Submitted:
27 July 2025
Posted:
28 July 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The aftermath of COVID-19, caused by the SARS-CoV-2 virus, has brought to light unexpected com-plications beyond the lungs including its connection to chronic kidney disease (CKD) and potentially, kidney stone disease (KSD). Kidney stones, classified as radiopaque or radiolucent, differ not only in composition but also in their biological origins. Radiopaque stones, which include calcium phosphate and oxalate types, are visible on X-rays, while radiolucent stones, such as uric acid and cystine, are not. Their formation is heavily influenced by urine pH: alkaline urine (pH ≥ 7) favors radiopaque stones, while acidic urine (pH ≤ 5.3) promotes radiolucent types. Because these stones stem from distinct molecular mechanisms, hence the “one size fits all” treatment approach has proven largely ineffective. Among the different categories of stones ranging from calcium-based to sulfur-containing types like those found in cystinuria or homocystinuria, the interplay of metabolic and epigenetic factors has been gaining attention. It appears that SARS-CoV-2 hijacks host methylation machinery, and by doing so leads to an increased production of homocysteine (Hcy), a non-protein amino acid that we believe is linked to stone formation. While cysteine can aid in calcium oxalate crystal aggregation, the elevated Hcy levels are often due to impaired mitochondrial sulfur metabolism (transsulfuration) that may be uniquely pathogenic in kidney stone development. Adding another layer of complexity, the COVID-19-associated gut dysbiosis, particularly involving Escherichia coli, can, in fact, de-grade citric acid (a key TCA cycle metabolite) into oxalic acid, thereby enhancing calcium oxalate stone formation. Moreover, SARS-CoV-2 induced succinylation of TCA cycle enzymes and disrupted mitochondrial bioenergetics further implicate defective transsulfuration as a bridge between acute kidney injury (AKI) and chronic stone disease. In essence, we opine that the COVID-19 may catalyze a shift from AKI to KSD through the host cell epigenetic reprogramming events, metabolic dysregulation, and mitochondrial sulfur pathway impairment revealing a novel link between viral infection, mitochondrial bioenergetics, and kidney stone pathogenesis.
Keywords:
spike protein
; acute kidney injury
; homocysteine
; trans-sulfuration pathway
; epigenetics
; kidney stone disease
1. Introduction
The outbreak of SARS-CoV-2, the etiological agent of COVID-19, has profoundly altered global health dynamics, leaving behind a persistent trail of long-term complications, collectively referred to as post COVID sequelae [1,2]. While the acute manifestations of Covid-19 have been widely studied, it is becoming increasingly evident that the virus’s impact on organ systems persists well beyond initial recovery, contributing to diverse chronic conditions [3,4,5,6,7]. Recent clinical reports and retrospective cohort studies have raised concerns about a potential rise in renal complications, including an unexpected uptick in kidney stone presentations among post-COVID patients [8,9,10]. Although large-scale population-based studies are still emerging, preliminary data suggest an increased incidence of urolithiasis in individuals recovering from SARS-CoV-2 infection, particularly those who experienced renal involvement during the acute phase [8]. This underscores the urgent need to investigate post-viral renal sequelae beyond the immediate window of AKI5, [11,12,13]. It is noteworthy that dietary supplementation during the COVID-19 pandemic in the form of vitamin C (ascorbic acid) gained attention as a possible treatment or preventive measure, and it is known that vitamin C can lead to elevated levels of urinary oxalate that increase the risk of hyperoxaluria and the formation of oxalate kidney stones [14,15,16].
It is important to mention that the trans-sulfuration pathway is a metabolic route that converts the non-proteinogenic amino acid Hcy into cysteine through a series of enzymatic steps, primarily involving the enzymes cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) [17]. This pathway plays a crucial role in regulating Hcy levels and producing cysteine, which is essential for synthesizing glutathione, a major antioxidant in the body [18,19,20,21]. In the kidneys, the proper function of the trans-sulfuration pathway supports redox balance, vascular tone, and cellular detoxification processes [22]. Disruption of this pathway leads to elevated Hcy, oxidative stress, and impaired nitric oxide (NO) signaling, all of which have been linked to endothelial dysfunction and renal injury [23,24]. In the context of KSD, such metabolic disturbances can create an environment that favors crystal formation and stone development [15,16,25].
Current therapeutic interventions focus primarily on immune modulation and RNA-based antiviral strategies, yet these approaches offer limited protection against long-term metabolic, vascular, and renal complications. Among the most concerning consequences is the link between Covid-19 and the progression of chronic kidney disease (CKD), including the development of KSD [26,27,28]. Despite growing clinical observations, the molecular mechanisms driving this transformation from AKI to chronic stone-forming nephropathy remain highly elusive. Our review proposes a novel, mitochondria-centered mechanism connecting Covid-19 infection to the onset of KSD [10]. We postulate that disruptions in mitochondrial sulfur metabolism, specifically within the trans-sulfuration pathway, play a key role in kidney stone pathogenesis, particularly in the context of viral infection and immune dysregulation. This pathway, essential for Hcy clearance and cysteine production, becomes compromised during Covid-19 due to viral interference with epigenetic and bioenergetic cellular machinery [28,29]. Elevated Hcy levels, a condition known as hyperhomocysteinemia (HHcy), may not only exacerbate systemic inflammation but can also contribute directly to stone formation by promoting the crystallization of sulfur-containing amino acids in the renal system.
Moreover, Covid-19–induced metabolic reprogramming characterized by enzyme succinylation, mitochondrial dysfunction, and microbial dysbiosis creates a powerful biochemical milieu that is favorable to nephrolithiasis [30,31]. For instance, gut microbiota such as Escherichia coli degrades tricarboxylic acid (TCA) intermediates into oxalate, thereby facilitating calcium oxalate stone formation [32,33]. Simultaneously, viral proteins and therapeutic antibodies have the potential to enhance immune activation and endothelial damage, thus setting the stage for micro-stone formation [8,10,15,16].
Mechanistically, viral entry into host cells is mediated by the spike (S) protein of SARS-CoV-2, which binds to angiotensin-converting enzyme 2 (ACE2) receptor [34,35] (Figure 1). This interaction is not merely a gateway for infection; it also initiates a cascade of cellular stress responses that can compromise mitochondrial function, redox balance, and renal epithelial integrity [36,37]. Hence, understanding the downstream effects of viral-host interactions is critical for designing mechanism-based therapeutic interventions that go beyond the acute phase and target the root causes of long-term renal complications, including kidney stone formation [10]. By integrating insights from virology, renal physiology, epigenetics, and mitochondrial biology, our review aims to chart a conceptual framework linking post-Covid metabolic disruptions to kidney stone disease. In doing so, we hope to pave the way for novel diagnostic markers and targeted therapies for patients at risk of long-term nephrological complications after Covid-19 infection. Unlike previous reviews that have focused either on viral-induced kidney injury or metabolic changes in isolation, this manuscript integrates epigenetic hijacking, sulfur metabolism impairment, and renal immune-metabolic crosstalk as a unified pathway from COVID-19 to kidney stone formation [5,10,15,16].
2. Epigenetics of COVID-19 Infection and KSD
2.1. Epigenetic Methylation Dysregulation and SARS-CoV-2
The epigenetic landscape plays a pivotal role in modulating host responses to SARS-CoV-2, particularly within renal tissues [38,39,40]. A central regulatory node involves DNA and histone methylation, which directly shapes gene expression, immune surveillance, and metabolic processing. Methionine adenosyl transferase (MAT) synthesizes S-adenosylmethionine (SAM), the principal methyl donor for DNA, RNA, protein, and histone methylation [41,42] . Viral infection disrupts this tightly regulated balance since SARS-CoV-2 encodes its own methyltransferase (MT) that hijacks the host methylation machinery, skewing normal methylation rhythms and leading to elevated production of Hcy as a toxic byproduct [43,44]. Impaired SAM availability altered DNA methyltransferase (DNMT) activity, and dysregulation of TET enzymes can collectively modify chromatin accessibility and inflammatory gene expression. Importantly, methylation changes such as increased H3K4me3 and RNA N6-methyladenosine (m⁶A) marks have been implicated in sustaining inflammatory loops and oxidative stress, both of which can promote tubular damage, fibrosis, and a stone-promoting milieu [15,16,42].
2.2. Hcy and Sulfur Metabolism: Implications for KSD
While cysteine promotes calcium oxalate (CaOx) crystal formation [45], and HHcy, that are common in severe or post-COVID patients, may exacerbate renal lithogenesis through distinct sulfur-mediated mechanisms [42]. HHcy arises from disrupted methylation cycles and impaired mitochondrial transsulfuration, leading to intracellular Hcy accumulation [43,44]. Although the precise role of Hcy in crystallization remains poorly defined, its chemical reactivity may foster calcium-phosphate and sulfur-based crystal formation, particularly in inflamed or hypoxic renal microenvironments. Hcy may also induce renal epithelial injury via oxidative stress, NO depletion, and endothelial dysfunction, each contributing to crystal adhesion and fibrotic remodeling [46]. These hypotheses underscore the need for targeted experimental studies evaluating Hcy’s contribution to crystal nucleation and retention in COVID-19 affected kidneys.
2.3. Immune-Mediated Endothelial Injury and Fibrosis
Following SARS-CoV-2 entry through ACE2 and TMPRSS2, ACE2 downregulation alters the renin–angiotensin system (RAS), increasing Ang II levels while depleting protective Ang-(1–7) peptides [47,48,49]. This imbalance contributes to systemic hypertension, endothelial stress, and AKI, which may transition to CKD and predispose to KSD [50,51]. COVID–19–induced coagulopathy, despite normal platelet counts, reflects microvascular injury rather than classical thrombosis [41,52]. A notable factor is reduced a Distntegrin and Metalloproteinase with a Thrombospondin Type 1 motif, member 13 (ADAMTS13) activity, a metalloproteinase that regulates von Willebrand factor and vascular homeostasis [53,54,55]. Its suppression promotes endothelial dysfunction, vascular stiffness, and ECM fragmentation, laying the groundwork for glomerular injury, proteinuria, and tubulointerstitial fibrosis. Our prior studies in hACE2 mice intranasally challenged with SARS-CoV-2 SP revealed upregulation of pro-inflammatory M1 macrophages and activation of proteolytic enzymes such as TMPRSS2, neutrophil gelatinase-associated lipocalin (NGAL), ADAMTS13, and matrix metalloproteinases (MMPs) [52,56,57]. These proteases participate in ECM remodeling and fibrosis, while Neopterin (NPT), a downstream product of M1 macrophage activity, depletes tetrahydrobiopterin (BH4), reducing endothelial nitric oxide synthase (eNOS) activity and vasodilation capacity. Subsequent peroxynitrite (ONOO⁻) formation drives further oxidative damage, metalloproteinase activation, and endothelial barrier dysfunction. Collectively, these pathways disrupt glomerular integrity and promote urinary stasis and lithogenesis [15,16].
2.4. Dysfunction and miRNA-Mediated Reprogramming
Mitochondrial dysfunction is a hallmark of COVID-19 and is linked to renal injury and fibrotic progression. Elevated levels of NGAL and FGF23, both implicated in tubular injury and vascular calcification, are observed in post COVID syndromes [58]. These changes are potentiated by neutrophil extracellular traps (NETs), which amplify renal damage via glycocalyx shedding and capillary rarefaction [59,60]. Single-cell transcriptomics confirm activation of ADAM17, a sheddase associated with focal segmental glomerulosclerosis. Male patients often show higher expression of TMPRSS2 and ADAM17, correlating with worse outcomes [61,62]. Hence, inhibitors targeting these proteases may hold therapeutic value. Furthermore, solute carriers such as SLC22A17, which complex with NGAL, regulate iron, cadmium, and zinc transport and are implicated in nephrotoxicity, osmotic stress adaptation, and renal inflammation [11,63]. Activation of MMP8 and release of microparticles during COVID-19 further damage the perivascular niche and glomerular architecture [64].
2.5. From Biomarkers to Therapeutics: Neopterin and iNOS
We propose a unified mechanistic model in which the SP primes ACE2/TMPRSS2 and activates M1 macrophages, driving iNOS upregulation, BH4 depletion, and peroxynitrite formation. These changes lead to endothelial dysfunction and trigger cascades involving NETs, NGAL, FGF23, MMPs, and ADAMTS proteases, the key mediators of glomerular leakage, vascular stiffness, and stone-promoting fibrosis [7]. The iNOS inhibition or genetic knockout (iNOSKO) significantly reduces these outcomes in experimental models, reinforcing the translational potential of anti-inflammatory therapeutics. In this context, neopterin evolves from a mere biomarker to a mechanistic driver of renal damage, suggesting its potential as a therapeutic target (Table 1).
2.6. Epigenetics, Viral Persistence, and Long-Term Risk of KSD
Beyond acute infection, SP persistence and chronic cytokine release sustain low-grade inflammation, mirroring features of autoimmune and fibrotic disease [65,66,67,68,69]. Epigenetic dysregulation continues post COVID, including histone methylation changes, miRNA activation (e.g., miR-21), and mitochondrial miRNA-2392 mediated suppression of oxidative phosphorylation [70,71]. These modifications reprogram host metabolism toward glycolysis and hypoxia, the conditions that impair renal repair and may favor crystal formation and retention [15,16].
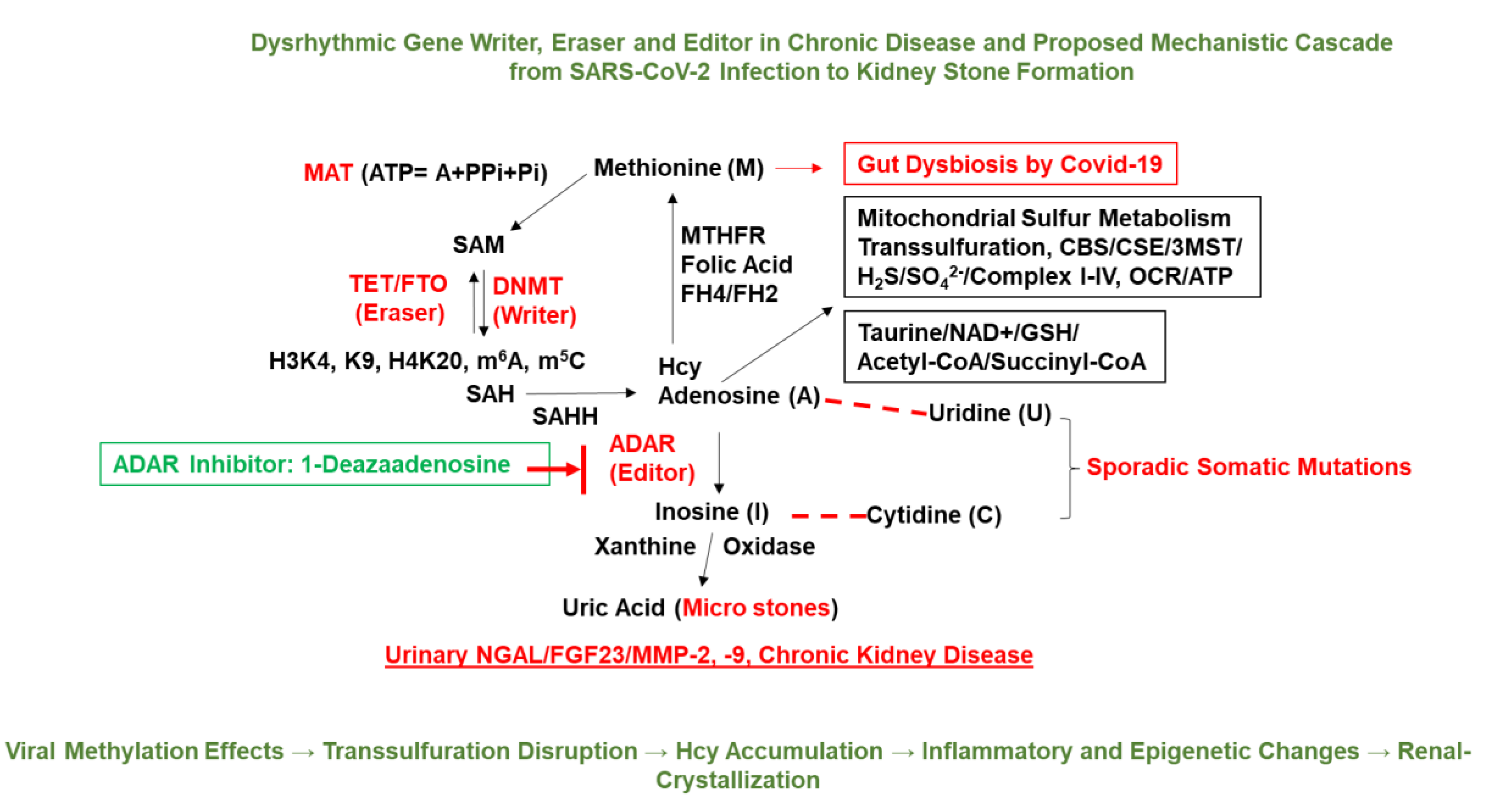
Figure 2.
Gut dysbiosis–induced epigenetic and metabolic reprogramming linking COVID-19 to renal pathology. The schematic illustrates the hypothesized cascade by which SARS-CoV-2 associated gut dysbiosis contributes to KSD and chronic kidney injury. SARS-CoV-2 infection disrupts the gut microbiota, impairing folate-mediated one-carbon metabolism and mitochondrial sulfur transsulfuration pathways. These changes lead to altered levels of key metabolites such as Hcy, SAM, and glutathione (GSH), which in turn influence the activity of epigenetic regulators: gene “writers” (e.g., DNMTs), “erasers” (e.g., TET demethylases, FTO), and “editors” (e.g., ADAR enzymes). Dysregulated epigenetic modifications such as aberrant histone methylation (H3K4me3, H3K9me3, H4K20me3) and RNA methylation (m⁶A, m⁵C) affect nuclear-mitochondrial crosstalk and downregulate mitochondrial oxidative phosphorylation components (Complexes I–IV), reducing oxygen consumption rate (OCR) and ATP production. This bioenergetic decline, coupled with increased oxidative stress and inflammation, contributes to renal tubular remodeling, crystal retention, and formation of uric acid microstones. The figure also highlights potential diagnostic biomarkers associated with this pathway such as NGAL, fibroblast growth factor 23 (FGF23), and MMP-2/-9 which are elaborated in Table 1. Together, this integrative model links gut dysbiosis, epigenetic reprogramming, and metabolic derangement to post COVID kidney stone disease [15,16].
Figure 2.
Gut dysbiosis–induced epigenetic and metabolic reprogramming linking COVID-19 to renal pathology. The schematic illustrates the hypothesized cascade by which SARS-CoV-2 associated gut dysbiosis contributes to KSD and chronic kidney injury. SARS-CoV-2 infection disrupts the gut microbiota, impairing folate-mediated one-carbon metabolism and mitochondrial sulfur transsulfuration pathways. These changes lead to altered levels of key metabolites such as Hcy, SAM, and glutathione (GSH), which in turn influence the activity of epigenetic regulators: gene “writers” (e.g., DNMTs), “erasers” (e.g., TET demethylases, FTO), and “editors” (e.g., ADAR enzymes). Dysregulated epigenetic modifications such as aberrant histone methylation (H3K4me3, H3K9me3, H4K20me3) and RNA methylation (m⁶A, m⁵C) affect nuclear-mitochondrial crosstalk and downregulate mitochondrial oxidative phosphorylation components (Complexes I–IV), reducing oxygen consumption rate (OCR) and ATP production. This bioenergetic decline, coupled with increased oxidative stress and inflammation, contributes to renal tubular remodeling, crystal retention, and formation of uric acid microstones. The figure also highlights potential diagnostic biomarkers associated with this pathway such as NGAL, fibroblast growth factor 23 (FGF23), and MMP-2/-9 which are elaborated in Table 1. Together, this integrative model links gut dysbiosis, epigenetic reprogramming, and metabolic derangement to post COVID kidney stone disease [15,16].
3. Kidney Stones
Kidney stones, or renal calculi, are mineralized deposits formed in the renal tubules and collecting system due to supersaturation of certain solutes in urine. They are broadly classified into two primary types based on their radiographic properties: radiopaque and radiolucent. Among the radiopaque stones, calcium-based calculi, particularly calcium oxalate and calcium phosphate stones, represent the most prevalent forms. In contrast, uric acid-containing calculi are the most common radiolucent stones [72]. The urinary pH plays a pivotal role in stone formation. Alkaline urine (pH ≥ 7.0) is associated with the precipitation of calcium phosphate and struvite stones, while acidic urine (pH ≤ 5.3) promotes the crystallization of uric acid and cystine stones [72,73]. Importantly, the biochemical and molecular pathogenesis of different stone types is distinct, involving diverse pathways of mineral metabolism, microbial dysbiosis, and epithelial dysfunction. A particularly compelling mechanism is the microbial degradation of the TCA cycle, which has implications in kidney stone pathophysiology. For instance, a pathogenic strain of Escherichia coli has been shown to degrade citrate (a TCA intermediate with anti-lithogenic properties) into dicarboxylic acids such as oxalate [73]. Oxalate is a key constituent of calcium oxalate stones, and such microbial-driven conversion from TCA to DCA intermediates provides a plausible mechanistic link between gut dysbiosis and lithogenesis. However, whether gut dysbiosis post COVID-19 infection plays a causative role in kidney stone formation remains to be systematically studied. Although these findings are compelling, direct clinical evidence linking E. coli mediated citrate degradation to increased urinary oxalate in post COVID patients is still limited. Most existing data are derived from in vitro microbial metabolism studies or animal models. Thus, the hypothesis that E. coli may contribute to post-infectious lithogenesis via citrate catabolism into oxalate requires further validation. This may be especially relevant in post COVID-19 individuals, where microbial dysbiosis and altered host-microbe metabolic interactions have been increasingly reported. Prospective studies examining urinary citrate and oxalate levels in relation to specific bacterial colonization patterns will be necessary to substantiate this mechanistic pathway.
Of additional concern is the observed increase in HHcy in patients suffering from severe COVID-19 and long COVID-19 symptoms, including pneumonia and renal complications [42,74,75]. Hcy is a sulfur-containing amino acid that, when elevated, exerts pro-inflammatory and pro-oxidative effects that may disrupt renal epithelial integrity, facilitate tubular injury, and increase crystalluria. Within the mitochondria, the transsulfuration pathway converts Hcy to hydrogen sulfide (H₂S), a gasotransmitter with protective vasodilatory and antioxidant effects. The 3-mercaptopyruvate sulfurtransferase (3MST), a mitochondrial enzyme, catalyzes this reaction, and its activity is vital for maintaining endothelial function and mitochondrial redox balance in renal tissues. The impairment of mitochondrial function has emerged as a central feature in post COVID-19 sequelae, including kidney disease and potentially kidney stone formation. Mitochondrial dysfunction, especially due to disrupted bioenergetics and altered post-translational modifications of key enzymes, is implicated in altered cellular homeostasis in the renal microenvironment. A notable modification is lysine succinylation, which occurs robustly on TCA cycle enzymes during COVID-19 infection [30]. This modification impairs enzymatic function and disrupts energy production and redox signaling. The mitochondrial desuccinylase SIRT5, which plays a key role in reversing lysine succinylation, is emerging as a critical target for restoring mitochondrial efficiency in the post-viral state [30]. Given the high energy demand and oxidative stress burden in renal epithelial cells, disrupted mitochondrial bioenergetics could create a milieu conducive to stone formation through cellular injury, impaired ion transport, and altered solute handling. Another potentially therapeutic strategy involves pyruvate supplementation, which enhances TCA cycle flux, mitigates oxidative stress, and suppresses viral replication [76]. In the context of COVID-19-induced mitochondrial dysfunction, pyruvate may also restore epithelial barrier integrity and reduce the risk of stone formation by normalizing energy metabolism in the renal tubular epithelium. In this context, KSD can no longer be viewed solely as a local renal pathology but rather as a systemic condition involving epigenetic reprogramming, microbial interactions, and mitochondrial bioenergetics, especially following COVID-19. It is therefore imperative to consider a multi-dimensional therapeutic approach, including restoring mitochondrial function, targeting HHcy through epigenetic and nutritional strategies, and exploring the role of microbiome modulation in the prevention of KSD post COVID-19.
4. Underlying Mechanisms Linking COVID-19 Infection to Kidney Stone Formation
The emergence of KSD following COVID-19 infection appears to involve a multifactorial pathophysiology, encompassing mitochondrial dysfunction, epigenetic alterations, inflammatory cascades, vascular and tubular injury, and disruptions in metabolic homeostasis, particularly within the sulfur amino acid pathway. Central to these processes is mitochondrial sulfur metabolism, which is regulated by enzymes such as 3MST. This enzyme integrates the transsulfuration pathway with the TCA cycle, epigenetic regulation, and redox balance [77,78].
4.1. Mitochondrial Sulfur Metabolism and Hcy Accumulation
The 3MST is involved in Hcy detoxification and H₂S generation, a gasotransmitter with anti-inflammatory, antioxidant, and vasodilatory properties in renal tissues [79]. In the context of COVID-19, mitochondrial stress and inflammation can impair 3MST activity, leading to elevated Hcy levels and diminished H₂S synthesis [80]. This state of HHcy is associated with increased oxidative stress, endothelial dysfunction, and thrombotic risk, all of which can contribute to renal microvascular injury and tubulointerstitial fibrosis [81,82]. Additionally, Hcy may crystallize in acidic urine, forming Hcy stones; however, this process requires further investigation in human studies. While Hcy accumulation is associated with renal injury, direct crystallization of Hcy in vivo has not yet been confirmed and warrants further investigation.
4.2. Transsulfuration Pathway Disruption and Epigenetic Consequences
The transsulfuration pathway comprising CBS, CSE, and 3MST is critical for converting methionine to cysteine and subsequently to H₂S. SARS-CoV-2 infection disrupts this pathway through mechanisms such as increased methylation demand from viral replication, which depletes SAM, alters methylation capacity, and affects gene regulation. Furthermore, dysfunction of sirtuins (e.g., SIRT5 and SIRT3) interferes with histone succinylation and acetylation, affecting mitochondrial enzyme activity and gene expression linked to renal tubular integrity [83]. These epigenetic shifts may promote fibrotic, inflammatory, and pro-thrombotic gene expression, potentially driving the progression from AKI to CKD [84].
4.3. COVID-19-Induced Mitochondrial Dysfunction
SARS-CoV-2 infection has been linked to mitochondrial alterations, including reduced oxidative phosphorylation, altered mitochondrial DNA expression (e.g., downregulation via miR-2392), increased reactive oxygen species (ROS), and mitochondrial membrane depolarization [36]. Reduced NAD⁺ levels also compromise sirtuin-dependent metabolic regulation. These changes impair TCA cycle activity, promote aerobic glycolysis, and support a pro-fibrotic and inflammatory renal microenvironment, especially in proximal tubular epithelial cells, which are central to solute regulation [85]. Such dysfunction may promote urinary supersaturation of calcium, oxalate, uric acid, and phosphate, favoring stone formation [14,15,16,86].
4.4. Oxidative and Nitrosative Stress
Excess ROS and peroxynitrite (ONOO⁻), generated in part through inducible nitric oxide synthase (iNOS), contribute to tetrahydrobiopterin (BH₄) depletion. This results in uncoupling of endothelial nitric oxide synthase (eNOS) and further exacerbates endothelial injury and renal ischemia. ROS and ONOO⁻ also activate MMPs, ADAMTS13, and NGAL, collectively leading to glycocalyx degradation, glomerular permeability, and renal fibrosis. MMP-driven remodeling may also alter renal architecture and facilitate crystal nidus formation [15,16].
4.5. Immune Activation, Macrophages, and NETosis
COVID-19 is characterized by heightened immune responses, including a cytokine storm and M1 macrophage polarization, which promotes NPT production and BH₄ depletion. Neutrophils in this context release NETs, which entrap urinary crystals and cellular debris, initiating inflammation and promoting stone matrix development [87,88]. The interplay between activated macrophages, NGAL, and ADAMTS in the kidney’s microvasculature contributes to COVID-19-associated coagulopathy (CAC) and may favor calcification and stone nucleation, particularly in distal tubules and collecting ducts [52,88].
4.6. Tubular Transport Dysfunction and Osmotic Stress
SARS-CoV-2 infection disrupts solute carriers and ion transporters, such as SLC22A17, which remains in complex with NGAL and is involved in the transport of metal ions (Fe, Cd, Zn) and tubular endocytosis [89,90]. Impaired transporter function can lead to osmotic imbalance, tubular cell injury, and urinary concentration of lithogenic solutes [88].
4.7. Role of Gut Microbiome and Uremic Toxins
Alterations in the gut microbiota following COVID-19 may elevate systemic levels of uremic toxins and oxalate. Loss of oxalate-degrading bacteria like Oxalobacter formigenes may increase the risk of calcium oxalate stone formation [91,92]. Although the precise impact of SARS-CoV-2 on gut microbiome composition and its role in enteric hyperoxaluria remains under investigation, emerging evidence suggests a potential link [93,94].
4.8. Systemic Hypoxia and Dehydration
Patients with COVID-19 commonly experience dehydration, hypoxemia, and immobility, especially during hospitalization [95,96]. These conditions concentrate urine, reduce urinary citrate excretion, and impair renal perfusion, which are well-established risk factors for uric acid and other types of stones, particularly in acidic environments [45,88]. In summary, SARS-CoV-2 infection initiates a cascade of interrelated molecular and cellular events that may contribute to a pro-lithogenic renal milieu. Central to this process is impairment of mitochondrial sulfur metabolism, particularly 3MST-mediated conversion of Hcy to H₂S. Disruption of this pathway results in HHcy, oxidative stress, epigenetic dysregulation, and metabolic alterations. These changes collectively induce glomerular and tubular injury, immune activation, and urinary solute supersaturation, ultimately facilitating renal crystal formation. We believe that a deeper understanding of these mechanisms may offer novel therapeutic strategies, including iNOS inhibition to mitigate nitrosative stress, pyruvate supplementation to support mitochondrial function, BH4 restoration to improve endothelial integrity, use of epigenetic modulators, and microbiome-based interventions [97]. Given the widespread prevalence of COVID-19 and its potential long-term renal effects, systematic investigation of its role in stone pathogenesis remains an important area of inquiry for nephrology and public health.
5. Potential Limitations and Future Directions
The global COVID-19 pandemic has illuminated numerous short and long-term health complications associated with SARS-CoV-2 infection. While the respiratory manifestations of COVID-19 are well characterized, there is an urgent need to expand our understanding of the virus's systemic consequences, particularly its renal effects and potential contribution to KSD. Current research primarily addresses viral infection mechanisms and related immune responses; however, much less attention has been paid to non-infective sequelae, such as the impact of S protein induced intracellular signaling, gut dysbiosis, and renal axis remodeling. These events may play a critical role in the pathophysiology of KSD post COVID-19 and hence deserve rigorous investigation. Our hypothesis for pathogenic remodeling by the S protein of SARS-CoV-2, particularly its interaction with angiotensin converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2), that helps initiate the viral entry into the host cells. However, this binding cascade may also trigger a pro-inflammatory and pro-oxidative signaling network independent of direct viral replication. Studies suggest that S protein alone is sufficient to elicit endothelial dysfunction, oxidative stress, and pro-fibrotic responses [30,52]. Thus, the S protein may act as a viral toxin, activating the inducible nitric oxide synthase (iNOS) resulting in the overproduction of nitric oxide (NO) and peroxynitrite (ONOO⁻), NPT production through M1 macrophage polarization, NETs formation, NGAL release from injured tubular cells, activation of MMPs and ADAMTS family members, including ADAM17, which is implicated in both shedding of ACE2 and pro-inflammatory cytokine activation [52,98,99]. This molecular cascade promotes disruption of the glomerular and tubular glycocalyx, epithelial leakage, collagen deposition, glomerulosclerosis, and tubulointerstitial fibrosis, all potential precursors to potential renal lithogenesis process [100]. Thus, these mechanisms offer a biologically plausible explanation for the emergence of KSD following COVID-19 and hence merit targeted experimental validation.
Emerging evidence highlights the essential role of the gut-kidney axis in renal health. SARS-CoV-2 infection significantly alters the composition and function of the intestinal microbiota, often leading to gut dysbiosis. This microbial imbalance can lead to increased oxalate absorption due to depletion of Oxalobacter formigenes, elevated production of uremic toxins such as p-cresol sulfate and indoxyl sulfate, compromised short-chain fatty acid (SCFA) biosynthesis that affects systemic inflammation and renal epithelial integrity [72,73]. These alterations influence renal stone formation both directly by increasing urinary oxalate, and indirectly, by promoting systemic inflammation and oxidative stress. Furthermore, dysbiosis-driven endotoxemia may exacerbate the inflammatory state in COVID-19, amplifying renal injury via the Toll-like receptor (TLR)-NFκB pathway, thereby intensifying cytokine storm effects and contributing to stone nucleation in a sensitized renal environment [100]. We opine that targeting the M1-iNOS pathway to mitigate renal injury might prove to be a novel and testable hypothesis emerging from this work because a targeted blockade of M1 macrophage-induced iNOS activity as a therapeutic intervention to prevent COVID-19-induced renal injury and lithogenesis. Further, inhibiting iNOS could restore BH4 availability and reduce oxidative/nitrosative stress, suppress NPT production and NETs formation, reduce NGAL expression and ADAMTS activation, preserve glycocalyx architecture and reduce endothelial permeability, prevent maladaptive fibrosis and crystal deposition [52,100,101,102]. The link between NET formation and renal lithogenesis in post-viral states remains speculative but represents an important direction for future research. Experimental models using selective iNOS inhibitors or BH4 supplementation could elucidate these mechanisms. Additionally, RNAi knockdown of TMPRSS2 and ADAM17, particularly in sex-differentiated models, may also provide insight into gender disparities observed in COVID-19 severity and renal complications [30,52]. Indeed, androgen-mediated upregulation of TMPRSS2 in males may underlie the increased severity and mortality reported during the pandemic, making TMPRSS2 inhibition a viable sex-specific strategy. To address the hypotheses mentioned above and bridge knowledge gaps, we would like to propose the following research directions:
5.1. Animal Models
Use S protein or pseudo viruses to simulate non-replicative renal injury. Assess iNOS, NPT, NGAL, and NETs expression in kidneys. Evaluate the efficacy of iNOS inhibitors and pyruvate supplementation [76]. Also, pone can use transgenic mice with renal cell-specific 3MST knockout to examine sulfur metabolism's role in post-viral injury.
5.2. Epigenetic Analyses
Perform ChIP-seq and mass spectrometry to profile histone succinylation and acetylation changes in kidneys post-infection. Investigate how SIRT5 modulation affects fibrosis, inflammation, and mitochondrial metabolism [30].
5.3. Clinical Biomarker Studies
Measure plasma and urinary levels of Hcy, NGAL, NPT, NETs, BH4, and ADAMTS13 in post COVID-19 patients with and without kidney stones. Correlate these biomarkers with renal function, imaging studies, and stone composition (Table 1).
5.4. Gut Microbiome Sequencing
Conduct longitudinal microbiota analyses pre- and post COVID-19 in KSD-prone individuals. Investigate restoration strategies such as probiotics, fecal microbiota transplantation, or oxalate degrading bacterial therapies.
5.5. Therapeutic Trials
Pilot trials using pyruvate, BH4 analogs, or SIRT activators in long COVID patients with early signs of renal dysfunction. Examine the effect of androgen receptor antagonists or TMPRSS2 inhibitors on male patients with recurrent stones or long COVID nephropathy.
5.6. Sex-Differentiated Renal Analysis
Evaluate gene and protein expression levels of ACE2, TMPRSS2, ADAM17, and related pathways in male versus female renal tissues. Study the protective role of estrogens or selective estrogen receptor modulators (SERMs) in modulating post COVID-19 renal injury.
6. Conclusions
While this conceptual framework is biologically plausible, several limitations must be acknowledged. Causality between COVID-19 and KSD remains speculative and primarily based on associative data, but emerging epidemiological reports underscore a notable uptick in nephrological complications, including kidney stones, among pos -COVID populations. For example, recent studies suggest increased incidence of renal colic, AKI, and recurrent nephrolithiasis in long COVID cohorts, particularly in patients with predisposing metabolic syndromes or immune dysregulation [9,103,104]. Large-scale health system data analysis and insurance claims databases have also begun to report higher than expected rates of new onset renal calculi following COVID-19 infection, especially among middle-aged and elderly individuals [105]. However, these findings remain under-validated and warrant prospective studies to confirm [106]. The longitudinal course of lithogenesis post COVID-19 is poorly understood due to lack of imaging follow-ups or urine biochemistry in post-viral cohorts. Animal models may not fully recapitulate human renal tubular physiology or immune responses to SARS-CoV-2 proteins. Biomarker assays (e.g., NETs, BH₄) lack standardization across clinical laboratories, hence limiting reproducibility (Table 1). The spike protein's off-target toxicity in non-infective settings, while mechanistically compelling, remains under-characterized. Despite these limitations, the proposed hypotheses and research avenues offer a transformative approach to understanding the renal sequelae of COVID-19. Given the chronic and recurrent nature of KSD and the expanding global population of long COVID patients estimated at over 65 million individuals worldwide as of recent WHO estimates [107,108], thus there is an urgent need to recognize and study non-traditional post-viral complications such as nephrolithiasis. Early identification of metabolic, immunological, and epigenetic drivers may help prevent irreversible kidney damage and improve long-term outcomes.
COVID-19 has challenged our understanding of viral diseases as primarily infective processes. Its ability to remodel host metabolic pathways, trigger lasting immune dysregulation, and reprogram epigenetic machinery elevates it to a disease of systems-level dysregulation. This important review highlights a novel interface between mitochondrial sulfur metabolism, Hcy biology, epigenetic remodeling, and renal lithogenesis, presenting a compelling case for broader investigations into non-traditional sequelae such as KSD. We propose that the next decade of nephrology must embrace this paradigm shift, integrating insights from virology, immunology, metabolism, and epigenetics to decode and mitigate the lingering effects of COVID-19 in vulnerable human beings. Furthermore, large-scale population studies, integration of KSD into long COVID registries, and targeted biomarker profiling in post-COVID cohorts should be prioritized to validate and operationalize these mechanistic insights in clinical care, Table 1.
Author Contributions
A.B., M.S., U.S., M.T., and S.C.T. wrote the very first draft of the manuscript together. M.S., and S.C.T conceived the review and finalized the manuscript. M.S., and S.C.T. drafted the figures. A.B., and S.C.T. searched for the reference papers. S.C.T., and U.S. acquired the funding. All authors have read and agreed to the published version of the manuscript.
Funding
A part of this study was supported by NIH grants AR-71789; HL139047; and DK116591.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This manuscript is a review article, so none of the data were generated or analyzed. All data discussed were sourced from previously published studies, which have been appropriately cited in the text.
Acknowledgments
During the preparation of our review manuscript, we used the premium version of Grammarly software to access features such as grammar and spell-check, as well as style suggestions, to improve clarity and language smoothness. All the authors reviewed and edited the final version of the manuscript and take full responsibility for the content of this publication.
Conflicts of Interest
No conflicts of interest; financial or otherwise; are declared by the author.
References
- Martin de Francisco Á, Fernández Fresnedo G: Long COVID-19 renal disease: A present medical need for nephrology. Nefrologia (Engl Ed) 2023, 43, 1–5. [CrossRef] [PubMed]
- Schiffl H, Lang SM: Long-term interplay between COVID-19 and chronic kidney disease. Int Urol Nephrol 2023, 55, 1977–1984. [CrossRef] [PubMed]
- Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY, Yi F, Yang HC, Fogo AB, Nie X, Zhang C: Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int 2020, 98, 219–227. [CrossRef]
- Kissling S, Rotman S, Gerber C, Halfon M, Lamoth F, Comte D, Lhopitallier L, Sadallah S, Fakhouri F: Collapsing glomerulopathy in a COVID-19 patient. Kidney Int 2020, 98, 228–231. [CrossRef]
- Caceres PS, Savickas G, Murray SL, Umanath K, Uduman J, Yee J, Liao TD, Bolin S, Levin AM, Khan MN, Sarkar S, Fitzgerald J, Maskey D, Ormsby AH, Sharma Y, Ortiz PA: High SARS-CoV-2 Viral Load in Urine Sediment Correlates with Acute Kidney Injury and Poor COVID-19 Outcome. J Am Soc Nephrol 2021, 32, 2517–2528.
- Tyagi SC, Singh M: Multi-organ damage by covid-19: congestive (cardio-pulmonary) heart failure, and blood-heart barrier leakage. Mol Cell Biochem 2021, 476, 1891–1895. [CrossRef]
- Homme RP, George AK, Singh M, Smolenkova I, Zheng Y, Pushpakumar S, Tyagi SC: Mechanism of Blood-Heart-Barrier Leakage: Implications for COVID-19 Induced Cardiovascular Injury. Int J Mol Sci 2021, 22.
- Gul M, Kaynar M, Yildiz M, Batur AF, Akand M, Kilic O, Goktas S: The Increased Risk of Complicated Ureteral Stones in the Era of COVID-19 Pandemic. J Endourol 2020, 34, 882–886. [CrossRef]
- Spooner J, Masoumi-Ravandi K, MacNevin W, Ilie G, Skinner T, Powers AL: Septic and febrile kidney stone presentations during the COVID-19 pandemic What is the effect of reduced access to care during pandemic restrictions? Can Urol Assoc J 2024, 18, E19–e25.
- Kasiri H, Moradimajd P, Samaee H, Ghazaeian M: The Increased Risk of Renal Stones in Patients With COVID-19 Infection. Pharmaceutical and Biomedical Research 2022, 8, 333–340. [CrossRef]
- Carollo C, Benfante A, Sorce A, Montalbano K, Cirafici E, Calandra L, Geraci G, Mulè G, Scichilone N: Predictive Biomarkers of Acute Kidney Injury in COVID-19: Distinct Inflammatory Pathways in Patients with and Without Pre-Existing Chronic Kidney Disease. Life (Basel) 2025, 15.
- Zhang W, Liu L, Xiao X, Zhou H, Peng Z, Wang W, Huang L, Xie Y, Xu H, Tao L, Nie W, Yuan X, Liu F, Yuan Q: Identification of common molecular signatures of SARS-CoV-2 infection and its influence on acute kidney injury and chronic kidney disease. Frontiers in immunology 2023, 14, 961642. [CrossRef]
- Marques F, Gameiro J, Oliveira J, Fonseca JA, Duarte I, Bernardo J, Branco C, Costa C, Carreiro C, Braz S, Lopes JA: Acute Kidney Disease and Mortality in Acute Kidney Injury Patients with COVID-19. J Clin Med 2021, 10.
- Kemble JP, Liaw CW, Alamiri JM, Ungerer GN, Potretzke AM, Koo K: Public Interest in Vitamin C Supplementation During the COVID-19 Pandemic as a Potential Risk for Oxalate Nephrolithiasis. Cureus 2025, 17, e79452.
- Abhishek A, Benita S, Kumari M, Ganesan D, Paul E, Sasikumar P, Mahesh A, Yuvaraj S, Ramprasath T, Selvam GS: Molecular analysis of oxalate-induced endoplasmic reticulum stress mediated apoptosis in the pathogenesis of kidney stone disease. Journal of physiology and biochemistry 2017, 73, 561–573. [CrossRef] [PubMed]
- Kaur M, Varanasi R, Nayak D, Tandon S, Agrawal V, Tandon C: Molecular insights into cell signaling pathways in kidney stone formation. Urolithiasis 2025, 53, 30. [CrossRef]
- Sbodio JI, Snyder SH, Paul BD: Regulators of the transsulfuration pathway. Br J Pharmacol 2019, 176, 583–593. [CrossRef]
- Zhu J, Berisa M, Schwörer S, Qin W, Cross JR, Thompson CB: Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab 2019, 30, 865–876.e5. [CrossRef] [PubMed]
- Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R: A functional transsulfuration pathway in the brain links to glutathione homeostasis. The Journal of biological chemistry 2006, 281, 35785–35793. [CrossRef]
- Mosharov E, Cranford MR, Banerjee R: The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 2000, 39, 13005–13011. [CrossRef] [PubMed]
- Perła-Kaján J, Jakubowski H: COVID-19 and One-Carbon Metabolism. International journal of molecular sciences 2022, 23.
- Scammahorn JJ, Nguyen ITN, Bos EM, Van Goor H, Joles JA: Fighting Oxidative Stress with Sulfur: Hydrogen Sulfide in the Renal and Cardiovascular Systems. Antioxidants (Basel) 2021, 10.
- Chen CJ, Cheng MC, Hsu CN, Tain YL: Sulfur-Containing Amino Acids, Hydrogen Sulfide, and Sulfur Compounds on Kidney Health and Disease. Metabolites 2023, 13.
- Kabil O, Banerjee R: Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal 2014, 20, 770–782. [CrossRef]
- Tavasoli S, Borumandnia N, Basiri A, Taheri M: Effects of COVID-19 pandemics on urinary metabolites in kidney stone patients: our kidney stone prevention clinic experience. Environ Health Prev Med 2021, 26, 112. [CrossRef]
- Chen IW, Chang LC, Ho CN, Wu JY, Tsai YW, Lin CM, Chang YJ, Hung KC: Association between COVID-19 and the development of chronic kidney disease in patients without initial acute kidney injury. Scientific reports 2025, 15, 10924.
- Lang SM, Schiffl H: Long-term renal consequences of COVID-19. Emerging evidence and unanswered questions. Emerging evidence and unanswered questions. Int Urol Nephrol 2025.
- du Preez HN, Lin J, Maguire GEM, Aldous C, Kruger HG: COVID-19 vaccine adverse events: Evaluating the pathophysiology with an emphasis on sulfur metabolism and endotheliopathy. Eur J Clin Invest 2024, 54, e14296. [CrossRef]
- du Preez HN, Aldous C, Hayden MR, Kruger HG, Lin J: Pathogenesis of COVID-19 described through the lens of an undersulfated and degraded epithelial and endothelial glycocalyx. Faseb j 2022, 36, e22052.
- Liu Q, Wang H, Zhang H, Sui L, Li L, Xu W, Du S, Hao P, Jiang Y, Chen J, Qu X, Tian M, Zhao Y, Guo X, Wang X, Song W, Song G, Wei Z, Hou Z, Wang G, Sun M, Li X, Lu H, Zhuang X, Jin N, Zhao Y, Li C, Liao M: The global succinylation of SARS-CoV-2-infected host cells reveals drug targets. Proc Natl Acad Sci U S A 2022, 119, e2123065119.
- Raj ST, Bruce AW, Anbalagan M, Srinivasan H, Chinnappan S, Rajagopal M, Khanna K, Chandramoorthy HC, Mani RR: COVID-19 influenced gut dysbiosis, post-acute sequelae, immune regulation, and therapeutic regimens. Front Cell Infect Microbiol 2024, 14, 1384939.
- An L, Wu W, Li S, Lai Y, Chen D, He Z, Chang Z, Xu P, Huang Y, Lei M, Jiang Z, Zeng T, Sun X, Sun X, Duan X, Wu W: Escherichia coli Aggravates Calcium Oxalate Stone Formation via PPK1/Flagellin-Mediated Renal Oxidative Injury and Inflammation. Oxid Med Cell Longev 2021, 2021, 9949697. [CrossRef]
- Li H, Xue X, Meng G, He C, Tong L, Lai Y: The roles of bacteria on urolithiasis progression and associated compounds. Biochem Pharmacol 2025, 237, 116958.
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X: Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [CrossRef]
- Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F: Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [CrossRef]
- Chen TH, Jeng TH, Lee MY, Wang HC, Tsai KF, Chou CK: Viral mitochondriopathy in COVID-19. Redox Biol 2025, 85, 103766.
- Diao B, Wang C, Wang R, Feng Z, Zhang J, Yang H, Tan Y, Wang H, Wang C, Liu L, Liu Y, Liu Y, Wang G, Yuan Z, Hou X, Ren L, Wu Y, Chen Y: Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat Commun 2021, 12, 2506. [CrossRef]
- Bhat S, Rishi P, Chadha VD: Understanding the epigenetic mechanisms in SARS CoV-2 infection and potential therapeutic approaches. Virus Res 2022, 318, 198853. [CrossRef] [PubMed]
- Rath S, Perikala V, Jena AB, Dandapat J: Factors regulating dynamics of angiotensin-converting enzyme-2 (ACE2), the gateway of SARS-CoV-2: Epigenetic modifications and therapeutic interventions by epidrugs. Biomed Pharmacother 2021, 143, 112095.
- Beacon TH, Delcuve GP, Davie JR: Epigenetic regulation of ACE2, the receptor of the SARS-CoV-2 virus(1). Genome 2021, 64, 386–399. [CrossRef]
- Singh M, Pushpakumar S, Bard N, Zheng Y, Homme RP, Mokshagundam SPL, Tyagi SC: Simulation of COVID-19 symptoms in a genetically engineered mouse model: implications for the long haulers. Mol Cell Biochem 2023, 478, 103–119. [CrossRef] [PubMed]
- Ponti G, Roli L, Oliva G, Manfredini M, Trenti T, Kaleci S, Iannella R, Balzano B, Coppola A, Fiorentino G, Ozben T, Paoli VD, Debbia D, De Santis E, Pecoraro V, Melegari A, Sansone MR, Lugara M, Tomasi A: Homocysteine (Hcy) assessment to predict outcomes of hospitalized Covid-19 patients: a multicenter study on 313 Covid-19 patients. Clin Chem Lab Med 2021, 59, e354–e7.
- Eslamifar Z, Behzadifard M, Zare E: Investigation of homocysteine, D-dimer and platelet count levels as potential predictors of thrombosis risk in COVID-19 patients. Mol Cell Biochem 2025, 480, 439–444. [CrossRef]
- Ponti G, Ruini C, Tomasi A: Homocysteine as a potential predictor of cardiovascular risk in patients with COVID-19. Med Hypotheses 2020, 143, 109859. [CrossRef]
- Martins MC, Meyers AA, Whalley NA, Rodgers AL: Cystine: a promoter of the growth and aggregation of calcium oxalate crystals in normal undiluted human urine. J Urol 2002, 167, 317–321. [CrossRef]
- Li S, Qiu B, Lu H, Lai Y, Liu J, Luo J, Zhu F, Hu Z, Zhou M, Tian J, Zhou Z, Yu S, Yi F, Nie J: Hyperhomocysteinemia Accelerates Acute Kidney Injury to Chronic Kidney Disease Progression by Downregulating Heme Oxygenase-1 Expression. Antioxid Redox Signal 2019, 30, 1635–1650. [CrossRef]
- Khezri MR, Ghasemnejad-Berenji M: Neurological effects of elevated levels of angiotensin II in COVID-19 patients. Hum Cell 2021, 34, 1941–1942. [CrossRef] [PubMed]
- Wang K, Gheblawi M, Nikhanj A, Munan M, MacIntyre E, O'Neil C, Poglitsch M, Colombo D, Del Nonno F, Kassiri Z, Sligl W, Oudit GY: Dysregulation of ACE (Angiotensin-Converting Enzyme)-2 and Renin-Angiotensin Peptides in SARS-CoV-2 Mediated Mortality and End-Organ Injuries. Hypertension 2022, 79, 365–378.
- Caputo I, Caroccia B, Frasson I, Poggio E, Zamberlan S, Morpurgo M, Seccia TM, Calì T, Brini M, Richter SN, Rossi GP: Angiotensin II Promotes SARS-CoV-2 Infection via Upregulation of ACE2 in Human Bronchial Cells. Int J Mol Sci 2022, 23.
- Lip S, Tran TQB, Hanna R, Nichol S, Guzik TJ, Delles C, McClure J, McCallum L, Touyz RM, Berry C, Padmanabhan S: Long-term effects of SARS-CoV-2 infection on blood vessels and blood pressure - LOCHINVAR. J Hypertens 2025, 43, 1057–1065. [CrossRef]
- Ravichandran B, Grimm D, Krüger M, Kopp S, Infanger M, Wehland M: SARS-CoV-2 and hypertension. Physiol Rep 2021, 9, e14800.
- Singh M, Pushpakumar S, Zheng Y, Smolenkova I, Akinterinwa OE, Luulay B, Tyagi SC: Novel mechanism of the COVID-19 associated coagulopathy (CAC) and vascular thromboembolism. Npj viruses 2023, 1.
- Belen Apak FB, Yuce G, Topcu DI, Gultekingil A, Felek YE, Sencelikel T: Coagulopathy is Initiated with Endothelial Dysfunction and Disrupted Fibrinolysis in Patients with COVID-19 Disease. Indian J Clin Biochem 2023, 38, 220–230.
- Ward SE, Fogarty H, Karampini E, Lavin M, Schneppenheim S, Dittmer R, Morrin H, Glavey S, Ni Cheallaigh C, Bergin C, Martin-Loeches I, Mallon PW, Curley GF, Baker RI, Budde U, O'Sullivan JM, O'Donnell JS: ADAMTS13 regulation of VWF multimer distribution in severe COVID-19. J Thromb Haemost 2021, 19, 1914–1921.
- Favaloro EJ, Henry BM, Lippi G: Increased VWF and Decreased ADAMTS-13 in COVID-19: Creating a Milieu for (Micro)Thrombosis. Semin Thromb Hemost 2021, 47, 400–418. [CrossRef]
- Chau CW, To A, Au-Yeung RKH, Tang K, Xiang Y, Ruan D, Zhang L, Wong H, Zhang S, Au MT, Chung S, Song E, Choi DH, Liu P, Yuan S, Wen C, Sugimura R: SARS-CoV-2 infection activates inflammatory macrophages in vascular immune organoids. Sci Rep 2024, 14, 8781. [CrossRef]
- Hönzke K, Obermayer B, Mache C, Fatykhova D, Kessler M, Dökel S, Wyler E, Baumgardt M, Löwa A, Hoffmann K, Graff P, Schulze J, Mieth M, Hellwig K, Demir Z, Biere B, Brunotte L, Mecate-Zambrano A, Bushe J, Dohmen M, Hinze C, Elezkurtaj S, Tönnies M, Bauer TT, Eggeling S, Tran HL, Schneider P, Neudecker J, Rückert JC, Schmidt-Ott KM, Busch J, Klauschen F, Horst D, Radbruch H, Radke J, Heppner F, Corman VM, Niemeyer D, Müller MA, Goffinet C, Mothes R, Pascual-Reguant A, Hauser AE, Beule D, Landthaler M, Ludwig S, Suttorp N, Witzenrath M, Gruber AD, Drosten C, Sander LE, Wolff T, Hippenstiel S, Hocke AC: Human lungs show limited permissiveness for SARS-CoV-2 due to scarce ACE2 levels but virus-induced expansion of inflammatory macrophages. Eur Respir J 2022, 60.
- Pode Shakked N, de Oliveira MHS, Cheruiyot I, Benoit JL, Plebani M, Lippi G, Benoit SW, Henry BM: Early prediction of COVID-19-associated acute kidney injury: Are serum NGAL and serum Cystatin C levels better than serum creatinine? Clin Biochem 2022, 102, 1–8. [CrossRef]
- Kim IS, Kim DH, Lee HW, Kim SG, Kim YK, Kim JK: Role of increased neutrophil extracellular trap formation on acute kidney injury in COVID-19 patients. Front Immunol 2023, 14, 1122510. [CrossRef]
- Henry BM, de Oliveira MHS, Cheruiyot I, Benoit J, Rose J, Favaloro EJ, Lippi G, Benoit S, Pode Shakked N: Cell-Free DNA, Neutrophil extracellular traps (NETs), and Endothelial Injury in Coronavirus Disease 2019- (COVID-19-) Associated Acute Kidney Injury. Mediators Inflamm 2022, 2022, 9339411.
- Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V: COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation Might the Double X-chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? Int J Mol Sci 2020, 21.
- Chanana N, Palmo T, Sharma K, Kumar R, Graham BB, Pasha Q: Sex-derived attributes contributing to SARS-CoV-2 mortality. Am J Physiol Endocrinol Metab 2020, 319, E562–e7. [CrossRef]
- Langelueddecke C, Roussa E, Fenton RA, Wolff NA, Lee WK, Thévenod F: Lipocalin-2 (24p3/neutrophil gelatinase-associated lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. J Biol Chem 2012, 287, 159–169. [CrossRef]
- Salomão R, Assis V, de Sousa Neto IV, Petriz B, Babault N, Durigan JLQ, de Cássia Marqueti R: Involvement of Matrix Metalloproteinases in COVID-19: Molecular Targets, Mechanisms, and Insights for Therapeutic Interventions. Biology (Basel) 2023, 12.
- Mobasheri L, Nasirpour MH, Masoumi E, Azarnaminy AF, Jafari M, Esmaeili SA: SARS-CoV-2 triggering autoimmune diseases. Cytokine 2022, 154, 155873. [CrossRef] [PubMed]
- Galipeau Y, Cooper C, Langlois MA: Autoantibodies in COVID-19: implications for disease severity and clinical outcomes. Front Immunol 2024, 15, 1509289.
- Brinkmann M, Traby L, Kussmann M, Weiss-Tessbach M, Buchtele N, Staudinger T, Gaidoschik E, Perkmann T, Haslacher H, Ratzinger F, Pickl WF, El-Gedawi K, Feichter M, Gelpi E, Höftberger R, Quehenberger P, Marculescu R, Mrak D, Kastrati K, Lechner-Radner H, Sieghart D, Aletaha D, Winkler S, Bonelli M, Göschl L: Autoantibody development is associated with clinical severity of COVID-19: A cohort study. Clin Immunol 2025, 274, 110471.
- Jansen J, Reimer KC, Nagai JS, Varghese FS, Overheul GJ, de Beer M, Roverts R, Daviran D, Fermin LAS, Willemsen B, Beukenboom M, Djudjaj S, von Stillfried S, van Eijk LE, Mastik M, Bulthuis M, Dunnen WD, van Goor H, Hillebrands JL, Triana SH, Alexandrov T, Timm MC, van den Berge BT, van den Broek M, Nlandu Q, Heijnert J, Bindels EMJ, Hoogenboezem RM, Mooren F, Kuppe C, Miesen P, Grünberg K, Ijzermans T, Steenbergen EJ, Czogalla J, Schreuder MF, Sommerdijk N, Akiva A, Boor P, Puelles VG, Floege J, Huber TB, van Rij RP, Costa IG, Schneider RK, Smeets B, Kramann R: SARS-CoV-2 infects the human kidney and drives fibrosis in kidney organoids. Cell Stem Cell 2022, 29, 217–231.e8.
- Reiser J, Spear R, Luo S: SARS-CoV-2 pirates the kidneys: A scar(y) story. Cell Metab 2022, 34, 352–354. [CrossRef]
- Larrue R, Fellah S, Van der Hauwaert C, Hennino MF, Perrais M, Lionet A, Glowacki F, Pottier N, Cauffiez C: The Versatile Role of miR-21 in Renal Homeostasis and Diseases. Cells 2022, 11.
- McDonald JT, Enguita FJ, Taylor D, Griffin RJ, Priebe W, Emmett MR, Sajadi MM, Harris AD, Clement J, Dybas JM, Aykin-Burns N, Guarnieri JW, Singh LN, Grabham P, Baylin SB, Yousey A, Pearson AN, Corry PM, Saravia-Butler A, Aunins TR, Sharma S, Nagpal P, Meydan C, Foox J, Mozsary C, Cerqueira B, Zaksas V, Singh U, Wurtele ES, Costes SV, Davanzo GG, Galeano D, Paccanaro A, Meinig SL, Hagan RS, Bowman NM, Wolfgang MC, Altinok S, Sapoval N, Treangen TJ, Moraes-Vieira PM, Vanderburg C, Wallace DC, Schisler JC, Mason CE, Chatterjee A, Meller R, Beheshti A: Role of miR-2392 in driving SARS-CoV-2 infection. Cell Rep 2021, 37, 109839.
- Valle A, Soto Z, Muhamadali H, Hollywood KA, Xu Y, Lloyd JR, Goodacre R, Cantero D, Cabrera G, Bolivar J: Metabolomics for the design of new metabolic engineering strategies for improving aerobic succinic acid production in Escherichia coli. Metabolomics : Official journal of the Metabolomic Society 2022, 18, 56. [CrossRef] [PubMed]
- Tong W, Hannou SA, Wang Y, Astapova I, Sargsyan A, Monn R, Thiriveedi V, Li D, McCann JR, Rawls JF, Roper J, Zhang GF, Herman MA: The intestine is a major contributor to circulating succinate in mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2022, 36, e22546.
- Kalan Sarı I, Keskin O, Seremet Keskin A, Elli Dağ HY, Harmandar O: Is Homocysteine Associated with the Prognosis of Covid-19 Pneumonia. Int J Clin Pract 2023, 2023, 9697871.
- Carpenè G, Negrini D, Henry BM, Montagnana M, Lippi G: Homocysteine in coronavirus disease (COVID-19): a systematic literature review. Diagnosis (Berl) 2022, 9, 306–310. [CrossRef]
- Lee SR, Roh JY, Ryu J, Shin HJ, Hong EJ: Activation of TCA cycle restrains virus-metabolic hijacking and viral replication in mouse hepatitis virus-infected cells. Cell & bioscience 2022, 12, 7.
- Stipanuk MH, Ueki I: Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis 2011, 34, 17–32. [CrossRef]
- Kimura Y, Koike S, Shibuya N, Lefer D, Ogasawara Y, Kimura H: 3-Mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (Cys-SSH and GSSH) together with signaling molecules H(2)S(2), H(2)S(3) and H(2)S. Sci Rep 2017, 7, 10459. [CrossRef]
- Sen U, Sathnur PB, Kundu S, Givvimani S, Coley DM, Mishra PK, Qipshidze N, Tyagi N, Metreveli N, Tyagi SC: Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am J Physiol Cell Physiol 2012, 303, C41–C51. [CrossRef] [PubMed]
- Agrawal R, Pal VK, K SS, Menon GJ, Singh IR, Malhotra N, C SN, Ganesh K, Rajmani RS, Narain Seshasayee AS, Chandra N, Joshi MB, Singh A: Hydrogen sulfide (H2S) coordinates redox balance, carbon metabolism, and mitochondrial bioenergetics to suppress SARS-CoV-2 infection. PLoS Pathog 2025, 21, e1013164.
- Han SJ, Noh MR, Jung JM, Ishii I, Yoo J, Kim JI, Park KM: Hydrogen sulfide-producing cystathionine γ-lyase is critical in the progression of kidney fibrosis. Free Radic Biol Med 2017, 112, 423–432. [CrossRef]
- Jung KJ, Jang HS, Kim JI, Han SJ, Park JW, Park KM: Involvement of hydrogen sulfide and homocysteine transsulfuration pathway in the progression of kidney fibrosis after ureteral obstruction. Biochim Biophys Acta 2013, 1832, 1989–1997. [CrossRef]
- Fabbrizi E, Fiorentino F, Carafa V, Altucci L, Mai A, Rotili D: Emerging Roles of SIRT5 in Metabolism, Cancer, and SARS-CoV-2 Infection. Cells 2023, 12.
- Sanchez-Russo L, Billah M, Chancay J, Hindi J, Cravedi P: COVID-19 and the Kidney: A Worrisome Scenario of Acute and Chronic Consequences. J Clin Med 2021, 10.
- Madsen HB, Durhuus JA, Andersen O, Straten PT, Rahbech A, Desler C: Mitochondrial dysfunction in acute and post-acute phases of COVID-19 and risk of non-communicable diseases. NPJ Metab Health Dis 2024, 2, 36. [CrossRef]
- Fong P, Wusirika R, Rueda J, Raphael KL, Rehman S, Stack M, de Mattos A, Gupta R, Michels K, Khoury FG, Kung V, Andeen NK: Increased Rates of Supplement-Associated Oxalate Nephropathy During COVID-19 Pandemic. Kidney Int Rep 2022, 7, 2608–2616. [CrossRef]
- Borczuk AC, Yantiss RK: The pathogenesis of coronavirus-19 disease. J Biomed Sci 2022, 29, 87.
- Karam A, Mjaess G, Younes H, Aoun F: Increase in urolithiasis prevalence due to vitamins C and D supplementation during the COVID-19 pandemic. J Public Health (Oxf) 2022, 44, e625–e6. [CrossRef]
- Thévenod F, Herbrechter R, Schlabs C, Pethe A, Lee WK, Wolff NA, Roussa E: Role of the SLC22A17/lipocalin-2 receptor in renal endocytosis of proteins/metalloproteins: a focus on iron- and cadmium-binding proteins. American journal of physiology Renal physiology 2023, 325, F564–f77. [CrossRef] [PubMed]
- Engström J, Koozi H, Didriksson I, Larsson A, Friberg H, Frigyesi A, Spångfors M: Plasma neutrophil gelatinase-associated lipocalin independently predicts dialysis need and mortality in critical COVID-19. Scientific reports 2024, 14, 6695.
- Kaufman DW, Kelly JP, Curhan GC, Anderson TE, Dretler SP, Preminger GM, Cave DR: Oxalobacter formigenes may reduce the risk of calcium oxalate kidney stones. J Am Soc Nephrol 2008, 19, 1197–1203. [CrossRef]
- Troxel SA, Sidhu H, Kaul P, Low RK: Intestinal Oxalobacter formigenes colonization in calcium oxalate stone formers and its relation to urinary oxalate. J Endourol 2003, 17, 173–176. [CrossRef]
- Mehta M, Goldfarb DS, Nazzal L: The role of the microbiome in kidney stone formation. Int J Surg 2016, 36, 607–612. [CrossRef]
- Pan Y, Su J, Liu S, Li Y, Xu G: Causal effects of gut microbiota on the risk of urinary tract stones: A bidirectional two-sample mendelian randomization study. Heliyon 2024, 10, e25704. [CrossRef]
- Serrano R, Corbella X, Rello J: Management of hypoxemia in SARS-CoV-2 infection: Lessons learned from one year of experience, with a special focus on silent hypoxemia. J Intensive Med 2021, 1, 26–30. [CrossRef]
- George CE, Scheuch G, Seifart U, Inbaraj LR, Chandrasingh S, Nair IK, Hickey AJ, Barer MR, Fletcher E, Field RD, Salzman J, Moelis N, Ausiello D, Edwards DA: COVID-19 symptoms are reduced by targeted hydration of the nose, larynx and trachea. Sci Rep 2022, 12, 4599. [CrossRef]
- Mafra D, Kemp JA, Cardozo L, Borges NA, Nerbass FB, Alvarenga L, Kalantar-Zadeh K: COVID-19 and Nutrition: Focus on Chronic Kidney Disease. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation 2023, 33, S118–s27. [CrossRef]
- Vuorio A, Raal F, Kovanen PT: Familial hypercholesterolemia: The nexus of endothelial dysfunction and lipoprotein metabolism in COVID-19. Curr Opin Lipidol 2023, 34, 119–125. [CrossRef] [PubMed]
- Camp TM, Smiley LM, Hayden MR, Tyagi SC: Mechanism of matrix accumulation and glomerulosclerosis in spontaneously hypertensive rats. J Hypertens 2003, 21, 1719–1727. [CrossRef] [PubMed]
- Grover PK, Kim DS, Ryall RL: The effect of seed crystals of hydroxyapatite and brushite on the crystallization of calcium oxalate in undiluted human urine in vitro: implications for urinary stone pathogenesis. Mol Med 2002, 8, 200–209. [CrossRef]
- Zhou S, Jiang S, Guo J, Xu N, Wang Q, Zhang G, Zhao L, Zhou Q, Fu X, Li L, Patzak A, Hultström M, Lai EY: ADAMTS13 protects mice against renal ischemia-reperfusion injury by reducing inflammation and improving endothelial function. American journal of physiology Renal physiology 2019, 316, F134–f45. [CrossRef] [PubMed]
- Al-Kuraishy HM, Al-Gareeb AI, Alzahrani KJ, Cruz-Martins N, Batiha GE: The potential role of neopterin in Covid-19: a new perspective. Molecular and cellular biochemistry 2021, 476, 4161–4166. [CrossRef]
- Anderson S, McNicholas D, Murphy C, Cheema I, McLornan L, Davis N, Quinlan M: The impact of COVID-19 on acute urinary stone presentations: a single-centre experience. Ir J Med Sci 2022, 191, 45–49. [CrossRef]
- Üntan İ: How did COVID-19 affect acute urolithiasis? An inner Anatolian experience. Ulus Travma Acil Cerrahi Derg 2023, 29, 780–785.
- Turney BW, Demaire C, Klöcker S, Woodward E, Sommerfeld HJ, Traxer O: An analysis of stone management over the decade before the COVID-19 pandemic in Germany, France and England. BJU Int 2023, 132, 196–201. [CrossRef]
- Shivakumar N, Nantha Kumar D, Joshi H: The Impact of Early COVID-19 Pandemic on the Presentation and Management of Urinary Calculi Across the Globe: A Systematic Review. J Endourol 2022, 36, 1255–1264. [CrossRef] [PubMed]
- The L: Long COVID: 3 years in. Lancet 2023, 401, 795.
- Makhluf H, Madany H, Kim K: Long COVID: Long-Term Impact of SARS-CoV2. Diagnostics (Basel) 2024, 14.
Figure 1.
Viral entry of SARS-CoV-2 and human coronaviruses via spike protein (SP)-mediated mechanisms. The SP of SARS-CoV-2, including emerging variants such as Omicron-α, Omicron-δ, and XBB.1.5, and other human coronaviruses (NL63, 229E, OC43, HKU1) enables viral entry into host cells by binding surface receptors and initiating fusion. After entry, SARS-CoV-2 hijacks the host’s methylation machinery, leading to epigenetic reprogramming and altered metabolic pathways. One key consequence is disruption of the mitochondrial sulfur transsulfuration pathway, which normally converts Hcy to cysteine via CBS and CSE enzymes. This disruption leads to Hcy accumulation, depletion of cysteine and glutathione, and impaired NO production, contributing to oxidative stress and renal endothelial dysfunction. These changes create a pro-stone environment in the kidney, linking COVID-19 pathogenesis to the development of KSD [10,15,16].
Figure 1.
Viral entry of SARS-CoV-2 and human coronaviruses via spike protein (SP)-mediated mechanisms. The SP of SARS-CoV-2, including emerging variants such as Omicron-α, Omicron-δ, and XBB.1.5, and other human coronaviruses (NL63, 229E, OC43, HKU1) enables viral entry into host cells by binding surface receptors and initiating fusion. After entry, SARS-CoV-2 hijacks the host’s methylation machinery, leading to epigenetic reprogramming and altered metabolic pathways. One key consequence is disruption of the mitochondrial sulfur transsulfuration pathway, which normally converts Hcy to cysteine via CBS and CSE enzymes. This disruption leads to Hcy accumulation, depletion of cysteine and glutathione, and impaired NO production, contributing to oxidative stress and renal endothelial dysfunction. These changes create a pro-stone environment in the kidney, linking COVID-19 pathogenesis to the development of KSD [10,15,16].
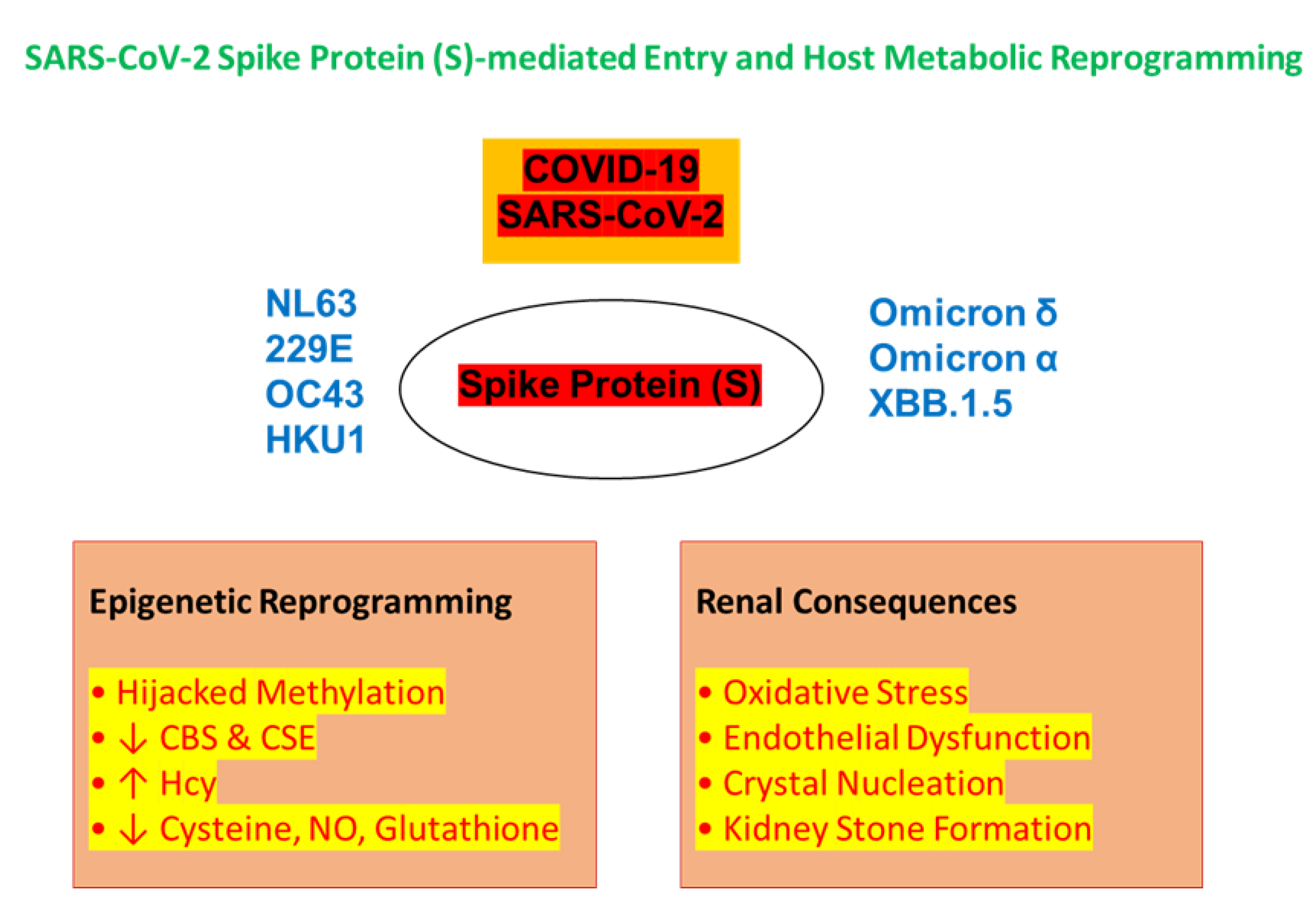

Table 1.
Key Biomarkers Implicated in COVID-19 Associated KSD. The candidate biomarkers relevant to the pathophysiological link between SARS-CoV-2 infection and kidney stone formation. Listed biomarkers include indicators of oxidative stress, immune activation, mitochondrial dysfunction, and impaired sulfur metabolism. Each marker is annotated with its biological source (e.g., urine, plasma), functional relevance, and potential utility in diagnosing or monitoring post COVID-19 renal complications.
Table 1.
Key Biomarkers Implicated in COVID-19 Associated KSD. The candidate biomarkers relevant to the pathophysiological link between SARS-CoV-2 infection and kidney stone formation. Listed biomarkers include indicators of oxidative stress, immune activation, mitochondrial dysfunction, and impaired sulfur metabolism. Each marker is annotated with its biological source (e.g., urine, plasma), functional relevance, and potential utility in diagnosing or monitoring post COVID-19 renal complications.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



