Submitted:
08 August 2025
Posted:
11 August 2025
You are already at the latest version
Abstract
Attention deficit/hyperactivity disorder (ADHD) is a neurodevelopmental condition marked by persistent inattention, hyperactivity, and impulsivity. Despite its considerable global prevalence, key gaps remain in our understanding of the structural and molecular changes underlying ADHD which complicate adult diagnosis, as symptoms present differently from those observed during childhood ADHD. On the other hand, while psychostimulants effectively mitigate some symptoms, significant controversy surrounds their long term effects on cognition, learning and memory, and day to day living. Moreover, our understanding of how various medications given to alleviate ADHD symptoms during pregnancy impact the developing fetal brain also remains largely unexplored. Here, we discuss the subtle differences between ADHD in children and adults and how these symptoms alter brain development and maturation. We further examine changes in monoamine signaling in ADHD and how psychostimulant and non pharmacological treatments modulate these neural networks. We evaluate and discuss findings as they pertain to the long term use of ADHD medications, including in utero exposure, on cognitive outcomes and contextualize these findings with mechanistic insights from animal models.
Keywords:
attention-deficit/hyperactivity disorder
; psychostimulants
; neurodevelopment
; monoamine signaling
; cognitive outcomes
; in utero exposure
; animal models
1. Introduction
The human brain undergoes remarkable changes from infancy through adolescence, shaping our cognitive, emotional, and behavioral traits. Neurodevelopment is a precisely timed and intricately orchestrated process, where perturbations and miscues in the formation of neural circuits can lead to neurodevelopmental disorders such as autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD), which are otherwise often blanketed as “neurodivergence” in cognition and behavior [1]. ADHD which affects approximately 5% of children [2], and 6.76% of adults [3], is diagnosed about twice as frequently in males as compared to females [4]. These higher numbers are likely attributable to behavioral symptoms of ADHD being more easily recognizable in males than females [5]. ADHD is generally characterized by inattentiveness, hyperactivity, and impulsivity [2]. The Diagnostic and Statistical Manual of Mental Disorders 5 (DSM-5) [6] defines the diagnosis of ADHD for children as the presence of at least 6 symptoms including inattentiveness, hyperactivity, and impulsivity, whereas in adults this decreases to five such indicators [7]. A fundamental question that remains unanswered is as to the underlying structural and functional anomalies in the brain that are likely responsible for various observed behavioral changes in ADHD. These alterations in the maturation of brain networks, notwithstanding synaptic plasticity, may underlie diverse behavioral traits, though the precise underlying causes of ADHD and other neurodevelopmental disorders – may they be genetic, cellular, molecular, or environmental, remain largely unknown. Moreover, various behavioral therapies and medications used to treat ADHD symptoms also remain controversial.
This review will first compare ADHD symptoms in children and adults, and how they relate to brain development during childhood and adolescence. Next, we will look at how neurotransmitters contribute to ADHD symptoms, and how stimulants and non-stimulants work to attenuate them. We will then discuss the mechanisms underlying the efficacy of ADHD medications, and how they impact long-term cognitive function in both humans and preclinical models. Subsequently, we will review the implications of ADHD medications when taken during pregnancy, and the use of transcranial brain stimulation as a possible future treatment. Lastly, we will finish by proposing a potential paradigm shift which could involve integrating synaptic level findings from animal models and linking them to more complex neuronal networks in humans to better understand the underlying causes of ADHD, and the impact of psychotherapies on long-term memory.
2. Diagnosing ADHD in Children and Adults: Similarities and Differences
The DSM-5 [6] considers the age-of-onset limit for ADHD in children to be 12 years, whereas adult ADHD with this reduced symptom threshold is considered for ages 17 and older. In children, ADHD symptoms are often more easily discernable, particularly those within the hyperactive domain. For example, a child with ADHD may fidget during class and struggle to remain seated, or miss social cues during conversations, such as frequently interrupting others [8]. Inattentiveness in children is typically observed with difficulties in planning and completing coursework, and consistently remaining unengaged during classroom lectures, often leading to poor academic performance [9]. In adults on the other hand, the hyperactive symptoms are often more subtle, transitioning from physical hyperactivity to more of an internal or emotional restlessness [10]. Due to these challenges, adult ADHD tends to be more challenging to diagnose, particularly because visible ADHD hyperactivity symptoms often decrease with age or are compensated for by cognitive overactivity, which can manifest itself in intrusive thoughts [11]. Thus, inattentiveness becomes the more persistent symptom of adult ADHD, which can lead to frequent employment changes, career instability, and challenges maintaining relationships [12].
The inattentiveness in adult ADHD patients appears to be more pronounced when performing complex tasks as compared to the simpler ones [13], which may limit performance in academic [14] and workplace settings [15]. Consequently, individuals diagnosed with adult ADHD often report a sense of relief from finally having an explanation provided for the challenges that they have been experiencing [16,17]. It is worth noting that ADHD does not necessarily perturb all aspects of cognitive function or behavior. For example, there has been a demonstrated link between ADHD and an increased creativity and “divergent” thinking, as defined by flexibility or originality when coming up with solutions for tasks at hand [18,19]. In fact, evidence suggests that ADHD medications may impair creativity in children [20,21] which has otherwise been attributed to creative storytelling [22], and in some cases greater achievement in artistic domains, such as music, writing, and theater [23]. Therefore, it is important to recognize that not all adults view their ADHD diagnosis as a limitation; rather some believe that it provides them with advantages from a neurodivergent perspective [16,24]. It however remains to be seen whether this augmentation in the above traits and achievements comes at the expense of other mental faculties.
The extent of inattention and hyperactivity symptoms can vary distinctly in both children and adults; thus, ADHD is classified into subtypes of primarily inattentive (ADHD-I), primarily hyperactive (ADHD-H), or combined (ADHD-C) based on the criteria provided in Figure 1.
Notwithstanding these established benchmarks, there are concerns regarding both the over- and under-diagnosis of ADHD, potentially arising from the fact that the criteria used are largely qualitative. Adding to the challenge, while ADHD itself is chronic, the symptoms in children often change in their presentation over time [25]. For adults with undiagnosed ADHD, it is possible that their symptoms may initially have gone unnoticed due to having greater structure to their day-to-day lives, alongside social support during childhood. Moreover, as compared to children, undiagnosed adults may have adapted to their circumstances better by trying to fit into social norms around them [26], thus masking their ADHD symptoms from being readily apparent. Additionally, several ADHD symptoms may exhibit overlap with ASD, such as an impaired response inhibition, challenges with sustained attention, and decreased social functioning [27,28]. One study provided evidence that the largest distinguishable characteristic between ASD and ADHD is the notion of social reciprocity, where ASD children struggle more with demonstrating both social enjoyment and guiding or directing attention during conversations [29]. Additionally, in this study ADHD children appeared to have externalized their problems through impulsive behavior and hyperactivity, whereas ASD children were more likely to withdraw from socialization. Therefore, while ADHD in children is comorbid with ASD in 9.8% of cases [30], ADHD does present unique symptoms that allow it to be distinguished from other neurodevelopmental disorders. Even on a day-to-day basis, the specific scenario that an individual with ADHD may experience can both improve or worsen their symptoms [31]. Thus, early childhood experiences are thought to be crucial for ADHD management, as social and educational support can lessen the progression of symptoms into adulthood which occurs in around 60% of cases [32,33,34].
As a case in point, support from parents and friends, participation in community traditions, and a sense of belonging during adolescence- all linked to greater emotional regulation for adults with ADHD [35]. Parental mental wellbeing, alongside using appropriate discipline strategies with their children, appears to be the best predictors of ADHD symptom persistence [36,37]. Additionally, it appears that for children with ADHD, having a caregiver who they feel could confide with them may be a strong predictor of better self-esteem and emotional wellbeing [38]. For both school-aged children and adolescents, social support from friends, parents, and teachers can act as a protective buffer against stress and improve self-perception [39,40].
On the other hand, adverse childhood experiences such as socioeconomic hardship, familial mental illness, and domestic violence are linked to more severe ADHD symptoms in adulthood [36,41]. Even when no significant differences in neurocognitive functioning are present, these adults with more adverse childhood experiences report greater perceived ADHD symptoms [41]. This reinforces the importance of positive early life experiences in shaping a child’s long-term perception of their ADHD, as well as the nature of their symptom progression as their brain continues to develop.
3. Brain Structural Changes Associated with ADHD
During critical periods of development in early childhood and adolescence, several structural differences have been observed in the ADHD brain. For instance, although a typical pattern of development is generally seen, there are temporal delays observed as early as five years after birth [42,43]. Specifically, children with ADHD have delayed prefrontal cortex (PFC) development of approximately 3 years, reaching 50% peak cortical thickness at around 10.4 years as compared to 7.5 years in typically developing individuals (TDIs) [43,44]. This delay in PFC development is linked to an overall reduction in brain volume in this region [45]. The PFC plays an integral role in planning, attention, and decision-making, which are likely impaired in ADHD, and receives more robust projections from the nucleus accumbens (NAc) [46], the reward center of the brain. Thus, this increased signaling from the NAc likely contributes to the impulsive symptoms seen in the ADHD, with reward-based decision making exerting greater influence over cognition and decision-making [46]. It has also been demonstrated that reduced connectivity from the NAc to the right paracingulate gyrus, which is involved in cognition and emotional processing [47], correlates with more severe ADHD symptoms during both childhood and adulthood [48]. On the other hand, reduced connectivity from the NAc to the right insular cortex, which is involved in broad functions including risk-reward behavior and sensorimotor processing [49], predicts better outcomes for ADHD symptoms [48]. This suggests that an imbalance within reward circuits between the NAc and its downstream connections likely plays an integral role in the expression of ADHD symptoms. Altogether, increased reward-related signaling to the PFC in ADHD may overwhelm the brain’s capacity for self-regulation and result in impulsive behavior. Unlike TDI, children with ADHD do not see an increase in middle and right PFC activity between ages 8 and 11 [50]. However, they do see an improvement in left PFC activity during this time which does not occur in TDIs [50]. Adolescents with ADHD have shown increased blood flow to this brain region during working memory tasks which did not occur in TDIs [51]. This increased activity of the left PFC may thus be a compensatory mechanism for the delayed maturation of the other PFC regions.
A similar developmental delay has been observed in the cerebellum, which reached 50% cortical maturation at 10.5 years in children with ADHD, as compared to 7.5 years in TDIs [43]. The cerebellum plays a key role in regulating movement, balance, coordination, alongside contributing to regulating attention, executive function, memory, language, and visuospatial abilities, and is implicated in ADHD symptoms pertaining to impaired motor control [52]. Loss of volume in the cerebellum has been demonstrated in the ADHD brain, which continues during adolescence and is associated with more severe ADHD symptoms [53,54]. ADHD symptoms regarding reduced coordination are also thought to arise from decreased volume of the corpus callosum (CC) which is observable in children [55,56]. The CC is responsible for interhemispheric communication, coordinating motor function across both sides of the body, and integrating complex cognitive processes such as problem-solving, language, and memory [57]. However, upon reaching adulthood, it appears that these reductions in CC size normalize to those of TDIs [58]. In males with ADHD around 70 years of age, inattention and hyperactivity were associated with decreased CC thickness, whereas age matched females demonstrated a thicker CC, which was linked to increased hyperactivity, suggesting the possibility that there may likely be sex-differences in the progression of hyperactivity into later stages of life [59]. It however remains unknown whether these ADHD symptoms in the elderly corroborate with age-related changes in various brain regions as observed during dementia and Alzheimer’s disease. Thus, it might be the case that there are likely sex-specific differences in CC thickness that may occur later in life for adults with ADHD. This would however be difficult to delineate due to progressive, age-related neurodegeneration which is not confined to just one brain region. The basal ganglia (BG) is another region of the brain where a loss of volume is thought to be associated with ADHD symptoms of hyperactivity, such as fidgeting [60,61,62]. Children with ADHD have a decreased surface area in the ventral striatal region of the BG from ages 8 to 18, atypical to the progressive increase in surface area seen in TDIs [61]. This region of the BG is involved in reward-based learning and decision-making [63], and thus loss of volume in this region may contribute to impulsivity in ADHD. This notion reinforces the narrative that the symptoms of ADHD are not only the result of delayed PFC development, rather there are widespread changes that occur across multiple brain regions including the frontal, temporal, parietal, cerebellar and limbic structures.
In general, it has been demonstrated that there is an overall decrease in total brain volume in ADHD [64,65]. This occurs predominantly through a loss of white matter volume (10.7%), alongside a decrease in grey matter volume (3.9%), leading to an approximately 3% total decrease in brain volume [66].
The brain structural changes associated with ADHD are summarized in Figure 2.
Structural anomalies aside, all brain functions rely upon synaptic connectivity between the neurons which often invoke either ion channel functions or neurotransmitter- receptor interactions. As such, a great deal of focus has been placed on fine tuning these neuronal pathways and modulating synaptic connectivity through either the use of chemical compounds or more novel therapies like transcranial magnetic stimulation.
4. Neurotransmitters and ADHD
Neurons communicate via neurotransmitters – both classical (dopamine, serotonin, acetylcholine, glutamate, GABA etc.) or peptides (norepinephrine, epinephrin, substance P etc.) released at the synaptic cleft or neuro-hormonally. Reduced levels of dopamine (DA) and norepinephrine (NE) have historically been hypothesized to be a major cause of ADHD [67]. Evidence supporting this hypothesis stems from the fact that psychostimulants, which are the first-line treatment for ADHD, increase levels of DA and NE, and as such are deemed effective treatment of ADHD symptoms [68]. These ADHD medications are classified as either stimulants, which include amphetamine (AMPH), or methylphenidate (MPH)-based pharmacotherapies, or non-stimulants (I.e. atomoxetine (ATX)) which do not directly increase DA release. Stimulant medications have common side effects including decreased appetite, insomnia, irritability, and increased blood pressure resulting from DA and NE release. However, stimulant medications like Adderall and Ritalin are often abused as cognitive enhancers in academic settings [69]. Additionally, the long-term effects of these medications on cognitive processes is not fully determined [70], and their long-term effects on the fetal brain during pregnancy are also not fully understood [71]. Here, we review the monoamine networks thought to be involved in the symptoms presented in ADHD.
Both stimulant and non-stimulant ADHD medications modulate the levels of monoamines that are available within the brain. This includes DA, NE, and serotonin (5-HT), which all form distinct pathways in the central nervous system (CNS). Monoamines have a fundamental role in modulating mood, cognition, and behavior [72] and perturbations in their transmission have been implicated in ADHD and the above mentioned symptoms [73]. Despite the historical focus on these transmitters in ADHD pathophysiology, the variation in ADHD phenotypes alongside case-by-case differences in brain region-specific transmitters, has made it difficult to deduce whether it is a reduction or an augmented monoaminergic transmission in the brain that causes ADHD symptoms.
4.1. Dopaminergic Signaling in the Brain
DA is involved in linking learning and reward-based decision-making [74] and reinforcement of repeated behavior [75]. At the heart of the DA system is the ventral tegmental area (VTA), which sends projections to form two key pathways (Figure 3A). The meso-cortical pathway connects the VTA to the PFC, in which DA signaling plays a key role in emotional and behavioral regulation, alongside working memory and decision-making [76]. Perturbed DA levels in the PFC are thought to contribute to deficits in habit formation and planning associated with ADHD [67]. The mesolimbic pathway contains projections from the VTA to limbic structures such as the hippocampus, amygdala, and NAc. Impaired DA signaling within these networks likely underlies impulsive decision-making observed in ADHD [77]. The nigrostriatal pathway bridges connectivity between the substantia nigra (SN) and the dorsal striatum (DS), and functions to regulate control of voluntary movement [78]. Lastly, the tuberoinfundibular pathway links the hypothalamus and pituitary gland, where dopamine can influence prolactin release [79]. Thus, the dopaminergic system may serve myriad functions linking various brain regions to influence attention, movement, hormones, and long-term decision-making.
DA receptors are G-protein-coupled-receptors (GPCRs), and contain 5 subtypes (D1-D5), which as classified as either D1-like (D1 and D5), or D2-like (D2, D3, D4) receptors [80]. D1-like receptors are stimulatory, where DA binding causes an elevation of cyclic adenosine monophosphate (cAMP) levels, leading to increased activity of protein kinase A (PKA). Activation of PKA leads to phosphorylation of the transcription factor cAMP response-binding protein (CREB), and molecular pathways linked to enhanced synaptic plasticity. On the other hand, D2-like receptors are inhibitory; a majority is found on non-dopaminergic neurons, whereas D2-like auto-receptors are found in dopaminergic neurons in the VTA and substantia nigra pars compacta (SNc) [81]. Activation of these receptors serves to inhibit cAMP levels and thus PKA activation, while also activating inward rectifying potassium channels (IRK) to decrease the likelihood of depolarization from occurring [82].
The historical view of ADHD as a disorder arising from hypodopaminergic signaling has been challenged in the past decade (as covered by MacDonald et al. [67] in a recent review). In humans, positron emission tomography (PET) scans can be used in conjunction with radioligands to assess the activity of dopamine transporter (DAT), DA receptors, and norepinephrine transporter (NET). Findings from Forssberg et al. [83] provided evidence for a reduced rate of DA synthesis in the midbrain of male adolescents with ADHD. This aligned well with previous studies that showed increased DAT activity in both children and adults within the striatum and basal ganglia [84,85]. A follow up study by Ludolph et al. [86] confirmed the presence of reduced DA synthesis in adults with ADHD. While Ernst et al. [87] provided evidence that children with ADHD had 48% greater accumulation of 3,4-dihydrophenylalanine (DOPA) in the right midbrain, the data provided was however not statistically significant across multiple comparisons.
Despite these findings suggesting that hypodopaminergic signaling underlies ADHD, these findings are not consistently observed in diverse cohorts of ADHD patients. Ample evidence in the past decade has accumulated to support the notion that hypo-dopaminergic signaling is not a homogeneous hallmark of ADHD as it has been purported to be. Jucaite et al. [88] have demonstrated reduced availability of DAT in the brains of children with ADHD. These findings have been validated by Volkow et al. [89], showing greater DAT binding in the NAc, midbrain, and left caudate. Their research team subsequently correlated increased DAT activity in the NAc and midbrain with increased motivation for adults with ADHD [90]. These same ADHD patients exhibited reduced DAT availability in the very same brain regions. Similar findings have been reported by Itagaki et al. [91] for reduced DAT availability in the NAc for ADHD adults that were not exposed to ADHD psychotherapies.
While the above studies may seem contradictory, a meta-analysis by Weber et al. [92] concluded that DA followed an inverted U-shaped relationship with working memory. That is to say, both hypodopaminergic and hyperdopaminergic signaling can impair cognitive processes. Therefore, when considering data obtained from PET scans in humans, it is important to consider DA transmission through the lens of functional signaling. For example, it has been shown that there are reduced D2 and D3 receptors in the midbrain and NAc for those with ADHD as compared to TDIs [89,90]. Given that these receptors inhibit DA transmission, one interpretation could be that their lower levels may result in increased DA levels in the ADHD brain. However, it is also possible that the downregulation of these receptors might be downstream of reduced DA signaling to begin with. Therefore, at this moment we can conclude that DA is likely involved in the pathophysiology of ADHD, and that there are synaptic level changes that occur in the ADHD brain. It also appears that it is most likely that ADHD may be linked to both excessive and/or perturbed DA signaling, which might help explain why ADHD medications do not work for all patients. These discrepancies regarding dopaminergic signaling and its involvement may arise from varying metabolic pathways that may be active in a diverse population of ADHD patients. As shown by Ludolph et al. [86], treatment history also appears to cause intragroup variability in DA metabolism for ADHD patients, hence it is unlikely that dysfunction in the DA system alone would be responsible for ADHD symptoms.
4.2. Noradrenergic Signaling in the Brain
DA metabolism involves the initial conversion of L-tyrosine into DOPA through tyrosine hydroxylase [80] (Figure 3B). Conversion of DOPA into DA is completed by aromatic L-amino acid decarboxylase and can then be converted into NE through dopamine β-hydroxylase (DBH). DBH is expressed inside the vesicles of norepinephrinergic neurons within the CNS [93]. The locus coeruleus (LC) is thought to be the primary site of NE synthesis in the brain, and innervates throughout the cerebral cortex, amygdala, cerebellum, and hippocampus [94] (Figure 3C). Perturbed NE signaling within the PFC contributes to poor concentration associated with ADHD. Additionally, NE signaling between the LC and CA3 region of the hippocampus is crucial for memory formation during new experiences [95]. NE is thought to enhance the long-term memory of emotional experiences through innervations between the LC, hippocampus, and amygdala [96]. Thus, impaired NE signaling in ADHD may also contribute to deficits in emotional memory, albeit exact mechanisms remain unclear.
Alongside DA, NE has also been historically implicated in the pathophysiology of ADHD. Until the late 2000s, methodology for assessing NET in the human brain using PET imaging was challenging due to the absence of effective radioligands [97]. Upon its availability however, Hannestad et al. [98] reported the first in vivo findings in humans that demonstrated MPH binding to NET to reduce the availability of this transporter in the brain. This verified that ADHD medications like MPH not only interact with DA signaling but also increase NE transmission. Vanicek et al. [99] assessed NET availability using PET imaging in adults with ADHD who had not been exposed to psychotherapies. They did not find differences in NET availability or distribution in the hippocampus, putamen, pallidum, thalamus, or midbrain, but did find a negative correlation between age and NET availability in the thalamus, midbrain, and LC. However, they were not able to assess cortical regions using their PET setup, where the PFC is one of the main regions exhibiting reduced NE and is believed to underlie ADHD symptoms. Ulke et al. [100] on the other hand, found reduced NET availability in the fronto-parietal-thalamic-cerebellar regions in adults with ADHD. Further evidence to suggest impaired NET function in ADHD was provided by Singurdardottir et al. [101], where methylation of NET was negatively correlated with the severity of hyperactivity-impulse symptoms in ADHD adults. This extended their previous findings which linked mutations in the NET SLC6A2 gene with reduced NET availability in the cerebellum [102]. Shang et al. [103] explored two specific mutations within the NET gene SLC6A2: rs36011 (T) and rs1566652 (G), which make up the TG haplotype. They found that children with this haplotype had reduced activity in brain regions responsible for visual memory and attention, and higher in regions linked to sensorimotor attention. Findings from Hohmann et al. [104] demonstrated an association between NET variants and ADHD diagnosis, and altogether suggest that genetic alterations of NET are likely implicated in ADHD.
The link between NE and ADHD was established by Barkley et al. [105], linking polymorphisms in the DBH gene to increased hyperactivity in childhood, and greater likelihood of behavioral problems in adolescence. This aligned well with previous findings of Smith et al. [106] which demonstrated a significant association between the TaqI A polymorphism in the DBH gene and ADHD using the same dataset. Kieling et al. [107] also provided evidence for polymorphisms within the promoter for the DBH gene corresponding to decreased performance in cognitive assessments for children with ADHD. These polymorphisms were also linked by Bellgrove et al. [108] to deficits in visual attention for ADHD children and adolescents. Altogether, this provides evidence for alterations in NE metabolism implicating it in ADHD, however it is still unclear whether there are either reductions or increases in NE production occurring in specific brain regions which could be a contributing factor.
4.3. Serotonergic Signaling the Brain
The majority of serotonergic input to the forebrain is provided by the dorsal raphe nucleus (DRn), which projects to the VTA [109] (Figure 3D). Inhibitory projections from the VTA and striatum innervate the DRn to link 5-HT and DA signaling [110], such as the DRn neurons inducing bursting pattern in the VTA to drive behavioral reinforcement [111]. The DRn also projects to the cerebral cortex, limbic system, striatum, thalamus, and hypothalamus to influence mood, cognition, attention, and emotional regulation. Therefore, decreased 5-HT levels in the ADHD brain likely underlie emotional symptoms due to perturbed signaling in these pathways. Serotonin is metabolized from tryptophan, and increased levels have been observed in the plasma, serum, and urine of ADHD patients [112]. This seems to suggest that tryptophan is not being effectively converted to 5-HT, and dysfunction of genes involved in the metabolic pathway has been associated with ADHD [113]. PET imaging has also been used to assess the levels of the serotonin transporter (SERT) in the brains of ADHD patients. Vanicek et al. did not find significant differences in SERT availability across brain regions between ADHD adults and TDIs [114]. These findings aligned with a previous study that also found no differences in SERT density linked to ADHD [115]. Therefore, it appears that 5-HT metabolism may be another contributing factor to ADHD pathophysiology, however the precise target sites and the underlying mechanisms remain poorly defined.
4.4. NMDAR and Dopamine Interactions
While ADHD medications primarily target monoamine signaling, it is important to discuss how this may contribute to other signaling pathways that are integrally involved in cognition. There have been well characterized interactions between D1-like and D2-like receptors with N-methyl-D-aspartate receptors (NMDARs) in the cortex, hippocampus, and striatum [141] (Figure 4). NMDAR is a glutamate receptor that plays a key role in synaptic plasticity, which is thought to form the basis of learning and memory [142]. Activation of NMDAR is dependent on both glutamate binding, alongside depolarization from α-amino-3-hydroxy-5-methyl-4-isoxazole propionic receptors (AMPARs), which removes the magnesium ions that block the NMDAR channel pore [142]. This enables calcium influx, activating a molecular cascade which can increase cAMP levels, leading to activation of PKA. Therefore, given that several of the molecules involved in NDMAR signaling are shared with DA receptor activation, there are many implications for ADHD medications and synaptic plasticity.
Modulation of NMDAR by these medications appears to be dependent on the NMDAR subunit composition. For instance, GluN2A-NMDAR predominant entorhinal-CA1 synapses show depressed NMDAR excitatory postsynaptic potentials (EPSCs), whereas GluN2B-NMDAR predominant CA3-CA1 inputs have potentiated NMDAR EPSCs from D1-like receptor activation [143]. For D2-like receptors, modulation of NMDAR appears more straightforward; activation of D2 receptors tends to depress NMDAR activity. Altogether, this produces a complex system by which ADHD medications can both enhance or depress NMDAR-mediated plasticity, which will be covered when we discuss the impact of these medications on cognition.
5. ADHD Medications
As outlined above, modulation of monoaminergic pathways is likely to underlie the efficacy of ADHD medications. The guidelines for prescribing ADHD medications differ between countries. For instance, in the United Kingdom, MPH is typically offered as the first-line pharmacological treatment for children of ages ≥ 5 or adolescents with ADHD under brand names like Ritalin or Concerta [144]. However, in Northern America both AMPH and MPH based medications can be considered as first-line treatments [145,146]. In general, assessments are performed to determine the cardiovascular health of the patient before stimulant medications are considered in addition to discussing preference for possible side effect profile of the different medications. This includes taking a family history of heart disease, as stimulant medications increase blood pressure by enhancing NE and DA signaling. A general overview of the prescription of these medications is provided in Figure 5.
6. Modes of Actions of Various ADHD Medications
While ADHD medications are somewhat similar in accomplishing the same end goal of elevating monoamine levels, their mechanism is unique and underlies their differences in potency and side-effects. These mechanisms for stimulants and non-stimulants are summarized in Figure 6.
6.1. Methylphenidate Based Medications
MPH-based medications function to inhibit DAT and NET, increasing the availability of DA and NE in the synaptic cleft (Figure 6A) [147]. This results in an increase in dopaminergic and adrenergic signaling within key brain regions such as the PFC, striatum, and NAc, to enhance memory, attention, motivation, motor control, and improve reward seeking behavior respectively [148,149]. However, MPH does not significantly impact the SERT, thus there is minimal change in the availability of serotonin within the brain as a direct effect of the drug [150,151]. Additionally, it appears that MPH also alters the distribution of dopamine vesicles, both in shifting them closer to active zones in the presynaptic terminal, or back to the cytoplasm for later release [152,153]. While this mechanism is not fully understood, it is possible that an elevation of DA through DAT inhibition may cause the recruitment of DA vesicles dependent on dopaminergic activity. This potentially identifies a mechanism by which MPH enables more controlled release of DA as compared to AMPH.
6.2. Amphetamine Based Medications
AMPH based medications differ from MPH and non-stimulants in that they enter the presynaptic neuron using DATs (Figure 6B). Unlike MPH, AMPH directly inhibits the vesicular monoamine transporter 2 (VMAT2), which impairs the packaging of monoamines into vesicles for subsequent release into the synaptic cleft. Concurrently, AMPH also impairs monoamine oxidase (MAO), preventing the breakdown of monoamines [154,155]. As a result of these two mechanisms, AMPH increases the concentration of monoamines in the presynaptic neuron, leading to a reversal of the monoamine transporters to shuttle DA, NE, and 5-HT into the synaptic cleft [156]. Like MPH, this results in increased alertness and concentration to reduce symptoms of ADHD. However, AMPH also increases 5-HT transmission by blocking the SERT [157], which could theoretically contribute to modulating DA release through RNc-VTA signaling [158] and in turn contribute to improving mood related symptoms of ADHD.
6.3. Non-Stimulant Medications
Non-stimulants on the other hand, do not directly increase DA levels. For example, ATX is a selective NE reuptake inhibitor, which blocks the NET and increases the levels of NE, increasing availability in the synaptic cleft (Figure 6C) [159]. As it does not directly increase DA levels, ATX often produces less severe side effects while reducing abuse risk linked to DA modulation of reward pathways [160]. Within the PFC, NET also partially contributes to reuptake of DA [161], thus ATX does still provide indirect benefits for increased DA transmission [162,163,164]. Viloxazine, a more recently approved ADHD medication, similarly inhibits the reuptake of NE which also increases DA levels in the PFC [165]. However, viloxazine differs from other non-stimulants in that it also increases 5-HT transmission through binding to 5HT2C receptors (Figure 6D). It has also been shown that viloxazine antagonizes 5HT2B receptors on GABA interneurons to increase the availability of 5-HT in the prefrontal cortex [166]. Guanfacine (Figure 6E) and clonidine (Figure 6F) are two other non-stimulant medications that function to modulate NE transmission. These medications bind to α2 adrenergic receptors, providing negative feedback thus reducing excessive NE release [167]. Guanfacine specifically targets α2A receptors located in the PFC and LC, which results in improved attention, working memory, and impulse control [168,169]. Clonidine on the other hand, binds to α2B receptors in the peripheral blood vessels to regulate vasoconstriction, and α2C receptors in the basal ganglia and limbic system which regulates mood and startle response [167]. Thus, clonidine calms hyperarousal, while improving sleep issues or anxiety and irritability.
7. Pharmacological Treatment for ADHD in Humans and Animal Models: Discrepancies and Possible Explanations
Animal studies form the backbone for drug discovery, for studying the effects of long-term exposure to various ADHD medications, and long-term toxicology at the pre-clinical stage. However, findings from animal studies do not always translate into clinical outcomes. This may be due to the heterogeneity of ADHD pathophysiology in humans, which is not fully represented in strain-controlled animal models that are devoid of several behavioral traits that are distinct to humans. The studies on “ADHD” animal models predominantly focus on modifying the genome to induce ADHD-like symptoms [170] whereby the genetic makeup and the behavioral repertoire may not match that of humans. While these studies may still be useful for testing the impact of specific molecular mechanisms in augmenting select “symptoms”, or testing the efficacy of ADHD medications, etiology of human ADHD itself is not always from a homogenous genetic cause. For example, DAT mutant mice do demonstrate “ADHD-like” symptoms, and present excessive dopamine levels [171]. However, while excessive DA levels are present for some ADHD patients, in others there is a decrease in DA signaling [67]. Similarly, the spontaneously hypertensive rat model has been used to model human ADHD. These animals demonstrate core ADHD symptoms in hyperactivity, impulsiveness, and inattention, and are more sensitive to delays in reinforcement like ADHD children [172]. While this model is effective for exploring pharmacological mechanisms in relation to behavior, hypertension itself is not a direct symptom of ADHD, and may produce confounding effects on behavior and cognition through influencing blood flow to the brain [173,174,175]. This is to say that it is unlikely that a single animal model of ADHD could recapitulate the behavioral repertoire that form the hallmarks of ADHD in humans. Even if this were the case, then the broad heterogeneity seen in human ADHD individuals will make the phenotype indiscernible.
For prenatal exposure specifically, it is also important to consider the significant differences between rodent and human development. Rodents are typically considered to have reached adulthood around ~70 days following birth [176]. Humans on the other hand continue brain development until around 25 years of age [177]. As a result, we argue that this expedited neurodevelopment in rodents relative to humans may increase the susceptibility to perturbations from prenatal ADHD medication exposure. While humans experience greater overall exposure to these medications throughout their lifetime, rodent models may instead involve a greater proportion of their developmental timespan. Moreover, the liver function and potential metabolic breakdown of various medications may also vary between rodents and humans, altering pharmacokinetics and pharmacodynamics of these drugs [178]. This might help explain why rodent models of prenatal exposure to ADHD medications demonstrate adverse effects not seen in humans. However, we also argue that this would make it even more challenging to ascertain the long-term effects of exposure to these medications in humans. Assessing the long-term effects of ADHD medications in humans would likely require longitudinal studies tracking cognitive function and MRI imaging from childhood to adulthood and older age, which raises feasibility concerns. There would also be a need to compare the relative change in cognition from timepoints between ADHD patients and TDIs, since it has been shown that ADHD medications do not always fully restore cognitive function [179]. Given that this is likely unrealistic from a cost and logistic perspective, animal models still provide us with valuable insights into the long-term effects of these medications. For example, there continues to be novel mechanisms being deduced that underlie the efficacy of amphetamine for treating ADHD, despite the medication being already prescribed for decades [180]. Even newer ADHD medications like Viloxazine, had their fundamental mechanisms deduced using animal models [181,182]. Animal models enable interrogation of molecular mechanisms to be made that would not otherwise be possible in humans, especially when considering in utero exposure. Here we will discuss the implications of treatment of human ADHD with AMPH- and MPH-based medications, alongside ATX for cognition, and discuss how these findings relate to what has been shown in animal models of ADHD.
7.1. ADHD Medications Generally Improve Cognition in Humans
Much of the controversy regarding ADHD medications pertains to concerns regarding their abuse as cognitive enhancers, alongside the potential for long-term side-effects when prescribed to children. In general, it appears that ADHD medications do not always produce an immediate improvement in cognitive performance for individuals with ADHD, where a long-term use of the medication over several weeks is required to see improvements to ADHD symptoms [183]. While these medications often improve memory for individuals with ADHD, they do not do so to the level of TDIs [184]. A systematic review which included 176 studies from 1980-2012 concluded that long-term academic outcomes for children with ADHD are improved with ADHD medication, but multimodal treatment provided the best results [185]. However, a study by Morell & Expόsito concluded long-term treatment with either stimulants or non-stimulants does not necessarily always improve all ADHD symptoms, such as delay aversion and risk taking behavior [179]. This suggests that it may still be worthwhile for medical practitioners to explore treatment plans that also include non-pharmacological approaches, to further reduce the gap in learning outcomes between children with ADHD and their classmates. Additionally, it is crucial to consider how the side-effects of ADHD medications may indirectly perturb learning, memory, attention, ability to multitask, and general cognition. For example, impaired sleep quality resulting from these medications [186], or stressful settings, might impair their encoding of memory [187]. Overall, ADHD medications, when taken as prescribed, do appear to improve performance in academic and work settings for individuals with ADHD. Additionally, stimulant-related ADHD medications have also been found to decrease the aforementioned alterations in brain structure associated with ADHD [188,189]. However, these effects appear to be age specific, for example, MPH has been shown to reduce cortical thinning in children with ADHD but not in adults [190]. This suggests that treatment with these medications in childhood may provide long-term benefits for brain maturation and trajectory and thus cognition. On the other hand, cumulative exposure to ADHD medications has been linked to decreased hippocampal CA1 region volumes in children [191] but it remains unclear why these side-effects appear to be region specific, and how this reduced hippocampal volume might correspond with learning and memory in ADHD children. Notwithstanding the potential beneficial effects of various medications, there exists a possibility of off-target effects of these compounds, and these are perhaps the least studied.
7.2. Amphetamine Effects on Cognition
For AMPH specifically, the effects of the drug on different types of cognition have had conflicting results, particularly when comparing human and animal studies. For individuals with ADHD, AMPH is often reported to enhance aspects of cognition, but these improvements appear to be highly individualized and context dependent. A systematic review from 2022 [192] highlighted that only one [193] out of the nine studies they looked at regarding the effect of AMPH on visual working memory (VWM) found improvements from the drug. This is likely due to differences between the studies in the time between participants taking AMPH and performing the VWM test. The study by de Wit et al. [193] had participants perform the VWM test around 3 hours after the taking AMPH, which would be the time period in which AMPH generally reaches the highest peak drug concentration in the bloodstream [194]. Additionally, participants within the de Wit et al. study conducted a baseline assessment on VWM on the same day in which they completed the treatment test for VWM; both factors might explain why their study found improvements in VWM whereas the other studies failed to see any significant improvements. These inconsistencies in the effect of AMPH were also true for spatial working memory (SWM). Within this systematic review [192], four out of 13 studies reported AMPH-based improvements in SWM [195,196,197,198], and three of these highlighted time-based effects on AMPH improvement of SWM [199,200,201]. These inconsistencies may be partly explained by factors such as that individuals with ADHD tend to have worse test-taking performance [202] while exhibiting increased test-anxiety [203]. Given that DA has been shown to have an inverse U-shaped action on human working memory and cognitive performance [204], it is possible that AMPH - through rapidly increasing extracellular DA may instead impair memory performance under stress. AMPH is shown to improve academic performance for children with ADHD [205], but not necessarily in all areas of cognition. Hence, it is likely that AMPH improvements in cognition are highly individualistic, and contingent upon specific brain chemistry of the individual in question. In general, it seems that these medications do not inherently improve academic performance. For example, non-prescribed use of ADHD medications has been linked to impaired academic habits, such as studying and motivation during cognitive testing [206]. Therefore, while AMPH does often enhance cognition, this will not always manifest itself in academic outcomes or assessments of cognitive abilities. It also remains unclear whether such improvements tap into short-term working memory or if they also culminate into long-term cognitive enhancement.
In contrast, animal studies offer mechanistic insights regarding how AMPH influences cognition at the cellular and synaptic level, particularly in the hippocampus, a key region for learning and memory processes. Acute AMPH exposure has been shown to enhance synaptic plasticity through an increased phosphorylation of CREB [207] and AMPAR GluA1 subunits in the CA1 and dentate gyrus regions [208], suggesting short-term enhancements in memory formation. However, chronic exposure to AMPH and sensitization to the drug appear to impair synaptic plasticity in animal models. Repeated AMPH administration has been linked to impairments in both short- and long-term memory in the hippocampus, reduced neuronal density in the CA1 region, and morphological changes such as longer dendrites with fewer mature spines [209]. Given that mature spines are thought to be associated with increased long term potentiation (LTP) and memory [210], the loss of these mature spines may be the mechanism by which AMPH sensitization impairs hippocampal based memory. Interestingly, AMPH can also promote neurite outgrowth in vitro through pathways independent of DA or nerve growth factor release [211], and yet chronic AMPH exposure appears to reduce both nerve growth factor and brain-derived neurotrophic factor (BDNF) levels in key brain regions, suggesting a potential downregulation of neurotrophic support over time [212].
AMPH has been shown to increase NMDAR-GluN2B synaptic currents in midbrain dopaminergic neurons [213]. This potentiation does not however occur when DAT is inhibited; thus, the enhancement of NMDAR mediated plasticity appears to be downstream of the established mechanism of AMPH entering the presynaptic terminal. Animal models have revealed that withdrawal from repeated AMPH administration can cause a downregulation in the expression of NMDAR [214], GluR1, GluR2 [215], and the GluN2B subunit [216]. This seems to suggest that withdrawal from AMPH-containing ADHD medications may cause a rebound effect in decreasing excitatory transmission. Moreover, the reduction in NMDAR expression may contribute to impairments in cognition that the ADHD individuals experience when stopping their medication [217].
Chronic AMPH exposure itself has been shown to cause downregulation of GluN2B, leading to long-term depression at cortico-accumbal glutamatergic synapses, resulting in behavioral sensitization to AMPH [216]. Interestingly, it appears that a loss of NMDAR signaling with specifically D1-like receptors may prevent AMPH sensitization [218]. When NMDAR signaling was lost with both D1R and D2R expressing neurons, sensitization to AMPH occurred [218]. This suggests that NMDAR contributes to synaptic plasticity with D2R expressing neurons, to ensure that they could provide sufficient inhibitory control over excessive DA to prevent addictive behaviors associated with AMPH.
Altogether, these findings from both human and animal studies converge on a central theme: the effects of AMPH on cognition are highly context dependent. Acute exposure may provide benefits for cognition through enhancing synaptic plasticity, particularly under optimal arousal conditions. However, chronic use and sensitization may instead induce neuroadaptive changes that impair memory and neural resistance. This underscores the importance of considering both neurobiological and contextual variables when evaluating the efficacy of AMPH for enhancing cognition, particularly in clinical settings.
7.3. Methylphenidate Effects on Cognition
MPH has been shown to exert inconsistent effects on different forms of cognition. While MPH improves spatial memory during an initial assessment, it paradoxically leads to decreased performance accuracy upon subsequent examination [219]. These findings are in line with other studies that have demonstrated MPH-based improvements in SWM, sustained attention, and working-memory tasks [220,221]. However, other studies have found that MPH does not improve spatial working memory nor the integration of sensory information [222,223]. Notably, despite MPH-induced increases in sympathetic arousal, emotional memory consolidation appears to remain unaltered [224].
Therefore, the question remains as to why, even with MPH treatment, individuals with ADHD still on average, do not reach the same level of academic achievement as compared to TDIs. One possibility is that MPH does not fully normalize intrinsic motivation, or accounts for the comorbidities of learning disabilities, anxiety, depression, or sleeping disorders associated with ADHD. Moreover, the side-effects of MPH which include reduced appetite and insomnia, may in some cases attenuate the functional benefits conferred by increased DA and NE signaling, thereby limiting improvements for some individuals with ADHD in academic and work settings.
At the molecular level, repeated MPH exposure produces sustained repression of CREB in the NAc when examined in an animal model [225]. This is likely linked to other findings that repeated MPH treatment represses PKA activity, alongside DA-induced adenyl cyclase activation in the DS [226]. These findings are further supported by evidence of downregulation of plasticity-related gene expression in the striatum following chronic MPH treatment [227]. Additionally, repeated MPH exposure has been shown to repress the expression of CREB, BDNF, PKA, MAPK3, and cFos in the amygdala [228], all of which contribute to molecular mechanisms governing synaptic plasticity. Together, these animal studies suggest that repeated MPH exposure may compromise plasticity in the amygdala and striatum, potentially posing risks for perturbed fear and reward responsiveness.
The above observed changes in plasticity from MPH exposure in animal models appear to be linked to NMDAR. MPH has also been demonstrated to alter NMDAR responses [229,230], and -rescue impaired synaptic plasticity in an NMDAR dependent manner [231]. This notion alludes towards an alteration of NMDAR expression similar to AMPH, where a single dose of MPH has been shown to significantly reduce protein levels of NMDAR subunits GluN1 and GluN2B [232]. Similarly, chronic MPH administration has been linked to widespread reductions in NMDAR expression across brain regions [233]. Given that the D1-like activation of GluN2B and predominant NMDAR contributes to synaptic plasticity, it would be important to investigate whether MPH may inhibit NMDAR plasticity for neurons expressing D1-like receptors. By downregulating these receptors, MPH may indirectly limit the capacity for plasticity in dopaminergic circuits, potentially contributing to reductions in impulsivity at a cost to cognitive flexibility or learning capacity.
The aforementioned molecular alterations are reflected in behavioral outcomes: chronic MPH exposure impairs object recognition in adolescent rats, but not in adults [234]. This same study reported the presence of depressive symptoms in adult rats, but not in adolescent rodents exposed to MPH. One explanation for this might be that early treatment of ADHD with MPH in human children is thought to normalize brain volumes [188]. Thus, it may be the case that repeated MPH exposure in young brain networks improves mesolimbic signaling in a manner that reduces depression symptoms, but the prolonged elevation of these transmitters would likely impair memory processes.
7.4. Atomoxetine Effects on Cognition
Like AMPH and MPH, ATX has generally been demonstrated to improve cognition for individuals with ADHD. This includes improvements in pattern and spatial recognition memory [235], reduced anxiety symptoms [236,237], and improved response inhibition [238]. Interestingly, ATX is known to reduce cognitive difficulties in perimenopausal and postmenopausal women with ADHD-like symptoms [239], suggesting that its utility may extend beyond traditional pediatric or young adult populations. However, several studies have shown that ATX does not always improve all cognitive challenges associated with ADHD, such as sustained attention [238] and visuospatial working memory [240]. Additionally, ATX has been shown to differ from MPH in how it modulates reward-based working memory. It appears that relative to MPH, ATX decreases the activation of working memory networks to reduce the typical enhancement of working memory when a reward is expected [241]. This might suggest that for individuals with ADHD who rely heavily on external motivation like rewards, ATX may not be as effective as MPH. On the other hand, it is also possible that ATX might be more effective than MPH in low-stimulation or low-reward environments, through regulating the brain without conflicting with motivational systems.
Animal models have thus helped elucidate the neurobiological mechanisms underlying these cognitive effects of ATX. For instance, repeated exposure to ATX has been demonstrated to increase mRNA levels in the hippocampus and PFC, while enhancing phosphorylation of AKT and GSK3B which are downstream components of BDNF signaling [242]. At the protein level, there was greater BDNF present in the PFC, which suggests ATX facilitates cognitive enhancement through enhancing synaptic plasticity via the CREB pathway.
Beyond BDNF-mediated plasticity, ATX also appears to enhance cholinergic neurotransmission within the PFC and hippocampus through α1 and D1 receptor activation [243]. These neurochemical changes are reflected behaviorally: whereby animals treated with ATX exhibit improved performance in memory tests [243], consistent with other studies demonstrating that ATX facilitates learning and memory in preclinical models [244,245,246]. Interestingly, ATX has been shown to increase histamine release, which improved performance in the Morris water maze test [246]. Given the role of histamine as a modulator of learning and memory [247], this might suggest another mechanism by which ATX improves symptoms of ADHD and enhances cognition.
There are fewer studies on ATX and NMDAR, ATX is shown to rescue LTP through a postsynaptic mechanism [244]. Similar findings to MPH and AMPH have been observed with decreased expression of GluN2B, alongside reduced mRNA levels for genes encoding for NMDAR [244,248]. ATX, MPH, and D-AMPH all increase firing of PFC neurons through potentiating NMDAR activation [249].
Together, these findings underscore the multifaceted mechanisms of ATX across various cognitive domains. Mechanistically, ATX appears to exert its effects through a variety of pathways involving NE, BDNF signaling, cholinergic modulation, and possibly histaminergic activity. Continued integration of human and animal studies will be essential to fully elucidate the therapeutic scope and neurobiological impact of ATX.
Given that chronic exposure to ADHD medications reduces NMDAR expression, it is crucial for future studies to investigate the extent to which this impacts long-term learning and memory outcomes. This decreased NMDAR plasticity might be an adverse side-effect of these medications, or it could simply be a mechanism by which they exert their therapeutic effects. It is possible that this reduced NMDAR plasticity if targeted towards reward circuits, could be protective against impulsive behavior.
Altogether, animal models have revealed that prolonged exposure to ADHD medications may impair cognition. However, it remains unclear whether these findings are translatable to humans since there are significant differences in human metabolism of these medications as compared to the rodent models. It is understandably challenging to perform long-term studies exploring the implications of prolonged ADHD medication use from adolescence to adulthood. There are many confounding variables that would need to be accounted for regarding environmental and social factors. As of right now, it appears that long-term exposure to ADHD medications in humans is potentially safe, and that these treatments do provide benefits to individuals. It is crucial that we continue to explore the implications of prolonged exposure through animal studies and pursue longitudinal studies in humans to evaluate how chronic use of these medications alters brain connectivity, activity, and behavior.
8. Implications of In Utero Exposure to ADHD Medications for Long-Term Cognition
8.1. Prescription of ADHD Medications During Pregnancy
One of the outstanding questions is whether ADHD medications are safe to use during pregnancy. Both AMPH and MPH have been shown to cross the placental barrier in humans and in rodents [250,251,252,253], and ATX in rats specifically [254]. Under most circumstances, pregnant individuals with ADHD are discouraged from taking these medications during pregnancy [255]. However, stopping psychostimulant treatment for ADHD during pregnancy has been shown to increase the risk of depressive symptoms in patients [256] - in addition to the potential cognitive and functional implications on productivity. Therefore, psychostimulants may still be prescribed during pregnancy, depending on the risk to benefit ratio, and these clinical considerations have been covered in a recent review [257]. ADHD medication use during pregnancy increased from 0.3% in 1998, to 1.3% during 2014 in North America [258]. Additionally, the percentage of privately insured reproductive-aged individuals who filled ADHD medication prescriptions increased from 0.9% in 2003 to 4.0% in 2015 in the United States [259]. Thus, with the increasing use of these medications during pregnancy and for woman of childbearing age, there is a dire need to validate their safety for developing fetal brain; and this would require a paradigm shift in our approach to ADHD medication.
8.2. Continued Evidence Suggests That ADHD Medications Are Safe During Pregnancy
Regarding concerns of birth complications, developmental defects, and risks of neurodevelopmental disorders, human studies have largely provided reassurance that ADHD medication use during pregnancy is safe [260,261]. Most recently, a population-based cohort study followed 2,257 children exposed to ADHD medications during pregnancy, for a mean follow-up time of 7 years [262]. Their findings highlight that AMPH, MPH, and ATX did not increase risks for long-term neurodevelopmental disorders. Some studies have however found a small increase in cardiac malformations associated with prenatal exposure to MPH, alongside decreased birth weights with both MPH and AMPH [263,264]. Moreover, prenatal exposure to AMPH has been linked to impaired academic outcomes [265], alongside increased symptoms of depression, anxiety, and ADHD [253]. However, much remains unknown regarding how in utero exposure to these medications may alter neurodevelopment from a molecular perspective. Animal models are often used to explore the impact of drug exposure on neurodevelopment, where postnatal day 0-14 in rodents is considered comparable to the third trimester in humans [266]. Here, we will review the cellular, molecular, and behavioral changes caused by prenatal exposure to ADHD psychostimulants in animal models, as few studies have been performed using non-stimulants.
8.3. In Utero Exposure to AMPH: Time Dependent Changes in Metabolism and Behavior in Animal Studies
Prenatal AMPH exposure is linked to decreased expression of D2-like inhibitory receptors in the NAc, alongside reduced expression of tyrosine hydroxylase [267]. Loss of these D2-like inhibitory receptors increases the risk of addictive behaviors in adulthood [268,269]. Thus, there is the possibility that prenatal exposure to AMPH may impair inhibitory signaling within reward pathways in the brain, posing risks for addiction behaviors later in life. Additionally, several studies have shown that prenatal AMPH exposure alters the metabolism and levels of monoamines in the brain [270,271,272]. One study which looked at whole brain concentrations, reported increased serotonin levels at birth that were normalized to those of controls over subsequent days [270]. This also included higher levels of GABA on postnatal day 21, which correlated with decreased locomotor activity in these prenatally exposed pubs. This reduction in motor activity from prenatal AMPH exposure has also been reported in 45-day old offspring [273]. Therefore, in animal models at least, prenatal AMPH exposure may have long-lasting effects on activity, likely resulting from increased inhibitory transmission.
On the other hand, reduced serotonin levels have been shown in the medial PFC with prenatal AMPH exposure [272]. These offspring also exhibited reduced neuronal density within the medial PFC shortly after birth, which normalized by day 30. This suggests that some perturbations caused by prenatal AMPH exposure may not be permanent, and recovery may ensue in the weeks following birth. For example, offspring of AMPH-exposed rats were shown to exhibit more memory errors in the Lashley III maze test 90 days following birth, but these errors normalized to that of controls in subsequent assessments [274]. The alterations in monoamine metabolism appear to extend towards increased conversion of tyrosine in DA and NE [271], which were linked with improved cognitive processes at 90 days post-birth [275]. It has also been shown that prenatal AMPH exposure in C. elegans causes increased expression of tyrosine hydroxylase paired with decreased expression of VMAT in the adult animal [276]. These same animals also demonstrated increased sensitivity to AMPH. One possibility for could be the loss of VMAT, which may have further potentiated AMPH-mediated reverse transport of monoamines. These findings have important potential implications for ADHD medication use during pregnancy. Given that ADHD can often be hereditary, if the pregnant individual is prescribed AMPH for their ADHD, it is possible that their child may be prescribed a similar medication in the future. Thus, these findings suggest that AMPH use during pregnancy may increase the offspring’s sensitivity to the drug, thus making them more susceptible to ADHD symptoms. Overall, it appears that the alterations in monoamine metabolism caused by prenatal AMPH exposure in animals are likely temporary, and do not produce life-long impairments in behavior.
However, prenatal exposure to AMPH is known to cause life-long impairments in glucose metabolism. Prenatal AMPH exposure was linked to a reduction in 5-HT within β-cells of the pancreas, alongside decreased expression of markers of pancreas maturation [277]. Mice of both sexes prenatally exposed to AMPH exhibited hypoglycemia 1 year after birth. However, female mice demonstrated sex differences with reduced β-cell islet sizes, alongside decreased bodyweight as compared to controls. This may stem from previous evidence pointing towards estrogen increasing the sensitivity of female rats to AMPH [278]. Therefore, the presence of sex-differences is interesting given that during prenatal exposure to the drug, both male and female animals are in a similar environment. It is plausible that sex-differentiation of the brain via estrogen in females, may modulate the effects of AMPH in a manner that does not occur in males. Therefore, it is worth exploring whether prenatal exposure to lower concentrations of AMPH in female animals produces similar effects to standard concentrations in male animals.
8.4. In Utero Exposure to Methylphenidate: Alterations in DA Pathways and Behavior in Animal Studies
Like AMPH, prenatal MPH exposure alters the expression of D2-like receptors. Adult male rats prenatally exposed to MPH present increased expression of tyrosine hydroxylase in the SNc, alongside NAc core and shell regions [279]. Both the dorsal and ventral regions of the striatum presented an increase in DAT density, whereas the SNc and VTA showed an increase in D2R density. This suggests that prenatal exposure to MPH increases DA metabolism paired with elevated regulatory control of DA reuptake and signaling, which manifested in reduced susceptibility to hyperactivity caused by cocaine. Similar findings have been reported with prenatal MPH exposure increasing time to seek rewards [280], suggesting reduced impulsivity. However, this was a sex-specific effect for males, whereas females demonstrated decreased time to seek rewards. It is unclear what causes this sex-specific effect, but one possibility is that prenatal MPH exposure may alter the development of the PFC in females to facilitate more impulsive decision-making. Again, it may be the case that even though same concentration of the drug was used across both male and female animals, estrogen may have potentiated the effects of MPH in females to cause these unintended symptoms. The evidence that increased impulsivity in observed in females is supported by the findings that fetal MPH exposure induces increased locomotor activity, and decreases anxiety behavior [281]. Therefore, at least in animal models, it appears that prenatal MPH exposure may induce ADHD-like behavior in the offspring and produce more pronounced effects in females as compared to males.
The impact of prenatal MPH exposure on behavior also appears to be dependent on when the drug is taken during pregnancy. MPH exposure on embryonic days 8-10 results in decreased anxiety-related behaviors and an increase in exploratory behavior that did not occur after exposures on embryonic days 12-14, and 16-18 [282]. This suggests that exposure to MPH during early fetal development can have more significant effects on behavior. When considering exposure even earlier in development, such as 0-5 days post-fertilization, it has been shown that MPH exposure can cause long-term impairments in spatial learning and predatory avoidance responses [283]. Altogether, evidence from animal models suggests that prenatal MPH exposure may cause ADHD symptoms with temporal and sex-specific influences. Given that prenatal MPH exposure in humans does not appear to increase the likelihood of developing ADHD [262], more research is required to determine whether these findings from animal models are applicable to the human fetal brain. It is possible that the observed effects in humans are either more subtle or take a longer time to manifest (such as adulthood) or potentially rectified through neuroplasticity and as such remain undetected in clinical studies.
Considering the side effects of many pharmacological agents on brain and behavior of ADHD patients, alternative and non-pharmacological approaches have come to light.
9. Transcranial Stimulation for ADHD Treatment
Given that ADHD pharmacotherapies are not always effective for all ADHD patients, non-pharmacological treatments are being explored. Transcranial stimulation has gained traction as a potential future treatment for ADHD. Modulation of monoamine pathways is believed to underlie the efficacy of repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS) (Figure 7). tDCS is a non-invasive procedure that utilizes weak electrical currents (typically 1-2mA) to modulate membrane excitability [284]. The cathode is used to hyperpolarize the membrane potential, whereas the anode is used to increase depolarization. On the other hand, rTMS involves the generation of an electric current in the brain through repeated magnetic pulses, which can directly trigger neuronal action potentials [285]. This can lead to alterations in synaptic plasticity, where generally low-frequency pulses (1 Hz) reduce cortical excitability, whereas high-frequency stimulation (5-20 Hz) produces the opposite effect [286]. Both tDCS and rTMS can thus influence neurotransmitter release, alongside the plasticity of networks that are thought to be perturbed in ADHD.
In the case of ADHD, these techniques are largely used to target the PFC [287], where reduced activity in ADHD patients has been documented [288,289]. In a cohort of 64 children with ADHD, Cao et al. [290] demonstrated that rTMS, when combined with ATX treatment, showed greater improvements in ADHD symptoms and executive function than ATX treatment alone. Subsequent studies have found similar improvements from rTMS for children with ADHD, which provided further evidence regarding the potential of this treatment for pediatric ADHD [291,292]. These improvements in cognition are thought to be linked to better blood flow, enhanced neuronal activity, and potentially improvements in sleep patterns for children with ADHD [293,294]. However, the precise mechanisms underlying the efficacy of rTMS remain unknown, and further studies are required to validate the efficacy of rTMS as a treatment option for ADHD.
tDCS is often used to target the left dorsolateral prefrontal cortex (dlPFC), which has been shown to improve response inhibition, attention, working memory, and cognitive flexibility in ADHD patients [295,296]. The outcome was not however consistent when tDCS was used to target the right dlPFC [297]. However, it does appear that tDCS targeting the dlPFC improves PFC activity during working memory tasks for adolescents with ADHD [298]. The left dlPFC is targeted based on its involvement with executive functioning, a key brain area where ADHD patients show deficits [297]. While still not fully understood, it is thought that the positive effects of stimulating the dlPFC occur due to the dlPFC being a crucial site for dopaminergic effects of cognition [295]. Improving dopaminergic signaling improves inhibitory control and decreases impulsive behaviors. tDCS stimulation of mouse motor cortex slices may enhance long-term potentiation in an NMDAR dependent manner, while increasing BDNF release within this brain region [299]. Like TMS, tDCS is more effective with repetition, as cognitive improvements from a single treatment last less than three days. Regarding safety, neither TMS nor tDCS have serious adverse effects reported to date, although common minor adverse effects were present, including itchy scalp, headache, and fatigue [297]. A word of caution here is that the human brain is a highly integrated system with its innate communication highways laid during development; the perturbation of one area using either TMS or tDSC may have lasting, and yet undiscerned effects on other regions of the brain.
As an alternative, the peripheral vagus nerve can be targeted by tDCS by surgically implanting an electrode with programmed frequency, duration, and charge [300]. VNS activates afferent fibers of the auricular branch of the vagus nerve, which leads to stimulation of the LC to increase NE release [301]. In animal models, vagus nerve stimulation (VNS) has also been shown to increase synaptic plasticity through increasing LC firing rates, modifying hippocampal activity, and altering gene expression leading to increased release of DA, NE, 5-HT, acetylcholine and brain-derived neurotrophic factor [302]. Common non-surgical adverse effects of VNS include hoarseness, cough, and headaches, though these tend to improve with time [303]. VNS is currently used to treat drug-resistant epilepsy but might show potential in future for ADHD treatment [300]. Because surgical implantation is invasive and poses serious risks, an alternative option is external trigeminal nerve stimulation (eTNS) which is done by placing electrodes on the forehead above the nerve. The trigeminal nerve projects to the LC, thereby eTNS appears to increase NE in the PFC and hippocampus. eTNS appears to act through stimulation of the brain stem, targeting the LC and raphe nucleus to increase NE and 5-HT release respectively [304,305]. McGough et al. [306] demonstrated that a 4-week long nightly treatment with eTNS improved ADHD symptoms in children with ADHD, which led to the FDA approval of the eTNS for pediatric ADHD [307]. These findings aligned with their previous study, which involved 24 ADHD children receiving eTNS for 8-weeks, and found improvements in measures of executive functioning [308]. Both studies showed improvements in impulsivity, hyperactivity, and attention that were statistically significant compared to a sham group. Common adverse effects were mild, consisting of headaches, fatigue, and an increased appetite [306,308]. While transcranial stimulation appears promising as a future avenue of treatment for ADHD, pharmacological therapies remain at the forefront of ADHD research and still require further validation in terms of their safety and efficacy.
10. A Paradigm Shift in ADHD Research: Integrating Synaptic and Systems Level Neuroscience
10.1. Reframing ADHD Treatment: From Disorder to Divergence
The classification of ADHD itself as a disorder reflects a framework that views deviation from normative behavior as inherently pathological. While this clinical model has been essential for securing support, services, and research, it can also reinforce stigma and negative self-perception in children diagnosed with ADHD. By defining ADHD primarily through its divergence from majority norms such as attention span, impulse control, and activity levels, there is the risk of portraying these traits as inherently defective rather than contextually challenging. This is intertwined with the controversy regarding concerns in the over prescription of ADHD medications. If ADHD is only viewed as a ‘disorder’, we then naturally gravitate heavily towards pharmacology without addressing the social context. Medication can help children regulate their attention and behavior, and there is evidence to support the notion that these treatments do help ameliorate brain structural changes in ADHD. However, these treatments should be complimented by furthering the integration of different learning styles in classroom settings, such as disseminating concepts through different forms of media and interactive materials. As Frolli et al. [309] demonstrated using the Universal Design for Learning framework [310], a more individual-centered approach to learning can enhance reading, writing, and mathematical skills for children with ADHD. If paired with educators who are equipped with greater awareness on how to recognize positive qualities of their students with ADHD, this could improve learning experiences in classroom settings. In the long-term, this may reduce the reliance on chronic use of pharmacological interventions for ADHD.
10.2. Improving ADHD Diagnosis and Interpretation of Human Data
Future ADHD diagnostic criteria should be refined further to better reflect the diverse presentations of the condition across sex and age, to enable personalized treatment plans only feasible when diagnoses are specific for ADHD subtypes. The discrepancy in diagnostic rates between males and females, as well as the difficulty in diagnosing ADHD in adulthood, suggest that an overemphasis on observable physical hyperactivity may limit diagnostic generalizability. Greater awareness for healthcare practitioners on internal hyperactivity symptoms such as restlessness, sensory overload, and mental overstimulation, especially in low-stimulation environments, may serve to improve accurate diagnosis of ADHD. Furthermore, distinguishing ADHD from other neurodevelopmental disorders like ASD requires greater attention to patients’ perceptions of social interaction and their ability to engage in reciprocal conversations, which are subtle yet revealing features [29] that standard assessments often overlook. Mechanistic research in humans, such as PET imaging studies of DAT availability, is frequently misinterpreted to infer dopaminergic tone. This same caution must apply to investigations of NET and SERT, as transporter availability does not directly equate to neurotransmitter levels. Monoamines act through diverse GPCR subtypes with distinct behavioral and cellular effects, and their availability alone cannot be assumed to reflect functional neurotransmission. Given that ADHD pharmacotherapy can normalize brain structure [188,190] and alter transporter availability [86], future studies must also account for treatment history, as grouping treated and untreated patients together risks obscuring clinically significant differences.
10.3. Further Longitudinal Studies Are Required to Assess the Impact In Utero Exposure to ADHD Medications on Cognition
When it comes to deducing the impact of in utero exposure to ADHD medications in humans, we believe continued longitudinal assessments will produce key findings. For example, Madsen et al. [262] conducted a longitudinal study on in utero exposure to ADHD medication, with children being followed up from age 3 to a mean follow-up time of 6.9 years. Since they found no increased risk of neurodevelopmental disorders for these children who would be around 10 years old, it would be interesting to see if this is still the case once these children reach adulthood. Additionally, it would be worthwhile where possible, to assess other parameters of cognition, such as academic outcomes, alongside social behaviors, particularly during adolescence.
10.4. Considerations for Animal Models and In Utero Exposure Studies
The use of ADHD animal models should be interpreted with subtype-specific context in mind. For example, DAT knockout mice model ADHD subtypes with excess extracellular dopamine [172], and because amphetamine relies on DAT to enter presynaptic neurons, its effects cannot be directly compared to controls. Regarding in utero exposure to ADHD medications, animal studies often lack clinical relevance due to inconsistent dosing and use of non-oral administration routes. As McDonnell-Dowling et al. [314] review, doses used in rodent studies frequently exceed human-equivalent exposures, and the Reagan-Shaw et al. [315] allometric scaling method could be used to improve translational accuracy. Additionally, the route of drug administration is crucial, where oral gavage, which more closely mimics human oral intake, is a more appropriate method for drug delivery in these models. Additionally, the impact of sex differences on drug effects must be better characterized. Estrogen, which enhances sensitivity to several ADHD medications [278], may modulate pharmacological effects during pregnancy. Ultimately, combining well-designed animal models with longitudinal human studies will provide a clearer picture of the developmental consequences of ADHD pharmacotherapy during pregnancy.
11. Conclusions
While there remains limited clinical evidence that long-term use of prescribed ADHD medications produces adverse effects, animal models have raised the possibility that molecular mechanisms involved in cognition may be perturbed by chronic use. These medications are generally effective in improving cognitive deficits in patients with ADHD and thus remain as the first-line treatment. Studies in humans continue to provide evidence that prescribed use of these medications during pregnancy does not produce noticeable cognitive impairments in children, but further studies are required to assess long-term cognitive outcomes for children exposed to these medications in utero. We believe that it is imperative that further animal research is dedicated to ascertaining potential molecular perturbations involved in cognition that result from in utero exposure. Within these future studies, it is crucial that appropriate methods of drug administration are leveraged, clinically relevant doses are used throughout, and the role of estrogen in enhancing the potency of AMPH during pregnancy is considered. Human studies should continue to be conducted to assess the consequences of Iin utero exposure, with an emphasis on considering the long-term cognitive outcomes of the child in adolescence and adulthood. The data obtained from humans is promising in terms of safety both during pregnancy and for general long-term prescribed use of these medications; however, the limited studies assessing long-term cognitive outcomes make it unclear whether there remain possible consequences for cognition.
Author Contributions
M.Y. and S.S. wrote the manuscript; M.Y., S.S., B.A., C.J., N.S., and F.I. conducted the literature search and data curation; M.Y., C.L., and N.I. S. conceptualized the idea and scope of the review; M.Y., S.S., and F.I. developed visual materials for manuscript; M.Y., S.S, B.A., C.J., N.S., F.I., C.L., and N.I. S. edited and revised manuscript; M.Y., S.S, B.A., C.J., N.S., F.I., C.L., and N.I. S. approved final version of manuscript.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Council of Canada (NSERC: #RT690332).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ADHD | Attention-deficit/hyperactivity disorder |
| ASD | Autism spectrum disorder |
| DSM-5 | Diagnostic and Statistical Manual of Mental Disorders 5 |
| ADHD-I | Primarily inattentive ADHD |
| ADHD-H | Primarily hyperactive ADHD |
| ADHD-C | Combined ADHD |
| PFC | Prefrontal cortex |
| TDIs | Typically developing individuals |
| NAc | Nucleus accumbens |
| CC | Corpus callosum |
| BG | Basal ganglia |
| DA | Dopamine |
| NE | Norepinephrine |
| AMPH | Amphetamine |
| MPH | Methylphenidate |
| ATX | Atomoxetine |
| 5-HT | Serotonin |
| CNS | Central nervous system |
| VTA | Ventral tegmental area |
| SN | Substantia nigra |
| DS | Dorsal striatum |
| GPCRs | G-protein-coupled-receptors |
| cAMP | Cyclic adenosine monophosphate |
| PKA | Protein kinase A |
| CREB | Cyclic adenosine monophosphate |
| SNc | Substantia nigra pars compacta |
| IRK | Inward rectifying potassium channels |
| PET | Positron emission tomography |
| DAT | Dopamine transporter |
| NET | Norepinephrine transporter |
| DOPA | Dihydrophenylalanine |
| DBH | Dopamine β-hydroxylase |
| LC | Locus coeruleus |
| DRn | Dorsal raphe nucleus |
| SERT | Serotonin transporter |
| NMDARs | N-methyl-D-aspartate receptors |
| AMPARs | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic receptors |
| EPSCs | Excitatory postsynaptic potentials |
| VMAT2 | Vesicular monoamine transporter 2 |
| MAO | Monoamine oxidase |
| VWM | Visual working memory |
| SWM | Spatial working memory |
| LTP | Long term potentiation |
| BDNF | Brain-derived neurotrophic factor |
| rTMS | Repetitive transcranial magnetic stimulation |
| tDCS | Transcranial direct current stimulation |
| dlPFC | Dorsolateral prefrontal cortex |
| VNS | Vagus nerve stimulation |
| eTNS | External trigeminal nerve stimulation |
References
- Astle DE, Bassett DS, Viding E. Understanding divergence: Placing developmental neuroscience in its dynamic context. Neuroscience & Biobehavioral Reviews 2024, 157, 105539. [Google Scholar] [CrossRef]
- Friedman LA, Rapoport JL. Brain development in ADHD. Current Opinion in Neurobiology 2015, 30, 106–111. [Google Scholar] [CrossRef]
- Song P, Zha M, Yang Q, Zhang Y, Li X, Rudan I. The prevalence of adult attention-deficit hyperactivity disorder: A global systematic review and meta-analysis. J Glob Health 2021, 11, 04009. [Google Scholar] [CrossRef]
- Willcutt, EG. The Prevalence of DSM-IV Attention-Deficit/Hyperactivity Disorder: A Meta-Analytic Review. Neurotherapeutics 2012, 9, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Young S, Adamo N, Ásgeirsdóttir BB, Branney P, Beckett M, Colley W, et al. Females with ADHD: An expert consensus statement taking a lifespan approach providing guidance for the identification and treatment of attention-deficit/ hyperactivity disorder in girls and women. BMC Psychiatry 2020, 20, 404. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders [Internet]. DSM-5-TR. American Psychiatric Association Publishing; 2022. Available online: https://psychiatryonline.org/doi/book/10.1176/appi.books.9780890425787 (accessed on 25 June 2025).
- Posner J, Polanczyk GV, Sonuga-Barke E. Attention-deficit hyperactivity disorder. Lancet 2020, 395, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Eiland LS, Gildon BL. Diagnosis and Treatment of ADHD in the Pediatric Population. J Pediatr Pharmacol Ther 2024, 29, 107–118. [Google Scholar]
- Jangmo A, Stålhandske A, Chang Z, Chen Q, Almqvist C, Feldman I, et al. Attention-Deficit/Hyperactivity Disorder, School Performance, and Effect of Medication. J Am Acad Child Adolesc Psychiatry 2019, 58, 423–432. [Google Scholar] [CrossRef]
- Weyandt LL, Iwaszuk W, Fulton K, Ollerton M, Beatty N, Fouts H, et al. The Internal Restlessness Scale: Performance of College Students With and Without ADHD. J Learn Disabil 2003, 36, 382–389. [Google Scholar] [CrossRef]
- Martel MM, von Eye A, Nigg J. Developmental differences in structure of attention-deficit/hyperactivity disorder (ADHD) between childhood and adulthood. Int J Behav Dev 2012, 36, 279–292. [Google Scholar] [CrossRef]
- Williams OC, Prasad S, McCrary A, Jordan E, Sachdeva V, Deva S, et al. Adult attention deficit hyperactivity disorder: a comprehensive review. Ann Med Surg (Lond) 2023, 85, 1802–1810. [Google Scholar] [CrossRef]
- Bálint S, Czobor P, Komlósi S, Mészáros Á, Simon V, Bitter I. Attention deficit hyperactivity disorder (ADHD): gender- and age-related differences in neurocognition. Psychol Med 2009, 39, 1337–1345. [Google Scholar] [CrossRef]
- Henning C, Summerfeldt LJ, Parker JDA. ADHD and Academic Success in University Students: The Important Role of Impaired Attention. J Atten Disord 2022, 26, 893–901. [Google Scholar] [CrossRef]
- Fuermaier ABM, Tucha L, Butzbach M, Weisbrod M, Aschenbrenner S, Tucha O. ADHD at the workplace: ADHD symptoms, diagnostic status, and work-related functioning. J Neural Transm (Vienna) 2021, 128, 1021–1031. [Google Scholar] [CrossRef]
- Fleischmann A, Fleischmann RH. Advantages of an ADHD Diagnosis in Adulthood: Evidence From Online Narratives. Qual Health Res 2012, 22, 1486–1496. [Google Scholar] [CrossRef] [PubMed]
- Young S, Bramham J, Gray K, Rose E. The Experience of Receiving a Diagnosis and Treatment of ADHD in Adulthood: A Qualitative Study of Clinically Referred Patients Using Interpretative Phenomenological Analysis. J Atten Disord 2008, 11, 493–503. [Google Scholar] [CrossRef]
- Girard-Joyal O, Gauthier B. Creativity in the Predominantly Inattentive and Combined Presentations of ADHD in Adults. J Atten Disord 2022, 26, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Stolte M, Trindade-Pons V, Vlaming P, Jakobi B, Franke B, Kroesbergen EH, et al. Characterizing Creative Thinking and Creative Achievements in Relation to Symptoms of Attention-Deficit/Hyperactivity Disorder and Autism Spectrum Disorder. Front Psychiatry 2022, 13, 909202. [Google Scholar] [CrossRef] [PubMed]
- Ten W, Tseng CC, Chiang YS, Wu CL, Chen HC. Creativity in children with ADHD: Effects of medication and comparisons with normal peers. Psychiatry Res 2020, 284, 112680. [Google Scholar] [CrossRef]
- González-Carpio Hernández G, Serrano Selva JP. Medication and creativity in Attention Deficit Hyperactivity Disorder (ADHD). Psicothema 2016, 28, 20–25. [Google Scholar] [CrossRef]
- Zentall, SS. Production deficiencies in elicited language but not in the spontaneous verbalizations of hyperactive children. J Abnorm Child Psychol 1988, 16, 657–673. [Google Scholar] [CrossRef]
- White HA, Shah P. Creative style and achievement in adults with attention-deficit/hyperactivity disorder. Personality and Individual Differences 2011, 50, 673–677. [Google Scholar] [CrossRef]
- Brod M, Pohlman B, Lasser R, Hodgkins P. Comparison of the burden of illness for adults with ADHD across seven countries: a qualitative study. Health Qual Life Outcomes 2012, 10, 47. [Google Scholar] [CrossRef]
- Lahey BB, Pelham WE, Loney J, Lee SS, Willcutt E. Instability of the DSM-IV Subtypes of ADHD from preschool through elementary school. Arch Gen Psychiatry 2005, 62, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Schrevel SJC, Dedding C, van Aken JA, Broerse JEW. “Do I need to become someone else?” A qualitative exploratory study into the experiences and needs of adults with ADHD. Health Expect 2016, 19, 39–48. [Google Scholar] [CrossRef]
- Schachar RJ, Dupuis A, Arnold PD, Anagnostou E, Kelley E, Georgiades S, et al. Autism Spectrum Disorder and Attention-Deficit/Hyperactivity Disorder: Shared or Unique Neurocognitive Profiles? Res Child Adolesc Psychopathol 2023, 51, 17–31. [Google Scholar] [CrossRef]
- Crisci G, Cardillo R, Mammarella IC. Social Functioning in Children and Adolescents with ADHD and Autism Spectrum Disorder: A Cross-Disorder Comparison. J Clin Child Adolesc Psychol 2024, 53, 489–502. [Google Scholar] [CrossRef] [PubMed]
- De Giacomo A, Craig F, Medicamento S, Gradia F, Sardella D, Costabile A, et al. Identifying Autistic-Like Symptoms in Children with ADHD: A Comparative Study Using ADOS-2. Neuropsychiatr Dis Treat 2024, 20, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Canals J, Morales-Hidalgo P, Voltas N, Hernández-Martínez C. Prevalence of comorbidity of autism and ADHD and associated characteristics in school population: EPINED study. Autism Res 2024, 17, 1276–1286. [Google Scholar] [CrossRef]
- Pedersen SL, Kennedy TM, Joseph HM, Riston SJ, Kipp HL, Molina BSG. Real-World Changes in Adolescents’ ADHD Symptoms within the Day and across School and Non-school Days. J Abnorm Child Psychol 2020, 48, 1543–1553. [Google Scholar] [CrossRef]
- Duh-Leong C, Fuller A, Brown NM. Associations Between Family and Community Protective Factors and Attention-Deficit/Hyperactivity Disorder Outcomes Among US Children. J Dev Behav Pediatr 2020, 41, 1–8. [Google Scholar] [CrossRef]
- Sibley MH, Swanson JM, Arnold LE, Hechtman LT, Owens EB, Stehli A, et al. Defining ADHD symptom persistence in adulthood: optimizing sensitivity and specificity. J Child Psychol Psychiatry 2017, 58, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Faraone SV, Biederman J, Mick E. The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol Med 2006, 36, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Lowe CT, Bath AC, Callahan BL, Climie EA. Positive Childhood Experiences and the Indirect Relationship With Improved Emotion Regulation in Adults With ADHD Through Social Support. J Atten Disord 2024, 28, 1615–1626. [Google Scholar] [CrossRef]
- Brown NM, Brown SN, Briggs RD, Germán M, Belamarich PF, Oyeku SO. Associations Between Adverse Childhood Experiences and ADHD Diagnosis and Severity. Academic Pediatrics 2017, 17, 349–355. [Google Scholar] [CrossRef]
- Roy A, Hechtman L, Arnold LE, Sibley MH, Molina BSG, Swanson JM, et al. Childhood Factors Affecting Persistence and Desistence of Attention-Deficit/Hyperactivity Disorder Symptoms in Adulthood: Results From the MTA. J Am Acad Child Adolesc Psychiatry 2016, 55, 937–944.e4. [Google Scholar] [CrossRef]
- Zhang X, Li Y, Xiao Y, Yu C, Pei Y, Cao F. Association of positive childhood experiences with flourishing among children with ADHD: A population-based study in the United States. Prev Med 2024, 179, 107824. [Google Scholar] [CrossRef] [PubMed]
- Mastoras SM, Saklofske DH, Schwean VL, Climie EA. Social Support in Children With ADHD: An Exploration of Resilience. J Atten Disord 2018, 22, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Atta MHR, Amin SM, Hamad NIM, Othman AA, Sayed YM, Sanad HS, et al. The Role of Perceived Social Support in the Association Between Stress and Creativity Self-Efficacy Among Adolescents With Attention Deficit Hyperactivity Disorder. J Psychiatr Ment Health Nurs.
- Alfonso D, Basurto K, Guilfoyle J, VanLandingham HB, Gonzalez C, Ovsiew GP, et al. The Effect of Adverse Childhood Experiences on ADHD Symptom Reporting, Psychological Symptoms, and Cognitive Performance Among Adult Neuropsychological Referrals. J Atten Disord 2024, 28, 43–50. [Google Scholar] [CrossRef]
- Bedford SA, Lai MC, Lombardo MV, Chakrabarti B, Ruigrok A, Suckling J, et al. Brain-Charting Autism and Attention-Deficit/Hyperactivity Disorder Reveals Distinct and Overlapping Neurobiology. Biological Psychiatry 2025, 97, 517–530. [Google Scholar] [CrossRef]
- Shaw P, Eckstrand K, Sharp W, Blumenthal J, Lerch JP, Greenstein D, et al. Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proc Natl Acad Sci U S A 2007, 104, 19649–19654. [Google Scholar] [CrossRef]
- Shaw P, Lerch J, Greenstein D, Sharp W, Clasen L, Evans A, et al. Longitudinal Mapping of Cortical Thickness and Clinical Outcome in Children and Adolescents With Attention-Deficit/Hyperactivity Disorder. Arch Gen Psychiatry 2006, 63, 540. [Google Scholar] [CrossRef]
- Emond V, Joyal C, Poissant H. Neuroanatomie structurelle et fonctionnelle du trouble déficitaire d’attention avec ou sans hyperactivité (TDAH). L’Encéphale 2009, 35, 107–114. [Google Scholar] [CrossRef]
- Costa Dias TG, Wilson VB, Bathula DR, Iyer SP, Mills KL, Thurlow BL, et al. Reward circuit connectivity relates to delay discounting in children with attention-deficit/hyperactivity disorder. Eur Neuropsychopharmacol 2013, 23, 33–45. [Google Scholar] [CrossRef]
- Smith SM, Fox PT, Miller KL, Glahn DC, Fox PM, Mackay CE, et al. Correspondence of the brain’s functional architecture during activation and rest. Proc Natl Acad Sci USA 2009, 106, 13040–13045. [Google Scholar] [CrossRef] [PubMed]
- Zaher A, Leonards J, Reif A, Grimm O. Functional connectivity of the nucleus accumbens predicts clinical course in treated and non-responder adult ADHD [Internet] 2024. Available online: http://medrxiv.org/lookup/doi/10.1101/2024.01.04.24300820 (accessed on 30 May 2025).
- Kortz MW, Lillehei KO. Insular Cortex. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK570606/ (accessed on 30 May 2025).
- Yasumura A, Omori M, Fukuda A, Takahashi J, Yasumura Y, Nakagawa E, et al. Age-related differences in frontal lobe function in children with ADHD. Brain and Development 2019, 41, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Sheridan MA, Hinshaw S, D’Esposito M. Efficiency of the Prefrontal Cortex During Working Memory in Attention-Deficit/Hyperactivity Disorder. Journal of the American Academy of Child & Adolescent Psychiatry 2007, 46, 1357–1366. [Google Scholar]
- Isaac V, Lopez V, Escobar MJ. Can attention-deficit/hyperactivity disorder be considered a form of cerebellar dysfunction? Front Neurosci 2025, 19, 1453025. [Google Scholar] [CrossRef]
- Wyciszkiewicz A, Pawlak MA, Krawiec K. Cerebellar Volume in Children With Attention-Deficit Hyperactivity Disorder (ADHD): Replication Study. J Child Neurol 2017, 32, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Mackie S, Shaw P, Lenroot R, Pierson R, Greenstein DK, Nugent TF, et al. Cerebellar Development and Clinical Outcome in Attention Deficit Hyperactivity Disorder. AJP 2007, 164, 647–655. [Google Scholar] [CrossRef]
- Hill DE, Yeo RA, Campbell RA, Hart B, Vigil J, Brooks W. Magnetic resonance imaging correlates of attention-deficit/hyperactivity disorder in children. Neuropsychology 2003, 17, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Cao Q, Sun L, Gong G, Lv Y, Cao X, Shuai L, et al. The macrostructural and microstructural abnormalities of corpus callosum in children with attention deficit/hyperactivity disorder: A combined morphometric and diffusion tensor MRI study. Brain Research 2010, 1310, 172–180. [Google Scholar] [CrossRef]
- Goldstein A, Covington BP, Mahabadi N, Mesfin FB. Neuroanatomy, Corpus Callosum. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK448209/ (accessed on 29 May 2025).
- Dramsdahl M, Westerhausen R, Haavik J, Hugdahl K, Plessen KJ. Adults with attention-deficit/hyperactivity disorder — A diffusion-tensor imaging study of the corpus callosum. Psychiatry Research: Neuroimaging 2012, 201, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Luders E, Kurth F, Das D, Oyarce DE, Shaw ME, Sachdev P, et al. Associations between corpus callosum size and ADHD symptoms in older adults: The PATH through life study. Psychiatry Research: Neuroimaging 2016, 256, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Qiu A, Crocetti D, Adler M, Mahone EM, Denckla MB, Miller MI, et al. Basal ganglia volume and shape in children with attention deficit hyperactivity disorder. Am J Psychiatry 2009, 166, 74–82. [Google Scholar] [CrossRef]
- Shaw P, De Rossi P, Watson B, Wharton A, Greenstein D, Raznahan A, et al. Mapping the Development of the Basal Ganglia in Children With Attention-Deficit/Hyperactivity Disorder. Journal of the American Academy of Child & Adolescent Psychiatry 2014, 53, 780–789.e11. [Google Scholar]
- Pasini A, D’agati E. Pathophysiology of NSS in ADHD. The World Journal of Biological Psychiatry 2009, 10, 495–502. [Google Scholar] [CrossRef]
- Haber, SN.; Neuroanatomy of Reward: A View from the Ventral Striatum. In: Gottfried JA, editor. Neurobiology of Sensation and Reward [Internet]. Boca Raton (FL): CRC Press/Taylor & Francis; 2011. (Frontiers in Neuroscience). Available online: http://www.ncbi.nlm.nih.gov/books/NBK92777/ (accessed on 29 May 2025).
- Greven CU, Bralten J, Mennes M, O’Dwyer L, Van Hulzen KJE, Rommelse N, et al. Developmentally Stable Whole-Brain Volume Reductions and Developmentally Sensitive Caudate and Putamen Volume Alterations in Those With Attention-Deficit/Hyperactivity Disorder and Their Unaffected Siblings. JAMA Psychiatry 2015, 72, 490. [Google Scholar] [CrossRef]
- Mooney MA, Bhatt P, Hermosillo RJM, Ryabinin P, Nikolas M, Faraone SV, et al. Smaller total brain volume but not subcortical structure volume related to common genetic risk for ADHD. Psychol Med 2021, 51, 1279–1288. [Google Scholar] [CrossRef]
- Castellanos, FX. Developmental Trajectories of Brain Volume Abnormalities in Children and Adolescents With Attention-Deficit/Hyperactivity Disorder. JAMA 2002, 288, 1740. [Google Scholar] [CrossRef]
- MacDonald HJ, Kleppe R, Szigetvari PD, Haavik J. The dopamine hypothesis for ADHD: An evaluation of evidence accumulated from human studies and animal models. Front Psychiatry 2024, 15, 1492126. [Google Scholar] [CrossRef]
- Cortese S, Adamo N, Del Giovane C, Mohr-Jensen C, Hayes AJ, Carucci S, et al. Comparative efficacy and tolerability of medications for attention-deficit hyperactivity disorder in children, adolescents, and adults: a systematic review and network meta-analysis. Lancet Psychiatry 2018, 5, 727–738. [Google Scholar] [CrossRef] [PubMed]
- McCabe SE, Knight JR, Teter CJ, Wechsler H. Non-medical use of prescription stimulants among US college students: prevalence and correlates from a national survey. Addiction 2005, 100, 96–106. [Google Scholar] [CrossRef]
- Bourgeois FT, Kim JM, Mandl KD. Premarket safety and efficacy studies for ADHD medications in children. PLoS One 2014, 9, e102249. [Google Scholar]
- Besag FMC. ADHD treatment and pregnancy. Drug Saf 2014, 37, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Jiang Y, Zou D, Li Y, Gu S, Dong J, Ma X, et al. Monoamine Neurotransmitters Control Basic Emotions and Affect Major Depressive Disorders. Pharmaceuticals (Basel) 2022, 15, 1203. [Google Scholar] [CrossRef]
- Nikolaus S, Mamlins E, Giesel FL, Schmitt D, Müller HW. Monoaminergic hypo- or hyperfunction in adolescent and adult attention-deficit hyperactivity disorder? Rev Neurosci 2022, 33, 347–364. [Google Scholar] [CrossRef]
- Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 2010, 68, 815–834. [Google Scholar] [CrossRef]
- Berke, JD. What does dopamine mean? Nat Neurosci 2018, 21, 787–793. [Google Scholar] [CrossRef]
- Ott T, Nieder A. Dopamine and Cognitive Control in Prefrontal Cortex. Trends in Cognitive Sciences 2019, 23, 213–234. [Google Scholar] [CrossRef]
- Elliott BL, D’Ardenne K, Mukherjee P, Schweitzer JB, McClure SM. Limbic and Executive Meso- and Nigrostriatal Tracts Predict Impulsivity Differences in Attention-Deficit/Hyperactivity Disorder. Biol Psychiatry Cogn Neurosci Neuroimaging 2022, 7, 415–423. [Google Scholar]
- Luo SX, Huang EJ. Dopaminergic Neurons and Brain Reward Pathways. The American Journal of Pathology 2016, 186, 478–488. [Google Scholar] [CrossRef]
- Qi-Lytle X, Sayers S, Wagner EJ. Current Review of the Function and Regulation of Tuberoinfundibular Dopamine Neurons. Int J Mol Sci 2023, 25, 110. [Google Scholar] [CrossRef]
- Klein MO, Battagello DS, Cardoso AR, Hauser DN, Bittencourt JC, Correa RG. Dopamine: Functions, Signaling, and Association with Neurological Diseases. Cell Mol Neurobiol 2019, 39, 31–59. [Google Scholar] [CrossRef]
- Ford, CP. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 2014, 282, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu JM, Gainetdinov RR. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacological Reviews 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed]
- Forssberg H, Fernell E, Waters S, Waters N, Tedroff J. Altered pattern of brain dopamine synthesis in male adolescents with attention deficit hyperactivity disorder. Behav Brain Funct 2006, 2, 40. [Google Scholar] [CrossRef]
- Cheon KA, Ryu YH, Kim YK, Namkoong K, Kim CH, Lee JD. Dopamine transporter density in the basal ganglia assessed with [123I]IPT SPET in children with attention deficit hyperactivity disorder. Eur J Nucl Med Mol Imaging 2003, 30, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Dougherty DD, Bonab AA, Spencer TJ, Rauch SL, Madras BK, Fischman AJ. Dopamine transporter density in patients with attention deficit hyperactivity disorder. The Lancet 1999, 354, 2132–2133. [Google Scholar] [CrossRef]
- Ludolph AG, Kassubek J, Schmeck K, Glaser C, Wunderlich A, Buck AK, et al. Dopaminergic dysfunction in attention deficit hyperactivity disorder (ADHD), differences between pharmacologically treated and never treated young adults: a 3,4-dihdroxy-6-[18F]fluorophenyl-l-alanine PET study. Neuroimage 2008, 41, 718–727. [Google Scholar] [CrossRef]
- Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D, Jons PH, Cohen RM. High midbrain [18F]DOPA accumulation in children with attention deficit hyperactivity disorder. Am J Psychiatry 1999, 156, 1209–1215. [Google Scholar] [CrossRef]
- Jucaite A, Fernell E, Halldin C, Forssberg H, Farde L. Reduced midbrain dopamine transporter binding in male adolescents with attention-deficit/hyperactivity disorder: association between striatal dopamine markers and motor hyperactivity. Biol Psychiatry 2005, 57, 229–238. [Google Scholar] [CrossRef]
- Volkow ND, Wang GJ, Kollins SH, Wigal TL, Newcorn JH, Telang F, et al. Evaluating Dopamine Reward Pathway in ADHD: Clinical Implications. JAMA 2009, 302, 1084. [Google Scholar] [CrossRef] [PubMed]
- Volkow ND, Wang GJ, Newcorn JH, Kollins SH, Wigal TL, Telang F, et al. Motivation deficit in ADHD is associated with dysfunction of the dopamine reward pathway. Mol Psychiatry 2011, 16, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Itagaki S, Ohnishi T, Toda W, Sato A, Matsumoto J, Ito H, et al. Reduced dopamine transporter availability in drug-naive adult attention-deficit/hyperactivity disorder. PCN Rep 2024, 3, e177. [Google Scholar] [CrossRef]
- Weber MA, Conlon MM, Stutt HR, Wendt L, Ten Eyck P, Narayanan NS. Quantifying the inverted U: A meta-analysis of prefrontal dopamine, D1 receptors, and working memory. Behav Neurosci 2022, 136, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lopez E, Vrana KE. Dopamine beta-hydroxylase and its genetic variants in human health and disease. Journal of Neurochemistry 2020, 152, 157–181. [Google Scholar] [CrossRef]
- Bari BA, Chokshi V, Schmidt K. Locus coeruleus-norepinephrine: basic functions and insights into Parkinson’s disease. Neural Regen Res 2020, 15, 1006–1013. [Google Scholar] [CrossRef]
- Wagatsuma A, Okuyama T, Sun C, Smith LM, Abe K, Tonegawa S. Locus coeruleus input to hippocampal CA3 drives single-trial learning of a novel context. Proc Natl Acad Sci USA [Internet] 2018 Jan 9; 115(2). Available online: https://pnas.org/doi/full/10.1073/pnas.1714082115 (accessed on 2 April 2025).
- Roozendaal B, Hermans EJ. Norepinephrine effects on the encoding and consolidation of emotional memory: improving synergy between animal and human studies. Current Opinion in Behavioral Sciences 2017, 14, 115–122. [Google Scholar] [CrossRef]
- Del Campo N, Chamberlain SR, Sahakian BJ, Robbins TW. The Roles of Dopamine and Noradrenaline in the Pathophysiology and Treatment of Attention-Deficit/Hyperactivity Disorder. Biological Psychiatry 2011, 69, e145–57. [Google Scholar] [CrossRef]
- Hannestad J, Gallezot JD, Planeta-Wilson B, Lin SF, Williams WA, Van Dyck CH, et al. Clinically Relevant Doses of Methylphenidate Significantly Occupy Norepinephrine Transporters in Humans In Vivo. Biological Psychiatry 2010, 68, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Vanicek T, Spies M, Rami-Mark C, Savli M, Höflich A, Kranz GS, et al. The norepinephrine transporter in attention-deficit/hyperactivity disorder investigated with positron emission tomography. JAMA Psychiatry 2014, 71, 1340–1349. [Google Scholar] [CrossRef]
- Ulke C, Rullmann M, Huang J, Luthardt J, Becker GA, Patt M, et al. Adult attention-deficit/hyperactivity disorder is associated with reduced norepinephrine transporter availability in right attention networks: a (S,S)-O-[11C]methylreboxetine positron emission tomography study. Transl Psychiatry 2019, 9, 301. [Google Scholar] [CrossRef]
- Sigurdardottir HL, Kranz GS, Rami-Mark C, James GM, Vanicek T, Gryglewski G, et al. Association of norepinephrine transporter methylation with in vivo NET expression and hyperactivity-impulsivity symptoms in ADHD measured with PET. Mol Psychiatry 2021, 26, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Sigurdardottir HL, Kranz GS, Rami-Mark C, James GM, Vanicek T, Gryglewski G, et al. Effects of norepinephrine transporter gene variants on NET binding in ADHD and healthy controls investigated by PET. Hum Brain Mapp 2016, 37, 884–895. [Google Scholar] [CrossRef]
- Shang CY, Lin HY, Gau SSF. The norepinephrine transporter gene modulates intrinsic brain activity, visual memory, and visual attention in children with attention-deficit/hyperactivity disorder. Mol Psychiatry 2021, 26, 4026–4035. [Google Scholar] [CrossRef]
- Hohmann S, Hohm E, Treutlein J, Blomeyer D, Jennen-Steinmetz C, Schmidt MH, et al. Association of norepinephrine transporter (NET, SLC6A2) genotype with ADHD-related phenotypes: Findings of a longitudinal study from birth to adolescence. Psychiatry Research 2015, 226, 425–433. [Google Scholar] [CrossRef]
- Barkley RA, Smith KM, Fischer M, Navia B. An examination of the behavioral and neuropsychological correlates of three ADHD candidate gene polymorphisms (DRD4 7+, DBH TaqI A2, and DAT1 40 bp VNTR) in hyperactive and normal children followed to adulthood. Am J Med Genet B Neuropsychiatr Genet.
- Smith KM, Daly M, Fischer M, Yiannoutsos CT, Bauer L, Barkley R, et al. Association of the dopamine beta hydroxylase gene with attention deficit hyperactivity disorder: genetic analysis of the Milwaukee longitudinal study. Am J Med Genet B Neuropsychiatr Genet.
- Kieling C, Genro JP, Hutz MH, Rohde LA. The −1021 C/T DBH polymorphism is associated with neuropsychological performance among children and adolescents with ADHD. American J of Med Genetics Pt B.
- Bellgrove MA, Mattingley JB, Hawi Z, Mullins C, Kirley A, Gill M, et al. Impaired Temporal Resolution of Visual Attention and Dopamine Beta Hydroxylase Genotype in Attention-Deficit/Hyperactivity Disorder. Biological Psychiatry 2006, 60, 1039–1045. [Google Scholar] [CrossRef]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 2012, 74, 858–873. [Google Scholar] [CrossRef]
- Pollak Dorocic I, Fürth D, Xuan Y, Johansson Y, Pozzi L, Silberberg G, et al. A Whole-Brain Atlas of Inputs to Serotonergic Neurons of the Dorsal and Median Raphe Nuclei. Neuron 2014, 83, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Grace AA, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci 2007, 30, 220–227. [Google Scholar] [CrossRef]
- Predescu E, Vaidean T, Rapciuc AM, Sipos R. Metabolomic Markers in Attention-Deficit/Hyperactivity Disorder (ADHD) among Children and Adolescents-A Systematic Review. Int J Mol Sci 2024, 25, 4385. [Google Scholar] [CrossRef]
- Kenna GA, Roder-Hanna N, Leggio L, Zywiak WH, Clifford J, Edwards S, et al. Association of the 5-HTT gene-linked promoter region (5-HTTLPR) polymorphism with psychiatric disorders: review of psychopathology and pharmacotherapy. Pharmgenomics Pers Med 2012, 5, 19–35. [Google Scholar]
- Vanicek T, Kutzelnigg A, Philippe C, Sigurdardottir HL, James GM, Hahn A, et al. Altered interregional molecular associations of the serotonin transporter in attention deficit/hyperactivity disorder assessed with PET. Hum Brain Mapp 2017, 38, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Karlsson L, Tuominen L, Huotarinen A, Leppämäki S, Sihvola E, Helin S, et al. Serotonin transporter in attention-deficit hyperactivity disorder – preliminary results from a positron emission tomography study. Psychiatry Research: Neuroimaging 2013, 212, 164–165. [Google Scholar] [CrossRef]
- Zhang Z, Cordeiro Matos S, Jego S, Adamantidis A, Séguéla P. Norepinephrine Drives Persistent Activity in Prefrontal Cortex via Synergistic α1 and α2 Adrenoceptors. Currie K, editor. PLoS ONE 2013, 8, e66122. [Google Scholar]
- Zaborszky, L. Sleep-wake mechanisms and basal forebrain circuitry. Front Biosci 2003, 8, d1146–d1169. [Google Scholar] [CrossRef]
- España RA, Berridge CW. Organization of noradrenergic efferents to arousal-related basal forebrain structures. J of Comparative Neurology 2006, 496, 668–683. [Google Scholar] [CrossRef]
- Galvez R, Mesches MH, McGaugh JL. Norepinephrine release in the amygdala in response to footshock stimulation. Neurobiol Learn Mem 1996, 66, 253–257. [Google Scholar] [CrossRef]
- Moreno-Castilla P, Pérez-Ortega R, Violante-Soria V, Balderas I, Bermúdez-Rattoni F. Hippocampal release of dopamine and norepinephrine encodes novel contextual information. Hippocampus 2017, 27, 547–557. [Google Scholar] [CrossRef]
- Stanley AT, Post MR, Lacefield C, Sulzer D, Miniaci MC. Norepinephrine release in the cerebellum contributes to aversive learning. Nat Commun 2023, 14, 4852. [Google Scholar] [CrossRef]
- Hoxha E, Tempia F, Lippiello P, Miniaci MC. Modulation, Plasticity and Pathophysiology of the Parallel Fiber-Purkinje Cell Synapse. Front Synaptic Neurosci [Internet] 2016 Nov 3; 8. Available online: http://journal.frontiersin.org/article/10.3389/fnsyn.2016.00035/full (accessed on 7 August 2025).
- Inoshita T, Hirano T. Norepinephrine Facilitates Induction of Long-term Depression through β-Adrenergic Receptor at Parallel Fiber-to-Purkinje Cell Synapses in the Flocculus. Neuroscience 2021, 462, 141–150. [Google Scholar] [CrossRef]
- Margules DL, Lewis MJ, Dragovich JA, Margules AS. Hypothalamic Norepinephrine: Circadian Rhythms and the Control of Feeding Behavior. Science 1972, 178, 640–643. [Google Scholar] [CrossRef]
- Greco AM, Gambardella P, Sticchi R, D’Aponte D, De Franciscis P. Circadian rhythms of hypothalamic norepinephrine and of some circulating substances in individually housed adult rats. Physiology & Behavior 1992, 52, 1167–1172. [Google Scholar] [CrossRef]
- Rodenkirch C, Liu Y, Schriver BJ, Wang Q. Locus coeruleus activation enhances thalamic feature selectivity via norepinephrine regulation of intrathalamic circuit dynamics. Nat Neurosci 2019, 22, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Af Bjerkén S, Stenmark Persson R, Barkander A, Karalija N, Pelegrina-Hidalgo N, Gerhardt GA, et al. Noradrenaline is crucial for the substantia nigra dopaminergic cell maintenance. Neurochemistry International 2019, 131, 104551. [Google Scholar] [CrossRef] [PubMed]
- Gu BM, Kim JG, Hossain A, Cron GO, Lee JH. Substantia nigra modulates breathing rate via locus coeruleus. iScience 2025, 28, 112423. [Google Scholar] [CrossRef]
- Sara, SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 2009, 10, 211–223. [Google Scholar] [CrossRef]
- Puig MV, Gulledge AT. Serotonin and Prefrontal Cortex Function: Neurons, Networks, and Circuits. Mol Neurobiol 2011, 44, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Sargin D, Jeoung HS, Goodfellow NM, Lambe EK. Serotonin Regulation of the Prefrontal Cortex: Cognitive Relevance and the Impact of Developmental Perturbation. ACS Chem Neurosci 2019, 10, 3078–3093. [Google Scholar] [CrossRef]
- Zangen A, Nakash R, Overstreet D, Yadid G. Association between depressive behavior and absence of serotonin-dopamine interaction in the nucleus accumbens. Psychopharmacology 2001, 155, 434–439. [Google Scholar] [CrossRef]
- Hon OJ, DiBerto JF, Mazzone CM, Sugam J, Bloodgood DW, Hardaway JA, et al. Serotonin modulates an inhibitory input to the central amygdala from the ventral periaqueductal gray. Neuropsychopharmacology 2022, 47, 2194–2204. [Google Scholar] [CrossRef] [PubMed]
- Balasubramani PP, Chakravarthy VS, Ravindran B, Moustafa AA. A network model of basal ganglia for understanding the roles of dopamine and serotonin in reward-punishment-risk based decision making. Front Comput Neurosci 2015, 9, 76. [Google Scholar]
- Portas CM, Bjorvatn B, Ursin R. Serotonin and the sleep/wake cycle: special emphasis on microdialysis studies. Progress in Neurobiology 2000, 60, 13–35. [Google Scholar] [CrossRef]
- Lin MT, Tsay HJ, Su WH, Chueh FY. Changes in extracellular serotonin in rat hypothalamus affect thermoregulatory function. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 1998, 274, R1260–7. [Google Scholar] [CrossRef]
- Kawashima, T. The role of the serotonergic system in motor control. Neuroscience Research 2018, 129, 32–39. [Google Scholar] [CrossRef]
- Yoshida K, Drew MR, Mimura M, Tanaka KF. Serotonin-mediated inhibition of ventral hippocampus is required for sustained goal-directed behavior. Nat Neurosci 2019, 22, 770–777. [Google Scholar] [CrossRef]
- Buhot MC, Martin S, Segu L. Role of serotonin in memory impairment. Ann Med 2000, 32, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Schwarting RKW, Thiel CM, Müller CP, Huston JP. Relationship between anxiety and serotonin in the ventral striatum. NeuroReport 1998, 9, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Jocoy E, Cepeda C, Levine M, André V. NMDA and Dopamine: Diverse Mechanisms Applied to Interacting Receptor Systems. In: VanDongen A, editor. Biology of the NMDA Receptor [Internet]. CRC Press; 2008. p. 41–57. (Frontiers in Neuroscience; vol 20085482). Available online: http://www.crcnetbase.com/doi/abs/10.1201/9781420044157.ch3 (accessed on 17 April 2025).
- Li F, Tsien JZ. Memory and the NMDA receptors. N Engl J Med 2009, 361, 302–303. [Google Scholar] [CrossRef]
- Varela JA, Hirsch SJ, Chapman D, Leverich LS, Greene RW. D1 /D5 Modulation of Synaptic NMDA Receptor Currents. J Neurosci 2009, 29, 3109–3119. [Google Scholar] [CrossRef]
- Attention deficit hyperactivity disorder: diagnosis and management [Internet]. London: National Institute for Health and Care Excellence (NICE); 2019. (National Institute for Health and Care Excellence: Guidelines). Available online: http://www.ncbi.nlm.nih.gov/books/NBK493361/ (accessed on 25 June 2025).
- Jerome D, Jerome L. Approach to diagnosis and management of childhood attention deficit hyperactivity disorder. Can Fam Physician 2020, 66, 732–736. [Google Scholar]
- Wolraich ML, Hagan JF, Allan C, Chan E, Davison D, Earls M, et al. Clinical Practice Guideline for the Diagnosis, Evaluation, and Treatment of Attention-Deficit/Hyperactivity Disorder in Children and Adolescents. Pediatrics 2019, 144, e20192528. [Google Scholar] [CrossRef]
- Quintero J, Gutiérrez-Casares JR, Álamo C. Molecular Characterisation of the Mechanism of Action of Stimulant Drugs Lisdexamfetamine and Methylphenidate on ADHD Neurobiology: A Review. Neurol Ther 2022, 11, 1489–1517. [Google Scholar] [CrossRef]
- Calipari ES, Jones SR. Sensitized nucleus accumbens dopamine terminal responses to methylphenidate and dopamine transporter releasers after intermittent-access self-administration. Neuropharmacology 2014, 82, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Grund T, Lehmann K, Bock N, Rothenberger A, Teuchert-Noodt G. Influence of methylphenidate on brain development--an update of recent animal experiments. Behav Brain Funct 2006, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Gatley SJ, Pan D, Chen R, Chaturvedi G, Ding YS. Affinities of methylphenidate derivatives for dopamine, norepinephrine and serotonin transporters. Life Sciences 1996, 58, PL231–9. [Google Scholar] [CrossRef]
- Stevens T, Sangkuhl K, Brown JT, Altman RB, Klein TE. PharmGKB summary: methylphenidate pathway, pharmacokinetics/pharmacodynamics. Pharmacogenet Genomics.
- Volz TJ, Farnsworth SJ, King JL, Riddle EL, Hanson GR, Fleckenstein AE. Methylphenidate Administration Alters Vesicular Monoamine Transporter-2 Function in Cytoplasmic and Membrane-Associated Vesicles. The Journal of Pharmacology and Experimental Therapeutics 2007, 323, 738–745. [Google Scholar] [CrossRef]
- Volz TJ, Farnsworth SJ, Rowley SD, Hanson GR, Fleckenstein AE. Methylphenidate-induced increases in vesicular dopamine sequestration and dopamine release in the striatum: the role of muscarinic and dopamine D2 receptors. J Pharmacol Exp Ther 2008, 327, 161–167. [Google Scholar] [CrossRef]
- Mantle TJ, Tipton KF, Garrett NJ. Inhibition of monoamine oxidase by amphetamine and related compounds. Biochemical Pharmacology 1976, 25, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Miller HH, Shore PA, Clarke DE. In vivo monoamine oxidase inhibition by d-amphetamine. Biochem Pharmacol 1980, 29, 1347–1354. [Google Scholar] [CrossRef]
- Sitte HH, Freissmuth M. Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol Sci 2015, 36, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Martin D, Le JK. Amphetamine. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK556103/ (accessed on 12 January 2025).
- Jones S, Kauer JA. Amphetamine depresses excitatory synaptic transmission via serotonin receptors in the ventral tegmental area. J Neurosci 1999, 19, 9780–9787. [Google Scholar] [CrossRef]
- Groom MJ, Cortese S. Current Pharmacological Treatments for ADHD. In: Stanford SC, Sciberras E, editors. New Discoveries in the Behavioral Neuroscience of Attention-Deficit Hyperactivity Disorder [Internet]. Cham: Springer International Publishing; 2022. p. 19–50. (Current Topics in Behavioral Neurosciences; vol. 57). Available online: https://link.springer.com/10.1007/7854_2022_330 (accessed on 20 May 2025).
- Long-Term Safety and Efficacy of Stimulant, vs.; Non-Stimulant Medications in ADHD Treatment: A Comparative Meta-Analysis Over Two Years. J Psychiatry Cogn Behav [Internet] 2023 Dec 16; 7(1). Available online: https://www.gavinpublishers.com/article/view/long-term-safety-and-efficacy-of-stimulant-vs-non-stimulant-medications-in-adhd-treatment-a-comparative-meta-analysis-over-two-years (accessed on 21 May 2025).
- Carboni E, Silvagni A, Vacca C, Di Chiara G. Cumulative effect of norepinephrine and dopamine carrier blockade on extracellular dopamine increase in the nucleus accumbens shell, bed nucleus of stria terminalis and prefrontal cortex. Journal of Neurochemistry 2006, 96, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Fedder D, Patel H, Saadabadi A. Atomoxetine. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK493234/ (accessed on 20 May 2025).
- Swanson CJ, Perry KW, Koch-Krueger S, Katner J, Svensson KA, Bymaster FP. Effect of the attention deficit/hyperactivity disorder drug atomoxetine on extracellular concentrations of norepinephrine and dopamine in several brain regions of the rat. Neuropharmacology 2006, 50, 755–760. [Google Scholar] [CrossRef]
- Bymaster, F. Atomoxetine Increases Extracellular Levels of Norepinephrine and Dopamine in Prefrontal Cortex of Rat A Potential Mechanism for Efficacy in Attention Deficit/Hyperactivity Disorder. Neuropsychopharmacology 2002, 27, 699–711. [Google Scholar] [CrossRef]
- Mathew BM, Pellegrini MV. Viloxazine. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK576423/ (accessed on 20 May 2025).
- Yu C, Garcia-Olivares J, Candler S, Schwabe S, Maletic V. New Insights into the Mechanism of Action of Viloxazine: Serotonin and Norepinephrine Modulating Properties. J Exp Pharmacol 2020, 12, 285–300. [Google Scholar] [CrossRef]
- Neuchat EE, Bocklud BE, Kingsley K, Barham WT, Luther PM, Ahmadzadeh S, et al. The Role of Alpha-2 Agonists for Attention Deficit Hyperactivity Disorder in Children: A Review. Neurol Int 2023, 15, 697–707. [Google Scholar] [CrossRef]
- Bello, NT. Clinical utility of guanfacine extended release in the treatment of ADHD in children and adolescents. Patient Prefer Adherence 2015, 9, 877–885. [Google Scholar] [CrossRef]
- Alamo C, López-Muñoz F, Sánchez-García J. Mechanism of action of guanfacine: a postsynaptic differential approach to the treatment of attention deficit hyperactivity disorder (adhd). Actas Esp Psiquiatr 2016, 44, 107–112. [Google Scholar]
- Russell VA, Sagvolden T, Johansen EB. Animal models of attention-deficit hyperactivity disorder. Behav Brain Funct 2005, 1, 9. [Google Scholar] [CrossRef]
- Gainetdinov, RR. Strengths and limitations of genetic models of ADHD. ADHD Atten Def Hyp Disord 2010, 2, 21–30. [Google Scholar] [CrossRef]
- Kim D, Yadav D, Song M. An updated review on animal models to study attention-deficit hyperactivity disorder. Transl Psychiatry 2024, 14, 187. [Google Scholar] [CrossRef]
- Schaare HL, Blöchl M, Kumral D, Uhlig M, Lemcke L, Valk SL, et al. Associations between mental health, blood pressure and the development of hypertension. Nat Commun [Internet] 2023 Apr 7; 14(1). Available online: https://www.nature.com/articles/s41467-023-37579-6 (accessed on 24 July 2025).
- Gąsecki D, Kwarciany M, Nyka W, Narkiewicz K. Hypertension, brain damage and cognitive decline. Curr Hypertens Rep 2013, 15, 547–558. [Google Scholar] [CrossRef]
- Fuemmeler BF, Østbye T, Yang C, McClernon FJ, Kollins SH. Association between attention-deficit/hyperactivity disorder symptoms and obesity and hypertension in early adulthood: a population-based study. Int J Obes (Lond) 2011, 35, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi A, Jeddi S, Kashfi K. The laboratory rat: age and body weight matter. EXCLI Journal; 20, Doc1431; ISSN 1611-2156 [Internet] 2021. Available online: https://www.excli.de/index.php/excli/article/view/4072 (accessed on 21 May 2025).
- Maldonado KA, Alsayouri K. Physiology, Brain. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK551718/ (accessed on 21 May 2025).
- Lin, JH. Species similarities and differences in pharmacokinetics. Drug Metabolism and Disposition 1995, 23, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Rubio Morell B, Hernández Expósito S. Differential long-term medication impact on executive function and delay aversion in ADHD. Applied Neuropsychology: Child 2019, 8, 140–157. [Google Scholar] [CrossRef] [PubMed]
- MacQueen DA, Minassian A, Kenton JA, Geyer MA, Perry W, Brigman JL, et al. Amphetamine improves mouse and human attention in the 5-choice continuous performance test. Neuropharmacology 2018, 138, 87–96. [Google Scholar] [CrossRef]
- Martin IL, Baker GB, Mitchell PR. The effect of viloxazine hydrochloride on the transport of noradrenaline, dopamine, 5-hydroxytryptamine and γ-amino-butyric acid in rat brain tissue. Neuropharmacology 1978, 17, 421–423. [Google Scholar] [CrossRef]
- Blackburn TP, Foster GA, Greenwood DT, Howe R. Effects of viloxazine, its optical isomers and its major metabolites on biogenic amine uptake mechanisms in vitro and in vivo. European Journal of Pharmacology 1978, 52, 367–374. [Google Scholar] [CrossRef]
- Lobato-Camacho FJ, López JC, Vargas JP. Enhancing spatial memory and pattern separation: Long-term effects of stimulant treatment in individuals with ADHD. Behavioural Brain Research 2024, 475, 115211. [Google Scholar] [CrossRef] [PubMed]
- Fuermaier ABM, Tucha L, Koerts J, Weisbrod M, Lange KW, Aschenbrenner S, et al. Effects of methylphenidate on memory functions of adults with ADHD. Applied Neuropsychology: Adult 2017, 24, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Arnold LE, Hodgkins P, Kahle J, Madhoo M, Kewley G. Long-Term Outcomes of ADHD: Academic Achievement and Performance. J Atten Disord 2020, 24, 73–85. [Google Scholar] [CrossRef]
- Whitehurst LN, Mednick SC. Psychostimulants may block long-term memory formation via degraded sleep in healthy adults. Neurobiology of Learning and Memory 2021, 178, 107342. [Google Scholar] [CrossRef]
- Ballard ME, Gallo DA, de Wit H. Amphetamine increases errors during episodic memory retrieval. J Clin Psychopharmacol 2014, 34, 85–92. [Google Scholar] [CrossRef]
- Spencer TJ, Brown A, Seidman LJ, Valera EM, Makris N, Lomedico A, et al. Effect of psychostimulants on brain structure and function in ADHD: a qualitative literature review of magnetic resonance imaging-based neuroimaging studies. J Clin Psychiatry 2013, 74, 902–917. [Google Scholar] [CrossRef]
- Wu F, Zhang W, Ji W, Zhang Y, Jiang F, Li G, et al. Stimulant medications in children with ADHD normalize the structure of brain regions associated with attention and reward. Neuropsychopharmacol 2024, 49, 1330–1340. [Google Scholar] [CrossRef]
- Walhovd KB, Amlien I, Schrantee A, Rohani DA, Groote I, Bjørnerud A, et al. Methylphenidate Effects on Cortical Thickness in Children and Adults with Attention-Deficit/Hyperactivity Disorder: A Randomized Clinical Trial. AJNR Am J Neuroradiol 2020, 41, 758–765. [Google Scholar] [CrossRef]
- Fotopoulos NH, Devenyi GA, Guay S, Sengupta SM, Chakravarty MM, Grizenko N, et al. Cumulative exposure to ADHD medication is inversely related to hippocampus subregional volume in children. Neuroimage Clin 2021, 31, 102695. [Google Scholar] [CrossRef]
- Kassim, FM. Systematic reviews of the acute effects of amphetamine on working memory and other cognitive performances in healthy individuals, with a focus on the potential influence of personality traits. Hum Psychopharmacol 2023, 38, e2856. [Google Scholar] [CrossRef] [PubMed]
- de Wit H, Enggasser JL, Richards JB. Acute administration of d-amphetamine decreases impulsivity in healthy volunteers. Neuropsychopharmacology 2002, 27, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Markowitz JS, Patrick KS. The Clinical Pharmacokinetics of Amphetamines Utilized in the Treatment of Attention-Deficit/Hyperactivity Disorder. Journal of Child and Adolescent Psychopharmacology 2017, 27, 678–689. [Google Scholar] [CrossRef]
- Barch DM, Carter CS. Amphetamine improves cognitive function in medicated individuals with schizophrenia and in healthy volunteers. Schizophrenia Research 2005, 77, 43–58. [Google Scholar] [CrossRef]
- Mintzer MZ, Griffiths RR. Triazolam-amphetamine interaction: dissociation of effects on memory versus arousal. J Psychopharmacol 2003, 17, 17–29. [Google Scholar] [CrossRef]
- Ward AS, Kelly TH, Foltin RW, Fischman MW. Effects of d-amphetamine on task performance and social behavior of humans in a residential laboratory. Experimental and Clinical Psychopharmacology 1997, 5, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Willson MC, Wilman AH, Bell EC, Asghar SJ, Silverstone PH. Dextroamphetamine causes a change in regional brain activity in vivo during cognitive tasks: A functional magnetic resonance imaging study of blood oxygen level-dependent response. Biological Psychiatry 2004, 56, 284–291. [Google Scholar] [CrossRef]
- Fleming K, Bigelow LB, Weinberger DR, Goldberg TE. Neuropsychological effects of amphetamine may correlate with personality characteristics. Psychopharmacol Bull 1995, 31, 357–362. [Google Scholar]
- Makris AP, Rush CR, Frederich RC, Taylor AC, Kelly TH. Behavioral and subjective effects of D-amphetamine and modafinil in healthy adults. Experimental and Clinical Psychopharmacology 2007, 15, 123–133. [Google Scholar] [CrossRef]
- Wardle MC, Yang A, De Wit H. Effect of d-amphetamine on post-error slowing in healthy volunteers. Psychopharmacology 2012, 220, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski L, Hendricks K, Gordon M. Test-Taking Performance of High School Students With ADHD. J Atten Disord 2015, 19, 27–34. [Google Scholar] [CrossRef]
- Dan O, Raz S. The Relationships Among ADHD, Self-Esteem, and Test Anxiety in Young Adults. J Atten Disord 2015, 19, 231–239. [Google Scholar] [CrossRef]
- Cools R, D’Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry 2011, 69, e113–e125. [Google Scholar] [CrossRef]
- Prasad V, Brogan E, Mulvaney C, Grainge M, Stanton W, Sayal K. How effective are drug treatments for children with ADHD at improving on-task behaviour and academic achievement in the school classroom? A systematic review and meta-analysis. Eur Child Adolesc Psychiatry 2013, 22, 203–216. [Google Scholar] [CrossRef]
- Ilieva IP, Farah MJ. Attention, Motivation, and Study Habits in Users of Unprescribed ADHD Medication. J Atten Disord 2019, 23, 149–162. [Google Scholar] [CrossRef]
- Choe, E.; Amphetamine Increases Phosphorylation of Extracellular Signal-regulated Kinase and Transcription Factors in the Rat Striatum via Group I Metabotropic Glutamate Receptors. Neuropsychopharmacology [Internet] 2002 Jun 9. Available online: https://www.nature.com/doifinder/10.1016/S0893-133X (accessed on day month year).
- Mao LM, Xue B, Jin DZ, Wang JQ. Dynamic increases in AMPA receptor phosphorylation in the rat hippocampus in response to amphetamine. J Neurochem 2015, 133, 795–805. [Google Scholar] [CrossRef]
- Arroyo-García LE, Tendilla-Beltrán H, Vázquez-Roque RA, Jurado-Tapia EE, Díaz A, Aguilar-Alonso P, et al. Amphetamine sensitization alters hippocampal neuronal morphology and memory and learning behaviors. Mol Psychiatry 2021, 26, 4784–4794. [Google Scholar] [CrossRef]
- Chidambaram SB, Rathipriya AG, Bolla SR, Bhat A, Ray B, Mahalakshmi AM, et al. Dendritic spines: Revisiting the physiological role. Progress in Neuro-Psychopharmacology and Biological Psychiatry 2019, 92, 161–193. [Google Scholar] [CrossRef] [PubMed]
- Park YH, Kantor L, Wang KKW, Gnegy ME. Repeated, intermittent treatment with amphetamine induces neurite outgrowth in rat pheochromocytoma cells (PC12 cells). Brain Research 2002, 951, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Angelucci F, Gruber SHM, El Khoury A, Tonali PA, Mathé AA. Chronic amphetamine treatment reduces NGF and BDNF in the rat brain. European Neuropsychopharmacology 2007, 17, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Li MH, Underhill SM, Reed C, Phillips TJ, Amara SG, Ingram SL. Amphetamine and Methamphetamine Increase NMDAR-GluN2B Synaptic Currents in Midbrain Dopamine Neurons. Neuropsychopharmacology 2017, 42, 1539–1547. [Google Scholar] [CrossRef]
- Lu W, Monteggia LM, Wolf ME. Withdrawal from repeated amphetamine administration reduces NMDAR1 expression in the rat substantia nigra, nucleus accumbens and medial prefrontal cortex. Eur J of Neuroscience 1999, 11, 3167–3177. [Google Scholar] [CrossRef] [PubMed]
- Lu W, Chen H, Xue CJ, Wolf ME. Repeated amphetamine administration alters the expression of mRNA for AMPA receptor subunits in rat nucleus accumbens and prefrontal cortex. Synapse 1997, 26, 269–280. [Google Scholar] [CrossRef]
- Mao LM, Wang W, Chu XP, Zhang GC, Liu XY, Yang YJ, et al. Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci 2009, 12, 602–610. [Google Scholar] [CrossRef]
- Lohr WD, Wanta JW, Baker M, Grudnikoff E, Morgan W, Chhabra D, et al. Intentional Discontinuation of Psychostimulants Used to Treat ADHD in Youth: A Review and Analysis. Front Psychiatry 2021, 12, 642798. [Google Scholar] [CrossRef]
- Beutler LR, Wanat MJ, Quintana A, Sanz E, Bamford NS, Zweifel LS, et al. Balanced NMDA receptor activity in dopamine D1 receptor (D1R)- and D2R-expressing medium spiny neurons is required for amphetamine sensitization. Proc Natl Acad Sci USA 2011, 108, 4206–4211. [Google Scholar] [CrossRef]
- Elliott R, Sahakian BJ, Matthews K, Bannerjea A, Rimmer J, Robbins TW. Effects of methylphenidate on spatial working memory and planning in healthy young adults. Psychopharmacology 1997, 131, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Agay N, Yechiam E, Carmel Z, Levkovitz Y. Methylphenidate enhances cognitive performance in adults with poor baseline capacities regardless of attention-deficit/hyperactivity disorder diagnosis. J Clin Psychopharmacol 2014, 34, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Hammerness P, Fried R, Petty C, Meller B, Biederman J. Assessment of cognitive domains during treatment with OROS methylphenidate in adolescents with ADHD. Child Neuropsychology 2014, 20, 319–327. [Google Scholar] [CrossRef]
- Linssen AMW, Vuurman EFPM, Sambeth A, Riedel WJ. Methylphenidate produces selective enhancement of declarative memory consolidation in healthy volunteers. Psychopharmacology (Berl) 2012, 221, 611–619. [Google Scholar] [CrossRef]
- Tucha O, Mecklinger L, Laufkötter R, Klein HE, Walitza S, Lange KW. Methylphenidate-induced improvements of various measures of attention in adults with Attention Deficit Hyperactivity Disorder. J Neural Transm 2006, 113, 1575–1592. [Google Scholar] [CrossRef]
- Maul J, Advokat C. Stimulant medications for attention-deficit/hyperactivity disorder (ADHD) improve memory of emotional stimuli in ADHD-diagnosed college students. Pharmacology Biochemistry and Behavior 2013, 105, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Andersen SL, Arvanitogiannis A, Pliakas AM, LeBlanc C, Carlezon WA. Altered responsiveness to cocaine in rats exposed to methylphenidate during development. Nat Neurosci 2002, 5, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Crawford CA, McDougall SA, Meier TL, Collins RL, Watson JB. Repeated methylphenidate treatment induces behavioral sensitization and decreases protein kinase A and dopamine-stimulated adenylyl cyclase activity in the dorsal striatum. Psychopharmacology 1998, 136, 34–43. [Google Scholar] [CrossRef]
- Beverley JA, Piekarski C, Van Waes V, Steiner H. Potentiated gene regulation by methylphenidate plus fluoxetine treatment: Long-term gene blunting (Zif268, Homer1a) and behavioral correlates. Basal Ganglia 2014, 4, 109–116. [Google Scholar] [CrossRef]
- Motaghinejad M, Motevalian M, Falak R, Heidari M, Sharzad M, Kalantari E. Neuroprotective effects of various doses of topiramate against methylphenidate-induced oxidative stress and inflammation in isolated rat amygdala: the possible role of CREB/BDNF signaling pathway. J Neural Transm (Vienna) 2016, 123, 1463–1477. [Google Scholar] [CrossRef]
- Di Miceli M, Omoloye A, Gronier B. Characterisation of methylphenidate-induced excitation in midbrain dopamine neurons, an electrophysiological study in the rat brain. Prog Neuropsychopharmacol Biol Psychiatry 2022, 112, 110406. [Google Scholar]
- Zhang CL, Feng ZJ, Liu Y, Ji XH, Peng JY, Zhang XH, et al. Methylphenidate enhances NMDA-receptor response in medial prefrontal cortex via sigma-1 receptor: a novel mechanism for methylphenidate action. PLoS One 2012, 7, e51910. [Google Scholar]
- Zhou BY, Li ZX, Li YW, Li JN, Liu WT, Liu XY, et al. Central Med23 deficiency leads to malformation of dentate gyrus and ADHD-like behaviors in mice. Neuropsychopharmacol [Internet] 2025 Mar 20. Available online: https://www.nature.com/articles/s41386-025-02088-1 (accessed on 29 April 2025).
- Urban KR, Li YC, Gao WJ. Treatment with a clinically-relevant dose of methylphenidate alters NMDA receptor composition and synaptic plasticity in the juvenile rat prefrontal cortex. Neurobiology of Learning and Memory 2013, 101, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Jalloh K, Roeder N, Hamilton J, Delis F, Hadjiargyrou M, Komatsu D, et al. Chronic oral methylphenidate treatment in adolescent rats promotes dose-dependent effects on NMDA receptor binding. Life Sciences 2021, 264, 118708. [Google Scholar] [CrossRef]
- Van Der Marel K, Bouet V, Meerhoff GF, Freret T, Boulouard M, Dauphin F, et al. Effects of long-term methylphenidate treatment in adolescent and adult rats on hippocampal shape, functional connectivity and adult neurogenesis. Neuroscience 2015, 309, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Shang CY, Gau SSF. Improving Visual Memory, Attention, and School Function with Atomoxetine in Boys with Attention-Deficit/Hyperactivity Disorder. Journal of Child and Adolescent Psychopharmacology 2012, 22, 353–363. [Google Scholar] [CrossRef]
- Kratochvil CJ, Newcorn JH, Arnold LE, Duesenberg D, Emslie GJ, Quintana H, et al. Atomoxetine Alone or Combined With Fluoxetine for Treating ADHD With Comorbid Depressive or Anxiety Symptoms. Journal of the American Academy of Child & Adolescent Psychiatry 2005, 44, 915–924. [Google Scholar] [CrossRef]
- Geller D, Donnelly C, Lopez F, Rubin R, Newcorn J, Sutton V, et al. Atomoxetine Treatment for Pediatric Patients With Attention-Deficit/Hyperactivity Disorder With Comorbid Anxiety Disorder. Journal of the American Academy of Child & Adolescent Psychiatry 2007, 46, 1119–1127. [Google Scholar]
- Griffiths KR, Leikauf JE, Tsang TW, Clarke S, Hermens DF, Efron D, et al. Response inhibition and emotional cognition improved by atomoxetine in children and adolescents with ADHD: The ACTION randomized controlled trial. Journal of Psychiatric Research 2018, 102, 57–64. [Google Scholar] [CrossRef]
- Epperson CN, Pittman B, Czarkowski KA, Bradley J, Quinlan DM, Brown TE. Impact of atomoxetine on subjective attention and memory difficulties in perimenopausal and postmenopausal women. Menopause 2011, 18, 542–548. [Google Scholar] [CrossRef]
- de Jong CGW, Van De Voorde S, Roeyers H, Raymaekers R, Allen AJ, Knijff S, et al. Differential effects of atomoxetine on executive functioning and lexical decision in attention-deficit/hyperactivity disorder and reading disorder. J Child Adolesc Psychopharmacol 2009, 19, 699–707. [Google Scholar] [CrossRef]
- Marquand AF, De Simoni S, O’Daly OG, Williams SC, Mourão-Miranda J, Mehta MA. Pattern Classification of Working Memory Networks Reveals Differential Effects of Methylphenidate, Atomoxetine, and Placebo in Healthy Volunteers. Neuropsychopharmacol 2011, 36, 1237–1247. [Google Scholar] [CrossRef]
- Fumagalli F, Cattaneo A, Caffino L, Ibba M, Racagni G, Carboni E, et al. Sub-chronic exposure to atomoxetine up-regulates BDNF expression and signalling in the brain of adolescent spontaneously hypertensive rats: Comparison with methylphenidate. Pharmacological Research 2010, 62, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Tzavara ET, Bymaster FP, Overshiner CD, Davis RJ, Perry KW, Wolff M, et al. Procholinergic and memory enhancing properties of the selective norepinephrine uptake inhibitor atomoxetine. Mol Psychiatry 2006, 11, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Piña R, Rozas C, Contreras D, Hardy P, Ugarte G, Zeise ML, et al. Atomoxetine Reestablishes Long Term Potentiation in a Mouse Model of Attention Deficit/Hyperactivity Disorder. Neuroscience 2020, 439, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Callahan PM, Plagenhoef MR, Blake DT, Terry AV. Atomoxetine improves memory and other components of executive function in young-adult rats and aged rhesus monkeys. Neuropharmacology 2019, 155, 65–75. [Google Scholar] [CrossRef]
- Liu L, Yang J, Lei G, Wang G, Wang Y, Sun R. Atomoxetine Increases Histamine Release and Improves Learning Deficits in an Animal Model of Attention-Deficit Hyperactivity Disorder: The Spontaneously Hypertensive Rat. Basic Clin Pharma Tox 2008, 102, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Nomura H, Shimizume R, Ikegaya Y. Histamine: A Key Neuromodulator of Memory Consolidation and Retrieval. In: Yanai K, Passani MB, editors. The Functional Roles of Histamine Receptors [Internet]. Cham: Springer International Publishing; 2021. p. 329–53. (Current Topics in Behavioral Neurosciences; vol. 59). Available online: https://link.springer.com/10.1007/7854_2021_253 (accessed on 7 May 2025).
- Ludolph A, Udvardi P, Föhr K, Henes C, Liebau S, Dreyhaupt J, et al. Atomoxetine affects transcription/translation of the NMDA receptor and the norepinephrine transporter in the rat brain – an in vivo study. DDDT.
- Di Miceli M, Gronier B. Psychostimulants and atomoxetine alter the electrophysiological activity of prefrontal cortex neurons, interaction with catecholamine and glutamate NMDA receptors. Psychopharmacology 2015, 232, 2191–2205. [Google Scholar] [CrossRef] [PubMed]
- Joya X, Pujadas M, Falcón M, Civit E, Garcia-Algar O, Vall O, et al. Gas chromatography–mass spectrometry assay for the simultaneous quantification of drugs of abuse in human placenta at 12th week of gestation. Forensic Science International 2010, 196, 38–42. [Google Scholar] [CrossRef]
- Li L, Sujan AC, Butwicka A, Chang Z, Cortese S, Quinn P, et al. Associations of Prescribed ADHD Medication in Pregnancy with Pregnancy-Related and Offspring Outcomes: A Systematic Review. CNS Drugs 2020, 34, 731–747. [Google Scholar] [CrossRef]
- Ornoy, A. Pharmacological Treatment of Attention Deficit Hyperactivity Disorder During Pregnancy and Lactation. Pharm Res 2018, 35, 46. [Google Scholar] [CrossRef]
- Ross EJ, Graham DL, Money KM, Stanwood GD. Developmental consequences of fetal exposure to drugs: what we know and what we still must learn. Neuropsychopharmacology 2015, 40, 61–87. [Google Scholar] [CrossRef]
- Sauer JM, Ring BJ, Witcher JW. Clinical Pharmacokinetics of Atomoxetine. Clinical Pharmacokinetics 2005, 44, 571–590. [Google Scholar] [CrossRef]
- McAllister-Williams RH, Baldwin DS, Cantwell R, Easter A, Gilvarry E, Glover V, et al. British Association for Psychopharmacology consensus guidance on the use of psychotropic medication preconception, in pregnancy and postpartum. J Psychopharmacol 2017, 31, 519–552. [Google Scholar] [CrossRef]
- Baker AS, Wales R, Noe O, Gaccione P, Freeman MP, Cohen LS. The Course of ADHD during Pregnancy. J Atten Disord 2022, 26, 143–148. [Google Scholar] [CrossRef]
- Scoten O, Tabi K, Paquette V, Carrion P, Ryan D, Radonjic NV, et al. Attention-deficit/hyperactivity disorder in pregnancy and the postpartum period. American Journal of Obstetrics and Gynecology 2024, 231, 19–35. [Google Scholar] [CrossRef]
- Louik C, Kerr S, Kelley KE, Mitchell AA. Increasing use of ADHD medications in pregnancy. Pharmacoepidemiol Drug Saf 2015, 24, 218–220. [Google Scholar] [CrossRef]
- Anderson KN, Ailes EC, Danielson M, Lind JN, Farr SL, Broussard CS, et al. Attention-Deficit/Hyperactivity Disorder Medication Prescription Claims Among Privately Insured Women Aged 15–44 Years — United States, 2003–2015. MMWR Morb Mortal Wkly Rep 2018, 67, 66–70. [Google Scholar] [CrossRef]
- Bang Madsen K, Robakis TK, Liu X, Momen N, Larsson H, Dreier JW, et al. In utero exposure to ADHD medication and long-term offspring outcomes. Mol Psychiatry 2023, 28, 1739–1746. [Google Scholar] [CrossRef]
- Cohen JM, Hernández-Díaz S, Bateman BT, Park Y, Desai RJ, Gray KJ, et al. Placental Complications Associated With Psychostimulant Use in Pregnancy. Obstetrics & Gynecology 2017, 130, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Bang Madsen K, Larsson H, Skoglund C, Liu X, Munk-Olsen T, Bergink V, et al. In utero exposure to methylphenidate, amphetamines and atomoxetine and offspring neurodevelopmental disorders – a population-based cohort study and meta-analysis. Mol Psychiatry [Internet] 2025 Mar 27. Available online: https://www.nature.com/articles/s41380-025-02968-4 (accessed on 7 May 2025).
- Huybrechts KF, Bröms G, Christensen LB, Einarsdóttir K, Engeland A, Furu K, et al. Association Between Methylphenidate and Amphetamine Use in Pregnancy and Risk of Congenital Malformations: A Cohort Study From the International Pregnancy Safety Study Consortium. JAMA Psychiatry 2018, 75, 167. [Google Scholar] [CrossRef] [PubMed]
- Ladhani NNN, Shah PS, Murphy KE. Prenatal amphetamine exposure and birth outcomes: a systematic review and metaanalysis. American Journal of Obstetrics and Gynecology 2011, 205, e1–e219. [Google Scholar] [CrossRef] [PubMed]
- Eriksson M, Jonsson B, Steneroth G, Zetterström R. Amphetamine abuse during pregnancy: environmental factors and outcome after 14-15 years. Scand J Public Health 2000, 28, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Chen VS, Morrison JP, Southwell MF, Foley JF, Bolon B, Elmore SA. Histology Atlas of the Developing Prenatal and Postnatal Mouse Central Nervous System, with Emphasis on Prenatal Days E7.5 to E18.5. Toxicol Pathol 2017, 45, 705–744. [Google Scholar] [CrossRef]
- Flores G, De Jesús Gómez-Villalobos M, Rodríguez-Sosa L. Prenatal Amphetamine Exposure Effects on Dopaminergic Receptors and Transporter in Postnatal Rats. Neurochem Res 2011, 36, 1740–1749. [Google Scholar] [CrossRef]
- Gong S, Fayette N, Heinsbroek JA, Ford CP. Cocaine shifts dopamine D2 receptor sensitivity to gate conditioned behaviors. Neuron 2021, 109, 3421–3435.e5. [Google Scholar] [CrossRef]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Ding YS, Sedler M, et al. Low Level of Brain Dopamine D2 Receptors in Methamphetamine Abusers: Association With Metabolism in the Orbitofrontal Cortex. AJP 2001, 158, 2015–2021. [Google Scholar] [CrossRef]
- Bigl V, Dalitz E, Kunert E, Biesold D, Leonard BE. The effect of d-amphetamine and amitriptyline administered to pregnant rats on the locomotor activity and neurotransmitters of the offspring. Psychopharmacology 1982, 77, 371–375. [Google Scholar] [CrossRef]
- Nasello AG, Ramirez OA. Brain catecholamines metabolism in offspring of amphetamine treated rats. Pharmacology Biochemistry and Behavior 1978, 9, 17–20. [Google Scholar] [CrossRef]
- Tavares MA, Silva MC, Silva-Araújo A, Xavier MR, Ali SF. Effects of prenatal exposure to amphetamine in the medial prefrontal cortex of the rat. Intl J of Devlp Neuroscience 1996, 14, 585–596. [Google Scholar] [CrossRef]
- Bell RW, Drucker RR, Woodruff AB. The effects of prenatal injections of adrenalin chloride and d- amphetamine sulfate on subsequent emotionality and ulcer-proneness of offspring. Psychon Sci 1965, 2, 269–270. [Google Scholar] [CrossRef]
- Nasello AG, Ramirez OA. Open-field and Lashley III maze behaviour of the offspring of amphetamine-treated rats. Psychopharmacology 1978, 58, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Nasello AG, Astrada CA, Ramirez OA. Effects on the acquisition of conditioned avoidance responses and seizure threshold in the offspring of amphetamine treated gravid rats. Psychopharmacologia 1974, 40, 25–31. [Google Scholar] [CrossRef]
- Ke T, Poquette KE, Amro Gazze SL, Carvelli L. Amphetamine Exposure during Embryogenesis Alters Expression and Function of Tyrosine Hydroxylase and the Vesicular Monoamine Transporter in Adult C. elegans. Int J Mol Sci 2024, 25, 4219. [Google Scholar] [CrossRef]
- Korchynska S, Krassnitzer M, Malenczyk K, Prasad RB, Tretiakov EO, Rehman S, et al. Life-long impairment of glucose homeostasis upon prenatal exposure to psychostimulants. EMBO J 2020, 39, e100882. [Google Scholar] [CrossRef]
- Plessinger, MA. PRENATAL EXPOSURE TO AMPHETAMINES. Obstetrics and Gynecology Clinics of North America 1998, 25, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Lepelletier FX, Tauber C, Nicolas C, Solinas M, Castelnau P, Belzung C, et al. Prenatal Exposure to Methylphenidate Affects the Dopamine System and the Reactivity to Natural Reward in Adulthood in Rats. International Journal of Neuropsychopharmacology [Internet] 2015 Feb;18(4). Available online: https://academic.oup.com/ijnp/article-lookup/doi/10.1093/ijnp/pyu044 (accessed on 2 May 2025).
- Lloyd SA, Oltean C, Pass H, Phillips B, Staton K, Robertson CL, et al. Prenatal exposure to psychostimulants increases impulsivity, compulsivity, and motivation for rewards in adult mice. Physiology & Behavior 2013, 119, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Aoki S, Kaizaki-Mitsumoto A, Hattori N, Numazawa S. Fetal methylphenidate exposure induced ADHD-like phenotypes and decreased Drd2 and Slc6a3 expression levels in mouse offspring. Toxicology Letters 2021, 344, 1–10. [Google Scholar] [CrossRef] [PubMed]
- McFadyen-Leussis MP, Lewis SP, Bond TLY, Carrey N, Brown RE. Prenatal exposure to methylphenidate hydrochloride decreases anxiety and increases exploration in mice. Pharmacology Biochemistry and Behavior 2004, 77, 491–500. [Google Scholar] [CrossRef]
- Levin ED, Sledge D, Roach S, Petro A, Donerly S, Linney E. Persistent behavioral impairment caused by embryonic methylphenidate exposure in zebrafish. Neurotoxicology and Teratology 2011, 33, 668–673. [Google Scholar] [CrossRef]
- Priori A, Hallett M, Rothwell JC. Repetitive transcranial magnetic stimulation or transcranial direct current stimulation? Brain Stimulation 2009, 2, 241–245. [Google Scholar] [CrossRef]
- Westwood SJ, Radua J, Rubia K. Noninvasive brain stimulation in children and adults with attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. J Psychiatry Neurosci 2021, 46, E14–33. [Google Scholar] [CrossRef]
- Dayan E, Censor N, Buch ER, Sandrini M, Cohen LG. Noninvasive brain stimulation: from physiology to network dynamics and back. Nat Neurosci 2013, 16, 838–844. [Google Scholar] [CrossRef]
- Han Y, Wei ZY, Zhao N, Zhuang Q, Zhang H, Fang HL, et al. Transcranial magnetic stimulation in attention-deficit/hyperactivity disorder: a systematic review and meta-analysis of cortical excitability and therapeutic efficacy. Front Psychiatry 2025, 16, 1544816. [Google Scholar] [CrossRef]
- Miao S, Han J, Gu Y, Wang X, Song W, Li D, et al. Reduced Prefrontal Cortex Activation in Children with Attention-Deficit/Hyperactivity Disorder during Go/No-Go Task: A Functional Near-Infrared Spectroscopy Study. Front Neurosci 2017, 11, 367. [Google Scholar] [CrossRef]
- Li Y, Ma S, Zhang X, Gao L. ASD and ADHD: Divergent activating patterns of prefrontal cortex in executive function tasks? Journal of Psychiatric Research 2024, 172, 187–196. [Google Scholar] [CrossRef]
- Cao P, Xing J, Cao Y, Cheng Q, Sun X, Kang Q, et al. Clinical effects of repetitive transcranial magnetic stimulation combined with atomoxetine in the treatment of attention-deficit hyperactivity disorder. Neuropsychiatr Dis Treat 2018, 14, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Nagy NAS, Amin GR, Khalil SA, Mahmoud DAM, Elkholy H, Shohdy M. The therapeutic role of repetitive transcranial magnetic stimulation in children with attention deficit/hyperactivity disorder in Egypt a randomized sham controlled clinical trial. Middle East Curr Psychiatry 2022, 29, 55. [Google Scholar] [CrossRef]
- Cao P, Wang L, Cheng Q, Sun X, Kang Q, Dai L, et al. Changes in serum miRNA-let-7 level in children with attention deficit hyperactivity disorder treated by repetitive transcranial magnetic stimulation or atomoxetine: An exploratory trial. Psychiatry Research 2019, 274, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wang J, Zou Z, Huang H, Zhang Y, He X, Su H, et al. Effects of repetitive transcranial magnetic stimulation on prefrontal cortical activation in children with attention deficit hyperactivity disorder: a functional near-infrared spectroscopy study. Front Neurol 2024, 15, 1503975. [Google Scholar] [CrossRef]
- Mendes Dos Santos R, Nunes M, Peres De Souza L, Nayara De Araújo Val S, Machado Santos Á, Cristina Vieira Da Costa A, et al. Hypothesis on the potential of repetitive transcranial magnetic stimulation to modulate neurochemical pathways and circadian rhythm in ADHD. Medical Hypotheses 2024, 189, 111411. [Google Scholar] [CrossRef]
- Allenby C, Falcone M, Bernardo L, Wileyto EP, Rostain A, Ramsay JR, et al. Transcranial direct current brain stimulation decreases impulsivity in ADHD. Brain Stimulation 2018, 11, 974–981. [Google Scholar] [CrossRef]
- Soff C, Sotnikova A, Christiansen H, Becker K, Siniatchkin M. Transcranial direct current stimulation improves clinical symptoms in adolescents with attention deficit hyperactivity disorder. J Neural Transm 2017, 124, 133–144. [Google Scholar] [CrossRef]
- Salehinejad MA, Nejati V, Mosayebi-Samani M, Mohammadi A, Wischnewski M, Kuo MF, et al. Transcranial Direct Current Stimulation in ADHD: A Systematic Review of Efficacy, Safety, and Protocol-induced Electrical Field Modeling Results. Neurosci Bull 2020, 36, 1191–1212. [Google Scholar] [CrossRef]
- Cachoeira CT, Leffa DT, Mittelstadt SD, Mendes LST, Brunoni AR, Pinto JV, et al. Positive effects of transcranial direct current stimulation in adult patients with attention-deficit/hyperactivity disorder - A pilot randomized controlled study. Psychiatry Res 2017, 247, 28–32. [Google Scholar] [CrossRef]
- Fritsch B, Reis J, Martinowich K, Schambra HM, Ji Y, Cohen LG, et al. Direct Current Stimulation Promotes BDNF-Dependent Synaptic Plasticity: Potential Implications for Motor Learning. Neuron 2010, 66, 198–204. [Google Scholar] [CrossRef]
- Wong HC, Zaman R. Neurostimulation in Treating ADHD. Neurostimulation in Treating ADHD. Psychiatr Danub 2019, 31 (Suppl. S3), 265–275. [Google Scholar]
- Zhi J, Zhang S, Huang M, Qin H, Xu H, Chang Q, et al. Transcutaneous auricular vagus nerve stimulation as a potential therapy for attention deficit hyperactivity disorder: modulation of the noradrenergic pathway in the prefrontal lobe. Front Neurosci 2024, 18, 1494272. [Google Scholar] [CrossRef]
- Aniwattanapong D, List JJ, Ramakrishnan N, Bhatti GS, Jorge R. Effect of Vagus Nerve Stimulation on Attention and Working Memory in Neuropsychiatric Disorders: A Systematic Review. Neuromodulation: Technology at the Neural Interface 2022, 25, 343–355. [Google Scholar] [CrossRef]
- Ben-Menachem, E. Vagus Nerve Stimulation, Side Effects, and Long-Term Safety. Journal of Clinical Neurophysiology 2001, 18, 415–418. [Google Scholar] [CrossRef]
- De Cicco V, Tramonti Fantozzi MP, Cataldo E, Barresi M, Bruschini L, Faraguna U, et al. Trigeminal, Visceral and Vestibular Inputs May Improve Cognitive Functions by Acting through the Locus Coeruleus and the Ascending Reticular Activating System: A New Hypothesis. Front Neuroanat 2017, 11, 130. [Google Scholar]
- Cook IA, Schrader LM, Degiorgio CM, Miller PR, Maremont ER, Leuchter AF. Trigeminal nerve stimulation in major depressive disorder: acute outcomes in an open pilot study. Epilepsy Behav 2013, 28, 221–226. [Google Scholar] [CrossRef] [PubMed]
- McGough JJ, Sturm A, Cowen J, Tung K, Salgari GC, Leuchter AF, et al. Double-Blind, Sham-Controlled, Pilot Study of Trigeminal Nerve Stimulation for Attention-Deficit/Hyperactivity Disorder. J Am Acad Child Adolesc Psychiatry 2019, 58, 403–411.e3. [Google Scholar] [CrossRef] [PubMed]
- Rubia K, Johansson L, Carter B, Stringer D, Santosh P, Mehta MA, et al. The efficacy of real versus sham external Trigeminal Nerve Stimulation (eTNS) in youth with Attention-Deficit/Hyperactivity Disorder (ADHD) over 4 weeks: a protocol for a multi-centre, double-blind, randomized, parallel-group, phase IIb study (ATTENS). BMC Psychiatry 2024, 24, 326. [Google Scholar]
- McGough JJ, Loo SK, Sturm A, Cowen J, Leuchter AF, Cook IA. An eight-week, open-trial, pilot feasibility study of trigeminal nerve stimulation in youth with attention-deficit/hyperactivity disorder. Brain Stimul 2015, 8, 299–304. [Google Scholar] [CrossRef]
- Frolli A, Cerciello F, Esposito C, Ricci MC, Laccone RP, Bisogni F. Universal Design for Learning for Children with ADHD. Children (Basel) 2023, 10, 1350. [Google Scholar]
- Moore, SL. David H. Rose, Anne Meyer, Teaching Every Student in the Digital Age: Universal Design for Learning: Association for Supervision and Curriculum Development, 1703 N. Beauregard St., Alexandria, VA 22311-1714, Product no 101042, $22.95 ASCD members, $26.95 nonmembers, ISBN-0-87120-599-8. Education Tech Research Dev 2007, 55, 521–525. [Google Scholar]
- Chen ZH, Pan TB, Zhang YH, Wang B, Sun XL, Gao M, et al. Transcriptional conservation and evolutionary divergence of cell types across mammalian hypothalamus development. Transcriptional conservation and evolutionary divergence of cell types across mammalian hypothalamus development. Developmental Cell 2025, S153458072500156X. [Google Scholar]
- Von Kügelgen N, Chekulaeva M. Conservation of a core neurite transcriptome across neuronal types and species. WIREs RNA 2020, 11, e1590. [Google Scholar] [CrossRef]
- Kauvar I, Richman EB, Liu TX, Li C, Vesuna S, Chibukhchyan A, et al. Conserved brain-wide emergence of emotional response from sensory experience in humans and mice. Science 2025, 388, eadt3971. [Google Scholar] [CrossRef]
- McDonnell-Dowling K, Kelly JP. Sources of variation in the design of preclinical studies assessing the effects of amphetamine-type stimulants in pregnancy and lactation. Behav Brain Res 2015, 279, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of ADHD subtypes and core symptom profiles. Differences between the subtypes of ADHD: (A) Primarily Inattentive (ADHD-I), (B) Primarily Hyperactive (ADHD-H), and (C) Combined (ADHD-C) using diagnostic criteria and symptoms as defined by the DSM-5 [6]. Diagnostic criteria require at least 6 symptoms (or 5 for individuals aged 17 and older) within each domain for ADHD-C. Diagnosis is characterized by several symptoms prior to age 12 years and those occurring in ≥ 2 settings and impede “normal” function or development. For diagnosis, these symptoms are not better explained by another mental disorder.
Figure 1.
Overview of ADHD subtypes and core symptom profiles. Differences between the subtypes of ADHD: (A) Primarily Inattentive (ADHD-I), (B) Primarily Hyperactive (ADHD-H), and (C) Combined (ADHD-C) using diagnostic criteria and symptoms as defined by the DSM-5 [6]. Diagnostic criteria require at least 6 symptoms (or 5 for individuals aged 17 and older) within each domain for ADHD-C. Diagnosis is characterized by several symptoms prior to age 12 years and those occurring in ≥ 2 settings and impede “normal” function or development. For diagnosis, these symptoms are not better explained by another mental disorder.
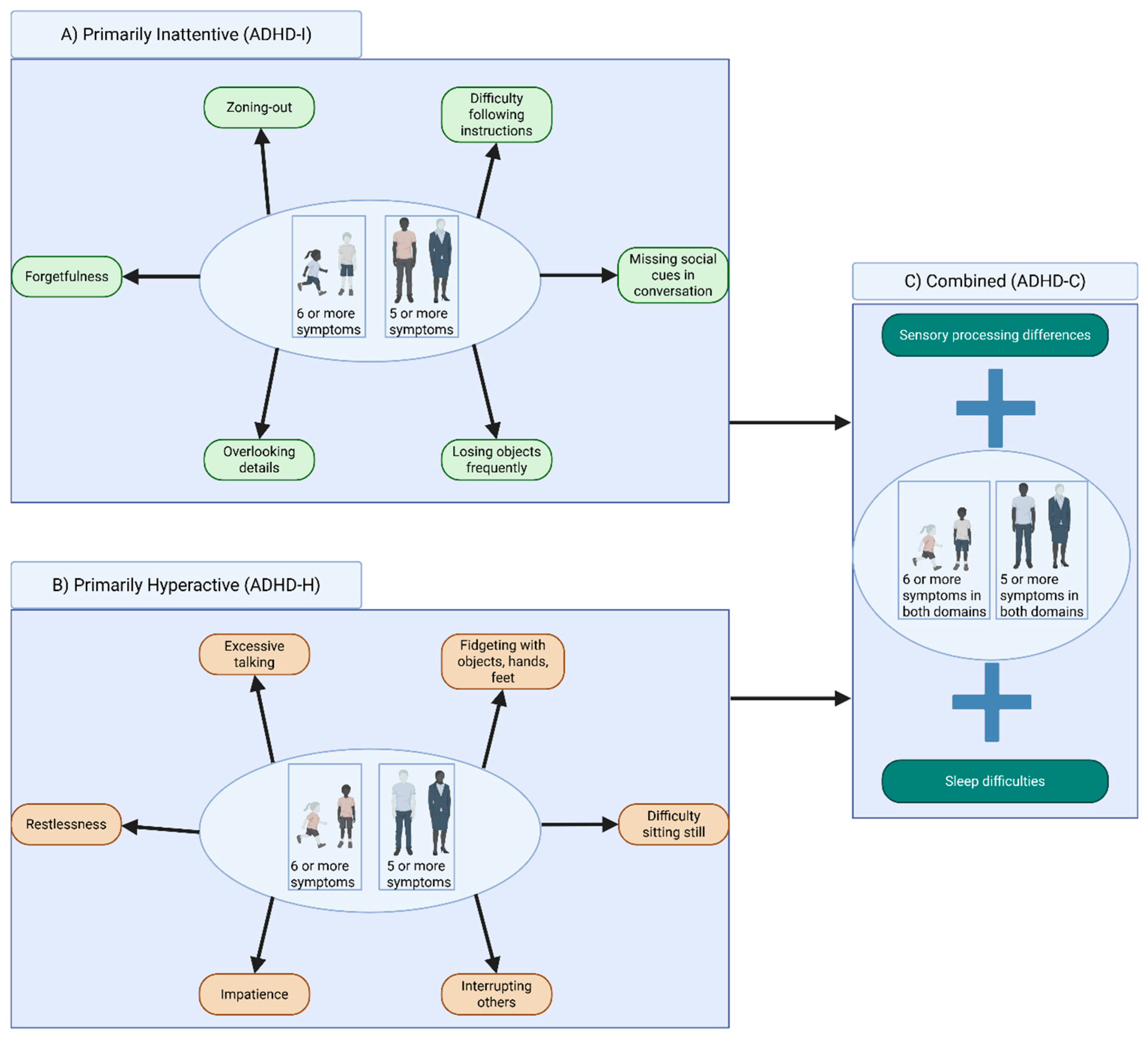
Figure 2.
Brain regions that are altered in ADHD. Symptoms pertaining to impulsiveness and inattention may involve delayed PFC size during development, alongside increased signaling from the NAc. Impairments to information integration during tasks are linked to a reduced size and thinning of the CC, whereas reductions in motor coordination are associated with decreased volume in the cerebellum. The motor restlessness and fidgeting associated with ADHD as indications of hyperactivity are thought to arise from reduced size of the BG, which is crucial for regulating voluntary motor movements. In general, patients with ADHD are thought to have an approximately 3% decrease in overall brain volume. .
Figure 2.
Brain regions that are altered in ADHD. Symptoms pertaining to impulsiveness and inattention may involve delayed PFC size during development, alongside increased signaling from the NAc. Impairments to information integration during tasks are linked to a reduced size and thinning of the CC, whereas reductions in motor coordination are associated with decreased volume in the cerebellum. The motor restlessness and fidgeting associated with ADHD as indications of hyperactivity are thought to arise from reduced size of the BG, which is crucial for regulating voluntary motor movements. In general, patients with ADHD are thought to have an approximately 3% decrease in overall brain volume. .
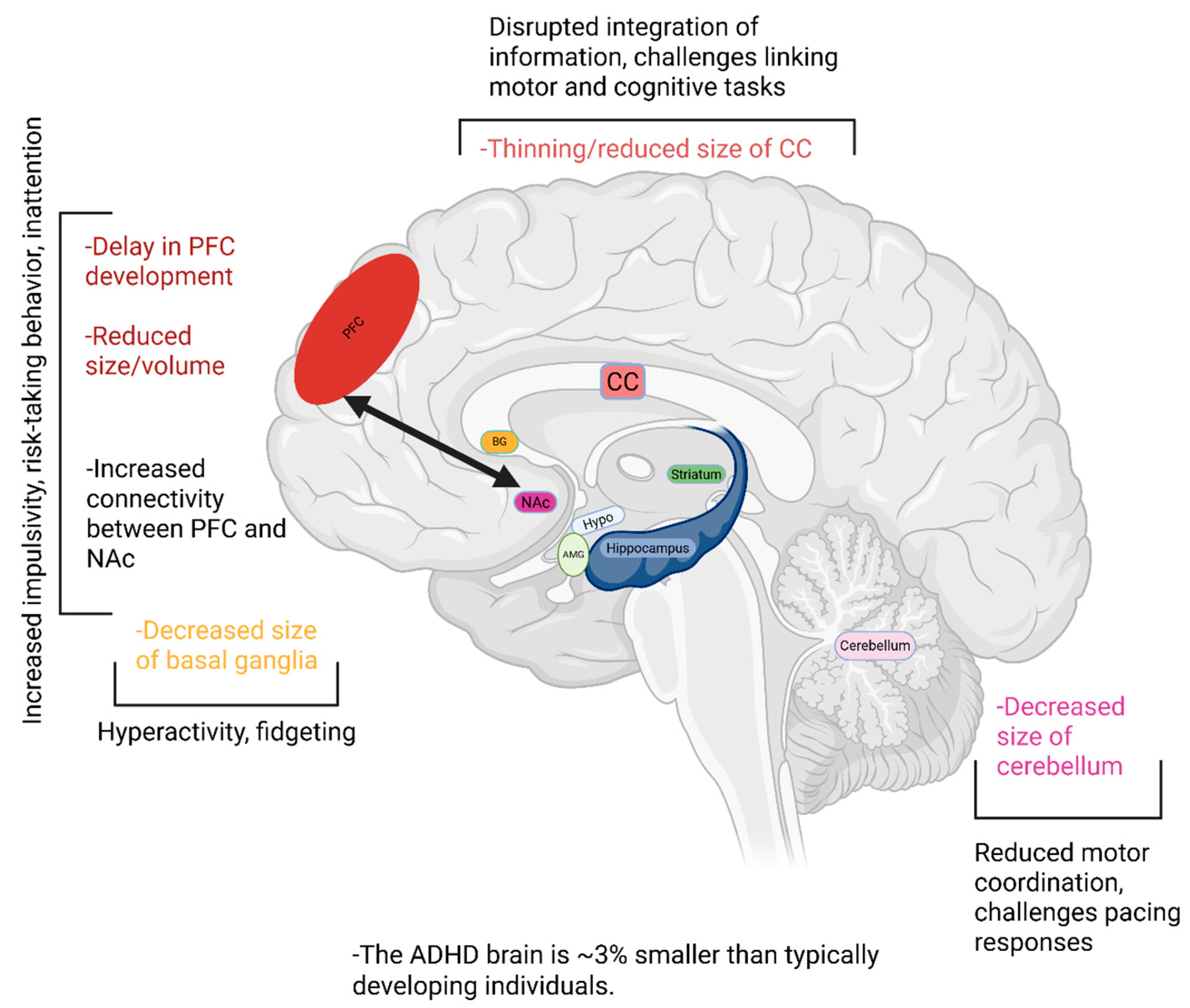
Figure 3.
Monoamine pathways in the CNS. (A) Dopaminergic pathways within the CNS [76,77,78,79]. Mesocortical pathway emanates via projections from the VTA innervating the PFC to modulate concentration, focus, working memory, and decision-making. The mesolimbic pathway contains projections originating from the VTA that innervate the AMG, hippocampus, and NAc emerge to influence motivation, emotional processing, and reward-based decision making. The tuberoinfundibular pathway involves innervations from DA neurons in the hypothalamus (hypo), which innervate neurons in the pituitary gland to provide negative feedback for the release of prolactin. Lastly, the nigrostriatal pathway emerges via projections between DA neurons in the SN to the striatum and is involved in regulating voluntary movement. (B) DA metabolism and conversion into NE [80,93,94]. (C) Noradrenergic signaling in the brain [116,117,118,119,120,121,122,123,124,125,126,127,128,129]. The LC is the primary site of NE synthesis from DA metabolism, and projects to the PFC alongside limbic structures of the brain. (D) Serotonergic signaling in the brain [126,130,131,132,133,134,135,136,137,138,139,140]. 5-HT transmission originates in the Rn and innervates limbic structures to modulate mood, hunger, and attention.
Figure 3.
Monoamine pathways in the CNS. (A) Dopaminergic pathways within the CNS [76,77,78,79]. Mesocortical pathway emanates via projections from the VTA innervating the PFC to modulate concentration, focus, working memory, and decision-making. The mesolimbic pathway contains projections originating from the VTA that innervate the AMG, hippocampus, and NAc emerge to influence motivation, emotional processing, and reward-based decision making. The tuberoinfundibular pathway involves innervations from DA neurons in the hypothalamus (hypo), which innervate neurons in the pituitary gland to provide negative feedback for the release of prolactin. Lastly, the nigrostriatal pathway emerges via projections between DA neurons in the SN to the striatum and is involved in regulating voluntary movement. (B) DA metabolism and conversion into NE [80,93,94]. (C) Noradrenergic signaling in the brain [116,117,118,119,120,121,122,123,124,125,126,127,128,129]. The LC is the primary site of NE synthesis from DA metabolism, and projects to the PFC alongside limbic structures of the brain. (D) Serotonergic signaling in the brain [126,130,131,132,133,134,135,136,137,138,139,140]. 5-HT transmission originates in the Rn and innervates limbic structures to modulate mood, hunger, and attention.
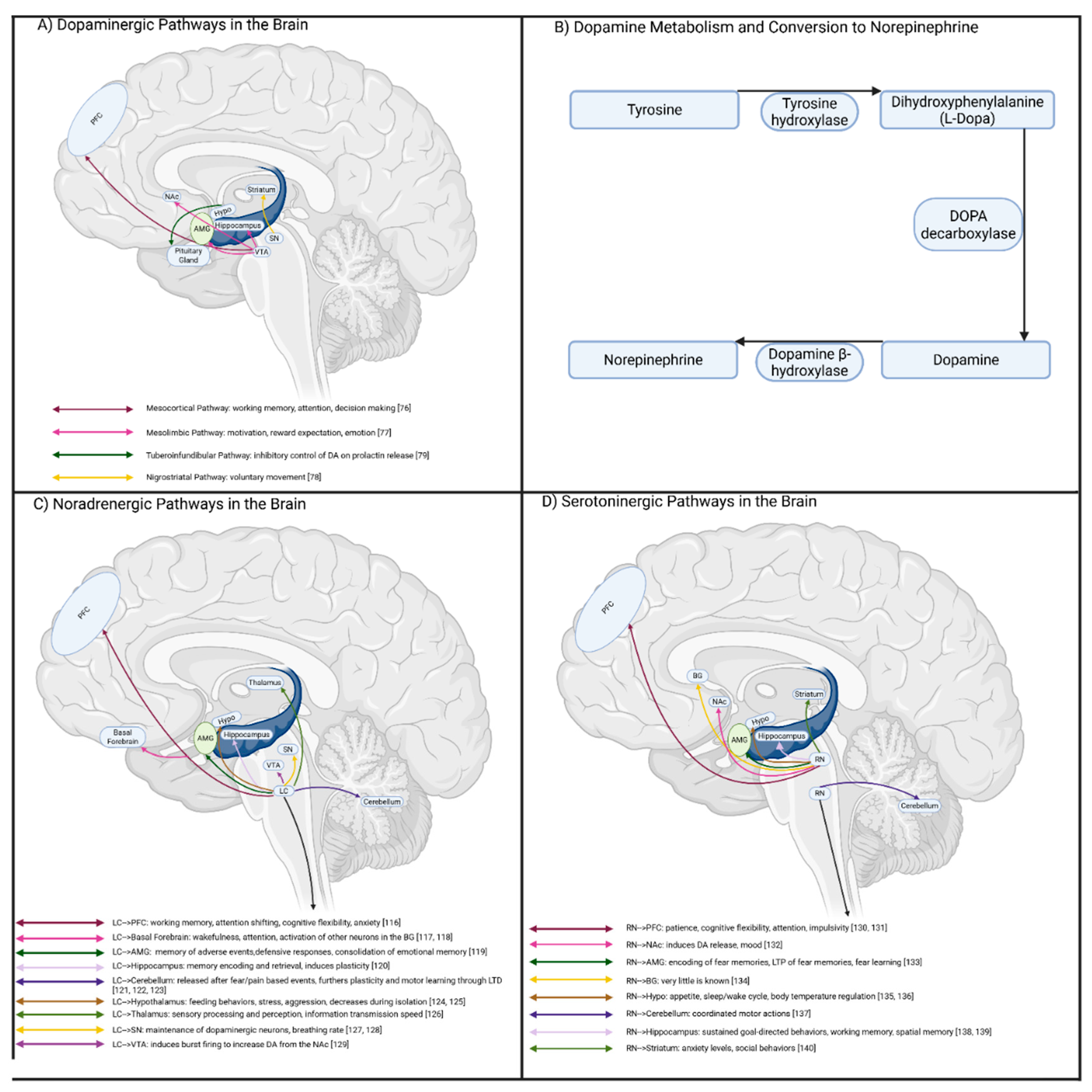
Figure 4.
Interactions between NMDAR and DA signaling within the CNS. DA receptors are GPCRs classified as either D1R-like or D2R-like. Activation of DA receptors initiates a molecular cascade that utilizes proteins that overlap with NMDAR signaling. D2R-like activation inhibits cAMP production and therefore PKA signaling, alongside opening IRK channels and reducing calcium-mediated release of neurotransmitters. Altogether, this reduces the likelihood of neurotransmission occurring, and functions to inhibit NMDAR activity. D1R-like activation generally functions to increase the activation of PKA, which can lead to increased phosphorylation of NMDAR subunits and trafficking of AMPAR to synaptic sites. Additionally, PKA can phosphorylate the transcription factor CREB to further the transcription of genes involved in synaptic plasticity that underlies learning and memory.
Figure 4.
Interactions between NMDAR and DA signaling within the CNS. DA receptors are GPCRs classified as either D1R-like or D2R-like. Activation of DA receptors initiates a molecular cascade that utilizes proteins that overlap with NMDAR signaling. D2R-like activation inhibits cAMP production and therefore PKA signaling, alongside opening IRK channels and reducing calcium-mediated release of neurotransmitters. Altogether, this reduces the likelihood of neurotransmission occurring, and functions to inhibit NMDAR activity. D1R-like activation generally functions to increase the activation of PKA, which can lead to increased phosphorylation of NMDAR subunits and trafficking of AMPAR to synaptic sites. Additionally, PKA can phosphorylate the transcription factor CREB to further the transcription of genes involved in synaptic plasticity that underlies learning and memory.
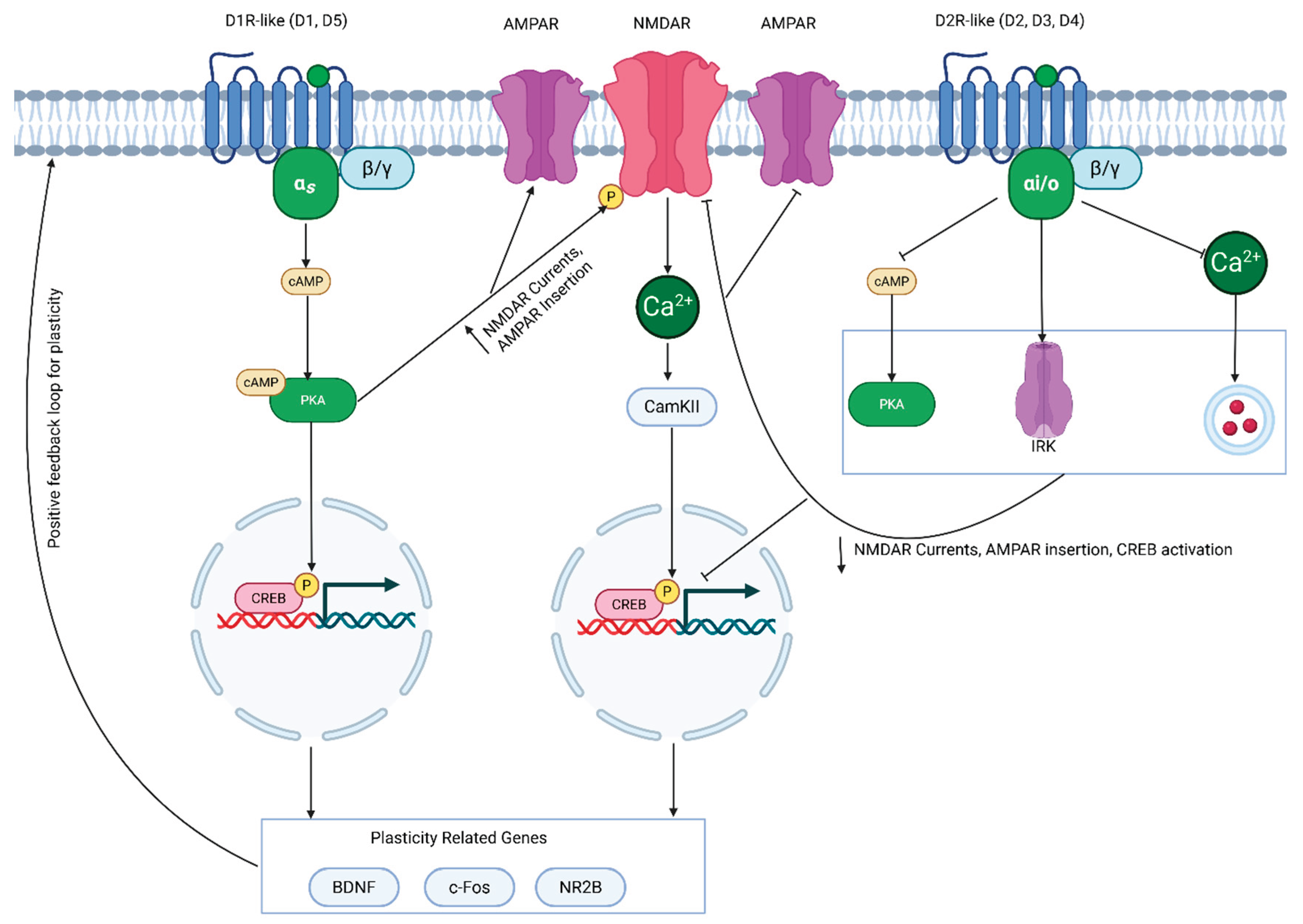
Figure 5.
Overview of prescribing stimulants and non-stimulants for ADHD. Following an ADHD diagnosis, a family history for cardiovascular health is taken, alongside assessments of the patient’s heart health. If the patient does not present with risk for cardiovascular disease, the physician then considers a first line treatment from either the AMPH- or MPH-based classes. In European countries, MPH-based medications are considered first, however in North America AMPH-based medications can also be considered. If the patient is a school-aged child, long-acting stimulants are preferred to ensure therapeutic benefits to the child throughout the entire school day. If first-line treatments prove ineffective, or present uncomfortable side-effects, second-line treatments are then considered. Should these second-line treatments continue to produce undesired side-effects, the patient may transition towards non-stimulants. If the patient does present cardiovascular risks following ADHD diagnosis, non-stimulants are prescribed instead.
Figure 5.
Overview of prescribing stimulants and non-stimulants for ADHD. Following an ADHD diagnosis, a family history for cardiovascular health is taken, alongside assessments of the patient’s heart health. If the patient does not present with risk for cardiovascular disease, the physician then considers a first line treatment from either the AMPH- or MPH-based classes. In European countries, MPH-based medications are considered first, however in North America AMPH-based medications can also be considered. If the patient is a school-aged child, long-acting stimulants are preferred to ensure therapeutic benefits to the child throughout the entire school day. If first-line treatments prove ineffective, or present uncomfortable side-effects, second-line treatments are then considered. Should these second-line treatments continue to produce undesired side-effects, the patient may transition towards non-stimulants. If the patient does present cardiovascular risks following ADHD diagnosis, non-stimulants are prescribed instead.
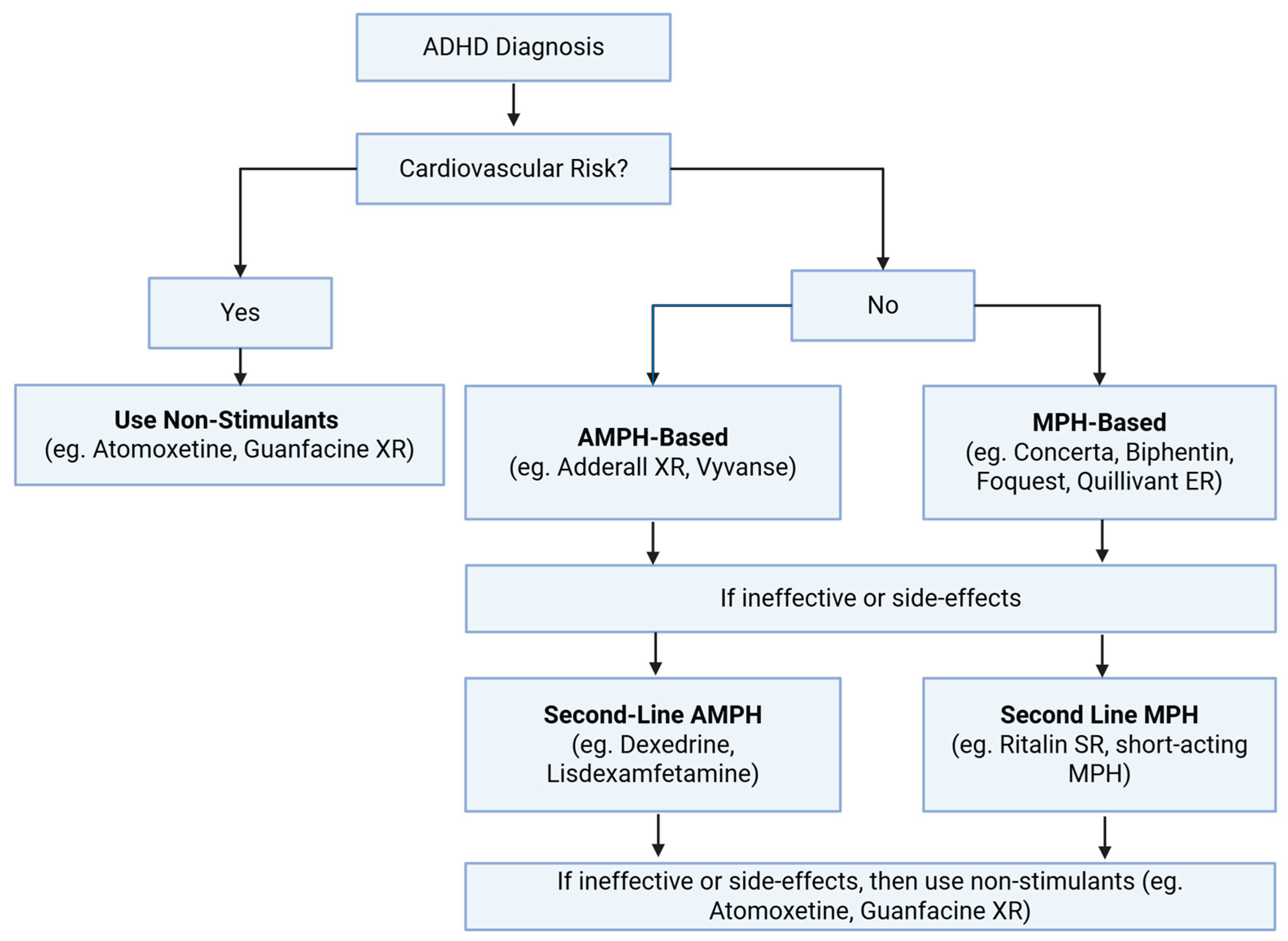
Figure 6.
Molecular mechanisms and receptor targets of ADHD medications. (A) MPH inhibits DAT and NET but does not show strong affinity for blocking SERT, leading to increased DA and NE transmission. Additionally, MPH has been demonstrated to alter the distribution of vesicles in the presynaptic terminal, which is thought to allow for more controlled release of monoamines. (B) AMPH enters the presynaptic neuron using monoamine transporters and inhibits MAO to prevent monoamine breakdown. The inhibition of VMAT-2 prevents packaging of monoamines into vesicles, leading to the reverse transport of monoamines into the synaptic cleft to increase DA, NE, and SE transmission. (C) ATX is a selective NET inhibitor, which causes an increase in the levels of NE available to bind to postsynaptic receptors. However, in the PFC these NET also contribute to reuptake of DA, thus ATX also increases DA transmission. (D) Viloxazine inhibits NET to increase NE signaling, alongside the availability of DA in the PFC. Additionally, viloxazine acts as an antagonist for 5-HT2B receptors, and an agonist for 5-HT2C receptors, which has been shown to increase 5-HT levels in the PFC. (E) Guanfacine binds to postsynaptic α2A receptors to inhibit cAMP activity, alongside PKA activation to reduce the opening of potassium channels. This enhances PFC neuronal activity to improve attention, working memory, and impulse control. (F) Similarly, Clonidine also binds to α2A receptors to exert similar effects, while binding to α2B and α2C receptors to increase vasodilation and regulation of mood and startle response respectively.
Figure 6.
Molecular mechanisms and receptor targets of ADHD medications. (A) MPH inhibits DAT and NET but does not show strong affinity for blocking SERT, leading to increased DA and NE transmission. Additionally, MPH has been demonstrated to alter the distribution of vesicles in the presynaptic terminal, which is thought to allow for more controlled release of monoamines. (B) AMPH enters the presynaptic neuron using monoamine transporters and inhibits MAO to prevent monoamine breakdown. The inhibition of VMAT-2 prevents packaging of monoamines into vesicles, leading to the reverse transport of monoamines into the synaptic cleft to increase DA, NE, and SE transmission. (C) ATX is a selective NET inhibitor, which causes an increase in the levels of NE available to bind to postsynaptic receptors. However, in the PFC these NET also contribute to reuptake of DA, thus ATX also increases DA transmission. (D) Viloxazine inhibits NET to increase NE signaling, alongside the availability of DA in the PFC. Additionally, viloxazine acts as an antagonist for 5-HT2B receptors, and an agonist for 5-HT2C receptors, which has been shown to increase 5-HT levels in the PFC. (E) Guanfacine binds to postsynaptic α2A receptors to inhibit cAMP activity, alongside PKA activation to reduce the opening of potassium channels. This enhances PFC neuronal activity to improve attention, working memory, and impulse control. (F) Similarly, Clonidine also binds to α2A receptors to exert similar effects, while binding to α2B and α2C receptors to increase vasodilation and regulation of mood and startle response respectively.
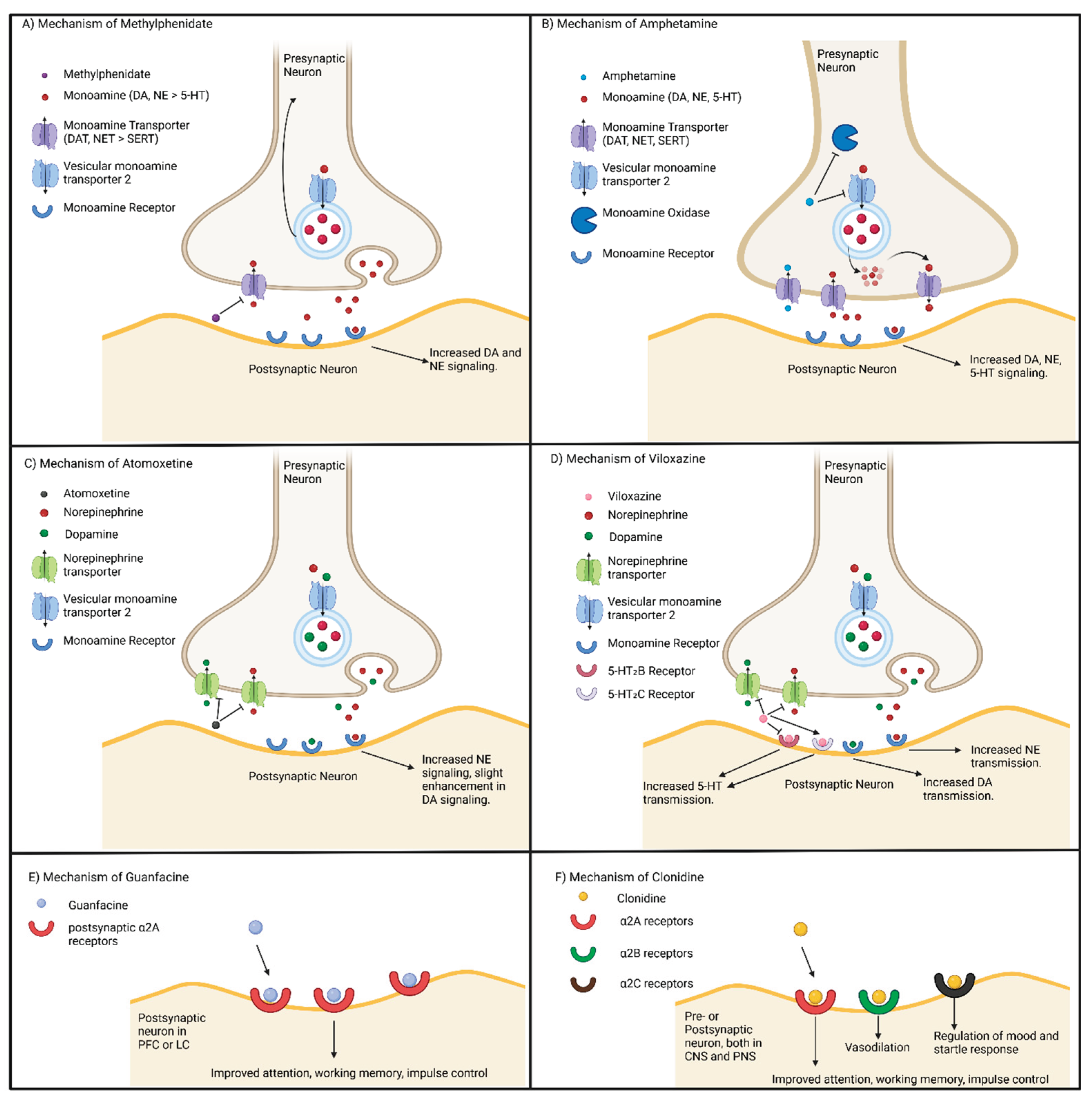
Figure 7.
Modes of transcranial stimulation. (A) An external electrode is applied to the forehead of the patient to stimulate the trigeminal nerve. The electrode is controlled by an external controller. (B) A current-carrying coil is attached to the scalp which creates magnetic field by the coil interacting with the membrane potential of neurons. (C) An electrode is surgically implanted next to the vagus nerve to stimulate it with an electric current controlled by a pre-programmed, surgically implanted controller. (D) Two electrodes are attached to the scalp, the current flows from the positively charged anode to the negatively charged cathode, which generates action potentials increasing neuronal firing in the brain. The electrodes are controlled by an external controller. .
Figure 7.
Modes of transcranial stimulation. (A) An external electrode is applied to the forehead of the patient to stimulate the trigeminal nerve. The electrode is controlled by an external controller. (B) A current-carrying coil is attached to the scalp which creates magnetic field by the coil interacting with the membrane potential of neurons. (C) An electrode is surgically implanted next to the vagus nerve to stimulate it with an electric current controlled by a pre-programmed, surgically implanted controller. (D) Two electrodes are attached to the scalp, the current flows from the positively charged anode to the negatively charged cathode, which generates action potentials increasing neuronal firing in the brain. The electrodes are controlled by an external controller. .
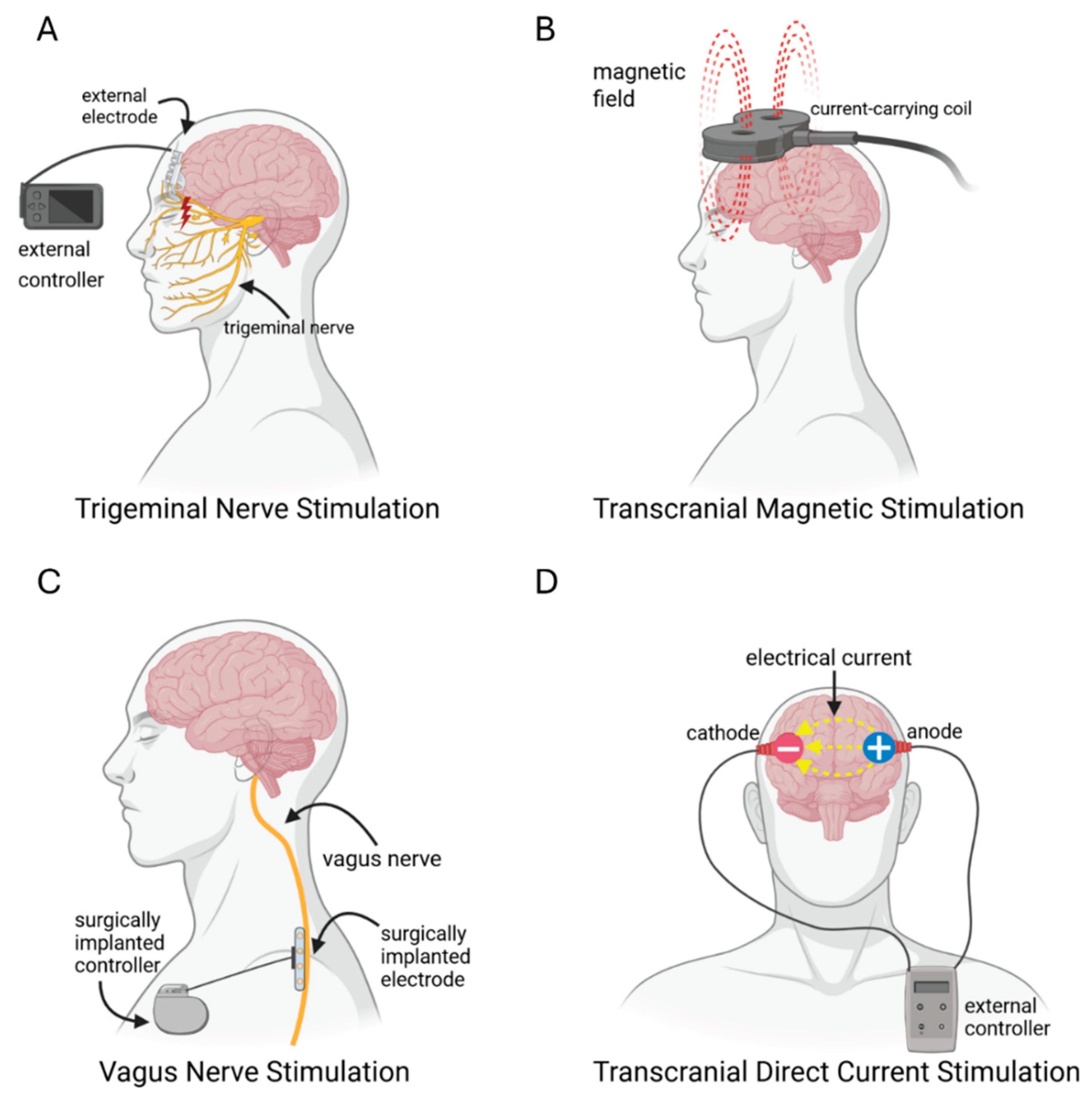
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



