Submitted:
15 September 2025
Posted:
15 September 2025
You are already at the latest version
Abstract
Dysregulated redox signalling, mitochondrial dysfunction and impaired autophagy are interconnected mechanisms that orchestrate inflammatory and immune responses during cardiovascular disease progression. Inflammation is largely modulated by altered redox signalling, which primarily involves reactive oxygen and nitrogen species (ROS/RNS). Mitochondria are essential for energy production and cellular homeostasis, but their dysfunction leads to the accumulation of excessive ROS, which triggers inflammation. This pro-oxidative milieu disrupts immune regulation by activating inflammasomes, promoting cytokine secretion, triggering immune cell infiltration and ultimately contributing to cardiovascular injury. Conversely, intracellular degradation processes such as mitophagy, alleviates these effects by selectively eliminating dysfunctional mitochondria, thereby decreasing ROS levels and maintaining immune homoeostasis. These interconnected processes influence myeloid cell function including macrophage polarization, dendritic cell activation, and neutrophil activity. The modulation of these immune responses is crucial for determining the severity and resolution of cardiac and vascular inflammation, and consequently the extent of cellular injury. This review examines the latest developments and understanding of the intricate relationships between redox signalling, mitochondrial dysfunction, autophagy and oxidative stress in modulating inflammation and immune responses in cardiovascular diseases. Understanding these interrelationships will inform future studies and therapeutic solutions for the prevention and treatment of cardiovascular diseases.
Keywords:
redox signalling
; oxidative stress
; mitochondrial dysfunction
; autophagy
; inflammation
; cardiovascular disease
1. Introduction
Cardiovascular disease (CVD) remains a major public health crisis worldwide, and its prevalence is expected to rise over the next few decades [1,2] A substantial risk of CVD-related mortality persists despite current conventional therapies, such as statins, angiotensin II converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), calcium channel blockers, beta-blockers, and lifestyle modifications, particularly in patients with obesity, diabetes, and chronic inflammation. The global number of cardiovascular deaths is estimated to increase from 20.5 million in 2025 to approximately 35.6 million by 2050 [2]. Conventional treatments primarily manage the symptoms instead of targeting the underlying cellular mechanisms that drive the onset and progression of CVD. Thus, there is an urgent need for more effective strategies for the management of CVD, to improve the quality of life, and reduce the burden on the healthcare system [3,4]
The hypothesis that oxidative stress contributes to CVD has gained traction ever since its first proposal by Daniel Steinberg and colleagues as a modifier of low-density lipoproteins (LDL) [5]. In the mid to late 1990s, oxidative stress became a mainstream mechanism in CVD research, however, clinical trials using antioxidants like vitamin E failed to show benefit [6,7,8]. These studies highlighted the complexity of redox biology.
The term redox is derived from the combination of “reduction” and “oxidation,” which defines the chemical processes associated with the transfer of electrons between reactants in chemical reactions [9,10]. Reactive oxygen species (ROS) are highly reactive molecular oxygen derivatives endogenously generated as a byproduct of cellular respiration [11]. ROS, including superoxide (O2•−), singlet oxygen (1O2), hydrogen peroxide (H2O2), and hydroxyl radicals (.OH), are synthesised through redox processes [10,12]. H2O2 is recognised as a major ROS that reversibly oxidizes critical redox-sensitive cysteine residues on target proteins [11,13]. Various signal transduction pathways are regulated by ROS either via direct modification of proteins or lipids, or by the coordinated transfer of electrons between molecules, forming a chain of redox reactions [14]. Redox biology regulates numerous crucial physiological processes, such as insulin signalling [11], regulation of vascular tone [15] and immunometabolism [16].
Extensive investigations have shown that redox imbalance, mitochondrial dysfunction, impaired autophagy, and unresolved inflammation are pivotal contributors to vascular and myocardial damage [17-20]. Dysregulated redox biology facilitates persistent ROS generation and pathological feedback loops. Therefore, a co[17–20mprehensive understanding of the contribution of redox signalling to cellular and molecular determinants of the various CVDs is critically needed for the development of novel therapeutic strategies.
Nonspecific antioxidants have not shown significant improvements in CVD outcomes in clinical trials [21,22]. These antioxidant approaches have largely failed due to lack of specificity, inability to target the main sources of ROS, and overlook the physiological roles of ROS in signalling and defence. Indeed, redox signalling is increasingly emerging as a pivotal player in metabolism and physiological processes, acting as a key mediator in the dynamic interactions between organisms and the external environment. A more nuanced approach to antioxidant interventions is needed, one that supports essential physiological redox processes yet affords protection against the onset and development of CVDs [10].
This review focuses on intracellular redox signalling and highlights the implications of redox imbalance in the pathophysiology of CVDs. Specifically, we focus on the root cause of redox imbalance, such as dysfunctional mitochondria, impaired endogenous antioxidant systems or impaired autophagy, and highlight their intricate interactions to discover novel therapeutic strategies. Furthermore, we highlight the complexity of redox signalling in CVDs with a focus on the spatial and temporal aspects of ROS signalling. We highlight how ROS that originate from different cellular compartments, or temporal fluctuations in ROS, may have different effects on redox-sensitive transcription factors and organelle crosstalk. Non-canonical redox modifications, such as S-glutathionylation, S-nitrosylation and redox-phosphorylation, remain an active area of investigation and are discussed in relation to their role in modulating key pathways such as mitochondrial metabolism, autophagy and inflammation. Additionally, we address the bidirectional nature of redox interactions. Providing detailed insight into these aspects is essential for advancing our understanding of redox-mediated CVDs and developing effective mechanism-based therapeutic strategies.
2. Mechanism of Intracellular ROS Generation
Mitochondrial complex I (NADH: ubiquinone oxidoreductase) and III (ubiquinol: cytochrome c oxidoreductase) are considered the major sources of mitochondrial ROS (mtROS) produced by the electron transport chain [23]. These complexes situated within the mitochondrial intermembrane space, generate O2•− and H2O2 from molecular oxygen [23,24]. Mitochondria-localized proteins, such as NADPH oxidase-4 (NOX4), p66shc, monoamine oxidase (MAO)-A and MAO-B have also been implicated in mitochondrial ROS production [23]. In response to stress and p66Shc activation, cytochrome c in the inner mitochondrial membrane generates H2O2 [25], which exacerbates pro-apoptotic ROS signalling and mitochondrial dysfunction to drive a variety of cardiovascular pathologies [26]. While the primary sites and mechanisms of mitochondrial ROS generation are well characterized, their regulation and relative contributions to disease, and the physiological significance of each site, are not yet clearly understood.
Cytosolic ROS (cytoROS) also play a crucial role in modulating numerous cellular signalling networks, whilst aberrant cytoROS disturb signalling pathways thereby promoting pathophysiological changes [27]. The NOXs are a family of transmembrane enzymes involved in generating cytoROS. NADPH oxidase-2 was the first source of ROS identified in macrophages and is the canonical isoform in this cell type [24]. NOXs facilitate the generation of superoxide by transferring a single electron from NADPH to oxygen [28]. Superoxide can be further converted to H2O2 either through spontaneous dismutation or by the activity of superoxide dismutase (SOD) [24]. In addition, xanthine metabolism, specifically through the enzyme xanthine oxidase (XO), produces H2O2 and O2•− in the cytoplasm.
Among the seven distinct isoforms of NOX, NOX-1, 2, 4, and 5 are expressed throughout the cardiovascular system [27,29,30]. Emerging data have revealed the crucial role of NOX2-derived O2•− as signalling molecules in autophagy [27,31]. Notably, a study reported that NOX2-derived ROS present in LC3-associated phagosomes promoted oxidative inactivation of the autophagic protease ATG4B, thereby regulating its stability and function [31]. Similarly, in palmitate-treated H9C2 cardiomyocytes and in the hearts of mice fed high-fat-diets, activation of NOX2 enhanced O2•− production, contributing to the inhibition of lysosomal enzymes and autophagosome turnover [29], suggesting that NOX-derived ROS play an important role in redox-dependent regulation of autophagy. Thus, modulating NOX activity and redox signalling to promote autophagy may offer a therapeutic avenue to restore cellular homeostasis and combat pathological remodelling of the heart.
Other sources of ROS include the endoplasmic reticulum (ER), peroxisomes and enzymes such as xanthine oxidase. The ‘redox triangle,’ formed by mitochondria, peroxisomes and the ER, acts as a central hub for redox signalling [32]. Excessive ROS within the redox triangle affects ER-mitochondria Ca2+ exchange, oxidative phosphorylation and protein folding within the ER [32]. However, the role of ROS is context-dependent and varies according to the cellular environment, compartmentalisation, exposure period and concentration [33]. In the human myocardium, the mitochondrial electron transport chain, NOX, xanthine oxidoreductase (XOR) and dysfunctional nitric oxide synthases (NOS) are the major sources of ROS [34].
3. Cellular Antioxidant System
The antioxidant system is a highly coordinated defence network that provides protection from oxidative damage caused by ROS and other free radicals. Enzymatic and non-enzymatic antioxidants work synergistically to maintain redox balance and cellular component integrity by modulating gene expression and associated signalling pathways. Thus, antioxidant therapeutics could provide an effective approach to preventing and treating many diseases where redox imbalance is a key pathological component, such as atherosclerosis, hypertension, ischemia–reperfusion injury, and diabetic cardiomyopathy (DCM) [35,36]. The major enzymatic antioxidants include SOD, catalase (CAT), and the glutathione peroxidases (GPx), with glutathione reductase (GR), peroxiredoxins (Prx) and the thioredoxins (TrxR) also maintaining the balance between oxidants and antioxidants. SOD catalyzes the dismutation of O2•− into H2O2 and molecular oxygen, while catalase converts H2O2 into water and oxygen. In turn, GPx reduces hydrogen peroxide and lipid peroxides using glutathione as a substrate [37]. The thioredoxin-peroxiredoxin system detoxifies H2O2 and organic hydroperoxides by transferring reducing equivalents from NADPH, via TrxR reductase and TrxR, to Prx. The non-enzymatic antioxidants such as glutathione (GSH), and vitamins C and E, actively scavenge free radicals and help regenerate oxidized antioxidants back to their active forms [37,38].
The transcription factor, nuclear factor erythroid 2–related factor 2 (Nrf2), acts as a central player in the regulation of the antioxidant system. Upon activation by oxidative stress, Nrf2 translocates to the nucleus and binds to antioxidant response elements (AREs) of a range of genes, enhancing the expression of numerous cytoprotective enzymes such as SOD, CAT, peroxiredoxins (Prx), heme oxygenase-1 (HO-1) and heat shock protein 70 (Hsp70) [39,40]. Together, these antioxidant defence systems help prevent oxidative stress-induced damage to DNA, proteins and lipids, and modulate redox sensitive signalling pathways involved in cell survival, inflammation, and metabolism. The production of excessive ROS and the cellular antioxidant defence system required for cellular homeostasis is illustrated in Figure 1. Disruptions to this system lead to oxidative stress, contributing to the pathogenesis of various diseases, including CVDs.
4. Physiological Role of ROS, Redox Signalling and Redox Homeostasis
Redox signalling involves the specific and usually reversible oxidation/reduction modification of molecules involved in cellular signalling pathways [10,41], consequently turning on or off various pathways [42]. At low to moderate levels, ROS such as O2•−, •OH and H₂O₂ contribute to normal cellular functions including proliferation, differentiation, migration and immune responses. Moderately increased levels of mitochondrial oxidants enhance systemic defences by inducing adaptive responses [43]. This is referred to as mitohormesis, the process where mitochondria signal in response to transient stress and activate adaptive cellular responses that increase cell survival, function and longevity [21,43,44]. Mitohormesis is increasingly viewed as an important aspect of normal physiology and a critical modulator of disease processes [44].
Physiological levels of H2O2 are in the range of 10 to 100 nanomolar (nM) [45]. H2O2 serves as a classical intracellular signalling molecule, modulating kinase-driven pathways at lower physiological levels [46]. The physiological steady state levels of H2O2 are controlled by balancing H2O2 production and scavenging by antioxidant enzymes such as CAT and GPx [13]. Understanding the physiological function of ROS, and the importance of maintaining redox homeostasis, is critical for distinguishing beneficial signalling from pathological oxidative stress in the context of CVDs.
The antioxidant system scavenges excess ROS and ensures that redox signalling remains within the physiological range. Redox homeostasis refers to the precise balance between the generation of ROS and antioxidant activity. When this balance is regulated, redox signalling sustains the normal function of cells and tissues. However, excessive ROS production or impaired antioxidant responses lead to persistent redox imbalance, resulting in oxidative stress. This perturbs cellular homoeostasis damages biomolecules such as proteins, lipids and DNA, and leads to the pathogenesis of various diseases, including CVDs.
5. Dysregulated Redox Biology: A Molecular Link to Inflammatory Pathways
Dysregulated redox biology refers to disruptions in the intricate network of redox reactions within a biological system, resulting in persistent oxidative stress and altered redox signalling [9,10]. In the context of cardiovascular diseases, disruptions in redox homeostasis drive inflammatory and immune responses that accelerate the activation and progression of CVDs. Broadly speaking, redox-mediated processes include the dysregulation of the endothelium, enhanced pyroptosis and inflammation, immune cell infiltration, cardiomyocyte injury and hypertrophy, and cellular proliferation, leading to tissue remodelling that ultimately contributes to CV dysfunction and disease progression (Figure 2) [47,48,49]. More specifically, dysregulated redox biology contributes to defective mitochondrial and autophagy pathways that amplify inflammatory signalling cascades such as Mitogen-Activated Protein Kinases (MAPKs), Nuclear factor kappa B (NF-κB) and the NLRP3 (Nucleotide-Binding Domain, Leucine-Rich–Containing Family, Pyrin Domain–Containing-3) inflammasome, whilst suppressing cytoprotective mechanisms including Nrf2-mediated antioxidant gene expression [27,50,51,52]. Multiple signalling cascades are activated or suppressed by interconnected redox-inflammatory regulators.
Evidence for the interconnectedness between redox biology and inflammation comes from various in vitro and in vivo studies including the following. Immunological signalling, including via the Toll-like receptor (TLR) and NLRP3 inflammasome assembly, has been shown to require transient ROS generation before initiation of downstream signalling pathways [53,54]. Furthermore, ROS have been identified as regulators of inflammasome assembly. For example, inhibition of mitophagy leads to the accumulation of damaged and impaired mitochondria, which exacerbates ROS generation, and consequently triggers NLRP3 inflammasome activation [55,56]. NLRP3 inflammasome activation leads to the production of Interleukin-1β (IL-1β), which further induces the secretion of interleukin-6 (IL-6) and is implicated in the chronic inflammation and progression of CVD [57,58]. A recent study reported that IL-6 facilitates mtROS production and reduces nitric oxide (NO) bioavailability in human aortic endothelial cells, contributing to the development of endothelial dysfunction [59].
Growth factor stimulation has been shown to activate P13K signalling via a redox-sensitive mechanism [49]. Tu, et al demonstrated that oxidative stress activates P13K and increases the activity of p70 S6 kinase-1, leading to enlargement of cardiomyocytes [60]. In vascular smooth muscle cells (VSMC), the ROS-sensitive kinase, p38 MAPK, and its substrate MAPKAPK-2, have been shown to mediate Akt activation, which contributes to VSMC hypertrophy [61].
Data from our laboratory demonstrated that dh404, a bardoxolone derivative and novel Nrf2 activator, ameliorates endothelial dysfunction in diabetic Akita mice by activating Nrf2, upregulating antioxidant enzymes, reducing ROS, and inhibiting redox-sensitive inflammatory pathways. In diabetic human aortic endothelial cell (HAECs), dh404 showed cytoprotective effects by significantly inhibiting inflammatory genes (VCAM-1 and the p65 subunit of NF-κB) and upregulating the Nrf2-responsive genes, NAD(P)H quinone oxidoreductase 1 (NQO1) and heme oxygenase-1 (HO-1), whilst decreasing the oxidative stress marker, nitrotyrosine and the ROS, O2•−and H2O2. In diabetic mice, dh404 decreased contraction in response to phenylephrine and suppressed the expression of inflammatory genes, including VCAM-1, ICAM-1, p65, IL-1β, as well as pro-oxidant genes, Nox1 and Nox2 [52]. We also showed that dh404 reduces inflammation and atherosclerosis in diabetic ApoE-/- mice [62]. Our data therefore highlight the interconnectedness between dysregulated redox pathways and inflammation, and suggest that specific targeted anti-oxidant therapy lessens CV burden via improvements in oxidative stress and inflammation.
In summary, accumulating evidence suggest that cellular redox imbalance plays a crucial role in driving a cascade of redox-sensitive signalling events and inflammatory pathways. This exacerbates cellular and tissue damage, ultimately leading to the development and progression of various CVDs. Advancing our understanding of how disrupted redox signalling exacerbates inflammatory and immune responses may assist in the discovery of novel therapeutic approaches to restore redox balance and regulate inflammation-associated pathologies.
6. The crosstalk Between Redox Signalling and Mitochondrial Function
Mitochondria are double membrane-bound organelles that are known to generate most of the energy needed to power biochemical reactions of the cell [63]. In 1966, Jensen initially reported that the mitochondrial respiratory chain generates ROS [64,65]. It was later established that H2O2 is produced from the dismutation of O2•− in the mitochondria [65,66,67]. Mitochondria constitute approximately 30-40% of the cardiomyocyte cell volume and play a crucial role in meeting the high metabolic and energy demand by primarily generating ATP through oxidative phosphorylation (OxPhos) [68]. A growing body of literature now supports the notion that mitochondria are both a major source and the target of ROS, positioning them at the centre of vital redox signalling networks. The bidirectional interaction between mitochondrial function and redox homeostasis forms a complex axis that regulates energy production, survival and stress responses. Disruptions of the interplay between mitochondrial function and redox homeostasis may contribute to the pathogenesis of numerous diseases, including cardiovascular and metabolic disorders [63,69].
Mitochondrial dysfunction leads to the dysregulation of mitochondrial dynamics, mitochondrial DNA (mtDNA) damage and impaired mitophagy [63]. Dysfunctional mitochondria also contribute to inflammation and an impaired immune response [70]. Dysfunctional mitochondria affect calcium homeostasis and cardiac energy supply, which causes changes in cardiac structure and function [63]. Therefore, dysfunctional mitochondria are associated with many cardiovascular diseases, including atherosclerosis, heart failure and myocardial infarction [20,63,71,72].
Accumulating evidence suggest that mtROS function as downstream effector molecules. mtROS can modulate various signalling pathways such as modulation of hypoxic signalling [73,74], cytosolic stress kinases [75], and activation of autophagy [76], thereby influencing cell metabolism and immune responses. In particular, mitochondrial oxidative stress has been shown to directly impact the IKKβ–RelA (NF-κB) pathway. Indeed, mitochondrial oxidative stress led to increased monocyte infiltration and exacerbated inflammatory responses in western diet fed Ldlr-/- mice. Conversely, decreasing mitochondrial stress in macrophages alleviated atherosclerosis by reducing monocyte infiltration and lesional inflammation in a mCAT transgenic (mCAT) Ldlr-/- mouse model of atherosclerosis [50]. Furthermore, NF-κB-induced oxidative stress contributed to mitochondrial and cardiac dysfunction in obese db/db mice, a model of type II diabetes. Notably, inhibition of NF-κB by an NF-κB inhibitor, pyrrolidine dithiocarbamate, reduced oxidative stress, restored mitochondrial integrity by decreasing ROS and increasing ATP synthesis, consequently improving cardiac function [77].
Mitochondrial ROS also cross-talk with the NLRP3-inflammasome to drive inflammatory responses. In addition to a direct effect on the activation of the NLRP3 inflammasome [78], a recent study demonstrated that cardiomyocyte-specific knockdown of a protein involved in autophagic flux, ATP6AP2, led to autophagy inhibition and activation of the NLRP3, further promoting maladaptive cardiac remodelling. In contrast, suppression of cellular and mitochondrial ROS in shR-ATP6AP2-transfected cardiomyocytes partially reversed NLRP3 upregulation, and mitigated mitochondrial impairment and dysfunction [76]. Thus, cellular and mitochondrial ROS promote activation of the NLRP3 inflammasome, which may contribute to cardiac dysfunction.
Mitochondrial ROS also act as upstream signals that promote Nrf2 activation by disrupting its interaction with KEAP1, thereby facilitating its nuclear translocation and transcriptional activation of antioxidant genes. A recent study by Luo et al demonstrated that in oxidized low-density lipoprotein (ox-LDL) injured macrophages, micheliolide (MCL), an active metabolite of parthenolide, reduced both total and mtROS level, increased SOD activity, improved mitochondrial function, modulated antioxidant responses and importantly, reduced atherosclerosis. Mechanistically, MCL binds to the Arg483 site of KEAP1, enhancing Nrf2 nuclear translocation and upregulating the transcription of GPX4 and xCT. These findings suggest that MCL ameliorates atherosclerosis by activating the Nrf2 signaling pathway and thereby reducing oxidative stress and the inflammatory response [79]. Furthermore, mtROS play a bidirectional role in regulating mitochondrial dynamics via modulation of mitochondrial fission and fusion, while these processes also influence mtROS production [27].
Excessive ROS can enhance mitochondrial fission by activating the major pro-fission protein dynamin-related protein 1 (DRP1) [63]. ROS-induced post-translational modifications such as phosphorylation, SUMOylation, S-nitrosylation, and O-GlcNAcylation play an important role in DRP1 activation [80,81]. Cytosolic DRP1 is recruited to mitochondrial membranes following post-translational modifications and interacts with the outer mitochondrial membrane protein Fis1 to initiate mitochondrial fission [80]. Additionally, three crucial GTPase proteins, Mitofusins 1 (MFN1), Mitofusins 2 (MFN2) on the outer membrane and atrophy 1 (OPA1) on the inner membrane mediate mitochondrial fusion [63]. Oxidative stress can inhibit mitochondrial fusion by impairing the function of key fusion proteins. In H9c2 cardiomyoblasts, H2O2-mediated oxidative stress disrupts OPA1-mediated mitochondrial dynamics via activation of OMA1, a key protease responsible for cleavage of OPA1, implicating a crucial role of ROS in mitochondrial dynamics [82]. Inhibition of mitochondrial fission promotes accumulation of dysfunctional mitochondria, which further exacerbate ROS generation. Similarly, impaired mitochondrial fusion in endothelial cells enhances superoxide production, which leads to atherosclerosis progression [83], highlighting the complex bidirectional link of ROS and mitochondrial dynamics and function.
In addition, mtROS play a role in mediating lytic cell death via oxidation of the pore forming protein, GSDMD, thereby promoting pyroptosis of macrophages [84,85]. The Regulator-Rag complex, a mediator of mTOR activities, has been shown to be involved in GSDMD pore formation and pyroptosis in macrophages [86]. The Regulator-Rag complex regulates mTORC1-dependent events to promote oligomerization of GSDMD and pore formation in the membrane by a mtROS-mediated process. However, the exact mechanism by which mtROS affects GSDMD oligomerization is not clearly understood [86]. Redox-regulation of proteins can be mediated by direct modification of thiol-containing amino acid residues such as cysteines [10]. Devant et al demonstrated that ROS enhances GSDMD activities through oxidative modification of multiple cysteine residues, with cysteine 192 (Cys192) being necessary and sufficient for ROS-mediated GSDMD pore formation and pyroptosis [85]. Thus, mtROS can activate the NLRP3-dependent pyroptosis pathway by inducing the oxidation of GSDMD, which damages cardiomyocytes and myocardial tissue, leading to various cardiovascular conditions, including cardiac hypertrophy, atherosclerosis and myocardial reperfusion injury [87,88,89].
Furthermore, oxidative post translational modification (Ox-PTM) of mitochondrial proteins can modulate ATP synthesis, electron transport efficiency and calcium handling [90,91]. For example, in the failing heart, ATP synthase undergoes oxidative modification at multiple cysteine residues via disulfide bond formation, S-glutathionylation and S-nitrosation. It has been shown that Cys294 of the ATP synthase α subunit acts as a redox switch that senses cellular redox status and modulates ATP synthase activity [91]. Importantly, cardiac resynchronization therapy has been shown to restore ATP synthase function, partially by reversing oxidative modifications on cysteine residues [91].
Redox signalling can also influence mitochondrial structure and function by regulating Ca2+ flux. Mitochondrial Ca²⁺ homeostasis is primarily maintained by Ca²⁺ influx into the matrix via the mitochondrial calcium uniporter (MCU), whilst the main efflux process is mediated by the Na⁺/Ca²⁺ exchanger (NCX) [92,93]. Recent studies demonstrated that redox modification of MICU3 regulates mitochondrial calcium influx [10,69,94]. Patron et al reported that the novel tissue-specific MCU modulator, MICU3, forms a disulfide bond with MICU1 at the Cys515 residue, which stimulates mitochondrial Ca2+ uptake [10,94]. Another study found that oxidation of MCU at cysteine 97 (Cys-97) also increased MCU activity. Cysteine 97 is a conserved thiol residue in human MCU, and has been shown to undergo S-glutathionylation, thereby increasing MCU activity [93,95]. This oxidative modification of MCU further enhances mtROS production, disrupts cellular bioenergetics, and sensitizes cells to mitochondrial calcium [Ca2+]m overload-induced cell death [95]. Thus, redox modifications directly regulate Ca2+ homeostasis, which can impact the development and progression of CVDs. Therefore, exploring the underlying mechanisms by which dysfunctional mitochondria contribute to oxidative imbalance within the cell, and how the redox-sensitive targets modulate mitochondrial function, may provide critical insights to discover more effective therapeutic targets for CVDs.
7. The Crosstalk Between Redox Signalling and Autophagy
The autophagy-lysosome system is a highly conserved cellular process that degrades damaged cellular content and maintains homeostasis [96,97]. The autophagy-lysosomal process is involved in three main types of autophagy: microautophagy, chaperone-mediated autophagy and macroautophagy. These processes provide the cell with a flexible degradative toolkit for different conditions. Microautophagy degrades cytoplasmic material through direct lysosomal membrane invaginations, while chaperone-mediated autophagy selectively transports proteins bearing a KFERQ motif across the lysosomal membrane via LAMP-2A [98,99]. Macroautophagy, commonly referred to as autophagy, involves the engulfment of cytoplasmic components within double-membrane vesicles called autophagosomes, which then fuse with the lysosome [97].Canonical autophagy comprises several sequential steps, mediated by an intricate interplay of multiple proteins and lipids derived from various membrane sources, including the endoplasmic reticulum, ER/mitochondria contact sites, the Golgi apparatus, recycling endosomes and the cell membrane [97]. More than 32 related proteins are associated with the autophagosome before fusion to the lysosome [100]. Mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) are important regulators of autophagy [101,102]. Under nutrient-rich conditions, active mTORC1 phosphorylates the ULK1 complex, specifically targeting ULK1 and ATG13, which is essential for autophagy initiation [102]. Conversely, under stress or low nutrient conditions, AMPK is activated, which promotes autophagy by direct phosphorylation of ULK1 and inhibition of mTORC1 activity. Downstream, proteins such as Beclin-1, part of the PI3K complex, drive phagophore nucleation, the process that initiates formation of autophagosome. Microtubule-associated protein 1A/1B-light chain 3 (LC3) is converted to LC3-II through the lipidation process driven by the action of ATG7 and ATG3. LC3-II anchors to the autophagosomal membrane, allowing it to facilitate cargo recruitment and enhance autophagosome formation [103]. These autophagosomes subsequently fuse with lysosomes, which allow lysosomal enzymes to recycle the sequestered material.
Under normal physiological conditions, macroautophagy plays a crucial role in cell survival and homeostasis. However, dysfunctional autophagy is associated with many diseases, including cardiovascular and metabolic diseases [104,105,106]. Autophagy can be activated by amino acid starvation, reduced insulin levels, and reduced ATP availability [107]. Excessive accumulation of ROS modulates autophagy via multiple pathways, including activation of AMPK signalling and the ULK1 complex, inhibition of Bcl-2/Beclin-1 and mTOR signalling, oxidation of Atg proteins and ultimately, perturbation of mitochondrial homeostasis thereby triggering mitophagy and degradation of dysfunctional mitochondria [108]. The redox-mediated regulation of autophagy facilitates cellular adaptations to stress and plays a key role in the inflammatory response, metabolic balance and cardiovascular pathophysiology [109,110].
Several studies report that AMPK is a highly conserved master regulator of metabolism, and plays a crucial role in regulating autophagy, particularly under oxidative and energy stress [111,112,113]. During nutrient deprivation and oxidative stress, the Atg1/ULK1 complex works as a key initiator of autophagy by receiving signals from the upstream sensors mTOR and AMPK, and directing them to the downstream autophagy effectors [114,115]. In a recent study, Tabata et al revealed the mechanism by which the ULK1 complex targets autophagosome formation and regulates autophagy initiation. They found that zinc finger DHHC type palmitoyltransferase 13 (ZDHHC13), palmitoylates ULK1 during autophagy induction, and enhances downstream events such as phosphorylation of Atg14L [115]. Notably, palmitoylation of ULK1 occurs specifically at cysteine residues, Cys927 and Cys1003 [115]. AMPK phosphorylates ULK1 and activates its function [111,113]. AMPK has also been found to de-repress the ULK1 complex by phosphorylating and inhibiting the mTOR complex [111].
Importantly, ROS regulate autophagy in a complex and context–dependent manner to either activate or suppress autophagy via multiple signalling pathways [113,116]. Excessive ROS typically induce autophagy through inhibition of mTORC1. However under certain conditions, ROS enhance mTORC1 activity and subsequently inhibit autophagy. Accumulation of H₂O₂ can induce autophagy by activating AMPK and inhibiting the mTORC1 signalling pathway [117]. Additionally, excessive ROS can activate multiple important transcription factors such as hypoxia-inducible factor-1α (HIF-1α), NRF2, p53 and forkhead box O-3 (FoxO3) which can activate the transcription of autophagy related genes including SQSTM1, LC3, and the mitophagy-associated genes BNIP3 and NIX [118,119]. In addition to activating upstream signalling pathways, redox stress can also directly modify key autophagy–related proteins such as ATG4, Beclin-1 and p62/SQSTM1 via oxidative posttranslational modifications, consequently affecting the efficiency and specificity of the autophagic process. For example, ATG4, a cysteine protease that processes LC3, is reversibly inhibited by ROS, acting as a redox-sensitive switch to regulate formation of the autophagosome [120,121]. Interestingly, under metabolic or oxidative stress conditions, phosphorylation of Bcl-2 at serine 70 (Ser70) disrupts its interaction with Beclin-1, enabling Beclin-1 to activate autophagy [122,123]. Redox modifications, particularly oxidation of the autophagy receptor p62, can also affect its oligomerization and cargo recognition ability, impacting selective autophagy [124]. Carrol et al., found that two oxidation-sensitive cysteine residues, C105 and C113, in the autophagy receptor SQSTM1/p62, facilitate the activation of pro-survival autophagy under stress [124]. These reversible redox modifications facilitate fine-tuning of the autophagic machinery in response to the changing redox state of the cell, thereby modulating autophagy. In addition to this, autophagy has been found to indirectly regulate ROS by p62-mediated selective degradation of Keap1, which results in the release and activation of Nrf2, and upregulation of antioxidant target genes, thereby reducing ROS levels [108,113]. A classic activator of NRF2, tBHQ, has been found to attenuate oxidative stress and suppress VSMCs calcification by inducing NRF2 nuclear translocation and increasing P62 and KEAP1 expression [125].
Autophagy, in turn, maintains redox homeostasis by eliminating ROS-producing dysfunctional mitochondria (via mitophagy) and degrading oxidized proteins and lipids, thereby maintaining mitochondrial functional integrity and cellular homeostasis [126,127]. This intricate interplay of autophagy and redox signalling limits oxidative damage in cells and tissues. A better understanding of the mechanism of autophagy in various diseases is crucial for therapeutic target design and the treatment of diseases [127]. Notably, ROS have been found to activate PINK1-Parkin-mediated mitophagy by inducing mitochondrial recruitment of Parkin [127,128]. SIRT3 also plays a key role in activating PINK1/Parkin-mediated mitophagy, by deacetylating PINK1 and Parkin directly or through the transcription factor FOXO3a [129]. Furthermore, the production of localized mtROS during metabolic stress or hypoxia serve as key upstream signalling molecules for the induction of mitophagy [130]. However, under certain conditions, mtROS are also elevated as a result of the induction of mitophagy [130].
From the aforementioned studies, it is clear that redox-dependent autophagic regulation is crucial for the adaptation to cellular stressors to maintain energy balance and quality control of proteins and organelles. In the cardiovascular system, cardiomyocytes and vascular endothelial cells have high metabolic demands and are frequently exposed to oxidative stress. Therefore, a more detailed mechanistic understanding of redox-sensitive checkpoints within the autophagic pathway, particularly in specific pathological contexts, could lead to the development of novel therapies for CVDs.
8. Interconnected Signalling and Feedback Loops: The Redox-Mitochondria-Autophagy-Inflammation Axis
The complex and bidirectional relationship between redox signalling, mitochondrial dysfunction, autophagy, mitophagy and inflammation, is gaining attention particularly in the context of cardiovascular and metabolic diseases. However, the comprehensive understanding of the interconnectedness of these processes in relation to immunometabolic regulation, and the onset and progression of CVDs is not completely understood. These interconnected signalling pathways create complex feedback loops that drive the progression and development of different cardiovascular conditions.
Research has shown that ROS function as important secondary messengers that activate transcription factors such as NF-ĸB and AP-1, promoting secretion of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α. These cytokines, in turn, trigger more ROS production by activating the NOX family of enzymes and disrupting mitochondrial electron transport, thereby facilitating a positive feedback loop between redox imbalance and inflammation. Similarly, increased ROS levels contribute to mitochondrial dysfunction, while impaired mitochondrial activity is associated with excessive ROS generation [131]. In particular, excess ROS disrupt mitochondrial integrity by damaging mitochondrial membranes, DNA and proteins, whereas impaired mitochondria become the main sources of further ROS production, triggering cellular stress and promoting damage to cardiomyocytes [131,132]. Furthermore, impaired autophagy is commonly observed in ageing and metabolic syndromes where damaged mitochondria accumulate, leading to persistent ROS production and chronic inflammation [133,134].
One of the major players regulating the cellular stress response is sirtuin1 (SIRT1), a class III histone deacetylase. SIRT1 is considered a crucial regulator of oxidative stress and has been shown to play a role in modulating CVDs, including atherosclerosis, myocardial infarction and heart failure [135,136,137]. SIRT1 mitigates inflammation by deacetylating NF-ĸB, p53, and PGC-1α during metabolic perturbations [138,139,140]. In oleic acid-treated vascular smooth muscle cells (VSMCs), SIRT1 deacetylases PGC-1α, restores mitochondrial dysfunction, and improves mitochondrial membrane potential [141]. SIRT1 promotes mitochondrial biogenesis, enhances the autophagy process, and reduces oxidative stress, thus it plays a crucial role in modulating redox-mediated cellular processes.
A further key regulator of this process is p62/SQSTM1. Multiple studies have demonstrated that p62/SQSTM1 sits at the intricate nexus of redox signalling, mitochondrial quality control, autophagy, and the inflammatory response [142,143,144,145]. However, it plays a context-dependent role in cellular homeostasis, acting both as a marker of impaired autophagy and as a mediator of protective responses. Specifically, p62 is an autophagic receptor/adaptor protein that shuttles damaged cargo into the autophagosome, but its accumulation reflects impaired autophagic flux and is typically associated with adverse cellular outcomes. With respect to complications of CVD, a recent study revealed that p62/SQSTM1 regulates oxidative and ER stress, and inflammation following cerebral I/R injury, with elevated p62 levels being associated with worse stroke outcomes. Mechanistically, the ZZ domain of p62 was shown to mediate dysregulated autophagy and cell death through the binding of specific substrates, especially those containing an N-terminal arginine (Nt-R). This interaction initiated p62 oligomerization, subsequent autophagosome formation, and yet the degradation of cargo was dysregulated [146]. In addition, Quan et al. found that p62 increased mitochondrial ROS in a NOX-independent manner in HEK293T cells after an I/R exposure. Importantly, accumulation of p62 under impaired autophagy conditions leads to prolonged inflammatory activation [147]. This underscores its importance as a therapeutic target in inflammation-driven cardiovascular diseases.
p62 has also been shown to mediate mitophagy by binding to ubiquitinated outer mitochondrial membrane proteins and recruiting autophagic machinery, thus degrading damaged and dysfunctional mitochondria and limiting the production of mitochondrial ROS [127,148]. Another study demonstrated that SQSTM1/p62 positively regulates mtDNA expression and mitochondrial OXPHOS [144]. In addition, SQSTM1/p62 induced the expression of mitochondrial ribosomal protein MRPL12 by activating p38/ATF2 signalling pathway, suggesting a new regulatory axis [144]. Thus, given its context-dependent roles in regulating autophagy, oxidative stress responses and inflammation, targeting p62 may offer a novel approach for CVD prevention, however, therapeutic strategies will need to carefully balance its protective functions with potential risks associated with p62 accumulation and impaired autophagic flux.
P66Shc, a key adapter protein, is yet another key regulator of oxidative stress with an impact on inflammatory outcomes. P66Shc controls the progression of various cardiac pathologies, including endothelial dysfunction, coronary artery disease (CAD), ischemia /reperfusion injuries, and cardiomyopathy [26]. P66Shc has been shown to regulate cardiac dysfunction and oxidative stress in a mouse model of pressure overload-induced heart failure (TAC model), with SOD and phosphodiesterase 5(PDE5) acting as downstream effectors of this pathway [149]. P66Shc is phosphorylated under oxidative stress and translocates to the mitochondria, where it enhances H2O2 generation and promotes mitochondrial permeability transition, thereby contributing to oxidative damage, apoptotic signalling, exacerbation of the inflammatory cascade and cellular dysfunction [150,151,152].
Importantly, these studies show the complex crosstalk between the pathways regulating redox biology, mitochondrial homeostasis, autophagy and inflammation, and how crosstalk may amplify pathological signalling. This challenges therapeutic targeting, since targeting one pathway may inadvertently affect others. However, by identifying and modulating key regulatory hubs within this network, such as Nrf2, the NOX enzymes, or mitochondrial quality control proteins, novel therapeutic strategies could restore multiple dysfunctional pathways, leading to broader cardioprotection.
9. Interplay of Autophagy, Mitochondrial Dysfunction and Cellular Redox States in the Context of CVDs
The intricate crosstalk between autophagy, mitochondrial dysfunction, ROS production and elevated inflammation is now increasingly being recognised as central to CVD progression (Figure 3). Clinically, these processes underpin key features of cardiovascular pathology, including endothelial dysfunction, cardiomyocyte death and adverse cardiac remodelling. The following section highlights the direct involvement of these interconnected pathways in the pathogenesis of atherosclerosis, cardiac hypertrophy, ischemia/reperfusion injury, heart failure and diabetic cardiomyopathy, and how an understanding of their interconnectivity might inform newer therapeutics to lessen disease burden.
9.1. Atherosclerosis
Atherosclerosis is a chronic immunometabolic disease that develops at multiple locations within the arterial system, and is the primary cause of CVD, including coronary artery disease (CAD), stroke, myocardial infarction (MI) and peripheral artery disease. It is also a major contributor to heart failure, especially heart failure with reduced ejection fraction (HFrEF) after MI. At a cellular level, endothelial cells, vascular smooth muscle cells and leukocytes, contribute to the development and progression of atherosclerotic lesions [153,154]. In response to a pro-atherogenic environment, an inflamed vascular endothelium attracts monocytes into the sub endothelial space of the vessel wall. It is within this space that monocytes internalize modified low-density lipoprotein (LDL) particles via scavenger receptors such as CD36, scavenger receptor-1 (SR-1) and lectin-like oxidized LDL receptor (LOX-1) to cause a build-up of plaque in the arterial wall [155].
Initial mechanistic investigations highlighted a critical role for ROS in the development of atherosclerosis. This led to the proposal of the oxidative modification hypothesis of atherosclerosis in 1989 [5]. This theory proposed that oxidized LDL (oxLDL) is a key driver of foam cell formation within the arterial wall. Subsequent studies proved that ROS function as mediators of multiple processes of atherosclerosis progression, including endothelial dysfunction, foam cell formation, and plaque destabilisation [156]. In the ensuing years, chronic inflammation was acknowledged as an additional key mediator [157], where an imbalance between pro-inflammatory and anti-inflammatory processes ultimately drive the onset and development of atherosclerotic plaques [158]. Furthermore, the importance of mtROS in mediating atherogenic processes is considered a nuanced advance in our understanding of ROS-mediated atherogenesis [159,160].
Over a decade ago, Wang et al. reported that mitochondrial oxidative stress is associated with the development of human atherosclerosis [50]. The crosstalk between mitochondrial dysfunction, ROS and inflammation in driving atherosclerosis was highlighted in their study by showing that mitochondrial oxidative stress in macrophages enhanced monocyte chemotactic protein-1 (MCP-1) production via the IκB-kinase β (IKKβ)–RelA NF-κB pathway. Notably, suppression of mitochondrial oxidative stress in myeloid cells inhibited early atherogenesis in Ldlr−/− mice overexpressing catalase in their mitochondria [50]. However, how cytoplasmic IKK was activated by mitochondrial oxidative stress remains unclear. More recent studies continue to support the interconnectedness of these pathways and draw attention to the importance of Nox4-driven mitochondrial ROS in pro-inflammatory macrophage reprogramming. For example, Vendrov et al., show that ablation or pharmacological inhibition of Nox4 reduced mitochondrial ROS, skewed macrophages towards a resolving M2 phenotype and attenuated plaque progression in the apolipoprotein E–deficient (ApoE-/-) mice [161].
In another study, Karnewar et al demonstrated that a mitochondria-targeted antioxidant, esculetin (Mito-Esc), significantly prevented atherosclerotic plaque formation, reduced serum pro-inflammatory cytokines and prevented dysregulation of mitochondrial biogenesis in the aorta of ApoE−/− mice. Furthermore, in human aortic endothelial cells and serum from ApoE−/− mice, Mito-Esc activated the metabolic and stress-sensing autophagy regulator, SIRT1, altered miR-19b and miR-30c, and significantly inhibited plasminogen activator inhibitor-1 (PAI-1), a key mediator of atherosclerosis [162]. These data suggest that via targeted reductions in mitochondria-mediated oxidative stress, it is possible to improve mitochondrial dysfunction and augment autophagy to reduce inflammation and cellular damage in CVDs.
Importantly, the interconnectedness of the autophagy and oxidative stress pathways is revealing novel therapeutic opportunities for atherosclerosis therapy. In a study by Xia et al., the significance of targeting AMPK/mTOR dependent autophagy in atherosclerosis was shown in ApoE−/− mice [163]. In these mice, inhibition of autophagy by U0126 resulted in an increase in aortic atherosclerosis with increased necrotic core and foam cell formation. Mechanistically, P62 was shown to accumulate together with a decrease in lactoferrin (LTF), an iron transport protein with anti-inflammatory, antioxidant, and antifibrotic properties, and mainly secreted by neutrophils. A decrease in autophagosomes was also noted, suggesting that autophagy was impaired. Using in vitro cell models, the study also showed that silencing the core autophagy protein, BECN1 or knocking down LTF, increased mTOR phosphorylation, inhibited the expression of LC3 II, and prevented the activation of AMPK, all indications that autophagy was impaired. This study suggests that dysregulated autophagy and high levels of oxidative stress are associated with the development of atherosclerosis, and that lactoferrin therapy might ameliorate atherosclerosis by accelerating the AMPK/mTOR signalling pathway [163]. Thus, advances in our understanding of the complex role of redox signalling can open avenues for novel therapeutic interventions for the treatment of atherosclerosis and aid in the development of strategies to prevent or slow plaque development.
9.2. Pathological Cardiac Hypertrophy
Pathological cardiac hypertrophy arises due to pressure overload, hypertension or aortic stenosis, and is an independent risk factor for cardiovascular diseases. It is a hallmark of heart failure and sudden death, and is typically characterized by an increase in cardiomyocyte size and left ventricular wall thickening [164,165]. A number of studies suggest that multiple signalling mediators contribute to the development of pathological cardiac hypertrophy by disrupting normal cellular functions, including mitochondrial respiration, calcium handling, metabolic regulation and autophagy. This manifests as alterations in oxidative stress and inflammation [165,166]. Indeed, ROS have been shown to play a crucial role in regulating multiple overlapping signalling pathways associated with the development and progression of pathological cardiac hypertrophy [166]. In particular, the role of mtROS and the interplay with a ROS modulator is clearly demonstrated in a recent study by Martens et al. Reactive oxygen species modulator 1 (ROMO1) is an inner mitochondrial membrane protein that influences mitochondrial dynamics and redox signalling. It facilitates ion flux via ion channel formation and affects mitochondrial membrane potential to drive ROS production. It is highly expressed in hypertrophic hearts resulting from transverse aortic constriction (TAC) surgery, and overexpressing ROMO1 is associated with developing hypertrophy in human AC16 cardiomyocytes. Notably, knockdown of ROMO1 markedly reduces ROS production and inhibition of NF-κB activity, suggesting the ROMO1-ROS-NF-κB signalling axis is involved in the regulation of pathological cardiomyocyte hypertrophy [167]. This study clearly highlights the interconnectivity between oxidative and inflammatory stress in pathological hypertrophy, and suggests that redox signalling acts as a central mediator in the development of cardiac hypertrophy. Advancing our knowledge of the critical role of redox signalling in hypertrophy may reveal potential therapeutic targets to prevent maladaptive remodelling and heart failure.
Although a role for autophagy has been implicated in pathological cardiac hypertrophy, conflicting data, as demonstrated below, suggest both a positive and a negative effect of autophagy on disease progression. A recent study demonstrated that solute carrier family 26 member 4 (SLC26A4), also known as pendrin, promotes autophagy and activation of the NLRP3 inflammasome in two cardiac hypertrophy models, the first a mouse model of phenylephrine (PE)-induced cardiomyocyte hypertrophy, and the second, a rat model of transverse aortic constriction (TAC) [168]. The mechanism most likely involves its anion exchange activity that influences cellular stress pathways to promote the development of cardiac hypertrophy. In isolated cardiomyocytes, protein levels of NLRP3 and IL-β were downregulated after treatment with the autophagy inhibitor 3-MA, or after silencing with a sh-lentivirus expressing SLC26A4. These data suggest that SLC26A4 mediates the activation of both autophagy and the NLRP3 inflammasome to promote the progression of cardiac hypertrophy both in vitro and in vivo.
On the other hand, a natural compound, thymoquinone decreased the levels of key hypertrophic markers, ANP and BNP and reduced type1 collagen expression in angiotensin II (AngII)-treated H9C2 cells and TAC mice, consequently mitigating cardiac hypertrophy. Importantly, the mechanism includedactivating adaptive autophagy through the PPAR-γ/14-3-3γ pathway [169]. Additionally, thymoquinone markedly decreased the level of ROS by upregulating NOX4 and SOD2 in both angiotensin II (AngII)-treated H9C2 cells and TAC mice, indicating a crucial role for autophagy and oxidative stress in pathological cardiac hypertrophy [169]. Taken together, these studies reveal that the role of autophagy is highly context-dependent, varying with the cellular environment, therefore targeting impaired autophagy and aberrant inflammasome activation may provide new therapeutic strategies for pathological cardiac hypertrophy. Furthermore, mitochondrial impairment is one of the major drivers of pathological cardiac hypertrophy [167,170,171]. In AngII-treated rat cardiomyocytes, overexpression of the resident mitochondrial protein, SBK3, reduced the level of mtROS and malonaldehyde, a marker of oxidative stress, in cardiomyocytes by increasing SOD2 activity. In addition, SBK3 overexpression restored the expression of mitochondrial dynamics-related proteins, including MFN1 and MFN2. Concurrently, SBK3 overexpression increased ATP production, improved the respiratory and oxygen consumption rate of cardiomyocytes, and consequently improved cardiac hypertrophy by regulating mitochondrial metabolism [171]. In addition, downregulation of the cardiac-specific mitochondrial fission-regulating protein, Drp-1, promotes accumulation of damaged and dysfunctional mitochondria and consequently increases in oxidative stress in the heart during pressure overload-induced cardiac hypertrophy, whereas Tat-Beclin 1 peptide treatment activates mitophagy and restores mitochondrial function thereby alleviating the progression of HF during pressure overload [172]. These data highlight the complex role of mitochondrial dysfunction, autophagy and oxidative stress in cardiac hypertrophy and the progression of heart failure.
9.3. Ischemia-Reperfusion (I/R) Injury
Ischemia-reperfusion (I/R) injury results in cardiac damage and dysfunction, which elevates the risk of heart failure. This occurs when blood flow to the heart is interrupted, resulting in excessive mitochondrial ROS generation upon reperfusion, largely due to the accumulation of TCA cycle intermediates, such as succinate driving HIF-1α mediated ROS production [11]. Myocardial cell death induced by myocardial I/R, plays a central role in the progression of acute myocardial infarction (AMI), mainly via necrosis, apoptosis, and autophagic death [173,174]. Myocardial I/R injury expands the infarct area, contributes to the aggregation of inflammatory cells in the ischemic myocardium, impairs vascular endothelial function, and causes metabolic dysfunction and apoptosis of myocardial cells, all of which exacerbate AMI [174,175].
Multiple studies underscore the protective role of autophagy in the cardiac response to ischemia via removal of damaged mitochondria and the reduction in oxidative stress [176,177]. A study demonstrated that I/R injury of the rat heart promote accumulation of ROS and metabolic dysfunction of mitochondria. In this study, mitochondrial sequesteration by the autophagasome was reduced in I/R rat hearts compared to control hearts, whilst this was improved by corosolic acid treatment. Mechanistically, it could be shown that corosolic acid exerted its protective effects by enhancing mitophagy through the PHB2/PINK1/Parkin signaling pathway, which facilitated elimination of damaged mitochondria, decreased oxidative stress and maintained mitochondrial function, consequently reducing infarct size and improving cardiac function post I/R injury in rats [176].
RhoA, a small G-coupled protein receptor and intracellular signal transducer, has been implicated in cardioprotective mechanisms post I/R injury. Activation of RhoA signaling reduces oxidative stress via suppression of mitochondrial death pathways [178,179,180]. Tu et al demonstrated that activation of RhoA upregulates exogenously expressed PINK1 and Parkin within the mitochondria. RhoA activation increased the level of LC3-II in mitochondria and this increase remained unaffected by Bafilomycin A1 treatment, indicating that RhoA promotes induction of mitophagy rather than affecting lysosomal degradation. This sustains mitochondrial quality control by modulating mitophagy [180]. Thus PINK1-mediated mitophagy contributes to the clearance of impaired mitochondria and safeguards cardiomyocytes from ischemic injury [180]. Another recent example of protection afforded by enhanced mitophagy comes from data investigating the protective effects of two xanthone derivatives isolated from Garcinia bracteata, Gerontoxanthone I (GeX1) and macluraxanthone (McX), that promote the activation of mitophagy through the PINK1-Parkin pathway and reduce the levels of ROS. Consequently, these compounds were shown to reduce injury and cell death of H9c2 cardiomyoblasts [181]. Collectively, these studies highlight the protection afforded by mitophagy against oxidative stress and I/R injury.
In contrast, other studies report that excessive or dysregulated mitophagy may exacerbate injury [182,183,184]. For example, during simulated ischemia reperfusion (SIR) in H9c2 monocytes, mitophagy was highly activated which exacerbated oxidative stress and mitochondrial dysfunction. Treatment with melatonin decreased the levels of mitophagy-associated proteins, including Beclin1, Parkin, Bcl-2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3), and NIX (BNIP3-like (BNIP3L), reduced the levels of ROS and restored mitochondrial function by reducing mitochondrial permeability transition pore (MPTP) opening and suppressing cyclophilin D (CypD) and voltage-dependent anion channel 1 (VDAC1) expression in H9c2 cells. These results suggest that melatonin protects H9c2 cells from SIR-induced injury by inhibiting excessive mitophagy [184]. These differential outcomes may reflect differences in the severity of the insult, cell type, or regulatory pathways involved in mitophagy activation, suggesting that mitophagy plays a context-dependent role in I/R injury.
In addition, there is evidence for a role for autophagy in mediating I/R injury. Depletion of the transcription factor ZNF143 has been shown to improve autophagic flux in myocardial I/R injury and decrease cardiomyocyte death, whereas overexpression of ZNF143 upregulates Raptor expression and inhibits autophagic activity, consequently exacerbating myocardial I/R injury. These data suggest that the regulation of impaired autophagic flux attenuates myocardial I/R injury [185]. Conversely, another study demonstrated that the DEP-domain containing mTOR-interacting protein (Deptor) ameliorates I/R-induced myocardial injury by inhibiting the mTOR pathway and by increasing cardiomyocyte autophagy [174]. Furthermore, in a setting of diabetes, disrupted autophagic flux leads to augmented I/R injury in streptozotocin (STZ)-induced hyperglycaemic mice [186], whilst I/R injury is aggravated in patients with diabetes [187,188].
The rate of ROS generation rapidly increases in the post-ischemic myocardium [189]. ROS generated from multiple sources have been implicated in ischemia–reperfusion injury. Mitochondria-localized circular RNAs (circRNAs), a newly identified class of noncoding RNAs, play an important role in regulating the production of mitochondria-derived ROS in cardiomyocytes [190]. Mitochondria-localized circRNA Samd4 decreases oxidative stress and regulates mitochondrial dynamics by inducing mitochondrial translocation of valosin-containing protein (Vcp) , consequently decreasing voltage-dependent anion channel 1 (Vdac1) expression and inhibiting mitochondrial permeability transition pore (mPTP) opening [190]. These results highlight how a mitochondrial non-coding circRNA regulates mitochondrial function and protects cells from oxidative stress, via a pathway that facilitates protein translocation into the mitochondria to alter gene expression and pore regulation. In addition, overexpression of circSamd4 induces cardiomyocyte proliferation and inhibits cardiomyocyte apoptosis, which results in improved cardiac function after AMI. By modulating circSamd4 or its downstream targets (Vcp or Vdac1), it may be possible to develop therapeutic strategies aimed at enhancing mitochondrial resilience and mitigating cellular damage [190].
Taken together, these studies provide strong evidence for the complex role of redox signalling, mitochondrial dysfunction and autophagy in the pathogenesis of AMI-induced cardiac injury, and suggest that considerations of dose, timing, cell type and disease stage are needed for effective strategies to treat I/R injury.
9.4. Heart Failure
Heart failure (HF), also known as congestive heart failure (CHF), is defined as a complex clinical syndrome where the heart is unable to pump blood effectively due to structural or functional impairments in ventricular filling [191]. HF affects at least 26 million people worldwide and contributes to high mortality and morbidity, poor quality of life, and increased healthcare costs. Increasing evidence suggest a close link between oxidative stress and heart failure [40,192]. In particular, ROS-mediated damage to cellular macromolecules such as lipids, proteins and DNA, lead to cell death and loss of cardiac contractile function [40]. Importantly, electron leakage from dysfunctional mitochondria leads to the formation of superoxide radicals [193], thereby amplifying oxidative stress and contributing to the development of heart failure [194]. Growing evidence from both animal studies and clinical observations reinforce the notion that excessive mtROS significantly exacerbate cardiac pathology in the failing heart [194,195,196]. Notably, excessive mitochondrial oxidative stress may act both as a cause and as a consequence of mitochondrial dysfunction during the progression to heart failure [196]
Strong evidence supports the interconnected role of ROS, mitochondrial dysfunction and autophagy in heart failure. Indeed, it has been found that the dopamine D5 receptor (D5R) reduces the production of mtROS through a cAMP and autophagy-dependent manner [197]. Notably, cardiac-specific dopamine D5R knockout mice (Drd5 myh6fl/fl-creERT2) develop hypertrophic cardiomyopathy and heart failure via mechanisms that lead to increased NADPH oxidase activity and ROS production and mitochondrial dysfunction, whilst antioxidant administration (Apocynin, Tempol, Mito-TEMPO) rescued the cardiac hypertrophy and fibrosis [198]. Interestingly, myeloid differentiation protein 1 (MD1) has been shown to enhance the rate of cardiomyocyte autophagy in heart failure with preserved ejection fraction (HFpEF) by activating the ROS-mediated MAPK signalling pathway [199].
Emerging evidence now highlights a complex interplay between the key regulators of the cellular stress response such as autophagy, hypoxic signalling and the regulation of oxidative stress, in driving cardiac fibrosis and the progression to heart failure [145,200,201]. In particular, a study by Ghosh et al., reported that the selective autophagy adaptor protein, p62, reduces hypoxia-induced cardiac dysfunction by stabilizing HIF-1α and Nrf2 [145]. In H9c2 rat cardiomyoblasts, depletion of p62 enhances proteasomal degradation of Nrf2, whereas overexpression of p62 stabilizes Nrf2 levels, suggesting a crucial role for p62 in Hif-1α and Nrf2 stabilization and transcriptional activity to maintain redox balance and protect the cell from hypoxic stress [145].
Moreover, a high level of oxidative stress can cause myocardial fibrosis by enhancing the proliferation of cardiac fibroblasts and collagen production, consequently stiffening the heart muscle and impairing its contractile and diastolic function, ultimately leading to heart failure [40,202]. A recent study described a mechanism by which ROS facilitate the proliferation of cardiac fibroblasts [202]. In a mouse model of cardiac fibrosis induced by Ang II or ischemia‒reperfusion injury, elevated levels of miRNAs with oxidized guanosine (O8G) modifications were observed. It was shown that treatment with Ang II or PDGF induced excess ROS, which resulted in oxidative modification of guanosine (G) to 8-oxoguanosine (O8G) in miR-30c. Modified miR-30c downregulated CDKN2C, a negative regulator of cardiac fibroblast proliferation, thereby enhancing proliferation of fibroblasts and excessive accumulation of extracellular matrix [202]. Collectively, redox imbalance, mitochondrial dysfunction and dysregulated autophagy, contribute to a deleterious feedback loop that amplifies cardiac remodelling and accelerates the progressive loss of cardiac function, which is characteristic of heart failure.
9.5. Diabetic Cardiomyopathy
Diabetic cardiomyopathy (DCM) is characterised by abnormal myocardial structure and function without the presence of additional cardiac risk factors such as coronary artery disease, hypertension and severe valve disease in diabetic patients [203]. DCM has emerged as the main cause of heart failure in diabetic individuals [204]. The underlying mechanisms of DCM are multifactorial and not yet fully understood. Oxidative stress is a key factor in the pathogenesis of DCM [205]. Numerous factors are implicated in the development of DCM including impaired insulin and metabolic signalling, impaired glucose uptake, oxidative stress, mitochondrial dysfunction, autophagy, mitophagy, imbalance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), impaired Ca2+ handling and inflammation [204,205].
Numerous preclinical and clinical studies have demonstrated the crucial role of oxidative stress in DCM [206,207,208,209]. Mechanistically, hyperglycemia accelerates mitochondrial electron transport chain activity, leading to electron leakage and excessive ROS generation, thereby driving oxidative stress [210]. In an experimental DCM model, metformin suppressed the expression of COL-I, III, TGF-β, CTGF, ICAM, and VCAM genes, reduced collagen deposition, and improved cardiac function by reversing DCM-associated damage [208]. Given its ability to activate AMPK, which in turn enhances mitochondrial efficiency and lessens mtROS, these effects are likely mediated by the known ability of metformin to mitigate oxidative stress [211]. Another study reported that in high glucose induced H9C2 cells, naringenin inhibits ROS generation, reduces inflammatory cytokine production and suppresses apoptosis. Similarly, in type 1 diabetic mice naringenin reduces oxidative stress and inflammation by inhibiting NF-κB and enhancing Nrf2 activity, consequently alleviating cardiac fibrosis and cardiomyocyte apoptosis [212]. With respect to diabetic patients, a clinical study reported high levels of mtROS and an increase in the inflammatory markers, NFκB-p65 and TNF-α in T2D leukocytes, which correlated with increased inflammatory and vascular complications, whilst MitoQ treatment enhanced antioxidant defences (GPX-1 levels) in T2D leukocytes and lessened chronic inflammation and the risk of CVD [213]. Furthermore, a naturally occurring flavonoid, Kaempferol, alleviated hyperglycemia-induced cardiac injury and apoptosis by inhibiting oxidative stress and inflammatory responses specifically through inhibition of NF-κB nuclear translocation and activation of Nrf2 in in vitro studies and in diabetic mice hearts [209]. Finally, myricitrin (Myr), another naturally occurring flavonoid, has been shown to reduce ROS and inflammatory cytokines, leading to reduced apoptosis in advanced glycation end products (AGE)-induced H9C2 cardiomyocytes [214]. Similarly, Myr treatment of streptozotocin-induced diabetic mice inhibited the production of inflammatory cytokines and apoptotic proteins, and downregulated the expression of enzymes associated with cardiomyopathy, as well as improved diastolic dysfunction. Mechanistically, it could be shown that Myr alleviated oxidative stress and inflammation via the Akt-dependent activation of Nrf2 signalling whilst inhibiting the NF-κB pathway [214].
Emerging evidence suggests that targeted modulation of T cells offers a promising strategy to attenuate DCM. In a recent study, regulatory T cells were shown to reduce oxidative stress, inflammation and apoptosis, thereby attenuating myocardial hypertrophy, fibrosis and improving cardiac dysfunction [215]. T cells also protected against the progression of DCM in db/db mice by regulating the PI3K–Akt and MAPK signalling pathways [215].
Collectively, across atherosclerosis, pathological cardiac hypertrophy, ischemia–reperfusion injury, heart failure and diabetic cardiomyopathy, strong preclinical and clinical evidence supports the notion that the convergence of oxidative stress, mitochondrial dysfunction, impaired autophagy and chronic inflammation initiates, amplifies and propagates CVDs, and that elucidation of the crosstalk relevant to each pathology may reveal mechanistically informed and specific targets for therapeutic intervention.
10. Refining Redox Approaches for CVD: from Vitamins to Precision Therapies
As discussed above, preclinical evidence strongly supports a role for redox imbalance as a critical mediator of cardiovascular diseases. The data suggest that redox signalling can be targeted for the prevention of CVDs and that modulating cellular redox state through lifestyle, dietary, and pharmacological interventions could be an important strategy to reduce the risk of onset and progression of CVDs, particularly in older adults who are more likely to develop symptoms. Unfortunately, clinical translation of antioxidant therapy with vitamins such as vitamin A or E have not proved efficacious in large scale clinical trials such as HOPE (Heart Outcomes Prevention Evaluation), HOPE-TOO and GISSI-Prevenzione [216,217,218]. HOPE-TOO evaluated long-term supplementation of vitamin E (400IU/day) in high-risk CV patients and found no reduction in major adverse CV events (MACE) and was instead associated with an increased risk of heart failure [217]. Similarly, the GISSI trial, which evaluated post AMI patients, showed no benefit from vitamin E therapy in reducing CV death or other MACE outcomes [218]. More recently a systematic review and meta-analysis of 38 studies showed that vitamin E did not prevent or reduce mortality of CVDs in most trials [8]. Additionally, some studies reported that high dose vitamin E may have detrimental effects on cardiovascular outcomes [8]. These studies question the translational validity of earlier preclinical findings. However, the failure of these trials may be multifactorial, namely that vitamin strategies often involve indiscriminate antioxidant activity; they may disrupt beneficial redox signalling, and paradoxically, through their modes operandi, vitamins generate further ROS. Vitamins may additionally fail to localize effectively to subcellular compartments where oxidative damage is most relevant. These limitations, along with the negative outcomes of the large-scale clinical trials, raise important questions about targeting ROS for CVD prevention, but importantly have highlighted that a more nuanced approach to antioxidant therapy is needed. Emerging evidence now suggests that pharmacologically targeting redox-sensitive pathways, such as the activation of Nrf2, the inhibition of NADPH oxidases, or directing therapies to the mitochondria, may offer a more precise and physiologically attuned approach to restoring redox balance.
Indeed, there is strong evidence supporting a role for mitochondria-targeted antioxidants, such as MitoQ or SkQ1, in improving cardiovascular outcomes. Effective delivery of therapeutic compounds to the mitochondria in vivo is challenging [219]. MitoQ is a chemically modified version of CoQ10 with an added triphenylphosphonium cation (TPP+) to assist with its translocation and accumulation in the mitochondria [219,220]. MitoQ has been shown to restore age-related decreases in endothelium-dependent dilation (EDD), to reduce aortic stiffness, and improve vascular function in both old mice and clinical studies of older adults without adverse effects [221,222]. Furthermore, in a randomized, placebo-controlled, double-blind crossover study, MitoQ was found to improve endothelial function partially by reducing mtROS in middle-aged and older adults [222]. In-depth mechanistic insights into the mode of action of CoQ10 and MitoQ are covered in section 11.2.1 of this review. With respect to Nrf2 activators such as bardoxylone methyl, clinical translation for CVDs has been limited by adverse CV events (most likely due to fluid overload), as seen in the BEACON trial [223].NADPH oxidase inhibitors such as GKT1378312 have shown promise in reducing vascular oxidative stress and fibrosis in animal studies [30], however, robust clinical data supporting their use in CVD prevention remain lacking.
11. Therapeutic Implications and Challenges
Considering the disconnect between preclinical and clinical data with respect to antioxidant therapies, future strategies need to address the type of antioxidant, the source of ROS production, the duration of treatment, the dosing regimen, and the population-specific requirements to avoid off-target effects, including modulation of physiological redox signalling essential for cellular homeostasis. A promising strategy might be to focus on preventing the production of reactive oxidants that damage cellular macromolecules such as DNA, proteins, and lipids by targeting key enzymatic sources such as NADPH oxidases, xanthine oxidase, and dysfunctional mitochondrial complexes. Moreover, inhibiting downstream redox-sensitive signalling pathways, such as NF-ĸB, MAPKs, and the NLRP3 inflammasome that drive inflammation, fibrosis and programmed cell death, should be the focus of newer antioxidant and anti-inflammatory strategies. Augmenting endogenous antioxidants by enhancing endogenous antioxidant enzymes, including SOD, catalase, and GPx, as well as modulating redox sensitive transcription factors, such as Nrf2, could provide protection against redox imbalance-associated diseases. Additionally, identifying specific small molecules or drug targets that improve mitochondrial dynamics, biogenesis and mitophagy processes, may facilitate the development of targeted therapeutic interventions for CVDs.
Therapeutic modulation of autophagy depends on disease context, since the role of autophagy is complex and context-dependent. Thus autophagy can be therapeutically targeted using agents such as AMPK activators, mTOR inhibitors, or transcription factor EB (TFEB) inducers to promote cellular clearance and stress adaptation. Similarly, when excessive autophagy contributes to cell damage, lysosomal blockers or autophagy initiation can restore or modulate impaired autophagy. The current understanding of these themes is explored below.
11.1. Targeting the Oxidative Stress/Mitochondria/Autophagy/Inflammation Axis
The integrated network of interactions that form the redox signalling-mitochondria-autophagy-inflammation axis has significant implications for the pathogenesis of CVDs. Targeting these pathways may prevent cardiovascular inflammation and tissue damage by suppressing oxidative stress, regulating mitochondrial dynamics and complex autophagic flux (Figure 4).
11.1.1. Targeting Oxidative Stress in Cardiovascular Disease
Numerous preclinical studies have demonstrated the protective role of the major antioxidant enzymes against oxidative stress and tissue damage in mouse models of cardiovascular disease [224,225,226,227]. For example, a deficiency of CuZnSOD enzyme activity in Sod1 KO mice augmented Nox2 levels in the heart and led to oxidative damage and dysfunctional cardiac function [226]. Extracellular superoxide dismutase (EC-SOD) has been shown to ameliorate hypoxia-induced epigenetic modifications of the tumor suppressor gene, RASSF1A, by modulating the Ras/ERK pathway, and decreasing fibrosis and tissue damage [226]. The clinically approved SOD mimic and redox-active drug, MnTnBuOE-2-PyP5+ (BMX-001), has been shown to inhibit human valve interstitial cell activation and extracellular matrix remodelling, in a murine model of aortic valve sclerosis [228].
Our lab and others have shown a protective role for GPx1 in the prevention of atherosclerosis [229]. We demonstrated a significant increase in plaque burden in diabetic ApoE/GPx1 double knockout (dKO) mice, which was accompanied by increased inflammation and oxidative stress. We also showed that the potent antioxidant and small molecule GPx1 mimetic, ebselen, prevented plaque formation under diabetic conditions in both ApoE-/- and ApoE/Gpx1 dKO mice, via a mechanism that included modulation of the inflammatory MAPK, JNK and p38 pathway [229].
Furthermore, compounds like N-acetylcysteine (NAC), which serve as substrates for antioxidant enzymes, have been investigated for their potential to treat heart failure (NCT00532688). In patients with cardiorenal syndrome, NAC treatment was associated with improved forearm blood flow and significant improvements in endothelial function [230]. Similarly, natural antioxidants such as berberine and urolithin A decrease oxidative stress and improve endothelial function, thereby preventing the development of atherosclerosis and other heart-related diseases such as AMI [40,231,232].
The transcription factor Nrf2 acts as a master regulator of antioxidant signalling and maintains redox balance in cells. Multiple potential Nrf2 activators, including dimethyl fumarate (DMF), bardoxolone methyl, resveratrol, quercitol and curcumin, have been shown to modulate the pathophysiology of various CVDs [68,233]. DMF treatment reduced the levels of serum and aortic ROS, as well as the expression of the oxidation-related protein gp91phox, also known as Nox2. It upregulated the expression of HO-1 and Nrf2, thereby reducing aortic atherosclerosis in diabetic ApoE−/− mice by activating the Nrf2/ARE signalling pathway [234]. Similarly, another potent Nrf2 activator, bardoxolone methyl, promoted Nrf2 binding to the transcriptional co-activator CREB-binding protein (CBP) and increased Nrf2 downstream targets, including NQO-1, HO-1, Catalase, and the Glutamate-Cysteine Ligase Catalytic (GCLC) subunit consequently attenuating myocardial inflammation and improving cardiac function in rats with chronic heart failure [235].
These newer preclinical antioxidant strategies offer alternate approaches to the vitamin strategies previously used in clinical trials. These newer approaches are more likely to be efficacious as they overcome some of the limitations of vitamin therapy such as their lack of target specificity [236,237,238]. Data from the UK Biobank and FinnGen databases further highlight the inherent risks of vitamin therapy, where a recent study found that elevated levels of specific circulatory antioxidants, particularly α-tocopherol, α-carotene, and retinol, were linked to increased risk of certain cardiovascular diseases [239]. Together, these insights underscore the urgent need for targeted, mechanism-based antioxidant therapies that not only avoid the pitfalls on non-specific vitamin supplementation but also hold genuine promise for reducing cardiovascular risk.
11.1.2. Targeting Mitochondrial Dysfunction and Autophagy in Cardiovascular Disease
Several therapeutic agents, including the AMPK activator metformin, CoQ10 and MitoQ, the potent mitochondrial-targeted peptide SS-31 (also known as elamipretide), and the mitophagy inducer urolithinA, modulate multiple components of the redox-mitochondria-autophagy axis simultaneously. As discussed below, these agents are proving to be important modulators of CVD outcomes. For ease of reference, pharmacological inhibitors targeting redox signalling, mitochondrial dysfunction and autophagy in various cardiovascular conditions are listed in Table 1.
Importantly, therapeutic enhancements of pathways that control mitochondrial quality have shown beneficial effects in preclinical models of cardiovascular disease. Mitochondrial quality control involves multiple tightly coordinated processes, including mitochondrial biogenesis, fusion/fission dynamics, proteostasis, and mitophagy. These processes collectively preserve mitochondrial function, control redox imbalance by regulating ROS generation, and regulate cellular homeostasis.
11.1.2.1. CoQ10
Coenzyme Q10 (CoQ10) or ubiquinone is an endogenously synthesized coenzyme and a key component of the ETC [240]. In particular, it is involved in the CoQ10–AMPK–OPA1 pathway where it activates AMPK, which in turn upregulates OPA1, thereby enhancing mitochondrial function by promoting ATP production [17]. It reduces oxidative stress by decreasing levels of lactate dehydrogenase (LDH) and malondialdehyde (MDA), while increasing antioxidants such as SOD and GSH. It is therefore implicated as a therapeutic agent in the treatment of CVDs [17,240]. In preclinical studies, CoQ10 administered to a rat model of I/R injury, ameliorated acute myocardial injury, reduced myocardial apoptosis and improved cardiac function by enhancing autophagy and reducing oxidative stress [241], suggesting that CoQ10 regulates redox signalling and improves mitochondrial function and autophagy to protect against CVD. A clinical study demonstrated that CoQ10 supplementation has potential prophylactic efficacy in reducing the incidence of fatal and non-fatal MI [242]. A systemic review and meta-analysis demonstrated that CoQ10 supplementation improved mitochondrial function, namely, ATP generation and respiratory capacity, and importantly, improved cardiovascular function suggesting that CoQ10 could be a beneficial adjunct therapy for CVD patients [243].
11.1.2.2. MitoQ
Mitochondria-targeted MitoQ plays a key role in mitochondrial quality control by triggering mitophagy, restoring mitochondrial membrane potential, and improving mitochondrial dynamics [244]. In addition, MitoQ has shown promising antioxidant and anti-inflammatory effects in preclinical and clinical trials for various cardiovascular conditions by reducing mitochondrial ROS and improving mitochondrial function [221,245,246,247]. More specifically, MitoQ improved cardiac function by enhancing PINK1/Parkin-mediated mitophagy in Type 2 diabetic rats [19]. MitoQ also markedly reduced ROS levels and mitigated triptolide-induced cardiotoxicity by activating the autophagy p62-Nrf2 signalling pathway in H9c2 cardiomyocytes [246]. In a rat pressure overload-induced heart failure model, MitoQ significantly improved mitochondrial dysfunction by decreasing hydrogen peroxide, improving mPTP opening, and enhancing mitochondrial respiration [244]. In addition, in a clinical trial of hypertensive patients, MitoQ supplementation administered together with moderate intensity endurance training, substantially reduced blood pressure, IL-6 levels and improved cardiac function in these patients [247]. One explanation involves the ability of MitoQ to downregulate MiR-21. Research has shown that MiR-21 is associated with ROS production, vascular remodeling, increases in the level of inflammatory C-reactive protein, and arterial stiffness [247,248]. In the hypertensive patients, MitoQ treatment led to a significant reduction in circulating miR-21 levels, accompanied by improvements in LV mass and systolic function [247].
11.1.2.3. Melatonin
Melatonin is a naturally occurring neurohormone primarily secreted by the pineal gland, and best known for its role in regulating circadian rhythm and promoting sleep. Accumulating evidence now suggests that it exerts far-reaching protective effects beyond the brain, including the CV system. For example, in neonatal mouse ventricular cardiomyocytes subjected to hypoxia and reoxygenation injury, treatment with melatonin enhanced mitochondrial metabolism, inhibited mitochondrial oxidative stress, induced mitochondrial fusion, and prevented mitochondria driven apoptosis. Mechanistically, melatonin improved mitochondrial biogenesis by activating the AMPK/PGC1α pathway and attenuated ischemia/reperfusion-induced myocardial damage [249]. Another study demonstrated that melatonin prevented the progression of atherosclerosis by inducing mitophagy and inhibiting activation of the NLRP3 inflammasome, which was mediated by the Sirt3/FOXO3a/Parkin signalling pathway [250]. Melatonin also suppressed galectin-3 (Gal-3), reduced the activity of the NF-κB signalling pathway, and promoted the nuclear translocation of transcription factor EB (TFEB), thereby enhancing autophagy and suppressing inflammation in atherosclerosis. Furthermore, melatonin enhanced autophagy via inhibition of the Gal-3/CD98/PI3K pathway in THP-1 macrophages, and alleviated inflammation, highlighting its potential as a therapeutic agent for the treatment of atherosclerosis [18]. Another study found that melatonin activated the autophagy process via the AMPK/mTOR/ULK1 signalling pathway and decreased vascular calcification of vascular smooth muscle cells isolated from the aortas of Sprague–Dawley rats [251]. Collectively, these data suggest a protective role for melatonin against cardiovascular injury and vascular calcification through modulation of autophagy.
11.1.2.4. Urolithin A
Urolithin A is a regulator of mitophagy and exhibits cardioprotective effects [70,126]. Several studies have revealed that urolithin A upregulates the expression of mitophagy-related genes and activates PINK1-Parkin-mediated mitophagy, thereby improving mitochondrial quality control [70,126,252]. Impaired mitochondrial function is also a key feature of cardiac aging in humans. A recent study demonstrated that Urolithin A improved cardiac function and mitochondrial health in aging mouse and rat models of heart failure with reduced ejection fraction (HFrEF). Urolithin A restored heart muscle ultrastructure and mitochondrial morphology, and improved cardiac and skeletal muscle function in non-diseased old C57BL/6RJ mice. Moreover, Urolithin A administered for 2 months improved systolic function, reduced end-systolic volume, and improved cardiac muscle contractility in rats with heart failure [20]. Mechanistically, in the hearts of AMI animals, Urolithin A increased the levels of mitochondrial oxidative phosphorylation associated genes, as well as the PINK1/parkin-mediated mitophagy marker, phospho-ubiquitin [20], suggesting that Urolithin A exerts its cardioprotective effects by activating mitochondrial recycling and enhancing the mitochondrial quality control system [20]. In clinical trials, 4 months of Urolithin A supplementation in healthy older adults significantly reduced plasma ceramide levels, which are associated with CVD risk in humans [20].
11.1.2.5. Elamipretide
Mitochondria-targeted elamipretide (SS-31) has been shown to preserve mitochondrial dynamics and restore energy production in ischemic mitochondria by binding to and stabilizing cardiolipin [253]. Remarkably, elamipretide reversed age-associated post-translational modifications such as S-glutathionylation of cysteine residues and phosphorylation of heart proteins [254]. Moreover, SS-31 significantly suppressed mitochondrial ROS production, decreased protein oxidation and cellular senescence, improved cardiac function and mitigated myocardial hypertrophy in aged mice [255]. These results support the therapeutic potential of SS-31 in alleviating mitochondrial dysfunction and redox-driven pathologies in cardiovascular disorders, particularly in senescence.
11.1.2.6. Metformin
Metformin, a widely used first-line therapy to improve glucose metabolism in type 2 diabetic patients, has emerged as a multifaceted agent that can simultaneously target redox signalling, mitochondrial function and autophagic flux, thereby maintaining cellular homeostasis [51,256]. A growing body of research demonstrates that metformin induces autophagy through multiple signalling pathways, including AMPK-dependent pathways such as AMPK/mTOR, AMPK/CEBPD, AMPK/ULK1 and AMPK/miR-221, where metformin directly and indirectly activates multiple autophagy-related proteins via inhibition of mTORC1 [257,258]. Additional autophagic pathways induced by metformin include Redd1/mTOR, STAT, and SIRT, TRIB3 as well as the PK2/PKR/AKT/GSK3β pathway and the Na+ /H+ exchangers [256]. Metformin also facilitates heart regeneration by enhancing autophagy in zebrafish [258]. Metformin has also shown protective effects against ischemic myocardial injury by reducing macrophage-driven inflammation via modulation of the autophagy–ROS–NLRP3 axis [51]. In Wistar rats, metformin reduced infarct size, cardiac arrhythmias and LV dysfunction by attenuating mitochondrial dynamic imbalance and apoptosis in cardiac ischaemia-reperfusion injury, most likely mediated in part, by the activation of the AMPK/PGC1α pathway [72]. However, clinical research investigating the use of metformin in CVD prevention remains limited and awaits more comprehensive and targeted studies to validate its cardioprotective potential.
11.1.2.7. Berberine
Berberine, an isoquinoline alkaloid found in plants, has shown promising cardioprotective properties against different CVDs [259,260]. In mice with HFpEF, berberine upregulated p-AMPK and PGC-1α, reduced mtROS, improved mitochondrial function and alleviated mitochondrial biogenesis disorders, thereby improving cardiac function [261]. Oxygen-glucose deprivation/re-oxygen (OGD/R) of human cardiomyocytes inhibited the production of GSH, GSH-Px and SOD, and increased the production of MDA, IL-1β, TNF-α and IL-6, whereas treatment with berberine markedly reduced indicators of inflammation and oxidative stress. In both human cardiomyocytes and a myocardial I/R rat model, berberine ameliorated inflammation, oxidative stress and ischemia–reperfusion injury by inducing miR-26b-5p and suppressing the PTGS2/MAPK signalling pathway [232]. Furthermore, in an animal model of carotid atherosclerosis, berberine reduced atherosclerotic plaque area, lipid accumulation, neointimal formation and cell apoptosis in carotid arteries by regulating the PI3K/AKT/mTOR signalling pathway, thereby improving carotid atherosclerosis [262]. A recent study further substantiated these findings in HFD-fed ApoE−/− mice, where administration of berberine mitigated atherosclerosis by promoting autophagy, suppressing inflammatory responses and maintaining vascular endothelial cell integrity, via modulations of the RAGE-NF-κB pathway [231]. With many of these multifaceted effects mediated via AMPK activation, inhibition of pro-inflammatory signalling (eg. NF-κB) and stimulation of Nrf2, berberine may offer an alternate approach to lessen CVDs, particularly as an adjunctive therapy for individuals with cardiometabolic disorders. However, numerous challenges remain regarding formulation and clinical standardization [263,264].

Table 1.
Pharmacological Inhibitors Targeting Redox Signalling, Mitochondrial Dysfunction and Autophagy in CVDs.
Table 1.
Pharmacological Inhibitors Targeting Redox Signalling, Mitochondrial Dysfunction and Autophagy in CVDs.
|
Therapeutic agent |
Signalling pathways and related mechanisms |
Treatment outcome |
Experimental models |
Disease context |
Ref. |
| CoQ10 | Inhibits Oxidative Stress. Improves Mitochondrial Function. Activates the AMPK-YAP-OPA1 Pathway. |
Increases SOD and GSH in serum in diseased mice. Suppresses the expression of IL-6, TNF-α, ICAM-1, VCAM-1 and NLRP3. Ameliorates Atherosclerosis. |
High fat diet (HFD) fed ApoE−/− mice | Atherosclerosis | [17] |
| Reduces Oxidative Stress. Enhances Autophagy. |
Increases GPx, GR, SOD, and GSH. Decreases TBARS in myocardial tissue in rats with AMI. Increases autophagy proteins beclin-1 and Atg5. Reduces infarct size. Improves cardiac function. |
AMI/R Sprague Dawley (SD) ratmodel | Acute Myocardial Ischemia-Reperfusion Injury(AMI) | [241] | |
| MitoQ | Reduces oxidative stress. Activates p62-Nrf2 signalling pathway. |
Decreases ROS accumulation. Improves cell viability. Reduces cardiotoxicity. |
Triptolide-induced cardiotoxicity in rat cardiomyocyte H9c2 cells | [246] | |
| Decreases oxidative stress. Regulates mitochondrial function. |
Restores mitochondrial membrane potential and respiration. Improves mitochondrial calcium retention capacity. Inhibits ROS production. Improves cardiac function. |
Rat model of heart failure induced by pressure overload | Heart failure | [244] | |
| Enhances mitophagy via PINK1/Parkin pathway. | Reduces myocardial infarction, myocardial pathological damage and cardiomyocyte apoptosis. Improves cardiac function. |
Myocardial ischemia–reperfusion injury in Type 2 diabetic rats | MIR injury in Type 2 diabetes (T2D) | [19] | |
| Melatonin | Suppresses oxidative stress. Enhances mitochondrial biogenesis via the AMPK/PGC1α pathway. |
Reduces mtROS production. Alters mitochondrial morphology of cardiomyocytes. Attenuates myocardial damage. |
Hypoxia/reoxygenation injury in cardiomyocytes. | Cardiac ischemia/reperfusion injury | |
| Reduces inflammation. Enhances autophagy. Promotes TFEB nuclear translocation. Inhibits NF-κB by inhibiting Gal-3. |
Inhibits secretion of IL-6, IL-18, IL-1β and TNF-α in arteries. Inhibits atherosclerotic plaque progression. |
HFD-fed ApoE−/− mice | Atherosclerosis | [18] | |
| Urolithin A | Restores mitochondrial dynamics proteins DRP1 and MFN1. Activates mitochondrial recycling and quality control (QC). |
Improves heart mitochondrial ultrastructure, morphology and function. Enhances cardiac function and skeletal muscle force in aging |
Non-diseased old C57BL/6RJ mice | Aging | [20] |
| Promotes mitochondrial QC pathways. | Improves systolic function. Improves cardiac function and mitochondrial health. |
Rat model of chronic heart failure (HFrEF) | Heart failure | [20] | |
| Elamipretide (SS-31) | Regulates age-associated post-translational modifications of heart proteins | Affects mouse heart function | Aged mouse hearts | Cardiac aging | [254] |
| Suppresses mtROS production. Inhibits protein oxidation and cellular senescence. |
Reduces cardiac hypertrophy. Improves cardiac function. |
Aged mice | Myocardial hypertrophy | [255] | |
| Metformin | Preserves mitochondrial function. | Alleviates mitochondrial dynamic imbalance and apoptosis. Reduces arrhythmia and infarct size. Improves cardiac function. |
Cardiac I/R injury in Wistar rats | Cardiac ischemia/reperfusion (I/R) injury | [72] |
| Induces autophagy. | Enhances epicardial, endocardial and vascular endothelial regeneration. Improves transient collagen deposition and resolution. Induces cardiomyocyte proliferation. Improves systolic function of the heart. |
Adult zebrafish model of heart cryoinjury | Myocardial infarction | [258] | |
| Berberine | Inhibits inflammatory responses and oxidative stress via miR-26b-5p-mediated PTGS2/MAPK. | Increases GSH, GSH-Px and SOD. Suppresses MDA, IL-1β, TNF-α, and IL-6. Preserves myocardial structure Improves cardiac function. |
OGD/R-treated cardiomyocytes. Rat model of myocardial ischemia-reperfusion (I/R) injury. |
Acute myocardial infarction model (AMI) | [232] |
| Activates autophagy and reduces inflammation. Modulates RAGE-NF-κB. |
Increases lipid accumulation and foam cell formation. Maintains vascular endothelial cell integrity. Reduces atherosclerotic inflammation. |
High fat diet ApoE−/− mouse model | Atherosclerosis | [231] | |
| Regulates PI3K/AKT/mTOR. | Improves intimal hyperplasia. Reduces carotid lipid accumulation. Promotes cell proliferation. |
High fat diet ApoE−/− mice | Carotid atherosclerosis | [262] | |
| Mdivi-1 | Suppresses mito-ROS/NLRP3 by inhibiting DRP1-dependent mitochondrial fission. | Decreases plaque area. Reduces foam cells. Inhibits M1 polarization. Inhibits activation of NLRP3. |
High fat diet ApoE−/− mice | Atherosclerosis | [71] |
| DMF | Exerts antioxidant effects by activating the Nrf2/ARE signalling pathway |
Reduces the area of aortic atherosclerosis. Decreases serum and aortic ROS, HO-1, NF-κB, ICAM-1 and gp91phox. Increases serum and aortic Nrf2, eNOS, and p-eNOS. |
ApoE−/− mice with streptozotocin-induced hyperglycemia | Atherosclerosis | [234] |
| Micheliolide (MCL) | Promotes KEAP1/NRF2 dissociation. Activates NRF2 pathway. |
Decreases inflammatory responses. Reduces oxidative stress. Inhibits macrophage ferroptosis. |
High fat diet ApoE−/− mice | Atherosclerosis | [79] |
| Bardoxolone- methyl | Increases Nrf2 binding to the CREB-binding protein. Increases Nrf2 downstream targets NQO1, HO-1 and CAT. |
Reduces myocardial oxidative stress and lipid peroxidation. Attenuates myocardial inflammation. |
Rat model of chronic heart failure | Chronic heart failure | [235] |
12. Discussion
This review highlights the opportunity to target the interconnected redox-mitochondria-autophagy-inflammation axis for the prevention and treatment of CVDs. Significant progress has been made in the understanding of the role of redox biology in regulating mitochondrial function and autophagy, and its associated link with inflammation and immune responses. However, one of the major challenges is maintaining context-dependent redox signalling, which if perturbed may contribute to the pathophysiology of CVDs. Depending on several factors e.g. the cellular context or the stage of the disease, the impact on these pathways may not always be cardio-protective. Indeed, targeting specific redox signalling pathways may inadvertently modulate other signalling networks, triggering off-target and unwanted side effects. For example, multiple studies demonstrate that Mitochondrial Division Inhibitor 1 (Mdivi-1), which reversibly inhibits Complex I of the ETC to modify mtROS production [240,265,266], reduces atherosclerosis in ApoE-/- mice [71]. However, the Mdivi-1 also altered AMPA receptor-activated Ca2+ signalling, and led to the loss of Ca2+ from the ER, which enhanced oligodendrocyte sensitivity to excitotoxic and ER stress. This subsequently resulted in oxidative stress and apoptosis [267]. Another study suggested that apart from inhibiting Drp-1 mediated mitochondrial fission, mdivi-1 affected ion channel function and altered the Rho kinase pathway, thereby affecting the regulation of vascular smooth muscle tone [268]. Thus, future studies should focus on therapies that target multiple nodes of the redox-mitochondria-autophagy-inflammation axis, with an emphasis on minimizing adverse effects.
As discussed in this review, the integrity of mitochondrial structure and function is key to maintaining redox homeostasis and mitigating the development of CVD. When considering the factors that contribute to CVD, the role that cellular senescence plays needs careful consideration. Age related decline in mitochondrial mass contributes to excessive mtROS with an associated depletion of ATP production. This drives endothelial and smooth muscle cell dysfunction, consequently contributing to CVDs such as atherosclerosis [162,194,269]. Removal of dysfunctional mitochondria should be a goal of CVD therapy. Indeed the removal of dysfunctional mitochondria by enhancing mitophagy improves cardiac contractile function, retards cardiomyocyte senescence and remodelling of heart tissue [70,270,271]. In particular, treatment with tetrahydroberberrubine, a derivative of berberine, is showing promise in preclinical studies where it promotes mitophagy in the aging heart, improves diastolic dysfunction, inhibits cardiac remodelling and suppresses cardiac senescence in aging mice [270]. Therefore, neutralising excessive mtROS by targeted delivery of ROS scavengers or improving mitochondrial function by enhancing mitochondrial biogenesis, may offer alternative therapies for age-related diseases where mitochondrial dysfunction is causal [70]. Furthermore, targeting mitochondrial DNA, mitochondrial microRNAs and associated proteins offers compelling future directions for therapies aimed at restoring mitochondrial function [69].
Newer technologies may assist in delivering a more targeted approach to therapy. Emerging evidence demonstrates that targeted nanotherapeutics can improve therapeutic efficacy and reduce systemic adverse events and off-target effects in CVDs [272]. Nano-drug delivery systems can be engineered to specifically target the cells and/or cellular compartments within the heart or the vasculature, thereby allowing for precise therapeutic intervention. In particular, targeting impaired mitochondria within pathological tissue has emerged as a promising strategy [240,272]. Nanoparticle-based drug delivery systems can also be effective tools for targeting atherosclerotic plaque. For example, lipoic acid nanoparticles passively target atherosclerotic plaque and exhibit better therapeutic efficacy than free lipoic acid. Specifically, lipoic acid nanoparticles reduced oxidative stress and inflammation, and inhibited lipid infiltration into plaques of HFD-ApoE-/- mice [273], suggesting that nanoparticle-based drug delivery systems offer a promising therapeutic strategy for CVDs.
In addition, to identify patient subgroups most likely to benefit from specific interventions, integrative omics and bioinformatics approaches are likely to bolster therapeutic strategies. System biology approaches and multi-omics profiling can be used to identify dysregulated pathways linking redox imbalance, mitochondrial dysfunction, dysregulated autophagy and inflammation.
13. Conclusions
In summary, redox signalling, mitochondrial function, autophagy and inflammation form a complex interconnected network required for maintaining cellular homeostasis. Depending on the cellular and physiological context, these processes influence each other such that dysregulated redox balance impairs mitochondrial function, while dysfunctional mitochondria exacerbate maladaptive autophagy that further enhances inflammatory responses. Unless therapeutically targeted, chronic inflammation perpetuates mitochondrial dysfunction and impaired autophagy, inducing oxidative stress that in combination, drives the progression of CVD. Clearly, perturbations such as hyperglycaemia, dyslipidaemia and hypertension, dysregulate this axis, which contributes to the onset and progression of various cardiovascular conditions. In highlighting the interconnectivity of this axis, this review has spotlighted a powerful avenue for therapeutic intervention in cardiovascular disease prevention and/or progression. Future research should focus on the identification of pharmacological interventions capable of modulating this axis. This would represent a promising and innovative approach to lessen the burden of CVDs.
Novelty
This review unravels the interplay of redox signalling, mitochondrial dysfunction, and the autophagy pathway in regulating cardiovascular inflammation and tissue damage in CVDs. This review contributes to a deeper understanding of the redox-mitochondria-autophagy-inflammation axis underlying CVDs. This review summarises recent findings and highlights the importance of focusing on redox pathways and associated signalling nodes to address the unmet clinical needs in CVD prevention.
Author Contributions
MP conceived, designed and wrote the manuscript. JDH wrote and revised the manuscript and supported the funding. All authors have read and approved the manuscript for submission.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work is supported by an Australian National Health and Medical Research (NHMRC) Ideas Grant to JdH, Project # 2029657.
Abbreviations
The following abbreviations are used in this manuscript:
| ACE | Angiotensin II converting enzyme |
| AGEs | Advanced glycation end products |
| AMI | Acute myocardial infraction |
| AMPK | AMP-activated protein kinase |
| ApoE-/- | Apolipoprotein E–deficient |
| ARBs | Angiotensin Receptor Blockers |
| AREs | Antioxidant response elements |
| BNIP3 | Bcl-2/adenovirus E1B 19-kDa-interacting protein 3 |
| CAD | Coronary artery disease |
| CAT | Catalase |
| CBP | Transcriptional coactivator CREB-binding protein |
| circRNAs | Circular RNAs |
| CoQ10 | Coenzyme Q10 |
| CVD | Cardiovascular Disease |
| CypD | Cyclophilin D |
| cytoROS | Cytosolic ROS |
| DCM | Diabetic cardiomyopathy |
| DMF | Dimethyl fumarate |
| DMPs | Damage-associated molecular patterns |
| DRP1 | Dynamin-related protein 1 |
| ER | Endoplasmic reticulum |
| Gal-3 | Galectin-3 |
| GCLC | Glutamate-Cysteine Ligase Catalytic |
| GeX1 | Gerontoxanthone I |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Glutathione |
| GSSG | Glutathione disulphide |
| H2O2 | Hydrogen peroxide |
| HAECs | Human aortic endothelial cells |
| HFD | High fat diet |
| HFpEF | Heart failure with preserved ejection fraction |
| HIF-1α | Hypoxia-inducible factor-1α |
| HO-1 | Heme oxygenase-1 |
| Hsp70 | Heat shock protein 70 |
| I/R | Ischemia-reperfusion |
| IKKβ | IκB-kinase β |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-16 |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LDH | Lactate dehydrogenase |
| LDL | Low density lipoproteins |
| LOX-1 | lectin-like oxidized LDL receptor |
| LTF | Lactoferrin |
| LV | Left ventricular |
| MAO-A | Monoamine oxidase A |
| MAPK | Mitogen-activated protein kinase |
| MCL | Micheliolide |
| MCL | Micheliolide |
| MCP-1 | Macrophages enhanced monocyte chemotactic protein-1 |
| MCU | Mitochondrial calcium uniporter |
| McX | Macluraxanthone |
| MD1 | Myeloid differentiation protein 1 |
| MDA | Malondialdehyde |
| Mdivi-1 | Mitochondrial Division Inhibitor 1 |
| MFN1 | Mitofusins 1 |
| MFN2 | Mitofusins 2 |
| MI | Myocardial infarction |
| Mito-Esc | Mitochondria-targeted esculetin |
| MMPs | Matrix metalloproteinases |
| mPTP | Mitochondrial permeability transition pore |
| mtDNA | Mitochondrial DNA |
| mtKATP | Mitochondrial adenosine triphosphate (ATP)-sensitive potassium K channel |
| mTOR | Mechanistic target of rapamycin |
| mtROS | Mitochondrial ROS |
| NAC | N-acetylcysteine |
| NF-κB | Nuclear factor kappa B |
| NLRP3 | Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3 |
| NOS | Nitric oxide synthases |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| Nt-R | N-terminal arginine |
| 1O2 | Singlet oxygen |
| O2•− | Superoxide |
| O8G | 8-oxoguanosine |
| .OH | Hydroxyl radicals |
| OGD/R | Oxygen-glucose deprivation/re-oxygen |
| OPA1 | Optic atrophy 1 |
| ox-LDL | Oxidized low-density lipoprotein |
| OxPhos | Oxidative phosphorylation |
| PAI-1 | plasminogen activator inhibitor-1 |
| PDE5 | Phosphodiesterase 5 |
| Prx | Peroxiredoxin |
| RNS | Reactive nitrogen species |
| ROMO1 | Reactive oxygen species modulator 1 |
| ROS | Reactive Oxygen Species |
| RV | Right venteicular |
| Sirt1 | Sirtuin 1 |
| SLC26A4 | Solute carrier family 26 member 4 |
| SOD | Superoxide dismutase |
| SR-1 | Scavenger receptor-1 |
| T2D | Type 2 diabetes |
| TAC | Transverse aortic constriction |
| TFEB | Transcription factor EB |
| TIMPs | Tissue inhibitors of metalloproteinases |
| TLR | Toll-like receptor |
| TPP+ | Triphenylphosphonium cation |
| TRXox | Oxidized thioredoxin |
| TRXred | Reduced thioredoxin |
| Vcp | Valosin-containing protein |
| VDAC1 | Voltage-dependent anion channel 1 |
| VSMC | Vascular smooth muscle cells |
| VSMCs | Vascular smooth muscle cells |
| XO | Xanthine oxidase |
| XOR | Xanthine oxidoreductase |
| ZDHHC13 | Zinc finger DHHC type palmitoyltransferase 13 |
| ΔΨm | Mitochondrial membrane potential |
References
- Goh, R.S.J.; Chong, B.; Jayabaskaran, J.; Jauhari, S.M.; Chan, S.P.; Kueh, M.T.W.; Shankar, K.; Li, H.; Chin, Y.H.; Kong, G.; et al. The burden of cardiovascular disease in Asia from 2025 to 2050: a forecast analysis for East Asia, South Asia, South-East Asia, Central Asia, and high-income Asia Pacific regions. Lancet Reg Health West Pac 2024, 49, 101138. [Google Scholar] [CrossRef]
- Chong, B.; Jayabaskaran, J.; Jauhari, S.M.; Chan, S.P.; Goh, R.; Kueh, M.T.W.; Li, H.; Chin, Y.H.; Kong, G.; Anand, V.V.; et al. Global burden of cardiovascular diseases: projections from 2025 to 2050. Eur J Prev Cardiol 2024. [Google Scholar] [CrossRef]
- Slavin, S.D.; Khera, R.; Zafar, S.Y.; Nasir, K.; Warraich, H.J. Financial burden, distress, and toxicity in cardiovascular disease. Am Heart J 2021, 238, 75–84. [Google Scholar] [CrossRef]
- Zhang, B.; Schmidlin, T. Recent advances in cardiovascular disease research driven by metabolomics technologies in the context of systems biology. NPJ Metab Health Dis 2024, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med 1989, 320, 915–924. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Khan-Merchant, N.; Penumetcha, M.; Khan, B.V.; Santanam, N. Did the antioxidant trials fail to validate the oxidation hypothesis? Curr Atheroscler Rep 2001, 3, 392–398. [Google Scholar] [CrossRef]
- Naidoo, N.; van Dam, R.M.; Koh, W.P.; Chen, C.; Lee, Y.P.; Yuan, J.M.; Ong, C.N. Plasma vitamin E and coenzyme Q10 are not associated with a lower risk of acute myocardial infarction in Singapore Chinese adults. J Nutr 2012, 142, 1046–1052. [Google Scholar] [CrossRef]
- Shah, S.; Shiekh, Y.; Lawrence, J.A.; Ezekwueme, F.; Alam, M.; Kunwar, S.; Gordon, D.K. A Systematic Review of Effects of Vitamin E on the Cardiovascular System. Cureus 2021, 13, e15616. [Google Scholar] [CrossRef]
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of redox regulation in biology. Nat Rev Mol Cell Biol 2024, 25, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ming, H.; Qin, S.; Nice, E.C.; Dong, J.; Du, Z.; Huang, C. Redox regulation: mechanisms, biology and therapeutic targets in diseases. Signal Transduct Target Ther 2025, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cocheme, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat Rev Mol Cell Biol 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Sies, H. Findings in redox biology: From H(2)O(2) to oxidative stress. J Biol Chem 2020, 295, 13458–13473. [Google Scholar] [CrossRef]
- Jennings, R.T.; Singh, A.K.; Knaus, U.G. Redox regulator network in inflammatory signaling. Current Opinion in Physiology 2019, 9, 9–17. [Google Scholar] [CrossRef]
- Galley, J.C.; Straub, A.C. Redox Control of Vascular Function. Arterioscler Thromb Vasc Biol 2017, 37, e178–e184. [Google Scholar] [CrossRef]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat Rev Immunol 2021, 21, 363–381. [Google Scholar] [CrossRef]
- Xie, T.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Wu, J.; Sun, H. CoenzymeQ10-Induced Activation of AMPK-YAP-OPA1 Pathway Alleviates Atherosclerosis by Improving Mitochondrial Function, Inhibiting Oxidative Stress and Promoting Energy Metabolism. Front Pharmacol 2020, 11, 1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, Z.; Zheng, Y.; Kou, J.; Song, D.; Yu, X.; Dong, B.; Chen, T.; Yang, Y.; Gao, X.; et al. Melatonin inhibits atherosclerosis progression via galectin-3 downregulation to enhance autophagy and inhibit inflammation. J Pineal Res 2023, 74, e12855. [Google Scholar] [CrossRef]
- Ji, Y.; Leng, Y.; Lei, S.; Qiu, Z.; Ming, H.; Zhang, Y.; Zhang, A.; Wu, Y.; Xia, Z. The mitochondria-targeted antioxidant MitoQ ameliorates myocardial ischemia-reperfusion injury by enhancing PINK1/Parkin-mediated mitophagy in type 2 diabetic rats. Cell Stress Chaperones 2022, 27, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Faitg, J.; Tissot, C.; Konstantopoulos, D.; Laws, R.; Bourdier, G.; Andreux, P.A.; Davey, T.; Gallart-Ayala, H.; Ivanisevic, J.; et al. Urolithin A provides cardioprotection and mitochondrial quality enhancement preclinically and improves human cardiovascular health biomarkers. iScience 2025, 28, 111814. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.C.; Case, A.J. Redox biology in physiology and disease. Redox Biol 2019, 27, 101267. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Canton, M.; Sanchez-Rodriguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front Immunol 2021, 12, 734229. [Google Scholar] [CrossRef]
- Cosentino, F.; Francia, P.; Camici, G.G.; Pelicci, P.G.; Luscher, T.F.; Volpe, M. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler Thromb Vasc Biol 2008, 28, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Haslem, L.; Hays, J.M.; Hays, F.A. p66Shc in Cardiovascular Pathology. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr Opin Struct Biol 2019, 59, 91–97. [Google Scholar] [CrossRef]
- Jaishy, B.; Zhang, Q.; Chung, H.S.; Riehle, C.; Soto, J.; Jenkins, S.; Abel, P.; Cowart, L.A.; Van Eyk, J.E.; Abel, E.D. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J Lipid Res 2015, 56, 546–561. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef]
- Ligeon, L.A.; Pena-Francesch, M.; Vanoaica, L.D.; Nunez, N.G.; Talwar, D.; Dick, T.P.; Munz, C. Oxidation inhibits autophagy protein deconjugation from phagosomes to sustain MHC class II restricted antigen presentation. Nat Commun 2021, 12, 1508. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis 2018, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.; de Oliveira, J.; da Silva Pontes, L.V.; de Souza Junior, J.F.; Goncalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, I.A. ROS: Basic Concepts, Sources, Cellular Signaling, and its Implications in Aging Pathways. Oxid Med Cell Longev 2022, 2022, 1225578. [Google Scholar] [CrossRef]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann Transl Med 2017, 5, 326. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Jena, A.B.; Samal, R.R.; Bhol, N.K.; Duttaroy, A.K. Cellular Red-Ox system in health and disease: The latest update. Biomed Pharmacother 2023, 162, 114606. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several lines of antioxidant defense against oxidative stress: antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch Toxicol 2024, 98, 1323–1367. [Google Scholar] [CrossRef]
- Chai, Y.C.; Mieyal, J.J. Glutathione and Glutaredoxin-Key Players in Cellular Redox Homeostasis and Signaling. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Chandimali, N.; Bak, S.G.; Park, E.H.; Lim, H.J.; Won, Y.S.; Kim, E.K.; Park, S.I.; Lee, S.J. Free radicals and their impact on health and antioxidant defenses: a review. Cell Death Discov 2025, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct Target Ther 2023, 8, 333. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Redox signaling in cardiovascular health and disease. Free Radic Biol Med 2013, 61, 473–501. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Liu, J.; Finkel, T. Mitohormesis. Cell Metab 2023, 35, 1872–1886. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis 2011, 2, e213. [Google Scholar] [CrossRef]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal 2008, 10, 1713–1765. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Yang, Y.; Zhou, K.; Deng, N.; Tian, Z.; Wu, Z.; Liu, X.; Zhang, F.; Jiang, Z. The role of ROS-induced pyroptosis in CVD. Front Cardiovasc Med 2023, 10, 1116509. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 2015, 116, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.Z.; Rabinovitch, P.S.; Tabas, I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ Res 2014, 114, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Fei, Q.; Ma, H.; Zou, J.; Wang, W.; Zhu, L.; Deng, H.; Meng, M.; Tan, S.; Zhang, H.; Xiao, X.; et al. Metformin protects against ischaemic myocardial injury by alleviating autophagy-ROS-NLRP3-mediated inflammatory response in macrophages. J Mol Cell Cardiol 2020, 145, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Rizky, L.; Stefanovic, N.; Tate, M.; Ritchie, R.H.; Ward, K.W.; de Haan, J.B. The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) activator dh404 protects against diabetes-induced endothelial dysfunction. Cardiovasc Diabetol 2017, 16, 33. [Google Scholar] [CrossRef]
- Xu, L.; Yang, X.; Liu, X.T.; Li, X.Y.; Zhu, H.Z.; Xie, Y.H.; Wang, S.W.; Li, Y.; Zhao, Y. Carvacrol alleviates LPS-induced myocardial dysfunction by inhibiting the TLR4/MyD88/NF-kappaB and NLRP3 inflammasome in cardiomyocytes. J Inflamm (Lond) 2024, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Ziehr, B.K.; MacDonald, J.A. Regulation of NLRPs by reactive oxygen species: A story of crosstalk. Biochim Biophys Acta Mol Cell Res 2024, 1871, 119823. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Pan, L.; Xiao, Z.; Rao, F.; Yakupu, W.; Rouzi, A.; Cheng, K.; Pang, Y.; Su, Y.; Wu, D. Kirenol attenuates pressure overload-induced heart failure by enhancing autophagy in macrophages. Int J Cardiol 2025, 440, 133681. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, H.; Hu, X.; Fu, W. Bone marrow NLRP3 inflammasome-IL-1beta signal regulates post-myocardial infarction megakaryocyte development and platelet production. Biochem Biophys Res Commun 2021, 585, 96–102. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Goldstein, D.R. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol 2021, 18, 58–68. [Google Scholar] [CrossRef]
- Velusamy, P.; Buckley, D.J.; Greaney, J.L.; Case, A.J.; Fadel, P.J.; Trott, D.W. IL-6 induces mitochondrial ROS production and blunts NO bioavailability in human aortic endothelial cells. Am J Physiol Regul Integr Comp Physiol 2025, 328, R509–R514. [Google Scholar] [CrossRef]
- Tu, V.C.; Bahl, J.J.; Chen, Q.M. Signals of oxidant-induced cardiomyocyte hypertrophy: key activation of p70 S6 kinase-1 and phosphoinositide 3-kinase. J Pharmacol Exp Ther 2002, 300, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Ushio-Fukai, M.; Hitomi, H.; Rocic, P.; Kingsley, M.J.; Pfahnl, C.; Weber, D.S.; Alexander, R.W.; Griendling, K.K. Role of p38 MAPK and MAPKAPK-2 in angiotensin II-induced Akt activation in vascular smooth muscle cells. Am J Physiol Cell Physiol 2004, 287, C494–499. [Google Scholar] [CrossRef]
- Tan, S.M.; Sharma, A.; Stefanovic, N.; Yuen, D.Y.; Karagiannis, T.C.; Meyer, C.; Ward, K.W.; Cooper, M.E.; de Haan, J.B. Derivative of bardoxolone methyl, dh404, in an inverse dose-dependent manner lessens diabetes-associated atherosclerosis and improves diabetic kidney disease. Diabetes 2014, 63, 3091–3103. [Google Scholar] [CrossRef]
- Xu, X.; Pang, Y.; Fan, X. Mitochondria in oxidative stress, inflammation and aging: from mechanisms to therapeutic advances. Signal Transduct Target Ther 2025, 10, 190. [Google Scholar] [CrossRef]
- Jensen, P.K. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles. I. pH dependency and hydrogen peroxide formation. Biochim Biophys Acta 1966, 122, 157–166. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem J 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Loschen, G.; Flohe, L.; Chance, B. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett 1971, 18, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Loschen, G.; Azzi, A.; Richter, C.; Flohe, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett 1974, 42, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Zuccolotto Dos Reis, F.H. Mitochondria and the heart. Eur Heart J 2024, 45, 1963–1964. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther 2024, 9, 124. [Google Scholar] [CrossRef]
- Ravindran, R.; Gustafsson, A.B. Mitochondrial quality control in cardiomyocytes: safeguarding the heart against disease and ageing. Nat Rev Cardiol 2025. [Google Scholar] [CrossRef]
- Su, Z.D.; Li, C.Q.; Wang, H.W.; Zheng, M.M.; Chen, Q.W. Inhibition of DRP1-dependent mitochondrial fission by Mdivi-1 alleviates atherosclerosis through the modulation of M1 polarization. J Transl Med 2023, 21, 427. [Google Scholar] [CrossRef]
- Palee, S.; Higgins, L.; Leech, T.; Chattipakorn, S.C.; Chattipakorn, N. Acute metformin treatment provides cardioprotection via improved mitochondrial function in cardiac ischemia / reperfusion injury. Biomed Pharmacother 2020, 130, 110604. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 2008, 294, H570–578. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cui, Y.J.; Liu, Y.; Li, H.X.; Su, Y.D.; Li, S.N.; Wang, L.L.; Zhao, Y.W.; Wang, S.X.; Yan, F.; Dong, B. ATP6AP2 knockdown in cardiomyocyte deteriorates heart function via compromising autophagic flux and NLRP3 inflammasome activation. Cell Death Discov 2022, 8, 161. [Google Scholar] [CrossRef]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc Res 2010, 85, 473–483. [Google Scholar] [CrossRef]
- Zhao, J.; Li, J.; Li, G.; Chen, M. The role of mitochondria-associated membranes mediated ROS on NLRP3 inflammasome in cardiovascular diseases. Front Cardiovasc Med 2022, 9, 1059576. [Google Scholar] [CrossRef]
- Luo, X.; Wang, Y.; Zhu, X.; Chen, Y.; Xu, B.; Bai, X.; Weng, X.; Xu, J.; Tao, Y.; Yang, D.; et al. MCL attenuates atherosclerosis by suppressing macrophage ferroptosis via targeting KEAP1/NRF2 interaction. Redox Biol 2024, 69, 102987. [Google Scholar] [CrossRef]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ Res 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Jin, J.Y.; Wei, X.X.; Zhi, X.L.; Wang, X.H.; Meng, D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin 2021, 42, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Garcia, I.; Innis-Whitehouse, W.; Lopez, A.; Keniry, M.; Gilkerson, R. Oxidative insults disrupt OPA1-mediated mitochondrial dynamics in cultured mammalian cells. Redox Rep 2018, 23, 160–167. [Google Scholar] [CrossRef]
- Chehaitly, A.; Guihot, A.L.; Proux, C.; Grimaud, L.; Aurriere, J.; Legouriellec, B.; Rivron, J.; Vessieres, E.; Tetaud, C.; Zorzano, A.; et al. Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol 2019, 11, 1069–1082. [Google Scholar] [CrossRef]
- Devant, P.; Borsic, E.; Ngwa, E.M.; Xiao, H.; Chouchani, E.T.; Thiagarajah, J.R.; Hafner-Bratkovic, I.; Evavold, C.L.; Kagan, J.C. Gasdermin D pore-forming activity is redox-sensitive. Cell Rep 2023, 42, 112008. [Google Scholar] [CrossRef]
- Evavold, C.L.; Hafner-Bratkovic, I.; Devant, P.; D'Andrea, J.M.; Ngwa, E.M.; Borsic, E.; Doench, J.G.; LaFleur, M.W.; Sharpe, A.H.; Thiagarajah, J.R.; Kagan, J.C. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 2021, 184, 4495–4511 e4419. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Dai, S.; Zhong, L.; Shi, X.; Fan, X.; Zhong, X.; Lin, W.; Su, L.; Lin, S.; Han, B.; et al. GSDMD (Gasdermin D) Mediates Pathological Cardiac Hypertrophy and Generates a Feed-Forward Amplification Cascade via Mitochondria-STING (Stimulator of Interferon Genes) Axis. Hypertension 2022, 79, 2505–2518. [Google Scholar] [CrossRef]
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ Res 2021, 129, 383–396. [Google Scholar] [CrossRef]
- Ye, X.; Zhang, P.; Zhang, Y.; Luan, J.; Xu, C.; Wu, Z.; Ju, D.; Hu, W. GSDMD contributes to myocardial reperfusion injury by regulating pyroptosis. Front Immunol 2022, 13, 893914. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res 2013, 112, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Foster, D.B.; Rucker, J.; O'Rourke, B.; Kass, D.A.; Van Eyk, J.E. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res 2011, 109, 750–757. [Google Scholar] [CrossRef]
- Campanella, M.; Pinton, P.; Rizzuto, R. Mitochondrial Ca2+ homeostasis in health and disease. Biol Res 2004, 37, 653–660. [Google Scholar] [CrossRef]
- Xie, A.; Song, Z.; Liu, H.; Zhou, A.; Shi, G.; Wang, Q.; Gu, L.; Liu, M.; Xie, L.H.; Qu, Z.; Dudley, S.C., Jr. Mitochondrial Ca(2+) Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy. J Am Heart Assoc 2018, 7. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ 2019, 26, 179–195. [Google Scholar] [CrossRef]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca(2+) Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol Cell 2017, 65, 1014–1028 e1017. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.E.; Wilson, N.; Son, S.M.; Obrocki, P.; Wrobel, L.; Rob, M.; Takla, M.; Korolchuk, V.I.; Rubinsztein, D.C. Autophagy, aging, and age-related neurodegeneration. Neuron 2025, 113, 29–48. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Rout, A.K.; Strub, M.P.; Piszczek, G.; Tjandra, N. Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J Biol Chem 2014, 289, 35111–35123. [Google Scholar] [CrossRef]
- Gomez-Virgilio, L.; Silva-Lucero, M.D.; Flores-Morelos, D.S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.M.; Zacapala-Gomez, A.E.; Luna-Munoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11. [Google Scholar] [CrossRef]
- Holczer, M.; Hajdu, B.; Lorincz, T.; Szarka, A.; Banhegyi, G.; Kapuy, O. Fine-tuning of AMPK-ULK1-mTORC1 regulatory triangle is crucial for autophagy oscillation. Sci Rep 2020, 10, 17803. [Google Scholar] [CrossRef]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res 2014, 24, 42–57. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol Biol 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Xu, C.; Cao, Y.; Liu, R.; Liu, L.; Zhang, W.; Fang, X.; Jia, S.; Ye, J.; Liu, Y.; Weng, L.; et al. Mitophagy-regulated mitochondrial health strongly protects the heart against cardiac dysfunction after acute myocardial infarction. J Cell Mol Med 2022, 26, 1315–1326. [Google Scholar] [CrossRef]
- Evans, S.; Ma, X.; Wang, X.; Chen, Y.; Zhao, C.; Weinheimer, C.J.; Kovacs, A.; Finck, B.; Diwan, A.; Mann, D.L. Targeting the Autophagy-Lysosome Pathway in a Pathophysiologically Relevant Murine Model of Reversible Heart Failure. JACC Basic Transl Sci 2022, 7, 1214–1228. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab 2019, 29, 803–826. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Interactions between reactive oxygen species and autophagy: Special issue: Death mechanisms in cellular homeostasis. Biochim Biophys Acta Mol Cell Res 2021, 1868, 119041. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhou, X.; Yang, T.; Wang, L.; Feng, L.; Wang, Z.; Xu, J.; Jing, W.; Wang, T.; Su, H.; et al. The role of autophagy in cardiovascular disease: Cross-interference of signaling pathways and underlying therapeutic targets. Front Cardiovasc Med 2023, 10, 1088575. [Google Scholar] [CrossRef]
- Dan, L.X.; Xie, S.P. Autophagy in cardiac pathophysiology: Navigating the complex roles and therapeutic potential in cardiac fibrosis. Life Sci 2025, 123761. [Google Scholar] [CrossRef]
- Park, J.M.; Lee, D.H.; Kim, D.H. Redefining the role of AMPK in autophagy and the energy stress response. Nat Commun 2023, 14, 2994. [Google Scholar] [CrossRef]
- Agostini, F.; Bisaglia, M.; Plotegher, N. Linking ROS Levels to Autophagy: The Key Role of AMPK. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Wible, D.J.; Bratton, S.B. Reciprocity in ROS and autophagic signaling. Curr Opin Toxicol 2018, 7, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef]
- Tabata, K.; Imai, K.; Fukuda, K.; Yamamoto, K.; Kunugi, H.; Fujita, T.; Kaminishi, T.; Tischer, C.; Neumann, B.; Reither, S.; et al. Palmitoylation of ULK1 by ZDHHC13 plays a crucial role in autophagy. Nat Commun 2024, 15, 7194. [Google Scholar] [CrossRef]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol 2020, 36, 101679. [Google Scholar] [CrossRef]
- She, C.; Zhu, L.Q.; Zhen, Y.F.; Wang, X.D.; Dong, Q.R. Activation of AMPK protects against hydrogen peroxide-induced osteoblast apoptosis through autophagy induction and NADPH maintenance: new implications for osteonecrosis treatment? Cell Signal 2014, 26, 1–8. [Google Scholar] [CrossRef]
- Kimball, S.R.; Gordon, B.S.; Moyer, J.E.; Dennis, M.D.; Jefferson, L.S. Leucine induced dephosphorylation of Sestrin2 promotes mTORC1 activation. Cell Signal 2016, 28, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Liu, P.F.; Chang, C.H.; Lin, Y.C.; Chen, Y.J.; Shu, C.W. The interplay of autophagy and oxidative stress in the pathogenesis and therapy of retinal degenerative diseases. Cell Biosci 2022, 12, 1. [Google Scholar] [CrossRef]
- Zheng, X.; Yang, Z.; Gu, Q.; Xia, F.; Fu, Y.; Liu, P.; Yin, X.M.; Li, M. The protease activity of human ATG4B is regulated by reversible oxidative modification. Autophagy 2020, 16, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, X.Y.; Liu, Y.J.; Feng, J.; Wu, Y.; Shen, H.M.; Lu, G.D. Full-coverage regulations of autophagy by ROS: from induction to maturation. Autophagy 2022, 18, 1240–1255. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Palabiyik, A.A. The role of Bcl-2 in controlling the transition between autophagy and apoptosis (Review). Mol Med Rep 2025, 32. [Google Scholar] [CrossRef]
- Carroll, B.; Otten, E.G.; Manni, D.; Stefanatos, R.; Menzies, F.M.; Smith, G.R.; Jurk, D.; Kenneth, N.; Wilkinson, S.; Passos, J.F.; et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat Commun 2018, 9, 256. [Google Scholar] [CrossRef]
- Wei, R.; Enaka, M.; Muragaki, Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci Rep 2019, 9, 10366. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.R.; Zhang, M.H.; Chen, Y.J.; Sun, Y.L.; Gao, Z.M.; Li, Z.J.; Zhang, G.P.; Qin, Y.; Dai, X.Y.; Yu, X.Y.; Wu, X.Q. Urolithin A ameliorates obesity-induced metabolic cardiomyopathy in mice via mitophagy activation. Acta Pharmacol Sin 2023, 44, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther 2023, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem 2017, 292, 16697–16708. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.K. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson's disease. Ageing Res Rev 2020, 62, 101107. [Google Scholar] [CrossRef]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid Redox Signal 2021, 34, 517–530. [Google Scholar] [CrossRef]
- Xu, M.; Wang, W.; Cheng, J.; Qu, H.; Xu, M.; Wang, L. Effects of mitochondrial dysfunction on cellular function: Role in atherosclerosis. Biomed Pharmacother 2024, 174, 116587. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.M. Mitochondrial Dysfunction in Cardiovascular Diseases. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef] [PubMed]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Michalak, K.P.; Michalak, A.Z. Understanding chronic inflammation: couplings between cytokines, ROS, NO, Ca(i) (2+), HIF-1alpha, Nrf2 and autophagy. Front Immunol 2025, 16, 1558263. [Google Scholar] [CrossRef]
- Ding, M.; Lei, J.; Han, H.; Li, W.; Qu, Y.; Fu, E.; Fu, F.; Wang, X. SIRT1 protects against myocardial ischemia-reperfusion injury via activating eNOS in diabetic rats. Cardiovasc Diabetol 2015, 14, 143. [Google Scholar] [CrossRef]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct Target Ther 2022, 7, 402. [Google Scholar] [CrossRef]
- Jia, D.; Ping, W.; Wang, M.; Wang, D.; Zhang, L.; Cao, Y. SIRT1 mediates the inflammatory response of macrophages and regulates the TIMP3/ADAM17 pathway in atherosclerosis. Exp Cell Res 2024, 442, 114253. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Wang, Y.; Chao, Y.; Zhang, J.; Jia, Y.; Tie, J.; Hu, D. Regulation of SIRT1 and Its Roles in Inflammation. Front Immunol 2022, 13, 831168. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Li, Y.; Yu, S.; Zhao, Y. SIRT1/PGC-1alpha Signaling Promotes Mitochondrial Functional Recovery and Reduces Apoptosis after Intracerebral Hemorrhage in Rats. Front Mol Neurosci 2017, 10, 443. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, D.; Wei, C.; Liu, L.; Xin, Z.; Gao, H.; Gao, R. The relationship between SIRT1 and inflammation: a systematic review and meta-analysis. Front Immunol 2024, 15, 1465849. [Google Scholar] [CrossRef]
- Sung, J.Y.; Kim, S.G.; Kang, Y.J.; Park, S.Y.; Choi, H.C. SIRT1-dependent PGC-1alpha deacetylation by SRT1720 rescues progression of atherosclerosis by enhancing mitochondrial function. Biochim Biophys Acta Mol Cell Biol Lipids 2024, 1869, 159453. [Google Scholar] [CrossRef]
- Jeong, S.J.; Zhang, X.; Rodriguez-Velez, A.; Evans, T.D.; Razani, B. p62/SQSTM1 and Selective Autophagy in Cardiometabolic Diseases. Antioxid Redox Signal 2019, 31, 458–471. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Q.; Li, Q.; Xing, S.; Liu, Y.; Liu, Y.; Chen, Y.; Liu, W.; Feng, F.; Sun, H. p62/SQSTM1, a Central but Unexploited Target: Advances in Its Physiological/Pathogenic Functions and Small Molecular Modulators. J Med Chem 2020, 63, 10135–10157. [Google Scholar] [CrossRef]
- Ma, Y.; Zhu, S.; Lv, T.; Gu, X.; Feng, H.; Zhen, J.; Xin, W.; Wan, Q. SQSTM1/p62 Controls mtDNA Expression and Participates in Mitochondrial Energetic Adaption via MRPL12. iScience 2020, 23, 101428. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Fatahian, A.N.; Rouzbehani, O.M.T.; Hathaway, M.A.; Mosleh, T.; Vinod, V.; Vowles, S.; Stephens, S.L.; Chung, S.D.; Cao, I.D.; et al. Sequestosome 1 (p62) mitigates hypoxia-induced cardiac dysfunction by stabilizing hypoxia-inducible factor 1alpha and nuclear factor erythroid 2-related factor 2. Cardiovasc Res 2024, 120, 531–547. [Google Scholar] [CrossRef]
- Quan, X.; Yang, Y.; Liu, X.; Kaltwasser, B.; Pillath-Eilers, M.; Walkenfort, B.; Voortmann, S.; Mohamud Yusuf, A.; Hagemann, N.; Wang, C.; et al. Autophagy hub-protein p62 orchestrates oxidative, endoplasmic reticulum stress, and inflammatory responses post-ischemia, exacerbating stroke outcome. Redox Biol 2025, 84, 103700. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front Cell Dev Biol 2022, 10, 793328. [Google Scholar] [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis 2022, 13, 444. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, H.; Liu, J. P66Shc Deletion Ameliorates Oxidative Stress and Cardiac Dysfunction in Pressure Overload-Induced Heart Failure. J Card Fail 2020, 26, 243–253. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Frijhoff, J.; Dagnell, M.; Augsten, M.; Beltrami, E.; Giorgio, M.; Ostman, A. The mitochondrial reactive oxygen species regulator p66Shc controls PDGF-induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic Biol Med 2014, 68, 268–277. [Google Scholar] [CrossRef]
- Mir, H.A.; Ali, R.; Mushtaq, U.; Khanday, F.A. Structure-functional implications of longevity protein p66Shc in health and disease. Ageing Res Rev 2020, 63, 101139. [Google Scholar] [CrossRef]
- Falk, E. Pathogenesis of atherosclerosis. J Am Coll Cardiol 2006, 47, C7–12. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Pratico, D.; Lin, L.; Mantzoros, C.S.; Bahijri, S.; Tuomilehto, J.; Ren, J. Inflammation in atherosclerosis: pathophysiology and mechanisms. Cell Death Dis 2024, 15, 817. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat Rev Dis Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Qian, C.; You, X.; Gao, B.; Sun, Y.; Liu, C. The Role of ROS in Atherosclerosis and ROS-Based Nanotherapeutics for Atherosclerosis: Atherosclerotic Lesion Targeting, ROS Scavenging, and ROS-Responsive Activity. ACS Omega 2025, 10, 22366–22381. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Engelen, S.E.; Robinson, A.J.B.; Zurke, Y.X.; Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat Rev Cardiol 2022, 19, 522–542. [Google Scholar] [CrossRef]
- Wang, Y.; Tabas, I. Emerging roles of mitochondria ROS in atherosclerotic lesions: causation or association? J Atheroscler Thromb 2014, 21, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Lozhkin, A.; Hayami, T.; Levin, J.; Silveira Fernandes Chamon, J.; Abdel-Latif, A.; Runge, M.S.; Madamanchi, N.R. Mitochondrial dysfunction and metabolic reprogramming induce macrophage pro-inflammatory phenotype switch and atherosclerosis progression in aging. Front Immunol 2024, 15, 1410832. [Google Scholar] [CrossRef]
- Karnewar, S.; Pulipaka, S.; Katta, S.; Panuganti, D.; Neeli, P.K.; Thennati, R.; Jerald, M.K.; Kotamraju, S. Mitochondria-targeted esculetin mitigates atherosclerosis in the setting of aging via the modulation of SIRT1-mediated vascular cell senescence and mitochondrial function in Apoe(-/-) mice. Atherosclerosis 2022, 356, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Liang, J.; Lu, Y.; Ding, J.; Peng, J.; Li, F.; Dai, J.; Liu, Y.; Wang, J.; Wan, C.; Luo, P. Lactoferrin influences atherosclerotic progression by modulating macrophagic AMPK/mTOR signaling-dependent autophagy. Sci Rep 2025, 15, 10585. [Google Scholar] [CrossRef]
- Leite-Moreira, A.M.; Lourenco, A.P.; Falcao-Pires, I.; Leite-Moreira, A.F. Pivotal role of microRNAs in cardiac physiology and heart failure. Drug Discov Today 2013, 18, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ Res 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Martens, M.D.; Holody, C.D.; Wells, L.; Silver, H.L.; Morales-Llamas, D.Y.; Du, W.W.; Reeks, C.; Khairy, M.; Chen, H.; Ferdaoussi, M.; et al. Reactive Oxygen Species Modulator 1 Plays an Obligate Role in Cardiomyocyte Hypertrophy. Circ Res 2024, 134, 114–116. [Google Scholar] [CrossRef]
- Tang, L.Q.; Wang, L.L.; Tang, Q.F.; Wang, W. SLC26A4 regulates autophagy and activates the NLRP3 inflammasome to mediate pathological cardiac hypertrophy. Sci Rep 2025, 15, 12511. [Google Scholar] [CrossRef]
- Qiu, R.B.; Zhao, S.T.; Xu, Z.Q.; Hu, L.J.; Zeng, R.Y.; Qiu, Z.C.; Peng, H.Z.; Zhou, L.F.; Cao, Y.P.; Wan, L. Thymoquinone mitigates cardiac hypertrophy by activating adaptive autophagy via the PPAR-gamma/14-3-3gamma pathway. Int J Mol Med 2025, 55. [Google Scholar] [CrossRef]
- Lu, X.; Tong, T.; Sun, H.; Chen, Y.; Shao, Y.; Shi, P.; Que, L.; Liu, L.; Zhu, G.; Chen, Q.; et al. ECSIT-X4 is Required for Preventing Pressure Overload-Induced Cardiac Hypertrophy via Regulating Mitochondrial STAT3. Adv Sci (Weinh) 2025, 12, e2414358. [Google Scholar] [CrossRef]
- Yang, A.; Cao, J.; Gu, J.; Zhu, X.; Qian, Y.; Qian, H.; Zhao, W.; Wang, Y.; Zhu, W. SBK3 suppresses angiotensin II-induced cardiac hypertrophy by regulating mitochondrial metabolism. Sci Rep 2025, 15, 22796. [Google Scholar] [CrossRef]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol Rev 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Duan, Q.; Yang, W.; Zhu, X.; Feng, Z.; Song, J.; Xu, X.; Kong, M.; Mao, J.; Shen, J.; Deng, Y.; et al. Deptor protects against myocardial ischemia-reperfusion injury by regulating the mTOR signaling and autophagy. Cell Death Discov 2024, 10, 508. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Y.; Yan, L.; Tan, M.; Jin, Y.; Yin, Y.; Han, L.; Ma, X.; Li, Y.; Yang, T.; et al. Corosolic acid attenuates cardiac ischemia/reperfusion injury through the PHB2/PINK1/parkin/mitophagy pathway. iScience 2024, 27, 110448. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, Z.; Guo, F.; Zhang, Y.; Peng, H.; Zhang, H.; Liu, Z.; Cao, C.; Xin, G.; Chen, Y.Y.; Fu, J. Mitophagy in ischemic heart disease: molecular mechanisms and clinical management. Cell Death Dis 2024, 15, 934. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.Y.; Vanhoutte, D.; Del Re, D.P.; Purcell, N.H.; Ling, H.; Banerjee, I.; Bossuyt, J.; Lang, R.A.; Zheng, Y.; Matkovich, S.J.; et al. RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Invest 2011, 121, 3269–3276. [Google Scholar] [CrossRef]
- Zhao, X.; Ding, E.Y.; Yu, O.M.; Xiang, S.Y.; Tan-Sah, V.P.; Yung, B.S.; Hedgpeth, J.; Neubig, R.R.; Lau, L.F.; Brown, J.H.; Miyamoto, S. Induction of the matricellular protein CCN1 through RhoA and MRTF-A contributes to ischemic cardioprotection. J Mol Cell Cardiol 2014, 75, 152–161. [Google Scholar] [CrossRef]
- Tu, M.; Tan, V.P.; Yu, J.D.; Tripathi, R.; Bigham, Z.; Barlow, M.; Smith, J.M.; Brown, J.H.; Miyamoto, S. RhoA signaling increases mitophagy and protects cardiomyocytes against ischemia by stabilizing PINK1 protein and recruiting Parkin to mitochondria. Cell Death Differ 2022, 29, 2472–2486. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Wu, M.; Zhang, L.; Fu, W.; Yang, J.; Zhang, B.; Zheng, Z.; Zhang, H.; Lao, Y.; Xu, H. Gerontoxanthone I and Macluraxanthone Induce Mitophagy and Attenuate Ischemia/Reperfusion Injury. Front Pharmacol 2020, 11, 452. [Google Scholar] [CrossRef]
- Wu, J.; Yang, Y.; Gao, Y.; Wang, Z.; Ma, J. Melatonin Attenuates Anoxia/Reoxygenation Injury by Inhibiting Excessive Mitophagy Through the MT2/SIRT3/FoxO3a Signaling Pathway in H9c2 Cells. Drug Des Devel Ther 2020, 14, 2047–2060. [Google Scholar] [CrossRef]
- Bai, Y.; Yang, Y.; Gao, Y.; Lin, D.; Wang, Z.; Ma, J. Melatonin postconditioning ameliorates anoxia/reoxygenation injury by regulating mitophagy and mitochondrial dynamics in a SIRT3-dependent manner. Eur J Pharmacol 2021, 904, 174157. [Google Scholar] [CrossRef]
- Wu, J.; Yang, Y.; Lin, D.; Wang, Z.; Ma, J. SIRT3 and RORalpha are two prospective targets against mitophagy during simulated ischemia/reperfusion injury in H9c2 cells. Heliyon 2024, 10, e30568. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.; Li, C.; Wang, L.; Jiang, Y.; Yang, L.; Li, H.; Yang, P.; Tao, J.; Lu, D.; Sun, L. ZNF143 regulates autophagic flux to alleviate myocardial ischemia/reperfusion injury through Raptor. Cell Signal 2022, 99, 110444. [Google Scholar] [CrossRef]
- Tang, J.; Yoon, N.; Dadson, K.; Sung, H.K.; Lei, Y.; Dang, T.Q.; Chung, W.Y.; Ahmed, S.; Abdul-Sater, A.A.; Wu, J.; et al. Impaired autophagy flux contributes to enhanced ischemia reperfusion injury in the diabetic heart. Autophagy Rep 2024, 3, 2330327. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, X.; Lee, S.H.; Chen, F.; Jiang, K.; Tu, Z.; Liu, Z.; Du, J.; Wang, L.; Yin, C.; et al. Diabetes Exacerbates Myocardial Ischemia/Reperfusion Injury by Down-Regulation of MicroRNA and Up-Regulation of O-GlcNAcylation. JACC Basic Transl Sci 2018, 3, 350–362. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, D. Research progress on the effects of novel hypoglycemic drugs in diabetes combined with myocardial ischemia/reperfusion injury. Ageing Res Rev 2023, 86, 101884. [Google Scholar] [CrossRef]
- Lefer, D.J.; Granger, D.N. Oxidative stress and cardiac disease. Am J Med 2000, 109, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Huang, S.; Wei, G.; Sun, Y.; Li, C.; Si, X.; Chen, Y.; Tang, Z.; Li, X.; Chen, Y.; et al. CircRNA Samd4 induces cardiac repair after myocardial infarction by blocking mitochondria-derived ROS output. Mol Ther 2022, 30, 3477–3498. [Google Scholar] [CrossRef]
- Shams, P.; Malik, A.; Chhabra, L. Heart Failure (Congestive Heart Failure). In StatPearls; Treasure Island (FL), 2025.
- D'Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid Med Cell Longev 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Osko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczynska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintron, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W., 2nd; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res 2010, 106, 1541–1548. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Lee, H.; Jiang, X.; Perwaiz, I.; Yu, P.; Wang, J.; Wang, Y.; Huttemann, M.; Felder, R.A.; Sibley, D.R.; Polster, B.M.; et al. Dopamine D(5) receptor-mediated decreases in mitochondrial reactive oxygen species production are cAMP and autophagy dependent. Hypertens Res 2021, 44, 628–641. [Google Scholar] [CrossRef]
- Li, G.; Liu, X.; Long, A.; Feng, J.; Sun, S.; Yang, Z.; Jiang, R.; Jiang, X. An inducible mouse model of heart failure targeted to cardiac Drd5 deficiency detonating mitochondrial oxidative stress. Int J Cardiol 2024, 396, 131560. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Kong, B.; Shuai, W.; Zhang, J.J.; Huang, H. MD1 deletion exaggerates cardiomyocyte autophagy induced by heart failure with preserved ejection fraction through ROS/MAPK signalling pathway. J Cell Mol Med 2020, 24, 9300–9312. [Google Scholar] [CrossRef] [PubMed]
- Chi, R.F.; Li, L.; Wang, A.L.; Yang, H.; Xi, J.; Zhu, Z.F.; Wang, K.; Li, B.; Yang, L.G.; Qin, F.Z.; Zhang, C. Enhanced oxidative stress mediates pathological autophagy and necroptosis in cardiac myocytes in pressure overload induced heart failure in rats. Clin Exp Pharmacol Physiol 2022, 49, 60–69. [Google Scholar] [CrossRef]
- Bielawska, M.; Warszynska, M.; Stefanska, M.; Blyszczuk, P. Autophagy in Heart Failure: Insights into Mechanisms and Therapeutic Implications. J Cardiovasc Dev Dis 2023, 10. [Google Scholar] [CrossRef]
- Chang, W.; Xiao, D.; Fang, X.; Wang, J. Oxidative modification of miR-30c promotes cardiac fibroblast proliferation via CDKN2C mismatch. Sci Rep 2024, 14, 13085. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Huo, J.L.; Feng, Q.; Pan, S.; Fu, W.J.; Liu, Z.; Liu, Z. Diabetic cardiomyopathy: Early diagnostic biomarkers, pathogenetic mechanisms, and therapeutic interventions. Cell Death Discov 2023, 9, 256. [Google Scholar] [CrossRef]
- Xu, N.; Liu, S.; Zhang, Y.; Chen, Y.; Zuo, Y.; Tan, X.; Liao, B.; Li, P.; Feng, J. Oxidative stress signaling in the pathogenesis of diabetic cardiomyopathy and the potential therapeutic role of antioxidant naringenin. Redox Rep 2023, 28, 2246720. [Google Scholar] [CrossRef]
- Ilkun, O.; Wilde, N.; Tuinei, J.; Pires, K.M.; Zhu, Y.; Bugger, H.; Soto, J.; Wayment, B.; Olsen, C.; Litwin, S.E.; Abel, E.D. Antioxidant treatment normalizes mitochondrial energetics and myocardial insulin sensitivity independently of changes in systemic metabolic homeostasis in a mouse model of the metabolic syndrome. J Mol Cell Cardiol 2015, 85, 104–116. [Google Scholar] [CrossRef]
- Hernandez-Mijares, A.; Rocha, M.; Rovira-Llopis, S.; Banuls, C.; Bellod, L.; de Pablo, C.; Alvarez, A.; Roldan-Torres, I.; Sola-Izquierdo, E.; Victor, V.M. Human leukocyte/endothelial cell interactions and mitochondrial dysfunction in type 2 diabetic patients and their association with silent myocardial ischemia. Diabetes Care 2013, 36, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Firgany, A.A.M.A.-H.a.A.E.L. Favorable outcomes of metformin on coronary microvasculature in experimental diabetic cardiomyopathy. J Mol Histol 2018, 49, 639–649. [Google Scholar] [CrossRef]
- Chen, X.; Qian, J.; Wang, L.; Li, J.; Zhao, Y.; Han, J.; Khan, Z.; Chen, X.; Wang, J.; Liang, G. Kaempferol attenuates hyperglycemia-induced cardiac injuries by inhibiting inflammatory responses and oxidative stress. Endocrine 2018, 60, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S. A Synopsis of the Associations of Oxidative Stress, ROS, and Antioxidants with Diabetes Mellitus. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Wu, Y. Metformin inhibits mitochondrial dysfunction and apoptosis in cardiomyocytes induced by high glucose via upregulating AMPK activity. Exp Biol Med (Maywood) 2023, 248, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, S.; Sun, H.; Li, Y.; Feng, J. Naringenin ameliorates myocardial injury in STZ-induced diabetic mice by reducing oxidative stress, inflammation and apoptosis via regulating the Nrf2 and NF-kappaB signaling pathways. Front Cardiovasc Med 2022, 9, 946766. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; Diaz-Morales, N.; Rovira-Llopis, S.; de Maranon, A.M.; Orden, S.; Alvarez, A.; Banuls, C.; Rocha, M.; Murphy, M.P.; Hernandez-Mijares, A.; Victor, V.M. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol 2016, 10, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shen, Q.; Chen, Y.; Pan, R.; Kuang, S.; Liu, G.; Sun, G.; Sun, X. Myricitrin Alleviates Oxidative Stress-induced Inflammation and Apoptosis and Protects Mice against Diabetic Cardiomyopathy. Sci Rep 2017, 7, 44239. [Google Scholar] [CrossRef]
- Zhang, K.; Li, Y.; Ge, X.; Meng, L.; Kong, J.; Meng, X. Regulatory T cells protect against diabetic cardiomyopathy in db/db mice. J Diabetes Investig 2024, 15, 1191–1201. [Google Scholar] [CrossRef]
- The HOPE (Heart Outcomes Prevention Evaluation) Study: the design of a large, simple randomized trial of an angiotensin-converting enzyme inhibitor (ramipril) and vitamin E in patients at high risk of cardiovascular events. The HOPE study investigators. Can J Cardiol 1996, 12, 127-137.
- Lonn, E.; Bosch, J.; Yusuf, S.; Sheridan, P.; Pogue, J.; Arnold, J.M.; Ross, C.; Arnold, A.; Sleight, P.; Probstfield, J.; et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA 2005, 293, 1338–1347. [Google Scholar] [CrossRef]
- Marchioli, R.; Schweiger, C.; Tavazzi, L.; Valagussa, F. Efficacy of n-3 polyunsaturated fatty acids after myocardial infarction: results of GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto Miocardico. Lipids 2001, 36 Suppl, S119-126. [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; de la Mata, M.; Villanueva-Paz, M.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Suarez-Carrillo, A.; Talaveron-Rey, M.; Munuera, M.; Sanchez-Alcazar, J.A. Atherosclerosis and Coenzyme Q(10). Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Gioscia-Ryan, R.A.; Battson, M.L.; Cuevas, L.M.; Eng, J.S.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. J Appl Physiol (1985) 2018, 124, 1194–1202. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; Seals, D.R. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; Meyer, C.J. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J Card Fail 2014, 20, 953–958. [Google Scholar] [CrossRef]
- Yoshida, T.; Maulik, N.; Engelman, R.M.; Ho, Y.S.; Das, D.K. Targeted disruption of the mouse Sod I gene makes the hearts vulnerable to ischemic reperfusion injury. Circ Res 2000, 86, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Kliment, C.R.; Suliman, H.B.; Tobolewski, J.M.; Reynolds, C.M.; Day, B.J.; Zhu, X.; McTiernan, C.F.; McGaffin, K.R.; Piantadosi, C.A.; Oury, T.D. Extracellular superoxide dismutase regulates cardiac function and fibrosis. J Mol Cell Cardiol 2009, 47, 730–742. [Google Scholar] [CrossRef]
- Varshney, R.; Ranjit, R.; Chiao, Y.A.; Kinter, M.; Ahn, B. Myocardial Hypertrophy and Compensatory Increase in Systolic Function in a Mouse Model of Oxidative Stress. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Rajgarhia, A.; Ayasolla, K.R.; Zaghloul, N.; Lopez Da Re, J.M.; Miller, E.J.; Ahmed, M. Extracellular Superoxide Dismutase (EC-SOD) Regulates Gene Methylation and Cardiac Fibrosis During Chronic Hypoxic Stress. Front Cardiovasc Med 2021, 8, 669975. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, W.; Branchetti, E.; Grau, J.B.; Li, G.; Ayoub, S.; Lai, E.K.; Rioux, N.; Tovmasyan, A.; Fortier, J.H.; Sacks, M.S.; et al. Porphyrin-Based SOD Mimic MnTnBu OE -2-PyP(5+) Inhibits Mechanisms of Aortic Valve Remodeling in Human and Murine Models of Aortic Valve Sclerosis. J Am Heart Assoc 2018, 7, e007861. [Google Scholar] [CrossRef]
- Sharma, A.; Yuen, D.; Huet, O.; Pickering, R.; Stefanovic, N.; Bernatchez, P.; de Haan, J.B. Lack of glutathione peroxidase-1 facilitates a pro-inflammatory and activated vascular endothelium. Vascul Pharmacol 2016, 79, 32–42. [Google Scholar] [CrossRef]
- Camuglia, A.C.; Maeder, M.T.; Starr, J.; Farrington, C.; Kaye, D.M. Impact of N-acetylcysteine on endothelial function, B-type natriuretic peptide and renal function in patients with the cardiorenal syndrome: a pilot cross over randomised controlled trial. Heart Lung Circ 2013, 22, 256–259. [Google Scholar] [CrossRef]
- Zhang, P.; Jin, M.; Zhang, L.; Cui, Y.; Dong, X.; Yang, J.; Zhang, J.; Wu, H. Berberine alleviates atherosclerosis by modulating autophagy and inflammation through the RAGE-NF-kappaB pathway. Front Pharmacol 2025, 16, 1540835. [Google Scholar] [CrossRef]
- Jia, X.; Shao, W.; Tian, S. Berberine alleviates myocardial ischemia-reperfusion injury by inhibiting inflammatory response and oxidative stress: the key function of miR-26b-5p-mediated PTGS2/MAPK signal transduction. Pharm Biol 2022, 60, 652–663. [Google Scholar] [CrossRef]
- Khan, S.U.; Khan, S.U.; Suleman, M.; Khan, M.U.; Khan, M.S.; Arbi, F.M.; Hussain, T.; Mohammed Alsuhaibani, A.; M, S.R. Natural Allies for Heart Health: Nrf2 Activation and Cardiovascular Disease Management. Curr Probl Cardiol 2024, 49, 102084. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Sun, Q.; Zhao, H.; Tao, J.; Yan, D. The Effects of Dimethyl Fumarate on Atherosclerosis in the Apolipoprotein E-Deficient Mouse Model with Streptozotocin-Induced Hyperglycemia Mediated By the Nuclear Factor Erythroid 2-Related Factor 2/Antioxidant Response Element (Nrf2/ARE) Signaling Pathway. Med Sci Monit 2019, 25, 7966–7975. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Gao, L.; Zhang, A.; Hackfort, B.T.; Zucker, I.H. Therapeutic Effects of Nrf2 Activation by Bardoxolone Methyl in Chronic Heart Failure. J Pharmacol Exp Ther 2019, 371, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am J Cardiol 2008, 101, 14D–19D. [Google Scholar] [CrossRef]
- A, M.D.; A, G.H. Why antioxidant therapies have failed in clinical trials. J Theor Biol 2018, 457, 1–5. [Google Scholar] [CrossRef]
- Bellanti, F.; Coda, A.R.D.; Trecca, M.I.; Lo Buglio, A.; Serviddio, G.; Vendemiale, G. Redox Imbalance in Inflammation: The Interplay of Oxidative and Reductive Stress. Antioxidants (Basel) 2025, 14. [Google Scholar] [CrossRef]
- Yang, R.; Lv, M.; Yang, X.; Zhai, S. A Mendelian randomized study of circulating antioxidants in the diet and risk of cardiovascular disease. Sci Rep 2025, 15, 10341. [Google Scholar] [CrossRef]
- Beg, M.A.; Huang, M.; Vick, L.; Rao, K.N.S.; Zhang, J.; Chen, Y. Targeting mitochondrial dynamics and redox regulation in cardiovascular diseases. Trends Pharmacol Sci 2024, 45, 290–303. [Google Scholar] [CrossRef]
- Liang, S.; Ping, Z.; Ge, J. Coenzyme Q10 Regulates Antioxidative Stress and Autophagy in Acute Myocardial Ischemia-Reperfusion Injury. Oxid Med Cell Longev 2017, 2017, 9863181. [Google Scholar] [CrossRef] [PubMed]
- Shah, I.A.; Memon, M.; Ansari, S.; Kumar, R.; Chandio, S.A.; Mirani, S.H.; Rizwan, A. Role of Coenzyme Q10 in Prophylaxis of Myocardial Infarction. Cureus 2021, 13, e13137. [Google Scholar] [CrossRef]
- Borges, J.Y.V. The Role of Coenzyme Q10 in Cardiovascular Disease Treatment: An Updated 2024 Systematic Review and Meta-Analysis of Prospective Cohort Studies (1990-2024). medRxiv 2024, 2024.2007. 2003.24309736.
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; KA, O.C.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic Biol Med 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Tan, G.; Qin, Z.; Jiang, S.; Zhang, L.; Zhang, G.; Huang, M.; Huang, Z.; Jin, J. MitoQ alleviates triptolide-induced cardiotoxicity via activation of p62/Nrf2 axis in H9c2 cells. Toxicol In Vitro 2023, 86, 105487. [Google Scholar] [CrossRef]
- Masoumi-Ardakani, Y.; Najafipour, H.; Nasri, H.R.; Aminizadeh, S.; Jafari, S.; Safi, Z. Moderate Endurance Training and MitoQ Improve Cardiovascular Function, Oxidative Stress, and Inflammation in Hypertensive Individuals: The Role of miR-21 and miR-222: A Randomized, Double-Blind, Clinical Trial. Cell J 2022, 24, 577–585. [Google Scholar] [CrossRef]
- Li, X.; Wei, Y.; Wang, Z. microRNA-21 and hypertension. Hypertens Res 2018, 41, 649–661. [Google Scholar] [CrossRef]
- Qi, X.; Wang, J. Melatonin improves mitochondrial biogenesis through the AMPK/PGC1alpha pathway to attenuate ischemia/reperfusion-induced myocardial damage. Aging (Albany NY) 2020, 12, 7299–7312. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; Cao, F. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid Med Cell Longev 2018, 2018, 9286458. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.R.; Yang, J.Q.; Liu, F.; Shen, X.Q.; Zhou, Y.J. Melatonin attenuates vascular calcification by activating autophagy via an AMPK/mTOR/ULK1 signaling pathway. Exp Cell Res 2020, 389, 111883. [Google Scholar] [CrossRef] [PubMed]
- Luan, P.; D'Amico, D.; Andreux, P.A.; Laurila, P.P.; Wohlwend, M.; Li, H.; Imamura de Lima, T.; Place, N.; Rinsch, C.; Zanou, N.; Auwerx, J. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci Transl Med 2021, 13. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Whitson, J.A.; Martin-Perez, M.; Zhang, T.; Gaffrey, M.J.; Merrihew, G.E.; Huang, E.; White, C.C.; Kavanagh, T.J.; Qian, W.J.; Campbell, M.D.; et al. Elamipretide (SS-31) treatment attenuates age-associated post-translational modifications of heart proteins. Geroscience 2021, 43, 2395–2412. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.H.; Campbell, M.D.; et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. Elife 2020, 9. [Google Scholar] [CrossRef]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The effects of metformin on autophagy. Biomed Pharmacother 2021, 137, 111286. [Google Scholar] [CrossRef]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an Anticancer Agent. Trends Pharmacol Sci 2018, 39, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xu, S.; Lu, Y.; Wong, K.F.; Sun, L.; Hasan, K.M.M.; Ma, A.C.H.; Tse, G.; Manno, S.H.C.; Tian, L.; et al. Metformin accelerates zebrafish heart regeneration by inducing autophagy. NPJ Regen Med 2021, 6, 62. [Google Scholar] [CrossRef]
- Chen, C.; Lin, Q.; Zhu, X.Y.; Xia, J.; Mao, T.; Chi, T.; Wan, J.; Lu, J.J.; Li, Y.; Cui, J.; et al. Pre-clinical Evidence: Berberine as a Promising Cardioprotective Candidate for Myocardial Ischemia/Reperfusion Injury, a Systematic Review, and Meta-Analysis. Front Cardiovasc Med 2021, 8, 646306. [Google Scholar] [CrossRef]
- Yang, H.; Cao, G.; Li, X.; Zhao, Z.; Wang, Y.; Xu, F. Berberine Intervention Mitigates Myocardial Ischemia-Reperfusion Injury in a Rat Model: Mechanistic Insights via miR-184 Signaling. Biologics 2025, 19, 31–42. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, X.; Zhao, Q.; Li, G.; Zhang, H.; Ma, Z.; Yu, H.; Zeng, Q.; Zhang, H.; Xu, D. Berberine improves cardiac insufficiency through AMPK/PGC-1alpha signaling-mediated mitochondrial homeostasis and apoptosis in HFpEF mice. Int Immunopharmacol 2025, 155, 114613. [Google Scholar] [CrossRef]
- Song, T.; Chen, W.D. Berberine inhibited carotid atherosclerosis through PI3K/AKTmTOR signaling pathway. Bioengineered 2021, 12, 8135–8146. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Yu, P.; Peng, L.; Luo, L.; Liu, J.; Li, S.; Lai, X.; Luan, F.; Meng, X. Berberine: A Review of its Pharmacokinetics Properties and Therapeutic Potentials in Diverse Vascular Diseases. Front Pharmacol 2021, 12, 762654. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Xin, Q.; Lu, J.; Miao, Y.; Lin, Q.; Cong, W.; Chen, K. A New Therapeutic Candidate for Cardiovascular Diseases: Berberine. Front Pharmacol 2021, 12, 631100. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell 2017, 40, 583–594. [Google Scholar] [CrossRef]
- Rexius-Hall, M.L.; Khalil, N.N.; Andres, A.M.; McCain, M.L. Mitochondrial division inhibitor 1 (mdivi-1) increases oxidative capacity and contractile stress generated by engineered skeletal muscle. FASEB J 2020, 34, 11562–11576. [Google Scholar] [CrossRef]
- Ruiz, A.; Quintela-Lopez, T.; Sanchez-Gomez, M.V.; Gaminde-Blasco, A.; Alberdi, E.; Matute, C. Mitochondrial division inhibitor 1 disrupts oligodendrocyte Ca(2+) homeostasis and mitochondrial function. Glia 2020, 68, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Trezza, A.; Gentile, M.; Paccagnini, E.; Lupetti, P.; Spiga, O.; Bova, S.; Fusi, F. The drp-1-mediated mitochondrial fission inhibitor mdivi-1 impacts the function of ion channels and pathways underpinning vascular smooth muscle tone. Biochem Pharmacol 2022, 203, 115205. [Google Scholar] [CrossRef]
- Akhigbe, R.; Ajayi, A. The impact of reactive oxygen species in the development of cardiometabolic disorders: a review. Lipids Health Dis 2021, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, X.Q.; Shi, Y.; Li, H.M.; Meng, Z.Y.; Chen, H.; Li, X.H.; Chen, Y.C.; Liu, H.; Hong, Y.; et al. Tetrahydroberberrubine retards heart aging in mice by promoting PHB2-mediated mitophagy. Acta Pharmacol Sin 2023, 44, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yan, J.Y.; Wang, Y.X.; Ling, Y.N.; Song, X.D.; Wang, S.Y.; Liu, H.Q.; Liu, Q.C.; Zhang, Y.; Yang, P.Z.; et al. Spermidine-enhanced autophagic flux improves cardiac dysfunction following myocardial infarction by targeting the AMPK/mTOR signalling pathway. Br J Pharmacol 2019, 176, 3126–3142. [Google Scholar] [CrossRef]
- Chen, W.; Schilperoort, M.; Cao, Y.; Shi, J.; Tabas, I.; Tao, W. Macrophage-targeted nanomedicine for the diagnosis and treatment of atherosclerosis. Nat Rev Cardiol 2022, 19, 228–249. [Google Scholar] [CrossRef]
- Li, X.; Zhang, M.; Chen, A.; Wang, X.; Yang, L.; Zhu, Y.; Li, Z. Lipoic Acid Nanoparticles Exert Effective Antiatherosclerosis Effects through Anti-Inflammatory and Antioxidant Pathways. ACS Omega 2024, 9, 48642–48649. [Google Scholar] [CrossRef]
Figure 1.
Intracellular reactive oxygen species (ROS) production and the cellular antioxidant defense system. ROS are produced by different cellular sources, primarily by: 1. the mitochondrial electron transport chain (ETC), 2. peroxisomes and 3. NADPH oxidase (NOX). Other sources of ROS are 4. endoplasmic reticulum (ER) and 5. cyclooxygenases (COXs). Mitochondrial O2•− is produced by the electron transfer system via mitochondrial complexes I (NADH: ubiquinone oxidoreductase) and III (ubiquinol: cytochrome c oxidoreductase). NOX4, located in the mitochondrial membrane, generates H2O2, which leads to a decrease in the mitochondrial membrane potential (↓ΔΨm) and ultimately results in mitochondrial dysfunction. H2O2 is also produced in the peroxisomes via β-oxidation and is eliminated by catalase (CAT). The transcription factors, Nrf2 and FOXO3, orchestrate the cellular antioxidant response via upregulation of antioxidant genes such as superoxide dismutase (SOD) and CAT, and enzymes involved in glutathione (GSH) synthesis. Enzymatic antioxidants (e.g. SOD, CAT and GPx) and non-enzymatic antioxidants (e.g. GSH) maintain redox balance and cellular integrity by modifying gene expression and associated signalling cascades. Created in https://BioRender.com.
Figure 1.
Intracellular reactive oxygen species (ROS) production and the cellular antioxidant defense system. ROS are produced by different cellular sources, primarily by: 1. the mitochondrial electron transport chain (ETC), 2. peroxisomes and 3. NADPH oxidase (NOX). Other sources of ROS are 4. endoplasmic reticulum (ER) and 5. cyclooxygenases (COXs). Mitochondrial O2•− is produced by the electron transfer system via mitochondrial complexes I (NADH: ubiquinone oxidoreductase) and III (ubiquinol: cytochrome c oxidoreductase). NOX4, located in the mitochondrial membrane, generates H2O2, which leads to a decrease in the mitochondrial membrane potential (↓ΔΨm) and ultimately results in mitochondrial dysfunction. H2O2 is also produced in the peroxisomes via β-oxidation and is eliminated by catalase (CAT). The transcription factors, Nrf2 and FOXO3, orchestrate the cellular antioxidant response via upregulation of antioxidant genes such as superoxide dismutase (SOD) and CAT, and enzymes involved in glutathione (GSH) synthesis. Enzymatic antioxidants (e.g. SOD, CAT and GPx) and non-enzymatic antioxidants (e.g. GSH) maintain redox balance and cellular integrity by modifying gene expression and associated signalling cascades. Created in https://BioRender.com.
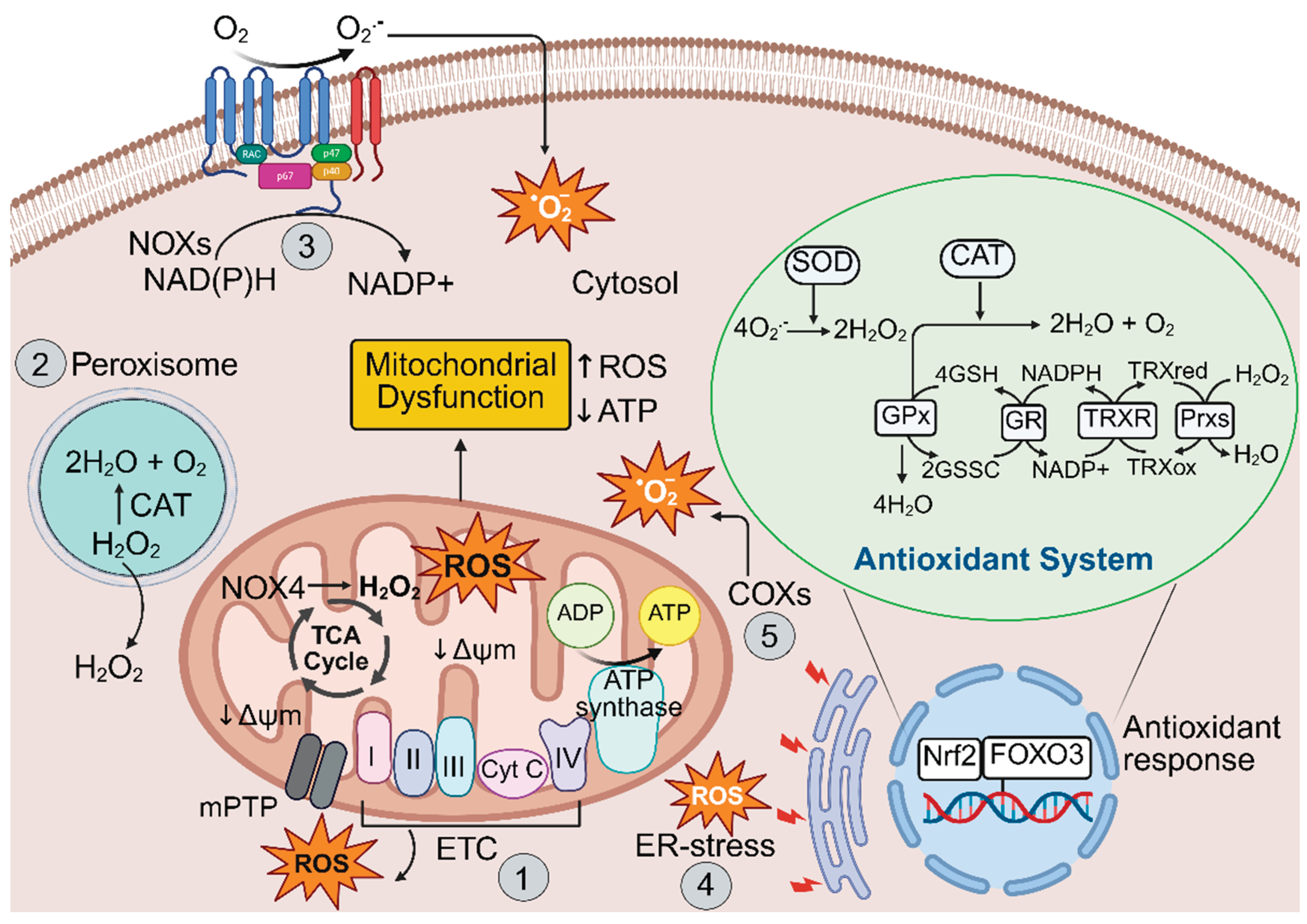
Figure 2.
Mechanistic overview of ROS generation, redox signaling and pathological signal transduction in cardiovascular diseases. Mitochondria function as the major site of ROS production and a central ROS-mediated signalling hub. ROS play an important role as second messengers in modulating multiple signalling molecules to regulate inflammation. ROS modulate various inflammatory signalling pathways, including the MAPKs, Nrf2, NF-κB, and the NLRP3 inflammasome. By modulating inflammatory pathways, ROS activate the expression of inflammatory genes, including IL-1β, TNF-α, VCAM-1 and ICAM-1, to enhance inflammatory and immune responses, ultimately accelerating the activation and progression of CVDs. Conversely, Nrf2 activation inhibits inflammation by cross talk with the NLRP3-inflammasome to lessen CVD. Created in https://BioRender.com.
Figure 2.
Mechanistic overview of ROS generation, redox signaling and pathological signal transduction in cardiovascular diseases. Mitochondria function as the major site of ROS production and a central ROS-mediated signalling hub. ROS play an important role as second messengers in modulating multiple signalling molecules to regulate inflammation. ROS modulate various inflammatory signalling pathways, including the MAPKs, Nrf2, NF-κB, and the NLRP3 inflammasome. By modulating inflammatory pathways, ROS activate the expression of inflammatory genes, including IL-1β, TNF-α, VCAM-1 and ICAM-1, to enhance inflammatory and immune responses, ultimately accelerating the activation and progression of CVDs. Conversely, Nrf2 activation inhibits inflammation by cross talk with the NLRP3-inflammasome to lessen CVD. Created in https://BioRender.com.
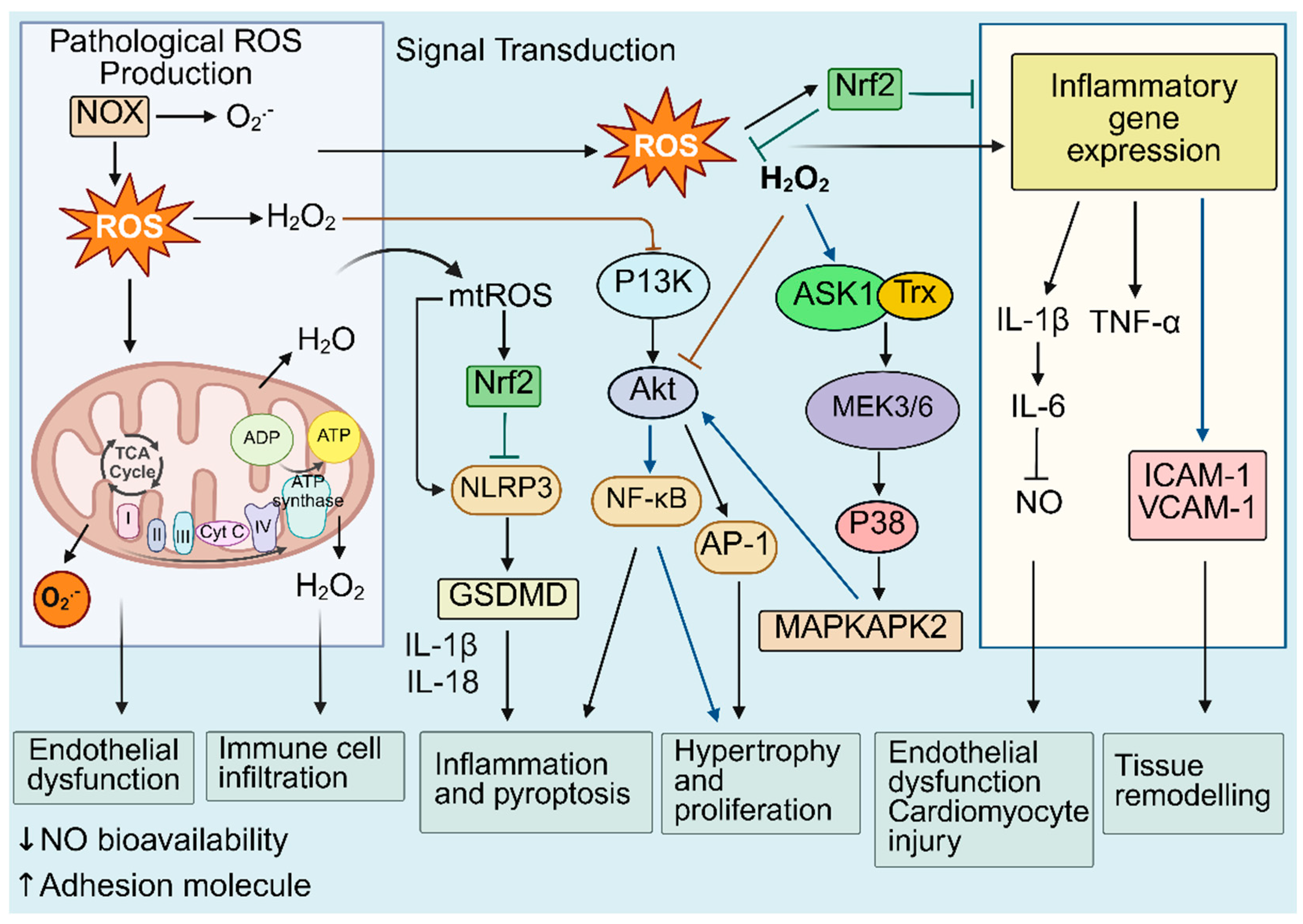
Figure 3.
Interconnectivity of the redox-mitochondria-autophagy-inflammation axis in the pathophysiology of CVDs. Dysregulation of these pathways leads to oxidative stress and metabolic imbalance, which ultimately contribute to the progression of CVDs, including ischemia/reperfusion(I/R) injury, diabetic cardiomyopathy (DCM), heart failure (HF), cardiac hypertrophy (CH) and atherosclerosis (AS). Created in https://BioRender.com.
Figure 3.
Interconnectivity of the redox-mitochondria-autophagy-inflammation axis in the pathophysiology of CVDs. Dysregulation of these pathways leads to oxidative stress and metabolic imbalance, which ultimately contribute to the progression of CVDs, including ischemia/reperfusion(I/R) injury, diabetic cardiomyopathy (DCM), heart failure (HF), cardiac hypertrophy (CH) and atherosclerosis (AS). Created in https://BioRender.com.
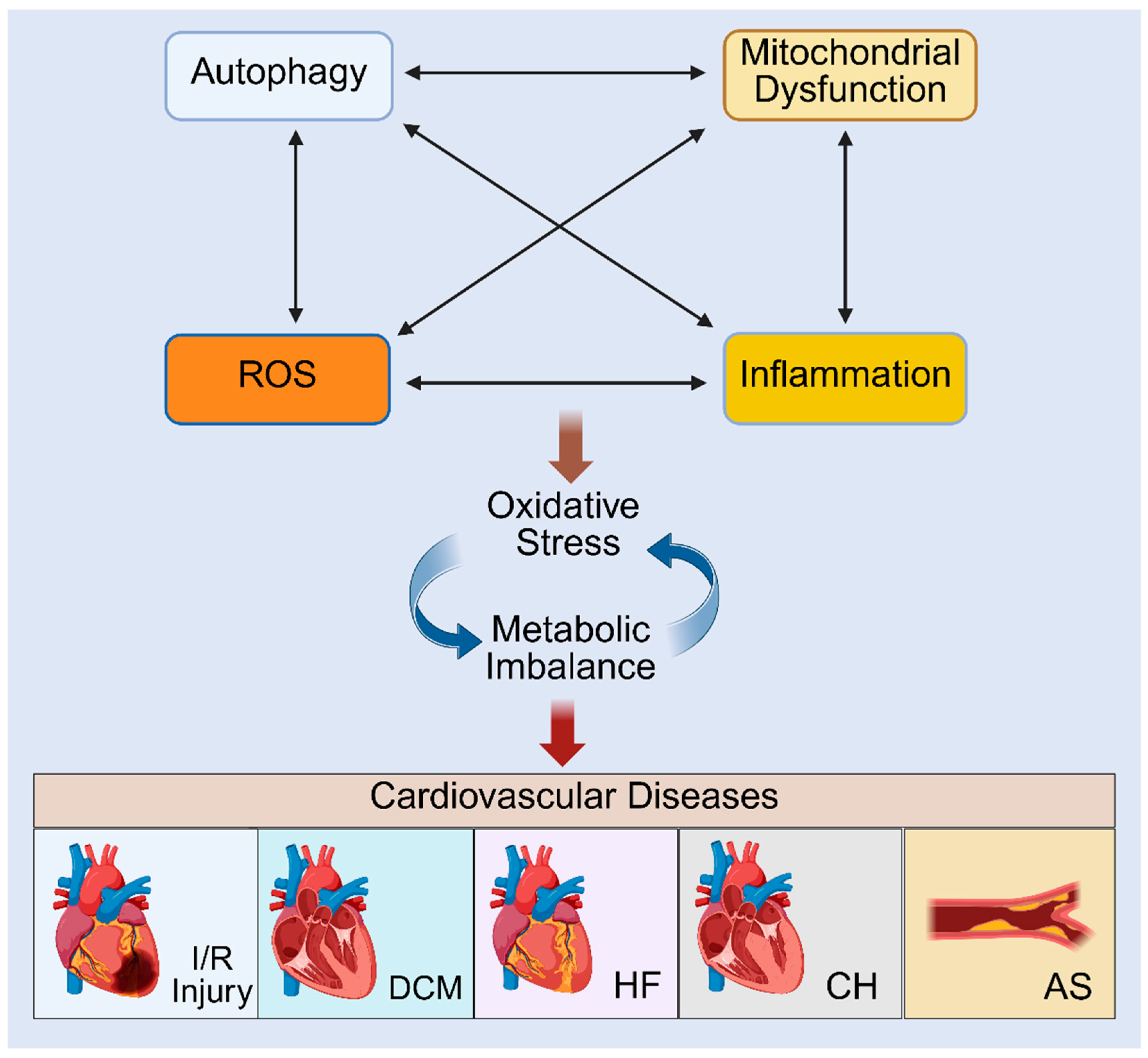
Figure 4.
Targeting the redox-mitochondria-autophagy-inflammation axis: novel strategies for the treatment of CVDs. Activating or inhibiting redox mediated signalling pathways provides a unique opportunity to develop novel therapeutic targets for the prevention and treatment of various CVDs. MitoQ and CoQ10 neutralize ROS such as superoxide (O2.-) and the hydroxyl radical (OH.) to prevent pathological redox signalling. Small molecules such as micheliolide, bardoxolone methyl and dimethyl fumarate (DMF) bolster Nrf2 levels to drive Nrf2-mediated responses that include upregulated antioxidant defences (superoxide dismutase (SOD), glutathione peroxide (GPx), thioredoxin (Trx) and heme-oxygenase-1 (HO-1). Additionally, metformin, berberine and melatonin stimulate autophagy via activation of AMPK, ULK1, HIF1a and LC3 to lessen inflammatory responses. SS-31 inhibits mitochondrial ROS production, thereby improving mitochondrial biogenesis, whilst Mdiv-1 blocks both inflammation and mitochondrial dysfunction. These are some of the newer strategies to lessen oxidative stress and inflammation in the quest for more targeted approaches to lessen CVD. Created in https://BioRender.com.
Figure 4.
Targeting the redox-mitochondria-autophagy-inflammation axis: novel strategies for the treatment of CVDs. Activating or inhibiting redox mediated signalling pathways provides a unique opportunity to develop novel therapeutic targets for the prevention and treatment of various CVDs. MitoQ and CoQ10 neutralize ROS such as superoxide (O2.-) and the hydroxyl radical (OH.) to prevent pathological redox signalling. Small molecules such as micheliolide, bardoxolone methyl and dimethyl fumarate (DMF) bolster Nrf2 levels to drive Nrf2-mediated responses that include upregulated antioxidant defences (superoxide dismutase (SOD), glutathione peroxide (GPx), thioredoxin (Trx) and heme-oxygenase-1 (HO-1). Additionally, metformin, berberine and melatonin stimulate autophagy via activation of AMPK, ULK1, HIF1a and LC3 to lessen inflammatory responses. SS-31 inhibits mitochondrial ROS production, thereby improving mitochondrial biogenesis, whilst Mdiv-1 blocks both inflammation and mitochondrial dysfunction. These are some of the newer strategies to lessen oxidative stress and inflammation in the quest for more targeted approaches to lessen CVD. Created in https://BioRender.com.
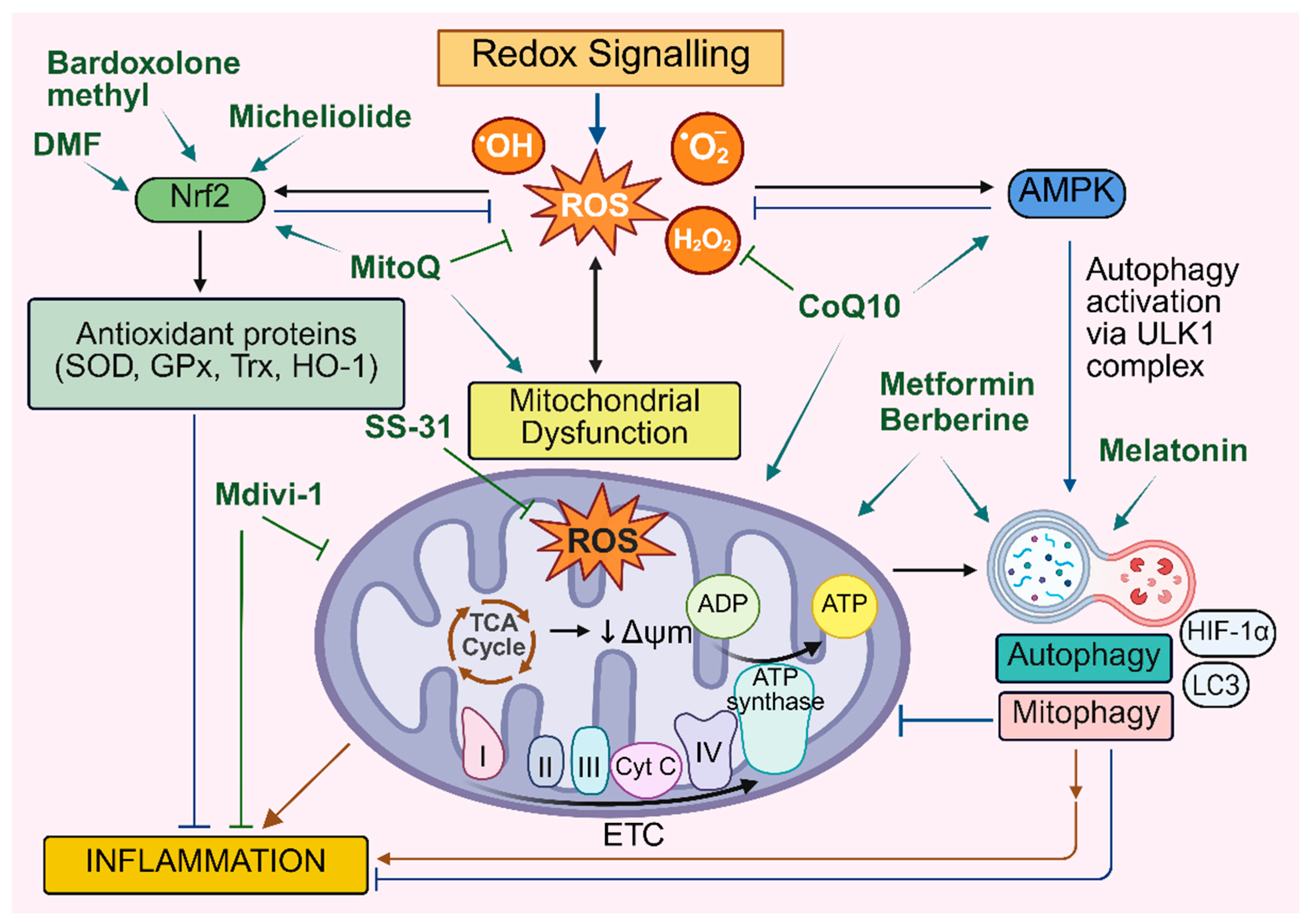
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



