Submitted:
15 October 2025
Posted:
15 October 2025
You are already at the latest version
Abstract
Dopamine signaling has long been framed through a neuron-centric lens, yet mounting evidence reveals that glial cells (astrocytes and microglia) serve as indispensable gatekeepers of dopaminergic tone, synaptic plasticity, and neuroimmune balance. Recent single-cell, spatial, and optical imaging studies have redefined dopamine circuits as multicellular ecosystems in which glial receptors, transporters, and gliotransmitters dynamically sculpt neuromodulation and behavior. Astrocytes fine-tune dopamine clearance, glutamate buffering, and metabolic coupling, while microglia integrate immune and stress cues recalibrate dopaminergic signaling across striatal and cortical circuits. Their bidirectional interactions, both glia–glia and glia–neuron, mediate resilience or vulnerability in contexts ranging from motivation and stress adaptation to Parkinson’s disease, depression, and post-viral fatigue syndromes. This review synthesizes emerging evidence that glial–dopamine crosstalk is a systems-level regulator of neuroinflammation and plasticity, bridging cellular metabolism, immune tone, and behavioral output. By integrating multi-omics, in vivo imaging, and computational models, we propose a translational framework for targeting astrocytic and microglial states to restore dopaminergic homeostasis. Understanding and manipulating these non-neuronal interfaces may open the next frontier in precision neuropsychiatry and neurodegeneration therapeutics.
Keywords:
glial–dopamine crosstalk
; astrocyte–microglia interactions
; neuroinflammation
; dopaminergic circuits
; motivation and stress
; Parkinson’s disease
; precision neurotherapeutics
; single-cell and spatial transcriptomics
1. Introduction
For decades, dopamine (DA) has been understood primarily through a neuronal lens. Synthesized and released by midbrain dopaminergic neurons—classically in the substantia nigra pars compacta and ventral tegmental area—DA was framed as a transmitter that sculpts basal ganglia, prefrontal cortex (PFC), amygdala, and interconnected circuits [1,2]. Within this model, dopamine mediated a wide spectrum of functions, from motor control and reinforcement learning to motivation, decision-making, and working memory [3]. Pathological perturbations in this system were accordingly linked to movement disorders such as Parkinson’s disease, as well as psychiatric syndromes including schizophrenia, addiction, and ADHD [4]. Glia, in contrast, were relegated to supportive or defensive roles: astrocytes as ionic and metabolic buffers, microglia as immune sentinels engaged in pruning and injury repair [5].
This classical dichotomy has increasingly eroded over the past decade, as mounting evidence has repositioned astrocytes and microglia from peripheral bystanders to active regulators of dopaminergic tone. Astrocytes not only express functional dopamine receptors but also directly modulate synaptic plasticity. For instance, activation of D1/D5 receptors in spinal astrocytes has been shown to induce non-Hebbian long-term potentiation at primary afferent inputs, thereby reshaping excitatory drive independently of canonical neuronal pathways [6]. Complementing this, recent work identified astrocytic Dop2R signaling as a potent regulator of neighboring dopaminergic neuron excitability, situating astrocytes within closed feedback loops that dynamically govern dopamine release [7]. Collectively, these findings converge on the recognition that astrocytic responses to dopamine are highly context-dependent: in certain environments, they exert anti-inflammatory and neuroprotective effects, whereas under other conditions they promote pro-inflammatory and neurotoxic outcomes, with gliotransmitter and neurotrophin release serving as critical modulatory levers [8,9]
Astrocytic influence also extends indirectly through crosstalk with other neuromodulators. Striatal astrocytes regulate extracellular GABA and adenosine, thereby constraining DA release through GABAA/B and adenosine A1 receptor pathways [10]. Notably, soma-to-soma configurations with cholinergic interneurons allow astrocytes to exert subsecond precision over dopamine dynamics, positioning them as fast integrators rather than slow homeostatic buffers.
Microglia have likewise emerged as indispensable architects of dopaminergic circuitry. Developmental studies demonstrate that microglia orchestrate DA axon growth, pruning, and synaptic connectivity, with early-life stress reprogramming microglial transcriptional states and destabilizing the maturation of dopaminergic projections [11,12]. In adulthood, dopamine itself exerts reciprocal control over microglial activity. Experimental evidence shows that DA exposure activates inflammasome signaling and upregulates (interleukin-1β) IL-1β expression in microglia and macrophages, with the magnitude and direction of these responses determined by the relative balance of D1-like versus D2-like receptor expression [13]. Under conditions of inflammatory comorbidity, such as HIV infection, these effects are markedly amplified, establishing bidirectional feedback loops in which dopamine modulates microglial state, while activated microglia, in turn, regulate DA synthesis, reuptake, and neuronal survival.
Together, these findings force a conceptual shift. The once-dominant tripartite synapse (neuron–astrocyte–presynaptic terminal) must be expanded into a quadripartite model of dopaminergic regulation, integrating microglial, vascular, and immune influences [14]. Cytokine release, blood–brain barrier dynamics, and immune trafficking emerge as indispensable modulators of DA physiology. High-resolution single-cell and spatial transcriptomics reveal marked heterogeneity across astrocytic and microglial populations in midbrain and striatal territories, some enriched for DA receptor expression, others correlated with selective neuronal vulnerability in aging and disease [15]. These findings dismantle the notion of dopamine as a purely neuronal currency, reframing it as a network-embedded signal embedded in glial, metabolic, and immune landscapes [16].
This reconceptualization yields three transformative implications. First, glial responses to DA are bidirectional and context-sensitive: receptor subtype, developmental window, and stress or disease state determine whether outcomes are neuroprotective or neurotoxic [17]. Second, DA–glia interactions exhibit striking temporal and spatial heterogeneity, differing across striatum, PFC, and hippocampus, and shifting from development to pathology [18]. Third, dopamine signaling is now inseparable from immune and metabolic states, embedding neuromodulation within vascular and systemic physiology [19].
The implications of these converging findings are profound and demand systematic synthesis. Recasting astrocytes and microglia as central gatekeepers of dopaminergic tone provides a novel conceptual lens through which to understand the mechanisms of selective vulnerability across a spectrum of disorders, including Parkinson’s disease, depression, and schizophrenia. At the translational interface, the modulation of glial dopamine receptors, the fine-tuning of receptor subtype ratios, and the targeting of dopamine-sensitive inflammatory cascades emerge as promising therapeutic strategies. In this context, the present review seeks to advance an integrative framework encompassing molecular, cellular, and systems-level perspectives within an expanded quadripartite synapse model designed to orient future research trajectories and inform the rational development of next-generation dopaminergic interventions.
2. Astrocytic Control of Dopamine Signaling
Astrocytes have increasingly been recognized as circuit-defining regulators of dopaminergic neurotransmission. Far from being passive support elements, they express functional dopamine receptors, release gliotransmitters that shape synaptic plasticity, and buffer neuromodulators such as adenosine and GABA with subsecond precision, particularly within striatal networks [20]. This dynamic control reframes dopaminergic function, positioning astrocytes as determinants of phasic versus tonic dopamine signaling and linking regional astrocytic heterogeneity to behavioral domains including movement, reward, affect, and nociception (Table 1) [9].
2.1. Astrocytic Dopamine Receptors: Distribution, Signaling Consequences, and Heteromers
Astrocytes express multiple subtypes of dopamine receptors, and their activation exerts direct consequences on neural circuit dynamics. Within nociceptive pathways of the dorsal horn, astrocytic D1/D5 receptor activity is indispensable for a form of non-Hebbian long-term potentiation (LTP), as astrocyte-specific receptor knockdown abolishes plasticity at primary afferent synapses whereas neuronal knockdown does not [6]. This establishes astrocytic dopamine receptors as causal determinants of synaptic gain and broadens the locus of dopaminergic influence beyond neurons.
Cortical investigations reveal region- and layer-specific gradients of receptor expression, with superficial astrocytes in layer I exhibiting strong immunoreactivity for D1R and D4R, moderate levels of D5R, and lower expression of D2R, whereas deeper protoplasmic astrocytes display substantially reduced expression [21]. These laminar differences suggest that astrocytic networks located near pyramidal apical dendrites are positioned as hubs for top-down cortical modulation of dopaminergic tone.
Beyond individual receptor subtypes, astrocytes form heteromeric receptor complexes that expand their computational repertoire. Assemblies such as D2–oxytocin receptor heteromers and higher-order A2A–D2–oxytocin receptor complexes have been identified in striatal astrocytic processes [22]. These complexes regulate intracellular Ca²⁺ signaling and glutamate release, creating receptor–receptor interactions (RRIs) that function as molecular logic gates for convergent neuromodulatory inputs. Through these RRIs, astrocytes integrate dopaminergic, adenosinergic, and oxytocinergic signals, highlighting their role as computational integrators of neuromodulation rather than passive relay stations [23,24].
2.2. Astrocytic Gliotransmission as an Upstream and Downstream Regulator of Dopamine
Astrocytic gliotransmission operates both upstream of dopamine release and downstream at postsynaptic sites [27]. In nociceptive networks, D1/D5 receptor activation in astrocytes drives non-Hebbian LTP at primary afferent synapses, even under conditions of minimal postsynaptic activity, indicating that astrocytes can independently set thresholds for synaptic potentiation [6]. This expands the classical framework of plasticity and positions astrocytes as active drivers of long-term information storage.
In the striatum, astrocytes modulate dopamine release indirectly by regulating extracellular adenosine and GABA, which in turn shape the excitability of cholinergic interneurons. In vivo imaging demonstrates that astrocytic depolarization can rapidly shift interneuron firing and thus sculpt dopamine release dynamics on subsecond timescales [10]. This astrocyte–interneuron axis reframes astrocytes as fast regulators of dopaminergic output, functioning with temporal precision previously attributed exclusively to neurons.
Astrocytes also act as cross-modal integrators at excitatory–dopaminergic interfaces. Dopamine lowers the threshold for glutamate-evoked Ca²⁺ waves in astrocytes, thereby amplifying and propagating intracellular signals [26]. This synergy creates a bidirectional dopamine–glutamate–astrocyte loop, enabling fine-tuning of excitatory integration and circuit output.
2.3. Astrocytic Uptake and Clearance in Dopamine Regulation
Astrocytes critically regulate neuromodulator tone through uptake and clearance mechanisms. In the striatum, astrocytic buffering of GABA and adenosine modulates the inhibitory control of dopaminergic terminals via GABAA/GABAB and A1 receptors. Transient depolarization or disruption of astrocytic buffering alters cholinergic interneuron excitability and dopamine release within hundreds of milliseconds, underscoring the role of astrocytes as real-time gatekeepers of dopaminergic signaling [10].
Although in vivo evidence for direct astrocytic involvement of EAAT1/2 transporters in dopamine regulation remains limited, single-cell and spatial transcriptomics reveal that astrocyte subtypes within dopamine-rich regions differentially express EAAT isoforms [28,29]. This suggests that glutamate clearance capacity indirectly modulates dopaminergic excitability, particularly where glutamate spillover from cortical or thalamic inputs could influence dopaminergic neurons or striatal projection neurons. By serving as buffers between excitatory drive and dopaminergic responsiveness, astrocytic EAATs provide a mechanistic substrate for glutamate–dopamine crosstalk [30,31].
Collectively, these insights establish astrocytic transporters and buffering systems as hidden regulators of dopamine tone, integrating GABAergic, adenosinergic, and glutamatergic dynamics into the dopaminergic system and expanding the framework of dopamine regulation beyond neuronal boundaries.
2.4. Astrocytic Modulation of Dopamine: Circuit-Specific Mechanisms in Striatum and Cortex
Astrocytic modulation of dopamine signaling displays striking regional specificity, reflecting the anatomical and computational demands of distinct circuits. In the striatum, astrocytes form specialized soma-to-soma “satellite” configurations with cholinergic interneurons, enabling direct influence over interneuron excitability. These structural interactions, combined with astrocytic buffering of adenosine and GABA, allow astrocytes to regulate dopamine release with remarkable temporal precision on subsecond timescales [10]. Striatal astrocytes are further enriched with heteromeric receptor complexes, including A2A–D2–oxytocin receptor assemblies, which couple to Ca²⁺ dynamics and glutamate gliotransmission [22,25]. This molecular machinery equips striatal astrocytes with multi-channel mechanisms for tuning phasic dopamine release, thereby shaping reinforcement learning, reward prediction error signaling, and motor control. Dysregulation of these astrocytic processes has been implicated in maladaptive dopaminergic states underlying Parkinson’s disease, substance use disorders, and compulsive habit formation.
By contrast, astrocytic contributions in the prefrontal cortex are defined by the laminar organization of cortical circuits. Superficial astrocytes in the pial and layer I zones display strong expression of D1R and D4R, moderate levels of D5R, and weak D2R enrichment, placing them in strategic proximity to pyramidal neuron apical dendrites [21]. These astrocytes are positioned to shape the apical integration of long-range inputs and are thought to engage Ca²⁺-dominated signaling cascades rather than classical cAMP-mediated pathways [32]. This lamina-specific dopaminergic responsiveness implicates cortical astrocytes in regulating higher-order processes such as working memory, attentional control, and cognitive flexibility. Perturbation in these astrocytic mechanisms may contribute to psychiatric pathophysiology, including schizophrenia and attention-deficit/hyperactivity disorder. Collectively, these findings underscore that astrocytic modulation of dopamine is spatially and functionally specialized, operating through distinct cellular principles in subcortical versus cortical territories.
2.5. Technological Innovations for Causal and Temporally Precise Dissection of Astrocyte–Dopamine Signaling
The recognition of astrocytes as active participants in dopaminergic signaling has been enabled by recent methodological advances that combine genetic precision, optical speed, and molecular resolution.
Genetic approaches such as conditional knockouts and astrocyte-specific receptor silencing have provided causal evidence that astrocytic dopamine receptors are indispensable for synaptic plasticity. The elimination of D1/D5 receptors selectively in astrocytes—but not neurons—abolishes non-Hebbian LTP in nociceptive pathways, establishing the astrocytic necessity for circuit-level plasticity [6].
Fast optical reporters now allow real-time monitoring of dopamine fluctuations and astrocytic dynamics in vivo. Genetically encoded GRAB-DA sensors, in conjunction with calcium indicators such as GCaMP, reveal astrocyte–interneuron interactions that control dopamine release with millisecond precision. These findings overturn the traditional notion of astrocytes as slow modulators, instead of situating them as rapid, temporally precise regulators of neuromodulation [10].
Molecular mapping techniques, including proximity ligation assays, super-resolution microscopy, and spatial transcriptomics, have delineated the subcellular organization of receptor heteromers in astrocytes. Complexes such as A2A–D2–oxytocin receptor assemblies localize to striatal astrocytic membranes, where they govern Ca²⁺ signaling and gliotransmitter release [22]. These techniques provide a structural and biochemical framework that links receptor topography to astrocytic neuromodulatory functions.
Together, these methodological innovations are transforming the field by enabling causal, temporally resolved, and molecularly precise dissection of astrocytic contributions to dopaminergic circuits. Such tools not only deepen mechanistic insight but also establish a foundation for translational strategies that target astrocytic pathways in dopamine-related disorders.
Despite the transformative advances that have repositioned astrocytes as dynamic and temporally precise regulators of dopaminergic signaling, several critical gaps remain. The direct contribution of astrocytes to dopamine transporter function is still unresolved; while anatomical proximity suggests potential interactions, definitive in vivo evidence is lacking. The full repertoire of gliotransmitters released under dopaminergic influence remains incompletely characterized, and the intracellular signaling pathways engaged by astrocytic dopamine receptors display considerable regional and state-dependent variability. Moreover, the behavioral significance of astrocytic dopamine signaling in higher-order cognition and psychiatric disorders remains insufficiently explored. Addressing these knowledge gaps will require systematic application of real-time dopamine sensors in combination with astrocyte-specific perturbations during behavior, alongside causal testing to link prefrontal astrocytic dopamine signaling with psychiatric phenotypes such as schizophrenia, attention-deficit/hyperactivity disorder, and mood disorders.
2.6. Therapeutic Horizons: Targeting Astrocytic Pathways in Dopaminergic Disorders
The recognition of astrocytes as active and temporally precise regulators of dopaminergic signaling has opened new translational frontiers, repositioning glial pathways as promising therapeutic entry points. Unlike traditional neuron-centric models, astrocytic targets provide opportunities for more nuanced and circuit-specific modulation of dopaminergic tone (Figure 1). Selective targeting of astrocytic D1/D5 receptors represents one avenue of intervention, offering the ability to influence non-Hebbian plasticity within nociceptive pathways. By modulating these receptors, it may be possible to normalize maladaptive plasticity underlying chronic pain syndromes, thereby extending dopaminergic therapies beyond canonical neuronal targets [6].
Another promising direction involves the manipulation of receptor heteromeric complexes. The identification of astrocytic A2A–D2–oxytocin receptor assemblies highlight novel druggable interfaces, with pharmacological ligands capable of stabilizing or disrupting these complexes to fine-tune Ca²⁺ dynamics and gliotransmitter release. Such strategies may provide higher specificity than single-receptor agents, with particular relevance to Parkinson’s disease, where aberrant adenosine–dopamine interactions drive motor fluctuations, and to compulsive or addictive disorders characterized by maladaptive reinforcement learning [16,22].
Astrocytic buffering of adenosine and GABA represents a further therapeutic axis. Pharmacological manipulation of these transporter systems may restore the phasic–tonic balance of dopamine release, with potential applications in conditions such as obsessive–compulsive disorder, habit pathology, and Parkinsonian dyskinesias [10]. Beyond pharmacology, astrocyte-directed neuromodulation technologies, including chemogenetic, optogenetic, and nanoparticle-based approaches—are emerging as powerful tools to achieve temporally precise and spatially selective control of astrocytic activity. These technologies provide both mechanistic insight and translational potential, enabling the development of targeted interventions within dopamine-related disorders.
Collectively, these strategies mark a paradigm shift from neuron-exclusive interventions toward glia–neuron co-modulation, underscoring astrocytic mechanisms as tractable and highly specific therapeutic nodes within dopaminergic circuitry. By leveraging receptor- and transporter-based strategies alongside next-generation neuromodulation platforms, astrocyte-centered therapeutics hold the potential to redefine the treatment landscape for disorders rooted in dopaminergic dysfunction.
3. Microglial–Dopamine Crosstalk and Neuroinflammation
Microglia are increasingly recognized as critical modulators of dopaminergic physiology. Beyond their classical roles in surveillance and synaptic pruning, microglia sense dopamine through receptor-mediated pathways, translate these signals into metabolic and inflammasome programs, and release cytokines that shape dopaminergic neuronal viability (Table 2). These bidirectional interactions are now implicated in prodromal Parkinson’s disease, systemic immune comorbidities such as HIV, and stress-related psychiatric syndromes, positioning microglia as context-dependent amplifiers—or brakes—of dopamine biology [13,33].
3.1. Dopamine Receptor Expression and Signaling in Microglia
Large-scale transcriptomic and proteomic studies confirm that microglia possess a selective repertoire of dopamine receptors. Across both human and rodent systems, DRD2 and DRD4 are consistently enriched, DRD1 and DRD3 are expressed at moderate levels, and DRD5 expression is minimal [34,35]. Crucially, these are not merely transcriptional traces but functional proteins capable of initiating intracellular cascades. DRD2 engagement couples to Gi/o signaling pathways, reducing cAMP levels and influencing downstream inflammasome activity, while DRD1 couples to Gs-cAMP-PKA signaling, thereby promoting opposing effects [36,37].
Functional assays in human primary microglia and immortalized lines reveal that dopamine signaling through DRD1 and DRD2 can suppress NLRP3 inflammasome activation, reducing IL-1β release under canonical (LPS/ATP), non-canonical (caspase-11), and proteinopathy-associated (α-synuclein) challenges [38,39]. This positions dopamine as an endogenous checkpoint regulator, constraining excessive immune activation and preventing runaway neuroinflammation. In this framework, dopamine emerges not only as a neuromodulator of synaptic transmission but also as an immunomodulatory signal critical for maintaining homeostatic balance.
3.2. Dopamine Control of Microglial Polarization and Inflammatory Tone
Microglia operate along a polarization spectrum spanning pro-inflammatory, M1-like states and reparative, M2-like states. Dopamine signaling exerts a powerful influence over this spectrum, but its effects are highly dependent on receptor balance, local immune context, and disease state. Under physiological conditions, DRD1/DRD2 activation promotes anti-inflammatory programs: attenuating NLRP3 activation, reducing IL-1β production, and upregulating reparative gene networks. Such actions are consistent with neuroprotective phenotypes observed in preclinical models of delirium and Parkinsonian pathology [40,41].
However, the ratio of DRD1-like to DRD2-like signaling appears to function as a regulatory switch. When DRD1 activity predominates, dopamine stimulation paradoxically amplifies IL-1β transcription, enhances inflammasome activity, and drives sustained pro-inflammatory responses, particularly under chronic immune challenge such as HIV infection [13]. By contrast, DRD2 signaling exerts counter-regulatory effects, restraining this pro-inflammatory cascade. Transcriptomic profiling of dopamine-exposed microglia further supports this duality, revealing coordinated induction of IL-1β pathway genes, inflammasome adaptors, and NF-κB targets under conditions of receptor imbalance [12,13,41].
Together, these findings establish dopamine as a context-sensitive modulator of microglial state, capable of functioning either as an anti-inflammatory brake or a pro-inflammatory accelerator. The outcome is determined not by dopamine alone, but by the dynamic balance of receptor subtype activation, immune context, and cellular state—a principle that has profound implications for neurodegeneration, neuroinflammation, and psychiatric disease.
3.3. Stress, Trauma, and Depression: Priming the Microglial–Dopamine Axis
Although direct investigations of dopamine–microglia interactions in stress-related and depressive disorders remain limited, converging mechanistic evidence implicates microglial metabolic priming as a central vulnerability factor. Chronic stress reshapes microglial physiology by altering lipid metabolism, mitochondrial respiration, and redox balance, thereby lowering the threshold for immune activation. Under these conditions, dopaminergic stimulation more readily engages inflammasome pathways, producing exaggerated pro-inflammatory responses in stressed neural circuits [42,43]. This metabolic reprogramming effectively converts dopamine from a homeostatic signal into a trigger for maladaptive immune amplification.
Clinical neuroimaging provides convergent evidence. In idiopathic REM sleep behavior disorder, widely regarded as a prodromal stage of Parkinson’s disease, translocator protein- positron emission tomography (TSPO-PET) imaging reveals heightened microglial activation in the substantia nigra and basal ganglia. These immune changes correlate with reduced dopamine transporter binding and diminished tyrosine hydroxylase activity, suggesting that immune activation may precede, or at minimum parallel, dopaminergic decline [44,45]. Such findings lend support to a feed-forward model in which microglial priming accelerates the trajectory from stress-linked vulnerability to neurodegenerative pathology.
Experimental models further illuminate the heterogeneity of stress-responsive microglia. Distinct inflammatory microglial subtypes have been identified in α-synucleinopathy and tauopathy contexts, enriched for gene signatures linked to lipid metabolism, immune cell trafficking, and inflammasome activation [46,47]. While direct characterization of dopamine receptor expression within these subtypes is still lacking, their transcriptional profiles strongly suggest increased sensitivity to dopaminergic modulation. This framework provides a mechanistic bridge between stress-induced microglial priming and the heightened susceptibility to dopamine-driven inflammatory cascades, with implications extending across psychiatric and neurodegenerative disorders.
3.4. Microglial Drivers of Dopaminergic Vulnerability in Parkinson’s Disease
The contribution of microglia to dopaminergic degeneration in Parkinson’s disease is now recognized as the outcome of converging processes involving protein aggregation, mitochondrial dysfunction, and dysregulated dopamine catabolism [48]. Aggregated α-synuclein interacts with pattern recognition receptors such as TLR2 and TLR4 on microglia, initiating NF-κB–NLRP3 inflammasome signaling and driving the release of pro-inflammatory cytokines including IL-1β, (tumor necrosis factor-α) TNF-α, and IL-6 [49,50]. This inflammatory cascade not only accelerates neuronal injury but also facilitates the cell-to-cell propagation of α-synuclein pathology. Post-mortem analyses consistently reveal microgliosis within the substantia nigra and striatum, while in vivo PET imaging with TSPO ligands demonstrates widespread microglial activation in Parkinson’s disease, closely correlating with both motor severity and dopaminergic loss [51,52].
Mitochondrial dysfunction further amplifies this inflammatory milieu. Damage to dopaminergic neurons and microglia generate excessive reactive oxygen species and releases mitochondrial DNA, cardiolipin, and other danger-associated molecular patterns, sustaining chronic inflammasome activity [53,54]. Transcriptomic profiling of inflammatory microglial states in α-synucleinopathy and tauopathy models highlights profound alterations in lipid metabolism and mitochondrial respiration, features that bias microglia toward pro-inflammatory polarization while diminishing their neuroprotective capacity [55].
In parallel, dopamine metabolism itself contributes toxic amplifiers of neurodegeneration. The reactive metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL) forms adducts with α-synuclein, enhances fibrillization, and exerts direct cytotoxic effects on dopaminergic neurons [56] . Impairments in detoxification pathways, such as aldehyde dehydrogenase deficiency, allow accumulation of dopamine-derived quinones and aldehydes, which impose oxidative and electrophilic stress that further bias microglia toward pro-inflammatory states [57]. Integrative analyses now emphasize how this metabolite-driven toxicity intersects with genetic susceptibility loci, including LRRK2 and GBA, to potentiate microglial activation and accelerate the trajectory of neurodegeneration [58].
Taken together, these findings establish Parkinson’s disease as a multifactorial disorder of microglia–dopamine interaction, in which aggregated α-synuclein, mitochondrial distress, and reactive dopamine metabolites converge on shared inflammasome and oxidative pathways. This convergence not only heightens dopaminergic vulnerability but also perpetuates a self-reinforcing cycle of neuroinflammation and neuronal loss, underscoring the centrality of microglial mechanisms in the pathophysiology of the disease.
3.5. Reciprocal Immune–Dopamine Feedback Loops
Microglia and dopaminergic neurons are engaged in reciprocal feedback circuits that, when dysregulated, amplify pathology across both neurodegenerative and psychiatric conditions. Activated microglia release cytokines such as IL-1β, TNF-α, and IL-6, which downregulate tyrosine hydroxylase expression, blunt dopamine release, and disrupt dopamine transporter trafficking [59]. These immune-mediated suppressive effects on dopamine synthesis are consistent with findings from preclinical toxin models as well as human neuroimaging. Longitudinal PET investigations in prodromal cohorts, such as idiopathic REM sleep behavior disorder, reveal that heightened microglial activation precedes measurable striatal dopaminergic decline, suggesting that immune activity is not a secondary consequence but an early driver of nigrostriatal dysfunction [60].
Peripheral immune comorbidities further recalibrate dopamine–microglia interactions. In states of chronic systemic inflammation or infection, such as HIV, dopamine signaling assumes a maladaptive profile. Human macrophage and microglial studies demonstrate that immune challenge amplifies dopamine-induced inflammasome activation and IL-1β release, an effect dependent on the relative balance of DRD1-like versus DRD2-like receptors [13]. This finding underscores how systemic immune states can reprogram microglial dopamine sensitivity, potentially explaining the heightened vulnerability to neuroinflammation in patients receiving dopaminergic therapies or psychostimulants in the context of comorbid infections. Thus, dopamine not only modulates microglial state but is itself reshaped by immune tone, creating a self-reinforcing loop wherein inflammation drives dopaminergic dysfunction and impaired dopamine signaling further potentiates microglial activation.
Despite these advances, several critical uncertainties remain. Most in vitro studies expose microglia to supraphysiologic dopamine concentrations, leaving unresolved how cells respond to physiologically relevant tonic versus phasic release across striatal and mesocortical circuits [12]. Conflicting reports of dopamine’s anti-inflammatory versus pro-inflammatory roles likely reflect differences in receptor subtype balance, microglial activation state, and systemic immune comorbidities, highlighting the need for receptor-specific in vivo perturbations through conditional knockouts or chemogenetics [40]. The impact of dopamine metabolites, particularly reactive species such as DOPAL and dopamine quinones, remains underexplored: while clearly cytotoxic to neurons, their direct influence on microglial receptor signaling, redox balance, and inflammasome priming has not been systematically defined [61].
Defining whether microglia sense and respond differently to dopamine versus its metabolites will be critical to understanding disease-specific pathophysiology. Regional and temporal specificity further complicate interpretation. Microglial responses to dopamine likely differ between substantia nigra, ventral tegmental area, striatum, and prefrontal cortex, and these trajectories may shift across disease stages [10,12]. Determining when and where microglia transitions from protective to deleterious roles will require harmonized approaches that integrate longitudinal TSPO-PET imaging, cerebrospinal fluid cytokine readouts, and single-cell transcriptomic profiling. Finally, stress and depression represent underexplored modulators of this axis. While chronic stress is known to prime microglia toward pro-inflammatory states, direct evidence linking dopamine receptor remodeling in microglia to stress-induced anhedonia or motivational deficits remains limited, underscoring the need for targeted molecular and imaging studies in affective disorders [12].
Collectively, these gaps highlight the urgent need for a mechanistic framework that accounts for dose, receptor balance, metabolite exposure, regional context, and systemic immune status. Only through such multidimensional integration can the field fully delineate how microglial–dopamine feedback loops shape vulnerability to neuroinflammation, neurodegeneration, and psychiatric disease.
4. Astrocyte–Microglia Interactions in Dopamine Circuits
4.1. From Tripartite to Network Synapses: Expanding the Framework of Dopaminergic Circuit Regulation
The classical tripartite synapse, originally described as the interplay between pre- and postsynaptic neurons with perisynaptic astrocytic processes, has been substantially revised. Accumulating evidence supports an expanded quadripartite model in which microglia act as active and contact-competent partners that influence synapse formation, elimination, and efficacy. Complement-dependent microglial pruning, first identified in development, is now recognized as a lifelong mechanism that contributes to activity-dependent remodeling of dopaminergic circuits in the striatum and prefrontal cortex [62,63,64]. Extending beyond this framework, the concept of the “network synapse” has emerged, integrating astrocytic endfeet, perivascular microglia, endothelial cells, and pericytes. This expanded view underscores the role of vascular and immune influences ranging from metabolic alterations to systemic inflammatory states in modulating dopaminergic plasticity [65,66]. By situating dopamine synapses within a glial–vascular–immune ecosystem, the field now conceptualizes dopaminergic signaling as dynamically embedded in systemic physiology and inflammatory tone.
4.2. Molecular Axes of Astrocyte–Microglia Crosstalk in Dopamine Regulation
Astrocytes and microglia engage in reciprocal signaling loops that exert direct influence on dopaminergic circuits. Cytokine- and complement-mediated communication is a central axis of this dialogue: microglia-derived IL-1β, TNF-α, and complement proteins drive astrocytes toward phagocytic and pro-inflammatory states, while astrocytic secretion of IL-6 and TGF-β conditions microglial polarization and establishes thresholds for synaptic engulfment (Table 3) [67,68,69,70]. Recent single-cell and spatial transcriptomic analyses demonstrate that astrocytic reactivity cannot be reduced to a binary A1/A2 paradigm but instead reflects heterogeneous and region-specific states that variably shape dopaminergic vulnerability [71,72].
Other communication channels reinforce this interglial interplay. Purinergic signaling synchronizes dopamine release with immune surveillance, as astrocytic regulation of adenosine and GABA tunes cholinergic interneurons—the principal gatekeepers of striatal dopamine release—while microglial purinergic receptor activity regulates motility and surveillance behaviors [73,74,75].
Chemokine-based signaling, particularly the CX3CL1–CX3CR1 axis, further coordinates astrocytic coverage with microglial process engagement, with shifts in fractalkine sources during pathology linked to maladaptive microglial activation and dopaminergic vulnerability [76,77]. Additionally, extracellular vesicles released by astrocytes, microglia, and neurons carry cytokines, miRNAs, and metabolic cargo that function both as biomarkers of glial state and as effectors of inflammatory propagation and synaptic remodeling [78,79,80].
Taken together, these reciprocal pathways form feed-forward and feedback networks in which astrocytic gliotransmission and microglial activation converge to regulate dopamine release dynamics, receptor composition, and long-term plasticity. In this framework, astrocyte–microglia interactions do not serve as a background modulatory system but constitute a computational layer of dopaminergic circuitry, integrating neuromodulatory, immune, and vascular signals to shape both resilience and disease vulnerability [81].
4.3. Spatial and Single-Cell Landscapes of Astrocytic and Microglial Heterogeneity in Dopaminergic Circuits
Recent advances in single-cell and spatial multi-omics have provided unprecedented resolution of astrocytic and microglial diversity in dopaminergic hubs. In the human substantia nigra, single-cell and single-nucleus atlases consistently identify oxidative–metabolic astrocyte subtypes enriched for genes involved in antioxidant defense, mitochondrial regulation, and lactate shuttle pathways (Table 3) [82]. In parallel, immune-primed microglial clusters have been mapped, characterized by interferon-stimulated genes, inflammasome components, and lipid metabolism programs, suggesting that local glial heterogeneity is a critical determinant of dopaminergic resilience versus vulnerability [83].
In Parkinson’s disease, integrated single-nucleus and spatial multi-omics analyses reveal the emergence of glial–immune niches defined by close apposition of reactive astrocytes, inflammatory microglia, and infiltrating T cells to degenerating dopamine neurons [84]. These niches exhibit coordinated transcriptomic programs involving antigen presentation, cytokine signaling, and oxidative stress responses, supporting the concept that selective dopamine neuron loss arises not from neuron-autonomous processes but from interglial–immune coupling within the microenvironment.
Spatial mapping in experimental models has further delineated inflammatory gradients across the substantia nigra, showing that astrocytic and microglial activation states are spatially orchestrated around dopamine territories. These gradients correlate with mitochondrial dysfunction and synaptic attrition in nearby neurons, highlighting the regional coordination of glial–neural interactions during degeneration [85,86]. In the striatum, longitudinal single-cell analyses across development and aging demonstrate protracted, lineage-specific transcriptional programs in astrocytes and microglia that shape the rules of dopamine receptor–specific plasticity. These studies define dynamic “glial neighborhoods” that interact with D1- and D2-receptor ensembles, embedding reinforcement learning mechanisms within a glial context [87,88].
An emerging integrative theme across datasets is that astrocytic diversity predominates under homeostatic conditions, while microglial heterogeneity expands dramatically under disease or inflammatory stress. This reciprocal choreography; astrocytic specialization in health and microglial diversification in pathology; recalibrates dopaminergic microenvironments, with direct implications for neuronal resilience and selective vulnerability [84,89].
4.4. Interglial Mechanisms as Architects of Dopaminergic Signaling and Plasticity
The molecular and cellular heterogeneity revealed by single-cell approaches has direct consequences for the rules of dopamine signaling and plasticity. Astrocytic regulation of extracellular adenosine and GABA determines the excitability of striatal cholinergic interneurons, which in turn set the timing and amplitude of sub-second dopamine release. Perturbations of this axis can flip the polarity of dopamine receptor modulation, altering interneuron firing and dopamine availability [10,73]. In addition, astrocytic G-protein–coupled receptor heteromers, such as A2A–D2, A2A–oxytocin receptor, and D2–oxytocin receptor complexes, act as molecular logic nodes that fine-tune release probability and plasticity thresholds, conferring context-dependent precision to dopamine signaling (Table 3) [25,90].
Microglial complement-dependent pruning further contributes to circuit refinement by selectively sculpting dopaminergic synapses. In coordination with astrocytic “eat-me” and “keep-me” signals, this process determines the balance between D1- and D2-biased ensembles and thereby the rules of corticostriatal plasticity and reinforcement learning [63,91]. Importantly, complement-tagged pruning persists into adulthood, implying that adaptive and maladaptive remodeling of dopamine circuits under stress, drug exposure, or disease arises from ongoing glial surveillance [92,93].
Metabolic and vascular coupling provides a third axis of regulation. Astrocytic endfeet and perivascular microglia integrate glucose and lactate shuttling, reactive oxygen species buffering, and blood–brain barrier integrity. Under mitochondrial stress or α-synuclein accumulation, these glial–vascular checkpoints become decisive determinants of dopamine neuron survival [84,94]. Disruption of astrocytic metabolic support or microglial vascular surveillance precipitates neuronal loss in both experimental models and human Parkinson’s disease tissue, underscoring the centrality of glial metabolism in dopaminergic neurodegeneration [44,95,96].
Together, these mechanisms demonstrate that astrocyte–microglia interactions not only modulate dopamine circuits but define the computational rules of plasticity. By setting the balance between phasic and tonic release, determining synaptic selection and remodeling, and calibrating metabolic resilience, interglial signaling emerges as a primary architect of dopaminergic circuit function. Recent evidence thus reframes plasticity as a glia-dependent property of the striatum and midbrain, with astrocytic and microglial crosstalk positioned upstream of dopaminergic computation itself.
4.5. Interglial Microenvironments as Determinants of Dopaminergic Degeneration and Motivational Dysfunction
Single-nucleus and spatial multi-omics have revealed that dopaminergic degeneration in PD unfolds within glia-centric niches of vulnerability. In the substantia nigra, microglial populations enriched in lipid metabolism, interferon-response, and inflammasome-related transcripts colocalize with astrocytic states characterized by oxidative stress, mitochondrial dysregulation, and disrupted glutamate handling (Table 3) [84]. These convergent programs suggest that dopaminergic cell loss is not solely the consequence of intrinsic neuronal fragility but reflects maladaptive microenvironments in which astrocytic and microglial states synergistically amplify vulnerability.
Meta-analyses of single-nucleus RNA-seq datasets reinforce this perspective, consistently highlighting microglial neuroinflammation and astrocytic metabolic/trophic dysregulation as recurrent axes of disease progression [97]. A striking example of this interglial remodeling is the observed shift in fractalkine (CX3CL1) signaling. Neuronal CX3CL1 expression diminishes in PD, while endothelial expression increases, thereby disrupting CX3CR1-mediated homeostatic signaling and biasing microglia toward maladaptive surveillance and heightened inflammatory activity [77].
EVs have also emerged as central mediators of PD pathophysiology. Glia-derived EVs carry α-synuclein, inflammatory factors, and microRNAs that propagate pathology across dopaminergic territories [80]. At the same time, engineered EVs are being investigated as therapeutic carriers for anti-inflammatory or pro-metabolic cargo, offering a translational pathway for restoring dopaminergic resilience [98].
Chronic stress and motivational pathology reveal parallel themes of interglial remodeling. Sustained inflammatory states whether systemic or centrally generated retune astrocyte–microglia cytokine and purinergic signaling loops within mesocorticolimbic circuits. This remodeling reduces phasic dopamine signaling and contributes to motivational deficits, including anergia, anhedonia, and apathy [99,100]. Transcriptomic studies show that prolonged stress expands the repertoire of reactive astrocytic and microglial states, many of which impair dopamine release and receptor-specific plasticity in the ventral striatum and prefrontal cortex [101]. These findings support a mechanistic framework in which glial heterogeneity mediates the link between inflammatory tone, dopaminergic dysfunction, and stress-related psychiatric comorbidities such as depression and apathy.
4.6. In Vitro Humanized Models of Interglial Signaling in Dopamine Circuits: Toward Mechanistic Precision and Translation
Recent progress in human iPSC-derived co-culture and tri-culture systems has enabled systematic dissection of astrocyte–microglia–neuron interactions under controlled conditions. These platforms faithfully recapitulate key inflammatory axes including IL-1β/TNF-α loops, complement signaling, and purinergic cascades while providing real-time readouts of dopaminergic endpoints such as synapse density, electrophysiological excitability, and extracellular vesicle cargo [102]. Critically, they bridge the gap between reductionist assays and in vivo complexity, offering scalable platforms for mechanistic perturbation and therapeutic discovery.
Interventions in these models have already demonstrated translational promise. For example, inhibition of complement pathways or modulation of P2Y12 signaling in tri-cultures normalizes dopaminergic neuron excitability, underscoring the causal role of specific interglial modules (Table 3) [103]. These advances highlight the utility of next-generation co-culture systems not only for mechanistic dissection but also as preclinical pipelines for identifying candidate therapies.
4.7. Computational Neuroscience of Glial–Dopamine Interactions: From Molecular States to Circuit-Level Algorithms
Theoretical neuroscience has begun to incorporate astrocytic and microglial states into models of dopamine circuits, reframing glia as computational rather than modulatory elements. Contemporary frameworks treat glial calcium dynamics, receptor occupancy, metabolic buffering, and gliotransmission as slow variables that define neuronal gain control, eligibility traces, and synaptic plasticity thresholds [104]. This reconceptualization positions interglial signaling as a “plasticity thermostat” that sets the computational boundaries within which dopaminergic teaching signals operate [19,105].
Embedding glial diversity into reinforcement-learning and network models now enables simulations that predict how inflammatory or metabolic perturbations bias behavioral strategies for example, shifting exploration–exploitation balance, altering habit consolidation, or impairing motivational drive. Such integrative approaches generate testable hypotheses linking molecular glial states to system-level dysfunction across PD, depression, and addiction.
Advancing this agenda requires a set of clearly defined priorities. Foremost is the need for causal mapping, which will demand simultaneous resolution of dopaminergic dynamics and glial states in vivo. This will require the integration of fast-scan cyclic voltammetry or comparable dopamine sensors with glial-specific optical reporters and spatial transcriptomic approaches in behaving animals, thereby enabling direct linkage between interglial signaling modules and dopaminergic output. Equally critical is the delineation of region-specific motifs, achieved by systematically comparing interglial signatures across the substantia nigra, ventral tegmental area, striatum, and prefrontal cortex using integrated single-cell, spatial, and proteogenomic atlases. Parallel translational pipelines should focus on therapeutic logic, rigorously testing targeted ligands for astrocytic GPCR heteromers, modulators of purinergic cascades, and engineered extracellular vesicles in both human iPSC-derived tri-culture systems and in vivo models. Finally, computational integration will be indispensable. Embedding glial states into decision-theoretic and reinforcement-learning models will provide a framework for predicting how inflammatory and metabolic set points reshape dopaminergic computations and downstream behavioral algorithms. By bridging molecular modules to system-level functions, these convergent strategies aim to construct a mechanistic continuum from interglial biology to the computational architecture of dopamine circuits.
5. Dopamine–Glia Interfaces in Motivation and Stress
Motivation and stress engage partially overlapping mesolimbic and mesocortical circuits, with DA signaling orchestrating key processes such as reward anticipation, effort allocation, reinforcement learning, and stress adaptation [106,107]. While neurons have traditionally been viewed as the primary drivers of these processes, recent evidence demonstrates that astrocytes and microglia exert active, phase-specific, and circuit-localized control over DA dynamics. These glial mechanisms shape cue encoding, regulate strategy selection between goal-directed and habitual actions, and contribute to susceptibility or resilience in stress-induced anhedonia (Table 4). Importantly, the convergence of causal manipulation tools (optogenetics, chemogenetics) with phase-resolved monitoring approaches (GRAB-DA fibre photometry, calcium imaging) has enabled precise dissection of how glial activity governs anticipatory versus consummatory epochs of reward processing [108,109].
5.1. Astrocytic Modulation of Reward-Seeking Behavior and Motivational States
A landmark advance has been the identification of activity-defined astrocytic ensembles within the posterior–ventral nucleus accumbens (NAc). Using a light-dependent transcriptional reporter, a sparsely distributed subset of astrocytes was shown to be selectively recruited during cue–reward learning [110]. Optogenetic reactivation of this ensemble alone without indiscriminate stimulation of the broader astrocytic population was sufficient to drive cue-motivated approach behavior, revealing an ensemble-level astrocytic code for motivational salience. These findings align with broader evidence that astrocytes exert input-specific, temporally precise neuromodulation rather than broad gain control, reframing them as active encoders of motivational signals [23].
Astrocytes in the external globus pallidus (GPe) play a regulatory role in motivational flexibility. Chemogenetic activation of GPe astrocytes has been shown to reduce habitual responding and enhance goal-directed actions in operant tasks [111]. More recent work has extended these findings, showing that GPe astrocytes are selectively recruited during reward-seeking action sequences and contribute to action-sequence refinement and strategic updating under repetitive conditioning paradigms [112]. Together, these observations highlight a circuit-level mechanism by which astrocytes influence striato-pallidal computations to tune the balance between motivational persistence and behavioral flexibility.
Stress paradigms emphasize the vulnerability of the anticipatory phase of DA signaling to glial modulation. In the chronic social stress (CSS) model, GRAB-DA fibre photometry recordings demonstrated selective attenuation of NAc DA activity during reward anticipation, while consummatory DA responses remained intact [113]. This anticipatory deficit was tightly correlated with impaired effort allocation in progressive-ratio tasks and delayed reward learning. Given their role in regulating cue encoding, extracellular glutamate/ATP/adenosine balance, and D-serine release, astrocytes are strong candidates for mediating this vulnerability. Failure of astrocytic support during stress likely disrupts terminal excitability and DA release probability, weakening motivational vigor and reward pursuit [113,114].
5.2. Microglial Contributions to Motivational Deficits, Anhedonia, and Stress
Microglia also play a critical role in shaping motivational circuits, particularly under chronic stress. Prolonged stress reliably activates microglia across mesocorticolimbic regions, inducing morphological hypertrophy, upregulation of immune-related genes, and remodeling of synaptic architecture in the NAc, PFC, and ventral tegmental area (VTA) [115]. These cellular and molecular changes correlate with impaired reward learning, diminished effort allocation, and increased vulnerability to stress-induced anhedonia. Moreover, psychostimulant exposure under prior stress conditions amplifies microglial reactivity, exacerbating motivational rigidity and addiction-like behaviors [116].
Inflammatory signaling provides a key mechanistic axis for these effects. In chronic unpredictable mild stress (uCMS) paradigms, animals exhibit elevated levels of IL-1β and TNF-α, changes that co-occur with anhedonic behaviors such as reduced sucrose preference [117,118]. These cytokine elevations are closely linked to dopaminergic pathway disruption, supporting a cytokine-to-DA signaling axis through which neuroinflammation undermines motivation. Clinical studies of major depressive disorder (MDD) reinforce these findings, consistently reporting microglial overactivation and elevated inflammatory markers in affected individuals [119,120]. Collectively, these insights converge on the view that microglia represent a tractable therapeutic target for restoring motivational drive by modulating dopaminergic signaling and network excitability.
Developmental perspectives further underscore the role of microglial priming in long-term motivational vulnerability. Early-life stress (ELS) is consistently associated with persistent elevations in pro-inflammatory cytokines, dysregulated hypothalamic–pituitary–adrenal (HPA) axis function, and blunted anticipatory reward responses, findings supported by both human neuroimaging and rodent models [121,122]. These data suggest that microglia undergo priming during sensitive developmental windows, lowering their threshold for inflammatory reactivity to later stressors. Complementary developmental studies demonstrate that microglia regulate NAc synaptogenesis during adolescence, sculpting excitatory–inhibitory balance in motivational circuits. Perturbations of this process through stress, immune activation, or genetic risk can durably reweight mesolimbic connectivity, biasing individuals toward long-term motivational deficits [123]. Together, these findings underscore the developmental origins of motivational pathology, with microglial priming serving as a key mechanistic link between early adversity and later impairments in reward processing.
5.3. Glial Mechanisms Modulating Dopamine-Dependent Motivational Circuits
Astrocytic gliotransmission and uptake exert a central influence on motivational encoding. Activity-defined astrocytic ensembles within the NAc, recruited during cue–reward associations, release gliotransmitters such as glutamate, ATP, and D-serine while simultaneously controlling their clearance from the extracellular space. These processes tune medium spiny neuron excitability and modulate dopamine terminal release probability, with particularly strong effects during anticipatory epochs of reward processing, thereby shaping motivational drive [81,110].
Within the external GPe, astrocytes regulate computations of the indirect pathway that determines strategy selection. Chemogenetic activation experiments demonstrate that astrocytic signaling in this region suppresses habitual action patterns and biases behavior toward flexible, outcome-sensitive strategies. This astrocytic influence represents a mechanism for balancing motivational persistence with adaptive flexibility under shifting reward contingencies [111].
Microglia likewise shape motivational states through cytokine–dopamine coupling. Reactive microglia release pro-inflammatory mediators such as IL-1β and TNF-α, which suppress dopamine synthesis in midbrain neurons, impair receptor expression in the NAc and PFC, and prune dendritic spines. These actions collectively blunt reward anticipation and promote anhedonia. Framing cytokines as neuromodulators of motivational valence highlights their dual role as both immune effectors and circuit-level regulators [124,125].
Developmental factors further determine the sensitivity of glial–dopamine interactions. Early-life stress and immune perturbations induce lasting microglial priming, biasing microglial states toward hyper-reactivity upon later stress exposure. This developmental priming alters the maturational trajectory of NAc connectivity and mesolimbic dopamine signaling, producing enduring phenotypes of reduced effort expenditure and diminished motivational resilience [12].
5.4. Behavioral Readouts and Experimental Toolchains for Dopamine–Glia Interactions
Recent advances in behavioral neuroscience and recording technologies have provided precise readouts of dopamine–glia interactions across motivational and stress paradigms. One of the most transformative has been phase-specific dopamine monitoring using genetically encoded GRAB-DA sensors in combination with fibre photometry. This approach enables discrimination between anticipatory and consummatory dopamine signals, revealing that chronic stress selectively attenuates anticipatory activity while sparing consummatory responses, a dissociation that identifies a mechanistic window for glial intervention [126,127].
Operant behavioral assays complement neurochemical recordings by indexing motivational strategy with high resolution. Progressive ratio schedules quantify effort-based responding, while outcome devaluation paradigms distinguish habitual from goal-directed action control [128]. These measures are highly sensitive to astrocytic manipulations within the GPe, where chemogenetic activation shifts behavior away from rigid habit formation toward flexible, outcome-driven strategies [111].
Causal approaches extend this framework by directly manipulating astrocytic ensembles. Optogenetic and chemogenetic interventions in activity-defined astrocytic populations of the NAc demonstrate that selective reactivation of these ensembles is sufficient to bias cue-driven approach behavior. Such findings establish that astrocytic ensembles encode motivationally salient information and can directly influence behavioral output [110,129].
Stress paradigms, including CSS, uCMS, and ELS, provide translationally relevant models for probing the intersection of immune activation with dopaminergic signaling. When combined with cytokine profiling and phase-resolved dopamine photometry, these paradigms map immune–dopamine–behavior triads, clarifying how systemic inflammation, glial reactivity, and motivational impairments converge [130,131].
Taking together, this expanding methodological toolchain integrates behavioral assays, real-time neurochemical monitoring, and causal manipulations to generate mechanistically precise and clinically relevant insights into glial regulation of motivation.
5.5. Glial–Dopamine Crosstalk Across Disorders: Shared Mechanisms of Motivation, Stress, and Neuroinflammation
Across psychiatric and stress-related disorders, convergent evidence implicates glial–dopamine interfaces as central mediators of motivational pathology. In major depressive disorders, both human neuroimaging and preclinical models consistently demonstrate attenuated anticipatory dopamine responses during reward expectation tasks. These deficits are paralleled by heightened microglial activation and amplified cytokine signaling, suggesting that neuroinflammatory cascades act as critical drivers of anhedonia. Translational studies now indicate that interventions targeting glial pathways, including cytokine inhibitors and adenosine A2A receptor modulators, may restore anticipatory dopamine signaling and thereby alleviate motivational impairments (Figure 2) [23,132,133].
In addiction and substance use disorders, psychostimulant exposure robustly engages microglial programs within the nucleus accumbens, disrupting the coupling between glutamate and dopamine and leading to maladaptive changes in synaptic plasticity and motivational salience. Under conditions of chronic stress, these drug-induced adaptations are exacerbated, creating a dual-hit scenario in which stress-driven microglial activation converges with drug-induced neuroplasticity to reinforce compulsive drug-seeking behaviors. This interplay underscores immune–glial signaling as a central mechanism regulating addiction vulnerability at the nexus of stress reactivity and reinforcement learning [134].
Immune-mediated suppression of anticipatory dopamine signaling has also been observed in conditions characterized by pathological fatigue and effort intolerance, including chronic fatigue syndrome, fibromyalgia, and stress-related medical syndromes. Elevated cytokines and sustained microglial activation in these contexts appear to blunt reward anticipation and disrupt effort valuation. This integrative framework provides a unifying account of motivational fatigue that bridges psychiatric symptomatology with somatic illness [135,136].
Emerging insights further suggest a role for glial modulation in attention-deficit/hyperactivity disorder, though causal evidence remains limited. Astrocytic gating of anticipatory dopamine signaling and microglial regulation of motivational flexibility are hypothesized to contribute to deficits in sustained effort, altered reward sensitivity, and impulsiveness. While speculative, these hypotheses position glial biology as a novel frontier in the mechanistic understanding of ADHD [137,138].
Despite these advances, fundamental questions remain regarding the precise dynamics by which glial–dopamine interfaces regulate motivation across stress and disease states. Temporal specificity constitutes a major unresolved dimension: it is unclear which phases of stress exposure—acute versus chronic, or anticipatory versus consummatory—are most susceptible to glial modulation. Closed-loop paradigms that integrate real-time dopamine monitoring with selective glial stimulation hold promise for resolving these dynamics with unprecedented precision. Another critical challenge involves subtype and state specificity. Single-cell and spatial multi-omics have revealed striking heterogeneity between nucleus accumbens shell and core astrocytes, as well as diverse microglial states ranging from homeostatic to pro-inflammatory. Yet, causal links between these transcriptional programs and motivational behaviors remain to be established.
Equally unresolved are the mechanisms by which gliotransmitters contribute to anticipatory gain control. The relative influence of adenosine, glutamate, and D-serine remains poorly defined, and advances will likely require transmitter-specific biosensors combined with astrocyte-restricted manipulations. From a translational standpoint, immune–dopamine bridges represent especially promising points of intervention. Strategies aimed at modulating IL-1β and TNF-α signaling or tuning adenosine A1/A2A pathways may restore anticipatory dopamine function and motivational vigor in stress-linked anhedonia, though rigorous clinical testing is still lacking.
Finally, developmental factors must be considered as early-life stress has been shown to prime microglial reactivity and disrupt the maturation of mesocorticolimbic circuitry, establishing long-term vulnerability to motivational pathology. Longitudinal studies integrating immune profiling, multimodal imaging, and behavioral phenotyping will be essential to identify critical preventive windows and to design strategies that recalibrate glial dopamine coupling before maladaptive trajectories become entrenched.
6. Dopamine–Glia Crosstalk in Neurodegeneration and Disease
Pathological states such as neurodegeneration, chronic infection, inflammation, psychological stress, and aging can progressively transform astrocytes and microglia from supportive partners into maladaptive phenotypes [139]. In the dopaminergic system, these shifts converge on mechanisms including redox imbalance, glutamate dysregulation, mitochondrial and ferroptotic vulnerability, extracellular matrix (ECM) remodeling, and the exosomal propagation of inflammatory signals, ultimately eroding neuronal resilience (Table 5) [140,141]. The advent of high-resolution multi-omics, in vivo imaging, and functional dissection has begun to delineate how these pathways collectively drive dopaminergic circuit fragility [142,143].
6.1. Glial–Dopamine Interfaces in Parkinson’s Disease: Inflammatory Reprogramming and Ferroptotic Stress
In PD, astrocytes undergo marked state transitions that critically influence dopaminergic neuron survival. Postmortem analyses and preclinical models consistently demonstrate the accumulation of A1-like, or “neurotoxic,” astrocytic programs within substantia nigra microenvironments [144,145,146]. These astrocytes exhibit heightened inflammatory and oxidative profiles, impaired EAAT2/GLT-1–mediated glutamate clearance, and diminished trophic and metabolic support [147]. Scientometric analyses of the PD–astrocyte literature highlight the rapid rise of A1 conversion as a conceptual anchor, reflecting growing recognition that astrocytic phenotype switching is mechanistically linked to dopaminergic vulnerability [148].
Microglia are principal drivers of this maladaptive transition. Cytokines such as IL-1α, TNF-α, and C1q induce A1 polarization, causing astrocytes to lose essential homeostatic functions, including glutamate buffering, potassium regulation, and metabolic shuttling—while gaining complement- and cytokine-mediated neurotoxicity [149]. These changes are especially damaging in the substantia nigra, where the oxidative burden of dopamine metabolism imposes intrinsic stress. Importantly, activated microglia is necessary for A1 induction, positioning microglial signaling upstream of astrocytic maladaptation [149,150].
Microglial state transitions themselves are central to PD progression. In α-synuclein overexpression models, inhibition of the colony-stimulating factor 1 receptor (CSF1R) with PLX5622 reduces microglial numbers, attenuates dopaminergic neurodegeneration, improves motor outcomes, and remodels ECM-associated transcriptional networks [151,152]. These findings implicate microglia–ECM crosstalk as a determinant of synaptic and circuit integrity [153]. In addition, microglia-derived exosomes function as carriers of inflammatory and pathological cargo, including molecules that convert astrocytes into A1-like states. Disrupting this vesicular axis through targets such as Peli1 has been proposed as a strategy to interrupt glia-to-glia amplification loops [154,155].
Converging evidence from seeded α-synuclein models further indicates that microglial depletion via CSF1R blockade attenuates α-syn propagation and protects dopaminergic neurons, reinforcing the concept that microglia act as gatekeepers of synucleinopathy spread [152] .
The intrinsic bioenergetic and metabolic architecture of dopaminergic neurons renders them acutely susceptible to ferroptotic death. Elevated iron turnover, catecholamine autoxidation, and the exceptionally high oxidative load of mitochondrial respiration collectively create a ferroptosis-permissive milieu [156]. Within this context, maladaptive astrocytic and microglial states act as critical amplifiers of ferroptotic stress. Experimental pharmacology underscores this vulnerability: treatment with ceftriaxone attenuates glial activation while suppressing ferroptosis through restoration of SLC7A11/GPX4 antioxidant defenses [157]. These effects translate into robust preservation of dopaminergic viability in both in vitro and in vivo models of Parkinson’s disease, thereby linking astrocytic glutamate clearance, redox homeostasis, and iron–lipid peroxidation dynamics to actionable therapeutic targets [158].
Recent multi-omics and network-level studies have further mapped iron homeostasis and lipid peroxidation cascades onto astrocytic and microglial gene regulatory programs, identifying glial populations as arbiters of ferroptotic thresholds in dopaminergic circuits [159,160]. This emerging framework provides a compelling rationale for glia-directed anti-ferroptotic interventions. Strategies under investigation include GPX4 activators, modulators of cystine–glutamate exchange, and approaches aimed at stabilizing astrocytic antioxidant and metabolic defenses. Together, these insights delineate a promising therapeutic frontier in the modification of Parkinson’s disease progression.
6.2. Immune–Glial Coupling and Dopamine Circuit Failure: Basal Ganglia Pathophysiology in Post-COVID States
Accumulating evidence indicates that viral infections, particularly SARS-CoV-2, exert lasting impacts on basal ganglia circuits, with significant consequences for dopaminergic signaling [161]. Multimodal neuroimaging studies consistently identify structural and functional alterations in the putamen, pallidum, and caudate nucleus, including reduced gray matter volume, disrupted metabolic activity, and impaired functional connectivity [162]. These abnormalities correlate with clinical phenotypes of Long COVID—fatigue, effort intolerance, motivational deficits, and cognitive slowing—symptoms that map directly onto dopamine-dependent computations of reward valuation and cognitive vigor.
Mechanistically, these outcomes converge on disruption of corticostriatal loops central to effort–reward integration. Sustained neuroinflammation, driven by persistent immune activation, is proposed to impair dopaminergic function through multiple suppression of dopamine synthesis, reduction in release probability, and downregulation of receptor sensitivity [163,164]. Such immune-driven imbalances establish a state of dopaminergic insufficiency under chronic inflammatory pressure.
Biomarker studies provide further support for this framework, consistently reporting elevated glial fibrillary acidic protein (GFAP), indicative of astrocytic injury, alongside pro-inflammatory cytokines including IL-6 and TNF-α in post-COVID cohorts [165,166]. Evidence of blood–brain barrier (BBB) disruption, mediated by inflammatory endothelial signaling and astrocytic endfoot dysfunction, reinforces the view of systemic–central immune coupling as a key driver of dopaminergic vulnerability. In this model, peripheral cytokine tone sustains central glial priming, which in turn alters basal ganglia homeostasis through tetrahydrobiopterin depletion, dysregulated dopamine transporter (DAT) kinetics, and reduced D1/D2 receptor availability. The outcome is a failure of neuromodulatory gain control within corticostriatal circuits, providing a mechanistic substrate for the motivational and cognitive deficits that characterize Long COVID [100,166].
6.3. Aging and the Glial–Dopamine Interface: Dynamic Amplification of Neuroinflammatory and Metabolic Stress
Aging functions not merely as a background risk factor but as a biological amplifier of glial–dopamine dysfunction. Microglia in aged brains adopt a “primed” phenotype, marked by lower activation thresholds and a pro-inflammatory bias. This state leads to exaggerated responses to secondary insults, such as viral infections or α-synuclein aggregates, and impairs resolution of inflammation, thereby prolonging dopaminergic stress exposure [43,167].
Astrocytic decline with age compounds this vulnerability. Aging astrocytes display impaired glutamate clearance, reduced lactate shuttling, diminished antioxidant buffering, and altered calcium signaling [96]. These deficits undermine metabolic and synaptic support for dopamine neurons, promoting excitotoxic and oxidative stress. Concurrently, age-related mitochondrial decline in glial populations enhances reactive oxygen species (ROS) accumulation and lipid peroxidation, amplifying damage to dopamine-rich circuits [168].
Recent scientometric analyses of Parkinson’s disease datasets emphasize the age–reactivity interaction, demonstrating that older cohorts exhibit more pronounced inflammatory astrocytic signatures and accelerated dopaminergic degeneration [169]. Together, these findings establish aging as a dynamic amplifier of maladaptive glial states, converging with neuroinflammation, mitochondrial stress, and systemic insults to erode dopaminergic resilience.
6.4. Glial Reprogramming as a Convergent Amplifier of Dopaminergic Dysfunction
Across Parkinson’s disease, viral insults such as Long COVID, and aging, several mechanistic axes converge to illustrate how glial reprogramming amplifies dopaminergic dysfunction.
A central pathway is the microglia-to-astrocyte signaling cascade, in which activated microglia secrete IL-1α, TNF-α, and C1q, driving the conversion of astrocytes into A1 neurotoxic states. Once reprogrammed, these astrocytes lose essential homeostatic functions, including glutamate buffering, potassium regulation, and trophic/metabolic support while acquiring neurotoxic complement and cytokine activities. This switch establishes a feed-forward inflammatory loop that accelerates dopaminergic degeneration, particularly in oxidative-stress–vulnerable regions such as the substantia nigra [149].
Another recurrent theme is ECM remodeling as a disease amplifier. In α-synuclein models, pharmacological depletion of microglia through CSF1R inhibition not only attenuated dopaminergic neurodegeneration but also reprogrammed ECM networks. These findings highlight that ECM–microglia crosstalk is a decisive determinant of synaptic stability and axonal survival, positioning ECM remodeling as a potential therapeutic axis to stabilize nigrostriatal architectures [170,171].
Glial communication is also propagated through exosomal transfer of pathological cargo. Microglia release extracellular vesicles enriched in pro-inflammatory proteins, miRNAs, and α-syn aggregates, which in turn induce astrocytic conversion toward A1-like states and propagate neuroinflammatory tone across local microenvironments. Targeting pathways of exosome biogenesis, trafficking, and uptake therefore offers a tractable opportunity to disrupt this glia–glia amplification loop and mitigate progressive dopaminergic vulnerability [172,173].
A further point of convergence lies in ferroptosis coupling to glial metabolism. Dopaminergic neurons are intrinsically ferroptosis-prone due to their high oxidative load, iron turnover, and catecholamine autoxidation. Glia modulates this vulnerability through SLC7A11/GPX4 antioxidant pathways and astrocytic GLT-1–mediated glutamate clearance, both of which serve as critical buffers against iron-lipid peroxidation. Pharmacological interventions such as ceftriaxone provide proof-of-concept that enhancing these pathways suppresses lipid peroxidation and ferroptotic stress, thereby conferring neuroprotection in dopaminergic circuits[174,175,176,177].
Finally, immune priming and BBB dysfunction emerge as unifying features in chronic inflammatory states. Biomarker studies in Long COVID consistently report elevated GFAP, increased systemic cytokine tone, and markers of BBB leak, collectively reflecting prolonged glial activation and impaired dopaminergic resilience [178,179]. BBB disruption thus represents both a mechanistic driver of central vulnerability and a measurable endpoint for risk stratification and therapeutic monitoring.
Taken together, these convergent mechanisms position glia as active amplifiers of dopaminergic stress across neurodegeneration, viral sequelae, and aging. By integrating cytokine signaling, ECM remodeling, exosomal transfer, ferroptotic coupling, and barrier dysfunction, recent work reframes dopaminergic vulnerability as a systems-level phenomenon in which glial states orchestrate the trajectory from resilience to degeneration.
6.5. Reframing Glial Therapeutics: Targeting Microglial and Astrocytic Programs in Parkinson’s Disease and Beyond
Therapeutic efforts targeting dopamine–glia interactions are moving beyond broad immunosuppression toward approaches that emphasize state reprogramming, pathway modulation, and resilience promotion (Figure 3). Preclinical depletion of microglia using CSF1R inhibitors such as PLX5622 has demonstrated robust neuroprotection and attenuation of α-synuclein pathology [180]. Yet, complete ablation is neither feasible nor desirable in humans, as microglia are indispensable for immune surveillance, synaptic remodeling, and debris clearance. Consequently, translational strategies now prioritize microglial reprogramming, seeking to bias these cells toward reparative, phagocytic, and anti-inflammatory phenotypes while suppressing inflammasome-driven and neurotoxic states [43,83].
Parallel efforts focus on preventing astrocytic conversion into maladaptive A1-like states, a consistent feature across Parkinsonian models. This transition, driven by microglia-derived cytokines such as IL-1α, TNF-α, and C1q, deprives astrocytes of critical homeostatic functions and confers neurotoxic activity. Therapeutic avenues under development include cytokine neutralization, selective inhibition of astrocytic inflammatory pathways, and reinforcement of metabolic and trophic programs that preserve homeostatic astrocyte functions [181].
Interventions at the level of extracellular vesicle signaling are also gaining prominence. Microglia-derived vesicles transfer α-syn aggregates, pro-inflammatory proteins, and miRNAs that trigger astrocytic A1 conversion and propagate neuroinflammation across dopaminergic territories [155]. Strategies aimed at disrupting vesicle biogenesis, such as targeting ESCRT machinery or the Peli1 axis, or blocking vesicle uptake at recipient astrocytes, represent novel ways to interrupt glia-to-glia amplification loops and contain the spread of pathology.
A further therapeutic channel involves controlling ferroptosis, an iron-dependent form of cell death exacerbated in dopaminergic neurons by high oxidative stress and catecholamine autoxidation. Glial regulation of glutamate homeostasis through GLT-1 and antioxidant buffering via the SLC7A11/GPX4 pathway provides critical protection against this process. Ceftriaxone has been shown to enhance astrocytic glutamate clearance and antioxidant activity, thereby reducing excitotoxicity and ferroptosis [177,182]. Beyond this, next-generation anti-ferroptotic compounds designed to stabilize glial redox capacity and iron handling are advancing as promising translational candidates [160].
Non-pharmacological interventions such as exercise add an important dimension, functioning as endogenous modulators of glial states. Aerobic and resistance training in experimental Parkinson’s models have been shown to downregulate pro-inflammatory astrocytic and microglial markers, enhance mitochondrial biogenesis, and improve motor outcomes [183,184]. Exercise thus acts as a physiological reprogrammer, shifting glia toward pro-homeostatic phenotypes and reinforcing dopaminergic resilience.
Post-viral syndromes, including Long-COVID, highlight another translational frontier. Patients consistently exhibit elevated GFAP, pro-inflammatory cytokines, and BBB disruption [179,185]. Targeting cytokine signaling pathways, such as IL-6 and TNF-α, together with strategies to stabilize astrocytic endfeet and endothelial barrier integrity, provides a rational approach to mitigating chronic glial priming and dopaminergic fragility under conditions of sustained immune activation.
Despite these promising advances, key challenges remain. A central priority is the identification of predictive biomarkers, including GFAP, neurofilament light chain (NfL), extracellular matrix fragments, and glia-derived vesicular cargo, capable of forecasting dopaminergic decline prior to irreversible nigrostriatal loss. Equally critical is the delineation of resilience programs, as astrocytic metabolic pathways such as lactate shuttling and antioxidant buffering and microglial phenotypes that preserve synaptic integrity without inflammasome activation may sustain long-term dopaminergic stability. Comparative multi-omics across Parkinson’s disease and Long-COVID cohorts are also required to determine whether a shared vulnerability transcriptome, spanning cytokine signaling, extracellular matrix remodeling, and vesicular pathways, underlies cross-disorder risk. Finally, therapeutic precision must be optimized, with the timing, dosing, and safety of interventions such as CSF1R modulators, anti-A1 conversion agents, and anti-ferroptotic compounds rigorously defined in clinical settings and supported by biomarker evidence demonstrating on-target glial reprogramming in humans.
Collectively, these emerging strategies redefine the therapeutic horizon for dopamine–glia biology. The field is shifting from indiscriminate suppression toward precision reprogramming, with the ultimate objective of stabilizing glial ecosystems that support dopaminergic resilience across stress, disease, and aging.
7. Glia-Directed Therapeutic Paradigms in Dopaminergic Neurodegeneration
Emerging evidence increasingly positions glial state transitions, rather than neuronal degeneration alone, as the primary inflection point for dopaminergic circuit vulnerability. This recognition is transforming therapeutic paradigms from neuron-centric rescue toward glia-directed interventions that normalize astrocytic metabolism, restrain maladaptive microglial reactivity, and enable longitudinal monitoring of glial dynamics through molecular imaging and fluid biomarkers (Figure 4) [12,186]. By reframing therapeutic entry points at the astrocyte–microglia interface, interventions can be positioned upstream of irreversible neuronal loss, thereby extending the window for disease modification.
7.1. Astrocytic Interventions in Glial–Dopamine Interfaces
Astrocytes occupy a central role in this therapeutic shift, given their regulation of synaptic glutamate concentrations via excitatory amino acid transporters (EAAT1/2) and their control of oxidative tone through monoamine oxidase-B (MAO-B) activity. In reactive states, MAO-B expression is markedly upregulated, functioning both as a driver of oxidative stress and as a tractable theranostic target [187,188]. Advances in astrocyte-selective PET imaging underscore this translational opportunity. The tracer [^18F]SMBT-1 exhibits highly selective and reversible binding to MAO-B with minimal nonspecific uptake, allowing longitudinal quantification of reactive astrogliosis [189]. Initial human studies confirmed favorable kinetic properties, and more recent preclinical applications extended their utility to amyloid and tau transgenic models, where tracer signals correlated strongly with histological indices of astrocytic reactivity [190]. Importantly, SMBT-1 is now being integrated into multi-tracer paradigms, where astrocytic imaging is combined with TSPO-PET (for microglial activation) and FDG-PET (for metabolic flux), generating multidimensional “glial fingerprints” of disease trajectory and therapeutic response [191].
Therapeutic efforts are proceeding in parallel with next-generation MAO-B inhibitors. Unlike classical agents that act solely on enzymatic suppression, newer compounds incorporate antioxidant scaffolds, iron-chelating groups, and mitochondrial-supportive motifs. These multifunctional designs simultaneously mitigate upstream enzymatic overactivity and downstream cascades of reactive oxygen species and lipid peroxidation [192,193]. Complementing pharmacologic inhibition, astrocytic metabolic reprogramming has emerged as a broader strategy, targeting mitochondrial dynamics, lipid metabolism, and glutathione-dependent antioxidant defenses. Modalities include viral vectors driving EAAT overexpression, small molecules enhancing GPX4 activity, and interventions that stabilize iron-handling proteins, collectively positioning astrocytes as pivotal hubs of dopaminergic neuroprotection [193].
Ferroptosis has become a particularly salient frontier, defined as an iron-dependent cell death pathway driven by glutathione depletion and phospholipid peroxidation. While dopaminergic neurons are intrinsically ferroptosis-prone due to their metabolic profile, maladaptive astrocytic and microglial states further amplify this vulnerability. Dysregulated iron sequestration and lipid metabolism within glia accelerate oxidative stress and destabilize nigrostriatal circuits [194]. Recent studies indicate that microglial cytokine signaling can disrupt astrocytic iron handling, leading to the accumulation of labile iron pools that precipitate peroxidative damage. This establishes a self-reinforcing loop in which iron-driven lipid peroxidation fuels neuroinflammation, further compounding dopaminergic fragility [195,196].
As a result, therapeutic paradigms are shifting toward combinatorial approaches that jointly target oxidative and inflammatory axes. Ferroptosis inhibitors, such as liproxstatin-1 analogues, are being paired with anti-inflammatory modulators to simultaneously suppress lipid peroxidation and restrain cytokine-driven propagation of neuroinflammation [197]. Such dual targeting is increasingly recognized as essential to disrupting the glia–iron–inflammation cycle that underlies Parkinsonian progression.
7.2. Microglial Reprogramming: Inhibitors, Modulators, and Immune Tuning
Therapeutic strategies increasingly recognize that targeting microglial states rather than broadly suppressing neuroinflammation offers a more refined approach to preserving dopaminergic resilience. Among the most extensively studied interventions are CSF-1R inhibitors, including PLX5622 and PLX3397 [198]. Sustained administration of these agents in preclinical Parkinsonian models induces near-complete depletion of resident microglia, which in turn attenuates α-synuclein accumulation, dopaminergic neuron loss, and motor dysfunction [151]. Notably, dosing regimens critically shape outcomes: while chronic depletion reduces pathology, shorter pulse exposures reorganize α-synuclein inclusions into fewer but larger aggregates, thereby altering clearance demands and circuit remodeling trajectories [199]. Following drug withdrawal, microglial repopulation arises from central nervous system progenitors, generating a population with reduced inflammatory tone and enhanced trophic support [200]. This “reset” phenotype suggests that temporally controlled depletion–repopulation cycles may function as a reprogramming strategy with long-term therapeutic relevance.
More recent combinatorial approaches have integrated CSF-1R inhibition with neuromodulatory interventions such as gamma entrainment using sensory stimulation (GENUS) and environmental enrichment. These paradigms demonstrate synergistic effects, restoring network synchrony, enhancing learning and memory, and accelerating recovery in preclinical models [201]. Such findings highlight the potential for immune modulation and circuit-level entrainment to act in concert, recalibrating dopaminergic networks through coordinated glial and neuronal plasticity.
Beyond CSF-1R signaling, a growing repertoire of microglial pathways has emerged as therapeutic entry points. Receptors such as TREM2, progranulin (PGRN), SORT1, LILRB4, P2Y6R, and TAM family members regulate phagocytosis, lipid metabolism, apoptotic clearance, and cytokine release [202]. Modulation of these axes can bias microglia toward reparative states that support synaptic integrity and metabolic homeostasis, while simultaneously restraining feed-forward astrocytic conversion into neurotoxic phenotypes. In parallel, pharmacological repurposing efforts have explored broad-spectrum agents such as minocycline, which reduces oxidative stress, dampens pro-inflammatory cytokine release, and disrupts maladaptive astrocyte–microglia feedback loops [203]. Yet, despite extensive preclinical efficacy, clinical translation has been limited by concerns regarding specificity, blood–brain barrier penetrance, and sustained efficacy. These limitations underscore the need for next generation immunomodulators designed with state selectivity and precise temporal control.
7.3. Next-Generation Glial Control: Gene Therapy, Optogenetics, and Chemogenetics
The convergence of molecular engineering and circuit neuroscience is enabling direct manipulation of glial physiology with unprecedented precision. Advances in viral vector technology, particularly those leveraging astrocyte-selective promoters such as GFAP and ALDH1L1, allow for targeted delivery of therapeutic transgenes to glial populations [204]. This approach has been applied to augment glutamate clearance through EAAT overexpression, bolster antioxidant defenses via GPX4, and stabilize iron homeostasis by enhancing expression of iron-handling proteins, thereby restoring astrocytic buffering capacity against excitotoxic and oxidative stress.
Complementing gene therapy, optogenetic and chemogenetic strategies provide causal control of glial contributions to dopaminergic circuitry. Manipulation of astrocytic Ca²⁺ dynamics can bidirectionally regulate synaptic plasticity, while DREADD-based modulation of microglia has been shown to reconfigure immune tone and alter dopaminergic responsiveness in vivo [205]. With the integration of genetically encoded dopamine sensors, calcium indicators, and closed-loop control systems, these tools are now capable of mapping and manipulating glial activity in temporally and spatially defined behavioral epochs [206]. Such approaches position astrocytic and microglial neuromodulation not only as experimental probes of causality but also as potential therapeutic modalities, capable of precision reprogramming of glial states in circuit-defined contexts relevant to Parkinson’s disease and related dopaminergic disorders.
7.4. Glial Biomarkers and Translational Monitoring in Dopaminergic Circuit Pathophysiology
Advances in molecular imaging and fluid biomarker development are redefining how glial dynamics in dopaminergic circuits are monitored and translated into clinical frameworks. Among these, astrocytic MAO-B PET tracers, particularly [^18F]SMBT-1, have emerged as highly selective and non-invasive tools for quantifying astrocyte reactivity in vivo. Clinical studies confirm favorable pharmacokinetics, binding specificity, and reversibility, supporting their integration into longitudinal interventional trials [189,207]. Preclinical investigations further validate SMBT-1 by demonstrating close correspondence between tracer uptake and histological burden in transgenic amyloid and tau models, establishing its face validity as a biomarker of glial engagement [208].
An increasingly powerful paradigm involves multimodal imaging, in which astrocytic MAO-B PET is combined with TSPO-PET for microglial activation and metabolic or connectivity measures such as FDG-PET and diffusion MRI. These integrated pipelines generate multicellular progression signatures that distinguish disease stages, identify vulnerable network nodes, and track therapeutic response [209,210]. Complementary neuropathological studies mapping GFAP and other astrocytic markers across Parkinsonian brains further anchor these imaging endpoints as validated measures of regional progression [211,212].
Beyond imaging, biofluid assays represent an essential translational layer. Cerebrospinal fluid (CSF) and plasma studies increasingly support soluble TREM2, inflammatory cytokine panels, and metabolic microglial signatures as reproducible indicators of disease activity and treatment response [213,214]. Standardization of biomarker panels and longitudinal sampling protocols is critical to ensure reproducibility across cohorts and facilitate trial stratification.
EVs have gained particular attention for their dual role as mechanistic mediators and biomarkers. Microglia-derived EVs carry α-synuclein seeds and pro-inflammatory cargo, directly inducing astrocytic conversion into neurotoxic phenotypes and amplifying dopaminergic stress. Recent findings identify Peli1-containing EV cargo as a driver of astrocytic reactivity, underscoring exosomal pathways as both actionable therapeutic targets and minimally invasive biomarkers for clinical monitoring [215,216].
7.5. Repurposed Therapeutics and Precision Small-Molecule Modulators of Glial–Dopamine Pathophysiology
The therapeutic landscape increasingly recognizes repurposed agents as glia-active neuromodulators capable of buffering dopaminergic vulnerability. Glucagon-like peptide-1 receptor agonists (GLP-1RAs), developed originally for diabetes, have shown striking promise in preclinical and early clinical studies. In experimental systems, GLP-1RAs suppress microglial cytokine release, reprogramming microglia toward homeostatic phenotypes, while simultaneously enhancing astrocytic metabolic support by stabilizing mitochondrial function, augmenting glucose uptake, and mitigating excitotoxic stress [217,218,219]. Clinical pilot data in Parkinson’s disease suggest improvements in motor and cognitive outcomes, spurring multiple Phase II/III trials aimed at clarifying efficacy and mechanistic underpinnings [220]. Mechanistically, GLP-1RAs reduce α-synuclein aggregation and suppress downstream pro-inflammatory cascades, reinforcing their position within a glial-centric therapeutic framework [221].
Other agents, including minocycline, remain within the therapeutic repertoire due to their capacity to attenuate oxidative stress, inhibit microglial pro-inflammatory cascades, and disrupt astrocyte–microglia amplification loops [222,223]. However, their translation has been constrained by limited specificity, modest long-term tolerability, and concerns over systemic immune suppression. These limitations highlight the urgency of advancing precision small molecules capable of targeting discrete glial sub-states or signaling axes.
Despite growing therapeutic momentum, several translational challenges persist. Timing is critical; both microglial modulation through CSF-1R targeting and astrocytic buffering of metabolic stress are most effective in prodromal or early disease phases, necessitating pre-symptomatic detection via PET imaging (MAO-B, TSPO) and fluid biomarkers (EV cargo, soluble TREM2, cytokine panels). Selectivity remains another hurdle, as broad microglial depletion suppresses pathogenic reactivity but risks compromising essential homeostatic functions such as synaptic refinement and debris clearance. Consequently, state-specific interventions and optimized dosing regimens are being prioritized to preserve beneficial roles while curbing maladaptive phenotypes.
Heterogeneity of glial states further complicates therapeutic design. Single-cell and spatial transcriptomics reveal striking diversity between astrocytic and microglial sub-states across brain regions, disease stages, and demographic backgrounds, underscoring the need for cell-resolved targeting strategies guided by molecular classifiers and regional imaging markers. Finally, biomarker validation remains a pressing bottleneck. Candidate markers, including MAO-B PET, exosomal cargo signatures such as Peli1, soluble TREM2, and cfDNA fragments, require rigorous cross-platform validation, longitudinal reproducibility testing, and confirmation in ancestrally and clinically diverse populations. Without such harmonization, translation risks stagnation despite strong preclinical foundations.
8. Future Directions: Emerging Priorities in Glial–Dopamine Biology
Glial–dopamine interactions are now recognized as systems-level regulators of behavior, plasticity, and disease vulnerability, yet critical gaps remain. Regional heterogeneity, demographic moderators, computational formalization, and translational applications represent key frontiers. Systematically addressing these dimensions will establish a mechanistic foundation for targeted interventions in both psychiatric and neurodegenerative disorders.
8.1. Regional Specificity: Striatum, PFC, Hippocampus, and Beyond
Large-scale transcriptomic deconvolution of healthy human brain tissue reveals striking region- and sex-specific differences in glial composition. Analyses of GTEx datasets (>1,600 samples) indicate that astrocytic fractions increase with age in males across select cortical regions, whereas microglial proportions rise in females, suggesting baseline gradients that shape DA–glia coupling. These patterns are particularly relevant to cortico-striatal and hippocampal loops, where DA dynamics are tuned by astrocytic buffering and microglial surveillance.
The disproportionate vulnerability of SNc neurons in PD relative to VTA neurons is increasingly attributed to regional differences in astrocytic metabolic support, microglial inflammatory thresholds, and vascular–oxidative load. However, causal human evidence remains limited. Similarly, DA–glia interactions in the PFC and hippocampus remain underexplored, despite their established roles in executive control, working memory, and contextual learning.
Future studies should combine region-resolved single-cell and spatial multi-omics with in vivo DA measurements to define glial receptor repertoires, metabolic programs, and inflammatory thresholds across nuclei. Behavioral paradigms selectively probing PFC (working memory), striatum (reward learning), and hippocampus (contextual encoding) could yield region-specific signatures, closing a major translational gap.
8.2. Sex, Age, and Ancestry Effects in Glial–Dopamine Coupling
Sex differences in glial biology are robust and multifaceted. Recent syntheses highlight sexually dimorphic microglial morphology and functional states across cortex, basal ganglia, and hippocampus, largely modulated by hormonal context. Astrocytic density and morphology also show sex-linked variation, with implications for synaptic support and neuromodulation. These cellular distinctions map onto DA dynamics: fast-scan cyclic voltammetry demonstrates greater DA release in the nucleus accumbens core of females, alongside differential regulation in the dorsolateral striatum, underscoring sex-dependent tuning of reward circuits.
Aging further reshapes glial–DA interactions. Declines in astrocytic glutamate clearance and metabolic support, coupled with microglial priming toward pro-inflammatory states, progressively erode DA signal fidelity. Yet longitudinal human datasets linking glial aging trajectories to DA function remain absent, marking an urgent research priority.
By contrast, ancestry-linked variability in glial–DA coupling is virtually unexplored. While population-level variation influences PD prevalence, drug response, and genetic architecture, no studies directly address ancestry-driven differences in astrocytic receptor expression, microglial activation thresholds, or DA–glia integration. Precision neuroscience must therefore prioritize integrative approaches that account for sex, age, and ancestry to improve generalizability and therapeutic equity.
8.3. Computational and AI-Driven Models
Computational neuroscience has only recently begun incorporating glial dynamics into normative frameworks. The 3M-Progress model, an embodied reinforcement learning paradigm, integrated neural–glial modules to reproduce neural and astrocytic signatures underlying futility-induced passivity in zebrafish, representing the first whole-brain normative model of behavior grounded in neural–glial coupling.
By contrast, most neuron–astrocyte models remain limited to Ca²⁺ dynamics, gliotransmitter buffering, or homeostatic regulation, with minimal consideration of reactive state transitions or DA-specific modulation. Future frameworks should explicitly incorporate glial state variables; astrocytic Ca²⁺ flux, extracellular metabolite concentrations, and microglial inflammatory activity into reinforcement learning architectures using eligibility traces and distributional value functions. Such integration would capture the slow, spatially diffuse modulatory effects of glia that shape DA thresholds, plastic windows, and motivational drive under stress or immune challenge.
Recent advances in large-scale neuron–astrocyte simulation platforms demonstrate the feasibility of embedding glia into whole-brain models. These tools could enable predictive simulations of DA decline across aging, PD, and stress-linked pathology, bridging mechanistic insights with clinical application.
8.4. Translational Roadmap: From Bench to Clinic
The translation of glial–dopamine research into clinical practice requires a multipronged strategy spanning imaging technologies, peripheral biomarker discovery, pharmacological innovation, and behavioral assay development. Progress has been most notable in the imaging domain, where astrocyte-selective PET tracers such as [^18F]SMBT-1, targeting MAO-B, have demonstrated favorable kinetics and reproducibility in both PD and Alzheimer’s disease cohorts. These tools represent a significant advance in the ability to non-invasively monitor astrocytic states in vivo. For microglial imaging, TSPO PET remains the most widely used approach; however, its interpretability is constrained by the rs6971 polymorphism. Despite this limitation, post-mortem validation studies have confirmed TSPO binding in progressive supranuclear palsy and Alzheimer’s disease, supporting its conditional clinical utility. The development of next-generation ligands targeting receptors such as P2X7 offers the promise of more specific inflammatory readouts, though large-scale clinical validation is still required.
Parallel to imaging efforts, the search for peripheral biomarkers of glial–dopamine coupling is accelerating. Brain-derived extracellular vesicles enriched in glial proteins and microRNAs, isolated from plasma, have shown diagnostic potential in PD, offering a minimally invasive window into central nervous system dynamics. Complementary approaches using cell-free DNA methylation signatures provide an additional means of inferring neural cell-type origins, enabling real-time monitoring of astrocytic and microglial turnover. The integration of peripheral assays with multimodal imaging platforms such as PET and MRI, anchored to single-cell and spatial transcriptomic datasets, represents a critical next step. Such convergence will allow for the precise mapping of glial states to dopaminergic vulnerability niches across the substantia nigra, ventral tegmental area, and striatal subdivisions, thereby refining both diagnostic accuracy and disease staging.
Pharmacological interventions targeting glial pathways in dopamine-related disorders are also progressing, though challenges remain. GLP-1 receptor agonists have emerged as promising agents with central anti-inflammatory actions and preliminary evidence of motor and cognitive benefits in PD. Yet, heterogeneity in clinical outcomes underscores the necessity for biomarker-guided evaluation to identify responsive subgroups. Conversely, broad-spectrum agents such as minocycline reliably attenuate microglial activation in preclinical models but have failed to deliver consistent disease-modifying benefits in clinical trials, emphasizing the importance of developing more selective compounds and robust markers of target engagement. Future trials will need to incorporate multimodal biomarker panels to ensure mechanistic validation and to facilitate precision stratification of patients.
Beyond pharmacology, behavioral paradigms are increasingly recognized as valuable translational tools for probing glial–dopamine interactions. Tasks indexing reward anticipation, effort-based decision-making, and fatigue provide ecologically relevant readouts that can be directly linked to underlying astrocytic and microglial modulation of dopaminergic tone. When integrated with glia-selective imaging or peripheral biomarker assays, these paradigms may serve as surrogate endpoints in clinical trials, enabling rigorous assessment of therapeutic effects on glial–dopamine coupling. Such approaches not only bridge mechanistic discovery with clinical application but also hold the potential to accelerate the development of targeted interventions aimed at restoring circuit-level function and improving patient outcomes.
9. Conclusions
In recent years, converging evidence from cellular, genetic, imaging, and systems-level studies has repositioned glial–dopamine crosstalk as a central regulator of behavior, plasticity, and disease, rather than a secondary adjunct to neuronal signaling. Astrocytes and microglia actively sense, gate, and sculpt dopaminergic circuits through receptor signaling, metabolic support, gliotransmission, and immunometabolic coupling. Mechanistic insights—such as astrocytic MAO-B expression visualized via [^18F]SMBT-1 PET imaging, microglial manipulation with CSF-1R inhibitors, and glial regulation of ferroptosis—demonstrate that glia directly calibrate dopaminergic tone, neuronal vulnerability, and resilience. These findings collectively redefine dopamine not as a purely neuronal signal but as a glia-conditioned output, shaped by local inflammatory states, metabolic buffering, and extracellular matrix dynamics.
This reframing underscores that the efficacy of dopamine signaling is not fixed but dynamically shaped by glial modulation. The selective vulnerability of dopaminergic populations, such as substantia nigra versus ventral tegmental area neurons, reflects regionally distinct glial phenotypes that determine differential resilience or susceptibility. Likewise, behavioral outcomes including motivation, anhedonia, and stress resilience are increasingly recognized as contingent on the functional state of glia rather than neuronal activity alone. Consequently, therapeutic strategies that focus exclusively on dopaminergic neurons are unlikely to be sufficient without parallel interventions addressing glial dysfunction. More integrative approaches that modulate glial inflammation, redox capacity, and receptor signaling hold greater promise for producing durable benefits across movement disorders, psychiatric conditions, and neurodegenerative disease.
Taken together, glial–dopamine crosstalk has matured from speculative hypothesis into a paradigm-shifting axis in neuroscience, reframing dopamine as a signal conditioned by astrocytic and microglial dynamics. The next frontier lies in harnessing this framework to devise glia-targeted or glia-informed interventions that restore the cellular milieu in which dopamine operates thereby promoting resilience, slowing degeneration, and advancing treatment of motivational and cognitive dysfunction across psychiatry and neurology.
Author Contributions
M.N. contributed to the design and writing of the main manuscript text.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Luo, S.X.; Huang, E.J. Dopaminergic Neurons and Brain Reward Pathways: From Neurogenesis to Circuit Assembly. Am J Pathol 2016, 186, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, H.; Paladini, C.A. Dynamic regulation of midbrain dopamine neuron activity: intrinsic, synaptic, and plasticity mechanisms. Neuroscience 2011, 198, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Haber, S.N.; Behrens, T.E. The neural network underlying incentive-based learning: implications for interpreting circuit disruptions in psychiatric disorders. Neuron 2014, 83, 1019–1039. [Google Scholar] [CrossRef]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci 2016, 17, 524–532. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson's Disease Pathogenesis. Trends Neurosci 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Li, J.; Serafin, E.K.; Koorndyk, N.; et al. Astrocyte D1/D5 Dopamine Receptors Govern Non-Hebbian Long-Term Potentiation at Sensory Synapses onto Lamina I Spinoparabrachial Neurons. J Neurosci 2024, 44. [Google Scholar]
- Guttenplan, K.A.; Maxwell, I.; Santos, E.; et al. GPCR signaling gates astrocyte responsiveness to neurotransmitters and control of neuronal activity. Science 2025, 388, 763–768. [Google Scholar] [CrossRef]
- Santos, D.E.; Silva Lima, S.A.; Moreira, L.S.; et al. New perspectives on heterogeneity in astrocyte reactivity in neuroinflammation. Brain Behav Immun Health 2025, 44, 100948. [Google Scholar] [CrossRef]
- Favetta, G.; Bubacco, L. Beyond neurons: How does dopamine signaling impact astrocytic functions and pathophysiology? Prog Neurobiol 2025, 251, 102798. [Google Scholar] [CrossRef]
- Stedehouder, J.; Roberts, B.M.; Raina, S.; et al. Rapid modulation of striatal cholinergic interneurons and dopamine release by satellite astrocytes. Nat Commun 2024, 15, 10017. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol 2016, 26, 587–597. [Google Scholar] [CrossRef]
- She, K.; Yuan, N.; Huang, M.; et al. Emerging role of microglia in the developing dopaminergic system: Perturbation by early life stress. Neural Regen Res 2026, 21, 126–140. [Google Scholar] [CrossRef]
- Matt, S.M.; Nolan, R.; Manikandan, S.; et al. Dopamine-driven increase in IL-1beta in myeloid cells is mediated by differential dopamine receptor expression and exacerbated by HIV. J Neuroinflammation 2025, 22, 91. [Google Scholar] [CrossRef]
- Gullotta, G.S.; Costantino, G.; Sortino, M.A.; et al. Microglia and the Blood-Brain Barrier: An External Player in Acute and Chronic Neuroinflammatory Conditions. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Hasel, P.; Aisenberg, W.H.; Bennett, F.C.; et al. Molecular and metabolic heterogeneity of astrocytes and microglia. Cell Metab 2023, 35, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Reyes-Resina, I.; Navarro, G. Dopamine in Health and Disease: Much More Than a Neurotransmitter. Biomedicines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Belujon, P.; Grace, A.A. Regulation of dopamine system responsivity and its adaptive and pathological response to stress. Proc Biol Sci 2015, 282. [Google Scholar] [CrossRef]
- Choi, H.; Lee, E.J.; Shin, J.S.; et al. Spatiotemporal characterization of glial cell activation in an Alzheimer's disease model by spatially resolved transcriptomics. Exp Mol Med 2023, 55, 2564–2575. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B.; Giachino, C.; Tirolo, C.; et al. "Reframing" dopamine signaling at the intersection of glial networks in the aged Parkinsonian brain as innate Nrf2/Wnt driver: Therapeutical implications. Aging Cell 2022, 21, e13575. [Google Scholar] [CrossRef]
- Sanz-Galvez, R.; Falardeau, D.; Kolta, A.; et al. The role of astrocytes from synaptic to non-synaptic plasticity. Front Cell Neurosci 2024, 18, 1477985. [Google Scholar] [CrossRef]
- Oda, S.; Funato, H. D1- and D2-type dopamine receptors are immunolocalized in pial and layer I astrocytes in the rat cerebral cortex. Front Neuroanat 2023, 17, 1111008. [Google Scholar] [CrossRef]
- Amato, S.; Averna, M.; Guidolin, D.; et al. Heteromerization of Dopamine D2 and Oxytocin Receptor in Adult Striatal Astrocytes. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Ma, Z.; Stork, T.; Bergles, D.E.; et al. Neuromodulators signal through astrocytes to alter neural circuit activity and behaviour. Nature 2016, 539, 428–432. [Google Scholar] [CrossRef]
- Guidolin, D.; Tortorella, C.; Marcoli, M.; et al. Modulation of Neuron and Astrocyte Dopamine Receptors via Receptor-Receptor Interactions. Pharmaceuticals (Basel) 2023, 16. [Google Scholar] [CrossRef]
- Pelassa, S.; Guidolin, D.; Venturini, A.; et al. A2A-D2 Heteromers on Striatal Astrocytes: Biochemical and Biophysical Evidence. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Bezerra, T.O.; Roque, A.C. Dopamine facilitates the response to glutamatergic inputs in astrocyte cell models. PLoS Comput Biol 2024, 20, e1012688. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S. On astrocyte-neuron interactions: Broad insights from the striatum. Neuron 2025. [CrossRef] [PubMed]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, I.A.G.; Hirokane, K.; Choi, W.; Graybiel, A. Striatal Astrocytes Influence Dopamine Dynamics and Behavioral State Transitions. bioRxiv 2024. [Google Scholar] [CrossRef]
- Petroccione, M.A.; D'Brant, L.Y.; Affinnih, N.; et al. Neuronal glutamate transporters control reciprocal inhibition and gain modulation in D1 medium spiny neurons. Elife 2023, 12. [Google Scholar]
- Martinez, D.; Rogers, R.C.; Hermann, G.E.; et al. Astrocytic glutamate transporters reduce the neuronal and physiological influence of metabotropic glutamate receptors in nucleus tractus solitarii. Am J Physiol Regul Integr Comp Physiol 2020, 318, R545–R564. [Google Scholar] [CrossRef] [PubMed]
- Requie, L.M.; Gomez-Gonzalo, M.; Speggiorin, M.; et al. Astrocytes mediate long-lasting synaptic regulation of ventral tegmental area dopamine neurons. Nat Neurosci 2022, 25, 1639–1650. [Google Scholar] [CrossRef]
- Qin, J.; Ma, Z.; Chen, X.; et al. Microglia activation in central nervous system disorders: A review of recent mechanistic investigations and development efforts. Front Neurol 2023, 14, 1103416. [Google Scholar] [CrossRef]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; et al. Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer's disease-related proteins. Mol Neurodegener 2018, 13, 34. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Martinez-Muriana, A.; Davis, E.; et al. Deep proteomic analysis of microglia reveals fundamental biological differences between model systems. Cell Rep 2024, 43, 114908. [Google Scholar] [CrossRef] [PubMed]
- Jayanti, S.; Dalla Verde, C.; Tiribelli, C.; et al. Inflammation, Dopaminergic Brain and Bilirubin. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Wong, T.S.; Li, G.; Li, S.; et al. G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders. Signal Transduct Target Ther 2023, 8, 177. [Google Scholar] [CrossRef]
- Han, Q.; Li, W.; Chen, P.; et al. Microglial NLRP3 inflammasome-mediated neuroinflammation and therapeutic strategies in depression. Neural Regen Res 2024, 19, 1890–1898. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer's Disease. Front Neurol 2020, 11, 570711. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Nowrangi, D.; Yu, L.; et al. Activation of dopamine D1 receptor decreased NLRP3-mediated inflammation in intracerebral hemorrhage mice. J Neuroinflammation 2018, 15, 2. [Google Scholar] [CrossRef]
- Pike, A.F.; Longhena, F.; Faustini, G.; et al. Dopamine signaling modulates microglial NLRP3 inflammasome activation: implications for Parkinson's disease. J Neuroinflammation 2022, 19, 50. [Google Scholar] [CrossRef]
- de Pablos, R.M.; Herrera, A.J.; Espinosa-Oliva, A.M.; et al. Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J Neuroinflammation 2014, 11, 34. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Lind-Holm Mogensen, F.; Seibler, P.; Grunewald, A.; et al. Microglial dynamics and neuroinflammation in prodromal and early Parkinson's disease. J Neuroinflammation 2025, 22, 136. [Google Scholar] [CrossRef] [PubMed]
- Theis, H.; Pavese, N.; Rektorova, I.; et al. Imaging Biomarkers in Prodromal and Earliest Phases of Parkinson's Disease. J Parkinsons Dis 2024, 14, S353–S365. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Bae, E.J.; Park, S.J.; et al. Microglia-driven inflammation induces progressive tauopathies and synucleinopathies. Exp Mol Med 2025, 57, 1017–1031. [Google Scholar] [CrossRef]
- Deyell, J.S.; Sriparna, M.; Ying, M.; et al. The Interplay between alpha-Synuclein and Microglia in alpha-Synucleinopathies. Int J Mol Sci 2023, 24. [Google Scholar]
- Badanjak, K.; Fixemer, S.; Smajic, S.; et al. The Contribution of Microglia to Neuroinflammation in Parkinson's Disease. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Miao, Y.; Meng, H. The involvement of alpha-synucleinopathy in the disruption of microglial homeostasis contributes to the pathogenesis of Parkinson's disease. Cell Commun Signal 2024, 22, 31. [Google Scholar] [CrossRef]
- Li, Y.; Xia, Y.; Yin, S.; et al. Targeting Microglial alpha-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson's Disease. Front Immunol 2021, 12, 719807. [Google Scholar] [CrossRef]
- Gerhard, A. TSPO imaging in parkinsonian disorders. Clin Transl Imaging 2016, 4, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Lavisse, S.; Goutal, S.; Wimberley, C.; et al. Increased microglial activation in patients with Parkinson disease using [(18)F]-DPA714 TSPO PET imaging. Parkinsonism Relat Disord 2021, 82, 29–36. [Google Scholar] [CrossRef]
- Brooks, N.A.H.; Riar, I.; Klegeris, A. Mitochondrial damage-associated molecular patterns: Neuroimmunomodulators in central nervous system pathophysiology. Neural Regen Res 2026, 21, 1322–1338. [Google Scholar] [CrossRef]
- Deus, C.M.; Tavares, H.; Beatriz, M.; et al. Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.X.; Wang, P.J.; Chao, S.; et al. Transcriptomic profiling identifies ferroptosis and NF-kappaB signaling involved in alpha-dimorphecolic acid regulation of microglial inflammation. J Transl Med 2025, 23, 260. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Lin, L.; Ma, C.; et al. Decoding crosstalk between neurotransmitters and alpha-synuclein in Parkinson's disease: pathogenesis and therapeutic implications. Ther Adv Neurol Disord 2025, 18, 17562864251339895. [Google Scholar] [CrossRef]
- Kang, S.; Noh, Y.; Oh, S.J.; et al. Neuroprotective Effects of Aldehyde-Reducing Composition in an LPS-Induced Neuroinflammation Model of Parkinson's Disease. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Bailey, H.M.; Cookson, M.R. How Parkinson's Disease-Linked LRRK2 Mutations Affect Different CNS Cell Types. J Parkinsons Dis 2024, 14, 1331–1352. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Sugino, Y.; Yamamuro, A.; et al. Dopamine inhibits the expression of proinflammatory cytokines of microglial cells through the formation of dopamine quinone in the mouse striatum. J Pharmacol Sci 2022, 148, 41–50. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Iranzo, A.; Ostergaard, K.; et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. Lancet Neurol 2017, 16, 789–796. [Google Scholar] [CrossRef]
- Burke, W.J.; Li, S.W.; Williams, E.A.; et al. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson's disease pathogenesis. Brain Res 2003, 989, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K. Stevens BThe "quad-partite" synapse: microglia-synapse interactions in the developing mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef]
- Huo, A.; Wang, J.; Li, Q.; et al. Molecular mechanisms underlying microglial sensing and phagocytosis in synaptic pruning. Neural Regen Res 2024, 19, 1284–1290. [Google Scholar] [CrossRef]
- Soteros, B.M.; Sia, G.M. Complement and microglia dependent synapse elimination in brain development. WIREs Mech Dis 2022, 14, e1545. [Google Scholar] [CrossRef]
- Loh, J.S.; Mak, W.Q.; Tan, L.K.S.; et al. Microbiota-gut-brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct Target Ther 2024, 9, 37. [Google Scholar] [CrossRef]
- Juarez Olguin, H.; Calderon Guzman, D.; Hernandez Garcia, E.; et al. The Role of Dopamine and Its Dysfunction as a Consequence of Oxidative Stress. Oxid Med Cell Longev 2016, 2016, 9730467. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liu, J.; Wang, B.; et al. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer's Disease and Related Therapeutic Targets. Front Immunol 2022, 13, 856376. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; et al. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct Target Ther 2023, 8, 359. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef]
- Sun, M.; You, H.; Hu, X.; et al. Microglia-Astrocyte Interaction in Neural Development and Neural Pathogenesis. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Spreng, A.S.; Brull, M.; Leisner, H.; et al. Distinct and Dynamic Transcriptome Adaptations of iPSC-Generated Astrocytes after Cytokine Stimulation. Cells 2022, 11. [Google Scholar] [CrossRef]
- Jiwaji, Z.; Tiwari, S.S.; Aviles-Reyes, R.X.; et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Ass pathology. Nat Commun 2022, 13, 135. [Google Scholar] [CrossRef]
- Naffaa, M.M. Mechanisms of astrocytic and microglial purinergic signaling in homeostatic regulation and implications for neurological disease. Exploration of Neuroscience 2025, 4, 100676. [Google Scholar] [CrossRef]
- Lindberg, D.; Shan, D.; Ayers-Ringler, J.; et al. Purinergic signaling and energy homeostasis in psychiatric disorders. Curr Mol Med 2015, 15, 275–295. [Google Scholar] [CrossRef]
- Carracedo, S.; Launay, A.; Dechelle-Marquet, P.A.; et al. Purinergic-associated immune responses in neurodegenerative diseases. Prog Neurobiol 2024, 243, 102693. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; et al. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Harms, A.S.; Boehringer, A.; et al. Decreased neuronal and increased endothelial fractalkine expression are associated with neuroinflammation in Parkinson's disease and related disorders. Front Cell Neurosci 2025, 19, 1557645. [Google Scholar] [CrossRef]
- Prada, I.; Gabrielli, M.; Turola, E.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: a new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol 2018, 135, 529–550. [Google Scholar] [CrossRef] [PubMed]
- Pistono, C.; Bister, N.; Stanova, I.; et al. Glia-Derived Extracellular Vesicles: Role in Central Nervous System Communication in Health and Disease. Front Cell Dev Biol 2020, 8, 623771. [Google Scholar] [CrossRef]
- Marchetti, B.; Leggio, L.; L'Episcopo, F.; et al. Glia-Derived Extracellular Vesicles in Parkinson's Disease. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Nagai, J.; Yu, X.; Papouin, T.; et al. Behaviorally consequential astrocytic regulation of neural circuits. Neuron 2021, 109, 576–596. [Google Scholar] [CrossRef]
- Agarwal, D.; Sandor, C.; Volpato, V.; et al. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat Commun 2020, 11, 4183. [Google Scholar] [CrossRef]
- Fornari Laurindo, L.; Aparecido Dias, J.; Cressoni Araujo, A.; et al. Immunological dimensions of neuroinflammation and microglial activation: exploring innovative immunomodulatory approaches to mitigate neuroinflammatory progression. Front Immunol 2023, 14, 1305933. [Google Scholar] [CrossRef]
- Ma, M.; Paryani, F.; Jakubiak, K.; et al. The spatial landscape of glial pathology and T cell response in Parkinson's disease substantia nigra. Nat Commun 2025, 16, 7146. [Google Scholar] [CrossRef]
- Gaertner, Z.; Oram, C.; Schneeweis, A.; et al. Molecular and spatial transcriptomic classification of midbrain dopamine neurons and their alterations in a LRRK2(G2019S) model of Parkinson's disease. Elife 2025, 13. [Google Scholar]
- Rademacher, K.; Doric, Z.; Haddad, D.; et al. Chronic hyperactivation of midbrain dopamine neurons causes preferential dopamine neuron degeneration. Elife 2025, 13. [Google Scholar]
- Gao, M.Y.; Wang, J.Q.; He, J.; et al. Single-Cell RNA-Sequencing in Astrocyte Development, Heterogeneity, and Disease. Cell Mol Neurobiol 2023, 43, 3449–3464. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.U.; Koulen, P.; Rubinstein, M.; et al. An astroglia-linked dopamine D2-receptor action in prefrontal cortex. Proc Natl Acad Sci U S A 2001, 98, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Rahimian, R.; Belliveau, C.; Chen, R.; et al. Microglial Inflammatory-Metabolic Pathways and Their Potential Therapeutic Implication in Major Depressive Disorder. Front Psychiatry 2022, 13, 871997. [Google Scholar] [CrossRef]
- Cervetto, C.; Maura, G.; Guidolin, D.; et al. Striatal astrocytic A2A-D2 receptor-receptor interactions and their role in neuropsychiatric disorders. Neuropharmacology 2023, 237, 109636. [Google Scholar] [CrossRef]
- Kopec, A.M.; Smith, C.J.; Ayre, N.R.; et al. Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nat Commun 2018, 9, 3769. [Google Scholar] [CrossRef]
- Furuyashiki T, Kitaoka S Neural mechanisms underlying adaptive and maladaptive consequences of stress: Roles of dopaminergic and inflammatory responses. Psychiatry Clin Neurosci 2019, 73, 669–675. [CrossRef]
- Jimenez-Gonzalez, A.; Gomez-Acevedo, C.; Ochoa-Aguilar, A.; et al. The Role of Glia in Addiction: Dopamine as a Modulator of Glial Responses in Addiction. Cell Mol Neurobiol 2022, 42, 2109–2120. [Google Scholar] [CrossRef]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat Rev Drug Discov 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Leonard, B.; et al. Microglial responses to dopamine in a cell culture model of Parkinson's disease. Neurobiol Aging 2009, 30, 1805–1817. [Google Scholar] [CrossRef]
- Mao, S.; Qiao, R.; Wang, Q.; et al. Activity and Heterogeneity of Astrocytes in Neurological Diseases: Molecular Mechanisms and Therapeutic Targets. MedComm (2020) 2025, 6, e70329. [Google Scholar] [CrossRef]
- Yoo, S.L.K.; Seo, J.; Choi, H.; Kim, S.; Chang, J.; Shim, Y.; Kim, J.; Won, J.; Park, S. Single-nucleus and spatial transcriptomic analysis identified molecular features of neuronal heterogeneity and distinct glial responses in Parkinson’s disease. biorxiv 2024.
- Hu, N.; Chen, L.; Hu, G.; et al. Advancements in extracellular vesicle therapy for neurodegenerative diseases. Explor Neuroprotective Ther 2025, 5. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Li, Z.; Haroon, E.; et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry 2016, 21, 1358–1365. [Google Scholar] [CrossRef]
- Felger, J.C.; Miller, A.H. Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol 2012, 33, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Sugama, S.; Takenouchi, T.; Hashimoto, M.; et al. Stress-induced microglial activation occurs through beta-adrenergic receptor: noradrenaline as a key neurotransmitter in microglial activation. J Neuroinflammation 2019, 16, 266. [Google Scholar] [CrossRef]
- Tujula, I.; Hyvarinen, T.; Lotila, J.; et al. Modeling neuroinflammatory interactions between microglia and astrocytes in a human iPSC-based coculture platform. Cell Commun Signal 2025, 23, 298. [Google Scholar] [CrossRef] [PubMed]
- Iring, A.T.A.; Baranyi, M.; Otrokocsi, L.; Módis, L.; Gölöncsér, F.; Varga, B.; Hortobágyi, T.; Bereczki, D.; Dénes, Á.; Sperlágh, B. Central inhibition of P2Y12R differentially regulates survival and neuronal loss in MPTP-induced Parkinsonism in mice. bioRxiv 2021. [Google Scholar]
- Murphy-Royal, C.; Ching, S.; Papouin, T. A conceptual framework for astrocyte function. Nat Neurosci 2023, 26, 1848–1856. [Google Scholar] [CrossRef]
- Stowell, R.; Wang, K.H. Dopaminergic signaling regulates microglial surveillance and adolescent plasticity in the mouse frontal cortex. Nat Commun 2025, 16, 7974. [Google Scholar] [CrossRef] [PubMed]
- Alcaro, A.; Huber, R.; Panksepp, J. Behavioral functions of the mesolimbic dopaminergic system: an affective neuroethological perspective. Brain Res Rev 2007, 56, 283–321. [Google Scholar] [CrossRef]
- Baik, J.H. Stress and the dopaminergic reward system. Exp Mol Med 2020, 52, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Ortinski, P.I.; Reissner, K.J.; Turner, J.; et al. Control of complex behavior by astrocytes and microglia. Neurosci Biobehav Rev 2022, 137, 104651. [Google Scholar] [CrossRef]
- Zhou, Z.C.; Gordon-Fennell, A.; Piantadosi, S.C.; et al. Deep-brain optical recording of neural dynamics during behavior. Neuron 2023, 111, 3716–3738. [Google Scholar] [CrossRef]
- Serra, I.; Martin-Monteagudo, C.; Sanchez Romero, J.; et al. Astrocyte ensembles manipulated with AstroLight tune cue-motivated behavior. Nat Neurosci 2025, 28, 616–626. [Google Scholar] [CrossRef]
- Kang, S.; Hong, S.I.; Kang, S.; et al. Astrocyte activities in the external globus pallidus regulate action-selection strategies in reward-seeking behaviors. Sci Adv 2023, 9, eadh9239. [Google Scholar] [CrossRef]
- Yang, M.A.; Kang, S.; Hong, S.I.; et al. Astrocytes in the External Globus Pallidus Selectively Represent Routine Formation During Repeated Reward-Seeking in Mice. eNeuro 2025, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dulinskas, R.; Ineichen, C.; et al. Chronic stress deficits in reward behaviour co-occur with low nucleus accumbens dopamine activity during reward anticipation specifically. Commun Biol 2024, 7, 966. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, S.J. Deciphering the star codings: astrocyte manipulation alters mouse behavior. Exp Mol Med 2020, 52, 1028–1038. [Google Scholar] [CrossRef]
- Nowak, D.B.; Taborda-Bejarano, J.P.; Chaure, F.J.; et al. Understanding Microglia in Mesocorticolimbic Circuits: Implications for the Study of Chronic Stress and Substance Use Disorders. Cells 2025, 14. [Google Scholar] [CrossRef]
- da Silva, M.C.M.; Iglesias, L.P.; Candelario-Jalil, E.; et al. Role of Microglia in Psychostimulant Addiction. Curr Neuropharmacol 2023, 21, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Avolio, E.; Fazzari, G.; Mele, M.; et al. Unpredictable Chronic Mild Stress Paradigm Established Effects of Pro- and Anti-inflammatory Cytokine on Neurodegeneration-Linked Depressive States in Hamsters with Brain Endothelial Damages. Mol Neurobiol 2017, 54, 6446–6458. [Google Scholar] [CrossRef]
- Farooq, R.K.; Isingrini, E.; Tanti, A.; et al. Is unpredictable chronic mild stress (UCMS) a reliable model to study depression-induced neuroinflammation? Behav Brain Res 2012, 231, 130–137. [Google Scholar] [CrossRef]
- Xia, X.; Li, K.; Zou, W.; et al. The central role of microglia in major depressive disorder and its potential as a therapeutic target. Front Behav Neurosci 2025, 19, 1598178. [Google Scholar] [CrossRef]
- Paganin, W. Signorini S Inflammatory biomarkers in depression: scoping review. BJPsych Open 2024, 10, e165. [Google Scholar] [CrossRef]
- Reemst, K.; Kracht, L.; Kotah, J.M.; et al. Early-life stress lastingly impacts microglial transcriptome and function under basal and immune-challenged conditions. Transl Psychiatry 2022, 12, 507. [Google Scholar] [CrossRef]
- Jamil, S.; Raza, M.L.; Moradikor, N.; et al. Early life stress and brain development: Neurobiological and behavioral effects of chronic stress. Prog Brain Res 2025, 291, 49–79. [Google Scholar] [PubMed]
- Gongwer, M.W.; Etienne, F.; Moca, E.N.; et al. Microglia regulate nucleus accumbens synaptic development and circuit function underlying threat avoidance behaviors. Res Sq. 2025.
- Smith, J.A.; Das, A.; Ray, S.K.; et al. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull 2012, 87, 10–20. [Google Scholar]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Zhuo, Y.; Luo, B.; Yi, X.; et al. Improved green and red GRAB sensors for monitoring dopaminergic activity in vivo. Nat Methods 2024, 21, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zhou, J.; Dai, B.; et al. Next-generation GRAB sensors for monitoring dopaminergic activity in vivo. Nat Methods 2020, 17, 1156–1166. [Google Scholar] [CrossRef]
- Thrailkill, E.A.; Daniels, C.W. The temporal structure of goal-directed and habitual operant behavior. J Exp Anal Behav 2024, 121, 38–51. [Google Scholar] [CrossRef]
- Delgado, L.; Navarrete, M. Shining the Light on Astrocytic Ensembles. Cells 2023, 12. [Google Scholar] [CrossRef]
- Shirokova, O.M.; Kuzmina, D.M.; Zaborskaya, O.G.; et al. The Long-Term Effects of Chronic Unpredictable Mild Stress Experienced During Adolescence Could Vary Depending on Biological Sex. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Bergamini, G.; Mechtersheimer, J.; Azzinnari, D.; et al. Chronic social stress induces peripheral and central immune activation, blunted mesolimbic dopamine function, and reduced reward-directed behaviour in mice. Neurobiol Stress 2018, 8, 42–56. [Google Scholar] [CrossRef]
- Nusslock, R.; Alloy, L.B.; Brody, G.H.; et al. Annual Research Review: Neuroimmune network model of depression: a developmental perspective. J Child Psychol Psychiatry 2024, 65, 538–567. [Google Scholar] [CrossRef]
- Cervetto, C.; Venturini, A.; Guidolin, D.; et al. Homocysteine and A2A-D2 Receptor-Receptor Interaction at Striatal Astrocyte Processes. J Mol Neurosci 2018, 65, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.J.; Dong, Y. Psychostimulant-Induced Adaptations in Nucleus Accumbens Glutamatergic Transmission. Cold Spring Harb Perspect Med 2020, 10. [Google Scholar] [CrossRef]
- Williams, D.M.; Cox, B.; Lafuse, D.W.; et al. Epstein-Barr Virus dUTPase Induces Neuroinflammatory Mediators: Implications for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Clin Ther 2019, 41, 848–863. [Google Scholar] [CrossRef] [PubMed]
- Dobryakova, E.; Genova, H.M.; DeLuca, J.; et al. The dopamine imbalance hypothesis of fatigue in multiple sclerosis and other neurological disorders. Front Neurol 2015, 6, 52. [Google Scholar] [CrossRef]
- MacDonald, H.J.; Kleppe, R.; Szigetvari, P.D.; et al. The dopamine hypothesis for ADHD: An evaluation of evidence accumulated from human studies and animal models. Front Psychiatry 2024, 15, 1492126. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.J.; Newcorn, J.H.; et al. Motivation deficit in ADHD is associated with dysfunction of the dopamine reward pathway. Mol Psychiatry 2011, 16, 1147–1154. [Google Scholar] [CrossRef]
- Vandenbark, A.A.; Offner, H.; Matejuk, S.; et al. Microglia and astrocyte involvement in neurodegeneration and brain cancer. J Neuroinflammation 2021, 18, 298. [Google Scholar] [CrossRef]
- Kanthasamy, A.; Jin, H.; Charli, A.; et al. Environmental neurotoxicant-induced dopaminergic neurodegeneration: a potential link to impaired neuroinflammatory mechanisms. Pharmacol Ther 2019, 197, 61–82. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Mai, D.; et al. Molecular Mechanisms of Glutamate Toxicity in Parkinson's Disease. Front Neurosci 2020, 14, 585584. [Google Scholar] [CrossRef] [PubMed]
- Kamath, T.; Abdulraouf, A.; Burris, S.J.; et al. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson's disease. Nat Neurosci 2022, 25, 588–595. [Google Scholar] [CrossRef]
- Sportelli, L.; Eisenberg, D.P.; Passiatore, R.; et al. Dopamine signaling enriched striatal gene set predicts striatal dopamine synthesis and physiological activity in vivo. Nat Commun 2024, 15, 3342. [Google Scholar] [CrossRef]
- Voicu, V.; Toader, C.; Serban, M.; et al. Systemic Neurodegeneration and Brain Aging: Multi-Omics Disintegration, Proteostatic Collapse, and Network Failure Across the CNS. Biomedicines 2025, 13. [Google Scholar] [CrossRef]
- Stevenson, R.; Samokhina, E.; Rossetti, I.; et al. Neuromodulation of Glial Function During Neurodegeneration. Front Cell Neurosci 2020, 14, 278. [Google Scholar] [CrossRef]
- Jiang, Q.; Liu, J.; Huang, S.; et al. Antiageing strategy for neurodegenerative diseases: from mechanisms to clinical advances. Signal Transduct Target Ther 2025, 10, 76. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; et al. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Lee, J.H.; Flausino, L.E.; et al. Neuroinflammation: An astrocyte perspective. Sci Transl Med 2023, 15, eadi7828. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front Neurosci 2022, 16, 824888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Niu, K.; Huang, T.; et al. Microglia depletion reduces neurodegeneration and remodels extracellular matrix in a mouse Parkinson's disease model triggered by alpha-synuclein overexpression. NPJ Parkinsons Dis 2025, 11, 15. [Google Scholar] [CrossRef]
- Thi Lai, T.; Kim, Y.E.; Nguyen, L.T.N.; et al. Microglial inhibition alleviates alpha-synuclein propagation and neurodegeneration in Parkinson's disease mouse model. NPJ Parkinsons Dis 2024, 10, 32. [Google Scholar] [CrossRef]
- Sunna, S.; Bowen, C.A.; Ramelow, C.C.; et al. Advances in proteomic phenotyping of microglia in neurodegeneration. Proteomics 2023, 23, e2200183. [Google Scholar] [CrossRef]
- Li, Z.; Xu, P.; Deng, Y.; et al. M1 Microglia-Derived Exosomes Promote A1 Astrocyte Activation and Aggravate Ischemic Injury via circSTRN3/miR-331-5p/MAVS/NF-kappaB Pathway. J Inflamm Res 2024, 17, 9285–9305. [Google Scholar] [CrossRef]
- Guo, M.; Guan, A.; Zhang, M.; et al. Exosome-mediated microglia-astrocyte interactions drive neuroinflammation in Parkinson's disease with Peli1 as a potential therapeutic target. Pharmacol Res 2025, 219, 107908. [Google Scholar] [CrossRef] [PubMed]
- Buoso, C.; Seifert, M.; Lang, M.; et al. Dopamine-iron homeostasis interaction rescues mitochondrial fitness in Parkinson's disease. Neurobiol Dis 2024, 196, 106506. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, K.J.; Gilliland, T.M.; Winkelstein, B.A. Upregulation of GLT-1 by treatment with ceftriaxone alleviates radicular pain by reducing spinal astrocyte activation and neuronal hyperexcitability. J Neurosci Res 2014, 92, 116–129. [Google Scholar] [CrossRef]
- Abulseoud, O.A.; Alasmari, F.; Hussein, A.M.; et al. Ceftriaxone as a Novel Therapeutic Agent for Hyperglutamatergic States: Bridging the Gap Between Preclinical Results and Clinical Translation. Front Neurosci 2022, 16, 841036. [Google Scholar] [CrossRef]
- Jiao, L.; Li, X.; Luo, Y.; et al. Iron metabolism mediates microglia susceptibility in ferroptosis. Front Cell Neurosci 2022, 16, 995084. [Google Scholar] [CrossRef]
- Ru, Q.; Li, Y.; Chen, L.; et al. Iron homeostasis and ferroptosis in human diseases: mechanisms and therapeutic prospects. Signal Transduct Target Ther 2024, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Rudroff, T. Decoding Post-Viral Fatigue: The Basal Ganglia's Complex Role in Long-COVID. Neurol Int 2024, 16, 380–393. [Google Scholar] [CrossRef]
- Khan, S.A.Y.; Kazi, N.; Sideeque, S.; Ansari, N.; Mohammed, H.; Byroju, V.; Caprara, A.; Rissardo, J. Brain structural and functional alteration in movement disorders: A narrative review of markers and their dynamic changes. NeuroMarkers 2025. [Google Scholar] [CrossRef]
- Mancini, M.; Natoli, S.; Gardoni, F.; et al. Dopamine Transmission Imbalance in Neuroinflammation: Perspectives on Long-Term COVID-19. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Adamu, A.; Li, S.; Gao, F.; et al. The role of neuroinflammation in neurodegenerative diseases: current understanding and future therapeutic targets. Front Aging Neurosci 2024, 16, 1347987. [Google Scholar] [CrossRef] [PubMed]
- Kanberg, N.; Simren, J.; Eden, A.; et al. Neurochemical signs of astrocytic and neuronal injury in acute COVID-19 normalizes during long-term follow-up. EBioMedicine 2021, 70, 103512. [Google Scholar] [CrossRef]
- Vrettou, C.S.; Vassiliou, A.G.; Keskinidou, C.; et al. A Prospective Study on Neural Biomarkers in Patients with Long-COVID Symptoms. J Pers Med 2024, 14. [Google Scholar] [CrossRef]
- Picca, A.; Ferri, E.; Calvani, R.; et al. Age-Associated Glia Remodeling and Mitochondrial Dysfunction in Neurodegeneration: Antioxidant Supplementation as a Possible Intervention. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, C.; Zhang, Y.; et al. Unraveling the role of neuregulin-mediated astrocytes-OPCs axis in the pathogenesis of age-related macular degeneration and Parkinson's disease. Sci Rep 2025, 15, 7352. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Lee, Y.J.; Horan-Portelance, L.; et al. Microglial and neuronal fates following inhibition of CSF-1R in synucleinopathy mouse model. Brain Behav Immun 2025, 123, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Stoll, A.C.; Kemp, C.J.; Patterson, J.R.; et al. Alpha-synuclein inclusion responsive microglia are resistant to CSF1R inhibition. J Neuroinflammation 2024, 21, 108. [Google Scholar] [CrossRef]
- Eo, H.; Kim, S.; Jung, U.J.; et al. Alpha-Synuclein and Microglia in Parkinson's Disease: From Pathogenesis to Therapeutic Prospects. J Clin Med 2024, 13. [Google Scholar] [CrossRef]
- Guo, M.; Wang, J.; Zhao, Y.; et al. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson's disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef]
- Jiao, D.; Yang, Y.; Wang, K.; et al. Ferroptosis: a novel pathogenesis and therapeutic strategies for Parkinson disease: A review. Medicine (Baltimore) 2025, 104, e41218. [Google Scholar] [CrossRef]
- Tang, S.; Zhang, J.; Chen, J.; et al. Ferroptosis in neurodegenerative diseases: potential mechanisms of exercise intervention. Front Cell Dev Biol 2025, 13, 1622544. [Google Scholar] [CrossRef]
- Zhi, H.; Wang, X.; Chen, Y.; et al. Ceftriaxone affects ferroptosis and alleviates glial cell activation in Parkinson's disease. Int J Mol Med 2025, 55. [Google Scholar] [CrossRef]
- Chotibut, T.; Meadows, S.; Kasanga, E.A.; et al. Ceftriaxone reduces L-dopa-induced dyskinesia severity in 6-hydroxydopamine parkinson's disease model. Mov Disord 2017, 32, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Connolly, R.; Brennan, D.; et al. Blood-brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nat Neurosci 2024, 27, 421–432. [Google Scholar] [CrossRef]
- Popa, E.; Popa, A.E.; Poroch, M.; et al. The Molecular Mechanisms of Cognitive Dysfunction in Long COVID: A Narrative Review. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, T.N.; Jamenis, A.S.; Abbas, M.; et al. A 14-day pulse of PLX5622 modifies alpha-synucleinopathy in preformed fibril-infused aged mice of both sexes. Neurobiol Dis 2023, 184, 106196. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Li, M.; Chen, M.; Li, H.; et al. Glial cells improve Parkinson's disease by modulating neuronal function and regulating neuronal ferroptosis. Front Cell Dev Biol 2024, 12, 1510897. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, G.; Liu, M.; et al. New perspectives on molecular mechanisms underlying exercise-induced benefits in Parkinson's disease. NPJ Parkinsons Dis 2025, 11, 256. [Google Scholar] [CrossRef]
- Luthra, N.S.; Mehta, N.; Munoz, M.J.; et al. Aerobic exercise-induced changes in fluid biomarkers in Parkinson's disease. NPJ Parkinsons Dis 2025, 11, 190. [Google Scholar] [CrossRef]
- Hein, Z.M.; Thazin Kumar, S.; et al. Immunomodulatory Mechanisms Underlying Neurological Manifestations in Long COVID: Implications for Immune-Mediated Neurodegeneration. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Huang, M.; Long, A.; Hao, L.; et al. Astrocyte in Neurological Disease: Pathogenesis and Therapy. MedComm (2020) 2025, 6, e70299. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.C.; Hardingham, G.E. The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Naffaa, M.M. Monoamine Oxidase B in Astrocytic GABA Synthesis: A Central Mechanism in Neurodegeneration and Neuroinflammation. Journal of Cellular Signaling 2025, 6. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Harada, R.; Dore, V.; et al. First-in-Humans Evaluation of (18)F-SMBT-1, a Novel (18)F-Labeled Monoamine Oxidase-B PET Tracer for Imaging Reactive Astrogliosis. J Nucl Med 2022, 63, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Bellaver, B.; Povala, G.; Ferreira, P.C.L.; et al. Astrocyte reactivity influences amyloid-beta effects on tau pathology in preclinical Alzheimer's disease. Nat Med 2023, 29, 1775–1781. [Google Scholar] [CrossRef]
- Kong, Y.; Cao, L.; Wang, J.; et al. In vivo reactive astrocyte imaging using [(18)F]SMBT-1 in tauopathy and familial Alzheimer's disease mouse models: A multi-tracer study. J Neurol Sci 2024, 462, 123079. [Google Scholar] [CrossRef] [PubMed]
- Chatzipieris, F.P.; Kokkalis, A.; Georgiou, N.; et al. New Prospects in the Inhibition of Monoamine Oxidase-B (MAO-B) Utilizing Propargylamine Derivatives for the Treatment of Alzheimer's Disease: A Review. ACS Omega 2025, 10, 26208–26232. [Google Scholar] [CrossRef]
- Zou, D.J.; Liu, R.Z.; Lv, Y.J.; et al. Chromone-deferiprone hybrids as novel MAO-B inhibitors and iron chelators for the treatment of Alzheimer's disease. Org Biomol Chem 2024, 22, 6189–6197. [Google Scholar] [CrossRef]
- Duta, C.; Muscurel, C.; Dogaru, C.B.; et al. Ferroptosis-A Shared Mechanism for Parkinson's Disease and Type 2 Diabetes. Int J Mol Sci 2024, 25. [Google Scholar]
- Liu, S.; Gao, X.; Zhou, S. New Target for Prevention and Treatment of Neuroinflammation: Microglia Iron Accumulation and Ferroptosis. ASN Neuro 2022, 14, 17590914221133236. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jia, B.; Li, J.; et al. The Interplay between Ferroptosis and Neuroinflammation in Central Neurological Disorders. Antioxidants (Basel) 2024, 13. [Google Scholar] [CrossRef]
- Feng, S.; Tang, D.; Wang, Y.; et al. The mechanism of ferroptosis and its related diseases. Mol Biomed 2023, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Ahn, H.; Jung, K.H.; et al. Evaluation of the Neuroprotective Effect of Microglial Depletion by CSF-1R Inhibition in a Parkinson's Animal Model. Mol Imaging Biol 2020, 22, 1031–1042. [Google Scholar] [CrossRef]
- Ho MS Clearance Pathways for alpha-Synuclein in Parkinson's Disease. J Neurochem 2025, 169, e70124. [CrossRef]
- Basilico, B.; Ferrucci, L.; Khan, A.; et al. What microglia depletion approaches tell us about the role of microglia on synaptic function and behavior. Front Cell Neurosci 2022, 16, 1022431. [Google Scholar] [CrossRef]
- Adaikkan, C.R.I.M.; Lorenzo Bozzelli, P.; Sears, M.; Parro, C.; Pao, P.; Sun, N.; Kim, T.; Abdelaal, K.; Sedgwick, M.; Kellis, M.; Tsai, L. A multimodal approach of microglial CSF1R inhibition and GENUS provides therapeutic effects in Alzheimer’s disease mice. bioRxiv 2025. [Google Scholar]
- Noh, M.Y.; Kwon, H.S.; Kwon, M.S.; et al. Biomarkers and therapeutic strategies targeting microglia in neurodegenerative diseases: current status and future directions. Mol Neurodegener 2025, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, D.; Ehara, A.; Kadowaki, T.; et al. Minocycline Alleviates Cluster Formation of Activated Microglia and Age-dependent Dopaminergic Cell Death in the Substantia Nigra of Zitter Mutant Rat. Acta Histochem Cytochem 2020, 53, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Fackelmeier, B.; Fong, D.M.; et al. Astrocyte-selective AAV gene therapy through the endogenous GFAP promoter results in robust transduction in the rat spinal cord following injury. Gene Ther 2019, 26, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Van Den Herrewegen, Y.; Sanderson, T.M.; Sahu, S.; et al. Side-by-side comparison of the effects of Gq- and Gi-DREADD-mediated astrocyte modulation on intracellular calcium dynamics and synaptic plasticity in the hippocampal CA1. Mol Brain 2021, 14, 144. [Google Scholar] [CrossRef]
- Zhong, X.; Gu, H.; Lim, J.; et al. Genetically encoded sensors illuminate in vivo detection for neurotransmission: Development, application, and optimization strategies. IBRO Neurosci Rep 2025, 18, 476–490. [Google Scholar] [CrossRef]
- Harada, R.; Hayakawa, Y.; Ezura, M.; et al. (18)F-SMBT-1: A Selective and Reversible PET Tracer for Monoamine Oxidase-B Imaging. J Nucl Med 2021, 62, 253–258. [Google Scholar] [CrossRef]
- Mishra, S.; Gordon, B.A.; Su, Y.; et al. AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: Defining a summary measure. Neuroimage 2017, 161, 171–178. [Google Scholar] [CrossRef]
- Zhang, M.; Qian, X.H.; Hu, J.; et al. Integrating TSPO PET imaging and transcriptomics to unveil the role of neuroinflammation and amyloid-beta deposition in Alzheimer's disease. Eur J Nucl Med Mol Imaging 2024, 51, 455–467. [Google Scholar] [CrossRef]
- Bonomi, C.G.; Chiaravalloti, A.; Camedda, R.; et al. Functional Correlates of Microglial and Astrocytic Activity in Symptomatic Sporadic Alzheimer's Disease: A CSF/(18)F-FDG-PET Study. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Ballweg, A.; Klaus, C.; Vogler, L.; et al. [(18)F]F-DED PET imaging of reactive astrogliosis in neurodegenerative diseases: preclinical proof of concept and first-in-human data. J Neuroinflammation 2023, 20, 68. [Google Scholar] [CrossRef]
- Lin, J.; Ou, R.; Li, C.; et al. Plasma glial fibrillary acidic protein as a biomarker of disease progression in Parkinson's disease: a prospective cohort study. BMC Med 2023, 21, 420. [Google Scholar] [CrossRef]
- Shi Q, Gutierrez RA, Bhat MA Microglia, Trem2, and Neurodegeneration. Neuroscientist 2025, 31, 159–176. [CrossRef]
- Zhang, L.; Xiang, X.; Li, Y.; et al. TREM2 and sTREM2 in Alzheimer's disease: from mechanisms to therapies. Mol Neurodegener 2025, 20, 43. [Google Scholar] [CrossRef]
- Gabrielli, M.; Raffaele, S.; Fumagalli, M.; et al. The multiple faces of extracellular vesicles released by microglia: Where are we 10 years after? Front Cell Neurosci 2022, 16, 984690. [Google Scholar] [CrossRef]
- Wang, P.; Lan, G.; Xu, B.; et al. alpha-Synuclein-carrying astrocytic extracellular vesicles in Parkinson pathogenesis and diagnosis. Transl Neurodegener 2023, 12, 40. [Google Scholar] [CrossRef]
- Kopp, K.O.; Glotfelty, E.J.; Li, Y.; et al. Glucagon-like peptide-1 (GLP-1) receptor agonists and neuroinflammation: Implications for neurodegenerative disease treatment. Pharmacol Res 2022, 186, 106550. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Yang, S.; Zhao, X.; et al. The Role of Glucagon-Like Peptide-1 Receptor Agonists (GLP-1 RA) in Diabetes-Related Neurodegenerative Diseases. Drug Des Devel Ther 2022, 16, 665–684. [Google Scholar] [CrossRef]
- Timper, K.; Del Rio-Martin, A.; Cremer, A.L.; et al. GLP-1 Receptor Signaling in Astrocytes Regulates Fatty Acid Oxidation, Mitochondrial Integrity, and Function. Cell Metab 2020, 31, 1189–1205. [Google Scholar] [CrossRef] [PubMed]
- Bayram, E.; Batzu, L.; Tilley, B.; et al. Clinical trials for cognition in Parkinson's disease: Where are we and how can we do better? Parkinsonism Relat Disord 2023, 112, 105385. [Google Scholar] [CrossRef] [PubMed]
- Diz-Chaves, Y.; Mastoor, Z.; Spuch, C.; et al. Anti-Inflammatory Effects of GLP-1 Receptor Activation in the Brain in Neurodegenerative Diseases. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Xu, F.; Previti, M.L.; et al. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J Neurosci 2007, 27, 3057–3063. [Google Scholar] [CrossRef] [PubMed]
- Sriram K, Miller DB, O'Callaghan JP Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem 2006, 96, 706–718. [CrossRef] [PubMed]
Figure 1.
Astrocyte-Targeted Therapeutic Pathways for Modulating Dopamine Circuits in Health and Disease.
Figure 1.
Astrocyte-Targeted Therapeutic Pathways for Modulating Dopamine Circuits in Health and Disease.
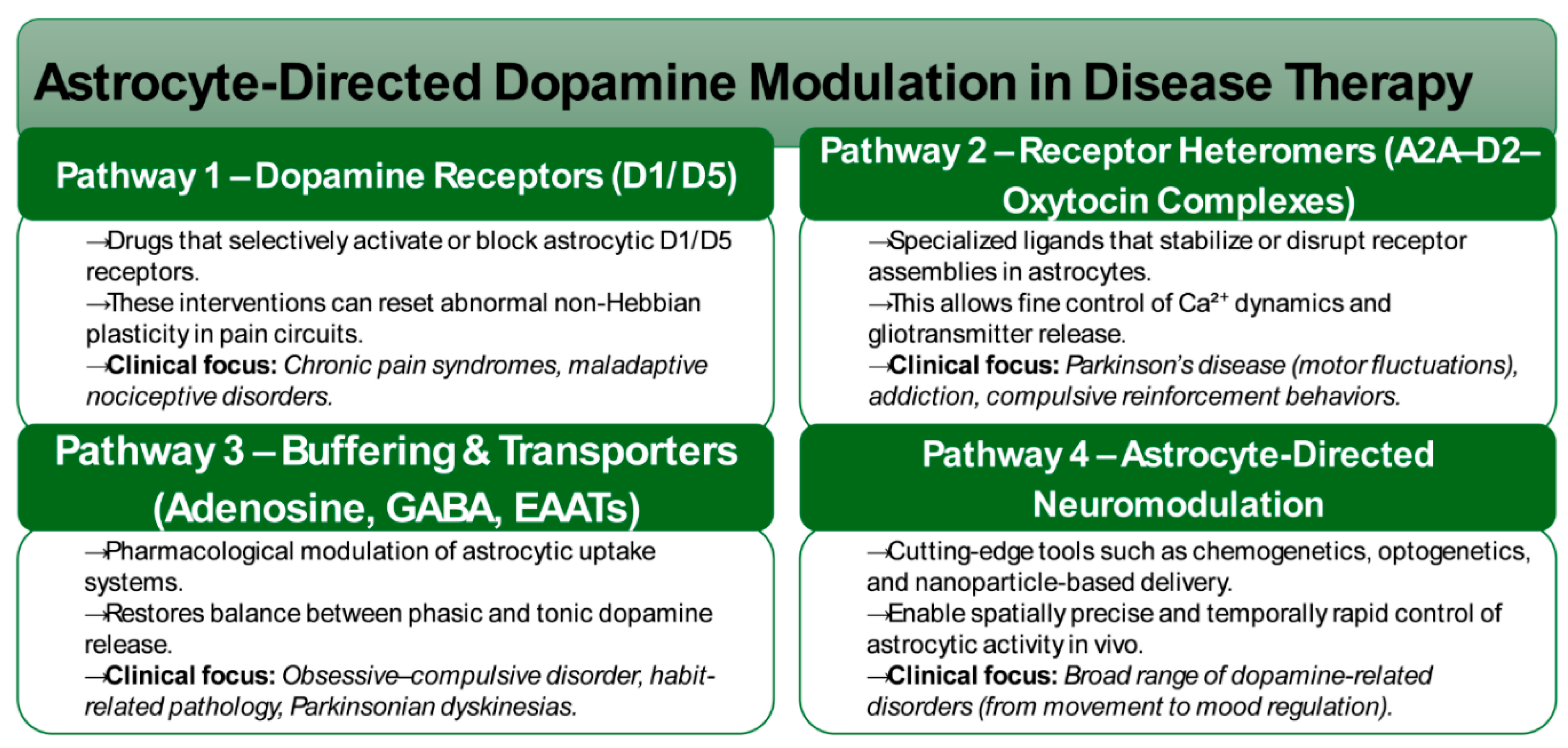
Figure 2.
Disorder-Specific Pathways of Glial–Dopamine Dysregulation and Motivational Dysfunction.
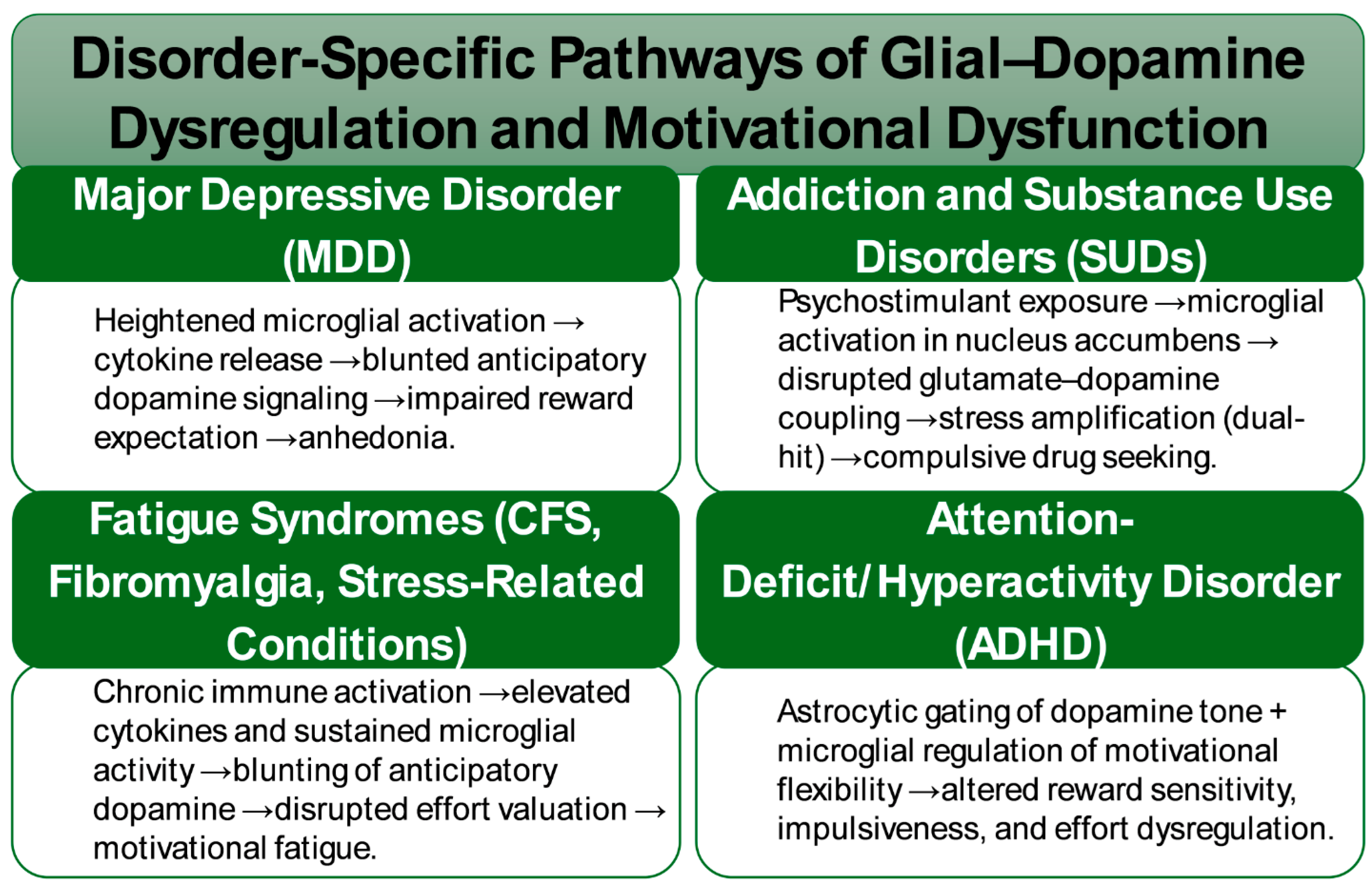
Figure 3.
Emerging Glial Therapeutic Pathways: Precision Modulation of Microglial and Astrocytic Programs in Parkinson’s Disease and Related Disorders.
Figure 3.
Emerging Glial Therapeutic Pathways: Precision Modulation of Microglial and Astrocytic Programs in Parkinson’s Disease and Related Disorders.

Figure 4.
Glia-Directed Therapeutic Paradigms in Dopaminergic Neurodegeneration.


Table 1.
Astrocytic Modulation of Dopaminergic Signaling.
| Focus | Astrocytic Mechanisms | Circuit Context | Key Implications | References |
|---|---|---|---|---|
| Dopamine Receptors | Expression of D1/D5, D2, D4; astrocytic D1/D5 required for non-Hebbian LTP; cortical laminar gradients (layer I > deep). | Spinal nociceptive pathways; cortical apical dendritic zones. | Establish astrocytic DA receptors as causal regulators of plasticity and cognition. | [6,21] |
| Receptor Heteromers | D2–OTR and A2A–D2–OTR complexes; regulate Ca²⁺ and glutamate release; integrate DA, adenosine, oxytocin. | Striatal astrocytes. | Provide multimodal integration; implicated in reward learning and habit pathology. | [22,23,24,25] |
| Gliotransmission | DA-induced glutamate release; lowered threshold for Ca²⁺ waves; DA–glutamate–astrocyte loop. | Spinal circuits (LTP induction); striatum (interneuron-driven DA release). | Astrocytes function upstream and downstream of DA, modulating plasticity and reinforcement. | [6,10,26,27] |
| Uptake & Clearance | Buffering of adenosine/GABA; EAAT1/2-mediated glutamate clearance. | Striatal DA terminals; cortico-thalamic inputs. | Real-time gating of DA tone; substrate for glutamate–DA crosstalk. | [10,28,29] |
| Regional Specificity | Striatal astrocytes: soma-to-soma contacts with interneurons, enriched heteromers. Cortical astrocytes: D1R/D4R-rich in layer I. |
Striatum: phasic DA release, motor/reward control. Cortex: working memory, attention. |
Dysregulation linked to PD, addiction, compulsive habits; schizophrenia, ADHD. | [10,21] |
Table 2.
Microglial Regulation of Dopamine Signaling and Neuroinflammation.
| Mechanistic Domain | Mechanistic Description | Representative Evidence | Pathological Relevance |
|---|---|---|---|
| Receptor-mediated signaling | Microglia express DRD1–DRD4 (DRD2/DRD4 enriched). DRD2 signaling suppresses inflammasome activity; DRD1 enhances pro-inflammatory cascades. Balance dictates outcome. | Transcriptomic and proteomic validation [3,4,5,6]; functional suppression or activation of NLRP3 inflammasome by DA [7,8]. | Dopamine acts as context-dependent brake (via DRD2) or amplifier (via DRD1) of neuroinflammation. |
| Inflammatory polarization | Dopamine tunes microglial states: DRD2 biases toward reparative, anti-inflammatory programs; DRD1 predominance amplifies IL-1β and NF-κB signaling. | Transcriptomic induction of cytokine and inflammasome genes in DA-exposed microglia [1,9,10,11]. | Determines switch between neuroprotection and neurotoxicity; central in chronic immune stress and HIV. |
| Stress and metabolic priming | Chronic stress alters lipid metabolism, mitochondrial respiration, and redox balance, lowering microglial activation thresholds. DA stimulation more readily triggers inflammasome activity. | GRAB-DA recordings in stress paradigms [12,13]; PET studies in prodromal PD showing immune activation preceding DA decline [14,15]. | Exaggerated immune reactivity; heightened vulnerability to depression, anhedonia, and neurodegenerative progression. |
| Proteinopathy and toxic metabolites | α-Synuclein activates TLR2/TLR4–NF-κB–NLRP3 axis; DA-derived metabolites (DOPAL, quinones) induce oxidative stress and toxic adduct formation. | α-synucleinopathy models show cytokine release and pathology propagation [19,20,21,22]; DOPAL–α-synuclein adducts accelerate fibrillization [26,27,28]. | Amplifies microglial pro-inflammatory bias; accelerates dopaminergic degeneration in Parkinson’s disease. |
| Reciprocal immune–dopamine loops | Microglial cytokines (IL-1β, TNF-α, IL-6) suppress TH, blunt DA release, and disrupt DAT trafficking. Systemic inflammation reprograms DA–microglia interactions. | Longitudinal PET studies in prodromal PD [29,30]; HIV and systemic immune models show maladaptive DA-driven inflammasome activation [1]. | Self-reinforcing cycle where inflammation impairs DA signaling and DA dysregulation further activates microglia. |
Table 3.
Mechanistic and Translational Dimensions of Astrocyte–Microglia Interactions in Dopamine Circuits.
Table 3.
Mechanistic and Translational Dimensions of Astrocyte–Microglia Interactions in Dopamine Circuits.
| Thematic Focus | Core Concept | Mechanistic Axes | Dopamine Circuit Effects | Clinical/Translational Relevance |
|---|---|---|---|---|
| Molecular Crosstalk | Reciprocal astrocyte–microglia signaling | - Cytokines: IL-1β, TNF-α ↔ IL-6, TGF-β - Complement: synaptic tagging & pruning - Purinergic: ATP/adenosine, P2Y12 - Chemokine: CX3CL1–CX3CR1 - Extracellular vesicles: cytokines, miRNAs, α-syn cargo |
- Sets thresholds for engulfment - Tunes release dynamics & receptor composition - Couples immune surveillance to dopamine signaling |
- Aberrant loops drive neuroinflammation & maladaptive pruning - EVs as biomarkers and therapeutic carriers |
| Spatial & Single-Cell Landscapes | Glial heterogeneity in dopamine hubs | - Oxidative–metabolic astrocyte subtypes (antioxidant, mitochondrial) - Immune-primed microglial clusters (ISGs, inflammasomes, lipid metabolism) - Pathological niches: astrocytes + microglia + infiltrating T cells |
- Defines “glial neighborhoods” embedded in D1/D2 ensembles - Orchestrates reinforcement learning & motivational plasticity |
- PD: emergence of glial–immune niches - Astrocytic diversity in health vs. microglial expansion in pathology - Biomarkers for selective vulnerability |
| Mechanisms of Plasticity | Glia as regulators of dopaminergic rules | - Astrocytic adenosine/GABA → cholinergic interneuron excitability - Astrocytic GPCR heteromers (A2A–D2, A2A–OTR, D2–OTR) - Microglial complement-mediated pruning - Vascular–metabolic coupling (ROS buffering, BBB integrity) |
- Determines phasic/tonic dopamine balance - Defines thresholds of corticostriatal plasticity - Shapes reinforcement learning algorithms |
- Disrupted metabolic support/vascular surveillance drives degeneration - Aberrant pruning and heteromer logic linked to PD, stress, addiction |
| Disease Microenvironments | Glia-centric niches of vulnerability | - Lipid- and inflammasome-enriched microglia + oxidative-stress astrocytes - Fractalkine signaling shift (neuronal → endothelial) - Glia-derived EVs propagate α-syn & inflammatory cargo |
- Maladaptive niches amplify dopaminergic vulnerability - Remodels mesocorticolimbic circuits under chronic stress |
- PD: neuroinflammation + metabolic collapse - Stress: impaired dopamine release, motivational deficits (anhedonia, apathy) - Engineered EVs as therapeutic vectors |
| Humanized In Vitro Models | iPSC-derived tri-cultures for mechanistic precision | - Recapitulate IL-1β/TNF-α loops, complement, purinergic cascades - Readouts: synapse density, excitability, EV cargo |
- Enable controlled dissection of astrocyte–microglia–neuron interactions - Define causal modules in dopamine regulation |
- Preclinical pipeline for drug discovery - Complement inhibition and purinergic modulation normalize excitability |
Table 4.
Astrocytic and Microglial Modulation of Dopamine Circuits in Motivation and Stress.
| Glial Mechanism | Circuit / Region | Experimental Evidence | Behavioral / Clinical Outcome |
|---|---|---|---|
| Astrocytic ensembles encoding reward cues | Nucleus accumbens (NAc, posterior–ventral) | Activity-defined astrocytic ensembles recruited during cue–reward learning; optogenetic reactivation drives approach behavior [5,6] | Astrocytes encode motivational salience; selective ensembles modulate cue-driven reward pursuit |
| Astrocytic regulation of strategy selection | External globus pallidus (GPe, indirect pathway) | Chemogenetic activation reduces habitual responding, enhances goal-directed actions; recruitment during action sequences [7,8] | Balances motivational persistence with behavioral flexibility |
| Stress-sensitive astrocytic modulation of DA | NAc (chronic social stress model) | GRAB-DA fibre photometry shows attenuated anticipatory DA under stress; astrocytic glutamate/ATP/adenosine/D-serine implicated [9,10] | Impaired effort allocation and reward learning; reduced motivational vigor |
| Microglial activation under chronic stress | NAc, PFC, VTA | Stress induces hypertrophy, immune gene upregulation, synaptic remodeling [11]; amplified with psychostimulants [12] | Impaired reward learning; vulnerability to anhedonia and addiction-like behaviors |
| Cytokine–dopamine coupling | Mesolimbic and mesocortical circuits | uCMS models show elevated IL-1β, TNF-α linked to anhedonia [13,14]; clinical studies confirm microglial activation and inflammation in MDD [15,16] | Cytokine-mediated suppression of DA signaling → reduced motivation |
| Microglial priming in early-life stress | Developmental NAc and mesolimbic circuits | ELS increases pro-inflammatory cytokines, disrupts HPA axis, blunts anticipatory reward [17,18]; microglia regulate synaptogenesis during adolescence [19] | Lasting motivational vulnerability; reduced resilience to later stressors |
| Astrocytic gliotransmission and DA release control | NAc, GPe | Astrocytic glutamate, ATP, and D-serine release regulate MSNs and DA terminal excitability [5,20]; GPe astrocytes bias flexible strategies [7] | Fine-tunes motivational encoding; shifts balance from habits to adaptive behaviors |
| Microglial cytokine release | NAc, PFC, VTA | IL-1β and TNF-α suppress DA synthesis, impair receptors, prune dendritic spines [21,22] | Blunted reward anticipation; anhedonia |
| Developmental trajectory of glial–DA coupling | NAc during adolescence | Microglial regulation of synaptogenesis disrupted by ELS, immune stress, genetic risk [23] | Long-term alterations in connectivity → chronic motivational deficits |
| Real-time DA monitoring with GRAB-DA sensors | NAc, mesolimbic DA terminals | Photometry distinguishes anticipatory vs. consummatory DA; stress selectively attenuates anticipatory responses [24,25] | Identifies anticipatory phase as window of vulnerability and therapeutic intervention |
| Behavioral assays linked to astrocytic manipulation | Operant paradigms (progressive ratio, outcome devaluation) | Chemogenetic GPe astrocyte activation biases goal-directed over habitual actions [7]; ensemble reactivation drives approach [5,27] | Establishes causal role of astrocytes in motivational computation |
| Integrated immune–DA–behavior paradigms | CSS, uCMS, ELS models | Cytokine profiling + DA photometry map inflammation–DA–behavior triads [28,29] | Clarifies convergence of neuroimmune signaling and motivational pathology |
Table 5.
Integrated Mechanisms of Dopamine–Glia Crosstalk in Neurodegeneration and Disease.
| Pathological Context | Core Mechanisms | Key Evidence | Clinical/Pathological Relevance |
|---|---|---|---|
| Parkinson’s disease: astrocytic and microglial reprogramming | Astrocytic A1-like conversion (loss of glutamate clearance, trophic support, ↑ oxidative/inflammatory signaling) induced by microglial cytokines (IL-1α, TNF-α, C1q). | Postmortem SN and preclinical models. | Microglia drive astrocytic maladaptation, weakening DA neuron resilience. |
| Microglial transitions, ECM, and exosomes | CSF1R inhibition (PLX5622) reduces microglia, protects DA neurons, remodels ECM; microglial exosomes carry inflammatory/α-syn cargo, induce A1 astrocytes. | α-syn models; CSF1R and Peli1 manipulations. | Microglia–ECM and vesicular signaling amplify synucleinopathy and degeneration. |
| Ferroptotic stress | DA neurons are highly vulnerable due to iron turnover, autoxidation, and mitochondrial load. Maladaptive glia exacerbates glutamate dysregulation, redox imbalance, lipid peroxidation. | Ceftriaxone restores SLC7A11/GPX4; multi-omics link glia to ferroptosis. | Identifies ferroptosis as glia-amplified death pathway; therapeutic targets include GPX4 activators and cystine–glutamate modulators. |
| Post-viral (Long COVID) basal ganglia dysfunction | Chronic inflammation disrupts corticostriatal DA loops: ↓ synthesis/release, ↓ receptor sensitivity; BBB disruption, astrocytic endfoot injury sustain glial priming. | Neuroimaging of basal ganglia; ↑ GFAP, IL-6, TNF-α in post-COVID cohorts. | Explains fatigue, effort intolerance, cognitive slowing via DA insufficiency and glial-driven basal ganglia failure. |
| Aging as amplifier | Aged microglia adopt primed pro-inflammatory states; astrocytes show impaired glutamate clearance, reduced lactate/antioxidant support, ↑ ROS/lipid peroxidation. | Microglial/astrocytic aging phenotypes; PD datasets show stronger inflammatory signatures in older cohorts. | Aging synergizes with inflammation and metabolic stress, accelerating dopaminergic degeneration. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



