Submitted:
01 September 2025
Posted:
02 September 2025
You are already at the latest version
Abstract
Viral metagenomic next-generation sequencing (vmNGS) has transformed our capacity for the untargeted detection and characterisation of (re)emerging zoonotic viruses, surpassing the limitations of traditional targeted diagnostics. In this review, we critically evaluate the current landscape of vmNGS, highlighting its integration within the One Health paradigm and its application to surveillance and discovery of (re)emerging viruses at the human–animal–environment interface. We provide a detailed overview of vmNGS workflows including sample selection, nucleic acid extraction, host depletion, virus enrichment, sequencing platforms, and bioinformatic pipelines, all tailored to maximise sensitivity and specificity for diverse sample types. Through selected case studies, including SARS-CoV-2, mpox, Zika virus, and a novel henipavirus, we illustrate the impact of vmNGS in outbreak detection, genomic surveillance, molecular epidemiology, and the development of diagnostics and vaccines. The review further examines the relative strengths and limitations of vmNGS in both passive and active surveillance, addressing barriers such as cost, infrastructure requirements and the need for interdisciplinary collaboration. By integrating molecular, ecological, and public health perspectives, vmNGS stands as a central tool for early warning, comprehensive monitoring, and informed intervention against (re)emerging viral threats, underscoring its critical role in global pandemic preparedness and zoonotic disease control.
Keywords:
(Re)emerging Viruses
; Metagenomic Next-Generation Sequencing
; One Health
; Surveillance
; Pandemic Preparedness
1. Introduction
The millions of fatalities and cases of morbidity resulting from the COVID-19, 2009/10 swine flu and AIDS pandemics, along with recurrent outbreaks of flaviviruses, filoviruses, poxviruses and others, illustrates the ongoing threat posed by (re)emerging viruses on human health [1]. The next pandemic may arise from an entirely unknown source, so-called Disease X in humans or Disease Y in animals, or rapidly evolving pathogens. Multiple recent outbreaks of novel aetiology have been caused by zoonotic spillover of viruses from animals to humans, which is driven by changes in animal, environmental and human health, aligning the Disease X/Y issue with the One Health paradigm introduced in Section 2.2. Timely detection and strategic surveillance of Disease X/Y causing viruses are critical components of pandemic preparedness, but the suitability of current PCR diagnostics is limited by the lack of genetic references. In these scenarios, untargeted approaches that do not require prior sequence knowledge such as metagenomic next generation sequencing (mNGS) have become increasingly valuable for both virus discovery and genomic surveillance. This review examines the current literature using mNGS for virus discovery and surveillance to propose a practical framework for integrating mNGS into (re)emerging virus surveillance strategies.
1.1. Metagenomic Next-Generation Sequencing: Transforming Virus Discovery and Surveillance
mNGS refers to the untargeted sequencing of all DNA and/or RNA present in a sample, the metagenome, enabling comprehensive identification of the diverse organisms from which nucleic acids originate. The integration of mNGS into pathogen discovery and surveillance is the culmination of technological advancements in molecular biology and NGS over the last 50 years (Figure 1).
Unlike first-generation sequencing pioneered by Fred Sanger in 1977 [18], second- and third-generation sequencing platforms generates much larger quantities of data and deliver whole genome sequences more rapidly and at substantially reduced costs. Critically, mNGS is sequence independent, allowing for unbiased detection of unknown or unexpected pathogens (Table 1).
The evolution of sequencing technologies is illustrated by comparing the discovery of three major zoonotic coronaviruses that have emerged in humans during the 21st century (Figure 1). In 2002/2003, SARS-CoV-1 was identified as the aetiological cause of a pneumonia epidemic using a combination of virus isolation, electron microscopy, serology, histology, PCR and partial genome sequencing via Sanger technology [11,12,22]. A decade later, the identification of MERS-CoV in 2012, leveraged similar methods but also incorporated whole genome sequencing (WGS) using viral enrichment methods, such as nuclease digestion and filtering as described in Section 3.2, and the Roche 454 short-read NGS platform [16]. More recently, in 2019, SARS-CoV-2 was directly identified from patient samples using short-read mNGS with the Illumina platform producing a complete viral genome sequence within days [17].
Continuous advancements in NGS technologies have greatly accelerated and broadened its practical applications, and its use for virus identification. As these technologies become more accessible, routine use of unbiased sequencing is poised to become central to rapid pathogen discovery and surveillance in the near future (Figure 1).
1.2. Pathogen Discovery and Surveillance: One Health Imperative
Approximately 60-80% of (re)emerging human viruses have zoonotic origins or circulate frequently between humans and animals [23,24,25,26]. Zoonosis, the transmission of a pathogen from non-human animals to humans [27], can also occur in the other direction (reverse zoonosis) and between different non-human species. Many (re)emerging threats are arboviruses, transmitted by arthropod vectors, which maintain both sylvatic (wildlife) and urban (domestic animal) cycles, before infecting people [28].
The ability of viruses to adapt to new host species is a key driver of their emergence in humans, as seen with SARS-CoV-1, where the virus evolved in civet cats (intermediate host) after spillover from bats (reservoir host) facilitating then human SARS-CoV-1 infections [29]. Concerns were raised when cattle-cattle transmission of highly pathogenic avian influenza virus H5N1 (HPAIV) was observed in 2024-25, as this could indicate a lowering of the evolutionary barrier to sustained human-human transmission [30,31]. Currently humans are dead-end hosts of HPAIV because virus is not transmitted from an infected human to any other species. Sequence-based surveillance of pathogens in animal populations enables early detection of mutations that may elevate zoonotic risk and helps prepare rapid response strategies.
The emergence of infectious diseases is driven by a complex interplay of human activities and environmental changes that intensify contact between species. Climate change driven by human activities including deforestation, intensive farming and encroachment on natural habitats can cause biodiversity loss, disrupt ecosystems and facilitate spillover events. Demographic trends, such as urbanisation, population growth and aging populations, along with globalisation via travel and trade accelerate virus dissemination. Following a spillover event, viruses can rapidly disseminate in immunologically naïve populations, with far reaching implications for conservation, agriculture, food security and human health [32,33,34].
These multidimensional challenges reinforce the importance of the One Health paradigm, which recognises the interdependencies of animal, environmental and human health (Figure 2). Coined by wildlife researchers [35], the “One Health” framework calls for interdisciplinary and cross-sectoral collaboration to generate equitable, sustainable and global solutions to emerging health threats. The World Health Organisation (WHO) Pandemic Agreement, adopted by WHO Member States in May 2025, is designed to strengthen global cooperation to promote the holistic One Health approach (Figure 2) to prevent or respond to future pandemics [36]. A central element of this agenda is enhanced surveillance of known or high-risk zoonotic and arboviral pathogens in animals and the environment. Regional authorities such as the EU also offer financial support to member states implementing One Health approaches with initiatives such as EU4Health funding research and programmes that strengthen pandemic preparedness through, for example, surveillance [37].
In this context, surveillance involves the monitoring and tracking of pathogens as they (re)emerge and spread to new locations. This requires pathogen identification, for which vmNGS can be used; knowledge of where and from whom the pathogen was isolated; and analysis to determine the relatedness of the identified pathogen to previous isolates. There are two forms of surveillance: active involving the proactive detection of pathogens before they cause disease in the at-risk populations and passive involving the responsive detection of pathogens after they cause disease.
This review focusses on the capabilities and applications of viral mNGS as a uniquely versatile tool for One Health pathogen discovery and surveillance. Its sequence-independent methodology enables rapid discovery of unknown pathogens, so-called Pathogen X or Y, without the need for prior genetic information, making it invaluable for outbreak investigations. At the same time, mNGS supports comprehensive viral genome surveillance enabling real-time monitoring of viral evolution, identification of origins and tracking of dissemination routes. To begin, the technical and practical considerations of vmNGS workflows are addressed by detailing each step from sample collection to nucleic acid extraction, sequencing and computational identification. The capabilities and limitations of vmNGS are then illustrated with practical examples and real-world applications, highlighting its deployment in the surveillance and discovery of zoonotic viruses for environmental and animal monitoring, and investigations of infectious cases of unknown aetiology. Finally, this review synthetises these insights to propose an integrated framework for effective emerging pathogen surveillance focusing on the prospective role of vmNGS in One Health Strategies.
2. Viral Metagenomic Next Generation Sequencing Workflows
Key steps of vmNGS workflows include sampling, extraction, virus enrichment, library preparation, sequencing, and bioinformatic analysis, which must be tailored to specific surveillance goals and sample matrices (Figure 3). Samples such as tissues, swabs, fluids, air and dust, demand distinct approaches due to variability in viral abundance, viral particle integrity, viral nucleic acid concentration and matrix composition (see Section 3.1). Clinical specimens typically follow optimised tissue or swab/fluid workflows, while environmental samples often require additional enrichment strategies to compensate for low viral loads (see Section 3.2). The two primary NGS platforms are short-read Illumina sequencing and long-read Oxford Nanopore Technologies (ONT), sequencing (see Section 3.3). Downstream, bioinformatic pipelines are essential for quality control, contaminant removal, assembly of overlapping reads into contigs and taxonomic identification (see Section 3.4).
2.1. Strategic Sample Selection and Quality Control in vmNGS
Thoughtful and targeted sample selection is fundamental to the success of vmNGS, directly influencing NA extraction and enrichment strategies, downstream processing, sequencing efficiency and the accuracy of pathogen detection. Whether in clinical or environmental surveillance, the choice of sample type and site collection should be informed by the likely site of pathogen presence, the properties of sampling matrix and relevant information such as disease symptoms or weather conditions at time of sample collection.
2.1.1. Environmental Settings
In environmental surveillance, location of sample collection significantly increases the likelihood of detecting circulating pathogens, thereby improving both sensitivity and cost-efficiency. For example, collecting water samples near agricultural, industrial or urban settings increase the likelihood of detecting a circulating pathogen as shown by the positive correlation between polio detection and population size for samples collected from multiple wastewater sites in Nigeria [38]. While air and dust samples should be collected from densely populated indoor environments [39]. Pathogen detection during environmental surveillance is also associated with seasonal fluctuations [39]. Working within the available resources is also challenging, especially when using mNGS as the detection tool, so periodic untargeted pathogen detection could be used to guide routine targeted environmental surveillance, which normally has reduced overall costs.
2.1.2. Clinical and Postmortem Context
Necropsy findings, clinical signs and reported symptoms should guide clinical sample selection to improve the likelihood of detecting the aetiological agents by vmNGS. For respiratory illness, nasopharyngeal swabs are appropriate, while cerebral spinal fluid (CSF) is most informative in cases of suspected neurological infection. Faecal samples or cloacal swabs are optimal for gastrointestinal diseases and suspected arbovirus infections supports collection of blood samples. Evidence-based sample selection had a direct impact on sensitivity during the 2024 HPAIV H5N1 outbreak in cattle in USA, where only 10% of nasal swabs, serum and blood buffy coat tested positive [31]. Subsequent analysis of milk samples, which aligned with clinical signs of mastitis, increased detection rate to 80% outlining the importance of logically matching sample collection with symptom presentation [31]. When symptoms are non-specific or when suitable sample is inaccessible, tissues or fluids with high likelihood of containing the pathogen, such as the spleen or blood, can be prioritised due to their roles in immunity and systemic infection. Equally important is the timing as virus abundance tends to peak during the acute phase of infection, and late samples collection could miss the pathogen due to immune clearance.
2.1.3. Sample Integrity and Contamination Management
Sample and nucleic acid integrity also play critical roles in workflow selection and success. Highly degraded samples may limit the applicability of certain enrichment or sequencing approaches, such as host depletion methods (see Section 3.2) that rely on intact viral particles and some long-read NGS technologies (see Section 3.3). There is greater flexibility and options available when sample and nucleic acid integrity is maintained, which begins with strict adherence to standard operating procedures for collection, transport and storage (see Box 1).
Box 1 . Sample Handling, Shipping and Storage
Nucleic acids and viral particles in clinical and environmental samples can be degraded by long term storage at non-freezing temperatures, acidic and alkaline pH and freeze-thaw cycles [261, 262]. Suitable storage buffers and maintaining the cold chain protects nucleic acids and viruses from degradation and loss of viability, respectively, while short-term storage (< 1 week) at 4°C avoids freeze-thaw cycles. Appropriate buffers used for sample storage depend on the intended outcomes. RNA is especially sensitive to degradation by RNases so samples should immediately be stored at < 4°C to reduce RNase activity or stored in buffers containing inhibitors of RNases, such as guanidium thiocyanate, which also inactivates virus [263]. If shipping samples across borders or virus isolation is not required, molecular transport media or lysis buffers from commercial RNA extraction kits can be used to inactivate virus while protecting nucleic acids [263]. Universal or virus transport media can be used if viable virus is required [261].
Sequencing the metagenome can detect all nucleic acids, so contamination poses a significant risk to specificity. Contamination can arise externally from the laboratory environment, such as from personnel or sampling equipment; and internally, such as from cross-contamination between samples. While, complete avoidance of contamination is not possible, it can be minimised by implementing standard operating procedures (see Box 1) and tracked by including negative controls [40].
2.2. Increasing Sensitivity with Host Depletion & Virus Enrichment
vmNGS faces sensitivity challenges due to the low abundance of viral nucleic acid, amidst high levels of host and environmental genetic material in most clinical and environmental samples. To address this, various strategies have been developed to increase the proportion of viral nucleic acid within the total nucleic acid (TNA) pool at different stages: pre-extraction, during extraction or during library preparation. These methods broadly fall in two categories: depletion of non-viral material and targeted virus enrichment (Figure 3).
2.2.1. Depletion of Non-Viral Material
Depletion of non-viral material focuses on selectively removing or excluding animal, plant, bacterial, fungal and parasitic material from the sample (Figure 3). Pre-extraction methods, including low-speed centrifugation and filtration, take advantage of the smaller size and lower density of viral particles compared to eukaryotic and prokaryotic cells enabling separation of virions from larger and denser components in biological fluids, swab fluid and some environmental samples.
Other depletion strategies are based on differences in viral and non-viral nucleic acid (Figure 3). Nuclease treatments, for instance, degrade unprotected host and bacterial nucleic acids, sparing DNA and RNA shielded within viral capsids [41,42,43,44]. Improved sensitivity for influenza virus detection was achieved using nuclease treatment [15] to deplete unprotected nucleic acid. However, these methods depend on sample integrity and have been shown to digest viral genomic DNA, which often lacks the protective capsid [45]. Antibody-based removal of methylated DNA can also be used because viral DNA is not methylated [46,47,48]. Post-extraction ribosomal RNA (rRNA) depletion is especially relevant as rRNA typically makes up over 90% of total RNA extracted from tissues [41,44,49,50,51]. Commercial rRNA depletion kits often use probes designed against human and rodent rRNA sequences for enzymatic or affinity-based depletion, but some kits are effective for avian rRNA as well as well [26,27]. Tissue sample TNA extracts are often treated with DNase, assuming actively replicating DNA and RNA viruses transcribe their genes and should be detected using a transcriptomics approach [41].
A balance must be found between enriching for viral nucleic acids and over-processing because the strategies described here can cause off-target depletion of viral nucleic acids and RNA degradation, which reduces sensitivity and generates false negatives [54]. Very low TNA concentrations resulting from host depletion often necessitate untargeted amplification to meet the minimum input requirement for library preparation and sequencing platforms (Box 2).
Box 2 . Sequence-independent amplification
Swabs and biological fluids often contain very low TNA concentrations following host depletion, so amplification of nucleic acid is required to increase the concentration. Many NGS library preparation methods and technologies have a minimal input of starting DNA/RNA. Sequence-independent, single-primer amplification (SISPA) non-specifically amplifies DNA or cDNA derived from RNA using a primer with 6-10 random bases at its 3’ end and a known sequence at its 5’ end, so PCR using a primer against the known sequence can be carried out [264]. This has been used for Illumina [265] and ONT NGS platforms (see section 3.3) [266, 267]. SISPA has also been modified to incorporate adapter sequences required for the Illumina flow cells, which avoids the need for expensive commercial library preparation kits [268].
2.2.2. Virus Enrichment
Hybrid capture (HC) is one form of virus nucleic acid enrichment, which uses biotinylated probes or probes conjugated to magnetic beads designed to bind sequences from viral families of interest. Following probe hybridisation, either pre- or post-library preparation, bound nucleic acids are captured while unbound material is washed away. If required, sequence-independent amplification can be performed to increase the input amounts before library preparation (Box 2).
Studies using artificially spiked samples have shown HC dramatically increases the sensitivity of vmNGS. The lower limit of detection for HC-vmNGS can reach as low as 10 virus particles/ml, compared to 103-104 virus particles/ml for untargeted approaches [34,35,36]. Similarly, genome coverage can exceed 90% with HC compared to less than 50% without [55,56,57]. Real-world applications have confirmed these advantages in various sample types, including plasma, respiratory secretions, and environmental matrices [55,56,58,59,60]. Notably, highly sensitive detection of zoonotic viruses such as Orthohantavirus and Coronaviridae in both animal and human samples has been achieved using custom enrichment probes sets [61,62]. In One Health surveillance studies, custom probe sets targeting 663 viral species identified 27 species of 11 families in bovine rectal swabs compared to 11 species from 6 families without HC [57].
Despite clear sensitivity benefits, HC-vmNGS has some limitations such as reduced performance with degraded samples due to impaired probe binding [62]. The method effectiveness is also reduced for viruses not represented in the probe set, making it less suitable for pathogen discovery [63]. Lists of known viral species are growing, allowing HC probe sets to be expanded. However, generating or purchasing probe sets can be expensive, although this can be offset by increased sample pooling per sequencing run thanks to reduced depth requirements. Using polyethylene glycol (PEG) to precipitate viral particles and then form a pellet by high-speed centrifugation can also be used to enrich for virus without the need for probe sets [64,65,66,67,68]. Effectiveness of PEG-vmNGS is dependent on intact viral particles, limiting its use, while a trade-off shared by HC- and PEG-vmNGS compared to untargeted vmNGS is the loss of dataset richness for non-viral, microbial and host identification; antimicrobial resistance gene surveillance; or host transcriptomic studies [26]. Nevertheless, for samples containing low concentrations of viral particles and nucleic acids, such as wastewater, the increased sensitivity of HC- or PEG-vmNGS is highly valuable. For broader pathogen discovery or characterisation of complex microbiomes, untargeted vmNGS remains the preferred strategy.
2.3. Next Generation Sequencing Technologies
NGS technologies enable comprehensive analysis of nucleic acids from complex samples by generating millions of nucleic acid sequence reads, which can be compared computationally to reference databases for pathogen identification (see section 3.4). NGS workflows begin with the preparation of sequencing libraries from DNA and RNA extracts. This process converts extracted nucleic acid into a format compatible with the NGS platform, typically involving fragmentation, adapter ligation and incorporation of barcode for multiplexing.
The Illumina instrument produces large volumes of short reads, typically in the range of 50-300 bp long [69]. To account for this read length, input DNA or cDNA must be fragmented during library preparation with fragmentation tailored to the nucleic acid integrity of the sample. Without adequate fragmentation, only the terminal fragments of long nucleic acids would be sequenced. ONT platforms permit sequencing of long reads reaching lengths of 10 – 1000 Kb with no strict upper limits [69]. This eliminates the need for fragmentation during library preparation. Both technologies require ligation of adapter sequences to the ends of nucleic acids, so they hybridise to flow-cells. When multiplexing and pooling samples on a single flow-cell, unique sequence barcodes are incorporated to differentiate sequencing reads and facilitate sample identification post-run.
Short-read sequencing, also known as shotgun sequencing, normally uses the fluorophore-based Illumina technology, while ONT uses disruption of electronic fields as nucleic acid strands move through a pore to achieve long-reads. Recent advances in ONT and Illumina technologies mean the latest instruments can produce terabytes of data per run. Illumina can yield billions of short reads, while ONT produces millions of long reads. Increased sensitivity would be expected due to the increased depth from Illumina, but in practice the two technologies have similar sensitivity and ONT achieves greater genome coverage [70,71,72,73]. Additional advantages of ONT include the portability of its Minion instrument, real-time sequence analysis and adaptive sequencing where reads corresponding to a predefined target are included and off-target reads are excluded in real-time. Illumina is advantageous when working with degraded or low-yield samples.
2.4. Bioinformatic Analysis for Virus Discovery, Characterisation and Molecular Epidemiology
The gigabytes or terabytes of data generated by an NGS run requires advanced computational methods for effective downstream processing of sequence data and meaningful interpretation in the context of complex clinical or environmental samples. Bioinformatic analysis is essential for both the identification and the molecular epidemiology of emerging and circulating viruses.
This process includes several interlinked steps starting with quality control to filter out low-quality, contaminant and host reads; de novo assembly of continuous sequences (contigs) based on read overlaps, a step particularly critical for short-read platforms but less so for long read systems like ONT. These assembled sequences or direct reads are then searched against reference databases to assign taxonomy and identify pathogens (Figure 4). Software packages incorporating these tools have been developed for streamlining and ease-of-use [74,75]. Specific tools have been developed for de novo assembly of reads obtained by mNGS [76] and vmNGS [77] enabling genome assembly of novel or highly variable viral genomes. More in-depth comparisons of bioinformatic tools for vmNGS can be found in these recent reviews [78,79].
After sequence assignment and pathogen identification, molecular epidemiological analyses are performed to understand patterns of variations, signature of host adaptation and molecular evolution (Figure 4). When necessary, tiled amplicon and NGS can be integrated in WGS workflows to ensure robust genome coverage and sensitivity. Phylogenetic analysis, which examines the genetic relationships and evolution of viral lineages is the cornerstone of molecular epidemiology. The rich genetic information and sequence variation obtained facilitate in-depth tracing of outbreak origins, transmission pathways and evolutionary trends and inform public health intervention [80].
Functional analysis and viral genes annotation is achieved using homology-based computational approaches to match new sequences with known gene functions, while powerful AI tools can be used to model protein structures given genetic sequences [81], then predict host-virus protein-protein interactions [82].
The breadth and depth of information derived from vmNGS bioinformatics demands significant expertise, computing power and data storage infrastructure and space, which present both opportunities and challenges for maximising the impact of mNGS in viral discovery and molecular surveillance.
3. Emerging Virus Discovery with vmNGS
Emerging zoonotic viruses have pandemic potential, as demonstrated by HIV, 2009/2010 IAV H1N1 and SARS-CoV-2, because they can spread through an immunologically naïve population who are often ill-prepared for countering the threat. Rapid identification of zoonotic viruses via passive surveillance of patients with suspected disease of unknown aetiology can accelerate the implementation of responses and tools to control spread. The lack of reference genomes and suitable targeted tests means vmNGS-based passive surveillance has been used to discover zoonotic virus when they first spill into humans. Genomic surveillance is another critical aspect as monitoring mutations occurring in virus isolates facilitates epidemiological tracking of virus spread, identification of novel variants and assessment of the continued effectiveness of currently used vaccines and antivirals based on mutations in immune epitopes and drug targets.
3.1. The Central Role of vmNGS in COVID-19 Discovery and Response
SARS-CoV-2 was identified as the aetiological agent of the COVID-19 pandemic when it was first detected using vmNGS and PCR in the lower respiratory tract of 41 patients suffering pneumonia in China December 2019 [83]. Details of the vmNGS methodology used were not included in this publication from 24th of January 2020 [83]. More details were included in a subsequent study on another of the first COVID-19 cases, a worker at Wuhan Live Food Market with pneumonia, which was published on 3rd of February 2020 [84]. RNA extracted from the bronchiolar lavage fluid underwent rRNA depletion and untargeted vmNGS on the Illumina platform to generate over 50 million 150 bp reads [84]. Over 99% coverage and approximately 6X depth (number of reads calling base in each position) were obtained for the 29,903 bp genome, which was the first complete and annotated SARS-CoV-2 genome deposited on Genbank (a public database of genetic sequences) [84].
Phylogenetic and recombination breakpoint analyses of the obtained SARS-CoV-2 genome identified its closest known relatives and predicted it arose from recombination between bat sarbecoviruses, which, along with early COVID-19 cases coming from the Wuhan Live Food Market, suggests a zoonotic origin [84]. Inferences of the SARS-CoV-2 replication cycle were made possible by modelling its spike protein structure using the genetic sequence, which showed homology with the receptor binding domain of SARS-CoV-1. This suggested SARS-CoV-2, like SARS-CoV-1, also used the ACE2 and TMPRSS2 as entry receptors, which was experimentally verified [84,85,86,87,88]. Having been used to identify early cases of SARS-CoV-2, complete genomes obtained by vmNGS were also used to infer phenotypic traits, which were often verified [84].
The value of publishing complete viral genomes is illustrated by the practical applications and research discoveries made possible by their availability through various sources including Genbank, Next Strain and the Global Initiative on Sharing All Influenza Data, with the latter two used for phylogenetic analysis and genomic surveillance of SARS-CoV-2 and other viruses [89,90]. The first sequences were used to design a robust, specific and sensitive PCR for the rapid diagnosis of SARS-CoV-2 infections, which was used to identify SARS-CoV-2 positive individuals who should quarantine as part of lockdown protocols implemented by most countries [91,92]. Genetic sequences obtained early in the pandemic led to rapid vaccine development with 115 candidates already in the pipeline by 8th of April 2020 – just 3 months after the first SARS-CoV-2 sequences were made available [93,94]. Phase II/III clinical trials for vaccine candidates using more established technologies, like the CanSino adenovirus-vector vaccine, began in April 2020 [95,96], while trials for the more novel mRNA vaccines started in May 2020, with vaccine rollout occurring within a year of the first detection of SARS-CoV-2 [93,97,98,99]. Vaccines were critical in preventing severe COVID-19 causing death or hospitalisation. Most likely, the timeframe for control of COVID-19 was accelerated and the number of fatalities reduced due to discoveries made possible by the SARS-CoV-2 sequences obtained using vmNGS.
3.2. A Henipavirus Emerging in the Shadow of COVID-19
The power of vmNGS for unbiased virus discovery was clearly established in 2022, when the cause of an acute febrile illness in a patient in China remained undiagnosed using conventional methods. Using only vmNGS, a novel Henipavirus, subsequently named Langya virus, was identified as the aetiological agent [100]. Genetic information gained from vmNGS from used to design PCR tests for the screening of additional patients with similar unexplained acute febrile illness, resulting in the identification of 34 more cases [100,101]. Studies to identify potential animal reservoirs revealed high seropositivity in shrews (27%) and lower rates in dogs (5%) and goats (2%) suggesting these animals may serve as reservoir or intermediate hosts highlighting potential sources of spillovers and avenues for viral evolution [100,101]. While all identified cases were non-fatal and showed no evidence of human-to-human transmission [100,101], the precedent of highly pathogenic Henipaviruses such as Nipah and Hendra viruses, underscores the need of close surveillance in line with the One Health approach [101,102].
4. Passive Surveillance of Reemerging Viruses using vmNGS
Reemergence occurs when a pathogen previously under control undergoes a resurgence in case numbers, which normally involves one or a combination of the following: spread to previously unaffected locations; altered transmission dynamics; loss of population-level immunity or cause of novel symptoms. Understanding and reacting to this phenomenon requires a One Health approach because animal, environmental, human and virological change can all give rise to reemergence. Targeted approaches are often unsuitable for reemerging virus detection because they are often unexpected causes of disease and genetic changes driving virus reemergence could reduce the sensitivity of tests targeting the sites of mutation [103,104]. Untargeted vmNGS is, therefore, often used for initial identification of reemerging viruses at the start of outbreaks (Table 2). The genetic information obtained by vmNGS can identify mutations causing novel phenotypes resulting in virus reemergence, while NGS platforms are used for WGS during genomic surveillance of reemerging disease outbreaks.
4.1. vmNGS to Monitor Mpox Epidemics and Global Spread
A striking example is the recent reemergence and geographical spread of mpox clade IIb (formally monkeypox virus) in 2022/2023 and mpox clade Ib since late 2023 [110,111]. Factors contributing to mpox reemergence include waning cross-protective population-level immunity since the cessation of smallpox vaccine in 1980 and increased human-wildlife interactions driven by human behaviours [112].
Mpox clade IIb descended from mpox IIa for which almost all human cases were from rural regions of West Africa and were contracted via zoonotic transmission [113,114]. This changed in 2017 when the ancestor of mpox IIb first emerged causing an outbreak in urban areas of Nigeria with HC-vmNGS used to obtain complete genome sequences for phylogenetic analysis, which demonstrated human-human transmission [115]. WGS of mpox isolated from travellers returning to the UK, Israel and Singapore in 2018/2019 showed they had contracted the same virus causing the Nigerian outbreak [116,117,118,119].
The global spread of mpox IIb might have been kickstarted by travellers returning from Nigeria, which led to the 2022/2023 global health emergency as it spread to at least 110 countries [113,114]. vmNGS played an important role in monitoring its global spread with the first complete mpox IIb genome obtained from a clinical sample by untargeted vm-NGS with the ONT platform in Portugal 2022 [120] and Illumina [121], ONT [122] and both [105] platforms were used to detect its introduction to previously mpox-free countries such as the Philippines and Brazil. Unlike the rare examples of suspected human-human mpox IIa transmission, which were mostly associated with direct or indirect contact with bodily fluids or skin pustules, a primary mode of mpox IIb transmission is homosexual sex shown by its high incidence in men who have sex with men, who were never previously associated with an increased risk of mpox infections [113,114]. A vaccine rollout targeting this at-risk community was implemented to protect the homosexual and bisexual community and control the spread of mpox.
Mpox Ib descended from mpox clade Ia, which has historically caused more severe infections with a higher fatality rate than clade II mpox. Historically clade I was a rare cause of disease in the Democratic Republic of Congo (DRC) and other countries the Congo Basin rainforest stretches into [123]. The geographic confinement and rarity of clade I mpox changed in late 2023 when clade Ib emerged from the DRC, which became endemic in the DRC, Burundi and Uganda, along with outbreaks in previously unaffected African nations [123]. Vulnerable populations to mpox Ib infection include sex workers and children indicating novel modes of transmission via heterosexual sex and close contact within the household or schools, respectively [124]. The first complete mpox Ib genome was obtained using the Illumina platform for HC-vmNGS from a patient sample in the DRC early 2024 [125], while vmNGS has continued to be used for WGS of DRC cases [126,127] and the first detected mpox Ib case in Europe [106].
The reemergence of mpox IIb and Ib was driven by the transition from primarily zoonotic to human-human transmission, with epidemiological evidence that sexual transmission contributed to both [112]. This phenotypic change could be driven by the high number of GA-AA mutations observed in mpox IIb and Ib genomes compared to their ancestors [124,128]. GA-AA mutations are characteristic of APOBEC3 cytosine deaminase modifications introduced as part of the human antiviral immune response suggesting undetected circulation in humans had driven the evolution of mpox clade IIb and Ib [125,129]. WGS was key to uncovering potential genetic drivers of the mpox reemergence. These genomic sequences were also used to design specific PCRs for diagnosis, which also contributed to control efforts and identified the at-risk populations requiring vaccination [130]. Similar symptoms caused by Herpesviruses and members of the Orthopoxvirus genus necessitates sensitive and highly specific molecular tests, which were designed by analysing complete viral genomes, while WGS of PCR positives is often used to verify results and facilitate epidemiological investigations [131,132,133,134]. vmNGS was important for mpox IIb detection in previously unaffected countries, while WGS contributed to technologies and studies that resulted in the containment of mpox IIb and protection of homosexual and bisexual communities. Hopefully pathogen identification and genomic surveillance using NGS will ensure mpox IIb remains under control and contribute to the response to contain mpox Ib outbreaks in Africa.
4.2. vmNGS in the Response and Understanding of Zika virus Reemergence
Prior to 2013, Zika virus was just another mosquito-borne arbovirus (virus transmitted by arthropods) normally associated with asymptomatic or rare cases of mild symptomatic illness in Africa and Asia, but this changed when it caused Guillain-Barré syndrome during a little-publicised outbreak in the French Polynesia [135]. The Brazilian epidemic marked a dramatic shift in public awareness of Zika virus when its unexpected geographic expansion, association with microcephaly and proof of vertical transmission was widely publicised one year after Brazil hosted the 2014 Football World Cup and one year before Rio de Janeiro hosted the 2016 Olympics [48].
Here too, vmNGS enabled direct identification from the amniotic fluids of Zika virus infected pregnant mothers during the Brazilian outbreak [107]. To track Zika virus spread, development of more rapid and sensitive diagnostics for screening less severe cases of disease was a priority, so efforts were made to design PCR assays, with specificity especially important due to the potential to cross-react with other Flavivirus species [136,137]. Zika virus RNA remained detectable by PCR significantly longer in urine (21 days post-symptom onset) compared to serum (5 days) [138,139]. NGS also contributed to Zika virus genomic surveillance where an amplicon-based WGS protocol was developed for Illumina and ONT platforms [140], while urine proved a more suitable clinical sample for obtaining complete sequencing in some studies but not others [141,142]. As well as its role in surveillance, WGS also enabled comparison of genetic sequences of Brazilian and pre-2015 isolates, which identified mutations linked to neurovirulence [143,144,145]. Increased retinoic acid response elements and phosphorylation sites in viral proteins were shown to reduce neurogenesis and increase apoptosis in neural progenitor cells [143,144,145,146,147].
Detection of Zika virus in mosquitos in 2014 and epidemiological investigations of viral genomes from the earliest known patients, have shown it was probably introduced to Brazil in 2013, suggesting timely application of untargeted sequencing could have allowed for earlier intervention and potentially limited the scope of the epidemic [148,149]. Active surveillance of arthropods can be used to identify (re)emerging arboviruses, as outlined in section 6.1.4, because infection of arthropods is a necessary stage of the infectious cycle that transmits arboviruses between different animal hosts.
4.3. vmNGS to Monitor the Spread and Reemergence of Arboviruses
Recently the threat posed by arboviruses has grown from a local to a global issue as climate change has expanded the geographic range of many arthropod vectors and the divide between the animals involved in sylvatic and urban cycles has crumbled as humans increasingly encroach on wildlife. With an estimated 400 million cases and 22,000 fatalities per annum, Dengue virus is the most prevalent arbovirus in humans, and its trajectory suggests cases numbers and geographic range will continue to grow [150]. Urbanisation and climate change have expanded the geographic range suitable for inhabitation by its main arthropod vectors, Aedes aegypti and Aedes albopictus [151], while globalisation has introduced these mosquitos to previously non-endemic countries [152,153,154]. The range of Dengue virus serotypes, which do not provide cross-reactive immunity, and the prevalence of asymptomatic infections hinders strategies to control its spread, while antibody dependent enhancement makes vaccine design challenging [155,156,157]. Dengue virus, along with Oropouche virus (OROV), Mayaro virus and Yellow Fever virus, is one of four arboviruses where human activities in the Amazon – including deforestation, mining and urban spread – have intensified human interactions with animals involved in sylvatic cycles and subsequent increases in human cases [158].
Viral genetic factors have also contributed to arbovirus reemergence, such as the reassortment event that gave rise to the 2023/2024 OROV epidemic in Brazil and neighbouring countries [159,160,161]. The 2023/2024 virus had increased replication kinetics in mammalian cell lines compared to ancestral OROV suggesting the reassortment gave rise to a strain better adapted to infection of animal hosts [162]. Viral mutations can also give rise to changes in the virus-arthropod dynamics that increase arbovirus transmission with the Chikungunya virus outbreak in 2005/2006 an example of this [163]. Previously Aedes aegypti was its main vector, but amino acid changes in the Chikungunya virus envelope glycoproteins enhanced virus replication in Aedes albopictus, so it was more readily spread by this mosquito species [164,165,166]. The broader geographic distribution of Aedes albopictus compared to Aedes aegypti enabled global spread of Chikungunya, including autochthonous transmission in Europe [153,154,167,168].
Untargeted vmNGS of patient samples was used to detect genetic changes associated with the 2023/2024 OROV outbreak [161], while targeted, amplicon-based WGS has been used for genomic surveillance of arbovirus epidemics [169,170,171]. Identifying the serotype responsible for Dengue virus infections is vital due to its association with antibody dependent enhancement and a multiplexed, amplicon-based NGS method has been developed to reveal the serotype and obtain complete genome sequence with a single test [172]. Point-of-care diagnostics are vital to strategies to control disease spread by rapidly identifying pathogens, and untargeted vmNGS was used for WGS of Dengue virus RNA recovered from positive antigen tests [173]. WGS is a critical required for phylogenetic characterisation of arboviruses, as conducted during epidemiological studies, and can reveal genetic differences contributing to novel phenotypic traits contributing to their (re)emergence.
vmNGS has also been used in the diagnosis of arboviral disease. Arbovirus infections often cause acute febrile illness with non-specific symptoms, while multiple species typically circulate in the same location and are spread by the same vector host. This can hinder selection of suitable targeted diagnostic tests [104,174], while the reemergence of arboviruses in novel locations is another barrier to targeted test selection [103,104,175]. When a syndromic approach to untargeted vmNGS surveillance of febrile patients in Uganda and Senegal was taken, Dengue, Rift Valley Fever and Yellow Fever were all identified [176,177], and a novel Orthobunyavirus was discovered in a separate study from Uganda [178]. Co-infections by multiple arbovirus species can also occur, which can be detected using vmNGS [174,179]. There is evidence untargeted vmNGS has applications for the passive surveillance of potential arboviral diseases, which provides additional advantages of obtaining genetic information.
5. Enhancing (Re)Emerging Virus Surveillance with vmNGS
Surveillance using mNGS of animals and environments generates knowledge of the infectious threats that could be incorporated into active surveillance strategies [180]. The previous sections described the effective use of passive surveillance for (re)emerging virus detection post zoonosis, but there are fewer success stories for the proactive detection of high-risk viruses before they reemerge in human or other animal populations. Targeted detection methods remain the most cost-effective and sensitive approach to active pathogen surveillance, but the benefits of building awareness of the unknown and reducing costs of NGS make its use in active surveillance important and increasingly practical.
5.1. Active and Passive (Re)Emerging Virus Surveillance in Animals
Active surveillance of animals for known or high-risk (re)emerging viruses is a One Health-based strategy to prevent and prepare for potential zoonoses before they threaten human and animal health. Zoonotic viruses typically have complex host dynamics with a mixture of reservoir, intermediate and dead-end hosts. Reservoirs hosts are typically asymptomatic carriers of the virus, which amplify and transfer the virus to new locations. Animals found at the human-animal interface and in which viruses adapt to humans are intermediate hosts [181]. Dead-end hosts cannot transfer the virus to other individuals in their population or to other species but can suffer symptomatic disease. Genetic monitoring of known or high-risk zoonotic viruses in reservoir and intermediate hosts can identify mutations that pre-dispose viruses to zoonotic spillover. Ideally, surveillance should cover other non-human animals because spillover of a virus to a new species can generate selective pressures driving the evolution of new traits such as adaptation to humans. For this reason, animals are often used as sentinels for tracking and monitoring known or high-risk zoonotic viruses as part of active surveillance efforts. While passive surveillance in response to suspected infectious disease of unknown aetiology in non-human animals will protect animal health in the short-term and generate knowledge that could protect human health in the long-term.
5.1.1. Farm Animals: The need for vmNGS Surveillance
The frequency of interactions between livestock and humans makes them a potential source of zoonoses in workplaces or live food markets, where spillover between non-human animals increases selective pressures driving antigenic drift, which could adapt a virus to infection of atypical hosts, and gives opportunities for recombination or reassortment. One example is common cold Human betacoronavirus OC43, which spilled over from cattle and might have caused a pandemic in the late 19th century [182,183].
In 2011, dairy farms in Central and Western Europe observed an unusual syndrome in adult cattle and sheep, characterised by reduced milk yield, diarrhoea, fever and spontaneous abortions and severe birth defects in offsprings. Conventional diagnostic tests failed to reveal the cause, prompting the use of vmNGS. This unbiased approach successfully identified a previously unrecognised Orthobunyavirus, now named Schmallenberg virus (SBV), as the aetiological agent [184]. Transmitted by the Culicoides midge, SBV rapidly spread west across Germany, the Netherlands, France, Belgium, the UK and Ireland between 2012-2014 [185], which garnered wide concern among veterinarians and public health officials wary of potential adaptation to humans. However, subsequent surveillance has found no evidence of human SBV infections [186,187]. WGS and epidemiological studies suggested a viral reassortment event between Sathuperi and Shamonda viruses gave rise to SBV [188], though its exact origin remains unresolved [189,190]. More recently, further evidence of genetic reassortment between Sathuperi and Shamonda viruses was gained following WGS of an orthobunyavirus isolated from cattle in Japan 2022 with the same genetic makeup as SBV [191]. SBV emergence is a prime example of the role vmNGS can play in One Health surveillance and virus discovery at the human-animal interface.

Table 1.
Known zoonotic viruses of livestock animals. 1 Shedding of Japanese encephalitis virus and swine-swine transmission has been demonstrated, with swine considered intermediate hosts that transmit the virus to mosquitos then onto humans.
Table 1.
Known zoonotic viruses of livestock animals. 1 Shedding of Japanese encephalitis virus and swine-swine transmission has been demonstrated, with swine considered intermediate hosts that transmit the virus to mosquitos then onto humans.
| Livestock | Zoonotic Virus | Location | Year | References |
| Cattle | Bovine coronavirus | Russia | Pre-1889 | [182,183,192] |
| Swine | Influenza virus H1N1 | Mexico | 2009 | [193] |
| HEV | Multiple (e.g. USA) | Multiple (e.g. 1998) | [194] | |
| Nipah virus | Malaysia & Singapore | 1998 | [195] | |
| Japanese encephalitis virus1 | Unknown | Unknown | [196] | |
| Poultry | Avian influenza A virus | USA or France | Pre-1918 | [193,197] |
| Newcastle Disease virus | USA | 1965 | [198] | |
| West Nile virus | Israel | 1998 | [199] | |
| Horses | Hendra virus | Australia | 1994 | [200] |
Animals farmed for their fur or medicine, including mink, foxes, rabbits and certain rodents, represent an important, often overlooked interface for zoonotic emergence. These species are not only susceptible to traditional animal viruses but have shown vulnerability to reverse zoonoses, acquiring infection from humans. Notably, mink have become infected with SARS-CoV-2 [201,202] and HEV [203] with outbreaks documented worldwide. In addition, HPAIV has been isolated from mink populations [204] raising concerns that they could serve as a reservoir, or even mixing vessels, where viral reassortment could occur. vmNGS has enabled comprehensive mapping of their virome, uncovering both known and unexpected viruses with potential zoonotic risks [49]. Mapping the virome of 461 small mammals farmed for fur or traditional medicines in China between 2021-2024, including foxes, mink, rabbits and rodents, revealed the presence of multiple high concern virus families (Coronaviridae, Flaviviridae, Hepeviridae, Orthomyxoviridae and Paramyxoviridae), and detected known zoonotic viruses such as Japanese encephalitis virus and Hepatitis E virus [205]. The detection of such viral diversity in a relatively small sample set underscores the global risk posed by fur farming. Active surveillance using mNGS is a powerful approach to monitor and pre-empt zoonotic spillover from this unique animal-human interface.
5.1.2. Wildlife Reservoirs and Metagenomic Surveillance: Preventing Zoonotic Spillover
Wild species, particular those in the orders Carnivora and Rodentia, are key reservoirs for zoonotic viruses and are often used as sentinels to assess spillover risk [206,207,208,209,210]. Although direct humans-wildlife contacts are generally lower than for farmed animals, episodic events such as natural disasters, or human activities, such as deforestation, can disrupt ecological boundaries and raise the risk of zoonotic spillover.
Bats exemplify the critical role of wildlife in zoonosis having been identified as original reservoirs for high-impact viruses such as EBOV in West Africa [211], Nipah virus in Southeast Asia [195] and sarbecoviruses in China [212]. Global vmNGS studies mapping bat viromes have uncovered a wide array of potential zoonotic viruses including members of Bunyaviridae, Coronaviridae, Hantaviridae and Picornaviridae [213,214,215,216,217]. Data mining and modelling have been used to identify the geographic hot spots – predominantly in Africa, South America, Southeast Asia, Subcontinental Asia and Eastern China – and animal taxa with highest zoonotic risk [218,219,220], which can be used to guide resource allocation and sampling strategies for maximum impact.
5.1.3. Companion Animals: A Critical Interface for One Health Surveillance
Companion animals, dogs and cats predominantly, occupy a unique position at the human-animal interface due to their ubiquitous presence in households and the frequency of close physical contact with their owners. The high level of attentiveness to companion animals makes them valuable candidate for both passive and active surveillance of (re)emerging viruses. Notable examples of zoonotic pathogens include Rabies virus and Norovirus from dogs to humans [107] with canine rabies consistently monitored via the integration of WGS and NGS into PCR-based surveillance programmes [221,222]. Zoonotic transmission from cats is comparatively rare, with occasional records of rabies infections following feline bites [223]. Cats are highly susceptible to often fatal IAV infections [224] and can be infected by SARS-CoV-2 via reverse-zoonosis [225].
Recognition of the role dogs and cats can play in zoonotic transmission is growing with vmNGS used to map their viromes [226,227]. A study from 2022/23 in China mapped the oralpharyngeal and rectal microbiomes and viromes of diseased and healthy cats and dogs, which identified animal pathogens not previously associated with disease and some zoonotic viruses and bacteria [226]. Another study mapping the gut virome of healthy dogs in China also showed they harbour known zoonotic viruses with alphacoronaviruses closely related to human common cold coronaviruses identified in both studies [226,227]. Awareness of the viruses harboured by companion animals is required to assess the risk they pose as sources of zoonotic pathogens.
5.1.4. Expanding the scope of Arbovirus Surveillance
Arthropod vectors transmit arboviruses, with mosquitoes being the most widely recognised group. However, a wide array of insects including midges, ticks, flies, fleas and lice also contributes to the spread of vector-borne pathogens at the human-animal-environmental interface. Mapping the arthropod virome using vmNGS identifies arboviruses most likely to cause disease in humans and enable their genomic surveillance, which is critical as many arboviral diseases are similar and their geographic ranges overlap [228,229,230,231]. The importance of knowing the Dengue virus serovars circulating was described in section 5.3, with their early identification possible through genomic surveillance of its mosquito vectors [232,233,234,235]. Applications of vmNGS for screening arthropod vectors includes gaining knowledge of arboviruses circulating in a specific location with potential to spill into humans [236,237]; identification of arthropod and viral species diversity [238,239,240]; and an understanding of the temporal and geographical dynamics influencing arbovirus circulation in insect populations [240]. The richness of arbovirus diversity; their spread to new locations; and their expanding seasonality hinders the selection of suitable targeted tests so vmNGS is an attractive alternative for their active surveillance.
5.2. Influenza: A Recurring Issue
There have been four pandemics caused by influenza A viruses (IAV) since 1918, making it the only virus species to cause more than one pandemic in the 20th and 21st centuries [241]. They all emerged in human populations following zoonotic spillover from swine and avian species [193,242,243]. Considering this, public health officials remain vigilant to the risk of HPAIV – responsible for the ongoing panzootic of birds and marine mammals – adapting to efficiently infect humans. Passive surveillance strategies for HPAIV in at-risk animals such as birds of prey and cattle have been implemented in the USA with PCR screens for initial detection followed by WGS using amplicon based or HC-vmNGS [244,245]. Active surveillance of HPAIV has also been conducted using environmental faecal samples and hunted ducks in Italy 2022-2024, which screened samples by PCR and carried out WGS to identify 5.26% positivity from 3,497 samples [246]. Influenza virus has also been detected following environmental surveillance of wastewater pointing to another avenue for active surveillance [247,248,249].
Beyond surveillance, vmNGS has already been used in IAV detection with retrospective detection of 2009/10 pandemic IAV H1N1 in 17/17 human infections following spillover from swine [15], while Illumina and ONT technologies have both been used to detect HPAIV in humans [250]. HPAIV WGS has been used to monitor mutations associated with human adaption with some arising in mammalian hosts [251,252]. Although sustained human-human HPAIV transmission has not yet been observed, surveillance of its multiple animal hosts is necessary to monitor zoonotic risk with vmNGS a useful tool should it spill into an unexpected host and for complete genome sequencing for epidemiological studies [253,254].
5.3. Rooting Through the Rubbish: Wastewater Surveillance
The principle behind environmental pathogen surveillance stems from infected hosts shedding virus particles and genetic material into their environments. As most environments are shared by multiple organisms, this approach enables the routine active surveillance of mixed populations with a single sample. Wastewater is the most studied environmental matrix for early detection and monitoring of reemerging viruses or ongoing outbreaks with SARS-CoV-2, IAV and mpox detected using HC- and PEG-vmNGS [64,65,66,67,255,256]. Additional reemerging or high-risk zoonotic viruses detected by HC- or PEG-vmNGS of wastewater include Hepatitis E virus [67,257]; Chikungunya virus, Jingmen tick virus and Rabies virus [66]; and species of Alphavirus, Flavivirus and Betacoronavirus [67]. From being on the cusp of eradication, polio virus is now reemerging, so its eradication programme is being stepped up. Wastewater surveillance is becoming an increasingly used component of the polio virus eradication programme with retrospective studies detecting polio virus in the wastewater of regions effected by polio, including in New York [68] and Israel in 2022/2023 [258]. Polio was also detected in London wastewater in 2022 before any cases were reported, which led to a vaccination drive among children in London with no cases subsequently reported, suggesting pathogen detection in wastewater can guide public health officials in real-world situations [259]. Opinions differ on the whether environmental wastewater surveillance can proactively detect pathogens and if mNGS is sufficiently specific and sensitive, so it appears further testing of its role from the academic sector is necessary to fully ascertain its effectiveness.
6. Discussion
The literature on vmNGS reveals a diverse array of targeted and untargeted workflows, and with no single approach meeting the needs of all sample types, surveillance goals or resource settings. Section 3 of this review outlined the tools available for vmNGS workflows from sample preparation choices and host depletion or viral enrichment strategies to sequencing platforms and bioinformatic pipelines, empowering stakeholders to select workflows best suited to their clinical or environmental surveillance objectives. Given the rapid pace of innovation in NGS technologies, periodic reassessment of these options will remain essential.
Current applications demonstrate that the workflows described in section 3 have already delivered substantial returns in both discovery and surveillance of (re)emerging viruses. While protocols differ between studies, consistent trends have emerged: HC- or PEG-based enrichment is often favoured for wastewater-based environmental surveillance [64,65,66,67,255,256], while untargeted approaches dominate in clinical settings, especially for diagnostics of unusual or unexplained cases such as SARS-CoV-2 [83,84] and Schmallenberg virus [260]. Even untargeted approaches frequently incorporate some level of host depletion most commonly rRNA removal to enhance viral signal.
Passive and syndromic surveillance has been the most impactful uses of vmNGS to date, enabling rapid detection and characterisation of novel threats like SARS-CoV-2 [83,84], and reemerging viruses like mpox clades IIb/Ib [105,106] (Figure 5A). In such scenarios, vmNGS is deployed when there are strong imperatives, such as public health urgency, that justify its higher cost, and its turnaround is markedly faster than alternative untargeted tools such as electron microscopy. Critically, the genomic data generated by vmNGS has informed the design of diagnostics and vaccines [91,93,94], and strengthened outbreak response by revealing viral evolution, transmission patterns and population vulnerability [113,114,124].
Incorporating vmNGS into ongoing genomic surveillance provide additional value during outbreaks. WGS data facilitates high-resolution genomic surveillance enabling public health agencies to track viral adaptation, origin and dissemination routes identifying mutations causing phenotypic changes, such as immune evasion, altered virulence profiles or novel transmission routes, resulting in virus reemergence [124,125,128,129,143,144,145,161,164]. Advances in sequencing platforms coupled with improved decision-making on when and where to deploy vmNGS could further shorten detection times, increase coverage and accuracy, and reduce costs.
By contrast, adoption of vmNGS into active surveillance remains comparatively rare. The expense of regularly screening large numbers of samples, combined with the need for specialised analytical expertise, constrains its routine use. Where it has been employed, semi-targeted vmNGS approaches, with HC- or PEG-, have proven effective in wastewater monitoring [64,65,66,67,255,256], while active surveillance of sentinel animals, such as arthropods, bats and small mammals farmed for fur or medicine, has identified potential zoonotic threats before they were detected in humans [49,208,209,214]. These findings can inform the targeting of more cost effective and strategic active surveillance programmes (Figure 5B). However, for routine active surveillance to become feasible, investment in local sequencing capacity, cost reduction, automated workflows and clear decision-making frameworks will be essential.
7. Conclusions
vmNGS offers a diverse tool kit that must be thoughtfully incorporated into workflows with careful attention to each step – sample selection and processing, host nucleic acid depletion and virus enrichment, sequencing and bioinformatic analysis – to ensure sensitive and specific viral detection. Workflow design should consider sample type (environmental or clinical), viral genome integrity, and contamination risks to maximise performance.
vmNGS has already proven valuable in passive surveillance, rapidly detecting (re)emerging viruses, especially for the discovery of unknown pathogens where targeted diagnostics are insufficient. Deployment of vmNGS for WGS has not only expedited vaccines and diagnostics development but also enabled real-time tracking of outbreak dynamics, to inform public health responses.
However, active surveillance applications remain relatively limited largely due to the costs and logistics of routine mass screening. The broader and more impactful use of vmNGS in both passive and active surveillance will depend on continued technological innovation, improved affordability, streamlining of workflows and more strategic decision-making around where and when to deploy the approach.
8. Future Directions
Progress in this field will depend on sustained collaboration between academia, industry and public health authorities to both drive the development of NGS and rigorously evaluate their impact within real-world surveillance frameworks. Surveillance is a key component of One Health approaches to pandemic preparedness with the rising threat of zoonotic and arboviral diseases demanding international and interdisciplinary cooperation and capacity building, particularly in resource-limited and rapidly changing environments. Community and stakeholder engagement is crucial to ensure surveillance strategies are contextually appropriate and equitable, while effective policy implementation by governments will be needed to mainstream and sustain these innovations.
By fully embracing a One Health perspective, future surveillance strategies powered by advanced sequencing technologies can facilitate early detection of (re)emerging and novel pathogens and enable more effective responses strengthening our collective ability to prevent or contain the next pandemic.
Author Contributions
Conceptualization, T.R., M.E., M.C., P.M. and V.G.; Investigation, T.R.; Resources, M.E., M.C., P.M. and V.G.; Writing – Original Draft Preparation, T.R.; Writing – Review & Editing, T.R., E.F. and V.G.; Visualization, T.R. and V.G.; Supervision, V.G.; Project Administration, M.E., M.C., P.M. and V.G.; Funding Acquisition, M.E., M.C., P.M. and V.G.
Funding
Co-funded by the European Union EU4Health Programme 2021-2027 (grant agreement No. 101132970, EU4H-2022-DGA-MS-IBA3), supporting the "One Health – ALL Ireland for European Surveillance (OH-ALLIES)" project. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Health and Digital Executive Agency, the granting authority. Neither the European Union nor the granting authority can be held responsible for them.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shanmugaraj B, Kothalam R, Tharik MS, Azeeze A. A brief overview on the threat of zoonotic viruses. Microbes and Infectious Diseases 2024;0:0–0.
- Burrell, C.J.; Mackay, P.; Greenaway, P.J.; Hofschneider, P.H.; Murray, K. Expression in Escherichia coli of hepatitis B virus DNA sequences cloned in plasmid pBR322. Nature 1979, 279, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-S.; Choo, Q.-L.; Weiner, A.J.; Ou, J.-H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (δ) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Chang, M.-F.; Shieh, C.-K.; Kamahora, T.; Vannier, D.M.; Govindarajan, S.; Lai, M.M.C. Molecular cloning and sequencing of a human hepatitis delta (δ) virus RNA. Nature 1987, 329, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.M.; Kalyanaraman, V.S.; Casey, J.M.; Srinivasan, A.; Andersen, P.R.; Devare, S.G. Molecular cloning and primary nucleotide sequence analysis of a distinct human immunodeficiency virus isolate reveal significant divergence in its genomic sequences. Proc. Natl. Acad. Sci. 1986, 83, 8380–8384. [Google Scholar] [CrossRef]
- Choo, Q.L.; Richman, K.H.; Han, J.H.; Berger, K.; Lee, C.; Dong, C.; Gallegos, C.; Coit, D.; Medina-Selby, R.; Barr, P.J. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. 1991, 88, 2451–2455. [Google Scholar] [CrossRef]
- Kato, N.; Hijikata, M.; Ootsuyama, Y.; Nakagawa, M.; Ohkoshi, S.; Sugimura, T.; Shimotohno, K. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc. Natl. Acad. Sci. 1990, 87, 9524–9528. [Google Scholar] [CrossRef]
- Henderson, W.W.; Monroe, M.C.; Jeor, S.C.S.; Thayer, W.P.; Rowe, J.E.; Peters, C.; Nichol, S.T. Naturally Occurring Sin Nombre Virus Genetic Reassortants. Virology 1995, 214, 602–610. [Google Scholar] [CrossRef]
- E Chizhikov, V.; Spiropoulou, C.F.; Morzunov, S.P.; Monroe, M.C.; Peters, C.J.; Nichol, S.T. Complete genetic characterization and analysis of isolation of Sin Nombre virus. J. Virol. 1995, 69, 8132–6. [Google Scholar] [CrossRef]
- Chew, M.H.L.; Arguin, P.M.; Shay, D.K.; Goh, K.; Rollin, P.E.; Shieh, W.; Zaki, S.R.; Rota, P.A.; Ling, A.; Ksiazek, T.G.; et al. Risk Factors for Nipah Virus Infection among Abattoir Workers in Singapore. J. Infect. Dis. 2000, 181, 1760–1763. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Lai, S.T.; Poon, L.L.M.; Guan, Y.; Yam, L.Y.C.; Lim, W.; Nicholls, J.; Yee, W.K.S.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Van Der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJM, et al. Identification of a new human coronavirus. Nature Medicine 2004 10:4 2004;10:368–373.
- Shinde, V.; Bridges, C.B.; Uyeki, T.M.; Shu, B.; Balish, A.; Xu, X.; Lindstrom, S.; Gubareva, L.V.; Deyde, V.; Garten, R.J.; et al. Triple-Reassortant Swine Influenza A (H1) in Humans in the United States, 2005–2009. New Engl. J. Med. 2009, 360, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Chen, E.C.; Sittler, T.; Scheinerman, A.; Roubinian, N.; Yu, G.; Kim, E.; Pillai, D.R.; Guyard, C.; Mazzulli, T.; et al. A Metagenomic Analysis of Pandemic Influenza A (2009 H1N1) Infection in Patients from North America. PLOS ONE 2010, 5, e13381. [Google Scholar] [CrossRef] [PubMed]
- Bhatt DL, Va Boston MPH, Harrington RA. Brief Report: Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. New England Journal of Medicine 2013;369:394–394.
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Correction: Satam et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. Biology 2024, 13, 286. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Morse, S.S.; Mazet, J.A.; Woolhouse, M.; Parrish, C.R.; Carroll, D.; Karesh, W.B.; Zambrana-Torrelio, C.; Lipkin, W.I.; Daszak, P. Prediction and prevention of the next pandemic zoonosis. Lancet 2012, 380, 1956–1965. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Wolfe, N.D.; Dunavan, C.P.; Diamond, J. Origins of major human infectious diseases. Nature 2007, 447, 279–283. [Google Scholar] [CrossRef]
- Jones, B.A.; Grace, D.; Kock, R.; Alonso, S.; Rushton, J.; Said, M.Y.; McKeever, D.; Mutua, F.; Young, J.; McDermott, J.; et al. Zoonosis emergence linked to agricultural intensification and environmental change. Proc. Natl. Acad. Sci. 2013, 110, 8399–8404. [Google Scholar] [CrossRef]
- Singh BB, Ward MP, Kostoulas P, Dhand NK. Zoonosis–Why we should reconsider “What’s in a name?” Front Public Health 2023;11:1133330.
- Figueiredo, L.T.M. Human Urban Arboviruses Can Infect Wild Animals and Jump to Sylvatic Maintenance Cycles in South America. Front. Cell. Infect. Microbiol. 2019, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.J.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Butt, K.M.; et al. Isolation and Characterization of Viruses Related to the SARS Coronavirus from Animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Kawaoka, Y. Highly pathogenic H5N1 avian influenza virus outbreak in cattle: the knowns and unknowns. Nat. Rev. Microbiol. 2024, 22, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Caserta, L.C.; Frye, E.A.; Butt, S.L.; Laverack, M.; Nooruzzaman, M.; Covaleda, L.M.; Thompson, A.C.; Koscielny, M.P.; Cronk, B.; Johnson, A.; et al. Spillover of highly pathogenic avian influenza H5N1 virus to dairy cattle. Nature 2024, 634, 669–676. [Google Scholar] [CrossRef]
- Thomson, T.N.; Marsland, M.J.; Minko, C.; Snow, K.J.; Friedman, N.D. Japanese encephalitis: A rapid review of reported prevalence of infection, clinical disease and sequelae in immunologically naive populations to inform Australia’s response. Aust. New Zealand J. Public Heal. 2023, 47, 1–4. [Google Scholar] [CrossRef]
- Das, S.; Smith, K.; Sarker, S.; Peters, A.; Adriaanse, K.; Eden, P.; Ghorashi, S.A.; Forwood, J.K.; Raidal, S.R. REPEAT SPILLOVER OF BEAK AND FEATHER DISEASE VIRUS INTO AN ENDANGERED PARROT HIGHLIGHTS THE RISK ASSOCIATED WITH ENDEMIC PATHOGEN LOSS IN ENDANGERED SPECIES. J. Wildl. Dis. 2020, 56, 896–906. [Google Scholar] [CrossRef]
- Pettersson JHO, Eldholm V, Seligman SJ, Lundkvist Å, Falconar AK, et al. Erratum for Pettersson et al., “How Did Zika Virus Emerge in the Pacific Islands and Latin America?” mBio 2018;9:e00386-18.
- Gibbs EPJ. The evolution of One Health: a decade of progress and challenges for the future. Veterinary Record 2014;174:85–91.
- Finch, A.; Vora, N.M.; Hassan, L.; Walzer, C.; Plowright, R.K.; Alders, R.; Suit-B, C.Y.; Amuasi, J.H.; Mulumba, M.; Loch-Temzelides, T.; et al. The promise and compromise of the WHO Pandemic Agreement for spillover prevention and One Health. Lancet 2025, 405, 1800–1802. [Google Scholar] [CrossRef]
- Samarasekera, U. New EU health programme comes into force. Lancet 2021, 397, 1252–1253. [Google Scholar] [CrossRef]
- Hamisu, A.W.; Blake, I.M.; Sume, G.; Braka, F.; Jimoh, A.; Dahiru, H.; Bonos, M.; Dankoli, R.; Bello, A.M.; Yusuf, K.M.; et al. Characterizing Environmental Surveillance Sites in Nigeria and Their Sensitivity to Detect Poliovirus and Other Enteroviruses. J. Infect. Dis. 2020, 225, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Minor, N.R.; Ramuta, M.D.; Stauss, M.R.; Harwood, O.E.; Brakefield, S.F.; Alberts, A.; Vuyk, W.C.; Bobholz, M.J.; Rosinski, J.R.; Wolf, S.; et al. Author Correction: Metagenomic sequencing detects human respiratory and enteric viruses in air samples collected from congregate settings. Sci. Rep. 2024, 14, 1–1. [Google Scholar] [CrossRef] [PubMed]
- Jurasz, H.; Pawłowski, T.; Perlejewski, K. Contamination Issue in Viral Metagenomics: Problems, Solutions, and Clinical Perspectives. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Fourgeaud, J.; Regnault, B.; Ok, V.; Da Rocha, N.; Sitterlé, É.; Mekouar, M.; Faury, H.; Milliancourt-Seels, C.; Jagorel, F.; Chrétien, D.; et al. Performance of clinical metagenomics in France: a prospective observational study. Lancet Microbe 2023, 5, e52–e61. [Google Scholar] [CrossRef] [PubMed]
- Yandle, Z.; Gonzalez, G.; Carr, M.; Matthijnssens, J.; De Gascun, C. A viral metagenomic protocol for nanopore sequencing of group A rotavirus. J. Virol. Methods 2022, 312, 114664. [Google Scholar] [CrossRef]
- Cebriá-Mendoza, M.; Arbona, C.; Larrea, L.; Díaz, W.; Arnau, V.; Peña, C.; Bou, J.V.; Sanjuán, R.; Cuevas, J.M. Deep viral blood metagenomics reveals extensive anellovirus diversity in healthy humans. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Ogunbayo, A.E.; Sabiu, S.; Nyaga, M.M. Evaluation of extraction and enrichment methods for recovery of respiratory RNA viruses in a metagenomics approach. J. Virol. Methods 2023, 314, 114677. [Google Scholar] [CrossRef]
- Edridge, A.W.D.; Deijs, M.; van Zeggeren, I.E.; Kinsella, C.M.; Jebbink, M.F.; Bakker, M.; van de Beek, D.; Brouwer, M.C.; van der Hoek, L. Viral Metagenomics on Cerebrospinal Fluid. Genes 2019, 10, 332. [Google Scholar] [CrossRef]
- Benoit, P.; Brazer, N.; de Lorenzi-Tognon, M.; Kelly, E.; Servellita, V.; Oseguera, M.; Nguyen, J.; Tang, J.; Omura, C.; Streithorst, J.; et al. Seven-year performance of a clinical metagenomic next-generation sequencing test for diagnosis of central nervous system infections. Nat. Med. 2024, 30, 3522–3533. [Google Scholar] [CrossRef]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. New Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef]
- Zhao, J.; Wan, W.; Yu, K.; Lemey, P.; Pettersson, J.H.-O.; Bi, Y.; Lu, M.; Li, X.; Chen, Z.; Zheng, M.; et al. Farmed fur animals harbour viruses with zoonotic spillover potential. Nature 2024, 634, 228–233. [Google Scholar] [CrossRef]
- He, X.; Wang, X.; Fan, G.; Li, F.; Wu, W.; Wang, Z.; Fu, M.; Wei, X.; Ma, S.; Ma, X. Metagenomic analysis of viromes in tissues of wild Qinghai vole from the eastern Tibetan Plateau. Sci. Rep. 2022, 12, 1–13. [Google Scholar] [CrossRef]
- Tan, J.K.; Servellita, V.; Stryke, D.; Kelly, E.; Streithorst, J.; Sumimoto, N.; Foresythe, A.; Huh, H.J.; Nguyen, J.; Oseguera, M.; et al. Laboratory validation of a clinical metagenomic next-generation sequencing assay for respiratory virus detection and discovery. Nat. Commun. 2024, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Parris, D.J.; Kariithi, H.; Suarez, D.L. Non-target RNA depletion strategy to improve sensitivity of next-generation sequencing for the detection of RNA viruses in poultry. J. Veter- Diagn. Investig. 2022, 34, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-L.; Li, W.-F.; Yuan, S.; Guo, J.-Y.; Li, Z.-L.; Chi, S.-H.; Huang, W.-J.; Li, X.-W.; Huang, S.-J.; Shao, J.-W. Meta-transcriptomic analysis reveals a new subtype of genotype 3 avian hepatitis E virus in chicken flocks with high mortality in Guangdong, China. BMC Veter- Res. 2019, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Prakoso, D.; Dark, M.J.; Barbet, A.F.; Salemi, M.; Barr, K.L.; Liu, J.J.; Wenzlow, N.; Waltzek, T.B.; Long, M.T. Viral Enrichment Methods Affect the Detection but Not Sequence Variation of West Nile Virus in Equine Brain Tissue. Front. Veter- Sci. 2018, 5, 318. [Google Scholar] [CrossRef]
- Piantadosi, A.; Mukerji, S.S.; Ye, S.; Leone, M.J.; Freimark, L.M.; Park, D.; Adams, G.; Lemieux, J.; Kanjilal, S.; Solomon, I.H.; et al. Enhanced Virus Detection and Metagenomic Sequencing in Patients with Meningitis and Encephalitis. mBio 2021, 12, e0114321. [Google Scholar] [CrossRef]
- Mourik, K.; Sidorov, I.; Carbo, E.C.; van der Meer, D.; Boot, A.; Kroes, A.C.M.; Claas, E.C.J.; Boers, S.A.; de Vries, J.J.C.; George, K.S. Comparison of the performance of two targeted metagenomic virus capture probe-based methods using reference control materials and clinical samples. J. Clin. Microbiol. 2024, 62, e0034524. [Google Scholar] [CrossRef]
- Mao, W.; Wang, J.; Li, T.; Wu, J.; Wang, J.; Wen, S.; Huang, J.; Shi, Y.; Zheng, K.; Zhai, Y.; et al. Hybrid Capture-Based Sequencing Enables Highly Sensitive Zoonotic Virus Detection Within the One Health Framework. Pathogens 2025, 14, 264. [Google Scholar] [CrossRef] [PubMed]
- Carbo, E.C.; Buddingh, E.P.; Karelioti, E.; Sidorov, I.A.; Feltkamp, M.C.; Borne, P.A.v.D.; Verschuuren, J.J.; Kroes, A.C.; Claas, E.C.; de Vries, J.J. Improved diagnosis of viral encephalitis in adult and pediatric hematological patients using viral metagenomics. J. Clin. Virol. 2020, 130, 104566. [Google Scholar] [CrossRef] [PubMed]
- Castellot, A.; Camacho, J.; Fernández-García, M.D.; Tarragó, D.; Liu, B.M. Shotgun metagenomics to investigate unknown viral etiologies of pediatric meningoencephalitis. PLOS ONE 2023, 18, e0296036. [Google Scholar] [CrossRef]
- Child, H.T.; Airey, G.; Maloney, D.M.; Parker, A.; Wild, J.; McGinley, S.; Evens, N.; Porter, J.; Templeton, K.; Paterson, S.; et al. Comparison of metagenomic and targeted methods for sequencing human pathogenic viruses from wastewater. mBio 2023, 14, e0146823. [Google Scholar] [CrossRef]
- Rosenbaum, W.; Ylitalo, E.B.; Castel, G.; Sjödin, A.; Larsson, P.; Byström, J.W.; Forsell, M.N.; Ahlm, C.; Pettersson, L.; Bäck, A.T. Hybrid capture-based next-generation sequencing of new and old world Orthohantavirus strains and wild-type Puumala isolates from humans and bank voles. J. Clin. Virol. 2024, 172, 105672. [Google Scholar] [CrossRef]
- Kuchinski, K.S.; Loos, K.D.; Suchan, D.M.; Russell, J.N.; Sies, A.N.; Kumakamba, C.; Muyembe, F.; Kingebeni, P.M.; Lukusa, I.N.; N’kAwa, F.; et al. Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats. eLife 2022, 11. [Google Scholar] [CrossRef]
- Nabel, C.S.; Sameroff, S.; Shilling, D.; Alapat, D.; Ruth, J.R.; Kawano, M.; Sato, Y.; Stone, K.; Spetalen, S.; Valdivieso, F.; et al. Virome capture sequencing does not identify active viral infection in unicentric and idiopathic multicentric Castleman disease. PLOS ONE 2019, 14, e0218660. [Google Scholar] [CrossRef]
- Shafer, M.M.; Bobholz, M.J.; Vuyk, W.C.; A Gregory, D.; Roguet, A.; Soto, L.A.H.; Rushford, C.; Janssen, K.H.; E Emmen, I.; Ries, H.J.; et al. Tracing the origin of SARS-CoV-2 omicron-like spike sequences detected in an urban sewershed: a targeted, longitudinal surveillance study of a cryptic wastewater lineage. Lancet Microbe 2024, 5, e335–e344. [Google Scholar] [CrossRef]
- Hassard, F.; Vu, M.; Rahimzadeh, S.; Castro-Gutierrez, V.; Stanton, I.; Burczynska, B.; Wildeboer, D.; Baio, G.; Brown, M.R.; Garelick, H.; et al. Wastewater monitoring for detection of public health markers during the COVID-19 pandemic: Near-source monitoring of schools in England over an academic year. PLOS ONE 2023, 18, e0286259. [Google Scholar] [CrossRef]
- Stockdale SR, Blanchard AA, Nayak A, Husain A, Nashine R, et al. RNA-Seq of untreated wastewater to assess COVID-19 and emerging and endemic viruses for public health surveillance. The Lancet Regional Health - Southeast Asia 2023;14:100205.
- Miyani, B.; Li, Y.; Guzman, H.P.; Briceno, R.K.; Vieyra, S.; Hinojosa, R.; Xagoraraki, I. Bioinformatics-based screening tool identifies a wide variety of human and zoonotic viruses in Trujillo-Peru wastewater. One Heal. 2024, 18, 100756. [Google Scholar] [CrossRef]
- Whitehouse, E.R.; Gerloff, N.; English, R.; Reckling, S.K.; Alazawi, M.A.; Fuschino, M.; George, K.S.; Lang, D.; Rosenberg, E.S.; Omoregie, E.; et al. Wastewater Surveillance for Poliovirus in Selected Jurisdictions, United States, 2022–2023. Emerg. Infect. Dis. 2024, 30, 2279–2287. [Google Scholar] [CrossRef]
- Zhang, T.; Li, H.; Jiang, M.; Hou, H.; Gao, Y.; Li, Y.; Wang, F.; Wang, J.; Peng, K.; Liu, Y.-X. Nanopore sequencing: flourishing in its teenage years. J. Genet. Genom. 2024, 51, 1361–1374. [Google Scholar] [CrossRef]
- Vandenbogaert, M.; Kwasiborski, A.; Gonofio, E.; Descorps-Declère, S.; Selekon, B.; Meyong, A.A.N.; Ouilibona, R.S.; Gessain, A.; Manuguerra, J.-C.; Caro, V.; et al. Nanopore sequencing of a monkeypox virus strain isolated from a pustular lesion in the Central African Republic. Sci. Rep. 2022, 12, 1–13. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, L.; Zhu, C.; Jin, J.; Song, C.; Dong, H.; Li, Z.; Wang, Z.; Chen, Y.; Yang, Z.; et al. Clinical value of metagenomic next-generation sequencing by Illumina and Nanopore for the detection of pathogens in bronchoalveolar lavage fluid in suspected community-acquired pneumonia patients. Front. Cell. Infect. Microbiol. 2022, 12, 1021320. [Google Scholar] [CrossRef]
- Hong, N.T.T.; Anh, N.T.; Mai, N.T.H.; Nghia, H.D.T.; Nhu, L.N.T.; Thanh, T.T.; Phu, N.H.; Deng, X.; van Doorn, H.R.; Chau, N.V.V.; et al. Performance of Metagenomic Next-Generation Sequencing for the Diagnosis of Viral Meningoencephalitis in a Resource-Limited Setting. Open Forum Infect. Dis. 2020, 7, ofaa046. [Google Scholar] [CrossRef] [PubMed]
- Horiba, K.; Torii, Y.; Aizawa, Y.; Yamaguchi, M.; Haruta, K.; Okumura, T.; Suzuki, T.; Kawano, Y.; Kawada, J.-I.; Hara, S.; et al. Performance of Nanopore and Illumina Metagenomic Sequencing for Pathogen Detection and Transcriptome Analysis in Infantile Central Nervous System Infections. Open Forum Infect. Dis. 2022, 9, ofac504. [Google Scholar] [CrossRef] [PubMed]
- Kalantar, K.L.; Carvalho, T.; A de Bourcy, C.F.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.-F.; King, R.; et al. IDseq—An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. GigaScience 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Pérot, P.; Bigot, T.; Temmam, S.; Regnault, B.; Eloit, M. Microseek: A Protein-Based Metagenomic Pipeline for Virus Diagnostic and Discovery. Viruses 2022, 14, 1990. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Antipov, D.; Raiko, M.; Lapidus, A.; A Pevzner, P.; Robinson, P. Metaviral SPAdes: assembly of viruses from metagenomic data. Bioinformatics 2020, 36, 4126–4129. [Google Scholar] [CrossRef]
- Ibañez-Lligoña, M.; Colomer-Castell, S.; González-Sánchez, A.; Gregori, J.; Campos, C.; Garcia-Cehic, D.; Andrés, C.; Piñana, M.; Pumarola, T.; Rodríguez-Frias, F.; et al. Bioinformatic Tools for NGS-Based Metagenomics to Improve the Clinical Diagnosis of Emerging, Re-Emerging and New Viruses. Viruses 2023, 15, 587. [Google Scholar] [CrossRef]
- Elrashedy, A.; Mousa, W.; Nayel, M.; Salama, A.; Zaghawa, A.; Elsify, A.; Hasan, M.E. Advances in bioinformatics and multi-omics integration: transforming viral infectious disease research in veterinary medicine. Virol. J. 2025, 22, 1–17. [Google Scholar] [CrossRef]
- Wadas, I.; Domingues, I. Systematic Review of Phylogenetic Analysis Techniques for RNA Viruses Using Bioinformatics. Int. J. Mol. Sci. 2025, 26, 2180. [Google Scholar] [CrossRef]
- Wee, J.; Wei, G.-W. Rapid response to fast viral evolution using AlphaFold 3-assisted topological deep learning. Virus Evol. 2025, 11, veaf026. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Chojnowski, G.; Rosenthal, M.; Kosinski, J.; Cowen, L. AlphaPulldown—a python package for protein–protein interaction screens using AlphaFold-Multimer. Bioinformatics 2022, 39. [Google Scholar] [CrossRef] [PubMed]
- Huang C, Wang Y, Li X, Ren L, Zhao J, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395:497.
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e889. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Samavati L, Uhal BD. ACE2, Much More Than Just a Receptor for SARS-COV-2. Front Cell Infect Microbiol 2020;10:554397.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Bogner, P.; Capua, I.; Lipman, D.J.; Cox, N.J. ; Others A global initiative on sharing avian flu data. Nature 2006, 442, 981–981. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed]
- team E editorial. Erratum for Euro Surveill. 2020;25(3). Eurosurveillance 2021;26:210204e.
- Sharma, O.; Sultan, A.A.; Ding, H.; Triggle, C.R. A Review of the Progress and Challenges of Developing a Vaccine for COVID-19. Front. Immunol. 2020, 11, 585354. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Andreadakis, Z.; Kumar, A.; Román, R.G.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Guan, X.-H.; Li, Y.-H.; Huang, J.-Y.; Jiang, T.; Hou, L.-H.; Yang, B.-F.; Wang, L.; Wang, W.-J.; Wu, S.-P.; et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 396, 479–488. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Li, Y.-H.; Guan, X.-H.; Hou, L.-H.; Wang, W.-J.; Li, J.-X.; Wu, S.-P.; Wang, B.-S.; Wang, Z.; Wang, L.; et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: Vaccine candidate may be more than 90% effective, interim results indicate. BMJ 2020, 371, m4347. [Google Scholar] [CrossRef]
- Mahase E. Covid-19: Moderna applies for US and EU approval as vaccine trial reports 94.1% efficacy. The BMJ;371. Epub ahead of print 2 December 2020. [CrossRef]
- Kirste, I.; Hortsch, S.; Grunert, V.P.; Legault, H.; Maglinao, M.; Eichenlaub, U.; Kashlan, B.; Pajon, R.; Jochum, S. Quantifying the Vaccine-Induced Humoral Immune Response to Spike-Receptor Binding Domain as a Surrogate for Neutralization Testing Following mRNA-1273 (Spikevax) Vaccination Against COVID-19. Infect. Dis. Ther. 2022, 12, 177–191. [Google Scholar] [CrossRef]
- Zhang, X.-A.; Li, H.; Jiang, F.-C.; Zhu, F.; Zhang, Y.-F.; Chen, J.-J.; Tan, C.-W.; Anderson, D.E.; Fan, H.; Dong, L.-Y.; et al. A Zoonotic Henipavirus in Febrile Patients in China. New Engl. J. Med. 2022, 387, 470–472. [Google Scholar] [CrossRef]
- Sah, R.; Mohanty, A.; Chakraborty, S.; Dhama, K. Langya virus: A newly identified zoonotic henipavirus. J. Med Virol. 2022, 94, 5621–5622. [Google Scholar] [CrossRef]
- Olamilekan Adesola R, Viola Miranda A, Shun Joshua Tran Y, Idris I, Lin X, et al. Langya virus outbreak: current challenges and lesson learned from previous henipavirus outbreaks in China, Australia, and Southeast Asia. Bulletin of the National Research Centre 2023 47:1 2023;47:1–6.
- Wise, E.L.; Márquez, S.; Mellors, J.; Paz, V.; Atkinson, B.; Gutierrez, B.; Zapata, S.; Coloma, J.; Pybus, O.G.; Jackson, S.K.; et al. Oropouche virus cases identified in Ecuador using an optimised qRT-PCR informed by metagenomic sequencing. PLOS Neglected Trop. Dis. 2020, 14, e0007897. [Google Scholar] [CrossRef]
- Wise, E.L.; Pullan, S.T.; Márquez, S.; Paz, V.; Mosquera, J.D.; Zapata, S.; Jackson, S.K.; Fejer, G.; Trueba, G.; Logue, C.H. Isolation of Oropouche Virus from Febrile Patient, Ecuador. Emerg. Infect. Dis. 2018, 24, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Isidro, J.; Borges, V.; Pinto, M.; Sobral, D.; Santos, J.D.; Nunes, A.; Mixão, V.; Ferreira, R.; Santos, D.; Duarte, S.; et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 2022, 28, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Treutiger, C.-J.; Filén, F.; Rehn, M.; Aarum, J.; Jacks, A.; Gisslén, M.; Sturegård, E.; Karlberg, M.L.; Lindsjö, O.K.; Sondén, K. First case of mpox with monkeypox virus clade Ib outside Africa in a returning traveller, Sweden, August 2024: public health measures. Eurosurveillance 2024, 29, 2400740. [Google Scholar] [CrossRef]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.O.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.M.; de Sequeira, P.C.; de Mendonça, M.C.L.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Coffey, L.L.; Murkey, J.; Symmes, K.; Sample, H.A.; Wilson, M.R.; Naccache, S.N.; Arevalo, S.; Somasekar, S.; Federman, S.; et al. Diagnosis of Fatal Human Case of St. Louis Encephalitis Virus Infection by Metagenomic Sequencing, California, 2016. Emerg. Infect. Dis. 2017, 23, 1964–1968. [Google Scholar] [CrossRef]
- Li, T.; Mbala-Kingebeni, P.; Naccache, S.N.; Thézé, J.; Bouquet, J.; Federman, S.; Somasekar, S.; Yu, G.; Martin, C.S.-S.; Achari, A.; et al. Metagenomic Next-Generation Sequencing of the 2014 Ebola Virus Disease Outbreak in the Democratic Republic of the Congo. J. Clin. Microbiol. 2019, 57, e00827–19. [Google Scholar] [CrossRef]
- Akingbola, A.; Abiodun, A.; Idahor, C.; Peters, F.; Ojo, O.; Jessica, O.U.; Alao, U.H.; Adewole, O.; Owolabi, A.; Chuku, J.; et al. Genomic Evolution and Epidemiological Impact of Ongoing Clade Ib MPox Disease: A Narrative Review. Glob. Heal. Epidemiology Genom. 2025, 2025, 8845911. [Google Scholar] [CrossRef]
- Americo, J.L.; Earl, P.L.; Moss, B. Virulence differences of mpox (monkeypox) virus clades I, IIa, and IIb.1 in a small animal model. Proc. Natl. Acad. Sci. 2023, 120. [Google Scholar] [CrossRef]
- Akingbola A, Adegbesan CA, Adewole O, Idahor C, Odukoya T, et al. Understanding the resurgence of mpox: key drivers and lessons from recent outbreaks in Africa. Trop Med Health 2025;53:1–12.
- Sharif, N.; Alzahrani, K.J.; Halawani, I.F.; Alzahrani, F.M.; Díez, I.D.l.T.; Lipari, V.; Flores, M.A.L.; Parvez, A.K.; Dey, S.K. Molecular epidemiology, transmission and clinical features of 2022-mpox outbreak: A systematic review. Heal. Sci. Rep. 2023, 6, e1603. [Google Scholar] [CrossRef]
- Van Dijck C, Hoff NA, Mbala-Kingebeni P, Low N, Cevik M, et al. Emergence of mpox in the post-smallpox era—a narrative review on mpox epidemiology. Clinical Microbiology and Infection 2023;29:1487–1492.
- Yinka-Ogunleye, A.; Aruna, O.; Dalhat, M.; Ogoina, D.; McCollum, A.; Disu, Y.; Mamadu, I.; Akinpelu, A.; Ahmad, A.; Burga, J.; et al. Outbreak of human monkeypox in Nigeria in 2017–18: a clinical and epidemiological report. Lancet Infect. Dis. 2019, 19, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.; Aarons, E.; Astbury, J.; Balasegaram, S.; Beadsworth, M.; Beck, C.R.; Chand, M.; O’cOnnor, C.; Dunning, J.; Ghebrehewet, S.; et al. Two cases of monkeypox imported to the United Kingdom, September 2018. Eurosurveillance 2018, 23, 1800509–6. [Google Scholar] [CrossRef] [PubMed]
- Erez N, Achdout H, Milrot E, Schwartz Y, Wiener-Well Y, et al. Diagnosis of Imported Monkeypox, Israel, 2018. Emerg Infect Dis 2019;25:980.
- Ng, O.T.; Lee, V.; Marimuthu, K.; Vasoo, S.; Chan, G.; Lin, R.T.P.; Leo, Y.S. A case of imported Monkeypox in Singapore. Lancet Infect. Dis. 2019, 19, 1166–1166. [Google Scholar] [CrossRef]
- Mauldin, M.R.; McCollum, A.M.; Nakazawa, Y.J.; Mandra, A.; Whitehouse, E.R.; Davidson, W.; Zhao, H.; Gao, J.; Li, Y.; Doty, J.; et al. Exportation of Monkeypox Virus From the African Continent. J. Infect. Dis. 2020, 225, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- First draft genome sequence of Monkeypox virus associated with the suspected multi-country outbreak, May 2022 (confirmed case in Portugal) - MPXV / Genome Reports - Virological. https://virological.org/t/first-draft-genome-sequence-of-monkeypox-virus-associated-with-the-suspected-multi-country-outbreak-may-2022-confirmed-case-in-portugal/799 (accessed ). 29 July.
- Ylaya, E.M.; Grande, P.G.; Dancel, L.L.; Nicolasora, A.D.; Polotan, F.G.; Pantoni, R.A.; Melo, E.; Ortia, S.P.; Manalo, J.I.; Abulencia, M.F.; et al. Case report: A comprehensive report on the first confirmed Mpox case in the Philippines during the 2022 Mpox global outbreak: from clinical presentation to shotgun metagenomic sequencing analysis. Front. Med. 2024, 11, 1387407. [Google Scholar] [CrossRef]
- Claro, I.M.; Romano, C.M.; Candido, D.d.S.; de Lima, E.L.; Lindoso, J.A.L.; Ramundo, M.S.; Moreira, F.R.R.; Barra, L.A.C.; Borges, L.M.S.; Medeiros, L.A.; et al. Shotgun metagenomic sequencing of the first case of monkeypox virus in Brazil, 2022. Rev. do Inst. de Med. Trop. de Sao Paulo 2022, 64, e48. [Google Scholar] [CrossRef]
- Tiwari, A.; Kalonji, T.; Miller, T.; Bossche, T.V.D.; Krolicka, A.; Muhindo-Mavoko, H.; Mitashi, P.; Tahita, M.C.; Lood, R.; Pitkänen, T.; et al. Emergence and Global Spread of Mpox Clade Ib: Challenges and the Role of Wastewater and Environmental Surveillance. J. Infect. Dis. 2025, 231, e825–e829. [Google Scholar] [CrossRef]
- Srivastava S, Laxmi, Sharma K, Sridhar SB, Talath S, et al. Clade Ib: a new emerging threat in the Mpox outbreak. Front Pharmacol 2024;15:1504154.
- Vakaniaki, E.H.; Kacita, C.; Kinganda-Lusamaki, E.; O’tOole, Á.; Wawina-Bokalanga, T.; Mukadi-Bamuleka, D.; Amuri-Aziza, A.; Malyamungu-Bubala, N.; Mweshi-Kumbana, F.; Mutimbwa-Mambo, L.; et al. Sustained human outbreak of a new MPXV clade I lineage in eastern Democratic Republic of the Congo. Nat. Med. 2024, 30, 2791–2795. [Google Scholar] [CrossRef]
- Brosius, I.; Vakaniaki, E.H.; Mukari, G.; Munganga, P.; Tshomba, J.C.; De Vos, E.; Bangwen, E.; Mujula, Y.; Tsoumanis, A.; Van Dijck, C.; et al. Epidemiological and clinical features of mpox during the clade Ib outbreak in South Kivu, Democratic Republic of the Congo: a prospective cohort study. Lancet 2025, 405, 547–559. [Google Scholar] [CrossRef]
- Masirika, L.M.; Udahemuka, J.C.; Schuele, L.; Nieuwenhuijse, D.F.; Ndishimye, P.; Boter, M.; Mbiribindi, J.B.; Kacita, C.; Lang, T.; Gortázar, C.; et al. Epidemiological and genomic evolution of the ongoing outbreak of clade Ib mpox virus in the eastern Democratic Republic of the Congo. Nat. Med. 2025, 31, 1459–1463. [Google Scholar] [CrossRef]
- Pan, D.; Nazareth, J.; Sze, S.; Martin, C.A.; Decker, J.; Fletcher, E.; Hollingsworth, T.D.; Barer, M.R.; Pareek, M.; Tang, J.W. Transmission of monkeypox/mpox virus: A narrative review of environmental, viral, host, and population factors in relation to the 2022 international outbreak. J. Med Virol. 2023, 95. [Google Scholar] [CrossRef] [PubMed]
- Marty, A.M. Why is there expanding community transmission of monkeypox in 2022? Lancet Microbe 2022, 3, e810–e811. [Google Scholar] [CrossRef] [PubMed]
- Srivastava S, Laxmi, Sharma K, Sridhar SB, Talath S, et al. Clade Ib: a new emerging threat in the Mpox outbreak. Front Pharmacol 2024;15:1504154.
- Schuele, L.; Masirika, L.M.; Udahemuka, J.C.; Siangoli, F.B.; Mbiribindi, J.B.; Ndishimye, P.; Aarestrup, F.M.; Koopmans, M.; Munnink, B.B.O.; Molenkamp, R.; et al. Real-time PCR assay to detect the novel Clade Ib monkeypox virus, September 2023 to May 2024. Eurosurveillance 2024, 29, 2400486. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.P.; Fogel, R.; Limson, J. Overview of Diagnostic Methods, Disease Prevalence and Transmission of Mpox (Formerly Monkeypox) in Humans and Animal Reservoirs. Microorganisms 2023, 11, 1186. [Google Scholar] [CrossRef]
- Ortins-Pina, A.; Hegemann, B.; Saggini, A.; Deml, K.; Wallerius, K.; Hörster, S.; Kraft, S.; Weyers, W. Histopathological features of human mpox: Report of two cases and review of the literature. J. Cutan. Pathol. 2023, 50, 706–710. [Google Scholar] [CrossRef]
- team E editorial. Authors’ correction for Euro Surveill. 2024;29(32). Euro Surveill 2024;29:240620c.
- Chan, J.F.; Choi, G.K.; Yip, C.C.; Cheng, V.C.; Yuen, K.-Y. Zika fever and congenital Zika syndrome: An unexpected emerging arboviral disease. J. Infect. 2016, 72, 507–524. [Google Scholar] [CrossRef]
- Singh, R.K.; Dhama, K.; Karthik, K.; Tiwari, R.; Khandia, R.; Munjal, A.; Iqbal, H.M.N.; Malik, Y.S.; Bueno-Marí, R. Advances in Diagnosis, Surveillance, and Monitoring of Zika Virus: An Update. Front. Microbiol. 2018, 8, 2677. [Google Scholar] [CrossRef]
- Landry, M.L.; George, K.S. Laboratory Diagnosis of Zika Virus Infection. Arch. Pathol. Lab. Med. 2016, 141, 60–67. [Google Scholar] [CrossRef]
- Gourinat, A.-C.; O’cOnnor, O.; Calvez, E.; Goarant, C.; Dupont-Rouzeyrol, M. Detection of Zika Virus in Urine. Emerg. Infect. Dis. 2015, 21, 84–86. [Google Scholar] [CrossRef]
- Rossini, G.; Gaibani, P.; Vocale, C.; Cagarelli, R.; Landini, M.P. Comparison of Zika virus (ZIKV) RNA detection in plasma, whole blood and urine – Case series of travel-associated ZIKV infection imported to Italy, 2016. J. Infect. 2017, 75, 242–245. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; A Beutler, N.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Quigley, J.E.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.H.; Song, D.H.; Lee, D.; Jang, J.; Kim, M.Y.; Jung, J.; Woo, K.I.; Kim, M.; Seog, W.; Oh, H.S.; et al. Whole-genome sequence analysis of Zika virus, amplified from urine of traveler from the Philippines. Virus Genes 2017, 53, 918–921. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, H.N.; Pareek, V.; Raza, K.; Dantham, S.; Kumar, P.; Mochan, S.; Faiq, M.A. A Possible Mechanism of Zika Virus Associated Microcephaly: Imperative Role of Retinoic Acid Response Element (RARE) Consensus Sequence Repeats in the Viral Genome. Front. Hum. Neurosci. 2016, 10, 403. [Google Scholar] [CrossRef]
- Jun SR, Wassenaar TM, Wanchai V, Patumcharoenpol P, Nookaew I, et al. Suggested mechanisms for Zika virus causing microcephaly: what do the genomes tell us? BMC Bioinformatics 2017;18:471.
- Faizan, I.; Abdullah, M.; Ali, S.; Naqvi, I.H.; Ahmed, A.; Parveen, S. Zika Virus-Induced Microcephaly and Its Possible Molecular Mechanism. Intervirology 2016, 59, 152–158. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.-S.; Lee, S.-A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Ayllón, T.; Campos, R.D.M.; Brasil, P.; Morone, F.C.; Câmara, D.C.P.; Meira, G.L.S.; Tannich, E.; Yamamoto, K.A.; Carvalho, M.S.; Pedro, R.S.; et al. Early Evidence for Zika Virus Circulation among Aedes aegypti Mosquitoes, Rio de Janeiro, Brazil. Emerg. Infect. Dis. 2017, 23, 1411–1412. [Google Scholar] [CrossRef]
- Naccache, S.N.; Thézé, J.; Sardi, S.I.; Somasekar, S.; Greninger, A.L.; Bandeira, A.C.; Campos, G.S.; Tauro, L.B.; Faria, N.R.; Pybus, O.G.; et al. Distinct Zika Virus Lineage in Salvador, Bahia, Brazil. Emerg. Infect. Dis. 2016, 22, 1788–1792. [Google Scholar] [CrossRef]
- Roy, S.K.; Bhattacharjee, S. Dengue virus: epidemiology, biology, and disease aetiology. Can. J. Microbiol. 2021, 67, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Hales, S.; de Wet, N.; Maindonald, J.; Woodward, A. Potential effect of population and climate changes on global distribution of dengue fever: an empirical model. Lancet 2002, 360, 830–834. [Google Scholar] [CrossRef]
- Chen, L.H.; Wilson, M.E. The Role of the Traveler in Emerging Infections and Magnitude of Travel. Med Clin. North Am. 2008, 92, 1409–1432. [Google Scholar] [CrossRef]
- Ding, F.; Fu, J.; Jiang, D.; Hao, M.; Lin, G. Mapping the spatial distribution of Aedes aegypti and Aedes albopictus. Acta Trop. 2018, 178, 155–162. [Google Scholar] [CrossRef]
- Abbasi, E. Global expansion of Aedes mosquitoes and their role in the transboundary spread of emerging arboviral diseases: A comprehensive review. IJID One Heal. 2025, 6. [Google Scholar] [CrossRef]
- Halstead, S.; O'ROurke, E. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 1977, 146, 201–217. [Google Scholar] [CrossRef]
- Grange, L.; Simon-Loriere, E.; Sakuntabhai, A.; Gresh, L.; Paul, R.; Harris, E. Epidemiological Risk Factors Associated with High Global Frequency of Inapparent Dengue Virus Infections. Front. Immunol. 2014, 5, 280. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.; Fernandez-Sesma, A. Challenges on the development of a dengue vaccine: a comprehensive review of the state of the art. J. Gen. Virol. 2023, 104, 001831. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, P.F.C.; da Rosa, A.P.A.T.; Rodrigues, S.G.; da Rosa, E.S.T.; Dégallier, N.; da Rosa, J.F.S.T. Inadequate management of natural ecosystem in the Brazilian Amazon region results in the emergence and reemergence of arboviruses. Cad. de Saude publica 2001, 17, S155–S164. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.R.R.; Dutra, J.V.R.; de Carvalho, A.H.B.; Reis, C.R.; Rios, J.S.H.; Ribeiro, M.d.O.; Arruda, M.B.; Alvarez, P.; Souza, R.P.; Voloch, C.; et al. Oropouche virus genomic surveillance in Brazil. Lancet Infect. Dis. 2024, 24, e664–e666. [Google Scholar] [CrossRef]
- Tilston-Lunel, N.L. Oropouche Virus: An Emerging Orthobunyavirus. J. Gen. Virol. 2024, 105, 002027. [Google Scholar] [CrossRef]
- Naveca, F.G.; de Almeida, T.A.P.; Souza, V.; Nascimento, V.; Silva, D.; Nascimento, F.; Mejía, M.; de Oliveira, Y.S.; Rocha, L.; Xavier, N.; et al. Human outbreaks of a novel reassortant Oropouche virus in the Brazilian Amazon region. Nat. Med. 2024, 30, 3509–3521. [Google Scholar] [CrossRef] [PubMed]
- Scachetti, G.C.; Forato, J.; Claro, I.M.; Hua, X.; Salgado, B.B.; Vieira, A.; Simeoni, C.L.; Barbosa, A.R.C.; Rosa, I.L.; de Souza, G.F.; et al. Re-emergence of Oropouche virus between 2023 and 2024 in Brazil: an observational epidemiological study. Lancet Infect. Dis. 2024, 25, 166–175. [Google Scholar] [CrossRef]
- Pialoux, G.; Gaüzère, B.-A.; Jauréguiberry, S.; Strobel, M. Chikungunya, an epidemic arbovirosis. Lancet Infect. Dis. 2007, 7, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; VanLandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Vazeille, M.; Moutailler, S.; Coudrier, D.; Rousseaux, C.; Khun, H.; Huerre, M.; Thiria, J.; Dehecq, J.-S.; Fontenille, D.; Schuffenecker, I.; et al. Two Chikungunya Isolates from the Outbreak of La Reunion (Indian Ocean) Exhibit Different Patterns of Infection in the Mosquito, Aedes albopictus. PLOS ONE 2007, 2, e1168. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; McGee, C.E.; Volk, S.M.; Vanlandingham, D.L.; Weaver, S.C.; Higgs, S.; Schneider, B.S. Epistatic Roles of E2 Glycoprotein Mutations in Adaption of Chikungunya Virus to Aedes Albopictus and Ae. Aegypti Mosquitoes. PLOS ONE 2009, 4, e6835. [Google Scholar] [CrossRef]
- Bonilauri, P.; Bellini, R.; Calzolari, M.; Angelini, R.; Venturi, L.; Fallacara, F.; Cordioli, P.; Angelini, P.; Venturelli, C.; Merialdi, G.; et al. Chikungunya Virus inAedes albopictus, Italy. Emerg. Infect. Dis. 2008, 14, 852–854. [Google Scholar] [CrossRef]
- Farooq, Z.; Segelmark, L.; Rocklöv, J.; Lillepold, K.; Sewe, M.O.; Briet, O.J.T.; Semenza, J.C. Impact of climate and Aedes albopictus establishment on dengue and chikungunya outbreaks in Europe: a time-to-event analysis. Lancet Planet. Heal. 2025, 9, e374–e383. [Google Scholar] [CrossRef]
- Adikari, T.N.; Riaz, N.; Sigera, C.; Leung, P.; Valencia, B.M.; Barton, K.; Smith, M.A.; Bull, R.A.; Li, H.; Luciani, F.; et al. Single molecule, near full-length genome sequencing of dengue virus. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Frumence E, Piorkowski G, Traversier N, Amaral R, Vincent M, et al. Genomic insights into the re-emergence of chikungunya virus on Réunion Island, France, 2024 to 2025. Eurosurveillance 2025;30:2500344.
- Deiana, M.; Malagò, S.; Mori, A.; Accordini, S.; Matucci, A.; Mantovani, R.P.; Gianesini, N.; Huits, R.; Piubelli, C.; Gobbi, F.G.; et al. Full Genome Characterization of the First Oropouche Virus Isolate Imported in Europe from Cuba. Viruses 2024, 16, 1586. [Google Scholar] [CrossRef]
- Vogels, C.B.F.; Hill, V.; Breban, M.I.; Chaguza, C.; Paul, L.M.; Sodeinde, A.; Taylor-Salmon, E.; Ott, I.M.; Petrone, M.E.; Dijk, D.; et al. DengueSeq: a pan-serotype whole genome amplicon sequencing protocol for dengue virus. BMC Genom. 2024, 25, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, F.-J.; Laubscher, F.; Chudzinski, V.; Kaiser, L.; Cordey, S. Direct Dengue Virus Genome Sequencing from Antigen NS1 Rapid Diagnostic Tests: A Proof-of-Concept with the Standard Q Dengue Duo Assay. Viruses 2023, 15, 2167. [Google Scholar] [CrossRef] [PubMed]
- Kabir, K.M.A.; Sigera, C.; Maduranga, S.; Weeratunga, P.; Rajapakse, S.; Fernando, D.; Lloyd, A.R.; Bull, R.A.; Rodrigo, C.; Harapan, H. Chikungunya masquerading as dengue infection in Sri Lanka uncovered by metagenomics. PLOS ONE 2025, 20, e0326995. [Google Scholar] [CrossRef]
- Fourgeaud, J.; Regnault, B.; Faury, H.; Da Rocha, N.; Jamet, A.; Stirnemann, J.; Eloit, M.; Perot, P.; Leruez-Ville, M.; Driessen, M.; et al. Fetal Zika virus infection diagnosed by metagenomic next-generation sequencing of amniotic fluid. Ultrasound Obstet. Gynecol. 2023, 61, 116–117. [Google Scholar] [CrossRef]
- Ashraf, S.; Jerome, H.; Bugembe, D.L.; Ssemwanga, D.; Byaruhanga, T.; Kayiwa, J.T.; Downing, R.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Shepherd, J.G.; et al. Uncovering the viral aetiology of undiagnosed acute febrile illness in Uganda using metagenomic sequencing. Nat. Commun. 2025, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.C.; Sene, A.; Mkandawire, W.; Deme, A.B.; Ndiaye, T.; Sy, M.; Gaye, A.; Diedhiou, Y.; Mbaye, A.M.; Ndiaye, I.M.; et al. Investigating the etiologies of non-malarial febrile illness in Senegal using metagenomic sequencing. Nat. Commun. 2024, 15, 1–13. [Google Scholar] [CrossRef]
- Ramesh, A.; Nakielny, S.; Hsu, J.; Kyohere, M.; Byaruhanga, O.; de Bourcy, C.; Egger, R.; Dimitrov, B.; Juan, Y.-F.; Sheu, J.; et al. Metagenomic next-generation sequencing of samples from pediatric febrile illness in Tororo, Uganda. PLOS ONE 2019, 14, e0218318. [Google Scholar] [CrossRef]
- Sardi, S.I.; Somasekar, S.; Naccache, S.N.; Bandeira, A.C.; Tauro, L.B.; Campos, G.S.; Chiu, C.Y.; Loeffelholz, M.J. Coinfections of Zika and Chikungunya Viruses in Bahia, Brazil, Identified by Metagenomic Next-Generation Sequencing. J. Clin. Microbiol. 2016, 54, 2348–2353. [Google Scholar] [CrossRef]
- Lipkin, W.I. The changing face of pathogen discovery and surveillance. Nat. Rev. Microbiol. 2013, 11, 133–141. [Google Scholar] [CrossRef]
- Zhao, J.; Cui, W.; Tian, B.-P. The Potential Intermediate Hosts for SARS-CoV-2. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.-M.; Van Ranst, M. Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef]
- Shaw, B.; Gatherer, D.; González-Domenech, C.M. Candidate historical events for the emergence of Human Coronavirus OC43: A critical reassessment of the molecular evidence. PLOS ONE 2023, 18, e0285481. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Scheuch, M.; Höper, D.; Jungblut, R.; Holsteg, M.; Schirrmeier, H.; Eschbaumer, M.; Goller, K.V.; Wernike, K.; Fischer, M.; et al. Novel Orthobunyavirus in Cattle, Europe, 2011. Emerg. Infect. Dis. 2012, 18, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Sedda, L.; Rogers, D.J. The influence of the wind in the Schmallenberg virus outbreak in Europe. Sci. Rep. 2013, 3, 3361. [Google Scholar] [CrossRef]
- Reusken, C.; van den Wijngaard, C.V.D; Van Beek, P.; Beer, M.; Bouwstra, R.; Godeke, G.-J.; Isken, L.; van den Kerkhof, H.; Van Pelt, W.; Van Der Poel, W.; et al. Lack of Evidence for Zoonotic Transmission of Schmallenberg Virus. Emerg. Infect. Dis. 2012, 18, 1746–1754. [Google Scholar] [CrossRef]
- Ducomble, T.; Wilking, H.; Stark, K.; Takla, A.; Askar, M.; Schaade, L.; Nitsche, A.; Kurth, A. Lack of Evidence for Schmallenberg Virus Infection in Highly Exposed Persons, Germany, 2012. Emerg. Infect. Dis. 2012, 18, 1333–5. [Google Scholar] [CrossRef]
- Yanase, T.; Kato, T.; Aizawa, M.; Shuto, Y.; Shirafuji, H.; Yamakawa, M.; Tsuda, T. Genetic reassortment between Sathuperi and Shamonda viruses of the genus Orthobunyavirus in nature: implications for their genetic relationship to Schmallenberg virus. Arch. Virol. 2012, 157, 1611–1616. [Google Scholar] [CrossRef]
- Collins, Á.B.; Doherty, M.L.; Barrett, D.J.; Mee, J.F. Schmallenberg virus: a systematic international literature review (2011-2019) from an Irish perspective. Ir. Veter- J. 2019, 72, 1–22. [Google Scholar] [CrossRef]
- Goller, K.V.; Höper, D.; Schirrmeier, H.; Mettenleiter, T.C.; Beer, M. Schmallenberg Virus as Possible Ancestor of Shamonda Virus. Emerg. Infect. Dis. 2012, 18, 1644–1646. [Google Scholar] [CrossRef]
- Tomochi, H.; Mekaru, Y.; Murota, K.; Konishi, M.; Ikeda, R.; Yanase, T. Emergence of a natural reassortant between Shamonda and Sathuperi viruses of the species Orthobunyavirus schmallenbergense in Japan. Arch. Virol. 2025, 170, 1–6. [Google Scholar] [CrossRef]
- Zhang, X.M.; Herbst, W.; Kousoulas, K.G.; Storz, J. Biological and genetic characterization of a hemagglutinating coronavirus isolated from a diarrhoeic child. J. Med Virol. 1994, 44, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Reid AH, Fanning TG, Hultin J V., Taubenberger JK. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc Natl Acad Sci U S A 1999;96:1651.
- Meng, X.-J.; Halbur, P.G.; Shapiro, M.S.; Govindarajan, S.; Bruna, J.D.; Mushahwar, I.K.; Purcell, R.H.; Emerson, S.U. Genetic and Experimental Evidence for Cross-Species Infection by Swine Hepatitis E Virus. J. Virol. 1998, 72, 9714–9721. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.-J.; et al. Nipah Virus: A Recently Emergent Deadly Paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, M.E.; García-Nicolás, O.; Brechbühl, D.; Python, S.; Zumkehr, B.; Nougairede, A.; Charrel, R.N.; Posthaus, H.; Oevermann, A.; Summerfield, A. Vector-free transmission and persistence of Japanese encephalitis virus in pigs. Nat. Commun. 2016, 7, 10832. [Google Scholar] [CrossRef]
- Agunos, A.; Pierson, F.W.; Lungu, B.; Dunn, P.A.; Tablante, N. Review of Nonfoodborne Zoonotic and Potentially Zoonotic Poultry Diseases. Avian Dis. 2016, 60, 553–575. [Google Scholar] [CrossRef]
- Hales, R.H.; Ostler, H.B. Newcastle disease conjunctivitis with subepithelial infiltrates. Br. J. Ophthalmol. 1973, 57, 694–697. [Google Scholar] [CrossRef]
- Bin H, Grossman Z, Pokamunski S, Malkinson M, Weiss L, et al. West Nile Fever in Israel 1999-2000. Ann N Y Acad Sci 2001;951:127–142.
- E Field, H. Hendra virus ecology and transmission. Curr. Opin. Virol. 2016, 16, 120–125. [Google Scholar] [CrossRef]
- Oreshkova, N.; Molenaar, R.J.; Vreman, S.; Harders, F.; Munnink, B.B.O.; Der Honing, R.W.H.-V.; Gerhards, N.; Tolsma, P.; Bouwstra, R.; Sikkema, R.S.; et al. SARS-CoV-2 infection in farmed minks, the Netherlands, April and May 2020. Eurosurveillance 2020, 25, 2001005. [Google Scholar] [CrossRef]
- team E editorial. Correction for Euro Surveill. 2020;25(23). Eurosurveillance 2021;26:210325c.
- Krog, J.S.; Breum, S.Ø.; Jensen, T.H.; Larsen, L.E. Hepatitis E Virus Variant in Farmed Mink, Denmark. Emerg. Infect. Dis. 2013, 19, 2028–2030. [Google Scholar] [CrossRef]
- Lindh, E.; Lounela, H.; Ikonen, N.; Kantala, T.; Savolainen-Kopra, C.; Kauppinen, A.; Österlund, P.; Kareinen, L.; Katz, A.; Nokireki, T.; et al. Highly pathogenic avian influenza A(H5N1) virus infection on multiple fur farms in the South and Central Ostrobothnia regions of Finland, July 2023. Eurosurveillance 2023, 28, 2300400. [Google Scholar] [CrossRef]
- Zhao, J.; Wan, W.; Yu, K.; Lemey, P.; Pettersson, J.H.-O.; Bi, Y.; Lu, M.; Li, X.; Chen, Z.; Zheng, M.; et al. Farmed fur animals harbour viruses with zoonotic spillover potential. Nature 2024, 634, 228–233. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, X.; Fan, G.; Li, F.; Wu, W.; Wang, Z.; Fu, M.; Wei, X.; Ma, S.; Ma, X. Metagenomic analysis of viromes in tissues of wild Qinghai vole from the eastern Tibetan Plateau. Sci. Rep. 2022, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Fan, K.; Liang, X.; Gong, W.; Chen, W.; He, B.; Chen, X.; Wang, H.; Wang, X.; Zhang, P.; et al. Virus diversity, wildlife-domestic animal circulation and potential zoonotic viruses of small mammals, pangolins and zoo animals. Nat. Commun. 2023, 14, 1–13. [Google Scholar] [CrossRef]
- Zhang, N.; Hu, B.; Zhang, L.; Gan, M.; Ding, Q.; Pan, K.; Wei, J.; Xu, W.; Chen, D.; Zheng, S.; et al. Virome landscape of wild rodents and shrews in Central China. Microbiome 2025, 13, 1–22. [Google Scholar] [CrossRef]
- Bodewes, R.; Ruiz-Gonzalez, A.; Schapendonk, C.M.E.; van den Brand, J.M.A.; Osterhaus, A.D.M.E.; Smits, S.L. Viral metagenomic analysis of feces of wild small carnivores. Virol. J. 2014, 11, 89–89. [Google Scholar] [CrossRef]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Délicat, A.; Paweska, J.T.; Gonzalez, J.-P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Ruiz-Aravena, M.; McKee, C.; Gamble, A.; Lunn, T.; Morris, A.; Snedden, C.E.; Yinda, C.K.; Port, J.R.; Buchholz, D.W.; Yeo, Y.Y.; et al. Author Correction: Ecology, evolution and spillover of coronaviruses from bats. Nat. Rev. Microbiol. 2022, 20, 315–315. [Google Scholar] [CrossRef]
- Geldenhuys, M.; Mortlock, M.; Weyer, J.; Bezuidt, O.; Seamark, E.C.J.; Kearney, T.; Gleasner, C.; Erkkila, T.H.; Cui, H.; Markotter, W.; et al. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLOS ONE 2018, 13, e0194527. [Google Scholar] [CrossRef]
- de Souza, P.J.; Fernandes, J.; Coelho, T.A.; Cosentino, M.; D’aRc, M.; Alves, P.D.G.; Guterres, A.; Vilar, E.M.; de Lemos, E.R.S.; Cordeiro-Estrela, P.; et al. A newly bat-borne hantavirus detected in Seba’s short-tailed bats (Carollia perspicillata) in the Brazilian Atlantic Rainforest. Mem. Do Inst. Oswaldo Cruz 2024, 119, e240132. [Google Scholar] [CrossRef]
- Crook, J.M.; Murphy, I.; Carter, D.P.; Pullan, S.T.; Carroll, M.; Vipond, R.; Cunningham, A.A.; Bell, D. Metagenomic identification of a new sarbecovirus from horseshoe bats in Europe. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Altan, E.; Reyes, G.; Halstead, B.; Deng, X.; Delwart, E.; Pfeiffer, J.K. Virome of Bat Guano from Nine Northern California Roosts. J. Virol. 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Hardmeier I, Aeberhard N, Qi W, Schoenbaechler K, Kraettli H, et al. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PLoS One 2021;16:e0252534.
- Allen, T.; Murray, K.A.; Zambrana-Torrelio, C.; Morse, S.S.; Rondinini, C.; Di Marco, M.; Breit, N.; Olival, K.J.; Daszak, P. Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Meisner, J.; Baines, A.; Ngere, I.; Garcia, P.J.; Sa-Nguansilp, C.; Nguyen, N.; Niang, C.; Bardosh, K.; Nguyen, T.; Fenelon, H.; et al. Mapping hotspots of zoonotic pathogen emergence: an integrated model-based and participatory-based approach. Lancet Planet. Heal. 2025, 9, e14–e22. [Google Scholar] [CrossRef] [PubMed]
- Desvars-Larrive, A.; Vogl, A.E.; Puspitarani, G.A.; Yang, L.; Joachim, A.; Käsbohrer, A. Author Correction: A One Health framework for exploring zoonotic interactions demonstrated through a case study. Nat. Commun. 2024, 15, 1–1. [Google Scholar] [CrossRef]
- Brunker K, Jaswant G, Thumbi SM, Lushasi K, Lugelo A, et al. Rapid in-country sequencing of whole virus genomes to inform rabies elimination programmes. Wellcome Open Res 2020;5:3.
- Huaman, C.; Paskey, A.C.; Clouse, C.; Feasley, A.; Rader, M.; Rice, G.K.; Luquette, A.E.; Fitzpatrick, M.C.; Drumm, H.M.; Yan, L.; et al. Genomic Surveillance of Rabies Virus in Georgian Canines. Viruses 2023, 15, 1797. [Google Scholar] [CrossRef]
- Peterson, B.; Barnes, A.N. Feline-Human Zoonosis Transmission in North Africa: A Systematic Review. Vector-Borne Zoonotic Dis. 2020, 20, 731–744. [Google Scholar] [CrossRef]
- Bonilla-Aldana, D.K.; Bonilla-Aldana, J.L.; Acosta-España, J.D.; Rodriguez-Morales, A.J. Highly Pathogenic Avian Influenza H5N1 in Cats (Felis catus): A Systematic Review and Meta-Analysis. Animals 2025, 15, 1441. [Google Scholar] [CrossRef]
- Doliff, R.; Martens, P. Cats and SARS-CoV-2: A Scoping Review. Animals 2022, 12, 1413. [Google Scholar] [CrossRef]
- Wu, W.-C.; Pan, Y.-F.; Zhou, W.-D.; Liao, Y.-Q.; Peng, M.-W.; Luo, G.-Y.; Xin, G.-Y.; Peng, Y.-N.; An, T.; Li, B.; et al. Meta-transcriptomic analysis of companion animal infectomes reveals their diversity and potential roles in animal and human disease. mSphere 2024, 9, e0043924. [Google Scholar] [CrossRef]
- Shi, Y.; Tao, J.; Li, B.; Shen, X.; Cheng, J.; Liu, H. The Gut Viral Metagenome Analysis of Domestic Dogs Captures Snapshot of Viral Diversity and Potential Risk of Coronavirus. Front. Veter- Sci. 2021, 8, 695088. [Google Scholar] [CrossRef]
- Patiño, L.; Benítez, A.D.; Carrazco-Montalvo, A.; Regato-Arrata, M. Genomics for Arbovirus Surveillance: Considerations for Routine Use in Public Health Laboratories. Viruses 2024, 16, 1242. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fang, Y.; Fujita, R.; Khater, E.I.M.; Li, Y.; Wang, W.; Qian, P.; Huang, L.; Guo, Z.; Zhang, Y.; et al. An Exploration of the Viral Coverage of Mosquito Viromes Using Meta-Viromic Sequencing: A Systematic Review and Meta-Analysis. Microorganisms 2024, 12, 1899. [Google Scholar] [CrossRef] [PubMed]
- Laiton-Donato, K.; Guzmán-Cardozo, C.; Peláez-Carvajal, D.; Ajami, N.J.; Navas, M.-C.; Parra-Henao, G.; Usme-Ciro, J.A. Evolution and emergence of mosquito-borne viruses of medical importance: towards a routine metagenomic surveillance approach. J. Trop. Ecol. 2023, 39, e13. [Google Scholar] [CrossRef]
- Cintra, A.M.; Noda-Nicolau, N.M.; Soman, M.L.d.O.; Affonso, P.H.d.A.; Valente, G.T.; Grotto, R.M.T. The Main Arboviruses and Virus Detection Methods in Vectors: Current Approaches and Future Perspectives. Pathogens 2025, 14, 416. [Google Scholar] [CrossRef]
- Castilho de Arruda LD, Giovanetti M, Fonseca V, Zardin MCSU, Lichs GG de C, et al. Dengue Fever Surveillance in Mato Grosso do Sul: Insights from Genomic Analysis and Implications for Public Health Strategies. Viruses 2023;15:1790.
- Carrazco-Montalvo, A.; Gutiérrez-Pallo, D.; Arévalo, V.; Ponce, P.; Rodríguez-Polit, C.; Echeverría-Garcés, G.; Coloma, J.; Nipaz, V.; Cevallos, V. Entomo-Virological Surveillance and Genomic Insights into DENV-2 Genotype III Circulation in Rural Esmeraldas, Ecuador. Pathogens 2025, 14, 541. [Google Scholar] [CrossRef]
- Neto, Z.; Martinez, P.A.; Hill, S.C.; Jandondo, D.; Thézé, J.; Mirandela, M.; Aguiar, R.S.; Xavier, J.; Sebastião, C.d.S.; Cândido, A.L.M.; et al. Molecular and genomic investigation of an urban outbreak of dengue virus serotype 2 in Angola, 2017–2019. PLOS Neglected Trop. Dis. 2022, 16, e0010255. [Google Scholar] [CrossRef]
- de Jesus, J.G.; Dutra, K.R.; Sales, F.C.d.S.; Claro, I.M.; Terzian, A.C.; Candido, D.d.S.; Hill, S.C.; Thézé, J.; Torres, C.; D’aGostini, T.L.; et al. Genomic detection of a virus lineage replacement event of dengue virus serotype 2 in Brazil, 2019. Mem. Do Inst. Oswaldo Cruz 2020, 115, e190423. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, X.; Li, C.; Liu, G.; Wang, S.; Chen, M.; Wei, X.; Wen, H.; Tao, Z.; Xu, Y.; et al. Metagenomic sequencing reveals viral diversity of mosquitoes from Shandong Province, China. Microbiol. Spectr. 2024, 12, e0393223. [Google Scholar] [CrossRef]
- Thannesberger, J.; Rascovan, N.; Eisenmann, A.; Klymiuk, I.; Zittra, C.; Fuehrer, H.P.; Scantlebury-Manning, T.; Gittens-St. HIlaire, M.; Austin, S.; Landis, R.C.; et al. Viral metagenomics reveals the presence of novel Zika virus variants in Aedes mosquitoes from Barbados. Parasites Vectors 2021, 14, 1–11. [Google Scholar] [CrossRef]
- Batovska, J.; Mee, P.T.; Sawbridge, T.I.; Rodoni, B.C.; Lynch, S.E. Enhanced Arbovirus Surveillance with High-Throughput Metatranscriptomic Processing of Field-Collected Mosquitoes. Viruses 2022, 14, 2759. [Google Scholar] [CrossRef] [PubMed]
- Mirza, J.D.; Guimarães, L.d.O.; Wilkinson, S.; Rocha, E.C.; Bertanhe, M.; Helfstein, V.C.; De-Deus, J.T.; Claro, I.M.; Cumley, N.; Quick, J.; et al. Tracking arboviruses, their transmission vectors and potential hosts by nanopore sequencing of mosquitoes. Microb. Genom. 2024, 10, 001184. [Google Scholar] [CrossRef] [PubMed]
- Pan YF, Zhao H, Gou QY, Shi PB, Tian JH, et al. Metagenomic analysis of individual mosquito viromes reveals the geographical patterns and drivers of viral diversity. Nature Ecology & Evolution 2024 8:5 2024;8:947–959.
- Harrington, W.N.; Kackos, C.M.; Webby, R.J. The evolution and future of influenza pandemic preparedness. Exp. Mol. Med. 2021, 53, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H. The beginning and ending of a respiratory viral pandemic-lessons from the Spanish flu. Microb. Biotechnol. 2022, 15, 1301–1317. [Google Scholar] [CrossRef]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef]
- Hall, V.; Cardona, C.; Mendoza, K.; Torchetti, M.; Lantz, K.; Bueno, I.; Franzen-Klein, D.; Ruiz-Saenz, J. Surveillance for highly pathogenic avian influenza A (H5N1) in a raptor rehabilitation center—2022. PLOS ONE 2024, 19, e0299330. [Google Scholar] [CrossRef]
- Stachler, E.; Gnirke, A.; McMahon, K.; Gomez, M.; Stenson, L.; Guevara-Reyes, C.; Knoll, H.; Hill, T.; Hill, S.; Messer, K.S.; et al. Establishing Methods to Monitor Influenza (A)H5N1 Virus in Dairy Cattle Milk, Massachusetts, USA. Emerg. Infect. Dis. 2025, 31, S70–S75. [Google Scholar] [CrossRef]
- Trogu, T.; Bellini, S.; Canziani, S.; Carrera, M.; Chiapponi, C.; Chiari, M.; Farioli, M.; Fusaro, A.; Savegnago, E.; Nucci, A.; et al. Surveillance for Avian Influenza in Wild Birds in the Lombardy Region (Italy) in the Period 2022–2024. Viruses 2024, 16, 1668. [Google Scholar] [CrossRef]
- Tiwari, A.; Meriläinen, P.; Lindh, E.; Kitajima, M.; Österlund, P.; Ikonen, N.; Savolainen-Kopra, C.; Pitkänen, T. Avian Influenza outbreaks: Human infection risks for beach users - One health concern and environmental surveillance implications. Sci. Total. Environ. 2024, 943, 173692. [Google Scholar] [CrossRef]
- Niu, Q.; Jiang, Z.; Wang, L.; Ji, X.; Baele, G.; Qin, Y.; Lin, L.; Lai, A.; Chen, Y.; Veit, M.; et al. Prevention and control of avian influenza virus: Recent advances in diagnostic technologies and surveillance strategies. Nat. Commun. 2025, 16, 1–7. [Google Scholar] [CrossRef]
- Louis S, Mark-Carew M, Biggerstaff M, Yoder J, Boehm AB, et al. Wastewater Surveillance for Influenza A Virus and H5 Subtype Concurrent with the Highly Pathogenic Avian Influenza A(H5N1) Virus Outbreak in Cattle and Poultry and Associated Human Cases — United States, –July 13, 2024. MMWR Morb Mortal Wkly Rep 2024;73:804–809. 12 May.
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J.; et al. Metagenomic Nanopore Sequencing of Influenza Virus Direct from Clinical Respiratory Samples. J. Clin. Microbiol. 2019, 58. [Google Scholar] [CrossRef]
- Leguia, M.; Garcia-Glaessner, A.; Muñoz-Saavedra, B.; Juarez, D.; Barrera, P.; Calvo-Mac, C.; Jara, J.; Silva, W.; Ploog, K.; Amaro, L.; et al. Highly pathogenic avian influenza A (H5N1) in marine mammals and seabirds in Peru. Nat. Commun. 2023, 14, 1–11. [Google Scholar] [CrossRef]
- Lindh, E.; Lounela, H.; Ikonen, N.; Kantala, T.; Savolainen-Kopra, C.; Kauppinen, A.; Österlund, P.; Kareinen, L.; Katz, A.; Nokireki, T.; et al. Highly pathogenic avian influenza A(H5N1) virus infection on multiple fur farms in the South and Central Ostrobothnia regions of Finland, July 2023. Eurosurveillance 2023, 28, 2300400. [Google Scholar] [CrossRef]
- Niu, Q.; Jiang, Z.; Wang, L.; Ji, X.; Baele, G.; Qin, Y.; Lin, L.; Lai, A.; Chen, Y.; Veit, M.; et al. Prevention and control of avian influenza virus: Recent advances in diagnostic technologies and surveillance strategies. Nat. Commun. 2025, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, M.P.G.; Behravesh, C.B.; A Cunningham, A.; Adisasmito, W.B.; Almuhairi, S.; Bilivogui, P.; A Bukachi, S.; Casas, N.; Becerra, N.C.; Charron, D.F.; et al. The panzootic spread of highly pathogenic avian influenza H5N1 sublineage 2.3.4.4b: a critical appraisal of One Health preparedness and prevention. Lancet Infect. Dis. 2024, 24, e774–e781. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.; Cregeen, S.J.; Avadhanula, V.; Zhang, P.; Ayvaz, T.; Feliz, K.; Hoffman, K.L.; Clark, J.R.; Terwilliger, A.; Ross, M.C.; et al. Wastewater sequencing reveals community and variant dynamics of the collective human virome. Nat. Commun. 2023, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, A.; Kantor, R.S.; Olm, M.R.; Whitney, O.N.; Al-Shayeb, B.; Lou, Y.C.; Flamholz, A.; Kennedy, L.C.; Greenwald, H.; Hinkle, A.; et al. Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. mBio 2021, 12. [Google Scholar] [CrossRef]
- Karatas, M.; Bloemen, M.; Swinnen, J.; Roukaerts, I.; Van Gucht, S.; Van Ranst, M.; Wollants, E.; Matthijnssens, J. Untapped potential of wastewater for animal and potentially zoonotic virus surveillance: Pilot study to detect non-human animal viruses in urban settings. Environ. Int. 2025, 199, 109500. [Google Scholar] [CrossRef]
- Zuckerman, N.S.; Bucris, E.; Morad-Eliyahu, H.; Weiss, L.; Vasserman, R.; Fratty, I.S.; Aguvaev, I.; Cohen-Said, Z.; Matar, R.; Erster, O.; et al. Environmental surveillance of a circulating vaccine-derived poliovirus type 2 outbreak in Israel between 2022 and 2023: a genomic epidemiology study. Lancet Microbe 2024, 5, 100893. [Google Scholar] [CrossRef]
- Hill, M.; Pollard, A.J. Detection of poliovirus in London highlights the value of sewage surveillance. Lancet 2022, 400, 1491–1492. [Google Scholar] [CrossRef]
- Hoffmann, B.; Scheuch, M.; Höper, D.; Jungblut, R.; Holsteg, M.; Schirrmeier, H.; Eschbaumer, M.; Goller, K.V.; Wernike, K.; Fischer, M.; et al. Novel Orthobunyavirus in Cattle, Europe, 2011. Emerg. Infect. Dis. 2012, 18, 469–472. [Google Scholar] [CrossRef]
- Dsa, O.C.; Kadni, T.S.; N, S. From cold chain to ambient temperature: transport of viral specimens- a review. Ann. Med. 2023, 55, 2257711. [Google Scholar] [CrossRef] [PubMed]
- Chheda, U.; Pradeepan, S.; Esposito, E.; Strezsak, S.; Fernandez-Delgado, O.; Kranz, J. Factors Affecting Stability of RNA – Temperature, Length, Concentration, pH, and Buffering Species. J. Pharm. Sci. 2023, 113, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Parmar S, Sridhar S, Forrest S, Kean I, Young J, et al. A blueprint for the implementation of a validated approach for the detection of SARS-Cov2 in clinical samples in academic facilities. Wellcome Open Res 2020;5:110.
- Reyes, G.R.; Kim, J.P. Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol. Cell. Probes 1991, 5, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Chrzastek, K.; Lee, D.-H.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef]
- Electron microscopy and a sequence-independent, single-primer amplification (SISPA) viromics approach for monkeypox virus genome determination - MPXV / Genome Reports - Virological. https://virological.org/t/electron-microscopy-and-a-sequence-independent-single-primer-amplification-sispa-viromics-approach-for-monkeypox-virus-genome-determination/899 (accessed ). 3 April.
- Peserico, A.; Marcacci, M.; Malatesta, D.; Di Domenico, M.; Pratelli, A.; Mangone, I.; D’aLterio, N.; Pizzurro, F.; Cirone, F.; Zaccaria, G.; et al. Diagnosis and characterization of canine distemper virus through sequencing by MinION nanopore technology. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Jothikumar, N.; Cromeans, T.; Shivajothi, J.; Vinjé, J.; Murphy, J. Development and evaluation of a ligation-free sequence-independent, single-primer amplification (LF-SISPA) assay for whole genome characterization of viruses. J. Virol. Methods 2022, 299, 114346–114346. [Google Scholar] [CrossRef]
Figure 1.
Milestones in Virus Genome Sequencing Technologies. The timeline illustrates the chronological progression of sequencing technologies and their impact on the discovery and characterisation of human viruses from 1979 to 2019. The evolution also emphasises how technological advances have transformed viral discovery and molecular epidemiology enhancing surveillance capabilities to enable more rapid responses to (re)emerging threats [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. Abbreviations: WGS, whole genome sequencing.
Figure 1.
Milestones in Virus Genome Sequencing Technologies. The timeline illustrates the chronological progression of sequencing technologies and their impact on the discovery and characterisation of human viruses from 1979 to 2019. The evolution also emphasises how technological advances have transformed viral discovery and molecular epidemiology enhancing surveillance capabilities to enable more rapid responses to (re)emerging threats [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. Abbreviations: WGS, whole genome sequencing.
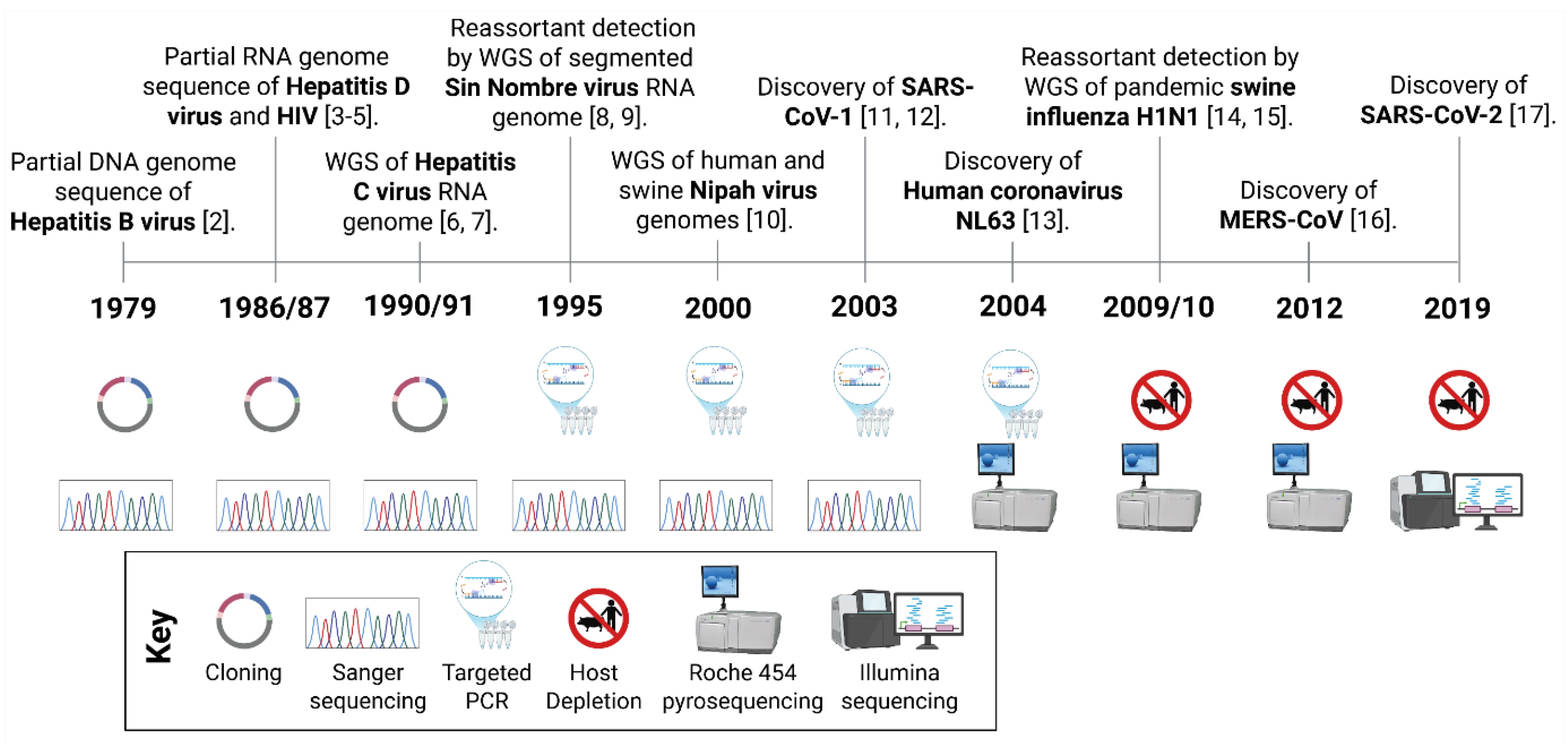
Figure 2.
The One Health Paradigm in (Re)Emerging Virus Investigation and Control. The One Health paradigm recognises the interconnectedness of animal, environmental and human health, promoting a holistic understanding of global health challenges and the pursuit of sustainable and equitable solutions with long-term benefits. This approach is particularly critical in the investigation and management of (re)emerging pathogens, as viral spillover events occur at the interface between these domains. Multiple ecological, biological and socio-economic factors influence spillover risk and the resulting impacts span public health, veterinary, agricultural and environmental sectors, making integration of a One Health perspective essential for effective research, prevention and control of (re)emerging viral threats.
Figure 2.
The One Health Paradigm in (Re)Emerging Virus Investigation and Control. The One Health paradigm recognises the interconnectedness of animal, environmental and human health, promoting a holistic understanding of global health challenges and the pursuit of sustainable and equitable solutions with long-term benefits. This approach is particularly critical in the investigation and management of (re)emerging pathogens, as viral spillover events occur at the interface between these domains. Multiple ecological, biological and socio-economic factors influence spillover risk and the resulting impacts span public health, veterinary, agricultural and environmental sectors, making integration of a One Health perspective essential for effective research, prevention and control of (re)emerging viral threats.
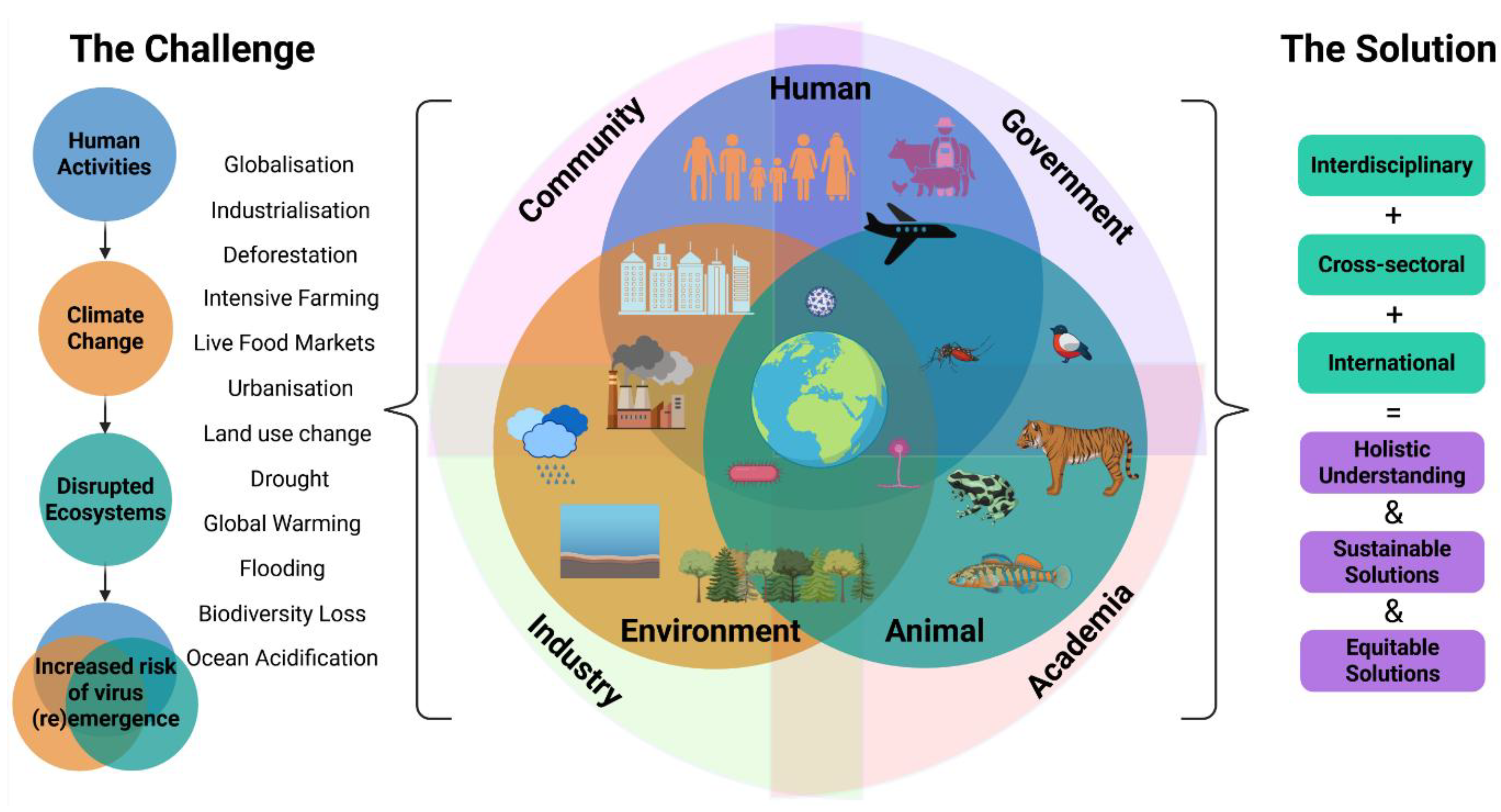
Figure 3.
Workflow Adaptations for Different Sample Matrices in Viral Metagenomic NGS (vmNGS). Illustrated workflows shows how vmNGS sample processing can be adapted based on the type of sample matrix, technological advancements and available resources. For tissue samples, RNA extraction is typically prioritised as actively replicating intracellular virus are expected to produce viral transcripts. For swabs, biological fluids, and environmental samples, both DNA and RNA should be analysed, since viruses are often extracellular, and may lack active transcription. Sequence-independent PCR amplification is often applied to extracts with low nucleic acid concentrations, particularly from swabs, fluids and environmental matrices. For environmental samples, viral particles may have lost capsid integrity, leaving nucleic acids unprotected; in such cases, nuclease digestions to remove host nucleic acids is not recommended.
Figure 3.
Workflow Adaptations for Different Sample Matrices in Viral Metagenomic NGS (vmNGS). Illustrated workflows shows how vmNGS sample processing can be adapted based on the type of sample matrix, technological advancements and available resources. For tissue samples, RNA extraction is typically prioritised as actively replicating intracellular virus are expected to produce viral transcripts. For swabs, biological fluids, and environmental samples, both DNA and RNA should be analysed, since viruses are often extracellular, and may lack active transcription. Sequence-independent PCR amplification is often applied to extracts with low nucleic acid concentrations, particularly from swabs, fluids and environmental matrices. For environmental samples, viral particles may have lost capsid integrity, leaving nucleic acids unprotected; in such cases, nuclease digestions to remove host nucleic acids is not recommended.
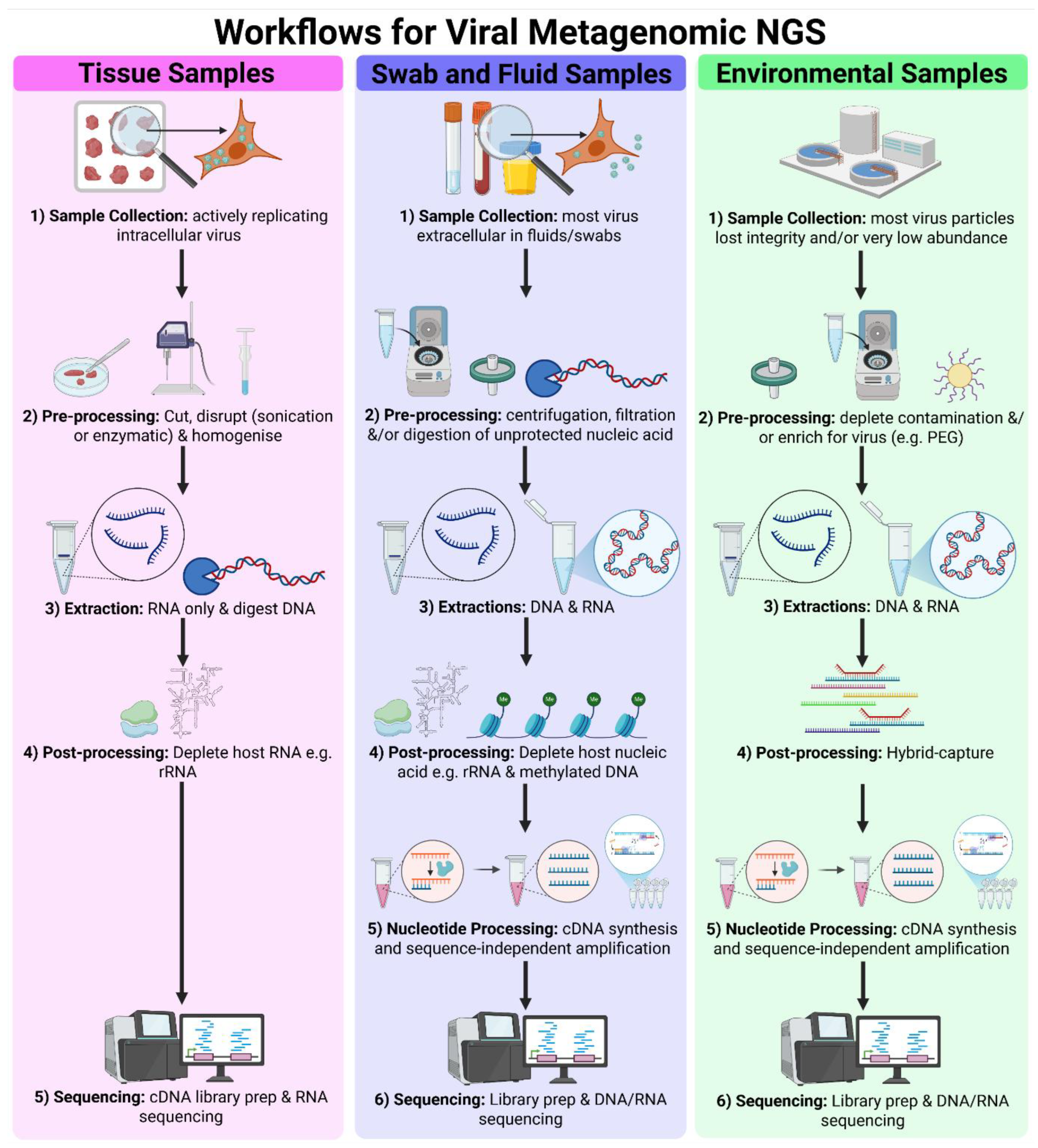
Figure 4.
Bioinformatics Pipelines for Viral Metagenomic NGS. vmNGS bioinformatics pipelines begin with raw sequence reads produced by NGS instruments, which require quality control steps to trim poor quality base calls, discard poor quality reads and filter out host reads. Overlapping reads then undergo de novo assembly to generate contigs. Nucleotide sequences and, following in silico translation of the three open reading frames, amino acid sequences are searched against viral nucleotide and amino acid databases to identify detected viruses. Reference genomes can be used to assemble whole or more complete genome sequences, which can be used for phylogenetic analysis, epidemiology and functional characterisation of viruses.
Figure 4.
Bioinformatics Pipelines for Viral Metagenomic NGS. vmNGS bioinformatics pipelines begin with raw sequence reads produced by NGS instruments, which require quality control steps to trim poor quality base calls, discard poor quality reads and filter out host reads. Overlapping reads then undergo de novo assembly to generate contigs. Nucleotide sequences and, following in silico translation of the three open reading frames, amino acid sequences are searched against viral nucleotide and amino acid databases to identify detected viruses. Reference genomes can be used to assemble whole or more complete genome sequences, which can be used for phylogenetic analysis, epidemiology and functional characterisation of viruses.
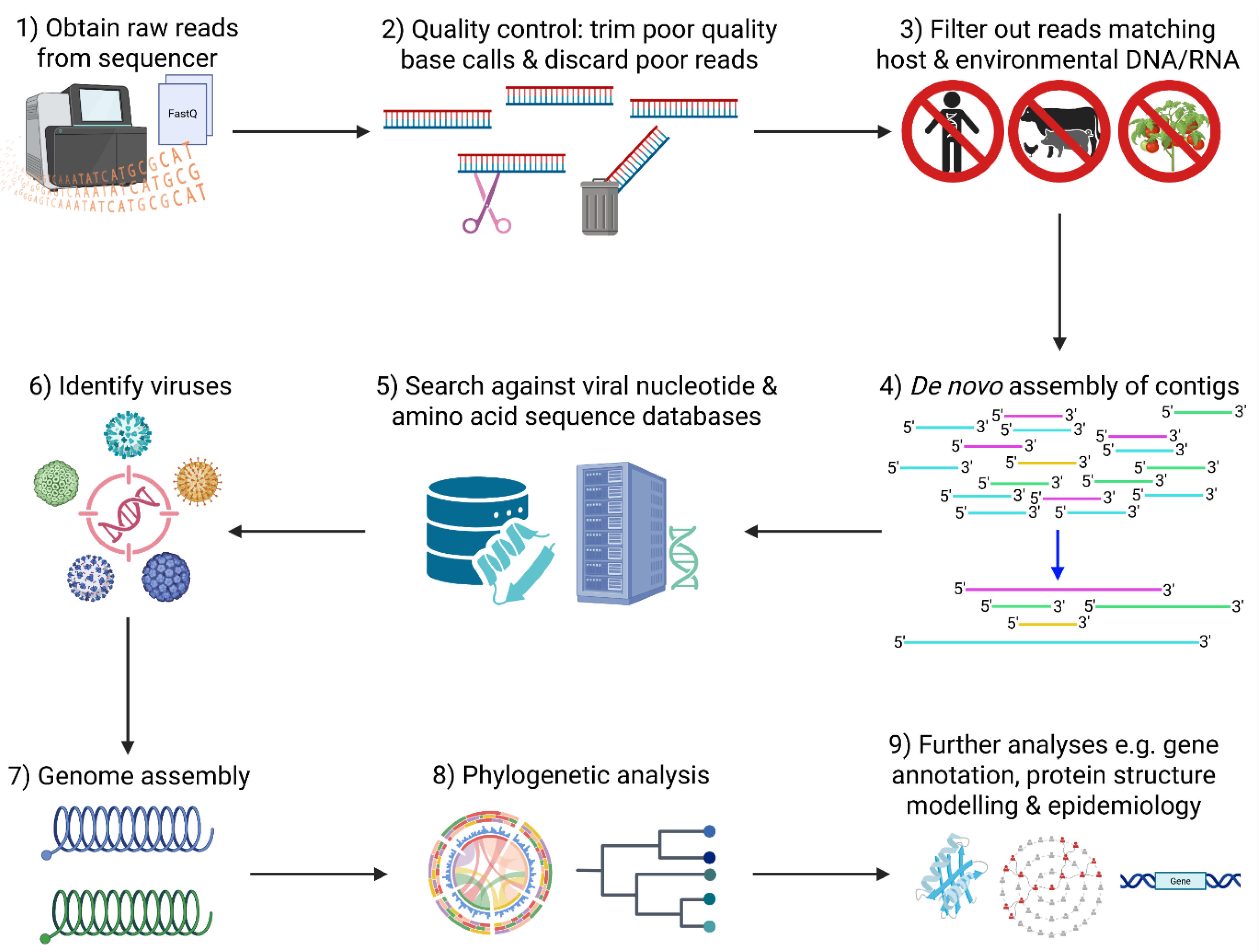
Figure 5.
Integration of vmNGS into Passive (A) and Active (B) Surveillance. A) Passive surveillance involves reacting to a disease outbreak. vmNGS should be deployed when infectious disease is suspected, aetiological agents are unknown, and risk of a larger or more severe outbreak is high. Targeted methods can then be designed to more cost-effectively and rapidly detect novel viruses. B) Active surveillance proactively screens high-risk animals and environments for potential (re)emerging pathogens before they cause an outbreak. The high number of samples required makes vmNGS too inefficient and expensive to be used routinely. It can be deployed when scoping out the viruses that should be included in active surveillance of a new region or to periodically update the viruses requiring inclusion. .
Figure 5.
Integration of vmNGS into Passive (A) and Active (B) Surveillance. A) Passive surveillance involves reacting to a disease outbreak. vmNGS should be deployed when infectious disease is suspected, aetiological agents are unknown, and risk of a larger or more severe outbreak is high. Targeted methods can then be designed to more cost-effectively and rapidly detect novel viruses. B) Active surveillance proactively screens high-risk animals and environments for potential (re)emerging pathogens before they cause an outbreak. The high number of samples required makes vmNGS too inefficient and expensive to be used routinely. It can be deployed when scoping out the viruses that should be included in active surveillance of a new region or to periodically update the viruses requiring inclusion. .
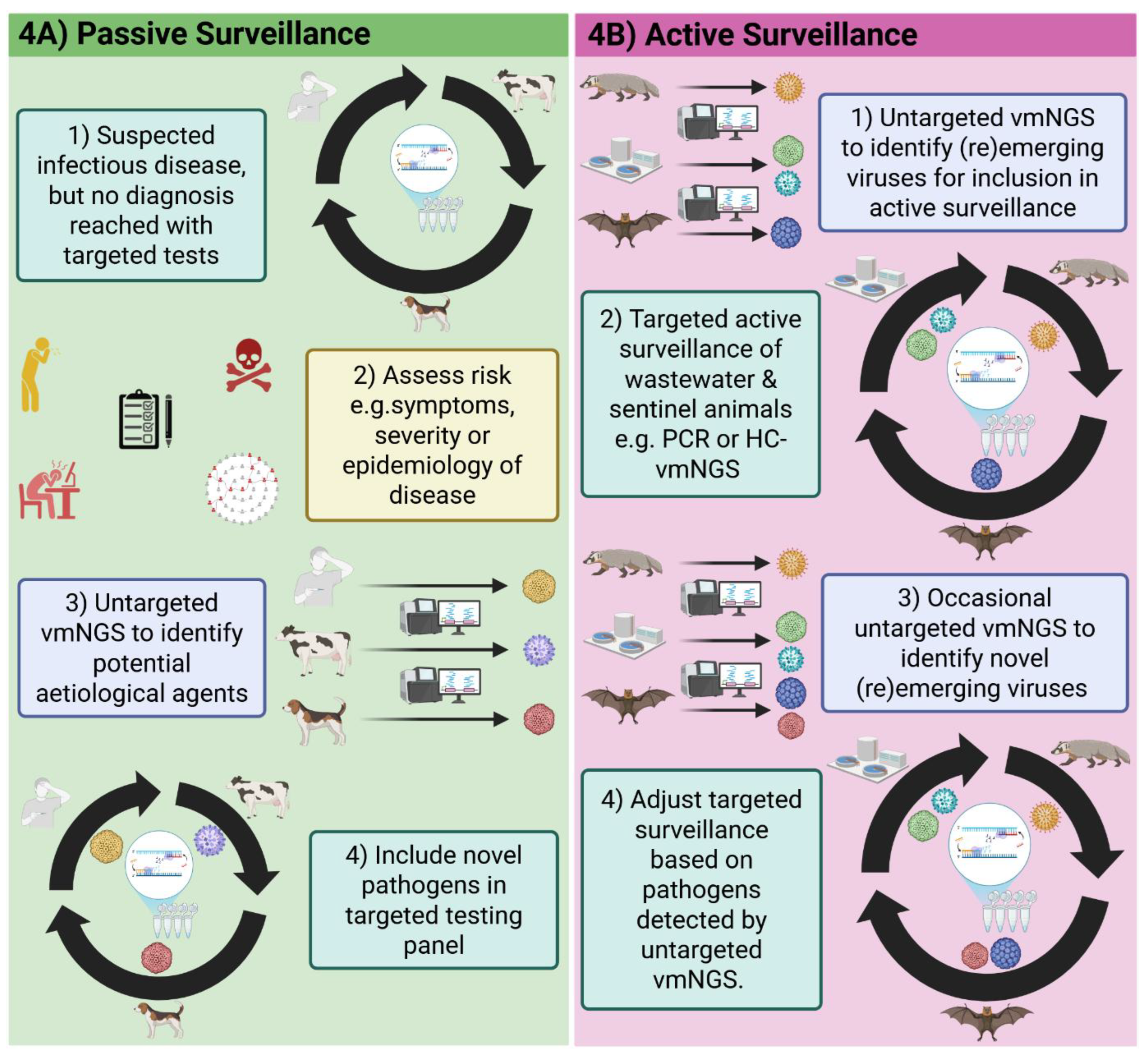
Table 1.
Practical and technical overview of sequencing technologies [19,20,21]. 1 Cost per base does not include initial costs for instrument purchase. 2 Adaptive sequencing enables the real-time selection of target sequences or exclusion of off-target sequences. Abbreviations: ONT, Oxford Nanopore Technologies; ds-cDNA, double stranded complementary DNA; SNP, single nucleotide polymorphism; WGS, Whole genome sequencing.
Table 1.
Practical and technical overview of sequencing technologies [19,20,21]. 1 Cost per base does not include initial costs for instrument purchase. 2 Adaptive sequencing enables the real-time selection of target sequences or exclusion of off-target sequences. Abbreviations: ONT, Oxford Nanopore Technologies; ds-cDNA, double stranded complementary DNA; SNP, single nucleotide polymorphism; WGS, Whole genome sequencing.
| Generation | First (Sanger) | Second (Illumina) | Third (ONT) |
|---|---|---|---|
| Other Platforms | Maxam-Gilbert | Roche 454, Ion Torrent, SOLiD | PacBio |
| Cost per Kb ($) 1 | 500-1,000 | 0.01-0.10 | 0.10-10.00 |
| Error Rate (%) | 0.001 | 0.1-1.0 | 1-15 |
| Output (bases per run) | 1,000 bp | 60 Gb – 6 Tb | 10 Gb – 10 Tb |
| Read Length | 1,000 bp | 50-300 bp | Up to 1+ Mb |
| Requirement for PCR | Yes (sequence-dependent to enrich target) | Yes (sequence-independent for cluster generation on flow cell, can also be used in library preparation and to enrich targets) | No (can be used to enrich targets) |
| Preparation Methods | PCR or clonal amplification of target | Generate library: fragmentation, ds-cDNA synthesis, adapter/barcode ligation and purification | Generate library: adapter/barcode ligation and purification |
| Data Storage Requirements | Low | High | High |
| Portable | No | No | Yes |
| Suitable for Metagenomics | No | Yes (including degraded samples) | Yes (unsuitable for degraded samples) |
| Other Applications | Genotyping and Targeted Sequencing | WGS, SNP Variant Calling and Transcriptomics | WGS, Splice Variant Detection, Real-time Sequencing, Direct RNA Sequencing, Epigenetic Modification Detection and Adaptive Sequencing 2 |
| Main Strengths | High sensitivity (for target only), high specificity and high accuracy | High accuracy, high sensitivity, high depth, high throughput and suitability for degraded samples | Long read length, high throughput, high sensitivity and high coverage |
| Main Weakness | Low throughput | Short read-length | Poor accuracy |
Table 2.
Overview of reemerging viruses where vmNGS contributed to their identification and characterisation. Abbreviations: DRC, Democratic Republic of Congo.
Table 2.
Overview of reemerging viruses where vmNGS contributed to their identification and characterisation. Abbreviations: DRC, Democratic Republic of Congo.
| Virus | Original Location | New Location | Year | Cause | Refs |
| Mpox IIb | Nigeria | Worldwide | 2022 | See main text | [105] |
| Mpox Ib | DRC | Worldwide | 2023 | See main text | [106] |
| Zika virus | Africa, Asia & French Polynesia | Brazil | 2015 | See main text | [107] |
| Oropouche virus | Brazil, Caribbean, Peru & Panama | Ecuador | 2016 | Unknown, PCR recognised mutated region | [103,104] |
| St Louis Encephalitis virus | Argentina | USA | 2015 | Migratory birds | [108] |
| Ebola virus | DRC | Guinea, Liberia, Sierra Leone | 2014 | Zoonotic spillover, probably bats | [109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



