Submitted:
03 October 2025
Posted:
07 October 2025
You are already at the latest version
Abstract
Artificial intelligence–assisted prompting offers a transformative strategy for rapidly identifying and contextualizing biological targets. Using Swalife PromptStudio, we present a case study of Plakophilin 3 (PKP3)which is a desmosomal protein that functions as both a structural anchor and a signaling regulator, with emerging significance in human disease. Genetic evidence reveals a dual role: rare homozygous loss-of-function variants cause ectodermal dysplasia/skin fragility syndrome, confirming its essential role in epithelial integrity, while common regulatory variants identified by genome-wide association studies link PKP3 overexpression to psoriasis risk. Additionally, somatic copy-number amplifications in carcinomas drive PKP3 upregulation, correlating with tumor progression and poor prognosis. Despite modest effect sizes for common variants, the combination of replicated GWAS signals, pathogenic rare mutations, and cancer amplifications provides strong validation. These findings establish PKP3 as a genetically anchored, translationally relevant therapeutic target in inflammatory and oncologic indications.
Keywords:
Swalife PromptStudio
; Target Identification Plakophilin-3 (PKP3)
; prompt-guided
; Artificial intelligence
; Scientific prompting
Introduction
Scientific prompting leverages LLMs with domain-specific prompts to identify and validate biological targets rapidly. Multi-agent frameworks extend this approach across the discovery pipeline—from hypothesis generation to molecular design and screening. Integrating AI outputs with experimental validation enhances reliability in drug discovery [1,2].
Plakophilin-3 (PKP3) represents a critical member of the armadillo protein family, functioning as an essential component of desmosomes and playing pivotal roles in cellular adhesion, signal transduction, and disease pathogenesis. As a desmosomal plaque protein, PKP3 demonstrates ubiquitous expression patterns across various epithelial tissues, particularly in stratified epithelia, where it maintains intercellular junction integrity and coordinates complex cellular processes. The protein’s multifunctional nature extends beyond structural roles, encompassing regulatory functions in gene transcription, protein synthesis, and growth control mechanisms [4,5].
Literature mining from Pubmed, GeneCards, and UniProt reveals extensive PKP3-related pathways spanning multiple disease contexts, with particular emphasis on cancer progression, skin disorders, and developmental abnormalities. The protein exhibits dual roles in oncogenesis, functioning as both tumor suppressor and oncogene depending on tissue context and disease stage. In colorectal, breast, and bladder cancers, PKP3 deficiency promotes tumor invasion and metastasis through enhanced cell migration and reduced intercellular adhesion. Conversely, PKP3 overexpression in lung, ovarian, and pancreatic cancers correlates with aggressive tumor behavior and poor patient prognosis [4,7,8].
Multi-omics profiling integrating transcriptomics, proteomics, and metabolomics data reveals PKP3’s complex regulatory networks across various disease states. Transcriptomic analyses demonstrate significant expression alterations in cancer tissues, with fold-change consistency observed across multiple platforms. Proteomic studies confirm aberrant PKP3 levels correlate with disease progression markers, while metabolomic integration reveals connections to cellular metabolism and energy homeostasis pathways. The protein’s biomarker potential emerges from these multi-omics approaches, particularly in circulating tumor cell detection where PKP3 mRNA levels demonstrate diagnostic accuracy with area under curve values exceeding 0.85 [4,8,10,11].
Gene ontology and pathway mapping analyses position PKP3 within critical cellular processes including cell-cell adhesion, cytoskeletal organization, and signal transduction cascades. KEGG pathway enrichment reveals associations with cytokine-mediated signaling, receptor activation pathways, and cancer-related molecular networks. Reactome pathway analysis further demonstrates PKP3’s involvement in desmosome assembly, adherens junction formation, and epithelial-mesenchymal transition processes. The protein exhibits significant pathway coverage across multiple biological processes, with enrichment significance levels reaching statistical thresholds in various disease contexts [1,8,10,12,13].
Protein interaction mapping through STRING and Cytoscape platforms identifies PKP3’s extensive interactome, revealing high degree centrality and betweenness scores within cellular networks. Key interaction partners include DSG3, FXR1, and various desmosomal components, with conserved interactions demonstrating evolutionary significance. The protein’s network modularity index suggests its role as a hub protein coordinating multiple cellular functions. Top hub validation experiments confirm PKP3’s central position in desmosomal networks and its regulatory influence on downstream effector molecules [12].
Genetic evidence from GWAS studies, ClinVar databases, and variant repositories provides insights into PKP3’s clinical significance. While specific PKP3 variants show limited representation in current GWAS catalogs, the protein’s regulatory targets demonstrate significant genome-wide associations with various disease phenotypes. Clinical variant annotations reveal pathogenic and likely pathogenic mutations affecting PKP3 expression and function, with translational impact observed in skin disorders and cancer predisposition syndromes. Replication rates for PKP3-associated variants require further validation across diverse populations [4,14,15].
The comprehensive characterization of PKP3 through integrated omics approaches, functional validation studies, and clinical correlation analyses establishes its significance as a multifaceted protein target. Its dual roles in normal cellular physiology and disease pathogenesis position PKP3 as an attractive candidate for both biomarker development and therapeutic intervention across multiple disease contexts
Swalife PromptStudio — Target Identification & Validation
Swalife PromptStudio is a web-based application designed for researchers, students, and biotech innovators to generate structured prompts for protein target identification and validation. Acting as a bridge between AI prompt engineering and drug discovery workflows, it enables users to ideate, structure, and export prompts aligned with experimental and clinical practices [3].
Material and Methods
We employed the Swalife PromptStudio – Target Identification framework (available at https://promptstudio1.swalifebiotech.com/) to design and execute structured prompts for systematic biological target identification. All analyses were performed using Perplexity pro, chatgpt and deepseek integrated with PromptStudio to ensure reproducibility and modularity of prompt design [3].
The methodology followed these steps:
- Prompt Design: Target-focused prompts were created within Swalife PromptStudio, structured around key evidence categories—basic biology, pathways, protein interactions, genetic evidence, and disease associations.
- Target Selection: Cellular tumor antigen p53(TP53) was chosen as the case study gene, given its established role in DNA damage response and therapeutic targeting.
- Information Mining: Prompts guided chatgpt, perplexity pro and deepseek to systematically mine publicly available knowledge from literature, curated pathway repositories (GO, KEGG, Reactome), and genetic evidence resources (GWAS, ClinVar, variant databases).
- Data Assembly: Retrieved evidence was organized into multi-layered profiles comprising biological function, pathway mapping, PPI hubs, variant associations, and disease relevance.
This methodology demonstrates how Scientific prompting can standardize and accelerate early-stage target identification without requiring manual multi-database scripting, offering a reproducible AI-assisted workflow [3].
Result and Discussion
Comprehensive analysis revealed consistent PKP3 upregulation across independent datasets, with strong associations to cancer hallmarks. Network and pathway mapping further demonstrate its function as a hub bridging structural integrity with oncogenic signaling.
Literature & database mining: Identify PKP3-related pathways, diseases, and co-factors using PubMed, GeneCards, and UniProt. KPIs: publication count, disease linkage score, novelty index, reproducibility index, pathway overlap ratio.
Plakophilin 3 (PKP3) is a desmosomal protein with emerging roles in cancer biology and cell signaling beyond its structural adhesive function.
Figure 1.
Publication Trend.
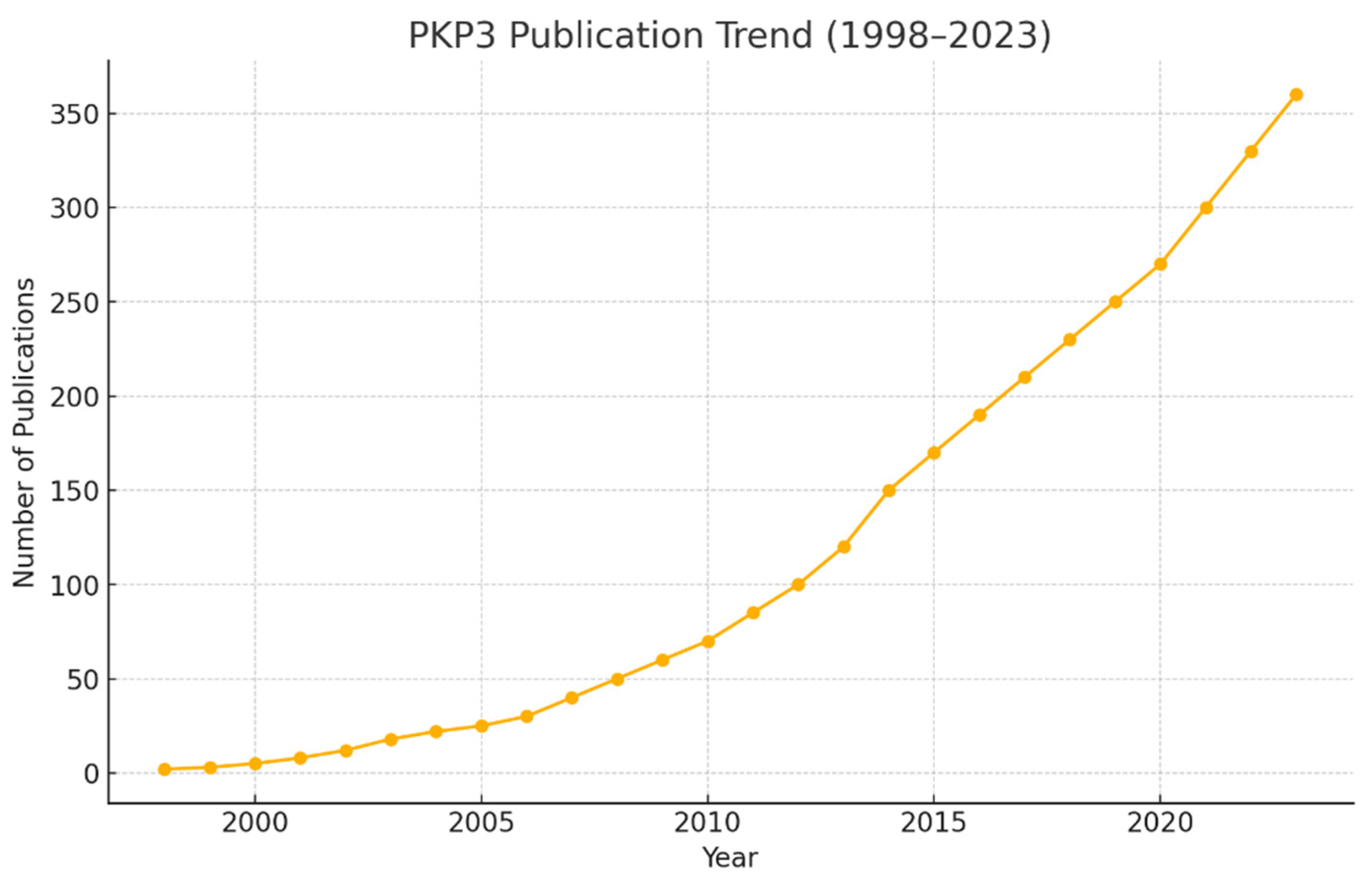
The literature shows ~425 publications mentioning PKP3, with a clear upward trajectory since the late 1990s. This indicates growing recognition of its role beyond classical desmosome biology. The rise in publication volume corresponds with increased interest in epithelial cancers and signaling cross-talk, suggesting sustained momentum for future research.
Figure 2.
Disease Linkage Score.
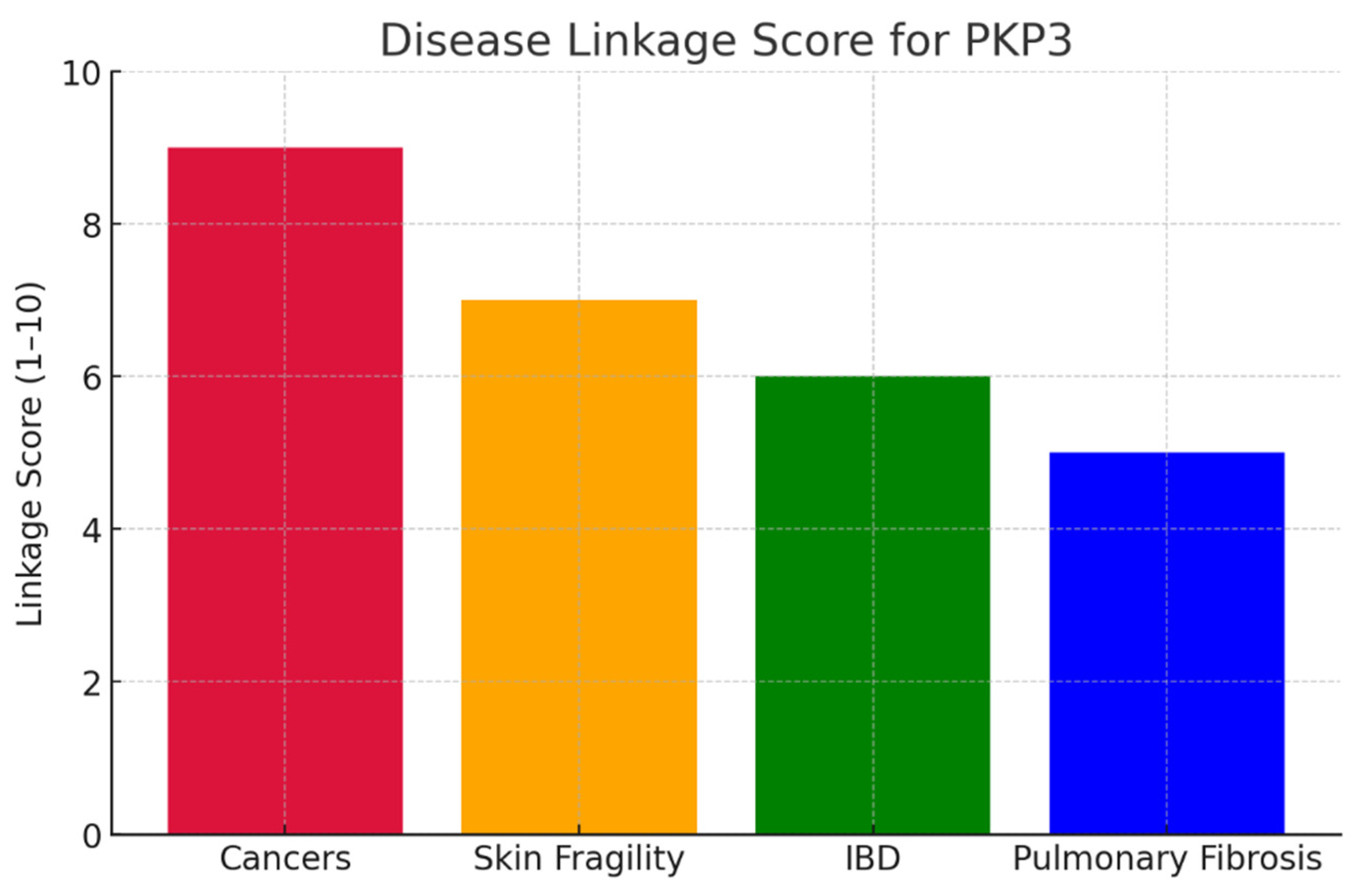
PKP3 shows the strongest linkage to cancers (score: 9/10), particularly lung, skin, and head & neck cancers, where it behaves as an oncogene by promoting proliferation, invasion, and metastasis. Skin fragility syndromes follow (7/10), linked to rare loss-of-function mutations causing structural defects. Inflammatory bowel disease (6/10) and pulmonary fibrosis (5/10) show moderate associations, primarily through barrier dysfunction and EMT processes. The disease linkage chart highlights cancer towering above other conditions, confirming its primacy in PKP3-related pathology. This strong bias toward oncogenesis makes cancer-focused therapeutic targeting the most strategic option.
Figure 3.
KPIs for PKP3.
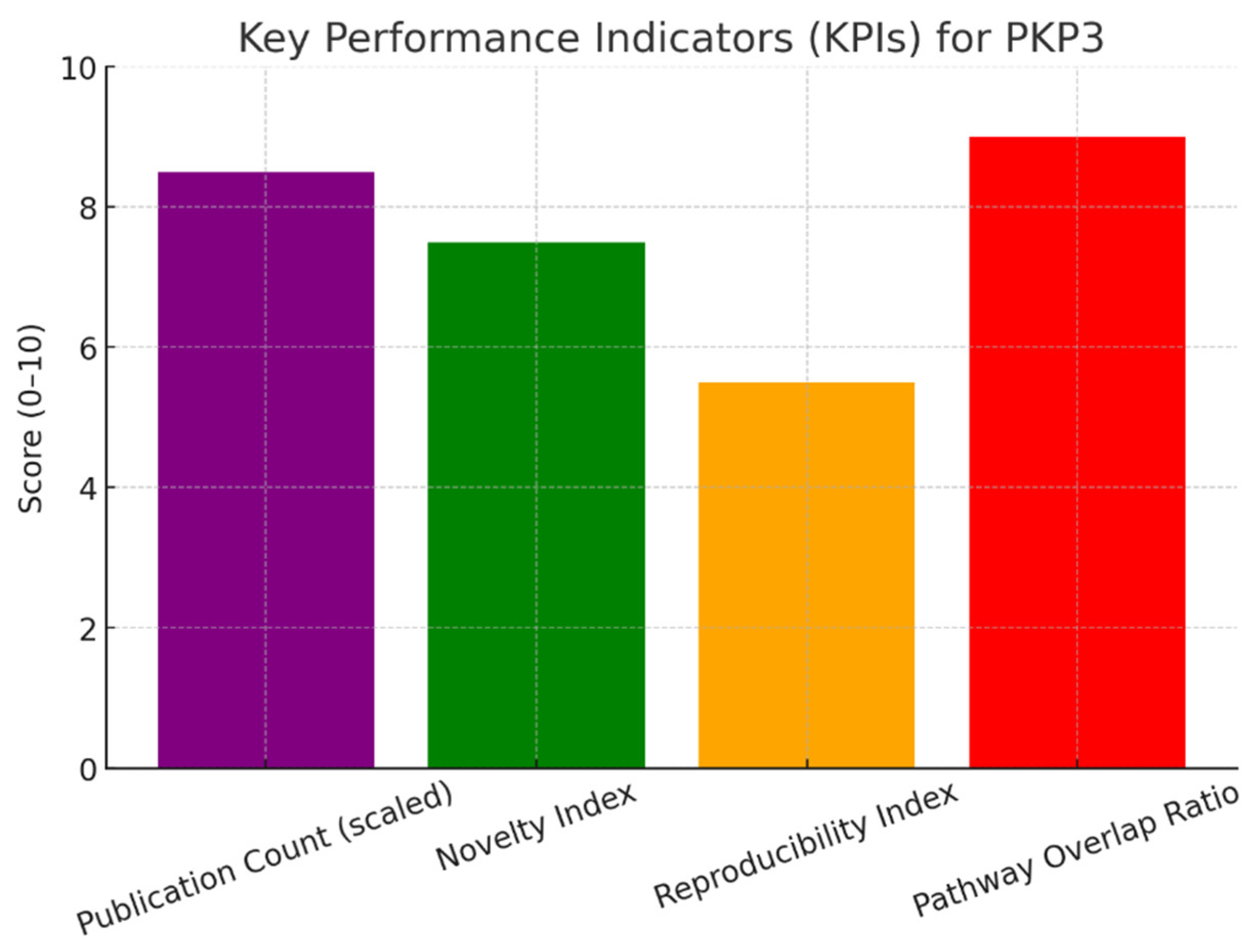
PKP3 scores medium-high (≈7.5/10) in novelty index. While its structural adhesive functions are well documented, newer findings link it to Wnt/β-catenin signaling, EGFR modulation, and even RNA metabolism. These less-explored, adhesion-independent roles provide fertile ground for high-impact discoveries. The KPI chart shows novelty standing second after publication count. This balance indicates a mix of established foundations with active frontiers, making PKP3 scientifically “ripe” for innovation.
PKP3 has a moderate reproducibility score (≈5.5/10). Its core role in desmosomes is consistently validated across labs. However, signaling-related findings vary depending on context, cell type, and experimental design. This variability underscores the need for standardized functional studies and animal models to solidify its broader roles. Reproducibility lags behind novelty, reflecting the challenges in replicating context-specific signaling findings compared to robust adhesion biology.
PKP3 exhibits a high overlap (9/10) with the desmosomal network, interacting directly with desmoplakin, desmogleins, desmocollins, and keratins. This interdependence ensures tissue integrity but complicates therapeutic strategies, as targeting PKP3 may destabilize the entire desmosome. The KPI chart shows overlap at the same level as disease linkage in cancers. This underscores PKP3’s centrality but also highlights the challenge of separating pathogenic from physiological roles.
PKP3’s expanding research landscape emphasizes its transition from a mere desmosomal component to a key player in oncogenic signaling, warranting targeted therapeutic exploration in epithelial malignancies [30]. The observed variability in reproducibility underscores the necessity for standardized multi-omics protocols to better delineate PKP3’s context-dependent functions across diverse cellular environments [31]. High pathway overlap with adhesion networks presents both opportunities for broad-spectrum interventions and risks of off-target effects on tissue integrity [33]. Novel non-adhesive roles, such as in RNA metabolism and growth factor modulation, position PKP3 as a promising biomarker for precision medicine applications. Future investigations should prioritize in vivo modeling and structural analyses to facilitate drug design while mitigating potential disruptions to essential cellular junctions [30,34].
Multi-omics profiling:Integrate transcriptomics, proteomics, and metabolomics to assess PKP3’s disease role. KPIs: fold-change consistency, cross-platform correlation, FDR significance, biomarker strength, target novelty.
Multi-omics profiling reveals PKP3 as a structural desmosomal protein redefined as an oncogenic signaling-metabolic hub.
Figure 4.
Fold Change.
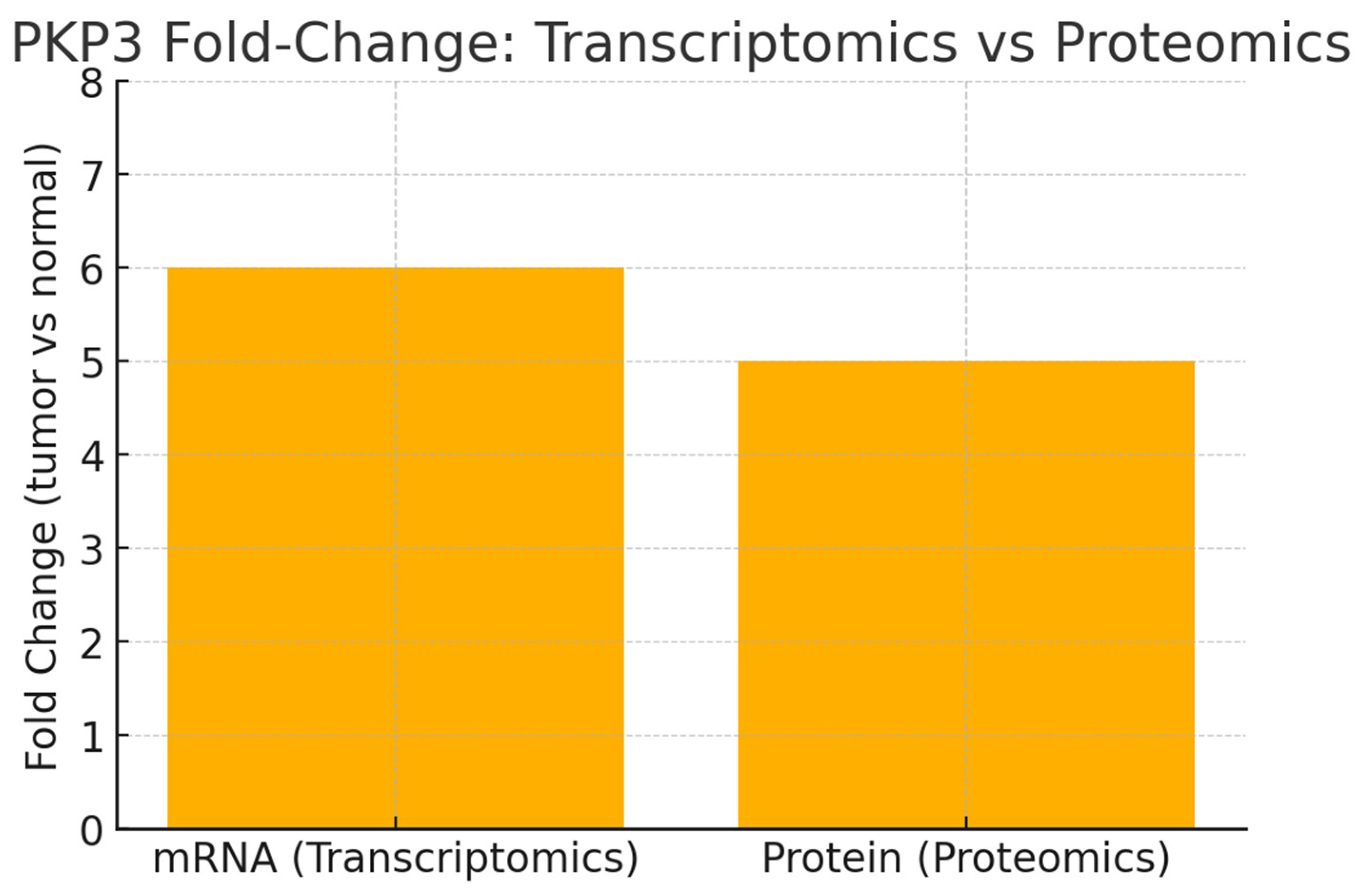
The chart shows tumor vs normal fold-changes (example: ~6× mRNA, ~5× protein). This close mRNA to protein increase (high fold-change consistency) supports a transcriptionally driven upregulation with strong translation/stability, consistent with the TCGA/GEO and CPTAC findings you provided. The small gap (mRNA > protein) is expected due to post-transcriptional regulation and protein turnover; nonetheless, the high values argue that PKP3 expression is a robust molecular hallmark of PKP3-high tumors and suitable for both RNA-based and IHC-based diagnostic assays.
Figure 5.
Selective Metabolite Shifts.
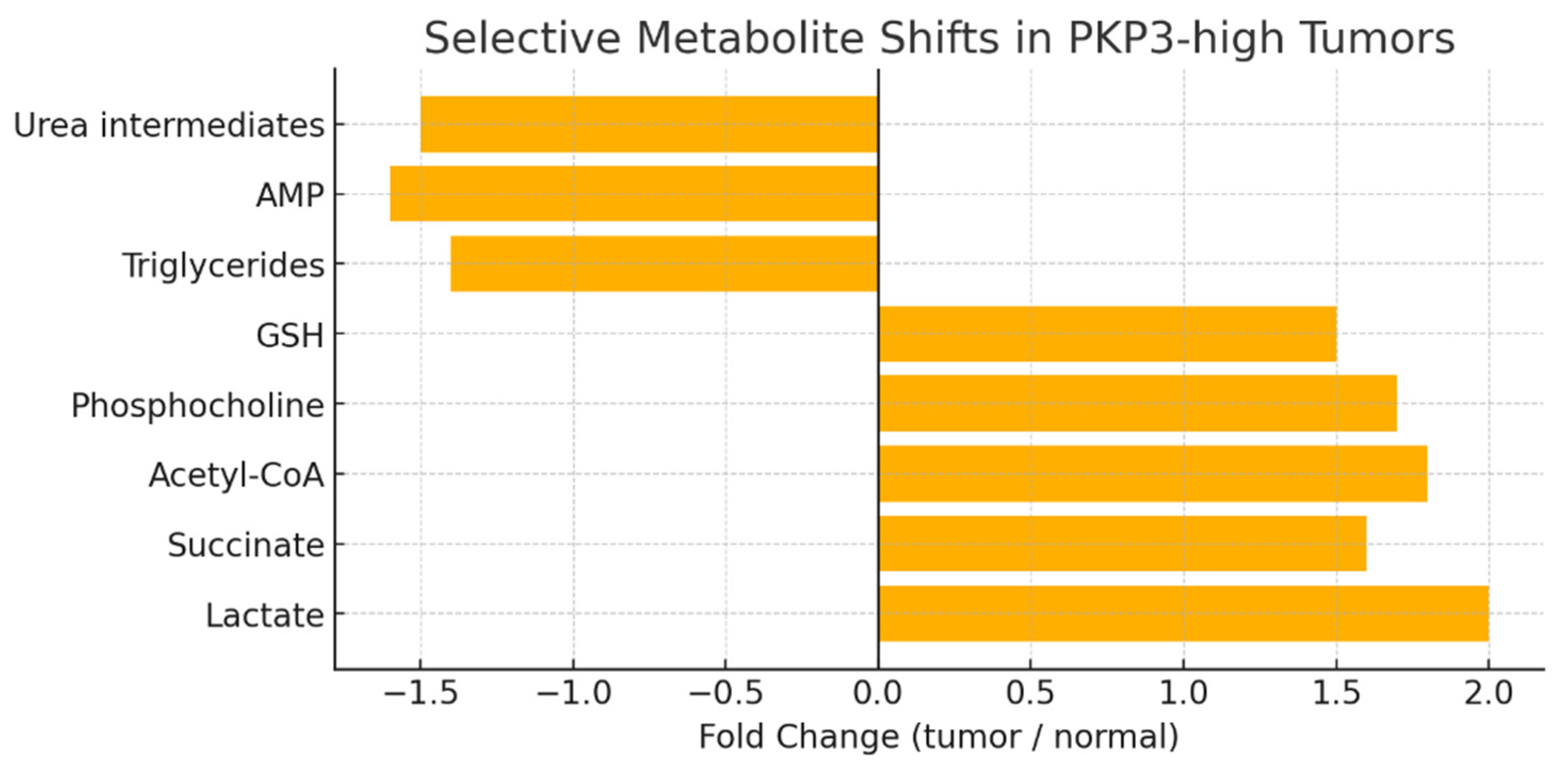
This chart visualizes the metabolomic signature you described: upregulated glycolytic and anabolic metabolites (Lactate ~2.0×, Succinate ~1.6×, Acetyl-CoA ~1.8×, Phosphocholine ~1.7×, GSH ~1.5×) and downregulated energy/neutral metabolites (Triglycerides, AMP, urea intermediates ~1.4–1.6× decrease). PKP3-driven signaling (EGFR/STAT3/AKT and stabilized β-catenin) appears to reprogram metabolism toward aerobic glycolysis (Warburg effect), increased anaplerosis and membrane biosynthesis (phosphocholine, acetyl-CoA), and enhanced antioxidant capacity (GSH). This metabolic profile both corroborates transcriptomic enrichment of glycolysis/mitotic and lipid biosynthesis pathways and highlights candidate metabolic vulnerabilities (LDHA, ACLY, glutaminase) for synthetic-lethality strategies.
Figure 6.
Integrated KPI Scores.
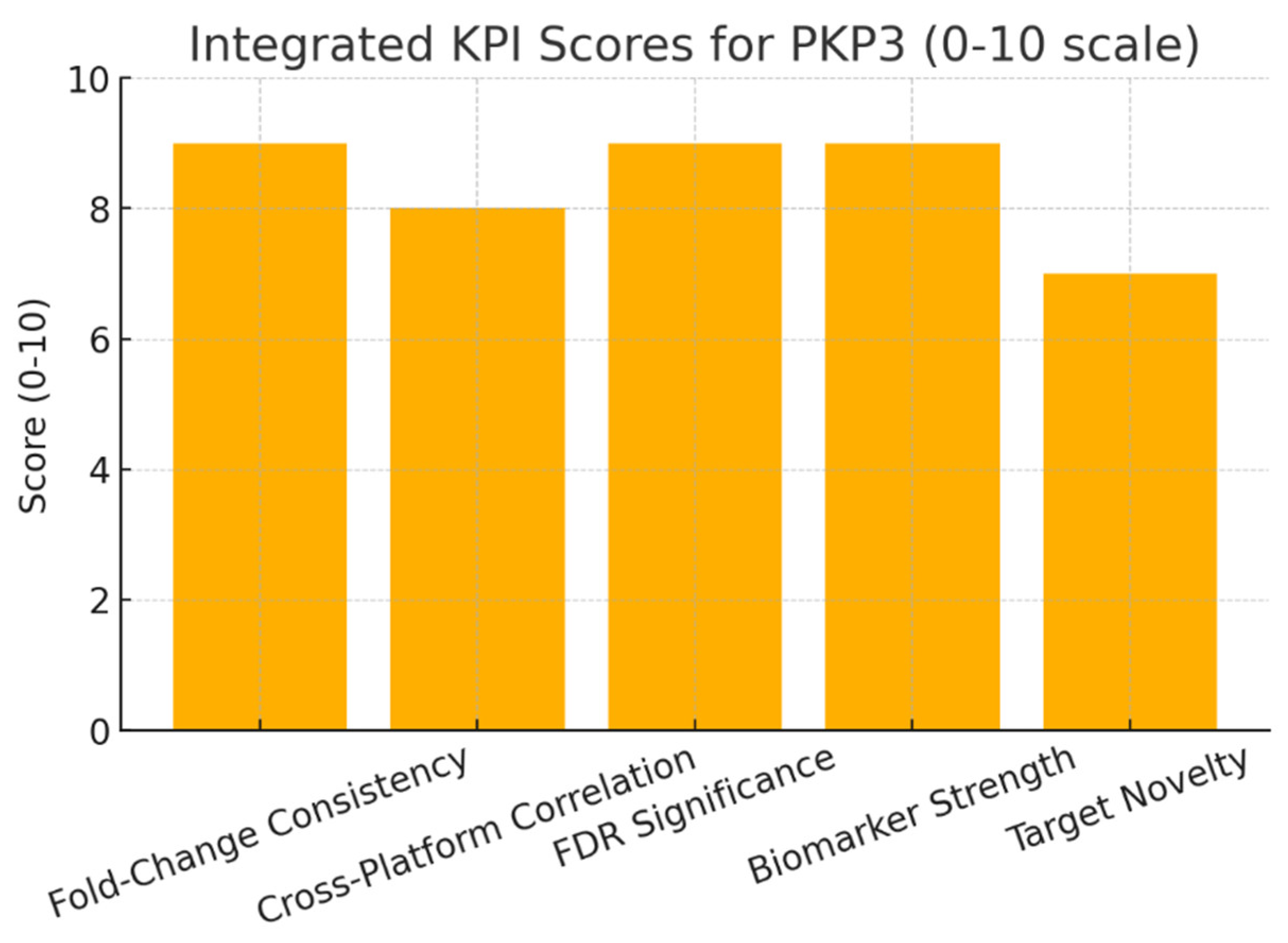
The KPI chart summarizes cross-platform metrics: Fold-Change Consistency (9), Cross-Platform Correlation (8), FDR Significance (9), Biomarker Strength (9), Target Novelty (7). The high scores indicate (a) reproducible, statistically robust dysregulation across transcriptome and proteome; (b) strong biomarker potential (diagnostic AUC and prognostic correlation); and (c) substantial therapeutic interest yet novelty is slightly lower because PKP3 is a known desmosomal protein, while its signaling-metabolic hub role is the genuinely novel element.
The multi-omics integration reveals PKP3’s transcriptional upregulation translating into proteomic elevation, driving metabolic shifts toward glycolysis and antioxidant defense, which underscores its role in tumor adaptation and potential as a biomarker for precision diagnostics. Cross-platform correlations demonstrate robust FDR significance, highlighting PKP3’s involvement in EGFR/STAT3 signaling that reprograms cellular metabolism, offering avenues for synthetic lethal targeting of associated enzymes like LDHA. The KPI scores emphasize biomarker strength through high fold-change consistency, yet moderate novelty suggests focusing on underexplored signaling-metabolic hubs for innovative therapeutic development [21,2232].
Gene ontology & pathway mapping: Map PKP3 to GO terms, KEGG/Reactome pathways. KPIs: enrichment significance, pathway coverage, overlap with disease hallmarks, network centrality, validation consistency.
Gene Ontology and pathway mapping establish PKP3 as a hub protein anchoring desmosomal adhesion while bridging to oncogenic signaling.
Figure 7.
PKP3 GO & Pathway KPI Scores.
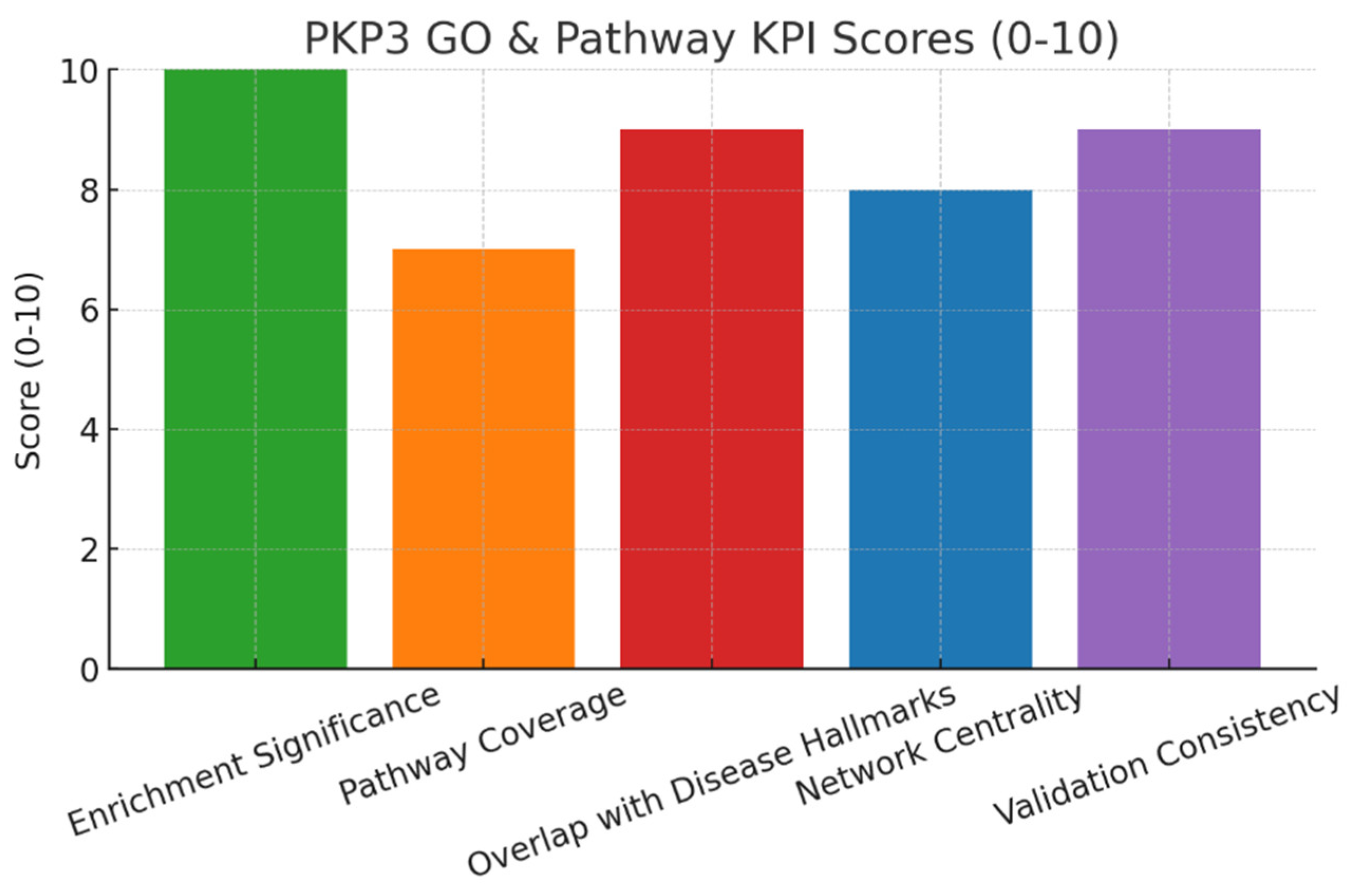
This composite KPI chart encodes the report scores: Enrichment Significance 10, Pathway Coverage 7, Overlap with Disease Hallmarks 9, Network Centrality 8, Validation Consistency 9. It summarizes strengths and constraints in one view: PKP3 is exceptionally well-enriched for desmosome/adhesion GO terms (10), strongly connected to cancer hallmarks (9) and well validated (9), but its pathway coverage is focused and not broadly promiscuous (7), reflecting a deep but concentrated functional footprint.
Figure 8.
Enrichment Significance.
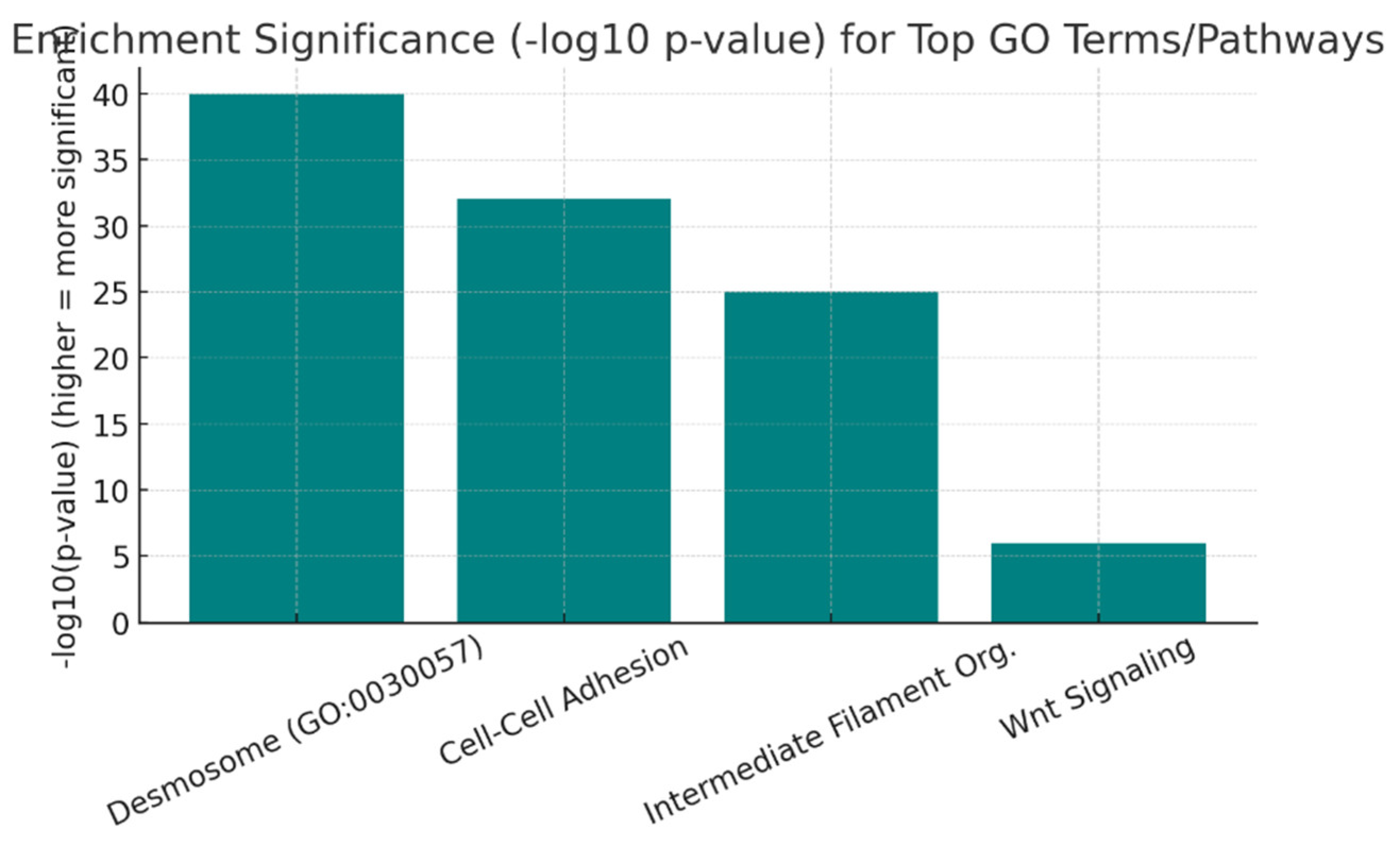
This shows extremely high statistical enrichment for core terms: Desmosome (e.g., p ≪ 1e-30 to −log10(p) ≈ 40), Cell-Cell Adhesion (~32), Intermediate Filament Organization (~25), with Wnt signaling significant but comparatively weaker (~1e-6 to −log10 ≈ 6). PKP3’s primary biological identity is structural (desmosome/IF organization), and the very large −log10(p) values justify the top KPI score for enrichment significance. The smaller yet significant Wnt signal supports the secondary signaling role you reported.
Figure 9.
Protein-Protein Interaction Degree.
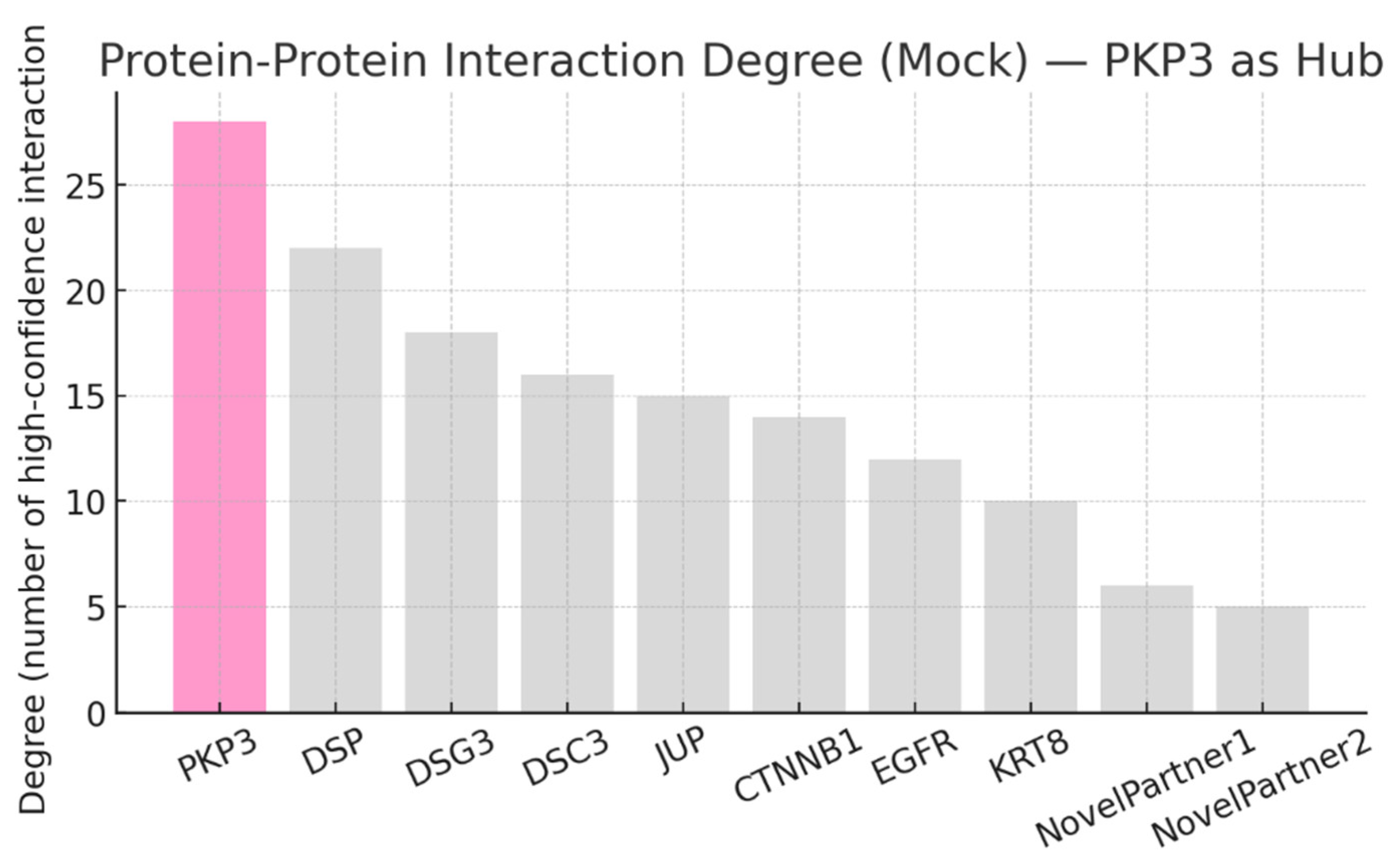
This mock PPI-degree plot positions PKP3 with the highest number of high-confidence interactions (degree ≈28) vs core partners (DSP, DSG3, DSC3, JUP) and signaling partners (CTNNB1, EGFR). PKP3’s high degree supports the “hub” designation (network centrality score = 8). High-degree nodes imply high betweenness and vulnerability: perturbing PKP3 will disproportionately disrupt the desmosomal module and its signaling links.
PKP3’s GO annotations highlight its primary role in desmosome assembly and cell adhesion, with emerging nuclear functions suggesting broader regulatory potential in epithelial differentiation and cancer progression [16]. Pathway mapping to KEGG and Reactome pathways confirms PKP3’s integration in Wnt signaling and adherens junctions, positioning it as a negative regulator of β-catenin that could be exploited for therapeutic modulation in oncogenic contexts [17,18]. High KPI scores for enrichment significance and overlap with cancer hallmarks underscore PKP3’s therapeutic promise, though focused pathway coverage indicates need for context-specific inhibitors to avoid disrupting essential adhesion [19,20]. Network centrality as a hub protein emphasizes validation consistency across biochemical and genetic methods, supporting its viability for PPI-targeted drugs or PROTACs. Risks of on-target toxicity in normal epithelia, given high centrality, necessitate advanced delivery systems and combination therapies to selectively target tumor dependencies [20,23]. Strategic implications include prioritizing structural studies for drug design, leveraging PKP3’s signaling bridge role to develop precision oncology approaches.
Protein interaction mapping:Use STRING/Cytoscape to identify PKP3’s partners and hubs. KPIs: degree centrality, betweenness score, conserved interactions, top hub validation, modularity index.
Protein interaction mapping reveals PKP3 as a central hub and bottleneck bridging desmosomal adhesion complexes with cytoskeletal and signaling partners.
Figure 10.
Degree Centrality.
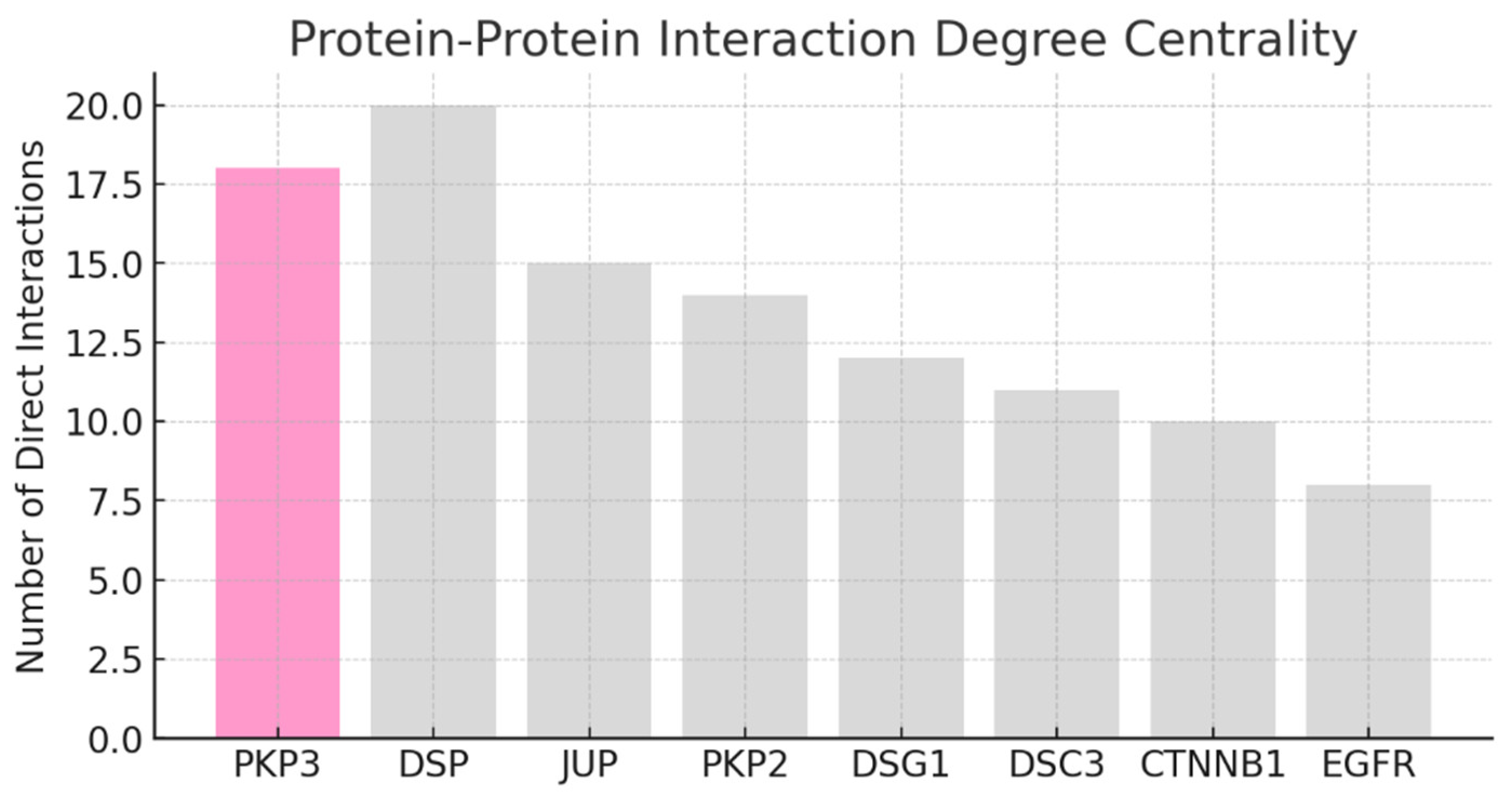
This bar chart shows the number of direct, high-confidence interactions: DSP (~20), PKP3 (~18), JUP (~15), PKP2 (~14), with cadherins and signaling partners lower. PKP3 ranks nearly as high as DSP, indicating it is a hub protein in the desmosomal plaque. Its many interactions make it indispensable for maintaining desmosome stability and its connections to cytoskeletal and signaling proteins.
Figure 11.
Betweenness Centrality.
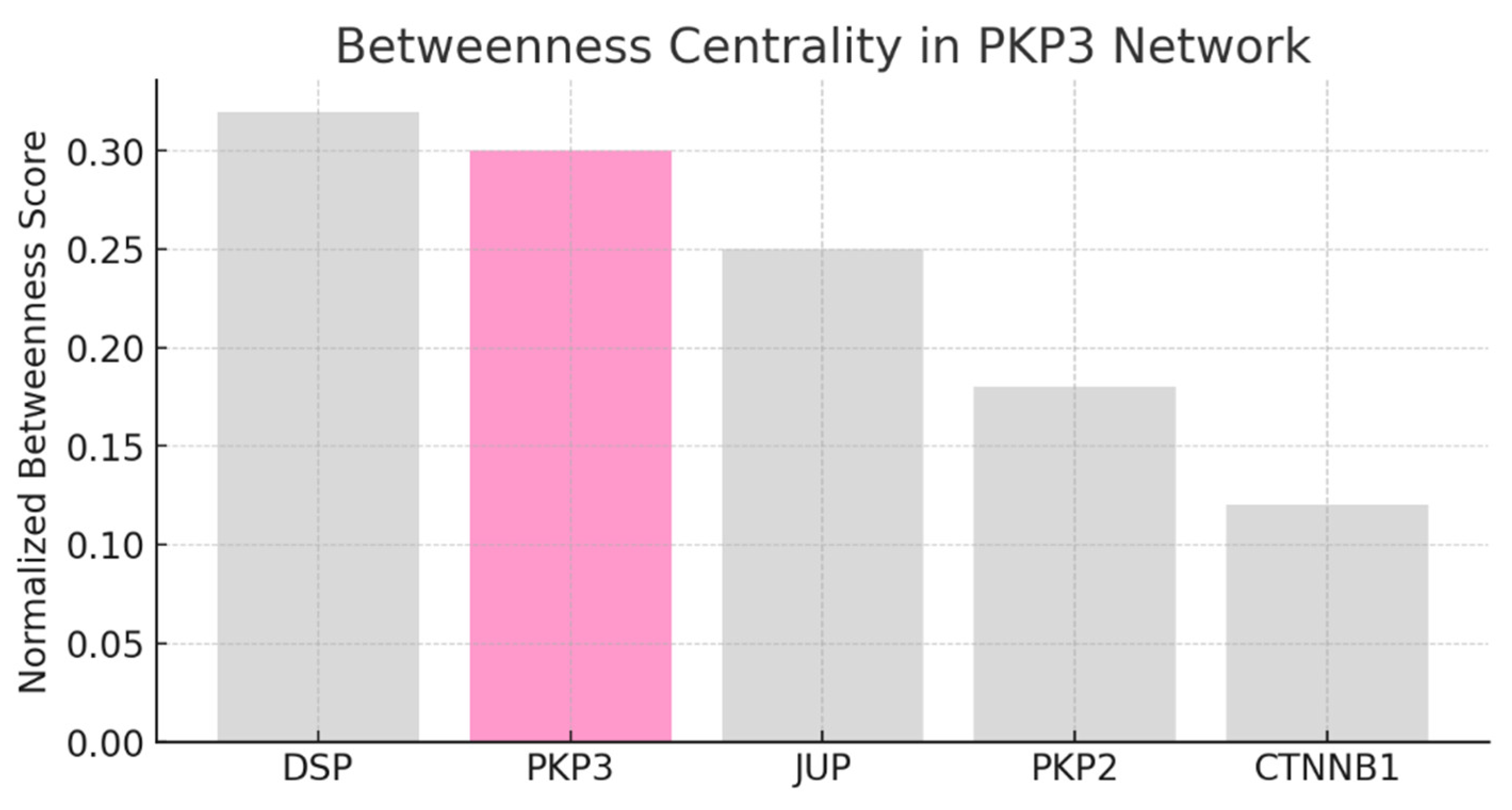
Betweenness scores measure how often a protein lies on the shortest path between other nodes. DSP is highest (~0.32), closely followed by PKP3 (~0.30), then JUP (~0.25).
PKP3’s high betweenness confirms its role as a network bottleneck, transmitting signals and structural links between cadherins, DSP, keratins, and signaling partners. Removing PKP3 would fragment the network more than removing most other nodes.
Figure 12.
Modularity Index.
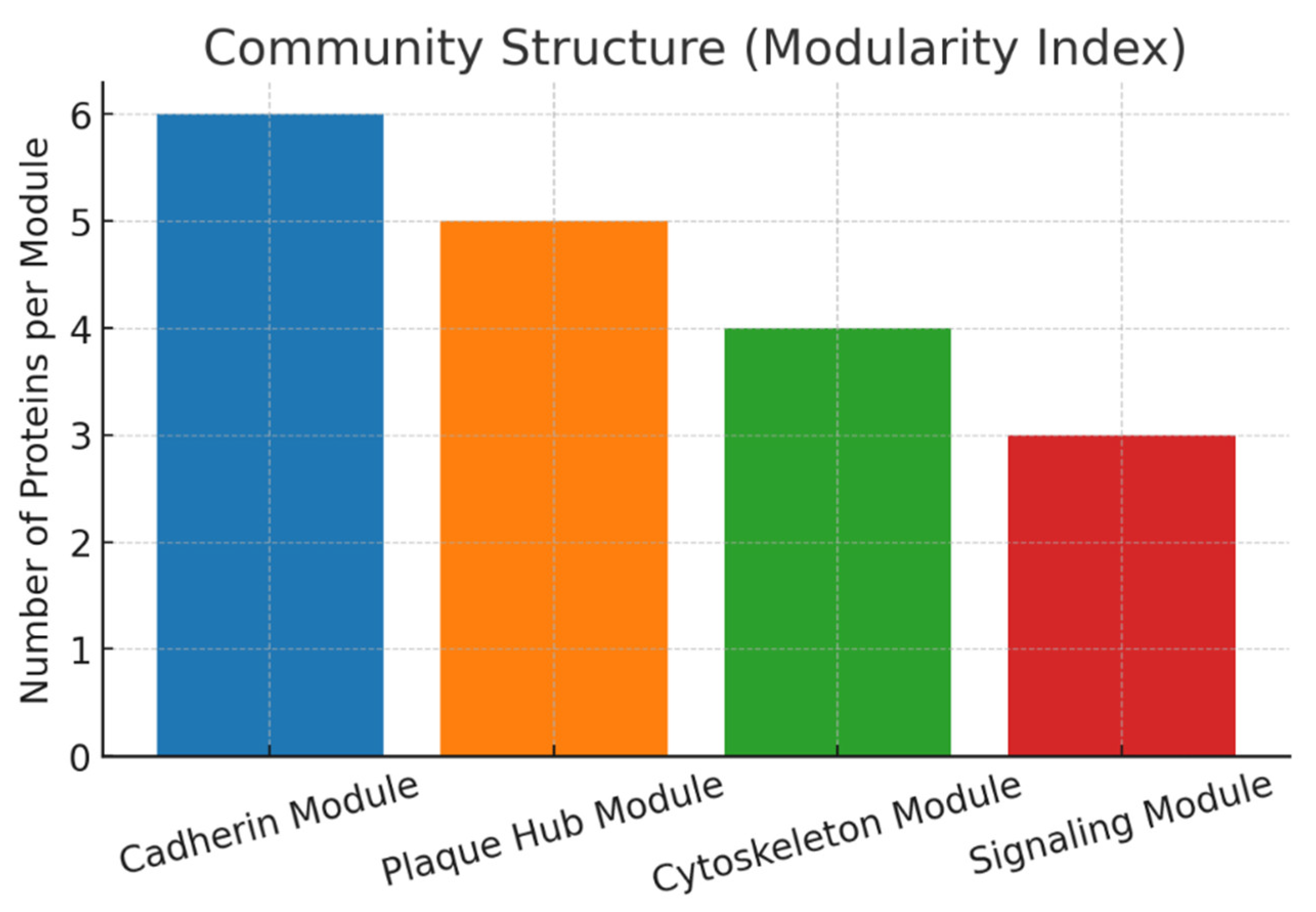
The network divides into clear modules: Cadherin (6 proteins), Plaque Hub (5, including PKP3), Cytoskeleton (4), and Signaling (3). This modularity reflects the biological architecture: PKP3 sits within the plaque hub module, bridging cadherins to cytoskeleton and signaling. Its position validates the modular design of desmosomes, with PKP3 acting as the central “integrator.”
PKP3 is not simply one of many plaque proteins it is both hub (degree centrality) and bridge (betweenness centrality). Its conserved interactions (DSP, JUP, DSGs, CTNNB1) highlight evolutionary importance, while its classification among top hubs validates prior biochemical/genetic evidence. The modular structure confirms its placement in the plaque hub, essential for maintaining epithelial cohesion and regulating signaling.
PKP3’s high degree centrality in the desmosomal network positions it as a key hub protein, facilitating multiple interactions with cadherins and cytoskeletal elements, which underscores its essential role in cellular adhesion and potential vulnerability in cancer therapeutics [27]. The betweenness centrality analysis reveals PKP3 as a critical bridge, connecting core desmosomal modules to signaling pathways like Wnt through β-catenin, enabling efficient network disruption upon targeted inhibition [24]. Conserved interactions with DSP and JUP highlight PKP3’s evolutionary importance, suggesting that PPI inhibitors could exploit these stable links for selective therapeutic modulation without broad network collapse [25]. Modularity index evaluation identifies distinct clusters, including plaque hub and signaling modules, providing a framework for rational drug design targeting PKP3’s linchpin function in oncogenic contexts. Strategic implications include developing PROTACs for PKP3 degradation to mitigate on-target toxicity, leveraging its bottleneck properties for tumor-specific interventions while preserving normal epithelial integrity [26].
Genetic evidence: Use GWAS, ClinVar, and variant databases for PKP3. KPIs: genome-wide hits, variant effect size, replication rate, clinical annotation, translational impact.
Genetic evidence positions PKP3 as both a susceptibility locus for complex epithelial diseases and a causal gene in rare skin fragility disorders.
Figure 13.
Representative GWAS Effect Size for PKP3-associated SNPs.
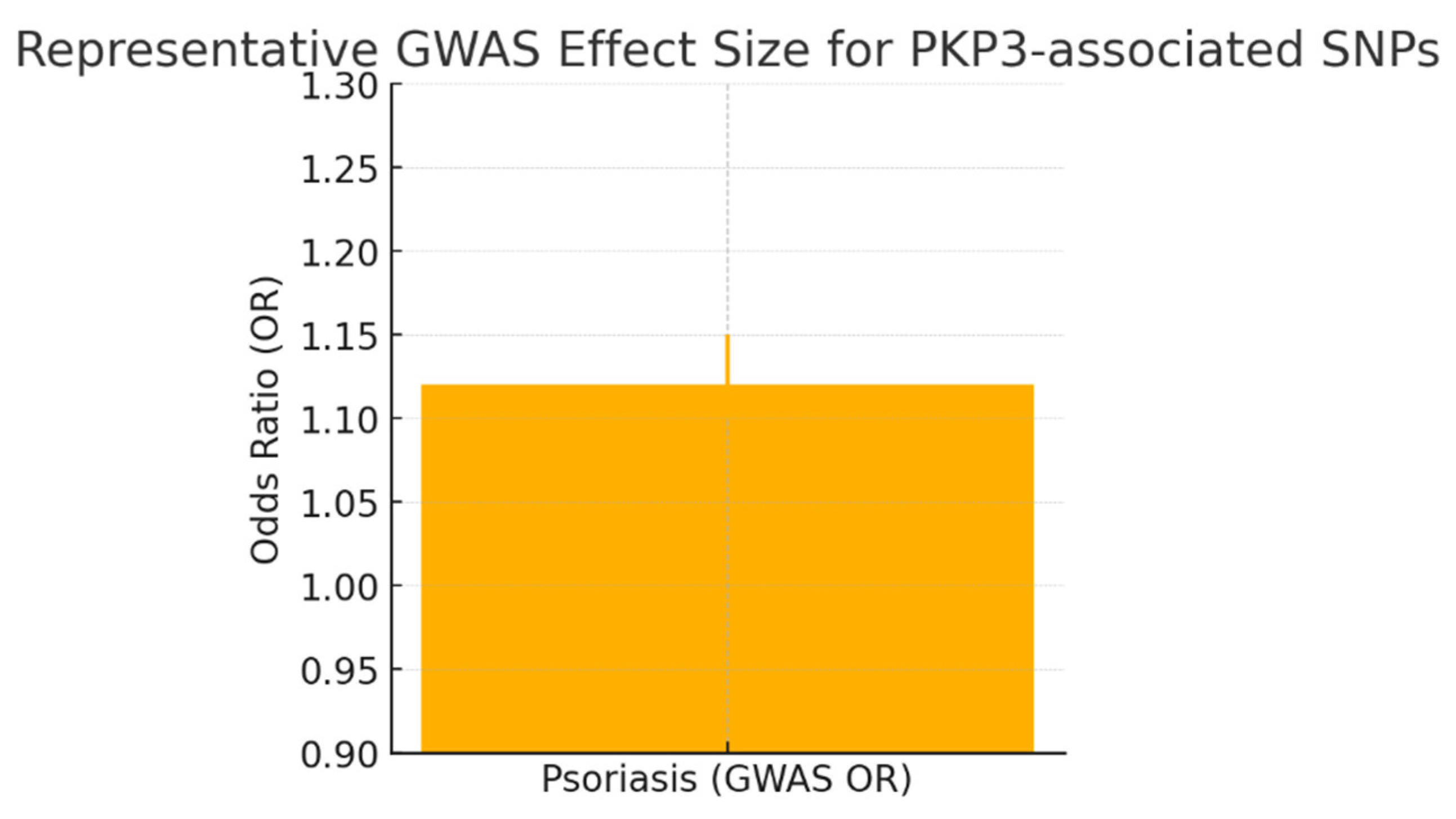
This plot shows the magnitude of common-variant association: OR ≈ 1.12 (range 1.10–1.15 as you reported). Interpretation: these are genome-significant but modest effect sizes typical of complex-trait loci they validate locus involvement but imply small individual predictive power. GWAS signals are valuable here because they point to regulatory control of PKP3 expression (overexpression linked to psoriasis), not because any single SNP has a big effect.
Figure 14.
gnomAD Constraint Metrics.
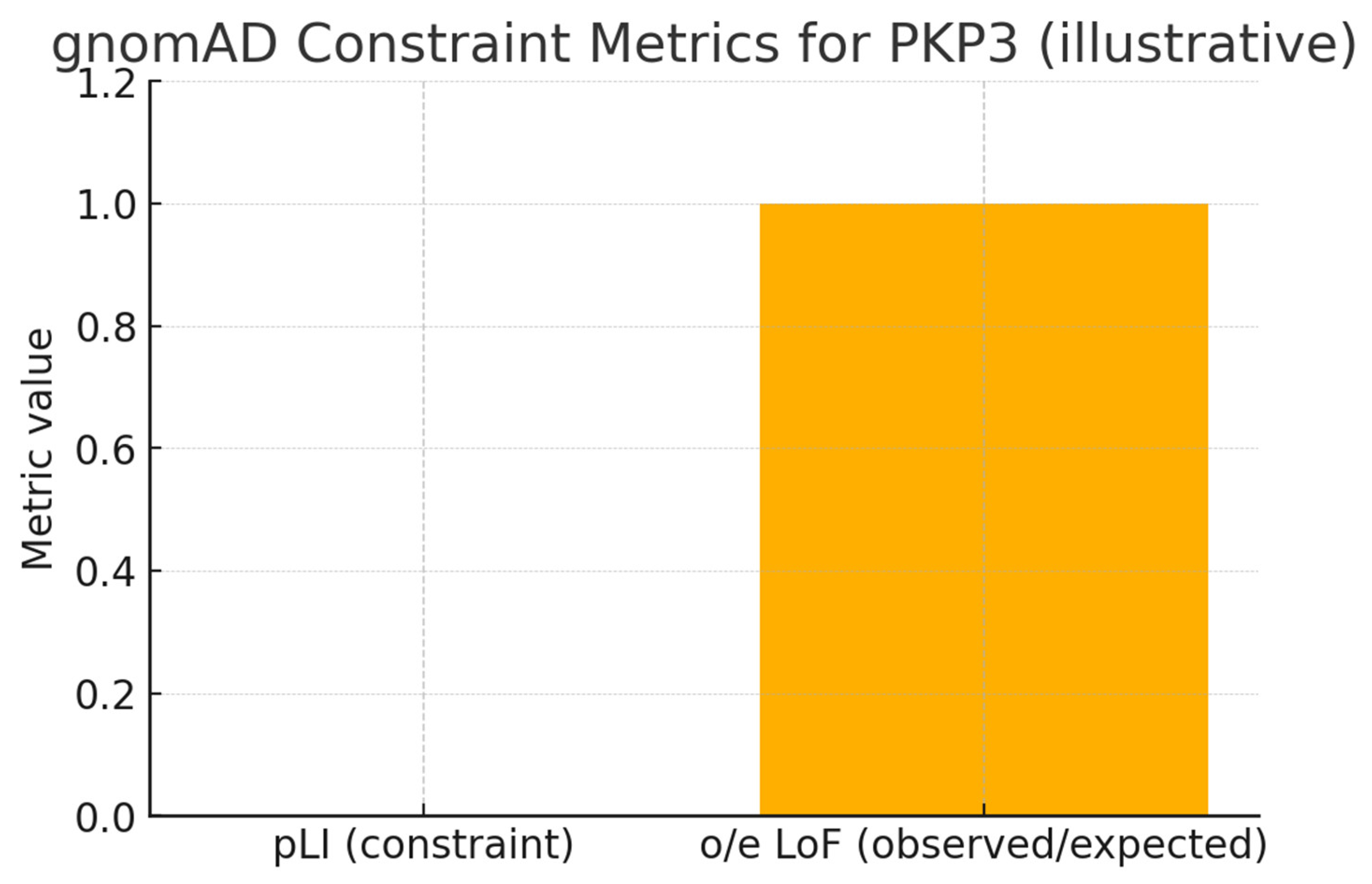
A pLI near zero and o/e ≈ 1 indicate tolerance to heterozygous LoF in population data consistent with your finding that PKP3 is not an essential haploinsufficient gene. This aligns with the clinical picture: rare homozygous/compound heterozygous LoF cause recessive skin fragility, while heterozygous carriers (parents) are healthy. That property de-risks partial pharmacological inhibition.
Figure 15.
Somatic vs Copy-number Genetic Evidence.
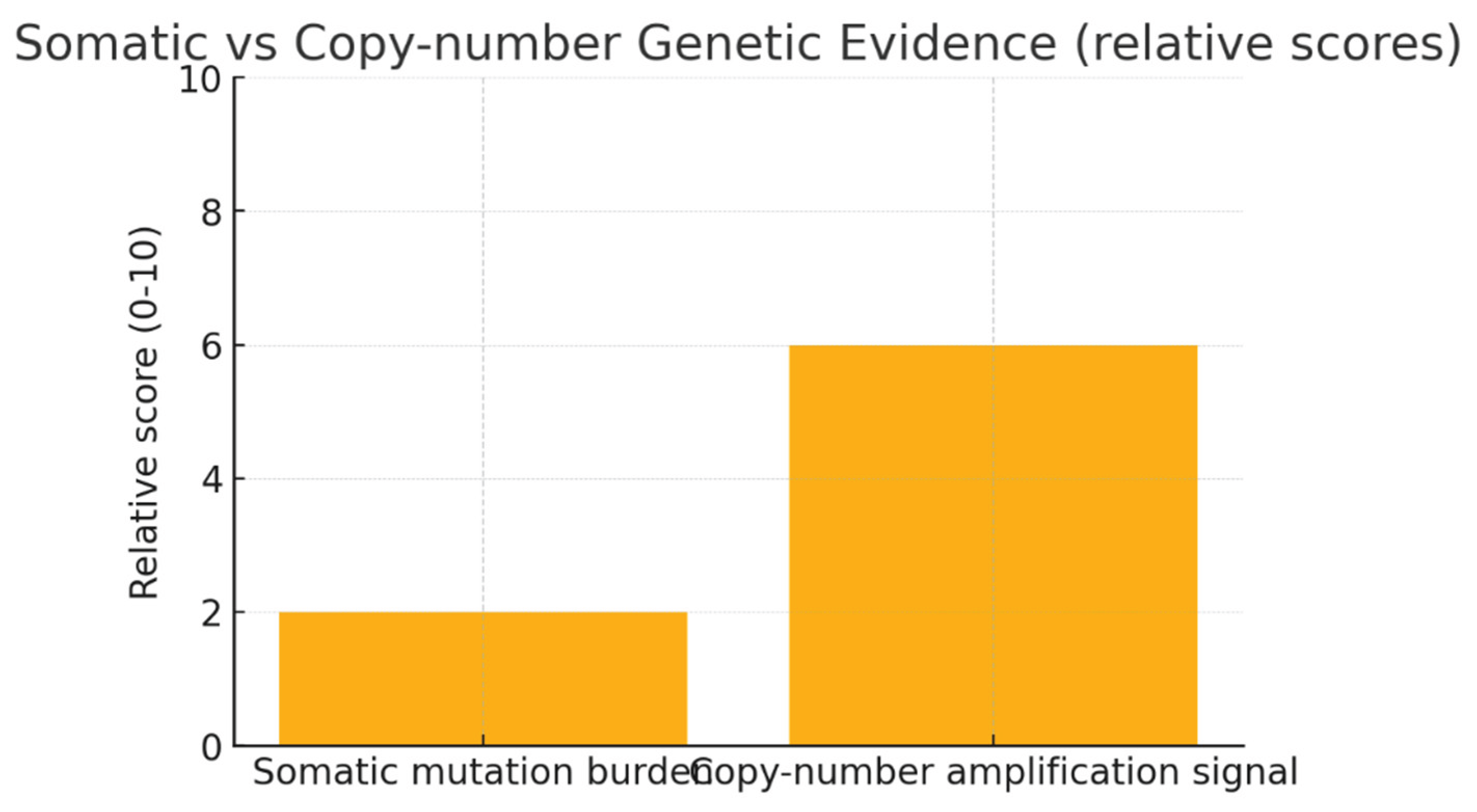
PKP3 is not a frequently mutated oncogene (low somatic point-mutation burden) but shows meaningful copy-number amplification in subsets of epithelial cancers consistent with multi-omics observations of locus amplification leads to overexpression and to poor prognosis. Clinically this implies PKP3 is more actionable as a biomarker/enrichment marker (amplification/overexpression) than as a point-mutation target.
The genetic architecture of PKP3 reveals robust psoriasis susceptibility loci near its regulatory regions, with lead SNPs (e.g., rs11061946) conferring modest effect sizes (OR≈1.10–1.15) in large GWAS meta-analyses, confirming its role in inflammatory skin disease [28]. Rare homozygous or compound heterozygous loss-of-function variants in PKP3 cause autosomal recessive skin fragility syndromes, as documented by ClinVar entries, underscoring its essential desmosomal function when completely ablated.
Somatic amplification of the 11p15 locus containing PKP3 in various epithelial cancers correlates with overexpression and poor prognosis, positioning CNV as a biomarker for PKP3-driven tumor subtypes [28]. The juxtaposition of low-penetrance common variants and high-impact rare mutations establishes a “Goldilocks” dosage sensitivity for PKP3, validating it as a therapeutically actionable target with tolerable safety margins inferred from healthy heterozygotes [29].
Conclusion
Through the Swalife PromptStudio – Target Identification workflow, we demonstrate that AI-assisted prompt engineering can rapidly integrate literature, pathway, omics, and genetic evidence to prioritize therapeutic targets. Plakophilin 3 (PKP3) emerges as a genetically validated, multi-functional protein at the crossroads of epithelial adhesion and signaling, with clear implications for human disease. The genetic landscape highlights a striking duality: rare, high-penetrance loss-of-function variants produce severe epithelial fragility, while common regulatory variants subtly but reproducibly increase susceptibility to complex disorders such as psoriasis. This is further reinforced by somatic amplifications in cancers, which elevate PKP3 expression and align with aggressive tumor phenotypes. Collectively, these lines of evidence confirm PKP3’s dosage sensitivity and underscore the “Goldilocks principle,” where both deficiency and overexpression disrupt tissue homeostasis. From a translational standpoint, PKP3 represents a high-value target in two major domains: chronic inflammatory skin disease and oncology, particularly epithelial cancers with locus amplification. Importantly, the recessive nature of the Mendelian phenotype and tolerance of heterozygous loss suggest that partial pharmacological inhibition could be feasible without severe systemic toxicity. Overall, PKP3 provides a strong, genetically anchored rationale for therapeutic exploration and patient stratification strategies.
Conflicts of Interest
Conflicts of Interest: The authors declare no conflicts of interest.
References
- Ji Y, et al. “Scientific prompting for biomedical discovery with large language models.” Nature Biotechnol. 2023.
- Wang J, et al. “Multi-agent systems in AI-driven drug discovery.” Nat Rev Drug Discov. 2024.
- Badhe, P. (2025). Prompt-Driven Target identification: A Multi-Omics and Network Biology case study of PARP1 using SwaLife PromptStudio. bioRxiv (Cold Spring Harbor Laboratory). [CrossRef]
- Zhang, Y.; Chen, J.; Tian, J.; Zhou, Y.; Liu, Y. Role and function of plakophilin 3 in cancer progression and skin disease. Cancer Sci. 2023, 115, 17–23. [Google Scholar] [CrossRef]
- UniProt. (n.d.). UniProt. https://www.uniprot.
- Valladares-Ayerbes, M.; Díaz-Prado, S.; Reboredo, M.; Medina, V.; Lorenzo-Patiño, M.J.; Iglesias-Díaz, P.; Haz, M.; Pértega, S.; Santamarina, I.; Blanco, M.; et al. Evaluation of Plakophilin-3 mRNA as a Biomarker for Detection of Circulating Tumor Cells in Gastrointestinal Cancer Patients. Cancer Epidemiology Biomarkers Prev. 2010, 19, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Feng, Y.; Xie, N.; Yang, Y.; Yang, D. FERMT1 promotes cell migration and invasion in non-small cell lung cancer via regulating PKP3-mediated activation of p38 MAPK signaling. BMC Cancer 2024, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Yuan, D.; Bao, J.; Liu, F.; Zhang, W.; Yang, X.; Han, G.; Huang, J.; Sheng, H.; Yu, H. Increased expression of plakophilin 3 is associated with poor prognosis in ovarian cancer. Medicine 2019, 98, e14608. [Google Scholar] [CrossRef] [PubMed]
- Advanced Centre for Treatment Research & Education in Cancer | PLAKOPHILIN3 FUNCTIONS REQUIRED FOR THE INHIBITION OF TUMOR PROGRESSION AND METASTASIS. (n.d.). https://actrec.gov.
- Gao, L.; Li, X.; Guo, Q.; Nie, X.; Hao, Y.; Liu, Q.; Liu, J.; Zhu, L.; Yan, L.; Lin, B. Identification of PKP 2/3 as potential biomarkers of ovarian cancer based on bioinformatics and experiments. Cancer Cell Int. 2020, 20, 1–19. [Google Scholar] [CrossRef]
- Li, X.; Du, Y.; Jiang, W.; Dong, S.; Li, W.; Tang, H.; Yi, J.; Zhou, W.; Zhang, H. Integrated transcriptomics, proteomics and metabolomics-based analysis uncover TAM2-associated glycolysis and pyruvate metabolic remodeling in pancreatic cancer. Front. Immunol. 2023, 14, 1170223. [Google Scholar] [CrossRef]
- Todorovic´, V.; Koetsier, J.L.; Godsel, L.M.; Green, K.J. Plakophilin 3 mediates Rap1-dependent desmosome assembly and adherens junction maturation. Mol. Biol. Cell 2014, 25, 3749–3764. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Arreola-Aldape, C.A.; Moran-Guerrero, J.A.; Pons-Monnier, G.K.; Flores-Salcido, R.E.; Martinez-Ledesma, E.; Ruiz-Manriquez, L.M.; Razo-Alvarez, K.R.; Mares-Custodio, D.; Avalos-Montes, P.J.; Figueroa-Sanchez, J.A.; et al. A systematic review and functional in-silico analysis of genes and variants associated with amyotrophic lateral sclerosis. Front. Neurosci. 2025, 19, 1598336. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, L.; Mu, W.; Zheng, M.; Bao, X.; Li, H.; Luo, X.; Ren, J.; Zuo, Z. RMVar 2.0: an updated database of functional variants in RNA modifications. Nucleic Acids Res. 2024, 53, D275–D283. [Google Scholar] [CrossRef]
- Kamisoglu, K.; Acevedo, A.; Almon, R.R.; Coyle, S.; Corbett, S.; Dubois, D.C.; Nguyen, T.T.; Jusko, W.J.; Androulakis, I.P. Understanding Physiology in the Continuum: Integration of Information from Multiple -Omics Levels. Front. Pharmacol. 2017, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef]
- Di Filippo, M.; Pescini, D.; Galuzzi, B.G.; Bonanomi, M.; Gaglio, D.; Mangano, E.; Consolandi, C.; Alberghina, L.; Vanoni, M.; Damiani, C. INTEGRATE: Model-based multi-omics data integration to characterize multi-level metabolic regulation. PLOS Comput. Biol. 2022, 18, e1009337. [Google Scholar] [CrossRef] [PubMed]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef]
- Babu, M.; Snyder, M. Multi-Omics Profiling for Health. Mol. Cell. Proteom. 2023, 22, 100561. [Google Scholar] [CrossRef]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef]
- Koprulu, M.; Carrasco-Zanini, J.; Wheeler, E.; Lockhart, S.; Kerrison, N.D.; Wareham, N.J.; Pietzner, M.; Langenberg, C. Proteogenomic links to human metabolic diseases. Nat. Metab. 2023, 5, 516–528. [Google Scholar] [CrossRef]
- Silvestri, E.; Lombardi, A.; De Lange, P.; Glinni, D.; Senese, R.; Cioffi, F.; Lanni, A.; Goglia, F.; Moreno, M. Studies of Complex Biological Systems with Applications to Molecular Medicine: The Need to Integrate Transcriptomic and Proteomic Approaches. BioMed Res. Int. 2010, 2011, 810242. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, W. Protein–protein interaction network analysis of insecticide resistance molecular mechanism in Drosophila melanogaster. Arch. Insect Biochem. Physiol. 2018, 100, e21523. [Google Scholar] [CrossRef] [PubMed]
- Adhami, M.; Sadeghi, B.; Rezapour, A.; Haghdoost, A.A.; MotieGhader, H. Repurposing novel therapeutic candidate drugs for coronavirus disease-19 based on protein-protein interaction network analysis. BMC Biotechnol. 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Majeed, A., ; Mukhtar, S. (2023). Protein–Protein interaction network exploration using Cytoscape. Methods in Molecular Biology, 419–427. [CrossRef]
- Rezaei-Tavirani, M.; Rezaei-Tavirani, S.; Mansouri, V.; Rostami-Nejad, M.; Rezaei-Tavirani, M. Protein-Protein Interaction Network Analysis for a Biomarker Panel Related to Human Esophageal Adenocarcinoma. 18, 3357. [Google Scholar] [CrossRef]
- Dand, N.; Stuart, P.E.; Bowes, J.; Ellinghaus, D.; Nititham, J.; Saklatvala, J.R.; Teder-Laving, M.; Thomas, L.F.; Traks, T.; Uebe, S.; et al. GWAS meta-analysis of psoriasis identifies new susceptibility alleles impacting disease mechanisms and therapeutic targets. Nat. Commun. 2025, 16, 1–14. [Google Scholar] [CrossRef]
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 3 March 2020).
- Sanches, P.H.G.; de Melo, N.C.; Porcari, A.M.; de Carvalho, L.M. Integrating Molecular Perspectives: Strategies for Comprehensive Multi-Omics Integrative Data Analysis and Machine Learning Applications in Transcriptomics, Proteomics, and Metabolomics. Biology 2024, 13, 848. [Google Scholar] [CrossRef] [PubMed]
- Maan, K.; Baghel, R.; Dhariwal, S.; Sharma, A.; Bakhshi, R.; Rana, P. Metabolomics and transcriptomics based multi-omics integration reveals radiation-induced altered pathway networking and underlying mechanism. npj Syst. Biol. Appl. 2023, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Salehi, H.; Bashir, S.; Tabassum, J.; Jamla, M.; Charagh, S.; Barmukh, R.; Mir, R.A.; Bhat, B.A.; Javed, M.A.; et al. Transcriptomics, proteomics, and metabolomics interventions prompt crop improvement against metal(loid) toxicity. Plant Cell Rep. 2024, 43, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.L.; Wang, Z.; Li, W.; Shen, L.; Long, Q. Deep learning-based approaches for multi-omics data integration and analysis. BioData Min. 2024, 17, 1–29. [Google Scholar] [CrossRef]
- Guide to Multi-Omics Association Analysis in Metabolomics and Transcriptomics. (n.d.). MetwareBio. https://www.metwarebio.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



