Submitted:
25 August 2025
Posted:
26 August 2025
You are already at the latest version
Abstract
Weaver syndrome is a rare congenital overgrowth disorder caused by heterozygous pathogenic variants in EZH2, a gene encoding a histone methyltransferase essential for epigenetic regulation. We describe a 4-year-old Taiwanese female who exhibited classical features of Weaver syndrome, including macrosomia (height and weight >97th percentile), macrocephaly, hypertelorism, prominent forehead, and developmental delay, along with atypical findings of severe bilateral camptodactyly and complex brain malformations. Neuroimaging revealed corpus callosum dysgenesis with rostral agenesis and genu hypoplasia, bilateral frontal lobe hypoplasia, and an arachnoid cyst. The patient had global developmental delay with marked gross and fine motor impairment but relatively preserved speech and cognition. Whole-exome sequencing identified a novel de novo pathogenic variant in EZH2: c.449T>C (p.Ile150Thr). This variant affects a highly conserved amino acid within the SANT domain and is predicted to be deleterious by computational analysis. Its de novo origin was confirmed by Sanger sequencing. This case broadens the clinical spectrum of Weaver syndrome by highlighting severe camptodactyly and complex brain malformations as possible EZH2-related manifestations. The corpus callosum dysgenesis and associated cerebral abnormalities suggest a wider role of EZH2 in neurodevelopment than previously recognized. These findings underscore the importance of comprehensive neuroimaging and molecular genetic testing in patients with suspected Weaver syndrome, particularly in atypical presentations.
Keywords:
weaver syndrome
; EZH2 variant
; corpus callosum dysgenesis
; camptodactyly
; overgrowth syndrome
1. Introduction
Weaver syndrome is a rare congenital overgrowth disorder characterized by prenatal and postnatal macrosomia, accelerated osseous maturation, distinctive craniofacial features, and variable intellectual disability [1]. First described by Weaver et al. [2] in 1974, the condition remained genetically undefined until 2011, when two independent studies identified heterozygous pathogenic variants in the EZH2 gene as the causative factor [3,4]. EZH2 encodes the catalytic subunit of Polycomb Repressive Complex 2 (PRC2), a key epigenetic regulator that catalyzes trimethylation of lysine 27 on histone H3 (H3K27me3), thereby establishing repressive chromatin states critical for developmental gene regulation [5].
The estimated incidence of Weaver syndrome is approximately 1 in 15,000 births, with over 90% of cases resulting from de novo mutations [6]. The phenotypic spectrum includes characteristic craniofacial features such as broad forehead, ocular hypertelorism, large fleshy ears, and micrognathia with a distinctive horizontal chin crease [7]. Skeletal findings consistently include advanced bone age, camptodactyly, and deep-set nails, whereas neurological features range from mild hypotonia to moderate intellectual disability, which is present in about 80% of affected individuals [5].
At the molecular level, the pathogenesis is linked to partial loss of EZH2 histone methyltransferase activity, leading to reduced H3K27 trimethylation and impaired developmental gene regulation [3]. Pathogenic variants are primarily missense mutations distributed throughout the gene, with notable clustering in the SET domain that mediates enzymatic activity [8]. Recent studies have expanded the recognized clinical spectrum to include neurological malformations such as corpus callosum dysgenesis and polymicrogyria as well as severe skeletal anomalies such as pronounced camptodactyly [9,10].
Despite advances in molecular understanding, Weaver syndrome continues to demonstrate significant phenotypic variability, and atypical presentations are increasingly reported. The identification of novel clinical features, particularly complex brain malformations and severe skeletal abnormalities, is crucial for refining the phenotypic spectrum and improving diagnostic accuracy in this rare disorder.
2. Case Presentation
2.1. Patient Demographics and Initial Presentation
The patient is a 4-year-old Taiwanese female (born September 18, 2020) who was first evaluated at 15 months of age for abnormal clinical phenotype and global developmental delay. She was born at 38 + 4 weeks’ gestation with a birth weight of 4,460 g (>97th percentile), length of 56 cm (>97th percentile), and head circumference of 35 cm (>97th percentile), consistent with prenatal macrosomia. Delivery was by normal spontaneous vaginal delivery, with Apgar scores of 8 at 1 min and 9 at 5 min. She is the second child of nonconsanguineous parents. Her older brother, born in 2018, demonstrates normal development.
Family history revealed paternal height of 175 cm and maternal height of 166 cm, with a predicted target height of 164.5 cm (75–85th percentile). The patient resides in Kinmen and receives follow-up care at Taipei Veterans General Hospital. Early clinical concerns included prolonged neonatal jaundice, generalized hypotonia, and distinctive craniofacial features, which prompted genetic evaluation.
2.2. Clinical Features and Physical Examination
Serial anthropometric assessments confirmed persistent overgrowth. At the most recent evaluation (4 yr 10 months), her height was 123 cm (>97th percentile), weight 26 kg (>97th percentile), and head circumference 52 cm (85–97th percentile). Body mass index was 17.2 kg/m², reflecting sustained overgrowth since infancy.
Physical examination revealed dysmorphic features consistent with Weaver syndrome (Figure 1). Craniofacial abnormalities included macrocephaly, prominent squared forehead, bilateral hypertelorism with epicanthal folds, downward-slanting palpebral fissures, broad nasal bridge, small mouth, and mild micrognathia without macroglossia. A horizontal chin crease—a pathognomonic feature of Weaver syndrome—was observed. The anterior fontanelle was nearly closed, and the ears were low-set and floppy.
Cardiovascular examination revealed normal heart sounds without murmurs. Abdominal examination showed hepatomegaly and a prominent umbilical hernia. Spinal assessment indicated mild thoracolumbar kyphoscoliosis. Extremity evaluation revealed bilateral curved tibiae, equinovalgus foot deformity (more pronounced on the left), possible hallux valgus, and severe bilateral camptodactyly with marked posterior finger flexion (Figure 2). Generalized mild hypotonia was noted in all extremities.
2.3. Developmental Assessment
Developmental evaluation demonstrated global delay, with a disproportionate pattern: severe motor impairment but relatively preserved cognitive and language abilities. Gross motor milestones were significantly delayed—sitting without assistance at 10 months, limited crawling, and absence of independent standing at 15 months. At 4 yr, she remained unable to run or jump, requiring ongoing Early Intervention Program support.
Fine motor skills were similarly impaired due to severe bilateral camptodactyly, which restricted dexterity and object manipulation. By contrast, speech and language developed more favorably, with progressive acquisition of verbal communication. Cognitive assessment suggested intellectual function within the mildly impaired range, consistent with typical Weaver syndrome presentations.
2.4. Neuroimaging Findings
Brain magnetic resonance imaging performed on March 31, 2021, at Taipei Veterans General Hospital revealed significant structural abnormalities extending the known neurological spectrum of Weaver syndrome. Findings included corpus callosum dysgenesis characterized by agenesis of the rostrum and hypoplasia/atrophy of the genu and anterior body. Additional abnormalities were suspected hypoplasia of the bilateral frontal lobes and an arachnoid cyst in the left quadrigeminal cistern. Bilateral middle-ear effusion was also noted.
These atypical neuroimaging findings suggest that EZH2-related disorders may encompass broader cerebral malformations than previously recognized. The corpus callosum anomalies likely contribute to impaired motor coordination and may affect long-term neurodevelopmental outcomes.
2.5. Genetic Testing and Molecular Diagnosis
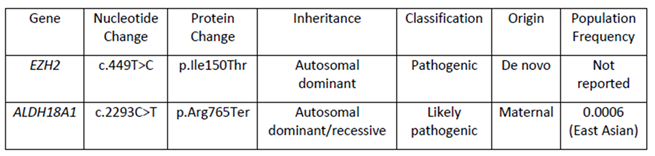
Initial chromosomal microarray analysis at Taipei Veterans General Hospital showed no pathogenic copy number variants. Whole-exome sequencing subsequently identified a novel de novo pathogenic variant in EZH2: c.449T>C (p.Ile150Thr) (Table 1). This variant affects a highly conserved residue within the SANT domain and was classified as pathogenic according to American College of Medical Genetics guidelines. It was absent from population databases and predicted to be deleterious by multiple computational tools.
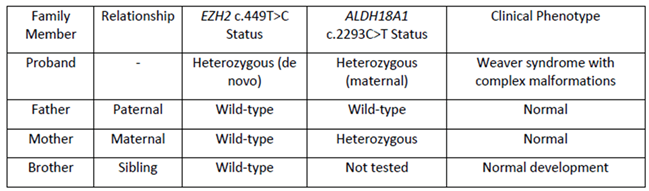
Family segregation analysis confirmed the de novo nature of the EZH2 variant. Sanger sequencing of both parents demonstrated wild-type sequences at the corresponding position, establishing this as a spontaneous mutation (Table 2). The patient’s father and older brother also underwent whole exome sequencing, which revealed no pathogenic variants related to the patient’ phenotype.
Additionally, whole exome sequencing identified a second variant of uncertain significance in the ALDH18A1 gene (c.2293C>T, p.Arg765Ter), inherited from the mother. This variant has been associated with connective tissue and neurological disorders but was considered less likely to explain the patient’s primary phenotype, given the dominant inheritance pattern and the characteristic features of Weaver syndrome.
Pedigree analysis revealed a nonconsanguineous, two-generation family, with the proband being the second child of healthy parents (Figure 3). The patient’s father, aged 35 yr, is 175 cm tall, and her mother, aged 33 yr, is 166 cm tall; both exhibit normal phenotypes. The proband has an older brother, born on April 6, 2018, who demonstrates normal growth and developmental milestones at 6 yr of age. Family segregation analysis through Sanger sequencing confirmed the de novo EZH2 variant, with both parents carrying wild-type sequences at the corresponding genomic position. The maternal inheritance of the ALDH18A1 variant was established through targeted sequencing, which showed the mother to be a heterozygous carrier of the c.2293C>T variant, while remaining phenotypically normal. The unaffected brother was not tested for the ALDH18A1 variant, given its uncertain clinical relevance to the proband’s phenotype.
This inheritance pattern strongly supports the pathogenic role of the de novo EZH2 variant as the primary cause of the patient’s Weaver syndrome, whereas the maternally inherited ALDH18A1 variant represents an incidental finding of uncertain clinical significance in this case.
2.6. Treatment and Management
The patient’s care is coordinated through a comprehensive multidisciplinary approach tailored to her complex medical needs. Ophthalmologic management includes corrective lenses for significant refractive errors, specifically hyperopia of +9.00 diopters with astigmatism of +2.25 diopters, along with amblyopia therapy. Orthopedic consultation has been initiated to address severe bilateral camptodactyly and equinovalgus foot deformities, which may require surgical intervention to optimize functional outcomes.
Nutritional support includes vitamin D3 replacement therapy (two drops daily of vitamin D3 solution) for documented deficiency, with dosing adjusted according to serum 25-hydroxyvitamin D levels. Zinc supplementation was also started to correct low serum zinc levels. Regular monitoring of liver function is performed due to persistent hepatomegaly observed on serial abdominal ultrasonography.
Early intervention services in Kinmen focus on enhancing gross and fine motor skills, with emphasis on adaptive strategies for severe camptodactyly. Speech therapy has been provided as needed, although language development has progressed more favorably than motor function.
The family has received comprehensive genetic counseling regarding the de novo EZH2 variant, inheritance patterns, and recommendations for long-term medical surveillance. This includes discussion of potential cancer screening protocols, given the reported increased risk of neuroblastoma in patients with Weaver syndrome.
3. Discussion
3.1. Genotype–Phenotype Correlation
The novel EZH2 variant c.449T>C (p.Ile150Thr) identified in our patient represents the first reported mutation affecting this specific residue within the SANT domain of the EZH2 protein. The SANT (SWI3, ADA2, N-CoR, and TFIIIB) domain functions as a critical histone reader that confers sensitivity to the modification state of the histone H4 tail [11]. Of the 48 confirmed EZH2 mutations reported in individuals with Weaver syndrome, only a small subset involve the SANT domain, yet these are often associated with more severe phenotypic presentations [1,3].
Recent functional studies indicate that pathogenic EZH2 variants in Weaver syndrome result in partial loss of histone methyltransferase activity, rather than complete haploinsufficiency [5,7]. The absence of early truncating mutations in reported cases supports this mechanism, as homozygous null mutations in EZH2 are incompatible with life in murine models [12]. The p.Ile150Thr substitution introduces polarity into a normally hydrophobic region of the SANT1 domain, likely impairing the protein’s ability to interact with histone H4 tails and maintain chromatin organization [11].
Despite extensive molecular characterization of EZH2 variants, robust genotype–phenotype correlations remain unclear [5]. The severity of clinical features does not consistently align with the degree of histone methyltransferase activity reduction observed in vitro [5], suggesting that additional factors—such as genetic background, epigenetic regulation, and stochastic developmental variation—contribute to phenotypic diversity. The severe bilateral camptodactyly and complex brain malformations in our patient align with the more severe phenotypic spectrum predicted for SANT domain mutations.
3.2. Expansion of the Phenotypic Spectrum
This case expands the phenotypic spectrum of Weaver syndrome by documenting severe corpus callosum dysgenesis and extreme bilateral camptodactyly. Although neurological malformations such as polymicrogyria and ventriculomegaly have been reported with EZH2 mutations [4,5], corpus callosum dysgenesis remains underrecognized and warrants systematic neuroimaging in all suspected cases.
The corpus callosum, the largest commissural white matter structure in the brain, contains approximately 200 million axons connecting the cerebral hemispheres [13]. Its development is particularly vulnerable between 11 and 20 weeks of gestation, when callosal fibers first cross the midline [13]. EZH2 plays a crucial role in cortical neuronal migration and regulation of genes involved in establishing interhemispheric connectivity [4]. The rostral agenesis and genu hypoplasia observed in our patient likely reflect disruption of commissural neuron specification and axon guidance during this critical window.
The patient’s severe bilateral camptodactyly requiring surgical consideration also represents an atypical manifestation. Camptodactyly affects approximately 1% of the general population but occurs with greater frequency and severity in Weaver syndrome [14]. The degree of bilateral involvement and functional impairment observed here highlights the potential for EZH2 mutations to markedly disrupt digit development and underscores the need for comprehensive orthopedic management.
3.3. Clinical Implications and Diagnostic Considerations
Recognition of corpus callosum dysgenesis as a potential feature of Weaver syndrome has important diagnostic implications. Current diagnostic criteria emphasize overgrowth, distinctive craniofacial features, and developmental delay but may not adequately capture neurological malformations [1,3]. Our findings suggest that comprehensive brain imaging should be incorporated into the diagnostic evaluation of suspected Weaver syndrome, especially in patients with unexplained developmental delays or motor dysfunction.
The subtle facial features of Weaver syndrome, particularly in older individuals, often complicate diagnosis [3,15]. Advanced bone age remains a consistent diagnostic marker and should prompt EZH2 testing when identified [3]. Intellectual disability, ranging from mild to moderate in approximately 80% of patients [3,8], is frequently the presenting concern.
The increased malignancy risk, particularly neuroblastoma during early childhood, remains an area of clinical uncertainty [16,17]. While lifetime malignancy risk is estimated at approximately 10%, the small patient population precludes evidence-based surveillance guidelines [16]. Current consensus emphasizes clinical vigilance over routine imaging, although some experts recommend periodic abdominal ultrasonography in early childhood [17].
3.4. Management Considerations
Management of Weaver syndrome requires a multidisciplinary approach tailored to diverse clinical manifestations. In our patient, severe bilateral camptodactyly necessitated early orthopedic evaluation and consideration of surgical correction. Conservative measures—such as progressive splinting, physical therapy, and occupational therapy—remain first-line treatments [18,19]. Surgery is generally reserved for persistent flexion contractures >60° or when functional limitations significantly impair activities of daily living [20].
Corpus callosum dysgenesis and associated frontal lobe hypoplasia highlight the need for comprehensive neurodevelopmental assessment and early intervention services. The patient’s motor impairment, contrasted with relatively preserved language function, underscores the benefit of targeted therapies addressing specific developmental domains. Regular neurological follow-up is essential to monitor for seizures and guide additional interventions.
Educational support through individualized education plans is critical, given the high prevalence of intellectual disability in Weaver syndrome [3]. Adaptive strategies focusing on gross and fine motor skill development may be especially beneficial, as verbal abilities are often relatively preserved.
The family received detailed genetic counseling regarding the de novo nature of the EZH2 variant, reassuring them of a low recurrence risk for future pregnancies while emphasizing the importance of long-term surveillance for the proband. Follow-up should include regular growth monitoring, developmental assessment, and vigilance for malignancy—particularly neuroblastoma risk during early childhood.
4. Conclusions
We report a novel de novo EZH2 variant, c.449T>C (p.Ile150Thr), in a 4-year-old Taiwanese female with Weaver syndrome. This represents the first documented mutation affecting this specific residue within the SANT domain. The case broadens the recognized phenotypic spectrum of EZH2-related overgrowth disorders by identifying severe bilateral camptodactyly and complex brain malformations, including corpus callosum dysgenesis with rostral agenesis and genu hypoplasia—features not previously emphasized in the classical Weaver syndrome phenotype. The severity of skeletal and neurological manifestations observed suggests that mutations within the SANT domain, a critical histone reader for chromatin targeting, may have particularly severe developmental consequences compared with SET domain mutations that primarily impair catalytic activity.
The identification of corpus callosum dysgenesis as a potential feature of Weaver syndrome has direct clinical implications for diagnosis and management. Comprehensive brain imaging should be incorporated into standard evaluation protocols, particularly given the potential impact on neurodevelopmental outcomes and seizure risk. Similarly, the extreme bilateral camptodactyly requiring surgical consideration highlights the importance of early orthopedic assessment and multidisciplinary intervention to optimize functional outcomes during critical developmental periods.
This case adds valuable molecular and phenotypic data to the international database of EZH2 variants, underscoring the importance of systematic case reporting from diverse populations in advancing rare disease knowledge. As one of the few molecularly confirmed Asian cases, it also highlights potential geographic and ethnic considerations in phenotypic expression and supports the implementation of standardized surveillance protocols, including neuroblastoma screening, cervical spine monitoring, and comprehensive neurodevelopmental support.
The documentation of this novel variant and its severe phenotype emphasizes the need for functional studies to clarify the pathogenic mechanisms underlying SANT domain mutations. Future research priorities should include the development of targeted therapies addressing the partial loss-of-function mechanism of EZH2 variants, establishment of collaborative international networks for systematic natural history studies, and implementation of precision medicine approaches based on molecular subtyping of EZH2-related disorders. While this single case provides important insights into phenotypic expansion and domain-specific effects, long-term follow-up and functional validation remain essential to guide clinical management and develop evidence-based treatment protocols for this rare but clinically significant disorder.
Author Contributions
C.-L.L. drafted the manuscript. S.-P.L. and H.-Y.L. participated in patient follow-up and contributed to manuscript drafting. C.-K.C., R.-Y.T., and Y.-T.L. performed biochemical analyses and revised the manuscript. H.-C.C. and Y.-H.C. were responsible for patient screening and critically revised the manuscript. All authors have read and approved the final version of the manuscript.
Data Availability Statement
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Ethics Statement: This study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of MacKay Memorial Hospital (Approval No. 21MMHIS109e, approved on 2021/10/01).
Informed Consent: Written informed consent for publication was obtained from the patient’s parents.
Acknowledgments
The authors sincerely thank the staff members who contributed to this study and extend their gratitude to the patient and family for their participation and cooperation.
Funding Information: This research was supported by multiple grants from MacKay Memorial Hospital (MMH-E-114-13, MMH-MM-113-13, MMH-E-113-13, MMH-MM-112-14, and MMH-E-112-13) and the Ministry of Science and Technology, Executive Yuan, Taiwan (NSTC-114-2811-B-195-002, NSTC-114-2314-B-195-001, NSTC-114-2314-B-715-001, NSTC-114-2314-B-195-002, NSTC-113-2314-B-195-003, NSTC-113-2314-B-195-004, NSTC-113-2314-B-715-002, NSTC-113-2314-B-195-021, NSTC-113-2811-B-195-001, NSTC-112-2314-B-195-014-MY3, NSTC-112-2811-B-195-001, and NSTC-112-2314-B-195-003,).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tatton-Brown, K.; Murray, A.; Hanks, S.; Douglas, J.; Armstrong, R.; Banka, S.; Bird, L.M.; Clericuzio, C.L.; Cormier-Daire, V.; Cushing, T.; Flinter, F.; Jacquemont, M.L.; Joss, S.; Kinning, E.; Lynch, S.A.; Magee, A.; McConnell, V.; Medeira, A.; Ozono, K.; Patton, M.; Rankin, J.; Shears, D.; Simon, M.; Splitt, M.; Strenger, V.; Stuurman, K.; Taylor, C.; Titheradge, H.; Van Maldergem, L.; Temple, I.K.; Cole, T.; Seal, S.; Childhood Overgrowth Consortium; Rahman, N. Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. Am. J. Med. Genet. A 2013, 161A, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.D.; Graham, C.B.; Thomas, I.T.; Smith, D.W. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J. Pediatr. 1974, 84, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Hanks, S.; Ruark, E.; Zachariou, A.; Duarte Sdel, V.; Ramsay, E.; Snape, K.; Murray, A.; Perdeaux, E.R.; Seal, S.; Loveday, C.; Banka, S.; Clericuzio, C.; Flinter, F.; Magee, A.; McConnell, V.; Patton, M.; Raith, W.; Rankin, J.; Splitt, M.; Strenger, V.; Taylor, C.; Wheeler, P.; Temple, K.I.; Cole, T.; Childhood Overgrowth Collaboration; Douglas, J. ; Rahman, N. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2011, 2, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.T.; Hood, R.L.; Zhan, S.H.; Bulman, D.E.; Fejes, A.P.; Moore, R.; Mungall, A.J.; Eydoux, P.; Babul-Hirji, R.; An, J.; Marra, M.A.; FORGE Canada Consortium; Chitayat, D. ; Boycott, K.M.; Weaver, D.D.; Jones, S.J. Mutations in EZH2 cause Weaver syndrome. Am. J. Hum. Genet. 2012, 90, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.S.; Yap, D.B.; Lewis, M.E.; Chijiwa, C.; Ramos-Arroyo, M.A.; Tkachenko, N.; Milano, V.; Fradin, M.; McKinnon, M.L.; Townsend, K.N.; Xu, J.; Van Allen, M.I.; Ross, C.J.; Dobyns, W.B.; Weaver, D.D.; Gibson, W.T. Weaver Syndrome-Associated EZH2 Protein Variants Show Impaired Histone Methyltransferase Function In Vitro. Hum. Mutat. 2016, 37, 301–307. [Google Scholar] [CrossRef] [PubMed]
- UK Genetic Testing Network. Proposal Form for the Evaluation of a Genetic Test for NHS Service Gene Dossier/Additional Provider; UK Genetic Testing Network: UK, 2012; Available online: https://web.archive.org/web/20191105191117/https://ukgtn.nhs.uk/uploads/tx_ukgtn/WVS_EZH2_GD_Sept_12.pdf (accessed on 5 November 2019).
- Lui, J.C.; Barnes, K.M.; Dong, L.; Yue, S.; Graber, E.; Rapaport, R.; Dauber, A.; Nilsson, O.; Baron, J. Ezh2 Mutations Found in the Weaver Overgrowth Syndrome Cause a Partial Loss of H3K27 Histone Methyltransferase Activity. J. Clin. Endocrinol. Metab. 2018, 103, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Ocansey, S.; Tatton-Brown, K. EZH2-Related Overgrowth. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 1993–2025; Available online: https://www.ncbi.nlm.nih.gov/books/NBK148820/ (accessed on 26 June 2025).
- Kendir-Demirkol, Y.; Yeter, B.; Jenny, L.A. Expanding the Phenotypic and Genotypic Spectrum of Weaver Syndrome: A Missense Variant of the EZH2 Gene. Mol. Syndromol. 2024, 15, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Bansal, A. Weaver syndrome: A report of a rare genetic syndrome. Indian J. Hum. Genet. 2009, 15, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Weaver, T.M.; Liu, J.; Connelly, K.E.; Coble, C.; Varzavand, K.; Dykhuizen, E.C.; Musselman, C.A. The EZH2 SANT1 domain is a histone reader providing sensitivity to the modification state of the H4 tail. Sci. Rep. 2019, 9, 987. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.J.; Sherr, E.H.; Barkovich, A.J.; Richards, L.J. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014, 137, 1579–1613. [Google Scholar] [CrossRef] [PubMed]
- Benson, L.S.; Waters, P.M.; Kamil, N.I.; Simmons, B.P.; Upton, J. , 3rd. Camptodactyly: classification and results of nonoperative treatment. J. Pediatr. Orthop. 1994, 14, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, F.; Wasiewski, W.; McCabe, E.R. Weaver syndrome: the changing phenotype in an adult. Am. J. Med. Genet. 1989, 33, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.E.; Alford, B.A.; Abel, M. Cervical spine anomalies and tumors in Weaver syndrome. Am. J. Med. Genet. 2000, 95, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Cyrus, S.; Burkardt, D.; Weaver, D.D.; Gibson, W.T. PRC2-complex related dysfunction in overgrowth syndromes: A review of EZH2, EED, and SUZ12 and their syndromic phenotypes. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 519–531. [Google Scholar] [PubMed]
- Wang, A.M.Q.; Kim, M.; Ho, E.S.; Davidge, K.M. Surgery and Conservative Management of Camptodactyly in Pediatric Patients: A Systematic Review. Hand (N Y) 2020, 15, 761–770. [Google Scholar] [PubMed]
- Kloc, J.; Dzula, B.; Varga, I.; Klein, M.; Steno, B. Camptodactyly: From Embryological Basis to Surgical Treatment. Medicina (Kaunas) 2023, 59, 966. [Google Scholar] [PubMed]
- Evans, B.T.; Waters, P.M.; Bae, D.S. Early Results of Surgical Management of Camptodactyly. J. Pediatr. Orthop. 2017, 37, e317–e320. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Facial dysmorphic features of the patient with Weaver syndrome. (a) Frontal view showing characteristic craniofacial features: macrocephaly, squared forehead, hypertelorism with epicanthal folds, downward-slanting palpebral fissures, broad nasal bridge, small mouth, and mild micrognathia. Note the horizontal chin crease, a pathognomonic feature. (b) Profile view showing a prominent forehead, low-set floppy ears, and facial profile consistent with Weaver syndrome. Age: 1 yr 6 months. Written informed consent was obtained from the patient’s parents for publication.
Figure 1.
Facial dysmorphic features of the patient with Weaver syndrome. (a) Frontal view showing characteristic craniofacial features: macrocephaly, squared forehead, hypertelorism with epicanthal folds, downward-slanting palpebral fissures, broad nasal bridge, small mouth, and mild micrognathia. Note the horizontal chin crease, a pathognomonic feature. (b) Profile view showing a prominent forehead, low-set floppy ears, and facial profile consistent with Weaver syndrome. Age: 1 yr 6 months. Written informed consent was obtained from the patient’s parents for publication.

Figure 2.
Severe bilateral camptodactyly in the patient. Clinical photograph showing marked bilateral camptodactyly with severe posterior flexion contractures of the fingers, representing an atypical and severe manifestation of skeletal abnormalities associated with Weaver syndrome. The degree of finger contracture observed here is more pronounced than typically reported in classical Weaver syndrome and results in significant functional impairment of fine motor skills. The patient was 18 months old at the time of photography. Written informed consent was obtained from the patient’s parents for publication of this image.
Figure 2.
Severe bilateral camptodactyly in the patient. Clinical photograph showing marked bilateral camptodactyly with severe posterior flexion contractures of the fingers, representing an atypical and severe manifestation of skeletal abnormalities associated with Weaver syndrome. The degree of finger contracture observed here is more pronounced than typically reported in classical Weaver syndrome and results in significant functional impairment of fine motor skills. The patient was 18 months old at the time of photography. Written informed consent was obtained from the patient’s parents for publication of this image.

Figure 3.
Pedigree of the Family Demonstrating Inheritance Patterns of EZH2 and ALDH18A1 Variants. The pedigree depicts a two-generation, nonconsanguineous family with the proband (indicated by arrow) affected by Weaver syndrome. Generation I shows unaffected parents: the father (square), 35 years old, 175 cm tall, and the mother (circle), 33 years old, 166 cm tall. Generation II includes the unaffected older brother (born in 2018) and the affected proband (born in 2020). Genetic testing results are indicated below each individual. The EZH2 c.449T>C variant occurred de novo in the proband (heterozygous, +/c.449T>C), while both parents carried wild-type alleles (+/+). The ALDH18A1 c.2293C>T variant was inherited from the phenotypically normal mother (heterozygous, +/c.2293C>T) to the proband, whereas the father carried wild-type alleles. The older brother was not tested for the ALDH18A1 variant (NT = not tested). Filled symbols represent affected individuals; open symbols represent unaffected individuals. This inheritance pattern confirms the de novo pathogenic role of the EZH2 variant as the cause of Weaver syndrome in the proband.
Figure 3.
Pedigree of the Family Demonstrating Inheritance Patterns of EZH2 and ALDH18A1 Variants. The pedigree depicts a two-generation, nonconsanguineous family with the proband (indicated by arrow) affected by Weaver syndrome. Generation I shows unaffected parents: the father (square), 35 years old, 175 cm tall, and the mother (circle), 33 years old, 166 cm tall. Generation II includes the unaffected older brother (born in 2018) and the affected proband (born in 2020). Genetic testing results are indicated below each individual. The EZH2 c.449T>C variant occurred de novo in the proband (heterozygous, +/c.449T>C), while both parents carried wild-type alleles (+/+). The ALDH18A1 c.2293C>T variant was inherited from the phenotypically normal mother (heterozygous, +/c.2293C>T) to the proband, whereas the father carried wild-type alleles. The older brother was not tested for the ALDH18A1 variant (NT = not tested). Filled symbols represent affected individuals; open symbols represent unaffected individuals. This inheritance pattern confirms the de novo pathogenic role of the EZH2 variant as the cause of Weaver syndrome in the proband.
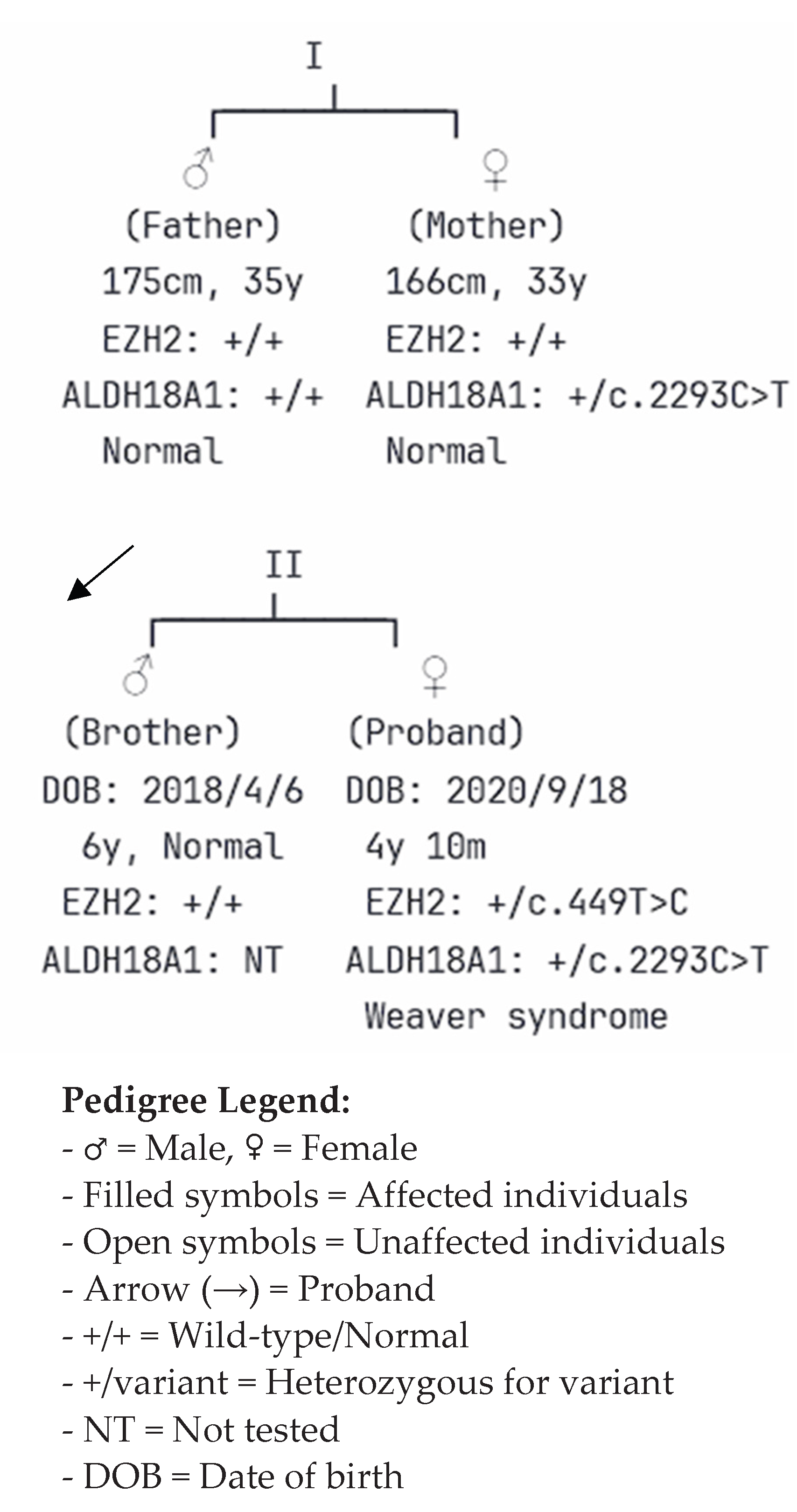

Table 1.
Molecular Findings and Genetic Analysis.
 |
Note. Molecular genetic findings were identified through whole exome sequencing. The EZH2 variant was confirmed to be de novo through family segregation analysis, while the ALDH18A1 variant was maternally inherited. Classification was performed according to ACMG guidelines.
Table 2.
Family Segregation Analysis.
 |
Note. Family segregation analysis confirmed the de novo origin of the EZH2 variant and the maternal inheritance of the ALDH18A1 variant. Sanger sequencing was used to validate the inheritance pattern.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



