Submitted:
14 August 2025
Posted:
15 August 2025
You are already at the latest version
Abstract
Acute human alpha-herpesvirus 1 (HSV-1) infection culminates in a latent infection of neurons in trigeminal ganglia (TG), and central nervous system. Following infection of mucosal epithelial cells, certain neurons survive infection and life-long latency is established. Periodically, stressful stimuli trigger reactivation from latency, which result in virus shedding, transmission to other people, and occasionally recurrent disease. The glucocorticoid receptor (GR) and Krüppel-like factor 15 (KLF15) comprise a feed-forward transcriptional loop that cooperatively transactivate key HSV-1 promoters that drive expression of infected cell protein 0 (ICP0), ICP4, and ICP27. Silencing KLF15 significantly reduces HSV-1 replication in cultured mouse neuroblastoma cells. Consequently, we hypothesized KLF15 mediates certain aspects of reactivation from latency. To test this hypothesis, we compared HSV-1 replication in KLF15–/– mice versus wild-type (wt) parental C57BL/6 mice. Virus shedding during acute infection was reduced in KLF15–/– mice. Male KLF15–/– mice shed higher titers of virus during late stages of reactivation from latency compared to KLF15–/– females and wt mice regardless of sex. At 15 d after explant-induced reactivation, virus shedding was higher in male KLF15-/- mice relative to wt mice and female KLF15-/- mice. These studies confirm KLF15 expression enhances viral replication during acute infection and reactivation from latency.
Keywords:
HSV-1
; KLF15
; latency
; trigeminal ganglia
; reactivation from latency
1. Introduction
Human alpha-herpesvirus 1 (HSV-1) infection of facial mucous membranes culminate in lifelong latency in sensory neurons in trigeminal ganglia (TG), brainstem neurons and other neurons in the central nervous system [1,2,3]. These infections can lead to recurrent disease including gingivostomatitis [4,5], herpes labialis [6] and stromal keratitis, which is the leading cause of infectious blindness in the world [7,8,9,10]. Furthermore, HSV-1 is the most common cause of sporadic, fatal encephalitis in which virus reactivation from latency accounts for over two-thirds of all cases [11,12,13,14]. There are no commercially available HSV-1 vaccines and current antiviral drugs (acyclovir and related compounds) do not significantly reduce reactivation from latency [15,16,17].
During productive HSV-1 infection, robust viral DNA replication and protein expression occurs, which culminates in high levels of infectious progeny for virus transmission and spread of disease [18,19,20]. In latently infected neurons, the locus that encodes the latency associated transcript (LAT), micro-RNAs, and other non-coding RNAs are the only abundantly expressed viral products during latency [21,22,23,24]. LAT gene products inhibit apoptosis and viral gene expression, thereby establishing and maintaining a reservoir of latently infected TG neurons, reviewed in [23,24]. During latency, infectious virus is not readily detected.
Periodically, various stimuli [19,25] including physical and psychological stress trigger reactivation from latency, reviewed in [25,26]. Stress leads to increased corticosteroid levels that bind and activate the glucocorticoid receptor (GR), which subsequently enters the nucleus, binds specific GR response elements in DNA, and induces gene expression [27]. The synthetic corticosteroid dexamethasone (DEX) consistently induces HSV-1 virus shedding during explant-induced reactivation from latency [28,29,30]. Stress also induces expression of certain cellular transcription factors, including several members of the Krüppel-like factor (KLF) family of transcription factors including KLF15 [31,32,33]. Notably, KLF15 plays a key role in amino acid, lipid, and glucose metabolism [34], catabolic metabolism of cardiomyocytes [35], adipogenesis (36), and maintenance of smooth muscle through interactions with GR [37,38]. Studies from our lab demonstrated GR and KLF15 cooperatively transactivate key HSV-1 immediate early promoters that drive expression of transcriptional viral regulatory proteins, including infected cell protein 0 (ICP0) [39,40], ICP4 cis-regulatory module [41], and an ICP27 cis-regulatory module [42] using transient transfection studies. Finally, silencing KLF15 impaired HSV-1 and bovine alpha-herpes virus 1 replication in permissive cultured cells [43].
Based on these observations, we hypothesized KLF15 plays a role during the HSV-1 latency and reactivation cycle. A KLF15-knockout (KLF15–/–) mouse model of ocular infection was used to test this hypothesis. Notably, virus shedding during acute infection was reduced in male and female KLF15–/– mice relative to wild-type (wt) age-matched littermates. In contrast, when latently infected TG are explanted in medium that contains DEX to stimulate reactivation, male KLF15–/– mice shed higher titers of virus at late stages of reactivation when compared to wt male and female littermates, and female KLF15–/– mice. Collectively, these studies revealed KLF15 is important but not required for HSV-1 acute infection following ocular infection of mice. Furthermore, sex-specific factors regulate virus production in KLF15–/– mice during explant-induced reactivation from latency.
2. Materials and Methods
2.1. Viruses and Cell Lines
Monkey kidney cells (Vero ATCC CCL-81) were grown in minimal essential medium (MEM; Corning) supplemented with 10% FBS (Atlas Biologicals), 2 mM L-glutamine (Corning), and antibiotics (100 IU/mL penicillin, 100 µg/mL Streptomycin, Cytiva Hyclone) at 37°C, 5% CO2. HSV-1 strain McKrae was obtained from the late Dr. Steven Wechsler (University of California, Irvine Medical School) and grown in Vero cells until >80% cytopathic effect was observed. Cells and supernatant were collected and serial freeze-thawed at 37°C/-80°C followed by centrifugation to remove cellular debris and titered on Vero monolayers to determine plaque-forming unit/mL (PFU/mL). Stocks were aliquoted and stored at -80°C prior to mouse infections.
2.2. Mouse Breeding and Infection Studies
All animal experiments were approved and performed in accordance with the Oklahoma State University Institutional Animal Care and Use Committee (protocol VM-24-58) and maintained in Techniplast Blue Line cages. Individually ventilated cages get 10–15 air changes/h within a 12 h-light/dark cycle, air-conditioned, temperature and humidity-controlled environment. Male and female mice heterozygous for KLF15/LacZ expression were kindly gifted by Dr. Anthony N. Gerber (National Jewish Health, Denver CO) [44] and fed high-fat diet (11% LabDiet, St. Louis, MO, USA) for enhanced fertility and reproduction [45]. Upon arrival, mice were paired for breeding while under quarantine for health testing and retained until either 5 months of age or 5 successful litters (gestational period ~21 days). Ten days after birth, pups were sexed, ear-tagged and < 2 mm of tail snipped, incubated with alkaline lysis buffer (25 mM NaOH / 0.2 mM EDTA) at 98°C for 1 h, and then neutralized with 40 mM Tris HCl (pH 5.5) [46]. Samples were centrifuged at 2,000 g for 1 min to pellet debris and supernatant isolated for PCR-based genotyping. Standard end-point PCR was used with primers designed to amplify the mouse KLF15 and LacZ insert. KLF15 forward: 5’-GCATCTGCTGTCCACCTATT-3’; KLF15 Reverse: 5’-GTTCGTCTATGCCTCACCTATTC-3’; LacZ forward 5’-ATCCTCTGCATGGTCAGGTC-3’; LacZ reverse: 5’-CCGTGGCCTGATTCATTCC-3’. Homozygous KLF15–/– male and female pups were weaned from parental breeding pairs 21 days after birth and used for subsequent breeding schemes. Between 6- and 8-weeks of age, KLF15–/– littermates were divided into disposable Techniplast cages (5 mice/cage) within a BSL-2 approved facility and C57BL/6 age-matched control animals were purchased from Jackson Labs (strain 000664). All mice were acclimated for a minimum of 7 days prior to infection. Mice were ocularly infected in both eyes without scarification using 105 PFU of HSV-1 strain McKrae as previously described [28,47,48]. All infection studies consisted of a minimum of two independent experiments with 4-5 mice/group unless otherwise indicated.
2.3. Infection of Primary Mouse Kidney Cells
Kidneys were aseptically dissected from 6–8-week-old uninfected KLF15–/– and wt C57BL/6 male and female mice, and single cells prepared as previously described [48]. Briefly, kidneys were dipped in 100% ethanol, minced into <3 mm pieces and incubated with 0.25% trypsin for 4-18 h with rocking at 4°C. At 4 h intervals, supernatant was removed, and single cells were pelleted via centrifugation. Remaining tissues were incubated with residual trypsin and incubated at 37°C for 30 min. Cells were pelleted via centrifugation at 200 g for 10 min then gently dispersed by pipetting. All cells were filtered through a 100 µm cell strainer, and viability was determined via trypan blue assay. Approximately, 105 cells were plated per well into 24-well plates and infected with HSV-1 at a multiplicity of infection of 1 at 37°C, 5% CO2 with rocking. Virus was removed and cells were incubated for 24 h prior to plaquing for virus titers on Vero cells as described above.
2.4. Ocular Swabs, TG Plaque Assays, and Explant-Induced Reactivation
Swabs from cornea and conjunctiva were collected from individual animals into 1 mL MEM with L-glutamine, antibiotics, and 10% FBS every other day for the first 10 days post-infection, and every 5 days until the remainder of the experiment. Samples were frozen at -80°C until ready for use. Serial dilutions of ocular swabs were used to infect Vero monolayers for 1 h at 37°C, 5% CO2 with rocking. Monolayers were overlayed with 1% methylcellulose in MEM with L-glutamine, 10% FBS, and antibiotics and monitored for visible plaques. If plaques were not observed after 72 h, samples were considered negative for the virus.
At 4- and 8-days post-infection, TG were dissected, minced into three to four pieces, and placed in 60 mm dishes with confluent Vero monolayers. If cytopathic effect was not observed, tissue and supernatant were removed and 1% methylcellulose in MEM with L-glutamine, 10% FBS, and antibiotics was overlayed onto the monolayer and allowed to incubate for 3 days prior to crystal violet staining for plaque visualization. If cytopathic effect was detected within 24 h, cells and supernatant were collected, serially frozen/thawed (−80 to 37°C), and titered on Vero monolayers..
For explant-induced reactivation, TG were collected in MEM with L-glutamine, antibiotics, 2% charcoal-stripped FBS, and 10 µM water-soluble DEX (Sigma; catalog no. D2915) to induce virus reactivation from latency 30 dpi and incubated in a CO2 incubator set at 37° C and 5% CO2. Daily, aliquots of supernatant were removed and used to plaque for infectious virus on Vero monolayers as previously described [28,47,48].
2.5. Reverse Transcription and Quantitative PCR (qPCR)
At the designated times, TG and kidney were prepared. Kidneys do not contain any HSV-1 DNA following ocular infection and serve as an internal reference tissue. The use of an internal reference tissue within the same animal as experimental samples provides a twofold reduction in technical variation and PCR efficiency [48,49,50]. Tissues were minced and incubated in alkaline lysis buffer as described for tail snips above. Lysis buffer was neutralized with 40 mM Tris-HCl pH 5 and briefly centrifuged to remove cell debris. Following cell lysis, DNA purification was performed as described [47,48,49,50]; briefly, two rounds of phenol:chloroform:isoamyl alcohol (25:24:1) extraction was performed followed by one round of chloroform:isoamyl alcohol (24:1 v/v) and ethanol precipitation. Two volumes of ice-cold 100% ethanol were added and incubated at −20°C overnight. DNA was pelleted via centrifugation (18,000 g for 30 min), washed twice with 100% ethanol twice and air-dried prior to dissolving in DEPC-water. A total of 10 ng of genomic DNA was used as a template for Sybr Green qPCR (Applied Biosciences) using primers for HSV-1 gB and mouse GAPDH. gB forward primer 5′- AACGCGACGCACATCAAG; gB reverse primer 5′- CTGGTACGCGATCAGAAAGC; GAPDH forward primer 5′- CATCACTGCCACCCAGAAGACTG; GAPDH reverse primer 5′- ATGCCAGTGAGCTTCCCGTTCAG.
For RT-qPCR, TG and kidney were dissected 30 dpi and homogenized in Trizol using GentleMACS M-tubes and program RNA_01. M Tubes were centrifuged at 2,000 g for 5 min prior to application with the Qiagen RNeasy Kit according to the manufacturer’s instructions. Samples were eluted with DEPC-water and analyzed for yield and quality. 100 ng RNA was used with the SuperScript III Platinum One-Step qRT-PCR Kit (Invitrogen) according to manufacturer’s instructions using primers for HSV-1 LAT and mouse GAPDH. LAT forward primer 5′- CCTTATCTAAGGGCCGGCTG-3’; LAT rev primer 5′- GGGACACATGCCTTCTTGGA-3’.
All primers were designed, synthesized, and purchased from IDT. A BioRad CFX Opus 96 PCR system was used along with CFX Maestro Analysis Software: Cq values between 20 and 35 were considered positive. Ratios of gB or LAT to GAPDH were calculated using the Delta-Delta CT method.
2.6. Statistical Analysis
All graphs and comparisons were performed using GraphPad Prism software (v10.5.p). P values less than 0.05 were considered significant for all calculations with specific tests and post-hoc analysis indicated in the respective figure legends.
3. Results
3.1. Generation and Validation of KLF15 Knockout Mice
To investigate the role KLF15 plays during HSV-1 infection, a mutant mouse model with a targeted deletion of the murine KLF15 gene was used. Heterozygous KLF15 knockout mice (KLF15+/–) contain a lacZ gene at the KLF15 ATG [34] and were bred to generate homozygous knockout (KLF15–/–) offspring. Endpoint PCR-based genotyping was performed on 10-day old pups to distinguish heterozygous from homozygous littermates using primers specific to the murine KLF15 and LacZ, as described in the materials and methods. As shown in Figure 1, KLF15 alleles were detected as ~483 bp bands (Figure 1: lanes 1 and 4), while the LacZ insertion yielded a 125 bp product (Figure 1: lanes 2 and 5). Pups expressing both genes are consistent with heterozygosity and were humanely euthanized; pups expressing only the 483 bp band (KLF15+/+) are referred to as wt. KLF15–/– mice lack the KLF15 PCR product but contain the 125 bp LacZ band, confirming successful deletion of KLF15. These mice were used for subsequent breeding and infection studies. Notably, breeding KLF15–/– males to KLF15–/– females resulted in runted pups relative to wt age-matched and heterozygous KLF15+/– mice (data not shown). KLF15–/– mouse litters weighted ~8 grams by 21 days of age: consequently, weaning was delayed until 28 days after birth. WT age-matched mice weighed between 10 and 12 grams and were weaned at the standard 21 days after birth. Each KLF15–/– litter contained approximately four males for every female and litters tended to be smaller.
3.2. Examination of HSV-1 Replication in Primary Kidney Cells from KLF15–/– Mice
Primary cells were prepared from kidneys dissected from uninfected KLF15–/– mice or age-matched wt controls. The neurovirulent HSV-1 strain McKrae was used to infect these cells at a multiplicity of infection (MOI) of 1. Twenty four hours post infection (hpi), virus replication was measured using plaque assays (Figure 2). For all groups, infection of primary kidney cells resulted in ~105 plaque-forming units (PFU)/mL. There were no differences detected between sexes (male versus female) or kidney cells from wt vs KLF15–/–
These data indicated KLF15 is not required for HSV-1 replication in kidney cells. These results were different compared to previously published studies because silencing KLF15 in a mouse neuroblastoma cell line (Neuro-2a) resulted in approximately 2-log reduction of HSV-1 virus production [43]. We suggest KLF15 is not important in certain cell-types because other transcription factors compensate for deletion of KLF15. For example, the ICP4 CRM was efficiently transactivated by GR and KLF15 in Vero cells but not in Neuro-2A cells [41].
3.3. HSV-1 Replication Is Impaired During Acute Infection of KLF15–/– Mutant Mice when Comapred to wt Mice
Eight week-old KLF15–/– or wt mice (males and females) were ocularly infected with HSV-1 strain McKrae as described in methods and materials. Swabs from the orbital cavity were collected every other day for the first 10 days to measure acute virus shedding and every 5 days from days 10 through 30 to test whether there were differences in the length of acute infection (Figure 3A). Viral titers from cornea and conjunctival surfaces were quantified using plaque assays. All groups of mice were shedding ~103 PFU/mL by 2 days post-infection (dpi, Figure 3B). Virus shedding from male and female wt mice peaked at 4 dpi with ~5x104 PFU/mL present in swabs. In contrast, virus shedding from ocular surfaces of KLF15–/– mice was significantly lower at 4 dpi compared to wt mice. For example, 2,500 PFU/mL were present in swabs from male and female KLF15–/– mice at 4 dpi. At 6 dpi, virus shedding in ocular surfaces of wt male and female mice was reduced and was not significantly different than KLF15–/– mice regardless of sex. Interestingly, HSV-1 virus shedding (~5000 PFU/mL) peaked at 6 dpi in KLF15–/– male and female mice. In contrast, the peak of HSV-1 shedding in ocular swab occurred at 4 dpi. As expected, infectious virus was not detected in ocular swabs of wt or KLF15–/– mice from 8 through 30 dpi indicating latency was established and maintained. Mortality did not occur following ocular infection with the MOI used for these studies in wt or KLF15–/– male and female mice.
3.4. Comparison of infectious virus and total viral DNA in TG of wt versus KLF15–/– mice after infection
TG neurons are a crucial site for establishing HSV-1 latency following infection of the ocular cavity, reviewed in [23,24,25,26]. TG from wt and KLF15–/– male and female mice were collected at 4 and 8 dpi and used to test for virus replication. At 4 dpi, wt male and female TG were shedding ~5,000 PFU/mL. (Figure 4A). In contrast, infectious virus present in TG from male and female KLF15–/– mice was significantly lower and contained less than 250 PFU/mL. By 8 dpi, virus shedding from TG of wt and KLF15–/– mice was not detected regardless of sex.
HSV-1 DNA levels in TG were measured by qPCR using primers specific for the highly conserved HSV-1 glycoprotein B [51,52]. TG collected from male and female KLF15–/– mice 4 dpi had significantly lower ratios of HSV-1 gB DNA relative to mouse GAPDH DNA compared to wt mice, with ratios of 500 for KLF15–/– males and only 25 for KLF15–/– females (Figure 4B). In comparison, wt male and female TG had gB:GAPDH ratios more than 1500. The trend of KLF15–/– mice containing lower HSV-1 DNA in TG was also present at 8 dpi when wt gB:GAPDH ratios dropped to ~25 for male and females. However, KLF15–/– male and female gB:GAPDH ratios were significantly decreased when compared to wt mice (Figure 4C).
By 30 dpi (operationally defined as latency), KLF15–/– DNA ratios (gB versus GAPDH) were significantly lower in female mice versus males (Figure 4D). DNA levels in wt male and female mice were significantly higher than female KLF15–/– mice but not male KLF15–/– mice (Figure 4D). These studies revealed that establishment of latency, as judged by HSV-1 DNA in TG 30 dpi, was similar for wt males, wt female mice, and KLF15–/– male mice. Conversely, female KLF15–/– mice contained significantly lower levels of viral DNA compared to the other groups.
3.5. Analysis of LAT RNA in Latently Infected wt and KLF15–/– Female Mice
Given the differences in gB:GAPDH DNA ratios for female KLF15–/– mice at 30 dpi compared to male KLF15–/– and wt mice of both sexes, LAT expression was measured. LAT is the only abundantly expressed viral gene during HSV-1 latency and accordingly serves as a hallmark of latency [21,23,24,53]. TG from the respective groups was collected at 30 dpi and RNA prepared for RT-qPCR with primers directed toward LAT (Figure 5A). As expected, LAT levels relative to GAPDH were equal for wt mice (male and female), and KLF15–/– male mice. In contrast, LAT RNA levels were significantly reduced (2-log) for female KLF15–/– mice relative to wt mice and KLF15–/– male mice. When comparing levels of HSV-1 gB DNA to LAT RNA expression, all groups had ratios of approximately 1 (Figure 5B) indicating LAT expression levels were the same when compared to viral DNA in TG of latently infected mice (Figure 4C). This study also revealed that female KLF15–/– mice contained similar levels of LAT/viral genomes. In summary, these studies suggest fewer TG neurons in female KLF15–/– mice were latently infected or fewer viral genomes were present in latently neurons.
3.6. Analysis of Explant-Induced Reactivation from Latency
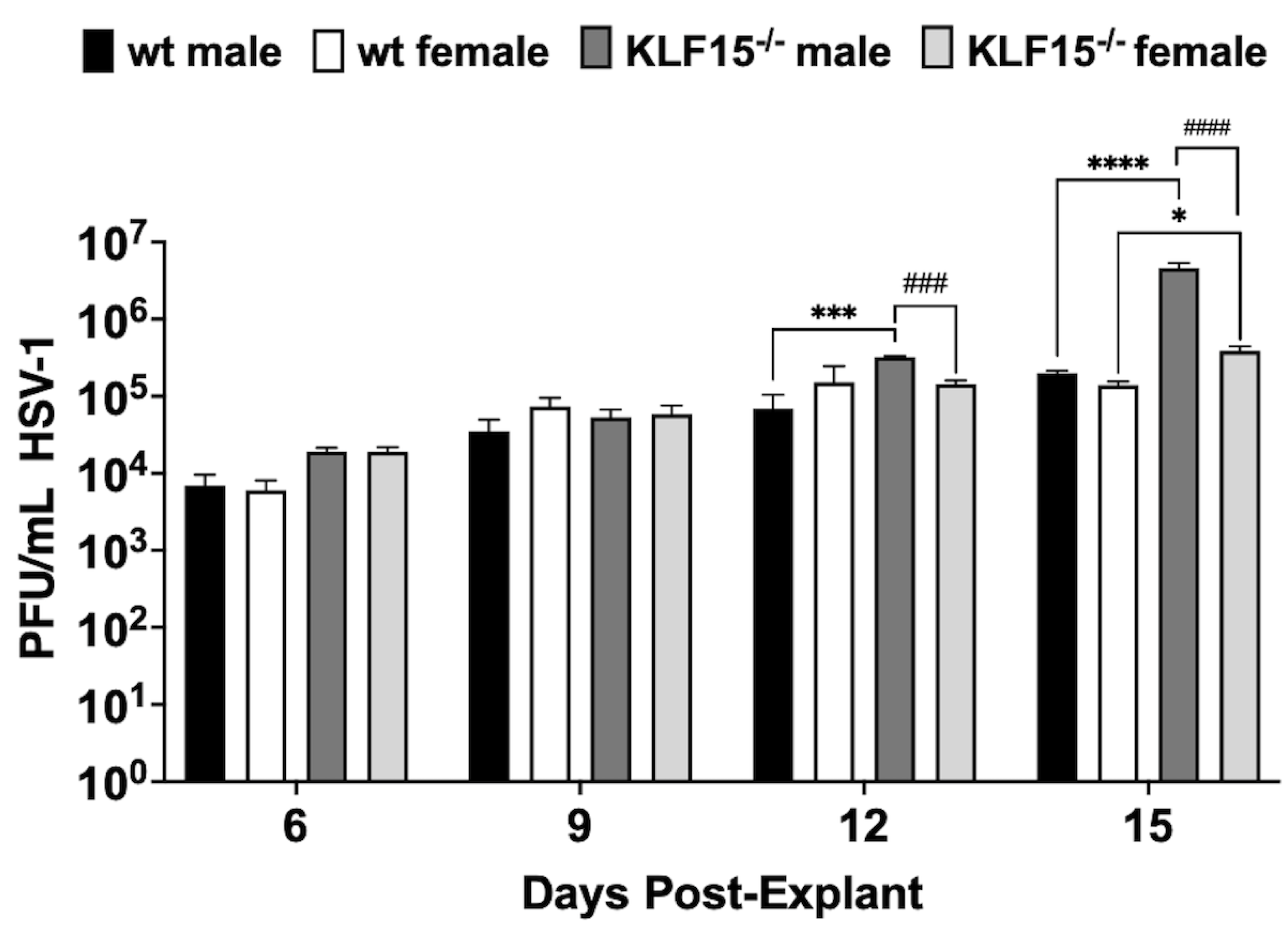
To investigate HSV-1 reactivation in latently infected KLF15–/– mice, TG were dissected at 30 dpi and explanted in minimal essential medium (MEM) + 2% charcoal-stripped fetal bovine serum (FBS) in the presence of 10 µM DEX to accelerate virus reactivation [28]. Passing FBS through an activated charcoal column removes hormonal factors and reduces the concentration of certain growth factors, vitamins, and metabolites while leaving salts, glucose, and most amino acids important for cellular growth [55,56,57]. Following TG explant, aliquots were collected after explant and used to plaque for infectious virus. Under the conditions of these studies, virus shedding is not readily detected until 4-6 days post-explant (dpe) [47,48]. All groups exhibited titer increases until 9 dpe with no significant differences (Figure 6). Between 12-15 dpe, virus shedding in TG of wt male and female mice was not significantly increased indicating virus production plateaued. Surprisingly, KLF15–/– male mice exhibited a significant increase in virus shedding relative to wt males at 12 or 15 dpi. By 15 days after explant, KLF15–/– male mice had over a 1-log increase in virus replication compared to wt mice regardless of sex. Conversely, TG from female KLF15–/– mice was not significantly different than wt females and males. Like wt mice 12 and 15 days after TG explant, KLF15–/– female TG was shedding significantly less virus than KLF15–/– males. For example, KLF15–/– male mice shed more than 3-fold HSV-1 compared to KLF15–/– female mice (males shed: ~3x105 PFU/mL vs females shed: ~105 PFU/mL) at 12 dpi. By 15 days after explant, KLF15–/– males shed more than 106 PFU/mL whereas KLF15–/– female mice remained significantly lower at ~4 x 105 PFU/mL. Despite lower levels of viral DNA in TG of KLF15–/– female mice, there was not a significant reduction of virus shedding compared to wt mice during explant-induced reactivation.
4. Discussion
These studies revealed HSV-1 replication was impaired in ocular swabs (Figure 3B) during acute infection. Furthermore, lower levels of HSV-1 virus shedding in TG were detected at 4- and 8-days post-infection regardless of sex (Figure 4A) whereas lower levels of viral DNA were detected only in TG of female KLF15-/- mice (Figure 4D). A previous study revealed silencing KLF15 in Neuro-2A cells reduced viral replication [43]. Since several studies demonstrated GR and KLF15 cooperatively transactivate HSV-1 immediate early promoter activity (ICP0, ICP4, and ICP27) [39,40,41,42], these results were expected. Conversely, the finding that HSV-1 grew as efficiently in primary kidney cells from KLF15–/– mice and wt C57Bl/6 mice (Figure 2) was unexpected. We predict primary kidney cells express certain cellular transcription factors and/or signaling pathways that compensate for lack of KLF15 expression. There are 18 human KLF family members suggesting one or more of these proteins compensated for lack of KLF15 expression [31,32,33].
Notably, sex-dependent differences were observed in KLF15–/– mice during establishment of latency and DEX-induced explant-induced reactivation from latency in TG. For example, male KLF15–/– mice contained higher levels of virus shedding for longer times after TG explant (12 and 15 days after explant) when compared to KLF15–/– females and wt mice regardless of sex. Conversely, female KLF15–/– mice contained lower levels of LAT and HSV-1 DNA levels at 30 dpi when compared to KLF15–/– male mice or wt mice. Despite LAT levels being lower in female KLF15–/– mice, the ratio of LAT compared to viral DNA was not significantly different than wt mice or male KLF15–/– mice. This may be important because LAT plays an essential role in maintaining latency by suppressing lytic gene expression and apoptosis [22,23,24,57]. Based on these observations in KLF15–/– mice, we suggest LAT promotes establishment and maintenance of latency in a small pool of latently infected TG neurons.
A previous study compared viral replication and reactivation from latency using a mouse model that contains a serine 229 to alanine mutation in GR (GRS229A) [48]. Serine 229 in the mouse GR must be phosphorylated for optimal GR-mediated transcription [58]. Like KLF15–/– mice, HSV-1 shedding from cornea and conjunctiva of infected GRS229A mice stopped prior to wt mice. In contrast to KLF15–/– mice, viral replication was significantly reduced in kidney cells from GRS229A mice when compared to wt mice. Furthermore, viral DNA levels in TG were not significantly different in GRS229A and wt mice during latency. Like KLF15–/– mice, HSV-1 viral titers during explant-induced reactivation were significantly reduced in female GRS229A mice versus male GRS229A mice or wt mice. To test whether a mouse that expresses the GRS229A protein but not KLF15, these two mutant mouse strains were bred to produce a double knockout. Double knockout pup frequencies were significantly less than the expected 6.25% [59]. Very few mice survived beyond 10 days postnatal; hence, this approach was abandoned. Since the GR and KLF15 feed-forward transcription loop is important for regulating brain and heart development [35,60,61], and amino acid, lipid and glucose metabolism [34,44,62], we suggest certain functions of GR and KLF15 expression were necessary for growth and mouse development after birth. Furthermore, GR and KLF15 cooperate to regulate growth and development of skeletal muscle, and transcriptional regulation following corticosteroid-induced muscle atrophy was more profound in male mice [63]. Collectively, these KLF15 functions may explain the observed breeding and developmental differences observed between male and female KLF15–/– mice.
Despite reduced viral replication in male KLF15–/– mice during acute infection, it was surprising to find that virus shedding increased during late stages of reactivation from latency in male KLF15–/– mice. We suggest other KLF family members or male-specific signaling pathways compensate for lack of KLF15 during late stages of explant-induced reactivation from latency. Recent studies revealed KLF15 transcriptionally activates expression of Serpina6, which encodes the corticosteroid-binding globulin [64]. In KLF15–/– mice, corticosteroid-binding globulin levels are 90% less than wt mice. Consequently, corticosterone levels are 3-4-fold higher in plasma of KLF15–/– mice, which may have allowed viral replication during late stages of reactivation from latency in male KLF15–/– mice. Furthermore, Serpina6 regulates a systemic response to inflammatory stress and KLF15 plays a role in clearing reactive oxygen species. We suggest multiple functions of KLF15 in mammals contributed to the unexpected effects on the HSV-1 latency-reactivation cycle in KLF15-/- mice. Future studies will test whether de novo HSV-1 protein expression during stress-induced reactivation and whether KLF15 interacts with other hormone receptors to provide insight into how KLF15 regulates viral replication and reactivation from latency. In addition to GR and KLF15 cooperatively transactivating the ICP0 promoter [40], ICP4 CRM sequences [41], and ICP27 CRM sequences [42], it is reasonable to predict that other important viral promoters are transactivated by GR and KLF15.
5. Conclusions
In conclusion, these studies revealed KLF15 expression was important during acute infection of mice but less important during explant-induced reactivation from latency. For example, despite reduced establishment of latency, explant-induced reactivation from latency was not dramatically affected in female KLF15–/– mice. The finding that male KLF15-–/– mice, but not wt mice or female KLF15–/– mice, continued to shed high levels of virus at 12 and 15 days after explant implies male-specific transcription factors or signaling pathways maintained reactivation or enhanced virus spread to adjacent TG neurons and viral replication occurred. Since KLF15 belongs to a large family of transcription factors [64,65,66,67], certain KLF family members may compensate for lack of KLF15 expression. In addition to DEX activating GR, KLF15 can cooperate with the progesterone receptor, and/or the androgen receptor to activate bovine herpesvirus (BoHV-1) immediate early gene expression, which enhances productive infection [68,69,70]. Hence, the progesterone receptor or androgen receptor may play a role in the sex-dependent HSV-1 effects that were observed during acute ocular infection and/or explant-induced reactivation in KLF15–/– mice. Since VP16 specifically transactivates IE promoters and is proposed to trigger reactivation from latency, reviewed in [23,24,25], it is reasonable to suggest GR and KLF family members transactivate VP16 promoter activity. However, a VP16 CRM reporter constructwas not transactivated by GR and KLF4 or KLF15 [70]. Finally, it is reasonable to predict that male-specific factors prolong the late effects that drove increased shedding virus in male KLF15-/- mice.
Author Contributions
Conceptualization, C.J. and K.S.H.; methodology, C.J. and K.S.H.; resources, C.J; writing—original draft preparation, K.S.H.; writing—review and editing, C.J and K.S.H.; project administration, C.J.; funding acquisition, C.J. and K.S.H. Both authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
All animal experiments were approved and performed in accordance with the Oklahoma State University Institutional Animal Care and Use Committee (protocol VM-24-58 approved 17 December 2024).
Informed Consent Statement
Not applicable.
Data Availability Statement
In accordance with MDPI’s data policies, the data presented in this study are available upon request from the corresponding author. Raw data supporting the findings of this research will be made available in compliance with the journal’s guidelines and ethical standards for data sharing
Acknowledgments
This research was supported by grants from the National Institute of Neurological Disorders and Stroke of the NIH under award number R01NS111167 (CJ), National Institute of Allergies and Infectious Diseases under award number NIH R21AI178282-01A1 (CJ), Oklahoma Center for Respiratory and Infectious Diseases (National Institutes of Health Centers for Biomedical Research Excellence Grant # P20GM103648), funds from the Sitlington Endowment (CJ), and a seed grant from the OSU CVM (KH). KLF15-/- mice were obtained from Dr. Anthony N. Gerber (National Jewish Health, Denver CO).
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
Abbreviations
The following abbreviations are used in this manuscript: TG (trigeminal ganglia); DEX (dexamethasone); HSV-1 (Herpes simplex virus-1); GR (glucocorticoid receptor); KLF15 (Krüppel-like factor 15); ICP (infected cell protein); wt (C57BL/6J mice); LAT (latency associated transcript); PCR (polymerase chain reaction); RT-qPCR (reverse transcription quantitative PCR); PFU (plaque forming units); dpi (days post-infection); dpe (days post-explant); MOI (multiplicity of infection); min (minutes); bp (base pair); SD (standard deviation); SEM (standard error of the mean); mL (milliliters); gB (glycoprotein B); GAPDH (Glyceraldehyde-3-phosphate dehydrogenase); ANOVA (analysis of variance); BoHV-1 (bovine herpesvirus-1).
References
- Fraser, N.W.; Lawrence, W.C.; Wroblewska, Z.; Gilden, D.H.; Koprowski, H. Herpes simplex virus type 1 DNA in human brain tissue. Proc Natl Acad Sci USA. 1981, 78, 6461–5. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, C.C.; Wohlenberg, C.; Openshaw, H.; Ray-Mendez, M.; Puga, A.; Notkins, A.L. Herpes simplex virus DNA sequences in the CNS of latently infected mice. Nature. 1980, 288, 228–90. [Google Scholar] [CrossRef]
- Rock, D.L.; Fraser, N.W. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature. 1983, 302, 523–5. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. Periodontal diagnoses and classification of periodontal diseases. Periodontol 2000, 34, 9–21. [Google Scholar] [CrossRef]
- Arduino, P.G.; Porter, S.R. Herpes Simplex Virus Type I infection: overview on relevant clinico-pathological features. J Oral Pathol Med. 2008, 37, 107–21. [Google Scholar] [CrossRef]
- Whitley, R.J.; Kimberlin, D.W.; Roizman, B. Herpes simplex viruses. Clin Infect Dis. 1998, 26, 541–53. [Google Scholar] [CrossRef]
- Smith, R.E.; McDonald, H.R.; Nesburn, A.B.; Minckler, D.S. Penetrating keratoplasty: changing indications, 1947 to 1978. Arch Ophthalmol. 1980, 98, 1226–9. [Google Scholar] [CrossRef]
- Pavan-Langston, D. Herpes simplex of the ocular anterior segment. Curr Clin Top Infect Dis. 2000, 20, 298–324. [Google Scholar]
- Barker, N.H. Ocular herpes simplex. BMJ Clin Evid. 2008, 2008, 0707. [Google Scholar]
- Cabrera-Aguas, M.; Robaei, D.; McCluskey, P.; Watson, S. Clinical translation of recommendations from randomized trials for management of herpes simplex virus keratitis. Clinical & Experimental Ophthalmology. 2018, 46, 1008–16. [Google Scholar]
- Skoldenberg, B. Herpes simplex encephalitis. Scand J Infect Dis Suppl. 1991, 80, 40–6. [Google Scholar] [PubMed]
- Stahl, J.P.; Mailles, A.; Dacheux, L.; Morand, P. Epidemiology of viral encephalitis in 2011. Medecine et maladies infectieuse. 2011, 41, 453–64. [Google Scholar] [CrossRef]
- Yamada, S.; Kameyama, T.; Nagaya, S.; Hashizume, Y.; Yoshida, M. Relapsing herpes simplex encephalitis: pathological confirmation of viral reactivation. J Neurol Neurosurg Psychiatry. 2002, 74, 262–4. [Google Scholar] [CrossRef]
- Rantalaiho TFårkkilå, N.; Vaheri, A.; Koskiniemi, M. Acute encephalitis from 1967 to 1991. J Neurol Sci. 2001, 184, 169–77. [Google Scholar] [CrossRef]
- Wald, A.; Johnston, C. Treatment and prevention of herpes simplex virus type 1 in immunocompetent adolescents and adults. The South African General Practitioner. 2024, 5, 25–32. [Google Scholar] [CrossRef]
- Sharma, D.; Sharma, S.; Akojwar, N.; Dondulkar, A.; Yenorkar, N.; Pandita, D.; et al. An Insight into Current Treatment Strategies, Their Limitations, and Ongoing Developments in Vaccine Technologies against Herpes Simplex Infections. Vaccines (Basel). 2023, 11, 206. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Wald, A.; Krantz, E.; Selke, S.; Warren, T.; Vargas-Cortes, M.; et al. Valacyclovir and Acyclovir for Suppression of Shedding of Herpes Simplex Virus in the Genital Tract. J Infect Dis. 2004, 190, 1374–81. [Google Scholar] [CrossRef]
- Harkness, J.M.; Kader, M.; DeLuca, N.A. Transcription of the Herpes Simplex Virus 1 Genome during Productive and Quiescent Infection of Neuronal and Nonneuronal Cells. J Virol. 2014, 88, 6847–61. [Google Scholar] [CrossRef]
- Koyuncu, O.O.; MacGibeny, M.A.; Enquist, L.W. Latent versus productive infection: the alpha herpesvirus switch. Future Virology. 2018/05/22 ed. 2018, 13, 431–43. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; Dekker, L.J.M.; van den Ham, H.J.; Lenac Rovis, T.; Haefner, E.S.; Jonjic, S.; Haas, J.; Luider, T.M.; Verjans, G.M. Analysis of Virus and Host Proteomes During Productive HSV-1 and VZV Infection in Human Epithelial Cells. Front Microbiol. 2020, 11, 1179. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thompson, R.L.; Sawtell, N.M. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J Virol. 1997, 71, 5432–40. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.L.; Sawtell, N.M. The herpes simplex virus type 1 latency associated transcript locus is required for the maintenance of reactivation competent latent infections. J Neurovirology. 2011, 17, 552–8. [Google Scholar] [CrossRef]
- Perng, G.C.; Jones, C. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis. 2010 Feb 02/20 ed. 2010, 2010, 1–18. [Google Scholar] [CrossRef]
- Phelan, D.; Barrozo, E.R.; Bloom, D.C. HSV1 latent transcription and non-coding RNA: A critical retrospective. J Neuroimmunology. 2017, 308(March):65, 67,71-66. [Google Scholar] [CrossRef] [PubMed]
- Jones, C. Intimate Relationship Between Stress and Human Alpha-Herpes Virus 1 (HSV-1) Reactivation from Latency. Curr Clin Microbiol Rep. 2023, 10, 236–245, Epub 2023 Jul 27. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jones, C. Reactivation from latency by alpha-herpesvirinae subfamily members: a stressful situation. Curr Topics Virol. 2014, 12, 99–118. [Google Scholar]
- Oakley RH and JA Cidlowski. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013, 132, 1033–44. [Google Scholar] [CrossRef]
- Harrison, K.S.; Zhu, L.; Thunuguntla, P.; Jones, C. Antagonizing the glucocorticoid receptor impairs explant-induced reactivation in mice latently infected with herpes simplex virus 1. J Virol. 2019, JVI.00418-19. [Google Scholar]
- Halford, W.P.; Gebhardt, B.M.; Carr, D.J. Mechanisms of herpes simplex virus type 1 reactivation. J Virol. 1996 Aug 08/01 ed. 1996, 70, 5051–60. [Google Scholar] [CrossRef]
- Du, T.; Zhou, G.; Roizman, B.; Du, G. Zhou, and B. Roizman T. Induction of apoptosis accelerates reactivation from latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A. 2012, 109, 14616–21. [Google Scholar] [CrossRef]
- Bieker, J.J. Krüppel-like factors: three fingers in many pies. J Biological Chemistry. 2001, 276, 34355–8. [Google Scholar] [CrossRef]
- McConnell, B.B.; Yang, V.W. Mammalian Krüppel-like factors in health and diseases. Physiol Rev. 2010, 90, 1337–81. [Google Scholar] [CrossRef] [PubMed]
- Yuce, K.; Ozkan, A.I. The krüppel-like factor (KLF) family, diseases, and physiological events. Gene. 2024, 895, 148027. [Google Scholar] [CrossRef]
- Sasse, S.K.; Mailloux, C.M.; Barczak, A.J.; Wang, Q.; Altonsy, M.O.; Jain, M.K.; et al. The glucocorticoid receptor and KLF15 regulate gene expression dynamics and integrate signals through feed-forward circuitry. Molec and Cellular Biol. 2013, 33, 2104–15. [Google Scholar] [CrossRef] [PubMed]
- Fisch, S.; Gray, S.; Heymans, S.; Haldar, S.M.; Wang, B.; Pfister, O.; et al. Krüppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2007, 104, 7074–9. [Google Scholar] [CrossRef]
- Mori, T.; Sakaue, H.; Iguchi, H.; Gomi, H.; Okada, Y.; Takashima, Y.; et al. Role of Kruppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem. 2005, 280, 12867–75. [Google Scholar] [CrossRef]
- Gray, S.; Kuo, C.; Watanabe, M.; Charles, H.; Feinberg, M.; Jain, M. Role of Klf15, a novel Kruppel-like factor, in regulating smooth muscle cell growth and differentiation. Circulation. 2001, 104, 90–90. [Google Scholar]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.; et al. Crosstalk between Glucocorticoid Receptor and Nutritional Sensor mTOR in Skeletal Muscle. Cell Metabolism. 2011, 13, 170–82. [Google Scholar] [CrossRef]
- Wijesekera, N.; Hazell, N.; Jones, C. Independent Cis-Regulatory Modules within the Herpes Simplex Virus 1 Infected Cell Protein 0 (ICP0) Promoter Are Transactivated by Krüppel-like Factor 15 and Glucocorticoid Receptor. Viruses. 2022, 14, 1284. [Google Scholar] [CrossRef] [PubMed]
- Ostler, J.B.; Harrison, K.S.; Schroeder, K.; Thunuguntla, P.; Jones, C. The glucocorticoid receptor (GR) stimulates Herpes Simplex Virus 1 productive infection, in part because the infected cell protein 0 (ICP0) promoter is cooperatively transactivated by the GR and Krüppel-like transcription factor 15. J Virol. 2019 Mar 01/04 ed. 2019.
- Ostler, J.B.; Thunuguntla, P.; Hendrickson, B.Y.; Jones, C. Transactivation of Herpes Simplex Virus 1 (HSV-1) Infected Cell Protein 4 Enhancer by Glucocorticoid Receptor and Stress-Induced Transcription Factors Requires Overlapping Krüppel-Like Transcription Factor 4/Sp1 Binding Sites. J Virol. 2021, 95. [Google Scholar] [CrossRef]
- Ostler, J.B.; Jones, C. Stress Induced Transcription Factors Transactivate the Herpes Simplex Virus 1 Infected Cell Protein 27 (ICP27) Transcriptional Enhancer. Viruses. 2021, 13. [Google Scholar] [CrossRef]
- El-Mayet, F.S.; Harrison, K.S.; Jones, C. Regulation of Krüppel-Like Factor 15 Expression by Herpes Simplex Virus Type 1 or Bovine Herpesvirus 1 Productive Infection. Viruses. 2021, 13. [Google Scholar] [CrossRef]
- Gray, S. M.W. Feinberg, S. Hull, C.T. Kuo, M. Watanabe, S.S. Banerjee, A. DePina, R. Haspel, and M.K. Jain. The Krüppel-like Factor KLF15 Regulates the Insulin-sensitive Glucose Transporter GLUT4. J of Biological Chemistry. 2002, 277, 34322–8. [Google Scholar] [CrossRef]
- Lecker, J.; Froberg-Fejko, K. Using environmental enrichment and nutritional supplementation to improve breeding success in rodents. Lab Animal. 2016, 45, 406–7. [Google Scholar] [CrossRef]
- Truett, G.E.; Heeger, P.; Mynatt, R.L.; Truett, A.A.; Walker, J.A.; Warman, M.L. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques. 2000, 29, 52–54. [Google Scholar] [CrossRef]
- Harrison, K.S.; Zhu, L.; Thunuguntla, P.; Jones, C. Herpes simplex virus 1 regulates β-catenin expression in TG neurons during the latency-reactivation cycle. PloS one. 2020, 15, e0230870. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.S.; Wijesekera, N.; Robinson, A.G.J.; Santos, V.C.; Oakley, R.H.; Cidlowski, J.A.; Jones, C. Impaired glucocorticoid receptor function attenuates herpes simplex virus 1 production during explant-induced reactivation from latency in female mice. J Virol. 2023, 97, e0130523. [Google Scholar] [CrossRef]
- Hildyard, J.C.W.; Wells, D.J.; Piercy, R.J. Identification of qPCR reference genes suitable for normalising gene expression in the developing mouse embryo. Wellcome Open Res. 2021, 6, 197. [Google Scholar] [CrossRef]
- Harrison, K.S.; Cowan, S.R.; Jones, C. Murine nasal-associated lymphoid tissue (NALT) harbors human alphaherpesvirus 1 (HSV-1) DNA during latency, and dexamethasone triggers viral replication. J Virol. 2025, 99, e0225124. [Google Scholar] [CrossRef]
- Lewandowski, G. Herpes simplex type 1 infects and establishes latency in the brain and trigeminal ganglia during primary infection of the lip in cotton rats and mice. Arch Virol. 2002, 147, 167–79. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.H.; Gu, B.; Person, S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J Virol. 1988, 62, 2596–604. [Google Scholar] [CrossRef] [PubMed]
- Jones, C. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv Virus Res. 1998, 51, 81–133. [Google Scholar]
- Cao, Z.; West, C.; Norton-Wenzel, C.S.; Rej, R.; Davis, F.B.; Davis, P.J.; et al. Effects of resin or charcoal treatment on fetal bovine serum and bovine calf serum. Endocr Res. 2009, 34, 101–8. [Google Scholar] [CrossRef]
- Tu, C.; Fiandalo, M.V.; Pop, E.; Stocking, J.J.; Azabdaftari, G.; Li, J.; et al. Proteomic Analysis of Charcoal-Stripped Fetal Bovine Serum Reveals Changes in the Insulin-like Growth Factor Signaling Pathway. J Proteome Res. 2018, 17, 2963–77. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.R.; Qu, L.H.; Ma, L.M. Differential impacts of charcoal-stripped fetal bovine serum on c-Myc among distinct subtypes of breast cancer cell lines. Biochem Biophys Res Commun. 2020, 526, 267–72. [Google Scholar] [CrossRef]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science. 2000, 287, 1500–3. [Google Scholar] [CrossRef] [PubMed]
- Galliher-Beckley, A.J.; Cidlowski, J.A. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 2009, 61, 979–86. [Google Scholar] [CrossRef]
- Hall, B.; Limaye, A.; Kulkarni, A.B. Overview: Generation of Gene Knockout Mice. Curr Protoc Cell Biol. 2009. Chapter:Unit-19.1217. [Google Scholar] [CrossRef]
- Kennedy, C.L.M.; Price, E.M.; Mifsud, K.R.; Salatino, S.; Sharma, E.; Engledow, S.; et al. Genomic regulation of Krüppel-like-factor family members by corticosteroid receptors in the rat brain. Neurobiology of Stress. 2023, 23, 100532. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, K.R.; Kennedy, C.L.M.; Salatino, S.; Sharma, E.; Price, E.M.; Haque, S.N.; et al. Distinct regulation of hippocampal neuroplasticity and ciliary genes by corticosteroid receptors. Nat Commun. 2021, 12, 4737. [Google Scholar] [CrossRef] [PubMed]
- Sasse, S.K.; Zuo, Z.; Kadiyala, V.; Zhang, L.; Pufall, M.A.; Jain, M.K.; et al. Response Element Composition Governs Correlations between Binding Site Affinity and Transcription in Glucocorticoid Receptor Feed-forward Loops. J Biol Chem. 2015, 290, 19756–69. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Schönke, M.; Buurstede, J.C.; Moll, T.J.A.; Gentenaar, M.; Schilperoort, M.; et al. Sexual Dimorphism in Transcriptional and Functional Glucocorticoid Effects on Mouse Skeletal Muscle. Front Endocrinol (Lausanne). 2022, 13, 907908. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Elsarrag, S.Z.; Duan, Q.; LaGory, E.L.; Wang, Z.; Alexanian, M.; et al. KLF15 cistromes reveal a hepatocyte pathway governing plasma corticosteroid transport and systemic inflammation. Science Advances. 2022, 8, eabj2917. [Google Scholar] [CrossRef]
- Black, A.R.; Black, J.D.; Azizkhan-Clifford, J. Sp1 and Krüppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001, 188, 143–60. [Google Scholar] [CrossRef] [PubMed]
- Suske, G. The Sp-family of transcription factors. Gene. 1999, 238, 291–300. [Google Scholar] [CrossRef]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Krüppel-like transcription factors. Genome Biology. 2003, 4, 206. [Google Scholar] [CrossRef]
- El-Mayet, F.S.; El-Habbaa, A.S.; D’Offay, J.; Jones, C. Synergistic Activation of Bovine Herpesvirus 1 Productive Infection and Viral Regulatory Promoters by the Progesterone Receptor and Krüppel-Like Transcription Factor 15. J Virol. 2018, 93, e01519–18. [Google Scholar] [CrossRef]
- El-mayet, F.S.; Sawant, L.; Thunuguntla, P.; Jones, C. Combinatorial Effects of the Glucocorticoid Receptor and Krüppel-Like Transcription Factor 15 on Bovine Herpesvirus 1 Transcription and Productive Infection. J Virol. 2017, 91, e00904–17. [Google Scholar] [CrossRef]
- Santos, V.C.; Ostler, J.B.; Harrison, K.S.; Jones, C. Slug, a stress-induced transcription factor, stimulates herpes simplex virus type 1 replication and transactivates a cis-regulatory module within the VP16 promoter. J Virol. 2023. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Identification of homozygous KLF15 knockout mice. Male and female mice heterozygous for KLF15/LacZ expression (KLF15+/–, lanes 1 and 4) were paired for breeding during and after they were quarantined for health testing. Pups were ear-tagged at postnatal day 10 and tail snips obtained for genotyping according to methods and materials. Male and female KLF15–/– pups positive only for LacZ (lanes 2 and 5) were used for breeding and infection studies.
Figure 1.
Identification of homozygous KLF15 knockout mice. Male and female mice heterozygous for KLF15/LacZ expression (KLF15+/–, lanes 1 and 4) were paired for breeding during and after they were quarantined for health testing. Pups were ear-tagged at postnatal day 10 and tail snips obtained for genotyping according to methods and materials. Male and female KLF15–/– pups positive only for LacZ (lanes 2 and 5) were used for breeding and infection studies.
Figure 2.
HSV-1 infection of KLF15–/– primary kidney cells. Primary kidney cells were prepared from 8-week-old male and female wt and KLF15–/– mice (n= 4–6 from two separate experiments). HSV-1 was used to infect these cells at a MOI of 1. Twenty-four hours post-infection, cells and supernatant were collected, freeze-thawed three times at 37°C/−80°C and subsequently used for plaque assays on Vero cell monolayers. Data shown are the mean + SD for triplicate wells of duplicate experiments in plaque forming units (PFU)/mL.
Figure 2.
HSV-1 infection of KLF15–/– primary kidney cells. Primary kidney cells were prepared from 8-week-old male and female wt and KLF15–/– mice (n= 4–6 from two separate experiments). HSV-1 was used to infect these cells at a MOI of 1. Twenty-four hours post-infection, cells and supernatant were collected, freeze-thawed three times at 37°C/−80°C and subsequently used for plaque assays on Vero cell monolayers. Data shown are the mean + SD for triplicate wells of duplicate experiments in plaque forming units (PFU)/mL.
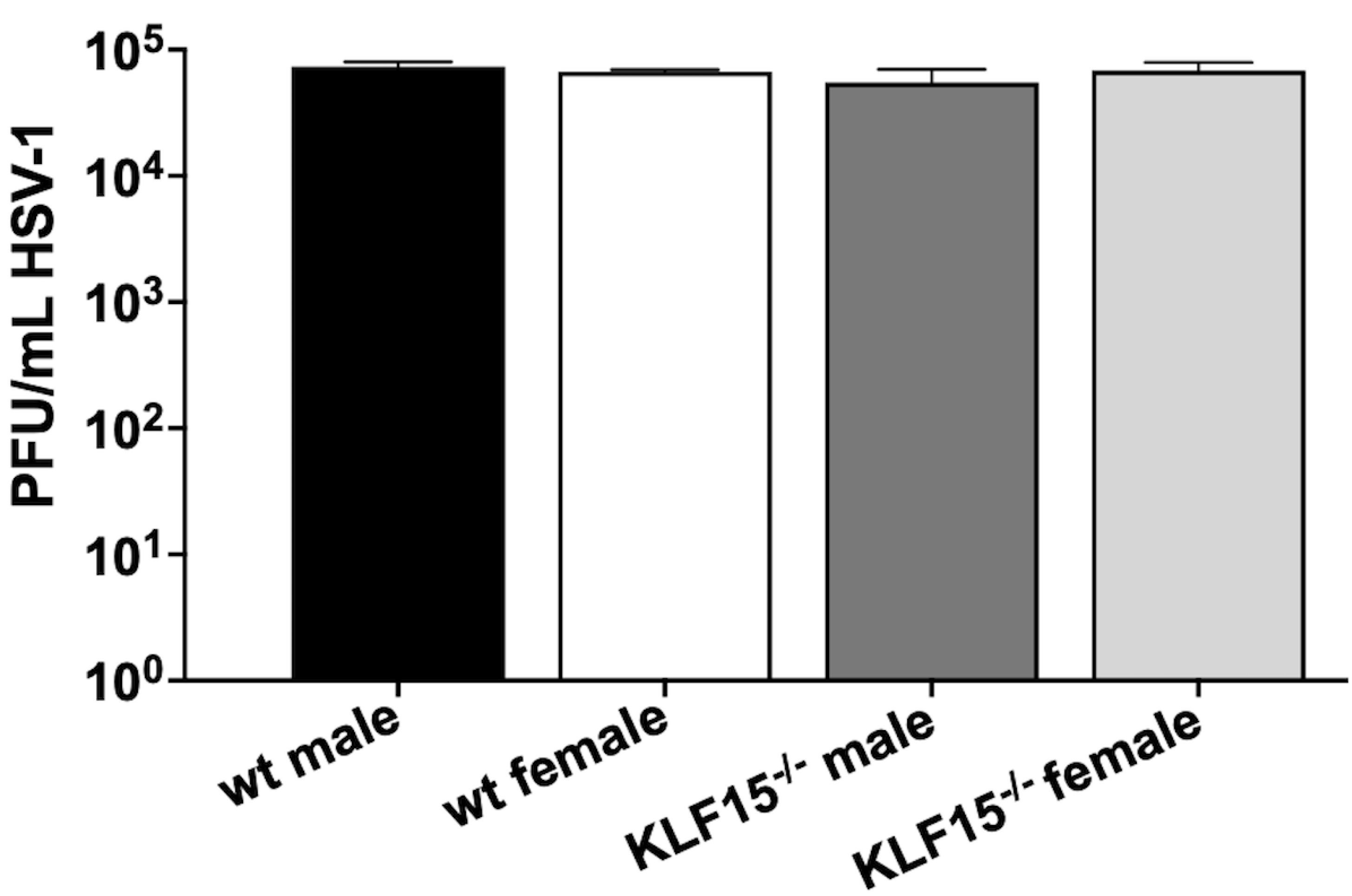
Figure 3.
Quantification of HSV-1 virus shedding in ocular surfaces of KLF15–/– and wt mice. Panel A: Schematic of animal infection studies. KLF15–/– or wt mice were ocularly infected with HSV-1 on day 0. Ocular swabs were collected every other day until day 10 when swabs were collected every 5 days. Panel B: Following ocular swabs of each group, infectious virus was measured at the denoted days after infection. Data are shown as time-plot mean ± SEM of individual animals (n = 5 mice/group, two separate experiments); #p<0.05 for wt mice compared to KLF15–/– mice using unpaired T-test. .
Figure 3.
Quantification of HSV-1 virus shedding in ocular surfaces of KLF15–/– and wt mice. Panel A: Schematic of animal infection studies. KLF15–/– or wt mice were ocularly infected with HSV-1 on day 0. Ocular swabs were collected every other day until day 10 when swabs were collected every 5 days. Panel B: Following ocular swabs of each group, infectious virus was measured at the denoted days after infection. Data are shown as time-plot mean ± SEM of individual animals (n = 5 mice/group, two separate experiments); #p<0.05 for wt mice compared to KLF15–/– mice using unpaired T-test. .
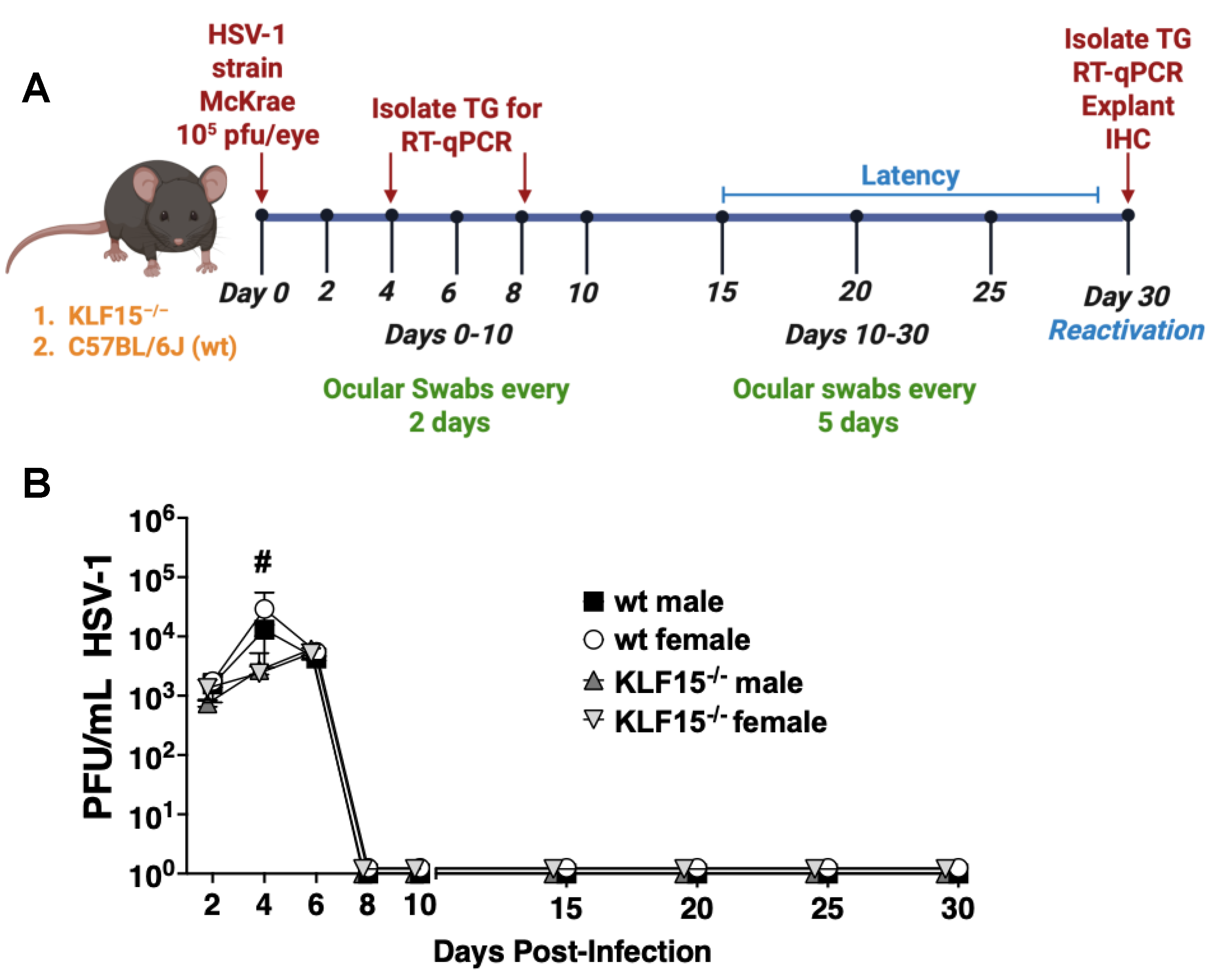
Figure 4.
Measurement of infectious virus in mouse TG during acute infection. Panel A: At 4- and 8 days post-infection, TG were dissected from mice (n=5), minced and incubated with Vero cell monolayers followed by plaque assays. Data are shown as mean + SEM for triplicate wells of duplicate experiments. Panels B–D: TG from 4-5 animals/group were collected at 4-, 8-, or 30-days post-infection (latency, 2 separate experiments). DNA was prepared from the respective samples and qPCR performed using primers for HSV-1 gB or mouse GAPDH. Ratio of gB:GAPDH DNA was calculated using the Delta-Delta CT method. Data are shown as box-and-whisker plots from max to min, with a line at the mean. **p<0.01; ***p< 0.005; ****p<0.0001 using one way ANOVA with unpaired multiple comparison post-test.
Figure 4.
Measurement of infectious virus in mouse TG during acute infection. Panel A: At 4- and 8 days post-infection, TG were dissected from mice (n=5), minced and incubated with Vero cell monolayers followed by plaque assays. Data are shown as mean + SEM for triplicate wells of duplicate experiments. Panels B–D: TG from 4-5 animals/group were collected at 4-, 8-, or 30-days post-infection (latency, 2 separate experiments). DNA was prepared from the respective samples and qPCR performed using primers for HSV-1 gB or mouse GAPDH. Ratio of gB:GAPDH DNA was calculated using the Delta-Delta CT method. Data are shown as box-and-whisker plots from max to min, with a line at the mean. **p<0.01; ***p< 0.005; ****p<0.0001 using one way ANOVA with unpaired multiple comparison post-test.
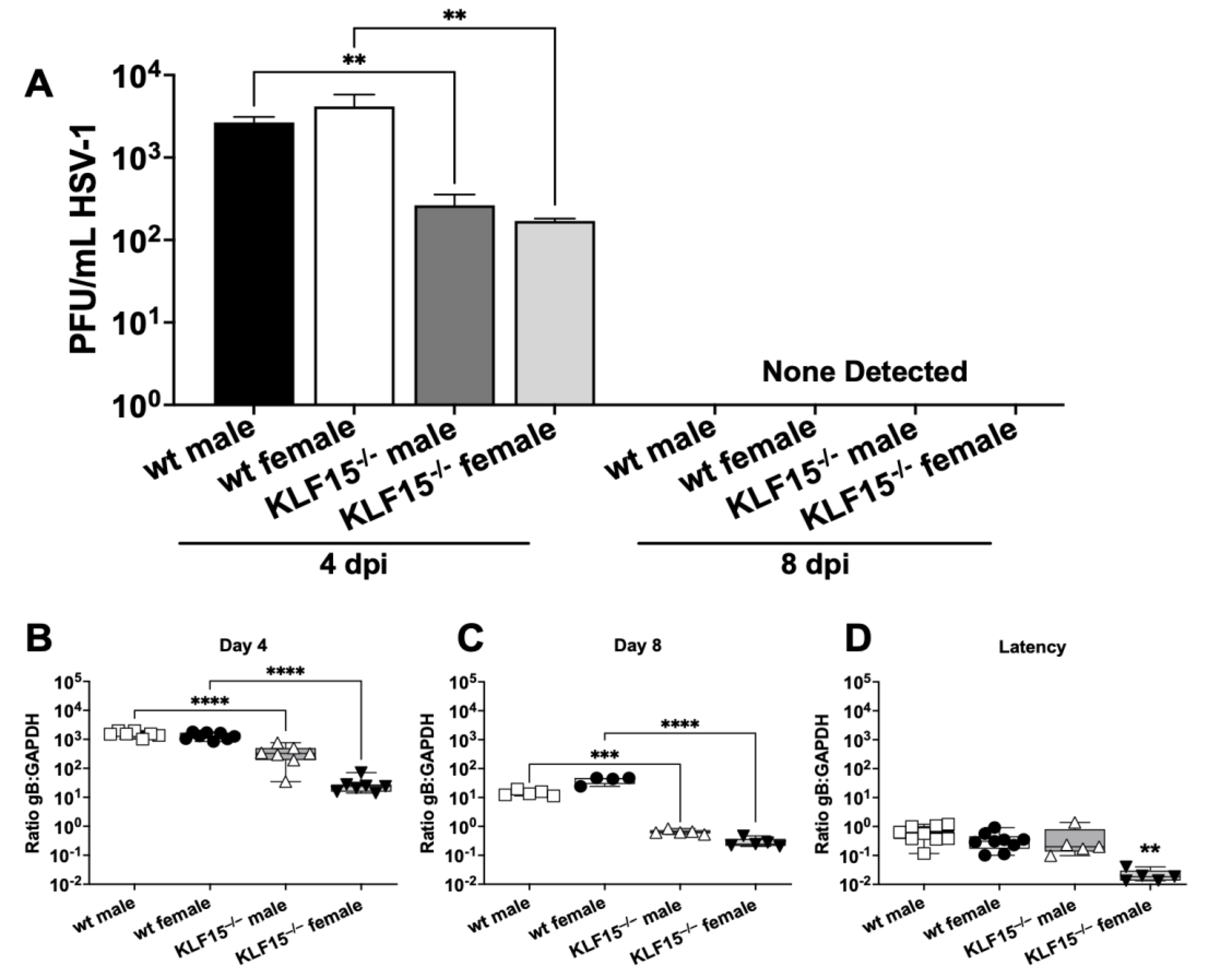
Figure 5.
LAT expression in TG of latently infected mice. Panel A: 30 dpi TG from latently infected mice were dissected from 5-6 mice/group from two separate experiments and then incubated in TRIzol. RNA was prepared as described in Materials and Methods. cDNA was synthesized using random hexamers, and qPCR performed using primers for HSV-1 LAT and mouse GAPDH. Panel B: Cq values for LAT expression (RT-qPCR) were compared to Cq values from gB (qPCR) to calculate the ratio of LAT expression to viral genomes. Data are shown as box and whisker plots from max to min with a line at the median for two separate experiments (n=3-5 mice each). **p<0.001 using one-way ANOVA with unpaired multiple comparison post-test. NS: not significant.
Figure 5.
LAT expression in TG of latently infected mice. Panel A: 30 dpi TG from latently infected mice were dissected from 5-6 mice/group from two separate experiments and then incubated in TRIzol. RNA was prepared as described in Materials and Methods. cDNA was synthesized using random hexamers, and qPCR performed using primers for HSV-1 LAT and mouse GAPDH. Panel B: Cq values for LAT expression (RT-qPCR) were compared to Cq values from gB (qPCR) to calculate the ratio of LAT expression to viral genomes. Data are shown as box and whisker plots from max to min with a line at the median for two separate experiments (n=3-5 mice each). **p<0.001 using one-way ANOVA with unpaired multiple comparison post-test. NS: not significant.
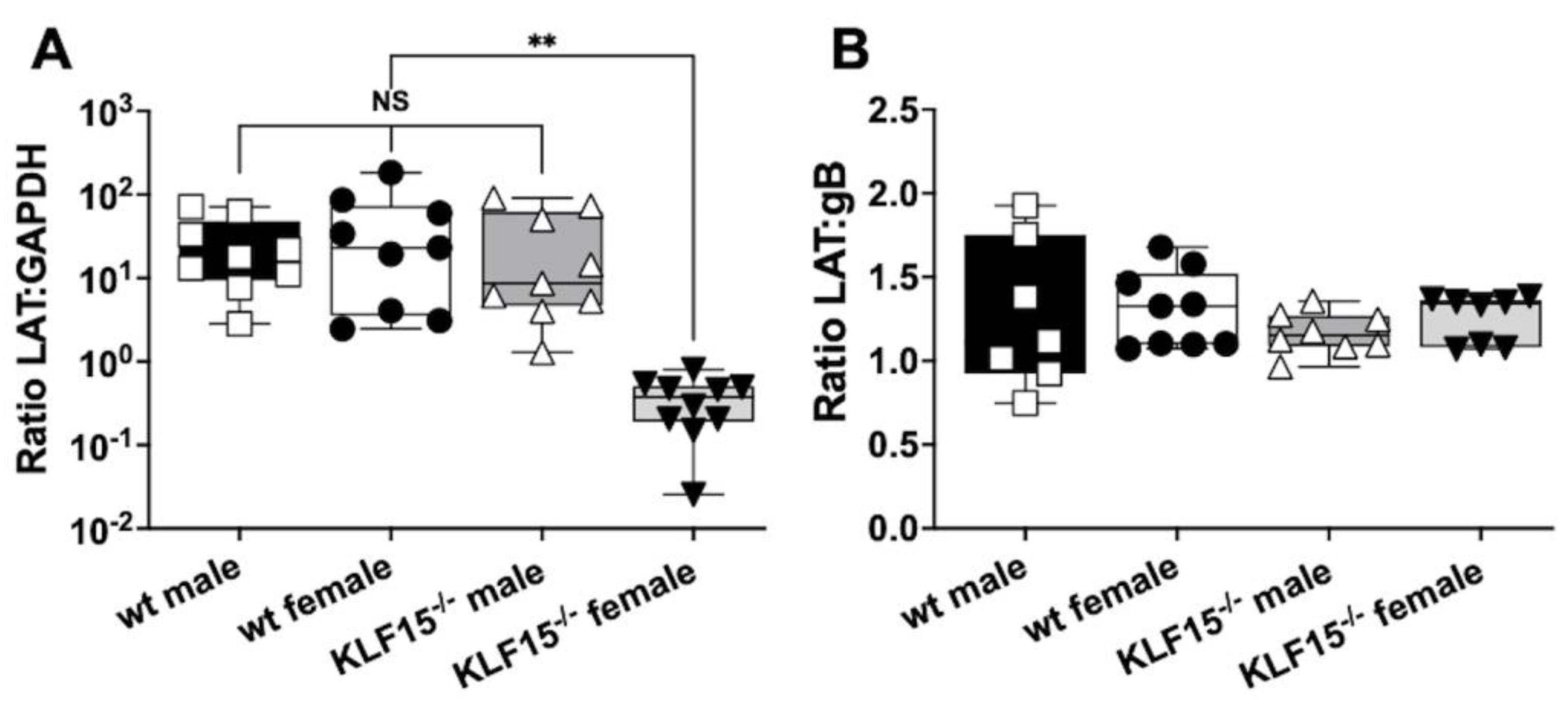
Figure 6.
Examination of HSV-1 explant-induced reactivation. TG from male or female wt and KLF15–/– mice (n=5 mice/group and three independent experiments) were dissected at 30 dpi (latency). TG was subsequently minced and incubated in MEM + 2% stripped FBS and 10 µM DEX. Aliquots of supernatant were collected as denoted and used to measure virus titers during HSV-1 explant-induced reactivation from latency. Data are shown as mean+SEM; *p< 0.05; ***p< 0.005; ****p<0.0001 for wt vs KLF15–/–; ####p<0.001 male vs female using one way ANOVA with unpaired multiple comparison post-test.
Figure 6.
Examination of HSV-1 explant-induced reactivation. TG from male or female wt and KLF15–/– mice (n=5 mice/group and three independent experiments) were dissected at 30 dpi (latency). TG was subsequently minced and incubated in MEM + 2% stripped FBS and 10 µM DEX. Aliquots of supernatant were collected as denoted and used to measure virus titers during HSV-1 explant-induced reactivation from latency. Data are shown as mean+SEM; *p< 0.05; ***p< 0.005; ****p<0.0001 for wt vs KLF15–/–; ####p<0.001 male vs female using one way ANOVA with unpaired multiple comparison post-test.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



