
Submitted:
04 August 2025
Posted:
05 August 2025
You are already at the latest version
Abstract
The role of vitamin D (VD) in cardiovascular health remains controversial. Observational studies have associated low serum 25(OH)D₃ levels with increased risk of cardiovascular events, while interventional trials and Mendelian randomization studies have largely failed to confirm causality. This inconsistency may arise from unrecognized interindividual differences in VD responsiveness, as well as from the poor correlation between circulating VD metabolites and tissue-specific biological activity. This review highlights the emerging paradigm of variable VD sensitivity, which spans from VD resistance (VDRES) to hypersensitivity (VDHY). Individuals with VDRES exhibit impaired responses to standard supplementation due to genetic or acquired factors affecting VD metabolism, transport, or receptor signaling. In contrast, those with VDHY may develop adverse effects—such as hypercalcemia or vascular calcification—even under normal VD exposure, particularly if mutations impair VD catabolism (e.g., CYP24A1 variants). These opposing phenotypes may account for the heterogeneous outcomes observed in clinical studies. Further, recent findings suggest that VD signaling intersects with cholesterol metabolism and vascular pathology. Locally dysregulated VD activation within vascular smooth muscle cells may promote calcification and plaque instability independent of systemic levels. By integrating insights from endocrinology, vascular biology, and genetics, this review argues for a shift away from one-size-fits-all supplementation strategies. A better understanding of the molecular determinants of VD responsiveness may improve cardiovascular risk assessment and allow for personalized therapeutic approaches. Until tools become available to assess tissue-level VD activity or predict individual responsiveness, clinicians should remain cautious—particularly in populations at risk of either insufficient effect or toxicity.
Keywords:
vitamin D
; vitamin D responsiveness
; cardiovascular diseases
; atherosclerosis
; vitamin D resistance
; vitamin D receptor
; IIH
; cholesterol
; vascular calcification
; atherosclerotic plaque
1. Introduction
The role of VD in the pathophysiology of cardiovascular (CV) disease remains controversial. Despite numerous studies, results have often been inconsistent, and no clear consensus has been reached. This review identifies two key factors that may contribute to the variability in findings: individual differences in VD responsiveness and the significance of local tissue-level effects that cannot be inferred solely from serum VD levels. Considering these elements may help generate more reliable conclusions regarding the benefits and risks of VD supplementation in cardiovascular contexts.
Atherosclerosis and Its Vascular Complications
Atherosclerotic diseases, such as myocardial infarction and stroke, remain the leading cause of death worldwide. Atherosclerosis is a multifactorial disease influenced by both genetic predisposition and environmental factors. Clinical manifestations develop gradually over decades. Significant risk factors include hypercholesterolemia, hypertension, diabetes, and smoking. However, atherosclerosis is increasingly recognized as a chronic inflammatory process initiated by injury to the vascular endothelium and perpetuated by metabolic and hemodynamic stressors. [1]
The vascular endothelium is a monolayer of endothelial cells (ECs) that lines all blood vessels and functions as a critical regulatory barrier between blood and tissues. [2]. In large and medium-sized arteries, ECs are in direct contact with the tunica media, which consists of vascular smooth muscle cells (VSMCs), elastic fibers, and collagen. Surrounding this is the tunica adventitia, mainly composed of connective tissue [3].
Mechanical forces acting on the vessel wall include tensile stress from blood pressure and wall shear stress (WSS), the tangential force from blood flow. Low or oscillatory WSS is associated with regions of disturbed flow, such as arterial bifurcations, and these sites are particularly prone to atherosclerotic plaque formation [5,6]. Hemodynamic stress alters endothelial cell signaling and promotes a pro-inflammatory state, setting the stage for lesion development [7]. Atherosclerosis begins with endothelial dysfunction, characterized by increased permeability, leukocyte adhesion, and prothrombotic surface changes [8]. The retention and modification of low-density lipoproteins (LDL) in the intima is a key early step [4,9]. Modified LDLs activate ECs and attract monocytes, which differentiate into macrophages [10]. These cells, along with VSMCs, engulf LDLs and transform into foam cells, forming the fatty streak—the earliest visible lesion of atherosclerosis [1,10]. As foam cells accumulate, they release cytokines and growth factors that recruit additional inflammatory cells and stimulate the proliferation of VSMCs and matrix deposition. This leads to plaque growth and formation of a necrotic core, rich in lipids and cell debris, surrounded by a fibrous cap. The cap is composed of collagen and VSMCs and serves to stabilize the lesion. However, persistent inflammation weakens the cap, increasing the risk of rupture [1]. Hemodynamic forces further exacerbate the process. Turbulent flow not only sustains inflammation but also facilitates LDL infiltration due to longer residence times and physical disruption of endothelial integrity [11]. In later stages, plaque calcification occurs, resembling the formation of bone. VSMCs and pericytes may differentiate into osteoblast-like cells, leading to calcium deposition. Microcalcifications coalesce into macrocalcifications, which can stiffen the vessel wall and contribute to plaque instability [12,13,14]. (Figure 1) Plaque rupture is a critical event in acute cardiovascular syndromes. Rupture exposes thrombogenic material to the bloodstream, triggering platelet aggregation and the formation of a thrombus. If the thrombus occludes the vessel, it can lead to a myocardial infarction or stroke. In some cases, a thrombus may detach, forming an embolus that obstructs smaller downstream arteries, leading to ischemia or infarction of the affected tissues. The transition from subclinical lesions to symptomatic disease highlights the importance of plaque stability. A stable plaque, characterized by a thick fibrous cap and minimal inflammation, may remain silent. In contrast, an unstable plaque, marked by a thin cap and active inflammation, is prone to rupture. Even in the absence of complete occlusion, growing plaques can reduce coronary perfusion and lead to chronic ischemic conditions such as angina pectoris or heart failure. [15,16,17]
Atherosclerosis is a complex disease driven by endothelial dysfunction, lipid accumulation, chronic inflammation, and hemodynamic stress. It evolves from fatty streaks to complex plaques that can calcify, rupture, and trigger life-threatening events. Understanding the molecular and biomechanical mechanisms of this process is essential for developing preventive and therapeutic strategies.
Endocrinology of Vitamin D
VD is a secosteroid hormone. It can be synthesized de novo in the skin by exposure to ultraviolet B light, which converts 7-dehydrocholesterol (7DHC) into cholecalciferol, or it can be absorbed through ingestion. Ingested or cutaneously synthesized cholecalciferol binds to the VD-binding protein and is transferred to the liver. The hepatic enzymes CYP2R1 and CYP27A1 (25-hydroxylases) transform VD to 25OHD. 25OHD has a long half-life and is usually used to determine VD status in the blood. The second hydroxylation step takes place in the kidneys. 1α-hydroxylase (CYP27B1) converts 25OHD into 1,25(OH)2D3 (active VD) in the proximal renal tubule. 1,25(OH)2D3 is released into the bloodstream and binds to the VD-binding protein. The VD receptor (VDR) is a ligand-activated transcription factor found in nearly every tissue. As a lipophilic hormone, 1,25(OH)2D3 can cross the cell membrane and bind to the VDR in target cells’ cytoplasm and/or nucleus, and. function as a critical regulatory barrier between blood and tissue. Genome-wide analyses have revealed thousands of VDR binding sites that influence the expression of hundreds of genes involved in immunity, inflammation, and cardiovascular function. This widespread genomic activity explains the pleiotropic effects of VD. [18]
VD metabolism is strongly regulated by calcium, phosphate, fibroblast growth factor 23 (FGF23), and parathyroid hormone (PTH) levels. FGF23 inhibits CYP27B1, downregulating 1,25(OH)2D3 production and promoting its catabolism [20]. On the other hand, PTH upregulates CYP27B1 expression in the kidneys, exerting the opposite effect. An increase in ionized blood calcium inhibits PTH secretion in the parathyroid glands, leading to lower 1,25(OH)2D3 production. Hyperphosphatemia also inhibits CYP27B1 activity in the kidneys. When present in excess, 1,25(OH)2D3 initiates harmful feedback mechanisms by downregulating the expression of the CYP27B1 gene in the kidney, downregulating the gene encoding PTH in the parathyroid glands, and upregulating FGF23 secretion in the skeleton. Due to the tight endocrine regulation of renal VD hormone production, circulating 1,25(OH)2D3 levels remain within physiological limits even in the presence of very low 25(OH)D3 levels associated with severe VD deficiency. [18,19,22].
VD can be activated extra-renally in tissues expressing CYP27B1 [21]. Hence, circulating 25(OH)D3 also serves as a substrate for local 1,25(OH)2D3 synthesis. Locally produced 1,25(OH)2D3 may have autocrine and paracrine effects in cells that express the VDR. Therefore, the biological effects of VD signaling in VDR-expressing target cells are presumably determined by the sum of circulating 1,25(OH)2D3 concentrations in addition to locally produced 1,25(OH)2D3. Hence, the blood concentrations of 25(OH)D3, which are routinely used to determine VD status, are not a direct readout for the activity of VD signaling within target cells. VD is essential for the survival of most vertebrates. Therefore, robust regulatory systems evolved during evolution that can maintain circulating concentrations of the active VD hormone, the active principle of the VD system, within narrow limits, despite variations in the circulating concentrations of the precursor molecule 25(OH)D3. Not so much is known about the regulation of local 1,25(OH)2D3 production within cardiovascular target cells, although what counts for the activity of VD signaling in a biological sense is the local concentration of 1,25(OH)2D3 within the target cell, regardless of its origin. (23)
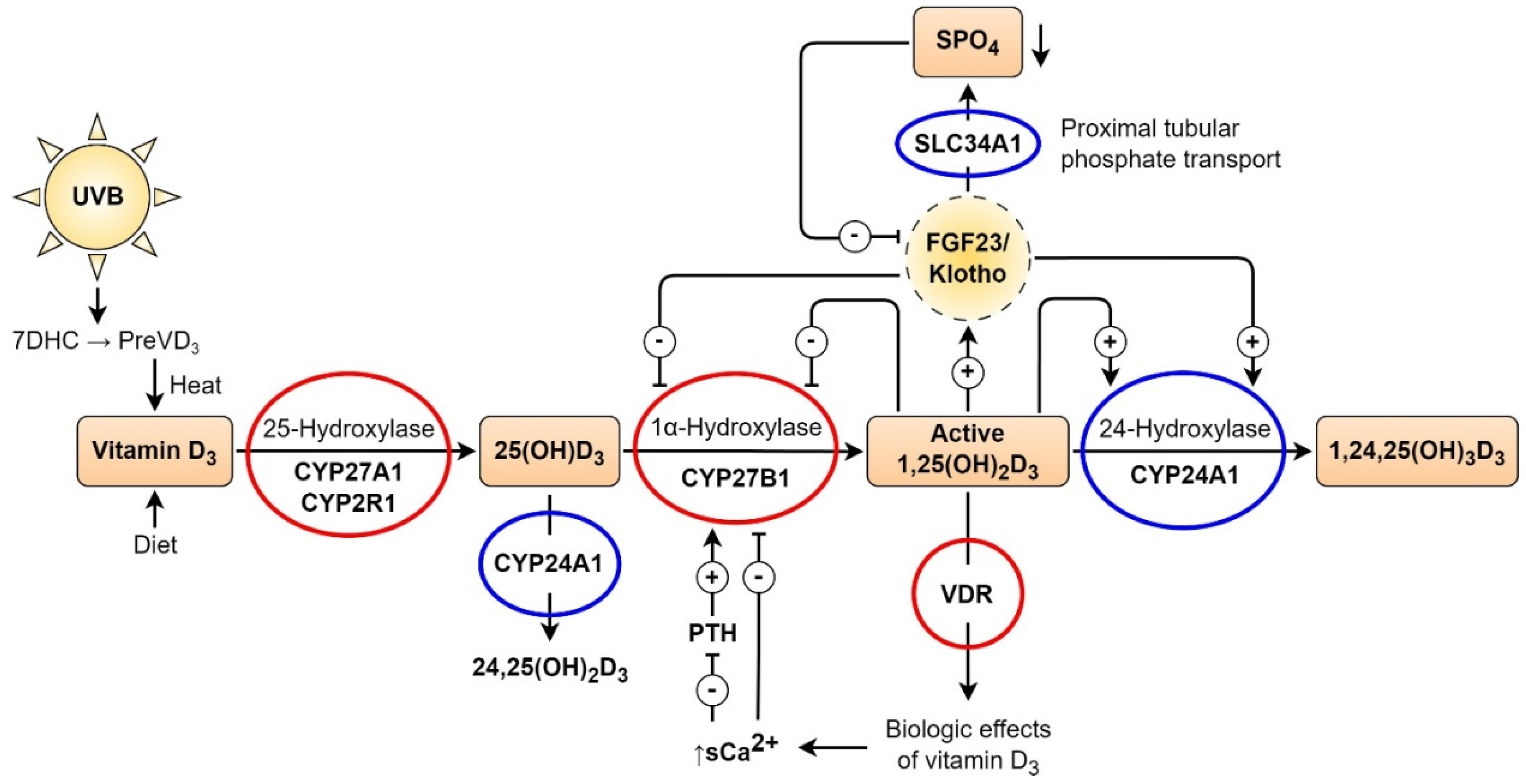
Figure 2.
Vitamin D (VD) synthesis and metabolism. Mutations of the VD pathway. The two-step hydroxylation mechanism of VD synthesis and metabolism transforms dietary or skin-produced VD into its active hormonal form, 1,25-(OH)2D3. VD binds to the vitamin D receptor, controlling the amounts of phosphate and calcium in the serum, which has various biological effects. The enzyme 1-hydroxylase (CYP27B1) is responsible for converting VD into its physiologically active form, 1,25-(OH)2D3, which is degraded by 24-hydroxylase (CYP24A1). Serum calcium, parathyroid hormone, and 1,25-(OH)2D3 levels control this process. Additionally, based on phosphate homeostasis, FGF23 affects VD metabolism by restricting the activity of 1,25-(OH)2D3 through the inhibition of 1-hydroxylase (CYP27B1) and activation of 24-hydroxylase (CYP24A1) activity. SLC34A1 regulates proximal tubule phosphate reabsorption from primary urine. Red circles denote proteins in the VD machinery that can cause rickets or autoimmune conditions when mutated. Mutations in CYP24A1 and SLC34A1 can cause nephrolithiasis, hypercalcemia, hypercalciuria, and decreased PTH levels. Hypophosphatemia occurs in patients with SLC34A1 mutations. These mutations are highlighted in blue circles. Renal insufficiency, vascular calcification, and calcification in other organs can result from mutations in the CYP24A1 and SLC34A1 genes. This figure is adapted by Kristian Järvelin from Glenville Jones’ article [24] with the permission of Glenville Jones and from Ulla Järvelin’s open-access article [25].
Figure 2.
Vitamin D (VD) synthesis and metabolism. Mutations of the VD pathway. The two-step hydroxylation mechanism of VD synthesis and metabolism transforms dietary or skin-produced VD into its active hormonal form, 1,25-(OH)2D3. VD binds to the vitamin D receptor, controlling the amounts of phosphate and calcium in the serum, which has various biological effects. The enzyme 1-hydroxylase (CYP27B1) is responsible for converting VD into its physiologically active form, 1,25-(OH)2D3, which is degraded by 24-hydroxylase (CYP24A1). Serum calcium, parathyroid hormone, and 1,25-(OH)2D3 levels control this process. Additionally, based on phosphate homeostasis, FGF23 affects VD metabolism by restricting the activity of 1,25-(OH)2D3 through the inhibition of 1-hydroxylase (CYP27B1) and activation of 24-hydroxylase (CYP24A1) activity. SLC34A1 regulates proximal tubule phosphate reabsorption from primary urine. Red circles denote proteins in the VD machinery that can cause rickets or autoimmune conditions when mutated. Mutations in CYP24A1 and SLC34A1 can cause nephrolithiasis, hypercalcemia, hypercalciuria, and decreased PTH levels. Hypophosphatemia occurs in patients with SLC34A1 mutations. These mutations are highlighted in blue circles. Renal insufficiency, vascular calcification, and calcification in other organs can result from mutations in the CYP24A1 and SLC34A1 genes. This figure is adapted by Kristian Järvelin from Glenville Jones’ article [24] with the permission of Glenville Jones and from Ulla Järvelin’s open-access article [25].
Vitamin D and Cardiovascular Diseases
VD exerts several regulatory effects on the cardiovascular system, mediated primarily through the VDR. In the heart, VDR expression has been demonstrated in both ventricular cardiomyocytes and cardiac fibroblasts [26,27]. Additional studies have identified VDR expression in cultured bovine aortic endothelial cells, and in the endothelial cells (ECs) lining of the rat aorta [28,29]. Furthermore, CYP27A1 is found to be expressed in human VSMCs, responsible for converting 25(OH)D3 into the active form 1,25(OH)₂D3 [30].
This suggests that cardiac tissues are not only responsive to VD but also capable of locally activating it. Indeed, local conversion of 25(OH)D3 to 1,25(OH)₂D3 has been observed in vitro in both ECs and vascular smooth muscle cells (VSMCs), further supporting the role of VD signaling in cardiovascular tissues [31].
Epidemiological studies have frequently reported an association between low serum 25(OH)D3 levels and increased cardiovascular risk, including ischemic heart disease, myocardial infarction, and early mortality [32,33,34,35,36,37]. Despite these findings, randomized controlled trials have largely failed to demonstrate consistent benefits of VD supplementation in reducing cardiovascular events. Major clinical trials such as VITAL, ViDA, and DO-HEALTH, which employed high-dose VD regimens over multiple years, did not show significant reductions in coronary events or cardiovascular mortality. Likewise, systematic reviews and meta-analyses encompassing tens of thousands of participants have generally reported neutral effects on coronary artery disease outcomes [38,39].
Mendelian randomization studies—designed to minimize confounding—have also yielded essentially null results regarding causality between VD status and cardiovascular disease [40]. These inconsistencies between observational and interventional data suggest that VD’s effects on cardiovascular health may not be uniform across all individuals.
Taken together, these findings underscore the need for a more refined approach to VD research in cardiovascular medicine. Rather than assuming a universal response to supplementation, future studies should investigate interindividual differences in VD sensitivity, metabolism, and genetic background. In particular, conditions such as VD resistance and hypersensitivity may serve as key modulators of cardiovascular outcomes, helping to explain the heterogeneity observed in clinical studies.
Vitamin D Responsiveness Range
Research led by Carsten Carlberg has revealed marked interindividual variability in the physiological and molecular response to VD supplementation. In the VitDmet study, conducted in Finland during the winter of 2015, 71 elderly participants with prediabetes received daily doses of 0, 1,600, or 3,200 IU of VD. The study assessed changes in the expression of 12 VD-regulated genes and various laboratory biomarkers. Surprisingly, even at the highest dose, approximately 25% of participants did not exhibit the expected molecular or biochemical changes, indicating low responsiveness to supplementation. Based on their responses, participants were categorized into low responders (24%), mid responders (51%), and high responders (25%). [41]
This categorization was later validated in the VitDbol study (2017), in which a group of healthy young adults received a single oral dose of 80,000 IU VD. The response patterns mirrored those observed in the earlier trial, suggesting that responsiveness is likely an intrinsic biological trait rather than one influenced primarily by age or metabolic status. [42]
In the VD endocrine system, parathyroid hormone (PTH) plays a central role. It enhances intestinal calcium absorption, promotes bone resorption, stimulates the conversion of 25(OH)D₃ to 1,25(OH)₂D₃, and inhibits phosphate reabsorption in the renal tubules. Under typical conditions, sufficient levels of 25(OH)D₃ suppress PTH into the lower tertile of the reference range. A failure to achieve this suppression may suggest VD resistance, even when serum 25(OH)D₃ levels appear adequate. [43]
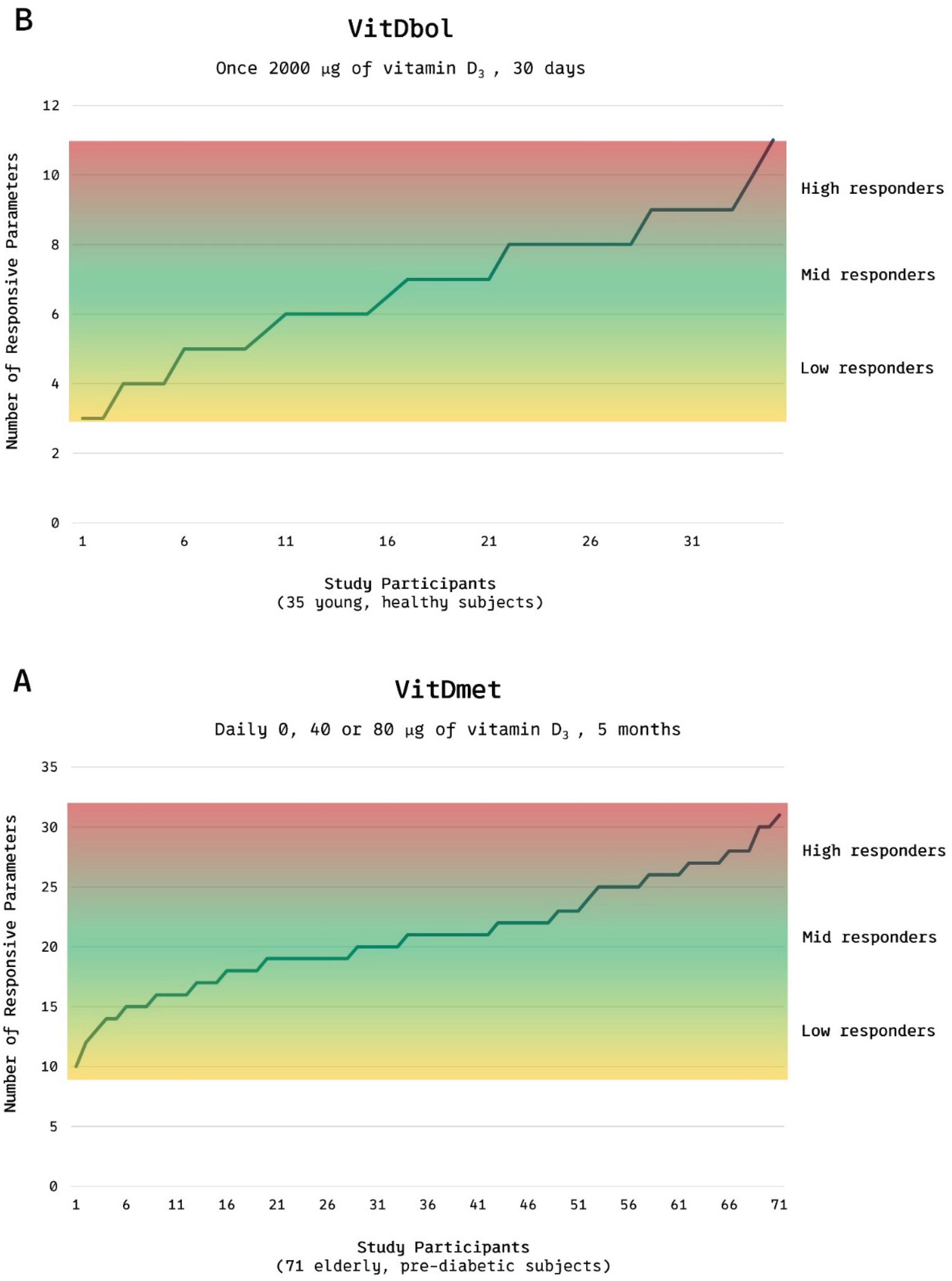
Figure 3.
VD responsiveness range Distribution of low, mid, and high responders to vitamin D₃ supplementation in (A) the VitDmet study (elderly pre-diabetic subjects; daily 0/1 600/3 200 IU for 5 months) and (B) the VitDbol study (healthy adults; single 80 000 IU bolus). Approximately one-quarter of participants showed minimal transcriptional or biochemical responses, and one-quarter of participants showed maximal responses, despite of similar intake of VD. Adapted from Carlberg C, Haq A. J Steroid Biochem Mol Biol 2018;175:12-17. © 2018 Elsevier, [44] by Kristian Järvelin.
Figure 3.
VD responsiveness range Distribution of low, mid, and high responders to vitamin D₃ supplementation in (A) the VitDmet study (elderly pre-diabetic subjects; daily 0/1 600/3 200 IU for 5 months) and (B) the VitDbol study (healthy adults; single 80 000 IU bolus). Approximately one-quarter of participants showed minimal transcriptional or biochemical responses, and one-quarter of participants showed maximal responses, despite of similar intake of VD. Adapted from Carlberg C, Haq A. J Steroid Biochem Mol Biol 2018;175:12-17. © 2018 Elsevier, [44] by Kristian Järvelin.
Vitamin D Resistance (VDRES)
VD resistance (VDRES) represents the low end of the responsiveness continuum, where standard or even high doses of VD fail to produce the expected physiological effects. This may be due to genetic mutations that impair VD signaling. For example, mutations in the VDR reduce its cellular function, while alterations in CYP27A1 or CYP27B1 affect VD activation. Such changes can contribute to autoimmune diseases and other chronic disorders, some of which may respond to high-dose VD protocols. [43]
The phenomenon of VD resistance was first described as early as 1937 in children with rickets that did not respond to VD therapy [45]. Later studies identified hereditary defects in CYP27B1 or VDR as causes of congenital VDRES, which is rare and presents with hypocalcemia, secondary hyperparathyroidism, and severe rickets.
However, more commonly, acquired VDRES arises over time, often due to environmental or immunological stressors. For instance, viral infections such as Epstein–Barr virus or cytomegalovirus have been shown to disrupt VDR function, either by direct receptor binding or by downregulating gene expression [46,47]. Inflammatory mediators and caspase-3 may also inactivate VDR signaling [48]. Additionally, glucocorticoid therapy can interfere with VDR gene expression and signaling pathways [49].
Aging further contributes to VDRES by impairing multiple components of the VD pathway. It reduces intestinal absorption of cholecalciferol [50], declines cutaneous production of VD [51], and decreases efficiency of hepatic and renal hydroxylation [52].
The most vulnerable link in this system appears to be the VDR itself. Mutations and polymorphisms affecting VDR function have been associated with autoimmune diseases and inadequate VD signaling despite normal serum levels [53].
In summary, individuals with VDRES either fail to activate VD effectively or are unable to utilize it at the tissue level. In these cases, standard supplementation may be insufficient, and personalized or high-dose regimens could be necessary to overcome this resistance.
Vitamin D Hypersensitivity (VDHY)
Compared to VD resistance (VDRES), VD hypersensitivity (VDHY) is the less understood end of the VD responsiveness spectrum. In individuals with VDHY, either excess amounts of active VD are present in the body, or the tissues exhibit heightened sensitivity to it. As a result, physiological effects occur at lower-than-average VD exposures, and supplementation may be unnecessary or even harmful. In such cases, regular sun exposure or standard therapeutic doses can lead to increased calcium absorption, bone resorption, and hypercalcemia. VDHY can be divided into two etiological categories: exogenous and endogenous. Exogenous VDHY is caused by excessive intake of pharmaceutical VD, often through high-dose supplementation. Endogenous VDHY, by contrast, refers to an exaggerated biological response to normal or low levels of VD, resulting from internal metabolic or genetic disturbances [54,55].
Historical cases of endogenous VDHY were first reported over 70 years ago, when children treated with high-dose VD for rickets developed symptoms of hypercalcemia. These outbreaks became endemic in three separate regions: Great Britain in the early 1950s [56], Poland in the 1970s [57], and East Germany in the 1980s [58].
In those historical cases, in some children, chronic moderate supplementation (e.g., 500 IU daily) led to delayed-onset toxicity. In contrast, in others, a single bolus of 600,000 IU caused symptoms resembling acute VD toxicity within days. Clinical features included hypercalcemia, hypercholesterolemia, cardiac murmurs, hypertension, neurological impairment, and renal dysfunction. In severe cases, children died, and post-mortem findings revealed extensive calcification of the heart, valves, and vasculature, including left ventricular hypertrophy and fragmentation of the arterial elastic lamina. [59].
Modern research has since clarified that endogenous VDHY is multifactorial in origin. One cause involves the ectopic synthesis of 1,25(OH)₂D₃ in granulomatous diseases such as sarcoidosis [60,61,62], tuberculosis [63], lymphomas [64,65], and fungal infections. In pregnancy, the placenta synthesizes 1,25(OH)₂D₃ [66]. Another cause is genetic mutations affecting
VD metabolism, particularly in CYP24A1 and SLC34A1, which impair the degradation or regulation of active VD [67,68,69,70]. Such mutations lead to the accumulation of 1,25(OH)₂D₃ and may result in persistent hypercalcemia, even under modest sun exposure or prophylactic VD dosing. These inherited defects may manifest as idiopathic infantile hypercalcemia in neonates or may remain clinically silent until triggered by environmental factors. [54,55]
The “gene-dose effect” is well established. Individuals with biallelic mutations often exhibit severe, early-onset disease characterized by hypercalcemia, nephrolithiasis, vascular calcification, suppressed PTH levels, and hypercalciuria. Monoallelic mutation carriers may remain asymptomatic unless exposed to additional triggers such as pregnancy, high-dose VD supplementation, or extended sun exposure. In these individuals, VD prophylaxis—even at standard doses—may precipitate toxicity. [71]
Importantly, VDHY may persist into adulthood, particularly in individuals with monoallelic mutations. These individuals constitute a genetic risk group who may appear healthy until exposed to otherwise safe levels of VD. This underscores the importance of tailoring VD dosing based on individual metabolic sensitivity
CYP24A1 and Cholesterol Metabolism
Beyond its well-established role in calcium and phosphate homeostasis, VD metabolism appears to intersect with lipid regulation—an underexplored and interesting area. Notably, some patients with CYP24A1 mutations, which are known to impair the catabolism of 1,25(OH)₂D₃, have also been reported to exhibit abnormalities in lipid metabolism, including hypercholesterolemia. Historical case reports of infantile idiopathic hypercalcemia have documented a co-occurrence of hypercholesterolemia and vascular lesions [59]. These observations raise the question of whether dysregulated VD signaling might disrupt cholesterol homeostasis through metabolic cross-talk or feedback.
To explore this hypothesis, researchers developed transgenic rats that constitutively overexpress the CYP24A1 gene. These animals exhibited significantly reduced circulating levels of 24,25(OH)₂D₃, reflecting enhanced catabolic activity. Interestingly, after weaning, the rats also developed albuminuria and hyperlipidemia, with lipid profiling revealing elevations across all lipoprotein fractions. Furthermore, they displayed atherosclerotic changes in the aorta, which were exacerbated by a high-fat, high-cholesterol diet [72]. These findings suggest that CYP24A1 may have broader biological roles than previously recognized, extending beyond VD catabolism to regulate lipid and vascular physiology.
From a biochemical perspective, 7-dehydrocholesterol (7DHC) serves as a critical metabolic branch point. It is a shared precursor for both cholesterol and VD synthesis. In the skin, 7DHC is photochemically converted to pre-VD under UVB radiation [18]. The metabolic competition or feedback regulation, in turn, occurs between VD and cholesterol biosynthesis. For example, excessive levels of 1,25(OH)₂D₃—either due to ectopic synthesis or impaired degradation from CYP24A1 mutations—could alter substrate flux or influence gene regulation. The reduction of cholesterol occurs via the enzyme 7-dehydrocholesterol reductase (DHCR7). [73]
Studies have shown that DHCR7 activity is sensitive to cholesterol levels. When cholesterol accumulates, it accelerates DHCR7 degradation via end-product inhibition, thereby reducing cholesterol synthesis and potentially directing 7DHC toward vitamin D production. [73] Interestingly, experiments in keratinocytes found that cholecalciferol rapidly suppresses DHCR7 activity without causing 7DHC accumulation. In contrast, 25(OH)D₃ suppressed DHCR7 activity modestly, and 1,25(OH)₂D₃ had no significant impact on 7DHC accumulation. It suggests that this regulation occurs independently of the VDR pathway. [74]
Altogether, these findings highlight DHCR7 as a potential regulatory switch between VD and cholesterol synthesis. The complex interplay between these pathways warrants further investigation, particularly in the context of VD hypersensitivity, where excessive active VD may disrupt metabolic balance, potentially contributing to hypercholesterolemia or vascular pathology.
Vascular Calcification and CYP24A1 Mutation
Mutations in the CYP24A1 gene, known for impairing the degradation of active VD metabolites, have been implicated not only in systemic hypercalcemia but also in vascular pathology. Elevated levels of 1,25(OH)₂D₃ can lead to cardiovascular complications, such as hypertension, arterial vasoconstriction, and notably, arterial calcification, particularly coronary artery calcification [73,74,75,76,77]. Recent genetic studies have identified associations between CYP24A1 variants and increased coronary artery calcification burden in independent populations [78,79], further suggesting that dysregulated VD metabolism contributes to coronary atherosclerosis. This is clinically relevant because coronary artery calcification is a well-established predictor of coronary heart disease and future cardiovascular events [80].
Although 25(OH)D3 is commonly used as a marker of VD status, it is the active metabolite 1,25(OH)₂D₃ that exerts the most potent biological effects. Importantly, circulating levels of 25(OH)D3 may not reflect tissue-specific activity, particularly in pathological conditions. In vascular tissues, extra-renal synthesis of 1,25(OH)₂D₃ can occur via CYP27B1 activity in ECs and immune cells. This local synthesis plays a role in atherosclerosis and plaque calcification, functioning independently of systemic VD levels [81].
The atherosclerotic plaque microenvironment provides an ideal setting for this “non-classical” VD activity. Within plaques, enzymes responsible for VD activation and the VDR are all expressed, enabling intracrine, autocrine, and paracrine signaling. In specific conditions—such as CYP24A1 mutations, or ectopic synthesis of VD—local VD metabolism becomes dysregulated, leading to elevated intraplaque 1,25(OH)₂D₃ concentrations that may remain undetected in serum yet promote vascular injury. [23,81]
Vascular calcification, particularly within atherosclerotic lesions, is now recognized as an active and regulated process that resembles osteogenesis. Calcified plaques contain bone morphogenetic proteins and structural proteins such as osteopontin, osteonectin, and osteocalcin, indicating a shared phenotype with bone tissue [82]. In vitro models using bovine vascular smooth muscle cells have demonstrated that the enzymes alkaline phosphatase and OPN are key mediators of vascular calcification [83].
Calcium-regulating hormones also influence this process. Parathyroid hormone (PTH) and parathyroid hormone-related peptide (PTHrP) suppress vascular calcification by downregulating AFOS activity [84]. In contrast, 1,25(OH)₂D₃ not only stimulates calcium influx [86] and inhibits smooth muscle proliferation [85,87] but also reduces PTHrP expression, removing a natural brake on vascular mineralization [82,83,84]. Excess VD—whether dietary or due to impaired catabolism—has been shown to induce vascular calcification in both animal models and experimental systems [87,88,89].
Taken together, these findings support the view that CYP24A1 mutations, by disrupting local VD catabolism, may contribute to increased vascular calcification and promote the development of unstable, calcified atherosclerotic plaques, particularly in individuals with genetic or inflammatory predispositions.
Future Research
Current research suggests that approximately 50% of individuals may exhibit altered responsiveness to VD—either heightened sensitivity or resistance. However, these estimates are based on small and relatively homogeneous study populations. To understand the broader public health implications, larger, multiethnic cohorts are needed.
Further in vitro and in vivo studies should investigate how VD—particularly in the context of CYP24A1 mutations—influences the expression of parathyroid hormone-related peptide (PTHrP) and the process of vascular calcification. Additionally, the observed relationship between CYP24A1 mutations and cholesterol metabolism deserves deeper mechanistic exploration.
Ultimately, the development of a clinical test to assess individual VD responsiveness could allow for safer and more effective supplementation strategies. Such a tool would help clinicians identify individuals who are at risk of either insufficient response or toxicity, minimizing potential harms such as atherosclerosis or hypercholesterolemia.
Discussion
VD exerts a wide spectrum of biological effects that extend far beyond its classical role in mineral metabolism. It modulates inflammation, immune function, and vascular health. This review highlights that inter-individual variability in VD responsiveness—whether due to genetic mutations, environmental factors, or disease states—is a critical but underrecognized determinant of cardiovascular outcomes.
In cases of VDRES, impaired response to VD may perpetuate chronic inflammation, autoimmune activation, or vascular dysfunction. In contrast, VDHY—whether due to CYP24A1 mutations, ectopic synthesis, or reduced catabolism—may lead to pathological calcification, even under standard supplementation or natural sun exposure.
Importantly, serum 25(OH)D levels may not reliably indicate true VD activity at the tissue level, especially in cells capable of local hormone synthesis. This is particularly relevant in VSMCs and macrophages, where dysregulated VD metabolism may directly contribute to vascular calcification and potentially influence cholesterol biosynthesis, given their shared metabolic pathways.
Taken together, these findings challenge the notion of universal VD sufficiency thresholds and support a paradigm shift toward individualized VD therapy, guided by molecular diagnostics and patient-specific risk profiles.
Conclusion
VD supplementation is neither inherently beneficial nor harmful. Instead, its cardiovascular impact depends on the individual’s biological response. In VDRES, supplementation may restore immune balance and protect vascular function. In VDHY, even modest doses may precipitate hypercalcemia, vascular calcification, or, perhaps, lipid disturbances.
Funding
This research received no external funding.
Data Availability Statement
No new data is collected.
Acknowledgements
Kristian Järvelin. It supports. Images. ChatGPT. Formatting the text, including the abstract, and reviewing the reference list. Help with IT problems. Grammarly Premium. Formatting the text.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| CYP224A1 | 24-hydroxylase |
| CYP27B1 | 1-hydoxylase |
| DHCR7 | 7-Dehydrocholesterol Reductase |
| FGF23 | Fibroblast growth factor 23 |
| PTH | Parathyreoidea Hormone |
| VDHY | Vitamin D Hypersensitivity |
| VDR | Vitamin D Receptor |
| VDRES | Vitamin D Resistance |
| VSMC | Vascular Smooth Muscle Cells |
References
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Virchow, R. Der Ateromatose Prozess der Arterien. Wien Med Wochenschr. 1856, 825–827. [Google Scholar]
- Malek, A.M.; Alper, S.L.; Izumo, S. Hemodynamic Shear Stress and Its Role in Atherosclerosis. JAMA 1999, 282, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.-J.; Chien, S. Effects of disturbed flow on vascular endothelium. Mol Cell Biomech. 2011, 8, 285–306. [Google Scholar] [CrossRef]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Mortensen, M.B.; Dzaye, O.; Bøtker, H.E.; Jensen, J.M.; Maeng, M.; Bentzon, J.F.; Kanstrup, H.; Sørensen, H.T.; Leipsic, J.; Blankstein, R.; et al. Low-Density Lipoprotein Cholesterol Is Predominantly Associated With Atherosclerotic Cardiovascular Disease Events in Patients With Evidence of Coronary Atherosclerosis: The Western Denmark Heart Registry. Circulation 2023, 147, 1053–1063. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pr. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Inflammatory, Metabolic, and Genetic Mechanisms of Vascular Calcification. Arter. Thromb. Vasc. Biol. 2014, 34, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Speer, M.Y.; Giachelli, C.M.; et al. Vascular calcification in atherosclerosis: calcifying vascular cells and osteogenic regulation. Trends in Cardiovascular Medicine 2009, 19, 132–137. [Google Scholar]
- Hutcheson, J.D.; et al. Calcification in atherosclerosis: bone biology and chronic inflammation at the arterial crossroads. Circ Res. 2014, 114, 1853–1863. [Google Scholar] [CrossRef]
- Doherty, T.M.; Asotra, K.; Fitzpatrick, L.A.; Qiao, J.-H.; Wilkin, D.J.; Detrano, R.C.; et al. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Circulation 1995, 92, 2346–2350. [Google Scholar] [CrossRef]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the Vulnerable Plaque. J Am Coll Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Virmani, R.; Burke, A.P.; Farb, A.; Weber, D.K.; Kutys, R.; et al. Pathologic assessment of the vulnerable human coronary plaque. Heart 2004, 90, 1385–1391. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Christakos, S.; Ajibade, D.V.; Dhawan, P.; Fechner, A.J.; Mady, L.J. Vitamin D: Metabolism. Endocrinol. Metab. Clin. North Am. 2010, 39, 243–253. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef]
- Bikle, D.D.; Patzek, S.; Wang, Y. Physiologic and pathophysiologic roles of extra renal CYP27b1: Case report and review. Bone Rep. 2018, 8, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.H.; May, B.K.; Morris, H.A. Vitamin D metabolism: New concepts and clinical implications. Clin. Biochem. Rev. 2003, 24, 13–26. [Google Scholar] [PubMed]
- Latic, N.; Erben, R.G. Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure. Int. J. Mol. Sci. 2020, 21, 6483. [Google Scholar] [CrossRef] [PubMed]
- Jones, G. 100 YEARS OF VITAMIN D: Historical aspects of vitamin D. Endocr. Connect. 2022, 11, e210594. [Google Scholar] [CrossRef] [PubMed]
- Järvelin, U.M.; Järvelin, J.M. Significance of vitamin D responsiveness on the etiology of vitamin D-related diseases. Steroids 2024, 207, 109437. [Google Scholar] [CrossRef]
- Tishkoff, D.X.; Nibbelink, K.A.; Holmberg, K.H.; Dandu, L.; Simpson, R.U. Functional Vitamin D Receptor (VDR) in the T-Tubules of Cardiac Myocytes: VDR Knockout Cardiomyocyte Contractility. Endocrinology 2008, 149, 558–564. [Google Scholar] [CrossRef]
- Chen, S.; Glenn, D.J.; Ni, W.; Grigsby, C.L.; Olsen, K.; Nishimoto, M.; Law, C.S.; Gardner, D.G. Expression of the Vitamin D Receptor Is Increased in the Hypertrophic Heart. Hypertension 2008, 52, 1106–1112. [Google Scholar] [CrossRef]
- Merke, J.; Milde, P.; Lewicka, S.; Hügel, U.; Klaus, G.; Mangelsdorf, D.J.; Haussler, M.R.; Rauterberg, E.W.; Ritz, E. Identification and regulation of 1,25-dihydroxyvitamin D3 receptor activity and biosynthesis of 1,25-dihydroxyvitamin D3. Studies in cultured bovine aortic endothelial cells and human dermal capillaries. J. Clin. Investig. 1989, 83, 1903–1915. [Google Scholar] [CrossRef]
- Wong, M.S.K.; Delansorne, R.; Man, R.Y.K.; Vanhoutte, P.M. Vitamin D derivatives acutely reduce endothelium-dependent contractions in the aorta of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H289–H296. [Google Scholar] [CrossRef]
- Somjen, D.; Weisman, Y.; Kohen, F.; Gayer, B.; Limor, R.; Sharon, O.; Jaccard, N.; Knoll, E.; Stern, N. 25-Hydroxyvitamin D 3 -1α-Hydroxylase Is Expressed in Human Vascular Smooth Muscle Cells and Is Upregulated by Parathyroid Hormone and Estrogenic Compounds. Circulation 2005, 111, 1666–1671. [Google Scholar] [CrossRef]
- Zehnder, D.; Bland, R.; Chana, R.S.; Wheeler, D.C.; Howie, A.J.; Williams, M.C.; Stewart, P.M.; Hewison, M. Synthesis of 1,25-dihydroxyvitamin D (3) by human endothelial cells is regulated by inflammatory cytokines: A novel autocrine determinant of vascular cell adhesion. J. Am. Soc. Nephrol. 2002, 13, 621–629. [Google Scholar] [CrossRef]
- Anderson, J.L.; May, H.T.; Horne, B.D.; Bair, T.L.; Hall, N.L.; Carlquist, J.F.; Lappé, D.L.; Muhlestein, J.B.; Intermountain Heart Collaborative (IHC) Study Group. Relation of Vitamin D Deficiency to Cardiovascular Risk Factors, Disease Status, and Incident Events in a General Healthcare Population. Am. J. Cardiol. 2010, 106, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Brøndum-Jacobsen, P.; Benn, M.; Jensen, G.B.; Nordestgaard, B.G. 25- hydroxyvitamin D levels and risk of ischemic heart disease, myocardial infarction, and early death: population-based study and meta-analyses of 18 and 17 studies. Arter. Thromb. Vasc. Biol. 2012, 32, 2794–2802. [Google Scholar] [CrossRef] [PubMed]
- Karakas, M.; Thorand, B.; Zierer, A.; Huth, C.; Meisinger, C.; Roden, M.; Rottbauer, W.; Peters, A.; Koenig, W.; Herder, C. Low Levels of Serum 25-Hydroxyvitamin D Are Associated with Increased Risk of Myocardial Infarction, Especially in Women: Results from the MONICA/KORA Augsburg Case-Cohort Study. J. Clin. Endocrinol. Metab. 2013, 98, 272–280. [Google Scholar] [CrossRef]
- Lee, J.H.; Gadi, R.; Spertus, J.A.; Tang, F.; O'KEefe, J.H. Prevalence of Vitamin D Deficiency in Patients With Acute Myocardial Infarction. Am. J. Cardiol. 2011, 107, 1636–1638. [Google Scholar] [CrossRef] [PubMed]
- Michos, E.D.; Melamed, M.L. Vitamin D and cardiovascular disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Shi, L.; Rimm, E.B.; Giovannucci, E.L.; Hu, F.B.; Manson, J.E.; Rexrode, K.M. Vitamin D intake and risk of cardiovascular disease in US men and women. Am. J. Clin. Nutr. 2011, 94, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Manousaki, D.; Rosen, C.; Trajanoska, K.; Rivadeneira, F.; Richards, J.B. The health effects of vitamin D supplementation: evidence from human studies. Nat. Rev. Endocrinol. 2022, 18, 96–110. [Google Scholar] [CrossRef]
- Barbarawi, M.; Kheiri, B.; Zayed, Y.; Barbarawi, O.; Dhillon, H.; Swaid, B.; Yelangi, A.; Sundus, S.; Bachuwa, G.; Alkotob, M.L.; et al. Vitamin D Supplementation and Cardiovascular Disease Risks in More Than 83,000 Individuals in 21 Randomized Clinical Trials: A Meta-analysis. JAMA Cardiol. 2019, 4, 765–776. [Google Scholar] [CrossRef]
- Bouillon, R.; LeBoff, M.S.; Neale, R.E. Health Effects of Vitamin D Supplementation: Lessons Learned From Randomized Controlled Trials and Mendelian Randomization Studies. J. Bone Miner. Res. 2023, 38, 1391–1403. [Google Scholar] [CrossRef]
- Saksa, N.; Neme, A.; Ryynänen, J.; Uusitupa, M.; de Mello, V.D.; Voutilainen, S.; Nurmi, T.; Virtanen, J.K.; Tuomainen, T.-P.; Carlberg, C. Dissecting high from low responders in a vitamin D3 intervention study. J. Steroid Biochem. Mol. Biol. 2015, 148, 275–282. [Google Scholar] [CrossRef]
- Seuter, S.; Virtanen, J.K.; Nurmi, T.; Pihlajamäki, J.; Mursu, J.; Voutilainen, S.; Tuomainen, T.-P.; Neme, A.; Carlberg, C. Molecular evaluation of vitamin D responsiveness of healthy young adults. J. Steroid Biochem. Mol. Biol. 2017, 174, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Lemke, D.; Klement, R.J.; Schweiger, F.; Schweiger, B.; Spitz, J. Vitamin D Resistance as a Possible Cause of Autoimmune Diseases: A Hypothesis Confirmed by a Therapeutic High-Dose Vitamin D Protocol. Front Immunol. 2021, 12, 655739. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Haq, A. The concept of the personal vitamin D response index. J. Steroid Biochem. Mol. Biol. 2018, 175, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Albright, F.; Butler, A.M.; Bloomberg, E. RICKETS RESISTANT TO VITAMIN D THERAPY. Arch. Pediatr. Adolesc. Med. 1937, 54, 529–547. [Google Scholar] [CrossRef]
- Yenamandra, S.P.; Hellman, U.; Kempkes, B.; Darekar, S.D.; Petermann, S.; Sculley, T.; Klein, G.; Kashuba, E. Epstein-Barr virus encoded EBNA-3 binds to the vitamin D receptor and blocks activation of its target genes. Cell. Mol. Life Sci. 2010, 67, 4249–4256. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.J.J.; Gröschel, C.; Kastner, M.T.; Kosulin, K.; Laengle, J.; Zadnikar, R.; et al. Human cytomegalovirus infection downregulates vitamin-D receptor in mammalian cells. J Steroid Biochem Mol Biol. 2017, 165, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Malloy, P.J.; Feldman, D. Inactivation of the Human Vitamin D Receptor by Caspase-3. Endocrinology 2009, 150, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, G.; Formenti, A.M.; Frara, S.; Doga, M.; Giustina, A. Vitamin D and Glucocorticoid-Induced Osteoporosis Front Horm Res. 2017; 50, 149–160. [Google Scholar]
- Barragry, J.M.; France, M.W.; Corless, D.; Gupta, S.P.; Switala, S.; Boucher, B.J.; Cohen, R.D. Intestinal Cholecalciferol Absorption in the Elderly and in Younger Adults. Clin. Sci. 1978, 55, 213–220. [Google Scholar] [CrossRef]
- MacLaughlin, J.; Holick, M.F.; Kasper, K. Aging Decreases the Capacity of Human Skin to Produce Vitamin D3. Nutr. Clin. Pr. 1986, 1, 57–58. [Google Scholar] [CrossRef]
- Rushton, C. VITAMIN D HYDROXYLATION IN YOUTH AND OLD AGE. Age and Ageing 1978, 7, 91–95. [Google Scholar] [CrossRef]
- Agliardi, C.; Guerini, F.R.; Bolognesi, E.; Zanzottera, M.; Clerici, M. VDR Gene Single Nucleotide Polymorphisms and Autoimmunity: A Narrative Review. Biology 2023, 12, 916. [Google Scholar] [CrossRef] [PubMed]
- Tebben, P.J.; Singh, R.J.; Kumar, R. Vitamin D-Mediated Hypercalcemia: Mechanisms, Diagnosis, and Treatment. Endocr. Rev. 2016, 37, 521–547. [Google Scholar] [CrossRef] [PubMed]
- Marcinowska-Suchowierska, E.; Urbańska, M.K.; Łukaszkiewicz, J.; Płudowski, P.; GJones, G. Vitamin D Toxicity–A Clinical Perspective Front Endocrinol (lausanne). 2018; 9, 550. [Google Scholar] [CrossRef]
- Samufel, H.S. Infantile Hypercalcaemia, nutritional rickets, and infantile scurvy in Great Britain. A British Paediatric Association Report. Br Med J. 1964, 1, 1659–1661. [Google Scholar] [CrossRef]
- Pronicka, E.; Ciara, E.; Halat, P.; Janiec, A.; Wójcik, M.; Rowińska, E. Biallelic mutations in CYP24A1 or SLC34A1 as a cause of infantile idiopathic hypercalcemia (IIH) with vitamin D hypersensitivity: molecular study of 11 historical IIH cases. J Appl Genet. 2017, 58, 349–353, 10. [Google Scholar] [CrossRef]
- Misselwitz, J.; Hesse, V.; Markestad, T. Nephrocalcinosis, hypercalciuria and elevated serum levels of 1,25-dihydroxyvitamin D in children. Possible link to vitamin D toxicity. Acta Paediatr Scand. 1990, 796-7, 637–643. [Google Scholar] [CrossRef]
- Coleman, E.N.; et al. Infantile hypercalcemia and cardiovascular lesions. Evidence, hypothesis, and speculation. Arch. Dis. Childhood, 1965/SCHLESINGER BE et al. Severe type of infantile hypercalcemia. BMJ 1956. [Google Scholar]
- Bell, N.H.; Stern, P.H.; Pantzer, E.; Sinha, T.K.; Deluca, H.F. Evidence that Increased Circulating 1α,25-Dihydroxyvitamin D is the Probable Cause for Abnormal Calcium Metabolism in Sarcoidosis. J. Clin. Investig. 1979, 64, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.S.; Frankel, T.; Chan, Y.-L.; Lissner, D.; Posen, S. Vitamin D Conversion by Sarcoid Lymph Node Homogenate. Ann. Intern. Med. 1984, 100, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Paramothayan, S.; Jones, P.W. Corticosteroid therapy in pulmonary sarcoidosis: a systematic review. JAMA 2002, 287, 1301–1307. [Google Scholar] [CrossRef]
- Adams, J.S.; Modlin, R.L.; Diz, M.M.; Barnes, P.F. Potentiation of the Macrophage 25-Hydroxyvitamin D1-Hydroxylation Reaction by Human Tuberculous Pleural Effusion Fluid*. J. Clin. Endocrinol. Metab. 1989, 69, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.; Gagel, R. Calcitriol: the central humoral mediator of hypercalcemia in Hodgkin’s disease and non-Hodgkin’s lymphomas. Blood 1993, 82, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Mudde, A.H.; van den Berg, H.; Boshuis, P.G.; Breedveld, F.C.; Markusse, H.M.; Kluin, P.M.; et al. Ectopic production of 1,25-dihydroxyvitamin D by B-cell lymphoma as a cause of hypercalcemia. Cancer 1987, 59, 1543–1559. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Cohen, W.R.; Silva, P.; Epstein, F.H. Elevated 1,25-dihydroxyvitamin D plasma levels in normal human pregnancy and lactation. J. Clin. Investig. 1979, 63, 342–344. [Google Scholar] [CrossRef]
- De Paolis, E.; Scaglione, G.L.; De Bonis, M.; Minucci, A.; Capoluongo, E. CYP24A1 and SLC34A1 genetic defects associated with idiopathic infantile hypercalcemia: from genotype to phenotype. Clin Chem Lab Med. 2019, 57, 1650–1667. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Kaufmann, M.; Weber, S.; Irwin, A.; Goos, C.; John, U.; Misselwitz, J.; Klaus, G.; Kuwertz-Bröking, E.; Fehrenbach, H.; et al. Mutations inCYP24A1and Idiopathic Infantile Hypercalcemia. New Engl. J. Med. 2011, 365, 410–421. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Ruminska, J.; Kaufmann, M.; Dursun, I.; Patti, M.; Kranz, B.; et al. Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol. 2016, 27, 604–614. [Google Scholar] [CrossRef]
- Gurevich, E.; Levi, S.; Borovitz, Y.; Alfandary, H.; Ganon, L.; Dinour, D.; et al. Childhood Hypercalciuric Hypercalcemia with Elevated Vitamin D and Suppressed Parathyroid Hormone: Long-Term Follow-Up. Front Pediatr. 2021, 9, 752312. [Google Scholar] [CrossRef]
- O’kEeffe, D.T.; Tebben, P.J.; Kumar, R.; Singh, R.J.; Wu, Y.; Wermers, R.A. Clinical and biochemical phenotypes of adults with monoallelic and biallelic CYP24A1 mutations: evidence of gene dose effect. Osteoporos. Int. 2016, 27, 3121–3125. [Google Scholar] [CrossRef]
- Kasuga, H.; Hosogane, N.; Matsuoka, K.; Mori, I.; Sakura, Y.; Shimakawa, K.; Shinki, T.; Suda, T.; Taketomi, S. Characterization of transgenic rats constitutively expressing the vitamin D-24-hydroxylase gene. Biochem. Biophys. Res. Commun. 2002, 297, 1332–1338. [Google Scholar] [CrossRef]
- Prabhu, A.V.; Luu, W.; Sharpe, L.J.; Brown, A.J. Cholesterol-mediated Degradation of 7-Dehydrocholesterol Reductase Switches the Balance from Cholesterol to Vitamin D Synthesis. J. Biol. Chem. 2016, 291, 8363–8373. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Porter, T.D. Rapid suppression of 7-dehydrocholesterol reductase activity in keratinocytes by vitamin D. J. Steroid Biochem. Mol. Biol. 2015, 148, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Campese, V.M. Calcium, Parathyroid Hormone, and Blood Pressure. Am. J. Hypertens. 1989, 2, 34S–44S. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-H.; Huang, C.-C.; Chang, Y.-Y.; Chen, Y.-F.; Chen, W.H.; Lai, S.-L. Vasoconstriction is the etiology of hypercalcemia-induced seizures. Epilepsia 2004, 45, 551–554. [Google Scholar] [CrossRef]
- Naganuma, T.; Takemoto, Y.; Uchida, J.; Nakatani, T.; Kabata, D.; Shintani, A. Hypercalcemia Is a Risk Factor for the Progression of Aortic Calcification in Kidney Transplant Recipients. Kidney Blood Press. Res. 2019, 44, 823–834. [Google Scholar] [CrossRef]
- Shen, H.; Bielak, L.F.; Ferguson, J.F.; Streeten, E.A.; Yerges-Armstrong, L.M.; Liu, J.; Post, W.; O'COnnell, J.R.; Hixson, J.E.; Kardia, S.L.; et al. Association of the Vitamin D Metabolism Gene CYP24A1 With Coronary Artery Calcification. Arter. Thromb. Vasc. Biol. 2010, 30, 2648–2654. [Google Scholar] [CrossRef]
- Qian, P.; Cao, X.; Xu, X.; Duan, M.; Zhang, Q.; Huang, G. Contribution of CYP24A1 variants in coronary heart disease among the Chinese population. Lipids Heal. Dis. 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Detrano, R.; Guerci, A.D.; Carr, J.J.; Bild, D.E.; Burke, G.L.; Folsom, A.R.; Liu, K.; Shea, S.; Szklo, M.; Bluemke, D.A.; et al. Coronary Calcium as a Predictor of Coronary Events in Four Racial or Ethnic Groups. N. Engl. J. Med. 2008, 358, 1336–1345. [Google Scholar] [CrossRef]
- Carbone, F.; Liberale, L.; Libby, P.; Montecucco, F. Vitamin D in atherosclerosis and cardiovascular events. Eur. Hear. J. 2023, 44, 2078–2094. [Google Scholar] [CrossRef]
- Jono, S.; Nishizawa, Y.; Shioi, A.; Morii, H. 1,25-Dihydroxyvitamin D3 increases in vitro vascular calcification by modulating secretion of endogenous parathyroid hormone-related peptide. Circulation 1998, 98, 1302–1306. [Google Scholar] [CrossRef]
- Shioi, A.; Nishizawa, Y.; Jono, S.; Koyama, H.; Hosoi, M.; Morii, H. β-Glycerophosphate Accelerates Calcification in Cultured Bovine Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 1995, 15, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; Nishizawa, Y.; Shioi, A.; Morii, H. Parathyroid hormone-related peptide as a local regulator of vascular calcification: its inhibitory action on in vitro calcification by bovine vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 1997, 17, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Kawashima, H. 1,25-dihydroxyvitamin D3 stimulates 45Ca2+-uptake by cultured vascular smooth muscle cells derived from rat aorta. Biochem. Biophys. Res. Commun. 1988, 152, 1388–1394. [Google Scholar] [CrossRef]
- Carthy, E.P.; Yamashita, W.; Hsu, A.; Ooi, B.S. 1,25-Dihydroxyvitamin D3 and rat vascular smooth muscle cell growth. Hypertension 1989, 13, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, G.S.; Morrison, L.M.; Ershoff, B.H. Induction of Aortic and Coronary Athero-arteriosclerosis in Rats Fed a Hypervitaminosis D, Cholesterol-Containing Diet. Exp. Biol. Med. 1971, 138, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Hines, T.G.; Jacobson, N.L.; Beitz, D.C.; Littledike, E.T. Dietary calcium and vitamin D: risk factors in the development of atherosclerosis in young goats. J. Nutr. 1985, 115, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Niederhoffer, N.; Bobryshev, Y.V.; Lartaud-Idjouadiene, I.; Giummelly, P.; Atkinson, J. Aortic Calcification Produced by Vitamin D3 plus Nicotine. J. Vasc. Res. 1997, 34, 386–398. [Google Scholar] [CrossRef]
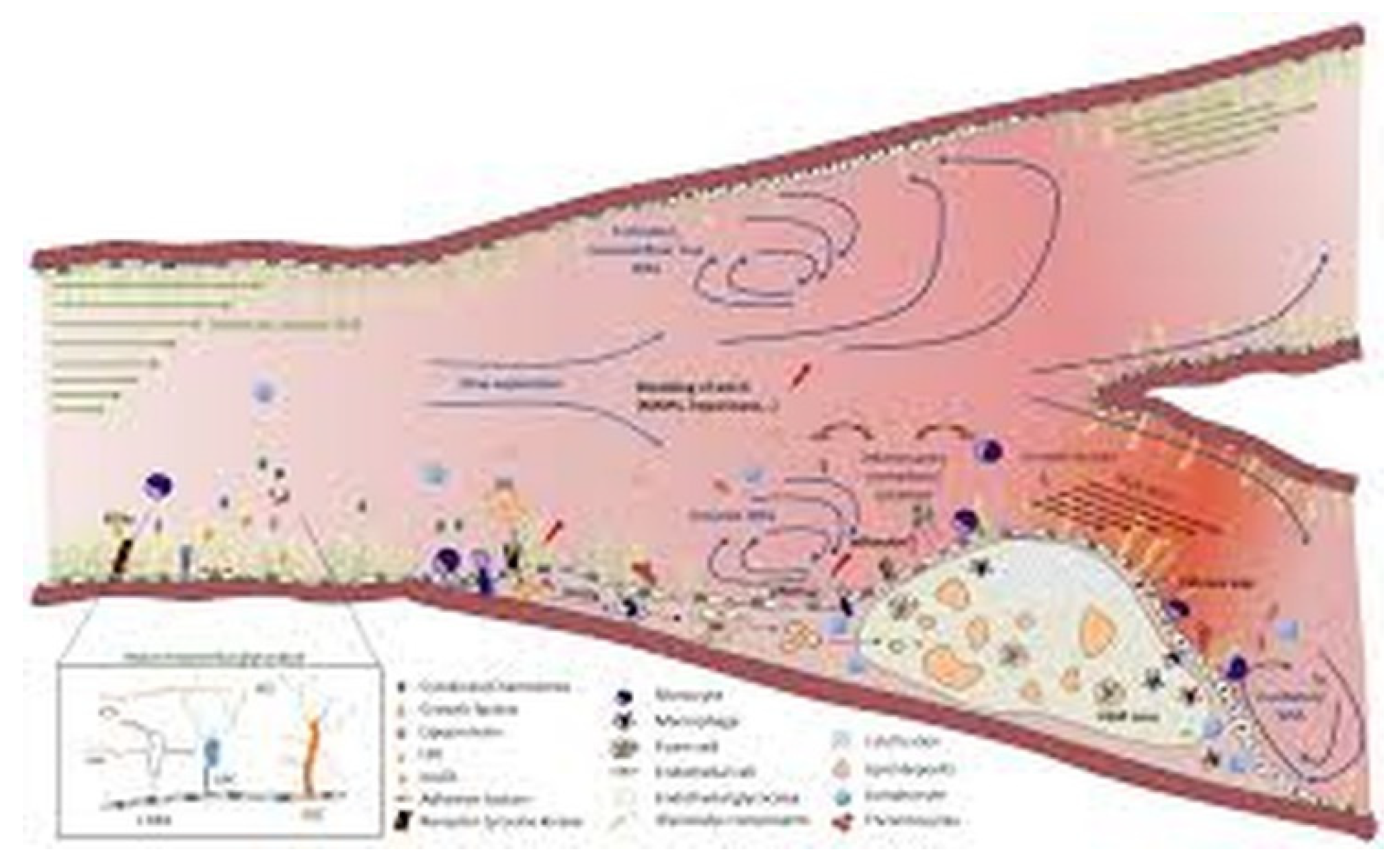
Figure 1.
Hemodynamic Forces and Atherogenesis. Moderate laminar wall shear stress supports vascular homeostasis, whereas low or oscillatory laminar wall shear stress promotes endothelial dysfunction, inflammation, lipid infiltration, and the formation of atherosclerotic plaques. Turbulent flow regions are especially prone to lesion development. Adapted from Int. J. Mol. Sci. 2021, 22(11), 5635. https://doi.org/10.3390/ijms22115635 (Open Access, CC BY).
Figure 1.
Hemodynamic Forces and Atherogenesis. Moderate laminar wall shear stress supports vascular homeostasis, whereas low or oscillatory laminar wall shear stress promotes endothelial dysfunction, inflammation, lipid infiltration, and the formation of atherosclerotic plaques. Turbulent flow regions are especially prone to lesion development. Adapted from Int. J. Mol. Sci. 2021, 22(11), 5635. https://doi.org/10.3390/ijms22115635 (Open Access, CC BY).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



