial texts through question answering. The study evaluates both LLMs and discriminative approaches, with the findings indicating the strong potential of generative LLMs, particularly in scenarios where only a few examples are available for learning. This suggests that LLMs can effectively reason about financial causality even with limited task-specific training data.
Recognizing the need for high-quality data to train and evaluate these systems, researchers have curated specialized datasets. SocialFinanceQA [413] introduces a benchmark dataset, comprising a vast collection of financial questions and answers extracted from Reddit’s finance-focused communities. The rationale behind this is that these real-world discussions reflect the actual language and types of questions that individuals have about finance, providing a valuable resource for fine-tuning and aligning LLMs to better serve user needs in this domain. A recent survey FinLLMs [458] also highlights financial QA as a primary benchmark task, and lists several benchmark datasets for financial QA task evaluation.
The practical application of LLMs in financial question answering is already evident in consumer-facing platforms. The Amplework blog [427] highlights NerdWallet as an example of a platform that utilizes AI to provide users with personalized answers to their financial questions related to investing, personal finance, and debt management in real-time. This demonstrates the ability of LLMs to make complex financial information more accessible and understandable to a wider audience. Furthermore, another article [414] mentions the testing of LLMs on CPA exam preparation materials. LLMs, such as ChatGPT-4, demonstrate strong capabilities in business analysis and reporting automation, which inherently involves the ability for complex financial QA.
Financial Knowledge Graph. Financial data is characterized by intricate relationships between various entities, including companies, financial instruments, market indicators, and economic events. Knowledge graphs offer a powerful way to represent these connections in a structured format, enabling more sophisticated analysis and information retrieval. LLMs are proving instrumental in automating the construction of these financial knowledge graphs from the vast amounts of unstructured textual data available [417]. For example, LLM Knowledge Graph Builder [419] allows users to transform unstructured data, including financial documents, into dynamic knowledge graphs without requiring specialized coding skills. It leverages LLMs to automatically identify and extract key entities and the relationships between them, subsequently converting this information into a graph structure that can be stored and queried efficiently.
This blog [420] further elaborates on the synergy between LLMs and knowledge graphs. LLMs excel at tasks like Named Entity Recognition (NER) and relationship extraction from unstructured text, which are fundamental steps in building knowledge graphs. By understanding the nuances of language and the context in which financial entities are mentioned, LLMs can accurately identify these entities and the connections between them, automatically populating and enriching financial knowledge graphs.
A practical application of this technology is detailed in the article [421], which provides a step-by-step guide on using the Neo4j LLM Knowledge Graph Builder to transform financial statements into knowledge graphs. This process involves uploading financial data, extracting key financial entities and their relationships (such as the relationship between revenue, expenses, and net income), and then querying the resulting knowledge graph using natural language via GraphRAG [422]. This allows financial analysts to gain deeper insights from financial statements more intuitively and efficiently.
While the examples above focus on specific tools and platforms, the underlying principles are broadly applicable. Furthermore, research has shown that knowledge graphs constructed by LLMs can enhance the performance of financial question answering systems. KG-RAG [417] uses knowledge graph triples (constructed using a fine-tuned small language model) as context for multi-document financial question answering, achieving better results than traditional RAG methods.
Robo-advisory. Robo-advisors have become a popular way for individuals to manage their investments through automated platforms that offer financial planning and investment management services [424]. Integrating LLMs into these platforms has the potential to significantly enhance their capabilities, making them more personalized, interactive, and effective.
Akira.ai [425] discusses in detail how AI agents powered by LLMs are transforming financial robo-advisory. These agents can engage in more natural and human-like conversations with clients, understand their financial goals and risk tolerance expressed in natural language, and provide highly personalized investment recommendations. Moreover, LLMs can analyze vast amounts of real-time market data to provide timely insights and support portfolio adjustments, and they can even assist with tasks like tax optimization by identifying tax-efficient investment strategies.
Several leading robo-advisor platforms are already leveraging AI models, including LLMs, to enhance their services. This blog [427] highlights platforms like Betterment and Wealthfront, which use AI-driven algorithms to offer personalized investment portfolios tailored to users’ financial situations and goals. These systems continuously monitor market conditions and rebalance portfolios as needed. LLMs also contribute to real-time market analysis, sentiment detection, and the automation of savings and investment strategies on these platforms. Additionally, robo-advisors like Ellevest use AI-powered tools to offer personalized financial education alongside investment management, catering to users’ specific financial literacy levels.
A fwe studies also indicate the broader potential of LLMs in various tasks relevant to robo-advisory. A recent survey [404] notes the increasing use of LLMs in finance for automating financial report generation, forecasting market trends, analyzing investor sentiment, and offering personalized financial advice. These capabilities can be directly integrated into robo-advisory platforms to provide more comprehensive and sophisticated services. While this article [426] emphasizes that human advisors still hold an edge in terms of personalization, it acknowledges the increasing sophistication of robo-advisors powered by AI and the use of techniques like retrieval-augmented generation (RAG) to improve their ability to provide relevant and accurate advice.
4.1.8. Benchmarks
Recent work, such as the FinLLMs survey [458], has thoroughly summarized the existing financial benchmarks and datasets for specific tasks. To provide a more intuitive understanding of the performance improvements brought by LLM-based methods over traditional approaches across various financial tasks, we further compile a comprehensive comparison, as shown in Table 18. However, with the rapid emergence of LLMs and autonomous agents in financial applications, existing benchmarks are increasingly inadequate for evaluating model performance. On one hand, the unified and powerful language capabilities of LLMs, combined with their extensive knowledge base, enable them to tackle a wide range of financial tasks, necessitating a more comprehensive assessment of their overall competencies in finance. On the other hand, as LLMs continue to advance, there is growing interest in deploying them in practical financial scenarios rather than isolated, idealized experimental settings. Therefore, this section addresses these two critical aspects: first, we introduce recent multi-perspective, multi-task benchmarks designed to evaluate LLMs’ comprehensive abilities in finance; second, we discuss how to construct datasets that better reflect real-world conditions using publicly available data sources.
Multi-Task Benchmarks
Table 19.
Summary of Multi-Task Financial Benchmarks for Evaluating Large Language Models
| Benchmark | Language | Size | Feature | Insights on LLMs |
|---|---|---|---|---|
| FinBen | English | 36 datasets | Broadest task range; includes forecasting and agent evaluations | LLMs perform best on IE/textual analysis, poorly on forecasting and reasoning-heavy tasks |
| R-Judge | English | 569 records | Multi-turn safety judgment for agents in real scenarios | LLMs lack behavioral safety judgment in interactive settings; fine-tuning helps significantly |
| FinEval | Chinese | 8,351 Qs | Covers academic, industry, security, and agent reasoning tasks | LLMs outperform average individuals but lag behind experts; complex reasoning and tool usage still weak |
| CFinBench | Chinese | 99,100 Qs | Career-aligned categories; diverse questions and rigorous filtering | Highlights knowledge gaps; current LLMs struggle with practical depth and legal reasoning |
| UCFE | English, Chinese | 330 data points | User-role simulation; dynamic multi-turn tasks | Human-like evaluations show LLMs align with users but fall short under dynamic, evolving needs |
| Hirano | Japanese | — | Domain-specific benchmark in Japanese | Domain-specific LLMs still underdeveloped in Japanese finance |
FinBen. FinBen [465] comprises 36 datasets covering 24 tasks across seven critical financial dimensions: information extraction, textual analysis, question answering, text generation, risk management, forecasting, and decision-making. Its key innovations include the introduction of stock trading evaluation, agent-based and retrieval-augmented generation (RAG) assessments, and the development of novel datasets specifically designed for summarization, regulatory QA, and trading. Evaluations of prominent LLMs reveal that while current models excel at tasks such as information extraction and sentiment analysis, they still struggle with complex reasoning and domain-specific challenges, particularly in forecasting and decision-making. These findings underscore the potential and current limitations of LLMs in financial applications.
R-Judge. R-Judge [466] is a comprehensive benchmark designed to assess the risk awareness of LLMs in agent-based environments, particularly focusing on their ability to detect and judge safety risks arising from multi-turn interactions. The dataset consists of 569 carefully curated agent interaction records, spanning 27 real-world scenarios across five application categories—programming, IoT, software, web, and finance—and covering ten distinct risk types, including financial loss, privacy leakage, and property damage. Within the financial domain, R-Judge introduces scenarios where LLM agents must make decisions that could lead to monetary losses or regulatory violations, thereby providing a practical testbed for evaluating LLMs’ judgment under real-world financial constraints. Experimental results reveal substantial performance gaps: most models perform at or below random baselines, struggling especially with the financial and high-risk categories. These findings highlight the significant challenges in equipping LLMs with robust safety reasoning and underscore the importance of domain-specific fine-tuning and richer contextual understanding for deploying LLMs in sensitive financial environments.
FinEval. FinEval [467] is a comprehensive benchmark designed to evaluate LLMs in the Chinese financial domain across both foundational knowledge and real-world application scenarios. The benchmark consists of 8,351 questions spanning four core areas: Financial Academic Knowledge, Financial Industry Knowledge, Financial Security Knowledge, and Financial Agent. It supports multiple evaluation settings—zero-shot, few-shot, and chain-of-thought prompting—and includes objective, subjective, and open-ended formats. Experimental results show that while models perform competitively in financial security and basic knowledge tasks, their performance drops significantly on agent tasks requiring dynamic planning and reasoning. These results underscore the current limitations of LLMs in simulating expert-level financial behavior and highlight the need for further advancement in domain adaptation and task complexity handling.
CFinBench. CFinBench [468] is the most comprehensive Chinese financial benchmark to date. It consists of 99,100 questions spanning 43 second-level categories across four core dimensions aligned with real-world financial career progression: Financial Subject, Financial Qualification, Financial Practice, and Financial Law. These categories test LLMs’ mastery of theoretical foundations (e.g., economics, auditing), professional certification knowledge (e.g., CPA, securities), applied job skills (e.g., tax consulting, asset appraisal), and legal compliance (e.g., banking and commercial law). The benchmark includes three question types (single-choice, multiple-choice, and judgment), enabling diverse and realistic assessment formats. Evaluations show that the best accuracy remains just over 60%, indicating substantial challenges and room for improvement in financial domain adaptation.
UCFE. UCFE [469] presents a user-centric financial expertise benchmark designed to evaluate LLMs in real-world financial scenarios through dynamic, multi-turn user interactions. It introduces 17 task types, encompassing both zero-shot and few-shot formats across domains such as stock prediction, risk assessment, financial consulting, and regulatory compliance. Grounded in a large-scale user study with 804 participants, including analysts, financial professionals, regulators, and the general public. The benchmark tests LLMs not only for factual accuracy but also for their adaptability, response depth, and alignment with user satisfaction. Results across 11 models demonstrate that domain-specific LLMs like Tongyi-Finance-14B and CFGPT2-7B outperform general-purpose counterparts in delivering context-aware, actionable financial insights. These findings highlight the critical importance of user alignment and task interactivity for advancing LLMs in finance.
Hirano. Hirano [470] constructs the first large-scale benchmark specifically designed to evaluate LLMs in the Japanese financial domain. It comprises five core tasks reflecting both professional knowledge and real-world financial practices in Japan: sentiment analysis of securities reports (chabsa), fundamental knowledge in securities analysis (cma_basics), auditing questions from the Certified Public Accountant exam (cpa_audit), multiple-choice questions from the national financial planner certification (fp2), and practice exams for securities broker representatives (security_sales_1). These tasks cover a range of difficulties and formats—binary, multiple-choice, and judgment-based—enabling a nuanced assessment of LLM capabilities. Benchmarking results across a wide array of models show that even top-performing models face challenges in complex, domain-specific tasks. The analysis further reveals that training data quality and domain relevance substantially impact performance, emphasizing the need for tailored model development.
Discussion. Based on the conclusions drawn from the aforementioned benchmarks, we summarize the following insights regarding the current capabilities and limitations of large language models (LLMs) in the financial domain:
- Challenges in complex financial tasks: Current LLMs still struggle with tasks that require deep domain knowledge, logical reasoning, and multi-step decision-making.
- Effectiveness of domain-specific fine-tuning: Fine-tuning LLMs on domain-specific corpora continues to yield notable performance gains, demonstrating its importance in enhancing model specialization.
- Benchmark coverage vs. real-world applicability: While these benchmarks effectively assess LLMs’ comprehensive capabilities in finance, they are primarily diagnostic and not tailored to specific application scenarios. Practical use cases often require the design of dedicated, task-specific benchmarks.
- Need for broader evaluation dimensions: Additional attention should be given to other meaningful evaluation perspectives, such as user alignment (e.g., UCFE) and risk awareness (e.g., R-Judge), which are crucial for safe and effective real-world deployment.
To provide an intuitive overview of the performance of mainstream LLMs on these benchmarks, we also present a selection of evaluation results from the aforementioned benchmarks, as shown in Table 20. Given the substantial data gaps and the continuous updates across different LLM providers, we recommend that interested readers conduct these benchmark evaluations independently to support their own assessments.
Financial Dataset Construction
Although LLM-based financial agents [364,365,375] are attracting increasing interest, standardized benchmarks [471] and datasets remain scarce. To support researchers in developing their own evaluation environments and datasets for trading and investment tasks, we present a curated list of raw data sources that can serve as the foundation for constructing custom benchmarks (Table 21). These include the following six categories of data:
- General Financial Data: Provides access to real-time and historical stock prices, fundamental financial indicators, and corporate financial statements. Such data are critical for simulating trading environments, developing investment strategies, conducting market forecasts, and evaluating algorithmic trading agents.
- Cryptocurrency Data: Offers market prices, trading volumes, and metadata for cryptocurrencies. These datasets are particularly useful for research on crypto trading strategies, market microstructure analysis, and portfolio optimization involving digital assets.
- Regulatory Filings: Includes official company disclosures, such as quarterly and annual reports (10-Q, 10-K), and other significant events (8-K filings). Regulatory filings are essential for fundamental analysis, event-driven trading, and financial sentiment extraction.
- Analyst Reports: Consists of investment opinions, earnings forecasts, and qualitative assessments from financial analysts. These resources are valuable for sentiment analysis, opinion aggregation, and modeling the impact of market expectations on asset prices.
- News Data: Covers financial news, press releases, and market commentary from a variety of media outlets. News data is critical for developing event-driven trading strategies, market volatility prediction, and detecting sentiment shifts in real-time.
- Social Media Data: Comprises user-generated content from platforms such as Twitter (X) and Reddit. Social media data enables the study of retail investor sentiment, information diffusion, and the dynamics of attention-driven market movements.
These raw data sources provide flexibility for researchers to create customized datasets, simulate trading scenarios, and design evaluation benchmarks tailored to specific financial tasks and market conditions. For instance, INVESTORBENCH [471] constructs a comprehensive benchmark environment for stock, cryptocurrency, and ETF trading tasks by aggregating multi-source data such as OHLCV market data from Yahoo Finance and CoinMarketCap, regulatory filings from the SEC EDGAR database, and news data from public datasets and Refinitiv. These data are further enriched with sentiment annotations generated by GPT-3.5 and structured into time-series segments for warm-up and testing phases, enabling rigorous simulation of real-world trading scenarios. TradingAgents [375] formulates its evaluation environment by collecting diverse financial signals—including historical stock prices, corporate fundamentals, and real-time news—from APIs and public financial repositories. FinMem [365] integrates high-frequency signals like stock price series, social media sentiment, and financial news into short-term memory, while storing earnings reports and macroeconomic indicators in long-term layers with decay-based retention. FinCon [364] constructs a multi-modal financial environment by combining text-based corporate disclosures, tabular time series, and audio transcripts of earnings calls.
4.1.9. Discussion
Opportunities and Impact. LLMs are ushering in a transformative era in financial research and applications, offering a unique combination of scalability, adaptability, and interpretability across diverse financial domains. As demonstrated in trading and investment [367,373,374], corporate finance [384,386], financial markets [389,391], financial intermediation and risk management [396,398], sustainable finance [402,404], and fintech innovation [417,425], LLMs provide new tools to process vast unstructured datasets, generate strategic insights, and support decision-making processes with unprecedented efficiency. Additionally, their ability to integrate multimodal inputs, perform natural language reasoning, and adapt across financial subdomains positions LLMs not just as supplementary tools, but as pivotal contributors to the future of finance.
Challenges and Limitations. Despite their promise, significant challenges remain. First, numerical reasoning remains a major limitation for LLMs [401], where tasks involving high-frequency trading, real-time credit scoring, and precision-focused financial modeling still favor traditional econometric or hybrid approaches. Second, the interpretability and explainability of LLM decisions, especially in black-box architectures, must be improved to meet the stringent compliance standards in regulated industries such as banking and insurance [393,398].
Another critical concern is robustness to distribution shifts. Financial markets are notoriously volatile and non-stationary; LLMs fine-tuned on historical data often struggle to generalize to unseen macroeconomic conditions or systemic shocks [388,393]. Similarly, context length limitations pose challenges for applications like ESG scoring, where processing long, detailed sustainability reports remains difficult [410].
Moreover, the fairness and bias inherent in LLM training [398] raises ethical concerns, particularly when models are deployed in high-stakes areas like credit scoring or fraud detection. Addressing these issues requires interdisciplinary collaboration between AI developers, financial domain experts, and regulators.
Research Directions. Several promising research directions emerge from current trends:
- Real-Time and Adaptive Learning. Future models must be capable of online learning and rapid adaptation to changing market conditions, with architectures that support dynamic knowledge updates and feedback-driven improvement [373].
- Ethics, Fairness, and Regulatory Compliance. Research must prioritize fairness audits, bias mitigation strategies, and interpretability mechanisms to ensure that LLM-based systems meet ethical and legal standards in finance [398].
Conclusion. LLMs offer a compelling expansion of the financial analytics toolkit, promising new capabilities in understanding, forecasting, and decision-making under uncertainty. However, realizing their full potential requires careful attention to their limitations, rigorous fine-tuning for financial contexts, and an ongoing commitment to ethical, robust, and interpretable AI development. As finance continues to evolve, the synergistic integration of LLMs with traditional quantitative methods, domain knowledge, and human oversight will likely define the next frontier in financial innovation.
4.2. Economics
4.2.1. Overview
Introduction. Economics, in its formal rigor, is the study of how agents allocate scarce resources to maximize utility or profits under constraints, guided by models rooted in mathematical logic and empirical testing [472]. More intuitively, it is the science of everyday choices—why people buy certain things, how firms decide what to produce, and how governments respond to inflation. Whether analyzing a shopper choosing between apples and oranges, or central banks managing interest rates, economics provides frameworks to understand behavior and institutional interactions.
Economics spans a wide range of subfields, each with its own research focus and methodological approach. Microeconomics studies individual decision-making and the functioning of markets [473], while macroeconomics looks at the economy as a whole, analyzing phenomena such as economic growth, inflation, and unemployment [474]. Specialized areas like labor economics [475], industrial organization [476], public finance [477], and behavioral economics [478] delve into specific aspects of economic activity—from how institutions shape labor markets to how cognitive biases influence individual choices. Across these domains, economists employ tools such as optimization theory, econometrics, and general equilibrium modeling [479,480]. Foundational frameworks like the IS-LM model [481], the Solow growth model [482], and Nash equilibrium in game theory [483] have not only advanced theoretical understanding but also played a pivotal role in shaping public policy and economic thought more broadly.
Traditional economic methods have made profound contributions to both scientific understanding and real-world decision-making. Empirical techniques like randomized controlled trials have redefined development economics—evident in Banerjee, Duflo, and Kremer’s field experiments [484] that earned the 2019 Nobel Prize and influenced global aid policy. Structural models, such as DSGE frameworks [485], are now central to monetary policy in institutions like the European Central Bank and the U.S. Federal Reserve. Game-theoretic tools underpin modern auction design [486], informing spectrum auctions worldwide and contributing to the 2020 Nobel Prize in Economics. Together, these traditional methods exemplify how rigorous economic modeling and empirical analysis have not only advanced scientific theory but also shaped impactful policies across global institutions.
Figure 11.
Overview of LLMs’ Applications in Economics Research.
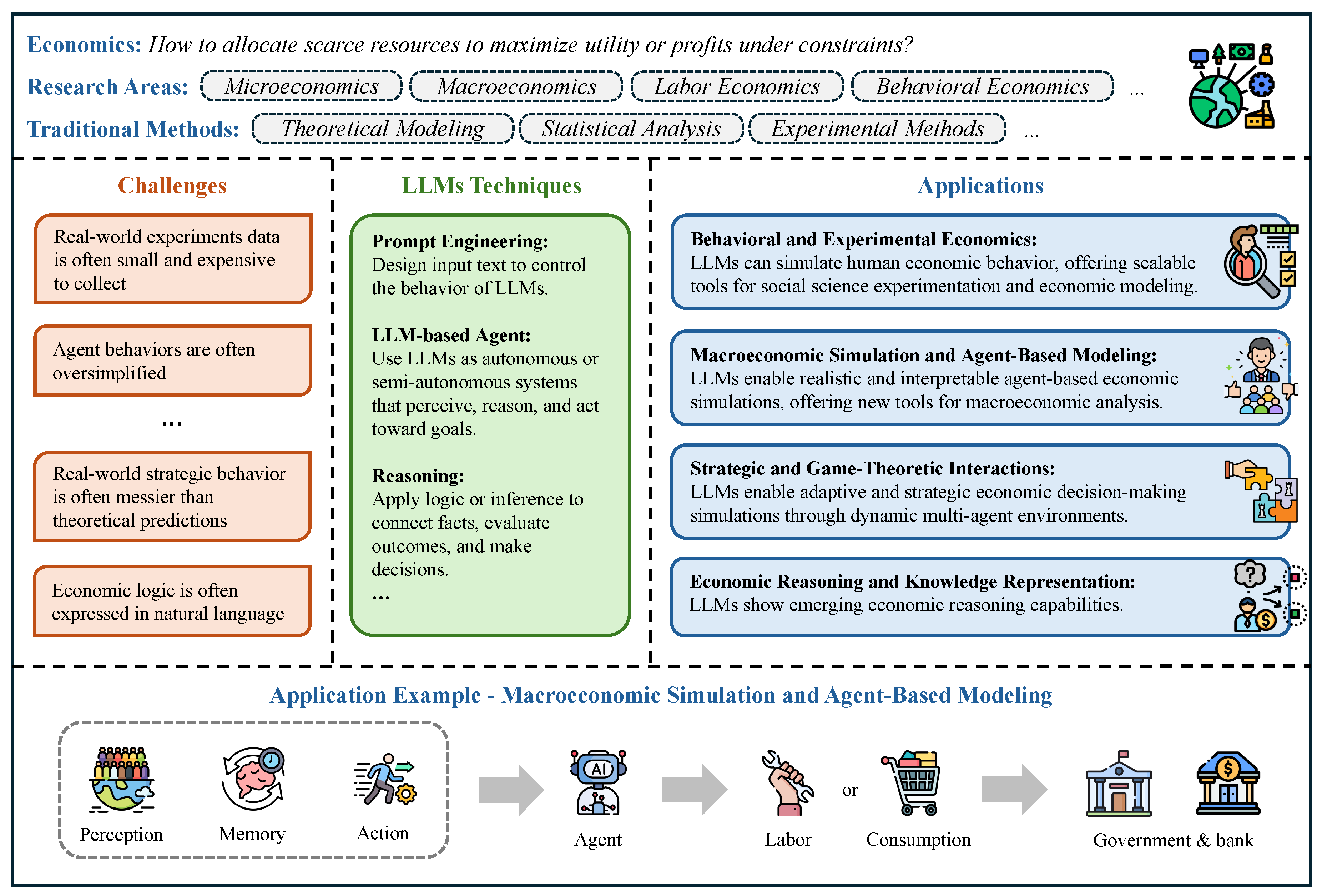
Despite the progress in economic research, significant challenges persist that limit the realism and applicability of traditional models. Real-world economic environments are inherently dynamic, characterized by constantly evolving institutions, shocks, and information flows. Economic agents are diverse in their preferences, constraints, information, and decision-making processes—yet conventional models often rely on simplifying assumptions such as representative agents, rational expectations, or static equilibria. These abstractions can obscure important heterogeneities and interactions that drive actual outcomes.
Furthermore, collecting high-quality experimental or observational data is frequently expensive, time-consuming, and limited in scale, which constrains empirical validation. Agent behaviors are often oversimplified, failing to capture bounded rationality, adaptive learning, or context-dependent strategies. Real-world strategic interactions—such as those seen in financial markets, policy negotiations, or consumer choices—tend to be far more nuanced and unpredictable than what classical theory anticipates.
In addition, economic reasoning is frequently articulated in natural language, making it challenging to formally encode into models or simulations. Problems like modeling evolving preferences, simulating large-scale economies with heterogeneous agents, or generating credible counterfactual scenarios remain analytically intractable or computationally intensive. These limitations highlight the need for more flexible, data-rich, and behaviorally informed approaches to economic analysis.
The Role of LLMs. LLMs, as text-based agents trained on vast human knowledge and capable of simulating reasoning and dialogue, offer a new toolset. Yet their capacity is domain-dependent: they may struggle with tasks demanding precise quantitative forecasting, consistent economic logic across contexts, or nuanced understanding of causality. Nonetheless, in specific domains, LLMs demonstrate potential. They can model human-like decisions in behavioral experiments [487], simulate agents in macroeconomic environments [488], and engage in strategic reasoning akin to game-theoretic thinking [489,490]. Their fluency in natural language allows them to interpret and generate policy scenarios, simulate negotiations, or even mimic human fairness preferences [491]. These affordances make them well-suited to complement existing tools in behavioral and experimental economics, agent-based modeling, and knowledge representation.
Table 22.
Applications and insights of LLMs in economics research
| Field | Key Insights and Contributions | Examples | Citations |
|---|---|---|---|
| Behavioral and Experimental Economics | LLMs can simulate human economic behavior by exhibiting rationality, personality traits, and behavioral biases, offering scalable tools for social science experimentation and economic modeling | Ross et al. [492]: Utility theory reveals LLMs’ behavioral biases across economic decision settings. Horton [493]: LLMs simulate economic agents, replicating human decisions in experiments. | [491,492,493,494] |
| Macroeconomic Simulation and Agent-Based Modeling | LLMs enable realistic, interpretable, and heterogeneous agent-based economic simulations by modeling complex decision-making, memory, perception, and policy responses, offering new tools for macroeconomic analysis and public policy evaluation | MLAB [495]: Multi-LLM agents simulate diverse economic responses for policy analysis. EconAgent [496]: LLM agents model macroeconomics with perception, memory, and decision modules. | [495,496,497] |
| Strategic and Game-Theoretic Interactions | LLMs enable robust, adaptive, and strategically nuanced economic decision-making simulations through dynamic multi-agent environments and standardized benchmarks | GLEE [489]: Economic game benchmarks evaluating LLM fairness, efficiency, and communication. Guo et al. [498]: LLM agents compete in dynamic games testing rationality and strategy. | [489,491,498] |
| Economic Reasoning and Knowledge Representation | LLMs show emerging economic reasoning capabilities through benchmarks and frameworks assessing causal, sequential, and logical inference | EconLogicQA [499]: Tests LLMs’ ability to sequence economic events logically, contextually. EconNLI [490]: Evaluates LLMs’ causal reasoning using premise-hypothesis economic event pairs. | [490,499,500] |
Taxonomy. To systematically explore these intersections, the following taxonomy organizes research at the nexus of economics and LLMs into four task domains:
- Behavioral and Experimental Economics. This field studies how real people make decisions, often deviating from the rational “homo economicus” model. Experiments with games like the dictator, ultimatum, and trust games reveal biases such as fairness concerns and the endowment effect. LLMs complement these methods by simulating diverse decision behaviors and allowing rapid pre-testing of economic experiments.
- Macroeconomic Simulation and Agent-Based Modeling. ABMs simulate how individual agents interact to shape aggregate outcomes like inflation or unemployment. Unlike equilibrium-based models, they capture dynamic, bottom-up processes but often lack realistic human behavior. LLMs enrich ABMs by powering adaptive, communicative agents, bringing greater realism and flexibility to macroeconomic simulations.
- Strategic and Game-Theoretic Interactions. Game theory examines how outcomes depend on the choices of multiple agents, requiring competition, cooperation, and anticipation. Traditional approaches rely on simplified assumptions, limiting realism. LLMs enable agents with recursive reasoning and natural language interaction, offering richer simulations of strategic scenarios.
- Economic Reasoning and Knowledge Representation. Economic reasoning analyzes trade-offs under scarcity, while knowledge representation encodes concepts for computational use. Rule-based methods struggle with complexity and scalability. LLMs simulate reasoning in natural language and generalize across contexts, though they remain sensitive to prompt design and prone to oversimplification.
Across economic research, LLMs do not replace traditional models or empirical methods, but extend the field’s analytical reach—by linking language with behavior, enabling simulation of human-like agents, and enhancing strategic reasoning. They bridge the gap between narrative and formal analysis, offering new tools for interpreting, modeling, and experimenting with economic decision-making.
4.2.2. Behavioral and Experimental Economics
Behavioral and Experimental Economics is a subfield of economics that studies how people actually make decisions, often deviating from the idealized “rational agent” model [501]. Unlike classical economics, which assumes individuals are perfectly rational, consistent, and self-interested (homo economicus), this field acknowledges that humans are prone to biases, emotions, and heuristics. Experimental economists design controlled lab or field experiments to observe behaviors such as fairness, cooperation, time inconsistency, or risk aversion. The vividness of this discipline comes from its attention to how real people respond to incentives and information—like how someone might overvalue an item they own (endowment effect) or hesitate to switch default options even if better alternatives exist (status quo bias) [502]. These insights are foundational to areas like policy design, marketing, and behavioral finance.
Traditionally, behavioral and experimental economics relies on human subjects in lab settings, often using games like the dictator game [503], ultimatum game [504], or trust game [505] to infer preferences and behaviors. But these methods are time-consuming, costly, and constrained by participant availability and ethical considerations. Moreover, running variations of an experiment—for example, tweaking the framing of a question or demographic profile of the subject—requires significant effort. Another challenge is that findings often suffer from limited external validity due to small sample sizes or artificial settings. Here is where LLMs open up a new frontier. Because LLMs can be prompted to act as decision-makers in structured economic tasks, researchers can simulate thousands of hypothetical agents with varying preferences or constraints, rapidly exploring behavioral responses across scenarios [492,493]. This opens the door to low-cost, scalable pre-testing of economic experiments, probing theoretical boundaries, or even generating novel hypotheses for subsequent testing with human subjects.
Recent research illustrates both the promise and nuance of using LLMs in behavioral and experimental economics. Horton positions LLMs as synthetic stand-ins for experimental participants [493]. He shows that GPT-3 can replicate results from classic behavioral experiments, like status quo bias in budget allocation or reactions to fairness in pricing scenarios, and that by simply altering the agent’s "social preferences" via prompts, one can elicit predictable shifts in decision-making. This mirrors how economists model preference heterogeneity, suggesting that LLMs might serve not just as simulation tools but as exploratory partners in theory development.
Another group of studies tests whether LLMs exhibit economic rationality in a formal sense. Chen et al. [494] apply revealed preference theory to GPT by placing it in structured budget allocation tasks across domains like risk, time, and social preferences. They find GPT’s choices to be remarkably consistent with utility maximization, often more so than actual human subjects. Yet the models show sensitivity to framing, indicating that context still matters—a pattern familiar from human experiments.
Complementing these findings, Ross et al. [492] take a utility-theoretic approach to map LLM biases. They assess the degree to which LLMs manifest behavioral regularities like loss aversion, risk aversion, and time discounting. While LLMs like GPT-4 show patterns that align with both rational agent models and human-like heuristics, the consistency and magnitude of these biases vary across models and prompting strategies. The implication is that LLMs, while powerful, are not monolithic agents. They behave in context-sensitive ways and can be shaped or nudged via design choices.
In sum, LLMs are emerging not just as tools for automating economic reasoning but as experimental agents in their own right. Their utility lies not in replacing human subjects but in extending the experimental toolkit of economists, providing new ways to generate hypotheses, test theories, and explore behavioral nuance at scale.
4.2.3. Macroeconomic Simulation and Agent-Based Modeling
Macroeconomic simulation and agent-based modeling (ABM) are computational tools used to study how individual economic decisions aggregate into macro-level phenomena [506]. Unlike traditional models that assume representative agents and equilibrium, ABM simulates economies from the bottom up, with heterogeneous agents, like households and firms, interacting in dynamic environments. Picture a digital society where agents choose whether to work, consume, or save based on personal traits and environmental cues. As these decisions ripple through the system, they give rise to emergent outcomes like inflation, unemployment, or economic cycles.
Traditional ABMs often rely on rule-based or statistical models that are rigid, require expert calibration, and struggle to represent real human behavior [507]. Behavioral and experimental economics aim to capture more realistic decision-making, but integrating these insights into scalable simulations is difficult. Agent heterogeneity and adaptive behavior remain major gaps. LLMs offer a powerful solution. With their capacity for reasoning, memory, and language understanding, LLMs can simulate more human-like agents that respond to complex environments, communicate, and evolve over time, reducing the need for hand-coded rules and improving realism.
Recent work illustrates this shift. EconAgent [496] uses LLM-powered agents to simulate macroeconomic activities like labor supply and consumption. These agents perceive their environment and reflect on past experiences, producing macro patterns that align with real-world phenomena such as the Phillips Curve. Woo et al. [497] develop a reinforcement learning-enhanced ABM where LLMs generate indices like perceived value and information spread, enabling agents to make context-sensitive purchasing decisions and model social influence. Hao and Xie [495] push heterogeneity further by using different LLMs to represent socioeconomically distinct agents. Their Multi-LLM-Agent-Based (MLAB) framework captures both cognitive and contextual diversity, allowing nuanced policy simulations, such as responses to taxation across income groups.
Together, these studies signal a new generation of economic modeling. By embedding LLMs into ABMs, researchers can build richer, more adaptive simulations that bring us closer to modeling the true complexity of economic behavior.
4.2.4. Strategic and Game-Theoretic Interactions
Strategic and game-theoretic interactions in economics involve decision-making where each agent’s payoff depends on the choices of others. Game theory provides formal tools to model such settings, capturing the logic of anticipation, competition, and cooperation [508]. Classic examples include auctions, bargaining, and coordination games. These environments often require recursive reasoning—thinking about what others think you will do—and can be challenging to model or simulate with traditional economic tools.
Standard approaches use mathematical models with strict assumptions about rationality or controlled human experiments [509], both of which struggle with complexity, communication, and scalability. LLMs, by contrast, offer a novel way to simulate agents in strategic settings. Their ability to process natural language and adapt strategies on the fly makes them uniquely suited for interactive, multi-agent environments.
Recent work shows both promise and limitations. Guo et al.[498] introduced EconArena, where LLMs compete in games like beauty contests and auctions. Results show that models like GPT-4 and Claude2 display bounded rationality and strategic adaptation, especially when given game history. However, none consistently reach Nash equilibrium, and rule-following varies by model. This suggests LLMs can reason about others’ strategies, but not perfectly. Similarly, Mei et al. [491] find through a behavioral Turing test that LLMs like ChatGPT-4 can exhibit behaviors remarkably close to human distributions in classic economic games, although their responses tend toward greater altruism and cooperation compared to typical human actions. Importantly, these models adapt their strategies based on context and past interactions, mirroring human learning and responsiveness to framing. Shapira et al. [489], using their GLEE framework, further emphasize the role of communication style in sequential strategic interactions such as bargaining and negotiation, highlighting that LLMs can realistically mimic human behavior, particularly where language nuances matter.
Together, these works highlight the potential of LLMs as tools for modeling strategic behavior, especially in dynamic or linguistically complex settings. Despite limitations regarding perfect rationality, their adaptability, expressiveness, and scale make them valuable complements to traditional economic methods.
4.2.5. Economic Reasoning and Knowledge Representation
Economic reasoning involves the structured analysis of how individuals, firms, and institutions make decisions under conditions of scarcity [510]. Knowledge representation in economics, on the other hand, refers to how economic facts, theories, and relationships are formally encoded so that they can be understood and utilized by computational systems. These two areas are interlinked: reasoning cannot happen without structured knowledge, and knowledge must be reasoned with to be useful.
Traditionally, economic reasoning and knowledge representation have relied on rule-based systems or statistical simulations. While these methods are methodologically rigorous, they often falter when faced with the ambiguity of natural language or the complexity of real-world causal chains. Encoding economic knowledge has typically involved labor-intensive manual curation, resulting in systems that are brittle and difficult to scale. LLMs, by contrast, offer a fundamentally different approach. They are capable of detecting patterns in economic text, simulating chains of reasoning, and generalizing across diverse contexts through zero-shot [511] and few-shot (in-context learning) [512] paradigms, as well as with Chain-of-Thought prompting [513]. Nonetheless, their effectiveness is highly sensitive to prompt design, and they remain prone to hallucinations or conceptual misapplications, particularly in more nuanced or domain-specific scenarios.
Recent work has begun to systematically explore these capabilities. For instance, the EconQA dataset [500] evaluates LLMs on multiple-choice questions sourced from economics textbooks, probing their ability to answer definitional, factual, and conceptual questions. Chain-of-thought (CoT) prompting—encouraging the model to reason step by step—improved performance modestly, especially in tasks requiring multi-step logic. However, results suggest that not all prompt formats yield consistent gains and that some models can reason effectively without explicit CoT cues.
Complementing this, EconNLI [490] evaluates models on their ability to infer causal relationships between economic events. This dataset requires not just linguistic understanding but familiarity with economic theories like the quantity theory of money. Models like GPT-4 performed better than random but still made frequent reasoning errors, particularly when the causal link wasn’t surface-level or intuitive. This highlights the gap between surface-level fluency and deep economic understanding. EconLogicQA [499] pushes LLMs further by asking them to sequence multiple interrelated economic events logically. This task mirrors real-world economic planning or policy analysis, where understanding the progression from cause to effect is crucial. Here, even advanced models like GPT-4 achieve only moderate accuracy, underscoring the complexity of sequential reasoning in economics. Unlike static inference, this task tests whether a model can integrate multiple facts into a coherent narrative, a skill essential for economic decision-making.
Together, these efforts illustrate both the promise and limitations of LLMs in economics. They can parse and process economic knowledge at scale and can support reasoning when guided by well-designed prompts. However, challenges remain in grounding their reasoning in robust theory and avoiding confident but incorrect inferences. As research progresses, combining LLMs with formal economic models or hybrid systems that inject domain knowledge may offer a fruitful path forward.
4.2.6. Benchmarks
GLEE [489] provides a unified framework and benchmark for studying language-based interactions in economic environments. It focuses on two-player sequential games, such as bargaining, negotiation, and persuasion, where communication occurs through natural language. GLEE standardizes the evaluation of LLM-based agents across multiple economic contexts by defining consistent parameterizations, degrees of freedom, and economic metrics such as efficiency and fairness. The benchmark includes extensive datasets from LLM vs. LLM and human vs. LLM interactions and enables controlled experimentation to study how language affects strategic behavior and outcomes in economic settings.
EconLogicQA [499] is a benchmark designed to assess the sequential reasoning abilities of LLMs within the domains of economics, business, and supply chain management. Unlike traditional benchmarks that evaluate models on isolated events, EconLogicQA requires models to logically sequence multiple interconnected events extracted from real-world business news articles. The benchmark presents multi-event multiple-choice questions, challenging LLMs to understand temporal and causal relationships in economic scenarios. A rigorous human review process ensures the quality and difficulty of the dataset, which serves as a tool for probing models’ reasoning depth in complex economic contexts.
EconNLI [490] introduces a natural language inference task specifically targeting economic event reasoning. EconNLI evaluates whether an LLM can correctly determine causal relationships between pairs of economic events or generate plausible consequent events based on a given premise. Unlike traditional NLI tasks based on semantic entailment, EconNLI requires understanding of economic theories and principles to infer causal links. Extensive experiments reveal that current LLMs struggle significantly with economic reasoning tasks, highlighting substantial gaps between surface-level language proficiency and domain-specific reasoning capabilities.
4.2.7. Discussion
Opportunities and Impact. The integration of LLMs into economics research offers a profound expansion of the traditional methodological toolkit. Across behavioral and experimental economics [492,493], macroeconomic simulation [496,497], strategic interaction modeling [491,498], and economic reasoning [490,500], LLMs provide new capacities: simulating human-like decision-making, enabling large-scale agent-based models with greater realism, supporting dynamic and adaptive strategic interactions, and offering scalable, language-driven representations of economic knowledge.
These tools open new frontiers for both theoretical exploration and empirical experimentation. Researchers can rapidly prototype behavioral experiments, simulate macroeconomic scenarios with heterogeneous agents, model strategic negotiations dynamically, and test causal reasoning across economic contexts at a scale and speed previously unattainable. For example, on the EconNLI dataset, even the best-performing traditional encoder-only model, fine-tuned BERT, achieved an F1 score of below 0.8. In contrast, relatively small-scale LLMs like LLAMA2-7B, after supervised fine-tuning, easily surpassed this benchmark with F1 score around 0.87, illustrating the superior reasoning capabilities of LLMs [490]. Additionally, LLMs allow economics to bridge formal models and narrative analysis, capturing nuances that traditional mathematical abstractions may miss. This flexibility is critical as economic phenomena increasingly span complex, dynamic, and information-rich environments.
Challenges and Limitations. Despite these promising advances, LLM-driven economics research faces notable limitations. First, economic consistency and coherence remain major concerns. While LLMs can mimic economic reasoning patterns, they are prone to hallucinations, oversimplifications, or logical inconsistencies, particularly in tasks requiring multi-step causal reasoning or deep theoretical grounding [490,499].
Second, prompt sensitivity and model variability introduce fragility into experimental results. Different prompt designs or minor variations in phrasing can lead to divergent outputs, complicating reproducibility and interpretation [492,500].
Third, rationality and strategic depth are bounded. While LLMs can engage in first-order strategic reasoning (anticipating others’ actions), they often struggle with deeper levels of recursive reasoning necessary for achieving equilibrium behaviors in complex games [498].
Additionally, the context alignment problem persists. LLMs are typically trained on broad internet data and may import biases or unrealistic assumptions when simulating economic agents, particularly when real-world institutional, cultural, or contextual factors are crucial.
Finally, ethical considerations emerge. Using LLMs as synthetic economic agents raises questions about bias propagation, external validity of simulated results, and the responsible interpretation of LLM-based experimental findings.
Research Directions. Several key research directions emerge to address these challenges:
- Structured Prompting and Experimental Design. Developing standardized, transparent prompting methodologies—analogous to experimental protocols—will be essential for ensuring replicability and robustness in LLM-driven economic experiments [492].
- Ethical Evaluation and External Validation. Establishing benchmarks, guidelines, and ethical frameworks for using LLMs as economic agents—alongside systematic validation against real human data—will be crucial for credible scientific practice.
Conclusion. LLMs represent a transformative tool for economics research, enabling novel forms of behavioral simulation, strategic modeling, macroeconomic exploration, and reasoning automation. Yet they are not substitutes for traditional theory or empirical grounding. Instead, when carefully validated and thoughtfully deployed, they can serve as powerful complements—extending economists’ ability to model, simulate, and interpret complex economic behavior in ways that were previously infeasible. Future progress will depend not only on technical advancements but also on establishing robust methodological foundations that blend linguistic fluency with economic rigor.
4.3. Accounting
4.3.1. Overview
Introduction. Accounting is formally defined as the systematic process of recording, classifying, summarizing, and interpreting financial information to support decision-making and ensure transparency and accountability [514,515]. It plays a central role in shaping how economic activity is communicated and regulated [516,517]. At its essence, accounting serves as the language of business: it tells the story of where money comes from, where it goes, and what it means. Similar to a medical professional analyzing vital signs, decision-makers rely on accounting to assess an organization’s financial health. It elucidates whether a company is profitable, solvent, or experiencing financial distress, thereby guiding strategic choices and fostering trust among stakeholders.
Accounting research spans several major areas that mirror professional practice. For example, financial reporting examines how firms communicate with investors and regulators, often evaluating the quality and usefulness of disclosures [518]; Auditing focuses on the verification of financial information, exploring how auditors assess risk, detect fraud, or maintain independence [519]; Managerial accounting centers on internal decision-making, providing tools for budgeting, cost control, and performance evaluation [520]; Taxation research investigates how firms respond to tax policies, design strategies, or manage compliance [521,522]. Each of these directions reflects accounting’s broader role in supporting economic coordination, regulatory oversight, and informed decision-making in markets and organizations.
Traditional research methods, e.g., econometrics, archival analysis, case studies, and content analysis, have significantly advanced both academic understanding and practice. Regression models, for instance, have helped uncover patterns in earnings management, shaping reforms in reporting standards and corporate governance [523,524]. These methods have yielded tangible benefits. Studies linking financial reporting quality to lower cost of capital have informed investor behavior and policy [525]. Improvements in audit procedures have reduced fraud and increased public trust in financial markets [519,526]. One study [527] estimates that the widespread preference among VC-backed startups for C-corporations over tax-advantaged LLCs has led to $43.9 billion in foregone tax savings—equivalent to 4.9% of total invested equity—highlighting how accounting-related decisions can carry substantial financial consequences. Though often resource-intensive and limited in scope, these methods form the empirical foundation of accounting knowledge. Their rigor has helped accounting evolve into a robust, evidence-based profession.
Modern accounting encounters a range of novel challenges, particularly in managing the vast and growing volumes of both structured and unstructured data [528]. Financial reports, audit notes, and regulatory disclosures are increasingly complex and voluminous, making them difficult to analyze using traditional tools [529,530,531]. Manual review and rule-based text analysis are not only labor-intensive but also brittle in the face of evolving language and formats. Furthermore, accounting professionals must navigate ever-changing legislation, which is difficult to track and interpret consistently across jurisdictions. Complex and evolving reporting standards like IFRS and GAAP introduce additional ambiguity, often leading to inconsistent application and interpretation. The pressure for faster, more accurate reporting adds urgency to these issues, especially as stakeholders demand real-time insights and greater transparency. Collectively, these challenges demand more adaptive, intelligent systems capable of understanding and processing the rich, nuanced content found in modern financial environments.
Figure 12.
Overview of LLMs’ Applications in Accounting Research.
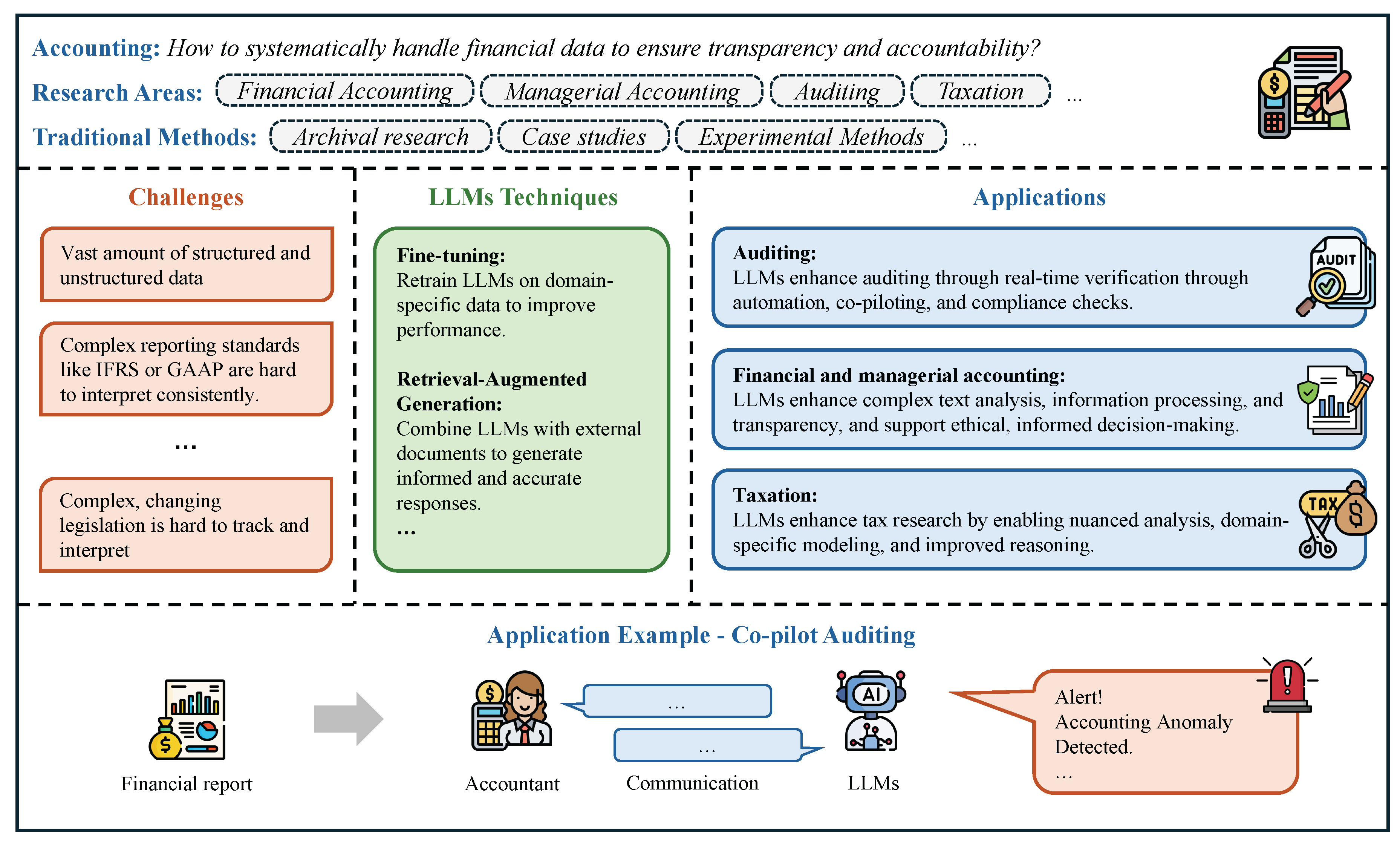
The Role of LLMs. LLMs provide an alternative. Their primary strength lies in interpreting and generating human-like text, rendering them effective for tasks such as summarizing disclosures, elucidating accounting standards, or extracting insights from audit reports. Their adaptability makes them valuable in both research and professional settings [532,533,534,535]. Nevertheless, LLMs also exhibit certain limitations. They can generate convincing yet erroneous statements, lack domain-specific reasoning, and pose risks when applied to sensitive data [303,536,537]. They cannot serve as replacements for expert judgment, particularly in tasks involving legal interpretation or ethical evaluation. Nevertheless, when employed judiciously, LLMs can augment human capabilities. They offer speed, scalability, and linguistic flexibility that can enhance productivity in tasks such as document review, preliminary analysis, or instructional support. Their value resides in complementing—rather than supplanting—traditional methods and expertise.
Taxonomy. To understand the potential application of LLMs in accounting research and practice, we propose a taxonomy that reflects the diversity of tasks across the field:
- Auditing. Traditionally reliant on manual, sample-based checks, auditing struggles with rising data volumes and fraud complexity. LLMs can automate text analysis, flag anomalies, and expand audit coverage, enabling smarter AI-assisted audits while underscoring the need for transparency and safeguards.
- Financial and Managerial Accounting. Both functions are central to decision-making but increasingly burdened by complex disclosures and fragmented systems. LLMs help extract insights, streamline reporting, and convert unstructured data into actionable analysis, strengthening transparency, accuracy, and strategic value.
- Taxation. Taxation involves intricate laws and resource-constrained enforcement, with traditional systems often missing nuances in legal texts. LLMs can interpret tax codes, analyze unstructured filings, and support compliance and enforcement, offering new efficiency while raising questions of trust and adaptability.
Across accounting tasks, LLMs do not replace traditional methods of measurement or verification, but instead unlock new forms of understanding—by interpreting complex language, streamlining routine processes, and expanding access to financial reasoning. They bridge the gap between narrative and numbers, enhancing clarity, efficiency, and insight in the accounting profession.
Table 23.
Applications and insights of LLMs in accounting research
| Field | Key Insights and Contributions | Examples | Citations |
|---|---|---|---|
| Auditing | LLMs enhance auditing by improving accuracy, efficiency, and real-time verification through automation, co-piloting, and compliance checks, while raising implementation and ethical challenges | Gu et al. [538]: Co-piloted auditing combines LLMs and humans for efficient audits. Berger et al. [539]: LLMs assess financial compliance, outperforming peers in regulatory audits. | [538,539,540,541,542,543,544,545,546] |
| Financial and Managerial Accounting | LLMs enhance accounting, reporting, and sustainability practices by automating complex text analysis, improving information processing, enabling transparency, and supporting ethical, informed decision-making across financial and ESG domains | De Villiers et al. [547]: AI reshapes sustainability reporting, raising greenwashing risks and governance questions. Föhr et al. [548]: LLMs audit sustainability reports using taxonomy-aligned prompt frameworks efficiently. | [547,548,549,550,551,552,553,554] |
| Taxation | LLMs enhance tax research, compliance, and enforcement by enabling nuanced analysis, firm-level measurement, domain-specific modeling, and improved reasoning through agent collaboration | PLAT [555]: PLAT tests LLMs’ tax reasoning under ambiguous penalty exemption scenarios. Alarie et al. [556]: LLMs assist tax research, but hallucinations limit reliable adoption. | [555,556,557,558,559] |
4.3.2. Auditing
Auditing is a cornerstone of financial accountability, designed to ensure that financial statements are accurate, complete, and compliant with regulatory standards [519]. At its core, auditing involves the systematic examination of a company’s financial records and transactions to assess their fairness and reliability [560,561]. In more practical terms, an audit can be visualized as a multi-layered process where auditors sift through mountains of structured and unstructured financial data, like journal entries, invoices, contracts, and disclosures, looking for inconsistencies, errors, or signs of fraud. This work demands high attention to detail, extensive knowledge of accounting standards, and a careful balance of skepticism and judgment [562]. Yet, the traditional tools at their disposal often rely on sampling and manual procedures, limiting coverage and efficiency [563,564].
Conventional audit methods have long faced limitations. Auditors typically work with samples rather than full datasets due to time and resource constraints, making audits prone to oversight [565,566]. They also contend with increasingly complex data types, regulatory changes, and mounting pressure for faster, higher-quality audits. Manual processing of financial texts and repetitive audit tasks remain time-consuming, while fraud detection often hinges on patterns too subtle for traditional rules-based systems.
LLMs, like ChatGPT, offer transformative potential for addressing these gaps. By understanding and generating human-like text, LLMs can assist auditors in a wide range of tasks, including analyzing financial narratives, extracting key indicators from disclosures, automating documentation, and even performing real-time risk assessments. For instance, Gu et al. [538] introduced the concept of "co-piloted auditing," where auditors work collaboratively with LLMs to analyze journal entries, perform ratio analysis, and identify anomalies using natural language prompts. The result is a more dynamic and scalable auditing process that goes beyond simple automation to offer contextual insights and proactive risk detection. Moreover, in continuous auditing settings, LLMs enable full population testing and real-time data cross-verification. Li et al. [540] showcased a real-world implementation in a government payroll audit where ChatGPT parsed regulatory text and matched it against accounting records with 96% accuracy, reducing verification time by 83%.
Nevertheless, LLMs are not without drawbacks. Their use introduces concerns around model hallucinations, data privacy, audit independence, and ethical accountability [544]. Ensuring model outputs are transparent and grounded in verifiable sources is essential to maintaining audit integrity.
Recent research reflects a surge in interest in applying AI and LLMs to auditing. Fedyk et al. [543] provide empirical evidence that audit firms investing in AI experience improved audit quality, reduced audit fees, and a shift in labor dynamics—particularly the displacement of junior audit staff. Gu et al. [538] advance this by conceptualizing AI not merely as a tool, but as a collaborative partner in the audit process. Their "co-pilot" model positions ChatGPT as a flexible assistant capable of reasoning through complex audit tasks using chain-of-thought prompting, showing how fine-tuned LLMs can contribute meaningfully to judgment-intensive activities.
Meanwhile, Wang et al. [546] propose AuditBench, a benchmark specifically designed to evaluate LLM performance in financial statement auditing, including tasks such as error identification, standards citation, and corrective action. The study highlights both the promise and current limitations of LLMs in reliably executing full audit cycles. Berger et al. [539] explore regulatory compliance verification through LLMs, focusing on whether models can assess conformity between financial text and legal mandates. The study by Föhr et al. [545] introduces a comprehensive framework for integrating deep learning and LLMs into risk-based auditing, emphasizing the need for organizational readiness and technical infrastructure. Meanwhile, Fotoh and Mugwira [544] explore ethical implications, emphasizing the importance of safeguarding independence, privacy, and professional judgment in an AI-augmented audit environment.
Other contributions expand the practical landscape. Emett et al. [542] document ChatGPT’s integration into the internal audit workflows of a multinational firm, reporting efficiency gains of 50–80% in audit preparation, fieldwork, and reporting. Similarly, Eulerich and Wood [541] demonstrate a wide array of LLM applications across the entire audit lifecycle, from planning and risk assessment to documentation and follow-up, highlighting its value for both professional and academic audiences.
These studies collectively illustrate a shifting paradigm in auditing—from static, manual procedures to dynamic, AI-assisted processes. While promising, the literature also cautions against over-reliance, stressing the need for robust controls, explainability, and updated ethical standards.
4.3.3. Financial and Managerial Accounting
Financial and managerial accounting form the backbone of modern business decision-making, albeit with different audiences and goals [567]. Financial accounting focuses on standardized reporting for external stakeholders—investors, regulators, and creditors—by presenting a historical snapshot of a firm’s financial health. In contrast, managerial accounting supports internal decision-making through more granular, often real-time data on costs, performance metrics, and forecasts [568].
Despite their centrality, both fields have long faced challenges. Financial accounting has become increasingly burdened by voluminous and complex disclosures that hinder rather than help decision-making [569,570]. Managerial accounting struggles with fragmented systems and the difficulty of converting raw figures into actionable insights [571,572]. These pain points are exacerbated by the rapid growth of unstructured data, requiring more than traditional tools to manage effectively [573].
LLMs, like ChatGPT, offer a new frontier for addressing these challenges. Their capacity to interpret, generate, and summarize complex text allows them to streamline both reporting and analysis. For financial accounting, LLMs can parse regulatory filings, reduce information overload, and improve transparency. A recent study by Kim et al. [549] demonstrates that LLM-generated summaries of financial disclosures not only condense content but often sharpen its informativeness, better aligning with market reactions. This ability to cut through “bloated” disclosures can enhance price efficiency and reduce information asymmetry.
In managerial contexts, LLMs bring value through integration with Robotic Process Automation (RPA), enabling them to not only interpret data but act on it. As shown in Li and Vasarhelyi’s [554] framework, such integrations automate tasks like transaction coding and report generation, overcoming limitations of standalone APIs or user interfaces. Beerbaum [553] further emphasizes the scalability of this approach, proposing RPA-LLM systems as ethically conscious and operationally robust tools for routine accounting tasks. The growing body of research also reflects this shift. One emerging theme centers on financial forecasting and performance estimation. For example, Comlekci et al. [552] used ChatGPT to project financial outcomes for publicly traded firms based on past data and sector developments. While accuracy varied by metric, the study highlighted LLMs’ potential in generating early-stage projections, especially when enhanced with external textual context.
Another important theme is the role of LLMs in auditing and assurance, especially in sustainability reporting. With the rise of ESG disclosures and regulatory initiatives like the EU’s CSRD, verifying qualitative statements has become more critical. Studies by Föhr et al. [548] and Ni et al. [551]introduce LLM-powered tools for benchmarking disclosures against frameworks like the TCFD, showing how domain-specific prompt engineering and retrieval mechanisms can produce traceable, high-quality evaluations.
At the same time, critical voices have emerged. De Villiers et al. [547] caution that generative AI may inadvertently replicate biased or "greenwashed" content if left unchecked. Their analysis calls for greater transparency and accountability in how LLMs are trained and deployed in reporting contexts, particularly in areas like sustainability, where narrative control is often strategic.
Another important line of work explores behavioral patterns embedded in financial texts. Harris [550] leverages LLMs to analyze how perceived competitive pressure influences managerial earnings manipulation, uncovering subtle textual signals in 10-K filings that correlate with discretionary accounting choices. This points to the growing role of LLMs in behavioral accounting research, where language not only describes but shapes economic decisions.
In sum, the integration of LLMs into financial and managerial accounting offers the potential to enhance efficiency, accuracy, and transparency, while also raising new questions about accountability and interpretation. As these models evolve, their role is not just as passive tools but as collaborators in reshaping how financial information is produced, analyzed, and understood.
4.3.4. Taxation
Taxation refers to the process through which governments collect financial contributions from individuals and organizations to fund public services, redistribute wealth, and support economic policy [574]. Taxes come in many forms, e.g., income tax, corporate tax, sales tax, and property tax, and their design involves balancing efficiency, equity, and administrative feasibility [575]. A fair and effective tax system is essential to a functioning society, yet its administration is often opaque and heavily reliant on expert interpretation.
Traditionally, the field of taxation has faced several persistent challenges. First, the tax code is extraordinarily complex, constantly evolving, and filled with exceptions, loopholes, and jurisdiction-specific nuances. For researchers, practitioners, and taxpayers alike, this complexity makes compliance difficult and error-prone [576,577]. Second, the enforcement of tax laws, such as through audits, is constrained by resource limitations and data opacity [578]. Third, tax research and policy analysis have long depended on structured data, which fails to capture the richness and nuance embedded in textual disclosures, court decisions, and regulatory interpretations [579].
This is where LLMs, such as GPT-4 and domain-specific models like TaxBERT [558], offer a compelling leap forward. LLMs can process vast amounts of unstructured textual data, understand nuanced language, and generate contextually rich responses—capabilities well-suited to the linguistically dense and interpretive domain of taxation. For example, Choi and Kim [559] leverage GPT-4 to develop a novel firm-level measure of tax audits by extracting insights from boilerplate narrative disclosures in 10-K filings. Their study reveals that tax audits significantly deter tax avoidance, with lasting effects post-audit, while also introducing corporate risks such as reduced investment and increased volatility. These insights, previously out of reach due to data limitations, underscore the transformative potential of LLMs in empirical tax research.
On the practitioner side, generative AI is gaining traction among tax professionals. According to a 2023 Thomson Reuters Institute survey [580], nearly three-quarters of tax professionals recognize the utility of ChatGPT and similar models for tasks like tax research, return preparation, and client advisory services. These models can help taxpayers understand complex tax codes, identify deductible expenses, and evaluate compliance risks without incurring the costs of professional services. However, concerns persist about model accuracy and transparency. As demonstrated in various evaluations, generic models such as ChatGPT-3.5 may "hallucinate" or produce legally incorrect advice when not properly grounded [536,537], emphasizing the need for specialized or fine-tuned models [556,557].
Recent academic contributions can be categorized into three major streams. First, empirical measurement using LLMs is exemplified by Choi and Kim [559], who construct a validated, firm-level proxy for tax audits using GPT-4, unlocking new research avenues into audit impacts and tax compliance behavior. Second, domain-specific model development is represented by Hechtner et al. [558], who introduce TaxBERT, a BERT-based model fine-tuned on tax-specific corpora. Their work demonstrates the superior performance of specialized models over generic LLMs in parsing nuanced tax disclosures. Third, model benchmarking and evaluation are advanced by Choi et al. [555], who present PLAT, a benchmark of complex tax penalty cases requiring reasoning beyond statutory interpretation. Their results show that while vanilla LLMs struggle with these tasks, multi-agent LLM architectures with retrieval and self-reflection mechanisms can substantially improve performance.
In sum, while the application of LLMs in taxation is still emerging, early evidence from both research and practice illustrates substantial promise. Future work should further explore how LLMs can enhance taxpayer support, assist regulators in enforcement, and drive more nuanced academic insights—all while addressing critical issues of reliability, interpretability, and domain adaptation.
4.3.5. Benchmarks
PLAT [555] is a benchmark designed to evaluate the ability of large language models to reason about complex taxation issues, specifically focusing on the legitimacy of additional tax penalties. Unlike earlier datasets that emphasize deductive reasoning from explicit tax statutes, PLAT challenges models to make nuanced judgments based on ambiguous legal standards and factual contexts drawn from Korean court precedents. The dataset requires comprehensive legal understanding and conflict resolution between competing principles, such as taxpayer responsibility versus the protection of legitimate expectations. Experiments show that while baseline LLMs struggle with these tasks, retrieval augmentation, self-reasoning, and multi-agent role-playing significantly enhance performance.
SARA [581] is an earlier dataset focused on computational statutory reasoning in U.S. tax law. It consists of simplified statutes from the U.S. Internal Revenue Code, along with natural language questions formulated as textual entailment and numerical prediction tasks. Each case requires the application of tax laws to specific fact patterns, emphasizing deductive reasoning based solely on statutory rules. SARA highlights the challenges of grounding language understanding in prescriptive legal norms and motivates the development of models capable of logical inference from structured legal texts.
4.3.6. Discussion
Opportunities and Impact. The emergence of LLMs presents a transformative opportunity across accounting research and practice. As demonstrated in auditing [538,546], financial and managerial accounting [549,554], and taxation [558,559], LLMs offer unprecedented capabilities for processing unstructured financial data, automating repetitive tasks, interpreting regulatory language, and assisting in judgment-intensive activities.
In auditing, LLMs enhance traditional practices by expanding coverage, reducing manual errors, and enabling real-time anomaly detection. In financial and managerial accounting, they streamline reporting, improve transparency, and support strategic analysis by synthesizing complex disclosures. In taxation, LLMs facilitate empirical research, regulatory compliance, and taxpayer support through efficient navigation of complex legal language. Demonstrating this potential at scale, Deloitte’s Zora AI aims to transform financial operations by saving up to 25% in costs and boosting productivity by up to 40%, freeing thousands of hours annually for finance teams [582]. This real-world example shows that LLMs are already powerful collaborators driving efficiency and better decision-making across accounting disciplines.
Challenges and Limitations. Despite their potential, significant challenges must be addressed before LLMs can be widely and responsibly deployed in accounting contexts.
First, accuracy and hallucination risks remain a persistent concern. LLMs can generate plausible yet incorrect outputs, posing serious risks in high-stakes settings such as audits, regulatory compliance, or tax advisory [536,544]. Second, lack of domain-specific reasoning limits their ability to consistently interpret complex accounting standards or apply nuanced legal doctrines, particularly when precise statutory interpretation is required [555,558].
Third, ethical and regulatory challenges arise from the integration of LLMs into sensitive accounting workflows. Issues such as audit independence, data privacy, confidentiality, and accountability for AI-assisted outputs must be carefully managed [544,547].
Moreover, prompt sensitivity and model opacity complicate the interpretability and reproducibility of LLM-based results [546], requiring the development of robust validation protocols and transparent deployment strategies.
Research Directions. To realize the full potential of LLMs in accounting while mitigating risks, several critical research directions emerge:
- Hybrid Human-AI Systems. Emphasizing "co-piloted" models where human expertise remains central will ensure that LLMs complement rather than replace professional judgment, particularly in high-risk domains like auditing and taxation [538].
Conclusion. LLMs are poised to become powerful allies in accounting research and practice, enhancing the ability to process, interpret, and reason over complex financial information. However, realizing this potential responsibly demands rigorous domain adaptation, human-centered oversight, transparent evaluation, and a strong ethical foundation. Rather than replacing traditional accounting methods and judgment, LLMs should be viewed as enablers—tools that expand the scope, depth, and inclusivity of financial reasoning in an increasingly complex and dynamic economic environment.
4.4. Marketing
4.4.1. Overview
Introduction. Marketing, in its formal sense, refers to the set of institutions, processes, and activities for creating, communicating, delivering, and exchanging offerings that have value for customers, clients, partners, and society at large [583]. At its core, marketing is a discipline rooted in understanding and influencing consumer behavior to fulfill organizational objectives. In simpler terms, marketing is about understanding what people need or want and finding effective ways to present and deliver products or services that meet those needs. It encompasses everything from identifying target audiences and researching their preferences, to designing products, setting prices, promoting offers, and ensuring smooth delivery.
Within the field of marketing, researchers address a range of tasks aimed at generating insights to inform managerial decision-making. These tasks typically fall under categories such as market segmentation, consumer behavior analysis, product positioning, pricing strategy, brand management, advertising effectiveness, and customer satisfaction measurement. Traditional research methods for approaching these tasks include qualitative techniques like interviews and focus groups [584,585], as well as quantitative approaches such as surveys, experiments, conjoint analysis, and econometric modeling [586,587]. These techniques have provided robust frameworks for testing hypotheses, identifying causal relationships, and quantifying consumer preferences [588].
The contribution of these traditional methods has been substantial. They have grounded the field of marketing in scientific rigor, enabling systematic inquiry into consumer psychology and market dynamics. Surveys, for instance, are a cornerstone of quantitative research, with online surveys being utilized regularly by 85% of market research professionals, followed by mobile surveys at 47% and proprietary panels at 32% [589]. Through this, researchers and practitioners alike have developed theories and models that remain foundational to both academia and industry. These approaches have also proven their value in real-world applications, informing strategies that drive competitive advantage, optimize customer experiences, and maximize return on investment. The field has benefitted immensely from its interdisciplinary nature, drawing from psychology, economics, sociology, and statistics, thereby enriching both theoretical developments and practical implementations [590,591,592].
Despite its strengths, marketing research faces a host of persistent challenges. One major difficulty lies in the inherent complexity and unpredictability of human behavior, which resists reduction into fixed, universal models. Consumers often act inconsistently, influenced by subtle emotional, social, and contextual cues that traditional research tools struggle to capture. Moreover, issues such as data sparsity, high dimensionality, and response biases, ranging from social desirability effects to non-response tendencies, undermine the reliability of collected data [593]. In today’s fast-moving digital landscape, the volume and velocity of information further complicate matters [594]. Researchers must grapple with monitoring vast amounts of unstructured data from social media, product reviews, forums, and industry publications, all of which demand new analytic capabilities. Manual tasks like survey coding and qualitative response analysis remain time-consuming and resource-heavy, while the growing demand for real-time, personalized content creates pressure to scale insights rapidly without sacrificing nuance. Together, these factors present a significant challenge for marketing professionals seeking to generate timely, accurate, and actionable insights.
Figure 13.
Overview of LLMs’ Applications in Marketing Research.
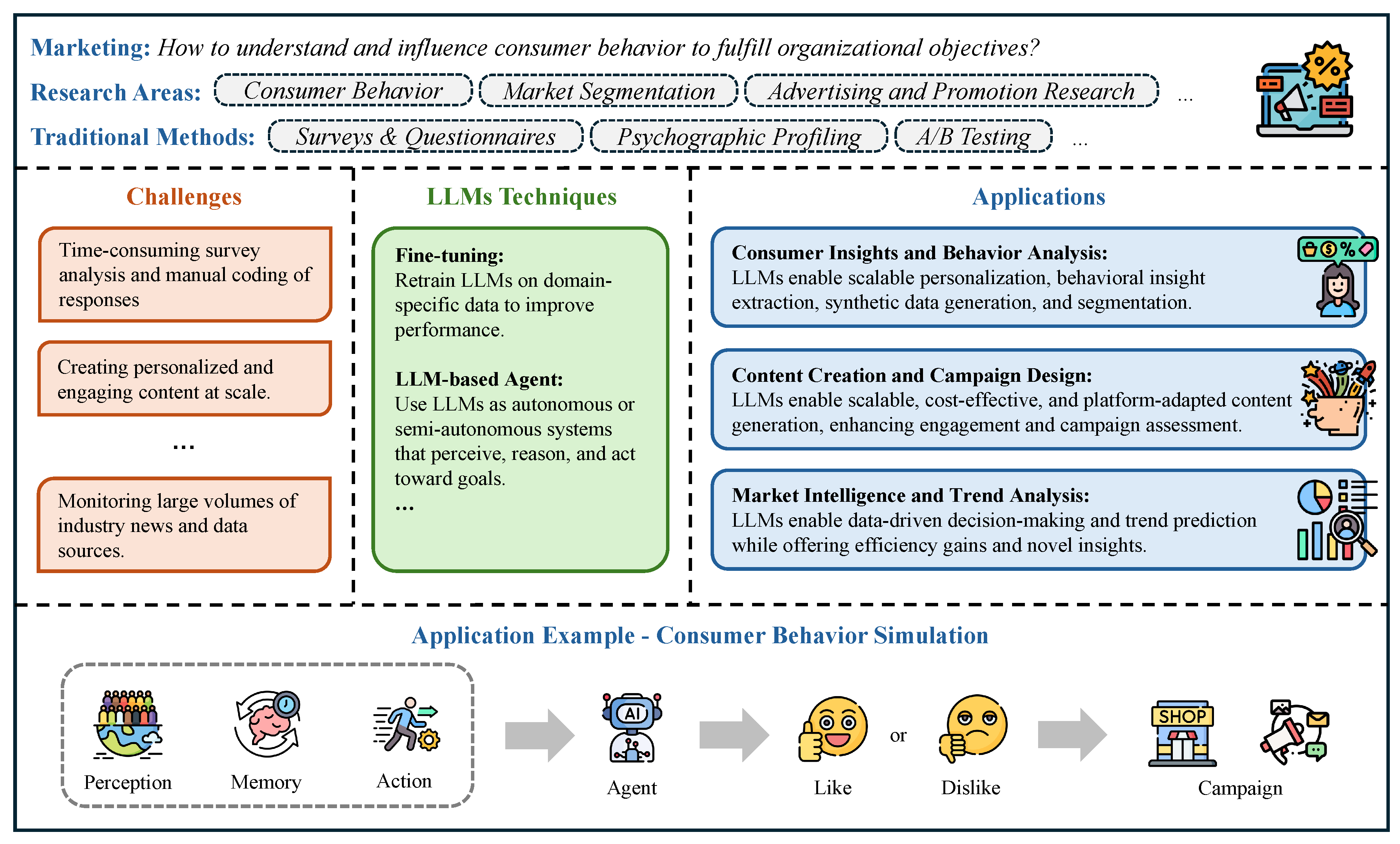
The Role of LLMs. Some of these challenges are beyond the reach of current LLMs. For instance, LLMs cannot autonomously conduct randomized controlled trials or derive causal inference with high reliability without carefully structured datasets and external validation. Nor can they fully replicate the depth of human empathy or ethical judgment necessary for designing sensitive interventions in consumer psychology [595]. However, LLMs are particularly well-suited for tasks where the core requirement is understanding, generating, or summarizing large volumes of unstructured text. They excel at automating tasks such as sentiment analysis, topic modeling, content generation, customer service automation, and text-based survey coding. Their ability to synthesize information across sources, generate coherent narratives, and respond adaptively to prompts positions them as powerful tools for both qualitative and quantitative marketing research [596,597,598,599]. Specifically, LLMs have shown utility in early-stage ideation, exploratory analysis, automated reporting, and even in the creation of synthetic respondents for preliminary hypothesis testing. The reason for their effectiveness lies in their training on vast corpora of language data, enabling them to generalize linguistic patterns, identify latent themes, and approximate conversational nuance with increasing sophistication.
Table 24.
Applications and insights of LLMs in marketing research
| Field | Key Insights and Contributions | Examples | Citations |
|---|---|---|---|
| Consumer Insights and Behavior Analysis | LLMs enable scalable personalization, behavioral insight extraction, synthetic data generation, and segmentation, though challenges remain in faithfully replicating human preferences and ensuring ethical, accurate application | Li et al. [600]: LLMs embed surveys, cluster consumers, simulate chatbots for marketing. Goli & Singh [601]: LLMs mimic preferences poorly; chain-of-thought aids segmentation hypotheses. | [600,601,602,603,604,605,606] |
| Content Creation and Campaign Design | LLMs enable scalable, cost-effective, and platform-adapted content generation, enhancing engagement and campaign assessment while requiring human oversight for quality, ethics, and strategic alignment | Kasuga & Yonetani [607]: CXSimulator simulates campaign effects using LLM embeddings and behavior graphs. Wahid et al. [608]: Generative AI reshapes content marketing, raising engagement and ethical concerns. | [607,608,609,610,611,612,613] |
| Market Intelligence and Trend Analysis | LLMs enable scalable content creation, personalized engagement, data-driven decision-making, trend prediction, and rapid research replication, while offering efficiency gains and novel insights across digital channels | Yeykelis et al. [614]: LLM personas replicate media experiments, accelerating marketing research validation. Saputra et al. [615]: ChatGPT improves Instagram marketing using AIDA model for engagement. | [602,614,615,616,617] |
Taxonomy. To understand the potential application of LLMs in marketing research, we propose a taxonomy that reflects the diversity of tasks across the field:
- Consumer Insights and Behavior Analysis. Consumer Insights and Behavior Analysis focuses on understanding the motivations behind consumer thoughts, feelings, and actions to inform effective marketing strategies. While traditional methods like surveys and interviews offer value, they often struggle with the scale and nuance of modern, unstructured data sources. LLMs are transforming this field by enabling scalable, nuanced analysis of language-rich data, offering deeper, real-time insights into consumer behavior.
- Content Creation and Campaign Design. Content Creation and Campaign Design are key pillars of marketing, combining creative storytelling with strategic planning to engage audiences and achieve business goals. Traditionally reliant on manual effort and intuition, this process has faced challenges in scalability, personalization, and real-time feedback. Today, LLMs are transforming how content is ideated, produced, and optimized, enhancing creativity, streamlining workflows, and enabling more dynamic, data-driven campaigns.
- Market Intelligence and Trend Analysis. Market Intelligence and Trend Analysis are essential for guiding strategic marketing decisions, helping businesses monitor competitors, anticipate consumer shifts, and navigate evolving market conditions. Traditionally grounded in surveys and expert insights, these methods often lag behind today’s fast-paced digital environment. LLMs are revolutionizing this space by enabling real-time analysis of vast, unstructured data sources, offering marketers faster, deeper, and more forward-looking insights to stay competitive and adaptive in a rapidly changing landscape.
Across marketing research tasks, LLMs do not replace established theories or empirical methods, but instead expand the field’s interpretive and creative capabilities by translating unstructured language into insight, automating content generation, and enhancing strategic awareness. They bridge the gap between human communication and data-driven analysis, enabling more personalized engagement, faster iteration, and richer understanding of consumer behavior and market dynamics.
4.4.2. Consumer Insights and Behavior Analysis
Consumer insights and behavior analysis lie at the heart of marketing strategy, aiming to understand why people think, feel, and act as they do in the marketplace [618]. These insights help businesses tailor products, messaging, and experiences to meet real consumer needs [619,620]. Traditionally, marketers have relied on surveys, interviews, and focus groups to collect this data. While useful, these tools often fall short in capturing the scale, complexity, and subtlety of modern consumer behavior, especially in a digital landscape teeming with unstructured data like reviews, chats, and social posts.
LLMs offer a powerful alternative. With their ability to understand and generate human-like text, LLMs can analyze vast amounts of consumer content, identifying sentiment, extracting themes, and revealing hidden patterns [606]. Unlike manual methods, LLMs can scale effortlessly and respond in real time, making them well-suited for decoding consumer reviews, personalizing communication, and even generating synthetic consumer data. For instance, marketers can use LLMs to simulate customer personas or segment audiences based on open-text responses, bridging gaps left by rigid demographic approaches [600,602,605].
Recent work highlights the breadth of LLM applications in this space. Praveen et al. [603] show that fine-tuned models outperform traditional tools in extracting emotion and sentiment from consumer reviews, especially in service industries. Li et al. [600] demonstrate how LLMs improve consumer segmentation by embedding and clustering open-ended survey data, while also enabling high-accuracy persona simulations. Sarstedt et al. [605] explore the use of “silicon samples”—synthetic respondents generated by LLMs—and suggest they are especially valuable in pretesting or pilot stages, though not always interchangeable with human data.
Wang et al. [602] address this concern by proposing a data augmentation approach that blends real and synthetic data to reduce bias in conjoint analysis. Their method enhances accuracy without the high costs of traditional data collection. Similarly, Goli and Singh [601] find that LLMs can struggle to fully replicate human preferences but show promise in uncovering heterogeneity when guided by structured prompting techniques like “chain-of-thought conjoint”.
Broader applications of LLMs also include content generation, hyperpersonalization, and dynamic customer interaction, as shown in research by Paul et al. [606] and Kumar et al. [604], further emphasizing their role as both analytical engines and interactive tools in modern marketing.
In sum, LLMs are reshaping how consumer insights are gathered and interpreted. While they are not without limitations, their ability to process complex language at scale marks a step-change in understanding and anticipating consumer behavior.
4.4.3. Content Creation and Campaign Design
In the realm of marketing, content creation and campaign design are central to how brands communicate their message and engage audiences. Content creation involves crafting valuable, relevant, and compelling digital materials, such as social media posts, blog entries, videos, or infographics, that resonate with the target audience [621]. Campaign design, on the other hand, is the strategic process of planning and organizing this content across various channels to achieve specific marketing objectives, such as brand awareness, lead generation, or customer loyalty [622,623]. One can think of a brand as a storyteller in a crowded marketplace. The content is the voice, tone, and story, while the campaign design is the stage, spotlight, and choreography that guide the narrative and make sure it reaches the right eyes and ears at the right moment.
Traditionally, content creation and campaign design have relied heavily on human creativity and intuition. Marketers would brainstorm ideas, conduct market research, and produce content manually—a process that is often time-consuming and resource-intensive. Challenges in this traditional approach include difficulties in consistently generating high-quality content, the substantial time and effort required for research and planning, and the complexities involved in tailoring content to diverse audience segments across various platforms. Additionally, measuring the effectiveness of campaigns and obtaining real-time feedback posed significant hurdles, making it challenging to optimize strategies promptly [624].
The advent of LLMs has introduced transformative solutions to these challenges. LLMs are advanced AI systems capable of understanding and generating human-like text, enabling marketers to automate and enhance various aspects of content creation and campaign design [608,612]. For instance, LLMs can generate drafts, headlines, social media captions, and even long-form articles within minutes, significantly boosting productivity and allowing marketers to focus more on strategy and creativity. Furthermore, LLMs can analyze vast amounts of data to personalize content for specific audience segments, ensuring that the messaging resonates more effectively with diverse groups. This personalization extends to creating engaging social media captions that enhance user interaction and brand presence [625].
A growing body of research explores this shift. Reisenbichler et al. [613] proposed a semiautomated content generation framework combining natural language generation (NLG) with human editing to produce SEO-optimized website content. Their findings show that machine-generated content, once refined by humans, can outperform human-written content in search rankings, while also reducing production costs by over 90%. Similarly, Ivanov [612] investigated audience perceptions of AI versus human-generated marketing content and found minimal differences, suggesting that consumers may not distinguish between the two when quality is high. In more experimental territory, Kasuga and Yonetani [607] introduced a CXSimulator that uses LLM embeddings to simulate user behavior and assess the likely outcomes of different marketing campaigns, offering a way to pre-test ideas without real-world deployment.
Meanwhile, Aldous et al. [609] demonstrated how ChatGPT-generated content could outperform human-created posts in terms of emotional appeal and engagement, particularly on platforms like Facebook, highlighting the model’s ability to adapt content to platform-specific norms. This adaptability and personalization have been emphasized as crucial in recent editorial work on generative AI in content marketing [610,611], where the focus is shifting from merely producing content to orchestrating entire advertising campaigns where AI helps strategize, generate, test, and iterate in rapid cycles.
Altogether, these developments point to a broader evolution: content creation and campaign design are becoming increasingly algorithmic, yet no less human. Rather than replacing creativity, LLMs are reconfiguring it—helping marketers shift from being sole creators to becoming skilled orchestrators of human-AI collaboration. As the boundaries blur, what emerges is a hybrid model of marketing practice, powered not only by technology but also by new workflows, new expectations, and new possibilities.
4.4.4. Market Intelligence and Trend Analysis
Market Intelligence (MI) and Trend Analysis play a vital role in modern marketing, serving as the compass by which organizations navigate competitive landscapes and shifting consumer demands. At its core, Market Intelligence involves collecting and analyzing data about market conditions, consumer behavior, competitors, and emerging trends to support strategic decision-making [626,627]. Trend Analysis, in tandem, looks beyond immediate patterns to identify longer-term movements in customer preferences, technology, and broader economic forces. One can think of MI and Trend Analysis as the business equivalent of weather forecasting: companies read the “climate” of the market to prepare for storms, seize sunshine, and stay ahead of seasonal changes.
Traditionally, businesses have relied on surveys, focus groups, expert panels, and basic statistical tools to conduct MI and Trend Analysis. These methods, though foundational, face significant limitations in speed, scalability, and responsiveness. For instance, survey-based data collection can be expensive and time-consuming, often reflecting stale consumer sentiments by the time analysis is complete. Moreover, as Soykoth et al. [616] highlight, the exponential growth of digital data has made it increasingly difficult to extract timely insights using manual or fragmented approaches. The inability to process unstructured data like social media posts, product reviews, or real-time feedback further constrains traditional methods.
This is where LLMs, such as ChatGPT, present a transformative opportunity. LLMs can process and generate human-like language, making them powerful tools for interpreting vast volumes of textual data, uncovering hidden patterns, and even simulating consumer responses. Unlike rigid models that require structured inputs, LLMs can parse unstructured content such as tweets, reviews, news, forum posts and distill meaningful insights with minimal setup. Mutoffar et al. [617] found that ChatGPT can enhance the ability of firms to predict market trends by offering nuanced interpretations of consumer sentiment and behavioral shifts.
Recent research and case studies further illuminate this evolution. For example, Saputra et al. [615] demonstrated that using ChatGPT to generate Instagram marketing content improved campaign effectiveness by driving higher user engagement under the AIDA (Attention, Interest, Desire, Action) framework. These cases suggest not only improved efficiency but also enhanced creativity and adaptability in digital marketing practices. A compelling narrative of progress also emerges from the literature around MarkBot [628], a chatbot framework powered by language models that leverages user-generated content to minimize the lead time for deployment in marketing environments. Similarly, Yeykelis et al. [614] demonstrated that LLMs could replicate human responses in marketing experiments with impressive fidelity, opening the door to AI-powered replication and prediction in media effects research.
These developments also resonate with the data-augmentation framework proposed by Wang et al. [602], who show that LLMs, when properly integrated with real-world data, can dramatically reduce the cost and time associated with conjoint analysis and preference modeling, albeit with careful consideration of biases and calibration.
Taken together, the literature suggests that we are witnessing a pivotal shift: from retrospective, human-labor-intensive MI and Trend Analysis to a forward-looking, AI-augmented ecosystem. While traditional methods still provide foundational rigor, LLMs offer a flexible and scalable supplement—or in some contexts, a powerful alternative. This hybrid approach may well define the next frontier in market intelligence.
4.4.5. Benchmarks
Although dedicated datasets and benchmarks specifically targeting LLM applications in marketing are still scarce, existing large-scale public datasets provide valuable resources for exploratory research and model evaluation. Google BigQuery, in particular, offers several comprehensive datasets related to user behavior, advertising, and public trends. These datasets serve as promising foundations for developing and benchmarking marketing-oriented LLM systems. Below, we list several notable examples (Table 25).
GA4 E-commerce Sample provides anonymized Google Analytics 4 event export data from the Google Merchandise Store. It captures detailed user behavior events such as pageviews, add-to-cart actions, and purchases, enabling analysis of customer journeys, conversion funnels, and marketing campaign effectiveness.
TheLook E-commerce is a synthetic e-commerce dataset designed for business intelligence and analytics training. It includes users, orders, products, inventory, and events data, suitable for customer segmentation, sales forecasting, and conversion analysis.
Google Trends contains search interest data over time for various keywords and regions. It enables marketers to analyze public interest dynamics, track emerging topics, and correlate search patterns with marketing efforts.
GDELT collects worldwide news event metadata, including actors, locations, event types, and sentiment. Marketers can use it to monitor brand exposure, public sentiment, and global events affecting market behavior.
Google Ads Transparency Report provides data on political and issue-based advertising on Google’s platforms, including ad content, spending, and targeting. It is valuable for studying advertising strategies, campaign transparency, and audience targeting trends.
These datasets significantly expand the opportunities for applying LLMs in marketing research. They offer rich, diverse, and real-world sources of structured and unstructured information that LLMs can effectively process to generate actionable insights. For example, LLMs can be used to perform large-scale customer journey analysis by synthesizing event-level data from GA4 exports, or to extract latent market trends by analyzing temporal patterns in Google Trends data. TheLook E-commerce dataset enables training and evaluation of LLM-based models for tasks such as automated customer segmentation, personalized recommendation generation, and sales forecasting. Similarly, the GDELT database allows LLMs to monitor and summarize brand mentions, sentiment shifts, and emerging crises across global news streams, supporting real-time market intelligence applications. The Ads Transparency Report can serve as a foundation for studying advertising strategies, targeting behaviors, and political messaging dynamics through LLM-driven content analysis and clustering. Together, these datasets provide a practical and scalable substrate for developing, fine-tuning, and benchmarking LLM systems in marketing contexts, advancing both academic research and industry practice.
4.4.6. Discussion
Opportunities and Impact. The application of LLMs in marketing research presents transformative opportunities across multiple domains. As seen in consumer insights [600,602,605], content creation [609,612,613], and market intelligence [602,617], LLMs greatly enhance scalability, speed, and analytical depth. In consumer insights, LLMs uncover latent sentiments and behavioral patterns from vast unstructured data. In content creation, they accelerate ideation and enable hyper-personalization. In market intelligence, they provide real-time synthesis of competitive landscapes and emerging trends.
A 2024 AMA survey reports that 90% of marketers now use generative AI, with 71% using it weekly and nearly 20% daily. Over 85% report productivity gains, and half cite improvements in both the quality and quantity of creative output [629]. These developments show that LLMs are not just automation tools but strategic partners redefining creativity and decision-making in marketing.
Challenges and Limitations. Despite their considerable promise, deploying LLMs in marketing research raises important challenges. First, bias and hallucination risks persist. LLMs trained on broad internet corpora can perpetuate stereotypes or generate plausible yet inaccurate outputs, which, if unrecognized, could misguide marketing strategies [605,606]. Second, lack of causal inference and experimental rigor limits LLMs’ ability to replace traditional empirical methods like randomized controlled trials [595]. Their outputs, while rich, are often correlational rather than causal, necessitating careful interpretation and external validation. Third, prompt sensitivity and reproducibility concerns complicate the use of LLMs for systematic research. Slight variations in prompting or task framing can yield inconsistent results [602,605], undermining reliability. Finally, ethical considerations arise regarding transparency, authorship, and authenticity, especially as AI-generated content becomes increasingly indistinguishable from human-created work [610].
Research Directions. Addressing these challenges points to several important research directions:
- Hybrid Approaches Combining LLMs with Human Oversight. Structuring workflows where human judgment complements AI output will help mitigate risks of error, bias, and unethical deployment.
- Domain-Specific Fine-Tuning and Contextualization. Fine-tuning LLMs on marketing-specific corpora and continuously updating them with domain-relevant data can improve accuracy, nuance, and relevance in marketing applications [617].
- Benchmarking and Standardized Evaluation. Creating rigorous benchmarks for LLM performance in marketing tasks, such as synthetic persona realism, content personalization efficacy, and market trend prediction accuracy, will be vital for advancing scientific rigor.
- Explainability and Interpretability. Incorporating methods such as chain-of-thought prompting, citation grounding, and attribution tracing will enhance transparency and user trust in LLM-assisted marketing insights [602].
- Ethical Guidelines and Best Practices. Marketing researchers must proactively develop frameworks for ethical AI use, covering disclosure norms for AI-generated content, fairness in segmentation, and protection against manipulative targeting.
Conclusion. LLMs are redefining the landscape of marketing research by unlocking new capacities for insight generation, content creation, and strategic analysis. However, their integration must be thoughtful, combining the creative power of human marketers with the linguistic and analytical capabilities of AI. Moving forward, marketing will likely evolve into a hybrid discipline, where the agility, scale, and personalization offered by LLMs complement traditional empirical rigor, creativity, and ethical responsibility to shape a more dynamic, insightful, and consumer-centered future.
5. LLMs for Science and Engineering
In this chapter, we chart how LLMs are utilized across science and engineering. The chapter spans mathematics; physics and mechanical engineering; chemistry and chemical engineering; life sciences and bioengineering; earth sciences and civil engineering; and computer science and electrical engineering. We open with mathematics—proof support, theoretical exploration and pattern discovery, math education, and targeted benchmarks. In physics and mechanical engineering, we cover documentation-centric tasks, design ideation and parametric drafting, simulation-aware and modeling interfaces, multimodal lab and experiment interpretation, and interactive reasoning, followed by domain-specific evaluations and a look at opportunities and limits. In chemistry and chemical engineering, we examine molecular structure and reaction reasoning, property prediction, materials optimization, test/assay mapping, property-oriented molecular design, and reaction-data knowledge organization, then compare benchmark suites. In life sciences and bioengineering section, we include genomic sequence analysis, clinical structured-data integration, biomedical reasoning and understanding, and hybrid outcome prediction, with emphasis on validation standards. In earth sciences and civil engineering section, we review geospatial and environmental data tasks, simulation and physical modeling, document workflows, monitoring and predictive maintenance, plus design/planning tasks, again with benchmarks. We close with computer science and electrical engineering: code generation and debugging, large-codebase analysis, hardware description language code generation, functional verification, and high-level synthesis, capped by purpose-built benchmarks and a final discussion of impacts and open challenges.
5.1. Mathematics
5.1.1. Overview
Introduction. Mathematics is the study of abstract structures and relationships conceived through axiomatic systems and logical deduction, focusing on quantities, shapes, patterns, and transformations [630,631,632]. It provides a framework for formulating hypotheses precisely, exploring them using consistent rule-based reasoning, and deriving theorems that remain valid whenever the initial assumptions hold. In other words, mathematics is a way of understanding and describing the world by looking at patterns, shapes, and numbers. Contemporary mathematical research encompasses a vast landscape, covering both the abstract realms of pure mathematics and the practical studies of applied mathematics [633,634,635]. Pure mathematical research focuses on the investigation and creation of original mathematical concepts, seeking to advance mathematical knowledge without immediate practical applications [636]. In contrast, applied mathematics is concerned with developing mathematical techniques for applications in science and other domains, or leveraging techniques from other fields to contribute to mathematics [637,638].
Mathematical research heavily relies on systematic approaches for problem-solving, i.e., Polya’s 4-step process [639]. Specifically, it introduces a general framework, including (i) understanding the problem; (ii) devising a plan to solve it; (iii) carrying out the plan, and (iv) looking back at the solution to review and reflect. Despite the success of this research method, mathematical research still faces several limitations. First, the nature of mathematical exploration often requires significant investments in time and resources. Solving a mathematical research problem can often extend over months or even years, requiring sustained effort and deep concentration [640]. For instance, reviewing relevant mathematical papers can be time-consuming and require considerable effort and in-depth knowledge of the corresponding field. Besides, traditional mathematical research can also present considerable barriers to entry and foster challenges in collaboration [641]. Acquiring the necessary background knowledge to contribute to a new field of mathematics often requires years of dedicated study. Even among senior researchers, effective collaboration can be hindered by the need for a shared understanding and expertise across different, often highly specialized, subdomains of mathematics. Moreover, the rigorous process of formalizing arguments into a mathematical format that can be verified by machines is often a laborious and long process. Furthermore, the vastness and complexity of certain mathematical spaces can also pose significant limitations for traditional research methods. Working with concepts involving infinite spaces and incredibly complex sets of equations can often stretch the limits of human intuition and comprehension. Similarly, pattern identification within extremely large datasets or highly complex mathematical systems can be extremely difficult for humans to perform manually [642]. Next, as a fundamental discipline, mathematical education has drawn significant attention recently. Traditional mathematics education, prevalent in the early to mid-20th century, typically involves direct instruction where teachers explicitly explain how to solve specific types of problems, with several standard methods [643]. This approach emphasizes procedural methods, formula memorization, and repetitive practice to master mathematical concepts. Skills and concepts are usually taught in a logical sequence, with a focus on building foundational arithmetic skills before progressing to more complex topics like algebra and geometry, which were traditionally reserved for high school. Standardized testing is frequently used to assess understanding and retention. Several studies have argued for the limitations and challenges of traditional mathematical education. For example, a study [644] mentions that traditional methods overemphasize memorization and rote learning, failing to promote a deep conceptual understanding of mathematical principles. Students may be able to perform calculations without understanding how the procedures work. In addition, Peter, E. E.(2012) [645] claims that focusing on procedural fluency can negate the development of critical thinking and problem-solving skills needed to apply mathematical knowledge in novel situations and real-world contexts.
Figure 14.
Overview of topics in Mathematics Discipline with LLMs solution.
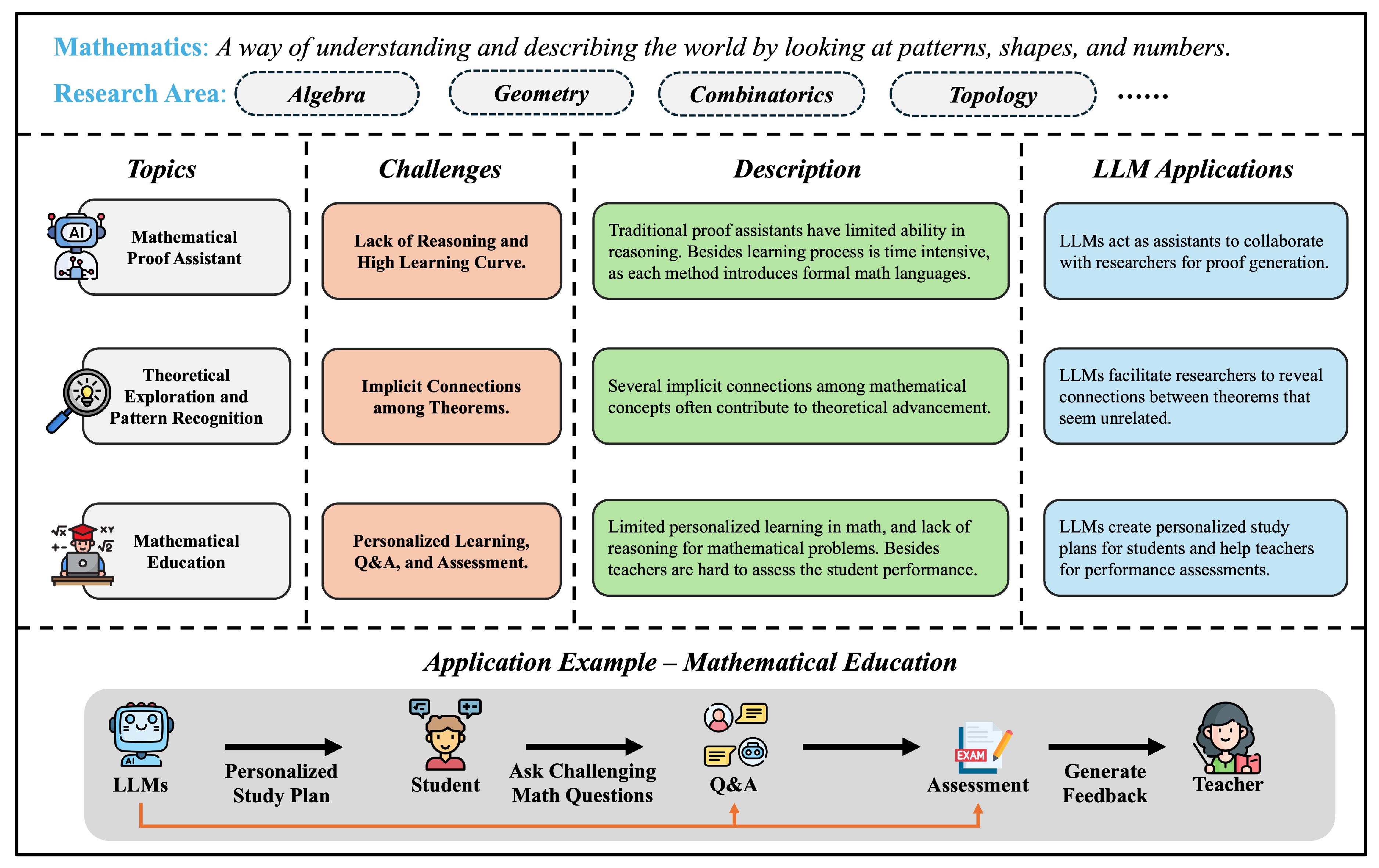
Role of LLMs. LLMs offer a promising avenue to address several limitations inherent in traditional mathematical research. One of the most significant potential contributions lies in assisting with proof development and verification [646]. LLMs can assist mathematicians in trivial tasks, such as identifying relevant lemmas and previously established theorems that might apply to the current proofs and formalizing mathematical arguments into rigorous proofs, allowing researchers to focus on more creative and conceptual tasks [647]. LLMs are also capable of generating novel conjectures and hypotheses [648]. By analyzing vast quantities of mathematical data and literature, LLMs can automatically identify patterns and propose new conjectures or relationships that might not be immediately apparent to human researchers. Another significant benefit of LLMs is their potential for lowering barriers to entry and enhancing accessibility within mathematics. LLMs are capable of accurately explaining core mathematical terms and fundamental intuitions, making specialized knowledge more accessible to researchers who are new to fields. Moreover, LLMs have potential in assisting teachers for mathematical education by providing personalized learning experiences, enhanced conceptual understanding, development of problem-solving skills, and automated assessment and feedback to students.
Limitations of LLMs. Despite the potential of LLMs to assist in mathematical research, several drawbacks and limitations must be carefully considered. Firstly, several studies [649,650] have discussed the limitations in genuine logical inference exhibited by LLMs. Consequently, the performance of LLMs on mathematical tasks can significantly decline as the complexity of the questions increases, particularly when dealing with unseen or novel problems [651]. LLMs may also face difficulties with abstract concepts and long-term reasoning [652,653,654], and may raise the risks of errors and hallucinations.
Taxonomy. To summarize the main applications of LLMs in mathematical research, we introduce the following taxonomy: (i) Mathematical Proof Assistant; (ii) Theoretical Exploration and Pattern Recognition; and (iii) Mathematic Education.
5.1.2. Mathematical Proof Assistant
The motivation for developing machine-assisted proof generation tools stemmed from the fact that proofs for some complex mathematical problems may be extremely lengthy, involving numerous logical steps or extensive case-by-case analysis (a.k.a. proof by exhaustion) [664,672]. Computer algorithms, embedded with rigorous deductive rules, could assist researchers by verifying the correctness of a proof or by automatically handling the proof-by-exhaustion steps. For a more detailed discussion about earlier automated reasoning and machine-assisted proof approaches, we refer readers to studies [673,674].
Traditional proof assistants, i.e., MetaMath [675], Isabelle/HOL [676], Rocq (Coq) [677], and Lean [678], aim to create mathematical proofs or verify the correctness of the proofs through structured logical frameworks. However, these tools cannot generate the reasoning steps and only verify the steps provided by users. Moreover, studying to utilize these systems may be time-consuming, as each method introduces a formal language to express mathematical statements. With the advances of LLMs, several studies [656,657] develop automated theorem-proving tools to facilitate researchers to generate reasoning steps or convert mathematical statements into formal languages. GPT-f [657], PACT [658], Thor [659] introduce language model-based automated theorem proving tools interfacing with Metamath, Lean, and Isabelle/HOL, respectively. Several advanced methods are further proposed to train LLMs through annotated datasets for proof assistant tasks, such as HyperTree Proof Search (HTPS) [660], Baldur [661], COPRA [662], LeanDojo [663], AlphaProof [655], and Lyra [679]. To further demonstrate the effectiveness of LLMs of mathematical proof assistants, compared with conventional methods, we provide the performance of several LLMs-baed methods and tree search methods in Table 27. According to the Table, we find that (i) Conventional approaches, i.e., tree search methods generally exhibit lower accuracies compared to the highest performing LLM-based methods, with hypertree proof search achieving 41.0% and curriculum learning methods ranging between 29.2% and 36.6%. (ii) LLM-based methods show variability in performance based on the sample budget, evidenced by DeepSeekMath’s performance improvement from 27.5% with 128 samples to 52.0% under a cumulative sampling strategy. (iii) Increasing the sample budget for methods such as DeepSeek-Prover (from 64 to 65,536 samples) leads to consistent accuracy gains, reaching up to 50.0% at the highest sample count.
Most recently, the breakthrough by Google DeepMind with the development of AlphaEvolve [680], highlights its significance as a transformative moment in the intersection of artificial intelligence and mathematics. Specifically, AlphaEvolve employs evolutionary programming and self-play, leveraging LLMs to autonomously generate and refine algorithms. Unlike FunSearch, which searches for single functions, AlphaEvolve is capable of optimizing entire codebases, including the interactions between different functions. However, AlphaEvolve still relies on human expertise in identifying interesting problems, defining clear evaluation metrics, and incorporating promising solutions into the iterative cycle. Several advancements of AlphaEvolve in the mathematical domain can be summarized as follows:
- Advancing the hexagon packing problem by finding better solutions for arranging 11 and 12 hexagons inside a larger hexagon, surpassing human achievements after 16 years of stagnation.
- Making progress on the kissing number problem, a mathematical challenge unsolved for over 300 years [683].
Table 27.
Performance of LLMs-based and traditional proof assistants over miniF2F [684].
Table 27.
Performance of LLMs-based and traditional proof assistants over miniF2F [684].
| Method | Model Size | Sample Budget | Accuracy |
| Traditional Proof Assistants | |||
| Curriculum Learning [685] | 837M | 34.5% | |
| 837M | 36.6% | ||
| Proof Artifact Co-Training [658] | 837M | 24.6% | |
| 837M | 29.2% | ||
| Hypertree Proof Search [660] | 600M | 41.0% | |
| LLMs-based Methods | |||
| DeepSeekMath [686] | 7B | 128 | 27.5% |
| 7B | Cumulative | 52.0% | |
| 7B | Greedy | 30.0% | |
| DeepSeek-Prover [687] | 7B | 64 | 46.3% |
| 7B | 128 | 46.3% | |
| 7B | 8,192 | 48.8% | |
| 7B | 65,536 | 50.0% | |
5.1.3. Theoretical Exploration and Pattern Recognition
Several major theoretical advances often revolve around revealing deep connections among mathematical concepts that seem unrelated [664]. A few studies have introduced leveraging LLMs to collaborate with researchers for theoretical exploration. For example, a recent study [665] leverages LLMs for conjecture generation within the Isabelle/HOL, and G-LED [666] explores the potential of combining transformer models and diffusion models to forecast complex dynamical systems. Their ability to process and understand external knowledge allows them to identify non-obvious connections and generate novel mathematical statements that can then be explored and potentially proven by mathematicians. A prominent example in this direction is FunSearch [648], an evolutionary procedure that pairs a pre-trained LLM with a systematic evaluator to discover new constructions and heuristics for established open problems.
5.1.4. Mathematical Education
LLMs present a dual-edged potential for mathematics education. They can offer personalized learning experiences by tailoring problems and explanations to individual student needs [667,669,670]. LLMs can also enhance conceptual understanding by generating diverse explanations and providing step-by-step solutions. A recent study [667] investigates the impact of LLMs, such as GPT-4, on mathematics learning. As discussed in the study, educators face concerns that students might use these tools to bypass genuine learning, but there is also hope that LLMs could serve as scalable, personalized tutors. The study aims to provide empirical evidence on whether and how LLM-generated explanations affect student learning in math. In Table 28, we present the performance of students in mathematical problem solving, as reported in the study [667]. The column Strategy refers to the method used to generate explanations for the questions, i.e., "Answer Only" indicates that no explanation is provided to the students, "Stock LLM" means the explanation is generated by an official LLM, and "Customized LLM" denotes that the explanation is tailored to the student. Moreover, "See Answer First" means that students receive both the question and the correct answer before attempting to solve it, while "Try It First" denotes that students initially attempt to solve the problem on their own before receiving the correct answer for practice. According to Table 28, we find that: (i) Participants who received LLM-generated explanations performed better on subsequent test questions than those who saw only correct answers. (ii) the greatest learning gains occurred when participants attempted problems, i.e., "Try It First", on their own before consulting the LLM explanations. Besides facilitating student in mathematic learning, they can assist teachers in creating engaging problems and automate assessment, potentially freeing educators for more direct student interaction [671]. However, LLMs are limited by their lack of true mathematical reasoning and conceptual understanding, often relying on pattern matching rather than genuine logic. This can lead to issues with reliability, accuracy, and the generation of incorrect information. Additionally, current LLMs struggle with visual and spatial reasoning, which are crucial in mathematics. Therefore, while LLMs can serve as valuable tools to augment mathematics education, they are not a replacement for human instruction and require careful oversight.
5.1.5. Benchmarks
Recent advances in LLMs have necessitated the development of sophisticated benchmarks to access mathematical reasoning capabilities. In Table 29, we list most existing mathematic datasets, which can categorized into four groups: comptition-level, education-focus, math word problem, and mathetmic reasoning. In the following paragraphs, we will introduce key benchmarks in detail.
MATH. The Mathematics Aptitude Test of Heuristics (MATH) dataset remains a foundational benchmark in examining the mathematic abilities of LLMs. MATH comprises 12,500 problems from prestigious competitions including AMC 10, AMC 12, and AIME. Each problem has a full step-by-step solution, which can be used for each model to generate answer derivations and explanations. Moreover, MATH contains the following features: (i) Five difficulty levels mirroring human problem-solving capabilities (ii) Detailed LaTeX-formatted solutions with boxed answers (iii) Coverage of seven mathematical domains including combinatorics and number theory. A smaller version of MATH, i.e., MATH 500, consists of 500 diverse problems from MATH, which serves as a standardized evaluation set for benchmarking and comparing the mathematical reasoning abilities of LLMs in a more manageable and reproducible way.
AIME. American Invitational Mathematics Examination (AIME) is a collection of problems for a prestigious high school mathematics competition in the US and Canada. Furthermore, each annual AIME examination comprises 15 questions, with each solution represented as a three-digit integer ranging from 000 to 999. The complexity of the problems escalates as the examination advances. The sample in AIME includes the year, problem number, the full problem statement, and the correct answer.
GSM8K. Grade School Math 8K (GSM8K) is a dataset of 8,500 high quality linguistically diverse grade school math word problems, designed for evaluating and training models on mathematical problem-solving tasks. These problems commonly require 2 to 8 steps to solve and solutions are provided in natural languages.
The performance of existing LLMs on the aforementioned datasets is provided in Table 30. According to the table, we find that: (i) DeepSeek R1 scores 97.30 in MATH, which appears to outperform several models. (ii) In MATH 500 benchmark, GPT-o1 is particularly strong here with , closely followed by DeepSeek R1 at approximately (iii) For AIME benchmarks, Grok 3 achieves the best performance in both years, i.e., 93.30%. (iv) Most models that display high accuracies on GSM8K. For instance, Qwen 2.5 and DeepSeek R1 are in the mid-to-hgih 90 percent range, with DeepSeek R1 at and Qwen 2.5 at . GPT-o1 and GPT-o3-mini also showcase robust performance with scores around and respectively. The performance of LLMs in GSM8K showcase that for elementary math and reasoning, most models achieve near ceiling performance.
Overall, models, DeepSeek R1 and certain GPT variants (especially GPT-o1 and GPT-o3-mini) demonstrate consistent performance across various benchmarks, making them attractive for applications that require a broad spectrum of math problem-solving capabilities. Although some models, such as GPT-o1, have a higher cost per token, their performance on certain benchmarks, i.e., MATH 500, might justify the expense for research or high-stakes applications. In contrast, DeepSeek R1 offers a very competitive performance at a much lower token cost. The performance variation across different benchmarks, i.e., MATH vs. AIME vs. GSM8K, suggests that each dataset poses unique challenges. For instance, most models perform exceptionally well on GSM8K, while the AIME benchmarks reveal differentiators in advanced problem-solving capabilities.
5.1.6. Discussion
Opportunities and Impact. LLMs offer significant opportunities to enhance both mathematical research and education. In mathematical research, LLMs are capable of processing and synthesizing large amounts of information to facilitate researchers in literature reviews, novel conjecture generation, and even assist in theorem proving. Frameworks like FunSearch demonstrate the potential for LLMs to contribute to mathematical discovery by identifying hidden patterns. This can free up human mathematicians to focus on higher-level conceptual work and tackle problems previously deemed intractable due to computational complexity. The impact could be a faster pace of discovery and a broader exploration of mathematical landscapes. As shown in Table 27, LLMs demonstrate better performance than traditional proof assistants in the miniF2F dataset, showcasing the potential of LLMs as assistants in mathematical research.
From the mathematical education perspective, LLMs can enhance personalized learning experiences by adapting to individual student’s needs and providing tailored reasoning. They can offer support for conceptual understanding by presenting information in multiple ways and aid in the development of problem-solving skills through engaging and relevant problems. As listed in Table 28, students who receiving the LLM-generate explanations performed better than these who receive only correct answers, demonstrating the potential of LLMs in personalized mathematical learning. Automated assessment and feedback can also streamline the educational process, allowing teachers to focus on individual student support. Ultimately, LLMs have the potential to make mathematics more accessible and engaging for a wider range of learners.
Challenges and Limitations. Despite the promising opportunities, significant challenges and limitations exist in the application of LLMs to mathematics. A primary concern is their lack of mathematical reasoning and deep conceptual understanding. LLMs primarily operate through pattern matching and may struggle with multi-step reasoning, especially when faced with irrelevant information. Moreover, it may face hallucinations which are still unknown the causes, indicating the necessity for human verifications. The inherent limitations in novelty and the potential for LLMs to merely reproduce existing knowledge raise questions about their ability to drive truly breakthrough discoveries without significant human guidance.
In education, these limitations translate to concerns about the reliability of LLM-generated explanations and solutions. LLMs struggle with visual and spatial reasoning poses a challenge for teaching, especially in geometric topics. Over-reliance on LLMs could also hinder the development of fundamental mathematical skills and critical thinking in students. Furthermore, ethical considerations regarding bias in training data and the potential impact on the integrity of mathematical knowledge need careful attention.
Future Research Directions. Future research should focus on addressing the limitations of LLMs in mathematics and exploring effective ways to integrate them into research and education. Here we summarize several key research directions:
- Enhancing Mathematical Reasoning. Developing novel architectures and training recipes that enable LLMs to move beyond pattern matching toward genetic models. This could involve incorporating symbolic computation capabilities or training on datasets designed to specifically test and improve reasoning skills.
- Improving Reliability and Accuarcy. Investigating methods to reduce hallucinations and errors in LLM outputs for mathematical tasks. This could involve techniques like self-verification, the use of external validators like theorem provers, or reinforcement learning from human feedback focused on accuracy.
- Effective Educational Tools. There is a need for designing and evaluating LLMs-empowered tools that effectively support mathematics learning without compromising conceptual understanding or fundamental skill development. This includes exploring the utilization of LLMs in personalized tutoring, generating diverse explanations, and creating engaging problem-solving activities.
- Integration Strategies Evaluation. Exploring optimal strategies for incorporating LLMs into conventional educational practices to develop hybrid learning environments that capitalize on the advantages of both methodologies is crucial. Additionally, it is vital to comprehend how to effectively instruct educators in the utilization of LLMs as pedagogical tools.
Conclusion. The integration of LLMs into the landscape of mathematical research and education presents a transformative potential, but it is crucial to approach this integration with a balanced perspective. While LLMs offer exciting opportunities to augment human capabilities, accelerate discovery, and personalize learning, they are not without significant limitations, particularly in the core areas of mathematical reasoning and reliability. Traditional methods in both research and education, with their emphasis on rigor, conceptual understanding, and human intuition, remain indispensable.
We want to emphasize that LLMs should serve as powerful tools to assist mathematicians and educators, rather than replacing them. By focusing on research that addresses the current limitations of LLMs and by thoughtfully integrating them into educational practices, we can harness their potential to advance mathematical knowledge and foster a deeper and more accessible understanding of mathematics for all. We believe the journey of integrating LLMs into mathematics is still in its early stages, and ongoing research, collaboration, and critical evaluation will be essential to navigate this evolving landscape effectively.
5.2. Physics and Mechanical Engineering
5.2.1. Overview
5.2.1.1 Introduction to Physics
Physics is a natural science that investigates the fundamental principles governing matter, energy, and their interactions through experimental observation and theoretical modeling [702]. It spans from the smallest subatomic particles to the largest cosmic structures, aiming to establish predictive, explanatory, and unifying frameworks for natural phenomena [703]. As the most fundamental natural science, physics provides conceptual foundations and methodological tools that support other sciences and engineering disciplines [704].
Simply put, physics is the science of understanding how the world works. It seeks to explain everyday phenomena, such as why apples fall, why lights turn on, or why we can see stars [702]. It not only provides explanations but also empowers us to harness these laws to develop new technologies [705].
For example, the detection of gravitational waves marked one of the most significant breakthroughs in 21st-century physics [706]. First predicted by Einstein’s theory of general relativity, gravitational waves had eluded direct observation for a century due to their extremely weak nature [707]. In 2015, the LIGO interferometer in the United States successfully captured signals produced by the collision of two black holes [706]. This discovery not only confirmed theoretical predictions but also launched a new field—gravitational wave astronomy [708]—with far-reaching impacts on astrophysics, cosmology, and quantum gravity [709,710].
The discipline of physics is vast and typically organized into three core domains, each addressing a class of natural phenomena and associated methodologies [711]:
Fundamental Theoretical Physics. This domain focuses on uncovering the basic laws of nature and forms the theoretical foundation of the entire field of physics [712]. It encompasses classical mechanics, electromagnetism, thermodynamics, statistical mechanics, quantum mechanics, and relativity [712]. The scope ranges from macroscopic motion (e.g., acceleration, vibration, and waves) to the behavior of subatomic particles (e.g., electron transitions, spin, self-consistent fields), and includes the structure of spacetime under extreme conditions such as near black holes [713]. Researchers in this domain employ abstract mathematical tools such as differential equations, Lagrangian and Hamiltonian mechanics, and group theory to construct theoretical models and derive predictions [714]. These models not only guide experimental physics but also provide essential frameworks for engineering applications [715].
Physics of Matter and Interactions. This domain explores the microscopic structure of matter, the interaction mechanisms across different scales, and how these determine macroscopic material properties [716]. Major subfields include condensed matter physics, atomic physics, molecular physics, nuclear physics, and particle physics [717,718]. Key research topics cover crystal structure, electronic band theory, spin and magnetism, superconductivity, quantum phase transitions, and the classification and interaction of elementary particles [716,718]. Common methodologies include first-principles calculations, quantum statistical modeling, and large-scale experiments involving synchrotron radiation, laser spectroscopy, or high-energy accelerators [719]. The findings from this domain have led to breakthroughs in semiconductor devices, materials development, quantum computing, and energy systems, driving significant technological innovation [720,721].
Cosmic and Large-Scale Physics. This domain addresses phenomena at scales far beyond laboratory conditions and includes astrophysics, cosmology, and plasma physics [722]. It investigates topics such as stellar evolution, galactic dynamics, gravitational wave propagation, early-universe inflation, and the nature of dark matter and dark energy [723]. In parallel, plasma physics explores phenomena like solar wind, magnetospheric dynamics, and the behavior of ionized matter in space environments [724]. Research in this domain typically relies on astronomical observations (e.g., telescopes, gravitational wave detectors, space missions), theoretical models, and large-scale simulations, forming a triad of “observation + simulation + theory” [725].
Together, these three domains constitute the intellectual architecture of modern physics [726]. From particle collisions in ground-based accelerators to observations of distant galaxies, physics consistently strives to understand the most fundamental laws of nature [727]. With the emergence of artificial intelligence, high-performance computing, and advanced instrumentation, the research landscape of physics continues to expand—becoming increasingly intelligent, interdisciplinary, and precise [728].
Figure 15.
The relationships between major research tasks between physics and mechanical engineering.
Figure 15.
The relationships between major research tasks between physics and mechanical engineering.
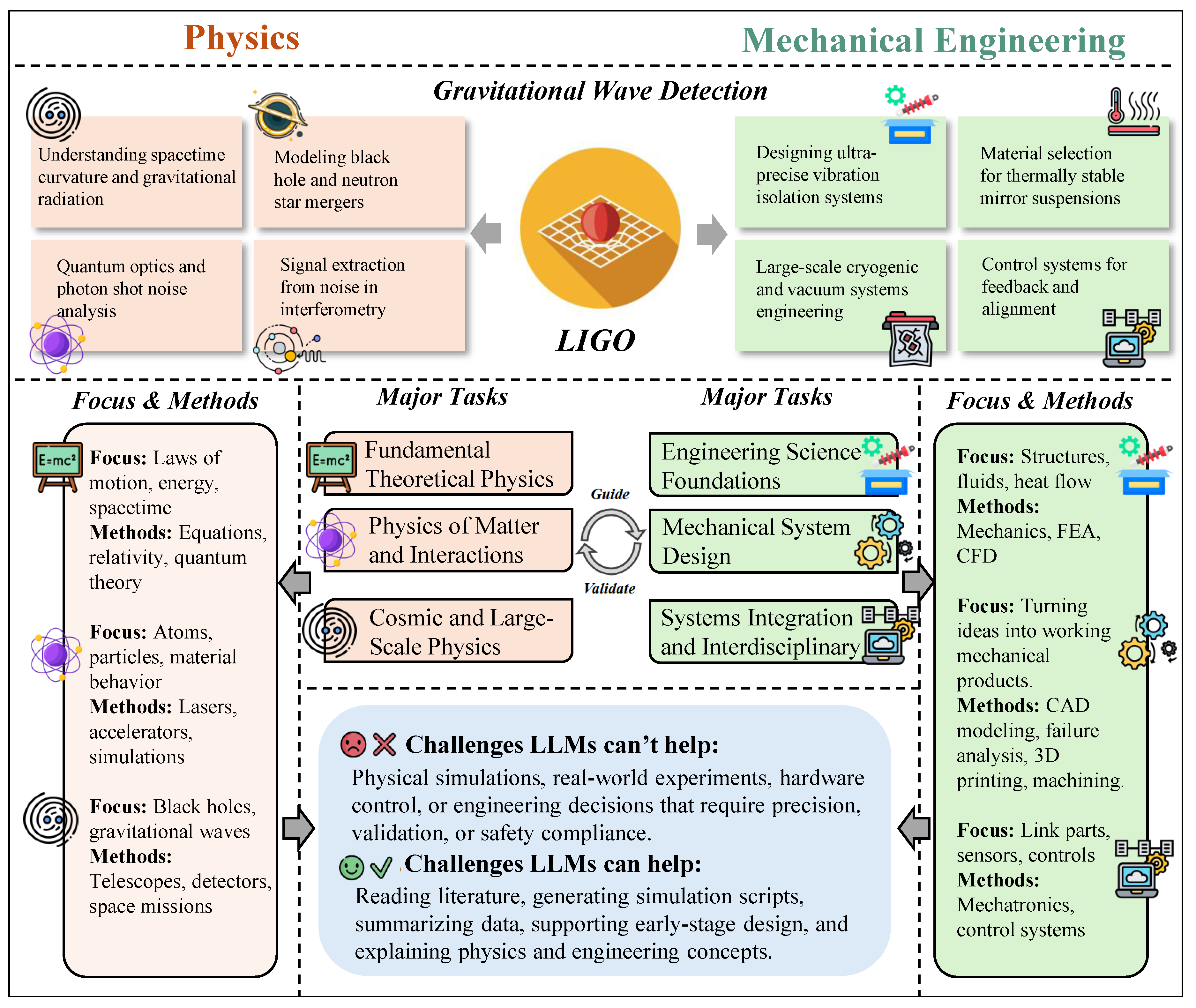
5.2.1.2 Introduction to Mechanical Engineering
Mechanical engineering is an applied discipline that focuses on the design, analysis, manufacturing, and control of mechanical systems driven by the principles of force, energy, and motion [729]. It integrates engineering mechanics, thermofluid sciences, materials engineering, control theory, and computational tools to solve problems across a wide range of industries. As a foundational engineering field, mechanical engineering provides the backbone for technological advancement in transportation, energy, robotics, aerospace, and biomedical systems [730].
In simple terms, mechanical engineering is the science and craft of making things move and work reliably. From engines and turbines to robots and surgical devices, it turns physical principles into functional products through design and manufacturing [730].
For example, the construction of the LIGO gravitational wave observatory represents not only a milestone in fundamental physics but also a triumph of mechanical engineering. LIGO’s ultrahigh-vacuum interferometers required vibration isolation at nanometer precision, thermally stable mirror suspensions, and large-scale structural systems integrated with active control. These engineering feats enabled the detection of gravitational waves, a task demanding extreme precision in structure, thermal regulation, and dynamic stability [706].
The field of mechanical engineering is vast and is typically categorized into three core domains, each covering a range of methodologies and applications:
Engineering Science Foundations. This domain forms the analytical and physical core of mechanical engineering. It encompasses: Mechanics: including statics, dynamics, solid mechanics, and continuum mechanics, used to model the deformation, motion, and failure of structures [731]. Thermal and fluid sciences: covering heat conduction, convection, fluid dynamics, thermodynamics, and phase-change phenomena [732]. Systems and control: involving system dynamics, feedback control theory, and mechatronic integration [733]. These fundamentals are implemented via tools such as finite element analysis (FEA), computational fluid dynamics (CFD), and system modeling platforms like Simulink and Modelica [734,735].
Mechanical System Design and Manufacturing. This domain addresses how ideas become real-world engineered products. It includes: Mechanical design: CAD modeling, mechanism design, stress analysis, failure prediction, and design optimization [736]. Manufacturing: traditional subtractive methods (e.g., milling, turning), additive manufacturing (3D printing), surface finishing, and process planning [737]. Smart manufacturing and Industry 4.0: integration of sensors, data analytics, automation, and cyber-physical systems to create responsive and intelligent production environments [738]. These technologies bridge the gap between virtual design and physical realization.
Systems Integration and Interdisciplinary Applications. Modern mechanical systems are often multi-functional and cross-disciplinary. This domain focuses on: Robotics and mechatronics: combining mechanics, electronics, and computing to build intelligent machines [739]. Energy and thermal systems: engines, fuel cells, solar collectors, HVAC systems, and sustainable energy technologies [740]. Biomedical and bioinspired systems: development of prosthetics, surgical tools, and biomechanical simulations [741]. Multiphysics modeling and digital twins: simulation of systems involving coupled fields (thermal, fluidic, mechanical, electrical) and virtual prototyping [742]. This integration-driven domain reflects mechanical engineering’s evolution toward intelligent, efficient, and adaptive systems. Together, these three domains define the scope of modern mechanical engineering. From high-speed trains and wind turbines to nanomechanical actuators and wearable exoskeletons, mechanical engineers shape the physical world with ever-growing precision and complexity [729,730].
5.2.1.3 Current Challenges
Physics and mechanical engineering are closely interwoven disciplines that form the foundation for understanding and shaping the material and technological world. Physics seeks to uncover the fundamental laws of nature that govern matter, motion, energy, and forces, while mechanical engineering applies these principles to design, optimize, and control systems that power modern life. Together, they enable critical innovations across transportation, energy, manufacturing, healthcare, and space exploration. These disciplines are indispensable for solving complex, cross-scale challenges such as energy efficiency, automation, sustainable mobility, and precision instrumentation. Despite rapid progress in theoretical modeling, simulation, and intelligent design tools, both fields continue to grapple with the intricacies of nonlinear dynamics, multiphysics coupling, and real-world uncertainties in physical systems.
Still Hard with LLMs: The Tough Problems.
- Complexity of Multiphysics Coupling and Governing Equations. Physical and mechanical systems are often governed by a series of highly coupled partial differential equations (PDEs), involving nonlinear dynamics, continuum mechanics, thermodynamics, electromagnetism, and quantum interactions [734,743]. Solving such systems requires professional numerical solvers, high-fidelity discretization techniques, and physics-informed modeling assumptions. Although LLMs can retrieve relevant equations or suggest approximate forms, they are incapable of deriving physical laws, ensuring conservation principles, or performing accurate numerical simulations.
- Simulation Accuracy and Model Calibration. Accurate mechanical design and physical predictions typically rely on high-fidelity simulations such as finite element analysis (FEA), computational fluid dynamics (CFD), or multiphysics modeling [744,745]. These simulations demand precise geometry input, boundary conditions, material models, and experimental validation. LLMs may assist in interpreting simulation reports or proposing modeling strategies, but they lack the resolution, numerical rigor, and feedback integration necessary to execute or validate such models.
- Experimental Prototyping and Hardware Integration. Engineering innovations ultimately require validation through physical experiments—building prototypes, tuning actuators, installing sensors, and measuring performance under dynamic conditions [746,747]. These tasks depend on laboratory facilities, fabrication tools, and hands-on experimentation, all of which are beyond the operational scope of LLMs. While LLMs can help generate test plans or documentation, they cannot replace real-world testing or iterative hardware development.
- Materials and Manufacturing Constraints. Real-world engineering designs must account for constraints such as thermal stress, fatigue life, manufacturability, and cost-efficiency [748]. Addressing these challenges often relies on materials testing, manufacturing standards, and domain experience in processes like welding, casting, and additive manufacturing. LLMs lack access to real-time physical data and material behavior, and thus cannot support tradeoff decisions in design or production.
- Ethical, Safety, and Regulatory Considerations. From biomedical devices to autonomous systems, mechanical engineers must weigh ethical impacts, user safety, and legal compliance [749]. Although LLMs can summarize policies or regulatory codes, they are not equipped to make decisions involving responsibility, risk evaluation, or normative judgment—elements essential for deploying certified, real-world systems.
Easier with LLMs: The Parts That Move.
- Although current LLMs remain limited in core tasks such as physical modeling and experimental validation, they have shown growing potential in assisting a variety of supporting tasks in physics and mechanical engineering—particularly in knowledge integration, document drafting, design ideation, and educational support:
- Literature Review and Standards Lookup. Both disciplines rely heavily on technical documents such as material handbooks, design standards, experimental protocols, and scientific publications. LLMs can significantly accelerate the literature review process by extracting key information about theoretical models, experimental conditions, or engineering parameters. For instance, an engineer could use an LLM to compare different welding codes, retrieve thermal fatigue limits of materials, or summarize applications of a specific mechanical model [750,751].
- Assisting with Simulation and Test Report Interpretation. In simulations such as finite element analysis (FEA), computational fluid dynamics (CFD), or structural testing, LLMs can help parse simulation logs, identify setup issues, or generate summaries of experimental findings. When integrated with domain-specific tools, LLMs may even assist in generating simulation input files, interpreting outliers in results, or recommending appropriate post-processing techniques [752,753].
- Supporting Conceptual Design and Parametric Exploration. During early-stage mechanical design or material selection, LLMs can suggest structural concepts, propose parameter combinations, or retrieve examples of similar engineering cases. For instance, given a prompt like “design a spring for high-temperature fatigue conditions,” the model might generate candidate materials, geometric options, and common failure modes [754,755].
- Engineering Education and Learning Support. Education in physics and mechanical engineering involves both theoretical understanding and hands-on application. LLMs can generate step-by-step derivations, support simulation-based exercises, or simulate simple lab setups (e.g., free fall, heat conduction, beam deflection). They can also assist with terminology explanation or provide example problems to enhance interactive and self-guided learning [756,757].
In summary, while physical modeling, engineering intuition, and experimental testing remain essential in physics and mechanical engineering, LLMs are emerging as effective tools for information synthesis, design reasoning, documentation, and education. The future of these disciplines may be shaped by deep integration between LLMs, simulation platforms, engineering software, and laboratory systems—paving the way from textual reasoning to intelligent system collaboration.
Figure 16.
The pipelines of physics and mechanical engineering.
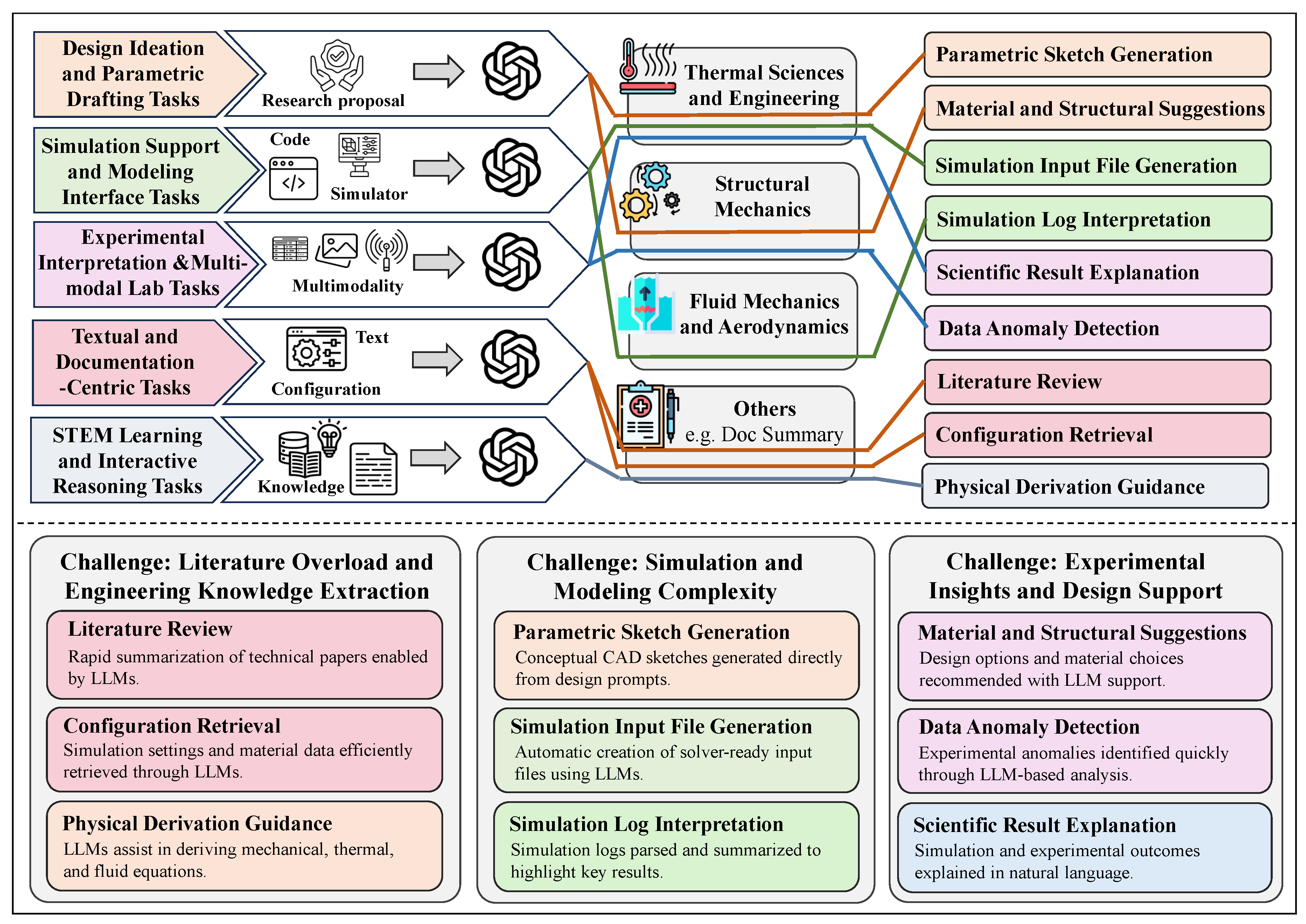
5.2.1.4 Taxonomy
Research in physics and mechanical engineering spans a broad spectrum of problems, from modeling fundamental laws of nature to designing and validating engineered systems. With the rapid development of LLMs, many of these tasks are being redefined through human-AI collaboration, automation, and intelligent assistance. Traditionally, physics and mechanical engineering are divided along disciplinary lines—e.g., thermodynamics, solid mechanics, control systems—but from the perspective of LLM-based systems, it is more productive to reorganize tasks based on their computational characteristics and data modalities.
This functional, task-driven taxonomy helps distinguish where LLMs can take on primary responsibilities, where they act in a supporting role, and where traditional numerical methods and expert reasoning remain indispensable. Based on this perspective, we propose five major categories that capture the current landscape of LLM-integrated research in physics and mechanical engineering:
- Textual and Documentation-Centric Tasks. LLMs are particularly effective in processing technical documents, engineering standards, lab reports, and scientific literature. For instance, Polverini and Gregorcic demonstrated how LLMs can support physics education by extracting and explaining key information from conceptual texts [758], while Harle et al. highlighted their use in organizing and generating instructional materials for engineering curricula [759].
- Design Ideation and Parametric Drafting Tasks. In early-stage design and manufacturing workflows, LLMs can transform natural language prompts into CAD sketches, material recommendations, and parameter ranges. The MIT GenAI group systematically evaluated the capabilities of LLMs across the entire design-manufacture pipeline [760], and Wu et al. introduced CadVLM, a multimodal model that translates linguistic input into parametric CAD sketches [755].
- Simulation-Support and Modeling Interface Tasks. Although LLMs cannot replace high-fidelity physical simulation, they can assist in generating model input files, translating specifications into solver-ready formats, and summarizing results. Ali-Dib and Menou explored the reasoning capacity of LLMs in physics modeling tasks [761], while Raissi et al.’s PINN framework demonstrated how language-driven architectures can help solve nonlinear partial differential equations by encoding physics into neural representations [762].
- Experimental Interpretation and Multimodal Lab Tasks. In experimental workflows, LLMs can support data summarization, anomaly detection, and textual explanation of multimodal results. Latif et al. proposed PhysicsAssistant, an LLM-powered robotic learning system capable of interpreting physics lab scenarios and offering real-time feedback to students and instructors [763].
- STEM Learning and Interactive Reasoning Tasks. LLMs are increasingly integrated into educational settings to guide derivations, answer conceptual questions, and simulate physical systems. Jiang and Jiang introduced a tutoring system that enhanced high school students’ understanding of complex physics concepts using LLMs [756], while Polverini’s work further confirmed the model’s utility in supporting structured, interactive learning [758].
5.2.2. Textual and Documentation-Centric Tasks
In physics and mechanical engineering, researchers and engineers routinely interact with large volumes of unstructured text: scientific papers, technical manuals, design specifications, test reports, and equipment logs. These documents are often dense, domain-specific, and heterogeneous in format. LLMs provide a promising tool for automating the extraction, summarization, and interpretation of this information.
One of the primary use cases is literature review and standards extraction. LLMs can parse multiple engineering reports or scientific articles to extract key findings, quantitative parameters, or references to specific standards, thereby reducing time-consuming manual review. For example, Khan et al. (2024) showed that LLMs can effectively assist in requirements engineering by identifying constraints and design goals from complex textual documents [764].
Another growing application is in log interpretation and structured report analysis. In mechanical systems testing and diagnostics, engineers often work with detailed experiment logs and operational narratives. Tian et al. (2024) demonstrated that LLMs can identify conditions, setup parameters, and key outcomes from such semi-structured text logs, making them useful in experiment-driven engineering workflows [765].
Furthermore, LLMs have been applied in sensor data documentation and matching. Berenguer et al. (2024) proposed an LLM-based system that interprets natural language descriptions to retrieve relevant sensor configurations and data, effectively bridging the gap between textual requirements and structured data sources [766].
These applications point to a broader role for LLMs as interfaces between human engineers and machine-readable engineering assets, enabling a smoother flow of information across documentation, modeling, and decision-making. While challenges remain—particularly in domain-specific precision and context disambiguation—the utility of LLMs in handling technical documentation is becoming increasingly evident.
5.2.3. Design Ideation and Parametric Drafting Tasks
In physics and mechanical engineering, the early stages of design—where ideas are generated and formalized into parameter-driven models—play a critical role in shaping the final product. These processes traditionally require both deep domain knowledge and creativity, often relying on iterative exploration using CAD tools, handwritten specifications, and physical prototyping. With the emergence of LLMs, this early design workflow is being significantly transformed. LLMs can help engineers rapidly generate, interpret, and modify design concepts using natural language, thus improving both accessibility and productivity in the drafting process.
Recent studies have shown that LLMs are capable of generating design concepts from textual prompts that describe functional requirements or contextual constraints. For instance, Makatura et al. (2023) introduced a benchmark for evaluating LLMs on design-related tasks, showing that these models can generate reasonable design plans and material suggestions purely based on natural language input [767]. This capability supports brainstorming and variant generation, especially in multidisciplinary systems where engineers must evaluate many trade-offs quickly.
Beyond concept generation, LLMs are increasingly used to support parametric drafting. This involves translating natural language into design specifications, such as dimensioned geometry, material choices, and assembly constraints. Wu et al. (2024) proposed CadVLM, a model capable of generating parametric CAD sketches from language-vision input, bridging LLMs with traditional CAD workflows [755]. Such models allow engineers to iterate on design through language-driven instructions (e.g., “Make the slot wider by 2 mm” or “Add a fillet at the bottom edge”), greatly simplifying the communication between design intent and digital geometry.
Some systems have also incorporated LLMs directly into CAD environments, allowing interactive, prompt-based drafting and editing. Tools like SketchAssistant and AutoSketch use LLMs to assist with geometry creation and layout proposals. These interfaces reduce the learning curve for non-expert users and open up early-stage design to a broader range of collaborators. However, challenges remain in aligning generated outputs with engineering standards, ensuring the manufacturability of outputs, and maintaining traceability between design versions and decision logic.
Overall, LLMs are becoming valuable collaborators in the ideation-to-drafting pipeline of physics and mechanical engineering design. While they are not yet substitutes for domain expertise or formal simulation, they significantly accelerate exploration, reduce iteration costs, and expand accessibility to design tools.
5.2.4. Simulation-support and Modeling Interface Tasks
In physics and mechanical engineering, simulations play a critical role in modeling complex systems, validating designs, and predicting behavior. Traditionally, configuring and running simulations requires significant domain expertise, specialized tools, and manual scripting. The integration of LLMs into simulation workflows is introducing new levels of efficiency and accessibility.
LLMs can translate natural language descriptions of physical setups into structured simulation code or configuration files. For example, FEABench evaluates the ability of LLMs to solve finite element analysis (FEA) tasks from text-based prompts and generate executable multiphysics simulations, showing encouraging performance across benchmark problems [752]. Similarly, MechAgents demonstrates how LLMs acting as collaborative agents can solve classical mechanics problems (e.g., elasticity, deformation) through iterative planning, coding, and error correction [753].
Beyond code generation, LLMs are being deployed as intelligent simulation interfaces. LangSim, developed by the Max Planck Institute, connects LLMs to atomistic simulation software, enabling users to query and configure simulations via natural language [768]. Such systems lower the barrier for non-experts to engage in simulation workflows, automate routine tasks, and reduce friction in setting up complex models.
Moreover, LLMs can help interpret simulation results, summarize outcome trends, and generate human-readable reports that connect raw numerical output with engineering reasoning. This interpretability is especially valuable in multi-physics scenarios where simulation logs and visualizations are often overwhelming.
While these advances are promising, there remain limitations in LLMs’ ability to ensure physical correctness, handle multiphysics coupling, and reason over temporal or boundary conditions. Nonetheless, their role as modeling assistants is becoming increasingly practical in early prototyping and parametric studies.
5.2.5. Experimental Interpretation and Multimodal Lab Tasks
In physics and mechanical engineering, laboratory experiments often generate complex datasets comprising textual logs, numerical measurements, images, and sensor outputs. Interpreting these multimodal datasets requires significant expertise and time. The advent of LLMs offers promising avenues to streamline this process by enabling automated analysis and interpretation of diverse data types.
LLMs can assist in translating experimental procedures and observations into structured formats, facilitating easier analysis and replication. For instance, integrating LLMs with graph neural networks has been shown to enhance the prediction accuracy of material properties by effectively combining textual and structural data [769]. This multimodal approach allows for a more comprehensive understanding of experimental outcomes.
Moreover, LLMs have demonstrated capabilities in interpreting complex scientific data, such as decoding the meanings of eigenvalues, eigenstates, or wavefunctions in quantum mechanics, providing plain-language explanations that bridge the gap between complex mathematical concepts and intuitive understanding [770]. Such applications highlight the potential of LLMs in making intricate experimental data more accessible.
Additionally, frameworks like GenSim2 utilize multimodal and reasoning LLMs to generate extensive training data for robotic tasks by processing and producing text, images, and other media, thereby enhancing the training efficiency for robots in performing complex tasks [771].
Despite these advancements, challenges remain in ensuring the accuracy and reliability of LLM-generated interpretations, especially when dealing with noisy or incomplete data. Ongoing research focuses on improving the robustness of LLMs in handling diverse and complex experimental datasets.
5.2.6. STEM Learning and Interactive Reasoning Tasks
LLMs are increasingly being integrated into STEM education to enhance learning experiences and support interactive reasoning tasks. Their ability to process and generate human-like text allows for more engaging and personalized educational tools.
LLMs can simulate teacher-student interactions, providing real-time feedback and explanations that adapt to individual learning needs. This capability has been utilized to improve teaching plans and foster deeper understanding in subjects like mathematics and physics [771]. Additionally, LLMs have been employed to interpret and grade student responses, offering partial credit and constructive feedback, which aids in the learning process [772].
Interactive learning environments powered by LLMs, such as AI-driven tutoring systems, have shown promise in facilitating inquiry-based learning and promoting critical thinking skills. These systems can guide students through problem-solving processes, encouraging them to explore concepts and develop reasoning abilities [770].
Despite these advancements, challenges remain in ensuring the accuracy and reliability of LLM-generated content. Ongoing research focuses on improving the alignment of LLM outputs with educational objectives and integrating multimodal data to support diverse learning styles.
Table 31.
Physics and Mechanical Engineering Tasks and Benchmarks
| Type of Task | Benchmarks | Introduction |
|---|---|---|
| CAD and Geometric Modeling | ABC Dataset [773] DeepCAD [774] Fusion 360 Gallery [775] CADBench [776] | The ABC Dataset, DeepCAD, and Fusion 360 Gallery together provide a comprehensive foundation for studying geometry-aware language and generative models. While ABC emphasizes clean, B-Rep-based CAD structures suitable for geometric deep learning, DeepCAD introduces parameterized sketches tailored for inverse modeling tasks. Fusion 360 Gallery complements these with real-world user-generated modeling histories, enabling research on sequential CAD reasoning and practical design workflows. CADBench further supports instruction-to-script evaluation by providing synthetic and real-world prompts paired with CAD programs. It serves as a high-resolution benchmark for measuring attribute accuracy, spatial correctness, and syntactic validity in code-based CAD generation. |
| Finite Element Analysis (FEA) | FEABench [? ] | FEABench is a purpose-built benchmark that targets the simulation domain, offering structured prompts and tasks for evaluating LLM performance in generating and understanding FEA input files. It serves as a critical testbed for bridging the gap between symbolic physical language and numerical simulation. |
| CFD and Fluid Simulation | OpenFOAM Cases [777] | The OpenFOAM example case library provides a curated set of fluid dynamics simulation setups, widely used for training models to understand solver configuration, mesh generation, and boundary condition specifications in CFD contexts. |
| Material Property Retrieval | MatWeb [778] | MatWeb is a widely-used material database containing thermomechanical and electrical properties of thousands of substances. It plays an essential role in supporting downstream simulation tasks such as material selection, constitutive modeling, and multi-physics simulation setup. |
| Physics Modeling and PDE Learning | PDEBench [779] PHYBench [780] | PDEBench and PHYBench collectively advance the evaluation of LLMs in physical reasoning and numerical modeling. PDEBench focuses on classical PDEs like heat transfer, diffusion, and fluid flow in the context of scientific machine learning, while PHYBench introduces a broader spectrum of perception and reasoning tasks grounded in physical principles. Together, they support benchmarking across symbolic reasoning, equation prediction, and simulation-aware generation. |
| Fault Diagnosis and Health Monitoring | NASA C-MAPSS [781] | NASA C-MAPSS provides real-world time-series degradation data from turbofan engines, serving as a benchmark for predictive maintenance, anomaly detection, and reliability modeling in aerospace and mechanical systems. |
5.2.7. Benchmarks
In physics and mechanical engineering, tasks such as computer-aided design (CAD), finite element analysis (FEA), and computational fluid dynamics (CFD) are characterized by strong physical constraints, structured representations, and deep reliance on geometry or numerical solvers. The development of benchmarks to support LLMs in these domains is still in its infancy. Although recent datasets have enabled initial exploration of LLMs in these fields, they present multiple challenges with respect to scale, accessibility, and alignment with language-based modeling.
In the CAD domain, several large-scale datasets have been developed to support geometric learning and generative modeling. For example, the ABC Dataset [773] provides over one million clean B-Rep (Boundary Representation) models, DeepCAD [774] offers parameterized sketches for inverse modeling, and the Fusion 360 Gallery [775] includes real-world design sequences from professional and amateur CAD users. However, most of these datasets represent geometry using numeric or parametric formats that lack symbolic or linguistic structure. Specifically, B-Rep trees and STEP files are low-level and require domain-specific parsers, making them difficult for LLMs to interpret or generate in a meaningful way.
While some recent efforts have attempted to represent CAD workflows through code-based formats such as FreeCAD Python scripts or Onshape feature code, these datasets are often small, sparse in supervision, and highly sensitive to syntactic or logical errors. Moreover, generating coherent and executable CAD programs remains a significant challenge due to the limited spatial reasoning capacity of current LLMs.
Recent advances, however, demonstrate that specialized instruction-to-code datasets and self-improving training pipelines can significantly improve LLM performance in CAD settings. For instance, BlenderLLM [776] is trained on a curated dataset of instruction–Blender script pairs and further refined through self-augmentation. As shown in Table 32, it achieves state-of-the-art results on the CADBench benchmark, outperforming models like GPT-4-Turbo and Claude-3.5-Sonnet across spatial, attribute, and instruction metrics, while maintaining a low syntax error rate. This indicates that domain-adapted LLMs, when paired with well-structured code-generation benchmarks, can overcome many of the geometric and syntactic limitations faced by general-purpose models.
To address these issues, several strategies can be explored. One direction involves decomposing full modeling workflows into modular sub-tasks, such as sketch creation, constraint placement, extrusion operations, and feature sequencing. This allows the LLM to focus on smaller, interpretable segments of the modeling pipeline. Another direction is to reframe CAD problems into geometric reasoning tasks—for instance, by translating design problems into 2D or 3D visual logic similar to those found in geometry exams. Prior studies have shown that LLMs such as GPT-4 perform surprisingly well on geometric puzzles when the problem is represented symbolically or visually. Furthermore, retrieval-augmented generation (RAG) can be employed to provide contextual examples from past designs or sketches, thus improving generation quality through analogy-based learning. Overall, bridging the gap between high-dimensional geometric information and language representation remains a central challenge in CAD-focused LLM research.
Similarly, simulation-based tasks in FEA and CFD also require structured input generation, including mesh topology, material properties, solver settings, and boundary conditions. These tasks often involve producing complete simulation decks compatible with engines such as CalculiX or OpenFOAM, followed by interpreting complex field outputs such as stress distributions or velocity gradients.
Benchmarks such as FEABench [? ] and curated OpenFOAM case libraries [777] provide valuable baselines for evaluating the simulation-awareness of LLMs. However, the availability of large-scale paired datasets—comprising natural language descriptions, simulation input files, and corresponding numerical results—remains limited, posing a bottleneck for supervised fine-tuning and instruction-based evaluation.
To address this gap, FEABench introduces structured tasks that assess LLMs’ ability to extract simulation-relevant information. Table 33 presents the performance of various LLMs across multiple physics-aware metrics, including interface factuality, feature recall, and dimensional consistency. Models like Claude 3.5 Sonnet and GPT-4o demonstrate strong results in retrieving factual and geometric descriptors, particularly in interface and feature extraction. However, all models show relatively low performance in recalling physical properties and structured feature attributes, reflecting ongoing challenges in capturing physical relationships from text. These results suggest that while LLMs can reliably recover high-level simulation inputs, deeper understanding of numerical structure and physical laws remains an open research problem.
A promising solution is to integrate LLMs with external numerical solvers in a simulator-in-the-loop framework. In this approach, an LLM is tasked with generating a complete simulation setup given a natural language prompt or design goal. The generated setup is then executed by a physics-based solver to produce ground-truth outputs. The input-output pairs, along with the original language prompt, can be stored as a triplet dataset and reused for supervised training. This method enables semi-automated dataset construction at scale, facilitates error correction via feedback from the simulator, and promotes the development of LLM agents that can reason across symbolic and physical domains. Additionally, by iterating through prompt refinement and result validation, such frameworks could enable reinforcement learning with human or physical feedback for high-fidelity simulation tasks.
Together, these benchmarks and emerging methodologies form the foundation of an evolving research area at the intersection of language modeling, geometry, and physics. As more domain-specific tools and datasets are adapted for LLM-compatible formats, we expect substantial progress in generative reasoning, simulation co-pilots, and data-driven modeling for engineering systems.
5.2.8. Discussion
Opportunities and Impact. LLMs are beginning to reshape workflows in physics and mechanical engineering, particularly in tasks such as CAD modeling, finite element analysis (FEA), material selection, simulation setup, and result interpretation. As demonstrated by models like CadVLM [755] which translates textual input into parametric sketches, FEABench [? ] which evaluates LLMs on FEA input generation, and LangSim [768] which enables natural language interaction with atomistic simulation tools, LLMs are emerging as intelligent intermediaries between domain experts and computational tools.
By converting natural language into structured engineering commands, LLMs greatly simplify early-stage design, parameter exploration, technical documentation, and preliminary simulation configuration. Through code generation, auto-completion, document retrieval, and example-based prompting, LLMs are becoming integral assistants in modern engineering workflows. As multimodal and multi-agent systems (e.g., MechAgents [753]) become more common, LLMs are poised to play a key role in the next generation of closed-loop “design–simulate–validate” engineering pipelines.
Challenges and Limitations. Despite these promising applications, multiple challenges persist. First, physical modeling tasks such as FEA and CFD involve highly coupled, nonlinear partial differential equations (PDEs) that require domain-specific inductive biases, numerical stability, and conservation principles—capabilities that current LLMs fundamentally lack.
Second, existing datasets in engineering domains present significant structural barriers. Most CAD datasets (e.g., B-Rep, STEP) are stored in numeric or parametric formats with minimal symbolic representation, making them difficult for LLMs to understand or generate. Code-based CAD datasets are more interpretable by LLMs, but are often limited in size, brittle in syntax, and sensitive to logical correctness.
Moreover, LLMs struggle with tasks requiring unit consistency, physical constraint enforcement, and boundary condition reasoning. In real-world engineering, even small errors in design parameters or simulation configurations can lead to system failure, safety risks, or structural inefficiencies. This makes it difficult to rely on LLMs for mission-critical design tasks without rigorous validation.
Research Directions. To further improve the effectiveness of LLMs in physics and mechanical engineering, several research directions are particularly promising:
- Simulation-Augmented Dataset Generation. Integrating LLMs with numerical solvers in a simulator-in-the-loop framework allows the generation of language-input–simulation-output triplets at scale. This enables supervised training, fine-tuning, and RLHF strategies grounded in physically valid feedback.
- Task Decomposition and Geometric Reformulation. Decomposing CAD workflows into modular sub-tasks (e.g., sketching, constraints, extrusion) and reformulating modeling problems as geometric reasoning tasks can align better with LLM capabilities and improve interpretability.
- Multimodal and Multi-agent Integration. Developing LLM systems that can call CAD tools, solvers, and databases autonomously—as seen in MechAgents or LangSim—will allow LLMs to reason, plan, and act across tools in complex design and simulation pipelines.
- Standardized Benchmarks and Evaluation. Creating large-scale, task-diverse, and format-unified benchmark datasets (e.g., combining natural language prompts, simulation files, and result summaries) will accelerate model evaluation and fair comparison in this field.
- Physics Validation and Safety Assurance. Embedding physical rule checkers and verification mechanisms into generation loops can help enforce unit consistency, structural validity, and simulation compatibility, ensuring that outputs are not just syntactically correct but physically plausible.
Conclusion. LLMs are becoming increasingly valuable assistants in physics and mechanical engineering, especially in peripheral tasks such as documentation, concept generation, parametric modeling, and simulation support. However, to deploy them in core workflows, future systems must integrate LLMs with symbolic reasoning, geometric logic, physics-based solvers, and expert feedback. This synergy will enable the transition from language-based assistance to trustworthy, intelligent co-creation in complex engineering design and modeling workflows.
5.3. Chemistry and Chemical Engineering
5.3.1. Overview
5.3.1.1 Introduction to Chemistry
Chemistry is the scientific discipline dedicated to understanding the properties, composition, structure, and behavior of matter. As a branch of the physical sciences, it focuses on the study of chemical elements and the compounds formed from atoms, molecules, and ions—their interactions, transformations, and the principles governing these processes [782,783]. Put simply, chemistry seeks to explain how matter behaves and how it changes [784]. We must acknowledge that the field of chemistry is vast and encompasses a variety of branches. Given the particularly rich application scenarios in areas such as organic chemistry, life sciences—especially in relation to LLM-related work—we will discuss these branches in the following chapter to provide a detailed introduction to works closely related to biology and life sciences.
In the field of chemistry, there are numerous sub-tasks, and many scientists have made significant contributions and achieved groundbreaking results over the past few hundred years. Before diving into LLM-related topics, we would like to provide an overview of major tasks and traditional methods in chemistry research. By integrating information from official websites [785] and literature across various branches of the field [786,787,788,789,790,791,792,793,794,795], we have summarized the research tasks in the domain of chemistry as follows:
Analysis and Characterization. This task involves identifying the substances present in a sample (qualitative analysis) and determining the quantity of each substance (quantitative analysis) [796]. In this section we emphasize experimental measurement and detection methods aimed at identifying which substances are present, as well as determining their composition, structure, and morphology; we do not here focus on how their properties change under varying conditions nor on prediction or modeling of those properties. It also includes elucidating the structure and properties of these substances at a molecular level [797]. Traditional methods for analysis and characterization include techniques such as observing physical properties (color, odor, melting point), performing specific chemical tests to identify certain substances (like the iodine test for starch or flame tests for metals), and classical quantitative analysis using precipitation, extraction, and distillation [798]. Modern research in this area heavily relies on sophisticated instruments. Spectroscopy, which studies how matter interacts with light, can provide significant insights into a molecule’s structure and composition [797]. Chromatography is employed to separate complex mixtures into their individual components for analysis [797]. Mass spectrometry (MS) is a powerful technique that can identify and quantify substances by measuring their mass-to-charge ratio with very high sensitivity and specificity [797,799].
Research on Properties. Research on properties in chemistry refers to the systematic exploration and analysis of the physical and chemical characteristics of substances, with the main objective being to reveal the behavior and reaction characteristics of substances under different conditions [800,801]. We take “Research on Properties” to include both experimental determination and prediction or modeling of physical and chemical properties, with a primary interest in how those properties behave or change under different conditions. Traditionally, researchers have employed experimental methods to determine these properties. For thermodynamic properties, calorimetry is a key technique used to measure heat flow during physical and chemical processes [802,803]. Equilibrium methods, such as measuring vapor pressure, can assist in determining energy changes during phase transitions [802]. For kinetic properties, traditional methods involve monitoring the changes in concentration of reactants or products over time [804].
Reaction Mechanisms. The primary objective of studying reaction mechanisms in chemistry is to reveal the specific processes and steps involved in chemical reactions, including the microscopic mechanisms by which reactants are converted into products. This research field focuses on the formation of various intermediates during the reaction, the reaction pathways, rate-determining steps, and their corresponding energy changes [800,805]. Traditional methods for investigating reaction mechanisms include kinetic studies, where the rate of a reaction is measured under different conditions to understand its progression [806,807,808]. Isotopic labeling involves using reactants with specific isotopes to trace the movement of atoms during the reaction [807]. Stereochemical analysis examines the spatial arrangement of atoms in reactants and products, providing insights into the reaction pathway [807]. Identifying the intermediate products formed during the reaction is also a crucial aspect of this research.
Chemical Synthesis. Chemical Synthesis refers to actually producing molecules in the laboratory or pilot-scale. The synthesis of natural products is an important task in chemistry, aimed at using chemical methods to synthesize complex organic molecules found in nature [809]. The realization of such synthesis in practice relies on several traditional experimental methods. Plant extraction separates compounds from plants using techniques like solvent extraction, cold pressing, or distillation, yielding various active ingredients. Fermentation technology utilizes microorganisms to produce natural products, commonly for antibiotics and bioactive substances [810]. Organic synthesis constructs chemical structures through multi-step synthesis and the introduction of functional groups [811]. Lastly, semi-synthetic methods modify simple precursors to create more complex natural compounds or their derivatives [812].
Molecule Generation. Molecule Generation involves computational chemistry and molecular modeling techniques to predict, optimize, or generate new molecular structures with desired functions or properties [813,814]. It includes computer-aided design, virtual screening, property prediction, structure optimization, and theoretical modelling of molecules, etc. [813,814]. Molecular synthesis and design encompass both experimental synthesis [814] and computer-aided design [815].
Applied Chemistry. Applied Chemistry refers to the branch of chemistry that focuses on practical applications in various fields such as industry, medicine, and environmental science. It involves using chemical principles to solve real-world problems and improve processes, including material chemistry and drug chemistry [816,817,818,819]. Traditionally, several key methods are relied upon, including structure-activity relationship (SAR) studies, computer-aided drug design [820], high-throughput screening [821], and synthetic chemistry [822].
5.3.1.2 Introduction to Chemical Engineering
Chemical engineering is an engineering field that deals with the study of the operation and design of chemical plants, as well as methods of improving production. Chemical engineers develop economical commercial processes to convert raw materials into useful products. Chemical engineering utilizes principles of chemistry, physics, mathematics, biology, and economics to efficiently use, produce, design, transport, and transform energy and materials [823]. According to the Oxford Dictionary, chemical engineering is a branch of engineering concerned with the application of chemistry to industrial processes, particularly involving the design, operation, and maintenance of equipment used to carry out chemical processes on an industrial scale [824]. In summary, it serves as the bridge that applies chemical achievements to industry.
Similar to chemistry, chemical engineering encompasses multiple fields, including not only chemistry, but also mathematics, physics, and economics. Through a comprehensive review of previous research [825,826,827,828,829], we have categorized the tasks in chemical engineering into the following types.
Chemical Process Engineering. Chemical process engineering includes chemical process design, improvement, control, and automation. Chemical process design focuses on the design of reactors, separation units, and heat exchange equipment to achieve efficient material conversion and energy utilization [827,830], typically employing computer-aided design software and process simulation tools [831,832]. Chemical process improvement involves the systematic analysis and optimization of existing chemical processes to enhance production efficiency, reduce resource consumption, and minimize environmental impact [830]. It primarily relies on quality management tools [833,834] and process simulation software [835]. Process control and automation aim to monitor and regulate chemical processes through control systems to ensure stable operation under set conditions, typically based on proportional–integral–derivative (PID) control systems [836], combined with advanced control technologies such as Model Predictive Control [837] to optimize processes. Distributed control systems and programmable logic controllers are also commonly used automation systems that can monitor and adjust process variables in real-time [838,839].
Equipment Design and Engineering. Equipment design and engineering focus on the design, selection, and maintenance of chemical engineering equipment to ensure its efficient and safe operation within specific processes. The reliability and functionality of the equipment directly impact overall efficiency and safety [840]. Equipment design is typically carried out in accordance with industry standards and regulations, such as American Society of Mechanical Engineers (ASME) and American Petroleum Institute (API). Engineers use computer-aided design (CAD) software for detailed design and simulation [841,842]. Additionally, strength analysis and fluid dynamics simulation are critical components, generally relying on computational fluid dynamics software to ensure the safety and efficiency of equipment under various operating conditions [840].
Sustainability and Environmental Engineering. Sustainability and environmental engineering focus on the impact of chemical processes on the environment and are dedicated to developing green chemical technologies to reduce pollution and resource consumption. This field emphasizes the importance of life cycle assessment and environmental impact assessment in achieving sustainability goals [843].
Scale-up and Technology Transfer. The task of translating chemical achievements into practical applications in chemical engineering involves bridging the gap between laboratory discoveries and industrial-scale implementation, ensuring that innovative chemical processes and materials are effectively integrated into real-world production systems to meet societal and industrial demands [844]. Traditionally, the application of chemical achievements employs methods such as pilot scale testing to validate the feasibility and stability of the technology [844], and process simulation and optimization (e.g., using tools like Aspen Plus and CHEMCAD) to model and optimize process flows, thereby reducing costs and improving efficiency. Simultaneously, factors such as economic viability, supply chain and market dynamics, and safety and environmental compliance are also evaluated and optimized [825,827].
From the definition, we can see that there is a strong logical relationship between the fields of chemical engineering and chemistry at the macroscopic level. The main battleground of chemical science is in the laboratory, while the main battleground of chemical engineering is in the factory. Chemical engineering aims to translate processes developed in the lab into practical applications for the commercial production of products, and then work to maintain and improve those processes [782,783,823,845].
At the microscopic level, chemistry and chemical engineering share many common technologies, such as CAD and computational simulation. Moreover, there are varying degrees of connections between the different sub-tasks within these two fields. We have summarized the relationships among them in the form of a diagram in Figure 17.
5.3.1.3 Contribution of Chemistry and Chemical Engineering
It is not difficult to imagine that chemistry, as a fundamental science, has profoundly impacted various aspects of human society, with its contributions evident in public health, materials innovation, environmental protection, and energy transition. Firstly, the contributions of chemistry to public health are significant. Through the synthesis and development of new pharmaceuticals, chemists have greatly improved human health [846,847]. For instance, the discovery of penicillin not only marked the beginning of the antibiotic era but also reduced the mortality risk associated with bacterial infections [846,848]. In recent years, the development of targeted therapies [847,849], such as drugs aimed at specific cancers, relies on a chemical understanding of the internal mechanisms of tumor cells, thereby significantly enhancing patient survival rates. Secondly, chemistry has a revolutionary impact on materials innovation. Through the development of polymers [850], alloys [851,852], and nanomaterials [853,854], chemists have not only enhanced material properties but also advanced technological progress. For example, the application of modern lightweight and high-strength composite materials has enabled greater energy efficiency and safety in the aerospace and automotive industries [850]. Moreover, the emergence of graphene and other nanomaterials has opened new possibilities for the development of electronic products [853,854].
In the realm of environmental protection, the contributions of chemistry cannot be overlooked. By developing efficient catalysts and clean technologies, chemists play a crucial role in reducing industrial emissions and tackling water pollution [855]. For example, selective catalytic reduction reactions effectively convert harmful gases emitted by vehicles, significantly improving urban air quality [856,857]. Furthermore, the role of chemistry in energy transition is becoming increasingly important [858,859]. The development of renewable energy storage and conversion is fundamentally supported by chemical technologies [860]. For instance, the research and development of lithium-ion batteries [861] and hydrogen fuel cells [862] depend on the optimization of chemical reactions and material innovations, making the use of clean energy feasible.
5.3.1.4 Challenges in the Era of LLMs
Despite the significant achievements in the fields of chemical science and chemical engineering, there remain unresolved challenges in these areas. The emergence of LLMs presents an opportunity to address these issues. We must acknowledge that, unfortunately, LLMs are not omnipotent; they cannot solve all the challenges within this field. However, for certain tasks, LLMs hold promise in assisting chemists in overcoming these challenges. We have listed the following difficulties that LLMs cannot solve:
The Irreplaceability of Time-consuming Chemical Experiments. LLMs-generated outcomes in chemical research still require experimental validation. Assessing the true utility of these generated molecules, such as evaluating their novelty in real-world applications, can be a time-consuming undertaking [863]. While LLMs have their advantages in data processing and information retrieval, solely relying on the results generated by the model may not accurately reflect the actual experimental conditions. Moreover, LLMs are trained on existing data and literature; if a specific field lacks sufficient data support, the outputs of the model may be inaccurate or unreliable [864].
Limitations in Learning Non-smooth Patterns. Traditional deep learning struggles to learn non-smooth target functions that map molecular data to labels, as these target functions are frequently non-smooth in molecular property prediction. This implies that minor alterations in the chemical structure of a molecule can lead to substantial changes in its properties [865]. Additionally, LLMs also find it difficult to solve this problem under the limited size of molecular datasets.
Dangerous Behaviors and Products. The field of chemistry carries certain inherent risks, as some products or reactions can be hazardous (e.g., flammable, explosive, toxic gases, etc.). LLMs may generate scientifically incorrect or unsafe responses, and in some cases, they may encourage users to engage in dangerous behavior [866]. Furthermore, LLMs can also be misused to create toxic or illegal substances [863]. At the current stage of development, LLMs still cannot be fully trusted to ensure complete reliability.
On the other hand, despite the aforementioned limitations, the potential of LLMs in the fields of chemistry and chemical engineering is undeniable, as they hold promise in addressing many challenges:
Decrease the Vast Chemical Exploration Space. Inverse design enables the creation of synthesizable molecules that meet predefined criteria, accelerating the development of viable therapeutic agents and expanding opportunities beyond natural derivatives [867]. However, this quest faces a combinatorial explosion in the number of potential drug candidates—the chemical space made up of all small drug-like molecules—which is unimaginably large (estimated at ) [867]. Testing any significant fraction of these molecules, either computationally or experimentally, is simply impossible [868]. This field has been revolutionized by machine learning methods, particularly generative models, which narrow down the search space and enhance computational efficiency, making it possible to delve deeply into the seemingly infinite chemical space of drug-like compounds [869]. LLMs, such as MolGPT [870], which employs an autoregressive pre-training approach, have proven to be instrumental in generating valid, unique, and novel molecular structures. The emergence of multi-modal molecular pre-training techniques has further expanded the possibilities of molecular generation by enabling the transformation of descriptive text into molecular structures [871].
Generation of 3D Molecular Conformations. Generating three-dimensional molecular conformations is another significant challenge in the field of molecular design, as the three-dimensional spatial structure of a molecule profoundly impacts its chemical properties and biological activity. Traditional computational methods are often resource-intensive and time-consuming, making it difficult for researchers to design and screen new drugs effectively. Unlike conventional approaches based on molecular dynamics or Markov chain Monte Carlo, which are often hindered by computational limitations (especially for larger molecules), LLMs based on 3D geometry exhibit remarkable superiority in conformation generation tasks, as they can capture inherent relationships between 2D molecules and 3D conformations during the pre-training process [871].
Automate Chemical Agents. Autonomous chemical agents combine LLM “brains” with planning, tool use, and execution modules to carry out experiments end-to-end. In the coscientist system, for example, GPT-4 first decomposes a high-level goal (“optimize a palladium-catalyzed coupling”) into sub-tasks (reagent selection, condition screening), retrieves relevant literature via a search tool, generates executable Python code for liquid-handling robots, and then interprets sensor feedback to iteratively refine the protocol—closing the loop between design and execution [872]. Similarly, Boiko et al. built an agent that plans ultraviolet–visible spectroscopy (UV–Vis) experiments by writing code to control plate readers and analyzers, automatically processing spectral data to identify optimal wavelengths, and even adapting to novel hardware modules introduced after the model’s training cutoff [873]. By leveraging LLMs for hierarchical task decomposition, self-reflection, tool invocation (e.g., search APIs, code execution, robotics control), and memory management, these systems drastically accelerate repetitive experimentation and free researchers to focus on hypothesis generation rather than manual protocol execution [873].
Enhance Understanding of Complex Chemical Reactions. The field of reaction prediction faces several key challenges that affect the accuracy of forecasting chemical reactions. A significant issue is reaction complexity, stemming from multi-step pathways and dynamic intermediates, which complicates product predictions, especially with varying conditions like different catalysts. Traditional models often struggle with these complexities, leading to biased outcomes. Utilizing advanced transformer architectures, LLMs can model complex relationships in chemical reactions and adjust predictions based on varying conditions. They excel in learning from unlabeled data through self-supervised pretraining, helping identify patterns in chemical reactions, particularly useful for rare reactions.
Multi-task Learning and Cross-domain Knowledge. The complexity of multi-task learning makes the simultaneous optimization of diverse prediction tasks difficult, while LLMs effectively handle this via shared representations and multi-task fine-tuning [874]. Traditional methods also struggle to integrate cross-domain knowledge from chemistry, biology, and physics, yet LLMs address this seamlessly through pre-training and knowledge graph enhancement.
5.3.1.5 Taxonomy
As summarized in Table 34, in efforts to integrate chemistry research with artificial intelligence, particularly LLMs, many chemists primarily focus on tasks such as property prediction, property-directed inverse design, and synthesis prediction [867,874,875]. However, other chemists highlight additional significant tasks, including data mining and predicting synthesis conditions [876,877]. By synthesizing insights from these studies along with other seminal works [878,879,880], we propose a more comprehensive classification method. This method accounts for both the rationality of chemical task classification and the characteristics of computer science.
From the chemistry perspective, our taxonomy echoes the field’s established research divisions—such as molecular property prediction, property-directed inverse design, reaction type and yield prediction, synthesis condition optimization, and chemical text mining—ensuring that each category corresponds directly to a recognized experimental or theoretical task in chemical science. Concurrently, from the computer science perspective, by mapping every task onto a unified input–output modality framework, we add a computationally consistent structure that facilitates model development, benchmarking, and comparative analysis across diverse tasks within a single formal paradigm. Together, these dual alignments guarantee that our classification remains both chemically and algorithmically meaningful.
Figure 18.
A taxonomy of chemical tasks enabled by LLMs, categorized by input-output types and downstream objectives.
Figure 18.
A taxonomy of chemical tasks enabled by LLMs, categorized by input-output types and downstream objectives.
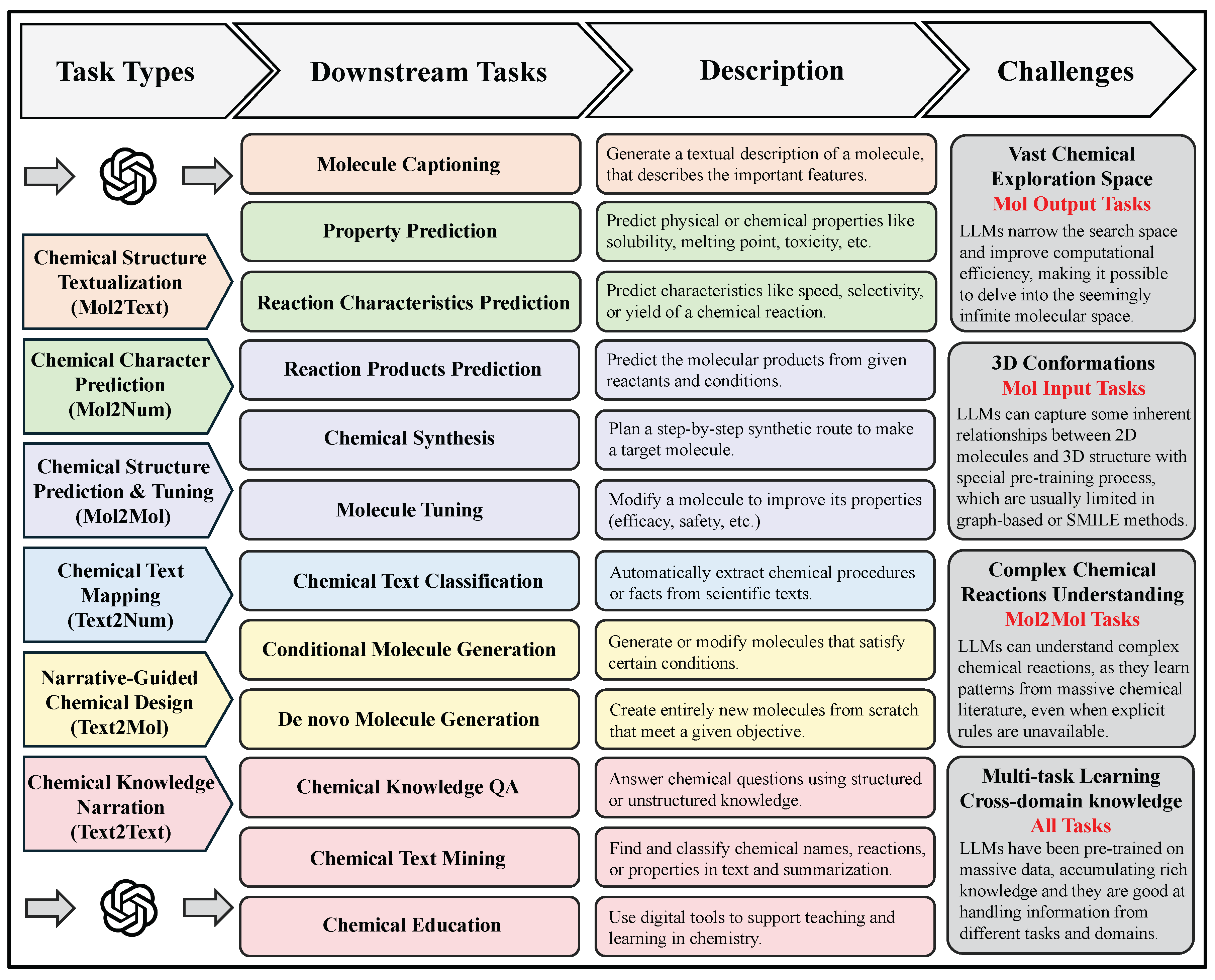
Chemical Structure Textualization. Chemical structure textualization is the process of taking a molecule’s SMILES sequence as input and producing a detailed textual depiction that highlights its structural features, physicochemical properties, biological activities, and potential applications. Here, SMILES (Simplified Molecular Input Line Entry System) encodes a molecule’s atomic composition and connectivity as a concise, linear notation—for example, “CCO” denotes ethanol (each “C” represents a carbon atom and “O” an oxygen atom), while “C1=CC=CC=C1” represents benzene (the digits mark ring closure and “=” indicates double bonds)—enabling computational models to capture meaningful structural patterns and relationships for downstream text generation. Subtasks include molecule captioning, which exemplifies the goal of generating rich, human-readable descriptions of molecules to give chemists and biologists rapid, accessible insights for experimental design and decision-making [881].
Chemical Characteristics Prediction. Nowadays, SMILES provides a standardized method for encoding molecular structures into strings [882]. This string-based representation enables efficient parsing and manipulation by computational models and underpins a variety of tasks in cheminformatics, drug discovery, and reaction prediction. Notably, many machine learning models, including large-scale language models like GPT, are pre-trained or fine-tuned using corpora of SMILES sequences. Among the tasks leveraging SMILES input are property prediction and reaction characteristics prediction, where the model takes a SMILES sequence as input and outputs numerical values, categorical labels, or multi-dimensional vectors representing chemical properties, reactivity, bioactivity, and other experimentally relevant quantities.
Chemical Structure Prediction & Tuning. Chemical structure prediction & tuning tasks represent a classical form of sequence-to-sequence modeling [883], where the goal is to transform an input molecular sequence into an output sequence. In chemistry, this formulation is particularly intuitive because molecules are often represented as SMILES strings, which encode structural information in a linear textual format. Given an input SMILES sequence, the model learns to generate another SMILES string corresponding to a chemically meaningful transformation. This input–output structure underlies a variety of chemical modeling tasks, including reaction product prediction, chemical synthesis planning, and molecule tuning. For instance, the input may describe reactants or precursors, and the output may represent reaction products or structurally modified molecules, making these tasks central to computational reaction modeling and automated molecular design.
Chemical Text Mapping. Chemical text mapping tasks refer to the process of transforming unstructured textual input into numerical outputs such as labels, scores, or categories. At their core, these tasks involve analyzing chemical text—ranging from scientific articles to experimental protocols—and mapping the extracted information to structured numerical values for downstream applications like classification, relevance scoring, or trend prediction [867,884]. A typical example is document classification, where the input is natural language text and the output is a discrete or continuous number representing, for example, a document’s category or relevance score. These tasks enable scalable analysis of chemical literature and facilitate integration of textual knowledge into data-driven modeling workflows.
Narrative-Guided Chemical Design. Narrative-Guided chemical design is a generative modeling paradigm extensively applied in chemistry and materials science, with the core objective of deriving molecular structures or material candidates that fulfill specific target properties or functional requirements [885,886]. Unlike conventional forward design—which predicts properties from a given structure—inverse design begins with the desired outcome and works backward to propose compatible structures. In this context, the input is a description of the target properties, which may take the form of numerical constraints, categorical labels, or free-text descriptions, and the output is a molecular structure—typically represented as a SMILES string—that satisfies those specified criteria. This framework encompasses tasks such as de novo molecule generation and conditional molecule generation, enabling applications like targeted drug design, property-driven material discovery, and personalized molecular synthesis.
Chemical Knowledge Narration. Chemical knowledge narration tasks in chemistry refer to the transformation of one form of textual input into another, with both input and output grounded in chemical knowledge and language use [867]. These tasks leverage Natural Language Processing (NLP) techniques to process, convert, or generate chemistry-related textual data, thereby facilitating a range of downstream applications in research, education, and industry. For instance, given a textual input such as a paragraph from a research paper, the model may extract key information, translate it into another language, generate a summary, or answer domain-specific questions. Such tasks—encompassing chemical text mining, chemical knowledge question answering, and educational content generation—typically operate on natural language input and produce human-readable textual output, making them essential tools for improving access to and understanding of chemical information.
Table 34.
Chemistry Tasks, Subtasks, Insights and References
| Type of Task | Subtasks | Insights and Contributions | Key Models | Citations |
|---|---|---|---|---|
| Chemical Structure Textualization | Molecular Captioning | LLMs, by learning structure–property patterns from data, generate meaningful molecular captions, thus improving interpretability and aiding chemical understanding. | MolT5: generates concise captions by mapping substructures to descriptive phrases; MolFM: uses fusion of molecular graphs and text for richer narrative summaries. | [887,888,889,890,891,892,893,894,895,896,897,898,899,900,901,902,892] |
| Chemical characteristics prediction | Property Prediction | LLMs, by capturing complex structure–property relationships from molecular representations, enable accurate property prediction, thereby providing mechanistic insights and guiding the rational design of molecules with desired functions. | SMILES-BERT: self-supervised SMILES pretraining for robust property inference; ChemBERTa: masked SMILES modeling boosting solubility and toxicity predictions. | [903,904,905,906,907,908,909,910,911,912,913,914] |
| Reaction Characteristics Classification | LLMs, by modeling the relationships between reactants, conditions, and outcomes from large reaction datasets, can accurately predict reaction types, yields, and rates, thereby uncovering hidden patterns in chemical reactivity and enabling chemists to optimize reaction conditions and select efficient synthetic routes with greater confidence. | RXNFP: fingerprint-transformer accurately classifies reaction types; YieldBERT: fine-tuned on yield data to predict experimental yields within 10% error. | [863,915,916,917,918,919,920,921,922,923,924,925,926,927] | |
| Chemical Structure Prediction & Tuning | Reaction Products Prediction | LLMs, by learning underlying chemical transformations from reaction data, can accurately predict reaction products, thus uncovering implicit reaction rules and supporting more efficient and informed synthetic planning. | Molecular Transformer: state-of-the-art SMILES-to-product translation; | [921,923,924,925,926,928,929,930,931,932,933] |
| Chemical Synthesis | LLMs, by capturing patterns in reaction sequences and chemical logic from large datasets, can suggest plausible synthesis routes and rationales, thereby enhancing human understanding of synthetic strategies and accelerating discovery. | Coscientist: GPT-4-driven planning and robotic execution. | [872,932,934,935,936,937,938,939,940,941,942,943,944,945,946,947,948,949,950,951] | |
| Molecule Tuning | LLMs, by modeling structure–property relationships across diverse molecular spaces, enable targeted molecule tuning to optimize desired properties, thereby providing insights into molecular design and accelerating the development of functional compounds. | DrugAssist: uses LLM prompts for ADMET property optimization; ControllableGPT: enables constraint-based molecular modifications. | [952,953,954,955,956,957,958,959,960,961,962,963,964,964] | |
| Chemical Text Mapping | Chemical Text Mining | LLMs, by capturing semantic and contextual nuances in chemical literature, enable accurate classification and regression in text mining tasks, thereby uncovering trends, predicting research outcomes, and transforming unstructured texts into actionable scientific insights. | Fine-tuned GPT: specialized for chemical classification and regression; ChatGPT: adapts zero-shot classification of chemical text. | [965,966,967,968,969,970,971,972,973,974,975,976,977,978] |
| Narrative-Guided Chemical Design | De Novo Molecule Generation | LLMs, by learning chemical syntax and patterns from large molecular corpora, enable de novo molecule generation with realistic and diverse structures, thus offering insights into unexplored chemical space and accelerating early-stage drug and material discovery. | ChemGPT: unbiased SMILES sampling for novel molecules; MolecuGen: scaffold-guided generative modeling for improved novelty. | [887,891,979,980,981,982,983,984,985,986,987,988,989,990,991,992] |
| Conditional Molecule Generation | LLMs, by conditioning molecular generation on desired properties or scaffolds, enable the design of compounds that meet specific criteria, thereby offering insights into structure–function relationships and streamlining the discovery of tailored molecules. | GenMol: multi-constraint text-driven fragment remasking. | [887,889,931,957,983,984,986,987,988,993,994,995,996] | |
| Chemical Knowledge Narration | Chemical Knowledge QA | LLMs, by integrating extensive chemical literature and diverse databases, can accurately address complex chemical knowledge questions, thereby uncovering valuable insights and enabling more informed, accelerated research and decision-making. | ChemGPT: conditional SMILES generation for property-specific tuning; ScholarChemQA: domain-specific QA fine-tuned on scholarly chemistry data. | [863,924,997,998,999,1000,1001,1002,1003,1004,1005,1006,1007,1008] |
| Chemical Text Mining | LLMs, by understanding and extracting structured information from unstructured chemical texts, enable efficient chemical text mining, thereby revealing hidden knowledge, facilitating data-driven research, and accelerating the discovery of relationships across literature. | ChemBERTa: BERT-based model fine-tuned for chemical text classification. SciBERT: pretrained on scientific text including chemical literature for robust retrieval. | [965,966,973,976,1009,1010,1011,1012,1013,1014] | |
| Chemical Education | LLMs, by generating intuitive explanations and answering complex queries in natural language, support chemical education by making abstract concepts more accessible, thereby enhancing student understanding and promoting more interactive, personalized learning experiences. | MetaTutor: LLM-based metacognitive tutor for chemistry learners. | [997,998,1015,1016,1017,1018,1019,1020,1021,1022] |
5.3.2. Chemical Structure Textualization
Chemical tasks that map molecular structures to text serve as a bridge between the structural world of chemistry and human language. Chemical structure textualization is essentially “describing molecules in words,” akin to how one might recognize a complex object (like a new gadget) and then explain it in a common language [863]. In everyday life, this is like looking at a detailed blueprint and verbally summarizing what it represents. In chemistry, such tasks are crucial: chemists often need to communicate structures verbally or in writing––for instance, by naming compounds or summarizing their features––so that others can understand them without seeing a drawing. Converting molecules to text makes chemistry more accessible and searchable (one can text-search a compound name in literature, but not a structure diagram). For example, a medicinal chemist might draw a novel molecule and want the International Union of Pure and Applied Chemistry (IUPAC) name or a simple description to include in a report. An environmental chemist might identify an unknown substance and need an AI to generate a description like “a chlorinated hydrocarbon solvent.” These translations from structure to language are essential for reporting, education, and integrating chemical information into broader databases.
Chemical Structure Textualization is fundamentally a Mol2Text task, mapping chemical structure representations (e.g., SMILES) as input to corresponding natural language descriptions as output. Mol2Text encompasses any scenario of “structure to language.” Key subtasks include chemical nomenclature generation (e.g. converting a molecular structure into its IUPAC name or common name), molecule captioning (generating a sentence describing the molecule’s class or properties), and even property or hazard annotations (predicting textual labels like “flammable” from a structure). For instance, nomenclature generation might take a SMILES string of a molecule and output “2-(4-Isobutylphenyl)propanoic acid” (the IUPAC name of ibuprofen). Molecule captioning could produce a phrase like “an aromatic benzene derivative with two nitro substituents” given a structure. They are LLM-friendly because large databases of molecules with names or descriptions exist (providing plenty of textual training data), and the output is structured text that follows rules (ideal for sequence generation models). We therefore highlight the area of molecule captioning in the following paragraph. These illustrate how LLMs evolved from simply reading text to “reading” chemistry and writing text.
Molecular Captioning. Beyond formal nomenclature, a growing chemical structure textualization application is molecular captioning––generating a natural-language description of a molecule’s structure or function. That is given a molecule’s representation (e.g. SMILES or graph), generating a coherent natural-language description that accurately captures its structural features, physicochemical properties, and potential biological activities. This is analogous to image captioning in computer vision (where an AI might look at a photo and say “a cat sitting on a sofa”). Here, the “image” is a chemical structure (which an LLM might ingest as a string like SMILES or another representation), and the output is a concise text description. For example, given the structure of caffeine, an ideal caption might be: “a bitter, white alkaloid of the purine class, commonly found in coffee and tea.” Or more straightforwardly: “Caffeine – a stimulant compound with a purine (xanthine) scaffold and multiple methyl groups.” This task is in its infancy, but its significance is clear: it would allow chemists to quickly get a textual summary of a molecule’s key features or known uses. In everyday life, this is akin to seeing a new plant and describing it as “a tall tree with waxy leaves and fragrant white flowers”––it communicates key identifying features in an intuitive way. MolT5 [887] first achieved end-to-end text-SMILES translation by jointly training on vast amount of SMILES and textual descriptions, but relying solely on sequence information led to captions lacking intuitive structural understanding. To address this, MolFM [891] introduced multimodal pretraining that combines SMILES, InChI, and text descriptions, significantly improving annotation accuracy and richness, yet it did not leverage molecular images for visual comprehension . Next, GIT-Mol [890] integrated molecular graphs, images, and text through cross-modal fusion, achieving higher fidelity captions but at the cost of large model size and high deployment and inference overhead . To improve efficiency and deployment, MolCA [899] designed a cross-modal projector and uni-modal adapters, greatly reducing fine-tuning and deployment costs while retaining multimodal capabilities, though its pretraining data coverage still needs expansion . Most recently, GraphT5 [897] employs multimodal cross-token attention to tightly integrate molecular graph structures with a language model, balancing caption quality and model scale, and providing an efficient and scalable foundation for molecule captioning
Problems Solved by LLMs. LLMs have dramatically improved both chemical nomenclature generation and molecular captioning by learning from vast corpora of paired data. In the case of nomenclature, Transformer-based models such as Struct2IUPAC have achieved accuracies of 98–99% in converting SMILES strings to formal IUPAC names—performance that rivals rule-based systems like Open Parser for Systematic IUPAC Nomenclature (OPSIN), which itself set the benchmark for open-source name-to-structure parsing over a decade ago [1023]. Simultaneously, proof-of-concept captioning studies have shown that LLMs can associate common substructures with descriptive terms (e.g., “–NO2” → “nitro compound”), enabling models like MolT5 to generate concise textual summaries of molecular features [887]. Together, these successes illustrate that LLMs can both “read” and “write” chemistry, transforming structural representations into human-readable language with high fidelity.
Remain Challenges. Despite these advances, challenges remain in both domains. Chemical naming models, while highly accurate, operate as black boxes; when they err on novel or highly complex molecules, it is difficult to trace the decision pathway or understand which structural elements led to a misnaming. Moreover, evolving IUPAC standards—such as recent organometallic nomenclature updates—require continual model retraining or fine-tuning to maintain correctness. In molecular captioning, the absence of a single “ground truth” means that errors are subtler and often manifest as partial truths or outright hallucinations (e.g., asserting a molecule is “used in perfumes” without evidence), and models struggle to calibrate the appropriate level of detail versus generalization [1024]. This fuzziness poses risks in scientific communication, as speculative or incorrect descriptors can mislead users.
Future Work. Looking ahead, integrating LLMs with external knowledge sources and multimodal inputs promises to address many of these limitations. Hybrid pipelines that combine neural proposals with rule-based validation could ensure 100% naming accuracy while preserving flexibility for new nomenclature conventions. Likewise, coupling captioning models with structured databases—so that, upon recognizing “glucose,” an LLM retrieves and incorporates the formula C6H12O6—would enhance factual correctness. Finally, multimodal architectures capable of ingesting both SMILES and 2D structural images, or embedding property predictors to append numerical descriptors (e.g., molecular weight, logP), will yield richer, more reliable textual outputs and usher in a new era of AI-driven chemical communication.
5.3.3. Chemical Characteristics Prediction
Chemical characteristics prediction task focuses on predictive modeling of molecular data, aiming to predict specific outcomes related to molecular structures using machine learning techniques. The primary input for this task is a detailed representation of molecules, typically in the form of SMILES strings, these representations allow models to learn meaningful patterns and relationships that inform predictions. Chemical Characteristics Prediction is fundamentally a Mol2Number task, taking molecular representations as input and producing quantitative property values as output.
In regression tasks, the goal is to predict continuous numerical values based on molecular features. For example, given the SMILES string "CCO" representing ethanol, a model might predict its boiling point as approximately 78.37°C. Similarly, for the SMILES "CC(=O)O" representing acetic acid, the model could predict a solubility of about 1000 g/L in water [1025]. In another case, the SMILES "C1=CC=CC=C1" for benzene might be used to predict its logP value as around 2.13. Similarly, in classification tasks, the objective is to determine discrete outcomes. For instance, given the SMILES "CC(=O)OC1=CC=CC=C1C(=O)O" for aspirin, the model might classify it as a non-steroidal anti-inflammatory drug (NSAID). Similarly, an example of reaction classification involves the reaction with the SMILES notation Brc1ccccc1.B(O)O >> c1ccccc1B(O)O, where the task is to classify the reaction type. The model classifies this reaction as a "Suzuki coupling" with a confidence of 98.2%.
The value of chemical characteristics prediction tasks in chemistry lies in their capacity to translate complex molecular and reaction information into quantitative predictions—enabling both experts and non-specialists to anticipate how molecules will behave under given conditions, including chemical properties, reaction types, yields, and reaction rates. For example, in property prediction, the SMILES string “CCO” succinctly encodes the structure of ethanol (“C” for carbon, “O” for oxygen), allowing a model to infer its physical and chemical properties without recourse to time-consuming quantum calculations. When we talk about reaction types, imagine mixing baking soda (NaHCO3) and vinegar (CH3COOH). A chemical characteristics prediction model would read the SMILES for each ingredient, see “NaHCOO” + “CC(=O)O,” and label it as an acid–base neutralization. That label helps chemists know that the reaction will produce CO2 gas. Predicting yields is like estimating how much carbon dioxide you’ll collect in a balloon when you mix your soda-and-yeast “volcano” experiment. A yield-prediction model might tell you, “Under these conditions, you’ll get about 80% of the maximum CO2 possible,” so you can plan ahead and avoid wasting ingredients. When discussing reaction-rate prediction, think of Alka-Seltzer fizzing in water. A model could predict how fast the tablet dissolves—does it take 30 seconds or three minutes?—based on temperature or how finely the tablet is crushed. In short, chemical characteristics prediction tasks let scientists—and even curious students—see, ahead of time, which “kitchen chemistry” will work best, how much product to expect, and how fast it will happen. This not only cuts down on costly trial-and-error in drug discovery or materials design, but also deepens our understanding of how molecules behave and reactions proceed.
Chemical characteristics prediction tasks encompass several categories: molecular property prediction (e.g., solubility — the maximum amount of a substance that can dissolve in a solvent, typically expressed in mol/L); bioactivity and binding-affinity prediction (e.g., IC50 — the concentration at which a compound inhibits 50% of a target’s biological activity); reaction characters prediction (e.g., reaction-type classification — categorizing a reaction by its mechanism, such as nucleophilic substitution or acid–base neutralization; and yield prediction — estimating the percentage of product obtained relative to the theoretical maximum under specified conditions); retrosynthesis-route scoring (e.g., feasibility scoring — assessing whether a proposed synthetic pathway can be practically executed in the laboratory; and cost scoring — predicting the total economic expense of reagents and operations); force-field and energy prediction (e.g., potential-energy surfaces — energy landscapes that map molecular geometry to potential energy; and interatomic forces — forces between atoms calculated as gradients on that energy surface); and molecular descriptor or embedding generation (e.g., low-dimensional embeddings — concise numerical vectors that capture key molecular characteristics in a reduced feature space).
Due to the intrinsic similarities among these tasks, we select two of the most representative ones—Property Prediction and Reaction Characters Prediction—for discussion in this paper. These two tasks benefit most from LLMs’ strengths: (1) they have abundant, well-structured textual and experimental data (e.g., MoleculeNet benchmarks, USPTO reaction corpora) that LLMs can readily learn from; (2) LLMs can provide both numerical predictions and human-readable rationales, enhancing interpretability over more opaque methods; and (3) improvements in these areas directly accelerate molecule prioritization and reaction planning in research and industry.
Property Prediction. The universal value of chemistry lies in accurately predicting compound properties to guide their practical use. Property Prediction is the task of predicting a molecule’s physicochemical or biological properties (e.g., solubility, binding affinity, toxicity) given its representation (such as a SMILES string, molecular graph, or descriptor vector). In pharmaceuticals, understanding how molecular structure influences bioactivity and toxicity enables the design of safer, more effective drugs; in materials science, predicting properties such as solubility, thermal stability, or mechanical strength from chemical structure accelerates the development of advanced polymers and functional materials. Traditional computational methods like quantum calculations and molecular dynamics offer high accuracy but demand extensive resources, whereas machine learning models can predict properties more efficiently. Recently, LLMs have demonstrated strong performance in molecular property and reaction-outcome prediction by leveraging vast textual and experimental datasets, achieving competitive accuracy without the heavy computational overhead of physics-based simulations. Combined with expert insight, AI-driven property prediction promises to revolutionize compound prioritization and materials design by focusing experimental efforts on the most promising candidates.
In early studies, LLMs such as BERT were applied to chemical reaction classification tasks. A representative work by Schwaller et al. achieved an impressive classification accuracy of up to 98.2%. The application focus then shifted from reaction classification to molecular property prediction, especially under scenarios with limited labeled data. Wang et al. proposed a semi-supervised model, SMILES-BERT [1026], which was pretrained on large-scale unlabeled data via a "masked SMILES recovery" task. It achieved state-of-the-art performance across multiple datasets and marked the first successful application of BERT in drug discovery tasks. During the early exploration of molecular language models, Chithrananda et al. introduced ChemBERTa [904], systematically examining the impact of pretraining dataset size, tokenization strategies (BPE vs. SmilesTokenizer), and molecular representations (SMILES vs. SELFIES) on model performance. Results showed that increasing the pretraining data from 100K to 10M led to significant improvements in downstream tasks such as BBBP and Tox21. Although ChemBERTa did not outperform the GNN baseline Chemprop, the authors suggested that further scaling could close this gap. Tokenization comparisons showed a slight advantage for the custom tokenizer. While no significant difference was observed between SMILES and SELFIES, attention head visualization using BertViz revealed neuron selectivity to functional groups, highlighting the importance of proper benchmarking and awareness of model carbon footprint. Building on this, Ahmad et al. developed ChemBERTa-2 [1027], aiming to create a general-purpose foundation model. With a multi-task regression head and pretraining on 77 million molecules, ChemBERTa-2 achieved comparable performance to state-of-the-art models on MoleculeNet tasks. The study emphasized that different pretraining strategies had varying effects on downstream tasks, suggesting that model performance depends not only on pretraining itself, but also on the specific chemical context and fine-tuning dataset. Further extending this direction, Yuksel et al. proposed SELFormer [1028], incorporating SELFIES to address concerns about the validity and robustness of SMILES. Pretrained on 2 million drug-like compounds and fine-tuned on a range of property prediction tasks (e.g., BBBP, SIDER, Tox21, HIV, BACE, FreeSolv, ESOL, PDBbind), SELFormer achieved leading performance in several cases. It demonstrated the ability to distinguish between molecules with varying structural properties, and suggested that future models should integrate structural data and textual annotations to build multimodal representations, enhancing generalizability and real-world utility.
To further improve molecular structure representation, Maziarka et al. introduced the MAT (Molecule Attention Transformer) [1029], incorporating atomic distances and molecular graph structure into the attention mechanism. This graph-structured self-attention led to performance gains in property prediction. Li and Jiang focused on capturing molecular substructures and proposed Mol-BERT [1030], pretrained on 4 million molecules from ZINC and ChEMBL. Treating fingerprint fragments as "words" and using Masked Language Modeling (MLM) to learn sentence-level molecular semantics, Mol-BERT outperformed both GNNs and sequence models on tasks like Tox21 and SIDER. Ross et al. developed MoLFormer, trained on over 1.1 billion SMILES from ZINC and PubChem. By introducing Rotary Position Embeddings, it more effectively captured atomic sequence relationships. MoLFormer not only surpassed GNNs on various benchmarks but also achieved 60x energy efficiency, representing progress toward environmentally sustainable AI.
On model generalization, Zhang et al. identified a bottleneck in the lack of correlation across different property datasets. They proposed MTL-BERT [1031], a multi-task learning model pretrained on large-scale unlabeled SMILES from ChEMBL. MTL-BERT improved prediction performance and enhanced interpretability of complex SMILES by extracting context and key patterns.
On the task-specific level, Yu et al. proposed SolvBERT [1025], a multi-task regression model designed to predict both solvation free energy and solubility. Despite the traditional reliance on 3D structural modeling for such tasks, SolvBERT—using only SMILES—achieved performance competitive with, or even superior to, GNN-based approaches, showcasing the potential of text-based modeling in physical chemistry.
While model performance continues to improve, limited labeled data remains a major challenge. In 2024, Jiang et al. introduced INTransformer [1032], which incorporated perturbation noise and contrastive learning to augment small datasets and improve global molecular representation, even under low-resource conditions. Similarly, MoleculeSTM [1033] used contrastive learning to align SMILES strings and textual molecular descriptions extracted from PubChem using a LLM. Extending this idea to proteins, Xu et al. proposed ProtST [1034], which models protein sequences using a protein language model and aligns them with protein descriptions encoded by LLMs, exploring multimodal fusion for biomacromolecule modeling.
Reaction Characters Prediction. Typical tasks in chemical reaction property prediction include reaction type classification (determining which type or mechanistic category a given reaction belongs to), reaction yield prediction (estimating the yield of the target product under specific conditions), and reaction rate prediction (assessing the kinetics of the reaction, such as activation energy or rate constant). These studies are of great importance in the fields of pharmaceuticals, materials science, and chemical engineering. For example, as early as 2018, Ahneman et al. demonstrated that machine learning could predict the yields of untested combinations in coupling reactions based on a limited amount of experimental data, successfully identifying previously unknown high-yield catalytic systems [1035]. Moreover, in the search for more efficient organic photovoltaic materials, it is often necessary to synthesize a series of candidate molecules. By using models to predict the yield of each reaction step, researchers can eliminate candidates with expected low yields and poor scalability early in the process, instead prioritizing synthetic routes that are predicted to be high-yielding and require fewer steps. This approach accelerates the screening process and conserves reagents.
Reaction type prediction aims to determine which category a given reaction belongs to—such as Suzuki coupling or Diels–Alder—based on its reactants and products. Traditionally, chemical reaction classification has relied on manually crafted rules or template libraries, but these approaches are poorly robust to new data and require complex atom-mapping preprocessing. To overcome this, Schwaller et al. introduced RXNFP [915], a Transformer-based encoder that learns fixed-length embeddings of entire reactions directly from unannotated SMILES in large datasets (e.g., USPTO) and then uses a simple k-NN or classifier to assign reaction classes. While RXNFP has been reported to achieve very high classification accuracy on reaction classification benchmarks (e.g. over 98% on some USPTO subsets) [915], it remains primarily a static feature extractor and is not designed for generative tasks like product generation or sequence-to-sequence modeling for continuous outputs. T5Chem [916] addresses many of these gaps by casting reaction tasks—classification, product prediction, retrosynthesis, and yield regression—as text-to-text problems. A single T5 model, pretrained on large molecular datasets from PubChem and fine-tuned on public reaction sets, achieves strong performance across multiple tasks with one shared architecture, improving multitask efficiency and generalization.
The prediction of reaction yields has long been a central challenge in synthesis planning and industrial optimization, owing to the complex interplay among substrate structures, reagents, catalysts, solvents, temperature, and other factors. Initially, Schwaller et al. leveraged their RXNFP reaction fingerprints by feeding the learned fixed-length reaction embeddings into a regression head to provide preliminary yield predictions for Buchwald–Hartwig and Suzuki–Miyaura coupling reactions, demonstrating the feasibility of end-to-end transformer embeddings for yield regression. However, RXNFP was not specifically designed for yield prediction, and its static fingerprints lacked sensitivity to changes in reaction conditions. To address this, T5Chem [928], a unified text-to-text multitask framework that, in addition to reaction classification and product prediction, incorporates a regression head for yield prediction. Pretrained on molecular data from PubChem and jointly fine-tuned on datasets such as USPTO, T5Chem matches or surpasses many baseline models across reaction prediction and yield tasks, showing that a single model can perform well on multiple reaction-related tasks. Building on this, the Schwaller team developed Yield-BERT [1036] in 2021 by fine-tuning ChemBERT on reaction SMILES to directly output yields; in high-throughput coupling reaction datasets Yield-BERT has been shown to achieve strong performance (e.g. values exceeding 0.90) compared to traditional methods using DFT-derived descriptors or handcrafted fingerprints. Yet Yield-BERT’s sensitivity to variations in catalysts, solvents, and other reaction conditions is limited, hindering its generalization across differing condition combinations. To enhance condition sensitivity, Yin et al. launched Egret [1037] in 2023, combining masked language modeling with condition-based contrastive learning to teach the model to distinguish yield differences for the same substrates under varying conditions; Egret achieved improved scores in several public benchmarks. Subsequently, Sagawa and Kojima’s ReactionT5 [925] employed a two-stage pretraining strategy—first training CompoundT5 on a molecular library, then pretraining on a reaction-level database—enabling the model, with limited fine-tuning data, to achieve good performance ( in challenging splits) in yield and product prediction tasks, highlighting the value of reaction-level pretraining. Most recently, the ReaMVP [1038] framework further incorporated 3D molecular conformations into pretraining alignment, aligning sequence and geometric views during a self-supervised stage, followed by fine-tuning on labeled yield data, resulting in modest boosts in on out-of-sample reactions and demonstrating the importance of multimodal information fusion for improving the generalizability of yield predictions.
Problems Solved by LLMs. LLMs have significantly advanced molecular property prediction by leveraging self-supervised learning on large unlabeled datasets to learn robust molecular representations that improve generalization to limited labeled data. They also enable effective multi-task learning through shared representations and task-specific fine-tuning strategies that mitigate interference between diverse prediction objectives. Furthermore, by integrating domain knowledge from chemistry, biology, and physics via pretraining on multimodal data and knowledge graph augmentation, these models can incorporate cross-domain insights seamlessly. Finally, LLMs excel at processing contextual molecular representations such as SMILES and International Chemical Identifier (InChI) codes, automatically learning high-dimensional features that capture complex structural interactions without the need for manual feature engineering.
Remain Challenges. Despite these advances, several challenges persist. The vastness of chemical space requires models that can reliably generalize to structurally novel molecules and unseen scaffolds, a task that remains difficult for current architectures . Activity cliffs, where minor structural modifications lead to dramatic changes in molecular properties, continue to undermine prediction accuracy and demand models that are sensitive to such subtle variations. Moreover, the inherently graph-structured nature of molecular data necessitates specialized neural architectures—such as graph neural networks and graph transformers—that can effectively capture both local and global structural patterns. Additionally, certain properties depend on three-dimensional conformations or quantum mechanical effects, which two-dimensional representations alone cannot fully capture, highlighting the need for methods that incorporate 3D structural information.
Future Work. Future work will focus on developing hybrid molecular representations that combine two-dimensional graph features with three-dimensional geometric descriptors—including conformer ensembles, steric effects, and electrostatic interactions—to more accurately model spatial relationships within molecules . Integrating molecular dynamics simulations and physics-informed neural networks can further enrich these representations by providing dynamic and mechanistic insights into molecular behavior over time. These advances are expected to enhance the generalization of models across diverse reaction conditions, improve the reliability of reaction yield predictions, and accelerate the discovery of novel compounds with desired properties.
5.3.4. Chemical Structure Prediction and Tuning
In many chemistry problems, the desired output is another molecule. These tasks can be seen as “translating one molecule into another” – hence chemical structure prediction & tuning. Chemical Structure Prediction & Tuning is inherently a Mol2Mol task. This category includes chemical reaction predictions, retrosynthesis planning, and molecule optimization, among others. An analogy from daily life would be cooking: you start with ingredients (molecules) and through a recipe (reaction), end up with a dish (a new molecule). Alternatively, think of it as solving a jigsaw puzzle: you have pieces (fragments of molecules) and want to put them together into a final picture (target molecule) – the input pieces and the output picture are both made of the same stuff, just rearranged. Chemical structure prediction & tuning tasks are central to chemistry because they essentially encompass chemical synthesis and design – predicting what will happen if molecules interact, or figuring out how to get from one molecule to another. For example, a forward reaction prediction might answer: “If I mix molecules A and B, what product will form?” A retrosynthesis task does the opposite: “I want molecule Z; what starting molecules could I use to make it?” These tasks directly assist chemists in the lab by suggesting likely outcomes or viable synthetic routes, thus speeding up research and discovery [1039].
Key subtasks under chemical structure prediction & tuning include reaction outcome prediction (given reactant molecules and possibly conditions, predict the product molecules), chemical synthesis (particularly retrosynthesis, given a target product, propose one or more sets of reactants that could produce it), and molecule-to-molecule optimization (propose a structural modification to an input molecule to improve some property, e.g. “suggest a similar molecule with higher potency”). Another subtask is chemical pathway completion (extending a partial sequence of reactions by suggesting the next molecule). All these involve generating molecules from molecules. Reaction prediction (input: reactants, output: products) is a prime example: e.g. input SMILES for ethanol + acetic acid, output SMILES for ethyl acetate (the esterification product). Chemical synthesis is similarly crucial: input a drug molecule, output a plausible precursor like an aromatic halide plus a coupling partner. Molecule tuning is essential for fine-adjusting a drug’s potency, selectivity, and pharmacokinetic properties while retaining its core active scaffold (e.g., introducing an amino group into the side chain of penicillin G produces ampicillin, thereby significantly improving oral bioavailability and antibacterial spectrum). We focus on these tasks because these have seen extensive development with LLM-like models and there are large datasets (like USPTO patent reactions) to train on.
Reaction Product Prediction. Reaction prediction is the task of predicting the products of a chemical reaction given the reactants (and sometimes reagents or conditions). It’s effectively a translation of a set of input molecules into a set of output molecules. In the language analogy, if reactants are words, the chemical reaction is a grammatical rule that rearranges those words into a new sentence (the product). A more concrete life analogy: consider mixing ingredients in cooking – if you combine flour, sugar, and butter and bake, you predict a cake as the outcome. Similarly, if you mix benzene with chlorine under certain conditions, you predict chlorobenzene (and hydrogen chloride as a byproduct) as the outcome. For decades, chemists approached this with rules (“if you see this functional group and add that reagent, you get this outcome”). The forward reaction prediction task asks an AI to learn those implicit rules from data. It’s significant because the space of possible products is enormous, and a human chemist’s intuition can be wrong or limited to known reactions. An accurate model can enumerate likely products, flagging surprises or confirming expectations. This can prevent wasted experiments and guide chemists toward successful reactions. It’s particularly useful in drug discovery or complex synthesis planning, where predicting side-products or main products can inform route selection.
Early machine learning models for reaction prediction often used template-based systems: large libraries of reaction templates were extracted, and algorithms matched reactants to these templates to propose products. While effective, those methods required expert-curated templates and struggled with reactions not in the template library. The turning point was realizing that chemical reactions could be encoded as strings (e.g. “CCO + O=C=O → CCC(=O)O” for esterification) and treated like a language translation problem. In 2016, Nam and Kim first applied a sequence-to-sequence RNN to reaction SMILES, showing that neural machine translation can predict organic reaction products directly from SMILES [1039]. However, RNNs had limitations – they sometimes forgot parts of the input and struggled with very long SMILES or complex rearrangements.
The real leap came with Transformers. In 2019, Schwaller et al. introduced the Molecular Transformer, a Transformer-based model for reaction outcomes that achieved 95% top-1 accuracy on the USPTO benchmark, significantly outperforming both template-based and RNN approaches [921]. By leveraging self-attention, the model considered all reactant tokens simultaneously, capturing reaction context more effectively. The Molecular Transformer also provided uncertainty estimates, enabling chemists to gauge prediction confidence. Despite its success, the first-generation Transformer had limitations. It tended to memorize frequent reaction patterns, leading to overconfidence on well-represented reactions and underperformance on rare or novel chemistries. It also handled only single-step reactions and did not predict yields or selectivities. To address memorization and improve generalization, Tetko et al. introduced SMILES augmentation—randomizing atom orders during training—which reduced overfitting and boosted top-accuracy metrics on large USPTO subsets [1040]. They also showed that beam-search decoding increased top-K accuracy (e.g. top-5) significantly. More recently, general-purpose LLMs have been fine-tuned on reaction data. For example, GPT-3 was adapted to reaction prediction tasks and achieved performance that is competitive with specialized Transformer models on some USPTO-style datasets [1041]. However, in zero-shot or few-shot settings, GPT-3.5 and GPT-4 still lag behind domain-trained models in tasks requiring precise structural prediction, with top-1 accuracies substantially lower in unconstrained prediction tasks [1042]. These findings underscore the continued importance of task-specific training and data augmentation for reliable reaction outcome prediction.
Chemical Synthesis. Once a target molecule has been identified, the next major challenge is to predict its optimal synthetic route—including per-step and overall yield—under realistic chemical constraints [1043]. While the demanding, elegant total syntheses of complex natural products have historically driven advances in organic chemistry, the past two decades have prioritized broadly applicable catalytic reactions [1043]; only recently has complex synthesis become relevant again as a digitally encoded knowledge source that can be mined by LLMs [1009]. Unlike single-molecule property prediction, reaction planning must account for the multi-body nature of synthesis—modifying one reactant often requires re-optimizing all others under different mechanisms or conditions—and must balance multiple objectives, such as maximizing overall yield, minimizing the number of steps and cost of readily available starting materials, and ensuring chemical compatibility at each stage. Planning can proceed forward—from simple substrates to the target—or, more commonly, via retrosynthesis, introduced by E. J. Corey [1044], which deconstructs the target molecule into fragments that are reassembled most effectively from inexpensive, commercially available reagents. Retrosynthesis is the reverse of reaction prediction: given a target molecule (the product), the task is to predict one or more sets of reactant molecules that could form it in a single step. It’s essentially “un-cooking” the dish to figure out the ingredients. In language terms, if a forward reaction is like forming a sentence from words, retrosynthesis is like taking a completed sentence and figuring out how to split it into two meaningful phrases that could combine to make that sentence. For example, the target “ethyl acetate” could be retrosynthesized to reactants “acetic acid + ethanol” (in the presence of an acid catalyst). Doing this well requires both creativity and extensive knowledge of known reactions, because the space of possible reactant combinations is enormous. For example, the first total synthesis of discodermolide required 36 individual steps (with a longest linear sequence of 24) to achieve only a 3.2 % overall yield, vividly illustrating the vast combinatorial explosion of possible routes and the reliance on expert intuition. By coupling structure–activity relationship predictions with synthesis planning, LLM-based approaches now promise to select or even design molecules not only for optimal properties but also for tractable, high-yielding synthetic accessibility—enabling both rapid route discovery and the creation of novel non-natural compounds chosen for their ease of synthesis and predicted performance [867].
Early computer-assisted synthesis planning began with recurrent architectures and handcrafted rules: Nam and Kim pioneered forward reaction prediction using a GRU-based translation model [1045], while Liu et al. applied an LSTM + attention seq2seq framework to retrosynthesis, achieving just 37.4 % accuracy on USPTO-50K [1046]. Schneider et al. then enhanced retrosynthesis by algorithmically assigning reaction roles to reagents and reactants [1047], and rule-based, template-driven systems such as Chematica and Segler & Waller [930] captured explicit atomic and bond transformations in reverse planning—training on millions of reactions to deliver 95 % top-10 retrosynthesis accuracy and 97 % reaction-prediction accuracy—yet remained limited by their reliance on manually curated template libraries and inability to propose truly novel transformations. Semi-template methods struck a balance: Somnath et al.’s synthon-based graph model decomposed products into fragments and appended relevant leaving groups, boosting top-1 accuracy to 53.7 % on USPTO-50K while retaining interpretability [1048].
While early synthesis planning methods relied on RNNs and handcrafted templates, the advent of LLMs has transformed the field by treating chemical synthesis as a data-driven “translation” task. Schwaller et al. [1049] first demonstrated this paradigm with a regex-tokenized LSTM-attention network that learned retrosynthetic rules directly from raw USPTO reactions—removing the need for explicit templates and uniquely tokenizing recurring reagents to distinguish solvents and catalysts. Building on that work, the Molecular Transformer applied the full Transformer encoder–decoder architecture to both forward reaction and retrosynthetic prediction, inferring subtle correlations between reactants, reagents, and products without any handcrafted rules and achieving state-of-the-art accuracy on USPTO-MIT, USPTO-LEF, and USPTO-stereo benchmarks. To extend LLMs beyond single-step predictions, Schwaller et al. introduced a hypergraph exploration strategy in their 2020 Molecular Transformer model [1050], dynamically expanding candidate routes using Bayesian-like scores and evaluating them with four new metrics—coverage (how much of chemical space is reachable), class diversity (variety of reaction types), round-trip accuracy (can predicted precursors regenerate the product), and Jensen–Shannon divergence (how closely the model’s predictions match real-world distributions). That same year, Zheng et al.’s SCROP [1051] model combined a template-free transformer with a neural syntax corrector to self-correct invalid SMILES, boosting top-1 retrosynthesis accuracy on USPTO-50K to 59.0%—over 6% better than template-based baselines.
More recently, pretrained encoder–decoder LLMs have further elevated performance and flexibility. Irwin et al.’s Chemformer [1052] used a BART backbone pretrained on millions of SMILES strings, then fine-tuned for sequence-to-sequence synthesis tasks and discriminative property predictions (ESOL, Lipophilicity, FreeSolv), demonstrating that task-specific pretraining is essential for efficiency and accuracy. In 2023, Toniato et al. [1053] introduced prompt-engineering into retrosynthesis by appending classification tokens to target SMILES, guiding the model toward diverse disconnection strategies and producing multiple viable routes “out of the box.” Finally, Fang et al.’s MOLGEN [1054] leveraged BART pretraining on 100 million SELFIES representations, domain-agnostic molecular prefix tuning, and an autonomous chemical feedback loop to ensure generated molecules are valid, non-hallucinatory, and retain their intended properties—foreshadowing autonomous LLM agents capable of end-to-end molecular design, synthesis planning, and iterative optimization.
Molecule Tuning. Molecule Tuning is the task of taking an initial molecule representation (e.g., SMILES string or graph) along with desired property modifications and generating structurally related analogs that optimize those specified properties while preserving the core scaffold. Molecule tuning leads compounds to simultaneously improve properties like potency, solubility, and safety—this is a cornerstone of drug design.
Early LLM-based approaches, such as DrugAssist [952], introduced an interactive, instruction-tuned framework that lets chemists iteratively “chat” with the model to optimize one or more properties at a time. DrugAssist has achieved leading results in both single- and multi-property optimization tasks, showing strong potential in transferability and iterative improvement [952]. However, it requires fine-tuning on task-specific datasets and its generalization to entirely new property combinations remains a challenge in practice. To advance further, Chemlactica and Chemma [954] were developed by fine-tuning language models on a large corpus of **110 million molecules with computed property annotations**. These models demonstrate strong performance in generating molecules with specified properties and predicting molecular characteristics from limited samples; relative improvements over prior methods (for example on the Practical Molecular Optimization benchmark) indicate they outperform earlier approaches in multi-property molecule optimization [954]. Despite these advances, fully zero-shot multi-objective optimization—where a model satisfies several new property constraints simultaneously without any additional training—remains difficult. Some approaches aim toward this goal (for example via prompt engineering, genetic methods, or sampling strategies), but no public model has yet been demonstrated to reliably achieve zero-shot control over wholly unseen property combinations. Finally, models that integrate richer structural context—such as using molecular graphs or fingerprint embeddings in addition to SMILES—are being explored. Early evidence suggests that these multimodal inputs can help propose chemically valid and synthetically accessible modifications under complex objectives, though again systematic evaluation under all multi-objective criteria is still emerging.
Problems Solved by LLMs. Thanks to LLM-based models such as the Molecular Transformer, many routine reaction predictions are now essentially solved: medicinal chemists can predict likely metabolites and process chemists can foresee side products with high confidence [921].
Remain Challenges. However, challenges remain in handling multi-step or “one-pot” reactions where sequential transformations occur, since current one-step models lack a mechanism to decompose complex cascades. Quantitative prediction of yields and selectivities is still out of reach, as these models only output the major product qualitatively. Additionally, out-of-domain reactions—those involving novel catalytic cycles or exotic reagents absent from training sets—often confound existing models [1041].
Future Work. Future LLMs for reaction prediction may incorporate mechanistic reasoning, internally decomposing reactions into elementary steps akin to chain-of-thought prompting [1055]. There is growing interest in multi-modal architectures that integrate text with molecular graphs or images, enabling a richer understanding of bond connectivity changes. Enhancing uncertainty quantification and explainability—such as highlighting which bonds form or break—will empower chemists to assess prediction confidence. Finally, embedding reaction prediction LLMs within autonomous laboratory systems could enable closed-loop experimentation, where AI proposes, executes, and learns from chemical reactions in real time. Future retrosynthesis LLMs will likely integrate external knowledge bases indicating which building blocks are inexpensive or readily available, biasing suggestions toward practical routes. We also anticipate multi-step planning architectures, where a higher-level agent orchestrates sequential calls to a one-step model, effectively planning entire synthetic routes. Finally, more interactive human–AI retrosynthesis tools may emerge, capable of asking clarifying questions or presenting alternative routes with pros and cons—transforming retrosynthesis from a static prediction task into a dynamic, collaborative design process.
5.3.5. Chemical Text Mapping
Chemical text mapping tasks take free-form chemical text as input and output either discrete labels (classification) or continuous values (regression). Chemical Text Mapping is fundamentally a Mol2Num task. For example, in a document classification scenario, given the safety note “During the reaction, hydrogen gas evolved rapidly and ignited upon contact with air,” the model outputs the hazard label “flammable.” In a text-based regression example, given the procedure description “1.0 g of reactant A yielded 0.8g of product B under standard conditions,” the model predicts a yield of 80%. By automating the extraction of critical information—hazard classes, reaction types, success/failure flags, yields, rate constants, temperatures, solubilities, pKa values, and more—chemical text mapping dramatically reduces manual curation, accelerates the creation of structured databases for downstream modeling, and empowers chemists and students to query “How dangerous is this step?” or “How much product did they actually get?” at scale.
Common tasks of this form include hazard classification, reaction-type classification, procedure outcome classification, yield and rate constant regression, temperature and solubility prediction, etc. In this work, we concentrate on chemical text mining within the chemical text mapping framework—harnessing LLMs to transform narrative chemical descriptions into actionable categorical and numerical data.
Chemical Text Classification. Chemical Text Classification is the task of categorizing chemical documents or text segments into predefined labels—such as reaction type, property mentions, or entity categories—based on their unstructured textual content.
Chemical text classification has matured through successive generations of chemistry-tuned LLMs, each addressing the gaps of its predecessors. ChemBERTa-2 [1027] was one of the first truly chemical-centric encoders—pretrained on 77 million SMILES strings (from PubChem) using masked-language modelling and multi-task regression—and it showed competitive performance on multiple downstream molecular property and classification benchmarks. However, as an encoder-only model, it requires separate fine-tuning heads for each task and lacks built-in generative or structured-output capabilities.
Recent studies, such as “Fine-tuning large language models for chemical text mining” [965], have explored unified frameworks that handle multiple chemical text mining tasks—compound entity recognition, reaction role labeling, MOF synthesis information extraction, NMR data extraction, and conversion of reaction paragraphs to action sequences. In these works, fine-tuned LLMs demonstrated exact match / classification accuracies in the range of approximately 69% to 95% across these tasks with only minimal annotated data [965]. Nevertheless, challenges remain in cross-sentence relations, complex numeric extractions, and the consistency / validation of structured or JSON-style output formats.
Problems Solved by LLMs. Firstly, they can achieve high-precision tasks such as hazard classification, reaction type annotation, yield and rate regression, with minimal or even no labeled data, through prompt engineering or a small number of examples, greatly reducing the cost of manually building domain dictionaries and rules; secondly, LLMs, with their powerful ability to understand context, can handle multiple information extraction subtasks (such as named entity recognition, relation extraction, and numerical prediction) simultaneously, thereby integrating the originally scattered pipeline processes into a unified end-to-end model; thirdly, combined with retrieval enhancement and chaining thinking technologies, LLMs also show robustness in long documents and cross-sentence dependency scenarios, laying the foundation for the automated construction of large-scale structured databases.
Remain Challenges. However, there are still several lingering issues that need to be addressed: LLMs sometimes generate confident but incorrect predictions (hallucination phenomenon), and their adaptability to extremely professional or the latest literature is limited; long text processing is limited by the size of the context window, making it difficult to complete global information integration across chapters or documents; in addition, support for multi-level nested entities and complex chemical ontologies is still not perfect.
Future Work. Future work can focus on introducing dynamic knowledge retrieval and knowledge graph fusion to build a continuously updated domain memory; exploring multimodal extraction (such as graph, spectrum, and text joint understanding); and combining uncertainty estimation and active learning strategies to improve the reliability and interpretability of the model, ultimately achieving a fully automated pipeline from laboratory notes to enterprise-level chemical knowledge bases.
5.3.6. Property-Directed Chemical Design
In property-directed inverse design, one begins with a set of target property criteria (for example, minimum thresholds for cell permeability, binding affinity, or solubility), optional domain priors encoded by pretrained generative LLMs, and a synthesizability filter to ensure practical feasibility. A LLM then directly generates candidate molecular structures—expressed as SMILES strings or molecular graphs—that are chemically valid, synthetically accessible, and predicted to meet the specified property requirements. Property-Directed Chemical Design is fundamentally a Text2Mol task. An everyday analogy might be describing a flavor or recipe you want (“something that tastes like chocolate but is spicier”) and having a chef create a new dish – here we describe a desired chemical property or scaffold, and the model devises a compound. The method’s objectives are to maximize compliance with target properties, promote novelty and diversity beyond natural-product scaffolds, and guarantee synthesizability via rule-based or learned retrosynthetic filters. Key constraints include chemical validity (proper valence and connectivity), synthesizability scores (e.g., predicted accessibility), and in-silico property feasibility. Analogous to random mutation screening but executed computationally at scale, only those molecules that satisfy both the validity/synthesizability filters and the predefined property thresholds are retained as viable candidates.
Key subtasks include conditional molecule generation, and text-conditioned molecule generation (where input is a description of desired properties or a prompt like “a molecule similar to morphine but non-addictive” and output is a new molecule suggestion). Another subtask is reaction-based text to molecule, such as “the product of acetone and benzaldehyde in aldol condensation” – where the input text implies a reaction and the output is the product structure. We will focus on (1) Conditional molecule generation and (2) De novo molecule generation, since these illustrate the spectrum from well-defined translation to open-ended generation.
Conditional Molecule Generation. Conditional molecule generation seeks to design novel compounds that satisfy user-specified criteria—whether a textual description, property targets, or structural constraints—directly in SMILES or 3D form.
The earliest text-conditioned methods relied on prompt-based sampling: both MolReGPT [889], which leverages GPT-3.5/4’s in-context few-shot learning to generate SMILES without any fine-tuning (albeit with variable chemical accuracy and prompt dependence), and Jablonka et al.’s GPT-3 adaptation [924], which fine-tuned the base model via prompt prefixes to produce valid SMILES matching property labels, showed that off-the-shelf LLMs can be repurposed for prompt-based conditional generation. In contrast, MolT5 [887] tackled text-to-SMILES translation via a T5 encoder–decoder fine-tuned on paired natural-language captions and SMILES, pioneering direct text-to-molecule mapping but without built-in multi-objective controls. To improve semantic alignment and diversity, TGM-DLM [986] introduced a diffusion-based language model conditioned on text embeddings, yielding molecules that more faithfully match user descriptions at the cost of extra compute. Recognizing the need for scaffold specificity, SAFE-GPT [957] adopted a fragment-based “SAFE” token representation, enforcing user-provided cores in each output while retaining peripheral variability. Extending conditioning into three dimensions, BindGPT [1056] embeds protein pocket geometries alongside sequence tokens to perform 3D structure-conditioned generation, enabling de novo ligand design tailored to binding-site shapes but requiring specialized 3D inputs. Beyond text and structure, target- and context-specific generation has been advanced by cMolGPT [1057], which integrates protein–ligand embedding vectors into a MOSES-pretrained transformer to produce candidate libraries for EGFR, HTR1A, and S1PR1 with QSAR-predicted activities correlating at Pearson r > 0.75, and by PETrans [1058], which couples a protein-sequence or 3D-pocket encoder with a SMILES decoder to generate ligands that respect detailed binding-site features. And ChemSpaceAL [1059] solves data-efficient, target-focused exploration by wrapping an uncertainty-driven active-learning acquisition loop around its transformer generator, iteratively sampling and scoring molecules against protein profiles to drastically reduce the need for large annotated inhibitor datasets while still uncovering high-affinity candidates.
De novo Molecule Generation. Here the task is: generate a molecule that fits a given textual description. This description could be very general (“a potent opioid painkiller that is less addictive”) or very specific (“a molecule with an ether linkage, and a molecular weight under 300 Da”). This is one of the holy grails of AI in drug discovery – allowing scientists to simply specify desired qualities and having the AI propose novel structures that meet them. It’s akin to an artist drawing a creature based on a myth description, except here it’s a chemist “drawing” a molecule based on a target profile. It could drastically speed up the brainstorming phase of drug design or materials design. Instead of manually tweaking structures, a researcher could ask the model for ideas: “Give me a drug-like molecule that binds to the serotonin receptor but doesn’t cross the blood-brain barrier” – a very high-level goal – and get some starting points.
Early work in de novo molecular design leveraged Adilov’s “Generative Pretraining from Molecules” [1060] , which adapted a GPT-2–style causal transformer to learn SMILES syntax via self-supervised pretraining and introduced adapter modules between attention blocks for minimal-change fine-tuning. This approach provided a resource-efficient generative backbone for both molecule creation and downstream property prediction. Scaling up, MolGPT [870] implemented a 6 million-parameter decoder-only model with masked self-attention to capture long-range SMILES dependencies, enforce valency and ring-closure rules for high-quality, chemically valid generation, and employ salience measures for token-level interpretability. MolGPT outperformed VAE-based baselines on MOSES and GuacaMol by metrics such as validity, uniqueness, Frechet ChemNet Distance, and KL divergence.
To better model global string context, Haroon et al. [1061] added relative attention heads to their GPT architecture, tackling the long-range dependency challenge and boosting validity, uniqueness, and novelty. ChemGPT [980] then systematically explored hyperparameter tuning and dataset scaling, revealing how pretraining corpus size and domain specificity drive generative performance. Subsequent work by Wang et al. further refined architectures and training strategies to surpass MolGPT benchmarks in de novo tasks. Departing from SMILES, Mao et al.’s iupacGPT [982] trained on IUPAC name sequences using SELFIES masking and adapters, producing human-interpretable outputs that align with chemists’ naming conventions and streamline validation, classification, and regression workflows. GraphT5 [897] first introduced multi-modal cross-token attention between 2D molecular graphs and text, enabling text-conditioned graph generation but lacking explicit control over scaffold or property constraints . MolCA [899] added uni-modal adapters and a cross-modal projector to improve robustness across representations, yet remained confined to 2D structures and did not follow complex textual instructions reliably.
To capture spatial information, 3D-MoLM [898] incorporated 3D molecular coordinates alongside text, allowing generation of conformers matching optical or binding-site descriptions, but it struggled with scaffold fidelity and multi-objective trade-offs. UTGDiff [987] addressed instruction fidelity by using a unified text-graph diffusion transformer that follows detailed prompts for substructure and property constraints. Addressing chirality, Yoshikai et al. [1062] coupled a transformer with a VAE and used contrastive learning from NLP to generate multiple SMILES representations per molecule—enhancing molecular novelty and validity while capturing stereochemical information. AutoMolDesigner [1063] wrapped de novo generation into an open-source pipeline for small-molecule antibiotic design, emphasizing domain-specific automation with heuristic filters and reaction-feasibility checks. Taiga [1064] introduced a two-stage approach—unsupervised SMILES → latent mapping followed by REINFORCE-based fine-tuning on metrics like QED, IC50, and anticancer activity—to achieve property-optimized design via reinforcement learning. Finally, cMolGPT [1057] demonstrated flexible mode switching, operating unconditionally to explore chemical space de novo and switching seamlessly to conditional, target-focused generation under the same architecture, thus unifying both paradigms in a single LLM framework.
Problems Solved by LLMs. LLMs have demonstrated that there exists a learnable mapping from natural language to chemical structures, allowing chemists to “draw” molecules with words instead of manually constructing SMILES strings. For example, MolT5—jointly trained on text and 100 million SMILES—can generate precisely “COc1ccccc1” in response to “Give me a molecule containing a phenyl ring, an ether linkage, and a molecular weight under 300Da.” MolReGPT goes further: using ChatGPT’s few-shot prompting, it can output valid candidate structures matching “phenyl ring + ether + MW<300” with no fine-tuning required. This capability drastically lowers the design barrier—researchers need only describe desired features to obtain testable structures. And more importantly, it dramatically narrows the chemical search space, focusing billions of possible molecules down to hundreds or thousands of the most relevant candidates and thereby greatly accelerating discovery. Moreover, LLMs support real-time, multi-objective, and multi-constraint generation—such as “increase hydrophilicity while retaining a hydrophobic ring” or “balance potency and synthetic accessibility”—and can explore chemical space rapidly even in low- or zero-data scenarios.
Remain Challenges. The imagination of LLMs is limited by their training. If certain property correlations were never seen, the model might not know how to fulfill a prompt. Also, validity of generated molecules is a concern – models like ChemGPT when generating freely can sometimes produce invalid SMILES or chemically impossible structures (though less often as training improves). When guided by text, the risk of hallucinating a molecule that meets text but is chemically nonsensical is real. For example, an LLM might attempt to satisfy “nonflammable gas” and produce something like “XeH” – which is not a stable molecule, but fits the prompt superficially (xenon is nonflammable, but xenon hydride is not a thing). Ensuring chemical validity often requires adding a post-check (like using cheminformatics software to validate and correct if needed). Another issue is the evaluation of success: if an AI generates a molecule from a prompt “potent opioid with less addiction,” how do we know it succeeded? We would have to test the molecule in silico or in lab. So often these models are used to generate candidates which are then fed into predictive models or experiments. It’s more of an ideation tool currently.
Future Work. We expect to see tighter integration of narrative-guided chemical design generation with other models and databases. One likely scenario is an interactive system: the user gives a prompt, the AI generates a molecule, then another AI (or the same with a different prompt) evaluates the molecule’s properties and explains why it might or might not meet the criteria, then the user or an automated agent refines the prompt or adds constraints, and the cycle continues – essentially an AI-driven design loop. Another direction is combining this with reinforcement learning or Bayesian optimization: use the text prompt to generate an initial population of molecules, then optimize them using property predictors (some recent work uses LLMs with in-context learning to do Bayesian optimization for catalysts) [1041], hinting at possibilities of optimization within the model’s latent space. Also, as these generative models improve, one can imagine integrating hard constraints (like no toxic substructures or obey Lipinski’s rules for drug-likeness) directly via prompt or via a filtering step in the generation process (some have tried using fragment-based control tokens, e.g., telling the model “include a benzene ring” or “avoid nitro groups”). Another interesting future aspect is diversity vs. focus: LLMs might have a bias to generate molecules that are similar to what they know (so-called mode collapse around familiar structures). Future models might include techniques to encourage more novelty (perhaps via lower sampling temperature or specialized training objectives) when novelty is desired. Conversely, if a very specific structure is needed, one might combine text prompt with a partial structure hint (like providing a scaffold SMILES and asking the model to complete it with substituents that confer certain properties).
5.3.7. Chemical Knowledge Narration
Chemical knowledge narration tasks take unstructured chemical text as input and produce another, more structured or user-friendly text output. It is fundamentally a Text2Text task. For example, given the free-form procedure “Add 5g of sodium hydroxide to 50mL of water, stir for 10 min, then slowly add 10g of benzaldehyde at ,” a model can generate a standardized protocol: “1. Dissolve 5g NaOH in 50mL H2O. 2. Cool to . 3. Add 10g benzaldehyde dropwise over 5 min. 4. Stir at for 10 min.” Likewise, from “The oxidation of cyclohexanol to cyclohexanone was performed using PCC in dichloromethane,” it can produce the concise summary “Cyclohexanol was oxidized to cyclohexanone with PCC in DCM,” and given the SMILES “CC(=O)OC1=CC=CC=C1C(=O)O” it can output the IUPAC name “2-acetoxybenzoic acid (aspirin).” These transformations standardize and clarify experimental descriptions, automate nomenclature and summarization, and enable seamless integration into electronic lab notebooks, saving researchers hours of manual editing.
Common chemical knowledge narration applications include protocol standardization, reaction summarization, SMILES-IUPAC conversion, literature summarization, question-answer generation, and explanatory paraphrasing. In our work, we harness these capabilities for chemical knowledge QA, chemical text mining, and chemical education.
Chemical Knowledge QA. Chemical Knowledge QA is the task of answering natural-language queries about chemical concepts, reactions, and properties by retrieving and reasoning over relevant information from unstructured or structured chemical sources.
Chemical knowledge QA first saw major gains with LlaSMol [1065], an instruction-tuned model using the SMolInstruct dataset (over three million samples, covering 14 chemistry tasks), which outperformed GPT-4 on several chemistry benchmarks such as SMILES-to-formula conversion and other canonical tasks [1065]. Nevertheless, it remains bounded by its training data cutoff, and it does not explicitly handle visual structure figure input in its evaluated tasks. To fill in more visual reasoning ability, ChemVLM [1007] introduces a multimodal model integrating chemical images and text, enabling it to perform tasks like chemical OCR, multimodal chemical reasoning, and molecule understanding from visual plus textual cues. It achieves competitive performance across these tasks [1007]. However, like many static QA models, its update of chemical knowledge is limited by its training corpus, and occasional incorrect or incomplete answers remain a concern.
Chemical Text Mining. Chemical Text Mining is the task of extracting and structuring relevant chemical information—such as reactions, molecular properties, or entity relationships—from unstructured textual sources (e.g., scientific articles, patents, and lab reports).
Chemical text mining has seen clear advances with models such as LlaSMol [1065], which uses the SMolInstruct dataset (over three million samples across 14 chemistry tasks) to fine-tune open-source LLMs, achieving substantial improvements over general-purpose models [1065]. More recently, the work “Fine-tuning Large Language Models for Chemical Text Mining” [965] demonstrated that fine-tuned models (including ChatGPT, GPT-3.5-turbo, GPT-4, and open-source LLMs such as Llama2, Mistral, BART) can handle five extraction tasks—compound entity recognition, reaction role labeling, MOF synthesis information extraction, NMR data extraction, and conversion of reaction paragraphs to action sequences—with exact accuracy in the range of approximately 69% to 95% using minimal annotated data [965]. There are also works which fine-tune pretrained LLMs (GPT-3, Llama-2) to jointly perform named entity recognition and relation extraction across sentences and paragraphs in materials chemistry texts, outputting structured or JSON-like formats with good performance in linking entities like dopants-host, MOF information, and general composition/phase/morphology/application extraction [976].
Chemistry Education LLMs. Chemistry education LLMs have evolved from generic chatbots to increasingly specialized, curriculum-aligned tutors. For instance, ChemLLM [926] is among the first LLMs dedicated to chemistry: trained on chemical literature, benchmarks, and dialogue interactions, it can provide explanations, perform interpretation of core concepts, and respond to student queries in a dialogue style with reasonable domain accuracy. Another relevant study[863], establishes a benchmark comprising eight chemistry tasks (e.g. explanation, reasoning, formula derivation), showing that models like GPT-4, GPT-3.5, Davinci-003 etc. outperform generic LLMs in zero-shot and few-shot settings on many such tasks. A further perspective by Du et al. [1019] discusses how LLMs can assist in lecture preparation, guiding students in wet-lab and computational activities, and re-thinking assessment styles, though it does not report a system that simultaneously generates new problems, offers misconception warnings, and supports dialogic tutoring. Each generation—from ChemLLM’s dialogue capability, to benchmark studies demonstrating explanation + reasoning, to educational perspectives exploring scalable assessment—shows how chemistry-tuned LLMs are gradually moving toward more capable teaching assistants, though fully interactive, curriculum-aligned AI tutors remain an open challenge.
Problems Solved by LLMs. LLMs have revolutionized chemical knowledge narration tasks by enabling end-to-end transformations—standardizing free-form procedures, summarizing complex reaction descriptions, and converting between SMILES and IUPAC names—all in a single, unified model without the need for multiple specialized tools or extensive manual intervention. Their strong contextual understanding and few-shot/in-context learning capabilities allow them to adapt quickly to new tasks with minimal examples, dramatically cutting the time researchers spend editing protocols, writing summaries, or updating electronic lab notebooks.
Remain Challenges. LLMs still occasionally “hallucinate” confidently incorrect details, struggle with long documents that exceed their context windows, and lack robust mechanisms for handling deeply nested or multi-step reaction descriptions.
Future Work. Future work must therefore focus on integrating retrieval-augmented generation to ground outputs in real literature, fusing text with experimental figures or spectra for more accurate multimodal summaries, incorporating chain-of-thought prompting to produce auditable reasoning paths, maintaining a dynamically updated chemical knowledge base to stay current with new findings, and developing specialized evaluation benchmarks for protocol standardization, reaction summarization, and nomenclature conversion. These advances will make LLMs even more reliable, explainable, and indispensable for chemical knowledge QA, text mining, and education.
5.3.8. Benchmarks
As summarized in Table 36, we have compiled a comprehensive list of datasets that have been employed across a broad range of chemistry tasks using LLMs. This table includes benchmarks for tasks such as molecular property prediction, reaction yield prediction, reaction type classification, reaction kinetics, molecule captioning, reaction product prediction, chemical synthesis planning, molecule tuning, conditional and de novo molecule generation, chemical knowledge question answering, chemical text mining, and chemical education.
Systematically cataloging current research benchmarks is essential to bridge the gap between generic language modeling advances and chemistry-specific tasks. By unifying evaluation protocols—standardizing data splits, SMILES preprocessing, and task formulations—we ensure that performance comparisons are both fair and reproducible. Moreover, a holistic survey reveals not only the breadth of existing benchmarks (from molecular property regression and reaction-type classification to molecule captioning and generative design) but also their inherent biases: most datasets focus on drug-like organic molecules or patent reactions, while domains such as inorganic chemistry, environmental pollutants, and negative-result reporting remain underrepresented. This comprehensive overview therefore lays the foundation for more rigorous model development and benchmarking, guiding researchers toward data curation and experimental designs that fully exploit LLM capabilities in chemical contexts.
In the sections that follow, we will select several of the most influential datasets from Table 36 for in-depth discussion. For each chosen dataset, we will describe its scope, annotation scheme, and typical use cases, and then survey the performance of representative LLMs on these benchmarks to elucidate current capabilities and remaining challenges.
MoleculeNet.(Mol2Num) MoleculeNet is a consolidated benchmark suite that currently bundles sixteen public datasets spanning quantum chemistry, physical chemistry, biophysics and physiology. All tasks share a uniform, text–first layout: each row of the .csv file starts with a canonical SMILES string followed by one or more property labels—binary for classification (e.g. BBBP, BACE, HIV, Tox21, SIDER) or floating–point for regression (ESOL, FreeSolv, Lipophilicity, the QM series). Where 3-D information is required, companion .sdf or NumPy archives store Cartesian coordinates and energies. Official JSON split files define random, scaffold and temporal partitions so that every study can reproduce the same train/validation/test folds. A typical classification row from BACE reads CC1=C(C2=C(N1)C(=O)N(C(=O)N2)Cc3ccccc3)O,1, the trailing "1" indicating an active -secretase inhibitor. In the regression task ESOL a sample line might be OC1=CC=CC=C1,-2.16, pairing a phenol SMILES with its experimental log-solubility in mol L−1.
Table 35.
Performance of large (chemical) language models and strong baselines on MoleculeNet (↑ = higher is better for ROC-AUC; ↓ = lower is better for RMSE). Best per column in blue. Values are taken from original papers/model cards; “—” means not reported.
Table 35.
Performance of large (chemical) language models and strong baselines on MoleculeNet (↑ = higher is better for ROC-AUC; ↓ = lower is better for RMSE). Best per column in blue. Values are taken from original papers/model cards; “—” means not reported.
| Model (source) | Classification (ROC-AUC ↑) | Regression (RMSE ↓) | ||||||
|---|---|---|---|---|---|---|---|---|
| BACE | BBBP | HIV | Tox21 | SIDER | ESOL | FreeSolv | Lipo | |
| MolBERT | 0.866 | 0.762 | 0.783 | — | — | 0.531 | 0.948 | 0.561 .[1] |
| ChemBERTa-2 | 0.799 | 0.728 | — | — | — | 0.889 | 1.363 | 0.798[2] |
| BARTSmiles | 0.705 | 0.997† | 0.851 | 0.825 | 0.745 | — | — | —[3] |
| MolFormer-XL | 0.690 | 0.948 | 0.847 | — | 0.882† | — | — | 0.937†[3] |
| ImageMol | — | — | 0.814 | — | — | — | — | —[4] |
| SELFormer | 0.832 | 0.902 | 0.681 | 0.653 | 0.745 | 0.682 | 2.797 | 0.735[5] |
Table 36.
Chemistry Tasks, Benchmarks, Introduction and Cross tasks
| Type of Task | Benchmarks | LLM Tested | Introduction | Cross tasks |
|---|---|---|---|---|
| Property Prediction | MoleculeNet [1066] | ✓ | 16 public datasets (SMILES + labels; quantum, phys-chem, physiology; 130k–400k samples) | – |
| OGB-PCQM4M-v2 [1067] | × | 3.8M molecular graphs with 3-D coords + HOMO–LUMO gaps | Reaction Rate | |
| Therapeutics Data Commons [1068] | ✓ | 30+ ADMET/bioactivity CSVs, leaderboard splits | Chemical Synthesis | |
| PDBbind [1069] | × | 22k protein–ligand complexes; PDB/mol2 + | – | |
| BindingDB [1070] | × | 3M structure–target Ki/IC50 pairs (SMILES, FASTA) | – | |
| Open Catalyst OC22 [1071] | × | Adsorption geometries + barriers for 1.3 M configs | Rate | |
| Reaction Yield Prediction | Buchwald–Hartwig HTE [1035] | × | 3955 C–N couplings; CSV (yield, ligands, bases) | – |
| Suzuki–Miyaura HTE [1072] | × | 5760 C–C couplings; yield matrix | – | |
| USPTO-Yield | ✓ | 1M patent reactions with numeric yields | Reaction type | |
| Open Reaction Database (ORD) [1073] | × | JSON records: reactants / products / conditions / yield | Reaction type | |
| ORDerly-Yield [1074] | × | Clean ORD + USPTO split, reproducible splits | Reaction Product Prediction | |
| AstraZeneca ELN [1075] | × | 25k ELN entries, diverse chemistries (CSV) | Conditions optimisation | |
| Reaction Type Classification | USPTO-50K [1076] | ✓ | 50036 atom-mapped reactions, 10 classes | Chemical Synthesis, Reaction Yield |
| USPTO-Full / MIT [1076] | ✓ | 400k–1.3M reactions; 60 coarse classes | Reaction Product, Chemical Synthesis | |
| Reaxys Reaction [1077] | × | 40M literature reactions, multi-level class labels | Reaction Rate | |
| ORDerly-Class [1074] | × | ORD subset with curated type labels | – | |
| Reaction Rate / Kinetics | NIST SRD-17 [1078] | × | 38k gas-phase rate constants, Arrhenius params (XML) | – |
| RMG Kinetics DB [1079] | × | 50k gas+ surface elementary steps (YAML) | Mechanism generation | |
| NDRL/NIST Solution DB | × | 17k solution-phase rate constants | – | |
| Combustion FFCM | × | Curated small-fuel flame kinetics set (YAML) | – | |
| Molecule Captioning | ChEBI-20 [1080] | ✓ | 33k SMILES–natural-language pairs (JSON) | Retrieval |
| SMolInstruct (Caption) [1065] | ✓ | 3M multi-task instructions incl. caption pairs | – | |
| MolGround [1081] | ✓ | 20k captions with atom-level grounding tags | – | |
| Reaction Product Prediction | USPTO-MIT Forward [921] | ✓ | 400k one-step reactions; SMILES input → product | Reaction Type |
| ORDerly-Forward [1074] | × | ORD/USPTO split, non-USPTO OOD set | Reaction Yield | |
| Chemical Synthesis | USPTO-50K Retro [1082] | ✓ | 50k product → reactant pairs, 10 classes | Reaction class |
| PaRoutes [1083] | × | 20k multi-step routes, JSON graphs | – | |
| ORDerly-Retro [1074] | × | ORD split with non-USPTO OOD test | Reaction Product | |
| TDC Retrosynthesis [1068] | ✓ | Wrappers for USPTO-50K + PaRoutes | – | |
| AiZynthFinder test [1084] | × | 100 difficult drug targets, MOL files | – | |
| Molecule Tuning | GuacaMol Goal-Dir. [1085] | ✓ | 20 oracle tests (SMILES, property calls) | Molecule Generation |
| PMO Suite [1086] | ✓ | 23 tasks, score-limited oracle calls (JSON) | – | |
| MOSES Opt [1086] | ✓ | Scaffold-constrained optimisation splits | De novo Molecule Generation | |
| TDC Docking [1068] | × | AutoDock/Vina scoring tasks (SDF) | Conditional Protein Generation | |
| LIMO Affinity [1087] | × | Gradient VAE optimisation toward nM affinity | Conditional Generation | |
| Chemical Text Classification | ChemProt [1088] | × | 1820 PubMed abstracts with 5 CP-relation labels | QA, Chemical text mining |
| BC5-CDR [1089] | × | 1500 abstracts; chemical–disease relations | NER | |
| CHEMDNER / BC4CHEMD [1090] | × | 10k abstracts, 84k chemical mentions | NER | |
| NLM-Chem [968] | × | 150 full-text articles, gold chemical tags | Chemical text mining | |
| ChEMU Patents [1091] | × | 1.5k patent excerpts with entity + event labels | Chemical text mining | |
| ChemNER 62-type [1092] | × | Fine-grained 62-label NER corpus | – | |
| Cond. Mol Generation | MOSES Scaffold [1086] | × | Bemis-Murcko prompts → molecules | De novo molecule generation |
| MOSES-2 [1086] | × | Adds stereo + logP/MW targets (JSON) | – | |
| GuacaMol Cond. [1085] | ✓ | Similarity + property dual constraints | Molecule Tuning | |
| LIMO [1087] | × | Latent inversion with docking-affinity oracle | – | |
| De Novo Mol Gen | MOSES (dist.) [1086] | ✓ | 1.9M ZINC clean-leads SMILES, train/test/scaffold | Conditional generation |
| GuacaMol Dist. [1085] | ✓ | 10 distribution-learning metrics tasks | Optimisation | |
| GEOM-Drugs [1093] | × | 100k drug-like molecules + 3-D conformers | Conformer generation | |
| QMugs [1094] | × | 665k drug-like molecules with QM labels | Property prediction | |
| TDC MolGeneration [1095] | × | Unified wrapper for MOSES/GuacaMol | – | |
| Chemical Knowledge QA | ScholarChemQA [1001] | ✓ | 40 k yes/no/maybe QAs from research abstracts | Chemical text mining |
| ChemistryQA [1096] | × | 4500 high-school calc-heavy MCQs | Education | |
| MoleculeQA [1000] | ✓ | 12k molecule-fact QAs (SMILES + text) | Molecule Captioning | |
| MolTextQA [1097] | × | MC-QA over PubChem descriptions | – | |
| Chemical text mining | CHEMDNER [1090] ChEMU [1091] NLM-Chem [968] | See classification rows (NER + event extraction) | NER / IE suites | |
| ChemBench [1098] | × | 7059 curated curriculum QAs; JSON | QA | |
| Chemical Education | ChemistryQA [1096] | × | High-school MCQ dataset (LaTeX problems) | QA |
USPTO.(Mol2Num, Mol2Mol) Starting from Daniel Lowe’s 1.8-million raw patent dump—which stores un-mapped SMILES triples like O=C=O.OCCN>>O=C(O)NCCO—researchers have carved out several task-specific subsets. The USPTO-MIT forward-prediction split keeps 479035 atom-mapped reactions and is the de-facto benchmark for single-step product prediction, where the input string CC(=O)Cl.O=C(O)c1ccccc1>>? asks the model to regenerate the ester product. USPTO-50K retains 50014 lines and appends a ten-class label, enabling reaction-type classification exemplified by CCBr.CC(=O)O>[base]>CCOC(=O)C,7, where “7” represents an acylation class. Building on the same patent source, USPTO-Yield merges textual yield phrases so that a row such as C1=CC=CC=C1Br.CC(=O)O>[Cu]>C1=CC=CC=C1CO,72 allows numeric yield regression, while USPTO-Stereo preserves wedge-bond chirality, demanding stereochemically exact output for inputs like C[C@H](Cl)Br.O>>?. Beyond single steps, PaRoutes links 450000 Lowe reactions into multistep route graphs so a model must recreate the full path ending in C1=CC=C(C=C1)C(=O)O rather than just its terminal disconnection. Finally, ORDerly re-formats selected USPTO lines into Open-Reaction-Database JSON with timestamped splits—each entry like "inputs":["smiles":"CCOC(=O)Cl"],"outputs":["smiles":"CCOC(=O)O"], "temperature": 298—so that forward prediction, condition inference and genuine time-splitting can be assessed simultaneously. Together these sub-corpora let large chemical language models be probed across product generation, type classification, yield regression, stereochemical accuracy and multistep planning without ever leaving the USPTO domain.
Table 37.
Transformer-scale LLM performance across USPTO sub-datasets. Columns report the main metric for each task: forward product Top-k accuracy on USPTO-MIT; reaction-type accuracy and Macro-F1 on USPTO-50K; yield regression and MAE on USPTO-Yield; stereochemical Top-1 accuracy on USPTO-Stereo; multi-step route success on PaRoutes; and forward Top-1 / condition accuracy on ORDerly. Best in each column is highlighted with blue.
Table 37.
Transformer-scale LLM performance across USPTO sub-datasets. Columns report the main metric for each task: forward product Top-k accuracy on USPTO-MIT; reaction-type accuracy and Macro-F1 on USPTO-50K; yield regression and MAE on USPTO-Yield; stereochemical Top-1 accuracy on USPTO-Stereo; multi-step route success on PaRoutes; and forward Top-1 / condition accuracy on ORDerly. Best in each column is highlighted with blue.
| Model | USPTO-MIT | USPTO-50K | USPTO-Yield | USPTO-Stereo | ||||
| Top-1↑ | Top-5↑ | Top-10↑ | Acc.↑ | F1↑ | ↑ | MAE | Top-1↑ | |
| Molecular Transformer | 0.875 | 0.937 | 0.954 | — | — | — | — | 0.825 |
| Augmented Transformer | 0.888 | 0.944 | 0.960 | 0.921 | 0.909 | — | — | 0.832 |
| MolFormer | 0.883 | 0.945 | 0.960 | 0.945 | 0.932 | — | — | 0.832 |
| ReactionBERT | 0.930 | 0.972 | 0.981 | 0.930 | 0.918 | — | — | 0.845 |
| Chemformer | 0.910 | 0.968 | 0.979 | 0.915 | 0.905 | — | — | 0.834 |
| ReactionT5 | 0.975 | 0.986 | 0.988 | — | — | — | — | 0.790 |
| CompoundT5 | 0.866 | 0.895 | 0.904 | — | — | — | — | — |
| ProPreT5 | 0.998 | 1.000 | 1.000 | — | — | — | — | — |
| ChemBERTa-2 | — | — | — | 0.880 | 0.865 | — | — | — |
| Yield-BERT | — | — | — | — | — | 0.41 | 13.2 | — |
| ReaLM | — | — | — | — | — | 0.52 | 10.5 | — |
ChEBI-20.(Mol2Text) ChEBI-20 is a medium-sized molecular–caption corpus that links 33,010 small-molecule SMILES strings to concise English sentences distilled from the ChEBI ontology. The data are released as a UTF-8 CSV whose first column stores the canonical SMILES and whose second column holds the free-text caption; a third column specifies the standard 8:1:1 train/validation/test split. Because each record couples structure and language, the collection naturally supports molecule-to-text caption generation, text-to-molecule retrieval and cross-modal representation learning. In the captioning task a model receives the input CC(C)OC(=O)C1=CC=CC=C1C(=O)O and is expected to output a sentence such as “Ibuprofen is a propionic acid derivative with an isobutyl side chain and an aromatic core.” In the inverse retrieval task the same caption is fed to the system, which must rank the correct SMILES ahead of thousands of distractors; the ground-truth pair above therefore serves both roles without modification.
Table 38.
Molecule captioning on ChEBI-20. Best per metric in blue.
| Model | BLEU-2↑ | BLEU-4↑ | METEOR↑ | ROUGE-1↑ | ROUGE-2↑ | ROUGE-L↑ |
|---|---|---|---|---|---|---|
| MolT5-base | 0.540 | 0.457 | 0.569 | 0.634 | 0.485 | 0.578 |
| MolReGPT (GPT-4-0314) | 0.607 | 0.525 | 0.610 | 0.634 | 0.476 | 0.562 |
| MolT5-large | 0.594 | 0.508 | 0.614 | 0.654 | 0.510 | 0.594 |
| Galactica-125M | 0.585 | 0.501 | 0.591 | 0.630 | 0.474 | 0.568 |
| BioT5 | 0.635 | 0.556 | 0.656 | 0.692 | 0.559 | 0.633 |
| ICMA (Galactica-125M) | 0.636 | 0.565 | 0.648 | 0.677 | 0.537 | 0.618 |
USPTO-MIT.(Mol2Mol, Mol2Num) The USPTO-MIT dataset, curated by MIT from Lowe’s extraction of original USPTO patent reactions and cleaned through atom mapping validation and SMILES normalization, comprises approximately 470,000 single-step forward reaction records split into training, validation, and test sets of 409,035, 30,000, and 40,000 examples respectively; each record encodes atom-mapped reactants, reagents, and products in SMILES (for example, CC(=O)O.CCCO>>CCCOC(=O)C denotes acetic acid and propanol yielding propyl acetate), and these high-quality, atom-mapped reactions support a variety of AI-driven chemistry tasks such as forward reaction prediction, single-step retrosynthesis, reaction classification, template extraction with atom mapping, and reagent prediction.
Table 39.
Forward reaction prediction performance of chemical LLMs and strong non-LLM baselines on the USPTO-MIT Separated dataset. Best per column in blue.
Table 39.
Forward reaction prediction performance of chemical LLMs and strong non-LLM baselines on the USPTO-MIT Separated dataset. Best per column in blue.
| Model | Top-1↑ | Top-2↑ | Top-3↑ | Top-5↑ |
|---|---|---|---|---|
| Molecular Transformer | 88.8 % | 92.6 % | — | 94.4 % |
| T5Chem | 90.4 % | 94.2 % | — | 96.4 % |
| CompoundT5 | 86.6 % | 89.5 % | 90.4 % | 91.2 % |
| ProPreT5 | 99.8 % | — | — | — |
| ReactionT5 | 97.5 % | 98.6 % | 98.8 % | 99.0 % |
Table 40.
Reaction type classification performance on USPTO-MIT for LLMs / transformer models and top non-LLM baselines. Best per metric in blue.
Table 40.
Reaction type classification performance on USPTO-MIT for LLMs / transformer models and top non-LLM baselines. Best per metric in blue.
| Model | Top-1 Accuracy↑ | Top-5 Accuracy↑ |
|---|---|---|
| Molecular Transformer | 90.4 % | 95.3 % |
| Augmented Transformer | 90.6 % | 96.1 % |
| ProPreT5 | 99.8 % | — |
GuacaMol.(Mol2Mol) GuacaMol is an open-source de novo molecular design benchmarking suite built from approximately 1.8 million deduplicated SMILES strings standardized from the ChEMBL database. The construction pipeline includes salt removal, charge normalization, element filtering (retaining only H, B, C, N, O, F, Si, P, S, Cl, Se, Br, I), truncation to under 100 characters, and removal of any compounds overly similar to the hold-out set(holdout_set_gcm_v1.smiles). GuacaMol defines 20 goal-directed tasks—ranging from simple property optimization (e.g., log P, TPSA) and rediscovery of known drugs to similarity-guided generation and scaffold-hopping—and, most centrally, molecule tuning multi-objective optimization tasks. These tuning tasks challenge models to perform fine-grained adjustments against scoring functions like QED, log P, and synthetic accessibility rather than merely reproducing the training distribution. For example, in the Cobimetinib multi-objective tuning task, generative models apply Pareto optimization strategies (such as NSGA-II or NSGA-III) to iteratively modify Cobimetinib’s SMILES substituents, maximizing a weighted combination of drug-likeness (QED) and solubility (log S) scores to produce novel candidates balanced across multiple property dimensions. This emphasis on molecule tuning not only tests a model’s ability to replicate known chemical spaces but also measures its practical value in accelerating lead optimization during early drug discovery by finely balancing multiple molecular properties.
GuacaMol not only supports goal-directed multi-property tuning tasks, but also provides two key generative scenarios: conditional molecule generation and de novo generation. In conditional generation, models must produce compounds that satisfy user-specified property or scaffold constraints. For example, MolGPT achieves strong control over QED and log P in GuacaMol’s conditional benchmarks, attaining validity ≈0.98, high uniqueness, and novelty close to 1.000, while cMolGPT extends these approaches by prepending target property values to the input, enabling precise conditional generation. More recently, LigGPT introduces flexible multi-constraint conditioning, allowing a single model to balance multiple property targets while retaining synthesizability and validity.
In the de novo setting, GuacaMol evaluates models on validity, uniqueness, novelty, Fréchet ChemNet Distance (FCD), and KL divergence. Here, MolGPT achieves validity 0.981, uniqueness 0.998, and novelty 1.000, LigGPT improves further with validity 0.986 and novelty 1.000, and SELF-BART excels on distributional similarity metrics such as FCD and KL divergence. Graph-based masked generation approaches also show competitive performance on these benchmarks, highlighting the impact of molecular representation on generation quality. Early generative frameworks such as ChemGAN and Entangled Conditional AAE serve as important references in the distribution-learning tasks, helping the community understand the strengths and limits of deep learning methods in exploring chemical space. Together, GuacaMol’s conditional and de novo tasks offer a comprehensive, rigorous testbed for chemical LLMs, driving continued innovation in model architectures and training strategies.
Table 41.
De novo molecule generation performance on GuacaMol for chemical-domain LLMs. Best per metric in blue.
Table 41.
De novo molecule generation performance on GuacaMol for chemical-domain LLMs. Best per metric in blue.
| Model | Validity↑ | Uniqueness↑ | Novelty↑ |
|---|---|---|---|
| MolGPT | 0.981 | 0.998 | 1.000 |
| LigGPT | 0.986 | 0.998 | 1.000 |
| GraphGPT | 0.975 | 0.999 | 1.000 |
| SmileyLlama (T=1.1) | 0.9783 | 0.9994 | 0.9713 |
| SmileyLlama (T=0.6) | 0.9968 | 0.9356 | 0.9113 |
MOSES.(Text2Mol) MOSES is a molecular generation benchmarking platform introduced by Polykovskiy et al. in 2020 in Frontiers in Pharmacology, derived from 4,591,276 SMILES in the ZINC Clean Leads collection and filtered by molecular weight (250–350 Da), rotatable bonds (≤ 7), XlogP (≤ 3.5), removal of charged/ non-C/N/S/O/F/Cl/Br/H atoms, PAINS, and medicinal chemistry filters to yield 1,936,962 drug-like molecules. These molecules are split into training (≈ 1,584,664), test (≈ 176,075), and scaffold-test (≈ 176,089 with unique Bemis–Murcko scaffolds) sets to assess model performance on both seen and unseen scaffolds. MOSES supports de novo generation, where the CharRNN baseline achieves validity 0.975, uniqueness 0.999, novelty 0.842, IntDiv1 0.856 and IntDiv2 0.850 on the test set; and conditional generation, for example SELF-BART applies property-conditioned decoding to generate molecules with desired constraints, attaining validity 0.998, uniqueness 0.999, novelty 1.000, and strong internal diversity scores. Consequently, MOSES serves as a unified and rigorous benchmark for core tasks in molecular generation, spanning distribution-learning to property-driven generation.
Table 42.
De novo (distribution-learning) generation performance on MOSES. Best per metric in blue.
| Model | Validity↑ | Unique@10k↑ | Novelty↑ | IntDiv1↑ | IntDiv2↑ |
|---|---|---|---|---|---|
| MolGPT | 0.994 | 1.000 | 0.797 | 0.857 | 0.851 |
| SELF-BART | 0.998 | 0.999 | 1.000 | 0.918 | 0.908 |
| MTMol-GPT | 0.845 | 0.993 | 0.984 | 0.835 | — |
| SF-MTMol-GPT | 1.000 | 0.955 | 0.932 | 0.850 | — |
Summary. Current benchmark datasets for LLMs in chemistry emphasize three main task categories. First, molecular property prediction dominates, with collections such as MoleculeNet and Therapeutics Data Commons providing numerous binary and regression targets (e.g. solubility, toxicity, binding affinity). Second, reaction outcome and classification tasks, primarily drawn from USPTO and Reaxys repositories, assess models on product prediction, reaction-type labeling, and yield regression. Third, molecule generation benchmarks (MOSES, GuacaMol) evaluate de novo and condition-driven design by measuring validity, novelty, and property optimization. By contrast, complex tasks like multi-step synthesis planning, reaction condition optimization, and 3D conformer reasoning remain underrepresented.
Compared to traditional cheminformatics and rule-based methods, domain-trained transformer models have demonstrated quantitative gains. Here, we illustrate such improvements using three representative tasks that are widely applied: Property Prediction, Reaction Prediction and Classification, and Molecule Generation. In property prediction, specialized SMILES-BERT variants outperform random forests on multiple ADMET assays by several percentage points. In single-step reaction tasks, sequence-to-sequence transformers surpass template-based systems, improving top-1 accuracy by 5–10%. In generative settings, LLM-based generators achieve near-perfect chemical validity (> 98 %) and higher diversity metrics than earlier recurrent or graph-based approaches, while also enabling multi-objective optimization of drug-like properties with average improvements of 10–20 % over heuristic baselines.
Despite encouraging results, current LLMs face important limitations when applied to chemistry domains.
A core challenge is that chemical data are highly structured (e.g. graphs), yet LLMs operate on linear token sequences. This mismatch means that transformers struggle to natively represent molecular topology and 3D geometry. For instance, an LLM given a SMILES string has no direct encoding of the molecule’s shape or stereochemistry, which can be crucial for many properties. This leads to errors when tasks fundamentally depend on spatial or structural reasoning – a known example is that models predicting quantum chemistry properties from SMILES (instead of actual 3D coordinates) perform poorly and misrepresent the true task. Another limitation is the knowledge cutoff of LLMs and their tendency to hallucinate. Without explicit chemical rules, an LLM may propose an impossible reaction or a nonsensical molecule, especially if it hasn’t seen similar examples in training. Ensuring validity and consistency in outputs remains non-trivial; even with grammar-constrained decoding, models might violate subtle chemical constraints or overlook rare elements.
Data scarcity and bias are additional concerns: many benchmark datasets are relatively small and biased toward drug-like molecules, so LLMs may generalize poorly to larger chemical space or unusual chemotypes. Researchers also report that LLM performance can be brittle – small changes in input format (SMILES vs another notation) or prompt phrasing can yield different results, reflecting an unstable understanding. From a practical standpoint, the resource requirements of large models pose a challenge: using cutting-edge LLMs (like GPT-4) can be orders of magnitude more costly and slower than using task-specific models. This makes it difficult for researchers to fine-tune or deploy the largest models on private data.
Finally, the interpretability of LLM decisions is limited – unlike human chemists or simpler models that can point to a mechanistic rationale, a transformer’s prediction is hard to dissect, which can erode trust in sensitive applications (e.g. drug discovery). In summary, today’s chemical LLMs are constrained by input representations, data quality, model transparency, and computational cost, highlighting the need for new strategies to realize their full potential.
From these observations we derive three core insights. First, dataset limitations in both scale and scope constrain LLM performance: public repositories often contain errors, inconsistent annotations, and limited chemical diversity—popular benchmarks such as QM9 can be misused and fail to represent realistic molecular spaces. To overcome this, a community effort should curate larger, cleaner datasets by aggregating high-quality experimental results (e.g. ADME assays, comprehensive reaction outcomes) and expanding initiatives like the Therapeutic Data Commons to include negative results and broader chemistries. Benchmarks must also incorporate crucial chemical information—3D conformations, stereochemistry, reaction conditions (catalysts, solvents, yields)—to foster deeper chemical reasoning beyond simple SMILES pattern matching. Second, model and methodology strategies require refinement. Treating chemical structures as linear text has merits but introduces tokenization and validity challenges, motivating exploration of alternative representations such as SELFIES or fragment-based vocabularies. Generic LLMs lack embedded chemical rules (valence, aromaticity) and benefit from domain-specific pretraining on extensive chemical corpora (molecules, patents, protocols). Hybrid architectures—integrating LLMs with graph neural networks, physics-based modules, or explicit inter-atomic distance matrices—can bridge the gap between sequence models and spatial structure. Finally, improving reliability and usability demands thoughtful task formulation and validation. Reformulating regression tasks as classification or ranking problems, developing chemistry-specific prompting (few-shot, chain-of-thought, multi-step retrosynthesis prompts), and embedding chemical validation loops (reinforcement learning with validity rewards or critic models) can reduce hallucinations and ensure chemical soundness. Coupling LLMs with external tools (“LLM + tools” paradigms) and advancing interpretability—via attention analysis, attribution methods, and standardized evaluation metrics—will build trust and utility. In conclusion, converging richer data, smarter representations, hybrid modeling, and robust benchmark design will propel LLMs toward becoming reliable, powerful instruments for chemical research., the convergence of richer data, smarter representations, hybrid modeling strategies, and thoughtful benchmark design will help overcome current limitations and guide LLMs to become more reliable and powerful tools for chemical research.
5.3.9. Discussion
Opportunities and Impact. LLMs are becoming transformative tools in chemistry and chemical engineering, bridging traditional chemical methods with cutting-edge computational advancements.
In molecular textualization (Mol2Text), the traditional rule-based naming system relies on manually written chemical naming rules, which often find it difficult to cover all cases of novel or complex molecules, while LLMs can learn naming patterns from large-scale chemical corpora, achieving better generalization and robustness. Transformer models such as Struct2IUPAC achieve 98.9 % correct SMILES-IUPAC conversions on a 100 k test set, halving the residual error ( % vs 3 %) observed for the long-standing rule-based parser OPSIN on comparable benchmarks [1099]. For example, when researchers discover a new antibiotic with a highly unusual structure, these models can immediately generate its official chemical name, saving chemists hours or days of manual naming efforts.
In property prediction (Mol2Number), traditional methods require the training of independent models for each property, making it difficult to share underlying chemical knowledge; while LLMs have absorbed the correlations between properties during the pre-training phase, achieving one-time multi-task predictions. LLM-driven approaches like ChemBERTa unify this process—after training on vast molecular datasets, a single model can simultaneously predict properties like solubility (e.g., “will the compound dissolve easily in water?”), toxicity (e.g., “is the compound safe?”), and expected chemical yields. Scaling ChemBERTa-2 pre-training from 0.1 M to 10 M molecules lifts average ROC-AUC across the MoleculeNet suite by +0.11 (e.g., 0.67 to 0.78) without training separate models per property, whereas classic GCN/GAT baselines plateau near 0.70 [1027]. For instance, pharmaceutical chemists designing new drugs can quickly assess multiple crucial drug properties simultaneously, significantly streamlining the early drug discovery phase.
For complex reaction planning (Mol2Mol), traditional reaction prediction software largely relies on manual reaction templates, which are difficult to capture complex mechanisms; while LLM based on Transformer can accurately plan synthetic routes by learning long-range dependencies between steps through self-attention mechanisms. LLM-based models, such as those inspired by Reaction Transformer, can break complex reactions into simpler, understandable stages. For example, when developing a complex cancer treatment molecule, these models can accurately suggest each intermediate step in synthesis, increasing the success rate of predicted synthetic routes by 10-20% compared to older rule-based methods. The Molecular Transformer attains 90.4 % top-1 accuracy on USPTO product prediction, while the best contemporary template-driven system (RetroComposer) reaches only 65.9 %, a 24-point jump that translates into far fewer manual template overrides during route design [921].
In chemical text classification (Text2Num), traditional text mining often relies on manual rules or shallow features, making it difficult to handle context-dependent chemical descriptions; while LLMs can accurately extract reaction conditions and results through deep semantic understanding. For example, researchers fine-tuned LLaMA-2-7B on 100,000 USPTO reaction processes, enabling it to directly generate structured records that comply with the Open Reaction Database (ORD) architecture—the model achieved an overall message-level accuracy of 91.25%, and a field-level accuracy of 92.25%, capable of stably identifying key numerical information such as temperature, time, and yield [1009]. In comparison, the best feature-engineered CRF/SSVM patent-NER pipeline achieved an F-measure of only 88.9% on the CHEMDNER-patents CEMP task, highlighting the significant performance improvement of LLM in chemical text information extraction [1100].
Inverse design (Text2Mol) benefits from LLMs’ ability to generate chemically valid candidates under user-specified property constraints, reducing trial-and-error cycles from weeks to minutes and expanding exploration of novel chemical space [870]. Traditional reverse engineering requires a large amount of trial and error and manual filtering, resulting in low efficiency; while LLM acquires distributed knowledge in the chemical space through pre-training and combines it with conditional generation, which can quickly output high-quality candidate molecules. With LLM-based generative models such as MolGPT [870] and ChemGPT [980], chemists can now simply describe the needed properties (e.g., "a molecule that lowers blood pressure but doesn’t cause dizziness") and instantly receive hundreds of suitable, chemically viable molecule suggestions. This dramatically shortens the molecule discovery process from potentially weeks of trial and error down to just minutes. Conditional generators such as Adapt-cMolGPT now yield 100 % syntactically valid molecules under SELFIES-based sampling, compared with 88–94 % validity for earlier SMILES-based GPT decoders—eliminating one in ten invalid proposals and narrowing medicinal-chemist triage [1101].
Finally, in chemical text mining (Text2Text), traditional text mining often relies on manual rules or shallow features, making it difficult to handle context-dependent chemical descriptions; while LLMs can accurately extract reaction conditions and results through deep semantic understanding. For example, researchers fine-tuned LLaMA-2-7B on 100,000 USPTO reaction processes, enabling it to directly generate structured records that comply with the Open Reaction Database (ORD) architecture—the model achieved an overall message-level accuracy of 91.25%, and a field-level accuracy of 92.25%, capable of stably identifying key numerical information such as temperature, time, and yield [1009]. In comparison, the best feature-engineered CRF/SSVM patent-NER pipeline achieved an F-measure of only 88.9% on the CHEMDNER-patents CEMP task, highlighting the significant performance improvement of LLM in chemical text information extraction [1100].
Challenges and Limitations. Despite these advances, LLM-driven chemical models still face critical hurdles.
First, the “experimental validation gap” persists: Despite impressive predictive power, LLM-driven chemical models still require extensive laboratory verification before their results can be trusted for critical decisions. For example, an LLM acts like an experienced chef who can predict delicious recipes based on prior knowledge, but ultimately you still have to cook the dish yourself and taste it to confirm whether it’s actually good [1102]. Without deeper integration with automated robotic experimental setups or closed-loop experimental cycles, this validation bottleneck remains a significant "last mile" barrier [1103].
Second, LLMs lack explicit mechanistic reasoning. Current LLM models predominantly learn associative patterns from large-scale datasets, often lacking explicit chemical reasoning or mechanistic insights. Imagine a student who memorizes a vast number of math solutions without understanding the underlying principles; they will struggle when encountering slightly altered problems. Similarly, predicting complex, multi-step chemical reactions (e.g., radical-based cascades) demands a mechanistic understanding that pure memorization from data cannot reliably provide, leading to frequent mistakes in subtle yet critical chemical details and limiting industrial adoption [932,1104].
Third, LLMs struggle to generalize to novel and sparsely represented chemical spaces. LLMs heavily depend on the breadth and quality of their training data, causing them to perform inadequately in chemically novel scenarios or sparsely represented reaction classes. For example, an LLM trained mostly on common organic reactions is like a chef proficient in everyday home cooking who suddenly faces preparing sophisticated French cuisine; the unfamiliar ingredients and methods may lead to frequent mistakes. This limitation restricts their predictive reliability in cutting-edge research or niche industrial applications, where innovation frequently occurs [1105].
Fourth, chemical “hallucinations” remain problematic: Generative chemical models powered by LLMs often produce chemically invalid or practically unsynthesizable molecules, a phenomenon known as chemical "hallucination." For example, an LLM could resemble an imaginative but inexperienced architect designing visually appealing buildings that are impossible to construct due to practical limitations of materials and construction methods. Although integrating rule-based filters partially mitigates this issue, systematic validation approaches remain inadequate, undermining trust in their use for real-world synthesis planning [1106].
Lastly, domain-specific fine-tuning demands high-quality annotated datasets, which are expensive or impractical to produce at scale for every niche subfield. Without robust few-shot or low-data learning methods, many specialized applications remain out of reach [1107].
Research Directions.
- Hybrid LLM–Mechanistic Frameworks. Combine LLMs with rule-based and physics-informed modules to integrate statistical language understanding with chemical theory.
- Multimodal Chemical Representations. Develop architectures that jointly process SMILES, molecular graphs, and spectroscopic or crystallographic data to capture 3D and electronic structure.
- Closed-Loop Experimental Pipelines. Integrate LLM outputs into automated synthesis and analysis platforms, enabling rapid hypothesis testing and feedback-driven model refinement.
- Data-Efficient Fine-Tuning. Leverage transfer learning, few-shot prompting, and synthetic augmentation of underrepresented reaction data to improve performance in sparse domains.
- Explainability and Uncertainty Quantification. Incorporate attribution methods and probabilistic modeling to provide confidence metrics and mechanistic rationales alongside predictions.
- Governance-First Deployment. Establish standards for model validation, transparent reporting (model cards), and ethical guidelines to ensure responsible use in chemical research and industry.
Conclusion. LLMs have significantly reshaped workflows in chemical discovery and engineering, but realizing their full potential requires innovations that marry linguistic intelligence with chemical reasoning, robust validation workflows, and ethical governance. By pursuing hybrid, multimodal, and closed-loop approaches, the community can overcome current limitations and drive the next wave of breakthroughs in chemical science and industrial application.
5.4. Life Sciences and Bioengineering
5.4.1. Overview
5.4.1.1 Introduction to Life Sciences
Life sciences refer to all branches of sciences that involve the scientific study of living organisms and life processes [1108,1109,1110]. In other words, they encompasses fields like biology, medicine, and ecology that explore how organisms (from micro-organisms to plants and animals) live, grow, and interact. Life scientists seek to understand the structure and function of living things, from molecules inside cells up to entire ecosystems, and to discover the principles that govern life [1111,1112]. In simple terms, life sciences are about studying living things (such as humans, animals, plants, and microbes) to learn how they work and affect each other and the environment [1109,1113,1114]. This knowledge not only satisfies human curiosity about nature but also underpins applications in health, agriculture, and environmental conservation.
Life sciences are vast domains, so their research tasks range from decoding genetic information to observing animal behavior. Traditionally, each type of task has relied on specific methods and tools developed over decades (or even centuries) of biological research [1108,1109]. Through its detailed subdivision into specific subfields, we have categorized research in life sciences based on investigations ranging from the microscopic to the macroscopic level [1115] and summaries of the review topics written by life sciences scientists [1116,1117,1118,1119,1120]. The main research tasks and their classic approaches include:
Deciphering Genetic Codes. Understanding heredity and gene function has been a core task. Early geneticists used breeding experiments to infer how traits are inherited [1121,1122]. In the 20th century, methods like DNA extraction [1123,1124,1125] and Sanger sequencing [1126,1127,1128] enabled reading the genetic code, while PCR (polymerase chain reaction) [1129,1130] revolutionized gene analysis by allowing DNA amplification. Today, high-throughput genome sequencing [1131,1132] and bioinformatics [1133] are standard for genetic research.
Studying Cells and Molecules. A fundamental task is to uncover how cells and their components (proteins, nucleic acids, etc.) function. Biochemists use centrifugation [1134,1135] and chromatography [1136] to separate molecules, and X-ray crystallography [1137,1138] or NMR to determine molecular structures [1139]. For instance, gel electrophoresis [1140] became a routine method to separate DNA or proteins by size, and later innovations like Western blots [1141] provided ways to detect specific molecules. These methods, combined with controlled experiments in test tubes, have traditionally powered discoveries in molecular and cell biology.
Physiology and Medicine. Life sciences also deals with whole organisms, how organ systems work and how to treat their maladies [1142,1143,1144,1145,1146,1147,1148]. Physiological experiments on model organisms (from fruit flies and mice to primates) have been crucial [1143,1144]. For example, testing organ function [1149] or disease processes [1150] often involves animal models where interventions can be done. In medicine, clinical observations and clinical trials (systematic testing of treatments in human volunteers) are standard for linking biological insights to health outcomes. Additionally, fields like immunology and neuroscience have developed specialized methods (e.g. antibody assays [1151,1152], brain imaging [1153]) to probe complex systems. Life scientists in these areas often use a combination of laboratory experiments, medical imaging (like MRI, X-rays), and longitudinal studies to unravel how the human body (and other organisms) maintain life and what goes wrong in diseases [1154,1155].
Ecology and Evolution. A broader task is understanding life at the population, species, or ecosystem level, how organisms interact with each other and their environment, and how life evolves over time [1156,1157,1158]. Field observations and experiments are the cornerstone of ecology – researchers might count and tag animals, survey plant growth, or manipulate environmental conditions in the wild [1159]. Long-term ecological research (e.g. observing climate effects on forests [1160]) and paleontological methods [1161] have illuminated evolutionary history. In evolution, apart from the fossil record, comparing DNA/protein sequences across species (made possible by sequencing methods) [1162,1163] is a modern approach, but historically, comparative anatomy and biogeography were used by Darwin and others to infer evolutionary relationships. Today, computational models and DNA analysis complement classical fieldwork to address ecological and evolutionary questions [1164].
Figure 19.
The relationships between major research tasks between biology and bio-engineering.
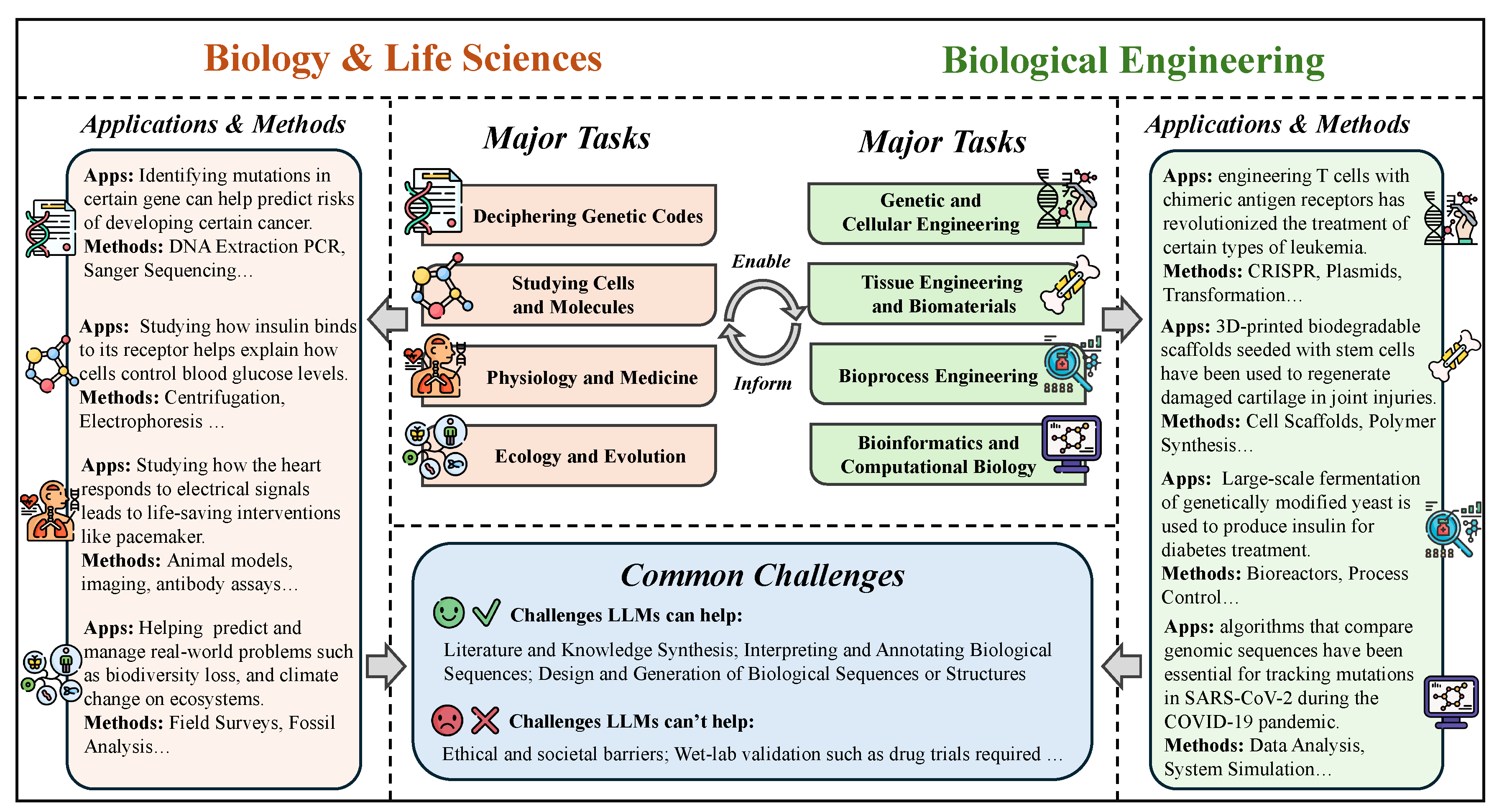
5.4.1.2 Introduction to Bioengineering
Bioengineering (also called biological engineering) is the application of biological principles and engineering tools to create usable, tangible, and economically viable products [1165,1166]. Essentially, bioengineering leverages discoveries from life sciences by applying engineering design to develop technologies addressing challenges in biology, medicine, or other fields involving living systems [1167,1168]. Put simply, bioengineering combines biological understanding with engineering expertise to design and build solutions, such as medical devices [1169], novel therapies [1170], or biomaterials [1171,1172]. It is inherently interdisciplinary: a bioengineer may employ mechanical engineering to construct artificial limbs, electrical engineering for biomedical sensors, chemical engineering for bioprocessing, and biological sciences across all applications [1173]. Thus, bioengineering bridges pure science with practical engineering, translating biological knowledge into innovations that enhance lives.
To better organize and understand bioengineering’s scope, traditional research tasks are categorized into well-established domains. This classification is based on historical developments and practical engineering workflows [1174,1175,1176], dividing the discipline according to how biological knowledge translates into engineering solutions and tangible products [1176,1177]. Each category corresponds to a major application domain within bioengineering, representing distinct pathways for integrating biology with engineering.
Genetic and Cellular Engineering. Many bioengineers modify biological cells or molecules for new functions—such as engineering bacteria to produce pharmaceuticals or editing genes to treat diseases [1178,1179,1180,1181]. Genetic engineering techniques from molecular biology are foundational. Since the 1970s, recombinant DNA technology (using restriction enzymes to manipulate genes) [1182,1183,1184,1185] and cell transformation [1186] have enabled scientists to insert genes into organisms. Practically, bioengineers often use plasmids to introduce genes into bacteria and employ fermentation bioreactors (borrowed from chemical engineering) for cultivating genetically modified microbes at scale. More recently, CRISPR-Cas9 gene editing (developed in the 2010s) has allowed precise genome modifications [1187,1188,1189,1190,1191]. Typical workflows include designing genetic constructs, altering cells, and scaling selected cell lines. This domain overlaps significantly with biotechnology and biomedical sciences [1167,1192].
Tissue Engineering and Biomaterials. A significant bioengineering area involves engineering or replacing biological tissues using methods such as cultivating cartilage, skin, or organs in laboratories [1193,1194,1195,1196]. Core techniques include cell culture and scaffold fabrication—bioengineers cultivate cells on biodegradable scaffolds (often polymers) to form tissues [1194]. Late 20th-century innovations demonstrated that seeding cells onto 3D scaffolds could produce artificial tissues (e.g., synthetic skin) [1197,1198]. Biomaterials science contributes materials (e.g., polymers, ceramics) designed to safely interact with the body, such as titanium or hydroxyapatite implants [1199,1200,1201]. This domain has produced synthetic skin grafts and advances toward lab-grown organs like bladders [1202] and blood vessels [1203].
Bioprocess Engineering. Bioengineers design processes to scale up biological products (e.g., mass-producing vaccines, biofuels, or fermented foods) [1204,1205,1206], drawing on chemical engineering principles adapted to biological contexts. Engineers design bioreactors, optimize conditions (temperature, pH, nutrients), and ensure sterile, efficient processes [1207]. Traditional methods include continuous culture [1208,1209] and process control systems. The large-scale production of penicillin in the 1940s exemplifies bioprocess engineering, involving optimizing Penicillium mold growth in industrial tanks [846,848,1210]. Today’s production of monoclonal antibodies or industrial enzymes similarly employs refined classical fermentation and purification techniques [1211,1212].
Bioinformatics and Computational Biology. Although occasionally considered separate fields, bioengineers frequently engage in computational modeling of biological systems or analyze biological data (genomic or protein structures) to guide engineering designs [1213,1214,1215,1216]. This domain involves algorithms and simulations—for example, modeling physiological systems using differential equations and computational methods from control theory. Computational approaches have long contributed to bioengineering, supporting prosthetic design optimization (via CAD and finite element analysis) and genomic analyses (software for DNA sequence analysis) [1217,1218,1219,1220,1221,1222]. This domain underscores bioengineering’s combination of wet-lab experimentation and computational methods.
5.4.1.3 Current Challenges
Life sciences and bioengineering are foundational disciplines that have transformed our understanding of life and significantly improved human health and well-being. Life sciences uncover the fundamental principles of biology, from Darwin’s theory of evolution and Mendel’s laws of inheritance to the germ theory of disease. These discoveries led to major advances such as vaccines, antibiotics like penicillin, and the molecular revolution sparked by the discovery of DNA’s double-helix structure. Tools like PCR and the Human Genome Project further deepened our ability to decode and manipulate genetic information, ushering in the era of personalized medicine.
Bioengineering complements these insights by applying them to solve real-world problems. The development of X-ray imaging allowed non-invasive diagnosis, while innovations like the implantable pacemaker and artificial organs expanded the scope of life-saving care. The production of human insulin through recombinant DNA technology marked a milestone in biopharmaceuticals. Later, tissue engineering demonstrated that lab-grown organs could be transplanted into humans, and gene-editing tools like CRISPR have opened new frontiers in treating genetic diseases.
Together, life sciences and bioengineering form a powerful synergy: the former provides deep biological insight, while the latter transforms that knowledge into tangible solutions. Their joint progress continues to revolutionize medicine, agriculture, and environmental science—improving quality of life and shaping a more advanced future.
Despite remarkable advancements in life sciences and bioengineering, numerous challenges persist due to the complexity and inter-connectivity inherent to biological systems. Intriguingly, many of the most formidable obstacles overlap between these fields, as they frequently address complementary facets of the same intricate biological phenomena. In this section, we systematically examine several critical common challenges, distinguishing between those that currently remain beyond the capability of artificial intelligence tools such as LLMs and those that can already benefit from LLM integration.
Still Hard with LLMs: The Tough Problems.
Here we analyze some key common challenges that are currently still beyond the reach of LLMs, we acknowledge LLMs’ limitations related to experimental design, interpretative complexity, and practical hands-on tasks, underscoring domains where human expertise remains indispensable.
- Ethical and Safety Challenges. Life sciences and bioengineering are inherently intertwined with ethical and societal considerations that transcend purely technical challenges [1223]. These fields routinely grapple with questions surrounding the responsible use of gene editing technologies like CRISPR [1224,1225], the long-term ecological effects of genetically modified organisms [1226,1227,1228], and the protection of sensitive patient data in genomics and biomedical research [1229]. While LLMs can assist in synthesizing scientific literature and outlining stakeholder perspectives, they lack the capacity for moral reasoning or normative judgment. Their outputs are constrained by the biases present in their training data, which poses risks when applied to ethically sensitive domains [1230,1231]. Ethical decision-making in bioengineering—such as determining the acceptability of human germline editing, setting standards for clinical trials, or regulating synthetic biology applications—remains the responsibility of human experts, policymakers, and the broader public. These decisions require inclusive debate, value alignment, and legal oversight that go beyond algorithmic capabilities [1232,1233]. As technologies advance, both the Life sciences and AI communities must collaboratively develop ethical frameworks that reflect societal values while fostering innovation.
- Needs for Empirical Validation. Both life sciences and bioengineering ultimately rely on physical experimentation [1234]. Scientific hypotheses must be empirically verified, and bioengineered solutions require testing under real-world conditions [1235]. However, such experiments often pose significant bottlenecks due to their inherent slowness, high costs, and ethical limitations—particularly in human studies, where experimentation is strictly constrained, and animal models often fail to fully replicate human biology. [1236,1237,1238] While computational models can alleviate some of these burdens, they cannot fully substitute for wet-lab or clinical experiments. Similarly, LLMs are incapable of conducting physical experiments or collecting new empirical data. Although they can assist in designing experimental protocols, they cannot implement or validate them [1239]. Consequently, research challenges that fundamentally require novel data acquisition—such as identifying new drug targets or evaluating biomaterials—remain beyond the scope of LLMs alone. The crucial last mile of validation in biology and engineering - demonstrating something works in actual living systems - remains dependent on laboratories, clinical trials, and real-world testing [1240].
- Complexity of Biological Systems. The overarching challenge is that living systems are astonishingly complex and multi-scale. Scientists struggle with this complexity as small changes can have cascading, unpredictable effects. For life scientists, this means incomplete understanding of many diseases and biological processes [1241,1242,1243]. For bioengineers, it means difficulty designing interventions without unintended consequences [1244,1245]. LLMs cannot reliably solve this because much of biological complexity stems from unknown factors requiring empirical observation and quantitative modeling beyond text-pattern recognition [1246]. While LLMs process information well, the emergent behavior of complex biological networks often requires specialized modeling that correlation-based systems can’t provide without explicit mathematical frameworks. Major challenges like understanding neural circuits or curing cancer remain unsolved because they require new scientific discoveries and experimental validation, not just knowledge retrieval [1247].
- Data Quality and Integration. Modern life sciences and bioengineering generate enormous volumes of data from genomic sequences, proteomics, patient records, and sensors. Making sense of this data reliably presents significant challenges because it’s often noisy, comes from disparate sources, and lacks integration [1248,1249]. While LLMs excel at processing text, they struggle with heterogeneous scientific data that includes numbers, images, and experimental measurements [8]. LLMs don’t have native capabilities to process raw experimental data like gene expression matrices or medical imaging unless specifically augmented with specialized tools [1250]. Challenges in biological big data - ensuring reproducibility, establishing causal relationships from observational data, or analyzing complex multi-modal datasets - still require specialized algorithms and human expertise in statistics and domain knowledge. LLMs might help report findings or suggest hypotheses, but they cannot replace the sophisticated analytical pipelines needed for rigorous scientific data analysis in these fields.
In summary, many of the grand challenges – decoding all the details of human biology, curing major diseases, sustainably engineering biology for the environment which are still open. LLMs, in their current state, are tools that can assist researchers but not solve these on their own, because the challenges often require new empirical discovery or involve complex systems and judgments beyond pattern recognition. An LLM might speed up literature review or suggest plausible theories, but it won’t automatically unravel the secrets of life that scientists themselves are still grasping at.
Easier with LLMs: The Parts That Move.
On a more optimistic note, there are challenges within life sciences and bioengineering where LLMs are already proving useful or have clear potential to contribute. These tend to be problems involving knowledge synthesis, pattern recognition in sequences/text, or generating hypotheses – tasks where handling language or symbolic representations is key. A few examples of such challenges that LLMs can tackle (and why they are suitable) include:
- Literature Overload and Knowledge Synthesis. One critical challenge uniquely pronounced in biology and bioengineering is managing the vast, rapidly growing, and fragmented body of research literature. Unlike fields such as mathematics, law, or finance, biological disciplines inherently encompass a multitude of interconnected subspecialties, each producing large volumes of highly specialized research. For example, understanding a complex disease like cancer may require integrating findings from genetics, immunology, cell biology, pharmacology, and bioinformatics—each field publishing detailed, specialized studies that must be synthesized for comprehensive insights. The complexity arises not only from the sheer volume but also from interdisciplinary connections, intricate experimental details, and extensive supplementary materials required for reproducibility. Consequently, researchers face significant difficulty in identifying relevant literature, extracting key insights, and synthesizing knowledge efficiently. This is precisely where LLMs show strong potential as intelligent literature reviewers [1251,1252,1253]. Advanced LLMs, such as GPT-4, can proficiently read, summarize, and contextualize complex biomedical texts, rapidly extracting relevant findings from extensive corpora [1253,1254]. For example, an LLM could swiftly provide researchers with an overview of current biomarkers for Alzheimer’s disease or consolidate recent advancements in biodegradable stent materials [1253]. By effectively navigating dense technical language and complex sentence structures inherent to biological literature, LLMs mitigate literature overload, facilitate interdisciplinary integration, and enable literature-based discovery—highlighting connections between seemingly disparate research findings [1255,1256,1257].
- Interpreting and Annotating Biological Sequences (Genomics/Proteomics). In both life sciences research and bioengineering applications like synthetic biology, understanding DNA, RNA, and protein sequences is crucial [1258,1259,1260]. These sequences can be thought of as strings of letters (A, T, C, G for DNA; amino acids for proteins) – in other words, a language of life. Recent work has shown that language models can be applied to these biological sequences, treating them like natural language, where “words” are motifs or codons and “sentences” are genes or protein domains. This is a challenge where LLM-like models shine [1261,1262,1263]. This means LLMs can help annotate genomes (predicting genes and their functions in a newly sequenced organism) or predict the effect of mutations (important for understanding genetic diseases) [1263]. In proteomics, models can suggest which parts of a protein are important for its structure or activity [1264,1265]. The advantage of LLMs here is their ability to handle long-range dependencies in sequences – biology often has context-dependent effects, and language models are designed to handle such context. Moreover, LLMs can generate sequences too, which leads to the next point.
- Design and Generation of Biological Sequences or Structures. In bioengineering, a cutting-edge challenge is designing new biological components – for instance, designing a protein that catalyzes a desired chemical reaction, or an RNA molecule that can serve as a therapeutic. Traditionally, this is very hard (the search space of possible sequences is astronomically large). However, LLMs have a generative capability that can be harnessed here. Already, models like ProGen [1266,1267] have shown they can generate novel protein sequences that have a predictable function across protein families. In simpler terms, an LLM trained on a vast number of protein sequences can be prompted to create a new sequence that looks like, say, an enzyme, and those sequences have been experimentally verified in some cases to fold and function [1266,1267,1268,1269,1270]. This is a remarkable development because it means LLMs can assist in protein engineering and drug discovery by proposing candidate designs that humans or simpler algorithms might not think of. Similarly, for DNA/RNA, an LLM could suggest a DNA sequence that regulates gene expression in a certain way (useful for gene therapy designs) [1259,1263] or propose improvements to a biosynthetic pathway by modifying enzyme sequences [1264,1265]. LLMs are suitable for these creative tasks because, much like with natural language, they can interpolate and extrapolate learned patterns to create new, coherent outputs (here, “coherent” means biologically plausible sequences). While any generated design still needs to be tested in the lab (to confirm it works as intended), LLMs can dramatically accelerate the ideation phase of bioengineering design.
Figure 20.
Our taxonomy for life sciences and bio-engineering.
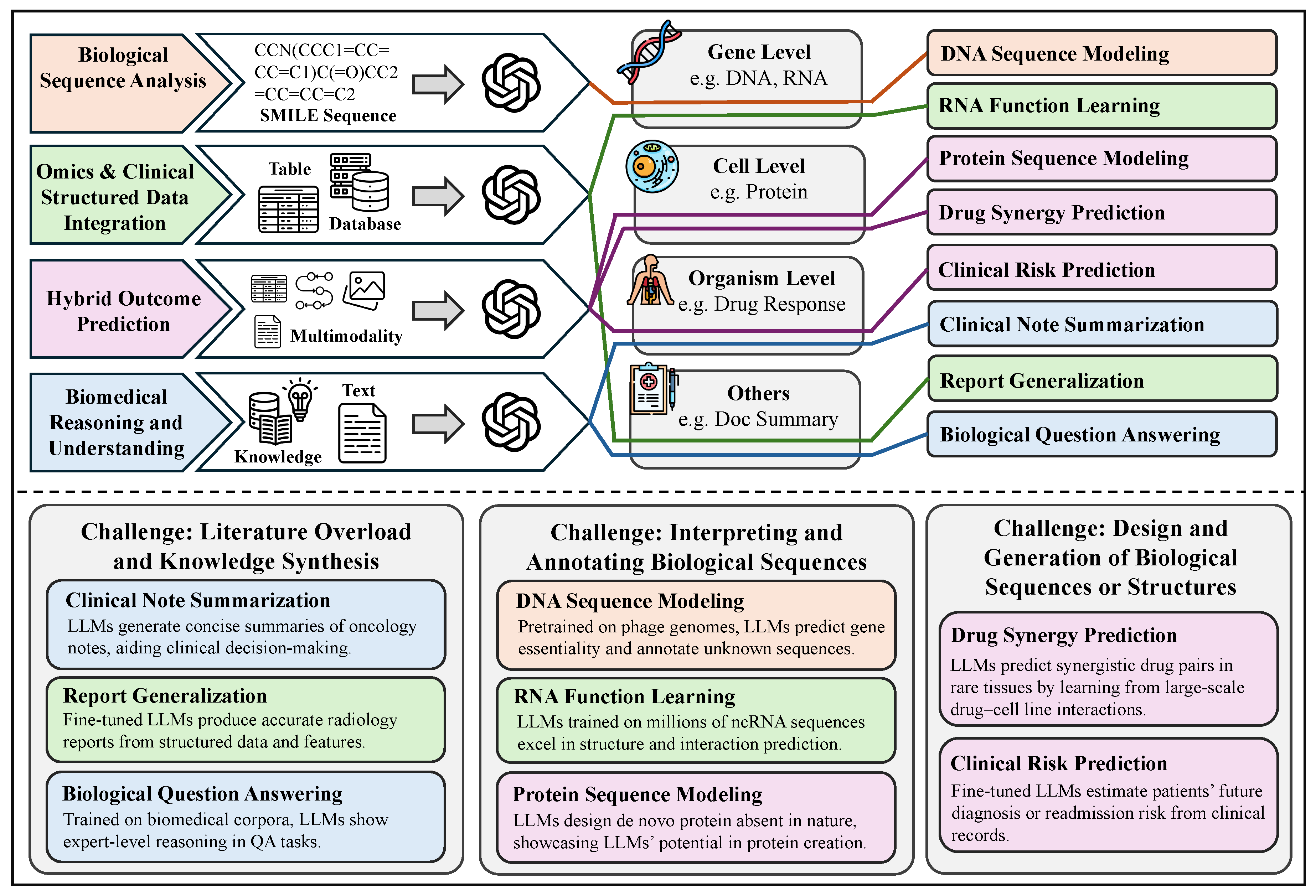
5.4.1.4 Taxonomy
Throughout the development of life sciences and bioengineering, research has gradually branched into increasingly specialized subfields. Nevertheless, many fundamental commonalities persist across domains. For instance, identifying potential folding targets within long protein sequences is methodologically similar to locating functional nucleotide fragments within DNA, both involve extracting biologically meaningful patterns from symbolic sequences [1239,1271,1272]. In addition, biological tasks often span multiple levels and modalities, encompassing data from molecular to system scales and ranging from unstructured text to structured graphs [1273,1274]. This inherent diversity renders traditional task-type-based classifications insufficient, as they obscure the subset of tasks where LLMs are particularly well-suited.
To address this, we propose a taxonomy that emphasizes the computational characteristics of tasks, enabling a more precise alignment with the capabilities and limitations of LLMs. This data-centric perspective offers three key advantages:
- Cross-domain transferability: Many life sciences tasks share structural similarities in their input data, making it easier to adapt and transfer methods across domains.
Sequence-Based Tasks. These tasks involve analyzing sequential biological data, such as DNA, RNA, or protein sequences, which are essentially strings of nucleotides or amino acids. Typical examples include genome annotation, mutation impact prediction, protein structure prediction from sequences, and the design of genetic circuits. The input for these tasks generally comprises one or multiple biological sequences, with outputs including sequence annotations or newly designed sequences. From an artificial intelligence standpoint, these tasks are analogous to language processing problems because biological sequences possess a form of syntax (e.g., motifs and domains) and semantics (functional implications). LLMs and other sequence-based AI models are thus particularly effective for these applications, interpreting biological sequences similarly to languages. For instance, predicting the pathogenicity of a DNA mutation is akin to detecting grammatical errors in a language, where biological viability parallels grammatical correctness [1277]. Recent advancements, such as protein language models, exemplify successful AI applications leveraging this analogy [1277,1278].
Structured and Numeric Data Tasks. These tasks involve handling structured datasets, numerical measurements, and graphs commonly encountered in physiology, biochemistry, and bioengineering. Examples include analyzing patient heart rate time-series data, interpreting metabolomics datasets, optimizing metabolic network models, and designing control systems for prosthetics. Inputs typically consist of numerical or tabular data (sometimes time-series), while outputs could involve predictions (e.g., forecasting patient adverse events) or control decisions. Such tasks traditionally rely on statistical methods or control theory, and they are generally less naturally suited for LLMs unless translated into textual or coded representations. A creative utilization of LLMs in this context is their ability to generate computational code from natural language descriptions, bridging descriptive problem statements to numeric analyses. For example, researchers can prompt an LLM to produce Python code to analyze specific datasets, utilizing the model as an intermediary tool between natural language and computational implementation [1279,1280,1281,1282]. However, purely numerical tasks involving complex calculations or optimizations typically remain better served by specialized algorithms.
Textual Knowledge Tasks. These tasks involve managing, interpreting, and generating text-based information prevalent in biology and engineering. Examples include literature searches and question-answering, proposal writing, extracting critical information from research articles, summarizing electronic health records, and analyzing biotechnology patents. Inputs here predominantly include unstructured textual documents (such as research articles, clinical notes, or patent filings), with outputs comprising synthesized textual summaries, detailed answers, or structured reports. This area represents the inherent strength of LLMs, encompassing various subtasks such as knowledge retrieval and question-answering, summarization and literature review, and protocol or technical report generation. Given their core competency in processing and synthesizing textual data, LLMs are exceptionally well-suited to these tasks.
Predictive Modeling Tasks (Hybrid). Many research questions in biology and bioengineering can be formulated as predictive problems, such as determining drug toxicity, predicting crop yields from genetic modifications, or forecasting protein folding success. These tasks frequently involve integrating multiple modalities—including sequences, structural data, textual descriptions—and require robust extrapolative capabilities. Inputs often combine diverse data formats, with outputs focusing on biological or engineering outcomes. Although many predictive tasks overlap with sequence-based analyses, this category explicitly emphasizes multidimensional predictions that leverage various feature sets. LLMs can contribute to these tasks through interpretive roles, qualitative reasoning, or as orchestrators within computational pipelines. For instance, an LLM could manage interactions between specialized bioinformatics tools, interpret computational outputs, and provide coherent explanations or qualitative predictions, highlighting their integrative and explanatory potential within complex predictive frameworks.
Table 43.
Life Science and Bioengineering Tasks, Subtasks, Insights and References
| Type of Task | Subtasks | Insights and Contributions | Key Models | Citations |
|---|---|---|---|---|
| Genomic Sequence Analysis | DNA Sequence Modeling | LLMs, by capturing regulatory grammars embedded in genomic sequences, enable accurate prediction of functional elements and variant effects, thus enhancing interpretability and advancing our understanding of gene regulation. | DNABERT: adapt BERT to human reference DNA and learn bidirectional representations;HyenaDNA: scale the context length and accelerate the model efficiency. | [1261,1283,1284,1285,1286,1287,1288,1289] |
| RNA Function Learning | LLMs, by modeling both sequence and structural contexts of RNA, uncover functional motifs and regulatory patterns, thereby improving interpretability and facilitating insights into post-transcriptional regulation. | RNA-FM: employing 23.7M deduplicated RNA sequences for training; 3UTRBERT: specialized in modeling 3’ untranslated regions (3’UTRs). | [1290,1291,1292,1293,1294,1295,1296] | |
|
Biomedical Reasoning and Understanding |
Question Answering | LLMs, by aligning domain-specific knowledge with natural language understanding, accurately interpret biomedical queries and texts, thereby enhancing information retrieval and supporting clinical and research decision-making. | Med-PaLM: achieved near-expert-level performance on the USMLE test; HuatuoGPT: proactively ask questions rather than respond passively. | [17,1297,1298,1299,1300,1301,1302,1303,1304,1305,1306,1307,1308,1309,1310,1311] |
| Language Understanding | LLMs, by learning semantic patterns and reasoning cues from biomedical texts, enable deep language understanding, thereby improving performance on tasks like inference, entity recognition, and document classification. | BioInstruct: covering multi understanding tasks; GPT-4: perform NLI without explicit fine-tuning, when prompted with queries. | [17,1297,1302,1305,1312,1313,1314,1315,1316] | |
|
Omics & Clinical Structured Data Integration |
Clinical Language Generation | LLMs, by capturing clinical language styles and contextual dependencies, generate coherent and context-aware narratives, thereby enhancing the automation and reliability of medical reporting and documentation. | ClinicalT5: pretraining on text-to-text tasks for long clinical narratives; GPT-4: perform good when prompted by specific queries. | [1301,1317,1318,1319,1320,1321] |
| EHR Based Prediction | LLMs, by integrating longitudinal and multimodal patient data from EHRs, model complex temporal and clinical dependencies, thereby enabling accurate prediction of outcomes and supporting personalized healthcare. | BEHRT: adapts BERT to longitudinal EHR data by encoding structured sequences; GatorTron: scaled up to 8.9B and was trained on over 90B clinical narratives and structured labels. | [1305,1322,1323,1324] | |
| Hybrid Outcome Prediction | Drug Synergy Prediction | LLMs, by jointly modeling chemical structures and cellular contexts, capture intricate drug–drug and drug–cell interactions, thereby enhancing the prediction of synergistic combinations and accelerating combination therapy design. | CancerGPT: fine-tuning GPT-3 to predict drug synergy in rare cancers; BAITSAO: a foundation model strategy that integrates multiple datasets and tasks. | [6,7,18,1325,1326,1327,1328] |
| Protein Modeling | LLMs, by learning evolutionary, structural, and functional signals from protein sequences, enable accurate modeling of folding, function, and interactions, thereby advancing protein engineering and therapeutic discovery. | ProLLaMA: achieving joint understanding and generation within a single framework; ProteinGPT: further supporting structure input, language interaction, and functional Q&A | [1267,1268,1269,1329,1330,1331,1332,1333,1334,1335,1336,1337,1338,1339,1340,1341,1342,1343,1344,1345] |
5.4.2. Genomic Sequence Analysis
Sequence-based tasks focus on the learning and modeling of sequential data in life science and bioengineering, aiming to assist researchers in extracting meaningful biological insights from sequence-encoded information. The primary inputs to these tasks are biological sequences, such as nucleotide sequences in DNA/RNA or amino acid sequences in peptides. For instance, DNA is composed of four nucleotides, adenine (A), guanine (G), thymine (T), and cytosine (C), which are inherently stored in organisms in a sequential format, requiring no additional transformation. These biologically grounded representations allow models to learn structural patterns and latent dependencies, thereby enabling effective downstream prediction and classification tasks.
In sequence-based tasks, the objective is to leverage learned representations to accomplish specific downstream goals. For example, given genomic DNA adjacent to genes that may contain enhancers, a binary label is predicted for each 128-base-pair segment to determine whether it belongs to an enhancer region. Enhancers are short, non-coding DNA elements that regulate gene expression and can exert influence across distances of thousands to over a million base pairs by physically interacting with gene promoters. Another representative task is predicting the gene-editing efficiency of a given single guide RNA (sgRNA) sequence when guided by Cas proteins in CRISPR-based applications.
The value of sequence-based tasks in life sciences and bioengineering lies in their capacity to capture long-range dependencies and hierarchical patterns in extremely long and sparse sequences—often in a semi-supervised or unsupervised manner. For instance, in the human genome, protein-coding regions account for only about 1% of the total DNA sequence. Likewise, functionally important non-coding regions such as promoters also constitute a very small fraction of the genome [1346]. By training on massive sequence datasets, models can learn to identify such regions with high accuracy. Researchers can then input unknown sequences into the model to annotate potential coding regions or predict functionally significant non-coding elements. In addition to classification, sequence-based models are also applied in predictive tasks. For example, they can estimate the likelihood and frequency of off-target mutations induced by CRISPR systems at unintended genomic loci, thereby improving the safety and efficacy of gene editing. In summary, sequence-based tasks significantly reduce the time and cost of sequence analysis, while deepening our understanding of the functional and regulatory roles encoded in biological sequences.
Although both DNA and RNA exist in the form of sequences, they differ significantly in biological function and modeling objectives [1347]. DNA sequences primarily encode genetic information, and related modeling tasks focus on identifying regulatory elements such as promoters, enhancers, and transcription factor binding sites, as well as capturing long-range dependencies across the genome. In contrast, RNA is more involved in functional execution, including splicing, modification, translational regulation, and interactions with proteins. Typical tasks involve RNA secondary structure prediction, modification site identification, and functional classification of non-coding RNAs [1348,1349]. Therefore, we categorize Sequence-Based Tasks into DNA Sequence Modeling and RNA Function Learning, and the subsequent discussion will be centered around these two directions. These two tasks share several common characteristics that make LLMs particularly well-suited for this domain. First, the abundance of unlabeled or sparsely labeled sequence data provides a rich resource for training. Second, LLMs not only deliver reliable predictions but also offer highly interpretable textual reasoning to support their outputs.
DNA Sequence Modeling. DNA Sequence Modeling refers to the task of computationally analyzing and learning patterns from nucleotide sequences—typically represented as strings composed of A, T, C, and G—to understand the underlying biological functions, regulatory mechanisms, and genetic variations encoded in the genome. In recent years, LLMs have rapidly emerged in the field of DNA, advancing our understanding of the information encoded within the genome. Transformer-like LLMs are now capable of reading, reasoning over, and even designing the 3.2Gb human genome, evolving from early convolutional neural networks (CNNs) that handled short windows to billion-parameter foundation models that process megabase-scale contexts. Early convolutional frameworks such as DeepSEA [1350] demonstrated that raw sequence alone is sufficient to predict chromatin features, inspiring a decade-long progression from hybrid CNN-RNN models and Transformer encoders to long-context state-space models. Today’s genomic LLMs often outperform traditional physics-based or motif-based methods across various tasks, including enhancer detection, cell type-specific expression, and non-coding variant effect prediction, while also providing saliency maps that highlight learned regulatory syntax.
DNABERT [1283] adapted the BERT architecture to human reference DNA by tokenizing sequences into 6-mers and using masked language modeling (MLM) to learn bidirectional representations, which proved transferable to promoter, enhancer, and splice site prediction tasks. Building on this, DNABERT-2 [1284] introduced byte-pair encoding (BPE) [1351], breaking the fixed k-mer limitation, reducing memory usage by 30%, and improving average MCC by 2 percentage points across 28 datasets. Techniques like self-distillation and adaptive masking further refined embeddings under limited data conditions.
Unlike typical LLMs, DNA inputs are significantly longer than those in standard NLP tasks. Even when models support large context windows, performance can degrade. To address these issues, Enformer [1285] combines ConvNet [1352] downsampling with 1D self-attention over 200kb regions, doubling the correlation with gene expression compared to prior models and significantly improving eQTL effect sign prediction. HyenaDNA [1261] scales context length to 1 million nucleotides using sub-quadratic implicit convolutions, enabling 160× faster training than FlashAttention Transformers [1353], while maintaining single-base resolution and outperforming Enformer on proximal promoter tasks.
GROVER [1286] uses frequency-balanced byte-pair encoding vocabularies to learn “sequence contextuality” directly from the human genome, outperforming prior k-mer baselines in next-token prediction and fine-tuning tasks like promoter detection and CTCF binding prediction. Its unsupervised embeddings can recover GC content, repeat categories, replication timing, and other functional signals purely from sequence, underscoring how tokenization design can unlock richer biological grammar.
With increased computational power and model scaling, larger models and training corpora have emerged. The Nucleotide Transformer [1287] with 2.5 billion parameters was pretrained on 3,202 human and 850 non-human genomes, generating general-purpose embeddings that improved performance across 18 downstream tasks—including pathogenicity scoring and enhancer–promoter linking—without requiring task-specific architectural changes. GenomeOcean [1288], a 4-billion-parameter model, extended this paradigm to 220TB of metagenomic data, capturing rare microbial taxa for better ecologically driven generalization.
LLMs have substantially advanced DNA sequence modeling by enabling scalable interpretation of genomic sequences, capturing long-range dependencies, and learning regulatory syntax directly from raw nucleotide strings. They have addressed core challenges such as integrating context across megabase-scale windows, improving prediction of non-coding variant effects, and providing transferable embeddings for diverse downstream tasks without extensive feature engineering. However, key challenges remain: performance often degrades with increasing sequence length despite architectural innovations, and current models still struggle with integrating multi-omic signals, rare variant generalization, and interpretability in clinical settings. Future directions include developing more efficient architectures for ultra-long sequences, incorporating cross-modal biological data (e.g., epigenomic or transcriptomic layers), and aligning model predictions with mechanistic biological knowledge to support hypothesis generation and therapeutic discovery.
RNA Function Learning. RNA Function Learning refers to the task of modeling RNA sequences to uncover their structural attributes and functional roles, often leveraging sequence-structure relationships to predict biological behaviors and interactions. Understanding RNA sequences and their structural-functional relationships is essential for numerous molecular biology applications, including splicing regulation, RNA-protein interactions, and non-coding RNA functional annotation. Traditional bioinformatics methods such as sequence alignment and thermodynamic folding models (e.g., RNAfold) provide accurate predictions but suffer from limitations like heavy computational demands and dependency on handcrafted features.
Recently, leveraging advances in natural language processing (NLP), large-scale pretrained language models adapted to RNA sequences have emerged, significantly improving our capacity to interpret biological information embedded in nucleotide sequences. Early efforts, such as RNABERT [1290], marked a shift toward learning biological grammar directly from data. RNABERT combined masked language modeling (MLM) with a structural alignment objective (SAL), enabling the model to internalize pairwise structural relationships by training on alignment scores derived from the Needleman-Wunsch algorithm [1354].
Subsequent models expanded on this foundation by enhancing both scale and methodological complexity. RNA-FM [1291] significantly scaled the training dataset, employing 23.7 million deduplicated RNA sequences from RNAcentral [1354], thus improving generalization capabilities for functional prediction tasks. RNA-MSM [1292] further advanced this approach by incorporating evolutionary context through homologous sequence modeling with multiple sequence alignments (MSAs), inspired by MSATransformer [1355]. Notably, RNA-MSM strategically excluded families with known structures during training, effectively reducing overfitting and enhancing performance on structure-aware tasks.
Parallel developments have addressed specific functional RNA elements, refining the modeling approach based on targeted biological contexts. For instance, SpliceBERT [1293] specifically targeted splicing regulation by training on 2 million vertebrate pre-mRNA sequences from UCSC [1356]. By focusing explicitly on pre-mRNA rather than mature transcripts, SpliceBERT captured sequence features critical for identifying splicing junctions, splice sites, and regulatory motifs (e.g., exonic splicing enhancers or silencers), aspects typically overlooked in traditional modeling frameworks [1294]. Consequently, this model supports tasks such as splice site prediction, detection of alternative splicing events, and the identification of novel, tissue-specific regulatory elements.
Complementing this targeted functional perspective, 3UTRBERT [1295] specialized in modeling 3’ untranslated regions (3’UTRs) to facilitate studies of post-transcriptional regulation. Building further upon the integration of structure into sequence modeling, UTR-LM [1296] explicitly incorporated structural supervision alongside MLM pretraining. It employed two biologically informed auxiliary tasks: secondary structure prediction and minimum free energy (MFE) regression. Secondary structures, predicted using the ViennaRNA toolkit [1357,1358], were utilized as local structural constraints during masking, while global thermodynamic stability (MFE) values were predicted from global contextual embeddings ([CLS] token). The training dataset included carefully curated natural and synthetic 5’UTR sequences from databases like Ensembl and high-throughput assays, ensuring robust learning of biologically relevant patterns [1359,1360,1361]. These methods strengthened the link between structural and functional RNA predictions, demonstrating applicability in translation efficiency prediction and synthetic RNA design.
Lastly, BEACON-B and BEACON-B512 expanded RNA language modeling into extensive datasets comprising over 500,000 human non-coding RNAs from RNAcentral [1354], exploring broader functional landscapes beyond coding transcripts [1294]. These models highlighted the importance of tailored training objectives, domain-specific masking strategies, and carefully curated datasets, all contributing to enhanced interpretability and biological accuracy.
LLMs have revolutionized RNA function learning by shifting the paradigm from alignment- or thermodynamics-based methods to data-driven, end-to-end models that learn both structural and functional features directly from sequences. These models have improved prediction accuracy for splicing patterns, RNA-protein interactions, and post-transcriptional regulatory elements, while also enabling interpretability through structural supervision and auxiliary tasks. Nonetheless, key challenges remain: many models still struggle with generalizing to novel RNA classes, integrating evolutionary and tertiary structural information, and explaining model decisions in biologically meaningful ways. Future research directions include scaling models to more diverse and comprehensive RNA datasets, incorporating multi-resolution structural priors, and aligning language model outputs with experimentally validated functional annotations to bridge the gap between sequence modeling and functional genomics.
5.4.3. Clinical Structured Data Integration
Clinical structured data integration focuses on the intelligent utilization of structured clinical information—such as Electronic Health Records (EHRs), laboratory test results, and coded diagnoses—to support and automate critical healthcare decision-making processes. The goal is to leverage artificial intelligence, particularly LLMs, to understand structured clinical inputs, build predictive or generative models, and produce meaningful outputs that improve clinical workflows, enhance patient care, and enable personalized medicine. These tasks primarily rely on structured or semi-structured datasets, including tabular EHR entries, time-series vital signs, coded diagnoses and treatments (e.g., ICD, CPT, LOINC), and structured questionnaire responses. Unlike free-form text, such data is inherently aligned with medical ontologies and clinical protocols, enabling models to reason with high factual precision and temporal awareness.
The primary objective of clinical structured data integration is to perform downstream tasks that generate clinically useful outputs based on structured patient data. For instance, given longitudinal EHRs containing timestamped diagnoses, prescriptions, and lab values, a model can forecast disease onset, stratify patient risk, or suggest treatment plans. In other scenarios, structured data is transformed into human-readable summaries—such as clinical progress notes or discharge instructions—to reduce the documentation burden on clinicians. A persistent challenge lies in bridging the gap between machine-readable formats and clinical narratives: generated content must be not only factually accurate but also contextually appropriate and linguistically coherent. Furthermore, since EHR data often contains missing values, noise, or institutional heterogeneity, models must be robust to irregular sampling and generalize across diverse healthcare settings.
In the broader context of life sciences and bioengineering, clinical structured data integration serves as a cornerstone of evidence-based medicine, offering scalable solutions for personalized care, automated documentation, and proactive health monitoring. By seamlessly connecting the structured backbone of clinical practice with the expressive power of language models, this work marks a critical step toward an intelligent, interoperable, and human-centered healthcare system.
To reflect the dual nature of this field, clinical structured data integration can be categorized into two main types: Clinical Language Generation and EHR-Based Prediction. The former focuses on converting structured clinical data into fluent, accurate natural language reports, enabling applications such as automated drafting of medical notes, radiology impression generation, and ICU event summarization. This task emphasizes controllable generation, temporal summarization, and medical factuality, requiring models to balance conciseness with informativeness. In contrast, EHR-based prediction aims to extract actionable insights from patient records to support tasks such as sepsis alerts, readmission prediction, and personalized risk scoring. These tasks demand strong temporal modeling, integration of clinical knowledge, and high interpretability, especially when informing critical medical decisions.
Across both task types, incorporating domain-specific inductive biases—such as hierarchical coding systems, medical knowledge graphs, or treatment ontologies—has been shown to enhance model performance. LLMs have demonstrated great potential in unifying diverse input modalities and producing clinically meaningful outputs, particularly when structured prompting or graph-aware architectures are employed. Moreover, the growing availability of publicly accessible, de-identified datasets such as MIMIC-III/IV [1362,1363] and eICU has fostered the development of standardized evaluation benchmarks. These advances not only enable rigorous comparison across methods but also promote the creation of generalizable and trustworthy AI systems for real-world clinical applications.
Clinical Language Generation. Clinical Language Generation refers to the use of natural language processing techniques to automatically produce coherent and clinically meaningful text from structured inputs such as electronic health records, diagnostic codes, or medical templates. Clinical Language Generation (CLG) is rapidly emerging as a foundational infrastructure in smart healthcare. By leveraging structured data or text recorded using templates, CLG models can automatically draft outpatient/inpatient notes, generate radiology report impressions, rewrite patient-friendly versions, and even transcribe real-time doctor-patient conversations. These capabilities significantly reduce the documentation burden on healthcare professionals, while improving the quality and readability of medical records, thereby supporting evidence-based decision-making and interdisciplinary collaboration.
With the maturation of large-scale pretraining corpora and instruction tuning techniques, CLG has evolved from early small-parameter models to multi-modal systems with tens of billions of parameters, offering unprecedented text generation capabilities in clinical settings.
One of the earliest representative works, ClinicalT5 [1317], adapted the T5 [1364] framework to hospital notes from datasets like MIMIC-III/IV [1362,1363], pretraining on text-to-text tasks for long clinical narratives. It achieved a 3.1 ROUGE-L improvement on discharge summary generation and outperformed long-text baselines such as BART [1365], demonstrating that generative models can effectively capture key information in complex, structured medical records. However, ClinicalT5’s training data was primarily composed of single-center English inpatient notes, which limits its generalization across languages and institutions.
In contrast, general-purpose LLMs are often trained on multilingual, multi-source datasets and inherently possess cross-domain generalization capabilities [1,6,7]. With the advancement of such models, GPT-4 [1] and Med-PaLM 2 [1301], through instruction tuning, can generate high-quality clinical drafts. GPT-4 achieved near-human accuracy in outpatient record analysis across three languages and can draft standardized clinical progress notes in zero-shot settings [1319]. Med-PaLM 2 excelled in the MultiMedQA evaluation framework, particularly in reasoning and safety dimensions, showcasing the strength of large decoder-based models in long-form clinical text generation.
For patient communication, Jonah Zaretsky et al. demonstrated that LLMs can rewrite structured discharge summaries to a 6–8th grade reading level, with readability scores 18% higher than physician-authored versions, greatly enhancing patient understanding of medication and follow-up instructions [1320]. Meanwhile, model scale and multimodal capabilities are also advancing. Me-LLaMA [1321], built on LLaMA 2 [52], integrates PubMed, clinical guidelines, and knowledge graphs, and supports 13–70B parameter ranges. With medical instruction tuning, it enables multimodal prompt-based generation for case summaries and diagnostic explanations.
In fact, many models designed for clinical or medical tasks possess some degree of CLG capabilities. However, as many focus more on medical QA or comprehension tasks, we will introduce those models in detail in the following sections.
LLMs have transformed Clinical Language Generation (CLG) by enabling automatic, fluent synthesis of complex clinical narratives from structured inputs, thereby alleviating documentation burdens and enhancing the accessibility of medical records for both professionals and patients. They have addressed key challenges such as adapting outputs to various clinical tasks, and generating patient-friendly text. Nonetheless, several challenges persist: generalization across institutions and languages remains limited due to training data biases; factual consistency and clinical safety must be rigorously validated; and integrating multimodal signals (e.g., images, vitals) into text generation is still nascent. Future work should prioritize domain adaptation techniques, fine-grained clinical factuality evaluation, multimodal integration, and collaborative frameworks involving clinicians to ensure that generated content is both medically reliable and practically useful in diverse healthcare environments.
EHR Based Prediction. An electronic health record (EHR) is the systematized collection of electronically stored patient and population health information in a digital format. EHRs play a critical role in modern healthcare systems. Beyond the advantages of digitization—such as easier storage and review—EHRs significantly improve the quality and efficiency of medical care. They provide comprehensive, accurate, and real-time patient information, enabling clinicians to make more informed and precise clinical decisions. Moreover, EHRs facilitate the sharing of patient health information across departments and institutions, enhancing collaborative efficiency and ensuring continuity of care across different healthcare settings. The vast amount of data accumulated in EHR systems also provides a solid foundation for training LLMs, as these records often contain structured annotations—such as specific diseases and severity levels—that typically require little to no transformation, making them highly suitable for LLM-based learning.
A foundational model in this domain is BEHRT [1322], a transformer-based model developed for healthcare representation learning. BEHRT adapts BERT to longitudinal EHR data by encoding structured sequences of medical codes (e.g., diagnoses, medications) along with age embeddings. By learning temporal dependencies, BEHRT achieved strong performance in tasks such as disease onset prediction (e.g., predicting diabetes based on early comorbidities), and it demonstrated robust performance in downstream stratification tasks with minimal fine-tuning.
However, BEHRT was trained on relatively small datasets, limiting its potential. In contrast, Med-BERT [1323], a context-based embedding model, was pre-trained on a large-scale structured EHR dataset comprising 28,490,650 patients and clinical coding systems like ICD-10 and CPT. Fine-tuning experiments showed that Med-BERT significantly improved prediction accuracy. Notably, Med-BERT performed exceptionally well with small fine-tuning datasets, achieving AUC scores that surpassed baseline deep learning models by over 20%, and even matched the performance of models trained on datasets ten times larger [1323].
Building on this trend, GatorTron [1324] scaled up the model size to 8.9 billion parameters and was trained on over 90 billion clinical narratives and structured labels. It demonstrated remarkable generalization capabilities in tasks such as phenotype prediction and cohort selection. Its scalability enables modeling of complex inpatient trajectories and supports patient-level reasoning even in low-resource scenarios.
In the multimodal space, models like MultiMedQA [1300] and Clinical Camel [1305] integrate structured EHR entries (e.g., vital signs, lab results) with textual prompts to generate clinical answers from tabular data. For example, given a prompt such as “Is this patient at risk for acute kidney injury?” and a series of lab values and medication records, the model outputs a response like “Yes, due to elevated creatinine levels and concurrent use of nephrotoxic drugs.”
LLMs have significantly advanced EHR-based prediction by leveraging structured medical codes, temporal information, and multimodal clinical signals to support personalized forecasting of disease onset, treatment outcomes, and patient risk stratification. These models, ranging from BEHRT and Med-BERT to the billion-parameter GatorTron, have demonstrated strong generalization across clinical tasks with minimal fine-tuning and are particularly effective even in low-resource settings. However, challenges remain in modeling long, sparse, and irregular patient timelines, ensuring clinical interpretability, and addressing domain shifts across institutions and EHR systems. Future work should focus on integrating heterogeneous modalities (e.g., imaging, genomics), improving temporal reasoning across fragmented records, and developing explainable frameworks that align model decisions with clinician expectations to foster trust and deployment in real-world healthcare settings.
5.4.4. Biomedical Reasoning and Understanding
Biomedical Reasoning and Understanding focus on the understanding and modeling of textual information in the fields of life sciences and bioengineering. The goal is to leverage artificial intelligence technologies to enhance the semantic parsing of natural language content such as scientific literature, clinical case records, and diagnostic reports. The primary inputs for these tasks are natural language texts—for example, research abstracts from PubMed or clinical notes from patient records. Similar to DNA or RNA sequences, natural language inherently contains rich semantic information and can be directly processed by language models without additional transformation. This representation allows models to learn semantic patterns, reasoning cues, and contextual dependencies embedded in the language, thereby providing strong semantic support and reasoning capabilities for downstream tasks such as disease diagnosis, clinical report generation, and biomedical literature question answering.
The main objective of Biomedical Reasoning and Understanding is to perform a variety of practically meaningful downstream tasks based on effective modeling of natural language texts. For example, given a research abstract from PubMed, the model needs to accurately identify and extract biomedical entities such as drug names, disease types, and gene symbols, and further uncover functional relationships among them, such as “Drug X treats Disease Y” or “Gene A is significantly associated with Disease B.” Additionally, the model can assist researchers in quickly understanding complex oncology reports and automatically answering questions such as “What treatment methods were used in this study?” or “Which patient subgroups benefited the most according to the results?” More generally, scenarios also include using medical examination questions (e.g., USMLE) as input to evaluate the model’s question-answering and reasoning capabilities across broad medical knowledge domains. These tasks rely on extensive biomedical knowledge found in literature, clinical notes, and databases, posing high demands on LLMs in terms of factual recall, domain-specific reasoning, and complex language interpretation.
In the life sciences and bioengineering domains, the value of Biomedical Reasoning and Understanding lies in their ability to extract critical information from massive volumes of text that is vital for research and clinical decision-making. Similar to the “information sparsity” seen in sequence-based tasks, biomedical texts also exhibit low information density but high-value key content—for instance, a clinical case report may be lengthy, yet the truly decisive content for diagnosis or treatment planning is often minimal. Therefore, models must possess robust capabilities in long-text modeling, information retrieval, and semantic compression to effectively accomplish the task objectives. Furthermore, in certain application scenarios, the model can also automatically generate clinical decision-making suggestions based on existing research findings, or explain treatment plans to patients in more accessible language—thus promoting research transparency and improving doctor-patient communication.
Although all these tasks involve text processing, they differ fundamentally in task structure, reasoning focus, and model design. Some subtasks revolve around retrieving or generating answers to biomedical questions—such as determining a diagnosis, choosing a treatment plan, or interpreting research outcomes—and typically require models to possess strong knowledge recall and evidence-based reasoning capabilities. In contrast, others focus on identifying semantic relationships, logical entailment, or classification problems within or across texts—for example, determining whether two sentences entail each other, or classifying text based on medical intent. These two categories reflect two long-standing paradigms in natural language processing: retrieval/generation and reasoning/classification, which also align with widely adopted benchmarking methods today. Therefore, we further subdivide Biomedical Reasoning and Understanding into two categories: Question Answering and Language Understanding.
Both categories benefit from the abundance of unlabeled or partially labeled biomedical text resources, including research papers, clinical notes, and medical examination datasets, which provide rich materials for self-supervised or weakly supervised pretraining. Furthermore, LLMs are not only capable of generating accurate answers or performing effective classification, but also excel at providing clear and interpretable reasoning, thereby significantly enhancing the transparency and trustworthiness of predictions. These characteristics enable LLMs to transcend individual task boundaries and provide robust technical support for knowledge-intensive biomedical reasoning.
Question Answering. Biomedical Question Answering focuses on enabling models to accurately extract or generate answers from scientific literature, clinical notes, or medical guidelines in response to domain-specific natural language queries. A series of LLMs and domain-specific models have been applied to biomedical question answering (QA). Early approaches employed transformer models such as BioBERT [17] and PubMedBERT [1297], BERT [9] based models pre-trained on biomedical corpora—and fine-tuned them for QA tasks. Compared to general-purpose language models, these domain-specific models demonstrated higher accuracy on biomedical QA benchmarks. For example, BioBERT [17] achieved higher F1 scores than baseline BERT in the BioASQ [1366] challenge tasks, owing to its domain-specific pretraining. Generative transformer models tailored to biomedicine have also been developed, such as BioGPT [1298] (a GPT-2-style [5] model trained on biomedical texts) and BioMedLM [1299] (also known as PubMedGPT 2.7B, a GPT-based model trained on PubMed abstracts). These models have achieved strong results in QA tasks.
Subsequently, instruction tuning and conversational LLMs entered the biomedical QA domain. Med-PaLM [1300] (and its successor Med-PaLM 2 [1301]) fine-tuned Google’s PaLM [1367] model on medical QA tasks and achieved near-expert-level performance on the United States Medical Licensing Examination (USMLE), with accuracy around 86.5%, approaching that of expert physicians (87%).
To move toward truly doctor-like LLMs—beyond simply answering questions—researchers have fine-tuned pretrained models on more novel datasets. For example, ChatDoctor [1302] was created by fine-tuning LLaMA [52] on medical dialogue data, enabling interactive QA in a patient-doctor chat format. HuatuoGPT [1303] posits that an intelligent medical advisor should proactively ask questions rather than respond passively. Huatuo-2 [1304] uses an innovative domain-adaptive approach to significantly improve its medical knowledge and conversational skills. It demonstrated optimal performance on several medical benchmark tests, notably surpassing the GPT-4 on the Specialist Assessment and the new version of the Physician Licensing Exam. Similarly, models such as Clinical Camel [1305] and DoctorGLM [1306] are LLM-based medical chatbots designed specifically to answer medical questions in a conversational style.
At the same time, thanks to LLMs’ inherent zero-shot capabilities, large general-purpose models like GPT-4 [1] remain competitive, which has demonstrated strong performance in medical QA and often outperforms smaller domain-specific models in zero-shot settings [1315].
Recently, reasoning has played an increasing role in this subtask. For example, Huatuo-o1 [1307] enhances complex reasoning ability by (1) guiding the search for complex reasoning trajectories using a verifier and fine-tuning the LLM accordingly, and (2) applying reinforcement learning (RL) with verifier-based rewards. FineMedLM-o1 [1308] further introduced Test-Time Training [1368] into the medical domain for the first time, promoting domain adaptation and ensuring reliable and accurate reasoning.
LLMs have substantially advanced biomedical question answering by enabling precise information extraction and fluent generation from diverse medical texts, ranging from clinical guidelines to patient dialogues. They have outperformed traditional domain-specific baselines by incorporating instruction tuning, conversational capabilities, and advanced reasoning techniques. However, several challenges remain: factual consistency and hallucination still pose risks in high-stakes clinical applications; models often struggle with ambiguous queries, underrepresented diseases, or multimodal reasoning; and real-world deployment requires careful alignment with clinical workflows and regulations. Future efforts should focus on integrating more life science and bio-engineering knowledge, enhancing traceable multi-step reasoning, and developing evaluation protocols that reflect real-world clinical utility, ensuring that QA systems can support clinicians safely and effectively.
Language Understanding. Language Understanding in the life science and bio-engineering involves modeling a system’s ability to comprehend, interpret, and reason over domain-specific texts, enabling accurate semantic inference and contextual judgment across diverse biomedical and scientific narratives. Beyond direct question answering, LLMs are increasingly applied to various language understanding tasks in the biomedical domain. These tasks require interpretation and reasoning over biomedical texts such as clinical narratives, scientific abstracts, or exam questions to support judgments or classifications. A typical example is natural language inference (NLI) in medicine: given a textual premise (e.g., a statement from a patient report) and a hypothesis, the model must determine whether the premise entails, contradicts, or is neutral with respect to the hypothesis. For instance, consider the premise “The patient denies any history of diabetes,” and the hypothesis “The patient has a history of diabetes.” A model with true understanding should correctly classify this as a contradiction, since the hypothesis directly conflicts with the premise. Language understanding is crucial for clinical decision support and information extraction, as it determines whether conclusions are genuinely grounded in clinical observations or life science facts. It also underpins tasks such as document classification, textual entailment, and reading comprehension. In essence, these tasks assess whether LLMs demonstrate deep comprehension of biomedical language, rather than mere memorization.
Many LLMs originally developed for QA have also been used for understanding tasks, often via fine-tuning for classification. Early models like BioBERT [17] and PubMedBERT [1297] pioneered performance improvements in biomedical text classification and NLI, achieving strong results on tasks such as MedNLI [1369]. Fine-tuned BioBERT on the MedNLI dataset significantly outperformed earlier RNN-based models in reasoning accuracy, because of its better grasp of clinical terminology and context. ClinicalBERT [1312], initialized from BioBERT [17] and further trained on electronic health records, proved particularly effective in clinical NLI and related tasks, as it captured domain-specific syntax and abbreviations from structured data. More recent domain-specific models, such as BioLinkBERT [1313] and BlueBERT [1314], report MedNLI accuracy in the mid-80% range—approaching human expert performance.
Meanwhile, large general-purpose LLMs have demonstrated capability in language understanding via prompting. For example, GPT-4 [1] can perform NLI without explicit fine-tuning, when prompted with queries like, “Does the following statement logically follow from the previous one?” [1315] Trained on a broad corpus—including some medical content—these models often achieve decent accuracy in zero-shot or few-shot settings.
However, instruction-finetuned biomedical models are pushing the boundaries further. A recent method, BioInstruct [1316], compiled around 25,000 biomedical instruction-response pairs, covering tasks such as NLI and QA, and used them to fine-tune a LLaMA model. This resulted in significant improvements across multiple benchmarks, indicating that targeted instruction tuning can effectively teach LLMs the reasoning patterns required for biomedical language understanding. Similarly, models like ChatDoctor [1302] and Clinical Camel [1305] (based on LLaMA [52]), which were introduced for QA, can also perform classification or inference in a dialogue format when guided appropriately through prompts or lightweight fine-tuning. In summary, a wide range of models—from domain-specific BERTs to large GPT-style models—have been leveraged for understanding tasks. The trend is moving away from training small task-specific models from scratch and toward adapting large foundation language models (e.g., LLaMA-7B or 13B) via fine-tuning or prompting, to better transfer their general knowledge and linguistic capability to the complex biomedical domain.
LLMs have significantly advanced language understanding in the biomedical and life science domains by enabling contextual reasoning, semantic inference, and classification across complex and specialized texts such as patient reports, scientific literature, and medical examinations. These models have proven effective in tasks like natural language inference (NLI), document classification, and reading comprehension—particularly through domain-adaptive pretraining and instruction tuning. However, key challenges remain: understanding nuanced clinical negations, reasoning over long and fragmented documents, and ensuring interpretability in high-stakes decision-making. Future work should focus on improving zero-shot generalization across clinical subdomains, integrating structured biomedical ontologies for more grounded reasoning, and developing explainable evaluation frameworks to assess whether models truly comprehend rather than memorize biomedical language.
5.4.5. Hybrid Outcome Prediction
Hybrid Outcome Prediction refers to a class of tasks where LLMs are employed to predict complex biological or therapeutic outcomes by integrating diverse types of biological, chemical, and contextual information. Unlike traditional sequence-only or structure-only modeling, hybrid prediction tasks often require models to simultaneously reason over multiple heterogeneous inputs—such as chemical structures, genetic profiles, and cellular environments—to forecast functional outcomes or treatment effects. These tasks are of paramount importance in life sciences and bioengineering, as many real-world biological phenomena—such as drug response, synergistic effects, or protein function—arise from the interplay of diverse molecular and cellular factors rather than from single-modality information.
Typical inputs to hybrid prediction tasks may include combinations of small molecules, amino acid sequences, gene expression profiles, mutation data, or even broader multi-omics signatures. The outputs range from continuous measurements (e.g., synergy scores, binding affinities) to categorical labels (e.g., synergistic vs. antagonistic drug pairs, functional vs. non-functional protein variants). Hybrid outcome prediction challenges models not only to capture complex intra- and inter-modality relationships but also to generalize across biological contexts that may differ substantially between training and deployment scenarios.
The importance of hybrid outcome prediction is amplified in translational research and therapeutic development, where accurate computational forecasts can dramatically reduce experimental costs, prioritize candidate interventions, and uncover novel biological mechanisms. However, this class of tasks poses unique challenges: input modalities are often high-dimensional and noisy; the relationships between features and outcomes can be nonlinear and context-dependent; and biological interpretability remains a significant hurdle. LLMs, with their ability to integrate multimodal data, model contextual dependencies, and adapt to new tasks through fine-tuning or prompting, are particularly well-suited to address these complexities.
In this section, we focus on two major sub-directions within Hybrid Outcome Prediction: Drug Synergy Prediction and Protein Modeling. Both represent critical applications where LLMs have demonstrated transformative potential, yet where significant challenges and opportunities for future development remain.
Drug Synergy Prediction. Drug synergy prediction involves forecasting the therapeutic efficacy of drug combinations. In many diseases—particularly cancer—combination therapies can improve treatment outcomes and prevent resistance. Drug synergy refers to a phenomenon where the combined effect of two (or more) drugs exceeds the effect of each drug administered individually. Identifying synergistic drug pairs is critical for accelerating the design of combination therapies while reducing the need for extensive laboratory testing. However, this task is highly challenging due to the combinatorial explosion of possible drug pairs and the complex biological mechanisms underlying their interactions. The synergy of a given drug pair can vary depending on the context—such as cell type or disease environment—making generalization difficult. Despite these challenges, accurate synergy prediction can dramatically narrow the search space for effective multi-drug treatment regimens.
Models designed for this task typically take two drugs as input—often represented by their chemical structures, such as SMILES strings or molecular fingerprints—along with contextual features like genomic profiles of the target cell line. The output is a synergy score or class label indicating whether the combination exhibits synergistic behavior. Specifically, the input may consist of a pair of SMILES strings {Drug A, Drug B} and a cell line ID, while the output could be a continuous synergy metric, such as a Bliss or Loewe additivity score, or a binary label (synergistic vs. non-synergistic). Some models use drug-pair dose-response matrices, though many modern approaches simplify the task to predicting a single synergy score per drug pair. Incorporating contextual information (e.g., gene expression or mutation data of the cell line) makes this a multimodal prediction task, as synergy is often conditional on biological context.
Early synergy prediction methods used feature-engineered machine learning models. DeepSynergy [1370], for example, employed deep neural networks to combine molecular descriptors with gene expression profiles. More recently, transformer-based and LLM-inspired models have emerged. One notable example is DFFNDDS [1325], which integrates a BERT-like language model [9] for encoding drug SMILES and introduces a dual-feature fusion attention mechanism to capture drug-cell interactions. The BERT module in DFFNDDS [1325] jointly attends to drug-drug and drug-cell features to learn non-linear synergy effects. This architecture helps discover subtle interaction patterns—such as complementary mechanisms of action—that may be missed by simpler models or naive feature concatenation.
CancerGPT [1326] introduced a few-shot approach using a GPT-style model, transforming tabular synergy data into natural language format and fine-tuning GPT-3 to predict drug synergy in rare cancers. This method leverages the prior knowledge embedded in the language model’s weights, enabling accurate predictions even with zero or few training samples in new tissue types. Another cutting-edge method, SynerGPT [1327], pretrains a GPT model to perform contextual learning of a “synergy function.” It is trained to take a personalized dataset of known synergistic pairs as a prompt and then predict new pairs under the same context. This context-based approach avoids reliance on fixed molecular descriptors or domain-specific biological knowledge, instead extrapolating from patterns embedded in the prompt—achieving competitive results.
Furthermore, LLMs can serve as foundation models to address a diverse set of tasks in this domain. One such approach, BAITSAO [1328], is a foundation model strategy that integrates multiple datasets and tasks. It uses context-rich embeddings from LLMs as initial representations of drugs and cell lines, and performs pretraining on large drug combination databases within a multitask learning framework. BAITSAO [1328] outperformed both classical models (like DeepSynergy [1370]) and more recent tabular or transformer-based models on benchmark datasets, thanks to its multitask training and transfer learning across drug combination contexts. Overall, these LLM-based strategies—from fine-tuned GPT models to transformer fusion networks—highlight the growing role of language model architectures in capturing the complex relationships underlying drug synergy.
LLMs have brought significant advances to drug synergy prediction by enabling the modeling of complex drug–cell line interactions through contextual embeddings, attention mechanisms, and prompt-based reasoning. These models reduce reliance on handcrafted features, generalize better across biological contexts, and support few-shot or even zero-shot inference, which is especially valuable for rare diseases or under-studied drug pairs. However, key challenges remain: biological interpretability is limited, especially in identifying mechanistic pathways; synergy predictions often lack consistency across datasets or experimental conditions; and integrating multi-omics data with chemical and pharmacological knowledge in a unified framework is still an open problem. Future work should focus on enhancing cross-dataset generalization, embedding biological priors into LLM architectures, and developing transparent, mechanistically grounded models that can support experimental design and clinical translation in combination therapy development.
Table 44.
Life Science and Bio-engineering Tasks, Benchmarks, Introduction and Cross tasks
| Type of Task | Benchmarks | Introduction | Cross tasks |
|---|---|---|---|
| DNA Sequence Modeling | BEND [1294] | A collection of realistic and biologically meaningful downstream tasks defined on the human genome | Gene finding, Enhancer annotation, Chromatin accessibility, Histone modification etc. |
| Genomic Benchmarks [1371] | Contains 8 datasets that focus on regulatory elements from 3 model organisms: human, mouse, and roundworm. | - | |
| GUE [1284] | A collection of 28 datasets across 7 tasks constructed for genome language model evaluation. | Promoter prediction, Splice site prediction, Covid variant classification, epigenetic marks prediction etc. | |
| NT [1287] | A collection of 18 datasets across 7 tasks constructed for genome language model evaluation. | Promoter prediction, Splice site prediction,Enhancer annotation etc. | |
| RNA Function Learning | RnaBench [1372] | Including 100 samples without any training and validation data. | Intra/Inter family prediction, Inverse RNA Folding. |
| BEACON [1373] | Containing 967k sequences with lengths ranging from 23 to 1182. | Structure Prediction, Contact Map Prediction, Modification Prediction, Mean Ribosome Loading etc. | |
| Clinical Language Generation | MIMIC-III [1362] | A large, freely-available database comprising deidentified health-related data associated with over forty thousand patients. | Report Summarization, Risk Prediction etc. |
| MIMIC-IV [1363] | A larger version including over 65,000 patients admitted to an ICU and over 200,000 patients admitted to the emergency department. | Report Summarization, Risk Prediction etc. | |
| IU X-Ray [1374] | A set of 7,470 chest X-ray images paired with their corresponding diagnostic reports. | Report Summarization, Image Caption etc. | |
| EHR Based Prediction | EHRSHOT [1375] | A collection of 6739 patients of EHR based benchmark in few-shot setting. | – |
| EHRNoteQA [1376] | A complex, multi-topic benchmark based on multiple patients’ electronic discharg records. | QA. | |
| Quastion Answering | MedQA [1377] | Consists of multiple-choice questions from the United States Medical Licensing Examination (USMLE). | – |
| MedMCQA [1378] | A large-scale multiple-choice QA dataset derived from Indian medical entrance examinations (AIIMS/NEET). | – | |
| PubMedQA [1379] | A closed-domain QA dataset, questions can be answered by looking at an associated context (PubMed abstract). | – | |
| MMLU Subsets [135] | For measuring multitask ability from various domains, including life science and Bio-engineering. | – | |
| MIMIC-IV [1363] | A collection of 1057 questions, answer could based on the referral letters. | – | |
| Language Understanding | BC5-Disease [1089] | Including three separate sets of articles with diseases, chemicals and their relations annotated. | Named Entity Recognition. |
| NCBI-Disease [1380] | Contains 6,892 disease mentions, which are mapped to 790 unique disease concepts | Named Entity Recognition. | |
| DDI [1381] | An annotated corpus with pharmacological substances and drug–drug interactions | Relation Extraction. | |
| GAD [1382] | A repository of molecular, clinical and study parameters for >5,000 human genetic association studies | Relation Extraction. | |
| HoC [1383] | Including 1852 PubMed publication abstracts manually annotated by experts. | Doc. Classification. | |
| Drug Synergy Prediction | CancerGPT [1326] | An framework involves testing LLMs’ performance in few/zero-shot learning scenarios across seven rare tissue types. | – |
| BAITSAO [1328] | A framework integrates both regression and classification, based on synergy scores and binary synergy labels derived from large-scale drug combination datasets. | – | |
| Protein Modeling | PEER [1384] | Comprising fourteen diverse protein sequence understanding tasks. | – |
| Type [1385] | Five biologically relevant protein tasks and evaluated self-supervised sequence models. | – |
Table 45.
Benchmark results on BEND. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 45.
Benchmark results on BEND. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| Model | Gene finding |
Enhancer annotation |
Chromatin accessibility |
Histone modification |
CpG methylation |
Variant effects (expression) |
Variant effects (disease) |
| ResNet (non-LLM) | 0.46 | 0.06 | - | - | - | - | - |
| CNN (non-LLM) | 0.00 | 0.03 | 0.75 | 0.76 | 0.84 | - | - |
| ResNet-LM | 0.36 | 0.02 | 0.82 | 0.77 | 0.87 | 0.55 | 0.55 |
| AWD-LSTM | 0.05 | 0.03 | 0.69 | 0.74 | 0.81 | 0.53 | 0.45 |
| NT-H | 0.41 | 0.05 | 0.74 | 0.76 | 0.88 | 0.55 | 0.48 |
| NT-MS | 0.68 | 0.06 | 0.79 | 0.78 | 0.92 | 0.54 | 0.77 |
| NT-1000G | 0.49 | 0.04 | 0.77 | 0.77 | 0.89 | 0.45 | 0.49 |
| NT-V2 | 0.64 | 0.05 | 0.80 | 0.76 | 0.91 | 0.48 | 0.48 |
| DNABERT | 0.20 | 0.03 | 0.85 | 0.79 | 0.91 | 0.60 | 0.56 |
| DNABERT-2 | 0.43 | 0.03 | 0.81 | 0.78 | 0.90 | 0.49 | 0.51 |
| GENA-LM BERT | 0.52 | 0.03 | 0.76 | 0.78 | 0.91 | 0.49 | 0.55 |
| GENA-LM BigBird | 0.39 | 0.04 | 0.82 | 0.78 | 0.91 | 0.49 | 0.52 |
| HyenaDNA large | 0.35 | 0.03 | 0.84 | 0.76 | 0.91 | 0.51 | 0.45 |
| HyenaDNA tiny | 0.10 | 0.02 | 0.78 | 0.76 | 0.86 | 0.47 | 0.44 |
| GROVER | 0.28 | 0.03 | 0.82 | 0.77 | 0.89 | 0.56 | 0.51 |
Table 46.
Benchmark results across various 13 RNA tasks. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 46.
Benchmark results across various 13 RNA tasks. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| Model | SSP | CMP | DMP | SSI | SPL | APA | NcRNA | Modif | MRL | VDP | PRS | CRI-On | CRI-Off |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 (%) | P@L (%) | (%) | (%) | ACC@K (%) | (%) | ACC (%) | AUC (%) | (%) | MCRMSE↓ | (%) | SC (%) | SC (%) | |
| CNN (non-LLM) | 49.95 | 43.89 | 27.76 | 34.36 | 8.43 | 50.93 | 88.62 | 70.87 | 74.13 | 0.361 | 45.40 | 29.69 | 11.40 |
| ResNet (non-LLM) | 57.26 | 59.59 | 30.26 | 37.74 | 21.15 | 56.45 | 88.33 | 71.03 | 74.34 | 0.349 | 55.21 | 28.55 | 11.50 |
| LSTM (non-LLM) | 58.61 | 40.41 | 44.77 | 35.44 | 36.66 | 67.03 | 88.78 | 94.83 | 83.94 | 0.329 | 55.45 | 26.83 | 8.60 |
| RNA-FM | 68.50 | 47.56 | 51.45 | 42.36 | 34.84 | 70.32 | 96.81 | 94.98 | 79.47 | 0.347 | 55.98 | 31.62 | 2.49 |
| RNABERT | 57.27 | 45.21 | 48.19 | 31.62 | 0.18 | 57.66 | 68.95 | 82.82 | 29.79 | 0.378 | 54.60 | 29.77 | 4.27 |
| RNA-MSM | 57.98 | 57.26 | 37.49 | 39.22 | 38.33 | 70.40 | 84.85 | 94.89 | 83.48 | 0.330 | 56.94 | 34.92 | 3.85 |
| Splice-H510 | 64.93 | 45.80 | 55.56 | 38.91 | 44.80 | 58.65 | 95.92 | 62.57 | 83.49 | 0.321 | 54.90 | 26.61 | 4.00 |
| Splice-MS510 | 43.24 | 52.64 | 10.27 | 38.58 | 50.55 | 52.46 | 95.87 | 55.87 | 84.98 | 0.315 | 50.98 | 27.13 | 3.49 |
| Splice-MS1024 | 68.26 | 47.32 | 55.89 | 39.22 | 48.52 | 60.03 | 96.05 | 53.45 | 67.15 | 0.313 | 57.72 | 27.59 | 5.00 |
| UTR-LM-MRL | 59.71 | 45.51 | 55.21 | 39.52 | 36.20 | 64.99 | 89.97 | 56.41 | 77.78 | 0.325 | 57.28 | 28.49 | 4.28 |
| UTR-LM-TE&EL | 59.57 | 60.32 | 54.94 | 40.15 | 37.35 | 72.09 | 81.33 | 59.70 | 82.50 | 0.319 | 53.37 | 32.49 | 2.91 |
| UTRBERT-3mer | 60.37 | 51.03 | 50.95 | 34.31 | 44.24 | 69.52 | 92.88 | 95.14 | 83.89 | 0.337 | 56.83 | 29.92 | 4.48 |
| UTRBERT-4mer | 59.41 | 44.91 | 47.77 | 33.22 | 42.04 | 72.71 | 94.32 | 95.10 | 82.90 | 0.341 | 56.43 | 23.20 | 3.11 |
| UTRBERT-5mer | 47.92 | 44.71 | 48.67 | 31.27 | 39.19 | 72.70 | 93.04 | 94.78 | 75.64 | 0.343 | 57.16 | 25.74 | 3.93 |
| UTRBERT-6mer | 38.56 | 51.56 | 50.02 | 29.93 | 38.58 | 71.17 | 93.12 | 95.08 | 83.60 | 0.340 | 57.14 | 28.60 | 4.90 |
| BEACON-B | 64.18 | 60.81 | 56.28 | 38.78 | 37.43 | 70.59 | 94.63 | 94.74 | 72.29 | 0.320 | 54.67 | 26.01 | 4.42 |
| BEACON-B512 | 58.75 | 61.20 | 56.82 | 39.13 | 37.24 | 72.00 | 94.99 | 94.92 | 72.35 | 0.320 | 55.20 | 28.17 | 3.82 |
Table 47.
Benchmark results on question answering. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 47.
Benchmark results on question answering. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| Model | MedQA | MedMCQA | MMLU | PubMedQA | Referal QA | Treat Recom. |
|---|---|---|---|---|---|---|
| Claude-2 | 65.1 | 60.3 | 78.7 | 70.8 | 80.5 | 9.1 |
| GPT-3.5-turbo | 61.2 | 59.4 | 73.5 | 70.2 | 81.1 | 7.3 |
| GPT-4 | 83.4 | 78.2 | 92.3 | 80.0 | 83.2 | 18.6 |
| Alpaca | 34.2 | 30.1 | 40.8 | 65.2 | 74.8 | 3.5 |
| Vicuna-7B | 34.5 | 33.4 | 43.4 | 64.8 | 76.4 | 2.6 |
| LLaMA-2-7B | 32.9 | 30.6 | 42.3 | 63.4 | 74.5 | 3.3 |
| Mistral | 35.7 | 37.8 | 46.3 | 69.4 | 77.7 | 5.0 |
| Vicuna-13B | 38.0 | 36.4 | 45.6 | 66.2 | 76.8 | 4.6 |
| LLaMA-2-13B | 38.1 | 35.5 | 46.0 | 66.8 | 77.1 | 4.8 |
| LLaMA-2-70B | 45.8 | 42.7 | 54.0 | 67.4 | 78.9 | 5.5 |
| LLaMA-3-70B | 78.8 | 74.7 | 86.4 | 71.4 | 82.4 | 10.2 |
| HuatuoGPT | 28.4 | 24.8 | 31.6 | 61.0 | 69.3 | 3.8 |
| HuatuoGPT-2-7B | 41.1 | 41.9 | - | - | - | - |
| HuatuoGPT-2-13B | 45.7 | 47.4 | - | - | - | - |
| HuatuoGPT-o1-8B | 72.6 | 60.4 | - | 79.2 | - | - |
| ChatDoctor | 33.2 | 31.5 | 40.4 | 63.8 | 73.7 | 5.3 |
| PMC-LLaMA-7B | 28.7 | 29.8 | 39.0 | 60.2 | 70.2 | 4.0 |
| Baize-Healthcare | 34.9 | 31.3 | 41.9 | 64.4 | 74.0 | 4.7 |
| MedAlpaca-7B | 35.1 | 32.9 | 48.5 | 62.4 | 75.3 | 4.8 |
| Meditron-7B | 33.5 | 31.1 | 45.2 | 61.6 | 74.9 | 5.8 |
| BioMistral | 35.4 | 34.8 | 52.6 | 66.4 | 77.0 | 7.6 |
| PMC-LLaMA-13B | 39.6 | 37.7 | 56.3 | 67.0 | 77.6 | 4.9 |
| MedAlpaca-13B | 37.3 | 35.7 | 51.5 | 65.6 | 77.4 | 5.1 |
| ClinicalCamel | 46.4 | 45.8 | 68.4 | 71.0 | 79.8 | 8.4 |
| Meditron-70B | 45.7 | 44.9 | 65.1 | 70.6 | 78.6 | 8.9 |
| HuatuoGPT-o1-70B | 83.3 | 73.6 | - | 80.6 | - | - |
| Med-PaLM 2 (5-shots) | 79.7 | 71.3 | - | 79.2 | - | - |
Table 48.
Benchmark results on language understanding. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 48.
Benchmark results on language understanding. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| Model | BC5 | NCBI | DDI | GAD | HoC | Pharma. QA | Drug Infer. |
|---|---|---|---|---|---|---|---|
| Claude-2 | 52.9 | 44.2 | 50.4 | 50.7 | 70.8 | 60.6 | 51.5 |
| GPT-3.5-turbo | 52.3 | 46.1 | 49.3 | 50.8 | 66.4 | 57.3 | 47.0 |
| GPT-4 | 71.3 | 58.4 | 64.6 | 68.2 | 83.6 | 63.8 | 56.5 |
| Alpaca | 41.2 | 36.5 | 37.4 | 36.9 | 52.6 | 41.3 | 47.5 |
| Vicuna-7B | 44.5 | 37.0 | 39.4 | 41.2 | 53.8 | 42.3 | 45.5 |
| LLaMA-2-7B | 40.1 | 34.8 | 37.9 | 39.3 | 48.6 | 46.5 | 48.0 |
| Mistral | 46.8 | 39.9 | 43.5 | 44.3 | 59.6 | 51.2 | 53.0 |
| Vicuna-13B | 46.2 | 39.0 | 41.3 | 43.5 | 56.7 | 45.1 | 46.0 |
| LLaMA-2-13B | 46.6 | 38.3 | 39.7 | 41.2 | 55.9 | 46.9 | 47.5 |
| LLaMA-2-70B | 47.8 | 41.5 | 45.6 | 44.7 | 63.2 | 49.3 | 51.5 |
| LLaMA-3-70B | 63.7 | 50.2 | 59.7 | 63.1 | 79.0 | 62.4 | 53.0 |
| PubMed-BERT-base | - | 87.8 | 82.4 | 82.3 | 82.3 | - | - |
| BioLink-BERT-base | - | 88.2 | 82.7 | 84.4 | 85.4 | - | - |
| HuatuoGPT | 43.6 | 37.5 | 40.1 | 38.2 | 50.2 | 44.1 | 49.5 |
| ChatDoctor | 45.8 | 40.9 | 41.2 | 40.1 | 55.7 | 42.7 | 48.5 |
| PMC-LLaMA-7B | 45.2 | 37.8 | 40.8 | 42.0 | 55.6 | 45.5 | 51.0 |
| Baize-Healthcare | 44.4 | 38.5 | 41.9 | 45.8 | 54.5 | 46.9 | 50.5 |
| MedAlpaca-7B | 47.3 | 39.0 | 43.5 | 44.0 | 58.7 | 47.9 | 52.0 |
| Meditron-7B | 46.5 | 39.2 | 42.7 | 43.3 | 57.9 | 50.7 | 52.0 |
| BioMistral | 48.8 | 40.4 | 46.0 | 48.5 | 64.3 | 54.5 | 54.0 |
| PMC-LLaMA-13B | 51.5 | 43.1 | 48.4 | 48.7 | 65.3 | 48.8 | 51.5 |
| MedAlpaca-13B | 49.2 | 41.6 | 44.1 | 44.5 | 59.4 | 51.6 | 50.0 |
| ClinicalCamel | 51.2 | 43.7 | 47.6 | 47.2 | 64.8 | 52.6 | 52.5 |
| Meditron-70B | 54.3 | 45.7 | 51.2 | 49.6 | 69.6 | 58.7 | 54.5 |
Table 49.
Benchmark results on EHRNoteQA. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 49.
Benchmark results on EHRNoteQA. Best are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| Model | Multi-Choice | Open-Ended | ||
|---|---|---|---|---|
| Level 1 | Level 2 | Level 1 | Level 2 | |
| GPT4 | 97.16 | 95.15 | 91.30 | 89.61 |
| GPT4-Turbo | 95.27 | 94.23 | 91.30 | 86.61 |
| GPT3.5-Turbo | 88.28 | 84.99 | 82.23 | 75.52 |
| Llama3-70b-Instruct | 94.33 | 91.92 | 89.04 | 86.84 |
| Llama2-70b-chat | 84.88 | – | 78.83 | – |
| qCamel-70 | 85.63 | – | 78.26 | – |
| Camel-Platypus2-70b | 89.79 | – | 78.83 | – |
| Platypus2-70b-Instruct | 90.36 | – | 80.53 | – |
| Mixtral-8x7b-Instruct | 87.52 | 86.61 | 88.28 | 81.52 |
| MPT-30b-Instruct | 79.96 | 75.52 | 67.11 | 62.59 |
| Llama2-13b-chat | 73.65 | – | 70.32 | – |
| Vicuna-13b | 82.04 | – | 70.51 | – |
| WizardLM-13b | 80.91 | – | 74.67 | – |
| qCamel-13 | 71.46 | – | 66.16 | – |
| OpenOrca-Platypus2-13b | 86.01 | – | 79.21 | – |
| Camel-Platypus2-13b | 78.07 | – | 67.86 | – |
| Synthia-13b | 79.21 | – | 74.48 | – |
| Asclepius-13b1 | – | – | 75.24 | – |
| Gemma-7b-it | 77.50 | 67.21 | 63.71 | 54.27 |
| MPT-7b-8k-instruct | 59.55 | 51.27 | 56.71 | 53.81 |
| Mistral-7b-Instruct | 82.04 | 64.90 | 72.97 | 53.81 |
| Dolphin-2.0-mistral-7b | 76.18 | – | 69.75 | – |
| Mistral-7b-OpenOrca | 87.15 | – | 79.58 | – |
| SynthIA-7b | 78.45 | – | 74.67 | – |
| Llama2-7b-chat | 65.78 | – | 58.98 | – |
| Vicuna-7b | 78.26 | – | 59.74 | – |
| Asclepius-7b | – | – | 66.92 | – |
Table 50.
AUPRC of k-shot learning on seven tissue sets. =total number of non-synergistic samples (not positive), =total number of synergistic samples (positive). Where XGBoost is non-LLM baseline method. Best are highlighted with dark blue.
Table 50.
AUPRC of k-shot learning on seven tissue sets. =total number of non-synergistic samples (not positive), =total number of synergistic samples (positive). Where XGBoost is non-LLM baseline method. Best are highlighted with dark blue.
| Tissue ( , ) | Methods | 0 | 2 | 4 | 8 | 16 | 32 | 64 | 128 |
|---|---|---|---|---|---|---|---|---|---|
| Pancreas (=38, =1) |
XGBoost | 0.026 | – | – | – | – | – | – | – |
| TabTransformer | 0.056 | – | – | – | – | – | – | – | |
| CancerGPT | 0.033 | – | – | – | – | – | – | – | |
| GPT-2 | 0.032 | – | – | – | – | – | – | – | |
| GPT-3 | 0.111 | – | – | – | – | – | – | – | |
| Endometrium (=36, =32) |
XGBoost | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | – | – | – |
| TabTransformer | 0.674 | 0.889 | 0.903 | 0.948 | 0.938 | 0.962 | – | – | |
| CancerGPT | 0.564 | 0.668 | 0.676 | 0.831 | 0.686 | 0.737 | – | – | |
| GPT-2 | 0.408 | 0.808 | 0.395 | 0.383 | 0.389 | 0.717 | – | – | |
| GPT-3 | 0.869 | 1.0 | 0.947 | 0.859 | 0.799 | 0.859 | – | – | |
| Liver (=192, =21) |
XGBoost | 0.132 | 0.132 | 0.132 | 0.132 | 0.132 | 0.132 | 0.12 | 0.12 |
| TabTransformer | 0.13 | 0.128 | 0.147 | 0.189 | 0.265 | 0.168 | 0.169 | 0.234 | |
| CancerGPT | 0.136 | 0.102 | 0.13 | 0.147 | 0.252 | 0.21 | 0.197 | 0.187 | |
| GPT-2 | 0.5 | 0.099 | 0.151 | 0.383 | 0.429 | 0.401 | 0.483 | 0.398 | |
| GPT-3 | 0.185 | 0.086 | 0.096 | 0.125 | 0.124 | 0.314 | 0.362 | 0.519 | |
| Soft tissue (=269, =83) |
XGBoost | 0.243 | 0.243 | 0.243 | 0.243 | 0.235 | 0.235 | 0.264 | 0.271 |
| TabTransformer | 0.273 | 0.287 | 0.462 | 0.422 | 0.526 | 0.571 | 0.561 | 0.64 | |
| CancerGPT | 0.314 | 0.315 | 0.338 | 0.383 | 0.383 | 0.403 | 0.464 | 0.469 | |
| GPT-2 | 0.259 | 0.298 | 0.254 | 0.262 | 0.235 | 0.297 | 0.254 | 0.206 | |
| GPT-3 | 0.263 | 0.194 | 0.28 | 0.228 | 0.363 | 0.618 | 0.638 | 0.734 | |
| Stomach (=1081, =109) |
XGBoost | 0.104 | 0.104 | 0.104 | 0.104 | 0.104 | 0.104 | 0.09 | 0.094 |
| TabTransformer | 0.261 | 0.371 | 0.396 | 0.383 | 0.294 | 0.402 | 0.45 | 0.465 | |
| CancerGPT | 0.3 | 0.297 | 0.316 | 0.325 | 0.269 | 0.308 | 0.297 | 0.312 | |
| GPT-2 | 0.116 | 0.124 | 0.099 | 0.172 | 0.165 | 0.107 | 0.152 | 0.131 | |
| GPT-3 | 0.078 | 0.106 | 0.17 | 0.37 | 0.1 | 0.19 | 0.219 | 0.181 | |
| Urinary tract (=1996, =462) |
XGBoost | 0.186 | 0.186 | 0.186 | 0.186 | 0.186 | 0.197 | 0.199 | 0.209 |
| TabTransformer | 0.248 | 0.264 | 0.25 | 0.278 | 0.274 | 0.249 | 0.293 | 0.291 | |
| CancerGPT | 0.241 | 0.226 | 0.246 | 0.239 | 0.256 | 0.271 | 0.266 | 0.269 | |
| GPT-2 | 0.191 | 0.192 | 0.188 | 0.156 | 0.193 | 0.185 | 0.183 | 0.185 | |
| GPT-3 | 0.27 | 0.228 | 0.222 | 0.201 | 0.206 | 0.2 | 0.24 | 0.272 | |
| Bone (=3732, =253) |
XGBoost | 0.064 | 0.064 | 0.064 | 0.064 | 0.064 | 0.064 | 0.064 | 0.064 |
| TabTransformer | 0.123 | 0.12 | 0.121 | 0.115 | 0.102 | 0.13 | 0.129 | 0.121 | |
| CancerGPT | 0.119 | 0.115 | 0.125 | 0.116 | 0.115 | 0.111 | 0.114 | 0.125 | |
| GPT-2 | 0.063 | 0.094 | 0.057 | 0.081 | 0.052 | 0.071 | 0.057 | 0.065 | |
| GPT-3 | 0.064 | 0.051 | 0.045 | 0.058 | 0.068 | 0.087 | 0.101 | 0.181 |
Table 51.
AUROC of k-shot learning on seven tissues sets. Where XGBoost is non-LLM baseline method.
| Tissue | Methods | 0 | 2 | 4 | 8 | 16 | 32 | 64 | 128 |
|---|---|---|---|---|---|---|---|---|---|
| Pancreas | XGBoost | 0.5 | – | – | – | – | – | – | – |
| TabTransformer | 0.553 | – | – | – | – | – | – | – | |
| CancerGPT | 0.237 | – | – | – | – | – | – | – | |
| GPT-2 | 0.211 | – | – | – | – | – | – | – | |
| GPT-3 | 0.789 | – | – | – | – | – | – | – | |
| Endometrium | XGBoost | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | – | – |
| TabTransformer | 0.694 | 0.857 | 0.878 | 0.939 | 0.939 | 0.959 | – | – | |
| CancerGPT | 0.489 | 0.693 | 0.714 | 0.735 | 0.612 | 0.612 | – | – | |
| GPT-2 | 0.265 | 0.816 | 0.224 | 0.184 | 0.204 | 0.612 | – | – | |
| GPT-3 | 0.837 | 1.0 | 0.949 | 0.898 | 0.878 | 0.898 | – | – | |
| Liver | XGBoost | 0.587 | 0.587 | 0.587 | 0.587 | 0.587 | 0.587 | 0.574 | 0.574 |
| TabTransformer | 0.535 | 0.506 | 0.526 | 0.535 | 0.609 | 0.647 | 0.702 | 0.804 | |
| CancerGPT | 0.615 | 0.468 | 0.59 | 0.641 | 0.782 | 0.776 | 0.737 | 0.737 | |
| GPT-2 | 0.731 | 0.449 | 0.558 | 0.66 | 0.679 | 0.763 | 0.731 | 0.731 | |
| GPT-3 | 0.615 | 0.49 | 0.542 | 0.583 | 0.474 | 0.731 | 0.737 | 0.91 | |
| Soft tissue | XGBoost | 0.491 | 0.491 | 0.491 | 0.491 | 0.454 | 0.476 | 0.542 | 0.552 |
| TabTransformer | 0.557 | 0.566 | 0.709 | 0.727 | 0.788 | 0.802 | 0.83 | 0.835 | |
| CancerGPT | 0.656 | 0.646 | 0.68 | 0.734 | 0.725 | 0.754 | 0.8 | 0.795 | |
| GPT-2 | 0.546 | 0.535 | 0.519 | 0.56 | 0.427 | 0.577 | 0.456 | 0.384 | |
| GPT-3 | 0.517 | 0.406 | 0.6 | 0.444 | 0.607 | 0.82 | 0.866 | 0.889 | |
| Stomach | XGBoost | 0.529 | 0.529 | 0.529 | 0.529 | 0.529 | 0.529 | 0.476 | 0.508 |
| TabTransformer | 0.804 | 0.863 | 0.855 | 0.853 | 0.812 | 0.85 | 0.885 | 0.869 | |
| CancerGPT | 0.794 | 0.792 | 0.796 | 0.794 | 0.785 | 0.787 | 0.824 | 0.804 | |
| GPT-2 | 0.551 | 0.569 | 0.521 | 0.516 | 0.589 | 0.538 | 0.469 | 0.566 | |
| GPT-3 | 0.419 | 0.575 | 0.724 | 0.769 | 0.534 | 0.69 | 0.742 | 0.724 | |
| Urinary tract | XGBoost | 0.494 | 0.494 | 0.494 | 0.494 | 0.494 | 0.526 | 0.53 | 0.544 |
| TabTransformer | 0.599 | 0.612 | 0.604 | 0.625 | 0.601 | 0.587 | 0.623 | 0.622 | |
| CancerGPT | 0.578 | 0.561 | 0.579 | 0.577 | 0.589 | 0.593 | 0.609 | 0.609 | |
| GPT-2 | 0.526 | 0.528 | 0.532 | 0.397 | 0.515 | 0.452 | 0.469 | 0.566 | |
| GPT-3 | 0.645 | 0.57 | 0.556 | 0.496 | 0.508 | 0.516 | 0.531 | 0.572 | |
| Bone | XGBoost | 0.499 | 0.499 | 0.499 | 0.499 | 0.499 | 0.499 | 0.499 | 0.499 |
| TabTransformer | 0.706 | 0.705 | 0.724 | 0.697 | 0.65 | 0.689 | 0.708 | 0.696 | |
| CancerGPT | 0.625 | 0.648 | 0.693 | 0.653 | 0.683 | 0.636 | 0.678 | 0.68 | |
| GPT-2 | 0.507 | 0.616 | 0.471 | 0.579 | 0.421 | 0.552 | 0.476 | 0.518 | |
| GPT-3 | 0.498 | 0.415 | 0.341 | 0.429 | 0.485 | 0.605 | 0.62 | 0.794 |
Table 52.
Benchmark Results on PEER. Best are highlighted with dark blue.
| Model | Flu | Sta | -lac | Sol | Sub | Bin | Cont | Fold | SSP | Yst | Hum | Aff | PDB | BDB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DDE | 0.638 | 0.652 | 0.623 | 59.77 | 49.17 | 77.43 | – | 9.57 | – | 55.83 | 62.77 | 2.908 | – | – |
| Moran | 0.400 | 0.322 | 0.375 | 57.73 | 31.13 | 55.63 | – | 7.10 | – | 53.00 | 54.67 | 2.984 | – | – |
| LSTM | 0.494 | 0.533 | 0.139 | 70.18 | 62.98 | 88.11 | 26.34 | 8.24 | 68.99 | 53.62 | 63.75 | 2.853 | 1.457 | 1.572 |
| Transformer | 0.643 | 0.649 | 0.261 | 70.12 | 56.02 | 75.74 | 17.50 | 8.52 | 59.62 | 54.12 | 59.58 | 2.499 | 1.455 | 1.566 |
| CNN | 0.682 | 0.637 | 0.781 | 64.43 | 58.73 | 82.67 | 10.00 | 10.93 | 66.07 | 55.07 | 62.60 | 2.796 | 1.376 | 1.497 |
| ResNet | 0.636 | 0.126 | 0.152 | 67.33 | 52.30 | 78.99 | 20.43 | 8.89 | 69.56 | 48.91 | 68.61 | 3.005 | 1.441 | 1.565 |
| ProtBert | 0.679 | 0.771 | 0.731 | 68.15 | 76.53 | 91.32 | 39.66 | 16.94 | 82.18 | 63.72 | 77.32 | 2.195 | 1.562 | 1.549 |
| ProtBert* | 0.339 | 0.697 | 0.616 | 59.17 | 59.44 | 81.54 | 24.35 | 10.74 | 62.51 | 53.87 | 83.61 | 2.996 | 1.457 | 1.649 |
| ESM-1b | 0.679 | 0.694 | 0.839 | 70.23 | 78.13 | 92.40 | 45.78 | 28.17 | 82.73 | 57.00 | 78.17 | 2.281 | 1.559 | 1.556 |
| ESM-1b* | 0.430 | 0.750 | 0.528 | 67.02 | 79.82 | 91.61 | 40.37 | 29.95 | 83.14 | 66.07 | 88.06 | 3.031 | 1.368 | 1.571 |
Protein Modeling. Protein Modeling refers to the task of learning structural, functional, or evolutionary patterns from amino acid sequences, enabling predictions of protein properties such as folding, function, or interaction based. The development of protein LLMs has been driven by the deepening integration of computational biology and natural language processing techniques. Early efforts focused on leveraging traditional deep learning architectures such as LSTMs for representation learning of protein sequences [1329,1330,1386]. Models like UniRep [1329] and Bepler & Berger [1330] made initial progress in constructing protein embedding vectors.
AlphaFold [1387] is a protein structure prediction model released by DeepMind, which revolutionized the long-standing protein folding problem through deep learning. The model integrates evolutionary homologous sequences, residue-pair geometric maps, and physical constraints using an attention-based network. In CASP14, it achieved an average GDT_TS of 92.4, marking the first time atomic-level accuracy was reached. The subsequent release of a database containing over 2 million predicted structures has significantly accelerated drug discovery, enzyme engineering, and pathogenic mutation annotation.
With the rise of Transformers [4] in natural language processing, this paradigm was rapidly transferred to protein modeling. Transformer-based models such as ProtTrans [1269] and ESM-1b [1331] emerged, offering enhanced capabilities in capturing long-range dependencies within sequences, significantly improving the accuracy of protein structure and function prediction.
The ESM series has since expanded in both model size and task scope—from ESM-1v [1332] to ESM-2 [1333] and the latest ESM-3 [1388]—achieving end-to-end sequence-to-structure prediction (e.g., ESMFold [1333]), incorporating multimodal information, and enabling complex reasoning and even generative design for protein function. These advancements signify a shift toward universal modeling and reasoning capabilities in protein LLMs.
Beyond foundational modeling capabilities, researchers have begun to explicitly inject structural information into the training process to enhance the models’ ability to capture 3D protein conformations. Models such as SaProt [1334] and ESM-GearNet [1335] integrate local or global structural features to enrich sequence representations, while approaches like OntoProtein [1336] and ProteinCLIP [1337] leverage knowledge graphs and contrastive learning with text to improve semantic understanding and generalization. These structure-informed and knowledge-enhanced strategies have not only improved model expressiveness on tasks such as mutation effect prediction, functional domain annotation, and binding site identification, but also extended the applicability of protein LLMs to drug target identification and molecular interaction prediction.
Building on foundational understanding and reasoning, protein LLMs have further evolved toward generative modeling. ProGen [1268] and ProtGPT2 [1338] were among the first to apply the autoregressive language modeling paradigm to protein sequence generation, capable of producing diverse, biologically active sequences conditioned on functional labels or species. ProGen2 [1267] scaled up both model size and training data, significantly enhancing its ability to model protein adaptability and diversity. Meanwhile, ProLLaMA [1339] incorporated protein sequence learning into the LLaMA architecture, achieving joint understanding and generation within a single framework and demonstrating the potential of multi-task and cross-modal pretraining. In contrast, models like Pinal [1340] and Ankh [1341] explore structure-guided, efficient encoder-decoder architectures to balance generation quality with parameter efficiency.
At the same time, several integrated frameworks have emerged to support protein design and engineering. For example, ProteinDT [1342] enables zero-shot generation of protein sequences from textual functional descriptions, while PLMeAE [1343] integrates with automated biological experimentation platforms to construct a “design-build-test-learn” loop for automated protein engineering. Innovative interactive tools such as ProteinGPT [1344] and ProteinChat [1345] have also appeared, supporting structure input, language interaction, and functional Q&A, further advancing protein language models toward intelligent agents with cognitive and interactive capabilities.
Overall, the evolution of protein LLMs has clearly progressed from small-scale LSTM-based semantic embeddings, to large-scale Transformer-based structural predictions, and toward multimodal-enhanced generative design. This trajectory has not only significantly expanded the frontiers of protein science but also laid a robust foundation for the next generation of biomolecular design, functional prediction, and clinical applications.
5.4.6. Benchmarks
The rapid adoption of LLMs in life sciences and bio-engineering has spurred the development of specialized benchmarks designed to systematically assess their performance across diverse biological and clinical tasks. Benchmarks such as BEND for DNA language models and BEACON for RNA language models rigorously evaluate the ability of LLMs to interpret complex genomic and transcriptomic information, encompassing tasks that range from functional element annotation in genomic sequences to predicting RNA secondary structures. Complementing these biological benchmarks, medical QA datasets like MedQA, MedMCQA, and PubMedQA focus on evaluating clinical knowledge, reasoning capabilities, and contextual understanding of biomedical literature. Together, these benchmarks offer a comprehensive framework to evaluate and drive progress in applying LLMs to real-world biomedical challenges.
BEND. BEND is a unified evaluation framework designed to systematically assess the performance of DNA language models (DNA LMs) on realistic biological tasks. The benchmark suite comprises seven tasks based on the human genome, covering functional elements at varying length scales, such as promoters, enhancers, splice sites, and transcription units. Each task provides input sequences and labels in a standardized format, supporting a range of downstream tasks including both classification and regression.
The task design of BEND reflects the core challenges of genome annotation: wide variation in sequence length, sparsity of functional regions, and low signal density. To evaluate the performance of DNA LMs on these tasks, BEND offers a scalable framework for generating embedding representations and training lightweight supervised models. Experimental results demonstrate that while certain DNA LMs can approach the performance of expert methods on specific tasks, they still face difficulties in capturing long-range dependencies (such as enhancer recognition). Moreover, different models display varying preferences for modeling gene structure and non-coding region features.
For example, in the enhancer annotation task, BEND formulates the problem as binary classification: for each 128-base-pair segment of gene-adjacent DNA, the model predicts whether it contains an enhancer. Data are sourced from CRISPR interference experiments and integrated with major transcription start site (TSS) information, with a 100,096-bp sequence extracted for each gene and annotated in 128-bp segments. The main challenge of this task lies in identifying distal regulatory relationships, which tests the model’s ability to capture long-range dependencies.
BEACON. BEACON is a comprehensive evaluation benchmark specifically designed for RNA language models, encompassing 13 tasks related to RNA structural analysis, functional studies, and engineering applications. All tasks adopt a unified data format and support both classification and regression evaluations, applicable to both sequence-level and nucleotide-level predictions.
For example, in the RNA secondary structure prediction task, the model is required to determine whether each pair of nucleotides forms a base pair, with the F1 score used as the evaluation metric. The data for this task is sourced from the bpRNA-1m database.
BEACON also includes a systematic evaluation of various models and finds that single-nucleotide tokenization and ALiBi positional encoding demonstrate superior performance across multiple tasks. Based on these findings, a lightweight baseline model named BEACON-B is proposed.
QA Benchmarks.
The landscape of biomedical and clinical QA benchmarks spans a diverse range of tasks, from licensing examination questions to domain-specific reasoning over scientific literature. These datasets challenge models not only on factual recall but also on higher-order reasoning, reading comprehension, and the ability to synthesize information from complex biomedical contexts. Together, they provide a comprehensive evaluation suite for assessing the medical knowledge, reasoning ability, and contextual understanding of AI systems in healthcare and biomedical research.
Current benchmarks are primarily concentrated within the English language domain, often based on medical licensure examinations from different English-speaking countries. There are also benchmarks in other languages, such as Chinese. These benchmarks fully simulate real-world exams, providing only the question stem and answer choices. In addition, there are benchmarks that supply LLMs with a reference document, requiring the model to combine its own knowledge with the provided context to generate a more informed answer.
- MedQA. The MedQA dataset consists of multiple-choice questions from the United States Medical Licensing Examination (USMLE). It covers general medical knowledge and includes 11,450 questions in the development set and 1,273 questions in the test set. Each question has 4 or 5 answer choices, and the dataset is designed to assess the medical knowledge and reasoning skills required for medical licensure in the United States.
- MedMCQA. MedMCQA is a large-scale multiple-choice QA dataset derived from Indian medical entrance examinations (AIIMS/NEET). It covers 2.4k healthcare topics and 21 medical subjects, with over 187,000 questions in the development set and 6,100 questions in the test set. Each question has 4 answer choices and is accompanied by an explanation. MedMCQA evaluates a model’s general medical knowledge and reasoning capabilities.
- PubmedQA. Different from MedQA and MedMCQA, PubMedQA is a closed-domain QA dataset, In which each question can be answered by looking at an associated context (PubMed abstract). It is consists of 1,000 expert-labeled question-answer pairs. Each question is accompanied by a PubMed abstract as context, and the task is to provide a yes/no/maybe answer based on the information in the abstract. The dataset is split into 500 questions for development and 500 for testing. PubMedQA assesses a model’s ability to comprehend and reason over scientific biomedical literature.
Drug Synergy Prediction
CancerGPT. CancerGPT [1326] assesses the capability of LLMs to predict drug pair synergy in rare cancer tissues with limited structured data. The evaluation framework involves testing LLMs’ performance in few-shot and zero-shot learning scenarios across seven rare tissue types, comparing the results to those of larger models like GPT-3 [7].
The evaluation process includes:
- Few-shot and Zero-shot Learning: Assessing the model’s ability to predict drug synergy with minimal or no training examples, highlighting the LLM’s capacity to generalize from limited data.
- Benchmarking Across Multiple Tissues: Testing the model’s predictive performance across seven different rare cancer tissue types to ensure robustness and generalizability.
This evaluation framework demonstrates that LLMs, even with fewer parameters, can effectively predict drug pair synergies in contexts with scarce data, offering a promising approach for biological inference tasks where traditional structured data is lacking.
BAITSAO. The benchmark framework integrates both regression and classification tasks, based on synergy scores (e.g., Loewe, Bliss, HSA, ZIP) and binary synergy labels derived from large-scale drug combination datasets such as DrugComb. Each sample consists of a drug pair and a cell line, with input features constructed from Large Language Model (LLM) embeddings of descriptive prompts about drugs and cell lines, standardized into numerical vectors.
The design of the BAITSAO evaluation suite reflects key challenges in drug synergy prediction: sparse synergy signals, heterogeneous data formats, and limited generalization to novel drug combinations. To evaluate model performance, BAITSAO pre-trains on large-scale synergy data under a multi-task learning (MTL) framework, capturing both single-drug inhibition and pairwise synergy. The model is then assessed on held-out datasets using metrics such as Pearson correlation, mean squared error, ROC-AUC, and accuracy. Ablation and sensitivity analyses are further conducted to study embedding strategies, training data scales, and model scaling laws.
For example, in the synergy classification task, BAITSAO formulates the problem as a binary prediction: given a pair of drugs and a cell line, predict whether the combination yields a synergistic effect. Inputs are constructed by averaging the embeddings of both drugs and concatenating with the cell line embedding, with synergy labels binarized using a threshold on the Loewe score. This setup evaluates the model’s ability to generalize to unseen drug-cell line combinations, including out-of-distribution (OOD) samples, and serves as a robust benchmark for multi-drug reasoning and zero-shot prediction.
Protein Modeling
TAPE. TAPE (Tasks Assessing Protein Embeddings) is a large-scale benchmark designed to evaluate transfer learning methods on protein sequences. It comprises five biologically relevant tasks, including protein structure prediction, remote homology detection, and protein engineering. Each task features carefully curated splits to assess models’ ability to generalize in biologically meaningful ways. TAPE evaluates various self-supervised learning approaches for protein representation and shows that pretraining significantly improves performance across nearly all tasks, although traditional non-neural methods still outperform in some cases. The benchmark promotes standardized evaluation and method comparison in protein modeling.
PEER. PEER (Protein sEquence undERstanding) is a comprehensive and multi-task benchmark designed to evaluate deep learning methods on protein sequences. It encompasses 17 biologically relevant tasks across five categories: protein function prediction, localization prediction, structure prediction, protein-protein interaction prediction, and protein-ligand interaction prediction. Each task includes carefully curated training, validation, and test splits to assess models’ generalization capabilities in real-world scenarios. PEER evaluates various sequence-based approaches, including traditional feature engineering methods, different sequence encoding techniques, and large-scale pre-trained protein language models.
Summary. Benchmarking efforts in life-science and bio-engineering LLMs now coalesce around four broad task families. First, sequence-based evaluation dominates DNA and RNA modeling. Suites such as BEND (DNA) and BEACON (RNA) probe classification and regression across functional-element annotation, secondary-structure inference, and variant-effect prediction. Second, clinical structured-data tasks assess models on Electronic Health Records, splitting into clinical language generation (e.g., ClinicalT5, GPT-4 hospital-note drafting) and EHR-based prediction (e.g., BEHRT, Med-BERT risk scoring). Third, textual knowledge tasks test biomedical reasoning and understanding via QA (MedQA, MedMCQA, PubMedQA) and natural-language inference benchmarks such as MedNLI, measuring factual recall, chain-of-thought reasoning, and long-context comprehension. Finally, hybrid outcome-prediction benchmarks—drug-synergy suites (e.g., DrugCombDB subsets) and protein-modeling challenges (ESMFold, ProGen)––demand multimodal integration across chemistry, omics and cellular context.
Across these categories, domain-trained transformers consistently outperform classical baselines. In sequence modeling, long-context LLMs (Enformer, HyenaDNA) improve enhancer or eQTL effect prediction correlations by 20–40% over CNN/RNN hybrids, while bidirectional masked models (DNABERT-2) raise MCC scores on promoter/enhancer detection by 2–5 pp versus 6-mer CNNs. In clinical language generation, instruction-tuned GPT-4 drafts discharge summaries that clinicians rate as equal or superior in accuracy and readability to human-written notes, and models like Med-PaLM 2 reach 86% accuracy on USMLE-style exams, narrowing the gap to licensed physicians. EHR-based predictors such as GatorTron boost AUROC for onset prediction tasks by 3–6 pp relative to GRU or logistic-regression baselines, even under low-data fine-tuning. In drug-synergy prediction, transformer fusion networks (DFFNDDS) and prompt-based few-shot GPT variants (CancerGPT) lift balanced accuracy by 5–12 pp over DeepSynergy, while LLM-generated protein sequences (ProGen2) exhibit in-vitro activities on par with natural enzymes in ≥50% of tested families.
Yet substantial limitations persist. Ultra-long genomic context still degrades accuracy despite linear-time attention variants; distal enhancer–promoter linkage and rare-variant generalization remain open. Multimodal fusion is ad-hoc: most benchmarks isolate a single modality, leaving cross-omics reasoning and image-augmented clinical tasks underexplored. Data quality and bias are acute—human-centric genomes, single-institution EHRs, and English-only QA corpora skew performance and hamper species-, population-, or language-level transfer. Safety and interpretability issues mirror those seen in chemistry: hallucinated diagnoses or biologically implausible sequence designs can slip through, and attention maps alone rarely satisfy domain experts’ need for mechanistic insight.
From these observations we derive three actionable insights. (1) Benchmark breadth and depth must expand. Community curation of larger, more diverse genomes (e.g., non-model organisms), multilingual clinical notes, and truly multimodal datasets (sequence+structure+phenotype+imaging) is essential. (2) Representation and architecture choices require re-thinking. Treating kilo- to megabase sequences as flat text overlooks 3-D chromatin contacts; integrating graph, spatial or physics-aware modules with transformers, and exploring alternative encodings (e.g., byte-pair k-mers, SELFIES-like bio-tokens) can bridge this gap. (3) Reliability hinges on task design and validation. Embedding biological priors, tool-augmented prompting (e.g., retrieval of wet-lab evidence), and post-hoc critic models can curb hallucination and enforce mechanistic plausibility; standardized factuality and safety metrics—analogous to clinical adjudication—should accompany benchmark scoreboards.
In sum, life-science–oriented LLM benchmarks have revealed impressive gains over traditional pipelines, but progress is gated by richer data, modality-aware architectures, and rigorous, biology-centric evaluation. Aligning these elements will accelerate LLMs from promising assistants to dependable engines for discovery and precision medicine.
5.4.7. Discussion
Opportunities and Impact. LLMs are now deeply integrated across the life sciences pipeline, supporting a broad spectrum of tasks ranging from genomic sequence interpretation to drafting clinical documentation. Their greatest impact has emerged in areas that align with their linguistic strengths—such as literature summarization, clinical note generation, and biomedical question-answering—where abundant data, low-cost supervision, and linguistic evaluation metrics have enabled rapid progress.
A major advantage lies in the tokenizable structure of biological data. Representations like k-mers for genomic sequences, SMILES for chemical compounds, and ICD codes in medical records are inherently suited to masked language modeling or autoregressive learning. As a result, LLMs like DNABERT-2, Nucleotide Transformer, BEHRT, and Med-PaLM 2 [1284,1287,1301] offer unified frameworks to model complex biological substrates. For example, RNA oligomers of various sizes require different experimental strategies—from NMR [1389] and FRET [1390] for small structures to cryo-EM and CLIP-seq for large complexes [1391,1392]. Traditional computational methods often rely on size-specific architectures with manual feature engineering, while LLMs can tokenize all scales using shard tokenizers and learn a size-agnostic representation space.
Furthermore, LLMs exhibit context extrapolation capabilities that allow modeling of long-range dependencies in genomic data. For example, predicting MYC expression traditionally required multiple CNN-based sliding windows, Hi-C loop assemblies, and handcrafted features [1393,1394], which struggled to capture distal interactions. In contrast, models like HyenaDNA [1261] process 1 Mb genomic windows in a single forward pass, leveraging sub-quadratic convolutions to directly learn enhancer–promoter logic, thereby eliminating the need for fragmented, manually-curated pipelines.
The instruction-following and multitask capabilities of LLMs further enable unified handling of diverse biomedical applications. For instance, Med-PaLM 2 can simultaneously draft clinical notes, generate ICD-10/LOINC codes, and rewrite instructions at a 6th-grade level—all in a single prompt. This integration replaces three siloed hospital systems and reduces development timelines from months to hours.
Challenges and Limitations. Despite these advancements, significant limitations persist. LLMs excel in symbolic and text-rich domains but underperform in tasks requiring deep experimental grounding or multi-scale biological reasoning.
First, the gap in empirical grounding remains a major bottleneck. Models such as ProGen2 can propose novel peptides, but their real-world efficacy is limited. For instance, the initial validation of ProGen2-generated incretin peptides showed an activity success rate of merely 7%, emphasizing the indispensable role of iterative wet-lab testing and retraining.
Second, life science problems often involve system-level complexity, where token-based reasoning is insufficient. Predicting off-target effects of CRISPR editing or long-term drug toxicity demands multiscale modeling across molecular, cellular, and organismal levels. Although models like CRISPR-GPT [1395] show promise, they still miss over 30% of off-target sites in whole-genome data with complex chromatin interactions [1187,1188,1230].
Third, the rise of powerful generative models introduces new ethical, safety, and provenance concerns. LLMs capable of generating accurate protocols may inadvertently facilitate dual-use research or propagate hallucinations. Open-source toxicity predictors like ToxinPred [1396,1397] can potentially be misused to design harmful biological sequences. Without clear traceability or accountability mechanisms, the risks of misuse escalate.
Research Directions. To address these challenges, we propose a forward-looking research agenda focused on hybrid architectures and responsible integration.
- First, future LLM systems should aim to unify diverse biological modalities—including genomic sequences, protein structures, cell images, clinical time-series, and textual notes—within a cohesive multimodal framework. Such models can enable integrated diagnosis and prediction by capturing complex biological correlations across data types.
- Second, LLMs should evolve from passive tools into active hypothesis-generating agents. This requires coupling with laboratory automation systems, real-time EHR streams, and high-throughput simulation platforms. For instance, an LLM-guided robotic lab could autonomously design, test, and refine molecular hypotheses in closed experimental loops, dramatically accelerating the discovery cycle.
- Third, the training of LLMs should incorporate biologically-informed learning techniques. Self-distillation improves interpretability through reasoning chains, contrastive alignment ensures consistency with biomedical knowledge bases, and physics-informed regularization grounds models in biophysical laws (e.g., thermodynamics in MD simulations), reducing hallucinations and enhancing trustworthiness.
- Finally, proactive governance must be embedded from the outset. Techniques such as differential privacy for sensitive patient data, watermarking synthetic DNA sequences to differentiate them from natural ones, and rigorous human oversight mechanisms are crucial for ensuring ethical deployment. Building responsible AI systems is not an afterthought—it must be integral to model development.
Conclusion. LLMs have transformed information processing in the life sciences, accelerating literature review, genomic annotation, and clinical documentation. Their most pronounced successes lie in tasks with high symbolic complexity but relatively low experimental demands. However, realizing their full potential in experimental biology and bioengineering will require overcoming structural limitations.
This transformation demands more than model scaling; it necessitates innovations in architecture, training paradigms, and system integration. Bridging the gap between computational prediction and empirical validation calls for hybrid systems that fuse LLMs with biological priors, experimental platforms, and domain-specific constraints.
When thoughtfully designed and ethically deployed, LLMs can serve not merely as intelligent assistants but as generative partners in hypothesis formation, experimental design, and therapeutic innovation—ultimately accelerating the transition from scientific discovery to clinical application.
5.5. Earth Sciences and Civil Engineering
5.5.1. Overview
Figure 21.
The relationships between major research tasks between earth sciences and civil engineering.
Figure 21.
The relationships between major research tasks between earth sciences and civil engineering.
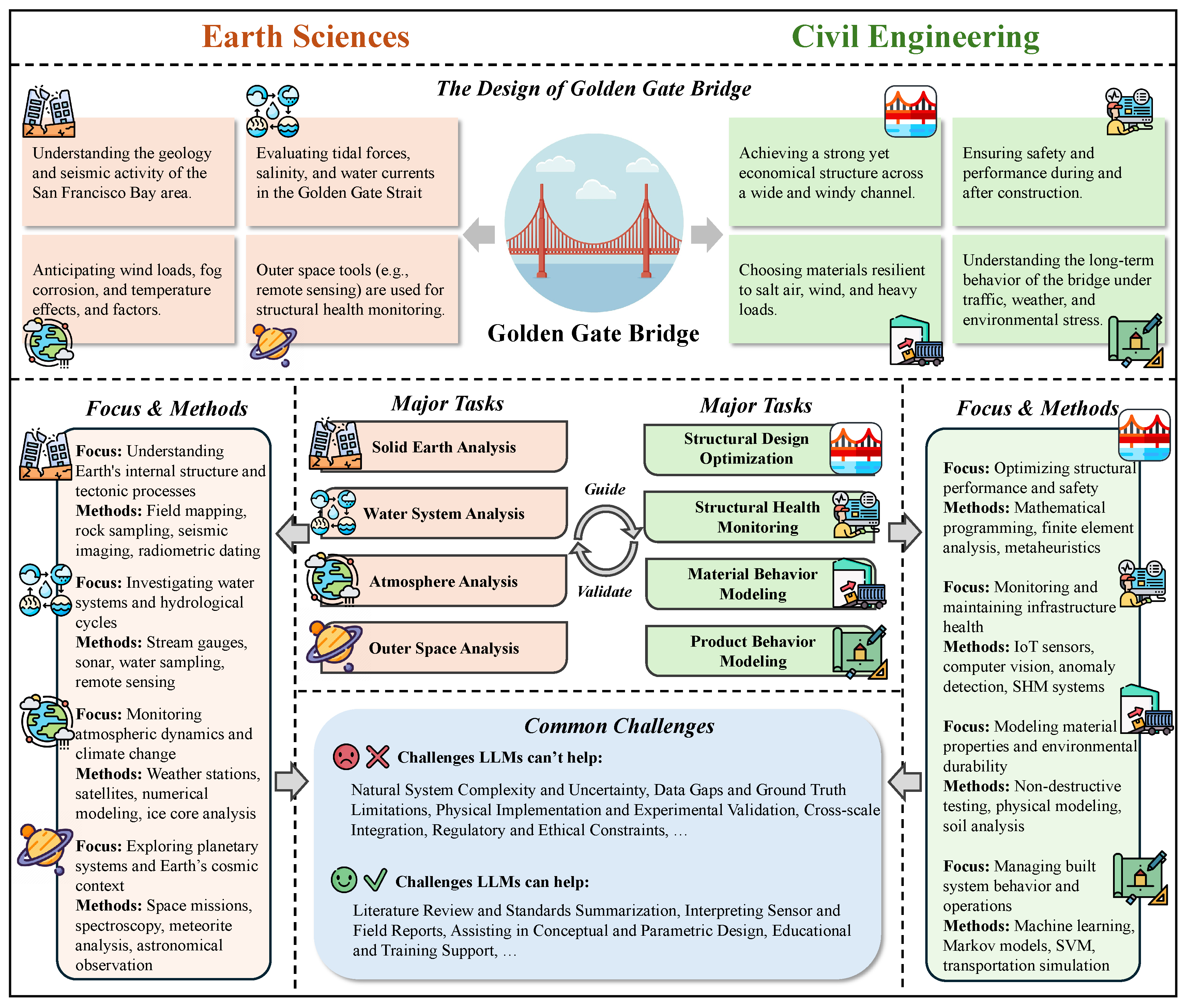
5.5.1.1 Earth Sciences Introduction
Earth sciences encompass the study of Earth’s physical structure, processes, and history, as well as its surrounding systems such as the atmosphere, oceans, and outer space [1398,1399,1400]. These sciences aim to understand how the Earth and related environments function and change over time [1401]. Researchers in this field investigate diverse phenomena ranging from earthquakes and volcanoes to ocean circulation, weather systems, and planetary formation. Earth science not only satisfies human curiosity about our planet but also supports practical applications such as natural disaster prediction [1402,1403], climate modeling [1404], resource exploration [1405], and environmental protection [1406].
For instance, the early detection of seismic waves through global monitoring networks has enabled the development of earthquake early warning systems, helping to save lives in vulnerable regions like Japan and California [1407]. Similarly, reconstructions of past climate from ice cores in Antarctica have revealed how small changes in atmospheric carbon dioxide were linked to major glacial cycles—insights that now guide our understanding of human-driven climate change [1408].
Given the interconnected nature of Earth’s systems, research in earth sciences is typically categorized by major environmental domains: the solid Earth, water systems, the atmosphere, and space. Each category relies on specialized observational and analytical techniques developed across centuries of geoscientific exploration [1409,1410,1411,1412]. Below, we outline the primary research tasks and traditional methods used in these four domains:
Solid Earth. This category involves studying Earth’s interior, crust, and surface features to understand geological processes such as mountain building, earthquakes, and volcanic activity. Geologists use tools like field mapping [1413,1414,1415], rock sampling [1416], and seismic imaging [1417,1418] to probe the planet’s subsurface. Techniques such as radiometric dating [1419] reveal the age of rocks and events, while plate tectonic theory [1420] provides a framework for explaining continental drift, subduction zones, and the formation of Earth’s landforms.
Water Systems. This area includes both freshwater and marine environments. Hydrologists and oceanographers study the movement, distribution, and quality of water using instruments like stream gauges, sonar systems, and deep-sea submersibles [1421,1422,1423]. Ocean circulation, sea level rise, and the global water cycle are key focus areas. Research methods include remote sensing, water sampling, and the deployment of autonomous floats to measure temperature, salinity, and current flow. These investigations are crucial for understanding climate change, water resource management, and marine ecosystems [1424].
Atmosphere. The atmospheric sciences examine weather patterns, climate dynamics, and the composition of the air enveloping Earth. Meteorologists use weather stations, balloons, radar, and satellites to observe atmospheric conditions [1425]. Climate scientists build numerical models based on physical laws [1426] to predict weather and analyze long-term climate trends. Historical data from sources like ice cores [1427] and tree rings [1428] help reconstruct past climates. Understanding the atmosphere is vital for forecasting extreme events, assessing air quality, and responding to global warming [1429,1430].
- Outer Space. Also called planetary or space science, this domain explores celestial bodies and their relevance to Earth. Techniques include astronomical observation, space probe missions, spectroscopy, and sample return analysis [1431]. Comparative studies of planets, moons, and asteroids help scientists infer Earth’s formation and evolutionary history. Data from missions like those to Mars or the Moon, along with meteorite analysis, deepen our understanding of planetary geology, habitability, and solar system dynamics [1432,1433,1434].
5.5.1.2 Introduction to Civil Engineering
Civil engineering is an engineering discipline dealing with the design, construction, and maintenance of physically or naturally built environment, including roads, bridges, residence and pipelines [1435,1436]. It is essentially the application of physical and scientific principles for addressing real-world challenges like climate crisis, encompassing fields like structures, material science, geology, soils, hydrology, environmental science, mechanics and other fields [1437]. In simpler terms, civil engineering is about the management of buildings and infrastructures.
Civil engineering is a broad field composed of various branches, ranging from geotechnical engineering to urban planning. To better organize and understand the scope of civil engineering, by integrating information from relevant literature [1438,1439], we have summarized the common research tasks and corresponding classic solutions in civil engineering as following categories:
Structural Design Optimization. Structural optimization aims to find the best arrangement of structural components to meet requirements in terms of size, shape, topology and other aspects [1440]. Early research on structural optimization proposed mathematical programming and numerical search techniques [1441,1442], which are still one of the most commonly applied approaches. Recently, metaheuristic methods have become popular due to their suitability in combinational optimization problems [1443]. However, they also suffer from high complexity and inadequacy for high-dimensional problems [1444,1445]. A substantial number of studies have been proposed to address these problems [1446,1447].
Structural Health Monitor (SHM). SHM refers to the process of structural data collection, assessment and damage detection in order to test the safety risks of new structures and the remaining time of existing structures [1448,1449]. With the rapid development of IoT and wireless communication techniques, civil engineers usually use smart sensors to access a variety of structural parameters, such as mechanical and optical parameters [1450,1451]. Anomaly detection methods are commonly adopted for structural assessment and detection, including response-based [1452,1453], reliability-based [1454], feature-based [1455] and computer vision methods [1456,1457].
Material Behavior Modeling. Based on historical data, the behavior of construction materials in different environments can be predicted, so that civil engineers can make informed decisions about material selection and construction arrangement [1438]. Different materials usually adopts different behavior modeling strategies. For example, non-destructive experimental assessment methods are commonly used for concrete strength estimation [1458,1459], numerical analysis methods and physical models are developed for soil behavior modeling [1460,1461], while finite element method is widely adopted to fill the heterogeneity gap among various composite materials [1462,1463].
Product Behavior Modeling. To ensure rational resource allocation and decision-making, it is also necessary to study the behavior of built systems, such as canal systems and transportation systems [1438]. Machine learning algorithms have been widely applied to product behavior modeling for a long time, where commonly used traditional methods include SVM, fuzzy logic, Markov chains and evolutionary computation [1464,1465,1466]. Recently, cutting-edge spatial-temporal network analysis techniques like Graph Neural Networks have received growing attention [1467,1468].
5.5.1.3 Current Challenges
Earth sciences and civil engineering are deeply interconnected disciplines essential for understanding and shaping the physical world we live in. Earth sciences seek to decipher the natural processes that govern our planet, while civil engineering applies this knowledge to construct resilient, functional infrastructure. These fields play a vital role in addressing global issues such as natural disasters, climate adaptation, urbanization, and sustainability. Despite major advances in observational tools, modeling, and design techniques, both domains continue to face substantial challenges stemming from the complexity, unpredictability, and dynamic nature of Earth systems and human-built environments.
Still Hard with LLMs: The Tough Problems.
- Natural System Complexity and Uncertainty. Earth systems are governed by highly nonlinear, interdependent physical processes—from tectonic shifts and groundwater flow to atmospheric circulation and sea-level dynamics [1404,1409]. Predicting these phenomena accurately requires not only long-term observational data but also high-resolution numerical simulations. Civil engineers must incorporate these uncertainties into infrastructure design—such as accounting for variable soil conditions or future climate extremes. However, LLMs, while capable of interpreting related literature or summarizing best practices, lack the ability to model dynamic systems governed by partial differential equations or simulate stochastic environmental behavior. Capturing such processes requires specialized physics-based modeling and empirical calibration, far beyond the capacity of current text-based AI systems.
- Data Gaps and Ground Truth Limitations. Earth and civil systems often involve inaccessible or hazardous environments (e.g., deep subsurface, remote ocean basins, or aging underground infrastructure), where direct measurement is difficult or incomplete [1416,1421]. As a result, both domains suffer from sparse and noisy datasets that hinder model calibration and decision-making. For example, limited geological borehole data may be insufficient for accurate subsurface mapping, and historical infrastructure records may be missing or outdated. While LLMs can help process available documents, they cannot compensate for missing sensor data, field measurements, or satellite coverage, which are essential for building reliable models or simulations.
- Physical Implementation and Experimental Validation. In civil engineering especially, the ultimate test of a solution lies in its physical realization—constructing structures, monitoring performance, and conducting stress tests under real-world conditions [1450,1459]. Earth scientists similarly depend on empirical fieldwork, such as drilling ice cores or deploying seismographs. These tasks involve tools, logistics, and materials that LLMs cannot access or control. While LLMs may assist in designing monitoring protocols or reviewing standards, they cannot carry out experiments or validate hypotheses in the field.
- Cross-scale Integration. A recurring challenge in both fields is integrating phenomena across vastly different spatial and temporal scales—for instance, linking microscale soil composition to large-scale slope stability, or connecting millennial-scale climate changes to today’s hydrological models [1405,1426]. This requires sophisticated multi-scale modeling, often coupling discrete and continuous frameworks, something well beyond the representational capacity of LLMs trained primarily on textual corpora. Such integration typically involves customized simulations and domain-specific algorithms informed by physics and engineering principles.
- Regulatory and Ethical Constraints. Civil engineers must design within strict regulatory, economic, and ethical frameworks—ensuring safety, sustainability, and community impact are considered [1448]. Earth scientists face ethical concerns in interventions like geoengineering or resource extraction. While LLMs can provide policy summaries or ethical perspectives from the literature, they cannot weigh trade-offs, assess context-specific risks, or make normative judgments. These decisions require human oversight, societal debate, and legal frameworks beyond what AI can resolve.
Easier with LLMs: The Parts That Move.
Despite these limitations, there are many areas in which LLMs can meaningfully assist researchers and practitioners in earth sciences and civil engineering—particularly in tasks related to knowledge synthesis, documentation, and early-stage design:
- Literature Review and Standards Summarization. Both fields rely on vast and highly technical bodies of knowledge, including regulatory documents, geological surveys, engineering design codes, and academic papers. LLMs can significantly streamline the literature review process by summarizing scientific reports, extracting key metrics, and comparing standards across regions [1251,1253]. For instance, an LLM could help an engineer quickly retrieve the seismic design requirements for bridges in a specific country or summarize recent advances in landslide risk prediction models.
- Interpreting Sensor and Field Reports. As structural health monitoring (SHM) and geoscience increasingly adopt IoT and sensor networks, LLMs can help translate raw sensor metadata or technician logs into structured, actionable insights [1448,1456]. They can assist in automatically annotating inspection reports, flagging anomalies in sensor readings, or identifying trends across multiple sources, especially when integrated with domain-specific tools.
- Assisting in Conceptual and Parametric Design. In civil engineering tasks such as structural layout or materials selection, LLMs can be useful in generating design suggestions, proposing parametric options, or reviewing existing case studies. For example, they can draft potential designs based on textual constraints or help identify suitable sustainable materials from engineering databases [1440,1447].
In conclusion, earth sciences and civil engineering face enduring challenges related to physical constraints, empirical validation, and systemic complexity. While LLMs are unlikely to replace domain experts or physical experimentation, they are emerging as valuable assistants in literature synthesis, document interpretation, early-stage design, and communication—accelerating workflows and enhancing accessibility. The future of these disciplines may lie in effective collaboration between human ingenuity, physical modeling, and intelligent AI support systems.
Figure 22.
The pipelines of Earth sciences and civil engineering.
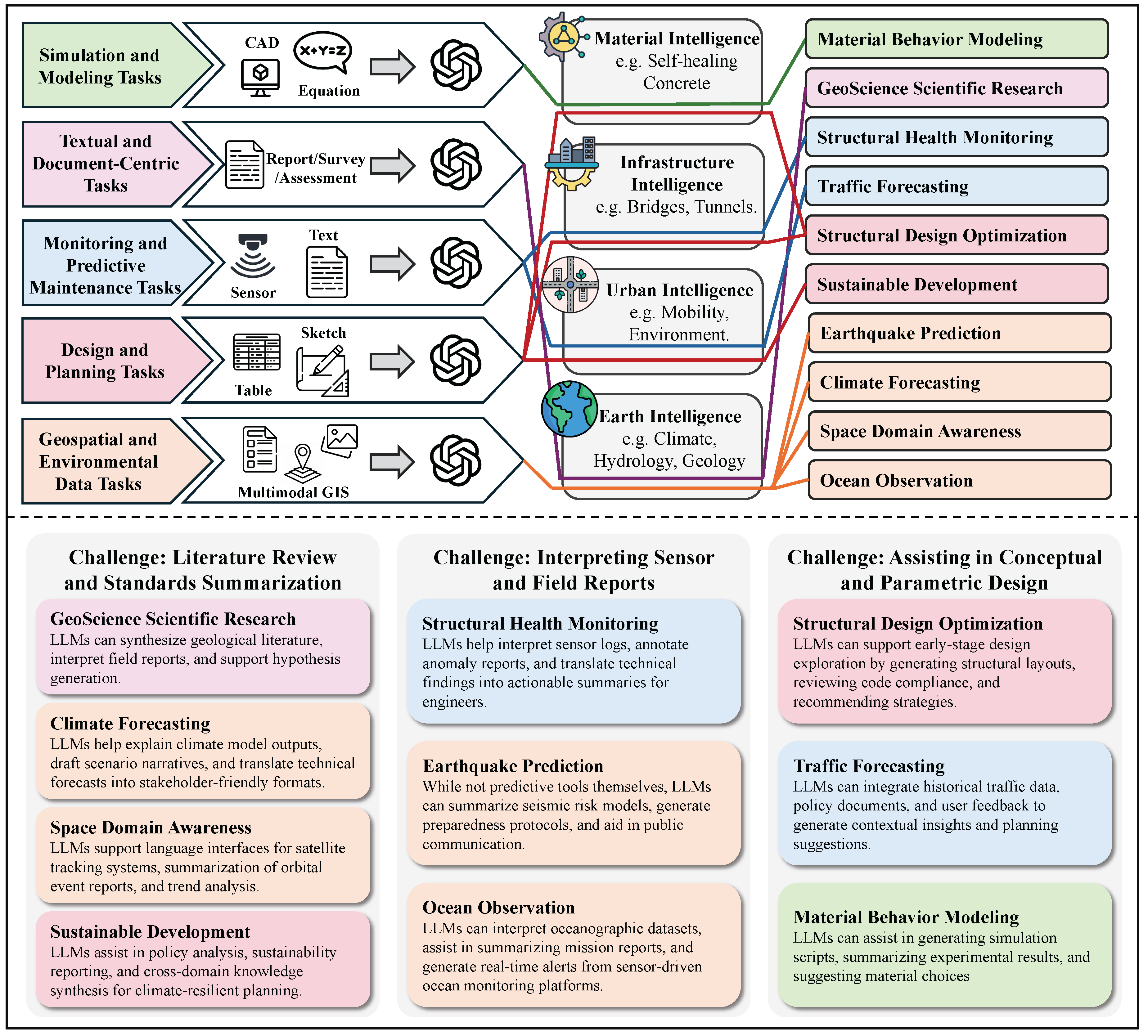
5.5.1.4 Taxonomy
Research in Earth sciences and civil engineering encompasses a broad array of problems, spanning from natural system modeling to infrastructure design and monitoring. While traditionally organized by disciplinary boundaries (e.g., geology, hydrology, structural engineering), these fields also share deep methodological similarities, particularly in their use of spatial data, physical modeling, and complex environmental measurements [1410,1438]. Importantly, many tasks require interpreting unstructured field reports, processing geospatial sensor data, or modeling physical systems under uncertainty—domains where the potential role of LLMs is still being explored. To support more effective use of LLMs in these fields, we propose a taxonomy grounded in computational characteristics rather than disciplinary labels. This perspective helps reveal where LLMs can directly contribute, where they serve supporting roles, and where traditional numerical modeling remains essential. It offers three practical advantages:
- Model compatibility: Clearly distinguishes tasks suitable for LLMs (e.g., text synthesis) versus those requiring numerical simulation or high-dimensional optimization [1276].
- Cross-domain transferability: Highlights common computational structures across environmental and infrastructure problems, facilitating the reuse of AI workflows.
- Pipeline orchestration: Supports hybrid modeling pipelines where LLMs coordinate, explain, or augment scientific and engineering workflows using diverse data types.
Geospatial and Environmental Data Tasks. These tasks involve interpreting and reasoning over georeferenced data from remote sensing, field instruments, or environmental simulations. Examples include analyzing satellite images to detect deforestation, processing LiDAR or GIS data to monitor urban growth, and interpreting topographic or hydrological maps for flood modeling [1421,1424]. Inputs typically consist of raster, vector, or tabular geospatial datasets; outputs may be spatial predictions (e.g., landslide zones), environmental classifications, or time-series trends. Although LLMs are not inherently designed for numerical geospatial data, they can assist in generating geospatial analysis scripts (e.g., in Python/ArcPy or QGIS), interpreting metadata or documentation, and explaining geostatistical outputs. Additionally, LLMs may serve as interfaces between users and GIS tools, enabling natural language-based queries about geospatial datasets [1279].
Engineering Simulation and Physical Modeling Tasks. This category includes tasks that rely on numerical simulations to model physical phenomena, such as seismic wave propagation, structural stress analysis, fluid flow in soil, or traffic modeling. Inputs often include CAD models, finite element meshes, or system equations; outputs range from displacement fields to safety margins or failure probabilities [1426,1459,1462]. These tasks are typically computationally intensive and governed by physics-based principles (e.g., Navier–Stokes or elasticity theory), which are poorly suited to LLMs. However, LLMs can still assist by auto-generating simulation code, translating natural-language specifications into solver-ready scripts (e.g., ANSYS or Abaqus macros), and summarizing simulation results for decision-makers. In this sense, they act as connective tissue between engineering intent and computational execution.
Textual and Document-Centric Tasks. A substantial portion of Earth science and civil engineering knowledge is encoded in unstructured documents: geological surveys, environmental impact assessments, building codes, inspection reports, and academic literature. Tasks in this domain include extracting key findings from regulatory reports, summarizing design guidelines, or generating technical proposals [1438,1448]. Inputs are typically plain text, PDFs, or semi-structured tables, with outputs including summaries, structured annotations, or natural language responses. These tasks represent the natural strength of LLMs. For instance, an LLM can assist an engineer in drafting a permit application by synthesizing local zoning codes and environmental constraints or help a geologist extract lithology descriptions from archival drilling logs.
Monitoring and Predictive Maintenance Tasks (Hybrid). This class includes tasks where sensor data and models are combined to anticipate failures or evaluate performance. Examples include structural health monitoring (e.g., identifying bridge fatigue), monitoring dam stability, predicting groundwater level fluctuations, or evaluating pavement degradation from satellite imagery and sensor inputs [1423,1451,1456]. Inputs span across time-series sensor data, weather models, images, and inspection text logs. Outputs typically include alerts, risk scores, or suggested interventions. While predictive modeling often depends on statistical learning or physics-based simulations, LLMs can play important interpretive roles: translating sensor anomalies into natural-language diagnostics, integrating multi-source logs into incident summaries, or suggesting maintenance schedules by referencing historical case reports.
Design and Planning Tasks. These tasks involve defining, reasoning about, and communicating high-level engineering plans—such as choosing structural layouts, evaluating site suitability, or balancing cost and safety trade-offs in urban planning. Inputs include sketches, specifications, tabular datasets, and design goals, while outputs may be schematic proposals, material selections, or scenario comparisons. Though the core design logic is domain-driven and often quantitative, LLMs can support rapid ideation, generate initial alternatives, or provide justifications based on case studies and standards [1440,1447]. They can also assist in multi-objective trade-off analysis, especially when paired with simulation tools and optimization libraries.
Table 53.
Applications and insights of LLMs in Earth Sciences and Civil Engineering
| Task Category | LLM Application Areas | Use Case-Inspired Research Question | Key Insights and Contributions | References |
|---|---|---|---|---|
| Geospatial and Environmental Data Tasks | Geospatial Data Analysis | Can LLMs help interpret and interact with complex geospatial datasets like GIS and remote sensing imagery? | LLMs and VLMs assist geospatial tasks; specialized models (e.g., RSGPT, GeoChat, EarthGPT) improve visual understanding and accessibility. | [1469,1470,1471,1472,1473] |
| Tool Selection and Program Generation | How can LLMs act as decision-makers to select appropriate geospatial tools and automate workflows? | Agents like GeoGPT, GeoLLM-Engine, and GeoAgent automate tool selection and program generation, enhancing task execution and reducing user burden. | [1474,1475,1476,1477] | |
| Engineering Simulation and Physical Modeling Tasks | Simulation Code Generation | Can LLMs generate accurate simulation scripts for civil and earth science modeling tasks? | Early efforts (e.g., Eplus-LLM, HydroSuite-AI) enable natural language-driven code generation; challenges remain in handling complex, interdependent systems. | [1478,1479,1480,1481] |
| Automated Simulation Interfaces | Can LLMs streamline simulation configuration and execution through natural language interaction? | Systems like ChatSUMO and GPT-based simulation platforms show early success, but complex modeling remains difficult. | [1482,1483] | |
| Textual and Document-Centric Tasks | Domain-Specific Knowledge Extraction | How can LLMs assist in extracting and structuring information from geoscientific and engineering texts? | Customized LLMs (e.g., GeoBERT, BB-GeoGPT) enhance information retrieval and QA; hallucination and data quality issues persist. | [1484,1485,1486] |
| Compliance Checking and Reporting | Can LLMs automate regulatory compliance checks and generate inspection reports? | Frameworks like AutoRepo and LLM-FuncMapper automate compliance interpretation; combining multimodal LLMs and ontology models enhances performance. | [1487,1488,1489] | |
| Monitoring and Predictive Maintenance Tasks (Hybrid) | O&M Data Processing and Optimization | Can LLMs support monitoring and maintenance through real-time data analysis and strategy optimization? | RAG-enhanced multi-agent systems and LLMs enrich BIM models, optimize energy usage, and support digital twin development. | [1490,1491,1492,1493] |
| Digital Twin Integration | How can LLMs contribute to the creation and operation of digital twins for infrastructure health monitoring? | LLMs facilitate real-time data collection and simulation in digital twins across sectors like transport, railways, and water networks. | [1494,1495,1496] | |
| Design and Planning Tasks | Building and Infrastructure Design | Can LLMs assist engineers and planners in creating optimized design solutions from natural language inputs? | Early systems like NADIA and PlanGPT use LLMs for building simulation and urban planning; still limited in complex layout and sequential action handling. | [1497,1498,1499] |
| Urban and Transportation Planning | How can LLMs support participatory urban design and traffic system optimization? | PlanAgent and TrafficGPT show LLMs’ potential in participatory planning and decision support; broader adoption needs further research. | [1500,1501,1502] |
5.5.2. Geospatial and Environmental Data Tasks
Geospatial and environmental data analysis focuses on data processing and interpretation, in order to assist researchers in data understanding. The primary inputs to these tasks are geospatial datasets, such as GIS data, satellite images and hydrological maps. These datasets can not be easily understood by humans. For instance, GIS data is usually in form of vector and raster, which is not as intuitive as natural language. Besides, despite the existence of APIs for data processing, practitioners are still plagued by challenges such as the accessibility of software and technological complexity. Interacting with these data through textual information can make them more accessible and understandable for researchers, which is essential for efficient and automated data processing.
Researchers have explored the incorporation of LLMs into the processing and analysis of geospatial data. The initial efforts mainly use LLMs to process text-based inputs to assist GIS tasks [1469,1470,1503,1504,1505]. For instance, [1469] introduced Autonomous GIS, an LLM-powered GIS to solve geospatial tasks including toponym recognition, location description and time series forecasting. [1470] introduced GEOGALACTICA to compensate geoscience-specific knowledge to general LLMs. These studies demonstrated the potential of LLM in geospatial data tasks. Considering that visual information is critical in the analysis of certain geospatial data like remote sensing data, the application of large vision-language models (VLMs) has emerged as a promising research direction. However, due to the unique characteristics of remote sensing images, such as high resolution, diverse scales, and complex acquisition angles, existing general VLMs perform poorly in remote sensing tasks. To address these issues, several studies have been conducted. Pioneering work RSGPT [1471] constructed a high-quality human-annotated Remote Sensing Image Captioning dataset (RSICap) and introduced RSGPT, a GPT-based model fine-tuned on RSICap with advanced performance in image captioning and visual question-answering tasks. RS-LLaVA [1472] improved LLaVA for remote sensing imagery and created the RS-instructions dataset by integrating four single-task datasets related to captioning and VQA. GeoChat [1506] further built a novel RS multimodal instruction-following dataset and introduced the first versatile remote sensing VLM with multitask conversational capabilities, which can answer image-level and region-specific queries and generate visually grounded responses. EarthGPT [1473] integrated a visual-enhanced perception mechanism, a cross-modal mutual comprehension approach, and a unified instruction tuning method to come up with a universal multimodal LLM for multisensor remote sensing image comprehension.
Despite the remarkable achievements of these models in various geospatial tasks, the challenges within geospatial and environmental data analysis lie not only in data comprehension, but dadata selection and the utilization of professional tools like APIs like APIs. Recent studies attempt to convert LLMs from an operator to a decision-maker [1474,1475]. In other words, they leverage the reasoning capability of LLMs to assess appropriate tools and generate executable programs and generate executable programs based on given requirements before addringessing downstream tasks. As one of the earliest studies, GeoGPT [1474] is designed to automate geospatial tasks. It uses an LLM to understand user demands from natural language descriptions and then selects and executes appropriate GIS tools from a pre-defined pool to conduct procedures like data collection, processing, and analysis. Change-Agent [1507] can follow user instructions to comprehensively interpret and analyze remote sensing change, which integrates a multilevel change interpretation (MCI) model as the toolbox and an LLM as the "brain". GeoLLM-Engine [1475] further equips more geospatial API tools and external knowledge bases, enabling agents to handle more complex tasks. [1508] designed a GIS agent framework for autonomous geospatial data retrieval, consisting of a data source index and a handbook inventory. It uses an LLM as the decision-maker to select appropriate data sources from a pre-defined list and generate programs to fetch data. To further enhance agents’ ability to process complex sequential tasks and avoid hallucination caused by the lack of domain knowledge, [1476] proposed a novel interactive framework GeoAgent, integrating a code interpreter, static analysis, and Retrieval-Augmented Generation (RAG) within a Monte Carlo Tree Search (MCTS) algorithm; [1477] proposed a Chain-of-Programming (CoP) framework, decomposing API selection and code generation process into five separate steps and incorporating external knowledge base and user feedback; [1509] proposed GeoCode-GPT specifically for geospatial data analysis code generation, which is pre-trained and fine-tuned on their built GeoCode geospatial code corpus.
In summary, the evolution of geospatial LLMs has clearly progressed from "workers" to "experts". They are required to formulate a complete solution specific to user demands, and meanwhile generate executable programs to obtain the expected outcomes, which significantly helps improve data processing efficiency. However, they still suffer from limited domain knowledge, inferior data quality and inability to handle complex sequential reasoning.
5.5.3. Engineering Simulation and Physical Modeling Tasks
Engineering simulation and physical modeling also focus on data understanding and interpretation. Compared with geoscience tasks, the primary inputs are in more complex forms, like CAD models and system equations. It is highly challenging to achieve automatic simulation with LLMs, and the reasons are presented as follows: (1) Numerical modeling of structures is complex, involving numerous inputs, simulation configurations and closely coupled sets of equations in calculation [1478,1479]. Even the slightest error can not be tolerated in this process. (2) General LLMs only have a vague understanding of the physical and dynamic rules of the real world [1510]. However, the translation between natural language and simulation code is essential for reducing modeling efforts and facilitating automated data and modeling workflow.
Although significant progress has been made in LLM-based code generation, LLM-assisted numerical simulation for earth science and civil engineering is still under-explored. Relevant research can be divided into two branches, simulation code generation and user interface for automatic simulation. Existing LLM-based simulation code generation are usually aimed at specific applications [1479,1480]. For instance, [1478] proposes an automated building modeling platform Eplus-LLM that uses a fine-tuned T5 to translate natural language descriptions of buildings into EnergyPlus models. HydroSuite-AI [1480] is designed for hydrology and environmental science research, which integrates HydroLang, HydroCompute, and HydroRTC open-source libraries to help researchers generate code snippets. GeoSim.AI [1481] designs a suite of AI assistants for numerical simulations in geomechanics, translating natural language and image inputs into simulation parameters and scripts. On the other hand, there are other researchers using natural language descriptions to guide LLM and conduct automatic simulation. [1482] proposes a GPT-based building performance simulation system that integrates simulation engines and data analytics in the GPT environment. [1483] designs an LLM-based agent ChatSUMO to simplify SUMO simulations for traffic modeling, which can convert user text inputs into relevant keywords for running Python scripts and generating traffic simulation scenarios.
While these advances are promising, only relatively simple modeling cases have been studied, and further enhancement is needed for more complex practical modeling. There are also concerns about model transparency and inability to handle interdependent user requirements. Nonetheless, the role of LLMs as simulation assistants is becoming increasingly practical in early prototyping studies.
5.5.4. Textual and Document-Centric Tasks
Textual and document-centric tasks mainly focus on knowledge extraction and translation for unstructured textual data. Knowledge extraction aims to extract required information from textual data like government documents and inspection reports, while knowledge translation aims to translate raw text data into other data forms to make them more understandable by humans or computers.
Although some studies have suggested that LLMs encoded preliminary geoscience knowledge from their training corpora [1511,1512,1513], the lack of domain knowledge still hinders the application of LLMs in earch science and civil engineering due to the unique properties of relevant textual data. Therefore, various strategies have been proposed to adapt general LLMs to achieve domain-specific question answering and knowledge extraction [1470,1503]. For instance, GeoBERT [1484] retrained BERT with geoscientific record dataset, which was further fine-tuned for geoscience question answering and query-based summarization task. [1485] proposed the first-ever LLM specialized for ocean science tasks. [1514] constructed a new pretraining dataset BB-GeoPT with GIS-specific knowledge, retrained an open-source LLM and obtained BB-GeoGPT, an LLM for GIS domain. [1486] customized an RAG-enhanced LLM to efficiently process geological document information.
Compliance check is one of the knowledge translation tasks that receives growing attention [1488,1489,1515], which aims to ensure that built structures or environment are free of safety risks and consistent with design regulations and industrial standards. The application of LLMs in automatic compliance check is still limited. Pioneering work [1487] proposes LLM-FuncMapper to identify predefined atomic functions for regulatory clauses interpretation with the help of LLMs. [1516] adopts GPT-based models and achieves automatic compliance check through prompt engineering. [1517] presents the AutoRepo framework, which combines unmanned vehicles and multimodal LLMs for automated construction inspection report generation. To further automate regulatory text processing and improve efficiency, [1488] integrates deep learning models, LLMs and ontology knowledge models, which are responsible for preliminary text classification, structured information extraction and compliance check respectively. Textual data only accounts for a small portion of all geoscience and urban science data, and there are several cross-modality knowledge translation tasks have been discussed, including urban profiling [1518] and traffic report summarization [1519].
In summary, the introduction of LLMs has made significant progress in textual and document-centric tasks, which has laid foundation for further development of domain-specific foundation models. However, existing methods still suffer from hallucination and the lack of high-quality training data.
5.5.5. Monitoring and Predictive Maintenance Tasks
Monitoring and maintenance tasks are responsible for the health management of environment and built structures, which is a critical stage of the lifecycle of bridges, residence and other research objects in civil engineering and earth science. Most of these tasks are within the scope of Operation and Maintenance (O&M) [1520,1521]. O&M is a complex systematic process, including data collection and analysis, performance prediction, condition assessment, strategy development and emergency response. There are also some other tasks within the construction stage, targeting for newly built structures. These procedures are used to be labor-intensive, which is inefficient and prone to errors. The introduction of LLMs into data processing and real-time inspection can significantly reduce the cost of time and data compared to traditional data mining methods.
For newly built structures, automatic compliance check is a critical step of construction inspection, in order to make sure the structures are free of safety risks and consistent with design regulations and industrial standards. Detailed introduction can be referred to Section 5.5.4. The application of LLMs is more extensive in O&M, which focuses more on system optimization. Some existing works tend to enhance traditional O&M tools with LLMs [1491,1522]. For example, [1523] proposes an automated data mining framework that combines maximal frequent itemset mining and GPT to detect energy waste patterns and optimize energy use. [1490] proposes an RAG-enhanced LLM-based multi-agent framework to deal with unstructured building data and achieve automatic energy optimization. [1491] using Semantic Textual Similarity and fine-tuned LLMs to automatically enrich Building Information Models (BIM) and assist building energy performance simulation. On the other hand, digital twin technologies have been widely applied to building maintenance and environmental monitoring [1524,1525] , where LLMs can potentially make significant contributions to the construction of digital twin [1492]. For instance, [1494] proposes an LLM-based digital twin solution to collect real-time human preference data and enhance the optimization of human-in-the-loop systems within the context of Cyber-Physical Systems and the Internet of Things (CPS-IoT). [1493] explores LLM-driven sensor data acquisition protocol to achieve automatic capturing of real-time sensor data based on the requirements of user inputs. LLMs can also contribute to digital twin construction for railway health monitoring [1495], smart transportation [1526], water distribution network management [1496] and many other applications.
Overall, researchers have made initial attempts to apply LLM techniques to monitoring and maintenance tasks, but current research still faces challenges like reliance on high-quality training data and limited transferability. The few-shot learning power of LLMs should be further exploited, and more application scenarios are worth exploring.
5.5.6. Design and Planning Tasks
Design and planning are the beginning of lifecycles of both individual buildings and complex systems. The core of design and planning is trade-off. For building structural design, engineers aim to achieve the trade-off among safety, cost, aesthetics and other constraints. As for more complex urban planning, more requirements need to be considered, such as functional section arrangement and commute. LLMs can function as both data analyzers and user interface in this stage [1478,1497,1527]. On the one hand, LLMs can analyze vast amount of collected data and come up with design verifications and recommendations. On the other hand, given user feedback and requirements, LLMs can generate inclusive solutions and conduct optimization accordingly.
The applications of LLMs in building structural design and optimization is still under-explored. LLMs have been applied to automatic design in this stage. In automatic design, attempts have been made to utilize LLMs as user interface, where LLMs accept user inputs and generate architectural details and simulation code. For instance, [1497] uses an LLM as the core controller, which can interpret engineers’ natural language descriptions and translate them into executable simulation code for shear wall structural design and optimization. [1498] proposes the NADIA framework, which integrates LLMs and BIM authoring tools to enable architectural design consulting and detailing via natural language. However, the application of LLMs in other design tasks like building layout planning and design rendering has not been explored [1528].
As for urban planning, researchers have shown growing interests in the development of urban foundation models to achieve urban general intelligence [1529]. Although general LLMs have difficulty in dealing with urban science related textual information due to their unique properties in text style and knowledge requirements [1499], recent works have proved that LLMs can provide urban planners with valuable insights through proper fine-tuning. To name a few, PlanGPT [1499] is the first specialized LLM for urban and spatial planning, which proposes a customized embedding model Plan-Emb and hierarchical search strategy Plan-HS for accurate information extraction and an LLM-based PlanAgent for tool invocation and information integration. [1500] explores the application of LLMs in participatory urban planning, proposing an LLM-based multi-agent collaboration framework and a fishbowl discussion mechanism to simulate the communication between planners and residents. [1501,1502,1530] explores the applications of LLMs in transportation system, utilizing natural language prompts and the reasoning ability of LLMs to analyze traffic data and provide decision support for traffic control. TrafficGPT [1501] integrates LLMs and traffic foundation models, while Open-ti [1502] bridges the gap between academic research and industry in traffic simulation and control via ChatZero control agent.
Overall, the introduction of LLMs into design and planning tasks have been proved to significantly reduce the cost of time and human labor. However, researchers have only conducted preliminary explorations on this topic. More application scenarios could be explored, and the agents’ inability to handle complex sequential actions need to be addressed in future research [1502].
5.5.7. Benchmarks
The application of LLMs in earth science and civil engineering is still quite limited, and available public benchmark datasets can not meet the requirements of various tasks in this field. Case studies and human evaluation are widely adopted for the evaluation of some domain-specific tasks, such as numerical simulation, compliance check, maintenance tasks and structural design. No effort has been made to construct a comprehensive benchmark for them. Therefore, in this section, we only summarize benchmarks for geoscientific understanding and urban planning. We will also select several representative benchmarks from Table 54 for in-depth discussion. For each chosen benchmark, we will describe its scope and data form, and then survey the performance of representative LLMs on it.
GeoBench. GeoBench is the first benchmark for evaluating the ability of LLMs to understand and utilize geoscience knowledge to solve geoscientific problems. The authors collected pure text questions related to geology, geography and environmental science from NPEE and APTest examinations, including 1,578 objective questions and 939 subjective questions, which are represented by multi-choice questions and essay questions respectively. Objective questions are evaluated based on accuracy, while subjective questions are evaluated based on perplexity and GPTScore [1531]. Benchmark results are listed in Table 55.
RSIEval. RSIEval is constructed to assess VLMs in multi-modal remote sensing tasks, especially remote sensing specific image captioning and VQA. Images from the validation set of DOTA-v1.5 are selected, divided into 512×512 patches, and 100 patches are picked for manual annotation. It contains 100 high-quality image-caption pairs and 936 diverse image-question-answer triplets. The questions used for VQA task are further divided into four categories. Object-related questions include presence, quantity, color, absolute position, relative position, area comparison, and road direction. Image-related questions involve high/low resolution and panchromatic/color images. Scene-related questions are about the main theme/scene and urban/rural scenes. Reasoning-related questions are like inferring the season of image capture, the dryness or wetness of the area, the moving speed of a ship, and the state of the water surface. Compared with previous RSVQA datasets, RSIEval provides a more diverse set of questions and answers, enabling a more comprehensive assessment. Benchmark results are listed in Table 56 and Table 57.
GeoCode. GeoCode benchmark is used to evaluate LLM-based geospatial data analysis method, measuring whether LLMs can follow complex task instructions and generate proper code for data analysis. It contains 18,148 single-turn tasks and 1,356 multi-turn tasks, involving 2,313 function calls from 28 widely used Python libraries across 8 key domains. These tasks and function calls reflect complex instructions and implementation logic, requiring models to have strong compositional reasoning abilities. The evaluation of single-turn tasks focuses on the accuracy of function calls and the success rate of individual tasks, while multi-turn task evaluation emphasizes the task completion rate. Benchmark results are listed in Table 58. Note that the authors did not conduct comprehensive evaluation for multi-turn tasks, and we only display single-turn task evaluation results here.
PlanGPT. PlanGPT benchmark is constructed to examine LLMs’ capabilities in urban planning from different dimensions. Evaluation data is collected from multiple sources, including spatial planning documents from different administrative levels and authoritative textbooks. Four downstream tasks are considered for evaluation, including text generation, text style transfer, information extraction, and text evaluation. Text generation aims to evaluate the quality of the urban planning documents generated by the model; Text style transfer aims to evaluate the model’s ability to convert text into the urban planning style; Information extraction aims to evaluate the model’s ability to extract key information from text; Text evaluation is designed to evaluate the model’s ability to evaluate urban planning proposals. Benchmark results are listed in Table 59.
Summary. In tasks related to textual data, ChatGPT and LLaMA consistently achieve satisfactory performance. In image-based tasks, LLaVA consistently outperforms other general LLMs. Besides, CodeGemma is capable of handling code generation tasks in the field of geoscience. However, the aforementioned benchmarks can not sufficiently evaluate more complex situations where LLMs are treated as user interface for knowledge retrieval, data analysis and numerical simulation. GeoLLM-Engine makes initial and valuable attempts by categorizing various downstream tasks based on user intents, while it is highly limited by the pre-defined system behavior functions. Further effort should be invested to achieve more extensive evaluation.
5.5.8. Discussion
Opportunities and Impact. LLMs are beginning to reshape workflows in earth sciences and civil engineering, offering new capabilities for knowledge extraction, document synthesis, conceptual design, and early-stage simulation support. As shown across geospatial data processing [1469,1474], engineering simulation [1478,1481], document-centric tasks [1486,1499], monitoring and predictive maintenance [1520,1524], and design and planning [1497,1529], LLMs significantly lower the barriers to access technical knowledge, accelerate tedious documentation tasks, and enhance preliminary design ideation.
By serving as interfaces between domain experts and computational tools, LLMs can democratize access to specialized workflows, enabling faster prototyping, enhanced interdisciplinary collaboration, and more accessible education and training pipelines. Their ability to interpret natural language specifications, summarize complex regulatory documents, and support tool-assisted reasoning holds particular promise for infrastructure resilience, environmental monitoring, and sustainable urban development. Moreover, the emergence of domain-specific foundation models (e.g., PlanGPT [1499], EarthGPT [1473]) suggests that specialized LLMs could become integral components of future scientific and engineering ecosystems.
- Challenges and Limitations. Despite these opportunities, fundamental limitations persist when applying LLMs to earth sciences and civil engineering. Physical system modeling—ranging from seismic simulation to hydrodynamic forecasting—requires accurate, high-dimensional numerical computations governed by partial differential equations [1426]. Current LLMs lack the inductive biases, precision, and physical fidelity needed for such tasks, relegating them to supporting roles in workflow orchestration rather than core simulation.
Similarly, data sparsity and uncertainty, particularly in hazardous or inaccessible environments [1416,1421], cannot be resolved through language-based reasoning alone. Real-world challenges like infrastructure resilience under extreme events or predictive maintenance of aging systems demand empirical validation and physical fieldwork beyond the reach of AI models.
Other notable concerns include the lack of transparency and verifiability in LLM outputs—particularly problematic for compliance verification and safety-critical applications [1489,1515]. Furthermore, issues such as regulatory complexity, ethical trade-offs in interventions (e.g., resource extraction, geoengineering), and societal value judgments require human oversight, collective deliberation, and domain-specific expertise that LLMs cannot replace.
- Research Directions. To maximize the impact of LLMs while respecting domain constraints, several promising research directions emerge:
- Domain-Specific Fine-Tuning and Hybridization. Building specialized LLMs for Earth sciences and civil engineering—such as those incorporating geospatial, structural, and regulatory corpora [1486,1529]—can enhance factual grounding and reasoning capabilities, particularly when paired with physics-based simulators.
- Ethical AI Frameworks for Infrastructure and Environmental Decisions. Future research must address bias mitigation, explainability, regulatory compliance, and human-centered design when deploying LLM-augmented systems in critical infrastructure and environmental governance [1489].
Conclusion. LLMs offer powerful new tools for supporting and accelerating workflows in earth sciences and civil engineering, particularly in domains related to knowledge synthesis, document understanding, and conceptual design. However, realizing their full potential requires carefully integrating them into hybrid pipelines alongside empirical observation, physical modeling, and human expertise. Moving forward, the synergy between specialized LLMs, domain-specific simulations, and expert-driven validation will define the next generation of scientific discovery and infrastructure innovation, ensuring that technological advances align with societal needs, environmental stewardship, and engineering excellence.
5.6. Computer Science and Electrical Engineering
5.6.1. Overview
Computer Science (CS). CS is the study of computers and the algorithmic processes that support their operation [1532,1533,1534]. In other words, CS studies all topics relevant to computers, which encompasses a broad range of focus, from fundamental computer principles to the design of hardware and software systems, along with diverse real-world applications. The interdisciplinary nature of CS [1535], explicitly drawing from mathematics, engineering, and logic, incorporating techniques from fields such as electronic circuit design, probability, and statistics, positions it as a central driving force behind innovation in numerous sectors and further enables CS to contribute to a wide array of applications, from scientific modeling to the development of intelligent systems.
Electrical Engineering (EE). EE focuses on the study and application of electricity, electronics, and electromagnetism [1536,1537], covers a multitude of areas, including power engineering, telecommunications, control systems, electronics, signal processing, and computer engineering. Fundamentally, EE are the architects and builders of the complex electrical and electronic infrastructure that sustains modern life [1538]. In particular, EE is about understanding and manipulating electricity to create practical solutions that benefit society. This involves a wide range of activities, from the generation and distribution of electrical power that lights our homes and powers industries to the design of intricate electronic circuits found in everyday devices such as smartphones, computers, and vehicles [1539].
As discussed above, CS and EE represent two distinct yet deeply interconnected disciplines. While CS primarily focuses on algorithms, software development, and computational theory, EE deals with the physical systems, hardware, and electronic components that enable computing. Their relationship is characterized by a complex interplay of shared foundations, complementary approaches, and evolving boundaries that continue to shape modern technology. Moreover, addressing every viewpoint in CS and EE is non-trivial, as these fields are pertinent to a wide range of areas. Therefore, in this section, we focus on two fundamental topics in CS and EE, i.e., software development and circuit design.
Figure 23.
The relationships between software development in computer science and circuit design in electrical engineering.
Figure 23.
The relationships between software development in computer science and circuit design in electrical engineering.
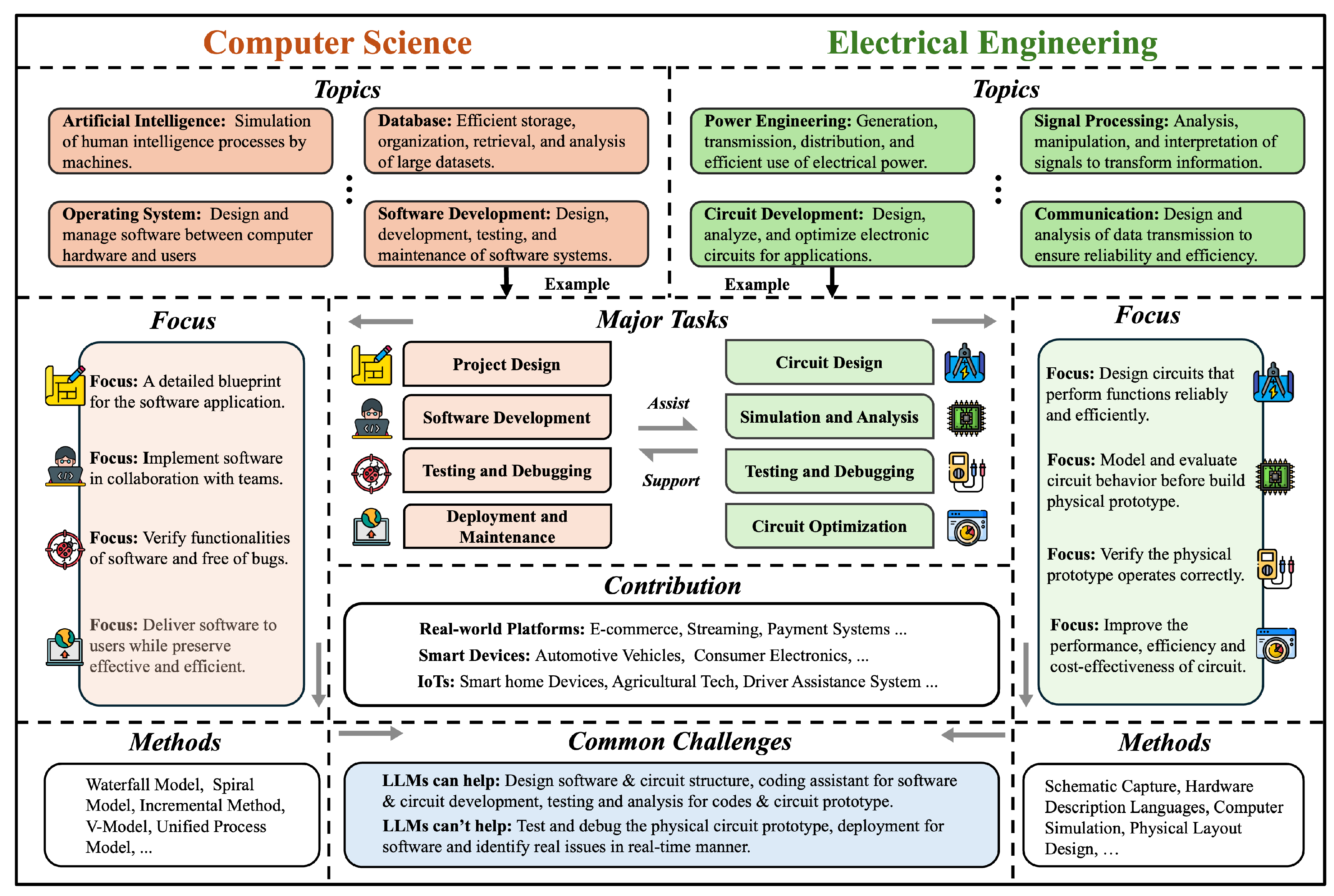
Conventional Software Development. Traditional programming approaches typically involve manual coding via programming languages, following the software development life cycle (SDLC). These methods have evolved from low-level machine code to high-level languages, but they generally rely on human developers to write, test, and debug code line by line [1540,1541]. Among traditional methodologies, several models have been widely adopted, including the Waterfall model, the Spiral model, the V-model, the Incremental Approach, and the Unified Process model [1542]. Next, we will highlight each model in detail:
- Waterfall Model: It represents a linear, sequential approach where each phase of the program development lifecycle, from requirements gathering to maintenance, must be completed before the next phase can begin [1543,1544,1545]. This model assumes that all requirements can be clearly defined and understood at the outset of the project. While straightforward and easy to manage for small projects with stable requirements, the Waterfall model struggles with complex projects or those where requirements are likely to change [1542]. Its rigidity makes this model difficult to accommodate new requirements or to revisit earlier phases once completed, leading to potential issues and increased costs if errors or omissions are discovered late in the development cycle [1546]. The lack of early prototypes also hinders client feedback, potentially resulting in a final product that does not fully meet user needs.
- Spiral Model: It offers a risk-driven approach that combines elements of the Waterfall model with iterative development [1545,1547]. Projects in the Spiral model progress through several iterations, with each iteration involving planning, risk analysis, engineering, and evaluation. By explicitly addressing risks at each stage, this model is better suited for complex projects with high potential risks compared to the linear Waterfall model [1544,1548]. The cyclical nature allows for revisiting and refining requirements and designs based on ongoing risk assessment, leading to a more adaptable outcome for projects where risks are a significant concern [1545]. The success of the Spiral Model heavily relies on accurate and thorough risk assessments at each iteration. If risk analysis is inadequate or incorrect, the project may suffer from poor planning and execution. Moreover, specialized knowledge in risk management is essential, but this expertise is not always available within every team
- V-Model: This is an extension of the Waterfall model that emphasizes the relationship between each development phase and a corresponding testing phase, focusing on verification and validation [1549,1550]. Executed in a V-shape sequence, this model ensures that testing activities are planned and executed in parallel with the development stages, highlighting the importance of quality assurance throughout the lifecycle. By linking verification and validation to each stage, the V-Model aims to improve software quality and ensure that the final product meets both the specified requirements and the user’s needs.
- Incremental Approach: This approach involves developing a program in a series of functional increments [1551]. Each increment adds more functionality to the previous ones, allowing for early delivery of working software to the client and the incorporation of feedback in subsequent increments. This approach offers greater flexibility compared to the Waterfall model, enabling the development team to adapt to changing requirements and reduce the risk associated with large, monolithic development efforts by delivering working software in smaller, manageable parts [1542].
- Unified Process Model: This model is an iterative and incremental program development framework driven by use cases, centered around architecture, and supporting component-based design [1551]. Utilizing the Unified Modelling Language to represent various phases and deliverables, the unified process model emphasizes iterative development, where the software evolves over multiple cycles, and is built piece by piece. This framework provides a flexible and adaptable approach suitable for a wide range of software projects, focusing on continuous improvement and active stakeholder involvement through use-case-driven development and an architectural focus that promotes a robust and scalable system design.
Beyond aforementioned conventional methodologies, the program development process commonly involves manually coding, where developers write source code in programming languages such as C/C++, Java, and Python, via tools like compilers, interpreters, and debuggers, while adhering to organizational coding standards [1542]. This manual translation of design specifications into executable code is inherently susceptible to human errors in logic, syntax, and implementation, particularly as the complexity of the program increases. Debugging relies on developers manually stepping through the code via debuggers, setting breakpoints to pause execution at specific points, and inspecting the variables to understand the program states and identify the causes of errors [1552,1553]. Techniques like inserting print statements at strategic locations in the code and conducting manual code reviews are also commonly employed to aid in the debugging process. However, these methods can prove particularly inefficient in large and complex systems, often requiring significant time and effort to trace the execution flow and pinpoint the source of bugs [1553]. Afterward, testing strategies in traditional SDLC models involve a distinct phase that occurs after or in parallel with the implementation of the program. This typically includes creating detailed test plans and test cases based on the software requirements, followed by the execution of these tests to identify defects and ensure that the program meets the specified standards. Testing in traditional methodologies is characterized as "difficult and late", implying that critical bugs are frequently discovered towards the end of the development cycle, making them more costly and time-consuming to rectify. Furthermore, clients are not typically involved in the coding and testing stages of traditional software development [1542].
Traditional Circuit Design. Next, we discuss the traditional circuit design in EE. The design of digital circuits has traditionally relied on a set of well-established methodologies to transform high-level specifications into physical hardware. These methods include schematic capture, the utilization of Hardware Description Languages (HDLs) [1554,1555,1556] such as VHDL [1554] and Verilog [1555,1557], simulation techniques, and the process of physical layout design:
- Schematic Capture: It involves the graphical creation of a circuit diagram using Electronic Design Automation (EDA) tools [1558,1559]. In this approach, designers place and connect symbols that represent electronic components, visually constructing the circuit’s architecture and interconnections [1560]. While schematic capture provides an intuitive way to represent smaller circuits, it becomes increasingly cumbersome and less scalable for complex designs containing a large number of components [1561]. Managing the intricate web of interconnections and ensuring accuracy through a purely graphical representation can be challenging for large integrated circuits.
- Hardware Description Languages (HDLs): HDLs, such as VHSIC HDL (VHDL) [1562] and Verilog [1557], are textual languages used to describe the behavior and structure of digital circuits at various levels of abstraction, ranging from the high-level Register Transfer Level down to the gate level. HDLs allow circuit designers to describe the flow of digital signals and the logical operations performed among them [1563,1564]. They offer a more scalable and manageable approach for designing complex digital systems compared to schematic capture, as the textual representation facilitates the use of automated EDA tools for simulation and synthesis. Historically, VHDL, initially developed for military applications, is known for its strong typing and structured syntax, making it suitable for large and critical projects. Verilog, on the other hand, gained widespread adoption in the semiconductor industry for the design and verification of Application-Specific Integrated Circuits (ASICs) and Field-Programmable Gate Arrays (FPGAs) due to its relative simplicity and ease of use.
- Simulation: Simulation techniques are crucial in traditional digital circuit design for verifying the functionality and timing of a circuit described in an HDL before it is physically implemented. This process involves creating test benches, which are sets of input stimuli applied to the circuit model, and observing the resulting output responses to ensure the design behaves as intended. While essential for detecting design errors early in the development cycle, simulating large and complex circuits can be computationally intensive and time-consuming. Thorough verification requires the careful planning and execution of extensive test cases to cover all possible operating conditions and scenarios, which can be a significant challenge for intricate designs.
- Physical Layout Design: At this stage, the abstract circuit design is transformed into a physical implementation on a substrate, such as a silicon die for an integrated circuit or a printed circuit board (PCB). This process involves determining the precise placement of components and the routing of the conductive interconnections between them, taking into account various factors such as signal integrity, power distribution, thermal management, and manufacturability. The physical layout significantly impacts the final performance, reliability, and cost of the circuit. Traditionally, achieving an efficient and effective physical layout, especially for high-performance designs, requires significant manual effort, expertise, and often involves iterative refinement to optimize for various conflicting requirements like area, speed, and power consumption.
Limitations and Challenges. Traditional methodologies in programming and circuit design, despite their long-standing use, face several limitations and challenges in the context of modern software development and increasing demands for complexity, performance and faster time-to-market. From a programming perspective, these limitations relate to scalability, the complexity of large codebases, the time-consuming nature of debugging, and the inherent potential for human error. For circuit design, these challenges include managing the increasing complexity of integrated circuits, the difficulty in verifying and testing these complex designs, the often long design cycles, and the high level of specialized experience required.
LLMs as solutions. LLMs offer potential solutions to several limitations inherent in traditional software programming methodologies. Their ability to generate and understand code, along with their natural language processing skills, can be leveraged to address issues related to routine coding tasks, debugging, code complexity, and human error. Moreover, the application of LLMs in digital circuit design is an emerging and rapidly evolving field. Researchers are exploring the potential of LLMs to tackle some of the limitations of traditional hardware design methodologies in areas such as HDL code generation, functional verification, and high-level synthesis. To summarize the main applications of LLMs in CSEE, we introduce the following taxonomy. (i) Code Generation Tasks; (ii) Code Assistant in Debugging; (ii) Code Analysis in Codebases; (iii) HDL Code Generation; (iv) Functional Verification; (v) High-Level Synthesis (HLS).
5.6.2. Code Generation Tasks
Several studies have demonstrated that LLMs excel at code generation tasks [1565,1566,1567]. By generating boilerplate code, standard data structures, common algorithms, and even entire functions based on natural language descriptions or specifications, LLMs can significantly reduce the amount of manual coding required from developers. Tools such as GitHub Copilot have already demonstrated the potential for substantial time savings by automating the creation of routine code elements [1568]. This automation allows developers to focus their efforts on more complex and creative problem-solving, rather than spending time on repetitive and often error-prone coding.
Several recent studies [1569,1570] have systematically conducted empirical experiments to demonstrate the effectiveness of LLMs in assisting developers with code generation tasks. Peng et al. [1569] conducted an experiment involving 95 developers from diverse backgrounds, dividing them into treatment and control groups. The results showed that, among those who completed the task, the average completion time was 71.17 minutes for the treatment group and 160.89 minutes for the control group—a 55.8% reduction in completion time. The t-test yielded a p-value of 0.0017, with a 95% confidence interval for the improvement ranging from [21%, 89%]. Notably, there were four outliers with completion times exceeding 300 minutes, all of whom were in the control group; the findings remain robust even when these outliers are excluded. These results indicate that Copilot significantly enhances average productivity in the studied population.
5.6.3. Code Assistant in Debugging
LLMs also play a crucial role in assisting with debugging [1571,1572,1573,1574]. For example, LDB [1571] introduced a step-by-step LLM debugger that mimics human reasoning by verifying runtime execution, showing that LLMs can systematically analyse program states to guide developers through complex bugs. By analysing code and error messages, LLMs can help developers understand the nature of the problem, identify potential causes, and even suggest fixes [1575,1576,1577]. ChatDBG [1572] demonstrated that an AI-powered assistant can interpret error logs, recommend debugging strategies, and interactively answer developer queries, significantly reducing the cognitive load during troubleshooting. Moreover, LLMs’ ability to understand the context of the code and the meaning of error messages can be particularly valuable in pinpointing the root cause of issues more quickly than traditional manual debugging techniques. Recent work by Yan et al. [1575] showed that combining static analysis with LLM reasoning leads to more explainable and accurate localisation of crashing faults, while LeDex [1576] trained LLMs to self-debug and explain their reasoning, further improving transparency and reliability in automated debugging. Furthermore, some LLMs are being developed with the capability to automatically identify and even repair certain types of bugs. By automating code generation and completion, LLMs can contribute to mitigating human error in software programming. For instance, UniDebugger [1573] proposed a multi-agent LLM-based system that leverages the synergy of several models to collaboratively diagnose and fix bugs, outperforming single-agent approaches. Another study [1574] provided a comprehensive evaluation of open-source LLMs, highlighting their growing competence in real-world debugging tasks. The real-time code suggestions and automated generation capabilities can reduce the likelihood of common mistakes such as typos, syntax errors, and simple logical errors. COAST [1577] further showed that refining LLM training data using communicative agent strategies can substantially boost the model’s ability to catch and correct subtle logical errors, expanding the practical utility of LLMs in everyday development. By predicting and generating code based on learned patterns and best practices, LLMs can help enforce consistency and reduce the introduction of errors during the coding process.
5.6.4. Code Analysis in Codebases
Addressing the complexity of large codebases is another area where LLMs can offer assistance [1578,1579,1580]. By generating natural language explanations of code snippets and providing summaries of code functionality, LLMs can make it easier for developers to understand and navigate complex or unfamiliar code. This can be particularly helpful when working with legacy systems or when onboarding new team members. Additionally, LLMs can potentially aid in refactoring code by suggesting improvements to its structure, identifying areas of high complexity, and even generating refactored code, thereby making large codebases more manageable and maintainable.
5.6.5. Hardware Description Language Code Generation
LLMs can potentially help in generating HDL code to manage the increasing complexity of digital circuits. By translating natural language descriptions of hardware functionality into Verilog or VHDL, LLMs could automate the creation of code for complex modules and systems, allowing designers to focus on higher-level architectural decisions. While challenges remain in ensuring the functional correctness and efficiency of the generated code, this application holds the promise of reducing manual coding effort and accelerating the design process. For example, AutoChip [1581] introduces an automated, feedback-driven method utilising LLMs to generate HDL. It combines LLMs with output from Verilog compilers to iteratively generate the design. An initial module is generated using design prompts. Afterwards, this module is corrected based on compilation errors and simulation messages. Besides desiging, many studies [1582,1583,1584,1585,1586,1587,1588,1589,1590,1591] have attempted to leverage LLMs for assistant code generation in circuit design. For example, MEIC [1591] employs a dual LLMs mechanism for automatic Verilog debugging. RTLFixer [1585] leverages Retrieval-Augmented Generation to address syntax errors in HDL. LLMs are further employed for debugging and automatic testing [1592,1593,1594,1595]. AutoSVA [1595] introduces an iterative framework that leverages formal verification to generate assertions from given hardware modules. VerilogReader [1592] leverages LLms to read Verilog code and coverage, aims at generate code coverage convergence tests.
5.6.6. Functional Verification
LLMs can also assist with functional verification by generating testbenches, assertions, and potentially aiding in formal verification. For example, AutoBench [1594] represents a significant advancement as the first LLM-based testbench generator for digital circuit design that requires only the description of the design under test (DUT) to automatically generate comprehensive testbenches. Their ability to understand design specifications could enable the automatic creation of test scenarios to ensure the circuit behaves as intended under various conditions, potentially addressing the difficulty in verifying complex designs and reducing the time required for this critical stage. Besides, assertion-based verification is critical for ensuring that design circuits comply with the architectural engineers. AssertLLM [1590] addresses this challenge by automatically processing complete specification files and generating functional verification assertions. It breaks down the complex task into three phases with customised LLMs-extracting structural specifications, mapping signal definitions, and generating assertions. Moreover, test stimuli generation is another crucial component of hardware verification, and has been transformed through frameworks like LLM4DV [1593] that harness the power of LLMs to create efficient test conditions. Specifically, LLM4DV introduces a prompt template for interactively eliciting test stimuli from LLMs along with innovative prompting improvements to enhance performance. When compared to traditional constrained-random testing (CRT), LLM4DV excels in efficiently handling straightforward DUT scenarios by leveraging basic mathematical reasoning and pre-trained knowledge. Although effectiveness decreases with more complex task settings, it still outperforms CRT in relative terms.
5.6.7. High-Level Synthesis
Furthermore, LLMs are being explored for their role in guiding High-Level Synthesis (HLS). For instance, HLSPilot [1596], the first LLM-enabled high-level synthesis framework, can fully automate high-level application acceleration on hybrid CPU-FPGA architectures. The framework applies a set of C-to-HLS optimisation strategies catering to various code patterns through in-context learning, which provides the LLMs with exemplary C/C++ to HLS prompts. Table 60 lists the performance of the LLMs-based and HLS-based methods in real-world application benchmarks. The results demonstrate that HLSPilot achieves comparable performance to manually crafted counterparts and can even outperform them in some cases. By understanding high-level language descriptions or natural language specifications of hardware algorithms, LLMs could help in translating these into HLS-compatible code or even directly guide the HLS tools to produce efficient hardware implementations. This could potentially make hardware design more accessible to engineers with software backgrounds and contribute to shorter design cycles. This capability addresses the challenge of the substantial semantic gap between natural language expressions and hardware design intent [1597]. Specifically, LLMs provide a more intuitive design process where engineers can specify hardware functionality in natural language, making hardware design more accessible to those without extensive HDL expertise.
Table 60.
Place & routing results for C→LLM→Verilog and C→HLS→Verilog approaches. EC (execution cycles) denotes the number of clock cycles required to complete a task or process, FF (Flip-Flops) represents the number of flip-flops used, i.e., basic storage elements in digital circuits, and LUT (Look-Up Tables) is the number of look-up tables used. Moreover, Slice denotes the number of FPGA slices used, DSP (Digital Signal Processors) is the number of dedicated DSP blocks used, and BRAM (Block RAM) represents the amount of block RAM used. Power counts the amount of power consumed by the design, and CP (Critical Path) denotes the longest combinational path between flip-flops, determining the maximum clock frequency.
Table 60.
Place & routing results for C→LLM→Verilog and C→HLS→Verilog approaches. EC (execution cycles) denotes the number of clock cycles required to complete a task or process, FF (Flip-Flops) represents the number of flip-flops used, i.e., basic storage elements in digital circuits, and LUT (Look-Up Tables) is the number of look-up tables used. Moreover, Slice denotes the number of FPGA slices used, DSP (Digital Signal Processors) is the number of dedicated DSP blocks used, and BRAM (Block RAM) represents the amount of block RAM used. Power counts the amount of power consumed by the design, and CP (Critical Path) denotes the longest combinational path between flip-flops, determining the maximum clock frequency.
| Benchmark | Approach | EC(↓) | FF(↓) | LUT(↓) | Slice (↓) | DSP (↓) | BRAM(↓) | Power(↓) | CP(↓) |
|---|---|---|---|---|---|---|---|---|---|
| syrk | LLM | 1,859,983 | 954 | 5300 | 1649 | 6 | 8 | 0.164 | 9.934 |
| HLS | 3,744,260 | 662 | 521 | 221 | 5 | 32 | 0.350 | 8.191 | |
| syr2k | LLM | 2,125,846 | 542 | 472 | 197 | 2 | 12 | 0.181 | 9.446 |
| HLS | 9,028,229 | 1042 | 960 | 1649 | 5 | 56 | 0.383 | 6.872 | |
| mvt | LLM | 44,996 | 8628 | 2663 | 4404 | 2 | 4 | 0.197 | 9.312 |
| HLS | 119,492 | 713 | 991 | 342 | 5 | 12 | 0.332 | 6.550 | |
| k3mm | LLM | 2,371,593 | 623 | 328 | 236 | 2 | 28 | 0.207 | 9.924 |
| HLS | 10,277,509 | 927 | 956 | 1649 | 5 | 56 | 0.398 | 6.646 | |
| k2mm | LLM | 1,863,816 | 537 | 311 | 202 | 2 | 20 | 0.189 | 9.967 |
| HLS | 7,963,269 | 929 | 659 | 313 | 5 | 56 | 0.400 | 6.814 | |
| gesummv | LLM | 65,991 | 437 | 288 | 170 | 2 | 16 | 0.176 | 9.253 |
| HLS | 148,805 | 795 | 561 | 228 | 5 | 20 | 0.316 | 6.855 | |
| gemm | LLM | 1,601,739 | 488 | 1702 | 300 | 2 | 16 | 0.178 | 9.697 |
| HLS | 4,542,980 | 1193 | 991 | 342 | 5 | 56 | 0.359 | 6.551 | |
| bicg | LLM | 46,478 | 1041 | 274 | 208 | 2 | 12 | 0.186 | 9.251 |
| HLS | 119,492 | 791 | 451 | 203 | 5 | 12 | 0.333 | 6.599 | |
| atax | LLM | 57,669 | 453 | 221 | 208 | 2 | 11 | 0.167 | 9.952 |
| HLS | 119,492 | 791 | 451 | 203 | 5 | 11 | 0.309 | 6.573 |
Table 61.
Applications of LLMs in computer science and electrical engineering.
| Taxonomy | Contributions | Representative Works | Citations |
| Code Generation Tasks. | LLMs facilitate developers in code generation (text to code), code completion (code snippet to code), and code comment (docstring). | EvalPlus [1598]: A code synthesis evaluation framework to rigorously benchmark the functional correctness of LLM-synthesized code. | [1565,1566,1567,1568,1598,1599,1600,1601] |
| Code Assistant in Debugging. | Automate to investigate the program for potential bugs statically and dynamically, and enable debugging programs efficiently. | Self-Debugging [1602]: A LLMs training framework to debug its predicted program via few-shot demonstrations. | [1571,1572,1573,1574,1575,1576,1577,1602,1603,1604,1605] |
| Code Analysis in Codebases. | LLMs assist developers for code summarization, searching, and translation in (large) codebases. | Paheli [1578] | [1578,1579,1580] |
| HDL Code Generation. | EAD for hardware design, code generation by translating natural language descriptions of hardware functionality into Verilog or VHDL | Verigen [1606], flipSyrup [1607], AutoChip [1581] | [1581,1582,1584,1585,1586,1587,1607,1608,1609,1610,1611,1612] |
| Functional Verification. | LLMs assist with functional verification by generating testbenches, assertions, and potentially aiding in formal verification. | AssertLLM [1590]: An automatic assertion generation framework that processes complete specification files of circuit designs. | [1589,1590,1591,1592,1593,1594,1595,1613,1614,1615] |
| High-Level Synthesis. | LLMs help in translating HLS-compatible code or guiding the HLS tools to produce efficient hardware implementations. | HLSPilot [1596]: The first LLM-enabled high-level synthesis framework that fully automate high-level application acceleration on hybrid CPU-FPGA architectures. | [1583,1588,1597,1616] |
5.6.8. Benchmarks
To evaluate the capabilities of LLMs in tasks related to coding and circuit design, a variety of benchmark datasets have been introduced. Table 62 enumerates the key benchmark datasets within the CSEE domain. Subsequently, we will delve into a more comprehensive discussion of the commonly utilized benchmark datasets, beginning with these pertaining to coding tasks.
HumanEval. It is a benchmark dataset developed by OpenAI specifically designed to evaluate an LLM’s code generation capabilities. HumanEval consists of 164 hand-crafted programming problems comparable to software interview questions, focusing on functional correctness rather than textual similarity to reference solution. Each problem includes a function signature, docstring, code body, and several unit tests, and the performance is measured via PASS@K [49]. HumanEval has recognized as a standard evaluation for many code-generation models.
LiveCodeBench. LiveCodeBench encompasses a comprehensive array of code-related functionalities, extending beyond mere code generation to include self-repair, code execution, and test output prediction. Notably, it places a significant emphasis on self-repair, wherein models, upon generating initial code, receive error feedback and are required to amend their solutions accordingly. This aspect aligns with the code debugging category within our taxonomy. Furthermore, LiveCodeBench necessitates that models predict the outcomes of code execution for specified inputs, which pertains to the code analysis category in our taxonomy. A pivotal innovation of LiveCodeBench lies in its methodology for mitigating data contamination, a critical concern in the evaluation of LLMs.
APPS. Automated Programming Progress Standard [1617] attempts to mirror how human programmers are evaluated by posting coding problems in natural langauge and testing solution correctness. The APPS dataset comprises 100,000 coding problems sourced from platforms such as Codeforces and Kattis, spanning a range from beginner to collegiate competition levels. Within this collection, there are 232,444 solutions crafted by human programmers. The average length of each problem is 203.2 words, reflecting the inherent complexity of the tasks.
CodeXGLUE. It stands for General Language Understanding Evaluation benchmark for CODE. CodeXGLUE offers an extensive array of code intelligence tasks and serves as a platform for the evaluation and comparison of models. It encompasses 14 datasets across 10 diverse programming language tasks, which include scenarios such as code-code (e.g., clone detection, defect detection), text-code (e.g., code search, text-to-code generation), code-text (e.g., summarization), and text-text (e.g., documentation translation).
CodeContests. It is a competitive programming dataset used for trianign AlphaCode [1619], DeepMind’s code generation system. It contains programming problems from sources like Aizu, AtCoder, CodeChef, Codeforces, and HackerEarth. Unlike other benchmarks that merely provide the gold solutions, it also contains incorrect human solutions in various languages.
Additional Benchmarks for Software Engineering. MBPP consists of around 1K crowd-sourced Python programming problems designed for entry-level programmers. It focuses on programming fundamentals adn standard library functionality. Each problem includes a task description, code solution, and three automated test cases. LeetCodeDataset is a high-quality benchmark for evaluating and trianing code-generation models, particularly focusesd on reasnoing abilities. LeetCodeDataset is sourced from LeetCode Python problems with rich metadata, and contains over a hundred test cases per problem. Moreover, it enables contamination-free evaluation and efficient supervised fine-tuning. WikiSQL is a corpus of hand-annotated SQL query and natural language question pairs for database-related tasks. WikiSQL encompasses 87,726 SQL query and natural language question pairs, split into training (61,297), development (9,145), and test (17,284) sets.
RTLLM. A benchmark for design RTL generation with LLMs. The latest version, i.e., RTLLM 2.0, expands on the original RTLLM benchmark, augmenting it to 50 hand-crafted designs across various applications: (i) Arithmetic Modules; (ii) Memory Modules; (iii) Control Modules and (iv) Miscellaneous Modules.
VerilogEval, A benchmark designed for evaluating LLMs in Verilog code generation for hardware design and verification. VerilogEval consists of 156 diverse problems from the Verilog instructional website HDLBits, ranging from simple combinational circuits to complex finite-state machines. Its evaluation framework uses simulation to verify the functional correctness of generated code by comparing outputs with golden solutions. The problem statements are provided in two types: machine-generated and human-written.
We further report several available performance report in Table 63. According to the table, we find that (i) Claude 3.7 Sonnet consistently delivers the strongest results wherever it is reported, topping three of the six benchmarks—CodeMMLU (67 %), Aider-Polyglot (64.9 %), and SWE-Bench (62.3 %). (ii) Claude 3.5 Sonnet remains competitive: although it trails its newer sibling on the knowledge-heavy benchmarks, it records the overall best HumanEval score (93.7 %) and the second-best EvalPlus (81.7 %). (iii) GPT-o1 places second on HumanEval (92.4 %) and first on EvalPlus (89 %), but its lead narrows or disappears on the more engineering-oriented datasets (CodeMMLU and SWE-Bench) (iv) Llama 3.3 matches proprietary giants on HumanEval (88.4 %) but lags on CodeMMLU (43.9 %), highlighting the gap that still exists for open models on reasoning-heavy evaluation.
In summary, in general, coding tasks, Claude 3.5 Sonnet and GPT-o1 give the best pass rates, with GPT-4o, Qwen 2.5, and Llama-3 70 B as cost-efficient runners-up. On analysis or multilingual engineering tasks, Claude 3.7 Sonnet is the clear leader, while Gemini 2.0 Flash and DeepSeek R1 provide solid mid-tier options. For large-scale debugging- and circuit desing-related tasks, Claude 3.7 Sonnet again excels, but GPT-4o delivers the top score on PICBench and remains the most balanced all-rounder.
5.6.9. Discussion
Opportunities and Impact. The integration of LLMs into both software programming and digital circuit design presents significant opportunities to improve production, streamlining workflow and fostering innovation [1635,1636]. In software programming, LLMs can automate repetitive coding tasks, leading to substantial time saving and allowing developers to concentrate on more complex jobs [1568]. Moreover, LLMs are capable of assisting developers in debugging in static and dynamic scenarios [1571,1572,1573,1574]. Furthermore, LLMs can improve code comprehension by generating human-readable explanations of complex codebases, facilitating maintenance and collaboration [1578,1579,1580]. The impace of these capabilities is a potentially accelerated software development lifecycle, improved code quality, and increased developer satisfaction.
Similarly, in digital circuit design, LLMs offer the potential to automate the generation of HDL code from natural language specifications, which can help manage the increasing complexity of modern integrated circuits [1582,1583,1584,1585,1586,1587,1588,1589,1590,1591]. They can also assist in functional verification by generating testbenches and assertions, potentially reducing the verification bottleneck [1590,1593,1594]. Moreover, LLMs are being explored for their role in high-level synthesis, potentially making hardware design more accessible to software engineers and shortening design cycles. The impact of LLMs in this domain could be faster time-to-market for hardware innovations and a more efficient design process.
Challenges and Limitations. Despite the promising opportunities, the application of LLMs in software programming and digital circuit design also entails challenges and limitations [1637]. In software, while LLMs can generate code, ensuring the correctness and reliability of complex logic remains a challenge, often requiring significant human oversight. LLMs may also struggle with maintaining state and reasoning about long-term dependencies in software systems. The issue of "hallucinations," where LLMs generate incorrect or non-sensical code, is a significant concern that needs to be addressed to ensure the trustworthiness of LLM-generated software.
In digital circuit design, the limitations of current LLMs are even more pronounced. Fundamental hardware architecture design, which requires a deep understanding of physics and microarchitectural innovations, is beyond the current capabilities of LLMs. Optimizing the physical layout for complex ICs demands specialized knowledge that LLMs do not yet possess. Verifying highly novel or safety-critical hardware designs often requires rigorous formal verification techniques that may exceed the reasoning abilities of current LLMs. Furthermore, the application of LLMs in analog circuit design is still in its infancy and presents unique challenges. Limited data sources in the hardware domain also poses a significant challenge for training effective LLMs for circuit design tasks.
Conclusion. Traditional methodologies have long served as the foundation for software programming and digital circuit design. The emergence of LLMs has opened a new pathway to address several limitations in these topics. In software programming, LLMs can automate repetitive coding tasks, assist developers in debugging, help manage the complexity of large codebases through explanation and refactoring suggestions, and mitigate human error through intelligent code generation and completion. In digital circuit design, LLMs offer promising solutions for generating HDL code, assisting with functional verification, and guiding high-level synthesis, potentially streamlining the design process and making it more accessible. Further research and development are crucial to improve the accuracy and reliability of LLM-generated code and verification components, particularly in critical applications. Seamlessly integrating LLMs into existing design workflows and EDA tools in hardware engineering, exploring their potential in more advanced design and optimization tasks, and addressing challenges like data scarcity in hardware design are essential areas for future investigation. As LLMs continue to evolve, they are poised to become valuable partners for engineers in both software and hardware, augmenting human capabilities and shaping the future of technological innovation.
References
- Achiam, J.; Adler, S.; Agarwal, S.; Ahmad, L.; Akkaya, I.; Aleman, F.L.; Almeida, D.; Altenschmidt, J.; Altman, S.; Anadkat, S.; et al. Gpt-4 technical report. arXiv 2023, arXiv:2303.08774. [Google Scholar] [CrossRef]
- Mesko, B. The ChatGPT (generative artificial intelligence) revolution has made artificial intelligence approachable for medical professionals. Journal of medical Internet research 2023, 25, e48392. [Google Scholar] [CrossRef]
- Yuan, L.; Chen, D.; Chen, Y.L.; Codella, N.; Dai, X.; Gao, J.; Hu, H.; Huang, X.; Li, B.; Li, C.; et al. Florence: A new foundation model for computer vision. arXiv 2021, arXiv:2111.11432. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Advances in neural information processing systems 2017, 30. [Google Scholar]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I.; et al. Improving language understanding by generative pre-training 2018.
- Radford, A.; Wu, J.; Child, R.; Luan, D.; Amodei, D.; Sutskever, I.; et al. Language models are unsupervised multitask learners. OpenAI blog 2019, 1, 9. [Google Scholar]
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language models are few-shot learners. Advances in neural information processing systems 2020. [Google Scholar]
- Zhao, W.X.; Zhou, K.; Li, J.; Tang, T.; Wang, X.; Hou, Y.; Min, Y.; Zhang, B.; Zhang, J.; Dong, Z.; et al. A survey of large language models. arXiv 2023, arXiv:2303.182231. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the Proceedings of the 2019 conference of the North American chapter of the association for computational linguistics: human language technologies, volume 1 (long and short papers); 2019. [Google Scholar]
- Li, X.L.; Liang, P. Prefix-tuning: Optimizing continuous prompts for generation. arXiv 2021, arXiv:2101.00190. [Google Scholar] [CrossRef]
- Sanh, V.; Webson, A.; Raffel, C.; Bach, S.H.; Sutawika, L.; Alyafeai, Z.; Chaffin, A.; Stiegler, A.; Scao, T.L.; Raja, A.; et al. Multitask Prompted Training Enables Zero-Shot Task Generalization. arXiv 2110, arXiv:2110.08207. [Google Scholar] [CrossRef]
- Lin, Z.; Madotto, A.; Fung, P. Exploring versatile generative language model via parameter-efficient transfer learning. arXiv 2020, arXiv:2004.03829. [Google Scholar] [CrossRef]
- Wei, J.; Tay, Y.; Bommasani, R.; Raffel, C.; Zoph, B.; Borgeaud, S.; Yogatama, D.; Bosma, M.; Zhou, D.; Metzler, D.; et al. Emergent abilities of large language models. arXiv 2022, arXiv:2206.07682. [Google Scholar] [CrossRef]
- Pahune, S.; Chandrasekharan, M. Several categories of large language models (llms): A short survey. arXiv 2023, arXiv:2307.10188. [Google Scholar] [CrossRef]
- Patil, A. Advancing Reasoning in Large Language Models: Promising Methods and Approaches. arXiv 2025, arXiv:2502.03671. [Google Scholar] [CrossRef]
- Kerner, T. Domain-Specific Pretraining of Language Models: A Comparative Study in the Medical Field. arXiv 2024, arXiv:2407.14076. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, W.; Kim, S.; Kim, D.; Kim, S.; So, C.H.; Kang, J. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2020, 36, 1234–1240. [Google Scholar] [CrossRef]
- Beltagy, I.; Lo, K.; Cohan, A. SciBERT: A pretrained language model for scientific text. arXiv 2019, arXiv:1903.10676. [Google Scholar] [CrossRef]
- Wei, J.; Wang, X.; Schuurmans, D.; Bosma, M.; Xia, F.; Chi, E.; Le, Q.V.; Zhou, D.; et al. Chain-of-thought prompting elicits reasoning in large language models. Advances in neural information processing systems 2022, 35, 24824–24837. [Google Scholar]
- OpenAI. Introducing OpenAI o1. OpenAI 2024. [Google Scholar]
- Guo, D.; Yang, D.; Zhang, H.; Song, J.; Wang, P.; Zhu, Q.; Xu, R.; Zhang, R.; Ma, S.; Bi, X.; et al. DeepSeek-R1 incentivizes reasoning in LLMs through reinforcement learning. Nature 2025, 645, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.X.; Zhou, K.; Li, J.; Tang, T.; Wang, X.; Hou, Y.; Min, Y.; Zhang, B.; Zhang, J.; Dong, Z.; et al. A Survey of Large Language Models. arXiv 2025, arXiv:2303.18223. [Google Scholar] [CrossRef]
- Jelinek, F. Statistical methods for speech recognition; MIT press, 1998. [Google Scholar]
- Rosenfeld, R. Two decades of statistical language modeling: Where do we go from here? Proceedings of the IEEE 2000, 88, 1270–1278. [Google Scholar] [CrossRef]
- Stolcke, A.; et al. SRILM-an extensible language modeling toolkit. In Proceedings of the Interspeech; 2002; Vol. 2002, p. 2002. [Google Scholar]
- Brown, P.F.; Della Pietra, S.A.; Della Pietra, V.J.; Mercer, R.L. The mathematics of statistical machine translation: Parameter estimation. Computational linguistics 1993, 19, 263–311. [Google Scholar]
- Brown, P.; Cocke, J.; Della Pietra, S.; Della Pietra, V.; Jelinek, F.; Mercer, R.; Roossin, P. A Statistical Approach to Language Translation. In Proceedings of the Coling Budapest 1988 Volume 1: International Conference on Computational Linguistics; 1988. [Google Scholar]
- Kilgarriff, A.; Grefenstette, G. Introduction to the special issue on the web as corpus. Computational linguistics 2003, 29, 333–347. [Google Scholar] [CrossRef]
- Resnik, P.; Smith, N.A. The web as a parallel corpus. Computational Linguistics 2003, 29, 349–380. [Google Scholar] [CrossRef]
- Banko, M.; Brill, E. Scaling to very very large corpora for natural language disambiguation. In Proceedings of the Proceedings of the 39th annual meeting of the Association for Computational Linguistics, 2001; pp. 26–33.
- Bengio, Y.; Ducharme, R.; Vincent, P.; Jauvin, C. A neural probabilistic language model. Journal of machine learning research 2003, 3, 1137–1155. [Google Scholar]
- Mikolov, T.; Karafiát, M.; Burget, L.; Cernockỳ, J.; Khudanpur, S. Recurrent neural network based language model. In Proceedings of the Interspeech. Makuhari; 2010; Vol. 2, pp. 1045–1048. [Google Scholar]
- Kombrink, S.; Mikolov, T.; Karafiát, M.; Burget, L. Recurrent neural network based language modeling in meeting recognition. In Proceedings of the Interspeech; 2011; Vol. 11, pp. 2877–2880. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural computation 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, J. Learning Natural Language Inference with LSTM. arXiv 2016, arXiv:1512.08849. [Google Scholar] [CrossRef] [PubMed]
- Tarwani, K.M.; Edem, S. Survey on recurrent neural network in natural language processing. Int. J. Eng. Trends Technol 2017, 48, 301–304. [Google Scholar] [CrossRef]
- Mienye, I.D.; Swart, T.G.; Obaido, G. Recurrent neural networks: A comprehensive review of architectures, variants, and applications. Information 2024, 15, 517. [Google Scholar] [CrossRef]
- Google. Found in translation: More accurate, fluent sentences in Google Translate. Goolgle Blog 2016.
- Google. Zero-Shot Translation with Google’s Multilingual Neural Machine Translation System. Goolgle Blog 2016.
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the limits of transfer learning with a unified text-to-text transformer. Journal of machine learning research 2020. [Google Scholar] [CrossRef]
- Lan, Z.; Chen, M.; Goodman, S.; Gimpel, K.; Sharma, P.; Soricut, R. Albert: A lite bert for self-supervised learning of language representations. arXiv 2019, arXiv:1909.11942. [Google Scholar] [CrossRef]
- Liu, Y.; Ott, M.; Goyal, N.; Du, J.; Joshi, M.; Chen, D.; Levy, O.; Lewis, M.; Zettlemoyer, L.; Stoyanov, V. Roberta: A robustly optimized bert pretraining approach. arXiv 2019, arXiv:1907.11692. [Google Scholar] [CrossRef]
- Weizenbaum, J. ELIZA—a computer program for the study of natural language communication between man and machine. Communications of the ACM 1966, 9, 36–45. [Google Scholar] [CrossRef]
- Brown, P.F.; Della Pietra, V.J.; Desouza, P.V.; Lai, J.C.; Mercer, R.L. Class-based n-gram models of natural language. Computational linguistics 1992, 18, 467–480. [Google Scholar]
- Johnson, M. PCFG models of linguistic tree representations. Computational Linguistics 1998, 24, 613–632. [Google Scholar]
- Mikolov, T.; Zweig, G. Context dependent recurrent neural network language model. In Proceedings of the 2012 IEEE Spoken Language Technology Workshop (SLT); 2012; pp. 234–239. [Google Scholar] [CrossRef]
- Sutskever, I.; Vinyals, O.; Le, Q.V. Sequence to sequence learning with neural networks. Advances in neural information processing systems 2014, 27. [Google Scholar]
- OpenAI. Introducing ChatGPT. OpenAI Blog, 2022.
- Chen, M.; Tworek, J.; Jun, H.; Yuan, Q.; Pinto, H.P.D.O.; Kaplan, J.; Edwards, H.; Burda, Y.; Joseph, N.; Brockman, G.; et al. Evaluating large language models trained on code. arXiv 2021, arXiv:2107.03374. [Google Scholar] [CrossRef]
- Chung, H.W.; Hou, L.; Longpre, S.; Zoph, B.; Tay, Y.; Fedus, W.; Li, Y.; Wang, X.; Dehghani, M.; Brahma, S.; et al. Scaling instruction-finetuned language models. Journal of Machine Learning Research 2024, 25, 1–53. [Google Scholar] [CrossRef]
- Bai, J.; Bai, S.; Chu, Y.; Cui, Z.; Dang, K.; Deng, X.; Fan, Y.; Ge, W.; Han, Y.; Huang, F.; et al. Qwen Technical Report. arXiv 2023, arXiv:2309.16609. [Google Scholar] [CrossRef]
- Touvron, H.; Lavril, T.; Izacard, G.; Martinet, X.; Lachaux, M.A.; Lacroix, T.; Rozière, B.; Goyal, N.; Hambro, E.; Azhar, F.; et al. Llama: Open and efficient foundation language models. arXiv 2023, arXiv:2302.13971. [Google Scholar] [CrossRef]
- Jiang, A.Q.; Sablayrolles, A.; Roux, A.; Mensch, A.; Savary, B.; Bamford, C.; Chaplot, D.S.; Casas, D.d.l.; Hanna, E.B.; Bressand, F.; et al. Mixtral of experts. arXiv 2024, arXiv:2401.04088. [Google Scholar] [CrossRef]
- Ouyang, L.; Wu, J.; Jiang, X.; Almeida, D.; Wainwright, C.L.; Mishkin, P.; Zhang, C.; Agarwal, S.; Slama, K.; Ray, A.; et al. Training language models to follow instructions with human feedback. arXiv 2022, arXiv:2203.02155. [Google Scholar] [CrossRef]
- Rae, J.W.; Borgeaud, S.; Cai, T.; Millican, K.; Hoffmann, J.; Song, F.; Aslanides, J.; Henderson, S.; Ring, R.; Young, S.; et al. Scaling language models: Methods, analysis & insights from training gopher. arXiv 2021, arXiv:2112.11446. [Google Scholar] [CrossRef]
- Kaplan, J.; McCandlish, S.; Henighan, T.; Brown, T.B.; Chess, B.; Child, R.; Gray, S.; Radford, A.; Wu, J.; Amodei, D. Scaling laws for neural language models. arXiv 2020, arXiv:2001.08361. [Google Scholar] [CrossRef]
- Kang, S.; Yoon, J.; Yoo, S. Large language models are few-shot testers: Exploring llm-based general bug reproduction. In Proceedings of the 2023 IEEE/ACM 45th International Conference on Software Engineering (ICSE). IEEE; 2023; pp. 2312–2323. [Google Scholar]
- Song, C.H.; Wu, J.; Washington, C.; Sadler, B.M.; Chao, W.L.; Su, Y. Llm-planner: Few-shot grounded planning for embodied agents with large language models. In Proceedings of the Proceedings of the IEEE/CVF international conference on computer vision, 2023; pp. 2998–3009.
- Wang, Y.; Zhong, W.; Li, L.; Mi, F.; Zeng, X.; Huang, W.; Shang, L.; Jiang, X.; Liu, Q. Aligning large language models with human: A survey. arXiv 2023, arXiv:2307.12966. [Google Scholar] [CrossRef]
- Wei, J.; Wang, X.; Schuurmans, D.; Bosma, M.; Ichter, B.; Xia, F.; Chi, E.; Le, Q.; Zhou, D. Chain-of-Thought Prompting Elicits Reasoning in Large Language Models. arXiv 2023, arXiv:2201.11903. [Google Scholar] [CrossRef]
- Wang, B.; Min, S.; Deng, X.; Shen, J.; Wu, Y.; Zettlemoyer, L.; Sun, H. Towards understanding chain-of-thought prompting: An empirical study of what matters. arXiv 2022, arXiv:2212.10001. [Google Scholar] [CrossRef]
- Huang, J.; Chang, K.C.C. Towards reasoning in large language models: A survey. arXiv 2022, arXiv:2212.10403. [Google Scholar] [CrossRef]
- Wang, X.; Wei, J.; Schuurmans, D.; Le, Q.; Chi, E.; Narang, S.; Chowdhery, A.; Zhou, D. Self-Consistency Improves Chain of Thought Reasoning in Language Models. arXiv 2023, arXiv:2203.11171. [Google Scholar] [CrossRef]
- OpenAI. GPT-4.5 System Card, 2025.
- OpenAI. GPT-5 System Card. Technical report, OpenAI, 2025.
- Claude. The Claude 3 Model Family: Opus, Sonnet, Haiku, 2024.
- Claude. Claude 3.7 Sonnet System Card, 2025.
- Koray Kavukcuoglu, CTO, Google DeepMind, on behalf of the Gemini team. Gemini 2.0 model updates: 2.0 Flash, Flash-Lite, Pro Experimental. Accessed: 2025-3-22.
- OpenAI. gpt-oss-120b & gpt-oss-20b Model Card. arXiv 2025, arXiv:2508.10925. [Google Scholar] [CrossRef]
- Yang, A.; Yang, B.; Hui, B.; Zheng, B.; Yu, B.; Zhou, C.; Li, C.; Li, C.; Liu, D.; Huang, F.; et al. Qwen2 Technical Report. arXiv 2024, arXiv:2407.10671. [Google Scholar] [CrossRef]
- Yang, A.; Yang, B.; Zhang, B.; Hui, B.; Zheng, B.; Yu, B.; Li, C.; Liu, D.; Huang, F.; Wei, H.; et al. Qwen2. 5 technical report. arXiv 2024, arXiv:2412.15115. [Google Scholar] [CrossRef]
- Team, Q. QwQ-32B: Embracing the Power of Reinforcement Learning, 2025.
- Liu, A.; Feng, B.; Xue, B.; Wang, B.; Wu, B.; Lu, C.; Zhao, C.; Deng, C.; Zhang, C.; Ruan, C.; et al. Deepseek-v3 technical report. arXiv 2024, arXiv:2412.19437. [Google Scholar] [CrossRef]
- Grattafiori, A.; Dubey, A.; Jauhri, A.; Pandey, A.; Kadian, A.; Al-Dahle, A.; Letman, A.; Mathur, A.; Schelten, A.; Vaughan, A.; et al. The llama 3 herd of models. arXiv 2024, arXiv:2407.21783. [Google Scholar] [CrossRef]
- Yenduri, G.; Ramalingam, M.; Selvi, G.C.; Supriya, Y.; Srivastava, G.; Maddikunta, P.K.R.; Raj, G.D.; Jhaveri, R.H.; Prabadevi, B.; Wang, W.; et al. Gpt (generative pre-trained transformer)–a comprehensive review on enabling technologies, potential applications, emerging challenges, and future directions. IEEE Access 2024. [Google Scholar] [CrossRef]
- Yenduri, G.; Srivastava, G.; Maddikunta, P.K.R.; Jhaveri, R.H.; Wang, W.; Vasilakos, A.V.; Gadekallu, T.R.; et al. Generative pre-trained transformer: A comprehensive review on enabling technologies, potential applications, emerging challenges, and future directions. arXiv 2023, arXiv:2305.10435. [Google Scholar] [CrossRef]
- Kaiser,,, L.; Gomez,, A.N.; Shazeer, N.; Vaswani, A.; Parmar, N.; Jones, L.; Uszkoreit, J. One model to learn them all. arXiv 2017, arXiv:1706.05137. [Google Scholar] [CrossRef]
- Finn, C.; Abbeel, P.; Levine, S. Model-agnostic meta-learning for fast adaptation of deep networks. In Proceedings of the International conference on machine learning. PMLR; 2017; pp. 1126–1135. [Google Scholar]
- McCann, B.; Keskar, N.S.; Xiong, C.; Socher, R. The natural language decathlon: Multitask learning as question answering. arXiv 2018, arXiv:1806.08730. [Google Scholar] [CrossRef]
- Ouyang, L.; Wu, J.; Jiang, X.; Almeida, D.; Wainwright, C.; Mishkin, P.; Zhang, C.; Agarwal, S.; Slama, K.; Ray, A.; et al. Training language models to follow instructions with human feedback. Advances in neural information processing systems 2022, 35, 27730–27744. [Google Scholar]
- OpenAI. Our Approach to Alignment Research. OpenAI Blog, 2022.
- Claude. Unified Model Context Interaction Protocol, 2025.
- OpenAI. GPT-4o System Card, 2024.
- OpenAI. GPT-4o mini: advancing cost-efficient intelligence, 2024.
- OpenAI. Reasoning models. Accessed: 2025-3-22.
- Claude. Calude’s Constitution, 2023.
- Anthropic. Introducing the next generation of Claude, 2024.
- Sundar Pichai, CEO of Google and Alphabet. Gemini: Google’s largest and most capable AI model. Accessed: 2025-3-22.
- xAI. Grok-3. Accessed: 2025-3-22.
- Ray, T. ChatGPT is ’not particularly innovative,’ and ’nothing revolutionary’, says Meta’s chief AI scientist, 2023.
- Badminton, N. Meta’s Yann LeCun on auto-regressive Large Language Models (LLMs), 2023.
- Ainslie, J.; Lee-Thorp, J.; De Jong, M.; Zemlyanskiy, Y.; Lebrón, F.; Sanghai, S. Gqa: Training generalized multi-query transformer models from multi-head checkpoints. arXiv 2023, arXiv:2305.13245. [Google Scholar] [CrossRef]
- Schulman, J.; Wolski, F.; Dhariwal, P.; Radford, A.; Klimov, O. Proximal policy optimization algorithms. arXiv 2017, arXiv:1707.06347. [Google Scholar] [CrossRef]
- Dauphin, Y.N.; Fan, A.; Auli, M.; Grangier, D. Language modeling with gated convolutional networks. In Proceedings of the International conference on machine learning. PMLR; 2017; pp. 933–941. [Google Scholar]
- Su, J.; Ahmed, M.; Lu, Y.; Pan, S.; Bo, W.; Liu, Y. Roformer: Enhanced transformer with rotary position embedding. Neurocomputing 2024, 568, 127063. [Google Scholar] [CrossRef]
- Baldacchino, T.; Cross, E.J.; Worden, K.; Rowson, J. Variational Bayesian mixture of experts models and sensitivity analysis for nonlinear dynamical systems. Mechanical Systems and Signal Processing 2016, 66, 178–200. [Google Scholar] [CrossRef]
- Yang, Z.; Dai, Z.; Yang, Y.; Carbonell, J.; Salakhutdinov, R.R.; Le, Q.V. Xlnet: Generalized autoregressive pretraining for language understanding. Advances in neural information processing systems 2019, 32. [Google Scholar]
- Keskar, N.S.; McCann, B.; Varshney, L.R.; Xiong, C.; Socher, R. Ctrl: A conditional transformer language model for controllable generation. arXiv 2019, arXiv:1909.05858. [Google Scholar] [CrossRef]
- Kojima, T.; Gu, S.S.; Reid, M.; Matsuo, Y.; Iwasawa, Y. Large language models are zero-shot reasoners. Advances in neural information processing systems 2022. [Google Scholar]
- Snell, C.; Lee, J.; Xu, K.; Kumar, A. Scaling llm test-time compute optimally can be more effective than scaling model parameters. arXiv 2024, arXiv:2408.03314. [Google Scholar] [CrossRef]
- Ji, Y.; Li, J.; Ye, H.; Wu, K.; Xu, J.; Mo, L.; Zhang, M. Test-time Computing: from System-1 Thinking to System-2 Thinking. arXiv 2025, arXiv:2501.02497. [Google Scholar] [CrossRef]
- Lewis, P.; Perez, E.; Piktus, A.; Petroni, F.; Karpukhin, V.; Goyal, N.; Küttler, H.; Lewis, M.; Yih, W.t.; Rocktäschel, T.; et al. Retrieval-augmented generation for knowledge-intensive nlp tasks. Advances in neural information processing systems 2020. [Google Scholar]
- Gao, Y.; Xiong, Y.; Gao, X.; Jia, K.; Pan, J.; Bi, Y.; Dai, Y.; Sun, J.; Wang, H.; Wang, H. Retrieval-augmented generation for large language models: A survey. arXiv 2023, arXiv:2312.10997. [Google Scholar] [CrossRef]
- Yang, R.; Song, L.; Li, Y.; Zhao, S.; Ge, Y.; Li, X.; Shan, Y. Gpt4tools: Teaching large language model to use tools via self-instruction. Advances in Neural Information Processing Systems 2023. [Google Scholar]
- Xiong, H.; Bian, J.; Li, Y.; Li, X.; Du, M.; Wang, S.; Yin, D.; Helal, S. When search engine services meet large language models: visions and challenges. IEEE Transactions on Services Computing 2024. [Google Scholar] [CrossRef]
- Socher, R.; Perelygin, A.; Wu, J.; Chuang, J.; Manning, C.D.; Ng, A.; Potts, C. Recursive Deep Models for Semantic Compositionality Over a Sentiment Treebank. In Proceedings of the Proceedings of the 2013 Conference on Empirical Methods in Natural Language Processing; 2013. [Google Scholar]
- Maas, A.L.; Daly, R.E.; Pham, P.T.; Huang, D.; Ng, A.Y.; Potts, C. Learning Word Vectors for Sentiment Analysis. In Proceedings of the Proceedings of the 49th Annual Meeting of the Association for Computational Linguistics: Human Language Technologies; 2011. [Google Scholar]
- Yelp Dataset Challenge. Yelp Open Dataset, 2015.
- Tjong Kim Sang, E.F.; De Meulder, F. Introduction to the CoNLL-2003 Shared Task: Language-Independent Named Entity Recognition. In Proceedings of the Proceedings of the Seventh Conference on Natural Language Learning at HLT-NAACL 2003; 2003. [Google Scholar]
- Zhang, Y.; Zhong, V.; Chen, D.; Angeli, G.; Manning, C.D. Position-aware Attention and Supervised Data Improve Slot Filling. In Proceedings of the Proceedings of the 2017 Conference on Empirical Methods in Natural Language Processing; 2017. [Google Scholar]
- Etzioni, O.; Banko, M.; Soderland, S.; Weld, D.S. Open information extraction from the web. Communications of the ACM 2008. [Google Scholar] [CrossRef]
- Levesque, H.J.; Davis, E.; Morgenstern, L. The Winograd schema challenge. KR 2012, 2012. [Google Scholar]
- Sakaguchi, K.; Bras, R.L.; Bhagavatula, C.; Choi, Y. Winogrande: An adversarial winograd schema challenge at scale. Communications of the ACM 2021. [Google Scholar] [CrossRef]
- Wang, A.; Pruksachatkun, Y.; Nangia, N.; Singh, A.; Michael, J.; Hill, F.; Levy, O.; Bowman, S. Superglue: A stickier benchmark for general-purpose language understanding systems. Advances in neural information processing systems 2019. [Google Scholar]
- Hermann, K.M.; Kocisky, T.; Grefenstette, E.; Espeholt, L.; Kay, W.; Suleyman, M.; Blunsom, P. Teaching machines to read and comprehend. Advances in neural information processing systems 2015. [Google Scholar]
- Narayan, S.; Cohen, S.B.; Lapata, M. Don’t give me the details, just the summary! topic-aware convolutional neural networks for extreme summarization. arXiv 2018, arXiv:1808.08745. [Google Scholar] [CrossRef]
- Gliwa, B.; Mochol, I.; Biesek, M.; Wawer, A. SAMSum Corpus: A Human-annotated Dialogue Dataset for Abstractive Summarization. In Proceedings of the Proceedings of the 2nd Workshop on New Frontiers in Summarization; 2019. [Google Scholar]
- Rajpurkar, P.; Zhang, J.; Lopyrev, K.; Liang, P. Squad: 100,000+ questions for machine comprehension of text. arXiv 2016, arXiv:1606.05250. [Google Scholar] [CrossRef]
- Kwiatkowski, T.; Palomaki, J.; Redfield, O.; Collins, M.; Parikh, A.; Alberti, C.; Epstein, D.; Polosukhin, I.; Devlin, J.; Lee, K.; et al. Natural Questions: A Benchmark for Question Answering Research. Transactions of the Association for Computational Linguistics 2019. [Google Scholar] [CrossRef]
- Joshi, M.; Choi, E.; Weld, D.S.; Zettlemoyer, L. Triviaqa: A large scale distantly supervised challenge dataset for reading comprehension. arXiv 2017, arXiv:1705.03551. [Google Scholar] [CrossRef]
- Yang, Z.; Qi, P.; Zhang, S.; Bengio, Y.; Cohen, W.W.; Salakhutdinov, R.; Manning, C.D. HotpotQA: A dataset for diverse, explainable multi-hop question answering. arXiv 2018, arXiv:1809.09600. [Google Scholar] [CrossRef]
- Clark, C.; Lee, K.; Chang, M.W.; Kwiatkowski, T.; Collins, M.; Toutanova, K. Boolq: Exploring the surprising difficulty of natural yes/no questions. arXiv 2019, arXiv:1905.10044. [Google Scholar] [CrossRef]
- Rao, S.; Tetreault, J. Dear Sir or Madam, May I Introduce the GYAFC Dataset: Corpus, Benchmarks and Metrics for Formality Style Transfer. In Proceedings of the Proceedings of the 2018 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long Papers); 2018. [Google Scholar]
- Paperno, D.; Kruszewski, G.; Lazaridou, A.; Pham, N.Q.; Bernardi, R.; Pezzelle, S.; Baroni, M.; Boleda, G.; Fernández, R. The LAMBADA dataset: Word prediction requiring a broad discourse context. In Proceedings of the Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Volume 1: Long Papers); 2016. [Google Scholar]
- Bojar, O.; Buck, C.; Federmann, C.; Haddow, B.; Koehn, P.; Leveling, J.; Monz, C.; Pecina, P.; Post, M.; Saint-Amand, H.; et al. Findings of the 2014 Workshop on Statistical Machine Translation. In Proceedings of the Proceedings of the Ninth Workshop on Statistical Machine Translation; 2014. [Google Scholar]
- Cettolo, M.; Girardi, C.; Federico, M. WIT3: Web Inventory of Transcribed and Translated Talks. In Proceedings of the Proceedings of the 16th Annual Conference of the European Association for Machine Translation; 2012. [Google Scholar]
- Goyal, N.; Gao, C.; Chaudhary, V.; Chen, P.J.; Wenzek, G.; Ju, D.; Krishnan, S.; Ranzato, M.; Guzmán, F.; Fan, A. The Flores-101 Evaluation Benchmark for Low-Resource and Multilingual Machine Translation. Transactions of the Association for Computational Linguistics 2022. [Google Scholar] [CrossRef]
- Austin, J.; Odena, A.; Nye, M.; Bosma, M.; Michalewski, H.; Dohan, D.; Jiang, E.; Cai, C.; Terry, M.; Le, Q.; et al. Program synthesis with large language models. arXiv 2021, arXiv:2108.07732. [Google Scholar] [CrossRef]
- Hendrycks, D.; Basart, S.; Kadavath, S.; Mazeika, M.; Arora, A.; Guo, E.; Burns, C.; Puranik, S.; He, H.; Song, D.; et al. Measuring coding challenge competence with apps. arXiv 2021, arXiv:2105.09938. [Google Scholar] [CrossRef]
- Lu, S.; Guo, D.; Ren, S.; Huang, J.; Svyatkovskiy, A.; Blanco, A.; Clement, C.; Drain, D.; Jiang, D.; Tang, D.; et al. Codexglue: A machine learning benchmark dataset for code understanding and generation. arXiv 2021, arXiv:2102.04664. [Google Scholar] [CrossRef]
- Jain, N.; Han, K.; Gu, A.; Li, W.D.; Yan, F.; Zhang, T.; Wang, S.; Solar-Lezama, A.; Sen, K.; Stoica, I. Livecodebench: Holistic and contamination free evaluation of large language models for code. arXiv 2024, arXiv:2403.07974. [Google Scholar] [CrossRef]
- Trivedi, H.; Balasubramanian, N.; Khot, T.; Sabharwal, A. MuSiQue: Multihop Questions via Single-hop Question Composition. Transactions of the Association for Computational Linguistics 2022. [Google Scholar] [CrossRef]
- Geva, M.; Khashabi, D.; Segal, E.; Khot, T.; Roth, D.; Berant, J. Did aristotle use a laptop? a question answering benchmark with implicit reasoning strategies. Transactions of the Association for Computational Linguistics 2021. [Google Scholar] [CrossRef]
- Mihaylov, T.; Clark, P.; Khot, T.; Sabharwal, A. Can a suit of armor conduct electricity? a new dataset for open book question answering. arXiv 2018, arXiv:1809.02789. [Google Scholar] [CrossRef]
- Hendrycks, D.; Burns, C.; Basart, S.; Zou, A.; Mazeika, M.; Song, D.; Steinhardt, J. Measuring massive multitask language understanding. arXiv 2020, arXiv:2009.03300. [Google Scholar] [CrossRef]
- Srivastava, A.; Rastogi, A.; Rao, A.; Shoeb, A.A.M.; Abid, A.; Fisch, A.; Brown, A.R.; Santoro, A.; Gupta, A.; Garriga-Alonso, A.; et al. Beyond the imitation game: Quantifying and extrapolating the capabilities of language models. arXiv 2022, arXiv:2206.04615. [Google Scholar] [CrossRef]
- Suzgun, M.; Scales, N.; Schärli, N.; Gehrmann, S.; Tay, Y.; Chung, H.W.; Chowdhery, A.; Le, Q.V.; Chi, E.H.; Zhou, D.; et al. Challenging big-bench tasks and whether chain-of-thought can solve them. arXiv 2022, arXiv:2210.09261. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Liu, H.; Huang, D.; Wang, Y.; Zhang, Y. Logiqa: A challenge dataset for machine reading comprehension with logical reasoning. arXiv 2020, arXiv:2007.08124. [Google Scholar] [CrossRef]
- Yu, W.; Jiang, Z.; Dong, Y.; Feng, J. ReClor: A Reading Comprehension Dataset Requiring Logical Reasoning. In Proceedings of the International Conference on Learning Representations; 2020. [Google Scholar]
- Cobbe, K.; Kosaraju, V.; Bavarian, M.; Chen, M.; Jun, H.; Kaiser, L.; Plappert, M.; Tworek, J.; Hilton, J.; Nakano, R.; et al. Training verifiers to solve math word problems. arXiv 2021, arXiv:2110.14168. [Google Scholar] [CrossRef]
- Chollet, F.; Knoop, M.; Kamradt, G.; Landers, B. Arc prize 2024: Technical report. arXiv 2024, arXiv:2412.04604. [Google Scholar] [CrossRef]
- Hendrycks, D.; Burns, C.; Kadavath, S.; Arora, A.; Basart, S.; Tang, E.; Song, D.; Steinhardt, J. Measuring mathematical problem solving with the math dataset. arXiv 2021, arXiv:2103.03874. [Google Scholar] [CrossRef]
- Talmor, A.; Herzig, J.; Lourie, N.; Berant, J. CommonsenseQA: A Question Answering Challenge Targeting Commonsense Knowledge. In Proceedings of the Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers); 2019. [Google Scholar]
- Zellers, R.; Holtzman, A.; Bisk, Y.; Farhadi, A.; Choi, Y. Hellaswag: Can a machine really finish your sentence? arXiv 2019, arXiv:1905.07830. [Google Scholar] [CrossRef]
- Bisk, Y.; Zellers, R.; Gao, J.; Choi, Y.; et al. Piqa: Reasoning about physical commonsense in natural language. In Proceedings of the Proceedings of the AAAI conference on artificial intelligence; 2020. [Google Scholar]
- Sap, M.; Rashkin, H.; Chen, D.; Le Bras, R.; Choi, Y. Social IQa: Commonsense Reasoning about Social Interactions. In Proceedings of the Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP); 2019. [Google Scholar]
- Kwiatkowski, T.; Palomaki, J.; Redfield, O.; Collins, M.; Parikh, A.; Alberti, C.; Epstein, D.; Polosukhin, I.; Kelcey, M.; Devlin, J.; et al. Natural Questions: a Benchmark for Question Answering Research. Transactions of the Association of Computational Linguistics 2019. [Google Scholar] [CrossRef]
- Berant, J.; Chou, A.; Frostig, R.; Liang, P. Semantic Parsing on Freebase from Question-Answer Pairs. In Proceedings of the Proceedings of the 2013 Conference on Empirical Methods in Natural Language Processing; 2013. [Google Scholar]
- Petroni, F.; Piktus, A.; Fan, A.; Lewis, P.; Yazdani, M.; De Cao, N.; Thorne, J.; Jernite, Y.; Karpukhin, V.; Maillard, J.; et al. KILT: a benchmark for knowledge intensive language tasks. arXiv 2020, arXiv:2009.02252. [Google Scholar] [CrossRef]
- Liang, P.; Bommasani, R.; Lee, T.; Tsipras, D.; Soylu, D.; Yasunaga, M.; Zhang, Y.; Narayanan, D.; Wu, Y.; Kumar, A.; et al. Holistic evaluation of language models. arXiv 2022, arXiv:2211.09110. [Google Scholar] [CrossRef]
- Lin, S.; Hilton, J.; Evans, O. Truthfulqa: Measuring how models mimic human falsehoods. arXiv 2021, arXiv:2109.07958. [Google Scholar] [CrossRef]
- Dua, D.; Wang, Y.; Dasigi, P.; Stanovsky, G.; Singh, S.; Gardner, M. DROP: A reading comprehension benchmark requiring discrete reasoning over paragraphs. arXiv 2019, arXiv:1903.00161. [Google Scholar] [CrossRef]
- Zhu, F.; Lei, W.; Huang, Y.; Wang, C.; Zhang, S.; Lv, J.; Feng, F.; Chua, T.S. TAT-QA: A Question Answering Benchmark on a Hybrid of Tabular and Textual Content in Finance. In Proceedings of the Proceedings of the 59th Annual Meeting of the Association for Computational Linguistics and the 11th International Joint Conference on Natural Language Processing (Volume 1: Long Papers); 2021. [Google Scholar]
- Xu, Q.; Hong, F.; Li, B.; Hu, C.; Chen, Z.; Zhang, J. On the tool manipulation capability of open-source large language models. arXiv 2023, arXiv:2305.16504. [Google Scholar] [CrossRef]
- Li, M.; Zhao, Y.; Yu, B.; Song, F.; Li, H.; Yu, H.; Li, Z.; Huang, F.; Li, Y. Api-bank: A comprehensive benchmark for tool-augmented llms. arXiv 2023, arXiv:2304.08244. [Google Scholar] [CrossRef]
- Reddy, S.; Chen, D.; Manning, C.D. CoQA: A Conversational Question Answering Challenge. Transactions of the Association for Computational Linguistics 2019. [Google Scholar] [CrossRef]
- Choi, E.; He, H.; Iyyer, M.; Yatskar, M.; Yih, W.t.; Choi, Y.; Liang, P.; Zettlemoyer, L. QuAC: Question Answering in Context. In Proceedings of the Proceedings of the 2018 Conference on Empirical Methods in Natural Language Processing; 2018. [Google Scholar]
- Bai, G.; Liu, J.; Bu, X.; He, Y.; Liu, J.; Zhou, Z.; Lin, Z.; Su, W.; Ge, T.; Zheng, B.; et al. Mt-bench-101: A fine-grained benchmark for evaluating large language models in multi-turn dialogues. arXiv 2024, arXiv:2402.14762. [Google Scholar] [CrossRef]
- Papineni, K.; Roukos, S.; Ward, T.; Zhu, W.J. Bleu: a method for automatic evaluation of machine translation. In Proceedings of the Proceedings of the 40th annual meeting of the Association for Computational Linguistics; 2002. [Google Scholar]
- Lin, C.Y. Rouge: A package for automatic evaluation of summaries. In Proceedings of the Text summarization branches out; 2004. [Google Scholar]
- Zhang, T.; Kishore, V.; Wu, F.; Weinberger, K.Q.; Artzi, Y. Bertscore: Evaluating text generation with bert. arXiv 2019, arXiv:1904.09675. [Google Scholar] [CrossRef]
- Rauh, M.; Mellor, J.; Uesato, J.; Huang, P.S.; Welbl, J.; Weidinger, L.; Dathathri, S.; Glaese, A.; Irving, G.; Gabriel, I.; et al. Characteristics of harmful text: Towards rigorous benchmarking of language models. Advances in Neural Information Processing Systems 2022. [Google Scholar]
- Wang, B.; Chen, W.; Pei, H.; Xie, C.; Kang, M.; Zhang, C.; Xu, C.; Xiong, Z.; Dutta, R.; Schaeffer, R.; et al. DecodingTrust: A Comprehensive Assessment of Trustworthiness in GPT Models. In Proceedings of the NeurIPS; 2023. [Google Scholar]
- Cui, S.; Zhang, Z.; Chen, Y.; Zhang, W.; Liu, T.; Wang, S.; Liu, T. Fft: Towards harmlessness evaluation and analysis for llms with factuality, fairness, toxicity. arXiv 2023, arXiv:2311.18580. [Google Scholar] [CrossRef]
- Lightman, H.; Kosaraju, V.; Burda, Y.; Edwards, H.; Baker, B.; Lee, T.; Leike, J.; Schulman, J.; Sutskever, I.; Cobbe, K. Let’s verify step by step. In Proceedings of the The Twelfth International Conference on Learning Representations; 2023. [Google Scholar]
- Li, Y.; Du, M.; Song, R.; Wang, X.; Wang, Y. A survey on fairness in large language models. arXiv 2023, arXiv:2308.10149. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, Y.; Ton, J.F.; Zhang, X.; Guo, R.; Cheng, H.; Klochkov, Y.; Taufiq, M.F.; Li, H. Trustworthy llms: a survey and guideline for evaluating large language models’ alignment. arXiv 2023, arXiv:2308.05374. [Google Scholar] [CrossRef]
- Liao, Q.V.; Vaughan, J.W. Ai transparency in the age of llms: A human-centered research roadmap. arXiv 2023, arXiv:2306.0194110. [Google Scholar] [CrossRef]
- Mishra, S.; Khashabi, D.; Baral, C.; Hajishirzi, H. Cross-task generalization via natural language crowdsourcing instructions. arXiv 2021, arXiv:2104.08773. [Google Scholar] [CrossRef]
- Gu, J.; Jiang, X.; Shi, Z.; Tan, H.; Zhai, X.; Xu, C.; Li, W.; Shen, Y.; Ma, S.; Liu, H.; et al. A survey on llm-as-a-judge. arXiv 2024, arXiv:2411.15594. [Google Scholar] [CrossRef]
- Zheng, L.; Chiang, W.L.; Sheng, Y.; Zhuang, S.; Wu, Z.; Zhuang, Y.; Lin, Z.; Li, Z.; Li, D.; Xing, E.; et al. Judging llm-as-a-judge with mt-bench and chatbot arena. Advances in Neural Information Processing Systems 2023. [Google Scholar]
- Li, D.; Jiang, B.; Huang, L.; Beigi, A.; Zhao, C.; Tan, Z.; Bhattacharjee, A.; Jiang, Y.; Chen, C.; Wu, T.; et al. From generation to judgment: Opportunities and challenges of llm-as-a-judge. arXiv 2024, arXiv:2411.16594. [Google Scholar] [CrossRef]
- Carr, E.H. What is history? Springer, 1961. [Google Scholar]
- Tosh, J. The pursuit of history: Aims, methods and new directions in the study of history; Routledge, 2015. [Google Scholar]
- Williams, R.C. The historian’s toolbox: A student’s guide to the theory and craft of history; Routledge, 2014. [Google Scholar]
- Jenkins, K. Rethinking history; Routledge, 2003. [Google Scholar]
- Gaddis, J.L. The landscape of history: How historians map the past; Oxford University Press, 2002. [Google Scholar]
- Howell, M.C.; Prevenier, W. From reliable sources: An introduction to historical methods; Cornell University Press, 2001. [Google Scholar]
- Marwick, A. The new nature of history: Knowledge, evidence, language 2001.
- Lloyd, C. The structures of history 1993.
- Wineburg, S. Historical thinking and other unnatural acts. Phi delta kappan 2010, 92, 81–94. [Google Scholar] [CrossRef]
- Jockers, M.L. Macroanalysis: Digital methods and literary history; University of Illinois Press, 2013. [Google Scholar]
- Moretti, F. Distant reading; Verso Books, 2013. [Google Scholar]
- Milligan, I. History in the age of abundance? How the web is transforming historical research; McGill-Queen’s University Press, 2019. [Google Scholar]
- Blevins, C. Paper trails: The US post and the making of the American West; Oxford University Press, 2021. [Google Scholar]
- Tian, Y.; Huang, T.; Liu, M.; Jiang, D.; Spangher, A.; Chen, M.; May, J.; Peng, N. Are Large Language Models Capable of Generating Human-Level Narratives? arXiv 2024, arXiv:2407.13248. [Google Scholar] [CrossRef]
- Piper, A.; Bagga, S. Using Large Language Models for Understanding Narrative Discourse. In Proceedings of the Proceedings of the The 6th Workshop on Narrative Understanding; Lal, Y.K.; Clark, E.; Iyyer, M.; Chaturvedi, S.; Brei, A.; Brahman, F.; Chandu, K.R., Eds., Miami, Florida, USA, 2024; pp. 37–46. [CrossRef]
- Varnum, M.E.; Baumard, N.; Atari, M.; Gray, K. Large Language Models based on historical text could offer informative tools for behavioral science. Proceedings of the National Academy of Sciences 2024, 121, e2407639121. [Google Scholar] [CrossRef]
- Zeng, Y. HistoLens: An LLM-Powered Framework for Multi-Layered Analysis of Historical Texts–A Case Application of Yantie Lun. arXiv 2024, arXiv:2411.09978. [Google Scholar] [CrossRef]
- Garcia, G.G.; Weilbach, C. If the sources could talk: Evaluating large language models for research assistance in history. arXiv 2023, arXiv:2310.10808. [Google Scholar] [CrossRef]
- Celli, F.; Spathulas, G. Language Models reach higher Agreement than Humans in Historical Interpretation. arXiv 2025, arXiv:2504.02572. [Google Scholar] [CrossRef]
- Blevins, C. A Large Language Model Walks Into an Archive..., 2024. Accessed: 2025-04-06.
- Hauser, J.; Kondor, D.; Reddish, J.; Benam, M.; Cioni, E.; Villa, F.; Bennett, J.S.; Hoyer, D.; Francois, P.; Turchin, P.; et al. Large Language Models’ Expert-level Global History Knowledge Benchmark (HiST-LLM). In Proceedings of the The Thirty-eight Conference on Neural Information Processing Systems Datasets and Benchmarks Track; 2024. [Google Scholar]
- Celli, F.; Mingazov, D. Knowledge Extraction from LLMs for Scalable Historical Data Annotation. Electronics 2024, 13, 4990. [Google Scholar] [CrossRef]
- Li, N.; Yuan, S.; Chen, J.; Liang, J.; Wei, F.; Liang, Z.; Yang, D.; Xiao, Y. Past Meets Present: Creating Historical Analogy with Large Language Models. arXiv 2024, arXiv:2409.14820. [Google Scholar] [CrossRef]
- Zheng, C.; Zhang, Y.; Huang, Z.; Shi, C.; Xu, M.; Ma, X. Co-Exploration. In Proceedings of the Proceedings of the 37th Annual ACM Symposium on User Interface Software and Technology, 2024; pp. 1–20.
- Green, A.; Troup, K. The houses of history: A critical reader in twentieth-century history and theory 1999.
- Ghaboura, S.; More, K.; Thawkar, R.; Alghallabi, W.; Thawakar, O.; Khan, F.S.; Cholakkal, H.; Khan, S.; Anwer, R.M. Time Travel: A Comprehensive Benchmark to Evaluate LMMs on Historical and Cultural Artifacts. arXiv 2025, arXiv:2502.14865. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, Y.; Wei, X.; Yang, S.; Zhu, Y.; Li, Y.; Liu, D.; Wu, B. AC-EVAL: Evaluating Ancient Chinese Language Understanding in Large Language Models. arXiv 2024, arXiv:2403.06574. [Google Scholar] [CrossRef]
- Sellars, W. Philosophy and the Scientific Image of Man. In Science, Perception and Reality; Routledge, 1963; pp. 1–40. [Google Scholar]
- Russell, B. The Problems of Philosophy; Oxford University Press, 1912. [Google Scholar]
- Kant, I. Critique of Pure Reason; Cambridge University Press, 1998. [Google Scholar]
- Aristotle. Metaphysics; Random House, 1941. [Google Scholar]
- Locke, J. Two Treatises of Government; Awnsham Churchill, 1689. [Google Scholar]
- Jefferson, T. The Declaration of Independence, 1776. U.S. National Archives.
- The United States Constitution, 1787. U.S. National Archives.
- Descartes, R. Meditations on First Philosophy; Cambridge University Press, 1996. [Google Scholar]
- Aristotle. Nicomachean Ethics; Cambridge University Press, 2000. [Google Scholar]
- Rawls, J. A Theory of Justice; Harvard University Press, 1971. [Google Scholar]
- Heidegger, M. Being and Time; Harper & Row, 1962. [Google Scholar]
- Nagel, T. What Is It Like to Be a Bat? The Philosophical Review 1974, 83, 435–450. [Google Scholar] [CrossRef]
- Frege, G. The Foundations of Arithmetic; Blackwell, 1953. [Google Scholar]
- Kuhn, T.S. The Structure of Scientific Revolutions; University of Chicago Press, 1962. [Google Scholar]
- Coeckelbergh, M. LLMs, Truth, and Democracy: An Overview of Risks. Science and Engineering Ethics 2024, 31, 4. [Google Scholar] [CrossRef]
- Overgaard, M.; Kirkeby-Hinrup, A. A clarification of the conditions under which Large Language Models could be conscious. Humanities and Social Sciences Communications 2024, 11, 1031. [Google Scholar] [CrossRef]
- Colombatto, C.; Fleming, S.M. Folk psychological attributions of consciousness to large language models. Neuroscience of Consciousness 2024, 2024, niae013. [Google Scholar] [CrossRef]
- Heersmink, R.; de Rooij, B.; Clavel Vázquez, M.J.; Colombo, M. A phenomenology and epistemology of large language models: transparency, trust, and trustworthiness. Ethics and Information Technology 2024, 26, 41. [Google Scholar] [CrossRef]
- Dillion, D.; Mondal, D.; Tandon, N.; Gray, K. AI language model rivals expert ethicist in perceived moral expertise. Scientific Reports 2025, 15, 4084. [Google Scholar] [CrossRef]
- Kempt, H.; Lavie, A.; Nagel, S.K. Towards a Conversational Ethics of Large Language Models. American Philosophical Quarterly 2024, 61, 339–354. [Google Scholar] [CrossRef]
- Wang, G.; Wang, W.; Cao, Y. Possibilities and challenges in the moral growth of large language models: a philosophical perspective. Ethics and Information Technology 2024. [Google Scholar] [CrossRef]
- Mugleston, J.; Truong, V.H.; Kuang, C.; Sibiya, L.; Myung, J. Epistemology in the Age of Large Language Models. Knowledge 2025, 5, 3. [Google Scholar] [CrossRef]
- Harnad, S. Language writ large: LLMs, ChatGPT, meaning, and understanding. Frontiers in Artificial Intelligence 2025, 7, 1490698. [Google Scholar] [CrossRef] [PubMed]
- Heimann, M.; Hübener, A.F. The extimate core of understanding: absolute metaphors, psychosis and large language models. AI & Society 2024. [Google Scholar]
- Gutenberg, P. Project Gutenberg, 1971. Available at https://www.gutenberg.org/.
- Foundation, P. PhilPapers: Online Research in Philosophy, 2009. Available at https://philpapers.org/.
- Paullada, A.; Raji, I.D.; Bender, E.M.; Denton, E.; Hanna, A. Data and its (dis) contents: A survey of dataset development and use in machine learning research. Patterns 2021, 2. [Google Scholar] [CrossRef] [PubMed]
- Dahl, R.A. Who Governs? Democracy and Power in an American City; Yale University Press, 1961. [Google Scholar]
- Easton, D. The Political System: An Inquiry into the State of Political Science; Alfred A. Knopf, 1953. [Google Scholar]
- Weber, M. Politics as a Vocation. In From Max Weber: Essays in Sociology; Originally delivered as a lecture in 1919; Gerth, H.H., Mills, C.W., Eds.; Oxford University Press, 1946; pp. 77–128. [Google Scholar]
- Skinner, Q. Visions of Politics, Volume 1: Regarding Method; Cambridge University Press, 2002. [Google Scholar]
- Lijphart, A. Comparative Politics and the Comparative Method. American Political Science Review 1971, 65, 682–693. [Google Scholar] [CrossRef]
- Converse, P.E. The Nature of Belief Systems in Mass Publics. In Ideology and Discontent; Apter, D.E., Ed.; Free Press, 1964; pp. 206–261.
- Downs, A. An Economic Theory of Democracy; Harper and Row, 1957.
- Grimmer, J.; Roberts, M.E.; Stewart, B.M. Text as Data: A New Framework for Machine Learning and the Social Sciences; Princeton University Press, 2021.
- Doe, J.; Smith, J. Large Language Models in Politics and Democracy: A Comprehensive Survey. Political Science Review 2024, 58, 123–145. [Google Scholar]
- Zhou, W.; Li, M. A Survey on Hallucination in Large Language Models: Principles, Taxonomy, and Open Challenges. ACM Computing Surveys 2023, 55, 1–38. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y. Algorithmic Bias in Large Language Models: Implications for Political Research. Journal of Computational Social Science 2025, 7, 210–230. [Google Scholar]
- Ornstein, J.T.; Blasingame, E.N.; Truscott, J.S. How to Train Your Stochastic Parrot: Large Language Models for Political Texts. Political Science Research and Methods 2024. [Google Scholar] [CrossRef]
- Le Mens, G.; Gallego, A. Positioning Political Texts with Large Language Models by Asking and Averaging. Political Analysis 2025. [Google Scholar] [CrossRef]
- O’Hagan, S.; Schein, A. Measurement in the Age of LLMs: An Application to Ideological Scaling. arXiv 2023, arXiv:2312.09203. [Google Scholar] [CrossRef]
- Liu, M.; Shi, G. PoliPrompt: A High-Performance Cost-Effective LLM-Based Text Classification Framework for Political Science. arXiv 2024, arXiv:2409.01466. [Google Scholar] [CrossRef]
- Törnberg, P. ChatGPT-4 Outperforms Experts and Crowd Workers in Annotating Political Twitter Messages with Zero-Shot Learning. arXiv 2023, arXiv:2304.06588. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, J. Performance and biases of large language models in public opinion simulation. Humanities and Social Sciences Communications 2024, 11, 231. [Google Scholar] [CrossRef]
- Yu, C.; Weng, Z.; Li, Z.; Hu, X.; Zhao, Y. A Large-Scale Simulation on Large Language Models for Decision-Making. SSRN Electronic Journal 2024. [Google Scholar] [CrossRef]
- Karanjai, R.; et al. Synthesizing Public Opinions with LLMs: Role Creation, Impacts, and Challenges. arXiv 2025, arXiv:2504.00241. [Google Scholar] [CrossRef]
- Lee, S.; Peng, T.Q.; Goldberg, M.H.; et al. Can large language models estimate public opinion about global warming? An empirical assessment of algorithmic fidelity and bias. PLOS Climate 2024, 3, e0000429. [Google Scholar] [CrossRef]
- Bradshaw, C.; Miller, C.; Warnick, S. LLM Generated Distribution-Based Prediction of US Electoral Results, Part I. ResearchGate 2024. [Google Scholar]
- Hackenburg, K.; Tappin, B.M.; Röttger, P.; Hale, S.; Bright, J.; Margetts, H. Evidence of a Log Scaling Law for Political Persuasion with Large Language Models. arXiv 2024, arXiv:2406.14508. [Google Scholar] [CrossRef]
- Aldahoul, N.; Ibrahim, H.; Varvello, M.; Kaufman, A.; Rahwan, T.; Zaki, Y. Large Language Models are Often Politically Extreme, Usually Ideologically Inconsistent, and Persuasive Even in Informational Contexts. arXiv 2025, arXiv:2505.04171. [Google Scholar] [CrossRef]
- Matz, S.C.; Teeny, J.D.; Vaid, S.S.; Peters, H.; Harari, G.; Cerf, M. The potential of generative AI for personalized persuasion at scale. Scientific Reports 2024, 14, 10444. [Google Scholar] [CrossRef]
- Williams, A.R.; Burke-Moore, L.; Chan, R.S.Y.; Enock, F.E.; Nanni, F.; Sippy, T.; Chung, Y.L.; Gabasova, E.; Hackenburg, K.; Bright, J. Large Language Models Can Consistently Generate High-Quality Election Disinformation. PLOS ONE 2025, 20, e0317421. [Google Scholar] [CrossRef] [PubMed]
- Šola, H.M.; Qureshi, F.H.; Khawaja, S. Human-Centred Design Meets AI-Driven Algorithms: Enhancing Political Campaign Materials. Informatics 2025, 12, 30. [Google Scholar] [CrossRef]
- Kulkarni, V.; Ye, J.; Skiena, S.; Wang, W.Y. Multi-view Models for Political Ideology Detection of News Articles. arXiv 2018, arXiv:1809.03485. [Google Scholar] [CrossRef]
- Libraries, S. Congressional Record for the 43rd–114th Congresses: Parsed Text, 2020. Available at https://data.stanford.edu/congress_text.
- Woolley, J.T.; Peters, G. The American Presidency Project, 1999. Available at https://www.presidency.ucsb.edu/.
- Check, M.B. Media Bias/Fact Check, 2025. Available at https://mediabiasfactcheck.com/.
- Lin, S.; Hilton, J.; Evans, O. TruthfulQA: Measuring How Models Mimic Human Falsehoods. In Proceedings of the Proceedings of the 60th Annual Meeting of the Association for Computational Linguistics (Volume 1: Long Papers). Association for Computational Linguistics; 2022; pp. 3214–3252. [Google Scholar]
- Davies, S. Definitions of art. In The Routledge companion to aesthetics; Routledge, 2005; pp. 187–198.
- Eisenman, P.D. Notes on conceptual architecture: Towards a definition. Design Quarterly 1970, 1–5. [Google Scholar] [CrossRef]
- Bravoco, R.R.; Yadav, S.B. Requirement definition architecture—an overview. Computers in Industry 1985, 6, 237–251. [Google Scholar] [CrossRef]
- Baxandall, M. Patterns of intention: On the historical explanation of pictures; Yale University Press, 1985.
- Panofsky, E.; Drechsel, B. Meaning in the visual arts; Penguin Books Harmondsworth, 1970.
- Denzin, N.K. Performance ethnography: Critical pedagogy and the politics of culture; Sage, 2003.
- Taylor, D. The archive and the repertoire: Performing cultural memory in the Americas; Duke University Press, 2003.
- Frampton, K. Studies in tectonic culture: the poetics of construction in nineteenth and twentieth century architecture; Mit Press, 2001.
- Hillier, B.; Hanson, J. The social logic of space; Cambridge university press, 1989.
- Groat, L.N.; Wang, D. Architectural research methods; John Wiley & Sons, 2013.
- Drucker, J. Graphesis: Visual forms of knowledge production. (No Title) 2014.
- Zhu, D.; Chen, J.; Shen, X.; Li, X.; Elhoseiny, M. Minigpt-4: Enhancing vision-language understanding with advanced large language models. arXiv 2023, arXiv:2304.10592. [Google Scholar] [CrossRef]
- Liu, H.; Li, C.; Wu, Q.; Lee, Y.J. Visual instruction tuning. Advances in neural information processing systems 2023, 36, 34892–34916. [Google Scholar]
- Yuan, Z.; He, Y.; Wang, K.; Ye, Y.; Sun, L. ArtGPT-4: Towards artistic-understanding large vision-language models with enhanced adapter. arXiv 2023, arXiv:2305.07490. [Google Scholar] [CrossRef]
- Bin, Y.; Shi, W.; Ding, Y.; Hu, Z.; Wang, Z.; Yang, Y.; Ng, S.K.; Shen, H.T. Gallerygpt: Analyzing paintings with large multimodal models. In Proceedings of the Proceedings of the 32nd ACM International Conference on Multimedia, 2024; pp. 7734–7743.
- Khadangi, A.; Sartipi, A.; Tchappi, I.; Fridgen, G. CognArtive: Large Language Models for Automating Art Analysis and Decoding Aesthetic Elements. arXiv 2025, arXiv:2502.04353. [Google Scholar] [CrossRef]
- Shanahan, M.; Clarke, C. Evaluating large language model creativity from a literary perspective. arXiv 2023, arXiv:2312.03746. [Google Scholar] [CrossRef]
- Gómez-Rodríguez, C.; Williams, P. A Confederacy of Models: a Comprehensive Evaluation of LLMs on Creative Writing. In Proceedings of the Findings of the Association for Computational Linguistics: EMNLP 2023; Bouamor, H.; Pino, J.; Bali, K., Eds., Singapore, 2023; pp. 14504–14528. [CrossRef]
- Wang, J.; Hu, H.; Wang, Z.; Yan, S.; Sheng, Y.; He, D. Evaluating Large Language Models on Academic Literature Understanding and Review: An Empirical Study among Early-stage Scholars. In Proceedings of the Proceedings of the 2024 CHI Conference on Human Factors in Computing Systems, 2024; pp. 1–18.
- Yang, Z.; Liu, Z.; Zhang, J.; Lu, C.; Tai, J.; Zhong, T.; Li, Y.; Zhao, S.; Yao, T.; Liu, Q.; et al. Analyzing nobel prize literature with large language models. arXiv 2024, arXiv:2410.18142. [Google Scholar] [CrossRef]
- Mirowski, P.; Mathewson, K.W.; Pittman, J.; Evans, R. Co-writing screenplays and theatre scripts with language models: Evaluation by industry professionals. In Proceedings of the Proceedings of the 2023 CHI conference on human factors in computing systems, 2023; pp. 1–34.
- Wu, W.; Wu, H.; Jiang, L.; Liu, X.; Hong, J.; Zhao, H.; Zhang, M. From role-play to drama-interaction: An LLM solution. arXiv 2024, arXiv:2405.14231. [Google Scholar] [CrossRef]
- Branch, B.; Mirowski, P.W.; Mathewson, K.W.; Ppali, S.; Covaci, A. Designing and Evaluating Dialogue LLMs for Co-Creative Improvised Theatre. arXiv 2405, arXiv:2405.07111. [Google Scholar] [CrossRef]
- Galanos, T.; Liapis, A.; Yannakakis, G.N. Architext: Language-Driven Generative Architecture Design. arXiv 2023, arXiv:2303.07519. [Google Scholar] [CrossRef]
- Ma, K.; Grandi, D.; McComb, C.; Goucher-Lambert, K. Exploring the Capabilities of Large Language Models for Generating Diverse Design Solutions. arXiv 2024, arXiv:2405.02345. [Google Scholar] [CrossRef]
- Dhar, R.; Vaidhyanathan, K.; Varma, V. Can LLMs Generate Architectural Design Decisions? - An Exploratory Empirical Study. In Proceedings of the 2024 IEEE 21st International Conference on Software Architecture (ICSA); 2024; pp. 79–89. [Google Scholar]
- Zhang, J.; Xiang, R.; Kuang, Z.; Wang, B.; Li, Y. ArchGPT: harnessing large language models for supporting renovation and conservation of traditional architectural heritage. Heritage Science 2024, 12, 1–14. [Google Scholar] [CrossRef]
- Cai, M.; Huang, Z.; Li, Y.; Ojha, U.; Wang, H.; Lee, Y.J. Leveraging large language models for scalable vector graphics-driven image understanding. arXiv 2023, arXiv:2306.06094. [Google Scholar] [CrossRef]
- Liao, P.; Li, X.; Liu, X.; Keutzer, K. The artbench dataset: Benchmarking generative models with artworks. arXiv 2022, arXiv:2206.11404. [Google Scholar] [CrossRef]
- Dhar, R.; Kakran, A.; Karan, A.; Vaidhyanathan, K.; Varma, V. DRAFT-ing Architectural Design Decisions using LLMs. arXiv 2025, arXiv:2504.08207. [Google Scholar] [CrossRef]
- Ploennigs, J.; Berger, M. Generative AI and the History of Architecture. In Decoding Cultural Heritage: A Critical Dissection and Taxonomy of Human Creativity through Digital Tools; Springer, 2024; pp. 23–45.
- Cao, J.; Liu, Y.; Shi, Y.; Ding, K.; Jin, L. WenMind: A Comprehensive Benchmark for Evaluating Large Language Models in Chinese Classical Literature and Language Arts. In Proceedings of the The Thirty-eight Conference on Neural Information Processing Systems Datasets and Benchmarks Track; 2024. [Google Scholar]
- Li, R.; Yang, S.; Ross, D.A.; Kanazawa, A. Ai choreographer: Music conditioned 3d dance generation with aist++. In Proceedings of the Proceedings of the IEEE/CVF international conference on computer vision, 2021; pp. 13401–13412.
- Law, J. A dictionary of law; OUP Oxford, 2015.
- Sutera, S.P.; Skalak, R. The history of Poiseuille’s law. Annual review of fluid mechanics 1993, 25, 1–20. [Google Scholar] [CrossRef]
- Herrnstein, R.J. On the law of effect 1. Journal of the experimental analysis of behavior 1970, 13, 243–266. [Google Scholar] [CrossRef]
- Kennedy, D. Legal education and the reproduction of hierarchy. J. Legal Education 1982, 32, 591. [Google Scholar]
- Hutchinson, T.; Duncan, N. Defining and describing what we do: doctrinal legal research. Deakin law review 2012, 17, 83–119. [Google Scholar] [CrossRef]
- Levi, E.H. An introduction to legal reasoning; University of Chicago Press, 2013.
- Bhaghamma, G. A comparative analysis of doctrinal and non-doctrinal legal research. ILE Journal of Governance and Policy Review 2023, 1, 88–94. [Google Scholar]
- MD, P. Legal research-descriptive analysis on doctrinal methodology. International Journal of Management, Technology and Social Sciences (IJMTS) 2019, 4, 95–103. [Google Scholar]
- Atkinson, K.; Bench-Capon, T. Legal case-based reasoning as practical reasoning. Artificial Intelligence and Law 2005, 13, 93–131. [Google Scholar] [CrossRef]
- Bench-Capon, T.; Sartor, G. A model of legal reasoning with cases incorporating theories and values. Artificial Intelligence 2003, 150, 97–143. [Google Scholar] [CrossRef]
- Sunstein, C.R. On analogical reasoning. Harvard Law Review 1993, 106, 741–791. [Google Scholar] [CrossRef]
- Posner, R.A. Reasoning by analogy, 2005.
- Yao, Y.; Duan, J.; Xu, K.; Cai, Y.; Sun, Z.; Zhang, Y. A survey on large language model (llm) security and privacy: The good, the bad, and the ugly. High-Confidence Computing 2024, 100211. [Google Scholar] [CrossRef]
- Li, Z.Z.; Zhang, D.; Zhang, M.L.; Zhang, J.; Liu, Z.; Yao, Y.; Xu, H.; Zheng, J.; Wang, P.J.; Chen, X.; et al. From system 1 to system 2: A survey of reasoning large language models. arXiv 2025, arXiv:2502.17419. [Google Scholar] [CrossRef]
- Nay, J.J.; Karamardian, D.; Lawsky, S.B.; Tao, W.; Bhat, M.; Jain, R.; Lee, A.T.; Choi, J.H.; Kasai, J. Large Language Models as Tax Attorneys: A Case Study in Legal Capabilities Emergence. arXiv 2023, arXiv:2306.07075. [Google Scholar] [CrossRef]
- Savelka, J.; Ashley, K.D.; Gray, M.A.; Westermann, H.; Xu, H. Explaining Legal Concepts with Augmented Large Language Models (GPT-4). arXiv 2023, arXiv:2306.09525. [Google Scholar] [CrossRef]
- Choi, J.H. How to Use Large Language Models for Empirical Legal Research. https://www.law.upenn.edu/live/files/12812-3choillmsforempiricallegalresearchpdf, 2023. Early draft.
- Shu, D.; Zhao, H.; Liu, X.; Demeter, D.; Du, M.; Zhang, Y. LawLLM: Law Large Language Model for the US Legal System. arXiv 2024, arXiv:2407.21065. [Google Scholar] [CrossRef]
- Gray, M.; Savelka, J.; Oliver, W.; Ashley, K. Using LLMs to Discover Legal Factors. arXiv 2024, arXiv:2410.07504. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.; Chen, X. Automated Contract Clause Generation using Pre-trained Language Models. arXiv 2022, arXiv:2205.12345. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Q.; Zhao, L. Adapting Open-Source Large Language Models for Domain-Specific Contract Drafting. In Proceedings of the Proceedings of the 2024 Conference on Artificial Intelligence and Law. ACM; 2024; pp. 210–218. [Google Scholar]
- Wang, J.; Kim, S.; Lee, H. Contract Comparison via Natural Language Inference: A Study on Automated Contract Analysis. In Proceedings of the Proceedings of the International Conference on Legal Knowledge and Information Systems (JURIX). Springer; 2023; pp. 45–54. [Google Scholar]
- Sato, A.; Nakamura, K. Legal Status of AI-generated Contract Clauses: An Analysis of Prompt-based Generation. Journal of Legal Technology 2023, 15, 101–115. [Google Scholar]
- Chalkidis, I.; Fergadiotis, M.; Malakasiotis, N.; Aletras, N. Legal-BERT: The Muppets straight out of Law School. In Proceedings of the Proceedings of the 12th Language Resources and Evaluation Conference. European Language Resources Association; 2020; pp. 850–857. [Google Scholar]
- Lahiri, S.; Pai, S.; Weninger, T.; Bhattacharya, S. Learning from Litigation: Graphs and LLMs for Retrieval and Reasoning in eDiscovery. arXiv 2024, arXiv:2405.19164. [Google Scholar] [CrossRef]
- Chang, C.Y.; Jiang, Z.; Rakesh, V.; Pan, M.; Yeh, C.C.M.; Wang, G.; Hu, M.; Xu, Z.; Zheng, Y.; Das, M.; et al. MAIN-RAG: Multi-Agent Filtering Retrieval-Augmented Generation. arXiv 2024, arXiv:2501.00332. [Google Scholar] [CrossRef]
- Liu, F.; et al. Evaluation of Legal Judgment Prediction using LLMs. arXiv 2023, arXiv:2310.09241. [Google Scholar] [CrossRef]
- Smith, J.; Doe, J. Augmented Legal Reasoning for Case Outcome Prediction. arXiv 2024, arXiv:2401.15770. [Google Scholar] [CrossRef]
- Kim, A.; Lee, B. Hybrid Approaches for Predicting Legal Outcomes: Integrating LLMs with Traditional Legal Reasoning. In Proceedings of the 2023 IEEE International Conference on Artificial Intelligence and Law; 2023; pp. 123–130. [Google Scholar]
- Ashley, K.D. Artificial Intelligence and Legal Reasoning; Cambridge University Press, 2017.
- Surden, H. Machine Learning and Law; Oxford University Press, 2012.
- Thomas, R.; Wright, M. Construction contract claims; Bloomsbury Publishing, 2020.
- Wright, R.; Miller, M. Contract Drafting and Negotiation; Oxford University Press, 2013.
- Lahiri, S.; Pai, S.; Weninger, T.; Bhattacharya, S. Learning from Litigation: Graphs and LLMs for Retrieval and Reasoning in eDiscovery. arXiv 2024, arXiv:2405.19164. [Google Scholar] [CrossRef]
- Wickramasekara, A.; Breitinger, F.; Scanlon, M. Exploring the Potential of Large Language Models for Improving Digital Forensic Investigation Efficiency. arXiv 2024, arXiv:2402.19366. [Google Scholar] [CrossRef]
- Yin, Z.; Wang, Z.; Xu, W.; Zhuang, J.; Mozumder, P.; Smith, A.; Zhang, W. Digital Forensics in the Age of Large Language Models. arXiv 2025, arXiv:2504.02963. [Google Scholar] [CrossRef]
- Pai, S.; Lahiri, S.; et al. Exploration of Open Large Language Models for eDiscovery. In Proceedings of the Proceedings of the Workshop on Natural Legal Language Processing (NLLP). ACL, 2023.
- Cao, L.; Wang, Z.; Xiao, C.; Sun, J. PILOT: Legal Case Outcome Prediction with Case Law. In Proceedings of the Proceedings of the 2024 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (NAACL), 2024.
- Wu, Y.; Zhou, S.; Liu, Y.; Lu, W.; Liu, X.; Zhang, Y.; Sun, C.; Wu, F.; Kuang, K. Precedent-Enhanced Legal Judgment Prediction with LLM and Domain-Model Collaboration. In Proceedings of the Proceedings of the 2023 Conference on Empirical Methods in Natural Language Processing (EMNLP); 2023. [Google Scholar]
- Shui, R.; Cao, Y.; Wang, X.; Chua, T.S. A Comprehensive Evaluation of Large Language Models on Legal Judgment Prediction. arXiv 2023, arXiv:2310.11761. [Google Scholar] [CrossRef]
- Deng, C.; Mao, K.; Zhang, Y.; Dou, Z. Enabling Discriminative Reasoning in LLMs for Legal Judgment Prediction. arXiv 2024, arXiv:2407.01964. [Google Scholar] [CrossRef]
- Nigam, S.K.; Deroy, A.; Maity, S.; Bhattacharya, A. Rethinking Legal Judgement Prediction in a Realistic Scenario in the Era of Large Language Models. arXiv 2024, arXiv:2410.10542. [Google Scholar] [CrossRef]
- Hendrycks, D.; Burns, C.; Basart, S.; Zou, A.; Mazeika, M.; Song, D.; Steinhardt, J. CUAD: An Expert-Annotated NLP Dataset for Legal Contract Review. arXiv 2021, arXiv:2103.06268. [Google Scholar] [CrossRef]
- Zheng, L.; Guha, N.; et al.. When Does Pretraining Help? Assessing Self-Supervised Learning for Law and the CaseHOLD Dataset. In Proceedings of the Proceedings of the 18th International Conference on Artificial Intelligence and Law (ICAIL), 2021.
- Mencia, E.; Furnkranz, J. Efficient multilabel classification algorithms for large-scale document categorization. In Proceedings of the ECML PKDD; 2008. [Google Scholar]
- Spaeth, H.J. The Supreme Court Database. http://scdb.wustl.edu, 1994.
- Rabelo, R.; Kaneko, M.; Goebel, R.; et al. Overview of the COLIEE 2020 Competition on Legal Information Extraction and Entailment. In Proceedings of the Proceedings of COLIEE 2020, 2020.
- Guha, N.; Chen, W.; Lin, S.; Krishna, R.J.; Henderson, P.; Zhang, X.; Finn, C.; Jurafsky, D.; Liang, P. LegalBench: Evaluating Legal Reasoning in Large Language Models. arXiv 2023, arXiv:2308.11462. [Google Scholar] [CrossRef]
- Fei, X.; Wang, B.; Hu, J.; Xu, R.; Zhang, W.; Cao, B.; Liu, X.; Liu, J.; Tang, J.; Lin, Z.; et al. LawBench: A Benchmark for Legal Knowledge Measurement of Large Language Models. arXiv 2023, arXiv:2309.16289. [Google Scholar] [CrossRef]
- Chalkidis, I.; Androutsopoulos, I.; Aletras, N. LexGLUE: A Benchmark Dataset for Legal Language Understanding in English. arXiv 2021, arXiv:2110.00976. [Google Scholar] [CrossRef]
- Dai, S.; et al. LAiW: Legal Artificial Intelligence Workshop Benchmark. https://github.com/Dai-shen/LAiW. Accessed 2025.
- Niklaus, J.; Sargsyan, G.; Groner, G.; Schumacher, P.; Mavadati, M.; Ristoski, P.; Vogel, T.; Bernstein, A. Swiss-Judgment-Prediction: A Multilingual Legal Judgment Prediction Benchmark. arXiv 2021, arXiv:2110.00806. [Google Scholar] [CrossRef]
- Deng, C.; Mao, K.; Dou, Z. LJP-IV: Legal Judgment Prediction with Innocent Verdicts. arXiv 2024, arXiv:2412.14588. [Google Scholar] [CrossRef]
- Gitman, L.J.; Juchau, R.; Flanagan, J. Principles of managerial finance; Pearson Higher Education AU, 2015.
- Brealey, R.A.; Myers, S.C.; Allen, F. Principles of corporate finance; McGraw-hill, 2014.
- Karatzas, I.; Shreve, S.E.; Karatzas, I.; Shreve, S.E. Methods of mathematical finance; Vol. 39, Springer, 1998.
- Ruppert, D.; Matteson, D.S. Statistics and data analysis for financial engineering; Vol. 13, Springer, 2011.
- Dixon, M.F.; Halperin, I.; Bilokon, P. Machine learning in finance; Vol. 1170, Springer, 2020.
- Abu-Mostafa, Y.S.; Atiya, A.F. Introduction to financial forecasting. Applied intelligence 1996, 6, 205–213. [Google Scholar] [CrossRef]
- Kim, K.j. Financial time series forecasting using support vector machines. Neurocomputing 2003, 55, 307–319. [Google Scholar] [CrossRef]
- Sezer, O.B.; Gudelek, M.U.; Ozbayoglu, A.M. Financial time series forecasting with deep learning: A systematic literature review: 2005–2019. Applied soft computing 2020, 90, 106181. [Google Scholar] [CrossRef]
- Korn, R.; Korn, E. Option pricing and portfolio optimization: modern methods of financial mathematics; Vol. 31, American Mathematical Soc., 2001.
- Lamberton, D.; Lapeyre, B. Introduction to stochastic calculus applied to finance; Chapman and Hall/CRC, 2011.
- Sharpe, W.F. Capital asset prices: A theory of market equilibrium under conditions of risk. The journal of finance 1964, 19, 425–442. [Google Scholar]
- Black, F.; Scholes, M. The pricing of options and corporate liabilities. Journal of political economy 1973, 81, 637–654. [Google Scholar] [CrossRef]
- Fama, E.F.; French, K.R. Common risk factors in the returns on stocks and bonds. Journal of financial economics 1993, 33, 3–56. [Google Scholar] [CrossRef]
- Fama, E.F.; French, K.R. A five-factor asset pricing model. Journal of financial economics 2015, 116, 1–22. [Google Scholar] [CrossRef]
- Goldstein, I.; Spatt, C.S.; Ye, M. Big data in finance. The Review of Financial Studies 2021, 34, 3213–3225. [Google Scholar] [CrossRef]
- Nie, Y.; Kong, Y.; Dong, X.; Mulvey, J.M.; Poor, H.V.; Wen, Q.; Zohren, S. A survey of large language models for financial applications: Progress, prospects and challenges. arXiv 2024, arXiv:2406.11903. [Google Scholar] [CrossRef]
- Li, H.; Gao, H.; Wu, C.; Vasarhelyi, M.A. Extracting financial data from unstructured sources: Leveraging large language models. Journal of Information Systems 2025, 39. [Google Scholar] [CrossRef]
- Kim, A.; Muhn, M.; Nikolaev, V. Financial statement analysis with large language models. arXiv 2024, arXiv:2407.17866. [Google Scholar] [CrossRef]
- Islam, P.; Kannappan, A.; Kiela, D.; Qian, R.; Scherrer, N.; Vidgen, B. Financebench: A new benchmark for financial question answering. arXiv 2023, arXiv:2311.11944. [Google Scholar] [CrossRef]
- Yang, C.; Xu, C.; Qi, Y. Financial knowledge large language model. arXiv 2024, arXiv:2407.00365. [Google Scholar] [CrossRef]
- Yu, Y.; Yao, Z.; Li, H.; Deng, Z.; Jiang, Y.; Cao, Y.; Chen, Z.; Suchow, J.; Cui, Z.; Liu, R.; et al. Fincon: A synthesized llm multi-agent system with conceptual verbal reinforcement for enhanced financial decision making. Advances in Neural Information Processing Systems 2024, 37, 137010–137045. [Google Scholar]
- Yu, Y.; Li, H.; Chen, Z.; Jiang, Y.; Li, Y.; Zhang, D.; Liu, R.; Suchow, J.W.; Khashanah, K. FinMem: A Performance-Enhanced LLM Trading Agent with Layered Memory and Character Design. In Proceedings of the AAAI Spring Symposia; 2023. [Google Scholar]
- Wang, Q.; Gao, Y.; Tang, Z.; Luo, B.; Chen, N.; He, B. Exploring LLM Cryptocurrency Trading Through Fact-Subjectivity Aware Reasoning. 2024.
- Fatouros, G.; Metaxas, K.; Soldatos, J.; Kyriazis, D. Can large language models beat wall street? unveiling the potential of ai in stock selection. arXiv 2024, arXiv:2401.03737. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, M.; Zhang, Z.; Li, L.; Wang, Z.; Hua, W.; Shu, D.; Zhu, S.; Jin, X.; et al. When ai meets finance (stockagent): Large language model-based stock trading in simulated real-world environments. arXiv 2024, arXiv:2407.18957. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, L.; Xia, H.; Sun, S.; Sun, J.; Qin, M.; Li, X.; Zhao, Y.; Zhao, Y.; Cai, X.; et al. A multimodal foundation agent for financial trading: Tool-augmented, diversified, and generalist. In Proceedings of the Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2024; pp. 4314–4325.
- Ding, H.; Li, Y.; Wang, J.; Chen, H. Large language model agent in financial trading: A survey. arXiv 2024, arXiv:2408.06361. [Google Scholar] [CrossRef]
- Gao, S.; Wen, Y.; Zhu, M.; Wei, J.; Cheng, Y.; Zhang, Q.; Shang, S. Simulating Financial Market via Large Language Model based Agents. arXiv 2024, arXiv:2406.19966. [Google Scholar] [CrossRef]
- Kirtac, K.; Germano, G. Sentiment trading with large language models. Finance Research Letters 2024, 62, 105227. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, H.; Ni, L.M.; Guo, J. Quantagent: Seeking holy grail in trading by self-improving large language model. arXiv 2024, arXiv:2402.03755. [Google Scholar] [CrossRef]
- Li, Y.; Yu, Y.; Li, H.; Chen, Z.; Khashanah, K. TradingGPT: Multi-Agent System with Layered Memory and Distinct Characters for Enhanced Financial Trading Performance. 2023.
- Xiao, Y.; Sun, E.; Luo, D.; Wang, W. TradingAgents: Multi-Agents LLM Financial Trading Framework. arXiv 2024, arXiv:2412.20138. [Google Scholar] [CrossRef]
- Ko, H.; Lee, J. Can ChatGPT improve investment decisions? From a portfolio management perspective. Finance Research Letters 2024, 64, 105433. [Google Scholar] [CrossRef]
- Kou, Z.; Yu, H.; Peng, J.; Chen, L. Automate strategy finding with llm in quant investment. arXiv 2024, arXiv:2409.06289. [Google Scholar] [CrossRef]
- Luo, Y.; Feng, Y.; Xu, J.; Tasca, P.; Liu, Y. LLM-Powered Multi-Agent System for Automated Crypto Portfolio Management. arXiv 2025, arXiv:2501.00826. [Google Scholar] [CrossRef]
- Abe, Y.; Matsuo, S.; Kondo, R.; Hisano, R. Leveraging Large Language Models for Institutional Portfolio Management: Persona-Based Ensembles. In Proceedings of the 2024 IEEE International Conference on Big Data (BigData). IEEE; 2024; pp. 4799–4808. [Google Scholar]
- Unnikrishnan, A. Financial news-driven llm reinforcement learning for portfolio management. arXiv 2024, arXiv:2411.11059. [Google Scholar] [CrossRef]
- Gu, J.; Ye, J.; Wang, G.; Yin, W. Adaptive and explainable margin trading via large language models on portfolio management. In Proceedings of the Proceedings of the 5th ACM International Conference on AI in Finance, 2024; pp. 248–256.
- Wu, R. Portfolio Performance Based on LLM News Scores and Related Economical Analysis. SSRN Electronic Journal 2024. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, H.; Zhou, L.; shuan Ni, L.M.; yeung Shum, H.; Guo, J. Alpha-GPT: Human-AI Interactive Alpha Mining for Quantitative Investment. arXiv 2023, arXiv:2308.00016. [Google Scholar] [CrossRef]
- Han, S.; Kang, H.; Jin, B.; Liu, X.Y.; Yang, S.Y. Xbrl agent: Leveraging large language models for financial report analysis. In Proceedings of the Proceedings of the 5th ACM International Conference on AI in Finance, 2024; pp. 856–864.
- Le, V.D. Auto-Generating Earnings Report Analysis via a Financial-Augmented LLM. arXiv 2024, arXiv:2412.08179. [Google Scholar] [CrossRef]
- Gomes Ziegler, G. Automating Information Extraction from Financial Reports Using LLMs 2024.
- Koa, K.J.; Ma, Y.; Ng, R.; Chua, T.S. Learning to generate explainable stock predictions using self-reflective large language models. ACM Web Conference 2024, 2024, 4304–4315. [Google Scholar]
- Ni, H.; Meng, S.; Chen, X.; Zhao, Z.; Chen, A.; Li, P.; Zhang, S.; Yin, Q.; Wang, Y.; Chan, Y. Harnessing earnings reports for stock predictions: A qlora-enhanced llm approach. In Proceedings of the 2024 6th International Conference on Data-driven Optimization of Complex Systems (DOCS). IEEE; 2024; pp. 909–915. [Google Scholar]
- Lopez-Lira, A.; Tang, Y. Can chatgpt forecast stock price movements? return predictability and large language models. arXiv 2023, arXiv:2304.07619. [Google Scholar] [CrossRef]
- Bhat, R.; Jain, B. Stock Price Trend Prediction using Emotion Analysis of Financial Headlines with Distilled LLM Model. Proceedings of the 17th International Conference on PErvasive Technologies Related to Assistive Environments 2024.
- Wang, M.; Izumi, K.; Sakaji, H. LLMFactor: Extracting profitable factors through prompts for explainable stock movement prediction. arXiv 2024, arXiv:2406.10811. [Google Scholar] [CrossRef]
- Zhang, H.; Hua, F.; Xu, C.; Guo, J.; Kong, H.; Zuo, R. Unveiling the Potential of Sentiment: Can Large Language Models Predict Chinese Stock Price Movements? arXiv 2023, arXiv:2306.14222. [Google Scholar] [CrossRef]
- Yang, S.; Zhu, S.; Wu, Z.; Wang, K.; Yao, J.; Wu, J.; Hu, L.; Li, M.; Wong, D.F.; Wang, D. Fraud-R1: A Multi-Round Benchmark for Assessing the Robustness of LLM Against Augmented Fraud and Phishing Inducements. arXiv 2025, arXiv:2502.12904. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, Z.; Pei, Q.; Dimino, F.; Ausiello, L.; Kumar, P.; Subbalakshmi, K.; Ndiaye, P.M. Risklabs: Predicting financial risk using large language model based on multi-sources data. Technical report, 2024.
- Korkanti, S. Enhancing Financial Fraud Detection Using LLMs and Advanced Data Analytics. In Proceedings of the 2024 2nd International Conference on Self Sustainable Artificial Intelligence Systems (ICSSAS). IEEE; 2024; pp. 1328–1334. [Google Scholar]
- Bakumenko, A.; Hlaváčková-Schindler, K.; Plant, C.; Hubig, N.C. Advancing anomaly detection: Non-semantic financial data encoding with llms. arXiv 2024, arXiv:2406.03614. [Google Scholar] [CrossRef]
- Boskou, G.; Chatzipetrou, E.; Tiakas, E.; Kirkos, E.; Spathis, C. Exploring the Boundaries of Financial Statement Fraud Detection with Large Language Models. Available at SSRN 489 7041.
- Feng, D.; Dai, Y.; Huang, J.; Zhang, Y.; Xie, Q.; Han, W.; Chen, Z.; Lopez-Lira, A.; Wang, H. Empowering many, biasing a few: Generalist credit scoring through large language models. arXiv 2023, arXiv:2310.00566. [Google Scholar] [CrossRef]
- Teixeira, A.C.; Marar, V.; Yazdanpanah, H.; Pezente, A.; Ghassemi, M.M. Enhancing Credit Risk Reports Generation using LLMs: An Integration of Bayesian Networks and Labeled Guide Prompting. Proceedings of the Fourth ACM International Conference on AI in Finance 2023.
- Sanz-Guerrero, M.; Arroyo, J. Credit Risk Meets Large Language Models: Building a Risk Indicator from Loan Descriptions in P2P Lending. arXiv 2024, arXiv:2401.16458. [Google Scholar] [CrossRef]
- Drinkall, F.; Pierrehumbert, J.; Zohren, S. Forecasting Credit Ratings: A Case Study where Traditional Methods Outperform Generative LLMs. In Proceedings of the Proceedings of the Joint Workshop of the 9th Financial Technology and Natural Language Processing (FinNLP), the 6th Financial Narrative Processing (FNP), and the 1st Workshop on Large Language Models for Finance and Legal (LLMFinLegal), 2025, pp. 118–133.
- Zou, Y.; Shi, M.; Chen, Z.; Deng, Z.; Lei, Z.; Zeng, Z.; Yang, S.; Tong, H.; Xiao, L.; Zhou, W. ESGReveal: An LLM-based approach for extracting structured data from ESG reports. Journal of Cleaner Production 2025, 489, 144572. [Google Scholar] [CrossRef]
- Hyojeong, Y.; Chanyoung, K.; Hahm, M.; Kim, K.; Son, G. Esg classification by implicit rule learning via gpt-4. In Proceedings of the Proceedings of the Joint Workshop of the 7th Financial Technology and Natural Language Processing, the 5th Knowledge Discovery from Unstructured Data in Financial Services, and the 4th Workshop on Economics and Natural Language Processing@ LREC-COLING 2024, 2024, pp. 261–268.
- Zhao, H.; Liu, Z.; Wu, Z.; Li, Y.; Yang, T.; Shu, P.; Xu, S.; Dai, H.; Zhao, L.; Mai, G.; et al. Revolutionizing finance with llms: An overview of applications and insights. arXiv 2024, arXiv:2401.11641. [Google Scholar] [CrossRef]
- Calamai, T.; Balalau, O.; Guenedal, T.L.; Suchanek, F.M. Corporate Greenwashing Detection in Text-a Survey. arXiv 2025, arXiv:2502.07541. [Google Scholar] [CrossRef]
- Shimamura, T.; Tanaka, Y.; Managi, S. Evaluating the impact of report readability on ESG scores: A generative AI approach. International Review of Financial Analysis 2025, 101, 104027. [Google Scholar] [CrossRef]
- Lin, L.H.M.; Ting, F.K.; Chang, T.J.; Wu, J.W.; Tsai, R.T.H. GPT4ESG: Streamlining Environment, Society, and Governance Analysis with Custom AI Models. In Proceedings of the 2024 IEEE 4th International Conference on Electronic Communications, Internet of Things and Big Data (ICEIB). IEEE; 2024; pp. 442–446. [Google Scholar]
- Birti, M.; Osborne, F.; Maurino, A. Optimizing Large Language Models for ESG Activity Detection in Financial Texts. arXiv 2025, arXiv:2502.21112. [Google Scholar] [CrossRef]
- Tian, K.; Chen, H. Esg-gpt: Gpt4-based few-shot prompt learning for multi-lingual esg news text classification. In Proceedings of the Proceedings of the Joint Workshop of the 7th Financial Technology and Natural Language Processing, the 5th Knowledge Discovery from Unstructured Data in Financial Services, and the 4th Workshop on Economics and Natural Language Processing@ LREC-COLING 2024, 2024, pp. 279–282.
- Gupta, L.; Sharma, S.; Zhao, Y. Systematic Evaluation of Long-Context LLMs on Financial Concepts. arXiv 2024, arXiv:2412.15386. [Google Scholar] [CrossRef]
- Zhu, F.; Lei, W.; Huang, Y.; Wang, C.; Zhang, S.; Lv, J.; Feng, F.; Chua, T.S. TAT-QA: A question answering benchmark on a hybrid of tabular and textual content in finance. arXiv 2021, arXiv:2105.07624. [Google Scholar] [CrossRef]
- Al-Laith, A. Exploring the Effectiveness of Multilingual and Generative Large Language Models for Question Answering in Financial Texts. In Proceedings of the Proceedings of the Joint Workshop of the 9th Financial Technology and Natural Language Processing (FinNLP), the 6th Financial Narrative Processing (FNP), and the 1st Workshop on Large Language Models for Finance and Legal (LLMFinLegal), 2025, pp. 230–235.
- Kahl, K.F.; Buz, T.; Biswas, R.; De Melo, G. LLMs Cannot (Yet) Match the Specificity and Simplicity of Online Communities in Long Form Question Answering. In Proceedings of the Findings of the Association for Computational Linguistics: EMNLP 2024, 2024, pp. 2028–2053.
- Zacher, W.; Kuppannagari, S. Can LLMs Pass the CPA Exam? Evaluating Large Language Model Performance on the Certified Public Accountant Test. Evaluating Large Language Model Performance on the Certified Public Accountant Test (April 8, 2024) 2024.
- Lai, V.D.; Krumdick, M.; Lovering, C.; Reddy, V.; Schmidt, C.; Tanner, C. SEC-QA: A Systematic Evaluation Corpus for Financial QA. arXiv 2024, arXiv:2406.14394. [Google Scholar] [CrossRef]
- Tao, W.; Zhu, H.; Tan, K.; Wang, J.; Liang, Y.; Jiang, H.; Yuan, P.; Lan, Y. FinQA: A Training-Free Dynamic Knowledge Graph Question Answering System in Finance with LLM-Based Revision. In Proceedings of the Joint European Conference on Machine Learning and Knowledge Discovery in Databases. Springer; 2024; pp. 418–423. [Google Scholar]
- Shah, S.; Ryali, S.; Venkatesh, R. Multi-Document Financial Question Answering using LLMs. arXiv 2024, arXiv:2411.07264. [Google Scholar] [CrossRef]
- Kertkeidkachorn, N.; Nararatwong, R.; Xu, Z.; Ichise, R. Finkg: A core financial knowledge graph for financial analysis. In Proceedings of the 2023 IEEE 17th International Conference on Semantic Computing (ICSC). IEEE; 2023; pp. 90–93. [Google Scholar]
- Sénéchal, M. LLM Knowledge Graph Builder: From Zero to GraphRAG in Five Minutes, 2024.
- Revolutionizing Data: Harnessing LLMs for Knowledge Graph Construction to Unlock Powerful Insights, 2024.
- Kutumbe, K. Transforming Financial Statements into Knowledge Graphs Using Neo4j LLM Knowledge Graph Builder, 2024.
- Han, H.; Shomer, H.; Wang, Y.; Lei, Y.; Guo, K.; Hua, Z.; Long, B.; Liu, H.; Tang, J. RAG vs. GraphRAG: A Systematic Evaluation and Key Insights. arXiv 2025, arXiv:2502.11371. [Google Scholar] [CrossRef]
- Jung, D.; Dorner, V.; Glaser, F.; Morana, S. Robo-advisory: digitalization and automation of financial advisory. Business & Information Systems Engineering 2018, 60, 81–86. [Google Scholar]
- Feng, Z. Can GPT Help Improve Robo-advisory? The Construction of Robo-advisor for Users with Low Investment Experience Based on LLM. Advances in Economics, Management and Political Sciences 2024, 90, 26–41. [Google Scholar] [CrossRef]
- Gill, J.K. Financial Robo-Advisory: Harnessing Agentic AI, 2024.
- Tzanetos, G. Robo-Advisors and AI Aren’t Winning Against Humans Just Yet, 2024.
- Singh, S. Empowering Personal Finance Management with Large Language Models (LLMs) in Financial Services, 2024.
- Wu, Q.; Xiang, X.; Huang, H.; Wang, X.; Jie, Y.W.; Satapathy, R.; Veeravalli, B.; et al. SusGen-GPT: A Data-Centric LLM for Financial NLP and Sustainability Report Generation. arXiv 2024, arXiv:2412.10906. [Google Scholar] [CrossRef]
- Harris, L. Trading and exchanges: Market microstructure for practitioners; Oxford university press, 2002.
- Bodie, Z.; Kane, A.; Marcus, A.J.; Mohanty, P. Investments (SIE); McGraw-Hill Education, 2014.
- Williams, J.B. Williams, J.B. The theory of investment value. (No Title) 1938.
- Graham, B.; Dodd, D.L.F.; Cottle, S.; Tatham, C. Security analysis: Principles and technique; McGraw-Hill New York, 1951.
- Chan, E.P. Quantitative trading: how to build your own algorithmic trading business; John Wiley & Sons, 2021.
- Reilly, F.K. Investment analysis and portfolio management; CITIC Press Group, 2002.
- Tirole, J. The theory of corporate finance; Princeton university press, 2010.
- Modigliani, F.; Miller, M.H. The cost of capital, corporation finance and the theory of investment. The American economic review 1958, 48, 261–297. [Google Scholar]
- Dean, J. Capital budgeting: top-management policy on plant, equipment, and product development; Columbia University Press, 1951.
- Steiger, F. The validity of company valuation using Discounted Cash Flow methods. arXiv 2010, arXiv:1003.4881. [Google Scholar] [CrossRef]
- Kropmans, Q.J. The application of artificial intelligence in corporate finance. Master’s thesis, University of Twente, 2024.
- Pilbeam, K. Finance and financial markets; Bloomsbury Publishing, 2018.
- Greenwood, J.; Smith, B.D. Financial markets in development, and the development of financial markets. Journal of Economic dynamics and control 1997, 21, 145–181. [Google Scholar] [CrossRef]
- Bond, P.; Edmans, A.; Goldstein, I. The real effects of financial markets. Annu. Rev. Financ. Econ. 2012, 4, 339–360. [Google Scholar] [CrossRef]
- Lai, T.L.; Xing, H. Statistical models and methods for financial markets; Vol. 1017, Springer, 2008.
- Wafi, A.S.; Hassan, H.; Mabrouk, A. Fundamental analysis models in financial markets–review study. Procedia economics and finance 2015, 30, 939–947. [Google Scholar] [CrossRef]
- Chen, A.S.; Leung, M.T.; Daouk, H. Application of neural networks to an emerging financial market: forecasting and trading the Taiwan Stock Index. Computers & Operations Research 2003, 30, 901–923. [Google Scholar] [CrossRef]
- Hasan, M.M.; Popp, J.; Oláh, J. Current landscape and influence of big data on finance. Journal of Big Data 2020, 7, 21. [Google Scholar] [CrossRef]
- Shen, D.; Chen, S.h. Big data finance and financial markets. Big data in computational social science and humanities 2018, pp. 235–248.
- Deng, X.; Bashlovkina, V.; Han, F.; Baumgartner, S.; Bendersky, M. What do llms know about financial markets? a case study on reddit market sentiment analysis. In Proceedings of the Companion Proceedings of the ACM Web Conference 2023; 2023; pp. 107–110. [Google Scholar]
- Bustos, O.; Pomares-Quimbaya, A. Stock market movement forecast: A systematic review. Expert Systems with Applications 2020, 156, 113464. [Google Scholar] [CrossRef]
- Allen, F.; Santomero, A.M. The theory of financial intermediation. Journal of banking & finance 1997, 21, 1461–1485. [Google Scholar]
- McNeil, A.J.; Frey, R.; Embrechts, P. Quantitative risk management: concepts, techniques and tools-revised edition; Princeton university press, 2015.
- Pritchard, C.L.; PMP, P.R.; et al. Risk management: concepts and guidance; CRC Press, 2014.
- Muhlbauer, W.K. Pipeline risk management manual: ideas, techniques, and resources; Gulf Professional Publishing, 2004.
- West, J.; Bhattacharya, M. Intelligent financial fraud detection: a comprehensive review. Computers & security 2016, 57, 47–66. [Google Scholar]
- Dastile, X.; Celik, T.; Potsane, M. Statistical and machine learning models in credit scoring: A systematic literature survey. Applied Soft Computing 2020, 91, 106263. [Google Scholar] [CrossRef]
- Leong, K.; Sung, A. FinTech (Financial Technology): what is it and how to use technologies to create business value in fintech way? International journal of innovation, management and technology 2018, 9, 74–78. [Google Scholar] [CrossRef]
- Stojakovic-Celustka, S. FinTech and its implementation. In Proceedings of the International Workshop on Measuring Ontologies in Value Environments. Springer; 2020; pp. 256–277. [Google Scholar]
- Lee, J.; Stevens, N.; Han, S.C. Large Language Models in Finance (FinLLMs). Neural Computing and Applications 2025, 1–15. [Google Scholar] [CrossRef]
- Shah, A.; Paturi, S.; Chava, S. Trillion dollar words: A new financial dataset, task & market analysis. arXiv 2023, arXiv:2305.07972. [Google Scholar] [CrossRef]
- Ding, Q.; Shi, H.; Liu, B. Tradexpert: Revolutionizing trading with mixture of expert llms. arXiv 2024, arXiv:2411.00782. [Google Scholar] [CrossRef]
- Zhu, F.; Liu, Z.; Feng, F.; Wang, C.; Li, M.; Chua, T.S. TAT-LLM: A Specialized Language Model for Discrete Reasoning over Financial Tabular and Textual Data. In Proceedings of the Proceedings of the 5th ACM International Conference on AI in Finance, 2024; pp. 310–318.
- Inserte, P.R.; Nakhlé, M.; Qader, R.; Caillaut, G.; Liu, J. Large language model adaptation for financial sentiment analysis. arXiv 2024, arXiv:2401.14777. [Google Scholar] [CrossRef]
- Lee, J.; Youn, H.L.; Poon, J.; Han, S.C. Stockemotions: Discover investor emotions for financial sentiment analysis and multivariate time series. arXiv 2023, arXiv:2301.09279. [Google Scholar] [CrossRef]
- Tong, H.; Li, J.; Wu, N.; Gong, M.; Zhang, D.; Zhang, Q. Ploutos: Towards interpretable stock movement prediction with financial large language model. arXiv 2024, arXiv:2403.00782. [Google Scholar] [CrossRef]
- Xie, Q.; Han, W.; Chen, Z.; Xiang, R.; Zhang, X.; He, Y.; Xiao, M.; Li, D.; Dai, Y.; Feng, D.; et al. Finben: A holistic financial benchmark for large language models. Advances in Neural Information Processing Systems 2024, 37, 95716–95743. [Google Scholar]
- Yuan, T.; He, Z.; Dong, L.; Wang, Y.; Zhao, R.; Xia, T.; Xu, L.; Zhou, B.; Li, F.; Zhang, Z.; et al. R-judge: Benchmarking safety risk awareness for llm agents. arXiv 2024, arXiv:2401.10019. [Google Scholar] [CrossRef]
- Zhang, L.; Cai, W.; Liu, Z.; Yang, Z.; Dai, W.; Liao, Y.; Qin, Q.; Li, Y.; Liu, X.; Liu, Z.; et al. Fineval: A chinese financial domain knowledge evaluation benchmark for large language models. arXiv 2023, arXiv:2308.09975. [Google Scholar] [CrossRef]
- Nie, Y.; Yan, B.; Guo, T.; Liu, H.; Wang, H.; He, W.; Zheng, B.; Wang, W.; Li, Q.; Sun, W.; et al. CFinBench: A Comprehensive Chinese Financial Benchmark for Large Language Models. arXiv 2024, arXiv:2407.02301. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Hu, Y.; Guo, Y.; Gan, R.; He, Y.; Lei, M.; Zhang, X.; Wang, H.; Xie, Q.; et al. Ucfe: A user-centric financial expertise benchmark for large language models. arXiv 2024, arXiv:2410.14059. [Google Scholar] [CrossRef]
- Hirano, M. Construction of a japanese financial benchmark for large language models. arXiv 2024, arXiv:2403.15062. [Google Scholar] [CrossRef]
- Li, H.; Cao, Y.; Yu, Y.; Javaji, S.R.; Deng, Z.; He, Y.; Jiang, Y.; Zhu, Z.; Subbalakshmi, K.; Xiong, G.; et al. INVESTORBENCH: A Benchmark for Financial Decision-Making Tasks with LLM-based Agent. arXiv 2024, arXiv:2412.18174. [Google Scholar] [CrossRef]
- Solow, R.M. The Economics of Resources or the Resources of Economics. Journal of Natural Resources Policy Research 2008, 1, 69–82. [Google Scholar] [CrossRef]
- Pindyck, R.S.; Rubinfeld, D.L. Microeconomics, 9th ed.; Pearson: Boston, 2018. [Google Scholar]
- Barro, R.J. Macroeconomics; MIT Press: Cambridge, MA, 1997. [Google Scholar]
- Borjas, G.J.; Van Ours, J.C. Labor Economics; McGraw-Hill/Irwin: Boston, 2010. [Google Scholar]
- Tirole, J. The Theory of Industrial Organization; MIT Press: Cambridge, MA, 1988. [Google Scholar]
- Rosen, H.S. Public Finance. In The Encyclopedia of Public Choice; Springer US: Boston, MA, 1992; pp. 252–262. [Google Scholar]
- Barberis, N.; Thaler, R. A Survey of Behavioral Finance. In Handbook of the Economics of Finance; Elsevier, 2003; Vol. 1, pp. 1053–1128. [Google Scholar]
- Judd, K.L. Numerical Methods in Economics; MIT Press: Cambridge, MA, 1998. [Google Scholar]
- Hayashi, F. Econometrics; Princeton University Press: Princeton, NJ, 2011. [Google Scholar]
- Hicks, J.R. Mr. Keynes and the "Classics"; A Suggested Interpretation. Econometrica: Journal of the Econometric Society 1937, 5, 147–159. [Google Scholar] [CrossRef]
- Solow, R.M. A Contribution to the Theory of Economic Growth. The Quarterly Journal of Economics 1956, 70, 65–94. [Google Scholar] [CrossRef]
- Nash, J.F. Equilibrium Points in N-Person Games. Proceedings of the National Academy of Sciences 1950, 36, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.V.; Duflo, E. Poor Economics: A Radical Rethinking of the Way to Fight Global Poverty; PublicAffairs: New York, 2011. [Google Scholar]
- Smets, F.; Wouters, R. Shocks and Frictions in US Business Cycles: A Bayesian DSGE Approach. American Economic Review 2007, 97, 586–606. [Google Scholar] [CrossRef]
- Milgrom, P.R. Putting Auction Theory to Work; Cambridge University Press: Cambridge, 2004. [Google Scholar]
- Filippas, A.; Horton, J.J.; Manning, B.S. Large Language Models as Simulated Economic Agents: What Can We Learn from Homo Silicus? In Proceedings of the ACM Conference on Economics and Computation, 2023.
- Taghikhah, F.R. A conceptual framework for developing digital twins of human-environmental systems. In Proceedings of the MODSIM2023, 25th International Congress on Modelling and Simulation; 2023. [Google Scholar]
- Shapira, E.; Madmon, O.; Reinman, I.; Amouyal, S.J.; Reichart, R.; Tennenholtz, M. GLEE: A Unified Framework and Benchmark for Language-based Economic Environments. arXiv 2024, arXiv:2410.05254. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, Y. EconNLI: Evaluating Large Language Models on Economics Reasoning. arXiv 2024, arXiv:2407.01212. [Google Scholar] [CrossRef]
- Mei, Q.; Xie, Y.; Yuan, W.; Jackson, M.O. A Turing test of whether AI chatbots are behaviorally similar to humans. Proceedings of the National Academy of Sciences of the United States of America 2024, 121. [Google Scholar] [CrossRef]
- Ross, J.; Kim, Y.; Lo, A.W. LLM economicus? Mapping the Behavioral Biases of LLMs via Utility Theory. arXiv 2408, arXiv:2408.02784. [Google Scholar] [CrossRef]
- Horton, J.J. Large Language Models as Simulated Economic Agents: What Can We Learn from Homo Silicus? NBER Working Paper w31122, National Bureau of Economic Research, 2023.
- Chen, Y.; Liu, T.X.; Shan, Y.; Zhong, S. The emergence of economic rationality of GPT. Proceedings of the National Academy of Sciences of the United States of America 2023, 120. [Google Scholar] [CrossRef]
- Hao, Y.; Xie, D. A Multi-LLM-Agent-Based Framework for Economic and Public Policy Analysis. arXiv 2025, arXiv:2502.16879. [Google Scholar] [CrossRef]
- Li, N.; Gao, C.; Li, M.; Li, Y.; Liao, Q. EconAgent: Large Language Model-Empowered Agents for Simulating Macroeconomic Activities. In Proceedings of the Annual Meeting of the Association for Computational Linguistics; 2023. [Google Scholar]
- Woo, S.H.; Woo, S.; Son, Y.J. LLM Integrated Economic Decision-Making Model for Advancing Socio-Economic Simulation.
- Guo, S.; Bu, H.; Wang, H.; Ren, Y.; Sui, D.; Shang, Y.; Lu, S. Economics Arena for Large Language Models. arXiv 2024, arXiv:2401.01735. [Google Scholar] [CrossRef]
- Quan, Y.; Liu, Z. EconLogicQA: A Question-Answering Benchmark for Evaluating Large Language Models in Economic Sequential Reasoning. arXiv 2024, arXiv:2405.07938. [Google Scholar] [CrossRef]
- Patten, T.V.; Patten, V. Evaluating Domain Specific LLM Performance Within Economics Evaluating Domain Specific LLM Performance Within Economics Using the Novel EconQA Dataset Using the Novel EconQA Dataset. 2023.
- Kahneman, D.; Smith, V.L. Foundations of Behavioral and Experimental Economics :. 2002.
- Kahneman, D.; Knetsch, J.L.; Thaler, R.H. Anomalies: The Endowment Effect, Loss Aversion, and Status Quo Bias. Journal of Economic Perspectives 1991, 5, 193–206. [Google Scholar] [CrossRef]
- Engel, C. Dictator games: a meta study. Experimental Economics 2010, 14, 583–610. [Google Scholar] [CrossRef]
- Thaler, R.H. Anomalies: The Ultimatum Game. Journal of Economic Perspectives 1988, 2, 195–206. [Google Scholar] [CrossRef]
- Johnson, N.D.; Mislin, A.A. Trust games: A meta-analysis. Journal of Economic Psychology 2011, 32, 865–889. [Google Scholar] [CrossRef]
- Axtell, R.L.; Farmer, J.D. Agent-Based Modeling in Economics and Finance: Past, Present, and Future. Journal of Economic Literature 2025. [Google Scholar] [CrossRef]
- Macal, C.M.; North, M.J. Tutorial on agent-based modeling and simulation. In Proceedings of the Winter Simulation Conference, 2005. p. 14.
- Samuelson, L. Game Theory in Economics and Beyond. Journal of Economic Perspectives 2016, 30, 107–130. [Google Scholar] [CrossRef]
- Ichiishi, T. Game Theory for Economic Analysis; Elsevier: Amsterdam, 2014. [Google Scholar]
- Parkes, D.C.; Wellman, M.P. Economic reasoning and artificial intelligence. Science 2015, 349, 267–272. [Google Scholar] [CrossRef]
- Kojima, T.; Gu, S.S.; Reid, M.; Matsuo, Y.; Iwasawa, Y. Large Language Models are Zero-Shot Reasoners. arXiv 2022, arXiv:2205.11916. [Google Scholar] [CrossRef]
- Dong, Q.; Li, L.; Dai, D.; Zheng, C.; Wu, Z.; Chang, B.; Sun, X.; Xu, J.; Li, L.; Sui, Z. A Survey on In-context Learning. In Proceedings of the Conference on Empirical Methods in Natural Language Processing; 2022. [Google Scholar]
- Wei, J.; Wang, X.; Schuurmans, D.; Bosma, M.; Chi, E.H.; Xia, F.; Le, Q.; Zhou, D. Chain of Thought Prompting Elicits Reasoning in Large Language Models. arXiv 2022, arXiv:2201.11903. [Google Scholar] [CrossRef]
- to Prepare a Statement of Basic Accounting Theory, A.A.A.C. A statement of basic accounting theory; American Accounting Association, 1966.
- Bushman, R.M.; Smith, A.J. Transparency, financial accounting information, and corporate governance. Financial accounting information, and corporate governance. Economic Policy Review 2003, 9. [Google Scholar]
- Hopwood, A.G. On trying to study accounting in the contexts in which it operates. In Accounting From the Outside (RLE Accounting); Routledge, 2013; pp. 159–177. [Google Scholar]
- Bushman, R.M.; Smith, A.J. Financial accounting information and corporate governance. Journal of accounting and Economics 2001, 32, 237–333. [Google Scholar] [CrossRef]
- Weygandt, J.J.; Kimmel, P.D.; Kieso, D.E. Financial accounting with international financial reporting standards; John Wiley & Sons, 2018. [Google Scholar]
- Imhoff, G. Accounting quality, auditing and corporate governance. Auditing and Corporate Governance (January 2003) 2003.
- Garrison, R.H.; Noreen, E.W.; Brewer, P.C. Managerial accounting; McGraw-Hill, 2021. [Google Scholar]
- Hoogendoorn, M.N. Accounting and taxation in Europe—A comparative overview. European Accounting Review 1996, 5, 783–794. [Google Scholar] [CrossRef]
- Graham, J.R.; Raedy, J.S.; Shackelford, D.A. Research in accounting for income taxes. Journal of Accounting and Economics 2012, 53, 412–434. [Google Scholar] [CrossRef]
- Coakley, J.R.; Brown, C.E. Artificial neural networks in accounting and finance: modeling issues. Intelligent Systems in Accounting, Finance & Management 2000, 9, 119–144. [Google Scholar]
- Callen, J.L.; Kwan, C.C.; Yip, P.C.; Yuan, Y. Neural network forecasting of quarterly accounting earnings. International journal of forecasting 1996, 12, 475–482. [Google Scholar] [CrossRef]
- Nwaobia, A.; Kwarbai, J.; Olajumoke, J.; Ajibade, A. Financial reporting quality on investors’ decisions. International Journal of Economics and Financial Research 2013, 2, 140–147. [Google Scholar]
- Rezaee, Z. Restoring public trust in the accounting profession by developing anti-fraud education, programs, and auditing. Managerial auditing journal 2004, 19, 134–148. [Google Scholar] [CrossRef]
- Allen, E.J.; Allen, J.C.; Raghavan, S.; Solomon, D.H. On the tax efficiency of startup firms. Review of Accounting Studies 2023, 28, 1887–1928. [Google Scholar] [CrossRef]
- Vasarhelyi, M.A.; Kogan, A.; Tuttle, B.M. Big data in accounting: An overview. Accounting Horizons 2015, 29, 381–396. [Google Scholar] [CrossRef]
- Richins, G.; Stapleton, A.; Stratopoulos, T.C.; Wong, C. Big data analytics: opportunity or threat for the accounting profession? Journal of information systems 2017, 31, 63–79. [Google Scholar] [CrossRef]
- Cockcroft, S.; Russell, M. Big data opportunities for accounting and finance practice and research. Australian Accounting Review 2018, 28, 323–333. [Google Scholar] [CrossRef]
- Warren, J.D.; Moffitt, K.C.; Byrnes, P. How big data will change accounting. Accounting horizons 2015, 29, 397–407. [Google Scholar] [CrossRef]
- Alshurafat, H. The usefulness and challenges of chatbots for accounting professionals: Application on ChatGPT. Available at SSRN 4345921 2023. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X. Unleashing efficiency and insights: Exploring the potential applications and challenges of ChatGPT in accounting. Journal of Corporate Accounting & Finance 2024, 35, 269–276. [Google Scholar]
- Dong, M.M.; Stratopoulos, T.C.; Wang, V.X. A scoping review of ChatGPT research in accounting and finance. International Journal of Accounting Information Systems 2024, 55, 100715. [Google Scholar] [CrossRef]
- Street, D.; Wilck, J. ’Let’s Have a Chat’: Principles for the Effective Application of ChatGPT and Large Language Models in the Practice of Forensic Accounting. Journal of Forensic and Investigative Accounting, July to December 2023. [Google Scholar] [CrossRef]
- Huang, L.; Yu, W.; Ma, W.; Zhong, W.; Feng, Z.; Wang, H.; Chen, Q.; Peng, W.; Feng, X.; Qin, B.; et al. A survey on hallucination in large language models: Principles, taxonomy, challenges, and open questions. ACM Transactions on Information Systems 2025, 43, 1–55. [Google Scholar] [CrossRef]
- Wang, C.; Liu, X.; Yue, Y.; Tang, X.; Zhang, T.; Jiayang, C.; Yao, Y.; Gao, W.; Hu, X.; Qi, Z.; et al. Survey on factuality in large language models: Knowledge, retrieval and domain-specificity. arXiv 2023, arXiv:2310.07521. [Google Scholar] [CrossRef]
- Gu, H.; Schreyer, M.; Moffitt, K.; Vasarhelyi, M. Artificial intelligence co-piloted auditing. International Journal of Accounting Information Systems 2024, 54, 100698. [Google Scholar] [CrossRef]
- Berger, A.; Hillebrand, L.; Leonhard, D.; Deußer, T.; De Oliveira, T.B.F.; Dilmaghani, T.; Khaled, M.; Kliem, B.; Loitz, R.; Bauckhage, C.; et al. Towards automated regulatory compliance verification in financial auditing with large language models. In Proceedings of the 2023 IEEE International Conference on Big Data (BigData). IEEE; 2023; pp. 4626–4635. [Google Scholar]
- Li, H.; Freitas, M.M.d.; Lee, H.; Vasarhelyi, M. Enhancing continuous auditing with large language models: AI-assisted real-time accounting information cross-verification. Available at SSRN 4692960 2024. [Google Scholar] [CrossRef]
- Eulerich, M.; Wood, D.A. A demonstration of how ChatGPT can be used in the internal auditing process. Available at SSRN 4519583 2023. [Google Scholar] [CrossRef]
- Emett, S.; Eulerich, M.; Lipinski, E.; Prien, N.; Wood, D.A. Leveraging ChatGPT for Enhancing the Internal Audit Process—A Real-World Example from Uniper, a Large Multinational Company. Accounting Horizons 1–11. [CrossRef]
- Fedyk, A.; Hodson, J.; Khimich, N.; Fedyk, T. Is artificial intelligence improving the audit process? Review of Accounting Studies 2022, 27, 938–985. [Google Scholar] [CrossRef]
- Fotoh, L.; Mugwira, T. Exploring Large Language Models (ChatGPT) in External Audits: Implications and Ethical Considerations. Available at SSRN 4453835 2023. [Google Scholar] [CrossRef]
- Föhr, T.L.; Schreyer, M.; Moffitt, K.; Marten, K.U. Deep Learning Meets Risk-Based Auditing: A Holistic Framework for Leveraging Foundation and Task-Specific Models in Audit Procedures. Available at SSRN 4488271 2023. [Google Scholar] [CrossRef]
- Wang, R.; Liu, J.; Zhao, W.; Li, S.; Zhang, D. AuditBench: A Benchmark for Large Language Models in Financial Statement Auditing. In Proceedings of the 2nd AI4Research Workshop: Towards a Knowledge-grounded Scientific Research Lifecycle.
- De Villiers, C.; Dimes, R.; Molinari, M. How will AI text generation and processing impact sustainability reporting? Critical analysis, a conceptual framework and avenues for future research. Sustainability Accounting, Management and Policy Journal 2024, 15, 96–118. [Google Scholar] [CrossRef]
- Föhr, T.L.; Schreyer, M.; Juppe, T.A.; Marten, K.U. Assuring sustainable futures: Auditing sustainability reports using AI foundation models. Available at SSRN 4502549 2023. [Google Scholar] [CrossRef]
- Kim, A.; Muhn, M.; Nikolaev, V. Bloated disclosures: can ChatGPT help investors process information? arXiv 2023, arXiv:2306.10224. [Google Scholar] [CrossRef]
- Harris, T. Managers’ perception of product market competition and earnings management: a textual analysis of firms’ 10-K reports. Journal of Accounting Literature 2024. [Google Scholar] [CrossRef]
- Ni, J.; Bingler, J.; Colesanti-Senni, C.; Kraus, M.; Gostlow, G.; Schimanski, T.; Stammbach, D.; Vaghefi, S.A.; Wang, Q.; Webersinke, N.; et al. aradigm shift in sustainability disclosure analysis: empowering stakeholders with chatreport, a language model-based tool 2023.
- Comlekci, İ.; Unal, S.; Ozer, A.; Oncu, M.A. Can AI Technologies Estimate Financials Accurately? A Research on Borsa Istanbul with ChatGPT. A Research on Borsa Istanbul with ChatGPT (April 8, 2023). Comlekci, İ., Unal, D., Ozer, a. & Oncu, Ma 2023, pp. 1–14.
- Beerbaum, D. Generative artificial intelligence (GAI) ethics taxonomy-applying chat GPT for robotic process automation (GAI-RPA) as business case. Available at SSRN 4385025 2023. [Google Scholar] [CrossRef]
- Li, H.; Vasarhelyi, M.A. Applying large language models in accounting: A comparative analysis of different methodologies and off-the-shelf examples. Journal of Emerging Technologies in Accounting 2024, 21, 133–152. [Google Scholar] [CrossRef]
- Choi, E.; Suh, Y.J.; Park, H.; Hwang, W. Taxation Perspectives from Large Language Models: A Case Study on Additional Tax Penalties. arXiv 2025, arXiv:2503.03444. [Google Scholar] [CrossRef]
- Alarie, B.; Condon, K.; Massey, S.; Yan, C. The Rise of Generative AI for Tax Research. Tax Notes Federal 2023, 1509. [Google Scholar]
- Zhang, L. Four tax questions for chatgpt and other language models 2023.
- Hechtner, F.; Schmidt, L.; Seebeck, A.; Weiß, M. How to design and employ Specialized Large Language Models for Accounting and Tax Research: The Example of TaxBERT. Available at SSRN 2025. [Google Scholar]
- Choi, G.Y.; Kim, A. Firm-level tax audits: A Generative AI-based measurement. Chicago Booth Research Paper 2024. [Google Scholar] [CrossRef]
- Boynton, W.C.; Johnson, R.N. Modern auditing: Assurance services and the integrity of financial reporting; John Wiley & Sons, 2005. [Google Scholar]
- Antipova, T. Auditing for financial reporting. In Global Encyclopedia of Public Administration, Public Policy, and Governance; Springer, 2023; pp. 656–664. [Google Scholar]
- Kumar, R.; Sharma, V. Auditing: Principles and practice; PHI Learning Pvt. Ltd., 2015. [Google Scholar]
- Coderre, D. Internal audit efficiency through automation; Wiley Online Library, 2009. [Google Scholar]
- Dittenhofer, M. Internal auditing effectiveness: an expansion of present methods. Managerial auditing journal 2001, 16, 443–450. [Google Scholar] [CrossRef]
- Christensen, B.E.; Elder, R.J.; Glover, S.M. Behind the numbers: Insights into large audit firm sampling policies. Accounting Horizons 2015, 29, 61–81. [Google Scholar] [CrossRef]
- Gepp, A.; Linnenluecke, M.K.; O’neill, T.J.; Smith, T. Big data techniques in auditing research and practice: Current trends and future opportunities. Journal of Accounting Literature 2018, 40, 102–115. [Google Scholar] [CrossRef]
- Warren, C.S.; Jones, J.P.; Tayler, W.B. Financial and managerial accounting; Cengage Learning, Inc., 2020. [Google Scholar]
- Williams, J.R.; Haka, S.F.; Bettner, M.S.; Carcello, J.V. Financial & managerial accounting: the basis for business decisions; McGraw-Hill, 2018. [Google Scholar]
- Krahel, J.P.; Titera, W.R. Consequences of big data and formalization on accounting and auditing standards. Accounting Horizons 2015, 29, 409–422. [Google Scholar] [CrossRef]
- Saha, A.; Morris, R.D.; Kang, H. Disclosure overload? An empirical analysis of international financial reporting standards disclosure requirements. Abacus 2019, 55, 205–236. [Google Scholar] [CrossRef]
- Heidmann, M.; Schäffer, U.; Strahringer, S. Exploring the role of management accounting systems in strategic sensemaking. Information Systems Management 2008, 25, 244–257. [Google Scholar] [CrossRef]
- Granlund, M.; Malmi, T. Moderate impact of ERPS on management accounting: a lag or permanent outcome? Management accounting research 2002, 13, 299–321. [Google Scholar] [CrossRef]
- Bhimani, A.; Willcocks, L. Digitisation,‘Big Data’and the transformation of accounting information. Accounting and business research 2014, 44, 469–490. [Google Scholar] [CrossRef]
- Besley, T.; Persson, T. Taxation and development. In Handbook of public economics; Elsevier, 2013; Vol. 5, pp. 51–110. [Google Scholar]
- Salanié, B. The economics of taxation; MIT press, 2011. [Google Scholar]
- Laffer, A.B.; Winegarden, W.H.; Childs, J. The economic burden caused by tax code complexity. The Laffer Center for Supply-Side Economics 2011, pp. 1–24.
- Brady, D. Tax Complexity 2024: It Takes Americans Billions of Hours to Do Their Taxes, 2024.
- Slemrod, J. Tax compliance and enforcement. Journal of economic literature 2019, 57, 904–954. [Google Scholar] [CrossRef]
- Hanlon, M.; Heitzman, S. A review of tax research. Journal of accounting and Economics 2010, 50, 127–178. [Google Scholar] [CrossRef]
- Reuters, T. While tax professionals recognize ChatGPT’s potential, they are aware of the risks, new report shows, 2023.
- Holzenberger, N.; Blair-Stanek, A.; Van Durme, B. A dataset for statutory reasoning in tax law entailment and question answering. arXiv 2020, arXiv:2005.05257. [Google Scholar] [CrossRef]
- Deloitte. Deloitte Unveils Zora AI, Agentic AI for Tomorrow’s Workforce, 2025. Accessed: 2025-05-13.
- Kotler, P.; Burton, S.; Deans, K.; Brown, L.; Armstrong, G. Marketing; Pearson Higher Education AU, 2015.
- Calder, B.J. Focus groups and the nature of qualitative marketing research. Journal of Marketing research 1977, 14, 353–364. [Google Scholar] [CrossRef]
- Mariampolski, H. Qualitative market research; Sage, 2001. [Google Scholar]
- Franses, P.H.; Paap, R. Quantitative models in marketing research; Cambridge University Press, 2001. [Google Scholar]
- Dzwigol, H. Innovation in marketing research: quantitative and qualitative analysis 2020.
- Malhotra, N.K.; Nunan, D.; Birks, D.F. Marketing research 2020.
- Backlinko. 23 Key Market Research Statistics for 2025, 2025. Accessed: 2025-04-06.
- Fligstein, N.; Dauter, L. The sociology of markets. Annu. Rev. Sociol. 2007, 33, 105–128. [Google Scholar] [CrossRef]
- Mazzocchi, M. Statistics for marketing and consumer research 2008.
- Glaeser, E.L. Psychology and the Market. American Economic Review 2004, 94, 408–413. [Google Scholar] [CrossRef]
- Williams, B. Limitations of Market Research: Common Challenges, 2024. Accessed: 2025-04-06.
- Pascucci, F.; Savelli, E.; Gistri, G. How digital technologies reshape marketing: evidence from a qualitative investigation. Italian Journal of Marketing 2023, 2023, 27–58. [Google Scholar] [CrossRef]
- Albrecht, J.; Kitanidis, E.; Fetterman, A.J. Despite" super-human" performance, current LLMs are unsuited for decisions about ethics and safety. arXiv 2022, arXiv:2212.06295. [Google Scholar] [CrossRef]
- Amini, R.; Amini, A. An overview of artificial intelligence and its application in marketing with focus on large language models. International Journal of Science and Research Archive 2024. [Google Scholar]
- Verma, S.; Sharma, R.; Deb, S.; Maitra, D. Artificial intelligence in marketing: Systematic review and future research direction. Int. J. Inf. Manag. Data Insights 2021, 1, 100002. [Google Scholar] [CrossRef]
- Jain, V.; Rai, H.; Parvathy, P.; Mogaji, E. The Prospects and Challenges of ChatGPT on Marketing Research and Practices. SSRN Electronic Journal 2023. [Google Scholar] [CrossRef]
- Aghaei, R.; Kiaei, A.; Boush, M.; Vahidi, J.; Zavvar, M.; Barzegar, Z.; Rofoosheh, M. Harnessing the Potential of Large Language Models in Modern Marketing Management: Applications, Future Directions, and Strategic Recommendations. arXiv 2025, arXiv:2501.10685. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Yu, M. Consumer segmentation with large language models. Journal of Retailing and Consumer Services. [CrossRef]
- Goli, A.; Singh, A. Frontiers: Can Large Language Models Capture Human Preferences? Mark. Sci. 2024, 43, 709–722. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, D.J.; Zhang, H. Large Language Models for Market Research: A Data-augmentation Approach. arXiv 2024, arXiv:2412.19363. [Google Scholar] [CrossRef]
- Praveen, S.; Gajjar, P.; Ray, R.; Dutt, A. Crafting clarity: Leveraging large language models to decode consumer reviews. Journal of Retailing and Consumer Services 2024. [Google Scholar] [CrossRef]
- Naik, N.; Bhat, R.; Kumar, A.; Bapat, G.S.; Kumar, A.; Hota, S.L.; Abishek, G.D.; Vaz, S. Unlocking Brand Excellence: Harnessing AI Tools for Enhanced Customer Engagement and Innovation. RAiSE-2023 2024.
- Sarstedt, M.; Adler, S.J.; Rau, L.; Schmitt, B. Using large language models to generate silicon samples in consumer and marketing research: Challenges, opportunities, and guidelines. Psychology & Marketing 2024. [Google Scholar]
- Paul, J.; Ueno, A.; Dennis, C. ChatGPT and consumers: Benefits, Pitfalls and Future Research Agenda. International Journal of Consumer Studies 2023. [Google Scholar] [CrossRef]
- Kasuga, A.; Yonetani, R. CXSimulator: A User Behavior Simulation using LLM Embeddings for Web-Marketing Campaign Assessment. In Proceedings of the International Conference on Information and Knowledge Management; 2024. [Google Scholar]
- Wahid, R.M.; Mero, J.; Ritala, P. Editorial: Written by ChatGPT, illustrated by Midjourney: generative AI for content marketing. Asia Pacific Journal of Marketing and Logistics 2023. [Google Scholar] [CrossRef]
- Aldous, K.K.; Salminen, J.O.; Farooq, A.; Jung, S.G.; Jansen, B.J. Using ChatGPT in Content Marketing: Enhancing Users’ Social Media Engagement in Cross-Platform Content Creation through Generative AI. In Proceedings of the 35th ACM Conference on Hypertext and Social Media; 2024. [Google Scholar]
- Golab-Andrzejak, E. The Impact of Generative AI and ChatGPT on Creating Digital Advertising Campaigns. Cybernetics and Systems 2025. [Google Scholar] [CrossRef]
- Huh, J.; Nelson, M.R.; Russell, C.A. ChatGPT, AI Advertising, and Advertising Research and Education. Journal of Advertising 2023, 52, 477–482. [Google Scholar] [CrossRef]
- Ivanov, S. Using Artificial Intelligence to Create Marketing Content – Opportunities and Limitations. Journal 2023. Accessed: 2025-04-06.
- Reisenbichler, M.; Reutterer, T.; Schweidel, D.A.; Dan, D. Frontiers: Supporting Content Marketing with Natural Language Generation. Mark. Sci. 2022, 41, 441–452. [Google Scholar] [CrossRef]
- Yeykelis, L.; Pichai, K.; Cummings, J.J.; Reeves, B. Using Large Language Models to Create AI Personas for Replication and Prediction of Media Effects: An Empirical Test of 133 Published Experimental Research Findings. arXiv 2024, arXiv:2408.16073. [Google Scholar] [CrossRef]
- Saputra, R.; Nasution, M.I.P.; Dharma, B. The Impact of Using AI Chat GPT on Marketing Effectiveness: A Case Study on Instagram Marketing. Indonesian Journal of Economics and Management 2023. [Google Scholar] [CrossRef]
- Soykoth, M.W.; Sim, W.; Frederick, S. Research trends in market intelligence: a review through a data-driven quantitative approach. Journal of Marketing Analytics 2024. [Google Scholar] [CrossRef]
- Mutoffar, M.M.; Kuswayati, S.; Sumarni, T.; Dewi, R.K.S.; Nurjanah, E. Role of ChatGPT as an Innovative Tool for Data Analysis and Market Trend Prediction in Business Information Systems. 2024, 13. [Google Scholar]
- Solomon, M.R. Consumer Behavior: Buying, Having, and Being. 1993.
- Hawkins, D.I.; Mothersbaugh, D.L. Consumer Behavior: Building Marketing Strategy. 1997.
- Peter, J.P.; Olson, J.C. Consumer Behavior and Marketing Strategy. 1990.
- Balteș, L.P. Content marketing - the fundamental tool of digital marketing. Bulletin of the Transilvania University of Brasov. Series V : Economic Sciences 2015, pp. 111–118.
- Dwivedi, Y.K. Social media marketing and advertising. The Marketing Review 2015, 15, 289. [Google Scholar] [CrossRef]
- Johnson, J.P.; Myatt, D.P. On the Simple Economics of Advertising, Marketing, and Product Design. IO: Theory 2005. [Google Scholar] [CrossRef]
- Flying V Group. Content Marketing vs Traditional Marketing: Which is Better?, n.d. Accessed: 2025-04-06.
- Sharma, A. How LLMs Become Your Marketing & Content Powerhouse. A3Logics Blog 2024. Accessed: 2025-04-06.
- Jenster, P.V.; Søilen, K.S. Market Intelligence: Building Strategic Insight. 2009.
- Wee, T.T.T. The use of marketing research and intelligence in strategic planning: key issues and future trends. Marketing Intelligence & Planning 2001, 19, 245–253. [Google Scholar] [CrossRef]
- Kushwaha, A.K.; Kar, A.K. MarkBot - A Language Model-Driven Chatbot for Interactive Marketing in Post-Modern World. Inf. Syst. Frontiers 2021, 26, 857–874. [Google Scholar] [CrossRef]
- Cashion, F.; O’Brien, J. Generative AI Takes Off with Marketers. American Marketing Association 2024. Accessed: 2025-05-12.
- Eves, H.W. Foundations and fundamental concepts of mathematics; Courier Corporation, 1997. [Google Scholar]
- Courant, R.; Robbins, H. What is Mathematics?: an elementary approach to ideas and methods; Oxford university press, 1996. [Google Scholar]
- Horsten, L. Philosophy of mathematics. Stanford Encyclopedia of Philosophy 2007. [Google Scholar]
- Council, N.R.; on Engineering, D.; Sciences, P.; on Mathematical Sciences, B.; Applications, T.; on the Mathematical Sciences in, C.; 2025. The mathematical sciences in 2025; National Academies Press, 2013.
- Treves, F. Topological Vector Spaces, Distributions and Kernels: Pure and Applied Mathematics, Vol. 25; Vol. 25, Elsevier, 2016.
- Lax, P.D.; Phillips, R.S. Scattering Theory: Pure and Applied Mathematics, Vol. 26; Vol. 26, Elsevier, 2016.
- Hersh, R. What is mathematics, really? Mitteilungen der Deutschen Mathematiker-Vereinigung 1998, 6, 13–14. [Google Scholar] [CrossRef]
- Holmes, M.H. Introduction to the foundations of applied mathematics; Vol. 56, Springer, 2009.
- Logan, J.D. Applied mathematics; John Wiley & Sons, 2013. [Google Scholar]
- Polya, G. How to solve it: A new aspect of mathematical method; Princeton university press, 1945. [Google Scholar]
- Etingof, P. PRIMES: How to Succeed in Mathematical Research.
- Ho, A.; Besiroglu, T. What is the Future of AI in Mathematics? Interviews with Leading Mathematicians, 2024. Accessed: 2025-04-04.
- ADVISER, M. Mathematicians use DeepMind AI to create new methods in problem-solving, 2021.
- Thurston, W.P. Mathematical education. arXiv 2005. [Google Scholar] [CrossRef]
- Kajander, A.; Boland, T. Mathematical models for teaching: Reasoning without memorization; Canadian Scholars’ Press, 2014. [Google Scholar]
- Peter, E.E. Critical thinking: Essence for teaching mathematics and mathematics problem solving skills. African Journal of Mathematics and Computer Science Research 2012, 5, 39–43. [Google Scholar] [CrossRef]
- Lama, V.; Ma, C.; Ghosal, T. Benchmarking Automated Theorem Proving with Large Language Models. In Proceedings of the Proceedings of the 1st Workshop on NLP for Science (NLP4Science), 2024; pp. 208–218.
- Song, P.; Yang, K.; Anandkumar, A. Lean Copilot: Large Language Models as Copilots for Theorem Proving in Lean. arXiv 2025, arXiv:2404.12534. [Google Scholar] [CrossRef]
- Romera-Paredes, B.; Barekatain, M.; Novikov, A.; Balog, M.; Kumar, M.P.; Dupont, E.; Ruiz, F.J.; Ellenberg, J.S.; Wang, P.; Fawzi, O.; et al. Mathematical discoveries from program search with large language models. Nature 2024, 625, 468–475. [Google Scholar] [CrossRef]
- Mirzadeh, I.; Alizadeh, K.; Shahrokhi, H.; Tuzel, O.; Bengio, S.; Farajtabar, M. Gsm-symbolic: Understanding the limitations of mathematical reasoning in large language models. In Proceedings of the ICLR; 2025. [Google Scholar]
- Cheng, F.; Li, H.; Liu, F.; van Rooij, R.; Zhang, K.; Lin, Z. Empowering LLMs with Logical Reasoning: A Comprehensive Survey. arXiv 2025, arXiv:2502.15652. [Google Scholar] [CrossRef]
- Agrawal, P.; Vasania, S.; Tan, C. Exploring the Limitations of Graph-based Logical Reasoning in Large Language Models. Openreview 2025. [Google Scholar]
- Li, Y.; Kuang, J.; Huang, H.; Xu, Z.; Liang, X.; Yu, Y.; Lu, W.; Li, Y.; Tan, X.; Qu, C.; et al. One Example Shown, Many Concepts Known! Counterexample-Driven Conceptual Reasoning in Mathematical LLMs. arXiv 2025, arXiv:2502.10454. [Google Scholar] [CrossRef]
- Davoodi, A.G.; Davoudi, S.P.M.; Pezeshkpour, P. Llms are not intelligent thinkers: Introducing mathematical topic tree benchmark for comprehensive evaluation of llms. arXiv 2024, arXiv:2406.05194. [Google Scholar] [CrossRef]
- Ferrag, M.A.; Tihanyi, N.; Debbah, M. Reasoning Beyond Limits: Advances and Open Problems for LLMs. arXiv 2025, arXiv:2503.22732. [Google Scholar] [CrossRef]
- AlphaProof.; teams, A. AI achieves silver-medal standard solving International Mathematical Olympiad problems, 2024.
- Yu, L.; Jiang, W.; Shi, H.; Yu, J.; Liu, Z.; Zhang, Y.; Kwok, J.T.; Li, Z.; Weller, A.; Liu, W. Metamath: Bootstrap your own mathematical questions for large language models. arXiv 2023, arXiv:2309.12284. [Google Scholar] [CrossRef]
- Polu, S.; Sutskever, I. Generative language modeling for automated theorem proving. arXiv 2020, arXiv:2009.03393. [Google Scholar] [CrossRef]
- Han, J.M.; Rute, J.; Wu, Y.; Ayers, E.W.; Polu, S. Proof artifact co-training for theorem proving with language models. arXiv 2021, arXiv:2102.06203. [Google Scholar] [CrossRef]
- Jiang, A.Q.; Li, W.; Tworkowski, S.; Czechowski, K.; Odrzygóźdź, T.; Miłoś, P.; Wu, Y.; Jamnik, M. Thor: Wielding hammers to integrate language models and automated theorem provers. In Proceedings of the NeurIPS; 2022. [Google Scholar]
- Lample, G.; Lacroix, T.; Lachaux, M.A.; Rodriguez, A.; Hayat, A.; Lavril, T.; Ebner, G.; Martinet, X. Hypertree proof search for neural theorem proving. Advances in neural information processing systems 2022, 35, 26337–26349. [Google Scholar]
- First, E.; Rabe, M.N.; Ringer, T.; Brun, Y. Baldur: Whole-Proof Generation and Repair with Large Language Models. In Proceedings of the Proceedings of the 31st ACM Joint European Software Engineering Conference and Symposium on the Foundations of Software Engineering, New York, NY, USA, 2023; ESEC/FSE 2023; pp. 1229–1241. [CrossRef]
- Thakur, A.; Wen, Y.; Chaudhuri, S. A Language-Agent Approach to Formal Theorem-Proving. OpenReview 2024. [Google Scholar]
- Yang, K.; Swope, A.; Gu, A.; Chalamala, R.; Song, P.; Yu, S.; Godil, S.; Prenger, R.J.; Anandkumar, A. LeanDojo: Theorem Proving with Retrieval-Augmented Language Models. In Proceedings of the Advances in Neural Information Processing Systems; Oh, A., Naumann, T., Globerson, A., Saenko, K., Hardt, M., Levine, S., Eds.; Curran Associates, Inc., 2023; pp. 21573–21612. [Google Scholar]
- Liang, S.; Zhang, W.; Zhong, T.; Liu, T. Mathematics and machine creativity: A survey on bridging mathematics with ai. arXiv 2024, arXiv:2412.16543. [Google Scholar] [CrossRef]
- Johansson, M.; Smallbone, N. Exploring Mathematical Conjecturing with Large Language Models. In Proceedings of the NeSy; 2023; pp. 62–77. [Google Scholar]
- Gao, H.; Kaltenbach, S.; Koumoutsakos, P. Generative learning for forecasting the dynamics of high-dimensional complex systems. Nature Communications 2024, 15, 8904. [Google Scholar] [CrossRef]
- Kumar, H.; Rothschild, D.M.; Goldstein, D.G.; Hofman, J.M. Math education with large language models: peril or promise? Available at SSRN 4641653 2023. [Google Scholar] [CrossRef]
- Wardat, Y.; Tashtoush, M.A.; AlAli, R.; Jarrah, A.M. ChatGPT: A revolutionary tool for teaching and learning mathematics. Eurasia Journal of Mathematics, Science and Technology Education 2023, 19, em2286. [Google Scholar] [CrossRef]
- Wang, S.; Xu, T.; Li, H.; Zhang, C.; Liang, J.; Tang, J.; Yu, P.S.; Wen, Q. Large language models for education: A survey and outlook. arXiv 2024, arXiv:2403.18105. [Google Scholar] [CrossRef]
- Xu, H.; Gan, W.; Qi, Z.; Wu, J.; Yu, P.S. Large language models for education: A survey. arXiv 2024, arXiv:2403.18105. [Google Scholar] [CrossRef]
- Hadzhikoleva, S.; Rachovski, T.; Ivanov, I.; Hadzhikolev, E.; Dimitrov, G. Automated Test Creation Using Large Language Models: A Practical Application. Applied Sciences 2024, 14, 9125. [Google Scholar] [CrossRef]
- De Bruijn, N.G. The mathematical language AUTOMATH, its usage, and some of its extensions. In Studies in Logic and the Foundations of Mathematics; Elsevier, 1994; Vol. 133, pp. 73–100.
- Harrison, J.; Urban, J.; Wiedijk, F. History of interactive theorem proving. In Handbook of the History of Logic; Elsevier, 2014; Vol. 9, pp. 135–214.
- Maric, F. A survey of interactive theorem proving. Zbornik radova 2015, 18, 173–223. [Google Scholar]
- Megill, N.; Wheeler, D.A. Metamath: a computer language for mathematical proofs; Lulu. com, 2019. [Google Scholar]
- Nipkow, T.; Wenzel, M.; Paulson, L.C. Isabelle/HOL: a proof assistant for higher-order logic; Springer, 2002. [Google Scholar]
- Coq, P. The coq proof assistant-reference manual. INRIA Rocquencourt and ENS Lyon, version 1996, 5. [Google Scholar]
- Moura, L.d.; Ullrich, S. The Lean 4 theorem prover and programming language. In Proceedings of the Automated Deduction–CADE 28: 28th International Conference on Automated Deduction, Virtual Event, July 12–15, 2021, Proceedings 28. Springer, 2021, pp. 625–635.
- Zheng, C.; Wang, H.; Xie, E.; Liu, Z.; Sun, J.; Xin, H.; Shen, J.; Li, Z.; Li, Y. Lyra: Orchestrating Dual Correction in Automated Theorem Proving, 2024.
- AlphaEvolve: A Gemini-powered coding agent for designing advanced algorithms, 2025.
- Huss-Lederman, S.; Jacobson, E.M.; Tsao, A.; Turnbull, T.; Johnson, J.R. Implementation of Strassen’s algorithm for matrix multiplication. In Proceedings of the Proceedings of the 1996 ACM/IEEE Conference on Supercomputing, 1996; pp. 32–es.
- Li, J.; Ranka, S.; Sahni, S. Strassen’s matrix multiplication on GPUs. In Proceedings of the 2011 IEEE 17th international conference on parallel and distributed systems. IEEE; 2011; pp. 157–164. [Google Scholar]
- Musin, O.R. The kissing number in four dimensions. Annals of Mathematics 2008, 1–32. [Google Scholar] [CrossRef]
- Zheng, K.; Han, J.M.; Polu, S. Minif2f: a cross-system benchmark for formal olympiad-level mathematics. arXiv 2021, arXiv:2109.00110. [Google Scholar] [CrossRef]
- Polu, S.; Han, J.M.; Zheng, K.; Baksys, M.; Babuschkin, I.; Sutskever, I. Formal mathematics statement curriculum learning. arXiv 2022, arXiv:2202.01344. [Google Scholar] [CrossRef]
- Shao, Z.; Wang, P.; Zhu, Q.; Xu, R.; Song, J.; Bi, X.; Zhang, H.; Zhang, M.; Li, Y.; Wu, Y.; et al. Deepseekmath: Pushing the limits of mathematical reasoning in open language models. arXiv 2024, arXiv:2402.03300. [Google Scholar] [CrossRef]
- Xin, H.; Guo, D.; Shao, Z.; Ren, Z.; Zhu, Q.; Liu, B.; Ruan, C.; Li, W.; Liang, X. Deepseek-prover: Advancing theorem proving in llms through large-scale synthetic data. arXiv 2024, arXiv:2405.14333. [Google Scholar] [CrossRef]
- Gao, B.; Song, F.; Yang, Z.; Cai, Z.; Miao, Y.; Dong, Q.; Li, L.; Ma, C.; Chen, L.; Xu, R.; et al. Omni-math: A universal olympiad level mathematic benchmark for large language models. arXiv 2024, arXiv:2410.07985. [Google Scholar] [CrossRef]
- AIME Problem Set: 1983-2024, 2024.
- Wei, T.; Luan, J.; Liu, W.; Dong, S.; Wang, B. Cmath: Can your language model pass chinese elementary school math test? arXiv 2023, arXiv:2306.16636. [Google Scholar] [CrossRef]
- Zhong, W.; Cui, R.; Guo, Y.; Liang, Y.; Lu, S.; Wang, Y.; Saied, A.; Chen, W.; Duan, N. Agieval: A human-centric benchmark for evaluating foundation models. arXiv 2023, arXiv:2304.06364. [Google Scholar] [CrossRef]
- Zhang, X.; Li, C.; Zong, Y.; Ying, Z.; He, L.; Qiu, X. Evaluating the performance of large language models on gaokao benchmark. arXiv 2023, arXiv:2305.12474. [Google Scholar] [CrossRef]
- Koncel-Kedziorski, R.; Roy, S.; Amini, A.; Kushman, N.; Hajishirzi, H. MAWPS: A math word problem repository. In Proceedings of the Proceedings of the 2016 conference of the north american chapter of the association for computational linguistics: human language technologies, 2016; pp. 1152–1157.
- Miao, S.y.; Liang, C.C.; Su, K.Y. A Diverse Corpus for Evaluating and Developing English Math Word Problem Solvers. In Proceedings of the Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics, 2020; pp. 975–984.
- Patel, A.; Bhattamishra, S.; Goyal, N. Are NLP Models really able to Solve Simple Math Word Problems? In Proceedings of the Proceedings of the 2021 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Online; 2021; pp. 2080–2094. [Google Scholar] [CrossRef]
- Ling, W.; Yogatama, D.; Dyer, C.; Blunsom, P. Program induction by rationale generation: Learning to solve and explain algebraic word problems. In Proceedings of the ACL; 2017. [Google Scholar]
- Amini, A.; Gabriel, S.; Lin, S.; Koncel-Kedziorski, R.; Choi, Y.; Hajishirzi, H. MathQA: Towards Interpretable Math Word Problem Solving with Operation-Based Formalisms. In Proceedings of the NAACL; 2019. [Google Scholar]
- Lightman, H.; Kosaraju, V.; Burda, Y.; Edwards, H.; Baker, B.; Lee, T.; Leike, J.; Schulman, J.; Sutskever, I.; Cobbe, K. Let’s Verify Step by Step. arXiv 2023, arXiv:2305.20050. [Google Scholar] [CrossRef]
- Chen, W.; Yin, M.; Ku, M.; Lu, P.; Wan, Y.; Ma, X.; Xu, J.; Wang, X.; Xia, T. Theoremqa: A theorem-driven question answering dataset. arXiv 2023, arXiv:2305.12524. [Google Scholar] [CrossRef]
- Mishra, S.; Finlayson, M.; Lu, P.; Tang, L.; Welleck, S.; Baral, C.; Rajpurohit, T.; Tafjord, O.; Sabharwal, A.; Clark, P.; et al. Lila: A Unified Benchmark for Mathematical Reasoning. In Proceedings of the Proceedings of the 2022 Conference on Empirical Methods in Natural Language Processing (EMNLP); 2022. [Google Scholar]
- Yue, X.; Qu, X.; Zhang, G.; Fu, Y.; Huang, W.; Sun, H.; Su, Y.; Chen, W. Mammoth: Building math generalist models through hybrid instruction tuning. arXiv 2023, arXiv:2309.05653. [Google Scholar] [CrossRef]
- Maxwell, J.C. Matter and Motion 1878.
- Wigner, E.P. The unreasonable effectiveness of mathematics in the natural sciences. Communications on Pure and Applied Mathematics 1960, 13, 1–14. [Google Scholar] [CrossRef]
- Einstein, A. Physics and Reality. Journal of the Franklin Institute 1936, 221, 349–382. [Google Scholar] [CrossRef]
- Feynman, R.P. The Character of Physical Law; MIT Press, 1965. [Google Scholar]
- Abbott, B.P.; Abbott, R.; Abbott, T.D.; Abernathy, M.R.; Acernese, F.; Ackley, K.; Adams, C.; Adams, T.; Addesso, P.; Adhikari, R.X.; et al. Observation of gravitational waves from a binary black hole merger. Physical review letters 2016, 116, 061102. [Google Scholar] [CrossRef]
- Einstein, A. Näherungsweise integration der feldgleichungen der gravitation. Sitzungsberichte der Königlich Preußischen Akademie der Wissenschaften 1916, 688–696. [Google Scholar]
- Sathyaprakash, B.S.; Schutz, B.F. Physics, astrophysics and cosmology with gravitational waves. Living reviews in relativity 2009, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Sathyaprakash, B.; Abernathy, M.; Acernese, F.; Ajith, P.; Allen, B.; Amaro-Seoane, P.; Andersson, N.; Aoudia, S.; Arun, K.; Astone, P.; et al. Scientific objectives of Einstein telescope. Classical and Quantum Gravity 2012, 29, 124013. [Google Scholar] [CrossRef]
- Yunes, N.; Siemens, X. Gravitational-wave tests of general relativity with ground-based detectors and pulsar-timing arrays. Living Reviews in Relativity 2013, 16, 1–124. [Google Scholar] [CrossRef]
- Halliday, D.; Resnick, R.; Walker, J. Fundamentals of physics; John Wiley & Sons, 2013. [Google Scholar]
- Halliday, D.; Resnick, R.; Walker, J. Principles of physics 2023.
- Misner, C.W.; Thorne, K.S.; Wheeler, J.A. Gravitation; Macmillan, 1973. [Google Scholar]
- Goldstein, H.; Poole, C.; Safko, J.; Addison, S.R. Classical mechanics, 2002.
- Arfken, G.B.; Weber, H.J.; Harris, F.E. Mathematical methods for physicists: a comprehensive guide; Academic press, 2011. [Google Scholar]
- Kittel, C.; McEuen, P. Introduction to solid state physics; John Wiley & Sons, 2018. [Google Scholar]
- Haken, H.; Wolf, H.C. The physics of atoms and quanta: introduction to experiments and theory; Springer Science & Business Media, 2006. [Google Scholar]
- Griffiths, D. Introduction to elementary particles; John Wiley & Sons, 2020. [Google Scholar]
- Martin, R.M. Electronic structure: basic theory and practical methods; Cambridge university press, 2020. [Google Scholar]
- Marder, M.P. Condensed matter physics; John Wiley & Sons, 2010. [Google Scholar]
- Zoller, P.; Beth, T.; Binosi, D.; Blatt, R.; Briegel, H.; Bruß, D.; Calarco, T.; Cirac, J.I.; Deutsch, D.; Eisert, J.; et al. Quantum information processing and communication: Strategic report on current status, visions and goals for research in Europe. The European Physical Journal D-Atomic, Molecular, Optical and Plasma Physics 2005, 36, 203–228. [Google Scholar] [CrossRef]
- Carroll, B.W.; Ostlie, D.A. An introduction to modern astrophysics; Cambridge University Press, 2017. [Google Scholar]
- Dodelson, S.; Schmidt, F. Modern cosmology; Elsevier, 2024. [Google Scholar]
- Kivelson, M.G.; Russell, C.T. Introduction to space physics 1995.
- of Sciences Engineering, N.A.; Medicine.; et al. Pathways to Discovery in Astronomy and Astrophysics for the 2020s; 2021.
- Heilbron, J.L. The Oxford guide to the history of physics and astronomy; Oxford University Press, 2005. [Google Scholar]
- Greene, B. The fabric of the cosmos: Space, time, and the texture of reality; Knopf, 2004. [Google Scholar]
- Carleo, G.; Cirac, I.; Cranmer, K.; Daudet, L.; Schuld, M.; Tishby, N.; Vogt-Maranto, L.; Zdeborová, L. Machine learning and the physical sciences. Reviews of Modern Physics 2019, 91, 045002. [Google Scholar] [CrossRef]
- Avallone, E.; Baumeister, I.; Sadegh, A. Marks’ Standard Handbook for Mechanical Engineers. 10; Citeseer, 2006. [Google Scholar]
- Wickert, J.; Lewis, K. An introduction to mechanical engineering; Cengage learning, 2013. [Google Scholar]
- Craig Jr, R.R.; Taleff, E.M. Mechanics of materials; John Wiley & Sons, 2020. [Google Scholar]
- Moran, M.J.; Shapiro, H.N.; Boettner, D.D.; Bailey, M.B. Fundamentals of engineering thermodynamics; John Wiley & Sons, 2010. [Google Scholar]
- Ogata, K.; et al. Modern control engineering; Prentice Hall India, 2009. [Google Scholar]
- Zienkiewicz, O.C.; Taylor, R.L.; Zhu, J.Z. The finite element method: its basis and fundamentals; Elsevier, 2005. [Google Scholar]
- Lomax, H.; Pulliam, T.H.; Zingg, D.W.; Pulliam, T.H.; Zingg, D.W. Fundamentals of computational fluid dynamics; Vol. 246, Springer, 2001.
- Budynas, R.G.; Nisbett, J.K.; et al. Shigley’s mechanical engineering design; Vol. 9, McGraw-Hill New York, 2011.
- Sharma, V.; Pandey, P.M. Additive and Subtractive Manufacturing Processes: Principles and Applications; CRC Press, 2022.
- Tyagi, A.K.; Tiwari, S.; Ahmad, S.S. Industry 4.0, Smart Manufacturing, and Industrial Engineering: Challenges and Opportunities 2024.
- Soori, M.; Dastres, R.; Arezoo, B.; Jough, F.K.G. Intelligent robotic systems in Industry 4.0: A review. Journal of Advanced Manufacturing Science and Technology 2024, 2024007. [Google Scholar] [CrossRef]
- Tian, Y.; Zhao, C.Y. A review of solar collectors and thermal energy storage in solar thermal applications. Applied energy 2013, 104, 538–553. [Google Scholar] [CrossRef]
- Gao, X.; Fraulob, M.; Haïat, G. Biomechanical behaviours of the bone–implant interface: a review. Journal of The Royal Society Interface 2019, 16, 20190259. [Google Scholar] [CrossRef] [PubMed]
- Thelen, A.; Zhang, X.; Fink, O.; Lu, Y.; Ghosh, S.; Youn, B.D.; Todd, M.D.; Mahadevan, S.; Hu, C.; Hu, Z. A comprehensive review of digital twin—part 1: modeling and twinning enabling technologies. Structural and Multidisciplinary Optimization 2022, 65, 354. [Google Scholar] [CrossRef]
- David, A.J.; et al. Computational fluid dynamics: the basics with applications. McGraw-Hill 1995, 547, 5. [Google Scholar]
- Bathe, K.J. Finite element procedures; Klaus-Jurgen Bathe, 2006. [Google Scholar]
- Mosavi, A.A.; Sedarat, H.; O’Connor, S.M.; Emami-Naeini, A.; Lynch, J. Calibrating a high-fidelity finite element model of a highway bridge using a multi-variable sensitivity-based optimisation approach. Structure and Infrastructure Engineering 2014, 10, 627–642. [Google Scholar] [CrossRef]
- Brooks, R.A. Elephants don’t play chess. Robotics and autonomous systems 1990, 6, 3–15. [Google Scholar] [CrossRef]
- Zagal, J.C.; Ruiz-del Solar, J.; Vallejos, P. Back to reality: Crossing the reality gap in evolutionary robotics. IFAC Proceedings Volumes 2004, 37, 834–839. [Google Scholar] [CrossRef]
- Forster, A.M.; Forster, A.M. Materials testing standards for additive manufacturing of polymer materials: state of the art and standards applicability 2015.
- American Society of Mechanical Engineers. ASME Code of Ethics of Engineers. ASME Official Publication, 2021. https://www.asme.org/getmedia/3e165b2b-f7e7-4106-a772-5f0586d2268e/p-15-7-ethics.pdf.
- Portenoy, J.; West, J.D. Constructing and evaluating automated literature review systems. Scientometrics 2020, 125, 3233–3251. [Google Scholar] [CrossRef]
- Accuris. Engineering Workbench: AI-powered platform for standards management. https://accuristech.com/solutions/engineering-workbench/, 2024. Accessed: 2025-04-21.
- Mudur, N.; Cui, H.; Venugopalan, S.; Raccuglia, P.; Brenner, M.P.; Norgaard, P. FEABench: Evaluating Language Models on Multiphysics Reasoning Ability. arXiv 2025, arXiv:2504.06260. [Google Scholar] [CrossRef]
- Ni, B.; Buehler, M.J. MechAgents: Large language model multi-agent collaborations can solve mechanics problems, generate new data, and integrate knowledge. arXiv 2023, arXiv:2311.08166. [Google Scholar] [CrossRef]
- Makatura, L.; Foshey, M.; Wang, B.; et al. Large Language Models for Design and Manufacturing. MIT GenAI 2024. [Google Scholar]
- Wu, S.; Khasahmadi, A.; Katz, M.; et al. CadVLM: Bridging Language and Vision in the Generation of Parametric CAD Sketches. arXiv 2024, arXiv:2409.17457. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, M. Beyond Answers: Large Language Model-Powered Tutoring System in Physics Education for Deep Learning and Precise Understanding. arXiv 2024, arXiv:2406.10934. [Google Scholar] [CrossRef]
- Abedi, M.; Alshybani, I.; Shahadat, M.R.B.; Murillo, M.S. Beyond Traditional Teaching: The Potential of Large Language Models and Chatbots in Graduate Engineering Education. arXiv 2023, arXiv:2309.13059. [Google Scholar] [CrossRef]
- Polverini, G.; Gregorcic, B. How understanding large language models can inform the use of ChatGPT in physics education. arXiv 2023, arXiv:2309.12074. [Google Scholar] [CrossRef]
- Harle, T.; et al. Large Language Models (LLMs) in Engineering Education. Information 2024, 15, 345. [Google Scholar] [CrossRef]
- Makatura, L.; Foshey, M.; Wang, B.; et al. Large Language Models for Design and Manufacturing. MIT GenAI 2023. [Google Scholar]
- Ali-Dib, M.; Menou, K. Physics simulation capabilities of LLMs. arXiv 2023, arXiv:2312.02091. [Google Scholar] [CrossRef]
- Raissi, M.; Perdikaris, P.; Karniadakis, G.E. Physics-informed neural networks: A deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. Journal of Computational Physics 2019, 378, 686–707. [Google Scholar] [CrossRef]
- Latif, E.; et al. PhysicsAssistant: An LLM-Powered Interactive Learning Robot for Physics Lab Investigations. arXiv 2024, arXiv:2403.18721. [Google Scholar] [CrossRef]
- Khan, J.A.; Qayyum, S.; Dar, H.S. Large Language Model for Requirements Engineering: A Systematic Literature Review. ResearchGate 2024. [Google Scholar]
- Tian, J.; Hou, J.; Wu, Z.; et al. Assessing Large Language Models in Mechanical Engineering Contexts: A Study on Experiment-Focused Log Interpretation. ResearchGate 2024. [Google Scholar]
- Berenguer, A.; Morejón, A.; Tomás, D.; Mazón, J.N. Leveraging Large Language Models for Sensor Data Retrieval. Applied Sciences 2024, 14, 2506. [Google Scholar] [CrossRef]
- Makatura, L.; Foshey, M.; Wang, B.; et al. Large Language Models for Design and Manufacturing. MIT GenAI 2023. [Google Scholar]
- for Iron Research, M.P.I. LangSim - Large Language Model Interface for Atomistic Simulation. https://www.mpie.de/5063016/LangSim, 2024.
- Landram, K. Multimodal machine learning model increases accuracy. Carnegie Mellon University News 2024. https://engineering.cmu.edu/news-events/news/2024/11/29-multimodal.html.
- Coders, M. Exploring the Role of Large Language Models (LLMs) in Physics. Metric Coders Blog 2024. https://www.metriccoders.com/post/exploring-the-role-of-large-language-models-llms-in-physics.
- Shipps, A. Multimodal and reasoning LLMs supersize training data for dexterous robotic tasks. MIT CSAIL News 2024. https://www.csail.mit.edu/news/multimodal-and-reasoning-llms-supersize-training-data-dexterous-robotic-tasks.
- Author, A. Grading explanations of problem-solving process and generating feedback using large language models. Physical Review Physics Education Research 2024, 21, 010126. [Google Scholar]
- Koch, S.; Matveev, A.; Jiang, Z.; Williams, F.; Artemov, A.; Burnaev, E.; Alexa, M.; Zorin, D.; Panozzo, D. Abc: A big cad model dataset for geometric deep learning. In Proceedings of the Proceedings of the IEEE/CVF conference on computer vision and pattern recognition, 2019; pp. 9601–9611.
- Rundi Wu, Chang Xiao, C.Z. DeepCAD: A Large-Scale CAD Dataset for Geometric Deep Learning. In Proceedings of the CVPR 2022, 2022.
- Lambourne, J.G.; Willis, K.D.; Jayaraman, P.K.; Sanghi, A.; Meltzer, P.; Shayani, H. BRepNet: A Topological Message Passing System for Solid Models. In Proceedings of the Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), June 2021; pp. 12773–12782.
- Du, Y.; Chen, S.; Zan, W.; Li, P.; Wang, M.; Song, D.; Li, B.; Hu, Y.; Wang, B. BlenderLLM: Training Large Language Models for Computer-Aided Design with Self-improvement. arXiv 2024, arXiv:2412.14203. [Google Scholar] [CrossRef]
- OpenFOAM Official Example Cases. https://www.openfoam.com/.
- MatWeb Material Property Data. http://www.matweb.com/.
- Takamoto, M.; Praditia, T.; Leiteritz, R.; MacKinlay, D.; Alesiani, F.; Pflüger, D.; Niepert, M. Pdebench: An extensive benchmark for scientific machine learning. Advances in Neural Information Processing Systems 2022, 35, 1596–1611. [Google Scholar]
- Qiu, S.; Guo, S.; Song, Z.Y.; Sun, Y.; Cai, Z.; Wei, J.; Luo, T.; Yin, Y.; Zhang, H.; Hu, Y.; et al. PHYBench: Holistic Evaluation of Physical Perception and Reasoning in Large Language Models. arXiv 2025, arXiv:2504.16074. [Google Scholar] [CrossRef]
- Saxena, A.; Goebel, K. Turbofan Engine Degradation Simulation Data Set. https://www.nasa.gov/content/prognostics-center-of-excellence-data-set-repository, 2008.
- Wikipedia. Chemistry. Wikipedia, The Free Encyclopedia 2023. [Accessed: 2025-03-22].
- Chemistry. Britannica. [Accessed: 2025-03-22].
- American Chemical Society. What is Chemistry?, 2023. Accessed: 2023-10-01.
- Nature. Chemistry, 2025. Accessed: [Insert Date Accessed].
- Carruthers, W.; Coldham, I. Modern methods of organic synthesis; Cambridge University Press, 2004. [Google Scholar]
- Harvey, D. Modern analytical chemistry; McGraw Hill, 2000. [Google Scholar]
- Christian, G.D.; Dasgupta, P.K.; Schug, K.A. Analytical chemistry; John Wiley & Sons, 2013. [Google Scholar]
- Levine, I.N.; Busch, D.H.; Shull, H. Quantum chemistry; Vol. 6, Pearson Prentice Hall Upper Saddle River, NJ, 2009.
- Jensen, F. Introduction to computational chemistry; John wiley & sons, 2017. [Google Scholar]
- Patrick, G.L. An introduction to medicinal chemistry; Oxford university press, 2023. [Google Scholar]
- Voet, D.; Voet, J.G. Biochemistry; John Wiley & Sons, 2010. [Google Scholar]
- Manahan, S.E. Environmental chemistry; CRC press, 2022. [Google Scholar]
- Ali, H.; Khan, E. Environmental chemistry in the twenty-first century. Environmental Chemistry Letters 2017, 15, 329–346. [Google Scholar] [CrossRef]
- Corey, E.J. The logic of chemical synthesis; 1991. [Google Scholar]
- Vedantu. Chemical Analysis - Methods, Techniques, and Applications. https://www.vedantu.com/chemistry/chemical-analysis, 2023. Accessed: 2023-10-05.
- RROIJ. Advancing Science: Exploring Modern Methods in Chemical Analysis. Research and Reviews: Open Access Journal 2023. [Google Scholar] [CrossRef]
- Britannica, E. Chemical Analysis - Classical Methods. https://www.britannica.com/science/chemical-analysis/Classical-methods, 2023. Accessed: 2023-10-05.
- Dutta, S.; Biswas, I.; Raghuwanshi, K.; Das, K.; Ahmed, A.; Srivastav, Y.; Kumar, A.; Jain, S.K.; Maiti, N.J. A Comprehensive review on Analytical Techniques for the Quantification of Pharmaceutical Compounds in Biological Matrices: Recent Advances and future directions. [CrossRef]
- Atkins, P.; De Paula, J. Elements of physical chemistry; Oxford University Press: USA, 2013. [Google Scholar]
- Brown, T.L.; LeMay, H.E.; Bursten, B.E. Chemistry: the central science; Pearson Educación, 2002. [Google Scholar]
- Robinson, J.; Barry, T. Experimental methods for the measurement of thermodynamic data and recommendation about future capability at NPL. 1997.
- Gill, P.; Moghadam, T.T.; Ranjbar, B. Differential scanning calorimetry techniques: applications in biology and nanoscience. Journal of biomolecular techniques: JBT 2010, 21, 167. [Google Scholar]
- Zheng, X.; Bi, C.; Li, Z.; Podariu, M.; Hage, D.S. Analytical methods for kinetic studies of biological interactions: A review. Journal of pharmaceutical and biomedical analysis 2015, 113, 163–180. [Google Scholar] [CrossRef]
- Silbey, R.J.; Alberty, R.A.; Papadantonakis, G.A.; Bawendi, M.G. Physical chemistry; John Wiley & Sons, 2022. [Google Scholar]
- Smith, I.W.; Rowe, B.R. Reaction kinetics at very low temperatures: laboratory studies and interstellar chemistry. Accounts of Chemical Research 2000, 33, 261–268. [Google Scholar] [CrossRef]
- Britannica, E. Reaction Mechanism. https://www.britannica.com/science/reaction-mechanism, 2023. Accessed: 2023-10-05.
- Kraka, E.; Cremer, D. Computational analysis of the mechanism of chemical reactions in terms of reaction phases: hidden intermediates and hidden transition states. Accounts of chemical research 2010, 43, 591–601. [Google Scholar] [CrossRef]
- Butler, M.S. The role of natural product chemistry in drug discovery. Journal of natural products 2004, 67, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Du, G.; Chen, J. Novel fermentation processes for manufacturing plant natural products. Current Opinion in Biotechnology 2014, 25, 17–23. [Google Scholar] [CrossRef]
- Gunstone, F.D. Fatty acid and lipid chemistry; Springer, 2012. [Google Scholar]
- Ikan, R. Natural products: a laboratory guide; Academic Press, 1991. [Google Scholar]
- Alkhzem, A.H.; Woodman, T.J.; Blagbrough, I.S. Design and synthesis of hybrid compounds as novel drugs and medicines. RSC advances 2022, 12, 19470–19484. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Campbell, C.M.; Hutchison, G.R. Computational design and selection of optimal organic photovoltaic materials. The Journal of Physical Chemistry C 2011, 115, 16200–16210. [Google Scholar] [CrossRef]
- Hassan Baig, M.; Ahmad, K.; Roy, S.; Mohammad Ashraf, J.; Adil, M.; Haris Siddiqui, M.; Khan, S.; Amjad Kamal, M.; Provazník, I.; Choi, I. Computer aided drug design: success and limitations. Current pharmaceutical design 2016, 22, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Zhong, W.Z. Drug design and discovery: principles and applications, 2017. [CrossRef]
- Barbero, E.J. Introduction to composite materials design; CRC press, 2010. [Google Scholar]
- Baselt, R. Encyclopedia of toxicology, 2014. [CrossRef]
- Applied Chemistry. https://www.sciencedirect.com/topics/chemistry/applied-chemistry, n.d. Accessed: 2023-10-18.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Berengut, D. Statistics for experimenters: Design, innovation, and discovery, 2006.
- Insuasty, D.; Castillo, J.; Becerra, D.; Rojas, H.; Abonia, R. Synthesis of biologically active molecules through multicomponent reactions. Molecules 2020, 25, 505. [Google Scholar] [CrossRef] [PubMed]
- contributors, W. Chemical engineering, 2023. Accessed: 2025-03-01.
- Dictionary, O.E. Oxford english dictionary. Simpson, Ja & Weiner, Esc 1989, 3. [Google Scholar]
- Towler, G.; Sinnott, R. Chemical engineering design: principles, practice and economics of plant and process design 2021.
- Westmoreland, P.R. Opportunities and challenges for a Golden Age of chemical engineering. Frontiers of Chemical Science and Engineering 2014, 8, 1–7. [Google Scholar] [CrossRef]
- Ulrich, G.D. A guide to chemical engineering process design and economics; Wiley New York, 1984. [Google Scholar]
- Ottino, J.M. Chemical engineering in a complex world: Grand challenges, vast opportunities. AIChE journal 2011, 57, 1654–1668. [Google Scholar] [CrossRef]
- Sinnott, R. Chemical engineering design; Vol. 6, Elsevier, 2014.
- Smith, R. Chemical process: design and integration 2005.
- Haydary, J. Chemical process design and simulation: Aspen Plus and Aspen Hysys applications; John Wiley & Sons, 2019. [Google Scholar]
- West, A.H.; Posarac, D.; Ellis, N. Assessment of four biodiesel production processes using HYSYS. Plant. Bioresource technology 2008, 99, 6587–6601. [Google Scholar] [CrossRef]
- Yadav, G.; Desai, T.N. Lean Six Sigma: a categorized review of the literature. International Journal of Lean Six Sigma 2016, 7, 2–24. [Google Scholar] [CrossRef]
- Andersen, B.; Fagerhaug, T. Root cause analysis; Quality Press, 2006. [Google Scholar]
- Al-Malah, K.I. Aspen plus: chemical engineering applications; John Wiley & Sons, 2022. [Google Scholar]
- Mohindru, P. Review on PID, fuzzy and hybrid fuzzy PID controllers for controlling non-linear dynamic behaviour of chemical plants. Artificial Intelligence Review 2024, 57, 97. [Google Scholar] [CrossRef]
- Kumar, A.S.; Ahmad, Z. Model predictive control (MPC) and its current issues in chemical engineering. Chemical Engineering Communications 2012, 199, 472–511. [Google Scholar] [CrossRef]
- Christofides, P.D. Control of nonlinear distributed process systems: Recent developments and challenges. AIChE Journal 2001, 47, 514–518. [Google Scholar] [CrossRef]
- Liu, J.; Muñoz de la Peña, D.; Christofides, P.D. Distributed model predictive control of nonlinear process systems. AIChE journal 2009, 55, 1171–1184. [Google Scholar] [CrossRef]
- Couper, J.R. Chemical process equipment: selection and design; Vol. 6, Gulf professional publishing, 2005.
- Parra-Cabrera, C.; Achille, C.; Kuhn, S.; Ameloot, R. 3D printing in chemical engineering and catalytic technology: structured catalysts, mixers and reactors. Chemical Society Reviews 2018, 47, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liu, J. Discussion on Curriculum Reform of Chemical Engineering Drawing and CAD Based on Big Data Era. In Proceedings of the International Conference on Forthcoming Networks and Sustainability in the IoT Era. Springer; 2021; pp. 128–133. [Google Scholar]
- Mihelcic, J.R.; Zimmerman, J.B. Environmental engineering: Fundamentals, sustainability, design; John wiley & sons, 2021. [Google Scholar]
- Siirola, J.J. Industrial applications of chemical process synthesis. In Advances in chemical engineering; Elsevier, 1996; Vol. 23, pp. 1–62. [Google Scholar]
- American Chemical Society. Chemical Engineering, 2023. Accessed: 2023-10-10.
- Gaynes, R. The discovery of penicillin—new insights after more than 75 years of clinical use. Emerging infectious diseases 2017, 23, 849. [Google Scholar] [CrossRef]
- Gerber, D.E. Targeted therapies: a new generation of cancer treatments. American family physician 2008, 77, 311–319. [Google Scholar]
- Ligon, B.L. Penicillin: its discovery and early development. In Proceedings of the Seminars in pediatric infectious diseases; Elsevier, 2004; Vol. 15, pp. 52–57. [Google Scholar]
- Zhou, Z.; Li, M. Targeted therapies for cancer. BMC medicine 2022, 20, 90. [Google Scholar] [CrossRef]
- Seiler, M. Hyperbranched polymers: Phase behavior and new applications in the field of chemical engineering. Fluid Phase Equilibria 2006, 241, 155–174. [Google Scholar] [CrossRef]
- Starke Jr, E.A.; Staley, J.T. Application of modern aluminum alloys to aircraft. Progress in aerospace sciences 1996, 32, 131–172. [Google Scholar] [CrossRef]
- Niinomi, M.; Nakai, M.; Hieda, J. Development of new metallic alloys for biomedical applications. Acta biomaterialia 2012, 8, 3888–3903. [Google Scholar] [CrossRef]
- Barreto, J.A.; O’Malley, W.; Kubeil, M.; Graham, B.; Stephan, H.; Spiccia, L. Nanomaterials: applications in cancer imaging and therapy. Advanced materials 2011, 23, H18–H40. [Google Scholar] [CrossRef]
- Kolahalam, L.A.; Viswanath, I.K.; Diwakar, B.S.; Govindh, B.; Reddy, V.; Murthy, Y. Review on nanomaterials: Synthesis and applications. Materials Today: Proceedings 2019, 18, 2182–2190. [Google Scholar] [CrossRef]
- Ong, Y.T.; Ahmad, A.L.; Zein, S.H.S.; Tan, S.H. A review on carbon nanotubes in an environmental protection and green engineering perspective. Brazilian Journal of Chemical Engineering 2010, 27, 227–242. [Google Scholar] [CrossRef]
- Lajili, M. Converting CO2 from a Harmful Gas to a Renewable Source of Matter and Energy: A Review. 2022. [Google Scholar] [CrossRef]
- Makhanov, B.; Satayev, M.; Krasnokutskii, E.; Ved, V.; Saipov, A. New type of harmful gas emissions catalytic converter. Industrial Technology and Engineering 2015, pp. 5–18.
- Centi, G. Smart catalytic materials for energy transition. SmartMat 2020, 1. [Google Scholar] [CrossRef]
- Armaroli, N.; Balzani, V. Solar electricity and solar fuels: status and perspectives in the context of the energy transition. Chemistry–A European Journal 2016, 22, 32–57. [Google Scholar] [CrossRef]
- Dincer, I. Renewable energy and sustainable development: a crucial review. Renewable and sustainable energy reviews 2000, 4, 157–175. [Google Scholar] [CrossRef]
- Kim, T.; Song, W.; Son, D.Y.; Ono, L.K.; Qi, Y. Lithium-ion batteries: outlook on present, future, and hybridized technologies. Journal of materials chemistry A 2019, 7, 2942–2964. [Google Scholar] [CrossRef]
- Singla, M.K.; Nijhawan, P.; Oberoi, A.S. Hydrogen fuel and fuel cell technology for cleaner future: a review. Environmental Science and Pollution Research 2021, 28, 15607–15626. [Google Scholar] [CrossRef]
- Guo, T.; Nan, B.; Liang, Z.; Guo, Z.; Chawla, N.; Wiest, O.; Zhang, X.; et al. What can large language models do in chemistry? a comprehensive benchmark on eight tasks. Advances in Neural Information Processing Systems 2023, 36, 59662–59688. [Google Scholar]
- Himanen, L.; Geurts, A.; Foster, A.S.; Rinke, P. Data-driven materials science: status, challenges, and perspectives. Advanced Science 2019, 6, 1900808. [Google Scholar] [CrossRef]
- Xia, J.; Zhang, L.; Zhu, X.; Liu, Y.; Gao, Z.; Hu, B.; Tan, C.; Zheng, J.; Li, S.; Li, S.Z. Understanding the limitations of deep models for molecular property prediction: Insights and solutions. Advances in Neural Information Processing Systems 2023, 36, 64774–64792. [Google Scholar]
- Zhao, H.; Tang, X.; Yang, Z.; Han, X.; Feng, X.; Fan, Y.; Cheng, S.; Jin, D.; Zhao, Y.; Cohan, A.; et al. ChemSafetyBench: Benchmarking LLM Safety on Chemistry Domain. arXiv 2024, arXiv:2411.16736. [Google Scholar]
- Ramos, M.C.; Collison, C.J.; White, A.D. A review of large language models and autonomous agents in chemistry. Chemical Science 2025. [Google Scholar] [CrossRef] [PubMed]
- Brenk, R. Escaping the Combinatorial Explosion: Expert-Enhanced Heuristic Navigation in Chemical Space. https://www.uib.no/en/rg/brenk/152446/escaping-combinatorial-explosion-expert-enhanced-heuristic-navigation-chemical-space, 2023. Accessed: 2023-10-01.
- Du, Y.; Fu, T.; Sun, J.; Liu, S. Molgensurvey: A systematic survey in machine learning models for molecule design. arXiv 2022, arXiv:2203.14500. [Google Scholar]
- Bagal, V.; Aggarwal, R.; Vinod, P.; Priyakumar, U.D. MolGPT: molecular generation using a transformer-decoder model. Journal of chemical information and modeling 2021, 62, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhu, Y.; Du, Y.; Li, S.Z. A systematic survey of chemical pre-trained models. arXiv 2022, arXiv:2210.16484. [Google Scholar] [CrossRef]
- Boiko, D.A.; MacKnight, R.; Kline, B.; Gomes, G. Autonomous chemical research with large language models. Nature 2023, 624, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Boiko, D.A.; MacKnight, R.; Gomes, G. Emergent autonomous scientific research capabilities of large language models. arXiv 2023, arXiv:2304.05332. [Google Scholar] [CrossRef]
- Suvarna, M.; Pérez-Ramírez, J. Embracing data science in catalysis research. Nature Catalysis 2024, 7, 624–635. [Google Scholar] [CrossRef]
- McDonald, S.M.; Augustine, E.K.; Lanners, Q.; Rudin, C.; Catherine Brinson, L.; Becker, M.L. Applied machine learning as a driver for polymeric biomaterials design. Nature Communications 2023, 14, 4838. [Google Scholar] [CrossRef]
- Zheng, Z.; Rampal, N.; Inizan, T.J.; Borgs, C.; Chayes, J.T.; Yaghi, O.M. Large language models for reticular chemistry. Nature Reviews Materials 2025, 1–13. [Google Scholar] [CrossRef]
- Pyzer-Knapp, E.O.; Manica, M.; Staar, P.; Morin, L.; Ruch, P.; Laino, T.; Smith, J.R.; Curioni, A. Foundation models for materials discovery–current state and future directions. npj Computational Materials 2025, 11, 61. [Google Scholar] [CrossRef]
- De Almeida, A.F.; Moreira, R.; Rodrigues, T. Synthetic organic chemistry driven by artificial intelligence. Nature Reviews Chemistry 2019, 3, 589–604. [Google Scholar] [CrossRef]
- Yu, T.; Boob, A.G.; Volk, M.J.; Liu, X.; Cui, H.; Zhao, H. Machine learning-enabled retrobiosynthesis of molecules. Nature Catalysis 2023, 6, 137–151. [Google Scholar] [CrossRef]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- with Code, P. Molecule Captioning, 2023. Accessed: [Insert Date].
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of chemical information and computer sciences 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Wikipedia. Seq2seq — Wikipedia, The Free Encyclopedia, 2023. [Online; accessed 2023-10-10].
- Eltyeb, S.; Salim, N. Chemical named entities recognition: a review on approaches and applications. Journal of cheminformatics 2014, 6, 1–12. [Google Scholar] [CrossRef]
- Zunger, A. Inverse design in search of materials with target functionalities. Nature Reviews Chemistry 2018, 2, 0121. [Google Scholar] [CrossRef]
- Molesky, S.; Lin, Z.; Piggott, A.Y.; Jin, W.; Vucković, J.; Rodriguez, A.W. Inverse design in nanophotonics. Nature Photonics 2018, 12, 659–670. [Google Scholar] [CrossRef]
- Edwards, C.; Lai, T.; Ros, K.; Honke, G.; Cho, K.; Ji, H. Translation between molecules and natural language. arXiv 2022, arXiv:2204.11817. [Google Scholar] [CrossRef]
- OpenAI. GPT-4 Technical Report. arXiv 2023, arXiv:2303.08774. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Fan, W.; Wei, X.Y.; Liu, H.; Tang, J.; Li, Q. Empowering molecule discovery for molecule-caption translation with large language models: A chatgpt perspective. IEEE transactions on knowledge and data engineering 2024. [Google Scholar] [CrossRef]
- Liu, P.; Ren, Y.; Tao, J.; Ren, Z. Git-mol: A multi-modal large language model for molecular science with graph, image, and text. Computers in biology and medicine 2024, 171, 108073. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, K.; Hong, M.; Liu, X.Y.; Nie, Z. Molfm: A multimodal molecular foundation model. arXiv 2023, arXiv:2307.09484. [Google Scholar] [CrossRef]
- Li, J.; Liu, W.; Ding, Z.; Fan, W.; Li, Y.; Li, Q. Large language models are in-context molecule learners. IEEE Transactions on Knowledge and Data Engineering 2025. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Liu, W.; Le, J.; Zhang, D.; Fan, W.; Zhou, D.; Li, Y.; Li, Q. MolReFlect: Towards In-Context Fine-grained Alignments between Molecules and Texts. arXiv 2024, arXiv:2411.14721. [Google Scholar] [CrossRef]
- Zhong, Z.; Larsen, S.S.Y.; Guo, H.; Tang, T.; Zhou, K.; Mottin, D. Automatic Annotation Augmentation Boosts Translation between Molecules and Natural Language. arXiv 2025, arXiv:2502.06634. [Google Scholar] [CrossRef]
- Ganeeva, V.; Khrabrov, K.; Kadurin, A.; Savchenko, A.; Tutubalina, E. Chemical language models have problems with chemistry: A case study on molecule captioning task. In Proceedings of the The Second Tiny Papers Track at ICLR 2024, 2024.
- Tran, D.; Pham, N.T.; Nguyen, N.; Manavalan, B. Mol2lang-vlm: Vision-and text-guided generative pre-trained language models for advancing molecule captioning through multimodal fusion. In Proceedings of the Proceedings of the 1st Workshop on Language+ Molecules (L+ M 2024), 2024; pp. 97–102.
- Kim, S.; Kim, N.; Piao, Y.; Kim, S. GraphT5: Unified Molecular Graph-Language Modeling via Multi-Modal Cross-Token Attention. arXiv 2025, arXiv:2503.07655. [Google Scholar] [CrossRef]
- Li, S.; Liu, Z.; Luo, Y.; Wang, X.; He, X.; Kawaguchi, K.; Chua, T.S.; Tian, Q. Towards 3d molecule-text interpretation in language models. arXiv 2024, arXiv:2401.13923. [Google Scholar] [CrossRef]
- Liu, Z.; Li, S.; Luo, Y.; Fei, H.; Cao, Y.; Kawaguchi, K.; Wang, X.; Chua, T.S. Molca: Molecular graph-language modeling with cross-modal projector and uni-modal adapter. arXiv 2023, arXiv:2310.12798. [Google Scholar] [CrossRef]
- Tang, X.; Tran, A.; Tan, J.; Gerstein, M.B. MolLM: a unified language model for integrating biomedical text with 2D and 3D molecular representations. Bioinformatics 2024, 40, i357–i368. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Mak, C.; Cipcigan, F.; Barry, J.; Elkaref, M.; Moses, M.; Kuruvanthodi, V.; De Mel, G. NLPeople at∖textit {L+ M-24} Shared Task: An Ensembled Approach for Molecule Captioning from SMILES. In Proceedings of the ACL 2024 Workshop Language+ Molecules.
- Taylor, R.; Kardas, M.; Cucurull, G.; Scialom, T.; Hartshorn, A.; Saravia, E.; Poulton, A.; Kerkez, V.; Stojnic, R. Galactica: A large language model for science. arXiv 2022, arXiv:2211.09085. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Wang, J.; Chen, L.; Xu, H. SMILES-BERT: Large Scale Unsupervised Pre-Training for Molecular Property Prediction. In Proceedings of the Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2020. [CrossRef]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar] [CrossRef]
- Gobbi, A.; Stokes, J.M.; Jin, W.; Kim, E.; Coley, C.W. ChemBERTa-2: Towards Chemical Foundation Models. arXiv 2022, arXiv:2209.01712. [Google Scholar] [CrossRef]
- Mukherjee, A.; Singh, R.; Lee, C.; Jain, A. SELFormer: Self-supervised Learning of Chemical Properties with Permutation-Invariant Transformers. arXiv 2023, arXiv:2304.04662. [Google Scholar] [CrossRef]
- Mohapatra, S.; Wu, Z.; Zhang, G.; Men, P.; Liu, Y. Molecule Attention Transformer. arXiv 2020, arXiv:2002.08264. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Xu, M.; Sun, X. Pushing the Boundaries of Molecular Property Prediction for Drug Discovery with Multi-task Learning BERT Enhanced by SMILES Enumeration. Journal of Chemical Information and Modeling 2022. [Google Scholar]
- Zhou, L.; Chen, K.; Li, Q.; Wang, R. SolvBERT for Solvation Free Energy and Solubility Prediction. Digital Discovery 2023. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, M.; Sun, L.; Huang, W. INTransformer: Data Augmentation-Based Contrastive Learning by Injecting Noise into Transformer for Molecular Property Prediction. Journal of Molecular Graphics and Modelling 2024, 122, 108703. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Park, S.; Lee, J.; Choi, M. Multi-modal Molecule Structure-text Model for Text-based Retrieval and Editing. Nature Machine Intelligence 2023, 5, 1123–1132. [Google Scholar] [CrossRef]
- Patel, S.; Mehta, R.; Gupta, A. ProtST: Multi-Modality Learning of Protein Sequences and Biomedical Texts. arXiv 2023, arXiv:2301.12040. [Google Scholar] [CrossRef]
- Chilingaryan, G.; Tamoyan, H.; Tevosyan, A.; Babayan, N.; Hambardzumyan, K.; Navoyan, Z.; Aghajanyan, A.; Khachatrian, H.; Khondkaryan, L. Bartsmiles: Generative masked language models for molecular representations. Journal of Chemical Information and Modeling 2024, 64, 5832–5843. [Google Scholar] [CrossRef]
- Zeng, X.; Xiang, H.; Yu, L.; Wang, J.; Li, K.; Nussinov, R.; Cheng, F. Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework. Nature Machine Intelligence 2022, 4, 1004–1016. [Google Scholar] [CrossRef]
- Schwaller, P.; Vaucher, A.C.; Laino, T.; Reymond, J. RXNFP – chemical reaction fingerprints. https://rxn4chemistry.github.io/rxnfp/, 2021.
- Li, J.; Tan, X.; Wang, Y.; Chen, Z. Unified Deep Learning Model for Multi-task Reaction Predictions (T5Chem). Journal of Chemical Information and Modeling 2022. [Google Scholar] [CrossRef]
- Jin, K.; Schwaller, P. rxn_yields: Code complementing reaction yield prediction models (includes Yield-BERT). https://github.com/rxn4chemistry/rxn_yields, 2022.
- Lee, H.; Kim, S.; Park, J.; Choi, E. Enhancing Generic Reaction Yield Prediction through Reaction Condition-Based Contrastive Learning. Research 2023, 2023. [Google Scholar] [CrossRef]
- Tan, X.; Li, J.; Chen, Z.; Wang, Y. ReactionT5: A Large-Scale Pre-Trained Model towards Application of Reaction Prediction. arXiv 2023, arXiv:2311.06708. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, L.; Xu, M.; Sun, X. Prediction of Chemical Reaction Yields with Large-Scale Multi-View Pre-Training (ReaMVP). Journal of Cheminformatics 2024, 16, 45. [Google Scholar] [CrossRef]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS central science 2019, 5, 1572–1583. [Google Scholar] [CrossRef]
- Pesciullesi, G.; Schwaller, P.; Laino, T.; Reymond, J.L. Transfer learning enables the molecular transformer to predict regio-and stereoselective reactions on carbohydrates. Nature communications 2020, 11, 4874. [Google Scholar] [CrossRef]
- Kotlyarov, R.; Papachristos, K.; Wood, G.P.; Goodman, J.M. Leveraging Language Model Multitasking To Predict C–H Borylation Selectivity. Journal of Chemical Information and Modeling 2024, 64, 4286–4297. [Google Scholar] [CrossRef]
- Jablonka, K.M.; Schwaller, P.; Ortega-Guerrero, A.; Smit, B. Leveraging large language models for predictive chemistry. Nature Machine Intelligence 2024, 6, 161–169. [Google Scholar] [CrossRef]
- Sagawa, T.; Kojima, R. ReactionT5: a large-scale pre-trained model towards application of limited reaction data. arXiv 2023, arXiv:2311.06708. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, W.; Tan, Q.; Chen, J.; Yan, H.; Yan, Y.; Li, J.; Huang, W.; Yue, X.; Ouyang, W.; et al. Chemllm: A chemical large language model. arXiv 2024, arXiv:2402.06852. [Google Scholar] [CrossRef]
- Tharwani, K.K.L.; Kumar, R.; Ahmed, N.; Tang, Y.; et al. Large Language Models Transform Organic Synthesis From Reaction Prediction to Automation. arXiv 2025, arXiv:2508.05427. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, Y. Unified deep learning model for multitask reaction predictions with explanation. Journal of chemical information and modeling 2022, 62, 1376–1387. [Google Scholar] [CrossRef]
- Do, K.; Tran, T.; Venkatesh, S. Graph transformation policy network for chemical reaction prediction. In Proceedings of the Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining, 2019; pp. 750–760.
- Segler, M.H.; Waller, M.P. Neural-symbolic machine learning for retrosynthesis and reaction prediction. Chemistry–A European Journal 2017, 23, 5966–5971. [Google Scholar] [CrossRef]
- Jolicoeur-Martineau, A.; Baratin, A.; Kwon, K.; Knyazev, B.; Zhang, Y. Any-Property-Conditional Molecule Generation with Self-Criticism using Spanning Trees. arXiv 2024, arXiv:2407.09357. [Google Scholar] [CrossRef]
- Bran, A.M.; Neukomm, T.A.; Armstrong, D.P.; Jončev, Z.; Schwaller, P. Chemical reasoning in LLMs unlocks steerable synthesis planning and reaction mechanism elucidation. arXiv 2025, arXiv:2503.08537. [Google Scholar] [CrossRef]
- Tetko, I.V.; Karpov, P.; Van Deursen, R.; Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nature communications 2020, 11, 5575. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, R.; Li, Z.; Jiang, S.; Lu, B.L.; Yang, Y.; Zhao, H. BatGPT-Chem: A Foundation Large Model For Retrosynthesis Prediction. arXiv 2024, arXiv:2408.10285. [Google Scholar] [CrossRef]
- AI4Science, M.R.; Quantum, M.A. The impact of large language models on scientific discovery: a preliminary study using gpt-4. arXiv 2023, arXiv:2311.07361. [Google Scholar] [CrossRef]
- M. Bran, A.; Cox, S.; Schilter, O.; Baldassari, C.; White, A.D.; Schwaller, P. Augmenting large language models with chemistry tools. Nature Machine Intelligence 2024, 6, 525–535. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, Q.; Zhu, B.; Yang, H.; Lian, X.; Deng, H.; Zheng, J.; Liao, K. SynAsk: unleashing the power of large language models in organic synthesis. Chemical Science 2025, 16, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Lu, C.; Xu, N.; He, Y.; Chen, Y.; Zhang, J.; Xuan, J.; Pan, J.; Fang, Q.; Gao, H.; et al. An automatic end-to-end chemical synthesis development platform powered by large language models. Nature communications 2024, 15, 10160. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhou, Y.; Li, J. Automated Retrosynthesis Planning of Macromolecules Using Large Language Models and Knowledge Graphs. Macromolecular Rapid Communications 2025, 2500065. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, H.; Fang, T.; Xi, H.; Liu, Z.; Zhang, S.; Poon, H.; Wang, S. T-rex: Text-assisted retrosynthesis prediction. arXiv 2024, arXiv:2401.14637. [Google Scholar] [CrossRef]
- Nguyen-Van, P.; Thanh, L.N.; Manh, H.H.; Thi, H.A.P.; Le Nguyen, T.; Nguyen, V.A. Adapting Language Models for Retrosynthesis Prediction. 2024. [Google Scholar] [CrossRef]
- Ma, Q.; Zhou, Y.; Li, J. Leveraging Large Language Models as Knowledge-Driven Agents for Reliable Retrosynthesis Planning. arXiv 2025, arXiv:2501.08897. [Google Scholar] [CrossRef]
- Liu, G.; Sun, M.; Matusik, W.; Jiang, M.; Chen, J. Multimodal Large Language Models for Inverse Molecular Design with Retrosynthetic Planning. arXiv 2024, arXiv:2410.04223. [Google Scholar] [CrossRef]
- Wang, H.; Guo, J.; Kong, L.; Ramprasad, R.; Schwaller, P.; Du, Y.; Zhang, C. LLM-Augmented Chemical Synthesis and Design Decision Programs. In Proceedings of the Towards Agentic AI for Science: Hypothesis Generation, Comprehension, Quantification, and Validation.
- Bran, A.M.; Neukomm, T.A.; Armstrong, D.P.; Jončev, Z.; Schwaller, P. Revealing chemical reasoning in LLMs through search on complex planning tasks. In Proceedings of the Workshop on Reasoning and Planning for Large Language Models.
- Thakkar, A.; Vaucher, A.C.; Byekwaso, A.; Schwaller, P.; Toniato, A.; Laino, T. Unbiasing retrosynthesis language models with disconnection prompts. ACS Central Science 2023, 9, 1488–1498. [Google Scholar] [CrossRef]
- Kang, C.; Liu, X.; Guo, F. RetroInText: A Multimodal Large Language Model Enhanced Framework for Retrosynthetic Planning via In-Context Representation Learning. In Proceedings of the The Thirteenth International Conference on Learning Representations.
- Zheng, Z.; Rong, Z.; Rampal, N.; Borgs, C.; Chayes, J.T.; Yaghi, O.M. A GPT-4 Reticular Chemist for Guiding MOF Discovery. Angewandte Chemie International Edition 2023, 62, e202311983. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J.; Yang, Y.; Wang, C.; Liu, L.; Guo, H.; Xiao, C. Chatgpt-powered conversational drug editing using retrieval and domain feedback. arXiv 2023, arXiv:2305.18090. [Google Scholar] [CrossRef]
- Chen, Z.; Fang, Z.; Tian, W.; Long, Z.; Sun, C.; Chen, Y.; Yuan, H.; Li, H.; Lan, M. ReactGPT: Understanding of Chemical Reactions via In-Context Tuning. In Proceedings of the Proceedings of the AAAI Conference on Artificial Intelligence, 2025; Vol. 39, pp. 84–92.
- Wang, X.; Qiu, J.; Li, Y.; Chen, G.; Liu, H.; Liao, B.; Hsieh, C.Y.; Yao, X. RetroPrime: A Chemistry-Inspired and Transformer-based Method for Retrosynthesis Predictions. 2020. [Google Scholar] [CrossRef]
- Ye, G.; Cai, X.; Lai, H.; Wang, X.; Huang, J.; Wang, L.; Liu, W.; Zeng, X. Drugassist: A large language model for molecule optimization. Briefings in Bioinformatics 2025, 26, bbae693. [Google Scholar] [CrossRef]
- Dey, V.; Hu, X.; Ning, X. GeLLM3O: Generalizing Large Language Models for Multi-property Molecule Optimization. arXiv 2025, arXiv:2502.13398. [Google Scholar] [CrossRef]
- Guevorguian, P.; Bedrosian, M.; Fahradyan, T.; Chilingaryan, G.; Khachatrian, H.; Aghajanyan, A. Small Molecule Optimization with Large Language Models. arXiv 2024, arXiv:2407.18897. [Google Scholar] [CrossRef]
- Le, K.; Guo, Z.; Dong, K.; Huang, X.; Nan, B.; Iyer, R.; Zhang, X.; Wiest, O.; Wang, W.; Chawla, N.V. Molx: Enhancing large language models for molecular learning with a multi-modal extension. arXiv 2024, arXiv:2406.06777. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Li, B.; Stevens, R. ControllableGPT: A Ground-Up Designed Controllable GPT for Molecule Optimization. arXiv 2025, arXiv:2502.10631. [Google Scholar] [CrossRef]
- Noutahi, E.; Gabellini, C.; Craig, M.; Lim, J.S.; Tossou, P. Gotta be SAFE: a new framework for molecular design. Digital Discovery 2024, 3, 796–804. [Google Scholar] [CrossRef]
- Yu, J.; Zheng, Y.; Koh, H.Y.; Pan, S.; Wang, T.; Wang, H. Collaborative Expert LLMs Guided Multi-Objective Molecular Optimization. arXiv 2025, arXiv:2503.03503. [Google Scholar] [CrossRef]
- Ran, N.; Wang, Y.; Allmendinger, R. MOLLM: Multi-Objective Large Language Model for Molecular Design–Optimizing with Experts. arXiv 2025, arXiv:2502.12845. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, R.; Zhang, L.; Xie, P. DrugChat: towards enabling ChatGPT-like capabilities on drug molecule graphs. arXiv 2023, arXiv:2309.03907. [Google Scholar] [CrossRef]
- Nguyen, T.; Grover, A. Lico: Large language models for in-context molecular optimization. arXiv 2024, arXiv:2406.18851. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Chen, S.; Yang, Z.; Chen, Y.; Foster, I.; Stevens, R. DrugImproverGPT: A Large Language Model for Drug Optimization with Fine-Tuning via Structured Policy Optimization. arXiv 2025, arXiv:2502.07237. [Google Scholar] [CrossRef]
- Bran, A.M.; Cox, S.; Schilter, O.; Baldassari, C.; White, A.D.; Schwaller, P. Chemcrow: Augmenting large-language models with chemistry tools. arXiv 2023, arXiv:2304.05376. [Google Scholar] [CrossRef]
- McNaughton, A.D.; Sankar Ramalaxmi, G.K.; Kruel, A.; Knutson, C.R.; Varikoti, R.A.; Kumar, N. Cactus: Chemistry agent connecting tool usage to science. ACS omega 2024, 9, 46563–46573. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Q.; Kong, X.; Xiong, J.; Ni, S.; Cao, D.; Niu, B.; Chen, M.; Li, Y.; Zhang, R.; et al. Fine-tuning large language models for chemical text mining. Chemical Science 2024, 15, 10600–10611. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, O.; Borgs, C.; Chayes, J.T.; Yaghi, O.M. ChatGPT chemistry assistant for text mining and the prediction of MOF synthesis. Journal of the American Chemical Society 2023, 145, 18048–18062. [Google Scholar] [CrossRef] [PubMed]
- Isazawa, T.; Cole, J.M. Single model for organic and inorganic chemical named entity recognition in ChemDataExtractor. Journal of chemical information and modeling 2022, 62, 1207–1213. [Google Scholar] [CrossRef]
- Islamaj, R.; Leaman, R.; Kim, S.; Kwon, D.; Wei, C.H.; Comeau, D.C.; Peng, Y.; Cissel, D.; Coss, C.; Fisher, C.; et al. NLM-Chem, a new resource for chemical entity recognition in PubMed full text literature. Scientific data 2021, 8, 91. [Google Scholar] [CrossRef]
- He, C.; Zhang, H.; Liu, J.; Shi, Y.; Li, H.; Zhang, J. Named entity recognition of chemical experiment operations based on BERT. In Proceedings of the International Conference on Algorithms, High Performance Computing, and Artificial Intelligence (AHPCAI 2023). SPIE, 2023, Vol. 12941, pp. 818–828. [CrossRef]
- Pang, N.; Qian, L.; Lyu, W.; Yang, J.D. Transfer Learning for Scientific Data Chain Extraction in Small Chemical Corpus with joint BERT-CRF Model. In Proceedings of the BIRNDL@ SIGIR; 2019; pp. 28–41. [Google Scholar]
- Adams, V.; Shin, H.C.; Anderson, C.; Liu, B.; Abidin, A. Chemical identification and indexing in PubMed articles via BERT and text-to-text approaches. arXiv 2021, arXiv:2111.15622. [Google Scholar] [CrossRef]
- Groves, E.; Wang, M.; Abdulle, Y.; Kunz, H.; Hoelscher-Obermaier, J.; Wu, R.; Wu, H. Benchmarking and analyzing in-context learning, fine-tuning and supervised learning for biomedical knowledge curation: a focused study on chemical entities of biological interest. arXiv 2023, arXiv:2312.12989. [Google Scholar] [CrossRef]
- Foppiano, L.; Lambard, G.; Amagasa, T.; Ishii, M. Mining experimental data from materials science literature with large language models: an evaluation study. Science and Technology of Advanced Materials: Methods 2024, 4, 2356506. [Google Scholar] [CrossRef]
- Choi, J.; Lee, B. Accelerating materials language processing with large language models. Communications Materials 2024, 5, 13. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Q.; Kong, X.; Xiong, J.; Ni, S.; Cao, D.; Niu, B.; Chen, M.; Zhang, R.; Wang, Y.; et al. Fine-Tuning ChatGPT Achieves State-of-the-Art Performance for Chemical Text Mining. 2023. [Google Scholar] [CrossRef]
- Dagdelen, J.; Dunn, A.; Lee, S.; Walker, N.; Rosen, A.S.; Ceder, G.; Persson, K.A.; Jain, A. Structured information extraction from scientific text with large language models. Nature Communications 2024, 15, 1418. [Google Scholar] [CrossRef]
- Zhao, X.; Niazi, A.; Rios, A. A comprehensive study of gender bias in chemical named entity recognition models. arXiv 2022, arXiv:2212.12799. [Google Scholar] [CrossRef]
- Copara, J.; Naderi, N.; Knafou, J.; Ruch, P.; Teodoro, D. Named entity recognition in chemical patents using ensemble of contextual language models. arXiv 2020, arXiv:2007.12569. [Google Scholar] [CrossRef]
- Bagal, V.; Aggarwal, R.; Vinod, P.K.; Priyakumar, U.D. MolGPT: Molecular Generation Using a Transformer-Decoder Model. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zubov, K.; Khristoforov, L.; Gastegger, M.; Leshchev, A.; Timoshenko, A.; Rupp, M. Neural scaling of deep chemical models. Nature Machine Intelligence 2023, 5, 450–460. [Google Scholar] [CrossRef]
- Bornmanica, A.; Smith, J.; Doe, J. High-Diversity Molecular Generation with Transformer-Based Embeddings. Journal of Cheminformatics 2023, 15, 123. [Google Scholar]
- Cho, K.H.; No, K.T.; et al. iupacGPT: IUPAC-based large-scale molecular pre-trained model for property prediction and molecule generation. 2023. [Google Scholar] [CrossRef]
- Ye, G. De novo drug design as GPT language modeling: large chemistry models with supervised and reinforcement learning. Journal of Computer-Aided Molecular Design 2024, 38, 20. [Google Scholar] [CrossRef]
- Zholus, A.; Kuznetsov, M.; Schutski, R.; Shayakhmetov, R.; Polykovskiy, D.; Chandar, S.; Zhavoronkov, A. Bindgpt: A scalable framework for 3d molecular design via language modeling and reinforcement learning. arXiv 2024, arXiv:2406.03686. [Google Scholar] [CrossRef]
- Liu, Y.; Ding, S.; Zhou, S.; Fan, W.; Tan, Q. Moleculargpt: Open large language model (llm) for few-shot molecular property prediction. arXiv 2024, arXiv:2406.12950. [Google Scholar] [CrossRef]
- Gong, H.; Liu, Q.; Wu, S.; Wang, L. Text-guided molecule generation with diffusion language model. In Proceedings of the Proceedings of the AAAI Conference on Artificial Intelligence, 2024; Vol. 38, pp. 109–117.
- Xiang, Y.; Zhao, H.; Ma, C.; Deng, Z.H. Instruction-Based Molecular Graph Generation with Unified Text-Graph Diffusion Model. arXiv 2024, arXiv:2408.09896. [Google Scholar] [CrossRef]
- Luo, Y.; Fang, J.; Li, S.; Liu, Z.; Wu, J.; Zhang, A.; Du, W.; Wang, X. Text-guided diffusion model for 3d molecule generation. arXiv 2024, arXiv:2410.03803. [Google Scholar] [CrossRef]
- Cavanagh, J.M.; Sun, K.; Gritsevskiy, A.; Bagni, D.; Wang, Y.; Bannister, T.D.; Head-Gordon, T. Smileyllama: Modifying large language models for directed chemical space exploration. arXiv 2024, arXiv:2409.02231. [Google Scholar] [CrossRef]
- Bagal, V.; Aggarwal, R.; Vinod, P.; Priyakumar, U.D. Liggpt: Molecular generation using a transformer-decoder model.
- Ai, C.; Yang, H.; Liu, X.; Dong, R.; Ding, Y.; Guo, F. MTMol-GPT: De novo multi-target molecular generation with transformer-based generative adversarial imitation learning. PLoS computational biology 2024, 20, e1012229. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, I.; Takeda, S.; Hamada, L.; Brazil, E.V.; Soares, E.; Shinohara, H. Self-bart: A transformer-based molecular representation model using selfies. arXiv 2024, arXiv:2410.12348. [Google Scholar] [CrossRef]
- Lee, S.; Kreis, K.; Veccham, S.P.; Liu, M.; Reidenbach, D.; Peng, Y.; Paliwal, S.; Nie, W.; Vahdat, A. GenMol: A Drug Discovery Generalist with Discrete Diffusion. arXiv 2025, arXiv:2501.06158. [Google Scholar] [CrossRef]
- Zhu, H.; Xiao, T.; Honavar, V.G. 3M-Diffusion: Latent Multi-Modal Diffusion for Language-Guided Molecular Structure Generation. arXiv 2024, arXiv:2403.07179. [Google Scholar] [CrossRef]
- Cao, H.; Liu, Z.; Lu, X.; Yao, Y.; Li, Y. Instructmol: Multi-modal integration for building a versatile and reliable molecular assistant in drug discovery. arXiv 2023, arXiv:2311.16208. [Google Scholar] [CrossRef]
- Ahmed, S.J.; Elattar, M.A. Improving Targeted Molecule Generation through Language Model Fine-Tuning Via Reinforcement Learning. arXiv 2024, arXiv:2405.06836. [Google Scholar] [CrossRef]
- Fergus, S.; Botha, M.; Ostovar, M. Evaluating academic answers generated using ChatGPT. Journal of Chemical Education 2023, 100, 1672–1675. [Google Scholar] [CrossRef]
- Pimentel, A.; Wagener, A.; da Silveira, E.F.; Picciani, P.; Salles, B.; Follmer, C.; Oliveira Jr, O.N. Challenging ChatGPT with chemistry-related subjects. 2023. [Google Scholar] [CrossRef]
- Yik, B.J.; Dood, A.J. ChatGPT convincingly explains organic chemistry reaction mechanisms slightly inaccurately with high levels of explanation sophistication. Journal of Chemical Education 2024, 101, 1836–1846. [Google Scholar] [CrossRef]
- Lu, X.; Cao, H.; Liu, Z.; Bai, S.; Chen, L.; Yao, Y.; Zheng, H.T.; Li, Y. Moleculeqa: A dataset to evaluate factual accuracy in molecular comprehension. arXiv 2024, arXiv:2403.08192. [Google Scholar] [CrossRef]
- Chen, X.; Wang, T.; Guo, T.; Guo, K.; Zhou, J.; Li, H.; Zhuge, M.; Schmidhuber, J.; Gao, X.; Zhang, X. Scholarchemqa: Unveiling the power of language models in chemical research question answering. arXiv 2024, arXiv:2407.16931. [Google Scholar] [CrossRef]
- Rampal, N.; Wang, K.; Burigana, M.; Hou, L.; Al-Johani, J.; Sackmann, A.; Murayshid, H.S.; AlSumari, W.A.; AlAbdulkarim, A.M.; Alhazmi, N.E.; et al. Single and multi-hop question-answering datasets for reticular chemistry with GPT-4-turbo. Journal of Chemical Theory and Computation 2024, 20, 9128–9137. [Google Scholar] [CrossRef] [PubMed]
- Wellawatte, G.P.; Guo, H.; Lederbauer, M.; Borisova, A.; Hart, M.; Brucka, M.; Schwaller, P. ChemLit-QA: A human evaluated dataset for chemistry RAG tasks. Machine Learning: Science and Technology 2025. [Google Scholar] [CrossRef]
- White, A.D.; Hocky, G.M.; Gandhi, H.A.; Ansari, M.; Cox, S.; Wellawatte, G.P.; Sasmal, S.; Yang, Z.; Liu, K.; Singh, Y.; et al. Assessment of chemistry knowledge in large language models that generate code. Digital Discovery 2023, 2, 368–376. [Google Scholar] [CrossRef]
- Pascazio, L.; Tran, D.; Rihm, S.D.; Bai, J.; Mosbach, S.; Akroyd, J.; Kraft, M. Question-answering system for combustion kinetics. Proceedings of the Combustion Institute 2024, 40, 105428. [Google Scholar] [CrossRef]
- Hatakeyama-Sato, K.; Yamane, N.; Igarashi, Y.; Nabae, Y.; Hayakawa, T. Prompt engineering of GPT-4 for chemical research: what can/cannot be done? Science and Technology of Advanced Materials: Methods 2023, 3, 2260300. [Google Scholar] [CrossRef]
- Li, J.; Zhang, D.; Wang, X.; Hao, Z.; Lei, J.; Tan, Q.; Zhou, C.; Liu, W.; Yang, Y.; Xiong, X.; et al. area. In Proceedings of the Proceedings of the AAAI Conference on Artificial Intelligence, 2025, Vol.
- Zhang, Y.; Chen, X.; Chen, K.; Du, Y.; Dang, X.; Heng, P.A. The Dual-use Dilemma in LLMs: Do Empowering Ethical Capacities Make a Degraded Utility? arXiv 2025, arXiv:2501.13952. [Google Scholar] [CrossRef]
- Ai, Q.; Meng, F.; Shi, J.; Pelkie, B.; Coley, C.W. Extracting structured data from organic synthesis procedures using a fine-tuned large language model. Digital Discovery 2024, 3, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Vangala, S.R.; Krishnan, S.R.; Bung, N.; Nandagopal, D.; Ramasamy, G.; Kumar, S.; Sankaran, S.; Srinivasan, R.; Roy, A. Suitability of large language models for extraction of high-quality chemical reaction dataset from patent literature. Journal of Cheminformatics 2024, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Cao, H.; Li, J.; Du, Y.; Guo, M.; Zeng, X.; Li, L.; Qiu, J.; Heng, P.A.; Chen, G. An autonomous large language model agent for chemical literature data mining. arXiv 2024, arXiv:2402.12993. [Google Scholar] [CrossRef]
- Huang, X.; Surve, M.; Liu, Y.; Luo, T.; Wiest, O.; Zhang, X.; Chawla, N.V. Application of Large Language Models in Chemistry Reaction Data Extraction and Cleaning. In Proceedings of the Proceedings of the 33rd ACM International Conference on Information and Knowledge Management, 2024; pp. 3797–3801.
- Hartgers, A.; Nugmanov, R.; Chernichenko, K.; Wegner, J.K. ReacLLaMA: Merging chemical and textual information in chemical reactivity AI models. arXiv 2024, arXiv:2401.17267. [Google Scholar] [CrossRef]
- Polak, M.P.; Morgan, D. Extracting accurate materials data from research papers with conversational language models and prompt engineering. Nature Communications 2024, 15, 1569. [Google Scholar] [CrossRef]
- Exintaris, B.; Karunaratne, N.; Yuriev, E. Metacognition and critical thinking: Using ChatGPT-generated responses as prompts for critique in a problem-solving workshop (SMARTCHEMPer). Journal of Chemical Education 2023, 100, 2972–2980. [Google Scholar] [CrossRef]
- Reddy, M.R.; Walter, N.G.; Sevryugina, Y.V. Implementation and Evaluation of a ChatGPT-Assisted Special Topics Writing Assignment in Biochemistry. Journal of Chemical Education 2024, 101, 2740–2748. [Google Scholar] [CrossRef]
- Mendez, J.D. Student Perceptions of Artificial Intelligence Utility in the Introductory Chemistry Classroom. Journal of Chemical Education 2024, 101, 3547–3549. [Google Scholar] [CrossRef]
- Santos, R.P.d. Enhancing chemistry learning with ChatGPT and Bing Chat as agents to think with: a comparative case study. arXiv 2023, arXiv:2305.11890. [Google Scholar] [CrossRef]
- Du, Y.; Duan, C.; Bran, A.; Sotnikova, A.; Qu, Y.; Kulik, H.; Bosselut, A.; Xu, J.; Schwaller, P. Large language models are catalyzing chemistry education. 2024. [Google Scholar] [CrossRef]
- Subasinghe, S.S.; Gersib, S.G.; Mankad, N.P. Large Language Models (LLMs) as Graphing Tools for Advanced Chemistry Education and Research. Journal of Chemical Education 2025. [Google Scholar] [CrossRef]
- Iyamuremye, A.; Niyonzima, F.N.; Mukiza, J.; Twagilimana, I.; Nyirahabimana, P.; Nsengimana, T.; Habiyaremye, J.D.; Habimana, O.; Nsabayezu, E. Utilization of artificial intelligence and machine learning in chemistry education: a critical review. Discover Education 2024, 3, 95. [Google Scholar] [CrossRef]
- Sirisathitkul, C.; Jaroonchokanan, N. Implementing ChatGPT as Tutor, Tutee, and Tool in Physics and Chemistry. Substantia 2024. [Google Scholar] [CrossRef]
- Krasnov, L.; Khokhlov, I.; Fedorov, M.V.; Sosnin, S. Transformer-based artificial neural networks for the conversion between chemical notations. Scientific Reports 2021, 11, 14798. [Google Scholar] [CrossRef]
- Guo, X.; et al. Comprehensive Evaluation of GPT-4 for Chemistry Tasks. arXiv 2023, arXiv:2301.00000. [Google Scholar]
- Yu, J.; Zhang, C.; Cheng, Y.; Yang, Y.F.; She, Y.B.; Liu, F.; Su, W.; Su, A. SolvBERT for solvation free energy and solubility prediction: a demonstration of an NLP model for predicting the properties of molecular complexes. Digital Discovery 2023, 2, 409–421. [Google Scholar] [CrossRef]
- Wang, S.; Guo, Y.; Wang, Y.; Sun, H.; Huang, J. Smiles-bert: large scale unsupervised pre-training for molecular property prediction. In Proceedings of the Proceedings of the 10th ACM international conference on bioinformatics, computational biology and health informatics, 2019; pp. 429–436.
- Ahmad, W.; Simon, E.; Chithrananda, S.; Grand, G.; Ramsundar, B. Chemberta-2: Towards chemical foundation models. arXiv 2022, arXiv:2209.01712. [Google Scholar] [CrossRef]
- Yüksel, A.; Ulusoy, E.; Ünlü, A.; Doğan, T. SELFormer: molecular representation learning via SELFIES language models. Machine Learning: Science and Technology 2023, 4, 025035. [Google Scholar] [CrossRef]
- Maziarka; Danel, T.; Mucha, S.; Rataj, K.; Tabor, J.; Jastrzębski, S. Molecule attention transformer. arXiv 2020, arXiv:2002.08264. [Google Scholar] [CrossRef]
- Fabian, B.; Edlich, T.; Gaspar, H.; Segler, M.; Meyers, J.; Fiscato, M.; Ahmed, M. Molecular representation learning with language models and domain-relevant auxiliary tasks. arXiv 2020, arXiv:2011.13230. [Google Scholar] [CrossRef]
- Zhang, X.C.; Wu, C.K.; Yi, J.C.; Zeng, X.X.; Yang, C.Q.; Lu, A.P.; Hou, T.J.; Cao, D.S. Pushing the boundaries of molecular property prediction for drug discovery with multitask learning BERT enhanced by SMILES enumeration. Research 2022, 2022, 0004. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Li, Y.; Zhang, R.; Liu, Y. INTransformer: Data augmentation-based contrastive learning by injecting noise into transformer for molecular property prediction. Journal of Molecular Graphics and Modelling 2024, 128, 108703. [Google Scholar] [CrossRef]
- Liu, S.; Nie, W.; Wang, C.; Lu, J.; Qiao, Z.; Liu, L.; Tang, J.; Xiao, C.; Anandkumar, A. Multi-modal molecule structure–text model for text-based retrieval and editing. Nature Machine Intelligence 2023, 5, 1447–1457. [Google Scholar] [CrossRef]
- Xu, M.; Yuan, X.; Miret, S.; Tang, J. Protst: Multi-modality learning of protein sequences and biomedical texts. In Proceedings of the International Conference on Machine Learning. PMLR; 2023; pp. 38749–38767. [Google Scholar]
- Ahneman, D.T.; Estrada, J.G.; Lin, S.; Dreher, S.D.; Doyle, A.G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 2018, 360, 186–190. [Google Scholar] [CrossRef]
- Schwaller, P.; Vaucher, A.C.; Laino, T.; Reymond, J.L. Prediction of chemical reaction yields using deep learning. Machine learning: science and technology 2021, 2, 015016. [Google Scholar] [CrossRef]
- Yin, X.; Hsieh, C.Y.; Wang, X.; Wu, Z.; Ye, Q.; Bao, H.; Deng, Y.; Chen, H.; Luo, P.; Liu, H.; et al. Enhancing generic reaction yield prediction through reaction condition-based contrastive learning. Research 2024, 7, 0292. [Google Scholar] [CrossRef]
- Shi, R.; Yu, G.; Huo, X.; Yang, Y. Prediction of chemical reaction yields with large-scale multi-view pre-training. Journal of Cheminformatics 2024, 16, 22. [Google Scholar] [CrossRef]
- Nam, J.; Kim, J. Linking the Neural Machine Translation and the Prediction of Organic Chemistry Reactions. arXiv 2016, arXiv:1612.09529. [Google Scholar] [CrossRef]
- Tetko, I.V.; Engkvist, O.; Koch, A.; Reymond, J.L.; Bjerrum, E.J. Augmented Transformer for Improved Chemical Reaction Prediction. Journal of Chemical Information and Modeling 2020, 60, 6253–6266. [Google Scholar] [CrossRef]
- Guo, K.; et al. Leveraging Large Language Models for Predictive Chemistry. Nature Machine Intelligence 2023. [Google Scholar] [CrossRef]
- Lin, J.; et al. Question Rephrasing for Quantifying Uncertainty in Large Language Models. arXiv 2024, arXiv:2408.03732. [Google Scholar] [CrossRef]
- Shenvi, R.A. Natural product synthesis in the 21st century: Beyond the mountain top. ACS Central Science 2024, 10, 519–528. [Google Scholar] [CrossRef]
- Corey, E. Robert Robinson lecture. Retrosynthetic thinking—essentials and examples. Chemical society reviews 1988, 17, 111–133. [Google Scholar] [CrossRef]
- Nam, J.; Kim, J. Linking the neural machine translation and the prediction of organic chemistry reactions. arXiv 2016, arXiv:1612.09529. [Google Scholar] [CrossRef]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Luu Nguyen, Q.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS central science 2017, 3, 1103–1113. [Google Scholar] [CrossRef]
- Schneider, N.; Stiefl, N.; Landrum, G.A. What’s what: The (nearly) definitive guide to reaction role assignment. Journal of chemical information and modeling 2016, 56, 2336–2346. [Google Scholar] [CrossRef]
- Somnath, V.R.; Bunne, C.; Coley, C.; Krause, A.; Barzilay, R. Learning graph models for retrosynthesis prediction. Advances in Neural Information Processing Systems 2021, 34, 9405–9415. [Google Scholar]
- Transformer, M. A Model for Uncertainty-Calibrated Chemical Reaction Prediction. Philippe Schwaller, Teodoro Laino, Théophile Gaudin, Peter Bolgar, Christopher A Hunter, Costas Bekas, Alpha A Lee ACS Central Science (2019-09-25) https://pubs. acs. org/doi/10.1021/acscentsci. 9b00576 DOI 2019, 10. [CrossRef]
- Schwaller, P.; Petraglia, R.; Zullo, V.; Nair, V.H.; Haeuselmann, R.A.; Pisoni, R.; Bekas, C.; Iuliano, A.; Laino, T. Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chemical science 2020, 11, 3316–3325. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Rao, J.; Zhang, Z.; Xu, J.; Yang, Y. Predicting retrosynthetic reactions using self-corrected transformer neural networks. Journal of chemical information and modeling 2019, 60, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Irwin, R.; Dimitriadis, S.; He, J.; Bjerrum, E.J. Chemformer: a pre-trained transformer for computational chemistry. Machine Learning: Science and Technology 2022, 3, 015022. [Google Scholar] [CrossRef]
- Toniato, A.; Vaucher, A.C.; Schwaller, P.; Laino, T. Enhancing diversity in language based models for single-step retrosynthesis. Digital Discovery 2023, 2, 489–501. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, N.; Chen, Z.; Guo, L.; Fan, X.; Chen, H. Domain-agnostic molecular generation with chemical feedback. arXiv 2023, arXiv:2301.11259. [Google Scholar]
- Wei, J.; Wang, X.; Schuurmans, D.; Bosma, M.; Ichter, B.; Xia, F.; Chi, E.H.; Le, Q.V.; Zhou, D. Chain-of-Thought Prompting Elicits Reasoning in Large Language Models. arXiv 2022, arXiv:2201.11903. [Google Scholar]
- Zholus, A.; Kuznetsov, M.; Schutski, R.; Shayakhmetov, R.; Polykovskiy, D.; Chandar, S.; Zhavoronkov, A. Bindgpt: A scalable framework for 3d molecular design via language modeling and reinforcement learning. In Proceedings of the Proceedings of the AAAI Conference on Artificial Intelligence, 2025; Vol. 39, pp. 26083–26091.
- Wang, Y.; Zhao, H.; Sciabola, S.; Wang, W. cMolGPT: a conditional generative pre-trained transformer for target-specific de novo molecular generation. Molecules 2023, 28, 4430. [Google Scholar] [CrossRef]
- Wang, X.; Gao, C.; Han, P.; Li, X.; Chen, W.; Rodríguez Patón, A.; Wang, S.; Zheng, P. PETrans: De novo drug design with protein-specific encoding based on transfer learning. International Journal of Molecular Sciences 2023, 24, 1146. [Google Scholar] [CrossRef]
- Kyro, G.W.; Morgunov, A.; Brent, R.I.; Batista, V.S. ChemSpaceAL: an efficient active learning methodology applied to protein-specific molecular generation. Biophysical Journal 2024, 123, 283a. [Google Scholar] [CrossRef]
- Adilov, S. Generative pre-training from molecules. 2021. [Google Scholar] [CrossRef]
- Haroon, S.; Hafsath, C.; Jereesh, A. Generative Pre-trained Transformer (GPT) based model with relative attention for de novo drug design. Computational Biology and Chemistry 2023, 106, 107911. [Google Scholar] [CrossRef]
- Yoshikai, Y.; Mizuno, T.; Nemoto, S.; Kusuhara, H. A novel molecule generative model of VAE combined with Transformer for unseen structure generation. arXiv 2024, arXiv:2402.11950. [Google Scholar] [CrossRef]
- Shen, T.; Guo, J.; Han, Z.; Zhang, G.; Liu, Q.; Si, X.; Wang, D.; Wu, S.; Xia, J. AutoMolDesigner for antibiotic discovery: an AI-based open-source software for automated design of small-molecule antibiotics. Journal of Chemical Information and Modeling 2024, 64, 575–583. [Google Scholar] [CrossRef]
- Mazuz, E.; Shtar, G.; Shapira, B.; Rokach, L. Molecule generation using transformers and policy gradient reinforcement learning. Scientific Reports 2023, 13, 8799. [Google Scholar] [CrossRef]
- Yu, B.; Baker, F.N.; Chen, Z.; Ning, X.; Sun, H. Llasmol: Advancing large language models for chemistry with a large-scale, comprehensive, high-quality instruction tuning dataset. arXiv 2024, arXiv:2402.09391. [Google Scholar]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: a benchmark for molecular machine learning. Chemical science 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Nakata, M.; Shimazaki, T. PubChemQC project: a large-scale first-principles electronic structure database for data-driven chemistry. Journal of chemical information and modeling 2017, 57, 1300–1308. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C.W.; Xiao, C.; Sun, J.; Zitnik, M. Therapeutics data commons: Machine learning datasets and tasks for drug discovery and development. arXiv 2021, arXiv:2102.09548. [Google Scholar] [CrossRef]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind database: Collection of binding affinities for protein- ligand complexes with known three-dimensional structures. Journal of medicinal chemistry 2004, 47, 2977–2980. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic acids research 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Tran, R.; Lan, J.; Shuaibi, M.; Wood, B.M.; Goyal, S.; Das, A.; Heras-Domingo, J.; Kolluru, A.; Rizvi, A.; Shoghi, N.; et al. The Open Catalyst 2022 (OC22) dataset and challenges for oxide electrocatalysts. ACS Catalysis 2023, 13, 3066–3084. [Google Scholar] [CrossRef]
- Perera, D.; Tucker, J.W.; Brahmbhatt, S.; Helal, C.J.; Chong, A.; Farrell, W.; Richardson, P.; Sach, N.W. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 2018, 359, 429–434. [Google Scholar] [CrossRef]
- Kearnes, S.M.; Maser, M.R.; Wleklinski, M.; Kast, A.; Doyle, A.G.; Dreher, S.D.; Hawkins, J.M.; Jensen, K.F.; Coley, C.W. The open reaction database. Journal of the American Chemical Society 2021, 143, 18820–18826. [Google Scholar] [CrossRef]
- Wigh, D.S.; Arrowsmith, J.; Pomberger, A.; Felton, K.C.; Lapkin, A.A. Orderly: data sets and benchmarks for chemical reaction data. Journal of Chemical Information and Modeling 2024, 64, 3790–3798. [Google Scholar] [CrossRef]
- Thakkar, A.; Kogej, T.; Reymond, J.L.; Engkvist, O.; Bjerrum, E.J. Datasets and their influence on the development of computer assisted synthesis planning tools in the pharmaceutical domain. Chemical science 2020, 11, 154–168. [Google Scholar] [CrossRef]
- Lowe, D. Chemical reactions from US patents (1976-Sep2016). (No Title) 2017.
- Reaxys®. https://www.elsevier.com/solutions/reaxys. Accessed: 2025-04-21.
- Mallard, W.G.; Westley, F.; Herron, J.; Hampson, R.F.; Frizzell, D. NIST chemical kinetics database; Vol. 126, National Institute of Standards and Technology Washington, DC, USA, 1992.
- Johnson, M.S.; Dong, X.; Grinberg Dana, A.; Chung, Y.; Farina Jr, D.; Gillis, R.J.; Liu, M.; Yee, N.W.; Blondal, K.; Mazeau, E.; et al. RMG database for chemical property prediction. Journal of Chemical Information and Modeling 2022, 62, 4906–4915. [Google Scholar] [CrossRef]
- de Matos, P.; Dekker, A.; Ennis, M.; Hastings, J.; Haug, K.; Turner, S.; Steinbeck, C. ChEBI: a chemistry ontology and database. Journal of cheminformatics 2010, 2, 1–1. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, T.; Chen, R.; Zhang, W.; Zhang, C.J.; Wei, X.; Qing, L. MolGround: A Benchmark for Molecular Grounding. arXiv 2025, arXiv:2503.23668. [Google Scholar] [CrossRef]
- Lowe, D.M. Extraction of Chemical Structures and Reactions from the US Patent Literature. Doctoral Thesis, University of Cambridge 2012.
- Genheden, S.; Bjerrum, E. PaRoutes: towards a framework for benchmarking retrosynthesis route predictions. Digital Discovery 2022, 1, 527–539. [Google Scholar] [CrossRef]
- Genheden, S.; Thakkar, A.; Chadimova, V.; Reymond, J.; Engkvist, O.; Bjerrum, E.J. AiZynthFinder: A Fast, Robust and Flexible Open-Source Software for Retrosynthetic Planning. Journal of Cheminformatics 2020, 12, 70. [Google Scholar] [CrossRef]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: benchmarking models for de novo molecular design. Journal of chemical information and modeling 2019, 59, 1096–1108. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular sets (MOSES): a benchmarking platform for molecular generation models. Frontiers in pharmacology 2020, 11, 565644. [Google Scholar] [CrossRef]
- Eckmann, P.; Sun, K.; Zhao, B.; Feng, M.; Gilson, M.K.; Yu, R. Limo: Latent inceptionism for targeted molecule generation. Proceedings of machine learning research 2022, 162, 5777. [Google Scholar] [PubMed]
- Taboureau, O.; Nielsen, S.K.; Audouze, K.; Weinhold, N.; Edsgärd, D.; Roque, F.S.; Kouskoumvekaki, I.; Bora, A.; Curpan, R.; Jensen, T.S.; et al. ChemProt: a disease chemical biology database. Nucleic acids research 2010, 39, D367–D372. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.; Johnson, R.J.; Sciaky, D.; Wei, C.H.; Leaman, R.; Davis, A.P.; Mattingly, C.J.; Wiegers, T.C.; Lu, Z. BioCreative V CDR task corpus: a resource for chemical disease relation extraction. Database 2016, 2016. [Google Scholar] [CrossRef]
- Krallinger, M.; Rabal, O.; Lourenço, A.; et al. The CHEMDNER Corpus of Chemicals and Drugs and Its Annotation Principles. Journal of Cheminformatics 2015, 7, S2. [Google Scholar] [CrossRef]
- Verspoor, K.; Nguyen, D.Q.; Akhondi, S.A.; Druckenbrodt, C.; Thorne, C.; Hoessel, R.; He, J.; Zhai, Z. ChEMU dataset for information extraction from chemical patents. Mendeley Data 2020, 2, 17632. [Google Scholar]
- Wang, X.; Hu, V.; Song, X.; Garg, S.; Xiao, J.; Han, J. ChemNER: fine-grained chemistry named entity recognition with ontology-guided distant supervision. In Proceedings of the Proceedings of the 2021 Conference on Empirical Methods in Natural Language Processing; 2021. [Google Scholar]
- Axelrod, S.; Gomez-Bombarelli, R. GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Scientific Data 2022, 9, 185. [Google Scholar] [CrossRef]
- Isert, C.; Atz, K.; Jiménez-Luna, J.; Schneider, G. QMugs, quantum mechanical properties of drug-like molecules. Scientific Data 2022, 9, 273. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C.W.; Xiao, C. Therapeutics Data Commons: Machine Learning Datasets and Tasks for Drug Discovery and Development. Scientific Data 2021, 8, 316. [Google Scholar] [CrossRef]
- Wei, Z.; Ji,W.; Geng, X.; Chen, Y.; Chen, B.; Qin, T.; Jiang, D. ChemistryQA: A Complex Question Answering Dataset from Chemistry 2020.
- Laghuvarapu, S.; Lee, N.; Gao, C.; Sun, J. MolTextQA: A Curated Question-Answering Dataset and Benchmark for Molecular Structure-Text Relationship Learning.
- Capuzzi, S.J.; Kim, I.S.; Lam, W.I.; Thornton, T.E.; Muratov, E.N.; Pozefsky, D.; Tropsha, A. Chembench: A Publicly-Accessible, Integrated Cheminformatics Portal. Journal of Chemical Information and Modeling 2017, 57, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, L.; Khokhlov, I.; Fedorov, M.; Sosnin, S. Struct2IUPAC–Transformer-Based Artificial Neural Network for the Conversion Between Chemical Notations. 2021. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Chen, H.; Wang, J.; Wu, Y.; Prakasam, M.; Xu, H. Chemical named entity recognition in patents by domain knowledge and unsupervised feature learning. Database 2016, 2016, baw049. [Google Scholar] [CrossRef]
- Yoo, S.; Kim, J. Adapt-cMolGPT: A Conditional Generative Pre-Trained Transformer with Adapter-Based Fine-Tuning for Target-Specific Molecular Generation. International Journal of Molecular Sciences 2024, 25, 6641. [Google Scholar] [CrossRef]
- Tom, G.; Schmid, S.P.; Baird, S.G.; Cao, Y.; Darvish, K.; Hao, H.; Lo, S.; Pablo-García, S.; Rajaonson, E.M.; Skreta, M.; et al. Self-driving laboratories for chemistry and materials science. Chemical Reviews 2024, 124, 9633–9732. [Google Scholar] [CrossRef] [PubMed]
- Seifrid, M.; Pollice, R.; Aguilar-Granda, A.; Morgan Chan, Z.; Hotta, K.; Ser, C.T.; Vestfrid, J.; Wu, T.C.; Aspuru-Guzik, A. Autonomous chemical experiments: Challenges and perspectives on establishing a self-driving lab. Accounts of Chemical Research 2022, 55, 2454–2466. [Google Scholar] [CrossRef]
- Dutta, S.; Singh, J.; Chakrabarti, S.; Chakraborty, T. How to think step-by-step: A mechanistic understanding of chain-of-thought reasoning. arXiv 2024, arXiv:2402.18312. [Google Scholar]
- Han, Y.; Wan, Z.; Chen, L.; Yu, K.; Chen, X. From Generalist to Specialist: A Survey of Large Language Models for Chemistry. arXiv 2024, arXiv:2412.19994. [Google Scholar] [CrossRef]
- Le, K.; Chawla, N.V. Utilizing Large Language Models in an iterative paradigm with domain feedback for molecule optimization. arXiv 2024, arXiv:2410.13147. [Google Scholar] [CrossRef]
- Sutanto, P.; Santoso, J.; Setiawan, E.I.; Wibawa, A.P. Llm distillation for efficient few-shot multiple choice question answering. arXiv 2024, arXiv:2412.09807. [Google Scholar] [CrossRef]
- Blumenthal, D.; Campbell, E.G.; Anderson, M.S.; Causino, N.; Louis, K.S. Withholding research results in academic life science: evidence from a national survey of faculty. Jama 1997, 277, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, D.; Causino, N.; Campbell, E.; Louis, K.S. Relationships between academic institutions and industry in the life sciences—an industry survey. New England Journal of Medicine 1996, 334, 368–374. [Google Scholar] [CrossRef]
- Louis, K.S.; Blumenthal, D.; Gluck, M.E.; Stoto, M.A. Entrepreneurs in academe: An exploration of behaviors among life scientists. Administrative science quarterly 1989, 110–131. [Google Scholar] [CrossRef]
- Geitmann, A.; Niklas, K.; Speck, T. Plant Biomechanics in the 21st Century. 2019, 70, 3435–3438. [Google Scholar] [CrossRef]
- Mondal, S.; Bagchi, B. From Structure and Dynamics to Biomolecular Functions: the Ubiquitous Role of Solvent in Biology. 2022, 77, 102462. [Google Scholar] [CrossRef]
- Lewis, W.A. Economic Survey; Routledge, 2013. [Google Scholar]
- Fisher, A.C.; Peterson, F.M. The environment in economics: a survey. Journal of Economic Literature 1976, 14, 1–33. [Google Scholar]
- Wikipedia. Life Science. Wikipedia, The Free Encyclopedia 2023. [Accessed: 2025-03-22].
- U.D.J.G.I.H.T..B.E..P.P..R.P..W.S..S.T..D.N..C.J.F..O.A..L.S..E.C..U.E..F.M.; 9, R.G.S.C.S.Y..F.A..H.M..Y.T..T.A..I.T..K.C..W.H..T.Y..T.T.; Genoscope.; 10, C.U..W.J..H.R..S.W..A.F..B.P..B.T..P.E..R.C..W.P.; Department of Genome Analysis, I.o.M.B.R.A..P.M..N.G..T.S..R.A..; 11, G.S.C.S.D.R..D.S.L..R.M..W.K..L.H.M..D.J.; 15, B.G.I.G.C.Y.H..Y.J..W.J..H.G..G.J.; et al. Initial sequencing and analysis of the human genome. nature 2001, 409, 860–921. [CrossRef]
- Lodish, H.F. Molecular cell biology; Macmillan, 2008. [Google Scholar]
- Carpenter, W. Principles of mental physiology; BoD–Books on Demand, 2023. [Google Scholar]
- Bayliss, W.M. Principles of general physiology; Longsmann Green, 1915. [Google Scholar]
- Darwin, C. Origin of the Species. In British Politics and the environment in the long nineteenth century; Routledge, 2023; pp. 47–55. [Google Scholar]
- Chen, Q.; Yan, W.; Duan, E. Epigenetic Inheritance of Acquired Traits Through Sperm RNAs and Sperm RNA Modifications. 2016, 17, 733–743. [Google Scholar] [CrossRef]
- Correns, C.E.; Correns, C.E. Gregor Mendel’s „Versuche über Pflanzen-Hybriden “und die Bestätigung ihrer Ergebnisse durch die neuesten Untersuchungen; Springer, 1924. [Google Scholar]
- Hearn, R.; Arblaster, K. DNA extraction techniques for use in education. Biochemistry and Molecular Biology Education 2010, 38, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Di Pinto, A.; Forte, V.; Guastadisegni, M.C.; Martino, C.; Schena, F.P.; Tantillo, G. A comparison of DNA extraction methods for food analysis. Food control 2007, 18, 76–80. [Google Scholar] [CrossRef]
- Rohland, N.; Hofreiter, M. Comparison and optimization of ancient DNA extraction. Biotechniques 2007, 42, 343–352. [Google Scholar] [CrossRef]
- Crossley, B.M.; Bai, J.; Glaser, A.; Maes, R.; Porter, E.; Killian, M.L.; Clement, T.; Toohey-Kurth, K. Guidelines for Sanger sequencing and molecular assay monitoring. Journal of Veterinary Diagnostic Investigation 2020, 32, 767–775. [Google Scholar] [CrossRef]
- Sikkema-Raddatz, B.; Johansson, L.F.; de Boer, E.N.; Almomani, R.; Boven, L.G.; van den Berg, M.P.; van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; Sijmons, R.H.; Jongbloed, J.D.; et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Human mutation 2013, 34, 1035–1042. [Google Scholar] [CrossRef]
- Beck, T.F.; Mullikin, J.C.; lesb@ mail. nih. gov, N.C.S.P.B.L.G. Systematic evaluation of Sanger validation of next-generation sequencing variants. Clinical chemistry 2016, 62, 647–654. [CrossRef]
- Erlich, H.A.; et al. PCR technology; Springer, 1989. [Google Scholar]
- Zhu, H.; Zhang, H.; Xu, Y.; Laššáková, S.; Korabečná, M.; Neužil, P. PCR past, present and future. Biotechniques 2020, 69, 317–325. [Google Scholar] [CrossRef]
- Kircher, M.; Kelso, J. High-throughput DNA sequencing–concepts and limitations. Bioessays 2010, 32, 524–536. [Google Scholar] [CrossRef]
- Lou, D.I.; Hussmann, J.A.; McBee, R.M.; Acevedo, A.; Andino, R.; Press, W.H.; Sawyer, S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proceedings of the National Academy of Sciences 2013, 110, 19872–19877. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, A.D.; Bader, G.D.; Wishart, D.S. Bioinformatics; John Wiley & Sons, 2020. [Google Scholar]
- Majekodunmi, S.O. A review on centrifugation in the pharmaceutical industry. Am. J. Biomed. Eng 2015, 5, 67–78. [Google Scholar]
- Brakke, M.K. Density gradient centrifugation: a new separation technique1. Journal of the American Chemical Society 1951, 73, 1847–1848. [Google Scholar] [CrossRef]
- Smith, I. Chromatography; Elsevier, 2013. [Google Scholar]
- Smyth, M.; Martin, J. x Ray crystallography. Molecular Pathology 2000, 53, 8. [Google Scholar] [CrossRef] [PubMed]
- Woolfson, M.M. An introduction to X-ray crystallography; Cambridge University Press, 1997. [Google Scholar]
- Schwieters, C.D.; Kuszewski, J.J.; Clore, G.M. Using Xplor–NIH for NMR molecular structure determination. Progress in nuclear magnetic resonance spectroscopy 2006, 48, 47–62. [Google Scholar] [CrossRef]
- Hames, B.D. Gel electrophoresis of proteins: a practical approach; Vol. 197, OUP Oxford, 1998.
- Kurien, B.T.; Scofield, R.H. Western blotting. Methods 2006, 38, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Lewin, G.R.; Barde, Y.A. Physiology of the neurotrophins. Annual review of neuroscience 1996, 19, 289–317. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiological reviews 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Saba, T.M. Physiology and physiopathology of the reticuloendothelial system. Archives of internal medicine 1970, 126, 1031–1052. [Google Scholar] [CrossRef]
- Boron, W.F.; Boulpaep, E.L. Medical Physiology E-Book: Medical Physiology E-Book; Elsevier Health Sciences, 2016. [Google Scholar]
- Hahnemann, S. Organon of medicine; B. Jain publishers, 2005. [Google Scholar]
- Castiglioni, A. A history of medicine; Routledge, 2019. [Google Scholar]
- Sigerist, H.E. A history of medicine; Vol. 2, Oxford University Press, 1987.
- Van Den Berg, A.; Mummery, C.L.; Passier, R.; Van der Meer, A.D. Personalised organs-on-chips: functional testing for precision medicine. Lab on a Chip 2019, 19, 198–205. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Honn, K.V. Inflammation and disease progression. Cancer and Metastasis Reviews 2006, 25, 481–491. [Google Scholar] [CrossRef]
- Yunginger, J.W.; Ahlstedt, S.; Eggleston, P.A.; Homburger, H.A.; Nelson, H.S.; Ownby, D.R.; Platts-Mills, T.A.; Sampson, H.A.; Sicherer, S.H.; Weinstein, A.M.; et al. Quantitative IgE antibody assays in allergic diseases. Journal of allergy and clinical immunology 2000, 105, 1077–1084. [Google Scholar] [CrossRef]
- Kricka, L.J. Human anti-animal antibody interferences in immunological assays. Clinical chemistry 1999, 45, 942–956. [Google Scholar] [CrossRef]
- Raichle, M.E.; Mintun, M.A. Brain work and brain imaging. Annu. Rev. Neurosci. 2006, 29, 449–476. [Google Scholar] [CrossRef] [PubMed]
- Kekelidze, M.; D’Errico, L.; Pansini, M.; Tyndall, A.; Hohmann, J. Colorectal cancer: current imaging methods and future perspectives for the diagnosis, staging and therapeutic response evaluation. World journal of gastroenterology: WJG 2013, 19, 8502. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, R.A.; Cidlowski, J.A. Apoptosis: the biochemistry and molecular biology of programmed cell death. Endocrine reviews 1993, 14, 133–151. [Google Scholar]
- Amaral-Zettler, L.A.; Zettler, E.R.; Mincer, T.J. Ecology of the plastisphere. Nature Reviews Microbiology 2020, 18, 139–151. [Google Scholar] [CrossRef]
- Polis, G.A.; Yamashita, T. Ecology. 1990.
- Odum, E.P.; Barrett, G.W.; et al. Fundamentals of ecology 1971.
- Sopinka, N.M.; Patterson, L.D.; Redfern, J.C.; Pleizier, N.K.; Belanger, C.B.; Midwood, J.D.; Crossin, G.T.; Cooke, S.J. Manipulating glucocorticoids in wild animals: basic and applied perspectives. Conservation Physiology 2015, 3, cov031. [Google Scholar] [CrossRef]
- Burt, T.; Howden, N.; McDonnell, J.; Jones, J.; Hancock, G. Seeing the climate through the trees: observing climate and forestry impacts on streamflow using a 60-year record. Hydrological processes 2015, 29, 473–480. [Google Scholar] [CrossRef]
- Hammer; Harper, D.A. Paleontological data analysis; John Wiley & Sons, 2024. [Google Scholar]
- Pearson, W.R.; Wood, T.; Zhang, Z.; Miller, W. Comparison of DNA sequences with protein sequences. Genomics 1997, 46, 24–36. [Google Scholar] [CrossRef] [PubMed]
- HUANG, X. Fast comparison of a DNA sequence with a protein sequence database. Microbial & comparative genomics 1996, 1, 281–291. [Google Scholar]
- Peltz, G.; Zaas, A.K.; Zheng, M.; Solis, N.V.; Zhang, M.X.; Liu, H.H.; Hu, Y.; Boxx, G.M.; Phan, Q.T.; Dill, D.; et al. Next-generation computational genetic analysis: multiple complement alleles control survival after Candida albicans infection. Infection and immunity 2011, 79, 4472–4479. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Ellis, T. Biological Engineered Living Materials: Growing Functional Materials with Genetically Programmable Properties. 2019, 8, 1–15. [Google Scholar] [CrossRef]
- Magin, R. Fractional calculus in bioengineering, part 1. Critical Reviews™ in Biomedical Engineering 2004, 32. [Google Scholar]
- Kumar, G.; Shekh, A.; Jakhu, S.; Sharma, Y.; Kapoor, R.; Sharma, T.R. Bioengineering of microalgae: recent advances, perspectives, and regulatory challenges for industrial application. Frontiers in Bioengineering and Biotechnology 2020, 8, 914. [Google Scholar] [CrossRef] [PubMed]
- Jaklenec, A.; Stamp, A.; Deweerd, E.; Sherwin, A.; Langer, R. Progress in the tissue engineering and stem cell industry “are we there yet?”. Tissue Engineering Part B: Reviews 2012, 18, 155–166. [Google Scholar] [CrossRef]
- Camara, C.; Peris-Lopez, P.; Tapiador, J.E. Security and privacy issues in implantable medical devices: A comprehensive survey. Journal of biomedical informatics 2015, 55, 272–289. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Koeller, M.; Weisman, M.H.; Emery, P. New therapies for treatment of rheumatoid arthritis. The lancet 2007, 370, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Ratner, B.D.; Bryant, S.J. Biomaterials: where we have been and where we are going. Annu. Rev. Biomed. Eng. 2004, 6, 41–75. [Google Scholar] [CrossRef]
- Tathe, A.; Ghodke, M.; Nikalje, A.P. A brief review: biomaterials and their application. Int. J. Pharm. Pharm. Sci 2010, 2, 19–23. [Google Scholar]
- Rahmati, M.; Mills, D.K.; Urbanska, A.M.; Saeb, M.R.; Venugopal, J.R.; Ramakrishna, S.; Mozafari, M. Electrospinning for Tissue Engineering Applications. 2020, 117, 100721. [Google Scholar] [CrossRef]
- Wikipedia. Biological Engineering. Wikipedia, The Free Encyclopedia 2023. [Accessed: 2025-03-22].
- Weidner, B.V.; Nagel, J.K.S.; Weber, H. Facilitation method for the translation of biological systems to technical design solutions. International Journal of Design Creativity and Innovation 2018, 6, 211–234. [Google Scholar] [CrossRef]
- Helms, M.E.; Vattam, S.; Goel, A.K. Biologically inspired design: process and products. Design Studies 2009, 30, 606–622. [Google Scholar] [CrossRef]
- Nagel, J.K.; Nagel, R.; Stone, R.; McAdams, D. Function-based, biologically inspired concept generation. Artificial Intelligence for Engineering Design, Analysis and Manufacturing 2010, 24, 521–535. [Google Scholar] [CrossRef]
- Nowakowski, A.; Andrzejewska, A.; Janowski, M.; Walczak, P.; Lukomska, B. Genetic engineering of stem cells for enhanced therapy. Acta neurobiologiae experimentalis 2013, 73, 1–18. [Google Scholar] [CrossRef]
- Zhou, Y.; Han, Y. Engineered bacteria as drug delivery vehicles: principles and prospects. Engineering Microbiology 2022, 2, 100034. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.U.; Park, J.H.; Park, J.M.; Kim, T.Y. Metabolic engineering of microorganisms: general strategies and drug production. Drug discovery today 2009, 14, 78–88. [Google Scholar] [CrossRef]
- Riglar, D.T.; Silver, P.A. Engineering bacteria for diagnostic and therapeutic applications. Nature Reviews Microbiology 2018, 16, 214–225. [Google Scholar] [CrossRef]
- Khan, S.; Ullah, M.W.; Siddique, R.; Nabi, G.; Manan, S.; Yousaf, M.; Hou, H. Role of recombinant DNA technology to improve life. International journal of genomics 2016, 2016, 2405954. [Google Scholar] [CrossRef]
- Wright, S. Recombinant DNA technology and its social transformation, 1972-1982. Osiris 1986, 2, 303–360. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.S. Human insulin from recombinant DNA technology. Science 1983, 219, 632–637. [Google Scholar] [CrossRef]
- Micklos, D.A.; Freyer, G.A. DNA science; a first course in recombinant DNA technology; 1990. [Google Scholar]
- Rubio, D.; Garcia-Castro, J.; Martín, M.C.; de la Fuente, R.; Cigudosa, J.C.; Lloyd, A.C.; Bernad, A. Spontaneous human adult stem cell transformation. Cancer research 2005, 65, 3035–3039. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 structures and mechanisms. Annual review of biophysics 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Redman, M.; King, A.; Watson, C.; King, D. What is CRISPR/Cas9? Archives of Disease in Childhood-Education and Practice 2016, 101, 213–215. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature protocols 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Wikipedia. Biology. Wikipedia, The Free Encyclopedia 2023. [Accessed: 2025-03-22].
- Ikada, Y. Challenges in tissue engineering. Journal of the Royal Society Interface 2006, 3, 589–601. [Google Scholar] [CrossRef]
- Chapekar, M.S. Tissue engineering: challenges and opportunities. Journal of Biomedical Materials Research: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials 2000, 53, 617–620. [Google Scholar] [CrossRef]
- Lanza, R.; Langer, R.; Vacanti, J.P.; Atala, A. Principles of tissue engineering; Academic press, 2020. [Google Scholar]
- Park, J.; Lakes, R.S. Biomaterials: an introduction; Springer Science & Business Media, 2007. [Google Scholar]
- Dar, A.; Shachar, M.; Leor, J.; Cohen, S. Optimization of cardiac cell seeding and distribution in 3D porous alginate scaffolds. Biotechnology and bioengineering 2002, 80, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Holy, C.E.; Shoichet, M.S.; Davies, J.E. Engineering three-dimensional bone tissue in vitro using biodegradable scaffolds: Investigating initial cell-seeding density and culture period. Journal of Biomedical Materials Research: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials 2000, 51, 376–382. [Google Scholar] [CrossRef]
- Ripamonti, U.; Roden, L.C.; Renton, L.F. Osteoinductive hydroxyapatite-coated titanium implants. Biomaterials 2012, 33, 3813–3823. [Google Scholar] [CrossRef] [PubMed]
- De Lange, G.; Donath, K. Interface between bone tissue and implants of solid hydroxyapatite or hydroxyapatite-coated titanium implants. Biomaterials 1989, 10, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.D.; Thomas, K.A.; Kay, J.F.; Jarcho, M. Hydroxyapatite-coated titanium for orthopedic implant applications. Clinical Orthopaedics and Related Research (1976-2007) 1988, 232, 225–243. [Google Scholar] [CrossRef]
- Ferber, D. Lab-grown organs begin to take shape. 1999. [Google Scholar] [CrossRef]
- Niklason, L.E.; Lawson, J.H. Bioengineered human blood vessels. Science 2020, 370, eaaw8682. [Google Scholar] [CrossRef]
- Doran, P.M. Bioprocess engineering principles; Elsevier, 1995. [Google Scholar]
- Harun, R.; Singh, M.; Forde, G.M.; Danquah, M.K. Bioprocess engineering of microalgae to produce a variety of consumer products. Renewable and sustainable energy reviews 2010, 14, 1037–1047. [Google Scholar] [CrossRef]
- Liu, S. Bioprocess engineering: kinetics, sustainability, and reactor design; Elsevier, 2020. [Google Scholar]
- Rio-Chanona, E.A.; Wagner, J.L.; Ali, H.; Fiorelli, F.; Zhang, D.; Hellgardt, K. Deep learning-based surrogate modeling and optimization for microalgal biofuel production and photobioreactor design. AIChE Journal 2018. [Google Scholar] [CrossRef]
- Herbert, D.; Elsworth, R.; Telling, R. The continuous culture of bacteria; a theoretical and experimental study. Microbiology 1956, 14, 601–622. [Google Scholar] [CrossRef]
- Andrews, J.F. A mathematical model for the continuous culture of microorganisms utilizing inhibitory substrates. Biotechnology and bioengineering 1968, 10, 707–723. [Google Scholar]
- Abraham, E.P.; Chain, E.; Fletcher, C.M.; Gardner, A.D.; Heatley, N.G.; Jennings, M.A.; Florey, H.W. Further observations on penicillin. The Lancet 1941, 238, 177–189. [Google Scholar] [CrossRef]
- Shukla, A.; Thömmes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends in biotechnology 2010, 28 5, 253–61. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, W.M. Production of Monoclonal Antibodies. Current Protocols in Cytometry 1999, 37. [Google Scholar] [CrossRef]
- Gentleman, R.; Carey, V.; Huber, W.; Irizarry, R.; Dudoit, S. Bioinformatics and computational biology solutions using R and Bioconductor; Springer Science & Business Media, 2005. [Google Scholar]
- Tang, B.; Pan, Z.; Yin, K.; Khateeb, A. Recent advances of deep learning in bioinformatics and computational biology. Frontiers in genetics 2019, 10, 214. [Google Scholar] [CrossRef]
- Ranganathan, S.; Nakai, K.; Schonbach, C. Encyclopedia of bioinformatics and computational biology: ABC of bioinformatics; Elsevier, 2018. [Google Scholar]
- Lio, P. Wavelets in bioinformatics and computational biology: state of art and perspectives. Bioinformatics 2003, 19, 2–9. [Google Scholar] [CrossRef]
- Bhavikatti, S. Finite element analysis; New Age International, 2005. [Google Scholar]
- Szabó, B.; Babuška, I. Finite element analysis: Method, verification and validation; 2021. [Google Scholar]
- Taylor, R.L. FEAP-A finite element analysis program, 2014.
- Marck, C. ‘DNA Strider’: a ‘C’program for the fast analysis of DNA and protein sequences on the Apple Macintosh family of computers. Nucleic acids research 1988, 16, 1829–1836. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic acids research 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Golding, M.P. Ethical issues in biological engineering. UCLA L. Rev. 1967, 15, 443. [Google Scholar]
- Dzau, V.J.; Cicerone, R.J. Responsible use of human gene-editing technologies. Human Gene Therapy 2015, 26, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Jasanoff, S.; Hurlbut, J.B.; Saha, K. CRISPR democracy: Gene editing and the need for inclusive deliberation. Issues in Science and Technology 2015, 32, 25–32. [Google Scholar]
- Tiedje, J.M.; Colwell, R.K.; Grossman, Y.L.; Hodson, R.E.; Lenski, R.E.; Mack, R.N.; Regal, P.J. The planned introduction of genetically engineered organisms: ecological considerations and recommendations. Ecology 1989, 70, 298–315. [Google Scholar] [CrossRef]
- Snow, A.A.; Andow, D.A.; Gepts, P.; Hallerman, E.M.; Power, A.; Tiedje, J.M.; Wolfenbarger, L. Genetically engineered organisms and the environment: Current status and recommendations 1. Ecological Applications 2005, 15, 377–404. [Google Scholar] [CrossRef]
- Wolfenbarger, L.L.; Phifer, P.R. The ecological risks and benefits of genetically engineered plants. Science 2000, 290, 2088–2093. [Google Scholar] [CrossRef]
- Karagyaur, M.; Efimenko, A.Y.; Makarevich, P.; Vasiluev, P.; Akopyan, Z.A.; Bryzgalina, E.; Tkachuk, V. Ethical and legal aspects of using genome editing technologies in medicine. 2019, 11, 117–132. [Google Scholar] [CrossRef]
- Piergentili, R.; Del Rio, A.; Signore, F.; Umani Ronchi, F.; Marinelli, E.; Zaami, S. CRISPR-Cas and its wide-ranging applications: From human genome editing to environmental implications, technical limitations, hazards and bioethical issues. Cells 2021, 10, 969. [Google Scholar] [CrossRef]
- of Sciences, N.A.; Medicine.; of Medicine, N.A.; on Human Gene Editing, C.; Scientific.; Medical.; Considerations, E. Human genome editing: science, ethics, and governance; National Academies Press, 2017.
- Floridi, L.; Cowls, J.; Beltrametti, M.; Chatila, R.; Chazerand, P.; Dignum, V.; Luetge, C.; Madelin, R.; Pagallo, U.; Rossi, F.; et al. AI4People—an ethical framework for a good AI society: opportunities, risks, principles, and recommendations. Minds and machines 2018, 28, 689–707. [Google Scholar] [CrossRef]
- Cath, C.; Wachter, S.; Mittelstadt, B.; Taddeo, M.; Floridi, L. Artificial intelligence and the ‘good society’: the US, EU, and UK approach. Science and engineering ethics 2018, 24, 505–528. [Google Scholar]
- Enderle, J.; Bronzino, J. Introduction to biomedical engineering; Academic press, 2012. [Google Scholar]
- Rabinow, P.; Bennett, G. Designing human practices: An experiment with synthetic biology; University of Chicago Press, 2012. [Google Scholar]
- Williams, S.M.; Haines, J.L.; Moore, J.H. The use of animal models in the study of complex disease: all else is never equal or why do so many human studies fail to replicate animal findings? Bioessays 2004, 26, 170–179. [Google Scholar] [CrossRef] [PubMed]
- McGonigle, P.; Ruggeri, B. Animal models of human disease: challenges in enabling translation. Biochemical pharmacology 2014, 87, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A. The flaws and human harms of animal experimentation. Cambridge Quarterly of Healthcare Ethics 2015, 24, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, Y.; Faraggi, E.; Zhan, J.; Zhou, Y. Direct prediction of profiles of sequences compatible with a protein structure by neural networks with fragment-based local and energy-based nonlocal profiles. Proteins: Structure, Function, and Bioinformatics 2014, 82, 2565–2573. [Google Scholar] [CrossRef]
- Berger, J. The last mile: how to sustain long-distance migration in mammals. Conservation biology 2004, 18, 320–331. [Google Scholar] [CrossRef]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.L. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Science 2013, 22, 693–724. [Google Scholar] [CrossRef]
- Cho, D.Y.; Kim, Y.A.; Przytycka, T.M. Chapter 5: Network biology approach to complex diseases. PLoS computational biology 2012, 8, e1002820. [Google Scholar] [CrossRef] [PubMed]
- Manojlovich, M.; Lee, S.; Lauseng, D. A systematic review of the unintended consequences of clinical interventions to reduce adverse outcomes. Journal of patient safety 2016, 12, 173–179. [Google Scholar] [CrossRef]
- Ni She, E.; Harrison, R. Mitigating unintended consequences of co-design in health care. Health Expectations 2021, 24, 1551–1556. [Google Scholar] [CrossRef]
- Riesselman, A.J.; Ingraham, J.B.; Marks, D.S. Deep generative models of genetic variation capture mutation effects. arXiv 2017, arXiv:1712.06527. [Google Scholar] [CrossRef]
- Mjolsness, E. Prospects for declarative mathematical modeling of complex biological systems. Bulletin of Mathematical Biology 2019, 81, 3385–3420. [Google Scholar] [CrossRef]
- Boughorbel, S.; Jarray, F.; Venugopal, N.; Elhadi, H. Alternating loss correction for preterm-birth prediction from ehr data with noisy labels. arXiv 2018, arXiv:1811.09782. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, W.; Peng, T.; Wang, Y.; Jin, M. Hard sample aware noise robust learning for histopathology image classification. IEEE transactions on medical imaging 2021, 41, 881–894. [Google Scholar] [CrossRef]
- Pal, S.; Mondal, S.; Das, G.; Khatua, S.; Ghosh, Z. Big data in biology: The hope and present-day challenges in it. Gene Reports 2020, 21, 100869. [Google Scholar] [CrossRef]
- Agarwal, S.; Laradji, I.H.; Charlin, L.; Pal, C. Litllm: A toolkit for scientific literature review. arXiv 2024, arXiv:2402.01788. [Google Scholar] [CrossRef]
- An, H.; Narechania, A.; Wall, E.; Xu, K. VITALITY 2: Reviewing Academic Literature Using Large Language Models. arXiv 2024, arXiv:2408.13450. [Google Scholar] [CrossRef]
- Glickman, M.; Zhang, Y. AI and generative AI for research discovery and summarization. arXiv 2024, arXiv:2401.06795. [Google Scholar] [CrossRef]
- McGinness, L.; Baumgartner, P.; Onyango, E.; Lema, Z. Highlighting Case Studies in LLM Literature Review of Interdisciplinary System Science. In Proceedings of the Australasian Joint Conference on Artificial Intelligence. Springer; 2025; pp. 29–43. [Google Scholar]
- Baek, J.; Jauhar, S.K.; Cucerzan, S.; Hwang, S.J. Researchagent: Iterative research idea generation over scientific literature with large language models. arXiv 2024, arXiv:2404.07738. [Google Scholar] [CrossRef]
- Wu, S.; Ma, X.; Luo, D.; Li, L.; Shi, X.; Chang, X.; Lin, X.; Luo, R.; Pei, C.; Du, C.; et al. Automated review generation method based on large language models. arXiv 2024, arXiv:2407.20906. [Google Scholar] [CrossRef]
- Matarazzo, A.; Torlone, R. A Survey on Large Language Models with some Insights on their Capabilities and Limitations. arXiv 2025, arXiv:2501.04040. [Google Scholar]
- Wang, Z.; Wang, Z.; Jiang, J.; Chen, P.; Shi, X.; Li, Y. Large Language Models in Bioinformatics: A Survey. arXiv 2025, arXiv:2503.04490. [Google Scholar] [CrossRef]
- Wu, W.; Li, Q.; Li, M.; Fu, K.; Feng, F.; Ye, J.; Xiong, H.; Wang, Z. GENERator: A Long-Context Generative Genomic Foundation Model. arXiv 2025, arXiv:2502.07272. [Google Scholar] [CrossRef]
- Zheng, Z.; Deng, Y.; Xue, D.; Zhou, Y.; Ye, F.; Gu, Q. Structure-informed language models are protein designers. In Proceedings of the International conference on machine learning. PMLR; 2023; pp. 42317–42338. [Google Scholar]
- Nguyen, E.; Poli, M.; Faizi, M.; Thomas, A.; Wornow, M.; Birch-Sykes, C.; Massaroli, S.; Patel, A.; Rabideau, C.; Bengio, Y.; et al. Hyenadna: Long-range genomic sequence modeling at single nucleotide resolution. Advances in neural information processing systems 2023, 36, 43177–43201. [Google Scholar]
- Ross, T.D.; Gopinath, A. Chaining thoughts and LLMs to learn DNA structural biophysics. arXiv 2024, arXiv:2403.01332. [Google Scholar] [CrossRef]
- Xiao, Y.; Sun, E.; Jin, Y.; Wang, W. RNA-GPT: Multimodal Generative System for RNA Sequence Understanding. arXiv 2024, arXiv:2411.08900. [Google Scholar]
- Fang, X.; Wang, F.; Liu, L.; He, J.; Lin, D.; Xiang, Y.; Zhang, X.; Wu, H.; Li, H.; Song, L. Helixfold-single: Msa-free protein structure prediction by using protein language model as an alternative. arXiv 2022, arXiv:2207.13921. [Google Scholar]
- Truong Jr, T.; Bepler, T. Poet: A generative model of protein families as sequences-of-sequences. Advances in Neural Information Processing Systems 2023, 36, 77379–77415. [Google Scholar]
- Madani, A.; McCann, B.; Naik, N.; Keskar, N.S.; Anand, N.; Eguchi, R.R.; Huang, P.S.; Socher, R. Progen: Language modeling for protein generation. arXiv 2020, arXiv:2004.03497. [Google Scholar] [CrossRef]
- Nijkamp, E.; Ruffolo, J.A.; Weinstein, E.N.; Naik, N.; Madani, A. Progen2: exploring the boundaries of protein language models. Cell systems 2023, 14, 968–978. [Google Scholar] [CrossRef]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos Jr, J.L.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large language models generate functional protein sequences across diverse families. Nature biotechnology 2023, 41, 1099–1106. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: towards cracking the language of life’s code through self-supervised learning. IEEE Transactions on Pattern Analysis and Machine Intelligence 2021, 44, 7112–7127. [Google Scholar] [CrossRef]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Liu, X.h.; Lu, Z.h.; Wang, T.; Liu, F. Large language models facilitating modern molecular biology and novel drug development. Frontiers in Pharmacology 2024, 15, 1458739. [Google Scholar] [CrossRef]
- Koonin, E.V.; Galperin, M.Y.; Koonin, E.V.; Galperin, M.Y. Principles and methods of sequence analysis. Sequence—Evolution—Function: computational approaches in comparative genomics 2003, pp. 111–192.
- Rajendran, S.; Pan, W.; Sabuncu, M.R.; Chen, Y.; Zhou, J.; Wang, F. Learning across diverse biomedical data modalities and cohorts: Challenges and opportunities for innovation. Patterns 2024, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ding, K.; Lv, T.; Wang, X.; Yin, Q.; Zhang, Y.; Yu, J.; Wang, Y.; Li, X.; Xiang, Z.; et al. Scientific large language models: A survey on biological & chemical domains. ACM Computing Surveys 2025, 57, 1–38. [Google Scholar] [CrossRef]
- de Carvalho, G.H.; Knap, O.; Pollice, R. Show, Don’t Tell: Evaluating Large Language Models Beyond Textual Understanding with ChildPlay. arXiv 2024, arXiv:2407.11068. [Google Scholar]
- Wang, Y.; Qi, J.; Gan, J. Accurate and Regret-aware Numerical Problem Solver for Tabular Question Answering. arXiv 2024, arXiv:2410.12846. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, W.; Wu, Y.; Petzold, L.; Wang, W.Y.; Chen, H. Dna-gpt: Divergent n-gram analysis for training-free detection of gpt-generated text. arXiv 2023, arXiv:2305.17359. [Google Scholar]
- Zhang, D.; Zhang, W.; Zhao, Y.; Zhang, J.; He, B.; Qin, C.; Yao, J. DNAGPT: a generalized pre-trained tool for versatile DNA sequence analysis tasks. arXiv 2023, arXiv:2307.05628. [Google Scholar] [CrossRef]
- Nascimento, N.; Guimaraes, E.; Chintakunta, S.S.; Boominathan, S.A. LLM4DS: Evaluating Large Language Models for Data Science Code Generation. arXiv 2024, arXiv:2411.11908. [Google Scholar] [CrossRef]
- Shen, L.; Yang, Q.; Zheng, Y.; Li, M. AutoIOT: LLM-Driven Automated Natural Language Programming for AIoT Applications. arXiv 2025, arXiv:2503.05346. [Google Scholar]
- Maddigan, P.; Susnjak, T. Chat2vis: Generating data visualizations via natural language using chatgpt, codex and gpt-3 large language models. Ieee Access 2023, 11, 45181–45193. [Google Scholar] [CrossRef]
- Nejjar, M.; Zacharias, L.; Stiehle, F.; Weber, I. Llms for science: Usage for code generation and data analysis. Journal of Software: Evolution and Process 2025, 37, e2723. [Google Scholar] [CrossRef]
- Ji, Y.; Zhou, Z.; Liu, H.; Davuluri, R.V. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef]
- Zhou, Z.; Ji, Y.; Li, W.; Dutta, P.; Davuluri, R.; Liu, H. DNABERT-2: Efficient Foundation Model and Benchmark For Multi-Species Genome. arXiv 2023, arXiv:2306.15006. [Google Scholar]
- Avsec, Ž.; Agarwal, V.; Visentin, D.; Ledsam, J.R.; Grabska-Barwinska, A.; Taylor, K.R.; Assael, Y.; Jumper, J.; Kohli, P.; Kelley, D.R. Effective gene expression prediction from sequence by integrating long-range interactions. bioRxiv [https://www.biorxiv.org/content/early/2021/04/08/2021.04.07.438649.full.pdf]. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sanabria, M.; Hirsch, J.; Joubert, P.M.; Poetsch, A.R. DNA language model GROVER learns sequence context in the human genome. Nature Machine Intelligence 2024, 6, 911–923. [Google Scholar] [CrossRef]
- Dalla-Torre, H.; Gonzalez, L.; Mendoza-Revilla, J.; Lopez Carranza, N.; Grzywaczewski, A.H.; Oteri, F.; Dallago, C.; Trop, E.; de Almeida, B.P.; Sirelkhatim, H.; et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nature Methods 2024, 1–11. [Google Scholar] [CrossRef]
- Zhou, Z.; Riley, R.; Kautsar, S.; Wu, W.; Egan, R.; Hofmeyr, S.; Goldhaber-Gordon, S.; Yu, M.; Ho, H.; Liu, F.; et al. GenomeOcean: An Efficient Genome Foundation Model Trained on Large-Scale Metagenomic Assemblies. bioRxiv 2025, 2025–01. [Google Scholar] [CrossRef]
- Fishman, V.; Kuratov, Y.; Petrov, M.; Shmelev, A.; Shepelin, D.; Chekanov, N.; Kardymon, O.; Burtsev, M. Gena-lm: A family of open-source foundational models for long dna sequences. bioRxiv 2023, 12, 2023. [Google Scholar] [CrossRef]
- Akiyama, M.; Sakakibara, Y. Informative RNA base embedding for RNA structural alignment and clustering by deep representation learning. NAR genomics and bioinformatics 2022, 4, lqac012. [Google Scholar] [CrossRef]
- Chen, J.; Hu, Z.; Sun, S.; Tan, Q.; Wang, Y.; Yu, Q.; Zong, L.; Hong, L.; Xiao, J.; Shen, T.; et al. Interpretable RNA foundation model from unannotated data for highly accurate RNA structure and function predictions. arXiv 2022, arXiv:2204.00300. [Google Scholar] [CrossRef]
- Zhang, Y.; Lang, M.; Jiang, J.; Gao, Z.; Xu, F.; Litfin, T.; Chen, K.; Singh, J.; Huang, X.; Song, G.; et al. Multiple sequence alignment-based RNA language model and its application to structural inference. Nucleic Acids Research 2024, 52, e3–e3. [Google Scholar] [CrossRef]
- Chen, K.; Zhou, Y.; Ding, M.; Wang, Y.; Ren, Z.; Yang, Y. Self-supervised learning on millions of pre-mRNA sequences improves sequence-based RNA splicing prediction. BioRxiv 2023, 2023–01. [Google Scholar] [CrossRef]
- Marin, F.I.; Teufel, F.; Horlacher, M.; Madsen, D.; Pultz, D.; Winther, O.; Boomsma, W. Bend: Benchmarking dna language models on biologically meaningful tasks. arXiv 2023, arXiv:2311.12570. [Google Scholar]
- Yang, Y.; Li, G.; Pang, K.; Cao, W.; Zhang, Z.; Li, X. Deciphering 3’UTR Mediated Gene Regulation Using Interpretable Deep Representation Learning. Advanced Science 2024, 11, 2407013. [Google Scholar] [CrossRef]
- Chu, Y.; Yu, D.; Li, Y.; Huang, K.; Shen, Y.; Cong, L.; Zhang, J.; Wang, M. A 5’UTR language model for decoding untranslated regions of mRNA and function predictions. Nature Machine Intelligence 2024, 6, 449–460. [Google Scholar] [CrossRef]
- Gu, Y.; Tinn, R.; Cheng, H.; Lucas, M.; Usuyama, N.; Liu, X.; Naumann, T.; Gao, J.; Poon, H. Domain-specific language model pretraining for biomedical natural language processing. ACM Transactions on Computing for Healthcare (HEALTH) 2021, 3, 1–23. [Google Scholar] [CrossRef]
- Luo, R.; Sun, L.; Xia, Y.; Qin, T.; Zhang, S.; Poon, H.; Liu, T.Y. BioGPT: generative pre-trained transformer for biomedical text generation and mining. Briefings in bioinformatics 2022, 23, bbac409. [Google Scholar] [CrossRef]
- Bolton, E.; Venigalla, A.; Yasunaga, M.; Hall, D.; Xiong, B.; Lee, T.; Daneshjou, R.; Frankle, J.; Liang, P.; Carbin, M.; et al. Biomedlm: A 2.7 b parameter language model trained on biomedical text. arXiv 2024, arXiv:2403.18421. [Google Scholar]
- Singhal, K.; Azizi, S.; Tu, T.; Mahdavi, S.S.; Wei, J.; Chung, H.W.; Scales, N.; Tanwani, A.; Cole-Lewis, H.; Pfohl, S.; et al. Large language models encode clinical knowledge. Nature 2023, 620, 172–180. [Google Scholar] [CrossRef]
- Singhal, K.; Tu, T.; Gottweis, J.; Sayres, R.; Wulczyn, E.; Amin, M.; Hou, L.; Clark, K.; Pfohl, S.R.; Cole-Lewis, H.; et al. Toward expert-level medical question answering with large language models. Nature Medicine 2025, 1–8. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Zhang, K.; Dan, R.; Jiang, S.; Zhang, Y. Chatdoctor: A medical chat model fine-tuned on a large language model meta-ai (llama) using medical domain knowledge. Cureus 2023, 15. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J.; Jiang, F.; Yu, F.; Chen, Z.; Li, J.; Chen, G.; Wu, X.; Zhang, Z.; Xiao, Q.; et al. HuatuoGPT, Towards Taming Language Models To Be a Doctor. arXiv 2023, arXiv:2305.15075. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Gao, A.; Jiang, F.; Chen, S.; Zhang, H.; Song, D.; Xie, W.; Kong, C.; Li, J.; et al. HuatuoGPT-II, One-stage Training for Medical Adaption of LLMs. arXiv 2023, arXiv:2311.09774. [Google Scholar]
- Toma, A.; Lawler, P.R.; Ba, J.; Krishnan, R.G.; Rubin, B.B.; Wang, B. Clinical camel: An open-source expert-level medical language model with dialogue-based knowledge encoding. CoRR 2023. [Google Scholar]
- Xiong, H.; Wang, S.; Zhu, Y.; Zhao, Z.; Liu, Y.; Wang, Q.; Shen, D. Doctorglm: Fine-tuning your chinese doctor is not a herculean task. arXiv 2023, arXiv:2304.01097. [Google Scholar]
- Chen, J.; Cai, Z.; Ji, K.; Wang, X.; Liu, W.; Wang, R.; Hou, J.; Wang, B. HuatuoGPT-o1, Towards Medical Complex Reasoning with LLMs. arXiv 2024, arXiv:2412.18925o1. [Google Scholar]
- Yu, H.; Cheng, T.; Cheng, Y.; Feng, R. Finemedlm-o1: Enhancing the medical reasoning ability of llm from supervised fine-tuning to test-time training. arXiv 2025, arXiv:2501.09213. [Google Scholar]
- Han, T.; Adams, L.C.; Papaioannou, J.M.; Grundmann, P.; Oberhauser, T.; Löser, A.; Truhn, D.; Bressem, K.K. MedAlpaca–an open-source collection of medical conversational AI models and training data. arXiv 2023, arXiv:2304.08247. [Google Scholar]
- Chen, Z.; Cano, A.H.; Romanou, A.; Bonnet, A.; Matoba, K.; Salvi, F.; Pagliardini, M.; Fan, S.; Köpf, A.; Mohtashami, A.; et al. Meditron-70b: Scaling medical pretraining for large language models. arXiv 2023, arXiv:2311.16079. [Google Scholar] [CrossRef]
- Labrak, Y.; Bazoge, A.; Morin, E.; Gourraud, P.A.; Rouvier, M.; Dufour, R. Biomistral: A collection of open-source pretrained large language models for medical domains. arXiv 2024, arXiv:2402.10373. [Google Scholar]
- Huang, K.; Altosaar, J.; Ranganath, R. ClinicalBERT: Modeling Clinical Notes and Predicting Hospital Readmission. arXiv 2019, arXiv:1904.05342. [Google Scholar]
- Yasunaga, M.; Leskovec, J.; Liang, P. LinkBERT: Pretraining Language Models with Document Links. In Proceedings of the Association for Computational Linguistics (ACL); 2022. [Google Scholar]
- Peng, Y.; Yan, S.; Lu, Z. Transfer Learning in Biomedical Natural Language Processing: An Evaluation of BERT and ELMo on Ten Benchmarking Datasets. In Proceedings of the Proceedings of the 2019 Workshop on Biomedical Natural Language Processing (BioNLP 2019), 2019; pp. 58–65.
- Nori, H.; King, N.; McKinney, S.M.; Carignan, D.; Horvitz, E. Capabilities of gpt-4 on medical challenge problems. arXiv 2023, arXiv:2303.13375. [Google Scholar] [CrossRef]
- Tran, H.; Yang, Z.; Yao, Z.; Bioinstruct, H.Y. Instruction tuning of large language models for biomedical natural language processing. arXiv 2023, arXiv:2310.19975. [Google Scholar] [CrossRef]
- Lu, Q.; Dou, D.; Nguyen, T. ClinicalT5: A generative language model for clinical text. In Proceedings of the Findings of the Association for Computational Linguistics: EMNLP 2022; 2022; pp. 5436–5443. [Google Scholar]
- Phan, L.N.; Anibal, J.T.; Tran, H.; Chanana, S.; Bahadroglu, E.; Peltekian, A.; Altan-Bonnet, G. Scifive: a text-to-text transformer model for biomedical literature. arXiv 2021, arXiv:2106.03598. [Google Scholar]
- Schwieger, A.; Angst, K.; De Bardeci, M.; Burrer, A.; Cathomas, F.; Ferrea, S.; Grätz, F.; Knorr, M.; Kronenberg, G.; Spiller, T.; et al. Large language models can support generation of standardized discharge summaries–a retrospective study utilizing ChatGPT-4 and electronic health records. International Journal of Medical Informatics 2024, 192, 105654. [Google Scholar] [CrossRef]
- Zaretsky, J.; Kim, J.M.; Baskharoun, S.; Zhao, Y.; Austrian, J.; Aphinyanaphongs, Y.; Gupta, R.; Blecker, S.B.; Feldman, J. Generative artificial intelligence to transform inpatient discharge summaries to patient-friendly language and format. JAMA network open 2024, 7, e240357–e240357. [Google Scholar] [CrossRef]
- Xie, Q.; Chen, Q.; Chen, A.; Peng, C.; Hu, Y.; Lin, F.; Peng, X.; Huang, J.; Zhang, J.; Keloth, V.; et al. Me-llama: Foundation large language models for medical applications. Research square 2024, rs–3. [Google Scholar]
- Li, Y.; Rao, S.; Solares, J.R.A.; Hassaine, A.; Ramakrishnan, R.; Canoy, D.; Zhu, Y.; Rahimi, K.; Salimi-Khorshidi, G. BEHRT: transformer for electronic health records. Scientific reports 2020, 10, 7155. [Google Scholar] [CrossRef]
- Rasmy, L.; Xiang, Y.; Xie, Z.; Tao, C.; Zhi, D. Med-BERT: pretrained contextualized embeddings on large-scale structured electronic health records for disease prediction. NPJ digital medicine 2021, 4, 86. [Google Scholar] [CrossRef]
- Yang, X.; Chen, A.; PourNejatian, N.; Shin, H.C.; Smith, K.E.; Parisien, C.; Compas, C.; Martin, C.; Costa, A.B.; Flores, M.G.; et al. A large language model for electronic health records. NPJ digital medicine 2022, 5, 194. [Google Scholar] [CrossRef]
- Xu, M.; Zhao, X.; Wang, J.; Feng, W.; Wen, N.; Wang, C.; Wang, J.; Liu, Y.; Zhao, L. DFFNDDS: prediction of synergistic drug combinations with dual feature fusion networks. Journal of Cheminformatics 2023, 15, 33. [Google Scholar] [CrossRef]
- Li, T.; Shetty, S.; Kamath, A.; Jaiswal, A.; Jiang, X.; Ding, Y.; Kim, Y. CancerGPT for few shot drug pair synergy prediction using large pretrained language models. NPJ Digital Medicine 2024, 7, 40. [Google Scholar] [CrossRef]
- Edwards, C.; Naik, A.; Khot, T.; Burke, M.; Ji, H.; Hope, T. Synergpt: In-context learning for personalized drug synergy prediction and drug design. arXiv 2023, arXiv:2307.11694. [Google Scholar]
- Liu, T.; Chu, T.; Luo, X.; Zhao, H. BAITSAO: Building A Foundation Model for Drug Synergy Analysis Powered by Language Models. bioRxiv [https://www.biorxiv.org/content/early/2024/04/12/2024.04.08.588634.full.pdf]. 2024. [Google Scholar] [CrossRef]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified rational protein engineering with sequence-based deep representation learning. Nature methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Bepler, T.; Berger, B. Learning protein sequence embeddings using information from structure. arXiv 2019, arXiv:1902.08661. [Google Scholar] [CrossRef]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.; Rao, R.; Verkuil, R.; Liu, J.; Sercu, T.; Rives, A. Language models enable zero-shot prediction of the effects of mutations on protein function. Advances in neural information processing systems 2021, 34, 29287–29303. [Google Scholar]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Su, J.; Han, C.; Zhou, Y.; Shan, J.; Zhou, X.; Yuan, F. Saprot: Protein language modeling with structure-aware vocabulary. bioRxiv 2023, 2023–10. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, C.; Xu, M.; Chenthamarakshan, V.; Lozano, A.; Das, P.; Tang, J. A Systematic Study of Joint Representation Learning on Protein Sequences and Structures. arXiv 2023, arXiv:2303.06275. [Google Scholar] [CrossRef]
- Zhang, N.; Bi, Z.; Liang, X.; Cheng, S.; Hong, H.; Deng, S.; Lian, J.; Zhang, Q.; Chen, H. Ontoprotein: Protein pretraining with gene ontology embedding. arXiv 2022, arXiv:2201.11147. [Google Scholar] [CrossRef]
- Wu, K.E.; Chang, H.; Zou, J. ProteinCLIP: enhancing protein language models with natural language. bioRxiv 2024, 2024–05. [Google Scholar] [CrossRef]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nature communications 2022, 13, 4348. [Google Scholar] [CrossRef]
- Lv, L.; Lin, Z.; Li, H.; Liu, Y.; Cui, J.; Chen, C.Y.C.; Yuan, L.; Tian, Y. ProLLaMA: A Protein Language Model for Multi-Task Protein Language Processing. arXiv 2024, arXiv:2402.16445. [Google Scholar] [CrossRef]
- Dai, F.; You, S.; Wang, C.; Fan, Y.; Su, J.; Han, C.; Zhou, X.; Liu, J.; Qian, H.; Wang, S.; et al. Toward de novo protein design from natural language. bioRxiv 2024, 2024–08. [Google Scholar] [CrossRef]
- Elnaggar, A.; Essam, H.; Salah-Eldin, W.; Moustafa, W.; Elkerdawy, M.; Rochereau, C.; Rost, B. Ankh: Optimized protein language model unlocks general-purpose modelling. arXiv 2023, arXiv:2301.06568. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Li, Z.; Gitter, A.; Zhu, Y.; Lu, J.; Xu, Z.; Nie, W.; Ramanathan, A.; Xiao, C.; et al. A text-guided protein design framework. Nature Machine Intelligence 2025, 1–12. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, W.; Qin, M.; Wang, Y.; Pu, Z.; Ding, K.; Liu, Y.; Zhang, Q.; Li, D.; Li, X.; et al. Integrating protein language models and automatic biofoundry for enhanced protein evolution. Nature Communications 2025, 16, 1553. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Sun, E.; Jin, Y.; Wang, Q.; Wang, W. Proteingpt: Multimodal llm for protein property prediction and structure understanding. arXiv 2024, arXiv:2408.11363. [Google Scholar] [CrossRef]
- Guo, H.; Huo, M.; Zhang, R.; Xie, P. Proteinchat: Towards achieving chatgpt-like functionalities on protein 3d structures. Authorea Preprints 2023. [Google Scholar]
- Mattick, J.; Amaral, P. The Human Genome. In RNA, the Epicenter of Genetic Information: A new understanding of molecular biology; CRC Press, 2022. [Google Scholar]
- Cheatham, T.E.; Kollman, P.A. Molecular dynamics simulations highlight the structural differences among DNA: DNA, RNA: RNA, and DNA: RNA hybrid duplexes. Journal of the American Chemical Society 1997, 119, 4805–4825. [Google Scholar] [CrossRef]
- Ozsolak, F.; Milos, P.M. RNA sequencing: advances, challenges and opportunities. Nature reviews genetics 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: the teenage years. Nature Reviews Genetics 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nature methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Gage, P. A new algorithm for data compression. The C Users Journal 1994, 12, 23–38. [Google Scholar]
- Liu, Z.; Mao, H.; Wu, C.Y.; Feichtenhofer, C.; Darrell, T.; Xie, S. A ConvNet for the 2020s. arXiv 2022, arXiv:2201.03545. [Google Scholar] [CrossRef]
- Dao, T.; Fu, D.Y.; Ermon, S.; Rudra, A.; Ré, C. FlashAttention: Fast and memory-efficient exact attention with IO-awareness. In Proceedings of the Advances in Neural Information Processing Systems (NeurIPS); 2022. [Google Scholar]
- RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic acids research 2021, 49, D212–D220. [CrossRef]
- Rao, R.M.; Liu, J.; Verkuil, R.; Meier, J.; Canny, J.; Abbeel, P.; Sercu, T.; Rives, A. MSA transformer. In Proceedings of the International Conference on Machine Learning. PMLR; 2021; pp. 8844–8856. [Google Scholar]
- Haeussler, M.; Zweig, A.S.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Hinrichs, A.S.; Gonzalez, J.N.; et al. The UCSC genome browser database: 2019 update. Nucleic acids research 2019, 47, D853–D858. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms for molecular biology 2011, 6, 1–14. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The vienna RNA websuite. Nucleic acids research 2008, 36, W70–W74. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic acids research 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Sample, P.J.; Wang, B.; Reid, D.W.; Presnyak, V.; McFadyen, I.J.; Morris, D.R.; Seelig, G. Human 5’UTR design and variant effect prediction from a massively parallel translation assay. Nature biotechnology 2019, 37, 803–809. [Google Scholar] [CrossRef]
- Cao, J.; Novoa, E.M.; Zhang, Z.; Chen, W.C.; Liu, D.; Choi, G.C.; Wong, A.S.; Wehrspaun, C.; Kellis, M.; Lu, T.K. High-throughput 5’UTR engineering for enhanced protein production in non-viral gene therapies. Nature communications 2021, 12, 4138. [Google Scholar] [CrossRef]
- Johnson, A.E.; Pollard, T.J.; Shen, L.; Lehman, L.w.H.; Feng, M.; Ghassemi, M.; Moody, B.; Szolovits, P.; Anthony Celi, L.; Mark, R.G. MIMIC-III, a freely accessible critical care database. Scientific data 2016, 3, 1–9. [Google Scholar] [CrossRef]
- Johnson, A.E.; Bulgarelli, L.; Shen, L.; Gayles, A.; Shammout, A.; Horng, S.; Pollard, T.J.; Hao, S.; Moody, B.; Gow, B.; et al. MIMIC-IV, a freely accessible electronic health record dataset. Scientific data 2023, 10, 1. [Google Scholar] [CrossRef]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the Limits of Transfer Learning with a Unified Text-to-Text Transformer. arXiv 2023, arXiv:1910.10683. [Google Scholar]
- Lewis, M.; Liu, Y.; Goyal, N.; Ghazvininejad, M.; Mohamed, A.; Levy, O.; Stoyanov, V.; Zettlemoyer, L. Bart: Denoising sequence-to-sequence pre-training for natural language generation, translation, and comprehension. arXiv 2019, arXiv:1910.13461. [Google Scholar]
- Krithara, A.; Nentidis, A.; Bougiatiotis, K.; Paliouras, G. BioASQ-QA: A manually curated corpus for Biomedical Question Answering. Scientific Data 2023, 10, 170. [Google Scholar] [CrossRef]
- Chowdhery, A.; Narang, S.; Devlin, J.; Bosma, M.; Mishra, G.; Roberts, A.; Barham, P.; Chung, H.W.; Sutton, C.; Gehrmann, S.; et al. PaLM: Scaling Language Modeling with Pathways. arXiv 2022, arXiv:2204.02311. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Liu, Z.; Miller, J.; Efros, A.; Hardt, M. Test-time training with self-supervision for generalization under distribution shifts. In Proceedings of the International conference on machine learning. PMLR; 2020; pp. 9229–9248. [Google Scholar]
- Romanov, A.; Shivade, C. Lessons from Natural Language Inference in the Clinical Domain. [1808.06752]. [CrossRef]
- Preuer, K.; Lewis, R.P.; Hochreiter, S.; Bender, A.; Bulusu, K.C.; Klambauer, G. DeepSynergy: predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 2018, 34, 1538–1546. [Google Scholar] [CrossRef]
- Grešová, K.; Martinek, V.; Čechák, D.; Šimeček, P.; Alexiou, P. Genomic benchmarks: a collection of datasets for genomic sequence classification. BMC Genomic Data 2023, 24, 25. [Google Scholar] [CrossRef]
- Runge, F.; Farid, K.; Franke, J.K.; Hutter, F. Rnabench: A comprehensive library for in silico rna modelling. bioRxiv 2024, 2024–01. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, Z.; Qiao, L.; Jing, H.; Cai, Y.; Xu, S.; Ye, P.; Ma, X.; Sun, S.; Yan, H.; et al. Beacon: Benchmark for comprehensive rna tasks and language models. Advances in Neural Information Processing Systems 2024, 37, 92891–92921. [Google Scholar]
- Demner-Fushman, D.; Kohli, M.D.; Rosenman, M.B.; Shooshan, S.E.; Rodriguez, L.; Antani, S.; Thoma, G.R.; McDonald, C.J. Preparing a collection of radiology examinations for distribution and retrieval. Journal of the American Medical Informatics Association 2016, 23, 304–310. [Google Scholar] [CrossRef]
- Wornow, M.; Thapa, R.; Steinberg, E.; Fries, J.; Shah, N. Ehrshot: An ehr benchmark for few-shot evaluation of foundation models. Advances in Neural Information Processing Systems 2023, 36, 67125–67137. [Google Scholar]
- Kweon, S.; Kim, J.; Kwak, H.; Cha, D.; Yoon, H.; Kim, K.; Yang, J.; Won, S.; Choi, E. Ehrnoteqa: An llm benchmark for real-world clinical practice using discharge summaries. Advances in Neural Information Processing Systems 2024, 37, 124575–124611. [Google Scholar]
- Jin, D.; Pan, E.; Oufattole, N.; Weng, W.H.; Fang, H.; Szolovits, P. What disease does this patient have. A Large-scale Open Domain Question Answering Dataset from Medical Exams. arXiv [cs. CL] 2020. [CrossRef]
- Pal, A.; Umapathi, L.K.; Sankarasubbu, M. Medmcqa: A large-scale multi-subject multi-choice dataset for medical domain question answering. In Proceedings of the Conference on health, inference, and learning. PMLR; 2022; pp. 248–260. [Google Scholar]
- Jin, Q.; Dhingra, B.; Liu, Z.; Cohen, W.; Lu, X. PubMedQA: A Dataset for Biomedical Research Question Answering. In Proceedings of the Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP); Inui, K.; Jiang, J.; Ng, V.; Wan, X., Eds., Hong Kong, China, 2019; pp. 2567–2577. [CrossRef]
- Doğan, R.I.; Leaman, R.; Lu, Z. NCBI disease corpus: a resource for disease name recognition and concept normalization. Journal of biomedical informatics 2014, 47, 1–10. [Google Scholar] [PubMed]
- Segura-Bedmar, I.; Martínez, P.; Herrero-Zazo, M. Semeval-2013 task 9: Extraction of drug-drug interactions from biomedical texts (ddiextraction 2013). In Proceedings of the Second Joint Conference on Lexical and Computational Semantics (* SEM), Volume 2: Proceedings of the Seventh International Workshop on Semantic Evaluation (SemEval 2013), 2013; pp. 341–350.
- Becker, K.G.; Barnes, K.C.; Bright, T.J.; Wang, S.A. The genetic association database. Nature genetics 2004, 36, 431–432. [Google Scholar] [CrossRef]
- Baker, S.; Silins, I.; Guo, Y.; Ali, I.; Högberg, J.; Stenius, U.; Korhonen, A. Automatic semantic classification of scientific literature according to the hallmarks of cancer. Bioinformatics 2016, 32, 432–440. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, Z.; Lu, J.; Zhu, Z.; Zhang, Y.; Chang, M.; Liu, R.; Tang, J. Peer: a comprehensive and multi-task benchmark for protein sequence understanding. Advances in Neural Information Processing Systems 2022, 35, 35156–35173. [Google Scholar]
- Rao, R.; Bhattacharya, N.; Thomas, N.; Duan, Y.; Chen, P.; Canny, J.; Abbeel, P.; Song, Y. Evaluating protein transfer learning with TAPE. Advances in neural information processing systems 2019, 32. [Google Scholar]
- Hie, B.; Zhong, E.D.; Berger, B.; Bryson, B. Learning the language of viral evolution and escape. Science 2021, 371, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.; Rao, R.; Akin, H.; Sofroniew, N.J.; Oktay, D.; Lin, Z.; Verkuil, R.; Tran, V.Q.; Deaton, J.; Wiggert, M.; et al. Simulating 500 million years of evolution with a language model. bioRxiv 2024. [Google Scholar] [CrossRef]
- Johnson, B.A.; Blevins, R.A. NMR View: A computer program for the visualization and analysis of NMR data. Journal of biomolecular NMR 1994, 4, 603–614. [Google Scholar]
- Jares-Erijman, E.A.; Jovin, T.M. FRET imaging. Nature biotechnology 2003, 21, 1387–1395. [Google Scholar] [CrossRef]
- Bai, X.C.; McMullan, G.; Scheres, S.H. How cryo-EM is revolutionizing structural biology. Trends in biochemical sciences 2015, 40, 49–57. [Google Scholar] [CrossRef]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS letters 2015, 589, 2570–2577. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—the grand orchestrator of cancer growth and immune evasion. Nature reviews Clinical oncology 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Roayaei Ardakany, A.; Gezer, H.T.; Lonardi, S.; Ay, F. Mustache: multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. Genome biology 2020, 21, 1–17. [Google Scholar] [CrossRef]
- Huang, K.; Qu, Y.; Cousins, H.; Johnson, W.A.; Yin, D.; Shah, M.; Zhou, D.; Altman, R.; Wang, M.; Cong, L. Crispr-gpt: An llm agent for automated design of gene-editing experiments. arXiv 2024, arXiv:2404.18021. [Google Scholar]
- Rathore, A.S.; Choudhury, S.; Arora, A.; Tijare, P.; Raghava, G.P. ToxinPred 3.0: An improved method for predicting the toxicity of peptides. Computers in Biology and Medicine 2024, 179, 108926. [Google Scholar] [CrossRef]
- Sharma, N.; Naorem, L.D.; Jain, S.; Raghava, G.P. ToxinPred2: an improved method for predicting toxicity of proteins. Briefings in bioinformatics 2022, 23, bbac174. [Google Scholar]
- Fu, Y.; Ma, Y.; Zhong, L.; Yang, Y.; Guo, X.; Wang, C.; Xu, X.; Yang, K.; Xu, X.; Liu, L.; et al. EARTH SCIENCES 2001.
- Council, N.R.; on Geosciences, C.; Resources.; on Earth Sciences, B.; Resources.; on Basic Research Opportunities in the Earth Sciences, C. Basic research opportunities in earth science 2001.
- Council, N.R.; on Engineering, D.; Sciences, P.; Board, S.S.; on Earth Science, C.; from Space, A.; Assessment, A.C.; for the Future, S. Earth science and applications from space: national imperatives for the next decade and beyond; National Academies Press, 2007.
- Von Engelhardt, W.; Zimmermann, J. Theory of earth science 1988.
- Keum, H.J.; Han, K.Y.; Kim, H.I. Real-time flood disaster prediction system by applying machine learning technique. KSCE Journal of Civil Engineering 2020, 24, 2835–2848. [Google Scholar] [CrossRef]
- Ge, X.; Yang, Y.; Chen, J.; Li, W.; Huang, Z.; Zhang, W.; Peng, L. Disaster prediction knowledge graph based on multi-source spatio-temporal information. Remote Sensing 2022, 14, 1214. [Google Scholar] [CrossRef]
- Neelin, J.D. Climate change and climate modeling; Cambridge University Press, 2010. [Google Scholar]
- Liu, H.F.; Luo, Z.C.; Hu, Z.K.; Yang, S.Q.; Tu, L.C.; Zhou, Z.B.; Kraft, M. A review of high-performance MEMS sensors for resource exploration and geophysical applications. Petroleum Science 2022, 19, 2631–2648. [Google Scholar] [CrossRef]
- Esty, D.C. Environmental protection in the information age. NYUL Rev. 2004, 79, 115. [Google Scholar] [CrossRef]
- Li, Z.; Meier, M.A.; Hauksson, E.; Zhan, Z.; Andrews, J. Machine learning seismic wave discrimination: Application to earthquake early warning. Geophysical Research Letters 2018, 45, 4773–4779. [Google Scholar] [CrossRef]
- Waters, C.N.; Zalasiewicz, J.; Summerhayes, C.; Barnosky, A.D.; Poirier, C.; Gałuszka, A.; Cearreta, A.; Edgeworth, M.; Ellis, E.C.; Ellis, M.; et al. The Anthropocene is functionally and stratigraphically distinct from the Holocene. Science 2016, 351, aad2622. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.M.R. The solid earth: an introduction to global geophysics; Cambridge University Press, 1990. [Google Scholar]
- Hoff, H.; Falkenmark, M.; Gerten, D.; Gordon, L.; Karlberg, L.; Rockström, J. Greening the global water system. Journal of Hydrology 2010, 384, 177–186. [Google Scholar] [CrossRef]
- Böhme, G. atmosphere. Online Encyclopedia Philosophy of Nature 2021. [Google Scholar]
- Tóth, G.; Sokolov, I.V.; Gombosi, T.I.; Chesney, D.R.; Clauer, C.R.; De Zeeuw, D.L.; Hansen, K.C.; Kane, K.J.; Manchester, W.B.; Oehmke, R.C.; et al. Space Weather Modeling Framework: A new tool for the space science community. Journal of Geophysical Research: Space Physics 2005, 110. [Google Scholar] [CrossRef]
- Hornak, J.P.; Szumowski, J.; Bryant, R.G. Magnetic field mapping. Magnetic resonance in medicine 1988, 6, 158–163. [Google Scholar] [CrossRef]
- Flick, U. Mapping the field. The SAGE handbook of qualitative data analysis 2014, 1, 3–18. [Google Scholar]
- Wandell, B.A.; Dumoulin, S.O.; Brewer, A.A. Visual field maps in human cortex. Neuron 2007, 56, 366–383. [Google Scholar] [CrossRef]
- Baecher, G.; Lanney, N.; Einstein, H. Statistical description of rock properties and sampling. In Proceedings of the ARMA US rock mechanics/geomechanics symposium. ARMA; 1977; pp. ARMA–77. [Google Scholar]
- Scales, J.A. Theory of seismic imaging; Vol. 2, Springer-Verlag Berlin, 1995.
- Biondi, B.L. 3D seismic imaging; Society of Exploration Geophysicists, 2006. [Google Scholar]
- Olsson, I.U. Radiometric dating. In Handbook of Holocene palaeoecology and palaeohydrology; 1986. [Google Scholar]
- Falvey, D.A. The development of continental margins in plate tectonic theory. The APPEA Journal 1974, 14, 95–106. [Google Scholar] [CrossRef]
- Sun, K.; Cui, W.; Chen, C. Review of underwater sensing technologies and applications. Sensors 2021, 21, 7849. [Google Scholar] [CrossRef]
- Purser, A.; Marcon, Y.; Dreutter, S.; Hoge, U.; Sablotny, B.; Hehemann, L.; Lemburg, J.; Dorschel, B.; Biebow, H.; Boetius, A. Ocean Floor Observation and Bathymetry System (OFOBS): a new towed camera/sonar system for deep-sea habitat surveys. IEEE Journal of Oceanic Engineering 2018, 44, 87–99. [Google Scholar] [CrossRef]
- Busby, R.F. Manned submersibles; Office of the Oceanographer of the Navy, 1976. [Google Scholar]
- Taylor, R.G.; Scanlon, B.; Döll, P.; Rodell, M.; Van Beek, R.; Wada, Y.; Longuevergne, L.; Leblanc, M.; Famiglietti, J.S.; Edmunds, M.; et al. Ground water and climate change. Nature climate change 2013, 3, 322–329. [Google Scholar] [CrossRef]
- Larom, D.; Garstang, M.; Payne, K.; Raspet, R.; Lindeque, M. The influence of surface atmospheric conditions on the range and area reached by animal vocalizations. Journal of experimental biology 1997, 200, 421–431. [Google Scholar] [CrossRef]
- McGuffie, K.; Henderson-Sellers, A. Forty years of numerical climate modelling. International Journal of Climatology: A Journal of the Royal Meteorological Society 2001, 21, 1067–1109. [Google Scholar] [CrossRef]
- Robin, G.d.Q. Ice cores and climatic change. Philosophical Transactions of the Royal Society of London. B, Biological Sciences 1977, 280, 143–168. [Google Scholar] [CrossRef]
- Tei, S.; Sugimoto, A.; Yonenobu, H.; Matsuura, Y.; Osawa, A.; Sato, H.; Fujinuma, J.; Maximov, T. Tree-ring analysis and modeling approaches yield contrary response of circumboreal forest productivity to climate change. Global Change Biology 2017, 23, 5179–5188. [Google Scholar] [CrossRef]
- Hou, P.; Wu, S. Long-term changes in extreme air pollution meteorology and the implications for air quality. Scientific reports 2016, 6, 23792. [Google Scholar] [CrossRef]
- He, C.; Kumar, R.; Tang, W.; Pfister, G.; Xu, Y.; Qian, Y.; Brasseur, G. Air pollution interactions with weather and climate extremes: Current knowledge, gaps, and future directions. Current Pollution Reports 2024, 10, 430–442. [Google Scholar] [CrossRef]
- Rast, M.; Painter, T.H. Earth observation imaging spectroscopy for terrestrial systems: An overview of its history, techniques, and applications of its missions. Surveys in Geophysics 2019, 40, 303–331. [Google Scholar] [CrossRef]
- Rossi, A.P.; Van Gasselt, S. Planetary geology; Springer, 2018. [Google Scholar]
- Horton, R.M.; De Sherbinin, A.; Wrathall, D.; Oppenheimer, M. Assessing human habitability and migration. Science 2021, 372, 1279–1283. [Google Scholar] [CrossRef]
- Murray, C.D.; Dermott, S.F. Solar system dynamics; Cambridge university press, 1999. [Google Scholar]
- Zavala, G.R.; Nebro, A.J.; Luna, F.; Coello Coello, C.A. A survey of multi-objective metaheuristics applied to structural optimization. Structural and Multidisciplinary Optimization 2014, 49, 537–558. [Google Scholar] [CrossRef]
- Mei, L.; Wang, Q. Structural Optimization in Civil Engineering: A Literature Review. Buildings 2021, 11. [Google Scholar] [CrossRef]
- Wikipedia. Civil Engineering. Wikipedia, The Free Encyclopedia 2025. [Accessed: 2025-04-18].
- Harle, S.M. Advancements and challenges in the application of artificial intelligence in civil engineering: a comprehensive review. Asian Journal of Civil Engineering 2024, 25, 1061–1078. [Google Scholar]
- Vadyala, S.R.; Betgeri, S.N.; Matthews, J.C.; Matthews, E. A review of physics-based machine learning in civil engineering. Results in Engineering 2022, 13, 100316. [Google Scholar] [CrossRef]
- Tsiptsis, I.N.; Liimatainen, L.; Kotnik, T.; Niiranen, J. Structural optimization employing isogeometric tools in Particle Swarm Optimizer. Journal of Building Engineering 2019, 24, 100761. [Google Scholar] [CrossRef]
- Kuhn, H.W.; Tucker, A.W. Nonlinear programming. In Traces and emergence of nonlinear programming; Springer, 2013; pp. 247–258. [Google Scholar]
- Aldwaik, M.; Adeli, H. Advances in optimization of highrise building structures. Structural and Multidisciplinary Optimization 2014, 50, 899–919. [Google Scholar] [CrossRef]
- Saka, M.P.; Hasançebi, O.; Geem, Z.W. Metaheuristics in structural optimization and discussions on harmony search algorithm. Swarm and Evolutionary Computation 2016, 28, 88–97. [Google Scholar] [CrossRef]
- Sörensen, K. Metaheuristics—the metaphor exposed. International Transactions in Operational Research 2015, 22, 3–18. [Google Scholar] [CrossRef]
- Mahdavi, S.; Shiri, M.E.; Rahnamayan, S. Metaheuristics in large-scale global continues optimization: A survey. Information Sciences 2015, 295, 407–428. [Google Scholar] [CrossRef]
- Mortazavi, A. A new fuzzy strategy for size and topology optimization of truss structures. Applied Soft Computing 2020, 93, 106412. [Google Scholar] [CrossRef]
- Zheng, S.; Tang, W.; Li, B. A new topology optimization framework for stiffness design of beam structures based on the transformable triangular mesh algorithm. Thin-Walled Structures 2020, 154, 106831. [Google Scholar] [CrossRef]
- Tokognon, C.A.; Gao, B.; Tian, G.Y.; Yan, Y. Structural health monitoring framework based on Internet of Things: A survey. IEEE Internet of Things Journal 2017, 4, 619–635. [Google Scholar] [CrossRef]
- Gharehbaghi, V.R.; Noroozinejad Farsangi, E.; Noori, M.; Yang, T.; Li, S.; Nguyen, A.; Málaga-Chuquitaype, C.; Gardoni, P.; Mirjalili, S. A critical review on structural health monitoring: Definitions, methods, and perspectives. Archives of computational methods in engineering 2022, 29, 2209–2235. [Google Scholar] [CrossRef]
- Hodge, V.J.; O’Keefe, S.; Weeks, M.; Moulds, A. Wireless sensor networks for condition monitoring in the railway industry: A survey. IEEE Transactions on intelligent transportation systems 2014, 16, 1088–1106. [Google Scholar] [CrossRef]
- Cai, J.; Qiu, L.; Yuan, S.; Shi, L.; Liu, P.; Liang, D. Structural health monitoring for composite materials. In Composites and their applications; IntechOpen, 2012. [Google Scholar]
- Huseynov, F.; Kim, C.; Obrien, E.J.; Brownjohn, J.; Hester, D.; Chang, K. Bridge damage detection using rotation measurements–Experimental validation. Mechanical Systems and Signal Processing 2020, 135, 106380. [Google Scholar] [CrossRef]
- Gomes, G.F.; Mendez, Y.A.D.; da Silva Lopes Alexandrino, P.; da Cunha, S.S.; Ancelotti, A.C. A review of vibration based inverse methods for damage detection and identification in mechanical structures using optimization algorithms and ANN. Archives of computational methods in engineering 2019, 26, 883–897. [Google Scholar] [CrossRef]
- Jamali, S.; Chan, T.H.; Nguyen, A.; Thambiratnam, D.P. Reliability-based load-carrying capacity assessment of bridges using structural health monitoring and nonlinear analysis. Structural Health Monitoring 2019, 18, 20–34. [Google Scholar] [CrossRef]
- Babajanian Bisheh, H.; Ghodrati Amiri, G.; Nekooei, M.; Darvishan, E. Damage detection of a cable-stayed bridge using feature extraction and selection methods. Structure and Infrastructure Engineering 2019, 15, 1165–1177. [Google Scholar] [CrossRef]
- Sari, Y.; Prakoso, P.B.; Baskara, A.R. Road crack detection using support vector machine (SVM) and OTSU algorithm. In Proceedings of the 2019 6th International Conference on Electric Vehicular Technology (ICEVT). IEEE; 2019; pp. 349–354. [Google Scholar]
- Sarrafi, A.; Mao, Z.; Niezrecki, C.; Poozesh, P. Vibration-based damage detection in wind turbine blades using Phase-based Motion Estimation and motion magnification. Journal of Sound and vibration 2018, 421, 300–318. [Google Scholar] [CrossRef]
- Breysse, D. Nondestructive evaluation of concrete strength: An historical review and a new perspective by combining NDT methods. Construction and Building Materials 2012, 33, 139–163. [Google Scholar] [CrossRef]
- Helal, J.; Sofi, M.; Mendis, P. Non-destructive testing of concrete: A review of methods. Electronic Journal of Structural Engineering 2015, 14, 97–105. [Google Scholar] [CrossRef]
- Mitchell, J.K.; Soga, K.; et al. Fundamentals of soil behavior; Vol. 3, John Wiley & Sons New York, 2005.
- Lou, M.; Wang, H.; Chen, X.; Zhai, Y. Structure–soil–structure interaction: Literature review. Soil dynamics and earthquake engineering 2011, 31, 1724–1731. [Google Scholar] [CrossRef]
- David Müzel, S.; Bonhin, E.P.; Guimarães, N.M.; Guidi, E.S. Application of the Finite Element Method in the Analysis of Composite Materials: A Review. Polymers 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Matthews, F.L.; Davies, G.; Hitchings, D.; Soutis, C. Finite element modelling of composite materials and structures; Elsevier, 2000. [Google Scholar]
- Yaseen, Z.M.; El-shafie, A.; Jaafar, O.; Afan, H.A.; Sayl, K.N. Artificial intelligence based models for stream-flow forecasting: 2000–2015. Journal of Hydrology 2015, 530, 829–844. [Google Scholar] [CrossRef]
- Lana, I.; Del Ser, J.; Velez, M.; Vlahogianni, E.I. Road Traffic Forecasting: Recent Advances and New Challenges. IEEE Intelligent Transportation Systems Magazine 2018, 10, 93–109. [Google Scholar] [CrossRef]
- Liang, B.; Liu, J.; You, J.; Jia, J.; Pan, Y.; Jeong, H. Hydrocarbon production dynamics forecasting using machine learning: A state-of-the-art review. Fuel 2023, 337, 127067. [Google Scholar] [CrossRef]
- Jiang, W.; Luo, J. Graph neural network for traffic forecasting: A survey. Expert Systems with Applications 2022, 207, 117921. [Google Scholar] [CrossRef]
- Belbute-Peres, F.D.A.; Economon, T.; Kolter, Z. Combining differentiable PDE solvers and graph neural networks for fluid flow prediction. In Proceedings of the international conference on machine learning. PMLR; 2020; pp. 2402–2411. [Google Scholar]
- Li, Z.; Ning, H. Autonomous GIS: the next-generation AI-powered GIS. International Journal of Digital Earth 2023, 16, 4668–4686. [Google Scholar] [CrossRef]
- Lin, Z.; Deng, C.; Zhou, L.; Zhang, T.; Xu, Y.; Xu, Y.; He, Z.; Shi, Y.; Dai, B.; Song, Y.; et al. Geogalactica: A scientific large language model in geoscience. arXiv 2023, arXiv:2401.00434. [Google Scholar]
- Hu, Y.; Yuan, J.; Wen, C.; Lu, X.; Liu, Y.; Li, X. Rsgpt: A remote sensing vision language model and benchmark. ISPRS Journal of Photogrammetry and Remote Sensing 2025, 224, 272–286. [Google Scholar] [CrossRef]
- Bazi, Y.; Bashmal, L.; Al Rahhal, M.M.; Ricci, R.; Melgani, F. Rs-llava: A large vision-language model for joint captioning and question answering in remote sensing imagery. Remote Sensing 2024, 16, 1477. [Google Scholar] [CrossRef]
- Zhang, W.; Cai, M.; Zhang, T.; Zhuang, Y.; Mao, X. Earthgpt: A universal multi-modal large language model for multi-sensor image comprehension in remote sensing domain. IEEE Transactions on Geoscience and Remote Sensing 2024. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, C.; Wu, S.; He, Z.; Yu, W. Geogpt: Understanding and processing geospatial tasks through an autonomous gpt. arXiv 2023, arXiv:2307.07930. [Google Scholar] [CrossRef]
- Singh, S.; Fore, M.; Stamoulis, D. Geollm-engine: A realistic environment for building geospatial copilots. In Proceedings of the Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, 2024; pp. 585–594.
- Chen, Y.; Wang, W.; Lobry, S.; Kurtz, C. An llm agent for automatic geospatial data analysis. arXiv 2024, arXiv:2410.18792. [Google Scholar] [CrossRef]
- Hou, S.; Jiao, H.; Shen, Z.; Liang, J.; Zhao, A.; Zhang, X.; Wang, J.; Wu, H. Chain-of-Programming (CoP): Empowering Large Language Models for Geospatial Code Generation. arXiv 2024, arXiv:2411.10753. [Google Scholar] [CrossRef]
- Jiang, G.; Ma, Z.; Zhang, L.; Chen, J. EPlus-LLM: A large language model-based computing platform for automated building energy modeling. Applied Energy 2024, 367, 123431. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Z.; Ford, V. Advancing building energy modeling with large language models: Exploration and case studies. Energy and Buildings 2024, 323, 114788. [Google Scholar] [CrossRef]
- Pursnani, V.; Ramirez, C.E.; Sermet, M.Y.; Demir, I. HydroSuite-AI: Facilitating Hydrological Research with LLM-Driven Code Assistance. 2024. [Google Scholar] [CrossRef]
- Bekele, Y.W. GeoSim. AI: AI assistants for numerical simulations in geomechanics. arXiv 2025, arXiv:2501.14186. [Google Scholar]
- Song, J.; Yoon, S. Ontology-assisted GPT-based building performance simulation and assessment: Implementation of multizone airflow simulation. Energy and Buildings 2024, 325, 114983. [Google Scholar] [CrossRef]
- Li, S.; Azfar, T.; Ke, R. ChatSUMO: Large Language Model for Automating Traffic Scenario Generation in Simulation of Urban MObility. IEEE Transactions on Intelligent Vehicles 2024, 1–12. [Google Scholar] [CrossRef]
- Denli, H.; Chughtai, H.A.; Hughes, B.; Gistri, R.; Xu, P. Geoscience language processing for exploration. In Proceedings of the Abu Dhabi International Petroleum Exhibition and Conference. SPE; 2021; p. D031S102R003. [Google Scholar]
- Bi, Z.; Zhang, N.; Xue, Y.; Ou, Y.; Ji, D.; Zheng, G.; Chen, H. Oceangpt: A large language model for ocean science tasks. arXiv 2023, arXiv:2310.02031. [Google Scholar] [CrossRef]
- Dong, T.; Subia-Waud, C.; Hou, S. Geo-RAG: Gaining insights from unstructured geological documents with large language models. In Proceedings of the Fourth EAGE Digitalization Conference & Exhibition. European Association of Geoscientists & Engineers, 2024; 2024; Vol. 2024, pp. 1–4. [Google Scholar]
- Zheng, Z.; Chen, K.Y.; Cao, X.Y.; Lu, X.Z.; Lin, J.R. Llm-funcmapper: Function identification for interpreting complex clauses in building codes via llm. arXiv 2023, arXiv:2308.08728. [Google Scholar] [CrossRef]
- Chen, N.; Lin, X.; Jiang, H.; An, Y. Automated Building Information Modeling Compliance Check through a Large Language Model Combined with Deep Learning and Ontology. Buildings 2024, 14. [Google Scholar] [CrossRef]
- Wan, H.; Zhang, J.; Chen, Y.; Xu, W.; Feng, F. Generative AI Application for Building Industry. arXiv 2024, arXiv:2410.01098. [Google Scholar] [CrossRef]
- Xiao, T.; Xu, P. Exploring automated energy optimization with unstructured building data: A multi-agent based framework leveraging large language models. Energy and Buildings 2024, 322, 114691. [Google Scholar] [CrossRef]
- Forth, K.; Borrmann, A. Semantic enrichment for BIM-based building energy performance simulations using semantic textual similarity and fine-tuning multilingual LLM. Journal of Building Engineering 2024, 95, 110312. [Google Scholar] [CrossRef]
- Qin, B.; Pan, H.; Dai, Y.; Si, X.; Huang, X.; Yuen, C.; Zhang, Y. Machine and Deep Learning for Digital Twin Networks: A Survey. IEEE Internet of Things Journal 2024, 11, 34694–34716. [Google Scholar] [CrossRef]
- Jia, F.; Fonsati, A.; Gudmundsson, K. Natural Language Communication with Sensor Data Through a LLM-Integrated Protocol: A Case Study. In Proceedings of the International Conference on Computing in Civil and Building Engineering. Springer; 2024; pp. 64–75. [Google Scholar]
- Yang, H.; Siew, M.; Joe-Wong, C. An LLM-Based Digital Twin for Optimizing Human-in-the Loop Systems. In Proceedings of the 2024 IEEE International Workshop on Foundation Models for Cyber-Physical Systems & Internet of Things (FMSys), 2024; pp. 26–31. [CrossRef]
- Ferdousi, R.; Hossain, M.A.; Yang, C.; Saddik, A.E. Defecttwin: When llm meets digital twin for railway defect inspection. arXiv 2024, arXiv:2409.06725. [Google Scholar] [CrossRef]
- Syed, T.A.; Muhammad, M.A.; AlShahrani, A.A.; Hammad, M.; Naqash, M.T. Smart Water Management with Digital Twins and Multimodal Transformers: A Predictive Approach to Usage and Leakage Detection. Water 2024, 16, 3410. [Google Scholar] [CrossRef]
- Qin, S.; Guan, H.; Liao, W.; Gu, Y.; Zheng, Z.; Xue, H.; Lu, X. Intelligent design and optimization system for shear wall structures based on large language models and generative artificial intelligence. Journal of Building Engineering 2024, 95, 109996. [Google Scholar] [CrossRef]
- Jang, S.; Lee, G.; Oh, J.; Lee, J.; Koo, B. Automated detailing of exterior walls using NADIA: Natural-language-based architectural detailing through interaction with AI. Advanced Engineering Informatics 2024, 61, 102532. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, W.; Huang, N.; Li, B.; Niu, L.; Fan, Z.; Lun, T.; Tao, Y.; Su, J.; Gong, Z.; et al. PlanGPT: Enhancing urban planning with tailored language model and efficient retrieval. arXiv 2024, arXiv:2402.19273. [Google Scholar] [CrossRef]
- Zhou, Z.; Lin, Y.; Jin, D.; Li, Y. Large language model for participatory urban planning. arXiv 2024, arXiv:2402.17161. [Google Scholar] [CrossRef]
- Zhang, S.; Fu, D.; Liang, W.; Zhang, Z.; Yu, B.; Cai, P.; Yao, B. Trafficgpt: Viewing, processing and interacting with traffic foundation models. Transport Policy 2024, 150, 95–105. [Google Scholar] [CrossRef]
- Da, L.; Liou, K.; Chen, T.; Zhou, X.; Luo, X.; Yang, Y.; Wei, H. Open-ti: Open traffic intelligence with augmented language model. International Journal of Machine Learning and Cybernetics 2024, 15, 4761–4786. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, T.; He, Z.; Chen, Q.; Shi, Y.; Xu, Y.; Fu, L.; Zhang, W.; Wang, X.; Zhou, C.; et al. utilization. In Proceedings of the Proceedings of the 17th ACM International Conference on Web Search and Data Mining, 2024, pp.
- Zhang, Y.; He, Z.; Li, J.; Lin, J.; Guan, Q.; Yu, W. MapGPT: an autonomous framework for mapping by integrating large language model and cartographic tools. Cartography and Geographic Information Science 2024, 51, 717–743. [Google Scholar] [CrossRef]
- Roberts, J.; Lüddecke, T.; Das, S.; Han, K.; Albanie, S. GPT4GEO: How a Language Model Sees the World’s Geography. arXiv 2023, arXiv:2306.00020. [Google Scholar] [CrossRef]
- Kuckreja, K.; Danish, M.S.; Naseer, M.; Das, A.; Khan, S.; Khan, F.S. Geochat: Grounded large vision-language model for remote sensing. In Proceedings of the Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, 2024; pp. 27831–27840.
- Liu, C.; Chen, K.; Zhang, H.; Qi, Z.; Zou, Z.; Shi, Z. Change-agent: Towards interactive comprehensive remote sensing change interpretation and analysis. IEEE Transactions on Geoscience and Remote Sensing 2024. [Google Scholar] [CrossRef]
- Ning, H.; Li, Z.; Akinboyewa, T.; Lessani, M.N. An autonomous GIS agent framework for geospatial data retrieval. International Journal of Digital Earth 2025, 18, 2458688. [Google Scholar] [CrossRef]
- Hou, S.; Shen, Z.; Zhao, A.; Liang, J.; Gui, Z.; Guan, X.; Li, R.; Wu, H. GeoCode-GPT: A large language model for geospatial code generation. International Journal of Applied Earth Observation and Geoinformation 2025, 104456. [Google Scholar] [CrossRef]
- Cherian, A.; Corcodel, R.; Jain, S.; Romeres, D. Llmphy: Complex physical reasoning using large language models and world models. arXiv 2024, arXiv:2411.08027. [Google Scholar] [CrossRef]
- Bhandari, P.; Anastasopoulos, A.; Pfoser, D. Are large language models geospatially knowledgeable? In Proceedings of the Proceedings of the 31st ACM International Conference on Advances in Geographic Information Systems; 2023; pp. 1–4. [Google Scholar]
- Haas, L.; Alberti, S.; Skreta, M. Learning generalized zero-shot learners for open-domain image geolocalization. arXiv 2023, arXiv:2302.00275. [Google Scholar] [CrossRef]
- Mooney, P.; Cui, W.; Guan, B.; Juhász, L. Towards understanding the geospatial skills of chatgpt: Taking a geographic information systems (gis) exam. In Proceedings of the Proceedings of the 6th ACM SIGSPATIAL international workshop on AI for geographic knowledge discovery, 2023; pp. 85–94.
- Zhang, Y.; Wang, Z.; He, Z.; Li, J.; Mai, G.; Lin, J.; Wei, C.; Yu, W. BB-GeoGPT: A framework for learning a large language model for geographic information science. Information Processing & Management 2024, 61, 103808. [Google Scholar]
- Kumar, S.; Ehtesham, A.; Singh, A.; Khoei, T.T. Architectural Flaw Detection in Civil Engineering Using GPT-4. arXiv 2024, arXiv:2410.20036. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.; Zhu, X. A GPT-based method of automated compliance checking through prompt engineering 2023.
- Pu, H.; Yang, X.; Li, J.; Guo, R. AutoRepo: A general framework for multimodal LLM-based automated construction reporting. Expert Systems with Applications 2024, 255, 124601. [Google Scholar] [CrossRef]
- Yan, Y.; Wen, H.; Zhong, S.; Chen, W.; Chen, H.; Wen, Q.; Zimmermann, R.; Liang, Y. When urban region profiling meets large language models. arXiv 2023, arXiv:2310.18340. [Google Scholar] [CrossRef]
- Da, L.; Gao, M.; Mei, H.; Wei, H. Llm powered sim-to-real transfer for traffic signal control. arXiv 2023, arXiv:2308.14284. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L. Revolutionizing Bridge Operation and maintenance with LLM-based Agents: An Overview of Applications and Insights. arXiv 2024, arXiv:2407.10064. [Google Scholar] [CrossRef]
- Li, Y.; Ji, M.; Chen, J.; Wei, X.; Gu, X.; Tang, J. A large language model-based building operation and maintenance information query. Energy and Buildings 2025, 334, 115515. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Z. Large language model-based interpretable machine learning control in building energy systems. Energy and Buildings 2024, 313, 114278. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, J.; Zhao, Y.; Lu, J. Automated data mining framework for building energy conservation aided by generative pre-trained transformers (GPT). Energy and Buildings 2024, 305, 113877. [Google Scholar] [CrossRef]
- Hosamo, H.H.; Imran, A.; Cardenas-Cartagena, J.; Svennevig, P.R.; Svidt, K.; Nielsen, H.K. A review of the digital twin technology in the AEC-FM industry. Advances in civil engineering 2022, 2022, 2185170. [Google Scholar] [CrossRef]
- Hamzah, A.; Aqlan, F.; Baidya, S. Drone-based digital twins for water quality monitoring: A systematic review. Digital Twins and Applications 2024, 1, 131–160. [Google Scholar] [CrossRef]
- Hong, Y.; Wu, J.; Morello, R. LLM-Twin: mini-giant model-driven beyond 5G digital twin networking framework with semantic secure communication and computation. Scientific Reports 2024, 14, 19065. [Google Scholar] [CrossRef]
- Zhang, W.; Han, J.; Xu, Z.; Ni, H.; Liu, H.; Xiong, H. Urban Foundation Models: A Survey. In Proceedings of the Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 2024; KDD ’24; pp. 6633–6643. [CrossRef]
- Jang, S.; Roh, H.; Lee, G. Generative AI in architectural design: Application, data, and evaluation methods. Automation in Construction 2025, 174, 106174. [Google Scholar] [CrossRef]
- Zhang, W.; Han, J.; Xu, Z.; Ni, H.; Liu, H.; Xiong, H. Urban foundation models: A survey. In Proceedings of the Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2024; pp. 6633–6643.
- Lai, S.; Xu, Z.; Zhang, W.; Liu, H.; Xiong, H. LLMLight: Large Language Models as Traffic Signal Control Agents. arXiv 2023, arXiv:2312.16044. [Google Scholar] [CrossRef]
- Fu, J.; Ng, S.K.; Jiang, Z.; Liu, P. Gptscore: Evaluate as you desire. arXiv 2023, arXiv:2302.04166. [Google Scholar] [CrossRef]
- Knuth, D.E. Computer science and its relation to mathematics. The American Mathematical Monthly 1974, 81, 323–343. [Google Scholar] [CrossRef]
- Shagrir, O. What is computer science about? The Monist 1999, 82, 131–149. [Google Scholar] [CrossRef]
- Cortina, T.J. An introduction to computer science for non-majors using principles of computation. Acm sigcse bulletin 2007, 39, 218–222. [Google Scholar] [CrossRef]
- Lehman, E.; Leighton, F.T.; Meyer, A.R. Mathematics for computer science; Massachusetts Institute of Technology Cambridge: Massachusetts, 2010. [Google Scholar]
- Dorf, R.C. The electrical engineering handbook; CRC press, 1997. [Google Scholar]
- Rizzoni, G.; Kearns, J. Fundamentals of electrical engineering; McGraw-Hill Higher Education, 2009. [Google Scholar]
- IEEE JSTSP Special Series on AI in Signal and Data Science - Toward Large Language Model (LLM) Theory and Applications, 2024.
- Abdollahi, M.; Yeganli, S.F.; Baharloo, M.A.; Baniasadi, A. Hardware Design and Verification with Large Language Models: A Literature Survey, Challenges, and Open Issues. 2024. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.; Bi, T.; Grundy, J.; Wang, Y.; Yu, J.; Chen, T.; Tang, Y.; Zheng, Z. An empirical study on low code programming using traditional vs large language model support. arXiv 2024, arXiv:2402.01156. [Google Scholar] [CrossRef]
- Thorne, S.; Ball, D.; Lawson, Z. Reducing error in spreadsheets: Example driven modeling versus traditional programming. International Journal of Human-Computer Interaction 2013, 29, 40–53. [Google Scholar] [CrossRef]
- Stoica, M.; Mircea, M.; Ghilic-Micu, B. Software development: agile vs. traditional. Informatica Economica 2013, 17. [Google Scholar] [CrossRef]
- Awad, M. A comparison between agile and traditional software development methodologies. University of Western Australia 2005, 30, 1–69. [Google Scholar]
- Risener, K. A study of software development methodologies 2022.
- Leong, J.; May Yee, K.; Baitsegi, O.; Palanisamy, L.; Ramasamy, R.K. Hybrid project management between traditional software development lifecycle and agile based product development for future sustainability. Sustainability 2023, 15, 1121. [Google Scholar] [CrossRef]
- Pawar, R.P. A comparative study of agile software development methodology and traditional waterfall model. IOSR Journal of Computer Engineering 2015, 2, 1–8. [Google Scholar]
- Boehm, B. A spiral model of software development and enhancement. ACM SIGSOFT Software engineering notes 1986, 11, 14–24. [Google Scholar] [CrossRef]
- Boehm, B.W. A spiral model of software development and enhancement. Computer 2002, 21, 61–72. [Google Scholar] [CrossRef]
- Balaji, S.; Murugaiyan, M.S. Waterfall vs. V-Model vs. Agile: A comparative study on SDLC. International Journal of Information Technology and Business Management 2012, 2, 26–30. [Google Scholar]
- Gracias, M.H.; Gallegos, E.E. Transitioning Perspectives: Agile and Waterfall Perceptions in the Integration of Model-Based Systems Engineering (MBSE) within Aerospace and Defense Industries. The ITEA Journal of Test and Evaluation 2024, 45. [Google Scholar] [CrossRef]
- Shaikh, S.; Abro, S. Comparison of traditional & agile software development methodology: A short survey. International Journal of Software Engineering and Computer Systems 2019, 5, 1–14. [Google Scholar] [CrossRef]
- Roychoudhury, A. Debugging as a Science, that too, when your Program is Changing. Electronic Notes in Theoretical Computer Science 2010, 266, 3–15. [Google Scholar] [CrossRef]
- Fitzgerald, S.; Lewandowski, G.; McCauley, R.; Murphy, L.; Simon, B.; Thomas, L.; Zander, C. Debugging: finding, fixing and flailing, a multi-institutional study of novice debuggers. Computer Science Education 2008, 18, 93–116. [Google Scholar] [CrossRef]
- Weste, N.H.; Harris, D. CMOS VLSI design: a circuits and systems perspective; Pearson Education India, 2015. [Google Scholar]
- Palnitkar, S. Verilog HDL: a guide to digital design and synthesis; Vol. 1, Prentice Hall Professional, 2003.
- Ashenden, P.J. Digital design (verilog): An embedded systems approach using verilog; Elsevier, 2007.
- Thomas, D.; Moorby, P. The Verilog® hardware description language; Springer Science & Business Media, 2008.
- Haas, A.; Stewart, J.C.; Kukal, T. Ensuring Reliable and Optimal Analog PCB Designs with Allegro AMS Simulator. Cadence Design Systems, Silicon Valley 2007. [Google Scholar]
- Lin, R.; Ramesh, R.; Iannopollo, A.; Sangiovanni Vincentelli, A.; Dutta, P.; Alon, E.; Hartmann, B. Beyond schematic capture: Meaningful abstractions for better electronics design tools. In Proceedings of the Proceedings of the 2019 CHI conference on human factors in computing systems, 2019; pp. 1–13.
- Nelson, B.; Riching, B.; Black, R. Using a Custom-Built HDL for Printed Circuit Board Design Capture. Technical report, Sandia National Lab.(SNL-NM), Albuquerque, NM (United States), 2012.
- Jones, B.A.; Moorhead, J.N.; Mohammadi-Aragh, M.J. Less is More: Developing complex designs using a minimal HDL subset in an introductory digital devices laboratory. In Proceedings of the 2015 ASEE Annual Conference & Exposition; 2015; pp. 26–1082. [Google Scholar]
- Dewey, A. VHSIC hardware description (VHDL) development program. In Proceedings of the 20th Design Automation Conference Proceedings. IEEE; 1983; pp. 625–628. [Google Scholar]
- Mano, M.M.R.; Ciletti, M.D. Digital Design: With an Introduction to the Verilog HDL, VHDL, and SystemVerilog, 5th ed.; Pearson, 2012. Fundamental textbook covering schematic capture and HDL-based design[1][10].
- Vemeko FPGA Team. What are Hardware Description Languages?, 2024.
- Hou, X.; Zhao, Y.; Liu, Y.; Yang, Z.; Wang, K.; Li, L.; Luo, X.; Lo, D.; Grundy, J.; Wang, H. Large language models for software engineering: A systematic literature review. ACM Transactions on Software Engineering and Methodology 2024, 33, 1–79. [Google Scholar] [CrossRef]
- Belzner, L.; Gabor, T.; Wirsing, M. Large language model assisted software engineering: prospects, challenges, and a case study. In Proceedings of the International Conference on Bridging the Gap between AI and Reality. Springer; 2023; pp. 355–374. [Google Scholar]
- Fan, A.; Gokkaya, B.; Harman, M.; Lyubarskiy, M.; Sengupta, S.; Yoo, S.; Zhang, J.M. Large language models for software engineering: Survey and open problems. In Proceedings of the 2023 IEEE/ACM International Conference on Software Engineering: Future of Software Engineering (ICSE-FoSE). IEEE; 2023; pp. 31–53. [Google Scholar]
- Ma, Q.; Wu, T.; Koedinger, K. Is ai the better programming partner? human-human pair programming vs. human-ai pair programming. arXiv 2023, arXiv:2306.05153. [Google Scholar] [CrossRef]
- Peng, S.; Kalliamvakou, E.; Cihon, P.; Demirer, M. The impact of ai on developer productivity: Evidence from github copilot. arXiv 2023, arXiv:2302.06590. [Google Scholar] [CrossRef]
- Song, F.; Agarwal, A.; Wen, W. The impact of generative AI on collaborative open-source software development: Evidence from GitHub Copilot. arXiv 2024, arXiv:2410.02091. [Google Scholar] [CrossRef]
- Zhong, L.; Wang, Z.; Shang, J. Debug like a human: A large language model debugger via verifying runtime execution step-by-step. arXiv 2024, arXiv:2402.16906. [Google Scholar] [CrossRef]
- Levin, K.; van Kempen, N.; Berger, E.D.; Freund, S.N. Chatdbg: An ai-powered debugging assistant. arXiv 2024, arXiv:2403.16354. [Google Scholar] [CrossRef]
- Lee, C.; Xia, C.S.; Yang, L.; Huang, J.t.; Zhu, Z.; Zhang, L.; Lyu, M.R. A unified debugging approach via llm-based multi-agent synergy. arXiv 2024, arXiv:2404.17153. [Google Scholar] [CrossRef]
- Majdoub, Y.; Ben Charrada, E. Debugging with open-source large language models: An evaluation. In Proceedings of the Proceedings of the 18th ACM/IEEE International Symposium on Empirical Software Engineering and Measurement, 2024; pp. 510–516.
- Yan, J.; Huang, J.; Fang, C.; Yan, J.; Zhang, J. Better debugging: Combining static analysis and llms for explainable crashing fault localization. arXiv 2024, arXiv:2408.12070. [Google Scholar] [CrossRef]
- Jiang, N.; Li, X.; Wang, S.; Zhou, Q.; Hossain, S.; Ray, B.; Kumar, V.; Ma, X.; Deoras, A. LeDex: Training LLMs to Better Self-Debug and Explain Code. Advances in Neural Information Processing Systems 2024, 37, 35517–35543. [Google Scholar]
- Yang, W.; Wang, H.; Liu, Z.; Li, X.; Yan, Y.; Wang, S.; Gu, Y.; Yu, M.; Liu, Z.; Yu, G. Enhancing the code debugging ability of llms via communicative agent based data refinement. arXiv 2024, arXiv:2408.05006. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Chakraborty, M.; Palepu, K.N.; Pandey, V.; Dindorkar, I.; Rajpurohit, R.; Gupta, R. Exploring large language models for code explanation. arXiv 2023, arXiv:2310.16673. [Google Scholar] [CrossRef]
- Arakelyan, S.; Das, R.J.; Mao, Y.; Ren, X. Exploring distributional shifts in large language models for code analysis. arXiv 2023, arXiv:2303.09128. [Google Scholar] [CrossRef]
- Li, D.; Shen, Y.; Jin, R.; Mao, Y.; Wang, K.; Chen, W. Generation-augmented query expansion for code retrieval. arXiv 2022, arXiv:2212.10692. [Google Scholar] [CrossRef]
- Thakur, S.; Blocklove, J.; Pearce, H.; Tan, B.; Garg, S.; Karri, R. Autochip: Automating hdl generation using llm feedback. arXiv 2023, arXiv:2311.04887. [Google Scholar] [CrossRef]
- Liu, M.; Tsai, Y.D.; Zhou, W.; Ren, H. CraftRTL: High-quality Synthetic Data Generation for Verilog Code Models with Correct-by-Construction Non-Textual Representations and Targeted Code Repair. arXiv 2024, arXiv:2409.12993. [Google Scholar] [CrossRef]
- Collini, L.; Garg, S.; Karri, R. C2HLSC: Can LLMs Bridge the Software-to-Hardware Design Gap? In Proceedings of the 2024 IEEE LLM Aided Design Workshop (LAD). IEEE; 2024; pp. 1–12. [Google Scholar]
- Xiao, W.; Putrevu, V.S.C.; Hemadri, R.V.; Garg, S.; Karri, R. PrefixLLM: LLM-aided Prefix Circuit Design. arXiv 2024, arXiv:2412.02594. [Google Scholar] [CrossRef]
- Tsai, Y.D.; Liu, M.; Ren, H. Automatically Fixing RTL Syntax Errors with Large Language Model. In Proceedings of the IEEE/ACM Design Automation Conference (DAC); 2024. [Google Scholar]
- Bai, Y.; Sohrabizadeh, A.; Qin, Z.; Hu, Z.; Sun, Y.; Cong, J. Towards a comprehensive benchmark for high-level synthesis targeted to FPGAs. Advances in Neural Information Processing Systems 2023, 36, 45288–45299. [Google Scholar]
- Sohrabizadeh, A.; Yu, C.H.; Gao, M.; Cong, J. AutoDSE: Enabling software programmers to design efficient FPGA accelerators. ACM Transactions on Design Automation of Electronic Systems (TODAES) 2022, 27, 1–27. [Google Scholar] [CrossRef]
- Abi-Karam, S.; Sarkar, R.; Seigler, A.; Lowe, S.; Wei, Z.; Chen, H.; Rao, N.; John, L.; Arora, A.; Hao, C. HLSFactory: A Framework Empowering High-Level Synthesis Datasets for Machine Learning and Beyond. In Proceedings of the Proceedings of the 2024 ACM/IEEE International Symposium on Machine Learning for CAD, 2024; pp. 1–9.
- Xu, K.; Zhang, G.L.; Yin, X.; Zhuo, C.; Schlichtmann, U.; Li, B. Automated c/c++ program repair for high-level synthesis via large language models. In Proceedings of the Proceedings of the 2024 ACM/IEEE International Symposium on Machine Learning for CAD, 2024; pp. 1–9.
- Fang, W.; Li, M.; Li, M.; Yan, Z.; Liu, S.; Xie, Z.; Zhang, H. Assertllm: Generating and evaluating hardware verification assertions from design specifications via multi-llms. arXiv 2024, arXiv:2402.00386. [Google Scholar] [CrossRef]
- Xu, K.; Sun, J.; Hu, Y.; Fang, X.; Shan, W.; Wang, X.; Jiang, Z. Meic: Re-thinking rtl debug automation using llms. arXiv 2024, arXiv:2405.06840. [Google Scholar] [CrossRef]
- Ma, R.; Yang, Y.; Liu, Z.; Zhang, J.; Li, M.; Huang, J.; Luo, G. VerilogReader: LLM-Aided Hardware Test Generation. In Proceedings of the 2024 IEEE LLM Aided Design Workshop (LAD). IEEE; 2024; pp. 1–5. [Google Scholar]
- Zhang, Z.; Chadwick, G.; McNally, H.; Zhao, Y.; Mullins, R. Llm4dv: Using large language models for hardware test stimuli generation. arXiv 2023, arXiv:2310.04535. [Google Scholar] [CrossRef]
- Qiu, R.; Zhang, G.L.; Drechsler, R.; Schlichtmann, U.; Li, B. Autobench: Automatic testbench generation and evaluation using llms for hdl design. In Proceedings of the Proceedings of the 2024 ACM/IEEE International Symposium on Machine Learning for CAD, 2024; pp. 1–10.
- Orenes-Vera, M.; Martonosi, M.; Wentzlaff, D. Using llms to facilitate formal verification of rtl. arXiv 2023, arXiv:2309.09437. [Google Scholar] [CrossRef]
- Xiong, C.; Liu, C.; Li, H.; Li, X. Hlspilot: Llm-based high-level synthesis. In Proceedings of the Proceedings of the 43rd IEEE/ACM International Conference on Computer-Aided Design, 2024; pp. 1–9.
- Liao, Y.; Adegbija, T.; Lysecky, R. Are llms any good for high-level synthesis? In Proceedings of the Proceedings of the 43rd IEEE/ACM International Conference on Computer-Aided Design; 2024; pp. 1–8. [Google Scholar]
- Liu, J.; Xia, C.S.; Wang, Y.; Zhang, L. Is your code generated by chatgpt really correct? rigorous evaluation of large language models for code generation. Advances in Neural Information Processing Systems 2023, 36, 21558–21572. [Google Scholar]
- Bareiß, P.; Souza, B.; d’Amorim, M.; Pradel, M. Code generation tools (almost) for free? a study of few-shot, pre-trained language models on code. arXiv 2022, arXiv:2206.01335. [Google Scholar] [CrossRef]
- Dong, Y.; Jiang, X.; Jin, Z.; Li, G. Self-collaboration code generation via chatgpt. ACM Transactions on Software Engineering and Methodology 2024, 33, 1–38. [Google Scholar] [CrossRef]
- Liu, C.; Bao, X.; Zhang, H.; Zhang, N.; Hu, H.; Zhang, X.; Yan, M. Improving chatgpt prompt for code generation. arXiv 2023, arXiv:2305.08360. [Google Scholar] [CrossRef]
- Chen, X.; Lin, M.; Schärli, N.; Zhou, D. Teaching Large Language Models to Self-Debug. arXiv 2023, arXiv:2304.05128. [Google Scholar] [CrossRef]
- Haque, M.A.; Li, S. The potential use of chatgpt for debugging and bug fixing. 2023. [Google Scholar] [CrossRef]
- Tian, R.; Ye, Y.; Qin, Y.; Cong, X.; Lin, Y.; Pan, Y.; Wu, Y.; Hui, H.; Liu, W.; Liu, Z.; et al. Debugbench: Evaluating debugging capability of large language models. arXiv 2024, arXiv:2401.04621. [Google Scholar] [CrossRef]
- Kang, S.; Chen, B.; Yoo, S.; Lou, J.G. Explainable automated debugging via large language model-driven scientific debugging. Empirical Software Engineering 2025, 30, 1–28. [Google Scholar] [CrossRef]
- Thakur, S.; Ahmad, B.; Pearce, H.; Tan, B.; Dolan-Gavitt, B.; Karri, R.; Garg, S. Verigen: A large language model for verilog code generation. ACM Transactions on Design Automation of Electronic Systems 2024, 29, 1–31. [Google Scholar] [CrossRef]
- Takamaeda-Yamazaki, S. Pyverilog: A python-based hardware design processing toolkit for verilog hdl. In Proceedings of the Applied Reconfigurable Computing: 11th International Symposium, ARC 2015, Bochum, Germany, April 13-17, 2015, Proceedings 11. Springer, 2015, pp. 451–460.
- Blocklove, J.; Garg, S.; Karri, R.; Pearce, H. Chip-chat: Challenges and opportunities in conversational hardware design. In Proceedings of the 2023 ACM/IEEE 5th Workshop on Machine Learning for CAD (MLCAD). IEEE; 2023; pp. 1–6. [Google Scholar]
- Nakkab, A.; Zhang, S.Q.; Karri, R.; Garg, S. Rome was Not Built in a Single Step: Hierarchical Prompting for LLM-based Chip Design. 2024. [Google Scholar] [CrossRef]
- Chang, K.; Wang, K.; Yang, N.; Wang, Y.; Jin, D.; Zhu, W.; Chen, Z.; Li, C.; Yan, H.; Zhou, Y.; et al. Data is all you need: Finetuning llms for chip design via an automated design-data augmentation framework. In Proceedings of the Proceedings of the 61st ACM/IEEE Design Automation Conference, 2024; pp. 1–6.
- Vijayaraghavan, P.; Nitsure, A.; Mackin, C.; Shi, L.; Ambrogio, S.; Haran, A.; Paruthi, V.; Elzein, A.; Coops, D.; Beymer, D.; et al. Chain-of-Descriptions: Improving Code LLMs for VHDL Code Generation and Summarization. In Proceedings of the Proceedings of the 2024 ACM/IEEE International Symposium on Machine Learning for CAD, 2024; pp. 1–10.
- Batten, C.; Pinckney, N.; Liu, M.; Ren, H.; Khailany, B. PyHDL-Eval: An LLM Evaluation Framework for Hardware Design Using Python-Embedded DSLs. In Proceedings of the Proceedings of the 2024 ACM/IEEE International Symposium on Machine Learning for CAD, 2024; pp. 1–17.
- Fu, Y.; Zhang, Y.; Yu, Z.; Li, S.; Ye, Z.; Li, C.; Wan, C.; Lin, Y.C. Gpt4aigchip: Towards next-generation ai accelerator design automation via large language models. In Proceedings of the 2023 IEEE/ACM International Conference on Computer Aided Design (ICCAD). IEEE; 2023; pp. 1–9. [Google Scholar]
- Latibari, B.S.; Ghimire, S.; Chowdhury, M.A.; Nazari, N.; Gubbi, K.I.; Homayoun, H.; Sasan, A.; Salehi, S. Automated Hardware Logic Obfuscation Framework Using GPT. In Proceedings of the 2024 IEEE 17th Dallas Circuits and Systems Conference (DCAS). IEEE; 2024; pp. 1–5. [Google Scholar]
- Wan, L.J.; Huang, Y.; Li, Y.; Ye, H.; Wang, J.; Zhang, X.; Chen, D. Software/hardware co-design for llm and its application for design verification. In Proceedings of the 2024 29th Asia and South Pacific Design Automation Conference (ASP-DAC). IEEE; 2024; pp. 435–441. [Google Scholar]
- Gai, J.; Chen, H.; Wang, Z.; Zhou, H.; Zhao, W.; Lane, N.; Fan, H. Exploring Code Language Models for Automated HLS-based Hardware Generation: Benchmark, Infrastructure and Analysis. In Proceedings of the Proceedings of the 30th Asia and South Pacific Design Automation Conference, 2025, pp.
- Hendrycks, D.; Basart, S.; Kadavath, S.; Mazeika, M.; Arora, A.; Guo, E.; Burns, C.; Puranik, S.; He, H.; Song, D.; et al. Measuring Coding Challenge Competence With APPS. NeurIPS 2021. [Google Scholar]
- Lu, S.; Guo, D.; Ren, S.; Huang, J.; Svyatkovskiy, A.; Blanco, A.; Clement, C.B.; Drain, D.; Jiang, D.; Tang, D.; et al. CodeXGLUE: A Machine Learning Benchmark Dataset for Code Understanding and Generation. CoRR 2021, arXiv:2102.04664. [Google Scholar] [CrossRef]
- Li, Y.; Choi, D.; Chung, J.; Kushman, N.; Schrittwieser, J.; Leblond, R.; Eccles, T.; Keeling, J.; Gimeno, F.; Lago, A.D.; et al. Competition-level code generation with AlphaCode. Science 2022, 378, 1092–1097. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, W.; Wang, Y.; Liu, J.K.; Sun, H.; Wu, S.; Hu, J.; Xu, X. LeetCodeDataset: A Temporal Dataset for Robust Evaluation and Efficient Training of Code LLMs. arXiv 2025, arXiv:2504.14655. [Google Scholar] [CrossRef]
- Zhong, V.; Xiong, C.; Socher, R. Seq2sql: Generating structured queries from natural language using reinforcement learning. arXiv 2017, arXiv:1709.00103. [Google Scholar] [CrossRef]
- Aider Blog Team. The Polyglot Benchmark. https://aider.chat/2024/12/21/polyglot.html#the-polyglot-benchmark, 2024.
- Jimenez, C.E.; Yang, J.; Wettig, A.; Yao, S.; Pei, K.; Press, O.; Narasimhan, K. Swe-bench: Can language models resolve real-world github issues? arXiv 2023, arXiv:2310.06770. [Google Scholar] [CrossRef]
- Just, R.; Jalali, D.; Ernst, M.D. Defects4J: A database of existing faults to enable controlled testing studies for Java programs. In Proceedings of the Proceedings of the 2014 international symposium on software testing and analysis, 2014; pp. 437–440.
- Lin, D.; Koppel, J.; Chen, A.; Solar-Lezama, A. QuixBugs: A multi-lingual program repair benchmark set based on the Quixey Challenge. In Proceedings of the Proceedings Companion of the 2017 ACM SIGPLAN international conference on systems, programming, languages, and applications: software for humanity, 2017; pp. 55–56.
- Widyasari, R.; Sim, S.Q.; Lok, C.; Qi, H.; Phan, J.; Tay, Q.; Tan, C.; Wee, F.; Tan, J.E.; Yieh, Y.; et al. Bugsinpy: a database of existing bugs in python programs to enable controlled testing and debugging studies. In Proceedings of the Proceedings of the 28th ACM joint meeting on european software engineering conference and symposium on the foundations of software engineering, 2020; pp. 1556–1560.
- Liu, S.; Chai, L.; Yang, J.; Shi, J.; Zhu, H.; Wang, L.; Jin, K.; Zhang, W.; Zhu, H.; Guo, S.; et al. Mdeval: Massively multilingual code debugging. arXiv 2024, arXiv:2411.02310. [Google Scholar] [CrossRef]
- Husain, H.; Wu, H.H.; Gazit, T.; Allamanis, M.; Brockschmidt, M. CodeSearchNet challenge: Evaluating the state of semantic code search. arXiv 2019, arXiv:1909.09436. [Google Scholar] [CrossRef]
- Nguyen, D.M.; Phan, T.C.; Le Hai, N.; Doan, T.T.; Nguyen, N.V.; Pham, Q.; Bui, N.D. CodeMMLU: A Multi-Task Benchmark for Assessing Code Understanding & Reasoning Capabilities of CodeLLMs. In Proceedings of the The Thirteenth International Conference on Learning Representations.
- Lu, Y.; Liu, S.; Zhang, Q.; Xie, Z. Rtllm: An open-source benchmark for design rtl generation with large language model. In Proceedings of the 2024 29th Asia and South Pacific Design Automation Conference (ASP-DAC). IEEE; 2024; pp. 722–727. [Google Scholar]
- Liu, M.; Pinckney, N.; Khailany, B.; Ren, H. Verilogeval: Evaluating large language models for verilog code generation. In Proceedings of the 2023 IEEE/ACM International Conference on Computer Aided Design (ICCAD). IEEE; 2023; pp. 1–8. [Google Scholar]
- Zhang, Y.; Yu, Z.; Fu, Y.; Wan, C.; Lin, Y.C. Mg-verilog: Multi-grained dataset towards enhanced llm-assisted verilog generation. In Proceedings of the 2024 IEEE LLM Aided Design Workshop (LAD). IEEE; 2024; pp. 1–5. [Google Scholar]
- Liu, S.; Lu, Y.; Fang, W.; Li, M.; Xie, Z. Openllm-rtl: Open dataset and benchmark for llm-aided design rtl generation. In Proceedings of the Proceedings of the 43rd IEEE/ACM International Conference on Computer-Aided Design, 2024; pp. 1–9.
- Wu, Y.; Yu, X.; Chen, H.; Luo, Y.; Tong, Y.; Ma, Y. PICBench: Benchmarking LLMs for Photonic Integrated Circuits Design. arXiv 2025, arXiv:2502.03159. [Google Scholar] [CrossRef]
- Jin, H.; Huang, L.; Cai, H.; Yan, J.; Li, B.; Chen, H. From llms to llm-based agents for software engineering: A survey of current, challenges and future. arXiv 2024, arXiv:2408.02479. [Google Scholar] [CrossRef]
- Kochar, D.V.; Wang, H.; Chandrakasan, A.; Zhang, X. LEDRO: LLM-Enhanced Design Space Reduction and Optimization for Analog Circuits. arXiv 2024, arXiv:2411.12930. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, F.; Shen, J.; Kim, S.; Kim, S. A survey on large language models for code generation. arXiv 2024, arXiv:2406.00515. [Google Scholar] [CrossRef]
- Chui, M.; Roberts, R.; Yee, L.; Hazan, E.; Singla, A.; Smaje, K.; Sukharevsky, A.; Zemmel, R. The Economic Potential of Generative AI: The Next Productivity Frontier. Technical report, McKinsey & Company, 2023.
- Mann, T. China’s DeepSeek just emitted a free challenger to OpenAI’s o1 – here’s how to use it on your PC. The Register, 2025.
- Hanbury, P.; Wang, J.; Brick, P.; Cannarsi, A. DeepSeek: A game changer in AI efficiency? Industry brief, Bain & Company, 2025.
- Masri, S.; Ashqar, H.I.; Elhenawy, M. Visual Reasoning at Urban Intersections: Fine-Tuning GPT-4o for Traffic Conflict Detection. arXiv 2025, arXiv:2502.20573. [Google Scholar] [CrossRef]
- Lai, W.; Mesgar, M.; Fraser, A. LLMs Beyond English: Scaling the Multilingual Capability of LLMs with Cross-Lingual Feedback. arXiv 2024, arXiv:2406.01771. [Google Scholar] [CrossRef]
- Warren, T. Microsoft makes OpenAI’s o1 reasoning model free for all Copilot users. The Verge, 2025.
- Garcez, A.d.; Lamb, L.C. Neurosymbolic ai: The 3 rd wave. Artificial Intelligence Review 2023, 56, 12387–12406. [Google Scholar] [CrossRef]
- Lee, H.; Phatale, S.; Mansoor, H.; Lu, K.R.; Mesnard, T.; Ferret, J.; Bishop, C.; Hall, E.; Carbune, V.; Rastogi, A. Rlaif: Scaling reinforcement learning from human feedback with ai feedback.
- Li, Z.; Fan, Z.; Tou, H.; Chen, J.; Wei, Z.; Huang, X. Mvptr: Multi-level semantic alignment for vision-language pre-training via multi-stage learning. In Proceedings of the Proceedings of the 30th ACM International Conference on Multimedia, 2022; pp. 4395–4405.
- Guo, D.; et al. DeepSeek-R1: Incentivizing reasoning capability in LLMs via reinforcement learning. arXiv 2025, arXiv:2501.12948. [Google Scholar] [CrossRef]
- Su, Y.; et al. Crossing the Reward Bridge: Expanding RL with Verifiable Rewards Across Diverse Domains. arXiv 2025, arXiv:2503.23829. [Google Scholar] [CrossRef]
- Pan, Y.; Feng, Y.; Zhuang, J.; Ding, S.; Zhou, Z.; Sun, B.; Xu, H.; Chou, Y.; Deng, A.; Hu, A.; et al. SpikingBrain Technical Report: Brain-Inspired Large Models for Efficient Long-Context Training and Inference. arXiv 2025, arXiv:2509.05276. [Google Scholar] [CrossRef]
- WhatAboutMyStar.; colleagues. The LLM Language Network: Functional Brain Networks in LLMs. In Proceedings of the Proceedings of the International Conference on Machine Learning (ICML), 2025.
- Alkhamissi, B.; Schrimpf, M.; et al. The LLM Language Network: A Neuroscientific Approach to Identifying Language-Selective Units in LLMs. In Proceedings of the Proceedings of the 2025 Conference of the North American Chapter of the Association for Computational Linguistics (NAACL); 2025. [Google Scholar]
- Wu, T.; et al. Continual Learning for Large Language Models: A Survey. arXiv 2024, arXiv:2402.01364. [Google Scholar] [CrossRef]
- Zhu, Y.; et al. MemoRAG: Boosting Long Context Processing with Global Memory-Augmented Retrieval. arXiv 2025, arXiv:2409.05591. [Google Scholar] [CrossRef]
- Pan, Y.; Feng, Y.; Zhuang, J.; Ding, S.; Zhou, Z.; Sun, B.; Xu, H.; Chou, Y.; Deng, A.; Hu, A.; et al. SpikingBrain Technical Report: Brain-Inspired Large Models for Efficient Long-Context Training and Inference. arXiv 2025, arXiv:2509.05276. [Google Scholar] [CrossRef]
- Gu, A.; Goel, K.; et al. Linear-Time Sequence Modeling with Selective State Spaces (Mamba). arXiv 2024, arXiv:2312.00752. [Google Scholar] [CrossRef]
- Hu, E.J.; Shen, Y.; Wallis, P.; Allen-Zhu, Z.; Li, Y.; Wang, S.; Wang, L.; Chen, W.; et al. Lora: Low-rank adaptation of large language models. ICLR 2022, 1, 3. [Google Scholar]
- Yang, H.; Yue, S.; He, Y. Auto-gpt for online decision making: Benchmarks and additional opinions. arXiv 2023, arXiv:2306.02224. [Google Scholar] [CrossRef]
- Salesforce. xLAM enters its next era: The evolution of Large Action Models. Salesforce Blog, 2025.
- Zhang, L.; He, S.; Zhang, C.; et al. SWE-bench Goes Live! arXiv 2025. [Google Scholar] [CrossRef]
- Gao, L.; Madaan, A.; Zhou, S.; Alon, U.; Liu, P.; Yang, Y.; Callan, J.; Neubig, G. Pal: Program-aided language models. In Proceedings of the International Conference on Machine Learning. PMLR; 2023; pp. 10764–10799. [Google Scholar]
- Meding, K. It’s Complicated: The Relationship of Algorithmic Fairness and Non-Discrimination Regulations in the EU AI Act. arXiv 2025, arXiv:2501.12962. [Google Scholar] [CrossRef]
- Board, E.D.P. AI Privacy Risks & Mitigations — Large Language Models (LLMs). Technical report, EDPB, 2025.
- LLP, C. The EU AI Act: Key Milestones, Compliance Challenges and the Road Ahead. Legal Commentary, 2025.
- OWASP. OWASP Top 10 for Large Language Model Applications 2025. OWASP GenAI Security Project, 2025.
- MacCormick, D.N.; Summers, R.S. Interpreting statutes: A comparative study; Routledge, 2016. [Google Scholar]
- Nicolini, D.; Mengis, J.; Swan, J. Understanding the role of objects in cross-disciplinary collaboration. Organization science 2012, 23, 612–629. [Google Scholar] [CrossRef]
- Brock, M.E.; Cannella-Malone, H.I.; Seaman, R.L.; Andzik, N.R.; Schaefer, J.M.; Page, E.J.; Barczak, M.A.; Dueker, S.A. Findings across practitioner training studies in special education: A comprehensive review and meta-analysis. Exceptional Children 2017, 84, 7–26. [Google Scholar] [CrossRef]
- Argyle, M.; et al. Out of One, Many: Using LMs to Simulate Human Samples. Political Analysis 2023, 31, 550–569. [Google Scholar] [CrossRef]
- Lane, C.; Rollnick, S. The use of simulated patients and role-play in communication skills training: a review of the literature to August 2005. Patient education and counseling 2007, 67, 13–20. [Google Scholar] [CrossRef]
- Tseng, Y.M.; Huang, Y.C.; Hsiao, T.Y.; Chen, W.L.; Huang, C.W.; Meng, Y.; Chen, Y.N. Two tales of persona in llms: A survey of role-playing and personalization. arXiv 2024, arXiv:2406.01171. [Google Scholar] [CrossRef]
- Louie, R.; Nandi, A.; Fang, W.; Chang, C.; Brunskill, E.; Yang, D. Roleplay-doh: Enabling domain-experts to create llm-simulated patients via eliciting and adhering to principles. arXiv 2024, arXiv:2407.00870. [Google Scholar] [CrossRef]
- Shao, Y.; Li, L.; Dai, J.; Qiu, X. Character-llm: A trainable agent for role-playing. arXiv 2023, arXiv:2310.10158. [Google Scholar] [CrossRef]
- Xiang, L.; Zhao, Y.; Zhang, Y.; Zong, C. A Survey of Large Language Models in Discipline-specific Research: Challenges, Methods and Opportunities. Studies in Informatics and Control 2025, 34, 1. [Google Scholar] [CrossRef]
- Wang, W.; et al. Python Symbolic Execution with LLM-empowered SMT Solving. arXiv 2024, arXiv:2409.09271. [Google Scholar] [CrossRef]
- Corbin, A.L. Legal analysis and terminology. Yale Lj 1919, 29, 163. [Google Scholar] [CrossRef]
- Shen, J.; Tenenholtz, N.; Hall, J.B.; Alvarez-Melis, D.; Fusi, N. Tag-LLM: Repurposing general-purpose LLMs for specialized domains. arXiv 2024, arXiv:2402.05140. [Google Scholar] [CrossRef]
- Singh, C.; Inala, J.P.; Galley, M.; Caruana, R.; Gao, J. Rethinking interpretability in the era of large language models. arXiv 2024, arXiv:2402.01761. [Google Scholar] [CrossRef]
- Cai, S.; Huang, A.; Li, J. The Impact of Embedded Question Prompts on Students’ Reflective Thinking and Learning Behaviors in AR Learning Environments. Journal of Science Education and Technology 2025, 1–19. [Google Scholar] [CrossRef]
- Chung, T.; Yang, C.J. Legal Document RAG: Multi-Graph Multi-Agent Recursive Retrieval through Legal Clauses. https://medium.com/enterprise-rag/legal-document-rag-multi-graph-multi-agent-recursive-retrieval-through-legal-clauses-c90e073e0052, 2024. Accessed: 2025-06-01.
- Xu, L.; et al. Better Aligned with Survey Respondents or Training Data? arXiv 2025, arXiv:2502.18282. [Google Scholar] [CrossRef]
- Potter, D.; et al. Hidden Persuaders: LLMs’ Political Leaning and Influence on Voters. In Proceedings of the Proceedings of EMNLP, 2024; pp. 47–63.
- Li, H.; Dong, Q.; Chen, J.; Su, H.; Zhou, Y.; Ai, Q.; Ye, Z.; Liu, Y. Llms-as-judges: a comprehensive survey on llm-based evaluation methods. arXiv 2024, arXiv:2412.05579. [Google Scholar] [CrossRef]
- Dai, J.; Pan, X.; Sun, R.; Ji, J.; Xu, X.; Liu, M.; Wang, Y.; Yang, Y. Safe rlhf: Safe reinforcement learning from human feedback. arXiv 2023, arXiv:2310.12773. [Google Scholar] [CrossRef]
- Institute of International Finance.; Ernst & Young. 2024 IIF-EY Survey Report on AI/ML Use in Financial Services. Technical report, Institute of International Finance, 2025. Accessed: 2025-05-12.
- Kamalnath, V.; Lerner, L.; Moon, J.; Sari, G.; Sohoni, V.; Zhang, S. Capturing the Full Value of Generative AI in Banking. McKinsey & Company 2023. Accessed: 2025-05-12.
- Maple, C.; Sabuncuoglu, A.; Szpruch, L.; Elliot, A.; Reinert, G.; Zemaitis, T. The Impact of Large Language Models in Finance: Towards Trustworthy Adoption. Technical report, The Alan Turing Institute, 2024. Accessed: 2025-05-12.
- Strickland, B. Gravitating to gen AI: CPA leaders show increased interest. Journal of Accountancy 2024. [Google Scholar]
- Sukharevsky, A.; Ess, A.; Emelyantsev, D.; Reasor, E.; Hürtgen, H.; Sokolov, O.; Kondratyuk, S. LLM to ROI: How to Scale Gen AI in Retail. McKinsey & Company 2024. Accessed: 2025-05-12.
- Tao, W.; Xing, X.; Chen, Y.; Huang, L.; Xu, X. TreeRAG: Unleashing the Power of Hierarchical Storage for Enhanced Knowledge Retrieval in Long Documents. In Proceedings of the Findings of the Association for Computational Linguistics: ACL 2025, 2025; pp. 356–371.
- Kiciman, E.; Ness, R.; Sharma, A.; Tan, C. Causal reasoning and large language models: Opening a new frontier for causality. Transactions on Machine Learning Research 2023. [Google Scholar]
- Chen, W.; Ma, X.; Wang, X.; Cohen, W.W. Program of thoughts prompting: Disentangling computation from reasoning for numerical reasoning tasks. arXiv 2022, arXiv:2211.12588. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, X.; Tian, Y.; Zhao, J. Deep think with confidence. arXiv 2025, arXiv:2508.15260. [Google Scholar] [CrossRef]
- Claridge, T.D. High-resolution NMR techniques in organic chemistry; Vol. 27, Elsevier, 2016.
- Aksenov, A.A.; da Silva, R.; Knight, R.; Lopes, N.P.; Dorrestein, P.C. Global chemical analysis of biology by mass spectrometry. Nature Reviews Chemistry 2017, 1, 0054. [Google Scholar] [CrossRef]
- Espenson, J.H.; et al. Chemical kinetics and reaction mechanisms; Vol. 2, Citeseer, 1995.
- Fogler, H.S. Essentials of chemical reaction engineering: essenti chemica reactio engi; Pearson education, 2010. [Google Scholar]
- Coley, C.W.; Jin, W.; Rogers, L.; Jamison, T.F.; Jaakkola, T.S.; Green, W.H.; Barzilay, R.; Jensen, K.F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chemical science 2019, 10, 370–377. [Google Scholar] [CrossRef]
- Xie, Z. Chemical Space Exploration via Large-Language-Model and Bayesian Optimization. PhD thesis, University of Liverpool, 2024.
- Liu, Y.; Kashima, H. Chemical property prediction under experimental biases. Scientific Reports 2022, 12, 8206. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, K.; Taly, A.; Monti, F.; Brenner, M.P.; Colwell, L.J. Using attribution to decode binding mechanism in neural network models for chemistry. Proceedings of the National Academy of Sciences 2019, 116, 11624–11629. [Google Scholar] [CrossRef] [PubMed]
- Berreziga, R.; Brahimi, M.; Kraim, K.; Azzoune, H. Combining GCN Structural Learning with LLM Chemical Knowledge for or Enhanced Virtual Screening. arXiv 2025, arXiv:2504.17497. [Google Scholar] [CrossRef]
- Xie, J.; Wang, W.; Gao, B.; Yang, Z.; Wan, H.; Zhang, S.; Fu, T.; Li, Y. QCBench: Evaluating Large Language Models on Domain-Specific Quantitative Chemistry. arXiv 2025, arXiv:2508.01670. [Google Scholar] [CrossRef]
- Krenn, M.; et al. SELFIES and the future of molecular string representations. Patterns 2022. [Google Scholar] [CrossRef]
- Zeng, Z.; Yin, B.; Wang, S.; Liu, J.; Yang, C.; Yao, H.; Sun, X.; Sun, M.; Xie, G.; Liu, Z. ChatMol: interactive molecular discovery with natural language. Bioinformatics 2024, 40, btae534. [Google Scholar] [CrossRef]
- Yan, C.; Fan, X.; Fan, J.; Yu, L.; Wang, N.; Chen, L.; Li, X. Hyformer: Hybrid transformer and cnn for pixel-level multispectral image land cover classification. International Journal of Environmental Research and Public Health 2023, 20, 3059. [Google Scholar] [CrossRef]
- Batzner, S.; et al. E(3)-equivariant graph neural networks for data-efficient interatomic potentials. Nature Communications 2022. [Google Scholar] [CrossRef]
- Xu, Y.; et al. Pretrained E(3)-equivariant message-passing neural networks for fast spectral prediction. npj Computational Materials 2025. [Google Scholar] [CrossRef]
- Anonymous. Instantiation-based Formalization of Logical Reasoning via Semantic Self-Verification. arXiv 2025, arXiv:2501.16961. [CrossRef]
- Tang, X.; Yu, Z.; Chen, J.; Cui, Y.; Shao, D.; Wang, W.; Wu, F.; Zhuang, Y.; Shi, W.; Huang, Z.; et al. CellForge: Agentic Design of Virtual Cell Models. arXiv 2025, arXiv:2508.02276. [Google Scholar] [CrossRef]
- Bunne, C.; Roohani, Y.; Rosen, Y.; Gupta, A.; Zhang, X.; Roed, M.; Alexandrov, T.; AlQuraishi, M.; Brennan, P.; Burkhardt, D.B.; et al. How to build the virtual cell with artificial intelligence: Priorities and opportunities. Cell 2024, 187, 7045–7063. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Key milestones in the development of LLMs.
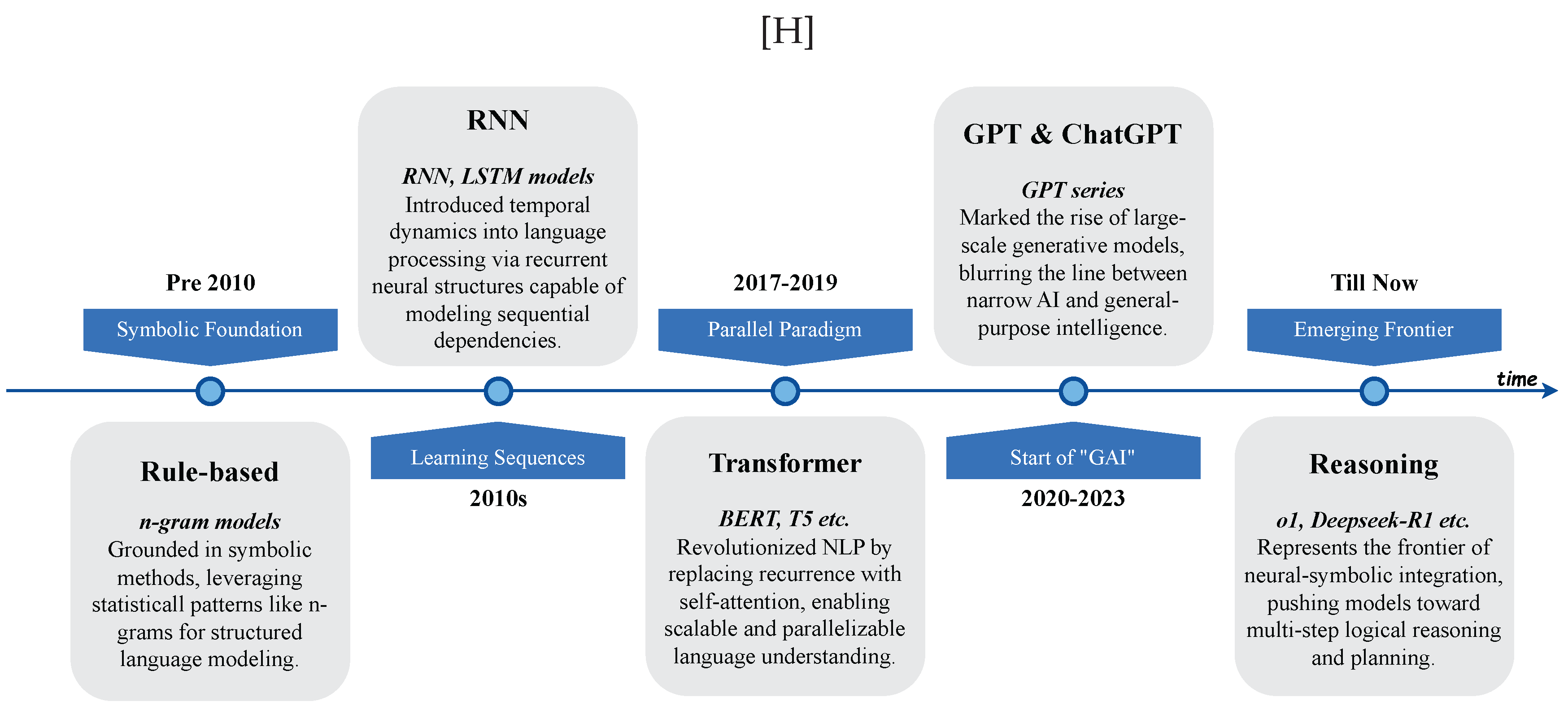
Figure 2.
The model architecture of Transformer (Figure source: the original paper [4]).
Figure 2.
The model architecture of Transformer (Figure source: the original paper [4]).
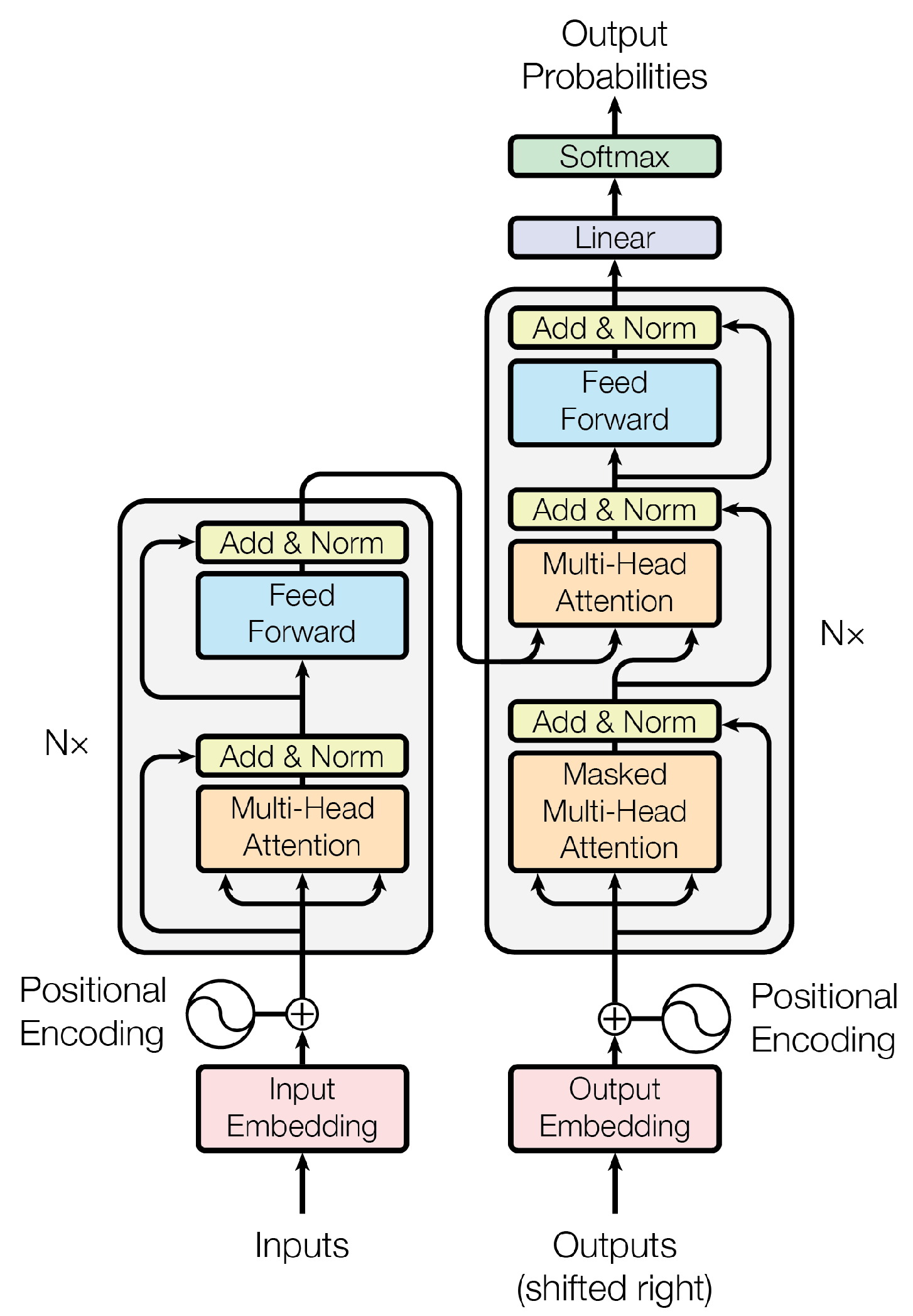
Figure 17.
The relationships between major research tasks in chemistry and chemical engineering.
Table 2.
Existing SOTA LLMs on benchmark datasets.
| Family | Model | Context | Output | Input | Cost ($/1M Tok) | Infer. Time | Strengths | Provider | ||
| Name | Window | Size | Modal | Input | Output | TPS | TFT | |||
| GPT 4 [1,64] | GPT-4o | 128K | 16,384 | 2.50 | 10.00 | 209.4 | 0.35 | High TPS, General-Purpose Tasks | OpenAI | |
| GPT-4o mini | 128K | 16,384 | 0.15 | 0.60 | 74.3 | 0.35 | Fine-tune / Distill, Focused Tasks | |||
| ChatGPT-4o | 128K | 16,384 | 5.00 | 15.00 | 214.0 | 0.81 | Chat-based Applications | |||
| GPT-4.5 | 128K | 16,384 | T, I | 75.00 | 150.00 | 11.5 | 1.47 | Complex / Creative Tasks | ||
| GPT 5 [65] | GPT-5 | 400K | 128K | 1.25 | 10.00 | 136.0 | 1.03 | Coding, Reasoning, and Agentic Tasks. | ||
| GPT-5-mini | 400K | 128K | 0.25 | 2.00 | 73.0 | 0.98 | Well-defined Tasks / Precise Prompts. | OpenAI | ||
| GPT-5-nano | 400K | 128K | 0.05 | 0.40 | 210.0 | 0.86 | Summarization / Classification Tasks. | |||
| OpenAI o1 | 200K | 100K | T, I | 15 | 60 | 108.9 | 25.56 | Scalable for Reasoning Tasks | OpenAI | |
| OpenAI | OpenAI o1-mini | 128K | 65,536 | T | 1.1 | 4.4 | 221.7 | 10.30 | Affordable Reasoning | |
| Reasoning | OpenAI o1-pro | 200K | 100K | T, I | 150 | 600 | – | – | – | |
| [20] | OpenAI o3-mini | 200K | 100K | T | 1.1 | 4.4 | 194.2 | 13.95 | Func. Call, Batch Process | |
| Claude 3 [66,67] | Claude 3 Opus | 200K | 4,096 | T, I, A | 15.00 | 75.00 | 26.8 | 1.22 | Multilingual, Tool Use | Anthropic |
| Claude 3.5 Haiku | 8,192 | T, I, A | 0.80 | 4.00 | 65.9 | 0.62 | Fast Response, Multilingual | |||
| Claude 3.5 Sonnet | 8,192 | T, I, A | 3.00 | 15.00 | 78.9 | 0.87 | General Tasks, Coding | |||
| Claude 3.7 Sonnet | 8,192 | T | 3.00 | 15.00 | 77.7 | 0.73 | Toggleable Extended Thinking | |||
| Gemini 2 [68] | Gemini 2.0 Flash | 1,000K | 8,192 | T, I, A | 0.10 | 0.40 | 257.0 | 0.34 | High TPS, Long Context Window | |
| Gemini 2.0 Flash Lite | 8,192 | T, I, A | 0.075 | 0.30 | 207.3 | 0.24 | Cost-Efficient, Fast TFT | |||
| Gemini 2.0 Flash TK | 8,192 | T, I, A | 0.10 | 0.40 | – | – | – | |||
| Gemini 2.5 Pro | 65,536 | T, I, A | – | – | 168.2 | 33.69 | Visual Reasoning | |||
| Family | Model Name | Model Size | Context Window | Input Modal | Strengths | License | Provider | |||
| GPT-OOS [69] | GPT-oos-20B | 21B | 131K | T | Reasoning and Agentic Tasks | Apache 2.0 | OpenAI | |||
| GPT-oss-120B | 117B | |||||||||
| Qwen 2 [70,71,72] | Qwen 2 | 0.5B - 72B | 131,072 | T | Previous Flagship Models | Alibaba | ||||
| Qwen 2.5 | 0.5B - 72B | 128K | T | General-Purpose Tasks | Apache | |||||
| Qwen 2.5-1M | 7B, 14B | 1M | T | Long Context Tasks | License | |||||
| QwQ | 32B | 131,072 | T | Reasoning and QA Tasks | ||||||
| DeepSeek [21,73] | DeepSeek-V3 | 671B | 128K | T | MoE, Math | MIT License | DeepSeek | |||
| DeepSeek-R1 | 671B | T | Complex Reasoning Tasks | |||||||
| DeepSeek-R1 Distill | 1.5B - 70B | T | Efficient Reasoning Inference | |||||||
| Llama 3 [74] | Llama 3.1 | 8B, 70B, 405B | 128K | T | Multilingual, Text Generation | Custom License | Meta AI | |||
| Llama 3.2 LW | 1B, 3B | T | Efficient Inference | |||||||
| Llama 3.2 MM | 11B, 90B | T, I | Multimodal, Image Tasks | |||||||
| Llama 3.3 | 70B | T | – | |||||||
Notes: T, I, and A denote Text, Image, and Audio, respectively. For time, we report the median time for 1K tokens prompt length. TPS is the output Tokens Per Second, and TFT is the Time to First Token, which is also called latency. Gemini 2.0 Flash TK represents Gemini 2.0 Flash Thinking. All the cost information are collected from LLM provider official websites, and inference time (Infer. Time) are obtained from Artificial Analysis. The strengths of each LLM are summarized according to the corresponding model technical report, Vellum LLM Leaderboard, and benchmark reports. Dash means the information is not available
Table 4.
Existing benchmarks on evaluating the capabilities of LLMs.
| Tasks | Subtask | Relevant Benchmarks | Domain | Data Source |
|---|---|---|---|---|
| Text Understanding | Sentiment Detection | SST-2 [106] | Movie Reviews | Rotten Tomatoes |
| IMDB [107] | Movie Reviews | IMDB | ||
| Yelp Reviews [108] | Business Reviews | Yelp | ||
| Information Extraction | CoNLL-2003 (NER) [109] | News Articles | Reuters Corpus | |
| TACRED [110] | Various Domains | News and Wikipedia | ||
| OpenIE [111] | General Text | Web Sources | ||
| Relationship Understanding | Winograd Schema Challenge [112] | Pronoun Resolution | Constructed Sentences | |
| Winogrande [113] | Commonsense Reasoning | Crowdsourced Sentences | ||
| SuperGLUE [114] | Various | Multiple Sources | ||
| Summarization | CNN/DailyMail [115] | News Articles | CNN and DailyMail | |
| XSum [116] | News Articles | BBC | ||
| SAMSum [117] | Dialogues | SAMSum Corpus | ||
| Text Generation | Question Answering | SQuAD v1/v2 [118] | Wikipedia | Stanford QA Dataset |
| NaturalQuestions [119] | Google Search | Real User Queries | ||
| TriviaQA [120] | Trivia | Trivia Websites | ||
| HotpotQA [121] | Wikipedia | Crowdsourced Questions | ||
| BoolQ [122] | Various | Wikipedia | ||
| Style Transferring | GYAFC [123] | Formality | Yahoo Answers | |
| Text Completion | LAMBADA [124] | Narrative Text | BookCorpus | |
| Machine Translation | WMT 14 [125] | News Articles | Various News Sources | |
| IWSLT [126] | TED Talks | TED | ||
| FLORES-101 [127] | Low-Resource Languages | Wikipedia | ||
| Complex Reasoning | Code Generation | HumanEval [49] | Programming | Handcrafted Problems |
| MBPP [128] | Python Programming | Crowdsourced Problems | ||
| APPS [129] | Programming | Competitive Programming | ||
| CodeXGLUE [130] | Programming | Multiple Sources | ||
| Live Code Bench [131] | Programming (multi-language) | Live code execution tasks | ||
| Multi-step Inference | HotpotQA [121] | Wikipedia | Crowdsourced Questions | |
| MuSiQue [132] | Wikipedia | Crowdsourced Questions | ||
| StrategyQA [133] | Various | Crowdsourced Questions | ||
| OpenBookQA [134] | Elementary Science | OpenBookQA Dataset | ||
| MMLU [135] | Multidomain (57 subjects) | Knowledge exams | ||
| BIG-Bench [136] | Multitask | Collaborative crowdsourced tasks | ||
| BIG-Bench Hard (BBH) [137] | Hard subset of BIG-Bench | Hard tasks from BIG-Bench | ||
| Logical Reasoning | LogiQA [138] | Logical Reasoning | National Civil Servants Examination | |
| ReClor [139] | Logical Reasoning | Standardized Tests | ||
| GSM8K [140] | Grade-school math | Crowdsourced word problems | ||
| ARC [141] | Middle-school science | Standardized test questions | ||
| MATH [142] | High school + competition math | Math contests and textbook problems | ||
| Commonsense Reasoning | CommonsenseQA [143] | Commonsense Knowledge | ConceptNet | |
| HellaSwag [144] | Commonsense Inference | Activity Descriptions | ||
| PIQA [145] | Physical Commonsense | Crowdsourced Scenarios | ||
| SocialIQA [146] | Social Interactions | ATOMIC | ||
| Knowledge Utilization | Open-Domain Question Answering | NaturalQuestions [147] | Google Search | Real User Queries |
| TriviaQA [120] | Trivia | Trivia Websites | ||
| WebQuestions [148] | Freebase | Web Queries | ||
| KILT [149] | Multiple Tasks | Wikipedia | ||
| HELM [150] | Broad NLP | Suite of 42 scenarios across domains | ||
| TruthfulQA [151] | General Knowledge | Crowdsourced and known misconceptions | ||
| Tool-Augmented Reasoning | DROP [152] | Reading + arithmetic | Wikipedia passages | |
| TATQA [153] | Table QA | Financial reports + tables | ||
| ToolBench [154] | Tool-use tasks | APIs + real-world tools | ||
| API-Bank [155] | Tool-use tasks | APIs + real-world tools | ||
| Conversational Search & Retrieve | CoQA [156] | Conversational QA | Text from diverse domains | |
| QuAC [157] | QA over Wikipedia | Wikipedia articles + dialogues | ||
| MultiHopHotpotQA [121] | Multi-hop QA | Wikipedia + multi-step chains | ||
| MT Bench [158] | Multi-domain dialogue | Human-crafted multi-turn prompts |
Table 5.
The performance of popular LLMs on widely used benchmarks. We use different colors to indicate best, second-best, and third-best performance. The first column is from Chatbot Arena and the remaining columns are from Vellum LLM leaderboard.
Table 5.
The performance of popular LLMs on widely used benchmarks. We use different colors to indicate best, second-best, and third-best performance. The first column is from Chatbot Arena and the remaining columns are from Vellum LLM leaderboard.
| Model | Chatbot | MMLU | GPQA | HumanEval | Math | BFCL | MGSM | Input Cost |
|---|---|---|---|---|---|---|---|---|
| Arena | (General) | (Reasoning) | (Coding) | (Tool Use) | (Multilingual) | / 1M Token | ||
| Qwen 2.5 | 1296 | 70.20% | 49% | 88% | 85% | 61.31% | - | - |
| Grok 2 | 1288 | 87.50% | 56% | 88.40% | 76.10% | - | - | $2.00 |
| LLAMA 3.3 | 1294 | 86% | 48% | 88.40% | 77% | 77.50% | 91.10% | - |
| DeepSeek V3 | 1318 | 88.50% | 59.10% | 82.60% | 90.20% | 57.23% | 79.80% | $0.27 |
| DeepSeek R1 | 1360 | 90.80% | 71.50% | - | 97.30% | - | - | $0.55 |
| Gemini 2.0 Flash | 1355 | 76.40% | 62.10% | - | 89.70% | - | - | $1.25 |
| Gemini 1.5 Pro | 1260 | 85.90% | 46.20% | 71.90% | 67.70% | 84.35% | 88.70% | $0.10 |
| Claude 3.5 Haiku | 1236 | 65% | 41.60% | 88.10% | 69.40% | 60% | 85.60% | $0.80 |
| Claude 3.5 Sonnet | 1283 | 88.30% | 65% | 93.70% | 78.30% | 90.20% | 91.60% | $3.00 |
| GPT-4o | 1374 | 88.70% | 53.60% | 90.20% | 76.60% | 83.59% | 90.50% | $2.50 |
| GPT-4.5 | 1398 | - | - | - | - | - | - | $75.00 |
| GPT-o1 | 1351 | 91.80% | 75.70% | 92.40% | 96.40% | 66.73% | 89.30% | $15.00 |
| GPT-o3-mini | 1304 | 86.90% | 70.70% | - | 97.90% | - | 92% | $1.10 |
Table 6.
The performance of popular reasoning LLMs on challenging benchmarks. We use colors to indicate best, second-best, and third-best performance. The first column is from Chatbot Arena and the remaining columns are from Vellum LLM leaderboard.
Table 6.
The performance of popular reasoning LLMs on challenging benchmarks. We use colors to indicate best, second-best, and third-best performance. The first column is from Chatbot Arena and the remaining columns are from Vellum LLM leaderboard.
| Model | Chatbot | GPQA Diamond | SWE-bench | Tool Use | Tool Use | MMMLU | MMMU | IFEval | MATH 500 | AIME 2024 | Input Cost |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Arena | (Reasoning) | (Agent coding) | (Retail) | (Airline) | (Multilingual) | (Visual) | (Math) | / 1M Token | |||
| DeepSeek R1 | 1360 | 71.50% | 49.20% | - | - | - | - | 83.30% | 97.30% | 79.80% | $0.55 |
| Claude 3.5 Sonnet | 1283 | 65.00% | 49.00% | 71.50% | 48.80% | 82.10% | 70.40% | 90.20% | 78.00% | 16.00% | $3.00 |
| Claude 3.7 Sonnet | 1296 | 68.00% | 70.30% | 81.20% | 58.40% | 83.20% | 71.80% | 90.80% | 82.20% | 23.30% | $3.00 |
| Claude 3.7 Sonnet thinking | 1302 | 84.80% | - | - | - | 86.10% | 75% | 93.20% | 96.20% | 80.00% | $3.00 |
| Grok 3 Beta | 1404 | 84.60% | - | - | - | - | 78.00% | - | - | 93.30% | $5.00 |
| OpenAI o3-mini (High) | 1325 | 79.70% | 49.30% | - | - | 79.50% | - | - | 97.90% | 87.30% | - |
| OpenAI o1 | 1351 | 78.00% | 48.90% | 73.50% | 54.20% | 87.70% | 78.20% | - | 96.40% | 83.30% | $15.00 |
Table 9.
Curated Benchmarks and Resources for Philosophy and Humanities
| Benchmark | Scope and Focus | Data Composition | Evaluation Tasks | Key Insights |
|---|---|---|---|---|
| Project Gutenberg [224] | Classical philosophical text access in the public domain | 75k of digitized philosophical and humanities books from major thinkers across eras | Long-form reasoning, document understanding, language modeling | Serves as foundational source for philosophy QA, argument parsing, and training philosophically informed LLMs |
| PhilPapers [225] | Bibliographic indexing and philosophical research discovery | Index of over 2.5M philosophy papers with abstracts, metadata, and categorization across subfields | Document retrieval, topic modeling, citation graph analysis | Widely used in research discovery and argument mining; useful for concept clustering and academic stance tracking |
Table 10.
Curated Benchmarks and Datasets for Political NLP
| Benchmark | Scope and Focus | Data Composition | Evaluation Tasks | Key Insights |
|---|---|---|---|---|
| POLITICS Dataset [253] | Ideology and stance detection from media coverage | 3.6M political news articles from diverse media sources (left, center, right) | Stance detection, ideology prediction, text classification | Highlights media bias and ideological polarization; useful for evaluating fairness and framing in political text models |
| U.S. Congressional Records [254] | Modeling formal political discourse and legislative language | Standardized collection of U.S. congressional transcripts and debates | Summarization, intent classification, dialogue analysis | Ideal for studying legal-parliamentary styles; enables models to learn structured argumentation in policymaking |
| American Presidency Project [255] | Presidential speech analysis and political rhetoric tracking | Public speeches, press conferences, and presidential communications from 1789 onward | Rhetorical style analysis, time-based policy comparison | Enables longitudinal study of U.S. executive discourse; supports leadership style profiling across administrations |
| Media Bias & Fact-check Dataset [256] | Detecting factual reliability and political bias in media | Fact-check ratings and bias scores from MediaBiasFactCheck and AllSides | Text labeling (bias/fact), misinformation detection | Supports training of bias-aware LLMs; widely used in political misinformation detection and source validation |
| TruthfulQA [257] | Evaluating truthful generation in politically sensitive contexts | Manually curated QA dataset focusing on false beliefs and politically sensitive facts | Question answering, hallucination detection | Used to stress-test LLMs on truthfulness and bias in politically charged content; benchmarks robustness to disinformation |
Table 15.
Overview of Benchmarks for Evaluating LLMs in the Legal Domain
| Benchmark | Scope and Focus | Data Composition | Evaluation Tasks | Key Insights |
|---|---|---|---|---|
| LegalBench [338] | Legal reasoning across 162 subtasks from diverse real-world legal contexts | Expert-authored prompts covering six reasoning types (e.g., rule recall, rule application, interpretation) | Zero-shot and few-shot reasoning, structured answer generation | Provides a fine-grained taxonomy of legal reasoning skills; reveals that even top-tier LLMs struggle with multi-step statutory logic and implicit legal inference, making it ideal for stress-testing general-purpose LLMs in high-reasoning settings |
| LawBench [339] | Legal knowledge probing for Chinese LLMs across memory, understanding, and application tiers | 20 distinct tasks, including classification, extraction, and generation; built from 90K+ QA pairs sourced from Chinese laws and judicial documents | Legal QA, clause classification, summarization, legal sentence rewriting | Designed to test hierarchical legal comprehension in Chinese LLMs; emphasizes generalization limitations across task types and uncovers alignment gaps in open-source vs. proprietary models |
| LexGLUE [340] | English legal language understanding and document classification across European legal systems | Seven curated datasets (e.g., ECtHR, EUR-Lex, SCOTUS) labeled with case outcomes, topics, and rulings | Multiclass and multilabel classification, case judgment prediction | Pioneered standardization of English-language legal NLP tasks; facilitates benchmarking across both general-purpose LMs and legal-domain-pretrained models; bridges legal and general NLP evaluation efforts |
| LAiW [341] | Comprehensive evaluation suite for Chinese legal LLMs across major task families | Tasks include judgment prediction, statute extraction, article matching, and legal QA based on Chinese court records and codes | Classification, matching, QA, retrieval, generation | Captures real-world judicial task structures in China’s legal system; reveals domain misalignment in Chinese LLMs and encourages community efforts in localized, high-quality legal datasets |
| Swiss-Judgment-Prediction [342] | Multilingual legal case outcome prediction in Swiss federal courts | Over 85K real-world legal cases in German, French, and Italian, annotated with ruling outcomes and metadata | Judgment prediction, multilingual classification, temporal generalization | Highlights challenges in cross-lingual legal learning and domain temporal drift; supports development of multilingual legal LLMs, especially for underrepresented legal languages |
| LJP-IV [343] | Fine-grained Chinese criminal verdict prediction with inclusion of “innocent” cases | Extended version of Chinese LJP dataset with new trichotomous labels (guilty, partially guilty, innocent) | Few-shot, multi-label classification, reasoning over multiple charges | Fills gap in prior LJP datasets that lacked nuanced outcome granularity; pushes models to reason over exoneration cases and subtle charge distinctions in realistic criminal scenarios |
Table 16.
LegalBench Benchmark Leaderboard (Top 10 Models)
| Rank | Model | Accuracy | Cost (In / Out) | Latency (s) |
|---|---|---|---|---|
| 1 | Gemini 2.5 Pro Exp | 83.6% | $1.25 / $10.00 | 3.51 |
| 2 | Gemini 2.5 Flash | 82.8% | $0.15 / $0.60 | 0.43 |
| 3 | o3 | 82.5% | $10.00 / $40.00 | 5.14 |
| 4 | Grok 3 Mini (High Reasoning) | 82.0% | $0.60 / $4.00 | 4.92 |
| 5 | Grok 3 Beta | 82.0% | $3.00 / $15.00 | 0.44 |
| 6 | GPT-4.1 | 81.9% | $2.00 / $8.00 | 0.42 |
| 7 | Gemini 2.5 Flash (Thinking) | 81.8% | $0.15 / $3.50 | 2.66 |
| 8 | o1 Preview | 81.7% | $15.00 / $60.00 | 10.33 |
| 9 | Grok 3 Mini (Low Reasoning) | 81.6% | $0.60 / $4.00 | 3.38 |
| 10 | DeepSeek V3 | 80.1% | $0.90 / $0.90 | 4.13 |
Table 18.
Comparison of Traditional Methods and LLMs Across Financial Tasks
| Task | Benchmark | Metric | Traditional | LLM-based | Reference | |
|---|---|---|---|---|---|---|
| Market Analysis | FOMC | F1-Score | Bi-LSTM: 53.9% | RoBERTa-Large: 71.7% | +17.8% | [459] |
| Quantitative Trading | TRADEXPERT | AR | DeepTrader: 32.5% | TradExpert: 49.8% | +17.3% | [460] |
| Financial QA | TAT-QA | EM Score | MVGE: 70.9% | TAT-LLM (70B): 81.4% | +11.4% | [461] |
| Sentiment Analysis | FLARE | Accuracy | XGBoost: 80.0% | FinMA-30B: 87.0% | +7.0% | [462] |
| Sentiment Analysis | StockEmotions | F1-Score | Bi-GRU: 76.0% | DistilBERT: 81.0% | +5.0% | [463] |
| Stock Movement Prediction | ACL18 | Accuracy | StockNet: 58.2% | PloutosGPT: 61.2% | +3.0% | [464] |
| Stock Movement Prediction | CIKM18 | Accuracy | DTML: 58.6% | PloutosGPT: 59.9% | +1.3% | [464] |
Table 20.
Collection of Evaluation Results. We use different colors to indicate best, second-best, and third-best performance.
Table 20.
Collection of Evaluation Results. We use different colors to indicate best, second-best, and third-best performance.
| Model | UCFE | FinEval | R-Judge | CFinBench | FinBen | Hirano |
|---|---|---|---|---|---|---|
| Claude 3.5-Sonnet | — | 72.90 | — | — | — | 77.02 |
| Gemini 1.5-Pro | — | 69.20 | — | — | — | 57.94 |
| Gemini 1.5-Flash | — | 65.60 | — | — | — | 63.10 |
| Gemini-Pro | — | — | — | — | — | 50.52 |
| GPT-4o | 1,117.68 | 71.90 | 74.45 | — | — | 65.26 |
| GPT-4o-mini | 901.75 | 66.20 | — | — | — | — |
| GPT-4-Turbo | — | — | — | — | 39.20 | 64.59 |
| GPT-4 | — | — | — | 55.80 | — | 66.07 |
| LLaMA-3.1-70B | 912.26 | — | — | — | 36.20 | — |
| LLaMA-3.1-8B | 1,046.87 | — | — | — | 34.30 | — |
| LLaMA-3-70B | — | — | — | 47.02 | — | 58.48 |
| LLaMA-3-8B | — | — | 61.01 | 26.61 | 25.80 | 42.13 |
| LLaMA-2-70B | — | — | — | 29.27 | — | 41.96 |
| LLaMA-2-13B | — | — | 54.80 | 30.12 | — | 40.29 |
| LLaMA-2-7B | — | — | 53.74 | 28.33 | 19.90 | 40.67 |
| Qwen2.5-72B | — | 69.40 | — | — | — | — |
| Qwen2.5-14B | 855.82 | — | — | — | — | — |
| Qwen2.5-7B | 814.48 | 62.30 | — | — | 28.30 | — |
| Qwen2-72B | — | — | — | 34.70 | — | 69.35 |
| Qwen1.5-72B | — | — | — | 56.47 | — | 59.62 |
| Qwen1.5-7B | — | — | — | 46.35 | — | 49.73 |
| Qwen-72B | — | — | — | 57.72 | — | 59.08 |
Table 21.
Raw data sources for financial tasks
| Category | Provider | Description | API | Link |
|---|---|---|---|---|
| General Financial Data | Alpaca | Provide API access to general financial data, e.g., real-time market data, historical market data, fundamental data, financial news. | 51 | https://docs.alpaca.markets/ |
| databento | 51 | https://databento.com/ | ||
| EODHD | 51 | https://eodhd.com/ | ||
| FMP | 51 | https://site.financialmodelingprep.com/ | ||
| yfinance | 51 | https://yfinance-python.org/ | ||
| Yahoo Finance | Official website of Yahoo Finance. | https://finance.yahoo.com/ | ||
| CRSP | Provide databases for economic forecasting, stock market research, and financial analysis. | https://www.crsp.org/ | ||
| Cryptocurrency Data | CoinMarketCap | Provide real-time and historical crypto market data. | 51 | https://coinmarketcap.com/api/ |
| SEC filings | SEC API | Provide SEC filings, e.g., 10-Q, 10-K, 8-K filings. | 51 | https://sec-api.io/ |
| Analyst Reports | Seeking Alpha | Provide news, analysis, and commentary from investors. | https://seekingalpha.com/ | |
| News | GNews | Provides an API to search for articles on Google News. | 51 | https://github.com/ranahaani/GNews |
| Bloomberg | Official website of Bloomberg. | https://www.bloomberg.com/ | ||
| CNBC | Official website of CNBC. | https://www.cnbc.com/ | ||
| WSJ | Official website of WSJ. | https://www.wsj.com/ | ||
| Social Media Data | X API | Provide programmatic access to X. | 51 | https://docs.x.com/x-api/introduction |
| Reddit API | Provide programmatic access to Reddit. | 51 | https://www.reddit.com/dev/api/ |
Table 25.
Marketing-related Public Datasets in Google BigQuery
| Dataset | Project Path | Description |
|---|---|---|
| GA4 E-commerce Sample | bigquery-public-data.ga4_obfuscated_sample_ecommerce | Anonymized GA4 event data for e-commerce customer behavior and marketing funnel analysis. |
| TheLook E-commerce | bigquery-public-data.thelook_ecommerce | Synthetic e-commerce data for customer segmentation, sales forecasting, and marketing analytics. |
| Google Trends | bigquery-public-data.google_trends | Keyword search interest data over time, useful for public interest and trend analysis. |
| GDELT | gdelt-bq | Global news event metadata, including sentiment analysis for brand exposure and market event tracking. |
| Google Ads Transparency | bigquery-public-data.google_ads_transparency | Data on political and issue ads, supporting research on advertising transparency and audience targeting. |
Table 26.
Applications and insights of LLMs in mathematic research and education
| Taxonomy | Contributions | Representative Works | Citations |
| Mathematical Proof Assistant | Automated theorem proving to generate reasoning steps or convert mathematical statements into formal languages. | AlphaProof [655]: A system developed by Google DeepMind that trains itself to prove mathematical statements in the formal language Lean. | [655,656,657,658,659,660,661,662,663,655] |
| Theoretical Exploration and Pattern Recognition | Theoretical Exploration and Pattern Recognition in the generation of conjectures, analysis of complex dynamical systems, or development of heuristics for solving open problems. | FunSearch [648]: An LLMs framework with a systematic evaluator for mathematical exploration. | [648,664,665,666] |
| Mathematical Education | LLMs for conceptual understanding, problem generation, and automate assessment. | Zhang et al [667]: A study that investigates whether LLMs can enhance learning outcomes in mathematics when used as tutoring tools. | [667,668,669,670,671] |
Table 28.
Performance of LLMs-based methods for mathematical education, as discussed in the study [667].
Table 28.
Performance of LLMs-based methods for mathematical education, as discussed in the study [667].
| Strategy | Answer Exposure Condition | Practice Phase | Test Phase |
| Answer Only | See Answer First | 85% - 90% | 50% - 52% |
| Try It First | 30% - 35% | 52% - 55% | |
| Stock LLM | See Answer First | 85% - 90% | 50% - 55% |
| Try It First | 35% - 40% | 65% - 70% | |
| Customized LLM | See Answer First | 85% - 90% | 55% - 60% |
| Try It First | 35% - 40% | 65% - 70% |
Table 29.
Benchmark dataset for mathematic.
| Cateogry | Benchmark | Description | Link |
| Competition | MATH [129] | High school & competition math | Github |
| Omini-MATH [688] | 4K competition-level problems | Github | |
| AIME [689] | American invitational mathematics examination | Kaggle | |
| Education | CMATH [690] | Chinese elemenentry school math problems. | Github |
| SAT-MATH [691] | Multiple-choice problem in SAT math exams. | Github | |
| GSM8K [140] | 8K grade school math questions | Github | |
| GAOKAO-Math [692] | Chinese high school mathematics questions from 2010 to 2022 | Github | |
| Math Word Problem | MAWPS [693] | 3K math word problems from web-sourced corpora | Github |
| ASDiv [694] | 2K elementary school level math word problems | Github | |
| SVAMP [695] | Advance version of ASDiv and MAWPS | Github | |
| Math Reasoning | AQuA-RAT [696] | 100K algebraic word problems | Huggingface |
| MathQA [697] | Enhanced version of AQuA-RAT | Huggingface | |
| PRM800K [698] | 800K math problems with step-wise labels. | Github | |
| TheoremQA [699] | Theorems-driven QA dataset | Github | |
| Lila [700] | Unified reasoning benchmark contains 20 existing datasets | Github | |
| MathInstruct [701] | Instruction-following style reasoning dataset. | Github |
Table 30.
Performance of LLMs over mathematic benchmarks.
| Cost ($/1M Tok) | MATH | MATH 500 | AIME 2024 | AIME 2025 | GSM8K | |
|---|---|---|---|---|---|---|
| Qwen 2.5 | – | 85.00% | 82.20% | – | – | 91.50% |
| Grok 3 | 5.00 | – | – | 93.30% | 93.30% | 89.30% |
| Llama 3.3 | – | 77% | – | – | – | – |
| DeepSeek R1 | 0.55 | 97.30% | 97.30% | 79.80% | 65.00% | 96.13% |
| Gemini 2.0 Flash | 1.25 | 89.70% | – | – | 30.00% | 95.53% |
| Claude 3.5 Sonnet | 3.00 | 78.30% | 78.00% | 16.00% | 3.33% | |
| Claude 3.7 Sonnet | 3.00 | – | 96.20% | 80.00% | – | |
| GPT-4o | 2.50 | 76.60% | – | – | 13.33% | 95.58% |
| GPT-o1 | 15.00 | 66.73% | 97.90% | 83.30% | 78.33% | 96.13% |
| GPT-o3-mini | 1.10 | – | 96.40% | 87.30% | 80.00% | 95.83% |
Table 32.
Instruction-to-Script generation results on CADBench [776]. Higher is better (↑), and best results are highlighted in dark blue, second-best in light blue. Syntax error rate is lower-the-better (↓).
Table 32.
Instruction-to-Script generation results on CADBench [776]. Higher is better (↑), and best results are highlighted in dark blue, second-best in light blue. Syntax error rate is lower-the-better (↓).
| Model | CADBench-Sim | CADBench-Wild | ||||||||
| Attr.↑ | Spat.↑ | Inst.↑ | Avg.↑ | ↓ | Attr.↑ | Spat.↑ | Inst.↑ | Avg.↑ | ↓ | |
| o1-Preview | 0.729 | 0.707 | 0.624 | 0.687 | 15.6% | 0.595 | 0.612 | 0.542 | 0.583 | 17.5% |
| GPT-4-Turbo | 0.658 | 0.621 | 0.488 | 0.589 | 18.2% | 0.526 | 0.541 | 0.478 | 0.515 | 24.5% |
| Claude-3.5-Sonnet | 0.687 | 0.608 | 0.482 | 0.593 | 15.6% | 0.529 | 0.508 | 0.430 | 0.489 | 14.3% |
| GPT-4o | 0.623 | 0.593 | 0.379 | 0.565 | 21.4% | 0.460 | 0.462 | 0.408 | 0.444 | 28.5% |
| BlenderGPT | 0.574 | 0.540 | 0.444 | 0.519 | 25.2% | 0.402 | 0.425 | 0.368 | 0.398 | 35.0% |
| Gemini-1.5-Pro | 0.535 | 0.483 | 0.387 | 0.468 | 30.2% | 0.375 | 0.404 | 0.361 | 0.380 | 38.0% |
| BlenderLLM | 0.846 | 0.767 | 0.626 | 0.747 | 3.2% | 0.717 | 0.614 | 0.493 | 0.608 | 5.0% |
Table 33.
Physics metrics on ModelSpecs benchmark from FEABench. Best results are highlighted in dark blue.
Table 33.
Physics metrics on ModelSpecs benchmark from FEABench. Best results are highlighted in dark blue.
| Model | Physics Extraction Metrics (↑) | Dimensionality | |||
| Interface Factuality | Interface Recall | Feature Recall | Feature Property Recall | Feature Dimension | |
| Claude 3.5 Sonnet | 0.85±0.10 | 0.71±0.13 | 0.80±0.10 | 0.22±0.10 | 0.95±0.05 |
| GPT-4o | 0.79±0.11 | 0.64±0.13 | 0.55±0.12 | 0.22±0.11 | 0.95±0.05 |
| Gemini-1.5-Pro | 0.54±0.14 | 0.43±0.14 | 0.39±0.10 | 0.15±0.09 | 0.86±0.14 |
| Gemma-2-27B-IT | 0.69±0.13 | 0.50±0.14 | 0.14±0.08 | 0.11±0.07 | - |
| Gemma-2-9B-IT | 0.70±0.15 | 0.43±0.14 | 0.06±0.04 | 0.07±0.07 | - |
| CodeGemma-7B-IT | 0.45±0.13 | 0.21±0.11 | 0.17±0.09 | 0.07±0.07 | - |
Table 54.
Earth Science and Civil Engineering Tasks, Benchmarks, Introduction and Cross Tasks.
| Type of Task | Benchmarks | Introduction | Cross Tasks |
|---|---|---|---|
| Geoscientific Understanding | GeoBench [1503] | A collection of pure text questions related to geology, geography and environmental science. | - |
| BB-GeoEval [1514] | Composed of 750 questions related to geoscience, including both objective and subjective questions. | - | |
| GeoChat [1506] | A collection of 6 datasets across 3 tasks constructed for remote sensing VLM evaluation. | - | |
| RSIEval [1471] | Self-collected image-caption pairs and image-question-answer triplets based on 100 Remote sensing images. | Remote sensing image captioning; Remote sensing VQA. | |
| GeoCode [1476] | Composed of 18k single turn tasks and 1.3k multi-turn tasks for data analysis code generation evaluation. | - | |
| GeoLLM-Engine [1475] | A collection of 7 datasets across 3 kinds of tasks constructed for the evaluation of geospatial copilots in earth observation capacity. | UI/Web navigation. | |
| Urban Planning | PlanGPT [1499] | A collection of 3 datasets across 4 tasks for urban and spatial planning evaluation. | Urban planning document generation & evaluation. |
Table 55.
Benchmark results on GeoBench integrated from [1470,1503]. Best results are highlighted with dark blue. Objective tasks are evaluated on two datasets separately based on accuracy, while subjective tasks adopt two evaluation metrics. If not emphasized, the metrics is considered the higher the better.
Table 55.
Benchmark results on GeoBench integrated from [1470,1503]. Best results are highlighted with dark blue. Objective tasks are evaluated on two datasets separately based on accuracy, while subjective tasks adopt two evaluation metrics. If not emphasized, the metrics is considered the higher the better.
| Model | Objective | Subjective | ||
| NPEE | APTest | Perplexity↓ | GPTScore | |
| Gal-6.7B | 25.7 | 29.9 | 34.57 | -2.3598 |
| LLaMA-7B | 21.6 | 27.6 | 40.07 | -1.9531 |
| MPT-7B | 28.4 | 26.0 | - | - |
| Vicuna-7B | 26.4 | 16.8 | - | - |
| GeoLLaMA-7B | - | - | 32.32 | -1.9457 |
| Alpaca-7B | 31.1 | 29.1 | 40.07 | -1.9536 |
| K2-7B | 39.9 | 29.3 | 32.32 | -1.9487 |
| ChatGPT | 48.8 | 20.0 | - | - |
| GeoGalactica-30B | 46.6 | 36.9 | - | - |
Table 56.
Image captioning results on RSIEval. Best results are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 56.
Image captioning results on RSIEval. Best results are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| Model | BLEU-1 | BLEU-2 | BLEU-3 | BLEU-4 | METEOR | ROUGE_L | CIDEr | CLAIR |
|---|---|---|---|---|---|---|---|---|
| BLIP2 | 0.15 | 0.07 | 0.03 | 0.01 | 3.40 | 11.57 | 0.09 | 27.40 |
| MiniGPT4 | 47.89 | 33.46 | 23.71 | 17.26 | 19.87 | 35.32 | 16.25 | 41.15 |
| LLaVA | 45.69 | 34.15 | 25.76 | 19.68 | 21.30 | 39.46 | 29.41 | 48.95 |
| RSGPT | 47.85 | 36.06 | 27.97 | 22.07 | 23.58 | 41.73 | 31.35 | 50.95 |
Table 57.
VQA results on RSIEval, with accuracy on the left and GPT-4 scoring on the right. Best results are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 57.
VQA results on RSIEval, with accuracy on the left and GPT-4 scoring on the right. Best results are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| Model | Presence | Quantity | Color | Absolute pos. | Relative pos. | Area comp. | Road dir. | Image | Scene | Reasoning | Avg accuracy |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BLIP2 | 39.48/39.74 | 10.83/7.50 | 20.69/25.86 | 1.10/4.40 | 2.50/2.50 | 64.58/64.58 | 60.00/60.00 | 45.83/45.83 | 8.89/8.89 | 29.09/29.09 | 31.44/32.04 |
| MiniGPT4 | 34.55/59.22 | 20.00/15.83 | 29.31/37.93 | 6.59/20.88 | 7.50/22.50 | 18.75/27.08 | 0.00/0.00 | 63.54/60.42 | 46.67/73.33 | 14.55/23.64 | 26.83/37.87 |
| LLaVA | 81.04/80.78 | 31.67/33.33 | 72.41/72.41 | 34.07/41.76 | 27.50/27.50 | 62.50/62.50 | 60.00/60.00 | 92.71/94.79 | 75.56/84.44 | 54.55/54.55 | 66.52/68.01 |
| RSGPT | 80.52/80.00 | 36.67/37.50 | 75.86/72.41 | 34.07/31.87 | 45.00/40.00 | 54.17/56.25 | 60.00/60.00 | 80.21/78.12 | 77.78/82.22 | 50.91/47.27 | 66.13/65.07 |
Table 58.
Single-turn evaluation results on GeoCode, including code generation pass rate (Pass@1) and function call performance (F1 score). Best results are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 58.
Single-turn evaluation results on GeoCode, including code generation pass rate (Pass@1) and function call performance (F1 score). Best results are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| GeoCode-GEE | GeoCode-Others | |||
|---|---|---|---|---|
| Model | Pass@1 | F1 | Pass@1 | F1 |
| LLaMA3.1-8B | 0.76 | 0.78 | 0.45 | 0.66 |
| CodeGemma-7B | 0.86 | 0.75 | 0.58 | 0.69 |
| Phi3.5 mini-3.8B | 0.66 | 0.70 | 0.5 | 0.66 |
| Qwen 2-7B | 0.61 | 0.76 | 0.39 | 0.63 |
Table 59.
Benchmark results on PlanGPT benchmark. Best results are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
Table 59.
Benchmark results on PlanGPT benchmark. Best results are highlighted with dark blue. If not emphasized, the metrics is considered the higher the better.
| Model | Generating | Style Transfer | Information Extraction | Text Evaluation | |
| PlanEval | PlanEval | Acc | Acc | F1 | |
| ChatGLM | 47.67 | 63.94 | 50.00 | 26.00 | 25.67 |
| Yi-6B | 16.00 | 15.41 | - | 20.00 | 8.33 |
| Baichuan2-13B-Chat | 62.67 | 43.90 | 50.32 | 33.00 | 17.42 |
| ChatGPT | 74.67 | 66.12 | - | 31.00 | 21.30 |
| PlanGPT | 60.33 | 66.80 | 65.18 | 41.00 | 35.28 |
Table 62.
Benchmark datasets for software programming and circuit design.
| Category | Benchmark | LLMs Tested | Description | Link |
| Code Generation | HumanEval [49] | ✓ | A comprehensive coding benchmark released by OpenAI. | Github |
| EvalPlus [1598] | ✓ | A code synthesis evaluation framework build upon HumanEval. | Github | |
| APPS [1617] | ✓ | 10k code generation problems | Huggingface | |
| CodeXGLUE [1618] | ✓ | Fourteen datasets from ten diversified programming language tasks. | Github | |
| CodeContests [1619] | ✓ | Competitive programming dataset released by DeepMind. | Github | |
| LeetCodeDataset [1620] | ✓ | LeetCode problems with a hundred test cases per problem; Supports contamination-free evaluation and SFT. | Github, Huggingface | |
| WikiSQL [1621] | × | A large crowd-sourced dataset for training relational database. | Github | |
| Aider Polyglot [1622] | ✓ | 697 coding problems in C++, Go, Java, JS, Python, and Rust. | Github | |
| Code Debugging | SWE-Bench [1623] | ✓ | Benchmark for evaluating LLMs on software issues from Github. | Website |
| DebugBench [1604] | ✓ | Evaluating Debugging Capability of Large Language Models. | Github | |
| Defects4J [1624] | × | A collection of reproducible Java bugs. | Github | |
| QuixBugs [1625] | × | A multi-lingual program repair benchmark based on Quixey. | Github | |
| BugsInPy [1626] | × | A Database of Existing Bugs in Python Programs to Enable Controlled Testing and Debugging Studies. | Github | |
| DebugEval [1577] | ✓ | A benchmark for LLMs debug tasks, i.e., bug localization and identification, code review and code repair. | Github | |
| MdEval [1627] | ✓ | multilingual debugging benchmark that contains eightteen programming languages. | Github | |
| Code Analysis | CodeSearchNet [1628] | ✓ | 2M code and comment pairs from open source libraries on Github. | Huggingface |
| CodeMMLU [1629] | ✓ | Benchmark to evaluate LLMs in coding and software knowledge. | Github | |
| LiveCodeBench [131] | ✓ | Five hundred coding problems in self-repair, code execution, and test ouotput prediction tasks. | Github | |
| Circuit Design | RTLLM [1630] | ✓ | An open-source benchmark for design RTL generation with LLMs | Github |
| VerilogEval [1631] | ✓ | Benchmark for Verilog code generation for hardware design and verification. | Github | |
| MG-Verilog [1632] | ✓ | A multi-grained dataset towards enhanced llm-assisted Verilog generation. | Github | |
| AssertEval [1633] | ✓ | Open-source benchmark for evaluating LLM’s assertion generation capabilities for RTL verification. | Github | |
| RTLCode [1633] | ✓ | 80K instruction-code dataset for LLMs RTL generation. | Github | |
| PICBench [1634] | ✓ | Benchmark for photonic integrated circuits design. | Github |
Table 63.
Performance of LLMs over CSEE benchmarks.
| HumanEval | EvalPlus | CodeMMLU | Aider Polyglot | SWE-Bench | PICBench | |
| Qwen 2.5 | 88.00% | 87.21% | 56.40% | 26.20% | – | – |
| Grok 3 | – | – | – | 53.30% | – | – |
| Llama 3.3 | 88.40% | – | 43.91% | – | – | – |
| DeepSeek R1 | – | – | – | 56.90% | 49.20% | – |
| Gemini 2.0 Flash | – | – | 59.81% | 35.60% | 52.20% | – |
| Claude 3.5 Sonnet | 93.70% | 81.70% | 61.65% | 51.60% | 49.00% | 13.33 |
| Claude 3.7 Sonnet | – | – | 67.00% | 64.90% | 62.30% | – |
| GPT-4o | 90.20% | 87.20% | – | 45.30% | – | 14.17 |
| GPT-o1 | 92.40% | 89.00% | 62.36% | – | 48.90% | 8.33 |
| GPT-o3-mini | – | – | – | 60.40% | 49.30% | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



