Submitted:
18 August 2025
Posted:
20 August 2025
You are already at the latest version
Abstract
Breast cancer is the most common cancer in women, depending on the sub-type of breast cancer treatment options are different. After completing advujant therapy, there may relapse even many years later. This review examines the tumour microenvironment, cancer cells do not exist alone but are a small part of the tumour microenvironment, described as an ecosystem. This includes stromal cells, immunosuppressive regulatory T-cells, myeloid derived suppression cells, cancer associated fibroblasts, tumour associated macrophages. The balance of the immunosuppressive tumour microenvironment and the anti-tumour immune response will determine if there is a future relapse. Most therapeutic options involve therapies directed against tumour cells, only in the last few years has there been attention on the effects of the tumour microenvironment on disease progression and the possibility of decreasing the risk of metastatic disease. This article reviews the latest development in preventing metastatic disease by influencing the tumour microenevironment; at best eliminating cancer cells or at least prolonging the latent period of cancer cell dormancy.
Keywords:
minimal residual disease
; breast cancer
; tumor microenvironment
; treatment
; metastasis
1. Introduction
Breast cancer is the most common cancer in women around the world [1], with approximately in the USA 342,000 women are diagnosed with breast cancer [2]. This signifies that during the women’s lifetime there is roughly one in eight who will be diagnosed with breast cancer [2]. Breast cancer has been divided into different sub-types. Each with a differing prognosis and treatment recommendations according to the 2025 NCCN guidelines [3]; those with ductal cancer in-situ, those whose tumour cells express the oestrogen receptor (ER) and the progesterone receptor (PgR) with a low proliferation rate as determined by the expression of Ki67 (<14%), known as Luminal A; Luminal B which express the ER but not the PgR and has a Ki67 (>14%). ER positivity has been defined as at least 1% of the tumour cells stain positive for the ER receptor using immunohistochemistry, or in other words up to 99% of the tumour cells the expression of ER is not detected. However, the evidence suggests that patients with 1-10% positive for ER may benefit from endocrine based treatments [4] and a better response as compared to ER negative tumours [5]. Conflicting evidence has been reported that in breast cancer patients with only a 1-10% of cells expressing the ER have more in common, both in the clinical and pathological settings, with ER negative tumours and showed little benefit from hormonal treatment [6,7]. In a retrospective analysis of 411 patients for the expression of ER y PgR showed a bimodal distribution. More than 20% of the cohort were negative from ER expression while two thirds had at least 80% of cells expressing the ER. For PgR expression one third were negative while only 38% had an expression of at least 80%. Tumours that did not express ER were of a higher grade in contrast to those tumours with a least 80% ER expression were of lower grade and earlier T-stage. With increasing ER expression there was a significant decrease in both local relapse and overall survival and a trend towards a decrease in distant relapse [8].
The subgroup of human epithelial growth factor receptor-2 (HER-2) breast cancer is defined as at least 10% of the cells express HER-2, based on immunohistochemistry with an intensity of 3+, independent of the expression of ER and PgR. Tumours which are only 1+ are classified as HER-2 low breast cancer, those primary tumours which do not express HER-2 as HER negative.
Tumours expressing HER-2 with an intensity of +2 must be reassessed using FISH. So, in other words even in HER-2 positive breast cancer patients may have up to 90% of the tumour cells which are not 3+ intensity stained. HER-2 positive patients form between 10-20% of all breast cancers [9].
Triple negative breast cancer (TNBC) is self-explaining, forming between 10-20% of breast cancer cases, in which none of the three receptors are expressed. Even so, each subtype of breast cancer shows intra-tumoral heterogeneity with differing subpopulations of tumour cells. Some are resistant to treatment, others may be able to disseminate to the circulation, survive the shear forces and implant in distant tissues. It has been reported that there are two cooperative mechanisms. Firstly, clonal heterogeneity which is defined by the variation of the different phenotypes and thus biological properties, which are influenced by spatial and temporal factors [10,11]. The clonal evolution of dominant or resistant breast cancer cells is one potential mechanism for tumour dissemination and/or resistance to treatment [12,13]. However, breast cancer is complex, with diverse cell types and different biological properties even within the same tumour or tumour subtype. This implies that not all subtypes of breast are the same and may require different treatments. More recently, RNA sequencing (scRNA) methods of single cell analysis have confirmed this complex cellular heterogeneity [14,15]. In studies of metastatic breast cancer analysing the types of cells in the TME have reported that both the immune and stromal environments are highly heterogeneous [16,17]. This dynamic and complex interplay between cancer cells and the TME have a critical role in the dissemination, implantation and development of micro-metastasis [18]. Using scRNA sequencing nine sub-clusters of breast cancer stem cells were reported to have the potential to form micro-metastasis in the draining lymph nodes [18].
2. The Tumour Microenvironment
Cancer cells do not exist alone but form an integral part of the tumour micro-environment. This TME is composed of host cells, fibroblasts, immune cells, blood vessels, endothelial cells, and the extracellular matrix for example. Paget in 1891 proposed the hypothesis of the idea of the soil and seed; those seeds (cancer cells) which fell on stony ground would not germinate or proliferate, while those that fell on fertile ground would germinate, proliferate, and eventually cause metastatic disease [19,20]. The interactions between the different TME components are dynamic and change with time. This multi-factorial process has been described as an ecosystem, classifying it as a “multi-dimensional, spatiotemporal unity of ecology and evolution” [21]. In this ecosystem there is an intraspecific relationship that is, communication between cells and an interspecific relationship between the cancer cells and host factors. It has further been described that this ecosystem represents the total of the primary, regional, distal, and systemic “onco-spheres”, each with its own local microenvironment, niches and immune, nervous and endocrine systems [22]. These ecosystems or onco-spheres may change with time or because of treatment for the primary tumour. At what stage this process in the metastatic cascade occurs is not known, core biopsies are only performed when there are imaging studies which raise the suspicion of a possible breast cancer. Without a positive image there are no tissue samples to analyse this process. The detection of mammaglobin expressing cells have been detected in the circulation immediately prior to core biopsy, even in intra ductal carcinoma in situ (Figure 1) [23]. Sanger et al [24] reported that even in ductal carcinoma in situ disseminated tumour cells could be detected in the bone marrow. This suggests that this ecosystem is functioning even before a suspicious lesion is detected on imaging studies.
The immunological response to tumour cells is comprised of two elements; firstly, the component that is responsible for eliminating cancer or abnormal cells. The innate immunological response is mediated by natural killer cells (NK-cells) which have cytotoxic properties to eliminate cancer cells. The second is the acquired immunological response, this involves cytotoxic T-lymphocytes (CTL) aided by dendritic cells which present neo-antigens to the CTL and thus enhance the immune response. Inversely, there is the inhibitory side of the immune system which inhibit these effector cells. The main cells involved are CD4, FOXP3 positive regulatory T-lymphocytes (Tregs), myeloid derived suppressor cells (MDSC), tumour associated macrophages and the cancer associated fibroblasts have a crucial role to play. These changes in the immune environment are related to cancer progression and the development of resistance to treatment [25]. In theory, more aggressive breast cancer subtypes should produce a more immunosuppressive TME. Agohozo et al [26] reported that the immune cell composition of the TME was dependent of the subtype of breast cancer, with a higher proportion of CTLs being present in less aggressive breast cancer subtypes. This onco-sphere is complex, dynamic, changes with time, disease progression and the effect of treatment. This complex microenvironment shows multiple interactions which balance between immunosuppression and the immune defence systems. To gain a better understanding of these components it is necessary to describe the role of each individual TME component.
Matrix metalloproteinase 2 (MMP-2) is a type IV collagenase; its expression in breast cancer cells is associated with a worse prognosis [27,28]. Not all breast cancer cells express MMP-2; those which do are able to pass through the basement membrane and the extracellular matrix and enter the circulation; cells that are MMP-2-negative may disseminate in a form of Indian file through the tract left by MMP-2-positive cells [29]. The multiple interactions between the tumour and stromal cells causes a re-programming of the phenotypic characteristics of both components [30,31]. MMP-2, in degrading the extracellular matrix, triggers the proteolysis of cytokines and their respective receptors such as tumoral necrosis factor receptor R, interleukin 6R and 2R [32]. In addition, MMP-2 causes TH2 polarisation of macrophages from the TH1 subtype, therefore restricting the anti-tumour immune response. It is also able to cleave the interleukin-2-R-alpha receptor which, in doing so, suppresses the proliferation of cytotoxic T-cells because of increased apoptosis [33]. MMP-2 in addition to causing local immunosuppression, it also causes immunosuppression at distant sites by the release of exosomes into the circulation.
3. Exosomes
Exosomes are released by a process of exocytosis, entering the circulation, and thus can reach distant tissues. Exosomes are membrane-bound vesicles with a diameter of approximately 50–100 nm; after fusing with the tumour cell membrane they are able to disseminate into the circulation by the process of exocytosis. Exosomes transport different bioactive components that are part of the metastatic cascade. They carry within their membrane, proteins, lipids. DNA, RNA and enzymes such as MMP-2 [34]. These exosomes have been defined as the key drivers of immunomodulation in patients with breast cancer. These extracellular exosomes alter CTL activity and as such affect the escape of cancer cells from immunomodulation [35]. There is a differential expression of integrins on the exosomes membrane which may play in part in the organotropic distribution of metastasis [36]. The exosomes fuse directly with the plasma membranes of target cells, releasing their contents into the normal cells and thus occupy the distant TME [37]. Those exosomes which contain miR-105 can disrupt the endothelial cell junctions facilitating their invasion of distant tissues [38]. The exosome membrane expresses different ligands; intergrin α6β1 and α6β4 are associated with lung metastasis, while αvβ4 with liver metastasis [36]. They also promote bone metastasis in breast cancer by the transfer of miR-21 to osteoclasts [39].
The exosomal contents cause immune dysfunction; firstly, they cause the differentiation of bone marrow monocytes into Myeloid Derived Suppressor Cells (MDSCs), causing local immunosuppression. Tumour cells are able produce chemokine CCL2 [40], which attracts MDSCs, Tregs, tumour associated macrophages and cancer associated fibroblasts into the primary tumour. It also increases the resistance of primary tumour cells against apoptosis and increases their proliferation and migration. It also interferes with the function of cytotoxic T-cells, NK-cells and dendritic cells and is associated with an increased proliferation of Tregs. This worsens the prognosis of the patient by enhancing the immunosuppressive of the primary TME [41,42,43]. These immunosuppressive cells also increase the conversion of immature B-cells into regulating B-cells (Bregs) causing further immunosuppression [44].
This not also affects the TME of the primary tumour, but the immunosuppressive nature of the pre-metastatic niche, that is one without tumour cells, creating a fertile soil for the future implantation of circulating tumour cells.
4. Cancer Associated Fibroblasts
Fibroblasts maintain the architecture of the TME. The definition of a fibroblast is somewhat difficult, they mostly are derived from the embryonic mesoderm, although some derive from the neural crest. As they lack cell specific biomarkers, they are often defined by their morphology, localisation in the tissue and a lack of epithelial, mesenchymal and white cell markers. Fibroblasts present in the primary tumour are activated to become cancer associated fibroblasts (CAFS) [45]. The phenotypic characteristics of the CAFs and activation is mediated by various stimuli including chemotherapy; this enables them to secrete cytokines and chemokines enabling them to remodel the TME, affecting cancer cell proliferation, invasion metastasis and angiogenesis [46,47,48]. CAFs are heterogeneous in nature producing differing activation patterns; they attract pro-tumorigenic myeloid cells such as MDSC macrophages and dendritic cells thus facilitating invasion and metastasis via the epithelial mesenchymal transition, cancer cell proliferation, resistance to cell death which facilitates cancer cell dissemination and promoting the metastatic cascade [48]. Finally, CAFs cause a stiffening of the extracellular membrane impeding the entrance of cytotoxic T-cells into the primary tumour. The effects of CAFs on the TME has recently been extensively reviewed by Kennel et al [47].
5. Tumour Associated Macrophages (TAMs)
TAMs may form up to 50% of the total of immune cells infiltrating the primary tumour causing, throughout different mechanisms, progression of the tumour [49,50]. There are basically two subtypes of macrophages; the activated M1 subtype which exerts a killing function of cancer or infected cells. The M2 sub-type, however, support the progression of the tumour by promoting cancer cell invasion, metastasis, angiogenesis, the formation of cancer stem cells and increasing the immunosuppressive nature of the TME. They play an important role in the formation of the pre-metastatic niche and secrete various metalloproteinases, including MMP-2 [51]. They also upregulate the expression of programmed cell death protein 1(PD-1), CTLs decreasing their effectiveness of eliminating tumour cells [52,53]. TAMs and MDSC cells cause immunosuppression in the TME in a cell-contact dependent manner producing a decrease or ineffective T-cell response [54]. This has been reviewed recently in greater depth by Huang et al [55]. Chemokines play an important role in the crosstalk between tumour and tumour-associated macrophages in the development of the immunosuppressive environment of the primary tumour. Creating therapies that antagonise this process may be one way of overcoming this immunosuppressive effect [56].
6. Cytotoxic CD8+ T-Cell
These cells are an essential part of the host immunological response to eliminate cancer cells. Antigen presenting cells interact with the cytotoxic T-cells to increase their activation and proliferation. This allows the T-cell to bind to the cancer cell to eradicate it, however for this mechanism the T-cell has to bind to the cancer cell. Thus, the stiffening of the extracellular membrane by CAFs may impede the entrance of CTLs into the primary tumour and thus limit their efficacy [47]. As previously mentioned, tumour cells con inhibit the function of cytotoxic T-cells. The use of antibody conjugates linked to cytotoxic T-cells may improve their efficacy by binding directly to the tumour and will be reviewed later in the article (Figure 2) [57].
7. Natural Killer Cells
These are part of the innate immune system eliminating infected and tumour cells by phagocytosis. As with cytotoxic T-cells the primary tumour can suppress their function, as previously mentioned. Figure 3 summarises these processes and their immunosuppressive effects.
The understanding of how tumour cells escape from the immune system by causing immunosuppression may help to develop new therapeutic agents. Each breast cancer subtype has a different TME and thus requires different treatment approaches. The more aggressive the subtype of breast cancer the more the TME exerts an immunosuppressive effect. In an article by Moura et al [58], they addressed the question of why Luminal B breast cancer causes an increased immunosuppressive TME than Luminal A type breast cancer. The report concluded that Luminal B type breast cancer had a higher percentage of Tregs, with lower levels of NK-cells and cytotoxic T-cells which may affect the patient’s prognosis and a possible development of targeting specific components of the TME (Figure 2).
This understanding is critically important if treatments of minimal residual disease are to be successful.
8. Dissemination of Tumour Cells to Distant Tissues
Tumour cells that escape from the primary tumour into the circulation must survive the shear forces in the blood and evade immune destruction. They express MMP-2 on the cell membrane permitting them to enter the pre-metastatic niche, converting it into the metastatic niche
Once there they interact with the new TME causing changes in their biological properties. Most of this micro-metastasis become MMP-2 negative (Figure 4) and this has recently been reviewed [59].
Circulating tumour cells target into the pre-metastatic niches, according to Paget a fertile soil for their future proliferation. The immunosuppression environment facilitating their survival, converting the pre-metastatic niche into the metastatic niche, these cancer cells can be detected in the bone marrow (Figure 4).
How this down regulation is achieved by the onco-sphere is not understood. Theoretically Tissue Inhibitor of Metalloproteinase 1 (TIMP-1) may inhibitor the ability of MMP-1 from converting the zymogen MMP-2 into its active form by affecting the cysteine switch [60] or directly inhibited by TIMP-2. The cysteine switch is the removal of a cysteine residue that protects the active site of MMP-2 being activated and thus causes down regulation of this enzyme. However, the loss of activated MMP-2 and the changes in the micro-metastasis caused by interactions with the new TME associated causes the tumour cells to go into a period of dormancy or latency.
During this time there is no net proliferation of the tumour cells, growth and the formation of macro-metastasis. The length of the period of dormancy varies between different types and subtypes of cancers.
Initially the circulating tumour cells, after leaving the circulation, enter the perivascular niche composed of perivascular cells and sinusoidal endothelial cells [61]. When the breast cancer cells enter the perivascular niche, they transform into breast cancer stem cells. This change to a stem like phenotype is mediated by the Wnt-β-catenin pathway [62]. Here they interact with exosomes released by mesenchymal stem cells, those exosomes released early in this process induce cycling quiescence, DNA repair and the formation of different subtypes of breast cancer cells [62]. Later, exosomes cause dedifferentiation of the breast cancer cells into a more homogenous stem cell population [62]. These cancer cells remain viable adapting to their new TME and by various mutations and epigenetic modifications are resistant to chemotherapeutic agents and immune surveillance [63]. The second is the endosteal niche, this differs in that it is composed of osteoclasts and osteoblasts [62]. Breast cancer cells in the bone marrow reside in specific bone marrow niches that can regulate their dissemination to and from the bone marrow [64] and act as a reservoir for the future development of metastatic disease [65]. The relatively hypoxic state of the bone marrow can promote the change to the stem cell like phenotype and dormancy [62]. These cancer stem cells are thought to be more “dormant” than non-stem cell like cancer cells and are more resistant to the actions of chemotherapy [61].
In the metastatic niche, as mentioned earlier micro-metastatic breast cancer cells may express a different pattern of the receptors, ER, PgR and HER-2. With respect to the expression of HER-2 a significant discordance was reported between the HER-2 status in the primary tumour as compared with tumour cells in the metastatic niche [66] the discordance between the expression of receptors, ER, PgR and HER-2 has been reported to reach 31.5% of cancer patients. In a second study this discordance reached 49% with HER-2 positive patients who had a worst prognosis as a result [67]. Konig et al [68] reported that in a study of 29 patients with Luminal A subtype of breast cancer patients HER-2 positive cells were detected in the metastatic niche. In cytokeratin negative tumour cells, they described that 76% of the cells were Ki-67 positive and 68% of the cells were HER-2 positive. In a meta-analysis of the discordance between ER, PgR and HER-2 status of the primary tumour the pooled proportions of tumours changing from positive expression of these biomarkers and the reverse were 24% and 14% respectively for the expression of ER, 46% and 15% for PgR and 13% and 5% for HER-2 [68]
Ditsch et al in a small study of 17 patients in was reported that while ER was detected in 65% of primary tumours only 12% were ER positive in the metastatic niche. They also reported that the expression of ER in the tumour cells found in the metastatic niche was heterogeneous revealing both ER positive and negative cells [69]. The authors concluded that there could be selective dissemination of ER negative tumour cells in the bone marrow or a negative impact of the TME on ER expression. It has been suggested that in the metastatic niche plasticity of receptor expression may change receptor status and thus have clinical implications on therapeutic options [68,70].
9. Subclassification of MRD
It has been reported that there are different types of MRD, in prostate and colon cancer [71,72,73]. The authors divided patients into those who were negative for both micro-metastasis in the bone marrow, those only positive for bone marrow micro-metastasis and those who were positive for CTCs independent of whether bone marrow micro-metastasis was present or not. In stage III colon cancer patients treated with FOLFOX adjuvant chemotherapy these three subgroups had differing times to relapse and the frequency of patients who relapsed (Figure 5). This action of chemotherapy is to eliminate proliferating tumour cells; however, these survival curves suggest that in micro-metastatic disease chemotherapy is less effective. In this study those patients with only bone marrow micro-metastasis had a similar survival curve as those patients negative for MRD. However, two years after completing chemotherapy there was an increased relapse rate suggesting that during the latency period cancer cells are resistant to the effects of chemotherapy due to their non-proliferative state. In breast cancer the simultaneous detection of bone marrow micro-metastasis and CTCs was associated with a shorter latent period and progression free survival [74,75]. In hormone receptor breast cancer patients, the presence of CTCs was associated with a response to chemotherapy while patients only positive for bone marrow micro-metastasis did not. Furthermore, the presence of bone marrow micro-metastasis has been reported to the source of further relapse in breast cancer patients [76,77].
10. The Effect of Treatment on the TME and MRD
The 2025 NCCN guidelines on the treatment of breast cancer vary according to the subtype of breast cancer [78]. Depending on the subtype of breast cancer, treatment includes neoadjuvant chemotherapy, surgical excision of the primary tumour, local radiotherapy and adjuvant therapy including chemotherapy, HER2 targeted therapy and hormonal therapy. Treatment options are based on the analysis of the primary tumour and as mentioned previously MRD may have different biological features, thus treatment selection based on the analysis of the tumour may not be optimal.
11. The Latency Period of MRD
The key questions are what causes micro-metastatic dormancy, what triggers the end of dormancy and finally how to eliminate MRD when present or at least maintain the latency period. The propensity that micro-metastasis remain in a non-proliferative state for an extended period provides the possible opportunity to maintain these cells in the latent state thus preventing them to become active, proliferate, grow and disseminate to form the metastatic disease.
Mechanisms that trigger dormancy and the end of the latent period in breast cancer patients are largely unknown. The TME and dormancy of tumour cells in different tissues are mediated by signalling from immune cells, stromal cells and the extracellular matrix. This introduces the possibility of using targeted therapies against MRD, as outlined in the NCCN guidelines, even though trials have been in the treatment of metastatic disease.
What induces dormancy? Intercellular communication between cancer cells and the immune system, lymphocytes, tumour associated myeloid cells, non-haematopoietic stromal cells impact dormancy and decrease cancer cell proliferation [79]. Exosomes play an important role in promoting cancer cell dormancy. Bone marrow mesenchymal stem cells can suppress BM2 human breast cancer cells in vitro, suppressing their proliferation, decreasing the expression of stem cell like surface markers, decreased the cancer cells invasive properties and decreased their sensitivity to taxanes. The bone marrow mesenchymal stem cells secrete exosomes that contain various miRNAs, especially the over-expression of miR-23b which induced dormancy in the cancer cells by the MARCKs gene. miR-23b decreases cell cycling and invasive properties by decreasing the expression of MARCKS. The authors suggested that the exosomal transfer of miRNAs may promote dormancy in breast cancer cells [80]. It has also been reported that mesenchymal stem cells release exosomes which are able to transform the cancer cells into dormant cells and impede DNA repair in the tumour cells. These bone marrow mesenchymal stem cells chemotactically migrate towards the implanted cancers cells causing an epigenome reorganization and this occurs early in the metastatic niche [81].
Less is known about what causes the end of the latency period, it has been hypothesized that changes in the micro-metastasis caused by clonal instability or changes in the TME with an increasing immune dysfunction or a combination of both. MDSCs are one possible candidate and promote cancer cell escape from cancer specific cytotoxic T-cells, escaping from both innate NK-cells, and acquired T and B lymphocytes [82]. In prostate cancer bone metastasis models, clusters of tumour cells were found to express myeloid markers which changed the pathways association with cancer progression and immune modulation. The authors suggested that the fusion of bone marrow cells with the cancers may be responsible for these hybrid cells [83]. The most significant changes, as shown by multi-omics were changes in cell adhesion and cancer cell proliferation leading in in vitro models an increased metastatic potential. Using single cell RNA sequencing it was determined that tumour associated neutrophils, macrophages and monocytes were significantly increased in the TME surrounding the hybrid cells, causing an increased immunosuppressive TME. There was enhanced epithelial –mesenchymal-transition with more aggressive biological properties and these cells were resistant to docetaxel and ferroptosis while remaining sensitive to radiotherapy [83].
It has also been suggested that osteoblasts and osteoclasts affect the dormant cancer cells, in a reversible form, like switching it on or off. Osteoblasts and osteoclasts can regulate this switch both by interacting with the cancer cells directly or by the secretion of cytokines. This switch involved the signalling pathways, TGFβ, Wnt axis and Notch2 [84].
12. The Detection of Minimal Residual Disease in Breast Cancer, What It Means and How Can It Be Used to Direct Treatment Options in Non-Metastatic Breast Cancer Patients
In terms of MRD, the three methods used are the detection of micro-metastasis in bone marrow samples, the detection of CTCs in the blood stream, and ctDNA. It is important to understand what these three markers of MRD signify and how they may be used to direct therapy before the appearance of macro-metastasis.
a) the analysis of the bone marrow can detect the presence or absence of micro-metastasis; however, it must be understood that a negative result may be due to sampling error and does not exclude the presence of visceral micro-metastasis.
It has been suggested that not all cells detected in bone marrow aspirates are micro-metastasis and may be CTCs passing through the bone marrow compartment [85]. Although, evaluation of the bone marrow is more invasive than a simple blood test the frequency of adverse events was low, 16/13,147 (0.08%) were reported 11/16 being haemorrhages [86]. This raises the question that while cancer treatment programmes are based on the clinical and pathological findings of the primary tumour, those patients with minimal residual disease found in the bone marrow after curative therapy may require a differing treatment as the surviving tumour cells may have differing biological characteristics than the primary tumour. Micro-metastasis can be classified using immunocytochemistry as being positive or negative for the expression of HER-2, ER, PD-1, PD-L1, Trop 2, CTLA-4 and PI3K, thus aiding the decision for treatment options. Using serial samples, the evolution of the micro-metastatic disease can be monitored in terms of changes in their biological properties and response to treatment. An increase in the expression of Ki-67, a marker indicating cellular proliferation, may indicate the end of the latency period, although this is just a hypothesis.
CTCs represent another tool for detecting MRD, their presence indicating that the latent period is over and tumour cells are disseminating to other tissues. The only FDA approved detection method is the Cellsearch® system (Veridex Corporation, Warren, NY) in patients with metastatic breast cancer. Due to the low frequency of CTCs in the circulation there must be an enrichment process, for which there are several methods each with its own pitfalls. Detection methods can be divided into those which use the biological properties of the CTCs and the other on the physical properties. Density gradient centrifugation is one such method of detection using the biological properties of the CTCs, as they are less dense and thus are separated from other blood components. The CTCs can be further analysed by immunocytochemical methods. Isolation by size of epithelial tumour cells (ISET), this method also allows the immunocytological and molecular characterization of the CTCs. Method involving the physical properties of CTCs using magnetic beads labelled with antibodies which target specific antigens. The may be achieved using negative selection whereby CD45 positive leukocytes are removed from the blood sample or positive selection whereby CTCs are detected using antibodies against epithelial markers and the different pitfalls of each methods has recently been reviewed [87].
Neither the detection of micro-metastasis nor CTCs have been incorporated into clinical guidelines. However, CTCs can be sub-classified equally as can micro-metastasis, for the expression of HER-2, ER, the expression of CD47 and PD-L1 in CTCs are associated with disease progression [88], whilst detecting variants of PIK3CA can indicate resistance to HER-2 directed therapies [89,90]. Similarly, detection of the oestrogen receptor variant 1 has a significant role in the development of resistance to hormonal therapy and can detected by liquid biopsy [91]. In first description of CTCs in metastatic breast was by Ashworth in 1869 [92], it was not until 2005 that the detection of CTCs was shown to be a prognostic factor in patients with newly diagnosed metastatic breast cancer [93]. A pooled analysis of CTC status in patients with metastatic breast cancer, provided similar results [94]. The PREDICT pooled analysis [95], reported a strong association between repeated CTC determination and overall survival independent of the breast cancer subtype and previous treatment. However, the results of using CTCs to decide treatment strategies have been conflicting. A retrospective analysis of metastatic breast patients showed that a cut-off point of at least 5 CTCs/blood sample at baseline could enable a method of risk stratification in patients with metastatic breast cancer. It was reported that this cut-off point could be used to classify patients into stage IV indolent cancer, that is with a number of CTCs < 5 cells/sample and stage IV aggressive cancer. Those patients with an aggressive subtype had a worse prognosis independent of breast cancer subtype or prior treatment [96]. However, the reported results of clinical trials have been contradictory. The STIC CTC trial reported that in a sub-analysis those patients with a high CTC count benefited from chemotherapy compared to those treated with hormonal therapy [97]. In contrast the SWOG SO500 trial failed to show an improvement in overall survival in continuing the same chemotherapy regime or switching to an alternative. In patients who had received at least two prior chemotherapy regimens failed to show an improved outcome in the CirCe01 trial [98]. In the DETECT III trial, phenotypic expression rather than the numbers of CTCs detected, comparing patients with HER-2 negative metastatic breast cancer with those who CTCs HER-2 positive detected were randomized to either standard treatment or randomized to receive lapatinib, a tyrosine kinase inhibitor. It was reported that the presence of HER-2 positive CTCs the use of lapatinib improved the overall survival compared with the expression of HER-2 in the metastatic tumour [99]. There conversion of the molecular subtypes of CTCs being approximately 82-90% between the primary tumour and CTCs, with the most frequent pattern changes was the conversion to unfavourable breast cancer variants. This may be partially explained by CTC heterogeneity and selection of specific CTC phenotypes and an indication of CTC plasticity. As most CTCs are cleared from the circulation within one to two hours, repeated blood sampling may be needed in patients testing negative for CTCs [100].
More recently circulating DNA and RNA also are providing new insights into breast cancer, prognosis and therapy options. This was recently reviewed by Ho et al [101], in summary circulating tumour DNA (ctDNA) and circulating tumour RNA (ctRNA) form a small fraction of circulating free DNA (cfDNA). cfDNA is released into the blood from cells that are undergoing apoptosis, necrosis, erythyroblastic enucleation and exosomes. The modifications in the genetic and epigenetic alterations in these molecules can be used to estimate the possible prognosis of a patient, the presence of MRD, options for treatment and the risk of relapse [102].
ctRNA is released in the same way as ctDNA and includes microRNA (miRNA) and long non-coding RNA (IncRNA). The patterns of expressions of miRNAs are tissue specific being essential for post-transcriptional regulation of gene expression. Differing from miRNA; IncRNA play a role in the regulation of cell growth, differentiation, proliferation and apoptosis [103]. In breast cancer the detection of a high level of ctDNA is associated with a more aggressive disease and relapse [104]. The two methods can be combined to direct targeted therapy against surviving cancer cells [105].
However, the combined analysis of micro-metastasis and ctDNA may not be concordant. In a case report of a patient with TNBC the use of neoadjuvant chemotherapy was stopped due to side-effects, although surgical resection of the primary tumour showed a complete pathologic response to treatment [106]. There was a discrepancy between tumour markers as revealed by ctDNA and micro-metastatic disease. In most patients with metastatic breast cancer frequency there are gene alterations which may differ between pre- and postmenopausal patients, being more common in patients postmenopausal. The most common mutations in ER positive disease were in the ESR1 and PIK3CA genes, whereby the use of fulvestrant and alpelisib may be more effective [107].
ER positive HER-2 negative metastatic breast cancers eventually become resistant to first and second hormonal therapy. Hormonal therapy resistance was associated with mutations in the TP53, PIK3CA, ESR1 and BRCA1/2 genes. The authors also reported that patients with ctDNA based alternative therapies had a longer progression free survival, 6.1 versus 4.6 months [108].
One pitfall of ctDNA is that it does not differentiate between apoptotic cells, necrotic cells or “living” cancer cells.
13. The Effect of Neoadjuvant, Surgical Excision, Adjuvant Treatment on the Immune System
The NCCN recommends chemotherapy, both neoadjuvant and adjuvant, surgical resection and depending on the subtype of breast cancer and if the breast cancer is operable or not [78]. Both neoadjuvant and adjuvant chemotherapy are based on three drugs, doxorubicin, cyclophosphamide with or without a taxane. Depending on the primary tumour analysis anti-HER2 or hormonal therapy are used as adjuvant treatment. These treatments are designed to target micro-metastatic disease to eradicate them. However, micro-metastatic disease is not just about the elimination of these cells, but the therapy also modulates the immune system, an integral part of the local and distant TME.
The use of combination of an anthracycline (doxorubicin or epirubicin), cyclophosphamide and a taxane are one of the standard treatments both in the adjuvant and neoadjuvant settings [78]. The combination of cytotoxic drugs affects the immune system in different modes [109]. Anthracyclines can potentiate the immune response against cancers. It has been reported they are able to sensitize cancer cells to immune driven cytotoxicity thus triggering the eradication of cancer cells. This occurs by enhancing immune effector cells and eliminate MDSCs, thus decreasing the immunosuppressive environment of the TME and restore the immune response against tumour cells [110]. In mice models the use of doxorubicin increased the sensitivity of tumour cells to NK-cells and cytotoxic T-cells and was dependent on the TRAIL signalling pathway. In xenogeneic model’s pre-treatment of cancer cells anthracyclines delayed tumour progression and overall survival when infused with NK-cells or cytotoxic T-cells as compared to non-anthracycline treatment [112]. Doxorubicin also sensitizes breast cancer tumour cells to the effects of NK-cells by increasing FAS receptors in tumour models [113]. In the MA.5 phase III trial, patients with node positive breast cancer were randomized to receive CMF (cyclophosphamide, methotrexate and 5-Florouracil) or CEF with the methotrexate being replaced by epirubicin. Using digital spatial profiling there was no significant difference between the two treatment regimes. However, in a secondary analysis, CEF proved to be superior to CMF in the cohort, especially in HER-2 enriched tumours. Using different biomarkers, the authors reported that low levels of T-cell immunoglobulin, TIM-3 and high levels of HLA-DR and PD-L1 were associated with an increased sensitivity to CEF, thus providing a method to assess the TME and especially tumour infiltrated lymphocyte levels in patients who could potentially benefit from immunogenic chemotherapy [114]. Epirubicin has a different effect on lymphocytes, B-cells are more affected, T-cells less so, CD4 T-cells being more affected that CD8 T-cells and to a lesser extent NK-cells [115].
Taxanes cause lymphocytopenia, however CD8 cytotoxic T-cells recuperate before Tregs, thus creating a less immunosuppressive TME and may help in eliminating micro-metastatic. They also cause the upregulation of PD1 and PD-L1 and thus potentially improve the response to PD1 and PD-L1 blockade [116]. In addition, exosomes secreted from M1-polarized macrophages enhanced paclitaxel antitumour activity by activating macrophages-mediated inflammation, the use of M1-exosomes to act as a carrier for paclitaxel enhanced the antitumour effects of paclitaxel in mouse xenograft studies [117].
Cyclophosphamide in low doses modulates T-cell subtypes and thus anti-tumour immunity: It selectively depletes Tregs and enhance cytotoxic T-lymphocyte function thus causing a decrease in the immunosuppressive TME. It also enhances cancer antibody immunotherapy by modulating the activity of macrophages to improve elimination of micro-metastasis in the bone marrow. In combination with trastuzumab it was reported to eliminate micro-metastasis in humanized mice [118]. Low dose cyclophosphamide modulates the TME, inhibiting tumour growth by decreasing Tregs and increasing CTL and NK-cells [119,120]. In combination with hormonal therapy has been reported to improve progression free survival in ER positive metastatic breast cancer [121].
The use of trastuzumab has been associated with changes in the immune environment. Although it has been reported that patients with increased immunosuppression have a worse outcome when treated with trastuzumab this has yet to be confirmed. The lack of neo-antigens in breast cancer implies that specific monoclonal antibodies have few targets to carry out their immune based elimination of tumour cells. The main target from monoclonal antibodies have been directed against HER-2 positive cancer cells. Trastuzumab, the first targeted anti-HER-2 antibody, was initially used post adjuvant chemotherapy in patients with a primary tumour expressing HER-2 3+ or HER-2 2+ as confirmed by FISH. Treatment duration was for one year and in ER positive HER-2 positive breast cancer combined with hormonal-therapy. HER-2 expression is based on its expression in the primary tumour may be different in paired recurrent metastatic breast cancer [122,123,124,125,126]. Metastasis from different tissues may differentially express HER2 [122]. In terms of MRD there may be different expression of HER-2 when compared to the primary tumour. It has been reported that after completing adjuvant chemotherapy 5/14 patients had HER-2 positive micro-metastasis in HER-2 negative primary tumours [127], those patients with discordant results were reported to have a worse prognosis [128]. The use of trastuzumab in these patients is effective in eradicating the tumour cells [129]. This is also seen in patients positive for CTCs; in vitro studies showed that the use of trastuzumab decreased or eliminated micro-metastasis and CTCs in patients with HER-2 negative primary tumours and those unresponsive to trastuzumab [130]. It has also been reported that the discordance rate between HER2 expression in the micrometastasis and the primary tumour was approximately 20%. The same report stated that even with therapy with trastuzumab the overexpression of HER-2 was 18% and lost in 19%, patients with an overexpression of HER-2 in CTCs during treatment with trastuzumab had a poorer prognosis [131]. The expression of HER-2 on CTCs is dynamic, in metastatic breast cancer CTCs acquired a HER-2 positive subpopulation after multiple courses of treatment in which can convert from HER-2 positive to HER-2 negative or vice-versa. Cells with one phenotype may after four cell doubling produce cells with the opposite phenotype [132]. Trastuzumab also affects the immune system increasing antibody dependent cell mediated cytotoxicity via the activity of NK cells [133]. The most recent studies have been conducted in patients with metastatic disease and not on the treatment of MRD. However, the implication is that if the anti-HER2 improves progressive free survival and overall survival in patients with metastatic disease it may have similar effects on MRD. However here is no reported evidence to support this hypothesis.
Hormonal therapy has been associated with changes in the immune system. Standard therapy for ER positive breast therapy is with hormonal therapy, in Liminal B it may be associated with adjuvant chemotherapy. Metastatic disease may express levels of ER different to that of the primary tumour, up to 26% of patients may undergo this conversion with loss of the ER and is associated with a worse prognosis [134] It occurs when sub lethal doses of cytotoxic treatments cause an upregulation of the so-called stress molecules and the subsequent increased activity of both NK-cells and CTL against cancer cells [135]. Studies in vitro using ER positive and ER negative cells were reportedly when treated with tamoxifen or an aromatase inhibitor in sub lethal doses sensitized the malignant cells to NK-cell proliferation and cytotoxicity regardless of the ER status [136]. However, the use of prolonged oestrogen blockage, such as recommended in the NCCN guidelines of up to ten years causes an immunosuppressive phenotype in breast cancer cells. In studies using MCF7 cells prolonged treatment with ER inhibition produced an activation of immune checkpoints, with the consequence that the antigen presenting system resulted in resistance to CTL killing [137].
Surgical excision of the primary tumour is an essential part of non-metastatic breast cancer treatment. The reduction of the tumour load, in theory, would reduce the immunosuppressive effects of breast cancer, thus improving effector cell activity against micro-metastatic disease. However, a recent study comparing minimal access and open breast surgery revealed differences in the immune response. In both surgical techniques there was a decrease in immune function, although immunosuppression was less and recovered faster in patients undergoing minimal access surgery. A significant suppression of immune function and activation of T-cells is seen after major surgery. In both these groups short term immunosuppression occurred but the recovery of immune function was quicker after minimal access breast surgery [138].
14. Immunosuppression in Breast Cancer
The mechanisms of immunosuppression classify breast cancer as immunologically “cold” with a TME that is essentially immunosuppressive [61]. This immunological TME changes with time, the primary tumour being “colder” than metastatic disease [96]. In metastatic bone disease the immunosuppression mechanisms and lack of cytotoxic cell activation results in a TME immunosuppressive and immunotherapy is much less effective than in other solid tumours [96]. Thus, the identification of biomarkers that characterize both the molecular, phenotypic, and biological properties that may predict the benefit of immunotherapy are required [96].
Relapse in nearly all solid tumours, including breast cancer, is the appearance of lesions visceral and/or skeletal in imaging studies, in other words patients with macro-metastatic disease.
15. Immunomodulation and Immunotherapy in Non-Metastatic Breast Cancer Patients
Neoadjuvant chemotherapy is designed to decrease the primary tumour size making the surgical field smaller and may influence MRD. In contrast adjuvant chemotherapy is designed to eliminate MRD after curative treatment. Although chemotherapy primarily is designed to eliminate proliferating cells, both cancer and benign cells it is also able to modify the immune environment of the TME.
There is now sufficient evidence that if neoadjuvant chemotherapy leads to complete pathologic response, the patient will enjoy a better outcome. Therefore, assessment of the degree of response to neoadjuvant chemotherapy has a major impact on patient selection and the follow-up management of each patient and defines patient outcome [140]. In a study of 231 patients DTCs were detected in 21% of patients before neoadjuvant chemotherapy and their continued presence 12 months after chemotherapy remained as poor prognostic factor in terms of disease-free survival, breast cancer specific survival and overall survival with hazard ratios of 2.2, 2.6 and 2.6 respectively [141]. The authors concluded that the presence of DTCs after neo-adjuvant chemotherapy indicated a high risk for relapse and death. Similarly, Hall et al [142] reported similar findings in a study of 95 patients with a median follow up of 2 years; although a pathological complete response was achieved in 26% of patients it was not predictive of the presence of DTCs post-treatment. Primary systemic therapy does not seem to eradicate DTCs, despite complete remission 36% of these patients had DTCs detected after treatment [143,144]. Similarly, in a study of 55 patients receiving chemotherapy for high-risk breast cancer, less than half of the initially DTC positive patients became negative, of 30 patients initially negative for DTCs 36.7% became positive, the presence of DTCs post chemotherapy was associated with a poor prognosis [145]. Later studies went on to classify detected DTCs, one report of 254 breast cancer patients found 107 (42%) had cytokeratin positive cells in the bone marrow. The determination of the oestrogen receptor status of the cells has been compared to the primary tumour. Positivity for the oestrogen receptor was only 12%, and in patients with more than one cell detected was heterogeneous. The primary tumours and DTCs showed a concordance of only 28% with most the DTCs being negative for the oestrogen receptor despite the presence of an oestrogen receptor positive primary tumour [145]. Similarly, for HER-2 status; using the Hercept®-test the expression of HER-2 was measured for matched primary tumour and detected DTCs. The frequency of HER-2 expression was higher in the DTCs with a concordance of HER-2 status between the primary tumour and DTCs of 62%. 12 of 20 patients with HER-2 negative primary tumours had HER-2 positive tumour cells, however there was heterogeneous expression of HER-2 in DTC positive patients [146,147,148]. The presence of HER-2 DTCs was associated with an increased risk of relapse, with only a minority being treated with trastuzumab [149]. in a mouse xenograft model trastuzumab significantly reduced the number of HER-2 positive DTCs even when the primary tumour was already unresponsive to the drug [149]. This raises the question that while cancer treatment programmes are based on the clinical and pathological findings of the primary tumour, those patients with minimal residual disease found in the bone marrow after curative therapy may require a different treatment as the surviving tumour cells may have differing biological characteristics than the primary tumour.
16. Immunotherapy Using Monoclonal Antibodies
The lack of neo-antigens in breast cancer implies that specific monoclonal antibodies have few targets to carry out their immune based elimination of tumour cells. The main target from monoclonal antibodies have been directed against HER-2 positive cancer cells. It has also been reported that the discordance rate between HER2 expression in micro-metastasis and the primary tumour was approximately 20%.
Pertuzumab a monoclonal anti-HER2 antibody differs from trastuzumab in that it binds to the HER-2 receptor subdomain II. In doing so it blocks signal transduction via the mitogen activated protein kinase (MARK) and phosphoinositide 3 kinase (PI3K), thus inhibiting tumour cell proliferation. Dual anti-HER-2 therapy with pertuzumab and trastuzumab plus docetaxel was superior to trastuzumab plus docetaxel and placebo in the CLEOPATRA trial [150]. In patients with metastatic HER-2 positive breast cancer not previously treated with anti-HER2 therapies or progressed at least six months after finishing trastuzumab therapy, the three-drug combination was approved by the EMA in 2013. In combination with hormonal therapy patient’s ER and HER-2 positive with or without systemic therapy for locally advanced metastatic breast cancer and treated with dual HER-2 therapy combined with an aromatase inhibitor showed a four-month benefit in progression free survival [151]. However, patients who had received induction chemotherapy had no benefit from the addition of pertuzumab.
Margetuximab is an anti-HER2 antibody which is similar to trastuzumab. It differs from trastuzumab in that it increases the affinity to the active receptor CD16A while decreasing the affinity for the inhibitor CD32B [152]. The combination of either HER-2 plus chemotherapy did not produce significant differences. From genotypic analysis, patients with tumour cells expressing CD16A-158FF had a better median overall survival, while those patients expressing CD16A-15VV had better results with the use of trastuzumab. This highlights the need for the molecular and genetic characterization of tumour to determine the best therapeutic option.
Trials using trastuzumab in patients classified as HER2 low (2+and FISH negative or 1+) or zero have produced conflicting results. After neoadjuvant chemotherapy there was no significant difference on the progression free and overall survival of the addition of trastuzumab [135,154]. Whereas Zhao et al [155] reported that after neoadjuvant chemotherapy 58% of HER2 zero patients changed to be HER2 low status and that 25% of HER2 low patients changed to HER2 zero or HER2 positive status. The authors suggested that patients that had stable or at least one HER2 low status may have less malignant biological characteristics with a long-term survival benefit.
17. Tyrosine Kinase Inhibitors of HER2
Lapatinib inhibits both HER1 and HER2 tyrosine kinases, combined with capecitabine or treated with capecitabine alone in patients with locally advanced or metastatic disease it significantly increased the time to disease progression. These patients had been previously treated with an anthracycline, a taxane and trastuzumab [156]. In combination with trastuzumab it was more effective in patients with HER-2 metastatic cancer which was resistant to trastuzumab [157]. In patients with ER positive metastasis the addition of lapatinib improved progression free survival [158,159].
Tucatinib is a selective inhibitor of the HER2 tyrosine kinase but with minimal effects of inhibiting HER1, while Neratinib is an irreversible inhibitor of HER1, HER2 and HER4 but both have shown limited success in heavily pre-treated patients and was recently reviewed [160].
18. Antibody Drug Conjugates
These are equivalent to BiTES used in prostate cancer and mentioned previously, consisting of trastuzmab linked covalently linked to a cytotoxic agent or more recently TROP2 linked to a cytotoxic agent. The ideal antibody conjugate is one that targets breast cancer cells and not benign breast cells, in the breast tissue left after surgery. Antibody drug conjugates (ADCs) initially targeted HER2 positive cells, the target antigen should be tumour specific and not expressed in non-cancer tissues. It is equally important that the ADC is directed against accessible antigens thus potentially allowing the cytotoxic component to act against the cancer cell [57], 161]. There are two antigens that can potentially target these cancer cells, firstly anti-HER2 which is overexpressed in up to 20% of breast cancers and TORP2 which in TNBC is overexpressed in more than 85% of the cancer cells [162]. The use of humanized antibodies in ADC reduces the possibility of production of antidrug antibodies by the host immune system [57].
The FDA approved the use in HER-2 positive breast cancer ado-trastuzumab- emtansine ADC as monotherapy in patients with early stage HER2 positive breast cancer after neoadjuvant treatment based on a taxane combined with trastuzumab [162]. Later ado-trastuzuab-emtansine was approved as monotherapy in patients with residual invasive disease after neoadjuvant therapy with trastuzumab and a taxane in high-risk HER-2 positive breast cancer [163]. Emtansine inhibits the polymerization of microtubules whilst the trastuzumab component retains its properties of cytotoxicity and signal inhibition [164]. Based on the results from the phase III EMILIA and KATHERINE trails [162,163], its use was approved. Its use proved to be superior to the combination of capecitabine with lapatinib as second line treatment after disease progression following trastuzumab and a taxane [163]. The phase III KATHERINE trial reported that the monoclonal combination improved the three- year disease free survival as compared to trastuzumab as monotherapy [163]. However, the use of ado-trastuzumab- emtansine without the use of prior chemotherapy combined with trastuzumab showed no definitive results [163].
Second generation ADCs include trastuzumab-deruxecan (T-DXd), deruxecan is a topoisomerase 1 inhibitor. In animal models it has shown efficacy in both HER2 positive and HER2 low breast cancer. T-DXd is able to enter the tumour before being degraded into its components. Deruxedan can pass through the tumour cell membrane and thus releases it causing cytotoxicity in the surrounding cancer cells, the so-called “bystander” effect [164]. It was first approved for the treatment of breast cancer patients with HER2 positive unresectable or metastatic disease and had failed with two prior anti-HER2 therapies [165]. Later it was approved for the treatment of metastatic HER2 positive breast cancer who had progressed after HER-2 targeted treatment [166] and finally in patients with unresectable or metastatic HER-2 low breast cancer after prior chemotherapy or recurrence [167].
Sacituzumab govitecan (SG) was approved in 2021, being firstly used in the treatment of metastatic TNBC and later it was evaluated in the therapy of ER/PgR positive tumours. It showed superior results in comparison with eribulin, vinorelbine or gemcitabine (TPC) in patients who had received at least two chemotherapy regimens [168]. SG also proved to be superior in ER/PgR positive advanced breast cancer patients who had received at least two chemotherapy regimens and progressed on CDK4/6 therapy and significantly improved patient outcomes [169,170], however the strategy of the sequence of therapies remains unknown. Ongoing phase III trials of SG as first line therapy versus TPC in advanced or TNBC in patients that are PD-L1 positive or negative and have progressed after receiving anti-PD-L1 therapy. Equally the ASCENT-04 trail aims to determine the progression free survival comparing SG and pembrolizumab against TPC plus pembrolizumab in patients with advanced or TNBC and are PD-L1 positive and who have been previously untreated.
Trophoblast agent 2 (TROP-2) is a transmembrane glycoprotein and its expression is increased in cancer cells as compared with normal cells [171], an elevated level is associated with a worse prognosis. [172]. TROP-2 has been detected in all subtypes of breast cancer, being higher in hormone receptor positive/HER2 negative and TNBC and less in HER2 positive breast cancer [173]. SG is a combination of anti-TROP2 combined with SN-38 an active form of irinotecan. The anti-TROP2 monoclonal antibody permits SG to bind to TROP2 positive cell membranes where it is transported to the cancer cell lysosomes [174] and as such is able to release SN-38. Being membrane permeable it also has a bystander effect. Previous treatment with an anthracycline or taxane did not affect the activity of SG. Datopatomab-deruxtecan (Dato-DXd) is an ADC directed against TROP-2 and contains a topoisomerase I inhibitor, it has no effect on cancer cell lines with a low level TROP2 expression [175]. The combination of different ADC has yet to be determined if combined therapy improves the clinical results or increases toxic side effects. The optimal duration of therapy is yet unknown and there are no predictive biomarkers with respect to the use of SG or Dato-DXd [176].
A further development has been treated targeting the HER-3 receptor with patritumab deruxtecan (HER3-DXd), again with inhibitor of topoisomersase I [177]. Phase I/II studies of treating advanced breast cancer and expressing HER-3 as well as a phase II trial have shown responses in tumours that express HER-3 or not, PAM50 subtypes and ER/PgR positive and negative tumours [178,179].
19. Check-Point Inhibitors in Breast Cancer
Immune check points maintain the equilibrium between the immunosuppressive and the immunological response to inflammation, infection and its imbalance may lead to autoimmune disorders when the immunosuppressive component is decreased. In patients with co-existent autoimmune disease these drugs may exacerbate the disease, more serious side-effects are interstitial lung disease and myocarditis. Tregs are important regulators of the immune system permitting self-tolerance, control of chronic infections and promoting the resolution of inflammation [180]. Two check point pathways have been well described: firstly, cytotoxic lymphocyte antigen 4 (CTLA-4) and second programmed cell death protein 1 (PD1).
CTLA-4 [181] is a transmembrane protein and is expressed on Tregs, CD4+ and CD8+ T-cells. It is located within cytoplasmic vesicles and after T-cell activation it migrates to the cell membrane where it is phosphorylated and remains bound to the cell membrane. It inhibits the T-cell costimulatory molecule CD28 by binding to CD80 and CD86, the transport by endocytosis into the cell decreases the levels of CD28 [182]. It is expressed in Tregs, T and B-lymphocytes, dendritic cells, stromal cells and breast cancer cells [183]. A significantly increased CTLA-4 level can also be detected in the serum of breast cancer patients compared to healthy subjects and thus has systemic effects. It inhibits the cytotoxic T-cell response to antigen presenting cells, inhibits the activation of T-cell signalling pathways decreasing cytotoxic T-cell activity and increasing the immunosuppressive TME [181]. Patients with a high expression of CTLA-4 and PD-L1 had the best response to immunotherapy, the transcriptomic expression of CTLA-4 RNA correlated with a high expression of other immune checkpoints [184].
PD1 is a transmembrane protein and is expressed in T and B-lymphocytes, NK cells and bone marrow cells. Check point inhibitors block the checkpoint proteins binding to their respective targets, thus increasing T-cell cytotoxicity against tumour cells [185]. This blocking of the binding of PD-L1 to PD-1 permits cytotoxic T-cells to recognize and eradicate breast cancer cells. This allows the patient’s immune system to eliminate tumour cells which were previously undetected or tolerated by the immune system. In combination with chemotherapy there is synergistic effect, releasing neoantigens from dying cancer cells, this results in the priming of dendritic and T-cells [186]. Thus, the cytotoxic killing of the cancer cells is improved potentiating the anti-tumour effect and improved response rates and prolonged survival [186]. Three categories of immune check inhibitors have been approved by the FDA. The PD-1 inhibitors, nivolumab, pembrolizmab, cemipliman and dosatatimab, the PD-L1 inhibitors durvalumab and avelumab and the CTLA-4 inhibitor ipilimumab. However, the response to these therapies differs in different patients, thus the selection of the appropriate checkpoint inhibitor is a difficult task.
In patients with TNBC the use of checkpoint inhibitors has shown to have a positive effect, possibly due to the high mutational burden not seen in other subtypes of breast cancer [187]. The early use of checkpoint inhibitors has its maximum effect when used early in the disease. This may be due to the less compromised immune system secondary to the effects of chemotherapy and/or the progression of immune escape with disease progression [188]. In the KEYNOTE 522 and I-SPY2 trials the addition of pembrolizumab as neoadjuvant and adjuvant co-therapy showed a decrease in the risk of relapse, complications or death by 37% [189,190]. The detection of circulating tumour cells in these patients had gene expression profiles suggestive of increased T-cell activity both in TNBC and ER positive patients. After four weeks of treatment the presence of PD-L1 positive CTCs was associated with a worse prognosis [191]. This implies that the use of CTC detection could be used as a potential biomarker in patients treated with checkpoint inhibitors.
20. Cyclin-Dependent Kinase (CDK) 4/6 Inhibitors
CDK 4/6 inhibitors improved progression free survival when combined with hormonal therapy but not an improvement in overall survival [192,193,194]. However, resistance to CDK4/6 inhibitors eventually develops. These resistant cancer cells have higher levels of Cyclin D1 and CDK4 proteins and the PI3K/mTOR showed increased activity which leads to resistance to CDK 4/6 inhibitors [197]. The combined use with PI3K/mTOR inhibitors can restore the sensitivity to CDK 4/6 inhibitors after resistance to CDK 4/6 inhibitors has developed [195]. The 2024 NCCN guidelines [78] recommends the use of ribociclib in combination with an aromatase inhibitor, while abemaciclib and ribociclib should be used in combination with fulvestrant. However, the use of CDK 4/6 inhibitors have significant side-effects, especially in older patients reducing the quality of life; 70% of patients require a dose reduction while in 15% of patient’s treatment was stopped [196]. The three approved CDK 4/6 inhibitors combined with hormonal therapy proved to be superior to hormonal monotherapy. However, a meta-analysis did not detect a significant difference between the three drugs, moreover the differences in overall survival between these CDK 4/6 drugs was not clinically significant, although statistically significant in patients with ER positive HER2 negative metastatic breast cancer [196]. The final results of the NATALEE three-year trial comparing ribociclib plus an aromatase inhibitor versus an aromatase inhibitor alone showed a benefit of the combined therapy [197].
21. Cancer Vaccines
Although the use of targeted therapies in treatment of breast cancer resistance eventually develops to these therapies. It has been reported that nearly one third of HER2 positive tumours develop resistance to all these treatments [198]. As such treatments based on passive immunotherapy has its limitations and it is recommended that continuous treatment being used until macroscopic progression. Differing from this, cancer vaccines based on cancer cell epitopes cause a prolonged activation of the immune system. With the activation of long-term immunological memory protection against various tumour antigens can develop and are comparatively safer than chemotherapy [198]. The most immunogenic breast cancers as TNBC and HER2 positive tumours. Vaccines maybe divided into passive and active; passive vaccines are for the prevention of disease whereas active vaccines are to treat people with disease. The immune system may be activated by various mechanisms such as HER2, carbohydrate antigens, telomerase reverse transcriptase 3 and mucin 1 [198]. The different types of vaccines have recently been reviewed with regards to peptide based, protein based, whole cell based, dendritic cell based and DNA based vaccines [198]. However, the use of anti-breast cancer vaccines has not shown clinical benefits as compared to immunotherapy. As mentioned earlier taxanes affect the immune system, the combination of docetaxel and a recombinant vaccine enhanced T-cell activity against cancer cells [199]. The immunosuppression environment of the TME causes a decreased efficacy or immunotherapy, as a result of the COVID epidemic vaccines based on neo-antigens have been seen as the preferred target of cancer vaccines [200]. These vaccines are more likely to be effective in early breast cancer than metastatic disease after multiple therapies [200]. The use of the immune system to eliminate cancer cells is a novel approach but anti-cancer vaccines have yet to shown to be effective [201]. In mice models the use of protein lysates to produce vaccines has shown promise but as yet this has to be confirmed [202].
22. Minimal Residual Disease, Clinical Utility, and a Guide to Treatment Options
Chemotherapy is a double-sided weapon; it has been reported that neoadjuvant cytotoxic therapy increases the detection of bone marrow micro-metastasis. Those patients positive for micro-metastasis had a worse prognosis and their presence predicted relapse in distant tissues [202]. Furthermore, the use of chemotherapy selected resistant cancer cells especially those with stem cell properties, as well as affecting tumour stromal cells. This occurs through autocrine and paracrine self-renewing and survival pathways of these cells. The resistant cancer stem cells and CTCs evolve to display a more mesenchymal and stem cell phenotypes increase the ability of them to disseminate and invade distant tissues. Cells in the perivascular niches favour the extravasation of these cancer cells and protects them from the effects of chemotherapy [203]. Cytotoxic therapy also damages normal benign tissues, which in mouse and human models potentiated metastatic invasion and proliferation [203]. Although targeting the anti-apoptotic BCL-xL effectively eliminated the fibroblasts, this was not seen in in vivo models, possibly due to these stromal cells depending on the other anti-apoptotic BCL-2 family members. This study demonstrates the role of the TME in the micro-metastatic environment and its part in the dissemination of cancer cells, indicating the need to limit the pro-tumorigenic effects of cytotoxic damage to normal cells [204]. Bone marrow derived haematopoietic, mesenchymal and immune cells are able to repair cytotoxic damage in the cancer cells permitting their dissemination to colonize distant tissues [205]. The other side of the coin is that the IBCSG trial 22-00 reported that metronomic use of low dose cyclophosphamide combined with methotrexate had a prognostic benefit on some subtypes of TNBC. A high expression of Tregs in immunomodulatory and basal-like immune subtypes of TNBC showed a significant survival benefit of metronomic cytotoxic therapy [206]. Those patients exhibiting the mesenchymal subtype of TNBC had a worse prognosis when treated with this combination [206]. Therefore, maintenance therapy with low dose cytotoxic agents may be an alternative strategy but it is important to identify the differing subtypes of not only TNBC but of the other subtypes of breast cancer.
In clinical trials the use of minnelide combined with cyclophosphamide significantly reduced tumour growth by reprogramming the TME and increasing CTL infiltration of the tumour. Minnelide selectively targets cancer stem cells via the Myc and HSP70 pathways, the resection of the primary tumour in mice eliminated MRD, thus this combination may result in durable responses in patients with the basal/TNBC subtype [207]. Again, this highlights the necessity to identify the different subtypes of breast cancers in order determine the optimal treatment combination. However, minnelide has adverse effects such as Grade 3 and 4 haematological side effects such as neutropenia and severe cerebellar toxicity [208]. All the previous mentioned therapies are directed against disseminated tumour cells; some treatments have beneficial collateral effects by decreasing the immunosuppressive nature of the TME. It is possible that by eliminating the majority of cancer cells the local and systemic immunosuppression caused by the tumour cells is deceased thus increasing the efficacy of immune effector cells permitting the further elimination of cancer cells or maintain them in a latent state. The use of simple peripheral blood parameters such as the neutrophil to lymphocyte ratio has been used a possible indirect marker of immune immunosuppression, it appears that the dynamic change in the ratio is a better predictor of prognosis as compared with pre- or post-treatment levels [208]. A low ratio is associated with an improved prognosis; however a definitive cut-off point has not established [209,210].
Targeting the immunosuppressive immunological mechanisms in the TME to increase the immunological effector cells may produce deleterious side effects as seen with the use of PD-1 and PD-L1 check point inhibitors. In patients with co-existent autoimmune disease these drugs may exacerbate the disease, more serious side-effects are interstitial lung disease and myocarditis [180].
Little has been focussed on therapies that could affect the TME especially and CAFs, MDSCs and TAMs. As mentioned previously CAFs play a multiple functional role in creating and maintaining the immunosuppressive TME.
CAFs can be targeted by cytotoxic drugs, talabostat an oral cytotoxic drug was reported that in mouse models it was able degrade the extracellular matrix showing some cancer control [211]. However, in patients with metastatic colorectal cancer it showed no therapeutic effect. [212]. The humanised anti-FAP antibody inhibitor, sibrotuzumab, was reported not to have activity against CAFs [213]. CAF cells express FAP, it has been reported that the use of anti-CAF prodrugs or protoxins when coupled with a FAP cleavage site are systemically administered. However, they are activated by the expression of FAP. Tumour lysis and inhibition of growth was seen when injected into human breast and prostate xenografts [214,215]. Antibody conjugates targeting FAP using immunotoxins have also been investigated. The anti-FAP-PE39 conjugate suppressed tumour growth and increased the infiltration of tumour infiltrating lymphocytes [216]. Another line of investigation used doxorubicin or anti-Tenacin C liposomes targeted at FAP both to deliver cytotoxic therapy and to remodel the TME [217,218]. The of use of a transinfected FAP mRNA dendritic cell vaccine showed a decreased proliferation of cancer cells [219]. A collateral effect was to increase NK-cell activity, increased the CTL response as well as an increased anti-tumoral humoral response [220]. The use of FAP specific chimeric antigen receptor T-cell therapy has been reported to be able to eliminate most FAP positive, both CAFs and decrease tumour stromal generation, however significant side effects were observed, especially with respect to bone marrow toxicity [221]. However, none of these strategies has been studied in clinical trials due to efficacy and safety side effects. Pirfenidone is an oral anti-fibrotic drug, used to treat idiopathic lung fibrosis. It has also been reported to target CAFs, producing a restriction of tumour cell proliferation, the immunosuppressive TME, the formation of metastasis, resistance to anti-tumour treatment and extracellular matrix stiffness. Used in combination with doxorubicin it produces synergistic anti-tumour effects while not damaging normal tissues [222]. The reduction in the number of CAFs has collateral effects, such as the reduction of MDSC recruitment into the TME, a decrease in TAMs, decreased intra-tumoral recruitment and survival of Tregs, as well as decreasing the secretion of cytokines and chemokines that favour the immunosuppressive environment [223].
There are reports of the suppression of MDSC recruitment, the addition of olaparib via the SDF1 alpha/CCCR4 signally pathway improved the efficacy of CAR-T cells in mouse breast cancer models [224]. A review of the mechanisms that MDSCs can be modulated by altering the myeloid cell component in the TME, their functional block, the acquirement of a pro-inflammatory phenotype, cytokine modulation, and the targeting of Siglec-15, TREM2, MARCO, LILRB2 and CLEVER 1 [225]. Tumour progression is accompanied by fibrosis due to the activity of CAFs resulting in the stiffening of the extracellular matrix.
23. Tumour Associated Macrophages
Circulating monocytes are the precursors of TAMs and due to their plasticity are able to infiltrate the TME where they form up to 50% of the TME [226]. Their phenotype is determined by the TME converting them into the M2 immunosuppressive subtype rather than the anti-tumour M1 subtype [227]. Thus they play a function in the immunosuppression of the patient’s immunological system, promote cancer cell proliferation, angiogenesis and finally metastasis [228]. The is a metabolic dialogue between the tumour cells and TAMs via exosomes which causes metabolic changes in the TME, enhancing tumour cell survival [229]. The Warburg effect is the upregulation of aerobic metabolism by tumour cells which inhibits apoptosis and increasing their survival rate, increasing the production of lactic acid and this increases the TAMs into the M2 phenotype [230]. Tumour cells also affect lipid metabolism with an increased need to produce lipids, biofilm construction and function maintenance. It also increases the conversion of TAMs into the M2 phenotype [231,232]. In addition, the increased levels of lactic acid promote the Warburg effect increasing TAMs into the M2 phenotype [233]. Thus, drugs that affect both tumour cell and TAM metabolism may aid in decreasing the immunosuppressive TME. Treatment with an inhibitor of lactate dehydrogenase can significantly improve the effect of paclitaxel and trastuzumab against resistant cancer cells, Galloflavin and Machilin A being two examples [234,235]. It has been reported that simvastatin, a cholesterol lowering drug, acts on the TAMs rather than the tumour cells, reverses the epithelial-mesenchymal-transition and promotes the conversion of TAMs to the M1 phenotype [236]. It has also been reported that chloroquine resets the Tam population to the M1 phenotype thus decreasing the immunosuppression of the TME [237]. PARP inhibitors also reprogram the metabolic features of the TME, activating M1 TAMs and CTL [237]. This was extensively reviewed by Liang et al [238]. Tumour progression is accompanied by fibrosis due to the activity of CAFs resulting in the stiffening of the extracellular matrix. This change in the extracellular matrix decreases the ability of immune effector cells from entering the tumour. It has been reported that TAMs, as a result of this stiffening, initiate the biosynthesis of collagen directed by transforming growth factor ß [239]. This changes the metabolic environment causing CTL exhaustion and an adverse TME for CTLs [239]. In the TME TAMs that do not express PD-L1 is associated with immunosuppression and allows them to co-localize with cancer cancers. Inversely TAMs that do express PD-L1 stimulate the anti- tumour immune system. During the monocyte to macrophage maturation PD-L1 is upregulated, these TAMs are mature cells and are able to stimulate CTL proliferation and their cytotoxic efficacy and thus are a good prognostic marker of outcome in breast cancer patients [240].
24. Myeloid Derived Suppressor Cells
MDSCs play a pivotal role in creating the immunosuppressive environment of the TME as previously mentioned. Contrasting with physiological maturation of haemopoietic cells the TME renders MDSCs incapable of differentiating into mature cells which results in a heterogeneous cell population [241]. TMEs with high concentrations of MDSCs are associated to a worse prognosis and decreased responses to immunotherapy [242]. The proliferation of MDSCs is driven mainly by tumour derived growth factors, including GM-CSF, G-CSF, M-CSF, VEGR and IL-6 [243]. In a mouse model the selective JAK/STAT3 inhibitor JSI-124 was able to reduce the number of MDSCs by promoting their differentiation [244]. However, in tumour sites the activity of STAT3 is lower than in the blood or spleen and that JSI-124 caused no significant change in number of MDSCs in the TME [244]. The immunosuppressive effect of MDSCs in the TME requires their activation. Via cytokines produced by tumour stromal cells or activated T-cells. These activated MDSCs in mouse models they specifically and effectively decrease CD8+ function by decreasing the levels of IFN gamma [245]. By activating the apoptotic cascade of T-cells the MDSCs reduce the generation of T-cells and thereby promoting immunosuppression through immune blocking rather than directly killing the T-cells [245] through a high expression of ARG1, iNOS and ROS in MDSC cells [246,247]. MDSCs show a great deal of plasticity in their biological properties, being able to stimulate the production of Tregs and inhibiting CTL activation [248,249] thus significantly reducing the effectivity of immunotherapy [250]. Targeting MDSCs is therefore a focus of research on how to eliminate MDSCs and decrease the immunosuppressive TME. There are four current lines of investigation to eliminate MDSCs are being followed. Firstly, their elimination using chemotherapy, the use of gemcitabine and 5-fluorouracil selectively eliminates MDSCs via the process of apoptosis and increases CD8+ CTL production of gamma IFN. In advanced cervical cancer the use of paclitaxel and carboplatin was able to decrease the number of circulating MDSCs. One drawback is these cytotoxic drugs are not specific for MDSCs and affect all cells that are proliferating including immune effector cells [251]. The CD33 targeted antibody-conjugate gemtuzumab ozogamicin is approved for use in patients with CD33+ acute myeloid leukaemia.It targets CD33 on the cell membrane and in doing so releases a derivative of the cytotoxic agent calicheamicin which affect internalization into the tumour cell causes cell death [252]. Independent of the subtype of MDSCs membrane CD33 is found to be expressed in all MDSCs and as a result increases the death of these cancer cells, thus it raises the question of it being effective in breast cancer MDSCs [253]. A second alternative is the differentiation of MDSCs into mature macrophages, although the mechanisms that regulate this is not well understood at present, the use of all transretinoic acid (ATRA) significantly decreasing the presence of MDSCs into mature macrophages [254]. HF1K16 which is a pegylated liposomal form of ATRA has a longer half-life and can deliver a higher dosage to the cancer cells and is ongoing phase I trials [255]. Low dose paclitaxel stimulated the maturation of MDSCs although did not affect apoptosis nor the generation of MDSCs [256]. Thirdly, there is the possibility of blocking the immunosuppression effect of MDSCs. In models on mouse breast cancer, phosphodiesterase-5 inhibitors such as sildenafil, tadalafil and vardenafil reversed the immunosuppressive effects of MDSCs and thus improving anti-tumour immune responses [257]. The use of histone deacetylase inhibitors is another type of therapy, the use of entinostat decreased the frequency of circulating MDSCs [258] and increased the efficacity of immune checkpoint inhibitors [259]. Thirdly, a further treatment variation is the use of the COX-2 inhibitor celecoxib which inhibits MDSC immunosuppressive and delays cancer cell proliferation as a result of decreased ARG1 expression [260]. Finally, the possible blockade of the accumulation of MDSCs may decrease their immunosuppressive effect on the TME. AZD9150 in combination with a PD-L1 inhibitor has shown some activity against MDSCs and is undergoing phase I/II trials [261]. In renal carcinoma sunitinib significantly reduced the number and efficacy of MDSCs but as yet there are no trials ongoing in breast cancer patients [262]. A new approach, at least in mice models is the use of self-assembled gemcitabine-celecoxib nano-twin drug therapy. The nanoparticles containing both drugs have a log half-life in the circulation enabling them to accumulate preferentially in the TME. The release of both drugs acts synergistically, in both inhibiting the proliferation and increases apoptosis in Tregs, depletion of MDSCs, activates both CTL and NK-cells and the conversion of TAMs to the M1 phenotype. This combination in mice with 4T1 breast tumours showed an increase in the anti-cancer and anti-metastasis capabilities as compared to placebo. [263].
25. Poly (ADP-Ribose) Polymerase Inhibitors (PARP)
Up to 10% of breast cancer patients have mutations in genes necessary for dNA repair and checkpoint activators of the cell cycle [266]. BRCA 1 and 2 genes have been the most studied, each impairing homologous recombination repair (HRR) which causes genetic instability. Breast cancers bearing these mutations are more aggressive, typically TNBC and high grade. Breast cancer cells with HRR have a higher response to certain treatments, especially those with homologous repair deficiencies (HRD). Two PARPs are approved for the treatment of metastatic HER2 negative, BRCA positive cancers, these are olaparib and talazoparib due to the results of the Olympi AD trial [265] and EMBRACA trial [266] which showed an increased progressive free survival and improved quality of life as compared to the use of chemotherapy, however, overall survival of these patients did not improve [267,268]. The results of the Olympia A phase III trial for the use of olaparib in high risk HER2 negative BRCA positive breast cancer after neoadjuvant and adjuvant chemotherapy plus local treatment showed an improved disease-free survival and recurrence free survival as compared with placebo and was approved by the FDA to be used in these patients [269]. However, PARP inhibitors have toxic effects especially myelosuppression including anaemia, neutropenia, thrombocytopenia, myelodysplasia and acute myeloid leukaemia [265,266,267]. It has been recommended that PARP inhibitors should be started after the myelosuppressive effects of chemotherapy has recovered. Interstitial lung disease is another serious adverse effect being reported more frequency in patients receiving olaparib [270]. Resistance to PARP inhibitors eventually develops via differing mechanisms such as a decreased intracellular drug accumulation and thus effectiveness via ABC transporters [271], decreased PARP trapping by PARP inhibitors via mutations in the PARP enzymes and thus decreasing the cytotoxic effects of PARP inhibitors [272]. Another mechanism is the reactivation of HRR in HRD cells, this often occurs after secondary mutations in the BRCA 1 or 2 genes. HRR can also be reactivated by epigenetic changes including the methylation of DNA and/or histone configurations [273]. By targeting TAMs and reducing their immunosuppressive effects in patients with BRCA1 associated triple-negative breast cancer the resistance to PARP inhibitors maybe overcome. This again highlights the close association between the immune system and effectiveness of anti-cancer therapy [274].
26. Chimeric Antigen Receptor (CAR) T-Cell Therapy
The patients’ T-cells are removed using leukopheresis and are then modified genetically to express synthetic receptors directed against tumour-specific epitopes. The T-cells are then cultured to expand their numbers and then are re-infused into the patient where in an MHC- independent mode they can potentially eliminate the tumour cells [275]. Second and third generation CATs contain intracellular costimulatory domains to further enhance both the activation and proliferation of CAR-T cells [276,277]. Third-generation CAR-T cells appear to have an increased activity against cancer cells as compared to second generation CAR-T cells, at least in pre-clinical trials [278]. The heterogeneity of tumour epitopes makes the design of CAR-T cells more difficult; plus, the fact that these epitopes must not be or must be very lowly expressed in normal cells to prevent tissue damage.
Thus different CAR-T cells must be generated for each subtype of breast cancer in order to target the most specific treatment. The use of CAR-T therapy in the Luminal A subtype is under investigated. The tumour associated antigen diganglioside GD2 has been found to be expressed in a high percentage of this breast cancer subtype [279]. The efficacy of CAR-T cells based on anti-GD2 has produced different results depending on the breast cancer type used. Cytolysis of the cell line MCF 7 showed significant activity, whereas in the T-47D cell line there was no cytotoxic effect [280]. The use of PD-L1 CAR-T cells eliminated MCF-7 cells that had a low expression of PD-L1. If combined with HER2 CAR-T cells this increased the expression of PD-L1 and thus was more effective in eliminating the tumour cells [281]. In breast cancer subtype Luminal B the use of CAR-T cells has been lacking, but merits investigation.
CAR-T cell therapy directed against HER-2 have been shown to inhibit tumour proliferation and growth both in vitro and in vivo models using HER2 positive
MCF-7 and SK-BR-3 cell lines [282]. HER-2 CAR-T therapy when combined with PD-L1 therapy showed a significantly improved response against breast cancer cells [283]. The importance of targeting tumour specific antigens is highlighted by the fact that in the normal lung cell there may be expression of HER-2, thus the use of HER-2 CAR-T cells can lead to multi-organ failure [284].
In TNBC the search for specific tumour associated antigens continues, from surface glycoproteins and effector antigens such as CD16 has been reviewed recently [285].
What is possibly more interesting is the use of CAR-T cells directed against specific components of the TME.
FAP is a therapeutic target expressed on CAFs found in HER2 positive breast cancer. In refractory HER2 positive breast the use of FAP targeted CAR-T cells overcame the resistance to trastuzumab [286]. It has also been reported that these same CAR-T cells with the elimination of CAFs increases the anti-tumour effect in TNBC. Beneficial collateral effects included decreased infiltration by MDSCs, increased CTL infiltration causing an enhanced response in TNBC [287]. By binding to TR2.41BB via TR-2 apoptosis in MDSCs occurs, HER2-targeted CAR-T cells that also the costimulatory TR2.41BB receptor increased the anti-cancer function in HER-2 positive breast cancer cells [288]. The efficacy of CAR-T cells may be reduced by the decreased infiltration and activation of CTL caused by MDSCs. On the other hand, CAR-T cells which target the EGFR combined with olaparib decreases MDSC recruitment into the tumour, there is a synergitic effect in that olaparib inhibits the activity of MDSCs and thus increases the efficacy of CAR-T cell treatment in TNBC [289,290].
TAMs as previously mentioned play an important role in the TME, in mouse models the use of CD19-targeted CAR-T cells combined with FA-TRL 7-1A repolarized the TAMs from the M2 phenotype to the M1 phenotype and increased the therapeutic effect against 4T1-mCD19 breast cell lines [291]. As yet there are no studies of CAR-T cell therapy against the immunosuppressive function of Tregs.
However, CAR-T cell therapy is not without serious side effects; approximately 60% of patients treated with CAR-T cell therapy develop the cytokine storm syndrome which can lead to organ failure and death [292]. CAR-T cells also kill normal cells which express the same antigen causing severe side effects such as pulmonary tissue [293]. A dysfunctional state due to CAR-T cell exhaustion is caused by an increase in inhibitory receptors as a result of continuous antigen stimulation [294]. To improve the effectiveness of CAR-T therapy chemotherapy to cause lymphodepletion has been investigated, normally using cyclophosphamide and/or fludarabine infusions for three days, starting five days before the infusion of the CAR-T cells. The hypothesis of this Phase I trial is that pre-treatment with chemotherapy may decrease the number of Tregs and there would be less inhibition of the CAR-modified T-cells [295].
27. Inhibition of MMP-2
As previously mentioned MMP-2 plays an important role in the metastatic cascade, however inhibitors of MMP-2 have shown to be unacceptably toxic and as such preclinical and clinical trials have been suspended both in first and second generation MMP-2 inhibitors [59].
28. The Use of Bisphosphonates in Breast Cancer
The bisphosphonates are used to treat osteopenia, osteoporosis and hypercalcemia in breast, prostate cancer and myeloma. The results of the use of bisphosphonates combined with standard adjuvant therapy has shown mixed results. Jallouk el al [296] reported that there was no benefit of the addition of a bisphosphonate when added to standard adjuvant treatment. A meta-analysis of the addition of zoledronic acid came to a similar conclusion [297]. Differing from these conclusions it has been reported that clodronate significantly reduced the risk of both skeletal and visceral metastasis [298,299]. In patients with non-amplified MAF gene clodronate significantly improved disease-free survival and overall survival [300]. Recent updated ASCO-OH (CCO) guidelines, based on a consensus recommendation, that in postmenopausal women (natural or therapy induced) should be treated early with a bisphosphonate as it showed a modest improvement in overall survival, irrespective of hormone receptor or HER2 status [301].
In addition, bisphosphonates of which zoledronic acid is the most potent, can modulate the immune system via various mechanisms. This change in the immunological balance in the TME may cause beneficial effects with minimal toxicity. Using breast cancer lines in vitro incubation with bisphosphonates produced irreversible inhibition of cancer proliferation, increased apoptosis and cancer cell necrosis. The authors proposed that this was caused by down-regulation of Bcl-2, the release of cytochrome c from mitochondria and activation of caspase [302,303]. Using the breast cell lines MCF-7, 747D and MDA.MB.231 the effects of clodronate, pamidronate, ibandronate and zoledronate all produced in a dose dependent manner an irreversible proliferation of the MCF-7 and 747D cell lines, although the MDA.MB.231 was more resistant to bisphosphonates. In the MCF-7 and 747D breast cancer cells apoptosis was induced. In the MCF-7 cell line this inhibition could be almost completely stopped by caspase inhibition, while in the 747D cell line inhibition of caspase activity was virtually unchanged [304]. Thus, at least in vitro bisphosphonates could induce cell death that could to a beneficial role in the management of breast cancer.
Zoledronate again was the most potent inhibitor of the invasion of breast cell lines, again in a dose dependent manner but required a lower concentration than that need to produce apoptosis [305]. Migration of macrophages is caused by the inhibition of RANK, reducing the RANK-L induced dissemination [306]. In breast cancer cells RANK is also expressed and the inhibition of this pathway may reduce cancer cell dissemination [307]. Zoledronate has a secondary effect in that in causes a repolarization from the M2 to M1 macrophage phenotype using an in vivo rat model [308]. As previously revised TAMs play an important role in the TME, zoledronate combined with chemotherapy can trigger the conversion of the pro-tumoural M2 TAM to the tumouricidal M1 phenotype [309]. Using a lipid coated nano-particle enveloping calcium zoledronate it was reported that TAMs were significantly reduced in a S180 mouse model [310,311]. This significantly decreased the immune suppressive TME, decreasing cancer cell growth as well as significantly decreasing angiogenesis without the appearance of systemic adverse effects. In patients with breast cancer the immunosuppressive TME inhibits NK-cell function by decreasing the expression of NKP46 and NKG2D which normally activate he NK-cells and increases the inhibitory KIR2DL1 receptors. Using RNA analysis extracted from blood of breast cancer and normal patients and cell cultures it was reported that zoledronate improved the expression of the NK activating receptors [312]. Using MDA-MB-231 cells zoledronate was able to decrease the activity of Tregs in a dose dependent manner [313]. In a prostate cancer mouse model zoledronate combined with thymosin alpha1 combined with androgen derivation therapy increased the infiltration of CTL into the tumour and increased their cytotoxic activity and also stimulated anti-tumour macrophages whether this occurs in breast cancer is unknown [314]. It also decreased HER2 receptor levels and the downstream signalling pathways in breast cancer, this effect was higher in hormone resistant cancer cells [315]. Bisphosphonates also decrease the activation of pro-MMP2 by MT-MMP-1, by decreasing MMP-2 expression the migration and invasion of tumour cells is significantly decreased [316,317].
29. Minimal Residual Disease, What It Means and How Do We Treat It
MRD requires a different approach, these are asymptomatic patients and as such to maintain their quality non-toxic or minimally toxic therapies are needed. As mentioned previously MRD negative patients have the best prognosis, those patients with only bone marrow micro-metastatic disease have an intermediate prognosis with a latent period before relapse is detected, while those with CTCs detected have the worst prognosis and a short latent period before relapse [71,72,73].
If it not possible to eradicate MRD, a second best would be to extend the latent period for as long as possible. The classification of the different subtypes of breast may need to be further sub-classification to determine the optimal therapy for each patient, the idea of precision medicine.
Micro-metastatic disease may be phenotypically and biologically different than the primary tumour, such an example would be HER2 positive MRD in patients with a primary tumour classified as HER2 negative [66,67,70]. The same can be said about CTCs with differing expression of HER-2 and may be negative even though the metastasis expresses HER-2 [100]. The use of ctDNA to detect the presence of CD16-A-158FF where the anti-HER2 antibody margetuximab produced a better median overall survival, whereas those expressing CD16-A-15VV showed better results with trastuzumab. Thus, monitoring using ctDNA may be useful to define optimal anti-HER2 therapy [152].
Similarly, for primary tumours classified as ER positive there may be a difference with the micro-metastasis. The interesting question is why cancer cells that are negative for HER-2 or ER do not proliferate, grow and disseminate while the patient is being treated with anti-HER-2 or hormonal therapy. The probable reason is the initial treatment of surgical removal of the primary cancer and axillary nodes plus neo- and/or adjuvant chemotherapy. After surgical removal of the tumour the only tumour cells left, if there are any, is in the context of MRD. The primary tumour and the TME was responsible for creating an immunosuppressive environment both locally and systemically. The pre-metastatic niche and metastatic niches resulting from the effects of the primary tumour interacting with the TME and causing a decreased effect of the anti-tumour immune response. The effect of chemotherapy as previously mentioned has the beneficial effect of modulating the immunological balance, affecting the immunosuppressive environment more than the anti-tumour immune response. Thus, it could be hypothesised that in the micro metastasis the immunological equilibrium is altered to favour the anti-tumour immune system. This change in the balance of positive and negative immunological effects may permit the anti-tumour immune response to eliminate resident tumour cells and/or maintain dormant. This is clearly seen when after finishing adjuvant therapy relapses may occur many years later, which raises the question how? When receiving no treatment, the implication is that the immune balance between immunosuppression and anti-tumour immune responses maintain the tumour cells dormant. However, due to clonal instability this balance may change to a more immunosuppressive environment allowing the tumour cells to leave their latency period, proliferate, grow and disseminate to eventually form the metastatic disease.
The goal of the treatment of the TME to provide a less immunosuppressive environment is ongoing using cell cultures and mouse models. In these patients it is important to evert potential serious side effects such as the cytokine storm with the use of CAR-Therapy or the induction of auto-immune disease with check-point inhibitors.
The use of CTC detection or ctDNA allows monitoring of breast cancer patients before imaging studies detect relapse. Wang et al [318] reported that in ER positive non-metastatic breast the change of hormonal therapy resulted in a significantly increased CTC clearance and lower CTC values, in those ER negative the use of capecitabine also produced similar effects. Although progression free survival was longer in those who changed treatment, there was no difference in overall survival rates between patients who changed treatment compared with those who did not.
While studies have reported the association of the absence of CTCs is associated with a better prognosis [95,112] it has been suggested that the detection of CTCs is not necessary for the formation of metastasis due to the unsuitability of the target organ [208]. Drug resistance in patients treated with hormonal therapy may occur, reported to be 31% after three years raising to 65% after 5 years of therapy. Endocrine resistance has been reported due to activation of HER2, EGFR, FGFR and receptor kinases [209].
Synergy with the addition of zoledronate was seen when gamma-delta CAR-T cells were used to target prostate cancer cells, the CAR-T cells recognising the phosphor- antigens in the TME [117]. It has been reported that CAR-T cells which are engineered to express cytokines, IL-12, IL15 or CCL19 give the CAR-T cells a higher proliferation rate, persistence in the animal model and higher antitumoral capacities than conventional CAR-T cells [117]. Pre-clinical studies using mice xenograft models to determine dosage and toxicity are ongoing. To improve the effectiveness of CAR-T therapy chemotherapy to cause lymphodepletion has been investigated, normally using cyclophosphamide and/or fludarabine infusions for three days, starting five days before the infusion of the CAR-T cells. The hypothesis of this Phase 1 trial is that pre-treatment with chemotherapy may decrease the number of Tregs and there would be less inhibition of the CAR-modified T-cells [118].
30. Treating the Tumour Microenvironment
As mentioned previously, tumour cells do not live alone but co-exist with the TME; the various treatments revised are directed against cancer cells although they may have collateral benefits on the TME. Tumour cells change the stromal tissue, but there are physiological processes that effect the stromal environment in benign tissues. One such effect is that of aging; these include changes in the extra-cellular matrix, changed in secreted factors and changes in the immune system [319]. In both the process of aging and similarly for cancer there is a time dependent accumulation of genetic and cellular damage. These changes include epigenetic changes, intracellular communication, proteostasis, dysfunction of the mitochondria and cellular senescence [320]. This aging effect in the normal cells of the TME are able to promote cancer progression and metastasis, with immune cells and fibroblasts being particularly being affected. With aging, immunosenescence decreases the effector immune system, causing a more immunosuppressive environment, increasing the presence of MDSC, Tregs and a switch to M2 macrophages while at the same time decreases the effectivity of NK-cells, CTL, macrophages and dendritic cells [321]. In aged cells there is a loss of extracellular matrix integrity, this dynamic process is affected by the aging of fibroblasts. These age related in the extracellular matrix’s physical properties are a result of a decrease in collagen density, thickness of the extracellular matrix and its mechanical properties which include stiffening and decreased crosslinking of fibulin, fibrillin and elastin further impairing the integrity of the extracellular matrix [322].
The use of zoledronate has been recommended as neo or adjuvant therapy [304]. It also affects the TME, using a mouse model the administration of zoledronate suppressed Treg activity, their proliferation, their migration and immunosuppressive effect in the TME [324] as well as blocking Treg-cancer cell interactions [316]. It also produced a selective decrease in the number of Tregs which significantly increased the proliferation of CTL and NK-cells in patients with metastatic breast cancer, as seen in blood samples. Zoledronate inhibited selectively essential signalling pathways reducing the expression of nuclear factor of activated T-cells and FOXP3 and as a consequence a reduced the expression of CD25, STAT and TFGβ. This leads to an increased anti-tumour immune response by CTL and NK-cells [323]. Using the MCF7 breast cell line the use of zoledronate to determine its effect of the induction and apoptosis on cancer stem cells. It caused a dose and time-dependent increase in the viability of cancer stem cells. It was determined that this was a result of increased apoptosis and S-phase cell cycle arrests in the cancer stem cells causing their death of drug resistant MCF-7 stem cells. This was achieved by the downregulation of anti-apoptopic genes and the up regulation of pro-apoptopic genes, with the implication that zoledronate may be effective in targeting chemoresistant cancer stem cells [324]. In addition, it has been reported that HER-2 overexpression is a major mechanism in hormonal therapy resistance which enables them to proliferate without the effects of oestrogen. Using breast cancer cell lines, the efficacy of zoledronate was higher in endocrine resistant breast cancer cells as compared with endocrine sensitive cells. It may circumvent resistance to fulvestrante in a synergistic fashion by decreasing the phosphorylation of the ERK signalling pathway and also by decreasing HER-2 and its downstream signalling pathways signalling in both tamoxifen and fulvestrant resistant cells. It also inhibited the migration and invasion of these resistant breast cells implying the possibility of inhibiting the formation of metastasis [316]. However, the use of bisphosphonates is not without adverse effects, the most serious being osteonecrosis of the jaw, though not difference was detected between patients with metastatic or non-metastatic disease. Their use in the treatment of osteoporosis has shown they have limited side effects and thus could be used in breast cancer patients before the appearance of metastasis [325].
TNBC is the more aggressive subtype of breast cancer and treatment has been based on the use of chemotherapy. However, in the last few years there has been interest in targeting the TME. Due to the poor prognosis of these patients it maybe that side-effects of the treatment are more acceptable to the patient. Studies have shown that the response to immune checkpoint blockade inhibitors, especially against PD-1/PD-L1 axis interact with the TME, involving immune cells, stromal and cancer cells. Although few in vitro or in vivo studies in mice were for other types of malignancy, such as melanoma, gastric cancer and non-small cell lung cancer cell lines [326]. Thus targeting the TME may improve the prognosis in triple negative breast [327]. TNBC is an immunogenic subtype of breast cancer with a high level of both mutational load and a higher presence of immune cell infiltrates. The expression of PD-L1 is increased in these tumour cells and therefore may be a biomarker for the response to immune check point blockade. These drugs have been associated with a better prognosis when combined with chemotherapy [328]. The approval of the use of pembrolizumab in combination with neoadjuvant chemotherapy in 2021 for high risk early stage TNBC improved the patient´s prognosis; however, the question of patient selection, management of side effects, identifying predictive biomarkers all need to be answered [329]. Although patients may suffer permanent immune related toxicities, it has been suggested that all node positive TNBC patients should receive an immune checkpoint inhibitor. It has also been suggested that some lower risk TNBC patients, that is stage I or II but with a high number of tumour infiltrating lymphocytes with or without a high PD-L1 expression may benefit from immune check point inhibitors and with the possibility of a lower dose of chemotherapy [330]. The same authors suggested this combination may improve progression free survival due to the quality and quantity of the anti-tumour T-cell response. More recently, using the gene expression of TNBC mRNA identified 18 differentially expressed genes that predicted patient outcome. A low expression of immune related genes was association with a worse prognosis and a significant difference in the number of infiltrating immune cells in the TME [333]. Furthermore, the expression of keratin 81 (KRT1) was found to be increased in TNBC, a low expression of KRT1 was associated with a better prognosis. A high expression of KRT1 in TNBC cancer cells was reported to be associated with increased cancer cell proliferation, migration, invasion and inhibiting CTL. Differing from this TNBC with a low KTR1 expression had increased CTL infiltration in the TME [332]. Potential future treatments may change the epigenetic dysregulation in TNBC cells; altered DNA methylation, histone modifications have a crucial role in the development of treatment resistance to TNBC cells. It is thought that an imbalance of histone methyl-transferases such as EZH2 and histone deacetylases produce changes in the TME leading to a more immunosuppressive TME [333].
In BRCA associated TNBC the use of PARP inhibitors may prolong the progression free survival of patients but its effects are usually transitory. PARP inhibitor resistance in patients with BRCA1 TNBC is associated with immunosuppressive tumour macrophages; the drug can both produce pro and anti-tumour responses. In vivo studios in mice reported that the associated use of a PARP inhibitor and a CSF-1R blocking antibody improved both the innate and acquired anti-cancer immune response, mediated by CTL [334]. Another mechanism of resistance to PARP inhibition is caused by osteoclasts synthesizing a higher level of glutamine. As bisphosphonates inhibit the activity of osteoclasts a combined therapy associated with PARP inhibits could potentially be of benefit in TNBC [335,336].
31. Conclusions
Cancer was originally seen as a single entity with treatment options aimed to eliminate all the tumour cells. Only patients receiving hormonal therapy received long-term adjuvant therapy. Patients were only re-treated when there was evidence of metastasis, again in an effort to eliminate the tumour. Orginally used in heaematological disease, the concept of MRD was applied to patients with solid tumours. Over the last decade there has a change in concept of cancer, although clinical guidelines have changed in concept although new drugs with differing mechanisms of action have been incorparated into the clinical recommendations. There is active ongoing research into new approaches to cancer treatment, with the concept that cancer cells only form a small of the TME in patients with MRD. This latent period before the appearance of metastasis gives a window of time to modify the TME and if not eliminate the tumour cells at least maintain them in a dormant state. The clasifiation of MRD allows more precision therapy, as the micrometasis may have differing biological phenotypic characteristics than the primary tumour. Treatments to decrease the immunosuppressive TME may potentially increase the anti-tumour immune response, but without causing serious adverse effects such as a cytokine storm ot autoimmune disease. The addition of bisphosphonates is one step in this direction, preclinical trials of other drugs are ongoing. Alternate strategies include the metronomic use of low dose cyclophosphamide combined with methotrexate in TNBC. Targeting CAFs is another possibility, such as the use of pirenidone, while simvastatin act aginst TAMs as does chloroquine. With regards to MDSCs the use of antibody-conjugate gmtuzumab-CD33 which is approved from CD33 positivie acute myeloid leukaemia. MDSCs all express CD33 and thus this drug decreases their number allowingthe anti-tumour immune system to be more effective. The use of all trans retinoic acid induces the maduration of MDSCs and HF 1K16 is undergoing phase 1 clinical trials. Blocking the immunosuppression effect of MDSCs the use of phosphodiesterase-5 inhibitors such as sildenafil improved the anti-tumour immune respose. The use of COX-2 inhibitors such as celecoxib also decreases MDSC activity. In mice models the use of nanoparticules, composed of gemcitabine and celecoxib act syngistically and cause a bystander effect. The release of both drugs decreases the immunosuppressive TME and activates the anti-tumour immune response. Thus by using the three markers to classify MRD and monitoring to detect early changes potentially could increase the latent period or eliminate tumour cells without serious side-effects and thus preventing metastasis.
References
- Siegel, R.; Miller, K.D.; Fuchs, H.E.; Jamal, A. Cancer statistics, 2022. CA A Cancer J Clin 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Giuquinto, N.; Colla, S.; Morandi, F.; Rizzoli, V. Breast cancer statistics, 2022. A Cancer J Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Makki, J. Diversity of breast carcinoma: Histological subtypes and clinical relevance. Pathology. 2015, 8, 23-3. [Google Scholar] [CrossRef]
- Anderson, W.F.; Chatterjee, N.; Ershler, T.; Brawley, O.W. Oestrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology and End Results database. Breast Cancer Res Treat. 2002, 76, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, J.S.; Korlimarla ADesai k Alexander, A.; Raghacvan, R.; Anupama, C.; Dendukuri, N.A.; Manjunath, S.; Correa, M.; Raman, N.; et al. majority of low (1-10%) ER positive breast cancer behave like hormone receptor negative tumors. J Cancer 2014, 5, 156–165. [Google Scholar] [CrossRef]
- Gloyeske, N.C.; Dabbs, D.J.; Bhargava, R. Low ER + breast cancer: Is this a distinct group? Am J Clin Pathol 2014, 141, 697–701. [Google Scholar] [CrossRef]
- Bouchard-Fortier, A.; Provencher, L.; Blanchette, C.; Diorio, C. Prognostic, and predictive value of low estrogen receptor expression in breast cancer. Curr Oncol. 2017, 24, e106–e114. [Google Scholar] [CrossRef]
- Sleightholm, R.; Nielson, B.K.; Elkhatib, S.; Flores, L.; Dukkipati, S.; Zhao, R.; Choudbury, S.; Gardner, B.; Carmichael, J.; Smith, L.; Bennion, N.; Wahl, A.; Baine, M. Percentage of hormone receptor positivity in breast cancer provides prognostic value: A single-institute study. J Clin Med Res 2021, 13, 9–19. [Google Scholar] [CrossRef]
- Marchio, C.; Annaratone, L.; Marques, A.; Casorzo, L.; Berrino, E.; Sapino, A. Evolving concepts in HER2 evaluation in breast cancer: Heterogeneity, HER2 low carcinomas and beyond. Semin Cancer Bio 2021, 72, 123–135. [Google Scholar] [CrossRef]
- Wahl, G.M.; Spike, B.T. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Caldas, C. The implications of clonal genome evolution for cancer medicine. N Eng J Med 2013, 368, 842–851. [Google Scholar] [CrossRef]
- Marusky, A.; Almendro, V.; Polyak, K. Intra-tumoral heterogeneity: a looking glass for cancer? Nat Rev Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal heterogeneity, and tumor evolution: past, present and future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Liu, T.; Liu, C.; Yan, M.; Zhang, L.; Zhang, J.; Xiao, M.; Li, Z.; Wei, X.; Zhang. Single cell profiling of primary and paired metastatic lymph node tumors in breast cancer patients. Nat Commun 2022, 13, 6823. [Google Scholar] [CrossRef]
- Mao, X.; Zhou, D.; Lin, K.; Zhang, B.; Gao, J.; Ling, F.; Zhu, L.; Yu, S.; Chen, P.; Zhang, C.; et al. Single cell and spatial transcriptome analysis analyses reveal cell heterogeneity and immune environment alterations in metastatic axillary lymph nodes in breast cancer. Cancer Immunol Immunother 2023, 72, 679–695. [Google Scholar] [CrossRef]
- Gulati, G.S.; Sikander, S.S.; Wesche, D.; Manjunath, A.; Bharadwaj, A.; Berger, M.J.; Ilagan, F.; Kuo, A.H.; Hsieh, R.W.; Cai, S.; et al. Single cell transcriptional diversity is a hallmark of development potential. Science 2020, 367, 405–411. [Google Scholar] [CrossRef]
- Van de Sande, B.; Flerin, C.; Davie, K.; De Weageneer, M.; Hulselmans, G.; Aibar, S.; Seurinck, R.; Saalens, W.; Cannoodt, R.; Rouchon, Q.; et al. A scalable SCENIC workflow for single cell gene regulatory network analysis. Nat Proc 2020, 15, 2247–2276. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wang, R.; Xie, H.; Hu, L.; Wang, C.; Xu, J.; Zhu, C.; Liu, Y.; Gao, F.; Li, X.; et al. Single-cell RNA sequencing reveals cell heterogeneity and transcriptome profile of breast cancer lymph node metastasis. Oncogenesis 2021, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Fidler, I.J.; Poste, G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008, 9, 8. [Google Scholar] [CrossRef]
- Luo, W. Nasopharyngeal carcinoma ecology theory: Cancer as a multidimension spatiotemporal “unity of ecology and evolution” pathological ecosystem. Theranostics 2023, 13, 1607–1631. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. The theory of tumour ecosystem. Cancer Commun. 2022, 42, 587–608. [Google Scholar] [CrossRef]
- Murray, N.P.; Miranda, R.; Ruiz, A.; Droguett, E. Diagnostic yield of primary circulating tumour cells in women suspected of breast cancer: the BEST (Breast Early Screening Test) Study. Asian Pac J Cancer Prev. 2015, 16, 1929–1934. [Google Scholar] [CrossRef]
- Sanger, N.; Effenberger, K.E.; Reithdorf, S.; Van Haasteren, V.; Gauwerky, J.; Wiegratz, I.; Strebhardt, K.; Kaufmann, M.; Pantel, K. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int J Cancer 2011, 129, 2522–2526. [Google Scholar] [CrossRef]
- Rodriguez-Bejerano, O.H.; Parra-Lopez, C.; Patarroyo, M.A. A review concerning the breast cancer-related tumour microenvironment. Crit Rev Oncol Hematol 2024, 199, 104389. [Google Scholar] [CrossRef] [PubMed]
- Agahozo, M.C.; van Boskstal, M.R.; Groenendijk, F.H.; van den Bosch, T.P.P.; Westenend, P.J.; van Deurzen, C.H.M. Ductal carcinoma in situ of the breast: immune cell composition according to subtype. Mod Pathol. 2020, 33, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, H. Prognostic values of tumoral MMP2 and MMP9 overexpression in breast cancer: a systematic review and meta-analysis. BMC Cancer 2012, 21, 149. [Google Scholar] [CrossRef]
- Yang, C.; Yu, H.; Chen, R.; Tao, K.; Jian, L.; Peng, M.; Li, X.; Liu, M.; Liu, S. CXCL1 stimulates migration and invasion in ER-negative breast cancer cells via activation of the ERK/MMP2/9 signalling pathway. Int J Oncol 2019, 55, 684–696. [Google Scholar] [PubMed]
- Kargozaran, H.; Yuan, S.Y.; Breslin, J.W.; Watson, K.D.; Gaudreault, N.; Breen, A.; Wu, M.H. A role for endothelial derived matrix metalloproteinase-2 in breast cancer cell transmigration across the endothelial-basement membrane barrier. Clin Exp Metastasis 2007, 24, 495–502. [Google Scholar] [CrossRef]
- Ross, J.S.; Kaur, P.; Sheehan, C.E.; Fisher, H.A.; Kaufman RAJr Kallakury, B.V. Prognostic significance of metalloproteinase 2 and tissue inhibitor of metalloproteinase 2 expression in prostate cancer. Mod. Pathol. 2003, 16, 198–205. [Google Scholar] [CrossRef]
- Trudel, D.; Fradet, Y.; Meyer, F.; Harel, F. Tetu, B. Significance of MMP-2 expression in prostate cancer: An immunohistochemical study. Cancer Res. 2003, 63, 8511–8515. [Google Scholar]
- Nissinen, L.; Kahari, V.M. MMPs in inflammation. Biochem. Biophys. Acta 2014, 1840, 2571–2580. [Google Scholar] [CrossRef]
- Lee, B.K.; Kim, M.J.; Jang, H.S.; Lee, H.R.; Ahn, K.M.; Lee, J.H.; Choung, P.H.; Kim, M.J. High concentrations of MMP-2 and MMP-9 reduce NK mediated cytotoxicity against oral squamous cell carcinoma line. In Vivo 2008, 22, 593–598. [Google Scholar]
- Li, J.; Yu, S.; Rao, M.; Cheng, B. Tumor-derived extracellular vesicles: key drivers of immunomodulation in breast cancer. Front Immunol 2025, 16, 1543585. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.; Gazinska, P.; Zhang, B.; Khiabany, A.; Sinha, S.; Alaguthuri, T.; Flores-Borja, F.; Vicencio, J.; Beuron, F.; Roxanis, I.; et al. Serum derived extracellular vesicles from breast cancer patients contribute to differential regulation of T-cell mediated immune-escape mechanisms in breast cancer subtypes. Front Immunol 2023, 14, 1204224. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatalo, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-mediated metastasis: From epithelial-mesenchymal transition to escape from immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O´Connor, S.T.F.; Chin, A.R.; et al. Cancer secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef]
- Yuan, X.; Qian, N.; Ling, S.; Li, Y.; Sun, W.; Li, J.; Du, R.; Zhong, G.; Liu, C.; Yu, G.; et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics 2021, 11, 1429–1445. [Google Scholar] [CrossRef]
- Korbecki, J.; Kojder, K.; Siminska, D.; Bohatyrewicz, R.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CC chemokines in a tumor. A review of pro-cancer and anti-cancer properties of the ligands of receptors od CCR1, CCR2, CCR3. And CCR4. Int J Mol Sci. 2020, 21, 8412. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef]
- Morrisey, S.M.; Zhang, F.; Ding, C.; Montoya-Durango, D.E.; Hu, X.; Yang, C.; Wang, Z.; Yuan, F.; Fox, M.; Zhang, H.G.; et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021, 33, 2020–2058. [Google Scholar] [CrossRef]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J. Whiteside. T.L. Tumor-derived micro-vesicles promote regulatory T-cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [PubMed]
- Figueiro, F.; Muller, L.; Funk, S.; Jackson, E.K.; Battastini, A.M.; Whiteside, T.L. Phenotypic and functional characteristics of CD39 high human regulatory B-cells (Breg). Oncoimmunology 2016, 5, e1082703. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer associated fibroblasts. Nat Rev Cancer 2020, 20, 174–86. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Cheng, P.; Pan, J.; Wang, S.; Ding, Q.; Jiang, Z.; Cheng, L.; Shao, X.; Huang, L.; Huang, J.; et al. An IL6-adenosine positive feedback loop between CD73 (+) Tregs and CAFs promotes tumor progression in human breast cancer. Cancer Immunol Res 2020, 8, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Kennel, K.B.; Bozlar, M.; De Valk, A.F.; Greten, F.R. Cancer associated fibroblasts in inflammation and antitumor immunity. Clin Cancer Res 2023, 29, 1009–1016. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Dis 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schroder, C.; et al. Tumor-associated macrophages in breast cancer: innocent bystanders or important player? Cancer Treat Rev 2018, 70, 178–189. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci 2019, 26, 78. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Soltani, A.; Ahmadi, S.; Abarghan, Y.J.; Rosengren, R.J.; Sahebkar, A. Tumor associated macrophages: role in cancer development and therapeutic implictions. Cell Oncol (Dordr) 2019, 42, 591–608. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8 (+) T-cell lymphocytes on cancer immunotherapy: a review. J Cell Physiol 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Hossain, M.A.; Liu, G.; Dai, B.; Si, Y.; Yang, Q.; Wazir, J.; Bimbauer, L.; Yang, Y. Reinvigorating exhausted CD8 (+) cytotoxic T-lymphocytes in the tumor microenvironment and current strategies in cancer immunotherapy. Med Res Rev 2021, 41, 156–201. [Google Scholar] [CrossRef]
- Ugel, S.; de Sanctis, F.; Mandruzzato. Tumor-induced myeloid deviation: when myeloide derived suppressor cells meet tumor associated macrophages. J Clin Invest 2015, 125, 3365–3376. [Google Scholar] [CrossRef]
- Huang, X.; Cao, J.; Zu, X. Tumor-associated macrophages: An important player in breast cancer progression. Thorac Cancer 2022, 13, 269–276. [Google Scholar] [CrossRef]
- Qin, R.; Ren, W.; Ya, G.; Wang, B.; He, J.; Ren, S.; Jiang, L.; Zhou, S. Role of chemokines in the crosstalk between tumor cells and tumor-associated macrophages. Clin Exp Med 2023, 23, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Marme, F. Antibody conjugates for breast cancer. Oncol Res Treat 2012, 45, 26.36. [Google Scholar] [CrossRef]
- Moura, T.; Caramelo, O.; Silva, I.; Silva, S.; Goncalo, M.; Portilha, M.A.; Moreira, J.N.; Gil, A.M.; Laranjiera, P. Early-stage Luminal B-like breast cancer exhibits a more tumor suppressive microenvironment than Luminal A type breast cancer. Biomolecules 2025, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.P. The role of matrix metalloproteinase-2 in the metastatic cascade: a review. Oncologie 2024, 26, 27–40. [Google Scholar] [CrossRef]
- Morgunova, E.; Tuuttila, A.; Bergmann, M.I.; Isupov, M.; Lindquist, Y.; Schneider, G.; Tryggvason, K. Structure of human pro-matrix metalloproteinase 2: activation mechanism revealed. Science 1999, 284, 1667–1670. [Google Scholar] [CrossRef]
- Mayhew, V.; Omokehinde, T.; Johnson, R.W. Tumor dormancy in bone. Cancer Rep 2020, 3, e1156. [Google Scholar] [CrossRef] [PubMed]
- Sandiford, O.A.; Donnelly, R.J.; El-Far, M.H.; Burgmeyer, L.M.; Sinha, G.; Pamarthi, S.H.; Sherman, L.S.; Ferrer, A.I.; de Vore, D.E.; Patel, S.A.; et al. Mesenchymal stem cell-secreted extracellular vesicles instruct stepwise dedifferentiation of breast cancer cells into dormancy at the bone marrow perivascular region. Cancer Res 2021, 81, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Tamamouna V, Pavlou E, Neophytou CM, Papageorgis P, Costeas P- Regulation of metastatic tumor dormancy and emerging opportunities for therapeutic intervention. Int J Mol Sci 2022, 23, 13931. [CrossRef] [PubMed]
- Byme, N.M.; Summers, M.A.; McDonald, M.M. Tumor cell dormancy and reactivation in bone: Skeletal biology and therapeutic opportunities. JBMR Plus 2019, 3, e10125. [Google Scholar]
- Muller, V.; Alix-Panabieres, C.; Pantel, K. Insights into minimal residual disease in cancer patients: implications for anti-cancer therapies. Cancer 2010, 46, 1189–1197. [Google Scholar] [CrossRef]
- Aurilio, G.; Mondardini, L.; Rizzo, S.; Sciandivasci, A.; Preda, L.; Bagnardi, V.; Disalvatore, D.; Pruneri, G.; Munzone, E.; Vigna, P.D.; et al. Discordant hormone receptor and human epidermal growth factor receptor 2 status in bone metastases compared to primary breast cancer. Acta Oncol 2013, 52, 1649–1656. [Google Scholar] [CrossRef]
- Aurilio, G.; Dislavatore, D.; Pruneri, G.; Bagnardi, V.; Viale, G.; Curigliano, G.; Adamoli, L.; Munzone, E.; Scianduivasci, A.; De Vita, F.; et al. A meta-analysis of oestroegen receptor, progesterone receptor and human epidermal growth factor receptor discordance between primary breast cancer and metastasis. Eur J Cancer 2014, 50, 277–289. [Google Scholar] [CrossRef]
- Konig, T.; Dogan, S.; Hohn, A.K.; Weydandt, L.; Aktas, B.; Nel, I. Multi-parameter analisis of disseminated tumor cells (DTCs) in early breast cancer patients with hormone receptor positive tumors. Cancer 2023, 15, 568. [Google Scholar] [CrossRef]
- 69Ditsch, N.; Meyer, B.; Rolle, M.; Untch, M.; Schildberg, F.W.; Funke, I. Estrogen receptor expression profile of disseminated epithelial tumor cells in bone marrow of breast cancer patients. Recent Results cancer Res. 2003, 162, 141–147. [Google Scholar]
- Jager, B.A.S.; Finkenzeller, C.; Bock, C.; Majunke, L.; Jueckstock, J.K.; Andergassen, U.; Neugebauer, J.K.; Pestka, A.; Friedl, T.W.P.; Jeschke, U.; et al. Estrogen receptor and HER-2 status on disseminated tumor cells and primary tumor in patients with early breast cancer. Transl Oncol 2015, 8, 509–516. [Google Scholar] [CrossRef]
- Murray, N.P.; Aedo, S.; Villalon, R.; Albarran, V.; Orrego, S.; Guzman, E. Subtypes of minimal residual disease and outcome for stage II colon cancer treated by surgery alone. Ecancermedicine 2020, 14, 1119. [Google Scholar]
- Murray, N.P.; Aedo, S.; Villalon, R.; Lopez, M.A.; Minzer, S.; Munoz, L.; Orrego, S.; Contreras, L.; Arzeno, L.; Guzman, E. Effect of FOLFOX on minimal residual disease in stage III colon cancer and risk of relapse. Ecancermedicine 2019, 13, 935. [Google Scholar] [CrossRef]
- Murray, N.P.; Aedo, S.; Fuentealba, C.; Reyes, E.; Salazar, A.; Lopez, M.A.; Minzer, S.; Orrego, S.; Guzman, E. Subtypes of minimal residual disease, association with Gleason score, risk and time to biochemical failure in pT2 prostate cancer treated with radical prostatectomy. Ecancermedicine 2019, 13, 934. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Yau, C.; Wolf, D.M.; Lee, J.S.; Chattopadhyay, A.; Scott, J.H.; Bowlby-Yoder, E.; Hwang, E.S.; Alvarado, N.; Ewing, AL.; et al. Synchronous detection in blood and disseminated tumor cells in breast cancer predicts adverse outcome in early breast cancer. Clin Cancer Res 2019, 25, 5388–5397. [Google Scholar] [CrossRef]
- Bilani, N.; Elson, L.; Liang, H.; Elimimian, E.B.; Arteta-Bulos, R.; Nahleh, Z. Prognostic and predictive value of circulating and disseminated tumor cells in breast cancer: A National Cancer Database (NCDB) analysis. Technol Cancer Res Treat 2020, 19, 1533033820980107. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabieres, C. Bone marrow as a reservoir for disseminated tumor cells: a special source for liquid biopsy in cancer patients. Bonekey Rep 2014, 3, 584. [Google Scholar] [CrossRef]
- Sai, B.; Xiang, J. Disseminated tumour cells in bone marrow are the source of cancer relapse after therapy. J Cell Mol Med 2018, 22, 5776–5786. [Google Scholar] [CrossRef] [PubMed]
- NCCN clinical practice guidelines in oncology. Breast cancer version 4.2025. www.nccn.org/professional/physician_gls/pdf/breastpdf accessed June 2025.
- Kabak, E.C.; Foo, S.L.; Rafaeva, M.; Martin, I.; Bentires-Alj, M. Microenvironment regulation of dormancy in breast cancer metastasis: “An ally that changes allegiances”. Adv Exp Med Biol 2025, 1464, 373–395. [Google Scholar]
- Lennart, N.A.; Rao, S.S. Cell-cell interactions mediating primary and metastatic breast cancer dormancy. Cancer Metastasis Rev 2024, 44, 6. [Google Scholar] [CrossRef] [PubMed]
- Sandiford, O.A.; Donnelly, R.J.; El-Far, M.K.; Burgmeyer, L.M.; Sinha, G.; Pamarthi, S.H.; Sherman, L.S.; Ferrer, A.I.; DeVore, D.E.; Patel, SA.; et al. Mesenchymal stem-cell secreted extracellular vesicles instruct stepwise dedifferentiation of breast cancer cells into dormancy at the bone marrow perivascular region. Cancer Res 2021, 81, 1567–1582. [Google Scholar] [CrossRef]
- Khadge, S.; Coler, K.; Talmadge, J.E. Myeloid derived suppressor cells and the release of micro-metastasis from dormancy. Clin Exp Metastasis 2021, 38, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Huang, X.; Fu, X.; Zhnag, X.; Lin, R.; Zhang, W.; Zhang, J.; Lu, Y. Myeloid-like tumor hybrid cells in bone marrow promote progression of prostate cancer bone metastasis. J Haematol Oncol 2023, 16, 46. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Liu, M.; Xiang, X.; Xi, Z.; Xu, H. Osteoblasts and osteoclasts: an important switch of tumour cell dormancy during bone metastasis. J Exp Clin Cancer Res 2023, 41, 316. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.P.; Reyes, E.; Tapia, P.; Badinez, L.; Orellana, N.; Fuentealba, C.; Olivares, R.; Porcell, J.; Duenas, R. Redefining micrometastasis in prostate cáncer, bone marrow disseminated tumour cells and micrometastasis: Implications in determining local or systemic treatment for biochemical failure after radical prostatectomy. Int J Mol Med 2012, 30, 896–904. [Google Scholar] [CrossRef]
- Bain, B. Bone marrow biopsy morbidity: review of 2003. J Clin Pathol 2005, 58, 406–40. [Google Scholar] [CrossRef]
- Galvis, M.M.; Ranschez-Romero, C.; Oliveira-Bueno, T.; Teng, Y. Toward a new era for the management of circulating tumour cells. Adv Exp Med Biol 2021, 1286, 125–134. [Google Scholar]
- Papadaki, M.A.; Koutsopoulos, A.V.; Tsoufas, P.G.; Lagoudaki, E.; Aggouraki, D.; Monastirrioti, A.; Koutoulaki, C.; Apostolopoulou, C.A.; Merodoulaki, A.C.; Papadaki, C. Clinical revelance of immune checkpoints on circulating tumor cells in breast cancer. Cancers 2020, 12, 376. [Google Scholar] [CrossRef]
- Kim, J.W.; Lim, A.R.; You, J.Y.; Lee, L.H.; Song, S.E.; Lee, N.K.; Jung, S.P.; Cho, K.R.; Kim, C.Y.; Park, H. PIK3CA mutation is associated with a poor response to HER-2 targeted therapy in breast cancer patients. Cancer Res Treat 2023, 55, 531–541. [Google Scholar] [CrossRef]
- Nakai, M.; Yamada, T.; Sekiya, K.; Sato, A.; Hankyo, M.; Kuriyama, S.; Takahashi, G.; Kurita, T.; Yanagihara, K.; Yoshida, H.; et al. PIK3CA detected by liquid biopsy in patients with metastatic breast cancer. J Nippon Med Sch 2021, 89, 66–71. [Google Scholar] [CrossRef]
- Urso, L.; Vernaci, G.; Carlet, J.; Lo Mele, M.; Fassan, M.; Zulato, E.; Faggioni, G.; Menichetti, A.; Di Liso, E.; Griguolo, G. ESR1 gene mutation in hormone receptor positive HER2 negative metastatic breast cancer patients. Concordance between tumour tissue and circulating tumor DNA analysis. Front Oncol 2021, 11, 625636. [Google Scholar] [CrossRef]
- Ashworth, T.R. A case of cancer in which cells similar to those in tumors were seen in the blood after death. Australian Med 1869, 14, 146–147. [Google Scholar]
- Cristofanilli, M.; Hayes, D.F.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Rueben, J.M.; Doyle, G.V.; Matera, J.; Allard, W.J.; Miller, MC.; et al. Circulating tumor cells: a novel prognostic factor for newly diagnostic metastatic breast cancer. J Clin Oncol. 2005, 23, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R. Mavroudis D, Grisanti S, Generali D., Garcia.Saenz R., Stebbing J et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient dsts. Lancet Oncol 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Janni, W.; Friedl, T.W.P.; Yab, T.C.; Bidard, F.C.; Cristofanilli, M.; Hayes, D.F.; Ignatiadis, M.; Regan, M.M.; Alix-Panabieres, C.; Barlow, WE.; et al. Clinical validity of repeated circulating tumor cell enumeration as an early treatment monitoring tool for metastatic breast cancer in the PREDICT global pooled analysis. Clin Cancer Res 2025, 31, 2196–2029. [Google Scholar] [CrossRef]
- Dvir, K.; Gordiano, S.; Leone, J.P. Immunotherapy in breast cancer. Int J Mol Sci 2024, 25, 7517. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Pierga, J.Y.; Rueban, J.; Rademaker, A.; Davis, A.A.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R.; Mavroudia, D.; et al. The clinical use of circulating tumor cells (CTCs) enumeration for staging of metastatic breast cancer (MBC): international expert consensus paper. Crit Rev Oncol Hematol 2019, 134, 39–45. [Google Scholar] [CrossRef]
- Bidard, F.C.; Kiavue, N.; Jacot, W.; Bachelot, T.; Dureau, S.; Bourgeois, H. Gonclaves A, Brain E, Ladoire S, Dalenc F et al. Overall survival with circulating tumor cell count-driven choice of therapy in advanced breast cancers: a randomized trial. J Clin Oncol 2024, 42, 383–389. [Google Scholar] [CrossRef]
- Cabal, L.; Berger, F.; Cottu, P.; Loirat, D.; Rampanou, A.; Brain, E.; Cyrille, S.; Bourgeois, H.; Klavue, N.; DeLuche, E.; et al. Clinical utility of circulating tumour cell-based monitoring of late-line chemotherapy for metastatic breast cancer: the randomized CirCe01 trial. Br J Cancer 2021, 124, 1207–1213. [Google Scholar] [CrossRef]
- Fehm, T.; Mueller, V.; Banys-Paluchowski, M.; Fasching, P.A.; Fried, T.W.P.; Hartkopf, A.; Huober, J.; Loehberg CRack, B.; Riethdorf, A.; et al. Efficacy of lapatinib in patients with HER2-negative metastatic breast cancer and HER2-positive circulating tumor cells: the DETECT III clinical trial. Clin Chem 2024, 70, 307–318. [Google Scholar] [CrossRef]
- Grigoryeva, E.S.; Tashireva, L.A.; Alifanov, V.V.; Savelieva, O.E.; Vtorsuhin, S.V.; Zavyalova, M.V.; Bragina, O.D.; Garbukov, E.Y.; Cherdyntyseva, N.V.; Choinzonov, E.L.; Perelmuter, V.M. Molecular subtype conversion in CTCs as indicator of treatment adequacy associated with metastasis-free survival in breast cancer. Sci Rep 2022, 12, 20949. [Google Scholar] [CrossRef]
- Ho, H.Y.; Chung, K.S.; Kan, C.M.; Wong, S.C. Liquid biopsy in the clinical management of cancer. Int J Med Sci 2024, 25, 8594. [Google Scholar] [CrossRef]
- de Miranda, F.S.; Barauna, V.G.; dos Santos, L.; Costa, G.; Vassallo, P.F.; Campos, L.C.G. Properties and application of cell free DNA as a clinical biomarker. Int J Mol Sci 2021, 22, 9110. [Google Scholar] [CrossRef] [PubMed]
- Kolenda, T.; Guglas, K.; Baranowski, D.; Sobocinska, J.; Kopcznska, M.; Teresiak, A.; Blizniak, R.; Ramperska, K. cfRNAs as biomarkers in oncology-still experimental or applied tool for personalized medicine already? Rep Pract Oncol Radiother 2020, 25, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Mazzitelli, C.; Santini, D.; Corradini, A.G.; Zamagni, C.; Trere, D.; Montanaro, L.; Taffurelli, M. Liquid biopsy in the management of breast cancer patients: Where are we now and where are we going? Diagnostics 2023, 13, 1241. [Google Scholar] [CrossRef]
- Shegekar T, Vodithala S, Juganavar A, The emerging role of liquid biopsies in revolutionizing cancer diagnosis and therapy. Cureus 2023, 15, e43650.
- Nel, I.; Herzog, H.; Atkas, B. Combined analysis of disseminated tumor cells (DTCs) and circulating tumour DNA (ctDNA) in a patient suffering from triple negative breast cancer revealed elevated risk. Front Biosci 2022, 27, 208. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Nguyen, E.T.; Pagano, I.; Fukui, J.A. Genomic landscape of circulating tumor DNA in a diverse cohort of metastatic breast cancer patients. Oncol Res Treat 2023, 46, 26–32. [Google Scholar] [CrossRef]
- 109 Tang, Y.; Li, J.; Lu, B.; Ran, J.; Hu, Z.Y.; Ouyang, Q. Circulating tumor DNA profile and its clinical significance in patients with hormone receptor positive and HER2 negative MBC. Front Endocrinol (Lausanne) 2022, 13, 1075830. [Google Scholar] [CrossRef]
- Wimmer, K.; Sachet, M.; Ramos, C.; Frantal, S.; Birnleitner, H.; Brostjan, C.; Exner, R.; Filipits, M.; Bago-Horvath, Z.; Rudas, M.; et al. Differential immunomodulatory effects of epirubicin/cyclophosphamide and docetaxel in breast cancer patients. J Exp Clin Cancer Res 2023, 42, 300. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, X.; Wang, Z.; Wu, P.; Huang, J. Anthracyclines potentiate anti-tumor immunity: A new opportunity for chemoimmunotherapy. Cancer Lett 2015, 369, 331–335. [Google Scholar] [CrossRef]
- Wennerberg, E.; Sarhan, D.; Carlsten MKaminskyy, V.O.; DÁrcy, P.; Zhivotovsky, B.; Childs, R.; Lundqvist, A. Doxorubicin sensitizes human tumor cells to NK-cell and T-cell mediating killing by augmented TRAIL receptor signalling. Int J Cancer 2013, 133, 1643–1652. [Google Scholar] [CrossRef]
- Sawasdee, N.; Wattanapanitch, M.; Thongsin, M.; Phanthaphol, N.; Chiawpanit, C.; Thuwajit, C.; Yenchitsomanus, P.T.; Panya, A. Doxorubicin sensitizes breast cancer cells to natural killer cells in connection with increased Fas receptors. Int J Mol Med 2022, 49. [Google Scholar] [CrossRef]
- Shenasa, E.; He, Y.; Wang, Z.; Dongsheng, T.; Gao, D.; Kos, Z.; Thornton, S.; O´Nielsen, T.O. Digital profiling of immune biomarkers in breast cancer: Relation to anthracycline benefit. Mod Pathol 2025, 38, 100718. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, W.; Dong, B.; Xin, Z.; Ji, Y.; Su, R.; Shen, K.; Pan, J.; Wang, Q.; Wie, W. Docetaxol remodels prostate cancer immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. Theranostics 2022, 12, 4965–4979. [Google Scholar] [CrossRef] [PubMed]
- Wang P, Wang H, Huang Q, Peng C, Yao L, Chen H, Qiu Z, Wu Y, Wang L, Chen W, Exosomes from M1-polarized macrophages enhance paclitaxel antitumor activity by activating macrophages-mediated inflammation. Theranostics 2019, 9, 1714–1727. [CrossRef] [PubMed]
- Maldondo, M.T.; Quinn, M.; Plebanski, M. Low dose cyclophosphamide: Mechanisms of T cell modulation. Cancer Treat Rev 2016, 42, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.Q.; Zhang, Y.; Wang, D.; Liu, H.Y.; Shen, D.; Luo, Y. Baseline lymphopenia: A predictor of poor outcomes in HER2 positive metastatic breast cancer treated with trastuzumab. Drug Des Devel Ther. 2019, 13, 3727–3734. [Google Scholar] [CrossRef]
- Roghanian, A.; Hu, G.; Fraser, C.; Singh, M.; Foxall, R.B.; Meyer, M.J.; Lees, E.; Huet, H.; Glennie, M.J.; Beers, R.B.; et al. Cyclophosphamide enhances cancer antibody immunotherapy in the resistant bone marrow niche by modulating macrophage FcR expression. Cancer Immunol Res 2019, 7, 1876–1890. [Google Scholar] [CrossRef]
- Tang, F.; Zhong, Q.; Yang, Z.; Li, H.; Pan, C.; Huang, L.; Ni, T.; Deng, R.; Wang, Z.; Tan, S.; et al. Low-dose cyclophosphamide combined with IL-2 inhibits tumor growth by decreasing regulatory TY cells and increasing CD8 + T cells and natural killer cells in mice. Immunobiol 2022, 227, 152212. [Google Scholar] [CrossRef]
- Zhong, H.; Lai, Y.; Zhang, R.; Daoud, A.; Feng, Q.; Zhou, J.; Shang, J. Low dose cyclophosphamide modulates tumor microenvironment by TGF-ß signalling pathway. Int J Mol Sci 2020, 21, 957. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, S.; Lv, S.; Liu, X.; Li, W.; Song, Y.; Rong, D.; Zheng, P.; Huang, H.; Zheng, H. Combined oral low-dose cyclophosphamide endocrine therapy may improve clinical response among patients with metastatic breast cancer via Tregs in TLSs. Sci Rep 2024, 14, 13432. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; O´Donovan, N.; McGowan, P.M.; O´Sullivan, F.; Duffy, M.J.; Crown, J. Trastuzumab induces antibody-dependent-cell-mediated cytotoxicity (ADCC) in HER2-non-amplified breast cancer cell lines. Ann Oncol 2012, 23, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Lower, E.E.; Glass, E.; Blau, R.; Harman, S. HER-2/neu expression in primary and metastatic breast cancer. Breast cancer Res Treat 2009, 113, 301–306. [Google Scholar] [CrossRef]
- Cai, M.; Ming, L.; Lv, H.; Zhou, S.; Xu, X.; Shui, R.; Yang, W. HER2-low breast cancer: evolution of HER2 expression from primary tumour to distant metastasis. BMC Cancer 2023, 23, 656. [Google Scholar] [CrossRef]
- Geukens, T.; de Schepper, M.; Richard, F.; Maetens, M.; Van Baelen, K.; Mahdami, A.; Nguyen, H.L.; Isnaldi, E.; Leduc, S.; Pabba, A.; et al. Intra-patient and inter-metastatic heterogeneity of HER-2 low status in metastatic breast cancer. Eur J Cancer 2023, 188, 152–160. [Google Scholar] [CrossRef]
- Lin, M.; Luo, T.; Jin, Y.; Zhong, X.; Zheng, D.; Zeng, C.; Guo, Q.; Wu, J.; Shao, Z.M.; Hu, X.; et al. Her-2 low heterogeneity between primary and paired recurrent/metastatic breast cancer: Implications in treatment and prognosis. Cancer 2024, 130, 851–862. [Google Scholar] [CrossRef]
- Krawczyk, N.; Banys, M.; Neubauer, H.; Solomayer, E.F.; Gall, C.; Hahn, M.; Becker, S.; Bachmann, R.; Wallwiener, D.; Fehm, T. HER2 status on persistent disseminated tumor cells after adjuvant therapy may differ from initial status on primary tumor. Anticancer Res 2009, 29, 4019–4024. [Google Scholar]
- Volmer, L.L.; Dannehl, D.; Matovina, S.; Taran, F.A.; Walter, C.B.; Wallwiener, M.; Brucker, S.Y.; Hartkopf, A.D.; Engler, T. Comparing the HER2 status of the primary tumor to that of disseminated tumor cells in early breast cancer. Int J Mol Sci 2024, 25, 5910. [Google Scholar] [CrossRef]
- Rack, B.; Juckstock, J.; Gunther-Biller, M.; Andergassen, U.; Neugebauer, J.; Hepp, P.; Schoberth, A.; Mayr, D.; Zwingers, T.; Schindlbeck, C.; et al. Trastuzumab clears HER2/neu-positive isolated tumor cells from bone marrow in primary breast cancer. Arch Gynecol Obstet 2012, 285, 485–492. [Google Scholar] [CrossRef]
- Barok, M.; Balazas, M.; Nagy, P.; Rokosy, Z.; Treszl, A.; Toth, E.; Juhasz, I.; Park, J.W.; Isola, J.; Vereb, G.; et al. Trastuzumab decreases the number of circulating tumor cells despite trastuzumab resistance of the primary tumor. Cancer Lett 2008, 260, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; O´Donovan, N.; McGowan, P.M.; O´Sullivan, F.; Duffy, M.J.; Crown, J. Trastuzumab induces antibody-dependent-cell-mediated cytotoxicity (ADCC) in HER2-non-amplified breast cancer cell lines. Ann Oncol 2012, 23, 1788–1795. [Google Scholar] [CrossRef]
- Munzone, E.; Nole, F.; Goldhirsch, A.; Botteri, E.; Espirito, A.; Zorzino, L.; Curiliano, G.; Minchella, I.; Adamoli, L.; Cassatella, M.C.; et al. Changes in HER2 status in circulating tumor cells compared with the primary tumor during treatment for advanced breast cancer. Clin Breast Cancer 2010, 10, 392–397. [Google Scholar] [CrossRef]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R.; et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef]
- Li, Z.; Lai, X.; Fu, S.; Ren, L.; Cai, H.; Zhang, H.; Gu, Z.; Ma, X.; Luo, K. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy effcieincy. Adv Sci (Weinh) 2022, 9, e2201734. [Google Scholar] [CrossRef]
- Meng, X.; Song, S.; Jiang, Z.; Sun, B.; Wang, T.; Zhang, S.; Wu, S. Receptor conversion in metastatic breast cancer: a prognosticator of survival. Oncotarget 2016, 7, 71887–17903. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Kwilas, A.R.; Ardiani, A.; Gameiro, S.R. Attacking malignant cells that survive therapy: exploiting immunogenic modulation. Oncoimmunology 2013, 2, e26937. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, B.; Padget, M.R.; Schlom, J.; Hodge, J.W. Exploiting off-target effects of estrogen deprivation to sensitize estrogen negative breast cancer to immune killing. J Immunother Cancer 2021, 9, e002258. [Google Scholar] [CrossRef]
- Huhn, D.; Marti-Rodrigo, P.; Mouron, S.; Hansel, C.; Tschapalda, K.; Porebski, B.; Haggblad, M.; Lidemalm, L.; Quintela-Fandino, M.; Carreras-Puigvert, J.; et al. Prolonged estrogen deprivation triggers a broad immunosuppressive phenotype in breast cancer cells. Mol Oncol 2022, 16, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Liao, J.; Tan, J.T.; Hu, H. Comparison of minimal access and open breast surgery: a propensity score-matched study on postoperative immune function in breast cancer. W J Surg Oncol 2024, 22, 183. [Google Scholar] [CrossRef]
- Michaels, E.; Chen, N.; Nanda, R. The role of immunotherapy in triple negative breast cancer (TNBC). Clin Breast cancer 2024, 24, 263–270. [Google Scholar] [CrossRef]
- Mathiesen, R.R.; Borgen, E.; Renolen, A.; Lokkevik, E.; Nesland, J.M.; Anker, G.; Ostenstad, B.; Lundgren, S.; Risberg, T.; Mjaaland, I.; et al. Persistence of disseminated tumor cells of neoadjuvent treatment for locally advanced breast cancer predicts poor survival. Breast Cancer Res 2012, 14, R117. [Google Scholar] [CrossRef]
- Hall, C.; Krishnamurthy, S.; Lodhi, A.; Bhattachaeryva, A.; Anderson, A.; Kuerer, H.; Bedrosian, I.; Singh, B.; Lucci, A. Disseminated tumor cells predict survival after neoadjuvant therapy in primary breast cancer. Cancer 2012, 118, 342–348. [Google Scholar] [CrossRef]
- Ven Becker Solomayer, E.; Becker-Pergola, G.; Wallwiener, D.; Fehm, T. Primary systemic therapy does not eradicate disseminated tumor cells in breast cancer patients. Breast Cancer Res Treat 2007, 106, 239–243. [Google Scholar]
- Braun, S.; Kentenich, C.; Janni, W.; Hepp, F.; de Waal, J.; Willgeroth, F.; Sommer, H.; Pantel, K. Lack of effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high risk breast cancer patients. J Clin Oncol 2000, 18, 80–86. [Google Scholar] [CrossRef]
- Fehm, T.; Krawczyk, N.; Solomayer, E.F.; Becker-Pergola, G.; Durr-Storzer, S.; Neubauer, H.; Seeger, H.; Staebler, A.; Wallwiener, D. ER alpha status of disseminated tumour cells in bone marrow of primary breast cancer patients. Breast Cancer Res 2008, 10, R76. [Google Scholar] [CrossRef]
- Lower, E.E.; Glass, E.; Blau, R.; Harman, S. HER-2/neu expression in primary and metastatic breast cancer. Breast cancer Res Treat 2009, 113, 301–306. [Google Scholar] [CrossRef]
- Cai, M.; Ming, L.; Lv, H.; Zhou, S.; Xu, X.; Shui, R.; Yang, W. HER2-low breast cancer: evolution of HER2 expression from primary tumour to distant metastasis. BMC Cancer 2023, 23, 656. [Google Scholar] [CrossRef]
- Geukens, T.; de Schepper, M.; Richard, F.; Maetens, M.; Van Baelen, K.; Mahdami, A.; Nguyen, H.L.; Isnaldi, E.; Leduc, S.; Pabba, A.; et al. Intra-patient and inter-metastatic heterogeneity of HER-2 low status in metastatic breast cancer. Eur J Cancer 2023, 188, 152–160. [Google Scholar] [CrossRef]
- Swain, S.M.; Baselga, I.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab and docetaxel in HER2 positive metastatic breast cancer. N Eng J Med 2015, 372, 724–734. [Google Scholar] [CrossRef]
- Aroino, G.; de la Haba-Rodriguez, J.; Ferrero, J.M.; De Placido, S.; Osborne, C.K.; Klingbiel, D.; Revelant, V.; Wohlfarth, C.; Poppe, R.; Rimawi, M.; et al. Pertuzumab, trastuzumab and an aromatase inhibitor for HER2 positive and hormone receptor positive metastatic/locally advanced breast cancer: PERTAIN final analysis. Clin Cancer Res 2023, CCR-22-1092. [Google Scholar]
- Rugo, H.; Im, S.A.; Cardoso, F.; Cortes, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Bachelot, T.; Wright, G.S.; Saura, C.; et al. Margetuximab versus trastuzumab in patients with previously treated HER2 positive advanced breast cancer (SOPHIA): final overall survival results from a randomized phase 3 trial. J Clin Oncol 2023, 41, 198–205. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Cecchini, R.S.; Geyer Jr, C.E.; Rastogi, P.; Costantino, J.P.; Atkins, J.N.; Crown, J.P.; Polikoff, J.; Boileau, J.F.; Provencer, L.; et al. NSABP B-47/NRG oncology phase III randomized trial comparing adjuvant chemotherapy with or without trastuzumab in high-risk invasive breast cancer negative for HER2 by FISH and with IHC 1+ or 2. J Clin Oncol 2020, 38, 444–453. [Google Scholar] [CrossRef]
- de Nonneville, A.; Houvenaeghel, G.; Cohen, M.; Sabiani, L.; Bannier, M.; Viret, F.; Goncalves, A.; Bertucci, F. Pathological complete response rate and disease-free survival after neoadjuvant chemotherapy in patients with HER2-low and HER2-0 breast cancers. Eur J Cancer 2022, 176, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, X.; Wang, Y.; Zhang, X.; Ye, Y.; Xu, S.; Zhou, L.; Lin, Y.; Lu, J.; Yin, W. Stable or at least once HER2-low status during neoadjuvant chemotherapy confers survival benefit in patients with breast cancer. Ann Med 2024, 56, 2409343. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, L.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2 positive advanced breast cancer. N Eng J Med 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.S.; Sledge, G.; Atkan, G.; Ellis, C.; Florance, A.; Vukelija, S.; Bischoff, J.; et al. Randomized study of lapatinib alone or in combination with trastuzumab in women with ErbB2 positive, trastuzimab refractory metastatic breast cancer. J Clin Cancer 2010, 28, 1124–1130. [Google Scholar]
- Johnson, S.; Pippen Jr, J.; Pivot, X.; Lichinitser, M.; Sadeghi, S.; Dieras, V.; Gomez, H.L.; Romieu, G.; Manikhas, A.; Kennedy, MJ.; et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor positive metastatic breast cancer. J Clin Oncol 2009, 27, 5538–5546. [Google Scholar] [CrossRef]
- Johnston, S.; Hegg, R.; Im, S.A.; Park, I.H.; Burdaeva, O.; Kurteva, G.; Press, M.F.; Tjulandin, S.; Iwata, H.; Simon, SD.; et al. Phase III randomized study of dual human epidermal growth factor receptor 2 (HER2) blockage with lapatinib plus trastuzumab in combination with an aromatase inhibitor in postmenopausal women with HER2 positive hormone receptor positive metastatic breast cancer: updated results of alternative treatment options. J Clin Oncol 2021, 39, 79–89. [Google Scholar]
- Stanowicka-Grada, M.; Senkus, E. Anti-HER2 drugs for the treatment of advanced HER2 positive breast cancer. Curr Treat Options Oncol 2023, 24, 1633–1650. [Google Scholar] [CrossRef]
- Li, L.; Zhang, D.; Liu, B.; Lv, D.; Zhai, J.; Guan, X.; Ji, Z.; Ma, F. Antibody-drug conjugates in HER2 positive breast cancer. Chin Med J 2021, 135, 261–262. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Stein, R.; Sharkey, R.M. The emergence of trophoblast cell surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget 2018, 9, 28989–29006. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau MBaselga, J.; Pegream, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab-emtansine for HER2 positive advanced breast cancer. N Eng J Med 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Huang, C.S. Mano MS, Loibl S, Mamounas EP, Untch M, Wolmark N, Rastogi P, Schneeweiss A, Redondo A et al. Trastuzumab emansine for residual invasive HER2 positive breast cancer. N Eng J Med 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.; Hill, E.; Al Degaither, D.; Weiner, L. Mechanisms of action of therapeutic antibodies for cancer. Mol Immunol 2015, 67, 28–45. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor-2 antibody-drug conjugate, in tumors with human epidermal factor receptor 2 heterogeneity. Cancer Sci 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in previously treated HER2 positive breast cancer. N Eng J Med 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Cortes, J.; Kim, S.B.; Chung, W.P.; Im, S.A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.M.; Petry, V.; Chung, C.F.; et al. Trastuzumab Deruxtecan versus trastuzumab emtansine for breast cancer. N Eng J Med 2022, 386, 1143–1154. [Google Scholar] [CrossRef]
- FDA FDA approves Fam-trastuzumab-Deruxtecan-Nxki for HER2 low breast cancer. FDA: Silver Spring, MD, USA 2022.
- Bardia, A.; Mayer, L.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond JRO´Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Goviectan-hziy in refractory metastatic triple negative breast cancer. N Eng J Med 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Bardia, A.; Marme, F.; Cortes, J.; Schmid, P.; Loirat, D.; Tredan, O.; Ciruelos, E.M.; Dalenc, F.; Gomez- Pardo, PG.; et al. Overall survival with Sacituzumab govitecan in hormone receptor positive and human epidermal growth factor negative metastatic breast cancer. (TROPiCS-02): A randomized open-label multicentre phase3 trail. Lancet 2023, 402, 1423–1433. [Google Scholar]
- Rugo, H.S.; Bardia, A.; Marme, F.; Cortes, J.; Schmid, P.; Loirat, D.; Tredan, O.; Ciruelos, E.; Delenc FPardo, PG.; et al. Overall survival (OS) with sacituzunab govitecan in hormone receptor positive and human epidermal growth factor receptor-2 negative metastatic breast cancer (TROPiCS-02) A randomized open-label multicentre phase 3 trial. Lancet 2023, 402, 1423–1433. [Google Scholar] [CrossRef]
- Trerotola; Cantanelli, P.; Guera, E.; Tripaldi, R.; Aloisi, A.L.; Bonasera, V.; Lattanzio, R.; de Lange, R.; Weidle, U.H.; Piantelli, M.; et al. Upregulation of Trop-2 quantitativly stimulates human cancer growth. Oncogene 2013, 32, 222–233. [Google Scholar] [CrossRef]
- Ambrogi, F.; Fornilli, M.; Boracchi, P.; Trerotola, M.; Relli, V.; Simone, P.; La Sorda, R.; Lattanzio, R.; Querzoli, P.; Pedriali, M.; et al. Trop-2 is a determinant of breast cancer survival. PLos One 2014, 9, e110606. [Google Scholar] [CrossRef] [PubMed]
- Vidula N, Yan C, Rugo H, Trophoblast cell surface antigen gene (TACSTD2) expression in primary breast cancer. Breast Cancer ResTreat 2022, 194, 569–571. [CrossRef]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop2 is a novel target for solid cancer therapy with Sacituzumab-govitcan (IMMU-132) an antibody drug conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef]
- Okajima, D.; Yasuda, S.; Maejima, T.; Karibe, T.; Sakurai, K.; Aida, T.; Toki, T.; Yamaguchi, J.; Kitamura, M.; Kamai, R.; et al. Datopotamab deruxtecan a novel TROP2 directed antibody drug conjugate, demonstrates potential antitumor activity by efficient drug delivery to tumor cells. Mol Cancer Therapeut 2021, 20, 2329–2340. [Google Scholar] [CrossRef]
- Krop, I.E.; Masuda, N.; Mukohara, T.; Takahashi, S.; Nakayama, T.; Inoue, K.; Iwata, H.; Toyoma, T.; Yamamoto, Y.; Hansra, DM.; et al. Results from the phase I/II study of patritumab deruxtecan, a HER3-directed antibody drug conjugate (ADC) in patients with HER3 expressing metastatic breast cancer (MBC). J Clin Oncol 2022, 40, 1002. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Dosunmu, O.; Sjastry, M.; Finney, L.; Sellami, D.B.; Sternberg, D.W.; Wright-Browne, V.; Toppmeyer, D.; Gwin, W.R.; Thaddeus, JT.; et al. A phase 2 study of HER3-DXd in patients (pts) with metastatic breast cancer (MBC). J Clin Oncol 2023, 41, 1004. [Google Scholar] [CrossRef]
- Plitas, G.Y.; Rudensky, A.Y. Regulatory T cells in cancer. Ann Rev Cancer Biol 2020, 4, 459–477. [Google Scholar] [CrossRef]
- Zhang, H.; Mi, J.; Xin, Q.; Cao, W.; Song, C.; Zhang, N.; Yuan, C. Recent research and clinical progress of CTLA-4 based immunotherapy for breast cancer. Front Oncol 2023, 13, 1256360. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shi, Y.; Kim, B.C.; Jo, M.H.; Cruz, L.O.; Gou, Z.; Ha, T.; Lu, L.F.; Reich, D.H.; Chen, Y. Force dependent trans-endocytosis by breast cancer cells depletes costimulatory CD80 and attenuates T-cell activation. Biosensors bioelectronics 2020, 165, 112389. [Google Scholar] [CrossRef]
- Jama, M.; Tabana, Y.; Barakat, K.H. Targeting cytotoxic lymphocyte antigen 4 (CTLA-4) in breast cancer. Eur J Med Res 2024, 29, 353. [Google Scholar] [CrossRef]
- Krishnamuethy N, Nishizaki D, Lippman SM, Miyahita H, Nesline MK, Pabla S, Conroy JM, DePietro P, Kato S, Kuurock. High CLTA-4 transcription expression correlates with high expression of other checkpoints and immunotherapy outcome. Ther Adv Med Oncol 2024, 16, 1758835923122050.
- Paucek, R.D.; Baltimore, D.; Li, G. Cancer immunotherapy using checkpoint inhibition. Science 2018, 359, 1350–1355. [Google Scholar]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O´Bryne, K.; Kulasinghe, A. Immune checkpoint inhibitors in cancer therapy. Curr Oncol 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Rad, H.S.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O´Bryne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med Res Rev 2021, 41, 1474–1498. [Google Scholar]
- O´Meara, T.A.; Tolaney, S.M. Tumor mutational burden as a predictor of immunotherapy response in breast cancer. Oncotarget 2021, 12, 394–400. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. VPT-2021: KEYNOTE-522. Phase III study of neoadjuvant pembrolizumab + chemotherapy vs placebo + chemotherapy followed by adjuvant pembrolizumab vs placebo in early stage TNBC. Annal Oncol 2021, 32, 1198–1200. [Google Scholar] [CrossRef]
- Nanda, R.; Liu, M.C.; Yau, C.; Asare, S.; Hylton, N.; Leer, L.V.; Perlmutter, J.; Wallace, A.M.; Chien, A.J.; Forero-Torreds, A.; et al. Pembrolizumab plus standard neoadjuvant therapy for high-risk breast cancer (BC): http://ascopubs.org/doi/abs/10.1200/JCO.20127.35.15_suppl.506 (accessed 23 June 2024).
- Andresen, N.K.; Rossevold, A.H.; Borgen, E.; Schirmer, C.B.; Gilje, B.; Garred, O.; Lomo, J.; Stensland, M.; Nordgard, O.; Falk, RS.; et al. Circulating tumour cells in metastatic breast cancer patients treated with immune checkpoint inhibitors- a biomarker analysis of the ALICE and ICON trials. Mol Oncol 2024, 8. [Google Scholar]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Van Bianchi, G.; Esteva, F.J. Martin M et al. Updated overall survival (OS) results from the phase III MONALESSA-3 trial of postmenopausal patients with HR+/HER2- advanced breast cancer (ABC) treated with fulvestrant (FUL) +/- ribociclib (RIB). J Clin Oncol suppl 2021, 39, 1001. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; et al. Overall survival with ribociclib plus letrozole in advanced breast cancer. N Eng J Med 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Lu, Y.S.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Cardoso, F.; Harbeck, N.; Hurvitz, S.; Chow, L.; Sohn, J.; et al. Updated overall survival of ribociclib plus endocrine therapy versus endocrine therapy alone in pre- and perimenopausal patients with HR+/HER” – advanced breast cancer in MONALESSA-7. A phase III trial. J Am Assoc Cancer Res 2022, 28, 851–859. [Google Scholar]
- Cai, Z.; Wang, J.; Li, Y.; Shi, Q.; Jin, L.; Li, S.; Zhu, M.; Wang, Q.; Wong, L.L.; Wang, W.; et al. Overexpression of cyclin D1 and CDK proteins are responsible for the resistance to CDK 4/6 inhibitor in breast cancer that can be reversed by PI3K7mTOR inhibitors. Sci China Life Sci 2023, 66, 94–109. [Google Scholar] [CrossRef]
- Kappel, C.; Elliott, M.J.; Kumar, V.; Nadler, M.B.; Desnoyers, A.; Amir, E. Comparative overall survival of CDK 4/6 inhibitors in combination with endocrine therapy in advanced breast cancer. SCI rep 2024, 14, 3129. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Lacko, A.; Sohn, J.; Cruz, F.; Borrego, M.R.; Manikhas, A.; Park, Y.H.; Stroyakovskiy, D.; Yardley, D.A.; Huang, C.S.; et al. A phase III trial of adjuvant ribociclib plus endocrine therapy versus endocrine therapy alone in patients with HR-positive/HER2 negative early breast cancer: final invasive disease-free survival results from the NATALEE trial. Ann Oncol 2025, 36, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Pallerla, S.; Abdul, A.R.M.; Comeau, J.; Jolis, S. Cancer vaccines, treatment of the future: with emphasis on HER2-positive breast cancer. Int J Med Sci 2021, 22, 779. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.T.; Schlom, J.; Hodge, J.W. Combination of docetaxel and recombinant vaccine enhances T-cell responses and antitumor activity: effects of docetaxel on immune enhancement. Clin Cancer Res 2008, 14, 3536–3544. [Google Scholar] [CrossRef]
- Corti, C.; Giachetti, P.P.M.B.; Eggermont, A.M.M.; Delaloge, S.; Curigliano, G. Therapeutic vaccines for breast cancer: Has the time finally come? Eur J Cancer 2022, 160, 150–174. [Google Scholar] [CrossRef]
- Davodabdia, F.; Sarhadi, M.; Arabour, J.; Sargazi, S.; Rahdar, A.; Diez-Pascual, A.M. Breast cancer vaccines: New insights into immunomodulatory and bano-therapeitic approaches. J Control Release 2022, 349, 844–875. [Google Scholar] [CrossRef]
- Zhoa, Y.; Song, D.; Wang, Z.; Huang, Q.; Huang, F.; Ye, Z.; Wich, D.; Chen, M.; Khirallah, J.; Goa, S.; et al. Antitumour vaccination vias targeted proteolysis of antigens isolated from tumour lysates. Nat Biomed Eng 2025, 9, 234–248. [Google Scholar] [CrossRef]
- Volmer, L.; Koch, A.; Matovina, S.; Dannehl, D.; Weiss, M.; Welker, G.; Hahn, M.; Engler, T.; Wallweiner, M.; Walter, CB.; et al. Neoadjuvant chemotherapy of patients with early breast cancer is associated with increased detection of disseminated tumor cells in the bone marrow. Cancers (Basel) 2022, 14, 635. [Google Scholar] [CrossRef]
- D´Alerio, S.; Scala, S.; Sozzi, G.; Roz, L.; Bertolini, G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin Cancer Biol 2020, 60, 351–361. [Google Scholar]
- Perkins, D.W.; Steiner, I.; Halder, S.; Robertson, D.; Buus, R.; O´Leary, L.; Isacke, C.M. Therapy induced normal tissue damage promotes breast cancer metastasis. iScience 2023, 27, 108503. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-induced metastasis, mechanisms and translational opportunities. Clin Exp Metastasis 2018, 35, 269–284. [Google Scholar] [CrossRef]
- Garcia, A.J.; Rediti, M.; Venet, D.; Majjaj, S.; Kammler, R.; Munzone, E.; Gianni, L.; Thurlmann, B.; Laang, I.; Colleoni, M.; et al. Differential benefit of metronomic chemotherapy among triple negative breast cancer subtypes treated in the IBCSG trial 22-00. Clin Cancer Res 2023, 29, 4908–4919. [Google Scholar] [CrossRef]
- Alkan, F.K.; Caglayan, A.B.; Alkan, H.K.; Benson, E.; Gunduz, Y.E.; Sensoy, O.; Durdagi, S.; Zarbaliyev, E.; Dyson, G.; Assad, H.; et al. Dual activity of minnelide chemosensitize basal/triple negative breast cancer stem cells and reprograms immunosuppressive tumor microenvironment. Sci Rep 2024, 28, 22487. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.; Tan, J.; Huang, J.; Zhang, X.; Guo, Y.; Huang, Y.; Yang, J. The dynamic change of neutrophil to lymphocyte ratio is predictive of pathological complete response after neoadjuvant chemotherapy in breast cancer patients. Breast Cancer 2020, 27, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Alshamsan, B.; Elshenawy, M.A.; Aseafan, M.; Fahmy, N.; Badran, A.; Elhassan, T.; Alsayed, A.; Suleman, K.; Al-Tweigeri, T. Prognostic significance of the neutrophil to lymphocyte ratio in locally advanced breast cancer. Oncel Lett 2024, 28, 429. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Li, H.; Liu, X.; Song, G.; Jiang, H.; Yan, Y.; Zhang, R.; Ran, R.; Zhang, J.; Liu, Y.; et al. The prognostic value of neutrophil to lymphocyte ratio in de novo stage IV breast cancer: a retrospective cohort study. Ann Transl Med 2023, 31, 45. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Miller, G.T.; Jesson, M.I.; Watanabe, T.; Jones, B.; Wallner, B.P. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumour effects and augments anti-body mediated cytotoxicity via a novel immune mechanism. Cancer Res 2004, 64, 5471–5480. [Google Scholar] [CrossRef]
- Narra, K.; Mullins, S.R.; Lee, H.O.; Strzemkowski-Brun, B.; Magalong, K.; Christiansen, V.J.; McKee, P.A.; Egleston, B.; Cohen, S.J.; Weiner, LM.; et al. Phase II trial of single agent Val.boroPro (Talabostat) inhibiting fibroblast activation patients in patients with metastatic colorectal cancer. Cancer Biol Ther 2007, 6, 1691–1699. [Google Scholar] [CrossRef]
- Hofheinz, R.D.; al-Batran, S.E.; Hartmann, F.; Hartung, G.; Jager, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal antigen targeting by a humanised monoclonal antibody: An early phase II of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Huang, S.; Fang, R.; Xu, J.; Qui, S.; Zhang, H.; Du, J.; Cai, S. Evaluation of the tumor targeting of a FAP alpha based doxorubicin prodrug. J Drug Target 2011, 19, 487–496. [Google Scholar] [CrossRef]
- Brennen, W.N.; Isaacs, J.T.; Denmeade, S.R. Rational behind targeting fibroblast activation protein-expressing carcinoma associated fibroblasts as a novel chemotherapeutic strategy. Mol Cancer Ther 2012, 11, 257–266. [Google Scholar] [CrossRef]
- Fang, J.; Xiao, L.; Joo, K.I.; Liu, Y.; Zhang, C.; Lui, S.; Conti, P.S.; Li, Z.; Wang, P.A. A potent immunotoxin targeting fibroblast activating protein for treatment of breast cancer in mice. Int J Cancer 2016, 138, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, Z.; Sun, J.; Song, Q.; He, B.; Zhang, H.; Wang, X.; Dai, W.; Zhang, Q. A tenascin C targeted nanoliposome with navitoclax for specifically eradicating of cancer associated fibroblasts. Nanomedicine 2016, 12, 131–141. [Google Scholar] [CrossRef]
- Truffi, M.; Mazzucchelli, S.; Bonizzi, A.; Sorentino, L.; Allevi, R.; Vanna, R.; Morasso, C.; Corsi, F. Nano-strategies to target breast cancer fibroblasts: Rearranging the tumor microenvironment to achieve antitumor efficacy. Int J Mol Sci 2019, 20, 1263. [Google Scholar] [CrossRef]
- Lee, J.; Fassnacht, M.; Nair, S.; Boczkowski, D.; Gilboa, E. Tumour immunotherapy targeting fibroblast activating protein, a product expressed in tumor-associated fibroblasts. Cancer Res 2005, 65, 11156–11163. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.D.; Duffy, M.R. Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, Campo L, Bryant RJ, Verril C, Lambert A et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res 2018, 78, 6852–6865. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- Rastegar-Pouyani, N.; Abdolvahab, M.H.; Farzin, M.A.; Zare, H.; Kesharwani, P.; Sahebkar, A. Targeting cancer-associated fibroblasts with pirfenidone: A novel approach for cancer therapy. Tissue cancer 2024, 91, 102624. [Google Scholar] [CrossRef]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-associated fibroblasts: Tumorigenicity and targeting for cancer therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef]
- Sun, R.; Luo, H.; Su, J.; Di, S.; Zhou, M.; Shi, B.; Sun, Y.; Du, G.; Zhang, H.; Jiang, H.; Li, J. Olaparib supresses MDSC recruitment via SDF1alpha/CXCR4 axis to improve the anti-tumor efficacy of CAR-T cells on breast cancer in mice. Mol Ther 2021, 29, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Johnson, K.C.C.; Gatti-Mays, M.E.; Li, Z. Emerging strategies in targeting tumor resident myeloid cells for cancer immunotherapy. J Haematol Oncol 2022, 15, 118. [Google Scholar] [CrossRef]
- Beatson, R.; Graham, R.; Grundland Freile, F.; Kannambath, S.; Pfiefer EWoodman, N.; Owen, J.; Nuamah, R.; Mandel, U.; Pinder, S.; et al. Cancer-associated hypersialylated MUC1 drives the differentiation of human monocytes into macrophages with a pathogenic phenotype. Commun Biol 2020, 3, 644. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur J Pharmacol 2020, 877, 173090. [Google Scholar] [CrossRef]
- Goswami, K.K.; Ghosh, T.; Ghosh, S.; Sarkar, M.; Bose, A.; Baral, R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol 2017, 316. [Google Scholar] [CrossRef]
- Giannone G, Ghisoni E, Genta S, Scotto G, Tuninetti V, Turinetto M, Valabrega. Immuno-metabolism and microenvironment in cancer. Int J Mol Sci 2020, 21, 4414.
- Vaupel, P.; Multhoff, G. Revisting the Warberg effect: historical dogma versus current understanding. J Physiol 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumour-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Bohn T, Rapp S, Luther N, Bruehl TJ, Kojima N, Lopez PA, Hahlbrock J, Muth S, Endo S, Pektor S et al Tumor immunoevasion via acidosis- dependent induction of regulatory tumor-associated macrophages. Nat Immunol 2018, 19, 1319–1329. [CrossRef]
- Jiang, H.; Wei, H.; Wang, H.; Wang, Z.; Li, J.; Ou, Y.; et al. Zeb1 induced metabolic reprogramming of glycolysis is essential for macrophage polarization in breast cancer. Cell Death Dis 2022, 13, 206. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino MManberba, M.; Fiume, I.; Roberti, M.; Din Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor induces death of human breast cancer cells with different glycolytic attitude by affecting distinct signalling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.W.; Kim, E.Y.; Han, C.W.; Park, S.Y.; Jeong, M.S.; Yoon, D.; Choi, H.J.; Jin, L.; Park, M.J.; Kwon, YJ.; et al. Macchilin A inhibits tumor growth and macrophage M2 polarization through the reduction of lactic acid. Cancers (Basel) 2019, 11. [Google Scholar]
- Jin H, He Y, Zhao P, Hu Y, Tao J, Chen J, Huang Y Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β/FAK pathway and tumor-associated macrophage repolarization using legumain activable delivery. Theranostics 2019, 9, 265–2378. [CrossRef] [PubMed]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Chloroquine modulates antitumor response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun 2018, 9, 873. [Google Scholar] [CrossRef]
- Liang, Y.; He, J.; Chen, X.; Lin, L.; Yuan, Q.; Zeng Zu, X.; Shen, Y. The emerging role of metabolism in the crosstalk between breast cancer cells and tumor-associated macrophages. Int J Biol Sci 2023, 9, 4915–4930. [Google Scholar] [CrossRef]
- Tharp, K.M.; Kersten, K.; Maller, O.; Timblin, G.A.; Stashko, C.; Canale, F.P.; Menjivar, R.E.; Hayward, M.K.; Berestjuk, I.; Ten Hoeve, J.; et al. Tumor associated macrophages restrict CD8+ T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat Cancer 2024, 5, 1045–1062. [Google Scholar] [CrossRef]
- Wang, L.; Guo, W.; Guo, Z.; Yu, J.; Tan, J.; Simons, D.L.; Hu, K.; Liu, X.; Zhou, Q.; Zheng, Y.; et al. PD-L1 expressing tumor-associated macrophages are immunostimulatory and associate with good clinical outcome in human breast cancer. Cell Rep Med 2024, 5, 101420. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumors. Nat Rev Immunol 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Hao, Z.; Li, R.; Wang, Y.; Li, S.; Hong, Z.; Han, Z. Landscape of myeloid derived suppressor cell in tumor immunotherapy. Biomark Res 2021, 9, 77. [Google Scholar] [CrossRef]
- Jin, S.; Yang, Z.; Hao, X.; Tang, W.; Ma, W.; Zong, H. Roles of HMGB1 in regulating myeloid-derived suppressor cells in the tumor microenvironment. Biomark Res 2020, 8, 21. [Google Scholar] [CrossRef]
- Nefedova, Y.; Nagaraj, S.; Rosenbauer, A.; Muro-Cacho, C.; Sebti, S.M.; Gabrilovich, D.I. Regulation of dendritic cell differentiation and antitumor response in cancer by pharmacological-selective inhibition of the janus-activate kinase/signal transducers and activators of transcription 3 pathway. Cancer Res 2005, 65, 9525–9535. [Google Scholar] [CrossRef]
- Apolloni, E.; Bronte, V.; Mazzoni, A.; Serfinin, P.; Cabrelle, A.; Segal, D.; Ypung, H.A.; Zanovello, P. Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated lymphocytes. J Immunol 2000, 165, 6723–6730. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, T.; Zhu, W.; Xie, S.; Zhao, Z.; Feng, B.; Guo, H.; Yang, R. Targeting MDSC for immune check-point blockade in cancer immunotherapy: current progress and new prospects. Clin Med Insights Oncol 2017, 15, 11795549211035540. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Sanseviero, E.; Timosenko, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells: a propitious road to clinic. Cancer Discov 2021, 11, 2693–2706. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, T.; Liu, J.; Tai, J.; Wang, B.; Chen, Z.; Quan, Z. Pan-cancer analysis reveals tumor-associated macrophage communication in the tumor microenvironment. Exp Hematol Oncol 2021, 10, 31. [Google Scholar] [CrossRef]
- Qui, Y.; Chen, T.; Hu, R.; Zhu, R.; Li, C.; Ruan, Y.; Xie, X.; Li, Y. Next frontier in tumor immunotherapy: macrophage mediated immune evasion. Biomark Res 2021, 9, 72. [Google Scholar]
- Tumino, N.; Weber, G.; Besi, F.; Del Bufalo, F.; Bertaina, V.; Paci, P.; Quatrini, L.; Antonucci, L.; Sinibaldi, M.; Quintarelli, C.; et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CART-cells in patients with neuroblastoma. J Hematol Oncol 2021, 14, 191. [Google Scholar] [CrossRef]
- Rivera Vargs, T.; Apetoh, L. Can immunogenic chemotherapies relieve cancer cell resistance to immune check point inhibitors. Front Immunol 2019, 10, 1181. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody conjugate: the “biological missile” for targeted cancer therapy. Signal Target Ther 2022, 7, 93. [Google Scholar] [CrossRef]
- Fultang, J.; Panetti, S.; Ng, M.; Collins, P.; Graef, S.; Rizkalla, N.; Booth, S.; Lenton, R.; Noyvert, B.; Shannon-Lowe, C.; et al. MDSC targeting with gemtuzumab ozogamicin restores T-cell immunity and immunotherapy against cancers. EBioMed 2019, 47, 235–246. [Google Scholar] [CrossRef]
- Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich. All-trans retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res 2003, 63, 4441–4449.
- Bauer, R.; Udonta, F.; Wroblewski, M.; Ben-Batalla, I.; Santos, I.M.; Taverna, T.; Kuhlencord, M.; Gensch, V.; Pasler, S.; Vinckier, S.; et al. Blockage of myeloid-derived suppressor cells expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res 2018, 78, 320–3230. [Google Scholar] [CrossRef]
- Michels, T.; Shirin, G.V.; Naiditch, H.; Sevko, A.; Umansky, V.; Shurin, M.R. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol 2012, 9, 292–300. [Google Scholar] [CrossRef]
- Serafini, P.; Menellsckel, K.; Kelso, M.; Noonan, K.; Caslifano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the effect of PD-1 inhibition in murine models of lung and renal carcinoma. Clin Cancer Res 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, NA.; et al. Entinostat converts immume-resistant breast and pancreatic cancers into checkpoint- responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res 2018, 6, 1561–1577. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Kohanbash, G.; Fellows-Mayle, W.; Hamilton, R.L.; Komohara, Y.; Decker, S.A.; Ohlfest, J.R.; Okada, H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res 2011, 71, 2664–2674. [Google Scholar] [CrossRef]
- Proia, T.A.; Singh, M.; Woessner, R.; Carnealli, L.; Bommakanti, G.; Magiera, L.; Srinivasan, S.; Grosskurth, S.; Collins, M.; Womack, C.; et al. STAT3 antisense oligonucleotide remodels the suppressive tumor microenvironment to enhance immune activation in combination with anti PD-L1. Clin Cancer Res 2020, 26, 6335–6349. [Google Scholar] [CrossRef]
- Ko, J.S.; Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 2009, 15, 2148–2157. [CrossRef] [PubMed]
- Zhang, X.; Liang, Q.; Cao, Y.; Yang, T.; An, M.; Liu, Z.; Yang, J.; Liu, Y. Dual depletion of myeloid derived suppressor ells and tumor cells with self-assembled gemcitabine-celecoxib nano-twin drug for cancer chemoimmunotherapy. J Nanotech 2024, 23, 319. [Google Scholar]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An overview of PARP inhibitors for the treatment of breast cancer. Target Oncol 2021, 16, 255–282. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with germline BRCA mutation. N Eng J Med 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yeruhalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Eng J Med 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Tung, N.; Garber, J.E. PARP inhibition in breast cancer: progress made and future hopes. NPJ Breast Cancer 2022, 8, 47. [Google Scholar] [CrossRef]
- De, K.; Jana, M.; Chowdhury, B.; Calaf, G.M.; Roy, D. Role of PARP inhibitors: A new hope for breast cancer therapy. Int J Mol Sci 2025, 26, 2773. [Google Scholar] [CrossRef]
- Tutt, A.N.J.; Garger, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmana, J.; et al. Adjuvant olaparib for patients with BRCA1 or BRCA 2 mutated breast cancer. N Eng J Med 2021, 384, 2394–2405. [Google Scholar] [CrossRef]
- Ma, Z.; Sun, X.; Zhao, Z.; Lu, W.; Guo, Q.; Wang, S.; You, J.; Zhang, Y.; Liu, L. Risk of pneumonitis in cancer patients treated with PARP inhibitors: a meta-analysis randomized controlled trials and a pharmacovigilance study of the FAERS database. Gyncol Oncol 2021, 162, 496–505. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derkson, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, J.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome wide and high density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun 2018, 9, 1849. [Google Scholar] [CrossRef]
- Wang, N.; Yang, Y.; Jin, D.; Zhang, Z.; Shen, K.; Yang, J.; Chen, H.; Zhao, X.; Yang, L.; Lu, H. PARP inhibitor resistance in breast and gynaecological cancer: resistance mechanisms and combination therapy. Front Pharmacol 2022, 13, 967633. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.R.; Hurst, K.E.; Taveira, M.O.; Johnson, NT.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat Cancer 2021, 2, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR-T therapy for solid tumors. Tumor associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- George, P.; Dasyam, N.; Giunti, G.; Mester, B.; Bauer, E.; Andrews, B.; Perera, T.; Ostapowicz, T.; Frampton, C.; Li, P. Third generation anti CD19 chimeric antigen receptor T-cells incorporating a TRL2 domain for relapsed or refractory B-cell lymphoma: A phase I clinical trial protocol (ENABLE). BMJ Open 2020, 10, e034629. [Google Scholar] [CrossRef] [PubMed]
- Roselli, E.; Boucher, J.C.; Li, G.; Kotani, H.; Spitler, K.; Reid, K.; Cervantes, E.V.; Bulliard, Y.; Tu, N.; Lee, S.B. 4-1BB optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J. Immunother. Cancer 2021, 9, e003354. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vys, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G. In vivo fate and activity of second versus third generation CD19 specific CAR-T cells in B cell non-Hodgkins lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Erber, R.; Kailayangiri, S.; Hueber, H.; Ruebner, M.; Hartmann, A.; Haberle, L.; Meyer, J.; Volkl, S.; Mackensen, A.; Landgraf, L.; et al. Variable expression of the diganglioside GD2 in breast cancer molecular subtypes. Cancers (Basel) 2021, 13, 5577. [Google Scholar] [CrossRef]
- Seitz, C.M.; Scroeder, S.; Knopf, P.; Krahl, A.S.; Hau, J.; Schleicher, S.; Martella, M. Quintanilla-Martinez L, Kneilling M, Pichler B et al. GD2 targeted chimeric antigen receptor T-cells prevent metastasis formation by elimination of breast cancer stem-like cells. Oncoimmunology 2020, 9, 1683345. [Google Scholar] [CrossRef]
- Bajor, M.; Graczyk-Jarznka, A.; Marhelava, K.; Burdzinska, A.; Muchowicz, A.; Gorel, A.; Zhylko, A.; Soroczynska, K.; Retecki, K.; Krawczyk M.; et al. J Immunother Cancer 2022, 10, e002500.
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat Commun 2019, 10, 4355. [Google Scholar] [CrossRef]
- Li, P.; Yang, L.; Li, T.; Bin, S.; Sun, B.; Huang, Y.; Yang, K.; Shan, D.; Gu, H.; Li, H. The third generation anti-HER2 chimeric antigen receptor mouse T-cells alone or together with anti-PD1 antibody inhibits the growth of mouse breast tumor cells expressing HER-2 in vitro and in immune competent mice. Front Oncol 2020, 10, 1143. [Google Scholar] [CrossRef]
- Duro-Sanchez, S.; Alonso, M.R.; Arribas, J. Immunotherapies against HER2 positive breast cancer. Cancer (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Niu, Z.; Wu, J.; Zhoa, Q.; Zhang, J.; Zhang, P.; Yang, Y. CAR-based immunotherapy for breast cancer: peculiarities, ongoing investigations and future strategies. Front Imunol 2024, 15, 1385571. [Google Scholar] [CrossRef] [PubMed]
- Rivas, E.I.; Linares, J.; Zwick, M.; Gomez-Llonin, A.; Guiu, M.; Labernadie, A.; Badia-Ramentol, J.; Llado, A.; Bardia, L.; Perez-Nunez, I.; et al. Targeted immunotherapy against distinct cancer-associated-fibroblasts overcomes treatment resistance in refractory HER2+ breast tumors. Nat Commun 2022, 13, 5310. [Google Scholar] [CrossRef]
- Das, S.; Valton, J.; Duchateau, P.; Poirot, L. Stromal depletion by TALEN-editied universal hipoimmunogenic FAP-CAR-T immunotherapy. Front Immunol 2023, 14, 1172681. [Google Scholar] [CrossRef]
- Nalawade, S.A.; Shafer, P.; Bajgain, P.; McKenna, M.K.; Ali, A.; Kelly, L.; Joubert, J.; Gottschalk, S.; Watanabe, N.; Leen, A.; et al. Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer. J Immunother Cancer 2021, 9, e003237. [Google Scholar] [CrossRef]
- Sun, R.; Luo, H.; Su, J.; Di, S.; Sun, R.; Chen, M.; Jiang, H.; et al. Olaparib suppresses MDSC recruitment via SDF1alpha/CXCR4 to improve the anti-tumor effect of CAR-T cells on breast cancer in mice. Mol Ther 2021, 29, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Di, S.; Zhu, M.; Pan, Z.; Sun, R.; Chen, M.; Jiang, H.; Shi, B. Luo H, Li Z. Adjuvant of poly LC improves antitumor effects of CAR-T cells. Front Oncol 2019, 9, 241. [Google Scholar] [CrossRef]
- Meng, Z.; Zhang, R.; Wu, X.; Zhang, M.; Jin, T. PD-L1 mediates triple-negative breast cancer evolution via the regulation of TAM/M2 polarization. Int J Oncol 2022, 61. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Porter, D. Cytokine release syndrome with chimeric antigen receptor T-cell therapy. Biol Blood Marrow Transplant 2018, 12, 756. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Z.; Ren, Z.; Li, Y. Reactions related to CAR-T cell therapy. Front Immunol 2021, 12, 663201. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse effect following the administration of T-cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: overcoming the challenges of T-cell exhaustion. EBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef]
- Zhong, H.; Lai, Y.; Zhang, R.; Daoud, A.; Feng, Q.; Zhou, J.; Shang, J. Low dose cyclophosphamide modulates tumor microenvironment by TGF-ß signalling pathway. Int J Mol Sci 2020, 21, 957. [Google Scholar] [CrossRef]
- Jallouk, A.P.; Paravastu, S.; Weilbaecher, K.; Aft, R.L. Long-term outcome of (neo)adjuvant zoledronic acid therapy in locally advanced breast cancer. Breast Cancer Res Treat 2021, 187, 135–144. [Google Scholar] [CrossRef]
- Liu, M.; Qian, S.; Wu, J.; Xiao, J.; Zeng, X. The effects of neoadjuvant zoledronic acid in breast cancer patients: A meta-analysis of randomized controlled trials. Asian J Surg 2023, 46, 4124–4130. [Google Scholar] [CrossRef]
- Powles, T.; Paterson, A.; McCloskey, E.; Schein, P.; Scheffler, B.; Tidy, A.; Ashley, S.; Smith, I.; Ottestad, L.; Kanis, J. Reduction in bone relapse and improved survival with oral clondronate for adjuvant treatment of operable breast cancer (ISRCTN83688026). Breast Cancer Res 2006, 8, R13. [Google Scholar] [CrossRef] [PubMed]
- Diel, I.J.; Solomayer, E.F.; Costa, S.D.; Gollan, C.; Goerner, R.; Wallweiner, D.; Kaufmann, M.; Bastert, G. Reduction in new metastases in breast cancer adjuvant clodronate treatment. N Eng J Med 1998, 339, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.G.; Lucas, P.C.; Anderson, S.J.; Mamounas, E.P.; Brufsky, A.; Baez-Diaz, L.; King, K.M.; Lad, T.; Robidoux, A.; Finigan, M.; et al. MAF amplification and adjuvant clodronate outcomes in early-stage breast cancer in NSABP B-34 and potential impact on clinical practice. JNCI Cancer Spectr 2021, 28, 5. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Somerfeld, M.R.; Accordine, M.K.; Blanchette, P.S.; Clemons, M.J.; Dhesy-Thind, S.; Dillmon, M.S.; D´Oronzo, S.; Fletcher, G.G.; Frank, ES.; et al. Use of adjuvant bisphosphonates and other bone-modifying agents in breast cancer: ASCO-OH (CCO) guideline update. J Clin Oncol 2022, 40, 787–800. [Google Scholar] [CrossRef]
- Senaratne, S.G.; Pirianov, G.; Mansi, J.L.; Arnett, T.R.; Colston, K.W. Bisphosphonates induce apoptosis in human breast cancer cell lines. Br J Cancer 2000, 82, 1459–1468. [Google Scholar] [CrossRef]
- Borutaite, V. Mitochondria as decision-makers in cell death. Environ Mol Mutagen 2010, 51, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Formigue, O.; Lagneaux, L.; Body, J.J. Bisphosphonates induce breast cancer cell death in vitro. J Bone Miner Res 2000, 15, 2211–2221. [Google Scholar] [CrossRef]
- Boissier, S.; Ferreras, O.; Peyuhaud, O.; Magnetto, S.; Ebetino, F.H.; Colombel, M.; Delmas, P.; Delaisse, J.M.; Clezardin, P. Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastasis. Cancer Res 2000, 60, 2949–2954. [Google Scholar] [PubMed]
- Denoyelle, C.; Hong, L.; Vannier, J.P.; Soria, J.; Soria, C. New insights into the action of bisphosphonate zoledronic acid in breast cancer cells by dual Rho-dependent and independent effects. Br J Cancer 2003, 88, 1631–1640. [Google Scholar] [CrossRef]
- Kimachi, K.; Kajiya, H.; Nakayama, S.; Ikebe, T.; Okabe, K. Zoledronic acid inhibits RANK expression and migration of osteoclast precursors during osteoclastogenesis. Naunyn-Schmiedeberg´s Arch of Pharmacology 2011, 383, 297–308. [Google Scholar] [CrossRef]
- Weber, M.; Homm, A.; Muller, S.; Frey, S.; Amann, K.; Ries, J.; Geppert, C.; Preidl, R.; Most, T.; Kammerer, PW.; et al. Zoledronate causes a systemic shift if macrophage polarization towards M1 in vivo. Int J Mol Sci 2021, 22, 1323. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J Transl Med 2011, 9, 177. [Google Scholar] [CrossRef]
- Zang, X.; Zhang, X.; Hu, H.; Qiao, M.; Zhao, X.; Deng, Y.; Chen, D. Targeted delivery of zoledronate to tumor-associated macrophages for cancer immunotherapy. Mol Pharm 2019, 16, 2249–2258. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, B.; Shen, Y.; Ren, J.; Chen, M.; Jiang, Y.; Wu, H.; Dai, W.; Zhang, H.; Wang, X.; et al. Bisphosphonate-mineralized nano-IFN gamma suppresses residual tumor growth caused by incomplete radiofrequency ablation through metabolically remodelling tumor-associated macrophages. Theranostics 2025, 15, 1057–1076. [Google Scholar] [CrossRef]
- Rahimian, L.; Khandani, B.K.; Nemati, M.; Hoseini-Shahrestanak, S.; Aminizadeh, N.; Jafarzedeh, A. Reduced expression of natural killer cell related activating receptors by peripheral blood mononuclear cells from patients with breast cancer and their improvement by zoledronic acid. Asian Pac J Cancer Prev. 2022, 23, 1661–1669. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.H.; Chen, S.C.; Chen, C.Y.; Lin, T.M. Zoledronic acid blocks the interaction between breast cancer cells and regulatory T-cells. BMC Cancer 2019, 19, 176. [Google Scholar] [CrossRef]
- Wang, S.; Huang, M.; Chen, M.; Sun, Z.; Jiao, Y.; Ye, G.; Pan, J.; Ye, W.; Zhao, J.; Zhang, D. Zoledronic acid and thymosin alpha1 elicit antumor immunity against prostate cancer by enhancing tumor inflammation and cytotoxic T cells. J Immunother Cancer 2023, 11, e006381. [Google Scholar] [CrossRef]
- Aschariyasakulchai, P.; Sakunrangsit, N.; Chokyakorn, S.; Suksanong, C.; Ketchart, W. Anticancer effect of zoledronic acid in endocrine-resistant breast cancer cells via HER-2 signalling. Biomed Pharmacother. 2024, 171, 116142. [Google Scholar]
- Li, X.Y.; Lin, Y.C.; Huang, W.L.; Hong, Q.C.; Chen, J.Y.; You, Y.; Li, W.B. Zoledronic acid inhibits proliferation and impairs migration and invasion through down regulation of VEGF and MMP expression in human nasopharyngeal carcinoma cells. Med Oncol 2012, 29, 714–720. [Google Scholar] [CrossRef]
- Dedes, P.G.; Gialelli, C.; Tsonis, A.I.; Kanakis, I.; Theocharis, A.D.; Kletsas, D.; Tzanakakis, G.N.; Karamanos, N.K. Expression of matrix macromolecules and functional properties of breast cancer cells are modulated by the bisphosphonate zoledronic acid. Biochem Biophys Acta 2012, 20, 1926.1939. [Google Scholar] [CrossRef] [PubMed]
- Fane, M.; Weeraratna, A.T. How the aging microenvironment influences tumour progression. Nat Rev Cancer 2019, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Aunan, J.R.; Cho, W.C.; Soreide, K. The biology of aging and cancer: a brief overview of shared and divergent molecular hallmarks. Aging Dis 2017, 8, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Panwar, P.; Lamour, G.; MacKenzie, N.C.W.; Yang, H.; Ko, F.; Li, H.; Bromme, D. Changes in structural-mechanical properties and degradability of collagen during aging-associated modifications. J Biol Chem 2015, 290, 23291–23306. [Google Scholar] [CrossRef]
- Liu, H.; Wang Chen, S.C.; Chen, C.Y.; Lo, J.L.; Lin, T.M. Immune modulation of CD4+ CD25+ regulatory T cells by zoledronic acid. BMC Immunol 2016, 17, 45. [Google Scholar] [CrossRef]
- Sarhan, D.; Leijonhufvud, C.; Murray, S.; Witt k Seitz, C.; Wallerius, M.; Xie, H.; Ullen, A.; Harmenberg, U.; Lidbrink, E.; et al. Zoledronic acid inhibits NFAT and IL-2 signalling pathways in regulatory T-cells and diminishes their suppressive function in patients with metastatic cancer. Oncoimmunology 2017, 6, e1338238. [Google Scholar] [CrossRef]
- Rouhrazi, H.; Turgan, N.; Oktem, G. Zoledronic acid overcomes chemoresistance by sensitizing cancer stem cells to apoptosis. Biotech Histochem 2018, 93, 77–88. [Google Scholar] [CrossRef]
- Jackson, C.; Freeman, A.L.J.; Szlamka, Z.; Spiegelhalter, D.J. The adverse effects of bisphosphonates in breast cancer: A systemic review and network meta-analysis. PLoS One 2021, 16, e246441. [Google Scholar] [CrossRef]
- Petitprez, F.; Meylan, M.; de Reynies, A.; Sautes-Fridman, C.; Fridman, W.H. The tumor microenvironment in the response to immune check point blockage therapies. Front Oncol 2020, 11, 784. [Google Scholar]
- Alharbi, M.; Roy, A.M.; Krishnan, J.; Kalinski, P.; Yao, S.; Gandhi, S. Targeting the tumor microenvironment to improve clinical outcomes in triple negative breast cancer patients and bridge the current disparity gap. Front Immunol 2024, 15, 1428118. [Google Scholar] [CrossRef]
- Ribeiro, R.; Carvalho, M.J.; Goncalves, J.; Moreira, J.N. Immunotherapy in triple-negative breast ancer: insights into tumor immune landscape and therapeutic opportunities. Front Med Biosci 2022, 9, 903065. [Google Scholar]
- Song, F.; Tarantino, P.; Garrido-Castro, A.; Lynce, F.; Tolaney, S.M.; Schlam, I. Immunotherapy for early-stage triple negative breast cancer: Is earlier better? Cur Oncol Rep 2024, 26, 21–33. [Google Scholar] [CrossRef]
- Dixon-Duglas, J.; Loi, S. Immunotherapy in early-stage-triple-negative breast cancer: Where are we now and where are we headed? Curr Treat Optyions Oncol 2023, 24, 1004–1020. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Miura, K.; Tamori, S.; Akimoto, K. Identification of a gene expression signature to predict the risk of early recurrence and the degree of immune cell infiltration in triple-negative breast cancer. Camcer Genomics Proteomics 2024, 21, 316–326. [Google Scholar] [CrossRef]
- Yan, Z.; Zhong, Z.; Shi, C.; Feng, M.; Feng, X.; Liu, T. The prognostic marker KRT81 is involved in suppressing CD8+ T cells and predicts immunotherapy response for triple-negative breast cancer. Cancer Biol Ther 2024, 25, 2355705. [Google Scholar] [CrossRef]
- Zhou, L.; Yu, C.W. Epigenetic modulations in triple-negative breast cancer: therapeutic implications for tumor microenvironment. Pharmacol Res 2024, 204, 107205. [Google Scholar] [CrossRef]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.R.; Hurst, K.E.; Taveira, M.O.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat Cancer 2021, 2, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Xu, Z.; Yao, K.; Zheng, B.; Zhang, Y.; Wang, X.; Zhang, T.; Lu, X.; Hu, H.; You, B.; et al. Osteoclast cancer cell metabolic cross-talk confers PARP inhibitor resistance in bone metastatic breast cancer. Cancer Res 2024, 84, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.P. Treatment of minimal residual disease in breast cancer: a longitudinal case study. Cureus 2017, 9, e1521. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Circulating tumour cell expressing mammaglobin (blue) [23].
Figure 1.
Circulating tumour cell expressing mammaglobin (blue) [23].
Figure 2.
Bivalent anti-HER-2 linked to CD3 binding to the breast cancer causing its elimination.
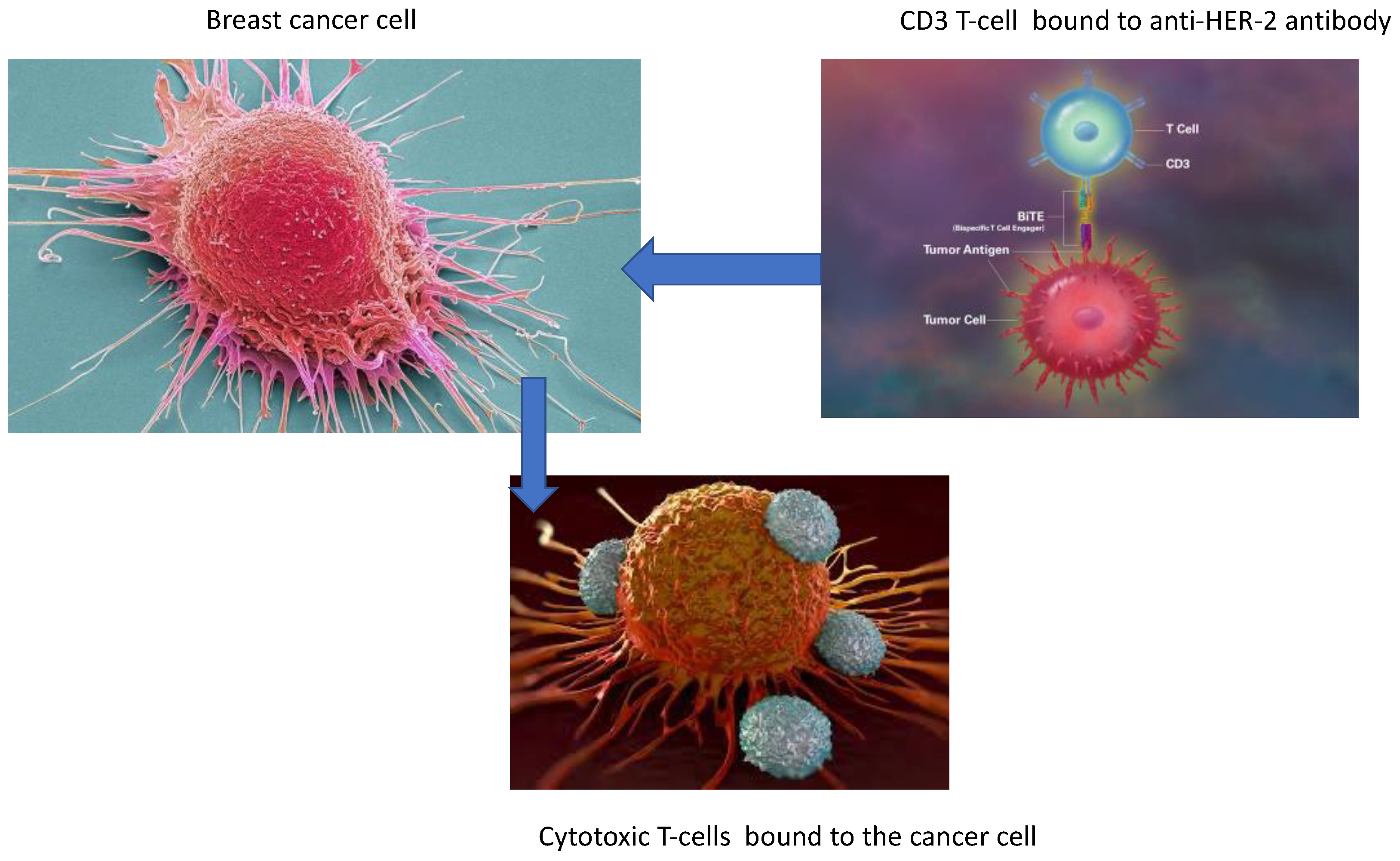
Figure 3.
The processes of cancer induced immunosuppression.
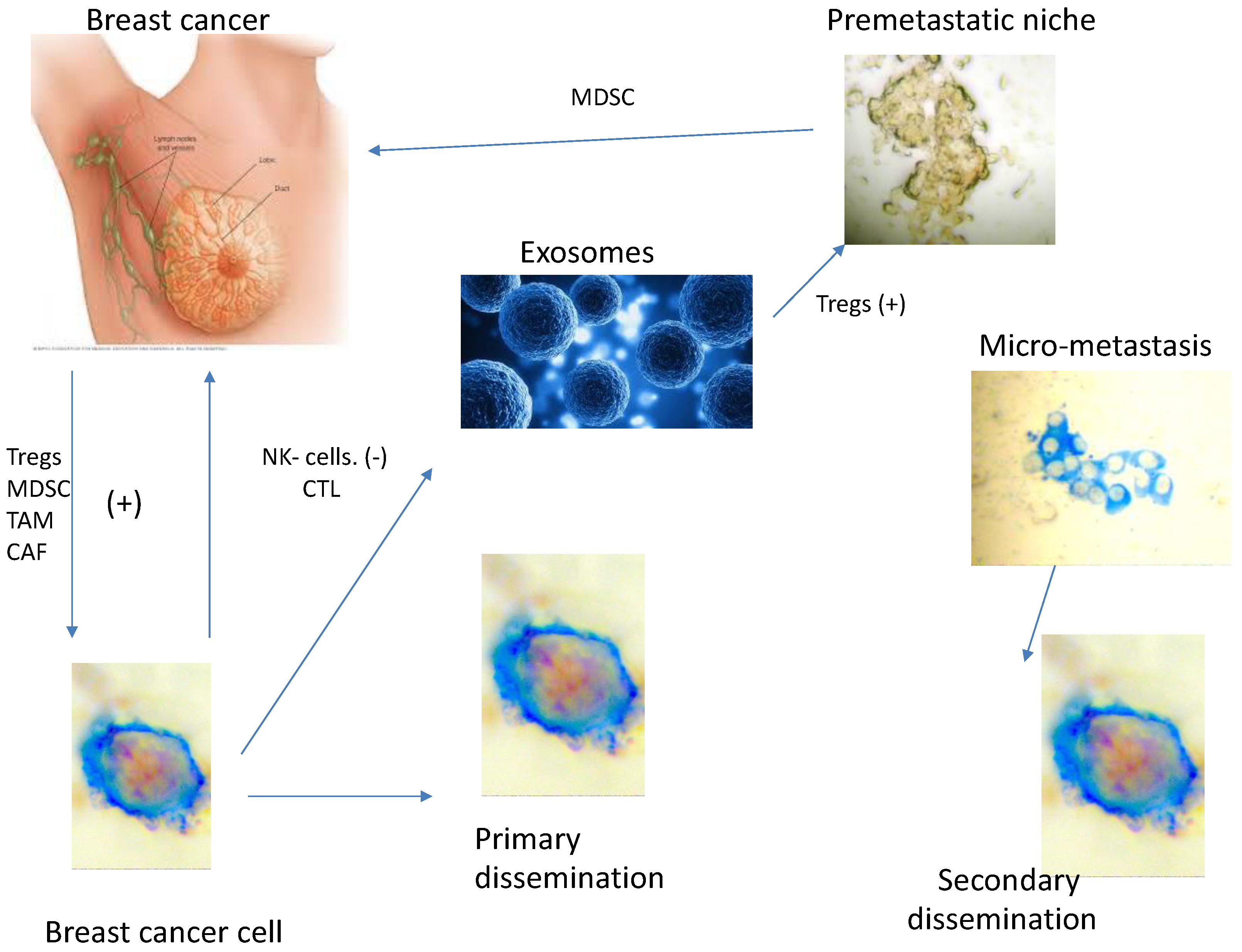
Figure 4.
Bone marrow micro-metastasis positive for mammoglobulin but negative for MMP-2 [337].
Figure 4.
Bone marrow micro-metastasis positive for mammoglobulin but negative for MMP-2 [337].
Figure 5.
Kaplan-Meier survival curves for patents with stage III colon cancer after adjuvant chemotherapy with FOLFOX. [72].
Figure 5.
Kaplan-Meier survival curves for patents with stage III colon cancer after adjuvant chemotherapy with FOLFOX. [72].
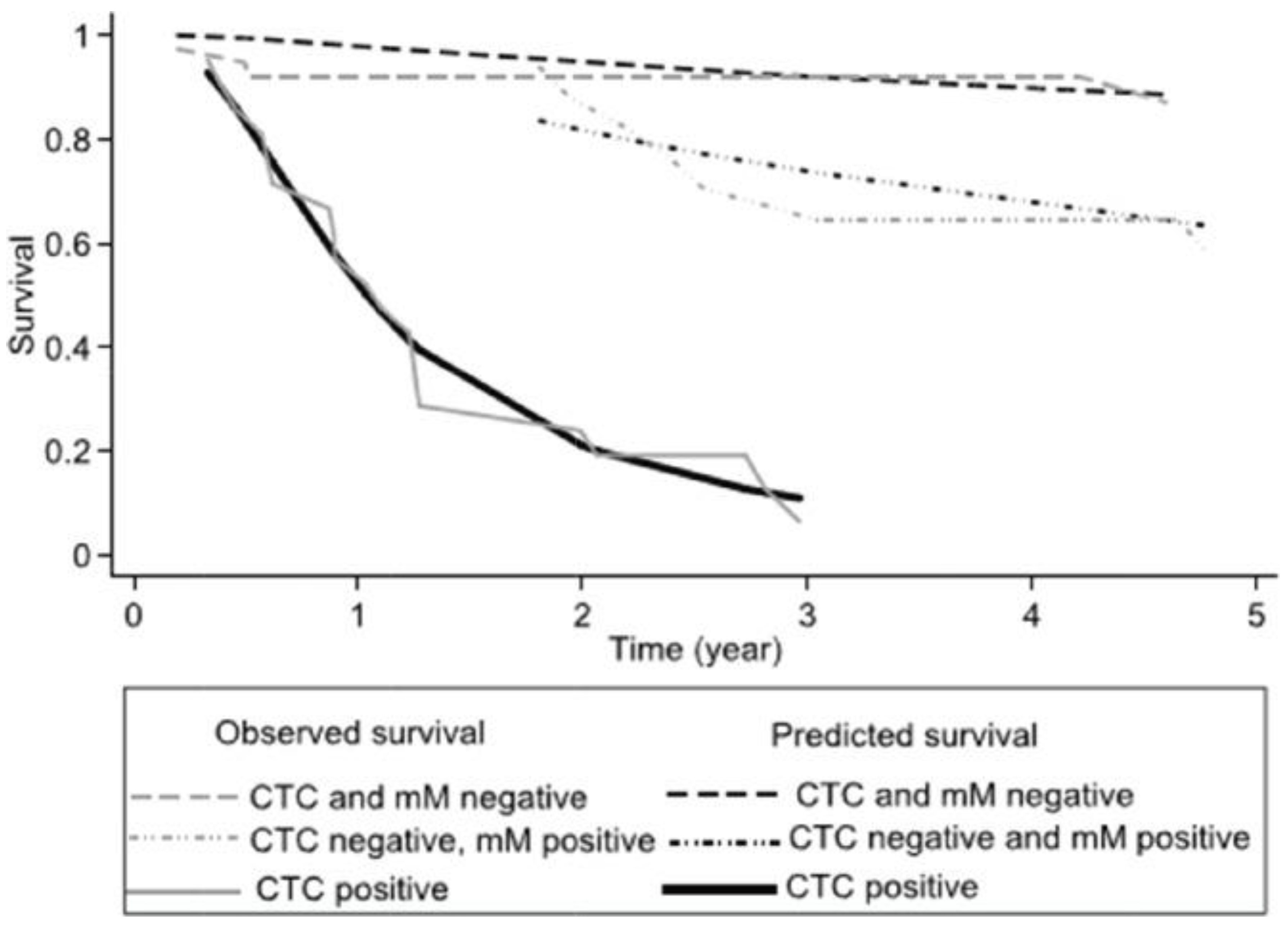
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



