
Submitted:
16 October 2025
Posted:
17 October 2025
You are already at the latest version
Abstract
HELIX syndrome is a rare autosomal recessive disorder characterized by Hypohidrosis, Electrolyte imbalance, Lacrimal gland dysfunction, Ichthyosis, and Xerostomia. We report an 8-year-old girl with long-standing anhidrosis and alacrimation who was found to carry a novel nonsense variant in the CLDN10 gene (c.138G>A; p.Trp46*), resulting in an early termination codon in exon 1. Brain MRI revealed bilateral, symmetrical periventricular and deep white-matter signal changes suggestive of demyelination. This case broadens the known phenotypic spectrum of HELIX syndrome and underscores the diagnostic value of genetic testing in children presenting with multisystem glandular dysfunction.
Keywords:
HELIX syndrome
; CLDN10
; anhidrosis
; alacrimation
; demyelination
; novel variant
Background
HELIX syndrome, first described by Hadj-Rabia et al. in 2018, results from biallelic pathogenic variants in CLDN10, encoding claudin-10, a tight-junction protein critical for paracellular ion transport and epithelial integrity. Disruption of claudin-10 function compromises epithelial ion permeability in tissues such as sweat glands, kidneys, and salivary and lacrimal glands, producing the characteristic clinical triad of anhidrosis, electrolyte imbalance, and glandular hypofunction. Given the extreme rarity of this condition, each new variant provides valuable insight into genotype–phenotype correlation and potential systemic manifestations.
Case Presentation
An 8-year-old girl was evaluated for lifelong absence of sweating and tearing. There was no parental consanguinity and no similar family history. Physical examination showed normal growth parameters and no dysmorphic features. Skin was mildly dry without scaling or erythema.
Laboratory Findings:
| Test | Patient Result | Reference Range |
| Serum Creatinine | 24.6 µmol/L | 60–115 µmol/L |
| Potassium | 4.4 mmol/L | 3.5–5.1 mmol/L |
| Sodium | 134 mmol/L | 135–145 mmol/L |
| Calcium | 2.32 mmol/L | 2.2–2.7 mmol/L |
| Hemoglobin | 11 g/dL | 12–14 g/dL |
| MCV | 83 fL | 78–98 fL |
| Vitamin D₃ | 7.34 ng/mL | 30–100 ng/mL |
Genetic Analysis
Next-generation sequencing revealed a novel nonsense variant in CLDN10 (NM_006984.4: c.138G>A; p.Trp46*), predicted to create a premature stop codon in exon 1. Although classified as a variant of uncertain significance (VUS), the genotype–phenotype correlation strongly supports pathogenicity.
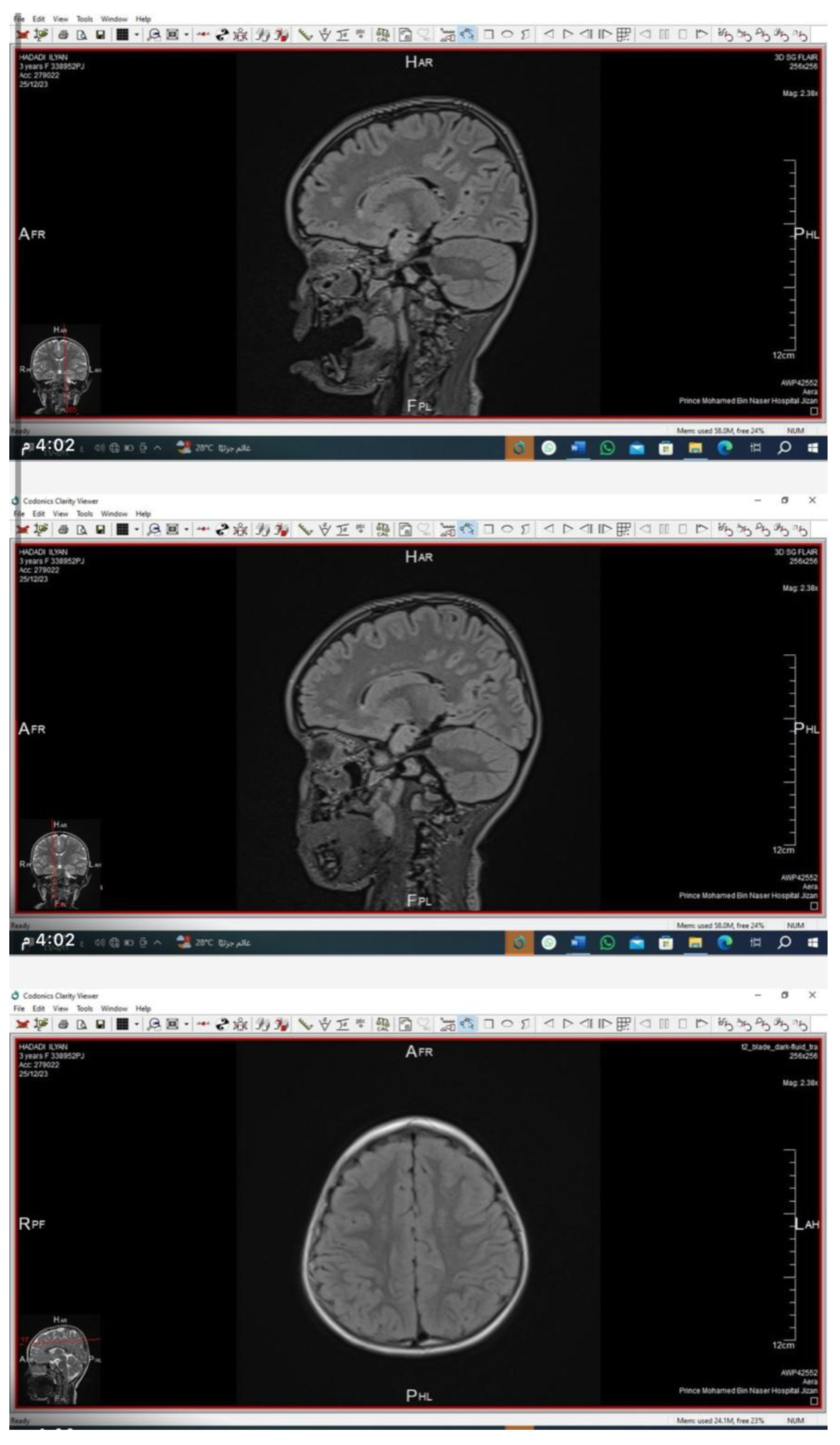
Neuroimaging
Brain and orbital MRI with contrast demonstrated bilateral, symmetrical, patchy hyperintensities in the periventricular and deep cerebral white matter on T2/FLAIR sequences, consistent with demyelination. The cerebellum and lacrimal glands appeared structurally normal.
Discussion
This case represents a previously undescribed CLDN10 variant associated with HELIX syndrome. The c.138G>A (p.Trp46*) mutation likely causes nonsense-mediated mRNA decay or truncation of claudin-10, leading to defective tight-junction ion selectivity. Our patient’s MRI findings are particularly noteworthy. Neurologic involvement is rarely reported in HELIX syndrome; thus, the observed white-matter changes may indicate a broader tissue distribution of claudin-10 or secondary metabolic effects.
Conclusion
We describe a novel CLDN10 nonsense variant (p.Trp46*) in a child presenting with the classical HELIX phenotype and previously unreported MRI abnormalities. This case expands the phenotypic and genotypic spectrum of HELIX syndrome and highlights the essential role of genetic analysis in unexplained multisystem glandular disorders.
Learning Points
1. HELIX syndrome should be suspected in children with concurrent anhidrosis and alacrimation.
2. Early genetic testing can identify novel variants and guide family counseling.
3. MRI abnormalities may suggest previously unrecognized neurological involvement.
4. Long-term multidisciplinary care is vital for optimal quality of life.
References
- Hadj-Rabia, S; et al. Genet Med. 2018;20(2):190–201. [CrossRef]
- Günzel D, Yu AS. Physiol Rev. 2013;93(2):525–69. [CrossRef]
- Boccara, O; et al. Eur J Med Genet. 2020;63(9):103992.
- Klar, J; et al. PLoS Genet. 2017;13(7):e1006897. [CrossRef]
- Milatz S, Breiderhoff T. Pflugers Arch. 2017;469(1):115–21. [CrossRef]
- Bondue, B; et al. J Clin Immunol. 2020;40(7):1055–8.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



