
Submitted:
22 September 2025
Posted:
23 September 2025
You are already at the latest version
Abstract
Alzheimer's disease (AD), the predominant dementia etiology, imposes a $1.5 trillion global burden in 2025, exacerbated by inexorable failures in amyloid-tau-centric monotherapies amid <1% trial success rates. This review synthesizes cross-disciplinary pathobiology underpinning oncology drug repurposing for AD, leveraging inverse epidemiological antagonism and convergent hallmarks: aberrant neuronal cell-cycle re-entry (CDK5/p25, ERK-MAPK), proteostasis overload (HSP90-UPS-autophagy nexus), mTORC1 hyperactivation impairing lysosomal flux, kynurenine pathway (IDO1/TDO)-driven immunometabolic decoupling, PARP1-mediated NAD+ parthanatos, maladaptive neuroinflammation (PD-1/PD-L1 checkpoints, TAM-microglial skewing), and senescence-associated secretory phenotype (SASP)-amplified gliosis. Computational nomination pipelines—transcriptomic signature reversal (CMap/LINCS), network proximity in multi-omic knowledge graphs, and ligand-based docking—prioritize pleiotropic oncology agents: c-Abl/Src TKIs (nilotinib, bosutinib) enhancing autophagic Aβ/tau clearance; multikinase modulators (masitinib) resolving mast cell-microglial dyshomeostasis; rapalogs restoring proteostasis; IDO1 antagonists normalizing astrocytic lactate shuttling; PARPi mitigating genomic instability; HDACi reinstating synaptic epigenomes; and senolytics (dasatinib+quercetin) purging senescent glia. Phase I/II trials evince biomarker modulation (↓CSF p-tau, amyloid-PET; ↑autophagic flux) with geriatric-adapted dosing, yet underscore translational hurdles: BBB penetration, off-target liabilities, and polypharmacy in APOE ε4-enriched cohorts. A precision roadmap advocates orthogonal combinations (e.g., TKI+IDO1i), delivery-first PBPK modeling, and biomarker-anchored adaptive platforms to recalibrate AD's networked trajectory toward disease-modifying efficacy.
Keywords:
Alzheimer’s disease
; oncology drug repurposing
; multi-omic networks
; immunometabolism
; senescent gliopathy
1. Introduction
Alzheimer’s disease (AD) stands as one of the most formidable biomedical challenges of our time, a disorder whose magnitude continues to escalate in parallel with demographic aging and increasing life expectancy. It is the most common cause of dementia and has emerged as the seventh leading cause of death worldwide and the sixth in the United States, reflecting its inexorable rise as mortality from other major diseases such as cardiovascular and cerebrovascular disorders has declined [1]. Current estimates indicate that approximately 60 million people are living with dementia globally, with AD accounting for the overwhelming majority of cases, and projections suggest that this figure will triple by 2050 in the absence of disease-modifying therapies [2]. In the United States alone, the prevalence exceeds 7 million among individuals over 65 years of age and is expected to approach 14 million by 2060 [3]. The clinical burden of AD extends beyond cognitive decline to encompass profound neuropsychiatric manifestations, including depression, agitation, psychosis, and progressive functional dependency, which collectively strip individuals of their autonomy and place extraordinary strain on families and healthcare systems. Pathologically, AD is characterized by extracellular amyloid-beta (Aβ) plaques, intracellular neurofibrillary tangles composed of hyperphosphorylated tau, synaptic degeneration, neuronal death, and widespread cortical and hippocampal atrophy, processes that accrue silently for decades before the emergence of overt dementia [4].
The societal consequences of this trajectory are staggering. The global economic cost of dementia care was estimated at $1.5 trillion in 2025, a figure that integrates direct medical expenditures, long-term care, and the immense burden of informal caregiving [5]. In high-income countries, AD now accounts for more than 15% of healthcare spending for older adults, while in low- and middle-income regions, the lack of institutional infrastructure shifts responsibility to families, amplifying financial, physical, and psychological strain. In the United States, healthcare and caregiving costs for AD reached $384 billion in 2025 and are projected to surpass $1 trillion annually by 2050 [3,5]. The hidden burden is borne disproportionately by caregivers: in 2022, over 11 million Americans provided an estimated 16 billion hours of unpaid care, valued at $272 billion, with associated risks of burnout, depression, and financial hardship [6]. The cumulative lifetime cost of caring for a single patient with AD has been estimated at more than $400,000, with families shouldering up to 70% of these expenses [7]. Moreover, the disease exacerbates health inequities, disproportionately affecting women, minorities, and socioeconomically disadvantaged populations, who face delayed diagnoses and reduced access to care [8]. This convergence of clinical devastation, social disruption, and economic unsustainability underscores the urgency of developing therapeutic strategies capable of altering the natural history of AD.
Yet, despite decades of intense research, the therapeutic landscape of AD remains remarkably barren. The failures of traditional drug development pipelines in this field are unparalleled in modern medicine [9,10]. Since 2002, over 99% of AD clinical trials have failed, yielding a therapeutic success rate of less than 1%—among the lowest across any disease area [11]. The few approved agents, such as cholinesterase inhibitors (donepezil, rivastigmine, galantamine) and the NMDA receptor antagonist memantine, offer only transient symptomatic relief without altering disease progression [12]. Even the more recent wave of monoclonal antibodies targeting Aβ, including aducanumab and lecanemab, have demonstrated at best modest slowing of cognitive decline, accompanied by significant safety concerns such as amyloid-related imaging abnormalities (ARIA) [13]. These repeated disappointments highlight the conceptual limitations of single-target interventions predicated largely on the amyloid cascade hypothesis. While amyloid clearance is achievable, clinical benefit remains elusive, underscoring the disconnect between biomarker modulation and patient-centered outcomes [14]. Parallel failures of tau-targeted therapies, γ-secretase and β-secretase inhibitors, and other single-pathway strategies reinforce this conclusion [15]. Indeed, the complex etiology of AD—wherein amyloid and tau interact with vascular dysfunction, neuroinflammation, mitochondrial failure, oxidative stress, synaptic dysregulation, and genetic susceptibility factors such as APOE ε4—renders monotherapies insufficient to arrest disease progression [16]. Moreover, the protracted preclinical phase of AD, often spanning decades, complicates therapeutic intervention, as treatments initiated after clinical onset may arrive too late to reverse entrenched neuropathology [17].
These realities mandate a paradigm shift away from the prevailing “one target, one drug” dogma toward more integrative, multi-target strategies. Here, the concept of drug repurposing emerges as a particularly rational and promising avenue. Drug repurposing, or repositioning, is the strategy of deploying existing, clinically approved drugs for new therapeutic indications. Its appeal lies in the ability to circumvent the prohibitive costs, timeframes, and attrition rates associated with de novo drug discovery [18]. Whereas traditional drug development requires 10–20 years and upwards of $2–3 billion, repurposing can compress this trajectory to 3–12 years at a fraction of the cost—averaging around $300 million—by leveraging existing pharmacokinetic, pharmacodynamic, and toxicology data [19]. Repurposed agents often bypass early-phase safety trials, allowing for rapid entry into Phase II and III efficacy studies. For a field like AD, where urgency is paramount and failure rates are intolerably high, such efficiencies represent not merely an advantage but a necessity.
Among the various classes of candidates for repurposing, oncology drugs stand out as particularly attractive [20]. This derives from both pragmatic and mechanistic considerations. Pragmatically, oncology drugs are among the most extensively studied therapeutics, with well-characterized pharmacological profiles, established manufacturing infrastructure, and decades of clinical experience across diverse populations, including older adults with comorbidities. Mechanistically, many of the molecular pathways targeted in cancer overlap with those implicated in neurodegeneration. Dysregulated kinase signaling, for example, not only drives malignant proliferation but also contributes to tau hyperphosphorylation in AD neurons [21,22]. Similarly, chronic inflammation—a hallmark of the tumor microenvironment—is mirrored by microglial activation and neuroinflammatory cascades in AD [23]. Autophagy, proteostasis, and mitochondrial metabolism, processes hijacked by tumor cells for survival, are also central to the pathogenesis of neurodegeneration, where their dysfunction accelerates protein aggregation and synaptic failure [21,22]. This convergence suggests that oncology-derived therapeutics, originally designed to modulate proliferative or survival pathways, may be repurposed to mitigate neuronal vulnerability in AD.
The epidemiological literature lends further weight to this rationale, revealing a fascinating inverse relationship between cancer and neurodegeneration. As early as the mid-20th century, clinicians observed reduced cancer incidence among patients with neurodegenerative diseases, including Parkinson’s and AD, and subsequent large-scale studies such as the Framingham Heart Study confirmed these associations [24,25,26]. Cancer survivors exhibit a 30–50% lower risk of developing AD, while patients with AD display reduced cancer prevalence independent of survival bias [27]. This inverse epidemiology likely reflects the antagonistic biology of these conditions: cancer thrives on uncontrolled proliferation and apoptosis evasion, whereas neurodegeneration is characterized by premature cell death, synaptic attrition, and aberrant cell cycle re-entry in post-mitotic neurons [28]. Shared molecular pathways—such as DNA damage responses, proteostasis mechanisms, metabolic reprogramming, and inflammatory signaling—manifest in opposite directions across the two diseases, further supporting the therapeutic potential of cross-domain pharmacology [29]. Genetic studies reinforce this perspective: variants in TP53, for instance, promote tumorigenesis while conferring resistance to neurodegeneration, exemplifying antagonistic pleiotropy [29]. Such findings provide not only epidemiological correlation but also mechanistic plausibility for oncology-to-neurodegeneration repurposing strategies.
In practical terms, oncology drugs offer several specific advantages as AD candidates. Tyrosine kinase inhibitors (TKIs) such as nilotinib and dasatinib, initially developed for hematologic malignancies, penetrate the central nervous system and have demonstrated the ability to modulate autophagy and enhance clearance of protein aggregates in preclinical models [30]. Immunomodulators targeting tumor-associated inflammation may be adapted to dampen chronic microglial activation in the AD brain. Epigenetic modulators, metabolic regulators, and DNA damage response inhibitors, long staples of oncology, likewise map onto key pathological processes in AD. Importantly, the pleiotropic nature of these drugs—often considered a liability in oncology due to off-target effects—may be an asset in AD, where simultaneous modulation of multiple nodes in the pathogenic network is desirable. Nonetheless, challenges remain, including the adaptation of oncology dosing regimens (often high and intermittent) to the requirements of long-term neurological therapy (low and sustained), as well as the careful navigation of safety profiles in elderly populations [31].
The scope of this review is to critically synthesize the evidence base supporting oncology drug repurposing in AD, bridging epidemiology, molecular biology, and translational pharmacology. We will examine the biological intersections between cancer and neurodegeneration, highlight oncology drug classes with preclinical or clinical promise, and explore emerging computational and experimental frameworks for systematic candidate identification. We will assess the landscape of completed and ongoing trials, including negative findings, and consider the translational hurdles that must be addressed to realize the potential of this strategy. Finally, we will outline a prioritized roadmap of oncology-derived candidates, discuss the potential for combination therapies and precision approaches tailored to genetic risk profiles such as APOE ε4, and articulate how drug repurposing can recalibrate the trajectory of AD therapeutics. The aim is not merely to propose incremental advances but to argue for a strategic redirection of resources and intellectual effort toward a paradigm that is both biologically grounded and pragmatically feasible. In doing so, this review positions oncology-derived drug repurposing not as a speculative adjunct but as a scientifically rigorous and urgently needed strategy to confront one of the greatest biomedical crises of the modern era.
2. Cross-Disciplinary Biology: Where Cancer and Alzheimer’s Intersect
The therapeutic rationale for repurposing oncology drugs for Alzheimer's disease is founded on a profound biological overlap between the two pathologies. While seemingly opposite—one characterized by uncontrolled cellular proliferation and the other by lethal neuronal loss—they share several key molecular and cellular pathways.
2.1. Cell-Cycle Dysregulation in Neurons
In cancer, oncogenic activation of cyclin–CDK modules (e.g., CDK4/6–Cyclin D, CDK2–Cyclin E) drives G1/S transition and sustained mitoses; in Alzheimer’s disease (AD), post-mitotic neurons aberrantly re-enter the cell cycle without completing division, a lethal, “abortive” process that presages synaptic failure and cell death [7,8]. Exposure to soluble Aβ oligomers and downstream stress kinase cascades (CDK5/p25, ERK1/2-MAPK, PI3K–AKT–mTOR) induce re-expression of cell-cycle proteins, S-phase entry, tetraploidization, and DNA replication markers in vulnerable hippocampal and cortical neurons [32,33]. Forced cell-cycle entry in neurons accelerates tau hyperphosphorylation and neurofibrillary tangle formation; conversely, experimental dampening of CDK activity reduces Aβ toxicity and preserves cognition in transgenic models[34]. These data frame cell-cycle machinery—and its oncologic drug armamentarium—as actionable in AD: small-molecule CDK inhibitors, ABL/SRC-family tyrosine kinase inhibitors (TKIs), and downstream MAPK modulators could blunt aberrant neuronal cycle signaling, preventing the cascade from genomic stress → tauopathy → apoptosis. Translational caveats include dose, schedule, and brain exposure; peripheral anti-proliferative effects must be minimized while achieving CNS target engagement.
2.2. Protein Homeostasis and Aggregation
Both malignancy and neurodegeneration stress the proteostasis network. Cancer cells exploit chaperones (HSP70/HSP90), the ubiquitin–proteasome system (UPS), and selective autophagy to buffer mutational burden and support high-flux protein synthesis. In AD, UPS and autophagic–lysosomal pathways are chronically overloaded or functionally impaired, promoting accumulation of misfolded Aβ, pathologic tau, and co-aggregating clients [9]. HSP90, which stabilizes numerous oncogenic kinases, also maintains tau in aggregation-competent states; HSP90 inhibition destabilizes tau and reduces client signaling in preclinical AD models [35,36,37]. Modulators of E3 ligases/deubiquitinases, proteasome activity, and ER-stress/UPR arms (PERK–eIF2α) represent bidirectional levers—but with asymmetry of risk: while proteasome inhibition is cytotoxic to tumors, it may worsen neuronal stress and neuropathy; by contrast, boosting chaperone capacity, enhancing ER-associated degradation, or restoring autophagic flux can be neuroprotective. Thus, oncology-derived HSP inhibitors, UPS regulators, and co-chaperone modulators merit selective exploration in AD where the therapeutic goal is restoration of clearance capacity rather than global proteolysis suppression.
2.3. Autophagy and mTOR Signaling
mTORC1 integrates nutrient and growth signals to promote anabolism and repress autophagy; it is hyperactive in many cancers, enabling biomass accrual and proliferation. In AD, sustained mTORC1 activation suppresses autophagic initiation and lysosomal biogenesis, impairs aggregate clearance, and intersects with aberrant cell-cycle re-entry [38,39,40]. In transgenic AD models, pharmacologic mTOR inhibition (rapamycin/rapalogs) reinstates autophagic flux, reduces Aβ and tau pathology, and improves synaptic and cognitive endpoints. Upstream oncogenic nodes (PI3K–AKT, Rheb) and downstream translational controls (p70S6K, 4E-BP) are dysregulated in both diseases, suggesting a shared intervention surface [38,39,40,41]. Repurposed rapalogs or dual mTORC1/2 inhibitors could provide disease modification by coupling proteostasis restoration with anti-proliferative signaling in neurons and glia. However, chronic mTOR suppression carries immunosuppressive and metabolic liabilities; brain-penetrant dosing paradigms, intermittent schedules, or autophagy-selective strategies (e.g., ULK1 activators, TFEB nuclearization enhancers) may optimize benefit-risk (with pharmacodynamic readouts from CSF/autophagy biomarkers and PET ligands) [40,42].
2.4. Metabolic Reprogramming and the Kynurenine Pathway
Metabolic remodeling is a hallmark of cancer (aerobic glycolysis/Warburg effect) and a signature of AD (neuronal hypometabolism on FDG-PET, astrocytic metabolic dysfunction). Convergent immunometabolic control emerges at the kynurenine pathway (KP) of tryptophan catabolism. Indoleamine-2,3-dioxygenase 1 (IDO1), upregulated by interferons and tumor inflammation, depletes tryptophan and generates kynurenine metabolites that suppress antitumor immunity; IDO1 is likewise induced in AD glia surrounding plaques, diverting astrocytic metabolism and diminishing lactate support to neurons [43]. In AD models, IDO1 inhibition restores hippocampal glucose handling, normalizes synaptic physiology, and improves memory, aligning with oncology’s clinical PK/PD knowledge of IDO1 antagonists [44]. More broadly, insulin/IGF signaling defects, mitochondrial Complex I/IV deficits, and lipidomic remodeling (ceramides, plasmalogens) are shared axes of vulnerability. Repurposable oncology-grade agents include IDO1/TDO inhibitors, biguanides (where appropriate), and FAO/mitochondrial modulators—with careful consideration of CNS penetration, glial–neuronal metabolic coupling, and genotype-specific modifiers (e.g., APOE ε4).
2.5. DNA Damage Response and PARP
Cancer therapy exploits DNA damage and repair vulnerabilities (synthetic lethality); neurons, although post-mitotic, accrue oxidative and replication-independent DNA lesions with age and AD. PARP1 senses single-strand breaks and catalyzes poly(ADP-ribosyl)ation (PARylation) using NAD+; chronic over-activation in AD depletes NAD+, impairs mitochondrial function, and triggers parthanatos, a caspase-independent form of cell death [45]. In oncology, PARP inhibitors (PARPi) incapacitate BER/SSBR, selectively killing HR-deficient tumors; in AD, calibrated PARP1 modulation could be neuroprotective by preventing futile NAD+ consumption, limiting PAR polymer toxicity, and stabilizing neuronal energetics [46]. Systems pharmacology and network analyses have repeatedly flagged PARP1 as an AD-relevant hub [47]. Key translational issues include timing (preventing chronic over-activation without impeding repair of physiological lesions), CNS exposure, and NAD+ salvage pathway status (NR/NMN utilization), with outcome markers spanning CSF PAR, brain NAD+/NADH ratios, γH2AX foci, and neuronal survival readouts.
2.6. Neuroinflammation and Tumor Immunology
Microglia in AD share features with tumor-associated macrophages (TAMs): chronic activation, phagocytic dysfunction, and an immunoregulatory cytokine milieu (IL-10, TGF-β) that both sustains pathology and blunts effective clearance [48,49,50]. Immune checkpoints (PD-1/PD-L1, CTLA-4) that restrain antitumor immunity are expressed in glia and infiltrating lymphocytes in AD lesions [51]; preclinical manipulation of PD-1/PD-L1 has variably enhanced microglial phagocytosis and reduced Aβ burden, with reports of improved circuit function, although results remain debated and protocol-dependent [52,53]. Oncology’s immunotherapy toolkit thus offers two conceptual levers: (i) checkpoint modulation to re-bias microglia toward a homeostatic, plaque-engulfing state (e.g., restoring P2RY12, TREM2 signaling) [51] and (ii) cytokine/chemokine axis re-wiring (e.g., IL-6/STAT3, CSF1R) to resolve maladaptive gliosis while preserving protective surveillance [54]. Translation to AD must navigate immune-related adverse events (irAEs), blood–brain barrier (BBB) transport, and the distinct set-points of CNS immune homeostasis [54,55]. Biomarker-guided strategies (soluble TREM2, TSPO-PET, microglial transcriptional states) are essential to titrate immunomodulation [54].
2.7. Cellular Senescence and SASP
Senescence couples’ durable cell-cycle arrest with a pro-inflammatory, matrix-remodeling secretome (senescence-associated secretory phenotype, SASP) [56,57]. In cancer, SASP can be tumor-promoting or suppressive depending on context; in brain aging and AD, senescent astrocytes, microglia, oligodendrocyte precursors, endothelial cells, and even neurons accumulate, secreting IL-6, IL-1β, TNF-α, MMPs, and complement factors that amplify neurotoxicity, impair synaptic plasticity, and compromise neurovascular integrity [56,57,58,59]. Senolytic combinations originating in oncology (e.g., dasatinib + quercetin, D+Q) clear senescent glia in AD models, lowering amyloid/tau pathology and improving behavioral outputs [57,59]. Early human pilot work shows CNS penetration and SASP biomarker modulation without short-term cognitive benefit over 12 weeks—consistent with the expectation that disease-modifying effects require longer exposure and earlier intervention [60]. The senotherapeutic space now spans senolytics (BCL-2/BCL-xL inhibitors, FOXO3a modulators) [56,57], senomorphics (JAK/STAT, NF-κB dampeners), and SASP-targeted interventions. Precision deployment in AD will likely integrate cell-type-specific delivery, periodic “hit-and-run” dosing, and composite biomarker panels (p16^INK4a^, p21^CIP1^, SASP proteomics, microglial state maps) to balance efficacy and safety [60,61]. These shared mechanisms and their therapeutic implications are summarized in Figure 2.
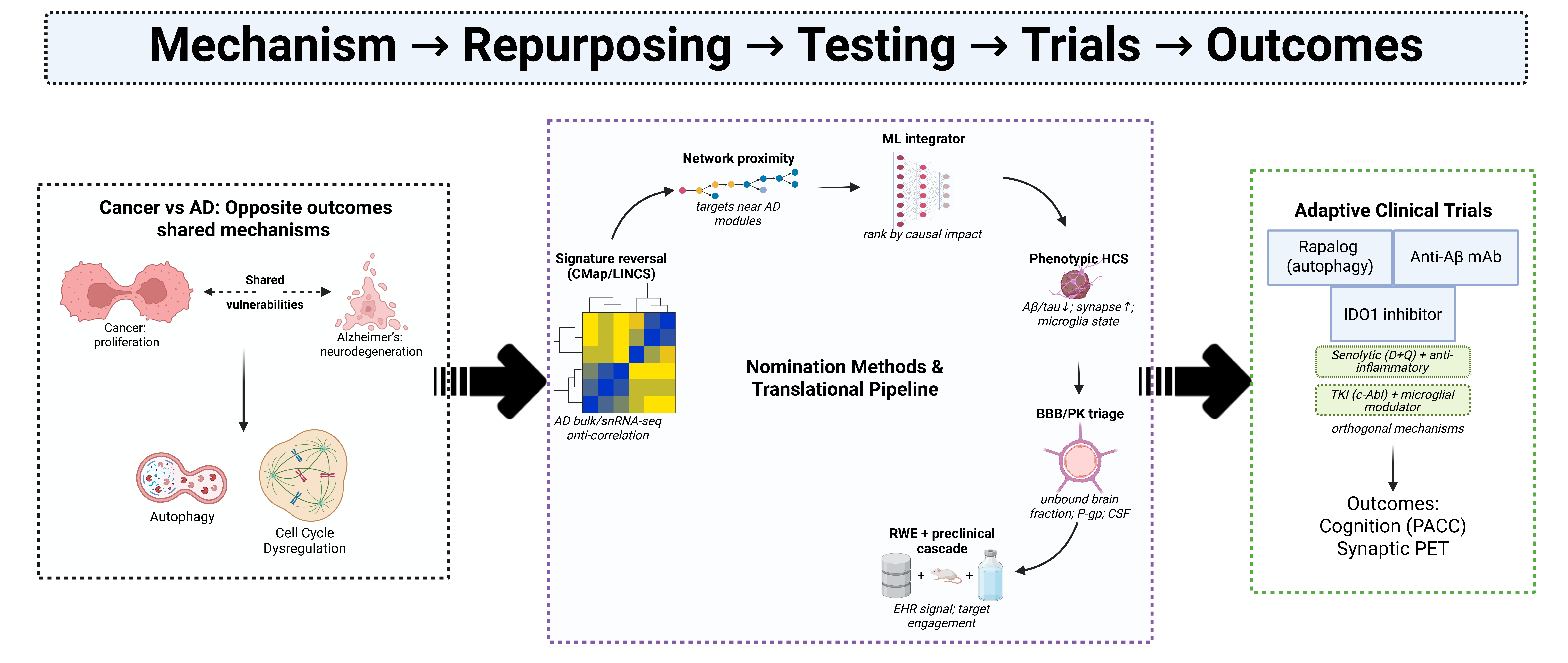
Figure 1.
Shared vulnerabilities and divergent outcomes in cancer and Alzheimer’s disease (AD). (A) Despite opposite phenotypes—uncontrolled proliferation in cancer versus neurodegeneration in AD—both conditions converge on dysregulated pathways including cell cycle, proteostasis, autophagy/mTOR, immune–metabolism (IDO1–kynurenine), DNA damage/PARP signaling, neuroinflammation, and cellular senescence. (B) Mechanistic modules illustrate how aberrant cell-cycle re-entry, proteostasis overload, impaired autophagy, and microglial immune checkpoints drive neuronal vulnerability, while corresponding oncology-derived inhibitors (e.g., CDK inhibitors, TKIs, rapalogs, PARP inhibitors, senolytics) provide repurposing opportunities. (C–D) Microglial senescence, SASP signaling, and DNA damage–induced PARP over-activation exemplify cross-disease mechanisms that can be therapeutically leveraged through precision repurposing.
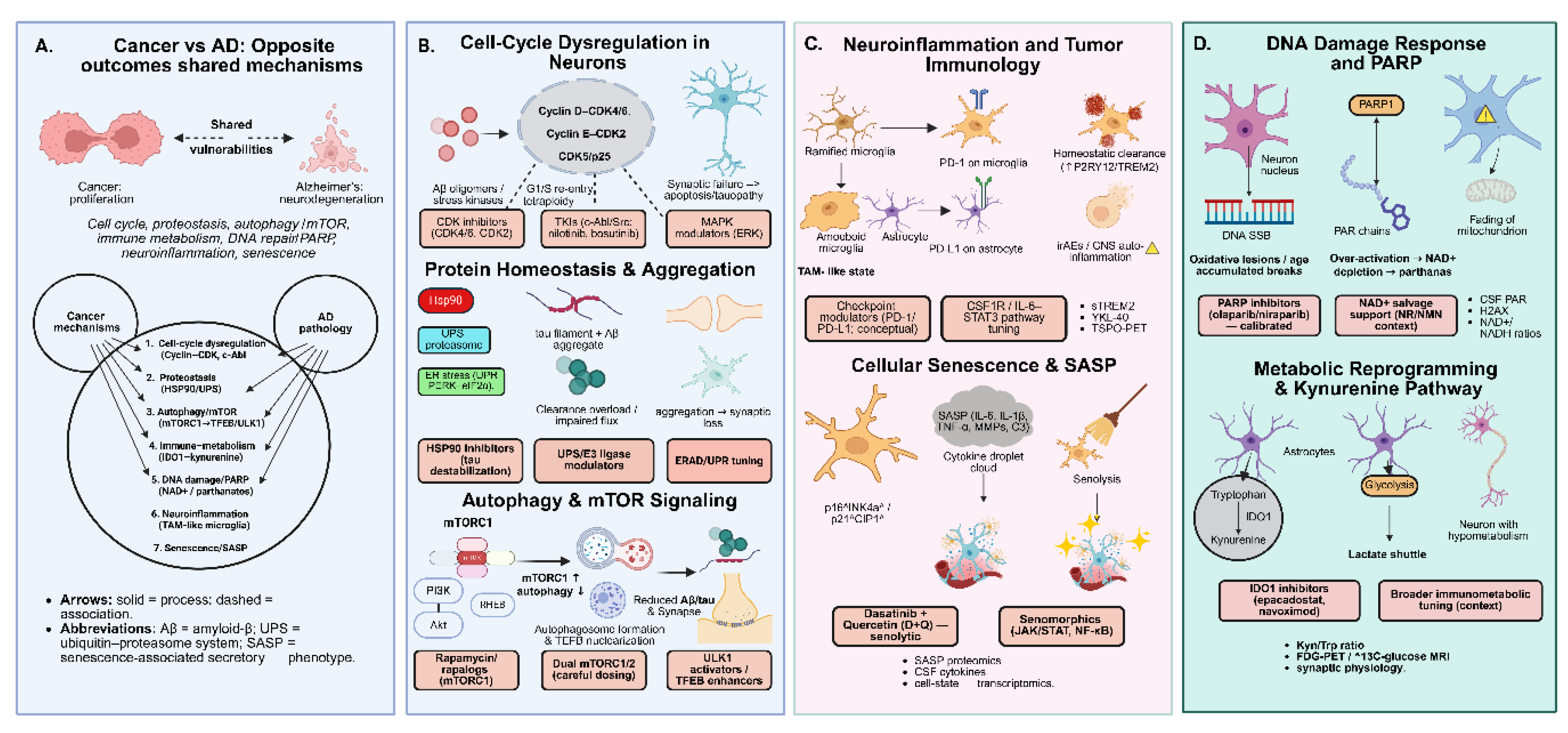
Figure 1.
Shared vulnerabilities and divergent outcomes in cancer and Alzheimer’s disease (AD). (A) Despite opposite phenotypes—uncontrolled proliferation in cancer versus neurodegeneration in AD—both conditions converge on dysregulated pathways including cell cycle, proteostasis, autophagy/mTOR, immune–metabolism (IDO1–kynurenine), DNA damage/PARP signaling, neuroinflammation, and cellular senescence. (B) Mechanistic modules illustrate how aberrant cell-cycle re-entry, proteostasis overload, impaired autophagy, and microglial immune checkpoints drive neuronal vulnerability, while corresponding oncology-derived inhibitors (e.g., CDK inhibitors, TKIs, rapalogs, PARP inhibitors, senolytics) provide repurposing opportunities. (C–D) Microglial senescence, SASP signaling, and DNA damage–induced PARP over-activation exemplify cross-disease mechanisms that can be therapeutically leveraged through precision repurposing.
The complex and multifaceted nature of AD, and its shared vulnerabilities with cancer, are summarized in the Table 1 below.
3. Oncology Drug Classes with Mechanistic/Translational Promise in AD
We now survey major oncology drug classes that have been evaluated (or proposed) for repurposing in AD. For each we outline mechanisms of action, preclinical evidence, and any clinical data, along with challenges.
3.1. Tyrosine Kinase Inhibitors (TKIs): Nilotinib, Bosutinib, etc.
Multi-target TKIs against c-Abl/Src/RTKs suppress aberrant neuronal cell-cycle signaling and upregulate autophagy; c-Abl hyperphosphorylates tau and promotes neuronal death [65,66,67]. TKIs (nilotinib, bosutinib) show BBB penetration [66,68]; masitinib (c-Kit/PDGFR/Fyn) modulates mast cells/microglia to dampen neuroinflammation [66,67,68]. Nilotinib enhances autophagy, clears Aβ/tau/α-synuclein, and improves memory in AD mice; bosutinib shows analogous effects in Lewy/Parkinson models; masitinib reduces inflammation and synaptic loss with cognitive benefit [66,67,69]. Phase II nilotinib (150–300 mg; NCT02921477) was safe with detectable exposure and biomarker shifts—reduced amyloid PET, lower CSF Aβ40/42 and p-tau, slower hippocampal atrophy—more favorable at 150 mg; 300 mg produced neuropsychiatric AEs [66,67,69]. Bosutinib is in early dementia with Lewy bodies trials with CSF penetration signals [66,67]; masitinib (4.5 mg/kg/day) slowed ADAS-Cog and ADL decline in phase 2B/3 (p<0.001); phase 3 confirmatory ongoing [68,70,71].
3.2. mTOR Inhibitors (Rapamycin and Rapalogs)
mTORC1 inhibition induces autophagy, reduces aberrant protein synthesis, and restores proteostasis; pathway is hyperactive in cancer and AD [42,72,73,74]. Multiple models show plaque/tangle reduction, improved synaptic function, and memory rescue with rapamycin/rapalogs, especially with early intervention [40,75,76]. Oral rapamycin 1 mg/day × 8 weeks in mild AD/MCI (NCT04200911) was safe but not CSF-detectable; CSF p-tau and GFAP rose, suggesting subtherapeutic CNS exposure or complex PD (37,68) [40]. Intranasal/low-dose paradigms and everolimus studies are ongoing [68]. BBB penetration is variable; chronic immunometabolic toxicity (infection, dyslipidemia, hyperglycemia) constrains dose. Precision delivery and early-stage enrollment may be decisive [40,42,74].
3.3. Retinoid X Receptor Agonists (Bexarotene) – A Cautionary Case
RXR activation was posited to boost ApoE-dependent Aβ clearance, cholesterol transport, and anti-inflammation. Initial 2012 report of rapid plaque clearance and cognitive rescue in transgenic mice spurred intense interest, but five independent groups failed to reproduce plaque-clearance; some observed modest soluble Aβ/cognition effects; outcomes varied with age, dosing, and formulation [69,77]. Toxicities (hypertriglyceridemia, hypothyroidism) were prominent [67,78]. BEAT-AD (NCT01782742) showed lipid/ApoE increases without clear cognitive or amyloid-PET benefit over 4–8 weeks; open-label work echoed tolerability issues [78,79]. APOE genotype context may matter, but safety at CNS-active exposure is poor. This case underscores rigorous replication and translational discipline.
3.4. Immune–Metabolism Targets: IDO1 Inhibitors and the Kynurenine Pathway
IDO1 diverts tryptophan to kynurenine, shaping immunometabolism. In AD, astrocytic/microglial IDO1 is induced by Aβ/tau, suppressing astrocyte glycolysis and lactate shuttling to neurons [80,81,82]. Brain-penetrant IDO1 inhibitors restored hippocampal glucose metabolism, normalized synaptic function, and fully rescued memory in AD mice [80,82]. Oncology-developed IDO1 inhibitors (e.g., epacadostat, navoximod) have human PK/PD and safety packages that can accelerate AD trials; metabolic imaging (FDG-PET, hyperpolarized ^13C) provides proximal biomarkers [83,84,85]. Immunomodulatory risks and CNS exposure heterogeneity require careful titration and biomarker-guided dosing [81,86].
3.5. Cytotoxic Agents Identified by Transcriptomic Signature Reversal (Letrozole + Irinotecan)
Connectivity- and network-based reversal of AD single-nucleus signatures across cell types nominated letrozole (neuronal modules) plus irinotecan (glial modules) [87,88]. In combined amyloid/tau mouse models, the combo outperformed monotherapy—improving memory, reducing plaques/p-tau, and normalizing snRNA-seq networks [87,88]. EHR analyses suggested reduced incident AD in patients exposed to these agents (confounded by cancer survivorship) [89,90,91]. Both drugs are FDA-approved chemotherapeutics; AD translation would require micro-dosed, brain-targeted regimens with stringent hematologic/GI safety oversight [89,92]. Cytotoxicity in frail elderly and chronic dosing feasibility are major hurdles; safer analogs or delivery vehicles may be needed.
3.6. PARP Inhibitors and DNA Damage Modulators
In AD, PARP1 over-activation depletes NAD+ and drives parthanatos; oncology PARP inhibitors could blunt futile PARylation, stabilize energetics, and protect neurons [46,93,94,95]. Preclinical and computational repurposing highlight PARP1 as a target; niraparib and related PARPi have theoretical neuroprotection potential [94,96]. Human PARPi PK/safety are well-characterized; AD trials are not yet planned. Neurons require basal PARP activity for physiological repair/transcription; chronic PARP inhibition risks impairing activity-dependent plasticity. Timing, dose, and NAD+ salvage status are critical [95,97,98].
3.7. Epigenetic Modulators (HDAC Inhibitors)
Pan- and selective HDAC inhibition rebalances chromatin acetylation, restoring synaptic gene programs and plasticity; oncology HDACi (vorinostat, panobinostat) provide precedence [99,100,101]. Vorinostat and related HDACi restore histone acetylation/LTP, reduce plaques and tau phosphorylation, and improve memory in AD mice [101,102]. Early human work shows CSF histone-acetylation changes without short-term cognitive benefit; toxicity (thrombocytopenia, fatigue, GI) and variable BBB penetration limit pan-HDACi [103](92). HDAC6-selective and BET-bromodomain strategies may enhance tolerability [99]. Isoform selectivity, CNS exposure, and chronic safety define the path forward [103,104].
3.8. Immune Checkpoint Modulators
Releasing PD-1/PD-L1 “brakes” could re-engage microglial phagocytosis and reset maladaptive neuroinflammation (TAM-to-microglia analogy) [51,105]. Mouse data are mixed: reports of plaque reduction/cognitive benefit via IFNγ-driven peripheral recruitment contrast with non-replications; protocol details and disease stage likely determine outcome [51,105,106]. No completed AD trials; oncology experience highlights serious irAEs (including CNS autoimmunity). Frailty, long treatment horizons, and BBB pharmacology argue for extreme caution; if attempted, early-stage AD with dense biomarker monitoring (sTREM2, TSPO-PET, microglial state signatures) is essential [105,106,107].
3.9. Other Oncology Agents (Anti-Angiogenics, Immunomodulators, Metabolic)
Thalidomide-class IMiDs (lenalidomide, pomalidomide) suppress TNFα/IL-6 [108,109]; in AD mice, lenalidomide reduced pro-inflammatory cytokines and improved behavior; a phase II low-dose lenalidomide trial in AD is underway [110]. Thalidomide itself reduced Aβ in mice but caused neuropathy in humans [108,109,111]. Anti-angiogenics (e.g., bevacizumab) and vascular-stabilizing TKIs (e.g., axitinib) have preclinical signals including BBB repair and plaque reduction; human AD efficacy data remain sparse [108,109,112]. Metabolic agents at the oncology–neuro interface (beyond IDO1) are conceptually attractive but currently under-evidenced for AD translation Class-specific toxicities, vascular fragility in elderly brains, and limited chronic safety windows necessitate conservative trial design [108,109,110].
4. Methods and Approaches Used to Nominate Oncology–AD Repurposing Candidates
A modern nomination pipeline for oncology-to-AD repurposing spans computational inference, high-content experimental screening, pharmacokinetic triage, and real-world validation, converging on mechanism-anchored candidates with measurable target engagement.
4.1. Transcriptomic Signature Reversal (CMap/LINCS)
AD bulk/snRNA-seq signatures are anti-correlated against large drug-perturbation matrices (CMap/LINCS L1000) to identify compounds that “reverse” disease programs [113,114]. The letrozole+irinotecan pair arose by partitioning neuronal vs. glial modules and selecting agents with complementary reversal across cell types, then validating multi-lineage rescue in vivo. Prioritization uses reversal magnitude, pathway coherence, and cell-type selectivity, with cross-checks against toxicity annotations [90,115,116].
4.2. Network Pharmacology and Knowledge Graph
Disease modules (genes, pathways, PPIs) are embedded in interactomes and overlaid with approved-drug targetomes; diffusion/proximity metrics rank drugs whose targets lie near or intersect the AD subnetwork [117]. Multi-omic knowledge graphs (genome/proteome/metabolome/chemistry) expose polypharmacology and rational combinations. These frameworks have repeatedly highlighted CDK4/6 and PARP inhibitors as proximal to AD modules, motivating mechanistic follow-up [118,119,120,121].
4.3. Cheminformatics, Docking, and Ligand-Based Screens
Structure-based docking and pharmacophore/similarity searches reveal off-target interactions of oncology agents with AD-relevant proteins (e.g., tau kinases, secretases, LRP1). Virtual screens of FDA-approved libraries yield tractable hits for biochemical IC₅₀ profiling, target engagement (e.g., tau-kinase p-substrates), and cellular phenotypes (aggregate load, neurite metrics), accelerating repurposing while retaining a favorable regulatory path [66,122].
4.4. AI/Machine-Learning Integrators
Supervised and deep multimodal models fuse omics, networks, chemical features, imaging, and clinical metadata to predict drug–disease links and estimate causal impact on AD modules. Generative/graph architectures (“DreamAI”-style) can rank proteostasis- and immune-metabolism–directed oncology agents, quantify uncertainty, and reweight toward human datasets to reduce animal→human translation gaps. Model outputs are gated by mechanistic plausibility and PK feasibility.
4.5. Single-Cell and Multi-Omics Integration
Cell-type resolution via sc/snRNA-seq, ATAC-seq, and proteomics maps druggable perturbation points in neurons, astrocytes, microglia, and oligodendroglia, and predicts combination logic across interacting cell populations. The letrozole–irinotecan example used single-cell reversal to correct neuron- and glia-specific networks; immune–metabolic targets such as astroglial/microglial IDO1 emerged from these layers [24]. Outputs guide cell-type-matched assays and biomarkers [123,124]. An overview of this integrative nomination-to-validation pipeline is shown in Figure 3.
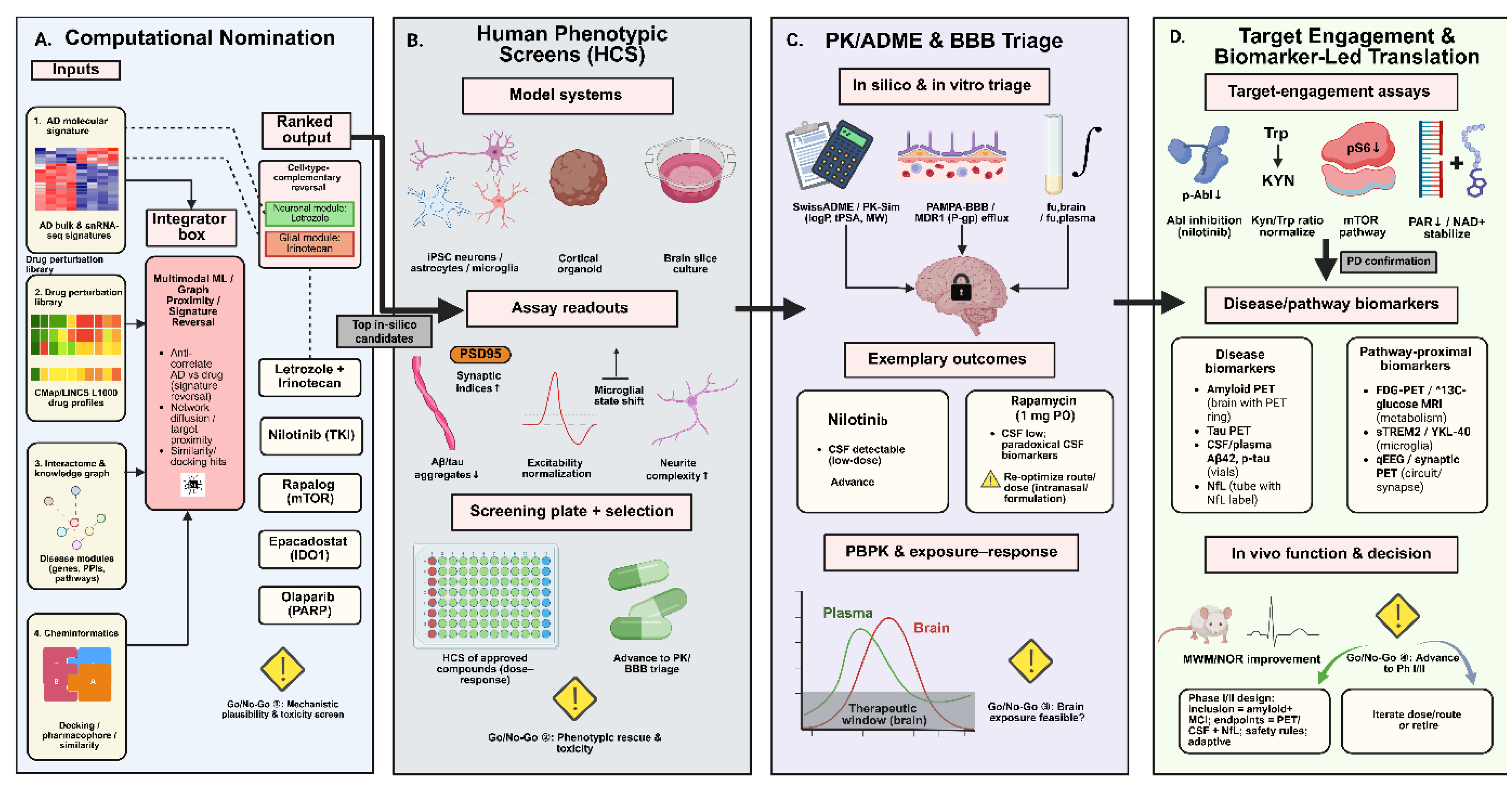
Figure 2.
Integrated pipeline for computational and experimental drug repurposing in Alzheimer’s disease (AD). (A) Computational nomination using AD transcriptomic signatures, drug perturbation libraries (CMap/LINCS), network-based interactome analyses, and cheminformatics approaches to identify candidate compounds through signature reversal, network proximity, and docking. (B) Phenotypic screening in patient-derived iPSC neurons, glia, organoids, and brain slice cultures, with readouts including synaptic integrity, excitability, and microglial state. (C) Pharmacokinetic and blood–brain barrier (BBB) triage via in silico and in vitro assays (SwissADME, PK-Sim, PAMPA-BBB), followed by exposure–response modeling. (D) Target engagement and biomarker-led translation using pathway-proximal markers (e.g., Abl inhibition, mTOR signaling, PARP/NAD⁺ stabilization) and disease biomarkers (amyloid/tau PET, CSF/plasma assays, neurofilament light chain). The pipeline enables iterative go/no-go decision points toward Phase I/II clinical trials.
Figure 2.
Integrated pipeline for computational and experimental drug repurposing in Alzheimer’s disease (AD). (A) Computational nomination using AD transcriptomic signatures, drug perturbation libraries (CMap/LINCS), network-based interactome analyses, and cheminformatics approaches to identify candidate compounds through signature reversal, network proximity, and docking. (B) Phenotypic screening in patient-derived iPSC neurons, glia, organoids, and brain slice cultures, with readouts including synaptic integrity, excitability, and microglial state. (C) Pharmacokinetic and blood–brain barrier (BBB) triage via in silico and in vitro assays (SwissADME, PK-Sim, PAMPA-BBB), followed by exposure–response modeling. (D) Target engagement and biomarker-led translation using pathway-proximal markers (e.g., Abl inhibition, mTOR signaling, PARP/NAD⁺ stabilization) and disease biomarkers (amyloid/tau PET, CSF/plasma assays, neurofilament light chain). The pipeline enables iterative go/no-go decision points toward Phase I/II clinical trials.
4.6. Network Pharmacology and Knowledge Graph
Disease modules (genes, pathways, PPIs) are embedded in interactomes and overlaid with approved-drug targetomes; diffusion/proximity metrics rank drugs whose targets lie near or intersect the AD subnetwork [121]. Multi-omic knowledge graphs (genome/proteome/metabolome/chemistry) expose polypharmacology and rational combinations. These frameworks have repeatedly highlighted CDK4/6 and PARP inhibitors as proximal to AD modules, motivating mechanistic follow-up [120].
4.7. Phenotypic High-Content Screening (iPSC, Organoids, Slices)
Human iPSC-derived neurons/glia, 3D organoids, and organotypic slices enable unbiased phenotypic discovery and validation of computational hits. High-content imaging quantifies Aβ/tau aggregates, synaptic indices (e.g., PSD95/Homer), firing/excitability, and microglial state transitions (homeostatic vs. DAM) [125]. Screens in Aβ-overexpressing neurons have identified oncology agents that reduce tau pathology or normalize synaptic readouts; co-culture/organoid systems clarify neuron–glia cross-talk and microglial phagocytosis [126,127].
4.8. PK/ADME and BBB Triage
CNS repurposing requires explicit early PK gating. In-silico tools (SwissADME, PK-Sim) and in-vitro assays (PAMPA-BBB, MDR1/P-gp efflux) prioritize favorable lipophilicity, polar surface area, and non-substrate status. Empirically, nilotinib’s properties and CSF detectability supported advancement [28], whereas low-dose oral rapamycin produced poor CSF exposure and paradoxical CSF biomarker shifts in a phase I AD study \[21]. Quantitative PBPK, unbound brain fraction, brain microdialysis, and transporter phenotyping de-risk translation [128,129].
4.9. Preclinical Validation Cascades and Translational Readouts
Converged candidates enter cascades that confirm target engagement (e.g., neuronal Abl inhibition by nilotinib; IDO1 flux via kynurenine/tryptophan ratios), proximal biomarker shifts (CSF soluble Aβ species, p-tau, neuroinflammatory cytokines, sTREM2), and functional rescue in vivo (MWM/NOR). Imaging biomarkers—amyloid/tau PET, FDG-PET for metabolism, synaptic PET—supply noninvasive pharmacodynamic readouts; electrophysiology and qEEG complement behavior. Exposure–response modeling (brain\:plasma, target occupancy) guides phase I/II design [130,131].

Table 2.
Repurposed oncology drugs in Alzheimer’s disease (AD): mechanisms, evidence, and trial status.
Table 2.
Repurposed oncology drugs in Alzheimer’s disease (AD): mechanisms, evidence, and trial status.
| Drug (Class) | Proposed mechanism in AD | Preclinical evidence | Trial (Phase; NCT) | Endpoints / Biomarkers | Key clinical status / comments |
|---|---|---|---|---|---|
| Nilotinib (TKI) | c-Abl/Src inhibition; ↑autophagy → Aβ/tau clearance | In AD mice: lowers Aβ and tau; cognition improved | Phase II; NCT02921477 [69] | Primary: ADAS-Cog; Secondary: amyloid PET, CSF Aβ/p-tau | Biomarker movement observed (↓brain amyloid; ↓CSF Aβ, p-tau) [18]. 150 mg tolerated; 300 mg linked to agitation. |
| Masitinib (TKI) | c-Kit/PDGFR/Fyn inhibition; mast-cell/microglial modulation | AD rodents: ↓inflammation, ↓plaques, memory rescue | Phase 2B/3; NCT00976118 [132] | ADAS-Cog, ADCS-ADL; MRI, CSF | 4.5 mg/kg qd significantly slowed cognitive/ADL decline (p=0.0003). Confirmatory Phase 3 ongoing. |
| Rapamycin (mTOR inhibitor) | mTORC1 inhibition → autophagy ↑; ↓Aβ/τ pathology [133] | Multiple AD rodent studies: rescue of memory; pathology reduction | Phase I; NCT04200911 [134] | CSF p-tau, GFAP; MRI volume | Safe, not detectable in CSF at tested dose; paradoxical ↑CSF p-tau, GFAP. Dose/route optimization needed. |
| Bexarotene (RXR agonist) | ↑ApoE/ABCA1; lipid transport; proposed Aβ clearance | Initial 2012 report of rapid plaque clearance not replicated across labs | Phase II (BEAT-AD, completed); NCT02061878 [135] | Amyloid PET; ADAS-Cog | Marked hypertriglyceridemia; no significant cognitive or PET benefit; tolerability limits CNS-relevant dosing. |
| Dasatinib + Quercetin (Senolytic) | Senescent cell clearance; ↓neuroinflammation | AD mice: preserved cognitive function; reduced senescent cells | Phase 1; NCT04063124 [136]; Phase 2 ongoing | Primary: Safety, CNS penetration; Secondary: CSF biomarkers | Dasatinib detected in CSF in most participants; signals for amyloid clearance and ↓inflammation; intermittent dosing well-tolerated. |
| Temsirolimus (mTOR inhibitor) | mTOR inhibition; ↑autophagy → Aβ clearance | APP/PS1 mice: ↓Aβ burden, ↓apoptosis, cognition improved | Preclinical only [137] | N/A | Promoted autophagic Aβ clearance; efficacy likely greatest in early disease windows. |
Abbreviations: AD = Alzheimer’s disease; MCI = mild cognitive impairment; ADAS-Cog = Alzheimer’s Disease Assessment Scale–Cognitive; ADCS-ADL = Alzheimer’s Disease Cooperative Study–Activities of Daily Living; PET = positron emission tomography; CSF = cerebrospinal fluid; TKI = tyrosine kinase inhibitor; RXR = retinoid X receptor; IMiD = immunomodulatory imide drug; HDAC = histone deacetylase; MM = multiple myeloma; SASP = senescence-associated secretory phenotype.
5. Clinical Evidence, Ongoing Trials, and Negative Studies
5.1. Summary of Trials
Systematic synthesis to 2021 catalogued 13 Alzheimer’s disease (AD) trials (phases I–III) testing 11 oncology agents: five tyrosine-kinase inhibitors (nilotinib, bosutinib, masitinib, dasatinib, abemaciclib), two retinoid X receptor (RXR) agonists (bexarotene, tamibarotene), two immunomodulators (lenalidomide, daratumumab) (129), one HDAC inhibitor (vorinostat), and one additional monoclonal antibody (daratumumab). Table 2 summarizes mechanisms, preclinical rationale, trial IDs, and endpoints. In brief, nilotinib and bosutinib have completed early-phase AD studies focused on safety and biomarkers; masitinib has reported positive phase 2B/3 cognitive/functional outcomes (130); bexarotene trials were negative; rapalogs are in phase I with mixed biomarker signals; IDO1 inhibitors and the letrozole/irinotecan combination remain preclinical; dasatinib (as part of senolysis) is in pilot studies; thalidomide/lenalidomide have early, mixed signals; vorinostat showed target engagement without efficacy. As of mid-2025, no repurposed oncology agent has yet shown definitive cognitive benefit in a large, confirmatory AD trial (131) (132).Systematic synthesis to 2021 catalogued 13 Alzheimer’s disease (AD) trials (phases I–III) testing 11 oncology agents: five tyrosine-kinase inhibitors (nilotinib, bosutinib, masitinib, dasatinib, abemaciclib), two retinoid X receptor (RXR) agonists (bexarotene, tamibarotene), two immunomodulators (lenalidomide, daratumumab) [138], one HDAC inhibitor (vorinostat), and one additional monoclonal antibody (daratumumab). Table 2 summarizes mechanisms, preclinical rationale, trial IDs, and endpoints. In brief, nilotinib and bosutinib have completed early-phase AD studies focused on safety and biomarkers; masitinib has reported positive phase 2B/3 cognitive/functional outcomes [139]; bexarotene trials were negative; paralogs are in phase I with mixed biomarker signals; IDO1 inhibitors and the letrozole/irinotecan combination remain preclinical; dasatinib (as part of senolysis) is in pilot studies; thalidomide/lenalidomide have early, mixed signals; vorinostat showed target engagement without efficacy. As of mid-2025, no repurposed oncology agent has yet shown definitive cognitive benefit in a large, confirmatory AD trial [140,141].
Nilotinib (TKI): Mechanistically, c-Abl/Src inhibition increases autophagy and facilitates Aβ/tau clearance. In AD mice, nilotinib reduces aggregates and improves cognition. Phase II NCT02921477 reported acceptable safety, detectable exposure, and favorable biomarker movements—reduced amyloid PET, lower CSF Aβ40/42 and p-tau, and slower hippocampal atrophy—with better tolerability and signal at 150 mg than 300 mg (the latter associated with agitation) [18,19]. Primary cognitive endpoints were exploratory; replication is needed [142].
Bosutinib (TKI): A c-Abl/Src inhibitor with anti-inflammatory effects (↑IL-10), bosutinib reduced α-synuclein in PD/DLB contexts and showed CSF penetration (~5% CSF:plasma) [case-series/early data]. Phase I/II NCT04670840 in AD/DLB emphasizes safety, CSF biomarkers (including α-synuclein), and cognition; definitive efficacy data are pending [143].
Masitinib (multikinase TKI): Targeting c-Kit/PDGFR/Fyn with actions on mast cells and microglia, masitinib reduced neuroinflammation and synaptic loss in rodent AD. In phase 2B/3 NCT00976118, 4.5 mg/kg once daily significantly slowed decline on ADAS-Cog and ADL (p = 0.0003) with acceptable safety; a confirmatory phase 3 is ongoing [138].
Rapamycin/rapalogs (mTOR inhibitors): Robust preclinical evidence supports aggregate clearance and synaptic rescue via mTORC1 inhibition [11]. In phase I NCT04200911 (1 mg/day, 8 weeks), rapamycin was safe but not CSF-detectable, with unexpected increases in CSF p-tau and GFAP—suggesting subtherapeutic CNS exposure or complex pharmacodynamics; dose/formulation optimization (including intranasal) is underway [138].
Bexarotene and tamibarotene (RXR agonists): Following a highly publicized but non-replicated preclinical report of rapid plaque clearance [22], the BEAT-AD phase II trial (NCT02061878) showed expected lipid increases without cognitive or amyloid-PET benefit; hypertriglyceridemia and hypothyroidism limited tolerability [23]. Tamibarotene (NCT03382147) remains under evaluation with preclinical signals of reduced insoluble Aβ and neuroinflammation; no efficacy readouts are yet available [140].
Immunomodulators (IMiDs) and anti-CD38: Thalidomide/lenalidomide reduce TNFα/IL-6 and improved behavior in AD mice [27]. A thalidomide trial (NCT02085265) was terminated for toxicity; lenalidomide (NCT03063686) is in phase II with biomarker-anchored endpoints. Daratumumab (anti-CD38; NCT04096217) is testing whether microglial/CD38 modulation translates to cognitive/functional benefit, supported by preclinical attenuation of amyloid pathology and inflammation [141].
HDAC inhibition: Vorinostat increased CSF acetylation markers but failed to improve cognition in mild–moderate AD (NCT01719861); class toxicities (cytopenias, fatigue) and CNS exposure remain limiting factors [141].
Senolysis: Dasatinib + quercetin (D+Q) cleared senescent glia, reduced plaques/tangles, and improved behavior in mice. Early human pilot work (e.g., NCT03430069) is primarily safety-focused in AD and Down syndrome; efficacy data are awaited [138].
Preclinical pipelines. IDO1 inhibition normalized hippocampal metabolism and rescued memory in AD models [12]; clinical translation is anticipated. The letrozole+irinotecan combination, nominated by transcriptomic signature reversal, outperformed monotherapies in aggressive amyloid/tau mice and rewired disease gene networks; human testing has not begun [138].
5.2. Lessons from Negative or Irreproducible Results
Bexarotene’s trajectory—from dramatic preclinical claims to multi-lab non-replication—illustrates the hazards of single-study over-interpretation, formulation differences, and model idiosyncrasies. More broadly, aggressive transgenic models frequently overpredict plaque/tangle clearance and underpredict human clinical response, reflecting AD’s multifactorial, slow-evolving biology. Negative or null outcomes emphasize the need for (i) convergent preclinical validation across models and species; (ii) genetic/contextual stratification (e.g., APOE genotype effects reported in subsets); and (iii) transparent methods and data sharing before resource-intensive trials. Vorinostat’s target engagement without efficacy cautions that proximal biomarker change is necessary but not sufficient; pathway choice, dosing, and CNS exposure must align with disease stage [140,141].
Dose re-engineering: Oncology paradigms aim for maximum tolerated dose and short courses; AD requires chronic, lower-intensity modulation in elderly patients. Nilotinib illustrates this gap (150–300 mg qd in AD vs oncology’s far higher dosing) and underscores the need for exposure–response mapping within CNS constraints. Rapamycin’s low oral dosing yielded poor CSF exposure and paradoxical biomarker shifts; alternative routes/formulations may be required [138,142].
Geriatric risk and polypharmacy: Frailty, comorbid cardiovascular–metabolic disease, and polypharmacy elevate risk for myelosuppression (IMiDs/HDACi/PARPi), infection/dyslipidemia (rapalogs), bleeding/arrhythmia and neuropsychiatric events (multi-TKIs). Drug–drug interactions via CYP3A4 (many TKIs) are common and mandate proactive management plans [142].
BBB and delivery: Brain exposure is a recurrent bottleneck. Strategies include medicinal chemistry (lipophilicity, P-gp avoidance), alternative routes (intranasal, intrathecal), and device-assisted delivery (focused ultrasound). Each introduces operational and safety complexity that must be justified by robust pharmacodynamic markers [138].
CNS off-targets: Multikinase profiles and epigenetic breadth can perturb protective signaling or synaptic programs; rigorous neurobehavioral monitoring and phased dose-finding are essential [138].
5.2.1. Clinical Trial Design Recommendations
Selection criteria: Prioritize compounds with replicated preclinical efficacy, human target engagement, and plausible CNS PK. Exclude agents with prohibitive geriatric risk. Require early demonstration of brain exposure (e.g., CSF drug levels) and pathway modulation [142].
Adaptive platforms: Multi-arm, multi-stage designs can evaluate several repurposed agents against shared controls, dropping non-signals and enriching promising arms. Seamless phase II/III transitions shorten timelines while preserving rigor [138].
Biomarker-anchored go/no-go: Use amyloid/tau PET, CSF/plasma Aβ42, p-tau, and NfL to confirm disease engagement; pathway-specific markers (e.g., kynurenine/tryptophan for IDO1; pS6 for mTOR; sTREM2 and YKL-40 for microglia) provide proximal readouts. FDG-PET or ^13C-MR spectroscopy can index metabolic rescue; qEEG/synaptic PET can capture circuit effects [138].
Enrichment: Enroll amyloid-positive MCI/mild AD; consider genotype (APOE ε4), inflammatory/metabolic signatures, or microglial activation (TSPO-PET) to match mechanism to biology and boost power [140].
Ethics/regulation: For cytotoxic/immune agents, risk-communication must be explicit. Accelerated approval based on robust biomarker modulation may be feasible, contingent on stringent safety surveillance and confirmatory trials [138].
A prioritized roadmap: Given current data, priority classes include c-Abl/Src TKIs (nilotinib/bosutinib) with biomarker signals and tolerable low-dose regimens; neuroimmune-targeted TKIs (masitinib) with phase 2B/3 efficacy; mTOR inhibitors contingent on brain-exposure solutions; senolytic strategies (dasatinib+quercetin) moving from mechanistic pilots to controlled trials; and IDO1 inhibitors with compelling preclinical rescue and clear translational biomarkers. Each candidate should advance only with verified CNS exposure and pathway-proximal modulation, leveraging adaptive designs and early biomarker gates to contain risk and concentrate resources on the most mechanistically coherent, clinically translatable leads [138,142,144].
6. Conclusions and Future Directions
Repurposing oncology agents for Alzheimer’s disease (AD) should proceed not as a speculative shortcut but as a rigorous, mechanism-led program that reflects the networked pathobiology of AD and leverages decades of human pharmacology from cancer therapeutics. The cumulative evidence across cell-cycle dysregulation, proteostasis failure, autophagy/mTOR signaling, immune–metabolic remodeling (notably the kynurenine pathway), DNA damage and PARP biology, neuroinflammation, and cellular senescence indicates a durable mechanistic bridge between cancer and AD that supports pleiotropic, multi-node interventions rather than single-target “magic bullets”. Future work must therefore embed precision repurposing into its foundations: stratifying patients by genotype (e.g., APOE ε4), molecular endophenotypes (neuroinflammatory or metabolic signatures), and cell-state atlases derived from single-cell and spatial transcriptomics to align mechanism with the most responsive subpopulations [145]. Human-anchored computational nomination should remain the front end of discovery, combining transcriptomic signature reversal at bulk and single-cell resolution with network proximity in knowledge graphs to prioritize agents whose targets lie within or adjacent to patient-derived AD modules; these in silico signals should be cross-validated with real-world evidence (RWE) from electronic health records and claims, recognizing both the inferential value and the biases inherent in observational datasets. A precision repurposing framework integrating patient stratification, delivery-first pharmacology, and prioritized portfolios is depicted in Figure 3.
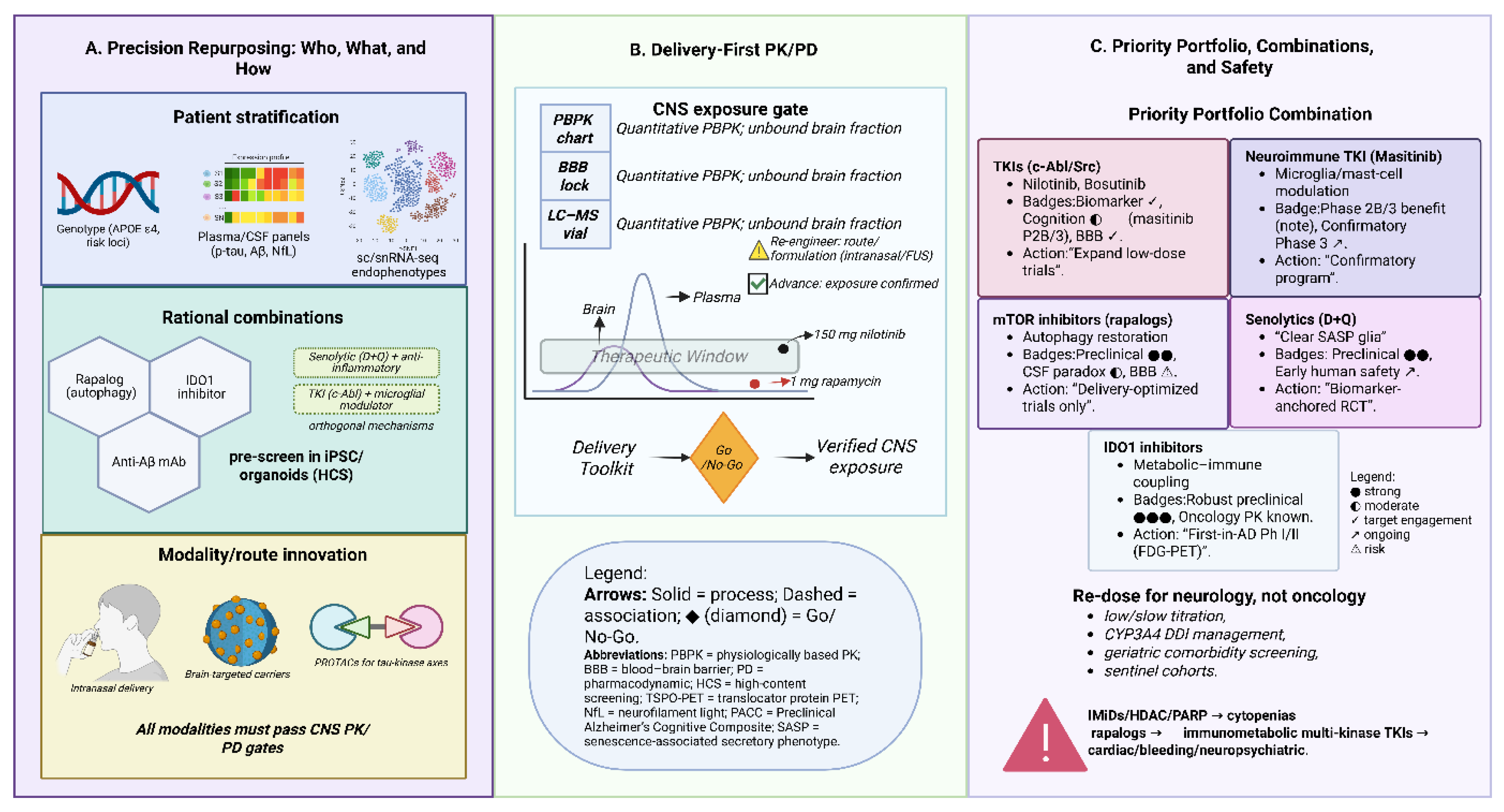
Figure 3.
Framework for precision repurposing in Alzheimer’s disease (AD). (A) Precision stratification integrates single-cell/snRNA-seq endophenotypes, APOE ε4 and risk genotypes, and CSF/plasma biomarkers (Aβ, p-tau, NfL) to identify responsive subgroups. Rational combinations are designed by pairing orthogonal mechanisms (e.g., IDO1 inhibition, mTOR/autophagy modulation, anti-amyloid antibodies, senolytics plus anti-inflammatories, or TKIs with microglial modulators), pre-screened in human iPSC/organoid models. (B) A delivery-first paradigm requires CNS pharmacokinetic/pharmacodynamic (PK/PD) validation through PBPK modeling, unbound brain fraction, and alternative routes (intranasal, focused ultrasound, carrier-mediated delivery). (C) A prioritized portfolio highlights senolytics, TKIs, neuroimmune TKIs, rapalogs, and IDO1 inhibitors, each with graded evidence (preclinical, biomarker, safety, cognition). The schematic emphasizes iterative go/no-go decision points, geriatric-adapted dosing, and safety gates to advance only candidates with verified brain exposure and pathway engagement.
Figure 3.
Framework for precision repurposing in Alzheimer’s disease (AD). (A) Precision stratification integrates single-cell/snRNA-seq endophenotypes, APOE ε4 and risk genotypes, and CSF/plasma biomarkers (Aβ, p-tau, NfL) to identify responsive subgroups. Rational combinations are designed by pairing orthogonal mechanisms (e.g., IDO1 inhibition, mTOR/autophagy modulation, anti-amyloid antibodies, senolytics plus anti-inflammatories, or TKIs with microglial modulators), pre-screened in human iPSC/organoid models. (B) A delivery-first paradigm requires CNS pharmacokinetic/pharmacodynamic (PK/PD) validation through PBPK modeling, unbound brain fraction, and alternative routes (intranasal, focused ultrasound, carrier-mediated delivery). (C) A prioritized portfolio highlights senolytics, TKIs, neuroimmune TKIs, rapalogs, and IDO1 inhibitors, each with graded evidence (preclinical, biomarker, safety, cognition). The schematic emphasizes iterative go/no-go decision points, geriatric-adapted dosing, and safety gates to advance only candidates with verified brain exposure and pathway engagement.
Given the multifactorial nature of disease progression, rational combination therapy—long the standard in oncology—should become a first-class strategy in AD. Proof-of-concept already exists: transcriptome-guided pairing of letrozole (neuronal module) with irinotecan (glial module) outperformed monotherapy, rewiring disease networks and improving cognition in aggressive amyloid–tau models . Mechanistically orthogonal pairings are equally compelling: autophagy enhancement (rapalogs) with anti-amyloid antibodies to couple aggregate clearance with proteostasis restoration; IDO1 inhibition to normalize astrocyte–neuron metabolic coupling alongside microglial modulators to rebias innate immunity; or senolytic regimens (dasatinib + quercetin) layered with anti-inflammatory agents to break SASP-amplified circuit toxicity. Such regimens should be pre-screened in human iPSC cocultures and organoids with high-content phenotyping, followed by adaptive clinical testing that is explicitly safety-gated.
The central operational barrier remains brain exposure at geriatric-tolerable dosing. Rapamycin’s low oral dose in mild AD/MCI yielded poor CSF detectability with paradoxical CSF biomarker shifts, illustrating how insufficient CNS pharmacokinetics can confound pharmacodynamics [21]. Conversely, nilotinib’s physicochemical profile and detectable human CSF exposure justified phase II evaluation and yielded multimodal biomarker movement at low doses [18,28]. Future programs must adopt delivery-first pharmacology: medicinal chemistry for BBB-permeant analogs and transporter-aware designs (avoiding P-gp substrates); quantitative PBPK modeling tied to unbound brain fraction; microdialysis in translational species; and exploration of alternative routes (intranasal formulations, focused ultrasound–assisted BBB modulation) with embedded pharmacodynamic readouts. Only candidates with verified CNS exposure and pathway-proximal modulation in humans should advance.
New modalities adapted from oncology expand the reachable target space. Proteolysis-targeting chimeras (PROTACs) can deliver event-driven degradation of tau-kinase axes or other AD-relevant proteins, potentially overcoming occupancy limits and conformational heterogeneity [57,58]. Antibody–drug conjugate–like shuttles and nanocarriers, matured in oncology, can be re-engineered for brain delivery of repurposed payloads, provided BBB shuttle design, immunogenicity control, and pharmacodynamic verification are achieved [146]. These modalities should enter the same nomination funnel—computational triage, human phenotypic validation, rigorous PK/PD gating—before clinical exploration.
Clinical development must match the biology in tempo and design. Multi-arm, multi-stage platform trials can evaluate several repurposed agents in parallel against shared controls, pruning futile arms early and expanding promising ones with seamless phase II→III transitions. Go/no-go criteria should be biomarker-led: pathway-proximal markers of target engagement (kynurenine/tryptophan ratios for IDO1; pS6 for mTOR; soluble TREM2 and YKL-40 for microglia; PAR polymers and NAD+-linked indices for PARP) paired with disease biomarkers (amyloid/tau PET; CSF/plasma Aβ and phospho-tau) and neuronal injury measures (NfL) [11,12,13,21,24]. Circuit-level rescue can be indexed by FDG-PET and ^13C-MR spectroscopy in metabolic trials or synaptic PET/qEEG when synaptic physiology is the focus. Enrichment should require amyloid positivity and mechanism-specific activity (e.g., TSPO-PET–defined neuroinflammation for microglial modulators), improving power and interpretability and mitigating the heterogeneity that has diluted past efforts.
Repurposed oncology agents must be re-dosed for neurology. The oncology paradigm of maximum tolerated dose over short horizons rarely suits chronic treatment in frail elders. Nilotinib’s AD regimen (150 mg daily) stands far below cancer dosing, with higher 300 mg exposure linked to neuropsychiatric agitation [19]. IMiDs, HDAC inhibitors, and PARP inhibitors carry hematologic risks; rapalogs impose immunometabolic liabilities; multikinase TKIs bring cardiac, bleeding, and neuropsychiatric concerns. Proactive drug–drug interaction management (notably CYP3A4 liabilities), conservative titration, geriatric comorbidity screening, and sentinel safety cohorts are essential. Safety signals should be adjudicated with domain expertise borrowed from oncology and geriatrics.
Reproducibility and transparency are preconditions for progress. The bexarotene episode—dramatic preclinical claims, multi-lab non-replication, and negative clinical trials—remains a cautionary template for rigorous preclinical standardization, formulation control, and cross-center replication before embarking on resource-intensive trials [22,23]. Open, precompetitive consortia that couple academic centers, industry, and federated EHR networks can accelerate validation of hits and early retirement of misses, while a shared registry of negative studies reduces duplication and publication bias [5,24,29].
A pragmatic near-term roadmap prioritizes classes with convergent mechanistic plausibility, early human target engagement, and tractable safety at CNS-relevant exposure. c-Abl/Src–directed TKIs (nilotinib, bosutinib) merit continued testing given biomarker shifts and tolerability at low doses [17,18,19]; neuroimmune-modulatory TKIs (masitinib) warrant confirmatory trials following phase 2B/3 cognitive and functional benefit [20]; mTOR inhibition should proceed only with delivery solutions and validated brain exposure [11,21]; senolytic strategies (dasatinib + quercetin) should transition from mechanistic pilots to controlled, biomarker-anchored efficacy studies [15]; and IDO1 inhibitors represent a high-priority metabolic–immune avenue with robust preclinical rescue and clear translational biomarkers [12]. Transcriptome-guided combinations (e.g., letrozole + irinotecan) deserve carefully engineered, dose-deescalated trials that prioritize brain targeting and safety [24].
In conclusion, oncology-to-AD repurposing is not a detour around scientific rigor but a disciplined redirection of existing pharmacology toward a disease that demands multi-target solutions. The field now has the tools—human-centric computation, single-cell atlases, advanced phenotyping, quantitative CNS PK/PD, RWE triangulation, and adaptive trial platforms—to execute this agenda. If applied with precision stratification, delivery innovation, biomarker-led decision-making, and uncompromising standards for reproducibility and geriatric safety, this program can compress timelines, contain risk, and yield mechanism-based treatments that begin to bend the trajectory of Alzheimer’s disease [11,12,13,15,17,18,19,20,21,22,23,24,28,29,30,57,58].
Author Contributions
Dyumn Dwivedi: Writing - Original Draft, Writing - Review & Editing, Visualization; Chandralekha Nair: Writing - Review & Editing; Dewang Singh: Visualization; Uddalak Das: Conceptualization, Project Administration, Supervision, Funding Acquisition, Review & Editing. All authors critically revised the paper and approved the final version
Funding
The author(s) acknowledge the funds received by Uddalak Das from the Department of Biotechnology (Grant No: DBTHRDPMU/JRF/BET-24/I/2024-25/376), the Council of Scientific and Industrial Research (Grant No: 24J/01/00130), and the Indian Council of Medical Research (Grant No: 3/1/3/BRET-2024/HRD (L1)).
Declaration of generative AI and AI-assisted technologies in the writing process
The writing of this review
paper involved the use of generative AI and AI-assisted technologies only to enhance the clarity, coherence, and overall quality of the manuscript. The authors acknowledge the contributions of AI in the writing process while ensuring that the final content reflects the author's own insights and interpretations of the literature. All
interpretations and conclusions drawn in this manuscript are the sole responsibility of the author.
Ethical Statement
None.
Acknowledgments
None.
Conflicts of Interest
The author(s) report no conflict of interest.
References
- Mamun M, Bin Shawkat S, Ahammed MS, Uddin MM, Mahmud MI, Islam AM. Deep Learning Based Model for Alzheimer’s Disease Detection Using Brain MRI Images. In: 2022 IEEE 13th Annual Ubiquitous Computing, Electronics & Mobile Communication Conference (UEMCON) [Internet]. New York, NY, NY, USA: IEEE; 2022 [cited 2025 Sept 2]. p. 0510–6. Available from: https://ieeexplore.ieee.org/document/9965730/.
- Rosende-Roca M, García-Gutiérrez F, Cantero-Fortiz Y, Alegret M, Pytel V, Cañabate P, González-Pérez A, De Rojas I, Vargas L, Tartari JP, Espinosa A, Ortega G, Pérez-Cordón A, Moreno M, Preckler S, Seguer S, Gurruchaga MJ, Tárraga L, Ruiz A, Valero S, Boada M, Marquié M. Exploring sex differences in Alzheimer’s disease: a comprehensive analysis of a large patient cohort from a memory unit. Alzheimers Res Ther. 2025 Jan 22;17(1):27. [CrossRef]
- 2025 Alzheimer’s disease facts and figures. Alzheimers Dement. 2025 Apr;21(4):e70235.
- C. Vickers J, Mitew S, Woodhouse A, M. Fernandez-Martos C, T. Kirkcaldie M, J. Canty A, H. McCormack G, E. King A. Defining the earliest pathological changes of Alzheimer’s disease. Curr Alzheimer Res. 2016 Feb 12;13(3):281–7.
- Wimo A, Seeher K, Cataldi R, Cyhlarova E, Dielemann JL, Frisell O, Guerchet M, Jönsson L, Malaha AK, Nichols E, Pedroza P, Prince M, Knapp M, Dua T. The worldwide costs of dementia in 2019. Alzheimers Dement. 2023 July;19(7):2865–73. [CrossRef]
- 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023 Apr;19(4):1598–695.
- Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary Costs of Dementia in the United States. N Engl J Med. 2013 Apr 4;368(14):1326–34. [CrossRef]
- 2024 Alzheimer’s disease facts and figures. Alzheimers Dement. 2024 May;20(5):3708–821.
- Paduvilan AK, Livingston GAL, Kuppuchamy SK, Dhanaraj RK, Subramanian M, Al-Rasheed A, Getahun M, Soufiene BO. Attention-driven hybrid deep learning and SVM model for early Alzheimer’s diagnosis using neuroimaging fusion. BMC Med Inform Decis Mak. 2025 July 1;25(1):219. [CrossRef]
- Das U, Chandramouli L, Uttarkar A, Kumar J, Niranjan V. Discovery of natural compounds as novel FMS-like tyrosine kinase-3 (FLT3) therapeutic inhibitors for the treatment of acute myeloid leukemia: An in-silico approach. Asp Mol Med. 2025 June;5:100058. [CrossRef]
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014 July 3;6(4):37. [CrossRef]
- Birks JS, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Dementia and Cognitive Improvement Group, editor. Cochrane Database Syst Rev [Internet]. 2018 June 18 [cited 2025 Sept 2];2018(6). [CrossRef]
- Van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, Froelich L, Katayama S, Sabbagh M, Vellas B, Watson D, Dhadda S, Irizarry M, Kramer LD, Iwatsubo T. Lecanemab in Early Alzheimer’s Disease. N Engl J Med. 2023 Jan 5;388(1):9–21.
- Gandy S, DeKosky ST. Toward the Treatment and Prevention of Alzheimer’s Disease: Rational Strategies and Recent Progress. Annu Rev Med. 2013 Jan 14;64(1):367–83. [CrossRef]
- Kim CK, Lee YR, Ong L, Gold M, Kalali A, Sarkar J. Alzheimer’s Disease: Key Insights from Two Decades of Clinical Trial Failures. J Alzheimers Dis. 2022 May 3;87(1):83–100. [CrossRef]
- Das U. Generative AI for drug discovery and protein design: the next frontier in AI-driven molecular science. Med Drug Discov. 2025 Sept;27:100213. [CrossRef]
- DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019 Dec;14(1):32.
- Das U, Chanda T, Kumar J, Peter A. Discovery of natural MCL1 inhibitors using pharmacophore modelling, QSAR, docking, ADMET, molecular dynamics, and DFT analysis. Comput Biol Chem. 2025 Aug;117:108427. [CrossRef]
- Grabowska ME, Huang A, Wen Z, Li B, Wei WQ. Drug repurposing for Alzheimer’s disease from 2012–2022—a 10-year literature review. Front Pharmacol. 2023 Sept 7;14:1257700.
- Das U, Uttarkar A, Kumar J, Niranjan V. In silico exploration natural compounds for the discovery of novel dnmt3a inhibitors as potential therapeutic agents for acute myeloid leukaemia. Silico Res Biomed. 2025 Apr;100006. [CrossRef]
- Weth FR, Hoggarth GB, Weth AF, Paterson E, White MPJ, Tan ST, Peng L, Gray C. Unlocking hidden potential: advancements, approaches, and obstacles in repurposing drugs for cancer therapy. Br J Cancer. 2024 Mar 23;130(5):703–15. [CrossRef]
- Xia Y, Sun M, Huang H, Jin WL. Drug repurposing for cancer therapy. Signal Transduct Target Ther. 2024 Apr 19;9(1):92.
- Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023 July 12;8(1):267. [CrossRef]
- Zabłocka A, Kazana W, Sochocka M, Stańczykiewicz B, Janusz M, Leszek J, Orzechowska B. Inverse Correlation Between Alzheimer’s Disease and Cancer: Short Overview. Mol Neurobiol. 2021 Dec;58(12):6335–49. [CrossRef]
- Driver JA, Beiser A, Au R, Kreger BE, Splansky GL, Kurth T, Kiel DP, Lu KP, Seshadri S, Wolf PA. Inverse association between cancer and Alzheimer’s disease: results from the Framingham Heart Study. BMJ. 2012 Mar 12;344(mar12 1):e1442–e1442.
- Okereke OI, Meadows ME. More Evidence of an Inverse Association Between Cancer and Alzheimer Disease. JAMA Netw Open. 2019 June 21;2(6):e196167. [CrossRef]
- Mezencev R, Chernoff YO. Risk of Alzheimer’s Disease in Cancer Patients: Analysis of Mortality Data from the US SEER Population-Based Registries. Cancers. 2020 Mar 26;12(4):796. [CrossRef]
- Nandakumar S, Rozich E, Buttitta L. Cell Cycle Re-entry in the Nervous System: From Polyploidy to Neurodegeneration. Front Cell Dev Biol. 2021 June 24;9:698661. [CrossRef]
- Seo J, Park M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell Mol Life Sci. 2020 July;77(14):2659–80. [CrossRef]
- Stevenson M, Algarzae NK, Moussa C. Tyrosine kinases: multifaceted receptors at the intersection of several neurodegenerative disease-associated processes. Front Dement. 2024 Aug 16;3:1458038.
- Valiukas Z, Tangalakis K, Apostolopoulos V, Feehan J. Microglial activation states and their implications for Alzheimer’s Disease. J Prev Alzheimers Dis. 2025 Jan;12(1):100013. [CrossRef]
- Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015 Feb;14(2):130–46.
- Gérard C, Goldbeter A. Temporal self-organization of the cyclin/Cdk network driving the mammalian cell cycle. Proc Natl Acad Sci. 2009 Dec 22;106(51):21643–8. [CrossRef]
- Denechaud M, Geurs S, Comptdaer T, Bégard S, Garcia-Núñez A, Pechereau LA, Bouillet T, Vermeiren Y, De Deyn PP, Perbet R, Deramecourt V, Maurage CA, Vanderhaegen M, Vanuytven S, Lefebvre B, Bogaert E, Déglon N, Voet T, Colin M, Buée L, Dermaut B, Galas MC. Tau promotes oxidative stress-associated cycling neurons in S phase as a pro-survival mechanism: Possible implication for Alzheimer’s disease. Prog Neurobiol. 2023 Apr;223:102386.
- Newton TM, Duce JA, Bayle ED. The proteostasis network provides targets for neurodegeneration. Br J Pharmacol. 2019 Sept;176(18):3508–14.
- Wellman R, Jacobson D, Secrier M, Labbadia J. Distinct patterns of proteostasis network gene expression are associated with different prognoses in melanoma patients. Sci Rep. 2024 Jan 2;14(1):198. [CrossRef]
- Tseng CS, Chao YW, Liu YH, Huang YS, Chao HW. Dysregulated proteostasis network in neuronal diseases. Front Cell Dev Biol. 2023 Feb 24;11:1075215. [CrossRef]
- Cai Z, Zhou Y, Xiao M, Yan LJ, He W. Activation of mTOR: a culprit of Alzheimer’s disease? Neuropsychiatr Dis Treat. 2015 Apr;1015.
- Panwar V, Singh A, Bhatt M, Tonk RK, Azizov S, Raza AS, Sengupta S, Kumar D, Garg M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther. 2023 Oct 2;8(1):375.
- Xie PL, Zheng MY, Han R, Chen WX, Mao JH. Pharmacological mTOR inhibitors in ameliorating Alzheimer’s disease: current review and perspectives. Front Pharmacol. 2024 May 30;15:1366061. [CrossRef]
- De D, Mukherjee I, Guha S, Paidi RK, Chakrabarti S, Biswas SC, Bhattacharyya SN. Rheb-mTOR activation rescues Aβ-induced cognitive impairment and memory function by restoring miR-146 activity in glial cells. Mol Ther - Nucleic Acids. 2021 June;24:868–87.
- Davoody S, Asgari Taei A, Khodabakhsh P, Dargahi L. mTOR signaling and Alzheimer’s disease: What we know and where we are? CNS Neurosci Ther. 2024 Apr;30(4):e14463. [CrossRef]
- Alzheimer’s: Novel cancer drug restores cognition, memory loss in mice [Internet]. 2024 [cited 2025 Sept 3]. Available from: https://www.medicalnewstoday.com/articles/cancer-drug-targets-brain-metabolism-alzheimers-treatment-study.
- Minhas PS, Jones JR, Latif-Hernandez A, Sugiura Y, Durairaj AS, Wang Q, Mhatre SD, Uenaka T, Crapser J, Conley T, Ennerfelt H, Jung YJ, Liu L, Prasad P, Jenkins BC, Ay YA, Matrongolo M, Goodman R, Newmeyer T, Heard K, Kang A, Wilson EN, Yang T, Ullian EM, Serrano GE, Beach TG, Wernig M, Rabinowitz JD, Suematsu M, Longo FM, McReynolds MR, Gage FH, Andreasson KI. Restoring hippocampal glucose metabolism rescues cognition across Alzheimer’s disease pathologies. Science. 2024 Aug 23;385(6711):eabm6131.
- Murata MM, Kong X, Moncada E, Chen Y, Imamura H, Wang P, Berns MW, Yokomori K, Digman MA. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol Biol Cell. 2019 Sept 15;30(20):2584–97. [CrossRef]
- Salech F, Ponce DP, Paula-Lima AC, SanMartin CD, Behrens MI. Nicotinamide, a Poly [ADP-Ribose] Polymerase 1 (PARP-1) Inhibitor, as an Adjunctive Therapy for the Treatment of Alzheimer’s Disease. Front Aging Neurosci. 2020 Aug 13;12:255.
- Gupta R, Kumar P. Computational Analysis Indicates That PARP1 Acts as a Histone Deacetylases Interactor Sharing Common Lysine Residues for Acetylation, Ubiquitination, and SUMOylation in Alzheimer’s and Parkinson’s Disease. ACS Omega. 2021 Mar 2;6(8):5739–53.
- Ispirjan M, Marx S, Freund E, Fleck SK, Baldauf J, Roessler K, Schroeder HWS, Bekeschus S. Markers of tumor-associated macrophages and microglia exhibit high intratumoral heterogeneity in human glioblastoma tissue. Oncoimmunology. 13(1):2425124. [CrossRef]
- Wang G, Zhong K, Wang Z, Zhang Z, Tang X, Tong A, Zhou L. Tumor-associated microglia and macrophages in glioblastoma: From basic insights to therapeutic opportunities. Front Immunol. 2022 July 27;13:964898. [CrossRef]
- Zhao W, Zhang Z, Xie M, Ding F, Zheng X, Sun S, Du J. Exploring tumor-associated macrophages in glioblastoma: from diversity to therapy. Npj Precis Oncol. 2025 May 2;9(1):126. [CrossRef]
- Park T, Chang L, Chung SW, Lee S, Bae S, Condello C, Kim YH, Lee J, Kim HJ, Kwon HK, Suh M. Targeting glial PD-1/PD-L1 restores microglial homeostasis and attenuates neuronal hyperactivity in an Alzheimer’s disease model [Internet]. bioRxiv; 2025 [cited 2025 Sept 3]. p. 2024.09.13.612389. Available from: https://www.biorxiv.org/content/10.1101/2024.09.13.612389v2.
- Liu Y, Wu J, Najem H, Lin Y, Pang L, Khan F, Zhou F, Ali H, Heimberger AB, Chen P. Dual targeting macrophages and microglia is a therapeutic vulnerability in models of PTEN-deficient glioblastoma. J Clin Invest [Internet]. 2024 Nov 15 [cited 2025 Sept 3];134(22). Available from: https://www.jci.org/articles/view/178628.
- Rosenzweig N, Dvir-Szternfeld R, Tsitsou-Kampeli A, Keren-Shaul H, Ben-Yehuda H, Weill-Raynal P, Cahalon L, Kertser A, Baruch K, Amit I, Weiner A, Schwartz M. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat Commun. 2019 Jan 28;10(1):465. [CrossRef]
- Mortada I, Farah R, Nabha S, Ojcius DM, Fares Y, Almawi WY, Sadier NS. Immunotherapies for Neurodegenerative Diseases. Front Neurol. 2021 June 7;12:654739.
- Immunotherapy of brain cancers and neurological disorders - MedCrave online [Internet]. [cited 2025 Sept 3]. Available from: https://medcraveonline.com/JCPCR/immunotherapy-of-brain-cancers-and-neurological-disorders.html.
- The Role of Glial Cell Senescence in Alzheimer’s Disease - Alshaebi - 2025 - Journal of Neurochemistry - Wiley Online Library [Internet]. [cited 2025 Sept 3]. [CrossRef]
- Han X, Zhang T, Liu H, Mi Y, Gou X. Astrocyte Senescence and Alzheimer’s Disease: A Review. Front Aging Neurosci. 2020 June 9;12:148. [CrossRef]
- Behfar Q, Ramirez Zuniga A, Martino-Adami PV. Aging, Senescence, and Dementia. J Prev Alzheimers Dis. 2022 July 1;9(3):523–31.
- Hudson HR, Sun X, Orr ME. Senescent brain cell types in Alzheimer’s disease: Pathological mechanisms and therapeutic opportunities. Neurotherapeutics. 2025 Apr 1;22(3):e00519. [CrossRef]
- Franco A de O, Schlickmann TH, Ferrari-Souza JP, de Bastiani MA, Castilhos RM, Zimmer ER. Senescence-associated secretory phenotype (SASP) index in individuals across the Alzheimer’s disease continuum. Alzheimers Dement. 2024;20(S2):e092727. [CrossRef]
- Yang J, Zhou Y, Wang T, Li N, Chao Y, Gao S, Zhang Q, Wu S, Zhao L, Dong X. A multi-omics study to monitor senescence-associated secretory phenotypes of Alzheimer’s disease. Ann Clin Transl Neurol. 2024 Apr 11;11(5):1310–24.
- Barrio-Alonso E, Hernández-Vivanco A, Walton CC, Perea G, Frade JM. Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci Rep. 2018 Sept 25;8(1):14316.
- Luo W, Rodina A, Chiosis G. Heat shock protein 90: translation from cancer to Alzheimer’s disease treatment? BMC Neurosci. 2008 Dec;9(S2):S7.
- Li VOK, Han Y, Kaistha T, Zhang Q, Downey J, Gozes I, Lam JCK. DeepDrug as an expert guided and AI driven drug repurposing methodology for selecting the lead combination of drugs for Alzheimer’s disease. Sci Rep. 2025 Jan 15;15(1):2093. [CrossRef]
- Das V, Miller JH, Alladi CG, Annadurai N, De Sanctis JB, Hrubá L, Hajdúch M. Antineoplastics for treating Alzheimer’s disease and dementia: Evidence from preclinical and observational studies. Med Res Rev. 2024 Sept;44(5):2078–111. [CrossRef]
- Ancidoni A, Bacigalupo I, Remoli G, Lacorte E, Piscopo P, Sarti G, Corbo M, Vanacore N, Canevelli M. Anticancer drugs repurposed for Alzheimer’s disease: a systematic review. Alzheimers Res Ther. 2021 May 5;13:96. [CrossRef]
- Eshraghi M, Ahmadi M, Afshar S, Lorzadeh S, Adlimoghaddam A, Rezvani Jalal N, West R, Dastghaib S, Igder S, Torshizi SRN, Mahmoodzadeh A, Mokarram P, Madrakian T, Albensi BC, Łos MJ, Ghavami S, Pecic S. Enhancing autophagy in Alzheimer’s disease through drug repositioning. Pharmacol Ther. 2022 Sept 1;237:108171. [CrossRef]
- Masitinib for the Treatment of Alzheimer’s Disease: Neurodegenerative Disease Management: Vol 11, No 4 [Internet]. [cited 2025 Sept 3]. [CrossRef]
- Jordan S. Open Label Study for the Use of Tyrosine Kinase Inhibitors for Treatment of Cognitive Decline Due to Degenerative Dementias [Internet]. clinicaltrials.gov; 2022 Sept [cited 2025 Sept 3]. Report No.: NCT02921477. Available from: https://clinicaltrials.gov/study/NCT02921477.
- AB Science. A Multicenter, Double-blind, Placebo-controlled, Randomized, Parallel-group Phase 3 Study to Evaluate the Safety and Efficacy of Masitinib in Patients With Mild to Moderate Alzheimer's Disease [Internet]. clinicaltrials.gov; 2023 Sept [cited 2025 Sept 3]. Report No.: NCT01872598. Available from: https://clinicaltrials.gov/study/NCT01872598.
- Dubois B, López-Arrieta J, Lipschitz S, Doskas T, Spiru L, Moroz S, Venger O, Vermersch P, Moussy A, Mansfield CD, Hermine O, Tsolaki M, AB09004 Study Group Investigators. Masitinib for mild-to-moderate Alzheimer’s disease: results from a randomized, placebo-controlled, phase 3, clinical trial. Alzheimers Res Ther. 2023 Feb 28;15(1):39.
- Shafei MA, Harris M, Conway ME. Divergent Metabolic Regulation of Autophagy and mTORC1—Early Events in Alzheimer’s Disease? Front Aging Neurosci [Internet]. 2017 June 2 [cited 2025 Sept 3];9. Available from: https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2017.00173/full.
- Xie PL, Zheng MY, Han R, Chen WX, Mao JH. Pharmacological mTOR inhibitors in ameliorating Alzheimer’s disease: current review and perspectives. Front Pharmacol [Internet]. 2024 May 30 [cited 2025 Sept 3];15. Available from: https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1366061/full. [CrossRef]
- Subramanian A, Tamilanban T, Alsayari A, Ramachawolran G, Wong LS, Sekar M, Gan SH, Subramaniyan V, Chinni SV, Izzati Mat Rani NN, Suryadevara N, Wahab S. Trilateral association of autophagy, mTOR and Alzheimer’s disease: Potential pathway in the development for Alzheimer’s disease therapy. Front Pharmacol. 2022 Dec 22;13:1094351. [CrossRef]
- Zhang W, Xu C, Sun J, Shen HM, Wang J, Yang C. Impairment of the autophagy–lysosomal pathway in Alzheimer’s diseases: Pathogenic mechanisms and therapeutic potential. Acta Pharm Sin B. 2022 Mar 1;12(3):1019–40. [CrossRef]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-β Levels in a Mouse Model of Alzheimer’s Disease. PLOS ONE. 2010 Apr 1;5(4):e9979.
- Tousi B. The emerging role of bexarotene in the treatment of Alzheimer’s disease: current evidence. Neuropsychiatr Dis Treat. 2015 Feb 5;11:311–5.
- Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, Joshi A, Pontecorvo M, Devous M, Tang A, Bena J. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res Ther. 2016 Jan 29;8:4.
- The Cleveland Clinic. A Double Blind Placebo Controlled Randomized Study to Evaluate the Efficacy and Safety of Bexarotene in Patients With Mild to Moderate Alzheimer's Disease [Internet]. clinicaltrials.gov; 2016 Feb [cited 2025 Sept 3]. Report No.: NCT01782742. Available from: https://clinicaltrials.gov/study/NCT01782742.
- Johnson LA, Macauley SL. Alzheimer’s and metabolism wed with IDO1. Science. 2024 Aug 23;385(6711):826–7.
- Crunkhorn S. Targeting the kynurenine pathway. Nat Rev Drug Discov. 2024 Sept 27;23(11):816–816.
- Duan Z, Shi L, He ZNT, Kuang C, Han T, Yang Q. The Protective Effect of IDO1 Inhibition in Aβ-Treated Neurons and APP/PS1 Mice. Am J Alzheimers Dis Other Demen. 2023 Nov 9;38:15333175231214861. [CrossRef]
- INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology | ACS Medicinal Chemistry Letters [Internet]. [cited 2025 Sept 3]. Available from: https://pubs.acs.org/doi/10.1021/acsmedchemlett.6b00391.
- Jiang K, Wang Q, Chen XL, Wang X, Gu X, Feng S, Wu J, Shang H, Ba X, Zhang Y, Tang K. Nanodelivery Optimization of IDO1 Inhibitors in Tumor Immunotherapy: Challenges and Strategies. Int J Nanomedicine. 2024 Aug 28;19:8847–82. [CrossRef]
- Jiang S, Li H, Piao L, Jin Z, Liu J, Chen S, Liu LL, Shao Y, Zhong S, Wu B, Li W, Ren J, Zhang Y, Wang H, Jin R. Computational study on new natural compound inhibitors of indoleamine 2,3-dioxygenase 1. Aging. 2020 June 22;12(12):11349–63. [CrossRef]
- Savonije K, Meek A, Weaver DF. Indoleamine 2,3-Dioxygenase as a Therapeutic Target for Alzheimer’s Disease and Geriatric Depression. Brain Sci. 2023 May 24;13(6):852. [CrossRef]
- Li Y, Serras CP, Blumenfeld J, Xie M, Hao Y, Deng E, Chun YY, Holtzman J, An A, Yoon SY, Tang X, Rao A, Woldemariam S, Tang A, Zhang A, Simms J, Lo I, Oskotsky T, Keiser MJ, Huang Y, Sirota M. Data-driven discovery of cell-type-directed network-correcting combination therapy for Alzheimer’s disease. bioRxiv. 2024 Dec 13;2024.12.09.627436.
- Li Y, Pereda Serras C, Blumenfeld J, Xie M, Hao Y, Deng E, Chun YY, Holtzman J, An A, Yoon SY, Tang X, Rao A, Woldemariam S, Tang A, Zhang A, Simms J, Lo I, Oskotsky T, Keiser MJ, Huang Y, Sirota M. Cell-type-directed network-correcting combination therapy for Alzheimer’s disease. Cell. 2025 July 15;S0092-8674(25)00737-8. [CrossRef]
- Alzheimer’s Mouse Model Shows Memory Boost from Anticancer Drug Combo [Internet]. [cited 2025 Sept 3]. Available from: https://www.genengnews.com/topics/drug-discovery/alzheimers-mouse-model-shows-memory-boost-from-anticancer-drug-combo/.
- Do These Two Cancer Drugs Have What It Takes to Beat Alzheimer’s? | UC San Francisco [Internet]. [cited 2025 Sept 3]. Available from: https://www.ucsf.edu/news/2025/07/430386/do-these-two-cancer-drugs-have-what-it-takes-beat-alzheimers.
- Alzheimer’s treatment: Cancer drug combo shows promise in mice [Internet]. [cited 2025 Sept 3]. Available from: https://www.medicalnewstoday.com/articles/might-a-combination-of-2-cancer-drugs-help-treat-alzheimers-disease.
- Cancer Drugs Could Treat Alzheimer’s: Drug Repurposing Win [Internet]. Medscape. [cited 2025 Sept 3]. Available from: https://www.medscape.com/viewarticle/cancer-drugs-could-treat-alzheimers-drug-repurposing-win-2025a1000kv4.
- Ordaz DA, Gupta K, Bota DA. The role of Poly-ADP ribose polymerase (PARP) enzymes in chemotherapy-induced cognitive impairments – parallels with other neurodegenerative disorders. Front Pharmacol. 2025 June 9;16:1615843. [CrossRef]
- Mao K, Zhang G. The role of PARP1 in neurodegenerative diseases and aging. FEBS J. 2022;289(8):2013–24. [CrossRef]
- Zeng J, Libien J, Shaik F, Wolk J, Hernández AI. Nucleolar PARP-1 Expression Is Decreased in Alzheimer’s Disease: Consequences for Epigenetic Regulation of rDNA and Cognition. Neural Plast. 2016;2016:8987928. [CrossRef]
- Martire S, Mosca L, d’Erme M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech Ageing Dev. 2015 Mar 1;146–148:53–64.
- Hu ML, Pan YR, Yong YY, Liu Y, Yu L, Qin DL, Qiao G, Law BYK, Wu JM, Zhou XG, Wu AG. Poly (ADP-ribose) polymerase 1 and neurodegenerative diseases: Past, present, and future. Ageing Res Rev. 2023 Nov 1;91:102078. [CrossRef]
- Abeti R, Abramov AY, Duchen MR. β-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain. 2011 June 1;134(6):1658–72. [CrossRef]
- Shukla S, Tekwani BL. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front Pharmacol [Internet]. 2020 Apr 24 [cited 2025 Sept 3];11. Available from: https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2020.00537/full.
- Joshi N, Khadse P, Jadhav S, Alavala RR. Histone Deacetylase Inhibition in Alzheimer’s Disease: Molecular Mechanisms, Therapeutic Potential, and Future Perspectives. Curr Top Med Chem. 2025 June 16; [CrossRef]
- Xu K, Dai XL, Huang HC, Jiang ZF. Targeting HDACs: A Promising Therapy for Alzheimer’s Disease. Oxid Med Cell Longev. 2011;2011:143269. [CrossRef]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of Class 1 Histone Deacetylases Reverse Contextual Memory Deficits in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology. 2010 Mar;35(4):870–80.
- Pereira M, Cruz MT, Fortuna A, Bicker J. Restoring the epigenome in Alzheimer’s disease: advancing HDAC inhibitors as therapeutic agents. Drug Discov Today. 2024 July 1;29(7):104052. [CrossRef]
- Zhang LY, Zhang SY, Wen R, Zhang TN, Yang N. Role of histone deacetylases and their inhibitors in neurological diseases. Pharmacol Res. 2024 Oct 1;208:107410. [CrossRef]
- Kummer MP, Ising C, Kummer C, Sarlus H, Griep A, Vieira-Saecker A, Schwartz S, Halle A, Brückner M, Händler K, Schultze JL, Beyer M, Latz E, Heneka MT. Microglial PD-1 stimulation by astrocytic PD-L1 suppresses neuroinflammation and Alzheimer’s disease pathology. EMBO J. 2021 Dec 15;40(24):e108662.
- Manenti S, Orrico M, Masciocchi S, Mandelli A, Finardi A, Furlan R. PD-1/PD-L Axis in Neuroinflammation: New Insights. Front Neurol [Internet]. 2022 June 9 [cited 2025 Sept 3];13. Available from: https://www.frontiersin.org/journals/neurology/articles/10.3389/fneur.2022.877936/full. [CrossRef]
- Linnerbauer M, Beyer T, Nirschl L, Farrenkopf D, Lößlein L, Vandrey O, Peter A, Tsaktanis T, Kebir H, Laplaud D, Oellinger R, Engleitner T, Alvarez JI, Rad R, Korn T, Hemmer B, Quintana FJ, Rothhammer V. PD-L1 positive astrocytes attenuate inflammatory functions of PD-1 positive microglia in models of autoimmune neuroinflammation. Nat Commun. 2023 Sept 9;14(1):5555. [CrossRef]
- Ito T, Vien PTX, Inaba M, Yoshimura H, Hotta M, Nakanishi T, Fujita S, Nakaya A, Satake A, Ishii K, Nomura S. Immunomodulatory Drugs (IMiDs), Lenalidomide and Pomalidomide, Suppress Th1-Inducing Capacity of Dendritic Cells but Enhance Th2-Mediated Allergic Response. Blood. 2016 Dec 2;128(22):2520. [CrossRef]
- Kotla V, Goel S, Nischal S, Heuck C, Vivek K, Das B, Verma A. Mechanism of action of lenalidomide in hematological malignancies. J Hematol OncolJ Hematol Oncol. 2009 Dec;2(1):36. [CrossRef]
- Decourt B, Wilson J, Ritter A, Dardis C, DiFilippo FP, Zhuang X, Cordes D, Lee G, Fulkerson ND, St Rose T, Hartley K, Sabbagh MN. MCLENA-1: A Phase II Clinical Trial for the Assessment of Safety, Tolerability, and Efficacy of Lenalidomide in Patients with Mild Cognitive Impairment Due to Alzheimer’s Disease. Open Access J Clin Trials. 2020;12:1–13. [CrossRef]
- Deng MY, Ahmad KA, Han QQ, Wang ZY, Shoaib RM, Li XY, Wang YX. Thalidomide alleviates neuropathic pain through microglial IL-10/β-endorphin signaling pathway. Biochem Pharmacol. 2021 Oct;192:114727. [CrossRef]
- Shortt J, Hsu AK, Johnstone RW. Thalidomide-analogue biology: immunological, molecular and epigenetic targets in cancer therapy. Oncogene. 2013 Sept;32(36):4191–202.
- Das U, Banerjee S, Sarkar M, Muhammad L F, Soni TK, Saha M, Pradhan G, Chatterjee B. Circular RNA vaccines: Pioneering the next-gen cancer immunotherapy. Cancer Pathog Ther. 2024 Dec;S2949713224000892. [CrossRef]
- Das U, Banerjee S, Sarkar M. Bibliometric analysis of circular RNA cancer vaccines and their emerging impact. Vacunas. 2025 Mar;500391.
- Team TB. Repurposing Cancer Drugs for Alzheimer’s Disease | Rutgers BHI [Internet]. BHI. 2025 [cited 2025 Sept 10]. Available from: https://brainhealthinstitute.rutgers.edu/2025/08/07/repurposing-cancer-drugs-for-alzheimers-insights/.
- Malesu VK. Dual cancer drugs restore memory and rewire brain cells in Alzheimer’s mouse models [Internet]. News-Medical. 2025 [cited 2025 Sept 10]. Available from: https://www.news-medical.net/news/20250722/Dual-cancer-drugs-restore-memory-and-rewire-brain-cells-in-Alzheimere28099s-mouse-models.aspx.
- Ganamurali N, Sabarathinam S. Network pharmacology-guided systems biology reveals β-Sitosterol’s multi-target role in reversing 7-ketocholesterol-induced oxidative and inflammatory stress. J Steroid Biochem Mol Biol. 2026 Jan;255:106863. [CrossRef]
- Ou YN, Tan L, Yu JT. Identification of Novel Drug Targets for Alzheimer’s Disease by Integrating Genetics and Proteomes from Brain and Blood (P7-9.012). Neurology. 2024 Apr 14;102(7_supplement_1):107.
- Podder A, Pandit M, Narayanan L. Drug Target Prioritization for Alzheimer’s Disease Using Protein Interaction Network Analysis. OMICS J Integr Biol. 2018 Oct;22(10):665–77.
- Funari A, De Smaele E, Paci P, Fiscon G. Prioritizing repurposable drugs for Alzheimer’s disease using network-based analysis with concurrent assessment of Long QT syndrome risk. Biotechnol Rep. 2025 July 29;47:e00909. [CrossRef]
- Peng Y, Yuan M, Xin J, Liu X, Wang J. Screening novel drug candidates for Alzheimer’s disease by an integrated network and transcriptome analysis. Bioinformatics. 2020 Nov 1;36(17):4626–32.
- Advani D, Kumar P. Therapeutic Targeting of Repurposed Anticancer Drugs in Alzheimer’s Disease: Using the Multiomics Approach. ACS Omega. 2021 May 19;6(21):13870–87. [CrossRef]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019 June;570(7761):332–7.
- Lin H, Su L, Mao D, Yang G, Huang Q, Lan Y, Zeng J, Song W, Liang G, Wei Q, Zou D, Li R, Zou C. Identification of altered immune cell types and molecular mechanisms in Alzheimer’s disease progression by single-cell RNA sequencing. Front Aging Neurosci [Internet]. 2024 Nov 14 [cited 2025 Sept 11];16. Available from: https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2024.1477327/full. [CrossRef]
- Das U. Emerging trends and research landscape of the tumor microenvironment in head-and-neck cancer: A comprehensive bibliometric analysis. Cancer Plus. 2025 Apr 8;0(0):025060008.
- Cai Y, Kanyo J, Wilson R, Bathla S, Cardozo PL, Tong L, Qin S, Fuentes LA, Pinheiro-de-Sousa I, Huynh T, Sun L, Mansuri MS, Tian Z, Gan HR, Braker A, Trinh HK, Huttner A, Lam TT, Petsalaki E, Brennand KJ, Nairn AC, Grutzendler J. Subcellular proteomics and iPSC modeling uncover reversible mechanisms of axonal pathology in Alzheimer’s disease. Nat Aging. 2025 Mar;5(3):504–27.
- Penney J, Ralvenius WT, Tsai LH. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol Psychiatry. 2020 Jan;25(1):148–67. [CrossRef]
- Pharmacokinetic (PK) Considerations For Central Nervous System (CNS) Drugs - Featured Stories - News [Internet]. [cited 2025 Sept 11]. Available from: https://www.prisysbiotech.com/news/pharmacokinetic-pk-considerations-for-centra-81209739.html.
- Caban A, Pisarczyk K, Kopacz K, Kapuśniak A, Toumi M, Rémuzat C, Kornfeld A. Filling the gap in CNS drug development: evaluation of the role of drug repurposing. J Mark Access Health Policy. 2017 Apr 10;5(1):1299833. [CrossRef]
- Pascoal TA, Aguzzoli CS, Lussier FZ, Crivelli L, Suemoto CK, Fortea J, Rosa-Neto P, Zimmer ER, Ferreira PCL, Bellaver B. Insights into the use of biomarkers in clinical trials in Alzheimer’s disease. eBioMedicine [Internet]. 2024 Oct 1 [cited 2025 Sept 11];108. Available from: https://www.thelancet.com/journals/ebiom/article/PIIS2352-3964(24)00358-X/fulltext. [CrossRef]
- Cummings J. The Role of Biomarkers in Alzheimer’s Disease Drug Development. Adv Exp Med Biol. 2019;1118:29–61.
- AB Science. A Multicenter, Double-blind, Placebo-controlled, Randomized, Parallel-group Study to Evaluate the Efficacy of Oral AB1010 in Adults Patients With Mild to Moderate Alzheimer-type Disease. [Internet]. clinicaltrials.gov; 2018 Dec [cited 2025 Sept 11]. Report No.: NCT00976118. Available from: https://clinicaltrials.gov/study/NCT00976118.
- Gonzales MM, Garbarino VR, Kautz TF, Song X, Lopez-Cruzan M, Linehan L, Van Skike CE, De Erausquin GA, Galvan V, Orr ME, Musi N, He Y, Bateman RJ, Wang CP, Seshadri S, Kraig E, Kellogg D. Rapamycin treatment for Alzheimer’s disease and related dementias: a pilot phase 1 clinical trial. Commun Med. 2025 May 20;5(1):189. [CrossRef]
- Trial | NCT04200911 [Internet]. [cited 2025 Sept 11]. Available from: https://cdek.pharmacy.purdue.edu/trial/NCT04200911/.
- Ghosal K, Haag M, Verghese PB, West T, Veenstra T, Braunstein JB, Bateman RJ, Holtzman DM, Landreth GE. A randomized controlled study to evaluate the effect of bexarotene on amyloid-β and apolipoprotein E metabolism in healthy subjects. Alzheimers Dement Transl Res Clin Interv. 2016 June;2(2):110–20. [CrossRef]
- Gonzales M. Pilot Study to Investigate the Safety and Feasibility of Senolytic Therapy to Modulate Progression of Alzheimer's Disease (SToMP-AD) [Internet]. clinicaltrials.gov; 2023 Feb [cited 2025 Sept 11]. Report No.: NCT04063124. Available from: https://clinicaltrials.gov/study/NCT04063124.
- Maiese K. Targeting molecules to medicine with mTOR, autophagy and neurodegenerative disorders. Br J Clin Pharmacol. 2016 Nov;82(5):1245–66. [CrossRef]
- Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement N Y N. 2021;7(1):e12179.
- Folch J, Petrov D, Ettcheto M, Pedrós I, Abad S, Beas-Zarate C, Lazarowski A, Marin M, Olloquequi J, Auladell C, Camins A. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev Neurother. 2015 June;15(6):587–96. [CrossRef]
- New AD drug development pipeline report is out. 2025 Aug 31 [cited 2025 Sept 11]; Available from: https://www.alzheimer-europe.org/news/new-ad-drug-development-pipeline-report-out?language_content_entity=en.
- Alzheimer’s Drug Development Pipeline: Promising Therapies, Pharma Investment Drive Momentum in Clinical Trials | UNLV [Internet]. 2023 [cited 2025 Sept 11]. Available from: https://www.unlv.edu/news/release/alzheimers-drug-development-pipeline-promising-therapies-pharma-investment-drive.
- van Bokhoven P, de Wilde A, Vermunt L, Leferink PS, Heetveld S, Cummings J, Scheltens P, Vijverberg EGB. The Alzheimer’s disease drug development landscape. Alzheimers Res Ther. 2021 Nov 11;13:186. [CrossRef]
- Mahdavi KD, Jordan SE, Barrows HR, Pravdic M, Habelhah B, Evans NE, Blades RB, Iovine JJ, Becerra SA, Steiner RA, Chang M, Kesari S, Bystritsky A, O’Connor E, Gross H, Pereles FS, Whitney M, Kuhn T. Treatment of Dementia With Bosutinib. Neurol Clin Pract. 2021 June;11(3):e294–302.
- Newton W. Alzheimer’s disease drug development: three major questions for 2023 [Internet]. Clinical Trials Arena. 2022 [cited 2025 Sept 11]. Available from: https://www.clinicaltrialsarena.com/features/alzheimers-2023/.
- Das U. Transforming Precision Medicine through Generative AI: Advanced Architectures and Tailored Therapeutic Design for Patient-Specific Drug Discovery. ChemistrySelect. 2025 Sept;10(36):e02448.
- Rajendran A, Rajan RA, Balasubramaniyam S, Elumalai K. Nano delivery systems in stem cell therapy: Transforming regenerative medicine and overcoming clinical challenges. Nano TransMed. 2025 Dec;4:100069. [CrossRef]
Table 1.
Shared biological hallmarks of cancer and Alzheimer’s disease, and their therapeutic implications.
Table 1.
Shared biological hallmarks of cancer and Alzheimer’s disease, and their therapeutic implications.
| Hallmark | Cancer Context | AD Context | Therapeutic Implications |
|---|---|---|---|
| Cell cycle control | Loss of cell-cycle checkpoints, uncontrolled proliferation | Aberrant re-entry of neurons into S-phase, leading to apoptosis [62] | Inhibitors of cell-cycle kinases (e.g. CDK/Abl inhibitors) |
| Proteostasis/chaperones | High demand on chaperones (HSPs) & UPS to stabilize mutated oncoproteins | Impaired UPS/autophagy, protein aggregates (Aβ, tau). Chaperones (Hsp90) stabilize toxic species [63] | HSP90 inhibitors, proteasome modulators, autophagy enhancers |
| Autophagy/mTOR pathway | mTORC1 often overactive (promoting growth), variable autophagy | mTORC1 hyperactivation blocks autophagy, contributing to tau/Aβ accumulation [40] | mTOR inhibitors (rapamycin/rapalogs) to restore autophagy |
| Metabolism (Warburg effect) | Aerobic glycolysis in tumors; high glutamine use | Neuronal glucose hypometabolism; astrocytic glycolysis deficits; tryptophan→kynurenine shift [44] | IDO1 inhibitors (e.g. epacadostat) to restore glucose metabolism |
| DNA damage/repair | DNA repair machinery often mutated; PARP inhibitors exploit BRCA-deficiency | Accumulation of DNA damage; PARP1 hyperactivation depletes NAD+ [64] | PARP inhibitors (e.g. niraparib) – potential neuroprotection, but needs caution |
| Chronic inflammation | Tumor microenvironment: TAMs, Tregs, immunosuppression | Microglial/astrocytic activation, elevated cytokines (IL-1β, TNFα); complement activation | Immune checkpoint blockade (PD-1/PD-L1), anti-inflammatory agents |
| Cellular senescence/SASP | Oncogenic stress leads to senescence; SASP fuels tumor progression | Age-induced senescence of neurons/glia; SASP factors contribute to neurodegeneration | Senolytic drugs (dasatinib+quercetin) to clear senescent cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



