
Submitted:
26 August 2025
Posted:
26 August 2025
You are already at the latest version
Abstract
Reactive oxygen species (ROS) function as critical signaling molecules in cancer biology, promoting proliferation, angiogenesis, and metastasis at controlled levels while inducing lethal damage when exceeding the cell’s buffering capacity. To survive under this state of chronic oxidative stress, cancer cells become dependent on a hyperactive antioxidant shield, primarily orchestrated by the Nrf2, glutathione (GSH), and thioredoxin (Trx) systems. These defenses maintain redox homeostasis and sustain oncogenic signaling, notably through the oxidative inactivation of tumor-suppressor phosphatases like PTEN, which drives the PI3K/AKT/mTOR pathway. Targeting this addiction to a rewired redox state has emerged as a compelling therapeutic strategy. Pro-oxidant therapies aim to overwhelm cellular defenses, with agents like high-dose vitamin C and arsenic trioxide (ATO) showing significant tumor-selective toxicity. Inhibiting the master regulator Nrf2 with compounds like Brusatol or ML385 dismantles the core antioxidant response. Disruption of the GSH system by inhibiting cysteine uptake with sulfasalazine or erastin potently induces ferroptosis, a non-apoptotic cell death driven by lipid peroxidation. Furthermore, the thioredoxin system is targeted by the repurposed drug auranofin, which irreversibly inhibits thioredoxin reductase (TrxR). Extensive preclinical data and ongoing clinical trials support the concept that this reliance on redox adaptation is a cancer-selective vulnerability. Pharmacologically tipping the redox balance beyond the threshold of tolerance offers a rational and powerful approach to eliminate malignant cells, defining a novel frontier for targeted cancer therapy.
Keywords:
Reactive Oxygen Species (ROS)
; Oxidative Stress
; Cancer
; Redox Signaling
; Nrf2
; Glutathione (GSH)
; Thioredoxin (Trx)
; Cancer Therapy
; Tumor Microenvironment (TME)
; Ferroptosis
; PI3K/AKT/Mtor
; PTEN
1. Introduction
Cellular life operates on a delicate balance of chemical reactions, chief among them being redox processes [1]. Redox (oxidation-reduction) reactions involve electron transfer reactions between chemical species and are fundamental processes in all living organisms, participating in numerous biological cellular functions in ageing, diseases, stress, and metabolism [2,3]. But, for a long time, the byproducts of redox reactions, ROS and reactive nitrogen species, were thought to cause damaging effects exclusively [4]. This perspective rapidly gave rise to the oxidative stress paradigm, a phenomenon caused by an overabundance of ROS overwhelming the cell’s ability to detoxify these reactive products, leading to indiscriminate damage to lipids, proteins, and DNA [5]. However, this damaging perspective of ROS has evolved significantly. In the past few decades, a trend for the appreciation of reactive species for their role in many signaling pathways has increased [6]. In normal physiological conditions, cells carefully maintain optimal concentration and distribution of intracellular reactive species, supporting necessary signaling pathways like “redox signaling” [7]. This dual nature of ROS is dramatically exploited in the context of cancer than anywhere else [1,8]. Bringing us to a central concept in modern biology: the Redox paradox. In cancer cells, heightened ROS levels act as pro-tumorigenic factors [7,9], including increased glucose metabolism, adaptation to hypoxic environments, and oncogenic mutations [10]. However, the toxic level of ROS production also has a beneficial outcome; they are anti-tumorigenic [11], causing an increased level of oxidative stress and induction of tumor cell death [12]. For this reason, therapies involving ROS production, either to eliminate or elevate, may be promising in cancer therapy.
2. Architects of the Malignant Redox State
2.1. ROS Sources
The malignant reprogramming by cancer cells to rewire their entire redox is a two-part process. First, cancer cells dramatically increase their endogenous ROS production through oncogenic signaling [13]. Second, to survive this self-inflicted oxidative stress, they must simultaneously build a powerful and hyperactive antioxidant defense system. ROS are reactive radicals or non-radicals generated from partial molecular oxygen metabolism [14]. Among them, free radicals contain one unpaired valence electron in their outer shell, making them highly reactive and unstable [15]. Widely known ROS include superoxide anion (O2.-), hydroxyl radical (·OH), hydrogen peroxide (H2O2), nitric oxide (·NO), and hypochlorous acid [16]. The O2- is short-lived, local, and does not cross the cellular membrane easily, generated from mitochondrial complexes 1,2, and 3 [17,18]. The cytosolic O2.- is rapidly converted to H2O2 by the enzymatic activity of superoxide dismutase 1 (SOD1) [19]. In mammalian cells, there are 41 sources of O2.- and H2O2-producing enzymes [20] and in cancer, O2.- and H2O2 are the most well-studied ROS, whereas H2O2 is the best-described ROS signaling molecule [10].
Mitochondria are central to cellular bioenergetics and, by extension, are an unintentional yet critical endogenous source of ROS [21]. During oxidative phosphorylation (OXPHOS), mitochondria generate approximately 90% of a cell's energy through the electron transport chain (ETC), serving as an indispensable channel for energy metabolism and cell survival [22]. This entire process involves redox reactions that continuously generate and consume high-energy molecules, such as NAD+, NADP+, and FAD+, to generate adenosine triphosphate (ATP) and reduce molecular oxygen (O2) to water in the ETC [23]. Electron leakage from ETC complex 1 (nicotinamide adenine dinucleotide (NADH): ubiquinone (Q) oxidoreductase) and complex 3 (ubiquinol–cytochrome c reductase) are the two prime sites for ROS production [24] as electron leakage from these two sites results in the production of O2.- Other than mitochondrial leakage as an active ROS producer, a small portion of consuming O2 in ETC is continuously reduced by a single step of one-electron reduction, thus again releasing some O2.- [25]. While this basal ROS production is a normal physiological byproduct, this scenario dramatically changes in the case of carcinomas. In cancer cells, the ROS is increased due to an increase in metabolic activity and higher ATP demand to aid rapid proliferation [26]. Cancer cells, despite possessing a fully functional mitochondria and adequate molecular oxygen, oxidize glucose to lactic acid, i.e, anaerobic respiration. This phenomenon, known as the Warburg effect, yields far less ATP than in OXPHOS in mitochondria, yet cancer cells manage to proliferate and grow rapidly [25]. Cancer cells’ mitochondria also rapidly generate ATP and aid in cell proliferation, and this high stress of energy production on the cell opens more paths for ROS generation and eventually ROS stress [27].
The next more important source of endogenous ROS is via an integral membrane enzyme family, NADPH Oxidases (NOXs) [28]. The NOX catalyze the reduction of O2 to O2.-, coupled to the oxidation of NADPH, and this enzyme family includes NOX1, NOX2, NOX3, NOX4, NOX5, and the dual oxidases Duox1 and Duox2 [29,30]. The generated O2.- is rapidly converted to H2O2 by the action of an antioxidant, Superoxide dismutase (SOD), by binding O2- to the active site of SOD, thereby transferring an electron to the SOD metal cofactor, reducing it. This transfer disrupts the bonds between the metal cofactor and nearby histidine, causing protonation of histidine and facilitating the release of molecular O2 as the first product. In the further step, a new O2.- binds to the SOD active site, receiving an electron from the previously reduced metal cofactor. This electron transfer promotes protonation of the new O2.-, ultimately generating H2O2 [28,31]. Interestingly, NOX-derived ROS and mitochondrial ROS amplify each other in a positive feedback loop. NOX-derived ROS increases mitochondrial ROS, and mitochondrial ROS stimulates NOX activation [32]. Research also suggests that NOX-dependent ROS generation is linked with oncogenic signaling of RAS and various other growth factors [33].
The endoplasmic reticulum (ER), a fine network of tubules, also contributes to ROS production in a eukaryotic cell. Apart from secretory pathways, the ER is also responsible for protein folding, biosynthesis, translocation, and post-translational modifications, including glycosylation, disulfide bond formation, and chaperone-mediated protein folding processes [34,35]. Evidence suggests that ER under stress undergoes protein misfolding, producing ROS, leading to oxidative stress [36]. ER also generates ROS, especially H2O2, while reoxidation of PDI active sites in the ER-associated degradation pathway [37]. Several NOXs are positioned in the ER membrane, catalyzing ROS generation. For instance, NOX4 produces H2O2 [38]. Increased ROS is also capable of causing ER stress and initiating the unfolded protein response [39]. Another major function ER plays are protein stabilization through the oxidative protein folding (OPF) reactions [40], for which O2 acts as a source of oxidizing equivalents necessary in intramolecular disulfide bond formation. This OPF is the major source of H2O2 [41].
Taken together, the combined ROS output from dysfunctional mitochondria, hyperactive NOX enzymes, and a stressed ER creates an immense oxidative intracellular environment. For a normal cell, such a massive and sustained ROS burden would be unsustainable, triggering apoptosis or senescence [42]. However, cancer cells adapt. To not only survive but thrive amidst this self-inflicted oxidative onslaught, they re-engineer their defensive capabilities [43]. This leads directly to the hyperactivation of the master antioxidant regulatory systems, which are co-opted from cellular components into key enablers of malignancy [44].
2.2. Antioxidant Defense of Cancer Cells
2.2.1. The Nrf2-Keap1 Axis
The cornerstone of the cellular antioxidant response is the Nrf2-Keap1 signaling axis [45]. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a master regulator of various cytoprotective genes and pathways, such as glutathione synthesis, ROS scavenging, drug detoxification, and NADP synthesis [46,47]. Under normal, homeostatic conditions, Nrf2 is held inactive in the cytoplasm by its negative regulator, Kelch-like ECH-associated protein 1 (Keap1). Keap1 acts as a sensor for oxidative stress, binding to Nrf2's Neh2 domain and targeting it for constant degradation by 26S proteasome, thus ensuring that the free Nrf2 in the cell is at appropriately low levels [48,49]. Hinge and latch theory suggests that when the cell is exposed to oxidative or electrophilic stress, cysteine residues in the IVR region on Keap1 dimer are modified. This modification causes a slight conformational modification which disrupts binding of the DLG motif of Nrf2’s Neh2 domain, all while keeping the ETGE motif intact with Keap1 [50]. While ETGE binding is intact to Keap1, the DLG binding is much weaker, resulting in quick disassociation, fine-tuning ubiquitination of Nrf2 [51]. Upon this modification of a specific cysteine residue, Nrf2 escapes from Keap1, translocating to the nucleus and binding and inducing expression of Antioxidant response elements (AREs)-containing cytoprotective genes [52]. This initiates a transient, protective transcriptional response. Traditionally, it was thought that this Nrf2 signaling provided cancer chemoprevention [53]. However, in recent years, the cytoprotective function of Nrf2 has suggested evidence that this activation might convey survival benefit to the cancer cells [54].
Cancer cells, especially lung cancer, seek persistent activation of the Nrf2 pathway to build a permanent antioxidant shield [55]. This is achieved through somatic mutations, epigenomic errors, exon skipping, etc. However, the most direct mechanism is through somatic mutations that disrupt the Keap1-Nrf2 interaction [56,57]. The occurrence of the genetic mutations in Nrf2 is documented concerning Loss-of-function (LOF) somatic mutation in the KEAP1 gene, elevating levels of transcription activity in tumor cells, presenting survival benefits to the cancer cells [58]. LOF mutations are documented in lung, gallbladder, ovary, breast, liver, and stomach carcinomas [59]. Being a master regulator of oxidative stress response, Nrf2 sits at the center of a regulatory network that leads to the initiation and development of diseases like cancer [60]. The transcriptional program activated by Nrf2 is vast, encompassing hundreds of genes that collectively form the cellular antioxidant shield. Central to this Nrf2-driven defense is the machinery responsible for the synthesis, function, and recycling of GSH. As the cell's most abundant non-protein thiol, the GSH system represents the primary pillar of the antioxidant response, and its fortification is a key consequence of constitutive Nrf2 activation in cancer.
2.2.2. The Glutathione (GSH) System
As the most abundant non-protein thiol in the cell, the glutathione (GSH) system serves as the primary and most versatile intracellular antioxidant shield, especially for cancer cells [61]. GSH, an important regulator of redox cell signaling, is biosynthesized in a well-managed pathway [62] in which the three precursor amino acids, namely L-glutamate, cysteine, and glycine, are combined to form the tripeptide GSH [63]. It is synthesized mainly in the cytosol of hepatocytes and then imported into the mitochondria and nucleus in a continuous two-step enzymatic reaction, which is ATP-dependent [64,65]. The first reaction in the biosynthesis of GSH is a ligation reaction involving glutamate and cysteine to form γ-glutamylcysteine, catalyzed by glutamate-cysteine ligase (GCL), which is a rate-limiting step [66]. This dipeptide then combines with glycine by GSH synthetase (GSS) to finally produce Glutathione [67]. Crucially, the availability of cysteine governs the entire rate of GSH production. And cancer cells, addicted to this antioxidant shield of GSH, drive the upregulation of cysteine/glutamine antiporter, system Xc- (encoded by the gene SLC7A11) [68], ensuring a continuous influx of crucial precursor, cystine, which is rapidly reduced to cysteine intracellularly, aiding in GSH production [69]. GSH acts by scavenging ROS directly in conjunction with enzymes, but its primary function is to serve as a co-substrate for powerful antioxidant enzymes. The most prominent of these are the cytosolic enzyme Glutathione Peroxidases (GPXs), belonging to the class of selenocysteine compound binding to four atoms of selenium, and using GSH as a co-substrate to reduce H2O2 and other organic peroxides to harmless water and alcohols [70]. A critical member of this family, GPX4, is the sole enzyme capable of neutralizing PLOOH within cellular membranes and inhibiting microsomal lipid peroxidation, thereby acting as the master guardian against the iron-dependent regulated cell death pathway known as Ferroptosis [71,72]. Another key family, the Glutathione S-Transferases (GSTs), although they have presence in membrane, mitochondria, and cytoplasm, in humans, the most diverse groups of GSTs are present as cytosolic enzymes, play a central role in the detoxification of reactive electrophile substances like carcinogens, mutagens, and tetragens [73,74]. GSTs catalyze the conjugation of GSH to a wide range of cytotoxic compounds, including many conventional chemotherapy agents, xenobiotics, and oxidative intermediates (DNA hydroperoxides and aldehydes), marking them for cellular efflux [75]. This GST-mediated drug clearance is a major mechanism contributing to acquired chemoresistance in cancer.
To maintain the antioxidant shield, the oxidized GSH - GSSG must be rapidly recycled back to GSH [76]. This vital task is performed by the enzyme Glutathione Reductase (GR), where nicotinamide adenine dinucleotide phosphate/H+ (NADPH/H+) acts as a crucial electron source [77]. To avail the benefit of this antioxidant system, cancer cells ensure a continuous supply of NADPH/H+ by upregulating the Pentose Phosphate Pathway (PPP), tightly coupling their metabolic state to their antioxidant capacity [78].
2.2.3. The Thioredoxin (Trx) System
Complementing the glutathione system, the thioredoxin system—comprising thioredoxin (Trx), thioredoxin reductase (TrxR), thioredoxin-interacting protein, and NADPH, represents the second major antioxidant and redox-regulating hub in the cell [79]. Its functions are diverse, ranging from scavenging ROS, transcription, DNA synthesis, cell growth stimulation, and repair proteins that have been oxidatively damaged, thereby maintaining protein function and redox homeostasis [80].
The central player is Trx, a small 12kDa redox protein containing a highly reactive dithiol active site that directly reduces oxidized cysteine residues on a vast number of target proteins [81]. In doing so, Trx itself undergoes oxidation. To complete the catalytic cycle, the oxidized Trx is reduced by the selenoenzyme Thioredoxin Reductase (TrxR), a reaction that is critically dependent on NADPH as the electron donor [80]. A major function of this system is to contribute to the peroxiredoxins (PRDXs), a family of highly abundant enzymes, by modulating the redox status, and act as primary sensors and scavengers of H2O2 [82]. PRDXs are rapidly oxidized by H₂O₂, and their constant regeneration by Trx makes them a major peroxide-detoxifying pathway in the cell [83].
The Trx system is profoundly implicated in tumor biology and progression at different levels, and many cancer cells exhibit clear dependency on its function. Several studies indicated upregulation of Trx expression in different type of cancers, such as breast, gastric, lung, and pancreatic, directly correlating with cancer cell growth [84]. Txr acts by reversing the oxidation of key signaling proteins; it ensures the activation of pro-survival pathways [85]. Dysregulated proliferation is a hallmark of tumor, and cancer cells have a high demand for ongoing DNA synthesis to maintain rapid proliferation and expand tumor mass [86]. Critically this demand is supplied as Trx the obligate electron donor for ribonucleotide reductase, an enzyme essential for synthesizing the deoxynucleotide needed for the DNA replication [87]. This indispensable role in both redox maintenance and proliferation make the Trx system, particularly the enzyme TrxR, an attractive target for anticancer drug development [81,87]
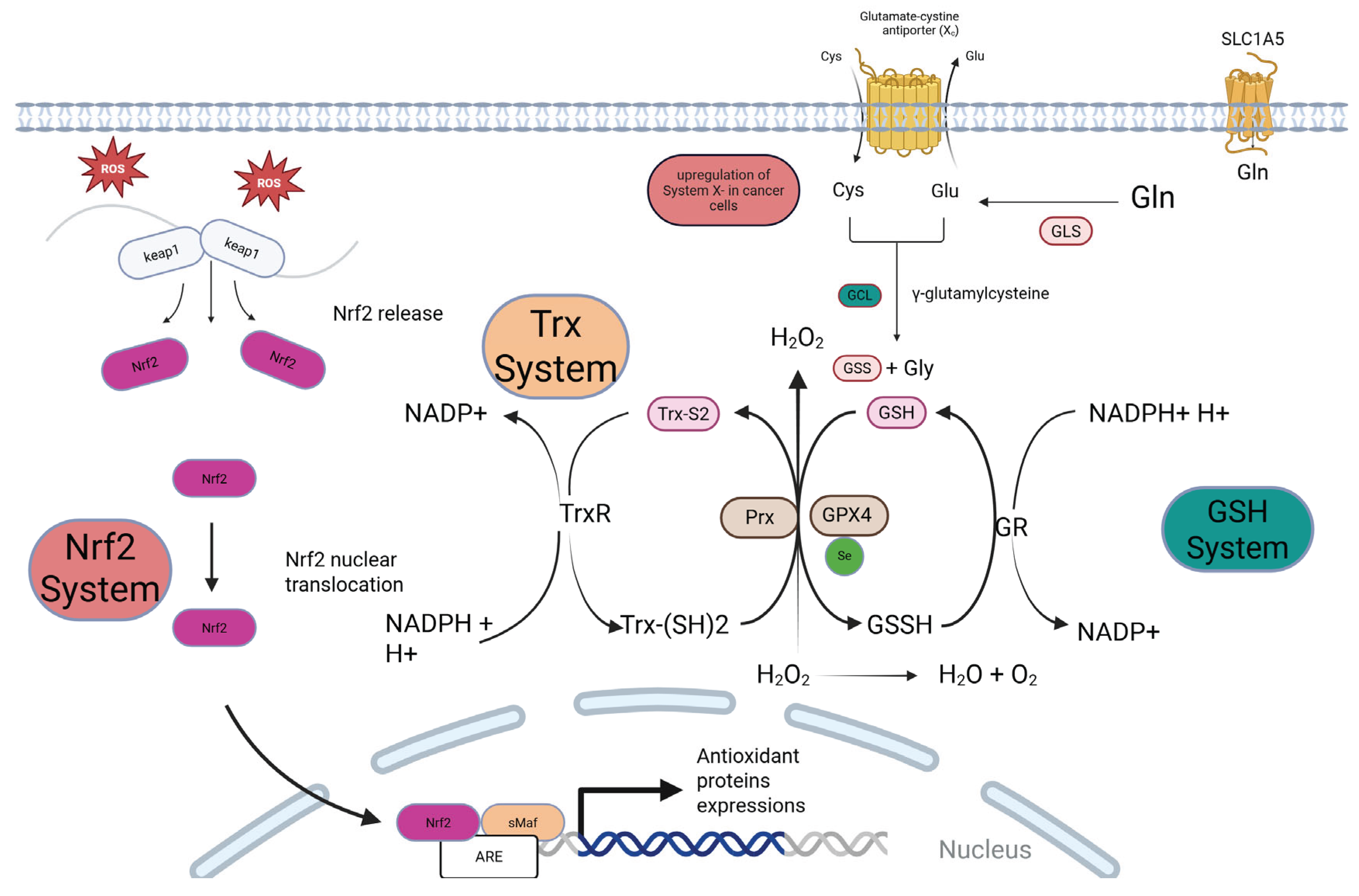
Figure 1.
The Antioxidant Systems in Cancer Cells. Cancer cells exhibit a hyperactive antioxidant defense system orchestrated by the Nrf2, Glutathione (GSH), and Thioredoxin (Trx) pathways. (A) Nrf2 Activation: High basal ROS triggers the release of the transcription factor Nrf2 from its inhibitor, Keap1. Nrf2 translocate to the nucleus, binds to the Antioxidant Response Element (ARE), and drives the expression of numerous antioxidant proteins. (B & C) The GSH and Trx Systems: The GSH system, synthesized from glutamate, cysteine, and glycine, is the major cellular antioxidant. It detoxifies peroxides via enzymes like Glutathione Peroxidase 4 (GPX4). The Trx system, centered on Thioredoxin (Trx) and Thioredoxin Reductase (TrxR), reduces oxidized proteins and regenerates peroxiredoxins (PRDXs). Both systems are critically dependent on NADPH for the regeneration of their active forms (GSH and reduced Trx). Nrf2 activation transcriptionally upregulates key components of both systems, creating a robust, interconnected shield against oxidative stress.
Figure 1.
The Antioxidant Systems in Cancer Cells. Cancer cells exhibit a hyperactive antioxidant defense system orchestrated by the Nrf2, Glutathione (GSH), and Thioredoxin (Trx) pathways. (A) Nrf2 Activation: High basal ROS triggers the release of the transcription factor Nrf2 from its inhibitor, Keap1. Nrf2 translocate to the nucleus, binds to the Antioxidant Response Element (ARE), and drives the expression of numerous antioxidant proteins. (B & C) The GSH and Trx Systems: The GSH system, synthesized from glutamate, cysteine, and glycine, is the major cellular antioxidant. It detoxifies peroxides via enzymes like Glutathione Peroxidase 4 (GPX4). The Trx system, centered on Thioredoxin (Trx) and Thioredoxin Reductase (TrxR), reduces oxidized proteins and regenerates peroxiredoxins (PRDXs). Both systems are critically dependent on NADPH for the regeneration of their active forms (GSH and reduced Trx). Nrf2 activation transcriptionally upregulates key components of both systems, creating a robust, interconnected shield against oxidative stress.
3. Redox Regulation of Cancer Hallmarks
ROS, apart from being just indiscriminate agents of damage, function as highly specific signaling molecules in redox biology [88]. This signaling is primarily achieved through the reversible oxidation of critical cysteine (Cys) residues within target proteins, a post-translational modification that is central to redox biology [89]. Compared to other ROS, H₂O₂ exhibits low overall reactivity but is highly sensitive to the thiol group of cysteine residues and therefore proves to be the ideal ROS for this role [1,90]. The sensitivity of a specific cysteine is governed by its low pKa, accessibility, and local microenvironment . When the finely tuned homeostasis is disrupted in cancer, sustained oxidative modification dysregulates the key signaling pathways that drive the acquisition of the malignant hallmarks.
3.1. Sustaining Proliferation and Evading Growth Suppressors via PTP Inactivation
A primary mechanism by which cancer cells achieve sustained proliferation is by dysregulating pro-proliferative signaling pathways. A paradigmatic example of this redox-driven dysregulation is the oxidative inhibition of protein tyrosine phosphatases (PTPs) [92]. PTP are the critical enzymes that dephosphorylate tyrosine residues on target proteins, thereby signaling and counterbalancing signals from Protein Tyrosine Kinases (PTKs), such as Receptor Tyrosine Kinases (RTKs) [33,93]. While RKTs regulate cellular signaling, which coordinates vital cellular processes such as proliferation, survival, growth, and metabolism when growth factors and chemokines interact with them [94].
The PTP family is vast, comprising classical PTPs and 63 dual-specificity phosphatases (DSPs), showcasing their diversity and complexity [95], but many members critically depends on a highly reactive cysteine in their signature motif (I/V)HCXAGXGR(S/T) for the catalytic function, this cysteine is responsible for nucleophilic attack on the phosphotyrosine substrate to generate an intermediate phospho-cysteine, after this the phosphate group is eliminated by hydrolysis via a conserved Asp residue within their WDP loop to reconstruct the native enzyme [96]. The low pKa of this cysteine, essential for its catalytic function, also makes it exceptionally susceptible to oxidation by ROS, which leads to its inactivation [97]. This oxidation converts the cysteine's thiol (-SH) to a sulfenic acid (-SOH), inactivating the enzyme. While this is often reversible through the action of the thioredoxin system, further oxidation to irreversible sulfinic (-SO₂H) or sulfonic (-SO₃H) acids can permanently disable the phosphatase [98]. In the high-ROS environment of a cancer cell, this creates a deep imbalance. RTKs remain hyper-phosphorylated while their opposing PTPs are constantly shut down, leading to sustained pro-growth signaling [99]. This directly drives the hallmark of sustained proliferative signaling.
3.2. Evading Growth Suppressors via Oxidation and Inactivation of Tumor Suppressors Like PTEN and p53.
This same mechanism allows cancer cells to evade growth suppressors, as key tumor suppressor phosphatases are prime targets of oxidative inactivation. The most notable example is PTEN (Phosphatase and Tensin Homolog). PTEN is a member of the PTP family, identified as a tumor-suppressor gene functioning via its lipid phosphatase activity, with a specific role in cell growth regulation [100]. The oxidative inactivation of PTEN has profound downstream consequences, most notably the hyperactivation of the PI3K/AKT/mTOR (PAM) pathway, a master regulator of cell growth, survival, and proliferation that is dysregulated in over half of human cancers [101,102]. PTEN is the primary brake on this pathway; its inactivation allows for the unchecked conversion of PIP2 to PIP3 by PI3K at the cell membrane, leading to the recruitment and constitutive activation of the kinase AKT [103]. Activated AKT then releases a cascade of pro-survival signals, principally through the mammalian target of rapamycin (mTOR). Operating via its mTORC1 and mTORC2 complexes, mTOR orchestrates the massive metabolic and biosynthetic programs required for tumor growth [104,105].
Beyond PTPs, ROS can also directly impact other major tumor suppressors. The redox-sensitive "guardian of the genome," p53, which can act both pro- and anti-apoptotically, can be oxidatively modified on specific cysteine residues, inhibiting its sequence-specific DNA binding and activating its tumor-suppressive transcriptional program [106]. This provides a direct, redox-mediated mechanism for neutralizing two of the most important tumor suppressor pathways in human cancer.
The Mitogen-Activated Protein Kinase (MAPK) cascades are also intensely regulated by redox signaling, although the outcomes are highly context-dependent. The Mitogen-Activated Protein Kinase (MAPK) signaling cascades—comprising the ERK, JNK, and p38 subgroups- are activated upon oxidative stress by ROS [107].The pro-proliferative ERK pathway is often potentiated by ROS through the oxidative inactivation of its endogenous negative regulators, the Dual-Specificity Phosphatases (DUSPs) [108]. The inactivation of ERK-specific phosphatases (e.g., DUSP6) prevents the dephosphorylation of ERK1/2, leading to a sustained signal for cell growth [109]
The oxidative control of the PTP-PTK balance is a powerful and representative example, but it is just one of many ways that cancer cells leverage redox signaling to achieve malignant transformation. Table 1. provide a comprehensive overview of this pervasive influence, the key redox-dependent mechanisms, molecular players, and functional outcomes for each major hallmarks.
4. The Tumor Microenvironment (TME):
4.1. Cancer-Associated Fibroblasts (CAFs)
While the intracellular redox reprogramming of cancer cells is foundational to their survival, the story of malignancy is incomplete without expanding the view to the surrounding tumor microenvironment (TME). A tumor is an organ of cancer cells, and is not merely a group of cells. It rather consists of heterogeneous infiltrating and resident host cells, secretory factors, and extracellular matrix, together called as TME [120]. TME has been shown to play a pivotal role in tumor initiation, development, and metastasis, actively manipulating the redox state of its neighbors [121]. This corruption transforms them into co-conspirators that support tumor growth, provide metabolic fuel, and suppress anti-tumor immunity . It is critical to note that within this ecosystem, redox signaling emerges as the primary language of intercellular crosstalk. Various cells and factors play a role in the TME, namely immune cells, natural killer (NK) cells, extracellular matrix (ECM), etc [123]. And amongst these cells, fibroblasts have been suggested to play a key role [124].
Normal fibroblast represents the majority of the stromal cells, and in the initial phase of tumor development, their role is inhibitory, acting via simple gap junctions between fibroblasts and IL-6 [125]. However, they are re-educated by signals from cancer cells, including persistent oxidative stress, to adopt and transform into pro-tumorigenic cancer-associated fibroblasts (CAFs) phenotype [126]. These activated fibroblast cells are particularly identified by expression of different biomarkers, such as smooth muscle actin, vimentin, desmin, and fibroblast activation protein [127]. CAF participates in the malignancy by actively secreting cytokines and chemokines, such as vascular endothelial growth factor A (VEGFA) and C-X-C motif chemokine ligand 12(CXCL12) [128]. As tumors are known to not follow conventional path in any aspect and so does not the cancer cells own metabolism [129].
The metabolic corruption within the TME give rise to a sophisticated strategy: the 'Reverse Warburg Effect'. In this model, the cancer cell, benefiting from ROS production, floods the TME with H2O2, this oxidative stress, coupled with loss of key regulator like cavelin-1 in the fibroblasts, drives the stabilization of HIF-1α [130]. The CAFs, following metabolic reprogramming, dramatically increases its rate of aerobic glycolysis, breaking down glucose into high-energy metabolites like lactate and pyruvate which is transported from CAF to TME via Monocarboxylate Transporter 4 (MCT4). This secreted lactate is a high-potency fuel source efficiently imported via Monocarboxylate Transporter 1 (MCT1) [131]. Once inside the cancer cell, the lactate is converted back to pyruvate and shuttled into mitochondria to oxidatively metabolize these energy-rich metabolites via TCA cycle and OXPHOS to produce large quantities of ATP from cancer cells [132]. Standard glycolysis alone is inefficient in providing such massive amount of ATP, this enormous energy reserves allows cancer cell in proliferation, invasion, and therapy resistance.
4.2. Immune Cells: The Redox-Mediated Suppression of Anti-Tumor Immunity
Beyond metabolic reprogramming of fibroblasts, redox signaling plays an equally crucial, if not more insidious, role in shaping the immune landscape of the tumor microenvironment. A fundamental requirement for tumor survival is the evasion of immune destruction, a hallmark achieved by creating a profoundly immunosuppressive local environment. Cancer cells orchestrate this by weaponizing ROS and RNS, using them not only for intracellular signaling but as paracrine agents to directly neutralize, exhaust, and suppress anti-tumor immune cells.
Central to the creation of an immunosuppressive TME are two key myeloid populations: Myeloid-Derived Suppressor Cells (MDSCs) and Tumor-Associated Macrophages (TAMs). Rather than attacking the tumor, these cells are co-opted and reprogrammed by cancer cells to promote disease progression [133], act as local enforcers, actively suppressing the immune function of T-cells and cause changes of B-cells in contact-dependent way [134]. They achieve this primarily through the enzymatic production of high levels of ROS and RNS [135]. Upon recruitment to the tumor, MDSCs and TAMs upregulate the expression of NOX2 and iNOS. The resulting flux of superoxide and NO has profound inhibitory effects on effector T-cells. For instance, these reactive species can nitrate the T-cell receptor (TCR), rendering it unable to recognize its antigen [136], and can also lead to the depletion of L-arginine, an amino acid essential for T-cell proliferation and function. This way, MDSCs and TAMs use redox weaponry to neutralize the body's primary anti-cancer defense.
5. Therapeutic Strategies Targeting Redox Vulnerabilities in Cancer
The extensive redox reprogramming described in the preceding sections, while a key driver of malignancy, paradoxically creates a unique and exploitable therapeutic vulnerability [137]. Unlike normal cells, which maintain low basal ROS levels and a large capacity to buffer oxidative damage, cancer cells exist in a state of chronic oxidative stress, operating perilously close to a toxic threshold beyond which cell death is inevitable [138]. Their survival is entirely dependent on their hyperactive antioxidant machinery (e.g., the Nrf2, GSH, and Trx systems). Cancer cells are 'addicted' to high ROS for signaling, but exquisitely sensitive to further increases, creating a heightened therapeutic window [139]. It allows for the development of strategies that can selectively target cancer cells by modulating their redox state.
5.1. Therapeutic ROS Induction
5.1.1. Conventional Therapies
Tumor radiotherapy or radiation therapy (RT) is a conventional technique used to inhibit and control the growth, proliferation, and even metastasis of the malignant tumor cells via various types of ionizing radiation. It is a primary cancer treatment modality, and the cancer cures account for at least 40% [140]. RT is now recognized as an essential element of effective cancer care and is frequently paired with surgical treatment as an adjuvant therapy [141]. Although also provided alone in cases of localized tumors, mainly via three modalities: external beam radiation therapy, brachytherapy, and radioisotope therapy. Although, in recent years many advances have been seen in RT [142].
Radiation therapies bring changes in the biological properties of the cancer cells, and the major effects on tumor tissues are apoptosis, necrosis, and senescence induced by DNA damage [143,144]. During radiotherapy, a massive burst of highly reactive ROS, most notably the hydroxyl radical (•OH) is generated [143]. These radicals indiscriminately attack cellular macromolecules, damaging tumor cell DNA, forming lesions, opening the deoxyribose ring, and causing single and double-stranded breaks (DBS) [145]. For cancer cells that are already operating at a high basal level of oxidative stress, this sudden, immense wave of ROS is sufficient to overwhelm its antioxidant defenses, pushing it past the toxic threshold and leading to cell death; therefore, targeting DNA damage is a promising approach for therapy [146]. Similarly, many classic chemotherapeutic agents, such as platinum-based drugs (e.g., cisplatin) and anthracyclines (e.g., doxorubicin), also exert a significant portion of their cytotoxic effects by disrupting mitochondrial function and promoting massive ROS production [147].
5.1.2. Targeted Pro-Oxidant Drugs
Building on the principle of ROS-induced cytotoxicity, a new generation of drugs has been developed to specifically and selectively elevate oxidative stress within cancer cells. A prominent, if once controversial, example is high-dose vitamin C (ascorbate). While dietary vitamin C functions as a classic antioxidant [148]. The pharmacological concentrations achievable only through intravenous administration cause it to act as a potent, tumor-selective pro-oxidant [149]. Vitamin C at a concentration ranging from 0.25-2.0 mM induces significant apoptosis in AML cell lines, as reported in an original study of 2013 [150]. The mechanism hinges on the Fenton reaction. In the iron-rich tumor microenvironment, ascorbate reduces ferric iron (Fe³⁺) to ferrous iron (Fe²⁺), which then reacts with O₂ to produce a substantial flux of H₂O₂ [151]. This creates a therapeutic window because, compared to normal cells, many cancer cells are deficient in H₂O₂-detoxifying enzymes, particularly catalase [152]. This relative catalase deficiency allows H₂O₂ to accumulate to cytotoxic levels, inducing oxidative damage and cell death [153]. Indeed, numerous studies have demonstrated this effect, with millimolar concentrations of ascorbate inducing apoptosis in AML and breast cancer cell lines [154,155] Consequently, high-dose intravenous ascorbate is now being rigorously investigated in clinical trials, typically in combination with standard chemotherapy or radiation, as a strategy to selectively poison cancer cells with ROS [156].
Another powerful clinical example of a pro-oxidant therapy is Arsenic Trioxide (ATO), a compound that has transformed the treatment of Acute Promyelocytic Leukemia (APL) and is approved by the U.S. Food and Drug Administration for use [157,158]. Study reported dose ranging from 1-300μM of ATO induces massive ROS production via mitochondria, initiating structural changes in DNA, including base-pair mutations, translocation, deletion, as well as DNA hype and hypomethylation [157]. In another study, promyelocytic leukemia (PML), ATO showed high binding affinity to cysteine residues, resulting in initiation of PML/RARα (retinoic acid receptor α) gene degradation [159] One separate study also showed that the ATO can induce inhibition of cell growth and apoptosis at 1-2 μM concentration, also in solid tumors such as colon cancer and neuroblastoma [160]. In case of lung cancer, ATO at low doses 1,2 and 4 μM has shown inhibition and cell cycle arrest [161].
A third, highly promising pro-oxidant strategy is represented by Piperlongumine (PL), a biological active alkaloid isolated from the long pepper plant (Piper longum) [162]. In recent years PL showed apoptotic cell death in bladder, colon, breast, pancreatic, osteosarcoma, and lung cancer cells [163]. It exhibits remarkable selectivity for cancer cells over normal cell; a specificity derived from its unique mechanism of action. Functioning as a ROS-activated pro-drug, it modulates and or inhibits redox enzymes essential for redox homeostasis [164].
5.2. Targeting Antioxidant Capacity
In a conceptually opposite yet complementary approach, the second major strategy for exploiting redox vulnerabilities involves disrupting the cancer cell's intrinsic antioxidant shield. As established previously, cancer cells are addicted to their hyperactive antioxidant systems—the Nrf2, glutathione, and thioredoxin pathways—to survive high levels of endogenous ROS [165]. By specifically inhibiting this antioxidant system, it is possible to trigger a catastrophic rise in oxidative stress, leading to selective cancer cell death. Furthermore, these inhibitors hold potential as chemo- and radio-sensitizers, capable of weakening a tumor's defenses and making them vulnerable to the effects of conventional therapies.
5.2.1. Nrf2 Inhibitors
As discussed earlier, transient activation of Nrf2 exhibits protective, anti-carcinogenic, and anti-mutagenic activities on non-malignant cells [166], but constitutive activation promotes therapeutic resistance and aggressive tumorigenic ability, driving malignant progression in cancer cells [167]. Hence, permanent inhibition of Nrf2 pathway shows therapeutic potential. One such promising drug for this role is Brusatol. It is a quassinoid compound with various pharmacological effects, mainly anti-inflammatory and anti-tumor activity, suppressing the Nrf2 signaling pathway [168]. In past studies it has shown tumor growth inhibition in pancreatic, colorectal, hepatocellular, and non-small cell lung cancer. [169]. Brusatol indirectly inhibits Nrf2 by inhibiting global protein translation and additionally through rapid and transient depletion of Nrf2 protein through posttranscriptional mechanism in mouse Hepa-1c1c7 hepatoma cells [170]. At concentration of 20-40 nM in cell culture line or in mice intraperitoneal administration at dose range of 1-2mg kg-, brusatol showed efficient Nrf2 protein reduction [45].
One main challenge with this drug acknowledged across studies remains about its specificity, Brusatol is proved to be a potent Nrf2 inhibitor, but several studies report different mechanism hence specificity of the drug remains unclear [47,171]. Additionally, specific small molecule inhibitor of Nrf2, ML385, specifically and directly interacts with Nrf2 protein blocking its transcriptional activity and marked tumor growth inhibition [172]. ML385 has also proven to inhibit cancer cell growth when administered in conjunction with an anti-tumor natural compound, Celastrol [173].
5.2.2. GSH & System Xc- Inhibitor
Given its role as the primary antioxidant buffer, the glutathione system is a logical and extensively studied target. The classic approach to depleting cellular GSH involves inhibiting its synthesis directly[174]. Buthionine sulfoximine (BSO) is an irreversible inhibitor of γ-glutamyl cysteine synthetase (γ-GCS), the rate-limiting enzyme in GSH biosynthesis [175]. In a study of BSO modified on hollow gold nanoparticle produced a dramatic loss of intracellular GSH levels in A549 cells in human lung cancer [176]. Although BSO showed depleted GSH in the tumor, but substantial therapeutic benefits were minimal; hence, with BSO therapy, sensitive cancer and patients selected using sensitivity markers might provide more clinical efficacy [177]. While showing limited efficacy as a monotherapy, BSO has been tested extensively in clinical trials as a potent chemo- and radio-sensitizer, demonstrating the principle of weakening the shield to enhance conventional therapies[178].
A more modern strategy focuses on starving the cell of cysteine, the essential precursor for GSH synthesis. This is achieved by inhibiting the System xc⁻ (SLC7A11) cystine/glutamate antiporter [179]. The tool compound Erastin and the repurposed FDA-approved drug Sulfasalazine (SAS) are well-known inhibitors of this transporter. Studies have shown that SAS decreases GSH levels and increases ROS accumulation by inhibiting xCT [68,180]. One study reported Erastin inhibits cystine uptake efficiently with an IC50 value of around 1.4µM [181]. This approach is particularly powerful as it delivers a dual blow: it depletes the GSH pool and, as previously discussed, is a potent inducer of ferroptosis, a non-apoptotic cell death pathway driven by lipid peroxidation [68,182]. This dual mechanism makes System xc⁻ inhibitors a highly attractive therapeutic avenue."
5.2.3. Trx System Inhibitor
The thioredoxin system, with its central role in protein repair and DNA synthesis, represents another critical node for therapeutic intervention [183]. A prime example of targeting this pathway is the repurposing of the drug Auranofin. Originally an FDA-approved gold-containing compound for treating rheumatoid arthritis, Auranofin was later discovered to be a potent and irreversible inhibitor of thioredoxin reductase (TrxR) [184]. By disabling TrxR, AF prevents the regeneration of active thioredoxin, leading to an accumulation of oxidized proteins and a dramatic increase in intracellular ROS [185]. This mechanism has proven effective in preclinical models of various cancers, particularly ovarian cancer and certain leukemias, and AF is being actively investigated in oncology clinical trials. It was demonstrated that AF could induce cell death in various breast cancer cell lines with IC50 values ranging between 0.5 and 2 µM in TNBC cells and impaired the growth of TNBC-derived spheroids [186]. Another study observed AF inhibits the NSCLS cell line with an IC50 value of less than 1.0µM [187]. In vitro evidence showed that in CRC, AF analogs exhibit cytotoxic effects [188].
6. Emerging Frontiers and Grand Challenges
One of the most exciting frontiers in redox-targeted therapy is the exploitation of ferroptosis, a non-apoptotic, iron-dependent form of regulated cell death first described in 2012 [189], . Ferroptosis is unique from other types of programmed cell death, depending on ROS derived from iron metabolism, lipid peroxidation, and ATP production for its induction [190]. It is long known that in comparison to non-malignant cells, malignant cells have a high demand for iron, allowing cancer cells to grow and proliferate rapidly [191,192]. A reaction between ferrous (Fe2+) or ferric (Fe3+) ions and H2O2 results in the Fenton reaction [193]. The hydroxyl radicals from this reaction trigger peroxidation of PUFA in membrane lipids, causing lethal accumulation of lipid hydroperoxide (L-OOH) products in the cell, a classical hallmark of ferroptosis, disrupting and breaking down the cell skeleton [194]. The primary defense against this process is GPX4. This places the GSH-System xc⁻-GPX4 axis as the master regulator of ferroptotic cell death. [195]. GSH is the key component of GPX4, synthesized from cysteine molecules [190]. Cysteine is transported into cells via System Xc- by exchange of glutamate at a 1:1 ratio. This antiporter system and the cysteine molecule serve as the backbone of GSH synthesis [196]. This discovery has unveiled a powerful therapeutic strategy: inducing ferroptosis in cancer cells, particularly those that have become resistant to traditional apoptosis-based therapies[197]. This can be achieved either by directly inhibiting GPX4 or by depleting its essential cofactor, GSH, by blocking cysteine import via the System xc⁻ antiporter [198,199].
Two grand challenges currently limit the broad clinical application of redox-based therapies: acquired resistance and lack of specificity. The same Nrf2-driven antioxidant reprogramming that promotes tumorigenesis is also a primary mechanism by which cancer cells become resistant to conventional therapies that rely on ROS for their efficacy [200]. Overcoming this requires a shift towards precision medicine, driven by the development of predictive biomarkers [201]. Characterizing a tumor's specific redox signature—through genetic sequencing of KEAP1/NRF2, immunohistochemistry for antioxidant proteins, or advanced imaging probes that can non-invasively map redox states—is essential for selecting the right patients for the right therapy [202,203,204]. Furthermore, achieving tumor specificity to avoid systemic toxicity remains a major hurdle [205]. Here, nanotechnology offers immense promise, enabling the targeted delivery of redox-modulating agents specifically to cancer cells via passive accumulation (the EPR effect) or active targeting of tumor-specific surface receptors [206].
The unprecedented success of immune checkpoint inhibitors (ICIs) has been tempered by the reality that many patients do not respond, often due to a highly immunosuppressive tumor microenvironment [207,208]. Emerging evidence now positions the redox state of the TME as a critical determinant of this resistance and, therefore, as a prime target for combination therapy. As discussed previously, the high-ROS environment within a tumor is profoundly immunosuppressive [209]. It directly impairs the function and survival of cytotoxic T-lymphocytes and empowers myeloid-derived suppressor cells (MDSCs) to further inhibit the anti-tumor response [210,211]. The oxidative shield effectively renders T-cells exhausted and dysfunctional, making them unable to mount an effective attack even when the PD-1/PD-L1 brake is released by ICIs. This provides a strong rationale for a new therapeutic paradigm: combining ICIs with redox-modulating agents to create a more permissive environment for a successful anti-tumor immune response [212,213].
7. Conclusions
The intricate relationship between cancer and cellular redox state is not one of mere stress, but of purposeful adaptation. As this review has detailed, cancer's grasp of redox biology represents a core dependency, fundamental to its survival and progression. Malignant cells systematically rewire their internal environment to maintain a heightened yet tightly controlled level of reactive oxygen species. This 'redox reset' is not a passive byproduct of transformation but an actively maintained state, where ROS is co-opted from a damaging agent into a critical signaling molecule that drives the acquisition of nearly every cancer hallmark, from unchecked proliferation to the exploitation of the tumor microenvironment. This deep-seated dependency, however, exposes a unique therapeutic vulnerability. The success of future redox-based cancer therapies will hinge on the ability to move beyond blunt, non-specific strategies and embrace a paradigm of Redox Precision Medicine. The development and clinical implementation of predictive biomarkers—from tissue-based analysis of Nrf2 activity to non-invasive imaging of a tumor's specific redox signature—are therefore paramount. Ultimately, cracking the cancer cell's 'redox code' is one of the great challenges and opportunities in modern oncology. The frontiers are clear: combining redox-modulating agents to reverse therapy resistance and synergizing them with immunotherapy to transform immunosuppressive microenvironments. The goal is to turn cancer cells’ most essential survival strategy into its catastrophic failure, exposing a new generation of personalized and effective cancer treatments.
Author Contributions
Conceptualization, literature search, J.S.R., M.K.S., S.J., S.H., and H.R.Y.; writing—original draft preparation, J.S.R.; figure preparation, J.S.R.; review and editing, J.S.R., M.K.S., S.J., S.H., and H.R.Y.; and supervision, J.S.R., I.K., and S.S.K.; project administration, I.K. and S.S.K.; funding acquisition, S.S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MEST) (grant NRF-2018R1A6A1A03025124).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the references are cited in the manuscript; however, we apologize for the omission of any primary citations.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lennicke, C. and H.M. Cochemé, Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Molecular Cell, 2021. 81(18): p. 3691-3707. [CrossRef]
- Chen, C. and G.E. Mann, Discussion on current topics in redox biology and future directions/ year in review. Free Radical Biology and Medicine, 2025. 233: p. S18. [CrossRef]
- Sies, H., R.J. Mailloux, and U. Jakob, Fundamentals of redox regulation in biology. Nature Reviews Molecular Cell Biology, 2024. 25(9): p. 701-719. [CrossRef]
- Di Meo, S., et al., Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Medicine and Cellular Longevity, 2016. 2016(1): p. 1245049.
- Pizzino, G., et al., Oxidative Stress: Harms and Benefits for Human Health. Oxid Med Cell Longev, 2017. 2017: p. 8416763. [CrossRef]
- Kong, H. and N.S. Chandel, Regulation of redox balance in cancer and T cells. Journal of Biological Chemistry, 2018. 293(20): p. 7499-7507. [CrossRef]
- Cheung, E.C. and K.H. Vousden, The role of ROS in tumour development and progression. Nature Reviews Cancer, 2022. 22(5): p. 280-297. [CrossRef]
- Huang, R., et al., Dual role of reactive oxygen species and their application in cancer therapy. Journal of Cancer, 2021. 12(18): p. 5543. [CrossRef]
- Kennel, K.B. and F.R. Greten, Immune cell-produced ROS and their impact on tumor growth and metastasis. Redox Biology, 2021. 42: p. 101891. [CrossRef]
- Moloney, J.N. and T.G. Cotter, ROS signalling in the biology of cancer. Seminars in Cell & Developmental Biology, 2018. 80: p. 50-64. [CrossRef]
- Zhao, Y., et al., Cancer metabolism: the role of ROS in DNA damage and induction of apoptosis in cancer cells. Metabolites, 2023. 13(7): p. 796. [CrossRef]
- de Sá Junior, P.L., et al., The Roles of ROS in Cancer Heterogeneity and Therapy. Oxidative Medicine and Cellular Longevity, 2017. 2017(1): p. 2467940. [CrossRef]
- Kirtonia, A., G. Sethi, and M. Garg, The multifaceted role of reactive oxygen species in tumorigenesis. Cellular and Molecular Life Sciences, 2020. 77(22): p. 4459-4483. [CrossRef]
- Marengo, B., et al., Redox homeostasis and cellular antioxidant systems: crucial players in cancer growth and therapy. Oxidative medicine and cellular longevity, 2016. 2016(1): p. 6235641. [CrossRef]
- Ahmad, G., et al., Overview and Sources of Reactive Oxygen Species (ROS) in the Reproductive System, in Oxidative Stress in Human Reproduction: Shedding Light on a Complicated Phenomenon, A. Agarwal, et al., Editors. 2017, Springer International Publishing: Cham. p. 1-16.
- Madkour, L.H., Function of reactive oxygen species (ROS) inside the living organisms and sources of oxidants. Pharm. Sci. Anal. Res. J, 2019. 2: p. 180023.
- Bardaweel, S.K., et al., Reactive Oxygen Species: the Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J Med, 2018. 50(3): p. 193-201. [CrossRef]
- Reczek, C.R. and N.S. Chandel, The Two Faces of Reactive Oxygen Species in Cancer. Annual Review of Cancer Biology, 2017. 1(Volume 1, 2017): p. 79-98. [CrossRef]
- Reczek, C.R. and N.S. Chandel, ROS-dependent signal transduction. Current Opinion in Cell Biology, 2015. 33: p. 8-13. [CrossRef]
- Averill-Bates, D., Reactive oxygen species and cell signaling. Review. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 2024. 1871(2): p. 119573.
- Sarniak, A., et al., Endogenous mechanisms of reactive oxygen species (ROS) generation. Postepy higieny i medycyny doswiadczalnej (Online), 2016. 70: p. 1150-1165. [CrossRef]
- Yang, Y., et al., Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. Journal of Cellular Physiology, 2016. 231(12): p. 2570-2581. [CrossRef]
- Okon, I.S. and M.-H. Zou, Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacological Research, 2015. 100: p. 170-174. [CrossRef]
- Xing, F., et al., The relationship of redox with hallmarks of cancer: the importance of homeostasis and context. Frontiers in oncology, 2022. 12: p. 862743. [CrossRef]
- Galaris, D., V. Skiada, and A. Barbouti, Redox signaling and cancer: The role of “labile” iron. Cancer Letters, 2008. 266(1): p. 21-29.
- Grasso, D., et al., Mitochondria in cancer. Cell Stress, 2020. 4(6): p. 114-146.
- Zhao, Y., et al., Cancer Metabolism: The Role of ROS in DNA Damage and Induction of Apoptosis in Cancer Cells. Metabolites, 2023. 13(7): p. 796. [CrossRef]
- Magnani, F. and A. Mattevi, Structure and mechanisms of ROS generation by NADPH oxidases. Current Opinion in Structural Biology, 2019. 59: p. 91-97. [CrossRef]
- Pecchillo Cimmino, T., et al., NOX Dependent ROS Generation and Cell Metabolism. International Journal of Molecular Sciences, 2023. 24(3): p. 2086. [CrossRef]
- Ogboo, B.C., et al., Architecture of the NADPH oxidase family of enzymes. Redox Biology, 2022. 52: p. 102298. [CrossRef]
- Quilaqueo-Millaqueo, N., et al., NOX proteins and ROS generation: role in invadopodia formation and cancer cell invasion. Biological Research, 2024. 57(1): p. 98. [CrossRef]
- Fukai, T. and M. Ushio-Fukai, Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells, 2020. 9(8): p. 1849. [CrossRef]
- Welsh, C.L. and L.K. Madan, Chapter Two - Protein Tyrosine Phosphatase regulation by Reactive Oxygen Species, in Advances in Cancer Research, D. Townsend and E. Schmidt, Editors. 2024, Academic Press. p. 45-74.
- Zeeshan, H.M., et al., Endoplasmic Reticulum Stress and Associated ROS. Int J Mol Sci, 2016. 17(3): p. 327. [CrossRef]
- Kritsiligkou, P., et al., Endoplasmic reticulum (ER) stress–induced reactive oxygen species (ROS) are detrimental for the fitness of a thioredoxin reductase mutant. Journal of Biological Chemistry, 2018. 293(31): p. 11984-11995. [CrossRef]
- Chen, X., et al., Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal Transduction and Targeted Therapy, 2023. 8(1): p. 352. [CrossRef]
- Yang, Y., et al., Stress Management: How the Endoplasmic Reticulum Mitigates Protein Misfolding and Oxidative Stress by the Dual Role of Glutathione Peroxidase 8. Biomolecules, 2025. 15(6): p. 847. [CrossRef]
- Bhattarai, K.R., et al., The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Experimental & Molecular Medicine, 2021. 53(2): p. 151-167. [CrossRef]
- Xiong, S., W.-J. Chng, and J. Zhou, Crosstalk between endoplasmic reticulum stress and oxidative stress: a dynamic duo in multiple myeloma. Cellular and Molecular Life Sciences, 2021. 78(8): p. 3883-3906. [CrossRef]
- Ellgaard, L., et al., Co-and post-translational protein folding in the ER. Traffic, 2016. 17(6): p. 615-638.
- Gao, L., et al., A possible connection between reactive oxygen species and the unfolded protein response in lens development: From insight to foresight. Frontiers in Cell and Developmental Biology, 2022. Volume 10 - 2022. [CrossRef]
- Snezhkina, A.V., et al., ROS generation and antioxidant defense systems in normal and malignant cells. Oxidative medicine and cellular longevity, 2019. 2019(1): p. 6175804. [CrossRef]
- Poillet-Perez, L., et al., Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox biology, 2015. 4: p. 184-192. [CrossRef]
- Kumari, S., et al., Reactive oxygen species: a key constituent in cancer survival. Biomarker insights, 2018. 13: p. 1177271918755391. [CrossRef]
- Zhang, D.D., Thirty years of NRF2: advances and therapeutic challenges. Nature Reviews Drug Discovery, 2025: p. 1-24. [CrossRef]
- Silva-Islas, C.A. and P.D. Maldonado, Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacological Research, 2018. 134: p. 92-99. [CrossRef]
- Cai, S.J., et al., Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell & Bioscience, 2019. 9(1): p. 45. [CrossRef]
- Lau, A., et al., Arsenic-Mediated Activation of the Nrf2-Keap1 Antioxidant Pathway. Journal of Biochemical and Molecular Toxicology, 2013. 27(2): p. 99-105.
- Suzuki, T. and M. Yamamoto, Molecular basis of the Keap1–Nrf2 system. Free Radical Biology and Medicine, 2015. 88: p. 93-100.
- Richardson, B.G., et al., Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorganic & Medicinal Chemistry Letters, 2015. 25(11): p. 2261-2268. [CrossRef]
- Katsuragi, Y., Y. Ichimura, and M. Komatsu, Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Current Opinion in Toxicology, 2016. 1: p. 54-61. [CrossRef]
- He, F., X. Ru, and T. Wen, NRF2, a transcription factor for stress response and beyond. International journal of molecular sciences, 2020. 21(13): p. 4777. [CrossRef]
- Menegon, S., A. Columbano, and S. Giordano, The Dual Roles of NRF2 in Cancer. Trends in Molecular Medicine, 2016. 22(7): p. 578-593. [CrossRef]
- Evans, J.P., et al., The Nrf2 inhibitor brusatol is a potent antitumour agent in an orthotopic mouse model of colorectal cancer. Oncotarget, 2018. 9(43): p. 27104-27116. [CrossRef]
- Stępkowski, T.M. and M.K. Kruszewski, Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radical Biology and Medicine, 2011. 50(9): p. 1186-1195. [CrossRef]
- Adinolfi, S., et al., The KEAP1-NRF2 pathway: Targets for therapy and role in cancer. Redox Biology, 2023. 63: p. 102726. [CrossRef]
- Taguchi, K. and M. Yamamoto, The KEAP1-NRF2 System in Cancer. Front Oncol, 2017. 7: p. 85.
- Evans, J.P., et al., The Nrf2 inhibitor brusatol is a potent antitumour agent in an orthotopic mouse model of colorectal cancer. Oncotarget, 2018. 9(43): p. 27104. [CrossRef]
- Sporn, M.B. and K.T. Liby, NRF2 and cancer: the good, the bad and the importance of context. Nature Reviews Cancer, 2012. 12(8): p. 564-571. [CrossRef]
- Wang, R., et al., Reactive oxygen species and NRF2 signaling, friends or foes in cancer? Biomolecules, 2023. 13(2): p. 353. [CrossRef]
- Balendiran, G.K., R. Dabur, and D. Fraser, The role of glutathione in cancer. Cell Biochemistry and Function, 2004. 22(6): p. 343-352.
- Balendiran, G.K., R. Dabur, and D. Fraser, The role of glutathione in cancer. Cell Biochemistry and Function: Cellular biochemistry and its modulation by active agents or disease, 2004. 22(6): p. 343-352.
- Kennedy, L., et al., Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules, 2020. 10(10): p. 1429. [CrossRef]
- Kalinina, E.V. and L.A. Gavriliuk, Glutathione Synthesis in Cancer Cells. Biochemistry (Moscow), 2020. 85(8): p. 895-907.
- Lv, H., et al., Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy. Oxidative Medicine and Cellular Longevity, 2019. 2019(1): p. 3150145. [CrossRef]
- Asantewaa, G. and I.S. Harris, Glutathione and its precursors in cancer. Current Opinion in Biotechnology, 2021. 68: p. 292-299.
- Bansal, A. and M.C. Simon, Glutathione metabolism in cancer progression and treatment resistance. Journal of Cell Biology, 2018. 217(7): p. 2291-2298. [CrossRef]
- Liu, M.-r., W.-t. Zhu, and D.-s. Pei, System Xc−: A key regulatory target of ferroptosis in cancer. Investigational New Drugs, 2021. 39(4): p. 1123-1131. [CrossRef]
- Bonifácio, V.D., et al., Cysteine metabolic circuitries: druggable targets in cancer. British journal of cancer, 2021. 124(5): p. 862-879. [CrossRef]
- Sarıkaya, E. and S. Doğan, Glutathione Peroxidase in Health. Glutathione system and oxidative stress in health and disease, 2020. 49.
- Seibt, T.M., B. Proneth, and M. Conrad, Role of GPX4 in ferroptosis and its pharmacological implication. Free Radical Biology and Medicine, 2019. 133: p. 144-152. [CrossRef]
- Maiorino, M., M. Conrad, and F. Ursini, GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxidants & Redox Signaling, 2017. 29(1): p. 61-74. [CrossRef]
- Chatterjee, A. and S. Gupta, The multifaceted role of glutathione S-transferases in cancer. Cancer Letters, 2018. 433: p. 33-42. [CrossRef]
- Averill-Bates, D.A., Chapter Five - The antioxidant glutathione, in Vitamins and Hormones, G. Litwack, Editor. 2023, Academic Press. p. 109-141.
- Fletcher, M.E., et al., Influence of glutathione-S-transferase (GST) inhibition on lung epithelial cell injury: role of oxidative stress and metabolism. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2015. 308(12): p. L1274-L1285. [CrossRef]
- Averill-Bates, D.A., The antioxidant glutathione, in Vitamins and hormones. 2023, Elsevier. p. 109-141.
- Wu, C., et al., Blocking glutathione regeneration: Inorganic NADPH oxidase nanozyme catalyst potentiates tumoral ferroptosis. Nano Today, 2022. 46: p. 101574. [CrossRef]
- Ju, H.-Q., et al., NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduction and Targeted Therapy, 2020. 5(1): p. 231. [CrossRef]
- Ghareeb, H. and N. Metanis, The Thioredoxin System: A Promising Target for Cancer Drug Development. Chemistry – A European Journal, 2020. 26(45): p. 10175-10184. [CrossRef]
- Matsuzawa, A., Thioredoxin and redox signaling: Roles of the thioredoxin system in control of cell fate. Archives of biochemistry and biophysics, 2017. 617: p. 101-105. [CrossRef]
- Ghareeb, H. and N. Metanis, The thioredoxin system: a promising target for cancer drug development. Chemistry–A European Journal, 2020. 26(45): p. 10175-10184. [CrossRef]
- Hasan, A.A., et al., The thioredoxin system of mammalian cells and its modulators. Biomedicines, 2022. 10(7): p. 1757. [CrossRef]
- Zhang, J., et al., Thioredoxin signaling pathways in cancer. Antioxidants & redox signaling, 2023. 38(4): p. 403-424.
- Mohammadi, F., et al., The thioredoxin system and cancer therapy: a review. Cancer Chemotherapy and Pharmacology, 2019. 84(5): p. 925-935. [CrossRef]
- Jovanović, M., et al., The role of the thioredoxin detoxification system in cancer progression and resistance. Frontiers in Molecular Biosciences, 2022. 9: p. 883297. [CrossRef]
- Hanahan, D. and Robert A. Weinberg, Hallmarks of Cancer: The Next Generation. Cell, 2011. 144(5): p. 646-674.
- Zhang, J., et al., Targeting the thioredoxin system for cancer therapy. Trends in pharmacological sciences, 2017. 38(9): p. 794-808. [CrossRef]
- Zhang, J., et al., ROS and ROS-mediated cellular signaling. Oxidative medicine and cellular longevity, 2016. 2016(1): p. 4350965.
- Mittler, R., ROS Are Good. Trends in Plant Science, 2017. 22(1): p. 11-19. [CrossRef]
- Rhee, S.G., H2O2, a necessary evil for cell signaling. Science, 2006. 312(5782): p. 1882-1883. [CrossRef]
- Liu, R., et al., Human protein tyrosine phosphatase 1B (PTP1B): from structure to clinical inhibitor perspectives. International Journal of Molecular Sciences, 2022. 23(13): p. 7027. [CrossRef]
- Monteiro, H.P., R.J. Arai, and L.R. Travassos, Protein tyrosine phosphorylation and protein tyrosine nitration in redox signaling. Antioxidants & redox signaling, 2008. 10(5): p. 843-890. [CrossRef]
- Östman, A., C. Hellberg, and F.D. Böhmer, Protein-tyrosine phosphatases and cancer. Nature Reviews Cancer, 2006. 6(4): p. 307-320.
- Spangle, J.M. and T.M. Roberts, Epigenetic regulation of RTK signaling. Journal of Molecular Medicine, 2017. 95(8): p. 791-798. [CrossRef]
- Lee, C. and I. Rhee, Important roles of protein tyrosine phosphatase PTPN12 in tumor progression. Pharmacological Research, 2019. 144: p. 73-78. [CrossRef]
- Dustin, C.M., et al., Redox regulation of tyrosine kinase signalling: more than meets the eye. The Journal of Biochemistry, 2019. 167(2): p. 151-163. [CrossRef]
- van der Vliet, A., C.M. Dustin, and D.E. Heppner, Chapter 16 - Redox regulation of protein kinase signaling, in Oxidative Stress, H. Sies, Editor. 2020, Academic Press. p. 287-313.
- Ray, P.D., B.-W. Huang, and Y. Tsuji, Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular Signalling, 2012. 24(5): p. 981-990. [CrossRef]
- Yang, Y., et al., Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal Transduction and Targeted Therapy, 2022. 7(1): p. 329. [CrossRef]
- Trinh, V.H., et al., Redox Regulation of PTEN by Reactive Oxygen Species: Its Role in Physiological Processes. Antioxidants, 2024. 13(2): p. 199. [CrossRef]
- Glaviano, A., et al., PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Molecular Cancer, 2023. 22(1): p. 138. [CrossRef]
- Yu, L., J. Wei, and P. Liu, Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Seminars in Cancer Biology, 2022. 85: p. 69-94. [CrossRef]
- Noorolyai, S., et al., The relation between PI3K/AKT signalling pathway and cancer. Gene, 2019. 698: p. 120-128. [CrossRef]
- Peng, Y., et al., PI3K/Akt/mTOR pathway and its role in cancer therapeutics: are we making headway? Frontiers in oncology, 2022. 12: p. 819128. [CrossRef]
- He, Y., et al., Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduction and Targeted Therapy, 2021. 6(1): p. 425. [CrossRef]
- Vurusaner, B., G. Poli, and H. Basaga, Tumor suppressor genes and ROS: complex networks of interactions. Free Radical Biology and Medicine, 2012. 52(1): p. 7-18. [CrossRef]
- Son, Y., et al., Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? Journal of signal transduction, 2011. 2011(1): p. 792639. [CrossRef]
- Sugiura, R., R. Satoh, and T. Takasaki, ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer. Cells, 2021. 10(10): p. 2509. [CrossRef]
- Ahmad, M.K., et al., Dual-specificity phosphatase 6 (DUSP6): a review of its molecular characteristics and clinical relevance in cancer. Cancer biology & medicine, 2018. 15(1): p. 14-28.
- Bellezza, I., et al., ROS-independent Nrf2 activation in prostate cancer. Oncotarget, 2017. 8(40): p. 67506. [CrossRef]
- Lingappan, K., NF-κB in oxidative stress. Current Opinion in Toxicology, 2018. 7: p. 81-86.
- Morgan, M.J. and Z.-g. Liu, Crosstalk of reactive oxygen species and NF-κB signaling. Cell research, 2011. 21(1): p. 103-115. [CrossRef]
- Yang, Y., et al., HIFs, angiogenesis, and cancer. Journal of cellular biochemistry, 2013. 114(5): p. 967-974. [CrossRef]
- Chio, I.I.C. and D.A. Tuveson, ROS in cancer: the burning question. Trends in molecular medicine, 2017. 23(5): p. 411-429. [CrossRef]
- Liu, G., et al., Redox signaling-mediated tumor extracellular matrix remodeling: pleiotropic regulatory mechanisms. Cellular Oncology, 2024. 47(2): p. 429-445. [CrossRef]
- Giannoni, E., M. Parri, and P. Chiarugi, EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxidants & redox signaling, 2012. 16(11): p. 1248-1263. [CrossRef]
- Khan, S.U., K. Fatima, and F. Malik, Understanding the cell survival mechanism of anoikis-resistant cancer cells during different steps of metastasis. Clinical & experimental metastasis, 2022. 39(5): p. 715-726. [CrossRef]
- Anastasiou, D., et al., Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science, 2011. 334(6060): p. 1278-1283. [CrossRef]
- David, S.S., V.L. O'Shea, and S. Kundu, Base-excision repair of oxidative DNA damage. Nature, 2007. 447(7147): p. 941-950. [CrossRef]
- Anderson, N.M. and M.C. Simon, The tumor microenvironment. Current Biology, 2020. 30(16): p. R921-R925.
- Arneth, B., Tumor microenvironment. Medicina, 2019. 56(1): p. 15.
- Wang, M., et al., Role of tumor microenvironment in tumorigenesis. J Cancer, 2017. 8(5): p. 761-773. [CrossRef]
- Vitale, M., et al., Effect of tumor cells and tumor microenvironment on NK-cell function. European journal of immunology, 2014. 44(6): p. 1582-1592. [CrossRef]
- Joshi, R.S., et al., The role of cancer-associated fibroblasts in tumor progression. Cancers, 2021. 13(6): p. 1399. [CrossRef]
- Kalluri, R. and M. Zeisberg, Fibroblasts in cancer. Nature reviews cancer, 2006. 6(5): p. 392-401.
- Li, Z., C. Sun, and Z. Qin, Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics, 2021. 11(17): p. 8322-8336. [CrossRef]
- Yuan, Y., et al., Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol Rep, 2016. 35(5): p. 2499-2515. [CrossRef]
- Nurmik, M., et al., In search of definitions: Cancer-associated fibroblasts and their markers. International Journal of Cancer, 2020. 146(4): p. 895-905. [CrossRef]
- Liberti, M.V. and J.W. Locasale, The Warburg Effect: How Does it Benefit Cancer Cells? Trends in Biochemical Sciences, 2016. 41(3): p. 211-218. [CrossRef]
- Roy, A. and S. Bera, CAF cellular glycolysis: linking cancer cells with the microenvironment. Tumor Biology, 2016. 37(7): p. 8503-8514. [CrossRef]
- Wilde, L., et al., Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Seminars in Oncology, 2017. 44(3): p. 198-203. [CrossRef]
- Liang, L., et al., ‘Reverse Warburg effect’ of cancer-associated fibroblasts (Review). Int J Oncol, 2022. 60(6): p. 67.
- Sica, A. and M. Massarotti, Myeloid suppressor cells in cancer and autoimmunity. Journal of Autoimmunity, 2017. 85: p. 117-125. [CrossRef]
- Li, L., M. Li, and Q. Jia, Myeloid-derived suppressor cells: Key immunosuppressive regulators and therapeutic targets in cancer. Pathology - Research and Practice, 2023. 248: p. 154711. [CrossRef]
- Law, A.M., F. Valdes-Mora, and D. Gallego-Ortega, Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells, 2020. 9(3): p. 561. [CrossRef]
- Ohl, K. and K. Tenbrock, Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Frontiers in Immunology, 2018. Volume 9 - 2018. [CrossRef]
- Parri, M. and P. Chiarugi, Redox molecular machines involved in tumor progression. Antioxidants & redox signaling, 2013. 19(15): p. 1828-1845. [CrossRef]
- Lee, B.W.L., P. Ghode, and D.S.T. Ong, Redox regulation of cell state and fate. Redox biology, 2019. 25: p. 101056. [CrossRef]
- Panieri, E. and M.M. Santoro, ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death & Disease, 2016. 7(6): p. e2253-e2253. [CrossRef]
- Sia, J., et al., Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Frontiers in Cell and Developmental Biology, 2020. Volume 8 - 2020. [CrossRef]
- Jaffray, D.A. and M.K. Gospodarowicz, Radiation Therapy for Cancer. 2015, The International Bank for Reconstruction and Development / The World Bank, Washington (DC).
- Rahman, W.N., et al., Enhancement of radiation effects by gold nanoparticles for superficial radiation therapy, in Nanomedicine in Cancer. 2017, Jenny Stanford Publishing. p. 737-752.
- Kim, W., et al., Cellular stress responses in radiotherapy. Cells, 2019. 8(9): p. 1105. [CrossRef]
- Wang, J.-s., H.-j. Wang, and H.-l. Qian, Biological effects of radiation on cancer cells. Military Medical Research, 2018. 5(1): p. 20. [CrossRef]
- Chen, H.H.W. and M.T. Kuo, Improving radiotherapy in cancer treatment: Promises and challenges. Oncotarget, 2017. 8(37): p. 62742-62758. [CrossRef]
- Baskar, R., et al., Biological response of cancer cells to radiation treatment. Frontiers in Molecular Biosciences, 2014. Volume 1 - 2014. [CrossRef]
- Wang, X. and Z. Guo, Targeting and delivery of platinum-based anticancer drugs. Chemical Society Reviews, 2013. 42(1): p. 202-224. [CrossRef]
- Carr, A.C. and S. Maggini, Vitamin C and Immune Function. Nutrients, 2017. 9(11): p. 1211. [CrossRef]
- Böttger, F., et al., High-dose intravenous vitamin C, a promising multi-targeting agent in the treatment of cancer. Journal of Experimental & Clinical Cancer Research, 2021. 40(1): p. 343. [CrossRef]
- Park, S., The Effects of High Concentrations of Vitamin C on Cancer Cells. Nutrients, 2013. 5(9): p. 3496-3505. [CrossRef]
- Ngo, B., et al., Targeting cancer vulnerabilities with high-dose vitamin C. Nature Reviews Cancer, 2019. 19(5): p. 271-282. [CrossRef]
- LEE, S.J., et al., Effect of High-dose Vitamin C Combined With Anti-cancer Treatment on Breast Cancer Cells. Anticancer Research, 2019. 39(2): p. 751-758. [CrossRef]
- Anwar, S., et al., Exploring Therapeutic Potential of Catalase: Strategies in Disease Prevention and Management. Biomolecules, 2024. 14(6): p. 697. [CrossRef]
- Suhail, N., et al., Effect of vitamins C and E on antioxidant status of breast-cancer patients undergoing chemotherapy. Journal of clinical pharmacy and therapeutics, 2012. 37(1): p. 22-26. [CrossRef]
- Giansanti, M., et al., High-Dose Vitamin C: Preclinical Evidence for Tailoring Treatment in Cancer Patients. Cancers, 2021. 13(6): p. 1428. [CrossRef]
- VOLLBRACHT, C., et al., Intravenous Vitamin C Administration Improves Quality of Life in Breast Cancer Patients during Chemo-/Radiotherapy and Aftercare: Results of a Retrospective, Multicentre, Epidemiological Cohort Study in Germany. In Vivo, 2011. 25(6): p. 983-990.
- Hoonjan, M., V. Jadhav, and P. Bhatt, Arsenic trioxide: insights into its evolution to an anticancer agent. JBIC Journal of Biological Inorganic Chemistry, 2018. 23(3): p. 313-329. [CrossRef]
- Jiang, Y., et al., An overview of arsenic trioxide-involved combined treatment algorithms for leukemia: basic concepts and clinical implications. Cell Death Discovery, 2023. 9(1): p. 266. [CrossRef]
- Wang, Q.Q., Y. Jiang, and H. Naranmandura, Therapeutic strategy of arsenic trioxide in the fight against cancers and other diseases. Metallomics, 2020. 12(3): p. 326-336. [CrossRef]
- Nakagawa, Y., et al., Arsenic trioxide-induced apoptosis through oxidative stress in cells of colon cancer cell lines. Life Sciences, 2002. 70(19): p. 2253-2269. [CrossRef]
- Huang, W. and Y. Zeng, A candidate for lung cancer treatment: arsenic trioxide. Clinical and Translational Oncology, 2019. 21(9): p. 1115-1126. [CrossRef]
- Parama, D., et al., The promising potential of piperlongumine as an emerging therapeutics for cancer. Exploration of Targeted Anti-Tumor Therapy, 2021. 2(4): p. 323. [CrossRef]
- Song, X., et al., Piperlongumine induces apoptosis in human melanoma cells via reactive oxygen species mediated mitochondria disruption. Nutrition and cancer, 2018. 70(3): p. 502-511. [CrossRef]
- Karki, K., et al., Piperlongumine induces reactive oxygen species (ROS)-dependent downregulation of specificity protein transcription factors. Cancer prevention research, 2017. 10(8): p. 467-477. [CrossRef]
- Ju, S., et al., Oxidative Stress and Cancer Therapy: Controlling Cancer Cells Using Reactive Oxygen Species. International Journal of Molecular Sciences, 2024. 25(22): p. 12387. [CrossRef]
- Pouremamali, F., et al., An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Communication and Signaling, 2022. 20(1): p. 100. [CrossRef]
- Kitamura, H. and H. Motohashi, NRF2 addiction in cancer cells. Cancer Science, 2018. 109(4): p. 900-911. [CrossRef]
- Xi, W., et al., Brusatol’s anticancer activity and its molecular mechanism: a research update. Journal of Pharmacy and Pharmacology, 2024. 76(7): p. 753-762. [CrossRef]
- He, T., et al., Brusatol: A potential sensitizing agent for cancer therapy from Brucea javanica. Biomedicine & Pharmacotherapy, 2023. 158: p. 114134. [CrossRef]
- Zhang, J., et al., Natural Nrf2 inhibitors: a review of their potential for cancer treatment. International Journal of Biological Sciences, 2023. 19(10): p. 3029. [CrossRef]
- Cai, S.J., et al., Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell Biosci, 2019. 9: p. 45. [CrossRef]
- Singh, A., et al., Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chemical Biology, 2016. 11(11): p. 3214-3225. [CrossRef]
- Xu, C., et al., ML385, an Nrf2 inhibitor, synergically enhanced celastrol triggered endoplasmic reticulum stress in lung cancer cells. ACS omega, 2024. 9(43): p. 43697-43705. [CrossRef]
- Kachadourian, R. and B.J. Day, Flavonoid-induced glutathione depletion: potential implications for cancer treatment. Free Radical Biology and Medicine, 2006. 41(1): p. 65-76. [CrossRef]
- Kalinina, E. and L. Gavriliuk, Glutathione synthesis in cancer cells. Biochemistry (Moscow), 2020. 85(8): p. 895-907.
- Liu, M., et al., Hollow Gold Nanoparticles Loaded with L-Buthionine-Sulfoximine as a Novel Nanomedicine for In Vitro Cancer Cell Therapy. Journal of Nanomaterials, 2021. 2021(1): p. 3595470. [CrossRef]
- Nishizawa, S., et al., Low tumor glutathione level as a sensitivity marker for glutamate-cysteine ligase inhibitors. Oncology Letters, 2018. 15(6): p. 8735-8743. [CrossRef]
- Wang, H., et al., Oxygen-deficient BiOCl combined with L-buthionine-sulfoximine synergistically suppresses tumor growth through enhanced singlet oxygen generation under ultrasound irradiation. Small, 2022. 18(9): p. 2104550. [CrossRef]
- Jyotsana, N., K.T. Ta, and K.E. DelGiorno, The role of cystine/glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Frontiers in oncology, 2022. 12: p. 858462. [CrossRef]
- Patel, D., et al., Novel analogs of sulfasalazine as system xc− antiporter inhibitors: Insights from the molecular modeling studies. Drug development research, 2019. 80(6): p. 758-777. [CrossRef]
- Sato, M., et al., The ferroptosis inducer erastin irreversibly inhibits system xc− and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Scientific reports, 2018. 8(1): p. 968. [CrossRef]
- Wang, H.-T., et al., Insights into ferroptosis, a novel target for the therapy of cancer. Frontiers in oncology, 2022. 12: p. 812534. [CrossRef]
- Ma, Q., Role of nrf2 in oxidative stress and toxicity. Annual review of pharmacology and toxicology, 2013. 53(1): p. 401-426. [CrossRef]
- Stafford, W.C., Elucidation of thioredoxin reductase 1 as an anticancer drug target. 2015: Karolinska Institutet (Sweden).
- Chmelyuk, N., et al., Inhibition of Thioredoxin-Reductase by Auranofin as a Pro-Oxidant Anticancer Strategy for Glioblastoma: In Vitro and In Vivo Studies. International Journal of Molecular Sciences, 2025. 26(5): p. 2084. [CrossRef]
- Gamberi, T., et al., Upgrade of an old drug: Auranofin in innovative cancer therapies to overcome drug resistance and to increase drug effectiveness. Medicinal Research Reviews, 2022. 42(3): p. 1111-1146. [CrossRef]
- Onodera, T., I. Momose, and M. Kawada, Potential anticancer activity of auranofin. Chemical and Pharmaceutical Bulletin, 2019. 67(3): p. 186-191. [CrossRef]
- Massai, L., et al., Auranofin and its analogs as prospective agents for the treatment of colorectal cancer. Cancer Drug Resistance, 2022. 5(1): p. 1. [CrossRef]
- Cao, J.Y. and S.J. Dixon, Mechanisms of ferroptosis. Cellular and Molecular Life Sciences, 2016. 73(11): p. 2195-2209.
- Xu, T., et al., Molecular mechanisms of ferroptosis and its role in cancer therapy. Journal of Cellular and Molecular Medicine, 2019. 23(8): p. 4900-4912. [CrossRef]
- Nie, Q., et al., Induction and application of ferroptosis in cancer therapy. Cancer Cell International, 2022. 22(1): p. 12. [CrossRef]
- Zhang, C., et al., Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Molecular cancer, 2022. 21(1): p. 47. [CrossRef]
- Dixon, S.J., Ferroptosis: bug or feature? Immunological reviews, 2017. 277(1): p. 150-157. [CrossRef]
- Wang, H., et al., Emerging mechanisms and targeted therapy of ferroptosis in cancer. Molecular Therapy, 2021. 29(7): p. 2185-2208. [CrossRef]
- Hassannia, B., P. Vandenabeele, and T. Vanden Berghe, Targeting ferroptosis to iron out cancer. Cancer cell, 2019. 35(6): p. 830-849. [CrossRef]
- Su, Y., et al., Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer letters, 2020. 483: p. 127-136. [CrossRef]
- Gout, P., et al., Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the xc− cystine transporter: a new action for an old drug. Leukemia, 2001. 15(10): p. 1633-1640. [CrossRef]
- Li, F.-J., et al., System Xc−/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Frontiers in pharmacology, 2022. 13: p. 910292. [CrossRef]
- Sun, S., et al., Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal transduction and targeted therapy, 2023. 8(1): p. 372. [CrossRef]
- Wu, S., H. Lu, and Y. Bai, Nrf2 in cancers: A double-edged sword. Cancer medicine, 2019. 8(5): p. 2252-2267.
- Collins, D.C., et al., Towards precision medicine in the clinic: from biomarker discovery to novel therapeutics. Trends in pharmacological sciences, 2017. 38(1): p. 25-40. [CrossRef]
- Malone, E.R., et al., Molecular profiling for precision cancer therapies. Genome medicine, 2020. 12(1): p. 8. [CrossRef]
- Gao, M., et al., Fluorescent chemical probes for accurate tumor diagnosis and targeting therapy. Chemical Society Reviews, 2017. 46(8): p. 2237-2271. [CrossRef]
- Gagan, J. and E.M. Van Allen, Next-generation sequencing to guide cancer therapy. Genome medicine, 2015. 7(1): p. 80.
- Chari, R.V., Targeted cancer therapy: conferring specificity to cytotoxic drugs. Accounts of chemical research, 2008. 41(1): p. 98-107. [CrossRef]
- Fan, D., et al., Nanomedicine in cancer therapy. Signal Transduction and Targeted Therapy, 2023. 8(1): p. 293.
- Gill, G.S., et al., Immune checkpoint inhibitors and immunosuppressive tumor microenvironment: current challenges and strategies to overcome resistance. Immunopharmacology and Immunotoxicology, 2025: p. 1-23. [CrossRef]
- Jia, Y., L. Liu, and B. Shan, Future of immune checkpoint inhibitors: focus on tumor immune microenvironment. Annals of Translational Medicine, 2020. 8(17): p. 1095. [CrossRef]
- Chen, X., et al., Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxidative Medicine and Cellular Longevity, 2016. 2016(1): p. 1580967. [CrossRef]
- Huang, J., et al., Function of reactive oxygen species in myeloid-derived suppressor cells. Frontiers in immunology, 2023. 14: p. 1226443. [CrossRef]
- Zhang, B., et al., CD8+ T cell exhaustion and its regulatory mechanisms in the tumor microenvironment: Key to the success of immunotherapy. Frontiers in Immunology, 2024. 15: p. 1476904. [CrossRef]
- Liu, R., et al., Oxidative stress in cancer immunotherapy: molecular mechanisms and potential applications. Antioxidants, 2022. 11(5): p. 853. [CrossRef]
- Li, H., et al., Nanomedicine embraces cancer radio-immunotherapy: mechanism, design, recent advances, and clinical translation. Chemical Society Reviews, 2023. 52(1): p. 47-96. [CrossRef]

Table 1.
Redox Regulation of the Hallmarks of Cancer.
| Hallmark of Cancer | Key redox-dependent mechanism(s) | Key molecular players | Downstream consequences | References |
|---|---|---|---|---|
| 3.3 Resisting Cell Death | 1. Nrf2-driven transcription: Constitutive activation of Nrf2 drives the direct transcriptional upregulation of anti-apoptotic genes. 2. NF-κB activation: ROS activates the IKK complex, leading to IκBα degradation and the release of the NF-κB transcription factor. |
1. Nrf2, BCL-2, BCL-xL 2. ROS, IKKβ, IκBα, NF-κB (p65/p50), cIAP, XIAP |
Increased threshold for apoptosis and expression of pro-survival factors, leading to resistance to both endogenous death signals and cancer therapies. | [110,111,112] |
| 3.4 Inducing Angiogenesis | HIF-1α stabilization (Pseudohypoxia): ROS oxidizes the Fe(II) cofactor in prolyl hydroxylase (PHD) enzymes, inactivating them. Blocking the VHL-mediated degradation of HIF-1α, leading to its stabilization even under normoxic conditions. | ROS (H₂O₂), PHDs, VHL, HIF-1α, ARNT, VEGF | Constitutive transcription and secretion of pro-angiogenic factors (e.g., VEGF), stimulating neovascularization to supply the growing tumor with oxygen and nutrients. | [113,114] |
| 3.5 Activating Invasion & Metastasis | 1. Matrix Remodeling: ROS-mediated activation of Matrix Metalloproteinases (MMPs) via the 'cysteine switch' mechanism. 2. EMT Induction: ROS acts as a second messenger for pro-metastatic pathways like TGF-β, driving the expression of EMT transcription factors. 3. Anoikis Resistance: The Nrf2-driven antioxidant shield protects detached cells from ROS-induced death, enabling survival during circulation. |
1. ROS, MMP-2, MMP-9 2. TGF-β, 3. Nrf2 |
Degradation of the basement membrane, acquisition of a migratory phenotype, and survival of circulating tumor cells, collectively promoting metastatic spread. | [115,116,117] |
| 3.6 Deregulating Cellular Metabolism | Enzymatic & transcriptional control: ROS directly oxidizes and modulates key metabolic enzymes (e.g., inhibiting PKM2 to divert flux to the PPP). Concurrently, ROS-stabilized HIF-1α transcriptionally upregulates glycolytic enzymes. | ROS, PKM2, HIF-1α, Glycolytic enzymes | Promotion of the Warburg Effect. This metabolic shift favors the production of biosynthetic precursors (for new cells) and NADPH (for antioxidant defense) over efficient ATP generation. |
[118] |
| 3.7 Genome Instability & Mutation | Direct DNA damage & repair inhibition: The hydroxyl radical (•OH) directly oxidizes DNA bases, creating mutagenic lesions like 8-oxoG. Concurrently, ROS can impair the function of DNA repair enzymes, preventing the correction of these lesions. | ROS (•OH), 8-oxoG, DNA bases, DNA repair enzymes | Increased somatic mutation rate and chromosomal instability, which fuels tumor evolution, intra-tumoral heterogeneity, and the development of drug resistance. | [119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



