
Submitted:
19 August 2025
Posted:
19 August 2025
You are already at the latest version
Abstract
Retinal neuronal dysfunction and degeneration emerge early in the pathogenesis of diabetic retinopathy (DR), preceding visible microvascular abnormalities. These neuronal alterations driven by chronic hyperglycemia initiate a cascade of mitochondrial failure, neuroinflammation, and synaptic disruption, which ultimately contribute to vascular breakdown and vision loss. A growing body of evidence identifies specific biomarkers that reflect these early neurodegenerative processes. Structural changes such as ganglion cell complex thinning and disorganization of the retinal inner layers (DRIL), functional impairments in flicker-induced vasodilation and oscillatory potentials, and molecular signals including caspase-3, Glial Fibrillary Acidic Protein (GFAP), and neurofilament light chain are quantifiable indicators of retinal neuronal injury. Emerging candidates such as miRNAs, retinal extracellular vesicle content, and semaphorin 3A provide additional insights into axonal transport defects and protein misfolding. Advances in artificial intelligence particularly in Optical Coherence Tomography (OCT), Optical Coherence Tomography Angiography (OCTA) image analysis, and pupillometry analysis have shown promise in enhancing the sensitivity and precision of early biomarker detection, enabling more objective risk prediction and monitoring. This review synthesizes current findings on neurodegeneration-related biomarkers in DR, emphasizing their potential for early diagnosis, risk stratification, and therapeutic monitoring. Broader clinical application will require non-invasive detection platforms, standardized protocols, and biomarker-driven trials aimed at shifting DR management toward proactive neuroprotection.
Keywords:
diabetic retinopathy
; neurodegeneration
; biomarkers of diabetic retinopathy
1. Introduction
Diabetic retinopathy (DR) is regarded as one of the leading cause of vision impairment and blindness worldwide, affecting millions of individuals with diabetes mellitus. As a progressive complication of diabetes, DR is characterized by damage to the retinal microvasculature and neural retina, ultimately leading to vision loss if untreated [1]. The global prevalence of DR is substantial, with estimates suggesting that approximately 35% of individuals with diabetes develop some form of retinopathy, and 7% progress to proliferative DR, a severe vision-threatening stage [2]. The socioeconomic burden of DR is significant, encompassing healthcare costs, reduced quality of life, and productivity losses, particularly in low and middle-income countries where access to screening and treatment is limited. Early detection of DR is important to prevent irreversible retinal damage, yet current diagnostic methods, such as fundus photography and optical coherence tomography (OCT), often detect changes only after significant vascular or structural damage has occurred. This delay presents a need for reliable, non-invasive biomarkers capable of identifying DR in its pre-symptomatic stages [3,4].
Recent research shows neurodegeneration to be an early event in diabetic retinopathy (DR), often preceding visible vascular changes and offering a critical window for early detection. This process involves progressive loss of retinal neurons, such as ganglion cells and photoreceptors, alongside glial dysfunction, driven by chronic hyperglycemia and metabolic stress, which disrupts retinal homeostasis5,6. Biomarkers, including molecular markers (e.g., glial fibrillary acidic protein [GFAP], neurofilament light chain [NfL]), imaging-based indicators (e.g., retinal layer thinning via optical coherence tomography [OCT]), and emerging candidates (e.g., microRNAs, proteomic profiles), hold promise for detecting DR before clinical symptoms manifest, enabling timely neuroprotective interventions [7,8].
Despite these insights, challenges persist in identifying and validating biomarkers for pre-symptomatic DR. Current diagnostic tools lack the sensitivity to detect early changes, and most studies focus on advanced DR stages. Heterogeneous detection methods and the absence of standardized validation protocols hinder clinical translation.
This review evaluates biomarkers of retinal neurodegeneration which may serve early indicators of DR. By synthesizing data on molecular, imaging-based, and novel biomarkers, it aims to categorize reported biomarkers, assess their diagnostic accuracy and clinical relevance, and highlight emerging candidates. The findings seek to inform early diagnostic strategies, shape clinical guidelines, and guide future research, ultimately reducing the global burden of diabetes-related vision loss.
2. Materials and Methods
This integrative review synthesizes findings from peer-reviewed journals, clinical reports, and studies and research articles on AI-assisted diagnostics in diabetic retinopathy (DR). Relevant literature was sourced from databases including PubMed, ScienceDirect, Scopus, Frontiers and Google Scholar using keywords such as “diabetic retinopathy,” “neurodegeneration,” “retinal biomarkers,” “artificial intelligence,” “microRNAs,” and “oxidative stress.” Inclusion criteria focused on studies investigating early neuronal changes in diabetic retinopathy such as inflammation or synaptic dysfunction preceding overt vascular pathology. Priority was given to research with focus on clinical, molecular insights, and AI-based diagnostic approaches.
3. Pathophysiology of Diabetic Retinopathy
This Diabetic retinopathy (DR) is a progressive and multifactorial complication of diabetes mellitus that affects the retina, which is the highly specialized, light-sensitive neural tissue lining the posterior segment of the eye [10]. Traditionally recognized as a microvascular disease, DR is now widely regarded as a complex neurovascular disorder, wherein both vascular dysfunction and neurodegeneration contribute to disease onset and progression. It remains one of the foremost causes of visual impairment and blindness among working-age adults globally, a trend that parallels the rising incidence of diabetes worldwide [11,12].
Chronic hyperglycemia is central to the pathogenesis of DR. Prolonged elevations in blood glucose initiate a cascade of metabolic and cellular disturbances that impair the structural and functional integrity of the retina. Among the detectable changes are microaneurysms, thickening of the capillary basement membrane, pericyte dropout, and increased vascular permeability, all of which compromise the blood–retinal barrier (BRB), allowing plasma components to leak into the retinal tissue. This vascular dysfunction leads to edema, capillary occlusion, and ischemia, setting the stage for neovascularization and proliferative retinopathy in later stages [13,14,15].
Beyond the vascular components, there is growing recognition of the crucial role played by retinal neurons and glial cells in the early stages of DR. The retina, being an extension of the central nervous system, contains neurons that are highly susceptible to metabolic insults [16]. Hyperglycemia triggers the activation of multiple biochemical pathways, including the polyol, hexosamine, protein kinase C, and advanced glycation end-product (AGE) pathways. These interconnected pathways enhance oxidative stress and inflammatory responses, disrupt mitochondrial function, and impair neurotrophic signaling [17].
Systemic factors such as dyslipidemia, insulin resistance, and low-grade chronic inflammation further exacerbate these effects. The resulting imbalance between oxidative injury and endogenous antioxidant defense mechanisms leads to glial cell activation (gliosis), microglial-mediated inflammation, and eventual neuronal apoptosis. This neuronal damage is often detectable before overt vascular abnormalities, as evidenced by early functional deficits like reduced contrast sensitivity, altered electroretinogram responses, and delayed visual processing [18,19,20].
Moreover, studies have highlighted the loss of neuroprotective agents such as brain-derived neurotrophic factor (BDNF) and pigment epithelium-derived factor (PEDF), both of which are vital for maintaining retinal homeostasis [21]. The deficiency of these protective molecules leaves retinal neurons more vulnerable to hyperglycemia-induced stress.
3.1. The Neurodegenerative Cascade
The onset of neurodegeneration in diabetic retinopathy is driven by sustained metabolic disturbances, with chronic hyperglycemia serving as a central initiator. This metabolic stress activates a series of interrelated biochemical pathways that converge to disrupt retinal neuronal integrity through mechanisms involving oxidative stress, mitochondrial dysfunction, inflammation, and glial cell activation. Over time, these insults compromise synaptic signaling, deplete neuroprotective factors, and lead to irreversible neuronal apoptosis. Among the most well-characterized contributors to this cascade are four major pathways that serve as upstream drivers of inflammation and neurodegeneration in the diabetic retina [17,22]:
- i.
- Polyol Pathway
One of the earliest and most significant metabolic pathways implicated in the development of diabetic retinopathy is the polyol pathway, catalyzed by the enzyme aldose reductase. Under euglycemic conditions, only a minor portion of intracellular glucose is metabolized through this pathway [23]. However, in the hyperglycemic milieu characteristic of diabetes, the surplus glucose is shunted into the polyol pathway. Aldose reductase reduces glucose to sorbitol, a sugar alcohol that is subsequently converted to fructose by sorbitol dehydrogenase [24]. The intracellular accumulation of sorbitol leads to osmotic stress, as sorbitol has limited membrane permeability and cannot freely diffuse out of cells. This effect is particularly deleterious to retinal neurons, pericytes, and Müller glial cells, leading to cellular edema, dysfunction, and death. Furthermore, the conversion of glucose to sorbitol consumes nicotinamide adenine dinucleotide phosphate (NADPH), an essential cofactor for regenerating reduced glutathione (GSH) a key intracellular antioxidant. NADPH depletion diminishes the retina’s antioxidant capacity, increasing its susceptibility to oxidative injury [23,25].
The polyol pathway also contributes to redox imbalance by promoting reactive oxygen species (ROS) production through secondary mechanisms such as increased mitochondrial superoxide generation and NADH accumulation, which further drives oxidative phosphorylation dysregulation. Moreover, the downstream metabolism of sorbitol to fructose generates advanced glycation end-products (AGEs), linking the polyol pathway to other deleterious metabolic routes. Collectively, these mechanisms osmotic stress, oxidative imbalance, mitochondrial dysfunction, and AGE formation initiate apoptosis in essential retinal cells, including pericytes, endothelial cells, and neurons. This contributes to the breakdown of the retinal neurovascular unit (NVU) and initiates the chronic degenerative changes observed in diabetic retinopathy [25,26].
- ii.
- Advanced Glycation End Products (AGEs)
Chronic hyperglycemia promotes the non-enzymatic glycation of proteins, lipids, and nucleic acids, leading to the accumulation of advanced glycation end products (AGEs), which are key contributors to the pathogenesis of diabetic complications, including DR. AGEs accumulate in the retinal extracellular matrix and intracellular compartments, where they disrupt normal cellular function and structure [27,28].
AGEs exert their effects primarily through binding to the receptor for advanced glycation end products (RAGE), a pattern-recognition receptor expressed on retinal neurons, glial cells, and endothelial cells. Engagement of RAGE by AGEs activates several downstream signaling cascades, particularly the nuclear factor-kappa B (NF-κB) pathway, which orchestrates the expression of pro-inflammatory cytokines, adhesion molecules, and chemokines such as TNF-α, IL-6, and ICAM-1. This amplifies chronic low-grade inflammation in the retina and facilitates leukostasis, a process that exacerbates capillary occlusion and BRB breakdown [29,30].
Additionally, AGE-RAGE signaling stimulates the generation of ROS via NADPH oxidase and mitochondrial sources, exacerbating oxidative stress and inducing further cellular damage. AGEs also cause cross-linking of extracellular matrix (ECM) proteins such as collagen and laminin, leading to basement membrane thickening and vascular stiffening, hallmark features of diabetic retinal microangiopathy [28,31,32]. Of particular concern is the AGE-induced apoptosis of retinal ganglion cells and photoreceptors, which shows the role of AGEs not only in vascular but also in neuronal degeneration [33]. The persistence and accumulation of AGEs in retinal tissue render them both pathogenic mediators and potential biomarkers for early retinal neurodegeneration in diabetes.
- iii.
- Protein Kinase C (PKC) Activation
Hyperglycemia induces de novo synthesis of diacylglycerol (DAG), a lipid second messenger that activates several isoforms of protein kinase C (PKC), particularly PKC-β and PKC-δ. This pathway is heavily implicated in vascular abnormalities observed in DR, but its role extends into neuroinflammation and glial activation [34]. PKC activation alters the phosphorylation of proteins involved in maintaining tight junction integrity, thereby increasing vascular permeability and contributing to BRB disruption. Additionally, PKC downregulates endothelial nitric oxide synthase (eNOS) activity, reducing nitric oxide (NO) bioavailability, which impairs retinal blood flow autoregulation and leads to localized ischemia [35].
In the neural retina, PKC promotes the activation of pro-inflammatory transcription factors and enhances the production of VEGF, a potent angiogenic and permeability factor. VEGF not only contributes to pathological neovascularization in proliferative DR but also exerts direct neurotoxic effects, particularly under hypoxic conditions. Studies have shown that excessive VEGF can induce apoptosis in retinal neurons and disrupt synaptic signaling. PKC is also involved in glial activation, particularly of Müller cells and microglia, which release additional pro-inflammatory mediators and propagate neuronal injury. Thus, PKC signaling contributes to both vascular and neurodegenerative processes, making it a crucial node in the pathophysiological network of DR [36].
- iv.
- Hexosamine Pathway
Hyperglycemia drives a fraction of excess fructose-6-phosphate into the hexosamine biosynthetic pathway (HBP) via the enzyme GFAT, generating elevated levels of UDP-N-acetylglucosamine (UDP-GlcNAc) the key substrate for post-translational modification of proteins by O-GlcNAc transferase (OGT). This modification, known as O-GlcNAcylation, is upregulated in both vascular and neural retinal compartments in diabetes, with UDP-GlcNAc levels significantly higher in the vitreous of proliferative DR patients compared to non-diabetics [37,38].
Similar to PKC-driven changes in DR, HBP activation alters endothelial function and neurovascular homeostasis. Elevated O-GlcNAcylation modifies tight-junction proteins like connexin-43 (Cx43) and occluding leading to impaired intercellular contact, increased permeability, and breakdown of the blood-retinal barrier. Suppressing O-GlcNAcylation restores Cx43 levels and tight junction function, highlighting direct involvement in vascular barrier disruption [39].
Within the neurovascular unit (NVU), heightened O-GlcNAcylation disturbs insulin/Akt signaling, reducing neuronal survival signaling and making retinal ganglion and other neurons more susceptible to apoptosis under chronic hyperglycemia40. Key transcription factors namely Sp1, NF-κB (p65), and p53 are modified by O-GlcNAcylation. For instance, Sp1 glycosylation enhances VEGF transcription; NF-κB’s activity is amplified by O-GlcNAc, promoting inflammatory cytokine expression, and p53 modification contributes to pericyte apoptosis, undermining vascular stability in early DR [41]. In parallel, O-GlcNAcylation aggravates oxidative stress: elevated HBP flux impairs NADPH utilization and glutathione regeneration, thereby elevating reactive oxygen species in retinal endothelial cells, neurons, and glia. The resulting oxidative milieu damages mitochondria and accelerates apoptosis and microvascular dysfunction [38]. Astrocytes and Müller cells also respond to HBP activation and O-GlcNAc modification such that translation regulators such as 4E-BP1 become glycosylated, altering protein synthesis in Müller glia and increasing expression of pro-inflammatory mediators like CD40. Concurrently, gap junction protein Cx43 becomes modified and dysregulated in Müller cells, potentiating glial activation and downstream neuronal injury [38,42].
These pathways are not isolated in their mechanism, they interact synergistically to enhance the generation of reactive oxygen species (ROS), deplete endogenous antioxidant reserves, and sustain a state of chronic inflammation. This redox imbalance leads to mitochondrial dysfunction, DNA fragmentation, and lipid peroxidation, undermining neuronal integrity. Antioxidant defense systems including superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH) are significantly diminished in the diabetic retina, failing to neutralize excessive free radicals and allowing oxidative damage to accumulate [43].
Alongside oxidative stress, chronic hyperglycemia promotes a sustained inflammatory state. The AGE-RAGE and PKC pathways upregulate pro-inflammatory cytokines and acute-phase proteins such as C-reactive protein (CRP) via activation of NF-κB signaling. This contributes to leukocyte adhesion, endothelial dysfunction, and microglial activation. Activated microglia release cytotoxic substances, including TNF-α, nitric oxide, and IL-1β, which not only disrupt neuronal signaling but also promote glial reactivity and further compromise the neurovascular microenvironment [44].
One of the earliest manifestations of neuronal distress is glutamate excitotoxicity. Impaired clearance of glutamate by reactive Müller glia leads to its extracellular accumulation, overactivating NMDA receptors on retinal neurons. In addition to this excitotoxic stress, γ-aminobutyric acid (GABA) signaling normally responsible for inhibitory neurotransmission and synaptic modulation is disrupted. This imbalance between excitatory and inhibitory inputs contributes to abnormal retinal circuitry, increased neuronal firing, and calcium-mediated apoptosis [45].
Furthermore, the diabetic retina experiences a significant loss of neurotrophic support. Levels of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF), both critical for neuronal survival, axonal integrity, and synaptic plasticity, are markedly reduced. The downregulation of pigment epithelium-derived factor (PEDF), a neuroprotective and anti-angiogenic glycoprotein further exacerbates the vulnerability of neurons to oxidative and ischemic stress. These deficits impair cellular resilience and regenerative capacity, accelerating the progression of neuronal degeneration [46]. Concurrently, Müller glia and astrocytes undergo reactive gliosis, as indicated by the upregulation of glial fibrillary acidic protein (GFAP). This reactive phenotype impairs their ability to regulate neurotransmitter homeostasis, maintain ionic balance, and support the blood-retinal barrier (BRB). As glial support functions decline, neuronal energy metabolism falters, synaptic connectivity is lost, and apoptotic cascades are triggered particularly affecting retinal ganglion cells, amacrine cells, and photoreceptors [47].
Functionally, these neurodegenerative events manifest as reduced visual performance and altered electrophysiological responses often detectable by electroretinography (ERG) before the onset of clinically apparent vascular lesions. Structurally, thinning of the retinal nerve fiber layer (RNFL) and inner plexiform layer can be visualized using optical coherence tomography (OCT), serving as early indicators of retinal injury [48].
As neurodegeneration progresses, it destabilizes the neurovascular unit (NVU), impairing autoregulation of retinal blood flow and promoting ischemia. Capillary occlusion due to endothelial dysfunction and leukostasis exacerbates tissue hypoxia, leading to the activation of hypoxia-inducible factors (HIFs). These, in turn, upregulate vascular endothelial growth factor (VEGF), increase vascular permeability, and initiate pathological neovascularization. Concurrent breakdown of the BRB, driven by inflammation and glial dysfunction, permits fluid and protein leakage into the retinal parenchyma, contributing to diabetic macular edema (DME)[43,49].
Figure 1.
Illustration of the Neurodegenerative Process in Diabetic Retinopathy.
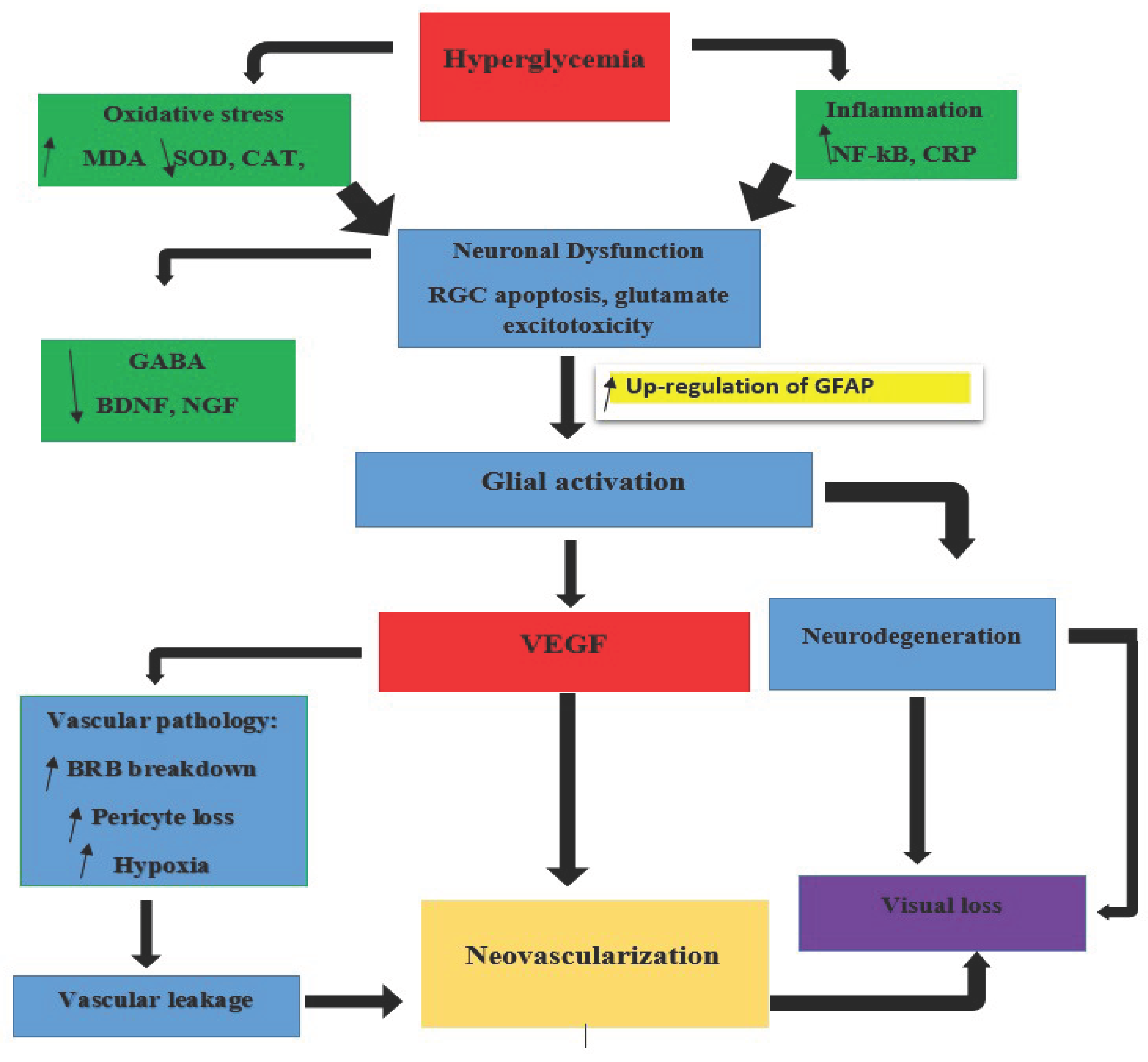
4. Biomarkers of Retina Neurodegeneration
Early identification of retinal neurodegeneration in diabetic retinopathy (DR) is important for preventing irreversible visual impairment. Since neurodegenerative changes often precede overt vascular damage, a growing body of research is now focused on the detection of sensitive and specific biomarkers that reflect early neuronal injury, oxidative stress, glial activation, and dysfunction of the neurovascular unit. These biomarkers may aid in early diagnosis and also serve as valuable tools for monitoring disease progression and evaluating therapeutic efficacy.
- i.
- Structural Biomarkers of Retinal Neurodegeneration
Ganglion Cell Complex Thinning
Ganglion cell complex thinning represents one of the earliest detectable structural changes in diabetic retinopathy, measurable via spectral-domain optical coherence tomography (SD-OCT). This thinning predominantly affects the macular region and reflects progressive loss of retinal ganglion cells and their axonal projections. Studies demonstrate that GCC thickness reductions exceeding 75 µm correlate with diminished contrast sensitivity and impaired color vision, detectable up to 5 years before clinical retinopathy diagnosis [50,51,52]. The pattern of thinning often shows a topographical gradient, with the superotemporal quadrant exhibiting the most significant changes. Assessment protocols should include macular cube scans with automated segmentation software to quantify the combined thickness of the retinal nerve fiber layer, ganglion cell layer, and inner plexiform layer. Progressive thinning rates exceeding 1.2 µm/year significantly predict future functional impairment independent of vascular abnormalities [51,53].
Disorganization of Retinal Inner Layers (DRIL)
Disorganization of retinal inner layers (DRIL) is a critical optical coherence tomography (OCT) derived biomarker defined by the loss of discernible boundaries between the ganglion cell-inner plexiform layer complex, inner nuclear layer, and outer plexiform layer. This structural disruption signifies advanced synaptic degeneration and neuronal dysfunction within the inner retina. When affecting over 500 µm in the central macula, DRIL correlates strongly with capillary non-perfusion on OCT angiography and serves as a predictor of treatment resistance to anti-VEGF therapies [52]. Spectral domain OCT studies in patients without overt DR consistently show thinning of macular ganglion cell–inner plexiform layer (GC IPL) and peripapillary RNFL, even when no microaneurysms or exudates are visible, indicating early neurodegeneration (systematic review/meta analysis; longitudinal RNFL studies)[54]. Measurement of retinal vessel caliber via fundus imaging reveals narrowing of arterioles and venules in early DR, which reflects endothelial dysfunction and impaired neurovascular coupling creating ischemic stress that accompanies neuronal loss [55]. The appearance of microaneurysms and exudates remains the earliest vascular markers, but thinning of neuronal layers and changes at the optic nerve head such as increased cup to disc ratio and neuroretinal rim loss often precede these visible lesions, correlating with ganglion cell death [56].
The assessment involves high-resolution line scans through the foveal center, with trained graders evaluating the extent of architectural disruption. Longitudinal studies show that baseline DRIL presence confers a 4-fold increased risk of suboptimal visual recovery following intervention, suggesting irreversible neuronal damage [51,52].
Hyperreflective Foci (HRF)
Hyperreflective Foci (HRF), manifest as punctate intraretinal dots (≤30 µm diameter) on OCT with reflectivity comparable to the nerve fiber layer. Histopathological studies indicate these structures represent lipid-laden macrophages, extravasated lipoproteins, or activated microglia migrating toward ischemic regions. Quantification exceeding 20 HRF in the inner retina strongly correlates with aqueous humor levels of microglial activation markers (CD14) and predicts macular edema development within 12 months. Their persistence following anti-VEGF treatment indicates sustained neuroinflammation and predicts diabetic macular edema recurrence. Automated quantification algorithms now enable standardized tracking of HRF burden as a metric of inflammatory neurodegeneration [57,58,59].
- ii.
- Functional Biomarkers of Neuronal Impairment
Flicker-Induced Vasodilation Deficits
Flicker-induced vasodilation deficits represent impaired neurovascular coupling, detectable before vascular pathology becomes clinically evident. This functional biomarker assesses the retina’s autoregulatory capacity through flicker-light stimulation, normally triggering nitric oxide-mediated vasodilation. In diabetic patients, attenuated vasodilatory responses (≤2% diameter change versus ≥4% in healthy controls) correlate with retinal ganglion cell dysfunction and predict subsequent microvascular damage. The assessment utilizes dynamic vessel analysis software to measure caliber changes in major retinal arterioles during precisely controlled flicker stimulation. This non-invasive functional test demonstrates 82% sensitivity for detecting preclinical retinopathy when combined with OCT parameters [60,61].
In vivo imaging with OCT angiography (OCT A) demonstrates reduced vessel density in the deep capillary plexus (DCP) and enlargement of the foveal avascular zone features seen in diabetics without DR and correlating with inner retinal thinning, supporting impaired neurovascular integrity [62].
Electroretinogram
Multifocal electroretinography (mfERG) reliably detects delayed implicit times and reduced amplitudes in retinal responses before vascular lesions emerge, reflecting early dysfunction of neuronal signal transmission especially in ganglion and amacrine cells [63]. Electroretinogram (ERG) abnormalities provide objective quantification of global retinal neuronal function. The most sensitive indicators include reduced oscillatory potential amplitudes (≥30% reduction) and prolonged implicit times (≥5 ms delay), reflecting inner retinal inhibitory circuit dysfunction [64,65]. These electrophysiological changes precede clinically detectable microvascular lesions by 2-4 years and correlate with glutamate excitotoxicity and GABAergic signaling deficits. Multifocal ERG further enables topographical mapping of localized neuronal dysfunction, with paracentral depression predicting future microaneurysm formation. Standardized testing protocols under photopic and scotopic conditions allow differentiation of specific neuronal pathway involvement, with bipolar cell dysfunction emerging as the earliest consistent abnormality [66,67,68].
Contrast Sensitivity Reduction
Contrast Sensitivity Reduction specifically at intermediate spatial frequencies (4-8 cycles/degree) demonstrates high sensitivity for early neurodegeneration. This psychophysical deficit correlates with GABAergic amacrine cell loss and precedes visual acuity deterioration by several years [68,69,70,71]. Quantitative assessment using sinusoidal gratings under standardized illumination (85 cd/m2) shows significant differences between diabetic patients without retinopathy and healthy controls. The deficit pattern shows strong correlation with inner plexiform layer thinning on OCT and serves as a functional biomarker for treatment response monitoring, with improvements following neuroprotective interventions often preceding structural changes [71,72,73].
Fundus autofluorescence (FAF)
Fundus autofluorescence (FAF) is a non-invasive imaging technique that detects natural fluorescence from lipofuscin and related fluorophores within the retinal pigment epithelium (RPE). In diabetic patients, FAF abnormalities can precede clinically detectable microvascular changes, making it a valuable early biomarker of retinal stress. The accumulation of lipofuscin a byproduct of photoreceptor outer segment degradation reflects oxidative damage and impaired metabolic turnover in RPE cells, often secondary to photoreceptor dysfunction and chronic hyperglycemia [74]. Hyperautofluorescent foci on FAF imaging signal increased metabolic stress, while hypoautofluorescent areas may represent RPE atrophy or dead zones of photoreceptor-RPE interaction. These changes have been shown to correlate with outer retinal thinning and ellipsoid zone disruption on OCT, confirming their link to neurodegenerative processes in diabetic retinopathy. FAF imaging also allows early detection of localized RPE dysfunction before the appearance of vascular lesions on fluorescein angiography or fundus photography, enabling timely intervention and monitoring of neuroprotective treatment responses [75,76,77].
Confocal scanning laser ophthalmoscopy (cSLO)
Confocal scanning laser ophthalmoscopy (cSLO) complements FAF by providing high-resolution, depth-resolved imaging of the retina and is capable of visualizing early cellular and inflammatory changes. In diabetic retinopathy models and early clinical cases, cSLO has been used to detect patterns of microglial activation in the inner retina manifesting as increased reflectivity or discrete cellular aggregates which serve as indirect markers of neuroinflammation and neuronal injury [78,79]. Experimental studies using transgenic mice and adaptive optics–enhanced cSLO have shown that activated microglia migrate toward regions of neural stress and adopt amoeboid morphology, often clustering near damaged ganglion cells and vasculature even before overt vascular pathology occurs [80,81]. These microglial changes, visualized in vivo, further reinforce the concept of diabetic retinopathy as a neurovascular disease and underscore the utility of cSLO and FAF as complementary, non-invasive tools for tracking early retinal neurodegeneration.
Additionally, quantitative analysis of outer retinal bands such as thinning between the inner segment ellipsoid and interdigitation zones on OCT demonstrates rod predominant photoreceptor degeneration in mild or no DR, indicating that neurodegeneration extends beyond inner retina [48].
- iii.
- Biochemical and Molecular Biomarkers
Hyperglycemia
Persistent elevations of HbA1c have been robustly linked to early neurodegenerative alterations in the diabetic retina, functioning as a systemic indicator that chronic hyperglycemia drives retinal neuronal stress. Alongside glycemic burden, abnormal lipid profiles particularly elevated triglycerides and reduced HDL cholesterol have been correlated with markers of inner retinal thinning as well as impaired electrophysiological responses in diabetic individuals without clinically visible retinopathy, suggesting that dyslipidemia accentuates oxidative and membrane damage in retinal neurons [63].
Dysregulated Neuroprotective Factors
Dysregulated neuroprotective factors including pigment epithelium-derived factor (PEDF) and somatostatin (SST) demonstrate significant alterations in diabetic retinopathy. Aqueous humor analysis reveals PEDF levels less than 8 ng/mL in early diabetes, reducing neuronal survival signals and permitting accelerated apoptosis. Conversely, vitreous VEGF levels above 500 pg/mL correlate with Müller cell activation and inner blood-retinal barrier breakdown. SST depletion specifically affects GABAergic amacrine cells, diminishing inhibitory neurotransmission and increasing excitotoxic vulnerability. Mass spectrometry of vitreous samples detects greater than 60% reduction in interphotoreceptor retinoid-binding protein (IRBP) years before vascular lesions appear. These biomarkers can be assessed through minimally invasive anterior chamber paracentesis during routine cataract surgery or via tear fluid analysis using advanced multiplex assays [53,83,84].
Other neurotrophic factors and anti angiogenic factors such as brain derived neurotrophic factor (BDNF), nerve growth factor (NGF) exhibit lower concentrations including somatostatin (SST) in ocular fluids of individuals with early DR. These reductions mirror loss of neuroprotective signaling and are associated with inner retinal thinning on OCT as well as poorer mfERG metrics. Altered expression of proteins such as apolipoprotein A1, fibrinogen A, IRBP, and semaphorin 3A have been observed in vitreous or plasma, and these changes correlate with markers of neuronal stress and early DR; an example, decreased IRBP reflects photoreceptor dysfunction, while elevated semaphorin 3A indicates disrupted axonal guidance and neural injury. VEGF, although traditionally viewed as a vascular mediator, is elevated in ocular fluids early in DR and correlates with both neural stress and future thinning of the GC IPL layer, suggesting its dual role as a marker of neurovascular disturbance [53,85,86,87].
Apoptotic Markers
Apoptotic Signaling Markers including activated caspase-3 and cleaved poly(ADP-ribose) polymerase (PARP) appear in ocular fluids before vascular histopathology develops. Caspase-3 cleaves tau proteins in ganglion cells, generating cytotoxic fragments detectable in vitreous humor. Increased activation of PARP (poly ADP ribose polymerase) is observed in diabetic retinas and is accompanied by overexpression of cleaved caspase 3, reduced BDNF, and impairment of synaptic markers findings confirmed in human tissue and animal models. PARP-related neuronal loss precedes visible microvascular changes and is reversible by PARP inhibitors in experimental settings [88]. Death-receptor pathway activation evidenced by Fas/FasL upregulation, cleaved caspase 8 and Bid cleavage, as well as imbalance in Bax/Bcl 2 ratios, have been demonstrated in neuroretinal tissues of diabetic individuals with minimal retinopathy, indicating engagement of both extrinsic and intrinsic apoptotic cascades early in the disease process [89].
PARP upregulation in Müller cells depletes NAD+, impairing glycolysis and promoting axonal demise. Clinical assessment now includes annexin V positron emission tomography for in vivo detection of retinal phosphatidylserine externalization, with signal intensity predicting 2-year progression risk. Vitreous samples obtained during vitrectomy enable quantification of cytochrome c and apoptosis-inducing factor through Western blot analysis [90,91].
Advanced glycation end-products (AGEs)
Advanced glycation end-products (AGEs) accumulate in retinal tissues in diabetes and promote neurodegeneration through oxidative injury and inflammatory signaling. AGE RAGE interactions stimulate NF κB–mediated cytokine release, increase ROS, and accelerate neuronal apoptosis. Studies examining vitreous and plasma levels of AGEs have demonstrated significant correlations with thinning of the ganglion cell–inner plexiform layer, even before microvascular lesions appear [27].
Advanced Glycation End Products (AGEs) can accumulate in retinal neurons within six months of hyperglycemia onset. Vitreous methylglyoxal hydroimidazolone (MG-H1) >300 nM disrupts insulin receptor signaling in ganglion cells, causing insulin resistance and synaptic failure. Skin autofluorescence measurements strongly correlate with retinal AGE levels (r=0.78) and predict DRIL development, enabling non-invasive risk stratification. The receptor for AGE (RAGE) shows increased expression in Müller cells, activating NF-κB pathways and amplifying inflammatory responses. Vitreous biopsy assessment now includes competitive ELISA for soluble RAGE isoforms, with low soluble RAGE predicting rapid neurodegeneration [31,92,93].
Inflammatory Mediators
Inflammatory mediators including C-reactive protein (CRP >3 mg/L) predict diabetic retinopathy incidence independent of glycemic control. CRP activates retinal microglia, which secrete IL-1β and TNF-α, damaging bipolar cells. C reactive protein (CRP), particularly high sensitivity CRP, is elevated in persons with early DR and correlates with neurodegenerative features on OCT and ERG. Elevated CRP is thought to reflect systemic inflammation that exacerbates retinal microglial activation and neuronal injury [94].
Aqueous humor CXCL13 >120 pg/mL recruits B-cells into the retina, forming autoantibodies against neuronal antigens like recoverin. Suppressing these mediators with corticosteroids preserves electroretinogram amplitudes even in established retinopathy. Novel ultrasensitive assays now detect IL-6, MCP-1, and IP-10 in tear fluid collected via absorbent matrices, enabling non-invasive monitoring [95,96].
Depletion of endogenous antioxidants superoxide dismutase (SOD), catalase (CAT), and reduced glutathione (GSH) has been documented in serum and vitreous fluid of diabetic individuals and is associated with increased oxidative stress markers and electrophysiological dysfunction, reflecting failure of neuronal redox protection [15].
- iv.
- Systemic Circulating Biomarkers
Neurofilament Light Chain (NFL)
Neurofilament Light Chain (NFL) elevation in plasma reflects axonal damage in the inner retina. Using single-molecule array (SiMoA) technology, NFL levels increase 1.8-fold in mild non-proliferative DR and 2.5-fold in proliferative DR/diabetic macular edema versus controls. This increase precedes microvascular lesion formation and correlates strongly with ganglion cell complex thinning (r=-0.63). Plasma NFL >15 pg/mL predicts 3-year progression to vision-threatening diabetic retinopathy with 89% sensitivity, outperforming traditional risk factors . The biomarker demonstrates excellent stability in plasma samples stored at -80°C, facilitating its incorporation into routine screening protocols [90,94].
Glial Fibrillary Acidic Protein (GFAP)
Glial Fibrillary Acidic Protein (GFAP) exhibits a biphasic response reflecting Müller cell dynamics. Early diabetes reduces plasma GFAP (fold difference 0.69) due to glial dysfunction, while advanced disease increases GFAP (fold difference 1.4) indicating reactive gliosis. Serial measurements identify rapid progressors, with rising GFAP over six months conferring a 5-fold increased diabetic macular edema risk. GFAP trajectories complement structural OCT assessments, with decreasing levels predicting treatment response to neuroprotective agents. Standardized collection protocols require avoidance of hemolyzed samples due to erythrocyte GFAP contamination [90,97,98].
- v.
- Emerging Biomarkers of Retinal Neurodegeneration
MicroRNA
MicroRNA regulate synaptic maintenance and vascular integrity through post-transcriptional gene silencing. MicroRNAs such as miR 146a, miR 21, miR 200b, and miR 497 have been consistently found dysregulated in ocular fluids or serum of early DR patients; these miRNAs regulate neuroinflammatory cytokines, VEGF expression, and neurotrophic factor levels [53,99].
Several microRNAs including miR-146a-5p, miR-21-3p, miR-200c, and miR-181a-5p are dysregulated in early diabetic retinopathy (DR), contributing to neuroinflammation, excitotoxicity, and oxidative stress. miR-146a-5p is elevated in proliferative DR and may impair neuronal glutamate clearance, promoting excitotoxic damage. Similarly, miR-21-3p enhances NLRP3 inflammasome activation in retinal microglia, while miR-200c disrupts antioxidant defense by inhibiting ZEB2. A combined panel of miR-21-3p, miR-181a-5p, and miR-342-3p has demonstrated high diagnostic accuracy for early DR with an area under the curve (AUC) of approximately 0.93. Therapeutically, locked nucleic acid inhibitors targeting these miRNAs have shown potential in restoring neuroretinal homeostasis in experimental models. Quantitative RT-PCR analysis of serum exosomes enriched for retinal-specific miRNAs, isolated using L1CAM antibodies, now enables non-invasive tracking of these molecular changes [100,101,102].
Exosomes
Exosomal Cargo shuttles neurodegenerative molecules between retinal cells. Plasma exosomes from diabetic retinopathy patients contain amyloid β42 (>1.8-fold increase) and phosphorylated tau-Thr181, inducing apoptosis in cultured ganglion cells. Exosomal heat shock protein 70 (HSP70) correlates with Müller cell stress and predicts progression within one year (hazard ratio=3.2). Retina-specific exosomes isolated via immunocapture targeting neuronal cell adhesion molecule (NCAM) or glutamate aspartate transporter (GLAST) provide enhanced specificity. Nanoparticle tracking analysis enables quantification of exosome concentration and size distribution as potential biomarkers themselves [103,104].
Metabolomic Profiles
Metabolomic Profiles reveal mitochondrial dysfunction through altered energy substrates. Increased retinal ketone bodies (β-hydroxybutyrate >150 μM) inhibit complex I respiration, while reduced succinate disrupts the electron transport chain. These alterations decrease ATP synthesis in inner retinal neurons, driving apoptosis. Corneal confocal microscopy detects early mitochondrial swelling in nerve fibers, with abnormal morphology predicting diabetic retinopathy development (sensitivity=91%). High-resolution mass spectrometry of plasma identifies carnitine esters and dicarboxylic acids as indicators of fatty acid β-oxidation defects in retinal neurons [103,105,106].
Semaphorin 3A
Semaphorin 3A (Sema3A) serves as an axonal guidance protein significantly upregulated in diabetic retinas. Vitreous Sema3A above 85 ng/mL causes growth cone collapse in ganglion cells via neuropilin-1 receptors, contributing to optic neuropathy. Anti-Sema3A antibodies preserve inner retinal thickness in primate models, and aqueous levels decrease following successful intravitreal therapy. The assessment employs electrochemiluminescence immunoassays of diluted vitreous samples, with levels showing diurnal variation requiring standardized collection timing [107,108].
- vi.
- AI-Driven Digital Biomarkers of Neurovascular Function
Dynamic pupillometry
Dynamic pupillometry analytics leverage AI to quantify neurovascular coupling through infrared pupillometry. Machine learning algorithms analyze pupillary light reflex latency and constriction velocity, which reflect autonomic nervous system dysfunction in early DR. Studies demonstrate that delayed latency (>2,100 ms) correlates with retinal ganglion cell apoptosis and predicts 3-year DR progression risk (AUC = 0.82). Microsaccade pattern mapping uses convolutional neural networks (CNNs) to detect fixational instability during visual tasks [109,110,111,112].
Retinal Hemodynamic Modeling
Retinal hemodynamic modeling employs AI to simulate blood flow dynamics from OCT angiography (OCTA). Deep learning models quantify temporal delays in arteriolar dilation post-flicker stimulation, a biomarker of neurovascular uncoupling. A delay greater than 4.2 seconds correlates with Müller cell gliosis and predicts macular edema development [113].
Multi-Modal AI Integration for Systemic Biomarkers
Liquid biopsy AI fusion approaches are emerging as powerful tools in early diabetic retinopathy (DR) detection. For example, mass spectrometry based profiling of tear fluid analyzing exosomal cargo such as miR 146a 5p or proteins like HSP70 can be integrated with deep learning classifiers. These neural network models effectively distinguish neuroinflammatory signatures in early-stage DR, improving detection beyond standard imaging risk factors [[95].
In the serum metabolomics domain, explainable AI models using graph neural network frameworks have been applied to map complex biochemical interactions. Studies show that perturbations in metabolites signaling mitochondrial dysfunction such as elevated β hydroxybutyrate and reduced succinate can be captured in AI-derived metabolic risk scores. These composite scores outperform HbA1c alone, achieving area under the curve (AUC) values around 0.87 for predicting DR progression [114,115].
5. Summary
This review identified a set of early biomarkers linked to neurodegeneration in diabetic retinopathy (DR). These biomarkers reflect molecular and functional impairments that precede overt vascular changes, including alterations in neuronal viability, synaptic signaling, oxidative stress, and neuroinflammation within the retina.
Biomarker categories encompassed circulating and ocular molecules, neuroinflammatory mediators, and neurosensory response metrics. Emerging evidence from both clinical and experimental studies supports their potential utility in identifying patients at risk for DR progression before clinical retinopathy becomes apparent.
Artificial intelligence (AI) technologies such as pupillometry analysis, visual tracking, and serum metabolomic modeling were shown to improve the sensitivity and specificity of biomarker-based detection. These tools facilitate the identification of subclinical neuronal deficits and enable risk stratification with greater precision than conventional methods.
Overall, the integration of neurodegeneration-based biomarkers with AI-assisted platforms holds promise for advancing early detection and preventive care in diabetic retinopathy.
6. Discussion
Diabetic retinopathy (DR) is increasingly recognized as a neurovascular disease rather than a condition confined to microvascular pathology. Mounting evidence suggests that retinal neurodegeneration not only precedes but also contributes to subsequent vascular damage. This review consolidates data from structural, molecular, functional, and systemic biomarker studies, demonstrating that early neuroglial impairment is a critical, and often underappreciated, component of DR pathogenesis.
Structural biomarkers such as retinal ganglion cell complex thinning and disorganization of retinal inner layers (DRIL), detectable via optical coherence tomography (OCT), offer quantifiable insights into early neuronal apoptosis and synaptic dysfunction. These alterations are often accompanied by deficits in flicker-induced vasodilation and reduced electroretinogram (ERG) oscillatory potentials both indicators of impaired neurovascular coupling and inner retinal cell dysfunction. The fact that such markers are measurable through routine ophthalmic imaging or electrophysiological assessments enhances their clinical relevance and accessibility.
Molecular biomarkers in ocular fluids, including reduced levels of pigment epithelium-derived factor (PEDF) and elevated caspase-3, reflect mitochondrial dysfunction and activation of intrinsic apoptotic pathways. Circulating systemic markers such as neurofilament light chain (NfL) further underscore the interconnectedness between retinal and central neurodegeneration, with elevated serum NfL levels correlating with accelerated DR progression irrespective of vascular status.
Disrupted neurotrophic signaling impairs the survival of retinal neurons. The downregulation of somatostatin in amacrine cells and interphotoreceptor retinoid-binding protein in photoreceptors exacerbates excitotoxicity, while advanced glycation end products (AGEs) compromise insulin receptor function in Müller glial cells. This cascade fosters a self-reinforcing cycle: neuroinflammation amplified by microglial activation markers like CXCL13, which compromises the blood-retinal barrier, allowing systemic neurotoxic mediators to penetrate retinal tissue. Novel biomarkers such as semaphorin 3A and exosomal tau proteins further delineate axonal transport impairment and protein misfolding mechanisms that parallel central neurodegenerative disorders.
Nonetheless, several challenges limit the immediate clinical implementation of these biomarkers. The invasive nature of sampling ocular fluids poses a barrier to routine molecular analysis, though non-invasive modalities such as tear fluid diagnostics and skin autofluorescence are showing promise. Artificial intelligence (AI) platforms are increasingly being used to integrate multimodal datasets including OCT angiography vessel density, circulating ceramides, and miRNA profiles for enhanced diagnostic accuracy. However, these approaches require further validation in diverse populations and settings. Moreover, emerging biomarkers such as hyperreflective foci or annexin V imaging lack standardized acquisition protocols, and therapeutic interventions targeting these neurodegenerative markers remain in experimental stages. While anti-VEGF agents provide partial neuroprotection, investigational therapies like caspase inhibitors or PEDF gene replacement are not yet clinically available.
The integration of neurodegenerative biomarkers into diagnostic frameworks presents a significant advancement in the early detection of diabetic retinopathy. Analytical models that incorporate structural, functional, and systemic markers may allow for the stratification of individuals based on the extent and dominance of neurodegenerative involvement. This stratification could enable earlier application of targeted neuroprotective strategies such as semaphorin 3A antagonists or miRNA inhibitors before irreversible neuronal damage occurs. As multi-omics technologies continue to develop, the neurobiological complexity of DR, embedding neurodegeneration-specific biomarkers into routine screening protocols has the potential to reorient clinical focus from late-stage vascular complications toward preclinical neurovascular preservation.
7. Conclusion
Diabetic retinopathy (DR) is increasingly understood as a neurovascular disorder, with neurodegeneration preceding overt vascular pathology. This review synthesizes current evidence on structural, functional, molecular, and emerging biomarkers that reflect early neuroretinal injury driven by hyperglycemia-induced metabolic stress, mitochondrial dysfunction, and neuroinflammatory responses. Advancing the clinical relevance of these biomarkers requires rigorous validation of non-invasive detection strategies, longitudinal studies to define progression thresholds, and controlled trials assessing biomarker-guided neuroprotective interventions. The integration of these multidimensional biomarkers into risk stratification models may redefine the clinical management of DR, shifting the paradigm from late-stage vascular intervention to early, targeted neuroprotection.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
Abbreviations
The following abbreviations are used in this manuscript:
| AGEs | Advanced Glycation End Products |
| AI | Artificial Intelligence |
| AUC | Area Under the Curve |
| BDNF | Brain-Derived Neurotrophic Factor |
| BRB | Blood-Retinal Barrier |
| CAT | Catalase |
| CNNs | Convolutional Neural Networks |
| CRP | C-Reactive Protein |
| cSLO | Confocal Scanning Laser Ophthalmoscopy |
| CXCL13 | Chemokine (C-X-C Motif) Ligand 13 |
| Cx43 | Connexin-43 |
| DAG | Diacylglycerol |
| DCP | Deep Capillary Plexus |
| DME | Diabetic Macular Edema |
| DR | Diabetic Retinopathy |
| DRIL | Disorganization of Retinal Inner Layers |
| ECM | Extracellular Matrix |
| eNOS | Endothelial Nitric Oxide Synthase |
| ERG | Electroretinogram |
| FAF | Fundus Autofluorescence |
| Fas/FasL | Fas Ligand |
| GFAP | Glial Fibrillary Acidic Protein |
| GFAT | Glutamine-Fructose-6-Phosphate Aminotransferase |
| GLAST | Glutamate Aspartate Transporter |
| GSH | Glutathione |
| GCC | Ganglion Cell Complex |
| GC-IPL | Ganglion Cell-Inner Plexiform Layer |
| HBP | Hexosamine Biosynthetic Pathway |
| HIFs | Hypoxia-Inducible Factors |
| HRF | Hyperreflective Foci |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| IL-1β | Interleukin-1 Beta |
| IL-6 | Interleukin-6 |
| IP-10 | Interferon Gamma-Induced Protein 10 |
| IRBP | Interphotoreceptor Retinoid-Binding Protein |
| L1CAM | L1 Cell Adhesion Molecule |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| MG-H1 | Methylglyoxal Hydroimidazolone |
| miRNA | MicroRNA |
| mfERG | Multifocal Electroretinogram |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NCAM | Neural Cell Adhesion Molecule |
| NF-κB | Nuclear Factor-Kappa B |
| NfL | Neurofilament Light Chain |
| NGF | Nerve Growth Factor |
| NO | Nitric Oxide |
| NVU | Neurovascular Unit |
| OCT | Optical Coherence Tomography |
| OCTA | Optical Coherence Tomography Angiography |
| OGT | O-GlcNAc Transferase |
| PEDF | Pigment Epithelium-Derived Factor |
| PARP | Poly(ADP-Ribose) Polymerase |
| PKC | Protein Kinase C |
| RAGE | Receptor for Advanced Glycation End Products |
| RPE | Retinal Pigment Epithelium |
| RNFL | Retinal Nerve Fiber Layer |
| ROS | Reactive Oxygen Species |
| RT-PCR | Reverse Transcription-Polymerase Chain Reaction |
| Sema3A | Semaphorin 3A |
| SiMoA | Single-Molecule Array |
| SOD | Superoxide Dismutase |
| SST | Somatostatin |
| TNF-α | Tumor Necrosis Factor-Alpha |
| UDP-GlcNAc | Uridine Diphosphate N-Acetylglucosamine |
| VEGF | Vascular Endothelial Growth Factor |
| ZEB2 | Zinc Finger E-Box Binding Homeobox 2 |
References
- Zhou, J.; Chen, B. Retinal Cell Damage in Diabetic Retinopathy. Cells. 2023, 12, 1342. [Google Scholar] [CrossRef]
- Ansari, P.; Tabasumma, N.; Snigdha, N.N.; Siam, N.H.; Panduru, R.V.N.R.S.; Azam, S.; Hannan, J.M.A.; Abdel-Wahab, Y.H.A. Diabetic Retinopathy: An Overview on Mechanisms, Pathophysiology and Pharmacotherapy. Diabetology. 2022, 3, 159–175. [Google Scholar] [CrossRef]
- Ong, C.J.T.; Wong, M.Y.Z.; Cheong, K.X.; Zhao, J.; Teo, K.Y.C.; Tan, T.-E. Optical Coherence Tomography Angiography in Retinal Vascular Disorders. Diagnostics. 2023, 13, 1620. [Google Scholar] [CrossRef]
- Herbort, C.P.; Papasavvas, I.; Tugal-Tutkun, I. Benefits and Limitations of OCT-A in the Diagnosis and Follow-Up of Posterior Intraocular Inflammation in Current Clinical Practice: A Valuable Tool or a Deceiver? Diagnostics 2022, 12, 2384. [Google Scholar] [CrossRef] [PubMed]
- Soni, D.; Sagar, P.; Takkar, B. Diabetic retinal neurodegeneration as a form of diabetic retinopathy. International Ophthalmology. 2021, 41, 3223–48. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Silva, P.S.; Stitt, A.W. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat Rev Endocrinol 2021, 17, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Simó-Servat, O.; Porta, M.; Grauslund, J.; Harding, S.P.; Frydkjaer-Olsen, U.; García-Arumí, J.; Ribeiro, L.; Scanlon, P.; Cunha-Vaz, J.; Simó, R. Serum glial fibrillary acidic protein and neurofilament light chain as biomarkers of retinal neurodysfunction in early diabetic retinopathy: results of the EUROCONDOR study. Acta Diabetologica. 2023, 60, 837–44. [Google Scholar] [CrossRef]
- Viggiano, P.; Vujosevic, S.; Palumbo, F.; Grassi, M.O.; Boscia, G.; Borrelli, E.; Reibaldi, M.; Sborgia, L.; Molfetta, T.; Evangelista, F.; Alessio, G. Optical coherence tomography biomarkers indicating visual enhancement in diabetic macular edema resolved through anti-VEGF therapy: OCT biomarkers in resolved DME. Photodiagnosis and Photodynamic Therapy. 2024, 46, 104042. [Google Scholar] [CrossRef]
- Smit-McBride, Z.; Morse, L.S. MicroRNA and diabetic retinopathy—biomarkers and novel therapeutics. Annals of translational medicine. 2021, 9, 1280. [Google Scholar] [CrossRef]
- Yapislar, H.; Gurler, E.B. Management of microcomplications of diabetes mellitus: Challenges, current trends, and future perspectives in treatment. Biomedicines 2024, 12, 1958. [Google Scholar] [CrossRef]
- Nian, S.; Lo, A.C.Y.; Mi, Y.; Ren, K.; Yang, D. Neurovascular unit in diabetic retinopathy: pathophysiological roles and potential therapeutical targets. Eye and Vision 2021, 8, 15. [Google Scholar] [CrossRef]
- Chen, J.; Yang, C.; Zheng, W.; Li, Z.; Huang, Y.; Yao, S.; Chen, X.; et al. Global, regional, and national epidemiology of visual impairment in working-age individuals, 1990-2019. JAMA ophthalmology 2024, 142, 25–32. [Google Scholar] [CrossRef]
- An, D.; Tan, B.; Yu, D.-Y.; Balaratnasingam, C. Differentiating microaneurysm pathophysiology in diabetic retinopathy through objective analysis of capillary nonperfusion, inflammation, and pericytes. Diabetes 2022, 71, 733–746. [Google Scholar] [CrossRef]
- Bora, K.; Kushwah, N.; Maurya, M.; Pavlovich, M.C.; Wang, Z.; Chen, J. Assessment of inner blood–retinal barrier: Animal models and methods. Cells 2023, 12, 2443. [Google Scholar] [CrossRef]
- Srejovic, J.V.; Muric, M.D.; Jakovljevic, V.L.; Srejovic, I.M.; Sreckovic, S.B.; Petrovic, N.T.; Todorovic, D.Z.; Bolevich, S.B.; Vulovic, T.S.S. Molecular and cellular mechanisms involved in the pathophysiology of retinal vascular disease—interplay between inflammation and oxidative stress. International Journal of Molecular Sciences 2024, 25, 11850. [Google Scholar] [CrossRef] [PubMed]
- Oshitari, T. The pathogenesis and therapeutic approaches of diabetic neuropathy in the retina. International journal of molecular sciences 2021, 22, 9050. [Google Scholar] [CrossRef]
- Lobanovskaya, N. Pathophysiology of Diabetic Retinopathy. In Diabetic Eye Disease-From Therapeutic Pipeline to the Real World. IntechOpen, 2022.
- Kovács-Valasek, A.; Rák, T.; Pöstyéni, E.; Csutak, A.; Gábriel, R. Three major causes of metabolic retinal degenerations and three ways to avoid them. International Journal of Molecular Sciences 2023, 24, 8728. [Google Scholar] [CrossRef] [PubMed]
- Sinclair Stephen, H.; Kiran, E.M.; Talekar, S.; Schwartz, S.S. Diabetes mellitus associated neurovascular lesions in the retina and brain: A review. Frontiers in Ophthalmology 2022, 2, 1012804. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, C.; Paulus, Y.M. Advances in structural and functional retinal imaging and biomarkers for early detection of diabetic retinopathy. Biomedicines 2024, 12, 1405. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Valasciuc, E.; Gosav, E.M.; Floria, M.; Buliga-Finis, O.N.; Ouatu, A.; Cucu, A.I.; Botoc, T.; Costea, C.F. Enhancing Retinal Resilience: The Neuroprotective Promise of BDNF in Diabetic Retinopathy. Life 2025, 15, 263. [Google Scholar] [CrossRef]
- Oshitari, T. Advanced glycation end-products and diabetic neuropathy of the retina. International journal of molecular sciences 2023, 24, 2927. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Permana, H.; Kartasasmita, A.S.; Hilmanto, D.; Hidayat, R. Epigenetic Regulation of Sorbitol Dehydrogenase in Diabetic Retinopathy Patients: DNA Methylation, Histone Acetylation and microRNA-320. Biologics: Targets and Therapy.
- Dănilă, A.-I.; Ghenciu, L.A.; Stoicescu, E.R.; Bolintineanu, S.L.; Iacob, R.; Săndesc, M.-A.; Faur, A.C. Aldose reductase as a key target in the prevention and treatment of diabetic retinopathy: a comprehensive review. Biomedicines 2024, 12, 747. [Google Scholar] [CrossRef]
- He, W.; Tang, P.; Lv, H. Targeting oxidative stress in diabetic retinopathy: mechanisms, pathology, and novel treatment approaches. Frontiers in Immunology 2025, 16, 1571576. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Ahmad, M.F.A.; Khan, S.; Alouffi, S.; Khan, M.; Khan, M.W.A.; Ansari, I.A. Inhibition of the polyol pathway by Ducrosia anethifolia extract: plausible implications for diabetic retinopathy treatment. Frontiers in Pharmacology 2024, 15, 1513967. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Dai, H.; Jiang, S.; Yu, L. Advanced glycation end products in diabetic retinopathy and phytochemical therapy. Frontiers in Nutrition 2022, 9, 1037186. [Google Scholar] [CrossRef]
- Peña, J.S.; Ramanujam, R.K.; Risman, R.A.; Tutwiler, V.; Berthiaume, F.; Vazquez, M. Neurovascular Relationships in AGEs-Based Models of Proliferative Diabetic Retinopathy” Bioengineering 2024, 11, 63. 11. [CrossRef]
- Lu, Z.; Fan, B.; Li, Y.; Zhang, Y. RAGE plays key role in diabetic retinopathy: a review. BioMedical Engineering OnLine 2023, 22, 128. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Meng, Z.; Li, Y.; Liu, S.; Hu, P.; Luo, E. Advanced glycation end products and reactive oxygen species: uncovering the potential role of ferroptosis in diabetic complications. Molecular Medicine 2024, 30, 141. [Google Scholar] [CrossRef]
- Taguchi, K.; Fukami, K. RAGE signaling regulates the progression of diabetic complications. Frontiers in Pharmacology 2023, 14, 1128872. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Lindner, C.; Schneider, I.; Gonzalez, I.; Uribarri, J. The RAGE Axis: A Relevant Inflammatory Hub in Human Diseases. Biomolecules 2024, 14, 412. [Google Scholar] [CrossRef]
- Mahmud, N.M.; Paraoan, L.; Khaliddin, N.; Kamalden, T.A. Thymoquinone in ocular neurodegeneration: modulation of pathological mechanisms via multiple pathways. Frontiers in Cellular Neuroscience 2022, 16, 786926. [Google Scholar] [CrossRef]
- Pan, D.; Xu, L.; Guo, M. The role of protein kinase C in diabetic microvascular complications. Frontiers in Endocrinology 2022, 13, 973058. [Google Scholar] [CrossRef]
- Robles-Osorio, M.L.; Sabath, E. Tight junction disruption and the pathogenesis of the chronic complications of diabetes mellitus: A narrative review. World Journal of Diabetes 2023, 14, 1013. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Z. Mechanistic pathogenesis of endothelial dysfunction in diabetic nephropathy and retinopathy. Frontiers in endocrinology 2022, 13, 816400. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Liu, Q.; Chen, B.; Wu, F.; Li, Y.; Dong, X.; Ma, N.; et al. Protein O-GlcNAcylation coupled to Hippo signaling drives vascular dysfunction in diabetic retinopathy. Nature Communications 2024, 15, 9334. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Eshwaran, R.; Beck, S.C.; Hammes, H.P.; Wieland, T.; Feng, Y. Contribution of the Hexosamine Biosynthetic Pathway in the Hyperglycemia-Dependent and -Independent Breakdown of the Retinal Neurovascular Unit. Molecular Metabolism 2023, 73, 101736. [Google Scholar] [CrossRef]
- Liu, C.; Dong, W.; Li, J.; Kong, Y.; Ren, X. O-GlcNAc modification and its role in diabetic retinopathy. Metabolites 2022, 12, 725. [Google Scholar] [CrossRef]
- Starace, V.; Battista, M.; Brambati, M.; Cavalleri, M.; Bertuzzi, F.; Amato, A.; Lattanzio, R.; Bandello, F.; Cicinelli, M.V. The role of inflammation and neurodegeneration in diabetic macular edema. Therapeutic Advances in Ophthalmology 2021, 13, 25158414211055963. [Google Scholar] [CrossRef]
- Seo, H.; Park, S.-J.; Song, M. Diabetic Retinopathy (DR): Mechanisms, Current Therapies, and Emerging Strategies. Cells 2025, 14, 376. [Google Scholar] [CrossRef]
- González-Casanova, J.; Schmachtenberg, O.; Martínez, A.D.; Sanchez, H.A.; Harcha, P.A.; Rojas-Gomez, D. An Update on Connexin Gap Junction and Hemichannels in Diabetic Retinopathy. International Journal of Molecular Sciences 2021, 22, 3194. [Google Scholar] [CrossRef]
- Hussain, A.; Ashique, S.; Afzal, O.; Altamimi, M.A.; Malik, A.; Kumar, S.; Garg, A.; Sharma, N.; Farid, A.; Khan, T.; et al. A correlation between oxidative stress and diabetic retinopathy: an updated review. Experimental eye research 2023, 236, 109650. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tao, L.; Jiang, Z. Alleviate oxidative stress in diabetic retinopathy: antioxidant therapeutic strategies. Redox Report 2023, 28, 2272386. [Google Scholar] [CrossRef]
- Fragiotta, S.; Pinazo-Durán, M.D.; Scuderi, G. Understanding neurodegeneration from a clinical and therapeutic perspective in early diabetic retinopathy. Nutrients 2022, 14, 792. [Google Scholar] [CrossRef]
- Callan, A.; Jha, S.; Valdez, L.; Tsin, A. Cellular and molecular mechanisms of neuronal degeneration in early-stage diabetic retinopathy. Current Vascular Pharmacology 2024, 22, 301–315. [Google Scholar] [CrossRef]
- Balzamino, B.O.; Cacciamani, A.; Dinice, L.; Cecere, M.; Pesci, F.R.; Ripandelli, G.; Micera, A. Retinal Inflammation and Reactive Müller Cells: Neurotrophins’ Release and Neuroprotective Strategies. Biology 2024, 13, 1030. [Google Scholar] [CrossRef] [PubMed]
- Le, D.; Son, T.; Lim, J.I.; Yao, X. Quantitative optical coherence tomography reveals rod photoreceptor degeneration in early diabetic retinopathy. Retina 2022, 42, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Haydinger, C.D.; Oliver, G.F.; Ashander, L.M.; Smith, J.R. Oxidative Stress and Its Regulation in Diabetic Retinopathy. Antioxidants 2023, 12, 1649. [Google Scholar] [CrossRef]
- Srinivasan, S.; Pritchard, N.; Sampson, G.P.; Edwards, K.; Vagenas, D.; Russell, A.W.; Malik, R.A.; Efron, N. Diagnostic capability of retinal thickness measures in diabetic peripheral neuropathy. Journal of optometry 2017, 10, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Chondrozoumakis, G.; Chatzimichail, E.; Habra, O.; Vounotrypidis, E.; Papanas, N.; Gatzioufas, Z.; Panos, G.D. Retinal Biomarkers in Diabetic Retinopathy: From Early Detection to Personalized Treatment” Journal of Clinical Medicine 2025, 14, 1343. 14. [CrossRef]
- Simó, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in diabetic retinopathy: does it really matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Joglekar, M.V.; Hardikar, A.A.; Keech, A.C.; O’Neal, D.N.; Januszewski, A.S. Biomarkers in diabetic retinopathy. The review of diabetic studies: RDS 2015, 12, 159. [Google Scholar] [CrossRef]
- Tang, Z.; Chan, M.Y.; Leung, W.Y.; Wong, H.Y.; Ng, C.M.; Chan, V.T.; Wong, R.; Lok, J.; Szeto, S.; Chan, J.C.K.; et al. Assessment of retinal neurodegeneration with spectral-domain optical coherence tomography: a systematic review and meta-analysis. Eye 2021, 35, 1317–1325. [Google Scholar] [CrossRef]
- Trivedi, A.; Desbiens, J.; Gross, R.; Gupta, S.; Dodhia, R.; Ferres, J.L. Retinal Microvasculature as Biomarker for Diabetes and Cardiovascular Diseases. arXiv arXiv:2107.13157, 2021. [CrossRef]
- Bianco, L.; Arrigo, A.; Aragona, E.; Antropoli, A.; Berni, A.; Saladino, A.; Parodi, M.B.; Bandello, F. Neuroinflammation and neurodegeneration in diabetic retinopathy. Frontiers in Aging Neuroscience 2022, 14, 937999. [Google Scholar] [CrossRef] [PubMed]
- Fragiotta, S.; Abdolrahimzadeh, S.; Dolz-Marco, R.; Sakurada, Y.; Gal-Or, O.; Scuderi, G.; Milani, P. Significance of hyperreflective foci as an optical coherence tomography biomarker in retinal diseases: characterization and clinical implications. Journal of ophthalmology 2021, 2021, 6096017. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Pihl-Jensen, G.; Torm, M.E.W.; Passali, M.; Larsen, M.; Frederiksen, J.L. Hyperreflective dots in the avascular outer retina in relapsing-remitting multiple sclerosis. Multiple Sclerosis and Related Disorders 2023, 72, 104617. [Google Scholar] [CrossRef]
- Midena, E.; Torresin, T.; Velotta, E.; Pilotto, E.; Parrozzani, R.; Frizziero, L. OCT hyperreflective retinal foci in diabetic retinopathy: a semi-automatic detection comparative study. Frontiers in Immunology 2021, 12, 613051. [Google Scholar] [CrossRef]
- Artemiev, D.; Valmaggia, C.; Tschuppert, S.; Kotliar, K.; Türksever, C.; Todorova, M.G. Retinal vessel flicker light responsiveness and its relation to analysis protocols and static and metabolic data in healthy subjects. Biomedicines 2025, 13, 1201. [Google Scholar] [CrossRef]
- Lott, M.E.J.; Slocomb, J.E.; Shivkumar, V.; Smith, B.; Gabbay, R.A.; Quillen, D.; Gardner, T.W.; Bettermann, K. Comparison of retinal vasodilator and constrictor responses in type 2 diabetes. Acta ophthalmologica 2012, 90, e434–e441. [Google Scholar] [CrossRef]
- Dadzie, A.K.; Le, D.; Abtahi, M.; Ebrahimi, B.; Son, T.; Lim, J.I.; Yao, X. Normalized blood flow index in optical coherence tomography angiography provides a sensitive biomarker of early diabetic retinopathy. Translational Vision Science & Technology 2023, 12, 3–3. [Google Scholar] [CrossRef]
- Sachdeva Mira, M. Retinal neurodegeneration in diabetes: an emerging concept in diabetic retinopathy. Current diabetes reports 2021, 21, 65. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.J.; Joglekar, M.V.; Hardikar, A.A.; Keech, A.C.; O’Neal, D.N.; Januszewski, A.S. Biomarkers in diabetic retinopathy. The review of diabetic studies: RDS 2015, 12, 159. [Google Scholar] [CrossRef] [PubMed]
- Dartt Darlene, A. Encyclopedia of the Eye. Vol. 1. Academic Press, 2010.
- Channa, R.; Wolf, R.M.; Simo, R.; Brigell, M.; Fort, P.; Curcio, C.; Lynch, S.; Verbraak, F.; Abramoff, M.D.; Brigell, M.; et al. A new approach to staging diabetic eye disease: staging of diabetic retinal neurodegeneration and diabetic macular edema. Ophthalmology Science 2024, 4, 100420. [Google Scholar] [CrossRef] [PubMed]
- Arias-Alvarez, M.; Tomas-Grasa, C.; Sopeña-Pinilla, M.; Orduna-Hospital, E.; Fernandez-Espinosa, G.; Bielsa-Alonso, S.; Acha-Perez, J.; Rodriguez-Mena, D.; Pinilla, I. Electrophysiological findings in long-term type 1 diabetes patients without diabetic retinopathy using different ERG recording systems. Scientific Reports 2024, 14, 3520. [Google Scholar] [CrossRef]
- Constable, P.A.; Lim, J.K.; Thompson, D.A. Thompson. Retinal electrophysiology in central nervous system disorders. A review of human and mouse studies. Frontiers in Neuroscience 2023, 17, 1215097. [Google Scholar] [CrossRef]
- Kaur, Kirandeep, and Bharat Gurnani. Contrast sensitivity. (2022). https://www.ncbi.nlm.nih.gov/books/NBK580542/.
- Alghwiri Alia, A. Balance and falls. In Geriatric physical therapy, p. 331. Elsevier, 2011.
- Andrade Luciana Cristina, O.; Souza, G.S.; Lacerda, E.M.C.B.; Nazima, M.T.S.; Rodrigues, A.R.; Otero, L.M.; Pena, F.P.S.; Silveira, L.C.L.; Côrtes, M.I.T. Influence of retinopathy on the achromatic and chromatic vision of patients with type 2 diabetes. BMC ophthalmology 2014, 14, 104. [Google Scholar]
- Safi, S.; Rahimi, A.; Raeesi, A.; Safi, H.; Amiri, M.A.; Malek, M.; Yaseri, M.; Haeri, M.; Middleton, F.A.; Solessio, E.; et al. Contrast sensitivity to spatial gratings in moderate and dim light conditions in patients with diabetes in the absence of diabetic retinopathy. BMJ open diabetes research & care 2017, 5. [Google Scholar]
- Zhang, Z. , Deng, C. and Paulus, Y.M. Advances in Structural and Functional Retinal Imaging and Biomarkers for Early Detection of Diabetic Retinopathy. Biomedicines 2024, 12, 1405. [Google Scholar] [CrossRef]
- Yoshitake, S.; Murakami, T.; Uji, A.; Unoki, N.; Dodo, Y.; Horii, T.; Yoshimura, N. Clinical relevance of quantified fundus autofluorescence in diabetic macular oedema. Eye 2015, 29, 662–669. [Google Scholar] [CrossRef]
- Dumitrescu, O.-M.; Zemba, M.; Brănișteanu, D.C.; Pîrvulescu, R.A.; Radu, M.; Stanca, H.T. Fundus Autofluorescence in Diabetic Retinopathy. Journal of Personalized Medicine 2024, 14, 793. [Google Scholar] [CrossRef]
- Freund, K.B.; Mrejen, S.; Jung, J.; Yannuzzi, L.A.; Boon, C.J.F. Increased fundus autofluorescence related to outer retinal disruption. JAMA ophthalmology 2013, 131, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Toh, H.; Jiang, P. Fluorescein Angiography Image-AI Based Early Diabetic Retinopathy Detection. bioRxiv 2025, 2025–03. [Google Scholar] [CrossRef]
- Mohankumar, Arthi, and Bharat Gurnani. Scanning laser ophthalmoscope. (2022).
- LaRocca, F.; Dhalla, A.-H.; Kelly, M.P.; Farsiu, S.; Izatt, J.A. Optimization of confocal scanning laser ophthalmoscope design. Journal of Biomedical Optics 2013, 18, 076015–076015. [Google Scholar] [CrossRef]
- Park, Y.G.; Lee, J.-Y.; Kim, C.; Cicinelli, M.V. Early Microglial Changes Associated with Diabetic Retinopathy in Rats with Streptozotocin-Induced Diabetes. Journal of Diabetes Research 2021, 2021, 4920937. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Reichhart, N.; Hernandez-Matas, C.; Zabulis, X.; Kociok, N.; Brockmann, C.; Joussen, A.M.; Strauß, O. In vivo analysis of the time and spatial activation pattern of microglia in the retina following laser-induced choroidal neovascularization. Experimental eye research 2015, 139, 13–21. [Google Scholar] [CrossRef]
- Zeng, S.; Zhang, T.; Madigan, M.C.; Fernando, N.; Aggio-Bruce, R.; Zhou, F. Interphotoreceptor retinoid-binding protein (IRBP) in retinal health and disease. Front Cell Neurosci. 2020, 14, 577935. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Joglekar, M.V.; Hardikar, A.A.; Keech, A.C.; O’Neal, D.N.; Januszewski, A.S. Biomarkers in diabetic retinopathy. The review of diabetic studies: RDS 2015, 12, 159. [Google Scholar] [CrossRef]
- Chen, Y.; Meng, Y.; Tan, M.; Ma, J.; Zhu, J.; Ji, M.; Guan, H. Changes in expression of inflammatory cytokines and ocular indicators in pre-diabetic patients with cataract. BMC ophthalmology 2025, 25, 119. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, Q.; Li, Y.; Zeng, L.; Liu, J.; Ou, K. On implications of somatostatin in diabetic retinopathy. Neural Regeneration Research 2024, 19, 1984–1990. [Google Scholar] [CrossRef] [PubMed]
- Soedarman, S.; Julia, M.; Gondhowiardjo, T.D.; Kurnia, K.H.; Prasetya, A.D.B.; Triyoga, I.F.; Sasongko, M.B. Serum Apolipoprotein B and B/A1 Ratio as Early Negative Biomarkers for OCT-and OCTA-Detected Retinal Changes in Diabetic Macular Edema. Clinical Ophthalmology 2025, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, E.R.; Mahmoud, D.A.; Ebrahim, G.E.; Al Anany, M.G.; Seliem, N.; Hassan, M.M. Serum metabolomic profiles and semaphorin-3A as biomarkers of diabetic retinopathy progression. Egypt. J. Immunol 2023, 30, 83–98. [Google Scholar] [CrossRef]
- Mohammad, G. , Siddiquei, M.M. and Abu El-Asrar, A.M. Poly (ADP-ribose) polymerase mediates diabetes-induced retinal neuropathy. Mediators of inflammation 2013, 2013, 510451. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Dralands, L.; Missotten, L.; Al-Jadaan, I.A.; Geboes, K. Expression of apoptosis markers in the retinas of human subjects with diabetes. Investigative ophthalmology & visual science 2004, 45, 2760–2766. [Google Scholar]
- Valverde, Angela M., Soledad Miranda, Marta García-Ramírez, Águeda González-Rodriguez, Cristina Hernández, and Rafael Simó. Proapoptotic and survival signaling in th Miyagishima, Kiyoharu Joshua, Francisco Manuel Nadal-Nicolás, Wenxin Ma, and Wei Li. Annexin-V binds subpopulation of immune cells altering its interpretation as an in vivo biomarker for apoptosis in the retina. International Journal of Biological Sciences 20, no. 15 (2024): 6073.e neuroretina at early stages of diabetic retinopathy. Molecular Vision 19 (2013): 47.
- Hajari, J.N.; Ilginis, T.; Pedersen, T.T.; Lønkvist, C.S.; Saunte, J.P.; Hofsli, M.; Schmidt, D.C.; Al-Abaiji, H.A.; Ahmed, Y.; Bach-Holm, D.; et al. Novel Blood-Biomarkers to Detect Retinal Neurodegeneration and Inflammation in Diabetic Retinopathy. International Journal of Molecular Sciences 2025, 26, 2625. [Google Scholar] [CrossRef] [PubMed]
- Klochkov, V.; Chan, C.-M.; Lin, W.-W. Methylglyoxal: A Key Factor for Diabetic Retinopathy and Its Effects on Retinal Damage. Biomedicines 2024, 12, 2512. [Google Scholar] [CrossRef]
- Bentata, R.; Cougnard-Grégoire, A.; Delyfer, M.N.; Delcourt, C.; Blanco, L.; Pupier, E.; Rougier, M.B.; Rajaobelina, K.; Hugo, M.; Korobelnik, J.F.; et al. Skin autofluorescence, renal insufficiency and retinopathy in patients with type 2 diabetes. Journal of Diabetes and its Complications 2017, 31, 619–623. [Google Scholar] [CrossRef]
- Qiu, F.; Ma, X.; Shin, Y.H.; Chen, J.; Chen, Q.; Zhou, K.; Wu, W.; Liang, W.; Wu, Y.; Song, Q.; et al. Pathogenic role of human C-reactive protein in diabetic retinopathy. Clinical Science 2020, 134, 1613–1629. [Google Scholar] [CrossRef]
- Amorim, M.; Martins, B.; Caramelo, F.; Gonçalves, C.; Trindade, G.; Simão, J.; Barreto, P.; Marques, I.; Leal, E.C.; Carvalho, E.; et al. Putative biomarkers in tears for diabetic retinopathy diagnosis. Frontiers in medicine 2022, 9, 873483. [Google Scholar] [CrossRef]
- Pan, X.; Tan, X.; McDonald, J.; Kaminga, A.C.; Chen, Y.; Dai, F.; Qiu, J.; Zhao, K.; Peng, Y. Chemokines in diabetic eye disease. Diabetology & Metabolic Syndrome 2024, 16, 115. [Google Scholar] [CrossRef]
- Thota, R.N.; Chatterjee, P.; Pedrini, S.; Hone, E.; Ferguson, J.J.; Garg, M.L.; Martins, R.N. Association of plasma neurofilament light chain with glycaemic control and insulin resistance in middle-aged adults. Frontiers in Endocrinology 2022, 13, 915449. [Google Scholar] [CrossRef]
- Balzamino, B.O.; Cacciamani, A.; Dinice, L.; Cecere, M.; Pesci, F.R.; Ripandelli, G.; Micera, A. Retinal Inflammation and Reactive Müller Cells: Neurotrophins’ Release and Neuroprotective Strategies. Biology 2024, 13, 1030. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ling, F.; Zhang, G.W.; Yu, N.; Yang, J.; Xin, X.Y. The correlation between MicroRNAs and diabetic retinopathy. Frontiers in immunology 2022, 13, 941982. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wan, J.; Tan, G. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in diabetic retinopathy. Frontiers in immunology 2023, 14, 1151185. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Su, G. Roles of miRNAs and long noncoding RNAs in the progression of diabetic retinopathy. Bioscience Reports 2017, 37, BSR20171157. [Google Scholar] [CrossRef]
- Zhao, H.; Cai, Y.; Pan, J.; Chen, Q. Role of MicroRNA in linking diabetic retinal neurodegeneration and vascular degeneration. Frontiers in Endocrinology 2024, 15, 1412138. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, W.; Zheng, X.; Yang, M.M. New insights of potential biomarkers in diabetic retinopathy: integrated multi-omic analyses. Frontiers in Endocrinology 2025, 16, 1595207. [Google Scholar] [CrossRef]
- Habib, S.; Mansour, A.M.; Baban, B.; Elmasry, K. Extracellular Vesicles and Diabetic Retinopathy: Nano-Sized Vesicles with Mega-Sized Hopes. In Diabetic Retinopathy-Advancement in Understanding the Pathophysiology and Management Strategies. IntechOpen, 2024.
- Peng, X.; Peng, Y.; Li, L.; Liu, L.; Zhou, G.; Cai, Y. Breakthroughs in diabetic retinopathy diagnosis and treatment using preclinical research models: current progress and future directions. Annals of Medicine 2025, 57, 2531251. [Google Scholar] [CrossRef]
- Carmichael, J.; Fadavi, H.; Tavakoli, M. Neurodegeneration of the cornea and retina in patients with type 1 diabetes without clinical evidence of diabetic retinopathy. Frontiers in Endocrinology 2022, 13, 790255. [Google Scholar] [CrossRef]
- Cerani, A.; Tetreault, N.; Menard, C.; Lapalme, E.; Patel, C.; Sitaras, N.; Beaudoin, F.; et al. Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell metabolism 2013, 18, 505–518. [Google Scholar] [CrossRef]
- Yuan, J.; Huang, R.; Nao, J.; Dong, X. The Role of Semaphorin 3 A in the Pathogenesis and Progression of Alzheimer’s Disease and Other Aging-Related Diseases: A Comprehensive Review. Pharmacological Research 2025, 107732. [Google Scholar] [CrossRef]
- Bista Karki, S.; Coppell, K.J.; Mitchell, L.V.; Ogbuehi, K.C. Dynamic pupillometry in type 2 diabetes: pupillary autonomic dysfunction and the severity of diabetic retinopathy. Clinical ophthalmology 2020, 3923–3930. [Google Scholar] [CrossRef] [PubMed]
- Chudzik, A.; Śledzianowski, A.; Przybyszewski, A.W. Przybyszewski. Machine learning and digital biomarkers can detect early stages of neurodegenerative diseases. Sensors 2024, 24, 1572. [Google Scholar] [CrossRef]
- Jain, M. , Devan, S.; Jaisankar, D.; Swaminathan, G.; Pardhan, S.; Raman, R. Pupillary abnormalities with varying severity of diabetic retinopathy. Scientific reports 2018, 8, 5636. [Google Scholar] [CrossRef]
- Hannan, A.; Mahmood, Z.; Qureshi, R.; Ali, H. Enhancing diabetic retinopathy classification accuracy through dual-attention mechanism in deep learning. Computer Methods in Biomechanics and Biomedical Engineering: Imaging & Visualization 2025, 13, 2539079. [Google Scholar] [CrossRef]
- Yao, J.; Lim, J.; Lim, G.Y.S.; Ong, J.C.L.; Ke, Y.; Tan, T.F.; Tan, T.E.; Vujosevic, S.; Ting, D.S.W. Novel artificial intelligence algorithms for diabetic retinopathy and diabetic macular edema. Eye and Vision 2024, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Yagin, F.H.; Colak, C.; Algarni, A.; Gormez, Y.; Guldogan, E.; Ardigò, L.P. Hybrid Explainable Artificial Intelligence Models for Targeted Metabolomics Analysis of Diabetic Retinopathy. Diagnostics 2024, 14, 1364. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, J.; Jin, E.; Zhong, Y.; Zhang, L.; Han, X.; Liu, J.; et al. Serum untargeted metabolomics reveal potential biomarkers of progression of diabetic retinopathy in asians. Frontiers in Molecular Biosciences 2022, 9, 871291. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



