Submitted:
21 July 2025
Posted:
21 July 2025
You are already at the latest version
Abstract
Systematic evaluation of spin orbit coupling (SOC) on models for superconducting LaH10 at pressure is based on ab initio DFT calculations with optimised experimental cell dimensions. Non-relativistic and relativistic DFT treatments of LaH10 band structures using either a cubic or primitive lattice reveal the presence of a cosine-shaped band crossing the Fermi level. Asymmetry of cosine-shaped bands is indicative of the superconducting gap and enables close matches of calculated Tc with experimental values for 135 GPa < P < 220 GPa. Using SOC to calculate DOS, van Hove singularities occur at −0.122(3) eV for pressures between 90 GPa and 250 GPa. DFT calculation with SOC reduces the optimised cell dimension non-linearly and with higher effect as pressure increases. We relate the inflection of reciprocal space cosine-shaped curves to intersecting planes of (h0l), (hk0) and (0kl) planes in the real space Fm3m structure. Electron density distributions associated with asymmetry of the LaH10 irregular chamfered cube is attributed to variations in hydrogen bonding with pressure and abrupt changes to lattice vibrations between 135 GPa and 220 GPa. Collective behaviour of hydrogen lattice vibrations and asymmetric changes to crystal structure are consistent with the dome-like format for experimental Tc values with pressure.
Keywords:
lanthanum hydride
; electronic band structure
; cosine-shaped bands
; spin orbit coupling
; density of states
; flat band
; van Hove singularity
; lattice vibrations
; interatomic distances
1. Introduction
The theoretical prediction and subsequent experimental verification of metal hydride superconductors at high pressure, such as sulphur hydride, H3S [1,2] and lanthanum hydride, LaH10 [2,3,4,5], have invigorated research in high temperature superconductors particularly in relation to the search for ambient pressure compounds [6]. Cubic LaH10 shows superconductivity transition temperatures (Tc) ranging from 230 K to 260 K between 135 GPa and 220 GPa pressure [2,5,7,8,9]. The structures of these metal hydride compounds implied potential to achieve high temperature superconductivity under conditions within the experimental capacity of a limited number of specialised laboratories. For LaH10, the experimental Tc and the isotope effect comply with the BCS theory of superconductivity [10,11].
Experimental verification of a face-centred cubic (fcc) structure, , for LaH10 at high pressures [2,3,7], enabled theoretical calculations to interpret superconductivity mechanisms using Allan-Dynes [12,13] and Migdal–Eliashberg [14,15] formalisms aided by, or compared with, density functional theory (DFT) [14,16,17]. These calculations provide estimates of a superconducting gap, Δ, or of a transition temperature, Tc. However, experimental and theoretical values for Tc with pressure (P), show a mismatch between theory and experiment [2,5,7,8] given allowances for systematic error [9]. Indeed, there is a better cluster of experimental Tc vs P values [2,9] despite the limited number and difficulty in conducting experiments.
An attribute of experimental data for cubic LaH10 noted by others [2,9,14] is a dome-like trend for data points between 135 GPa and 220 GPa. The choice of pseudo potential [12], phonon anharmonicity [14,17] and quantum effects [14] may account for differing calculated results for Tc. Nevertheless, a combination of ab initio Migdal-Eliashberg equations and an anisotropic energy gap calculation, has to date, provided a modest match with experimental values between 140 GPa and 220 GPa [14] but without a dome-like format.
Sun et al. [9] have re-visited the experimental determination of Tc for LaH10 between 120 GPa and 138 GPa. They show that the structure of LaH10 is monoclinic below 135 GPa (Space Group C2/m) albeit with a lower Tc of 189 K [9]. Pressure changes from 120 GPa to 138 GPa indicate a sharp, reversible structural transition at 135 GPa [9]. In this work, we focus on the cubic form of LaH10 and defer consideration of the monoclinic form to a later publication.
In calculations of LaH10 electronic properties, Liu et al. [17] consider spin-orbit coupling (SOC) and note the presence of an Electronic Topological Transition (ETT) near the Fermi level, EF. Errea et al. [14] used a proprietary version of DFT, called SCDFT, to calculate ab initio electronic properties of LaH10 and to evaluate phonon behaviour in superlattices of 2x and 3x the cubic unit cell. Both studies considered quantum effects noting their importance for stabilisation of compounds with high electron-phonon coupling at high pressure. Harmonic [12] and anharmonic [14,17] calculations, identify a peak in total electronic density of states (DOS) near EF; in some cases, across a broad pressure range from 100 GPa to 300 GPa [14].
We seek to extend earlier work [12,14,17] by considering SOC within the context of our ab initio DFT approach utilising superlattices and the identification of bonding-antibonding character [18,19,20]. Our premise for these and previous studies [21,22,23], is that DFT calculations can be used to precisely model electron, and some phonon, interactions based on quality crystallographic data of a given compound. This premise is inherent in machine learning evaluations for new materials prediction [24,25], the role of geometry in determining properties [26,27] and for complex interface chemistry [28,29]. With existing DFT software and computational capacity, this premise should also apply to superconductivity.
We have described the electronic properties of layered superconducting structures, such as MgB2 [18] and CaC6 [20], in terms of bonding–antibonding orbital character readily identified by asymmetric cosine-shaped band(s) near the Fermi level (EF) of an electronic band structure (EBS). Such asymmetric bands appear strongly linked to superconductivity, for example in CaC6 with change in pressure and in the presence of an ETT [19]. A further construct that can assist evaluation of such cosine-shaped bands is elucidation of a superlattice; validated experimentally [18,30] or by calculation [20]. Using superlattices of layered structures [18,19], we have shown that the superconducting gap can be directly determined from ab initio DFT calculation of band structures.
For this study, we optimise DFT parameters for cubic LaH10 with a focus on the structures at pressures shown by experiment that enable superconductivity. We extend our asymmetric cosine band approach, previously applied to rhombohedral symmetry layered structures [18,19], to the higher symmetry cubic structure of LaH10. Cubic symmetry can introduce substantive band degeneracy and/or multiple energy dispersion intersections compared to a lower symmetry structure. Accordingly, we explore computational methods to establish a superconducting gap value, based only on ab initio DFT calculations of EBSs including those reduced to their respective primitive cells.
2. Results
Key features of the LaH10 structure and calculated electronic band structures at pressure are presented in this section. Further detailed examples are provided in Supplemental.
2.1. LaH10 Structure
Figure 1 shows the LaH10 structure with calculated cell dimensions for 170 GPa normal to [101] highlighting sequential layers of La atoms and high densities of hydrogen atoms. In the LaH10 structure, hydrogen is in two different atomic positions, forming a H network with eight in H8c sites and a further 32 H atoms in the H32f sites as shown in Figure 1.
Two different hydrogen distances in Figure 1 are those between H8c and H32f sites and between H32f sites. For all LaH10 structures between 135 GPa and 220 GPa, these two shortest distances are less than 2.0 Å. As noted in Methods (Section 4), the lattice constant, a = 5.1019(5) from X-ray diffraction results for LaH10 at 150 GPa [2] is used as the basis for additional DFT optimised cell dimension calculations at other pressures. After optimisation, this cell dimension at 150 GPa refines to a = 5.11257 Å without SOC and to a = 5.05951 Å with SOC calculation.
Figure 2 shows a plot of experimentally determined cell dimensions for LaH10 with applied pressure. We note that the data obtained by Drozdov et al. [2] (diamond symbols) and Sun et al. [9] (square symbols) are our primary foci due to contemporaneous determination of Tc and cell dimension at the same applied pressures. Additional information on other LaH10 experiments is given in Methods (Section 4).
Figure 2 shows DFT optimized cell dimensions for a cubic LaH10 structure with and without spin orbit coupling (circles) using the LDA functional. The match of experimentally determined cell dimensions with LDA calculations for an optimized cubic LaH10 structure suggests that the choice of initial cell parameter used for DFT modelling in this study is reasonable. Figure 2 shows that these calculated trends for cubic cell dimension with applied pressure for LaH10 are not linear, as suggested by an earlier study [31]. A list of optimized cell dimensions calculated for 90 GPa < P < 300 GPa with and without SOC is provided in Supplemental Table S1.
2.2. Electronic Band Structures
Band structures for LaH10 at pressures 135 GPa < P < 220 GPa, as well as at pressures outside the experimental pressure regime for superconductivity, based on calculated cell dimensions shown in Figure 2, are exemplified below and in Supplemental.
2.2.1. Cosine-Shaped Electronic Bands
Bonding–antibonding orbital character is readily identified by asymmetric cosine-shaped band(s) near EF [18]. Cosine-shaped bands are discernible in layered compounds [18,19] and also in higher symmetry structures provided the structure contains approximate linear chains of atoms of suitable dimensions and appropriate combinations of reciprocal directions are included in EBS representations.
Figure 3 shows the EBS for LaH10 at 170 GPa calculated with the LDA functional for a cubic cell (a = 5.06223 Å) and for the corresponding primitive cell for which a = 3.53668 Å; in each case with and without SOC correction. For the EBS shown in Figure 3, a non-relativistic Schrödinger treatment is used. In all cases, a cosine-shaped band is identified as a red highlighted band. This cosine-shaped band is well defined for the primitive unit cell (Figure 3b and 3d) compared to the cubic form (Figure 3a and 3c).
The cosine band in Figure 3d shows minimal, to zero, intersection of other bands in the Γ–X direction although the band is degenerate for a part of this reciprocal direction. In all other cases, particularly Figure 3a and 3c, the cosine band shows multiple intersections, and/or “gaps”, with other bands. The number of band interferences is related to the high symmetry of the structure. However, the primitive calculation without SOC (Figure 3b) shows direct interaction of other bands with the highlighted cosine-shaped band. The band interaction shown in Figure 3b suggests that estimates of the cosine-band asymmetry may be inaccurate or affected by these other intersecting bands. Additional examples for cubic structures at 200 GPa and 220 GPa with and without SOC are shown in Supplemental Figure S1.
The amplitude and alignment of the cosine band relative to EF is affected by the choice of unit cell and the inclusion, or otherwise, of SOC. For primitive cell calculations, the center of the cosine band is above and below the Fermi level as shown in Figure 3b and 3d, respectively. Figure 3b and 3d show a clear difference in band structures at 170 GPa for an equivalent change in cell dimension of 0.06064 Å (Table S1) with calculation of SOC. Note that along the X–R and W–L directions, two bands occur close to the Fermi level at R and L, respectively (circled in Figure 3c and 3d). These bands, commonly referred to as “flat bands”, are not flat when shown in detail (Supplemental Figure S2).
Table 1 provides a summary of key parameters obtained from systematic DFT calculations for LaH10 at selected pressures using non-relativistic and relativistic treatments. Inspection of Table 1 shows that calculations with SOC result in a more stable LaH10 structure for which total enthalpy is substantially lower than that without SOC. Furthermore, for the primitive LaH10 cell, key derived parameters such as the H8c-H32f distances are equivalent for non-relativistic and relativistic calculations at the same pressure.
2.2.1. Cosine Band Asymmetry
Figure 4 shows an enlarged section of the Γ–X reciprocal direction for LaH10 calculated for the primitive cell at 170 GPa (as shown in Figure 3d) for a Schrödinger and a Koelling-Harmon (K-H) treatment, respectively. These figures detail one method to evaluate the energy asymmetry of the cosine band. In both cases, we measure the energy mid-point between the “anti-bonding” (A) and “bonding” (B) nodes of the cosine band designated as Emid in Figure 4.
Folding the band around ΓX/2 is equivalent to establishing a superlattice in the Γ–X direction [18]. Thus, the energy at which ΓX/2 intersects the cosine curve, denoted EΓX/2, is the notional energy of a symmetric cosine curve. The difference in energy between Emid and EΓX/2 is ΔET and represents the total energy asymmetry of the cosine curve between Γ and X. In Figure 4a, ΔET is 393.2 meV and in Figure 4b, the ΔET value is 391 meV. These different values for ΔET using non-relativistic and relativistic treatments (δ ~ 2.2 meV), have a direct influence on ab initio estimates of the superconducting gap but are within our estimated systematic error of ±6 meV.
Figure S3 (Supplemental) provides further detail on error estimates of band energy asymmetry for these calculations. Unlike our earlier estimates of error for MgB2 and CaC6 [18,19] which are much smaller (i.e. ± 1 meV), compute limitations (e.g. number of cores, available memory; number of atoms) also restrict the energy resolution we are able to achieve for convergent solutions of LaH10 band structures (see further detail in Figure S3). Additional examples of cosine curves along Γ–X at different pressures without SOC treatment are provided in Supplemental Figure S4 and with SOC treatment in Supplemental Figure S5.
In Figure 4, both cosine curves cross the Fermi level and both Emid and EΓX/2 points are below EF. In comparison, the cosine curve for the primitive calculation without SOC (Figure 3b) displays Emid and EΓX/2 points above EF and an estimate of band asymmetry, ΔET = 179 meV (shown in Supplemental Figure S4c). For the cosine curves in Figure 4, the degeneracy of the band(s) at Γ is 4. That is, four bands have the same energy value at the center of the primitive cell. Similar multiple degeneracy is observed for equivalent bands in DFT calculations of the cubic cell.
Figure 5 shows that Emid and EΓX/2 shift to lower energy (and below EF) with increased pressure. In addition, the range of energy, or amplitude, attributed to the cosine curve along Γ–X increases with additional pressure. K-H relativistic treatments for the primitive cell show a consistent change to ΔET with increased pressure. Other non-relativistic and relativistic treatments show similar trends for cosine curve characteristics.
The effect of SOC calculations applies to varying extents across the range of pressures attributed to superconductivity. The impact of SOC treatment on cell dimensions is highest at 220 GPa and lowest at 140 GPa reflecting an equivalent change in cell dimensions (compared with no SOC adjustment) of 0.07817 Å and 0.04919 Å, respectively (Supplemental Table S1). In addition, for a suite of calculations such as for a primitive cell with K-H treatment, the energy at the cosine inflection point, EΓX/2 reduces further below EF with increased pressure. This effect is commensurate with the cell dimension trend shown in Figure 2.
2.2.2. Estimates of Tc
We estimate values for the Tc of LaH10 at pressure using the value for ΔET obtained from DFT primitive structure calculations with SOC (i.e. as shown in Figure 4 and Figure 5). A primary reason for use of the primitive structure is the relative absence of other intersecting bands with the cosine curve. The superconducting gap, Δ, is based on the band degeneracy at Γ normalized to equivalent DFT calculated 2Δ values of ΔET for MgB2 [18].
Figure 6 compiles these calculated Tc values at selected pressures for comparison with experimentally determined values (shown as solid diamonds, triangles, hexagon and squares) [2,5,7,8,9]. Errors for experimental data are from the cited publications. Non-relativistic and relativistic DFT treatments from this work are also shown; in some cases, data points overlap. There is a good match of calculated values with experimental data given the systematic errors identified for both experimental and calculated data. As noted in earlier work [14], the calculated data also trend towards reduced values for Tc with P > 200 GPa. For comparison, we also plot previous theoretical calculations for the Tc of LaH10 with pressure (in Supplemental Figure S6), in addition to the data shown in Figure 6.
Table 2 shows parameters derived from DFT calculations including values for ΔET, Emid and EΓX/2 as shown in Figure 4 and Figure 5, the energy, ELmax, of the “flat band” at L, and the energy gap, ΔEL at L between bands close to EF. EBSs calculated for P < 140 GPa and for P > 220 GPa have also been obtained using the same method as that for 140 GPa < P < 220 GPa. Close inspection of the cosine curves shows changes in band character at pressures on either side of the experimentally determined superconducting regime. These changes in cosine band character at Γ and X reciprocal points are detailed in Supplemental Figure S7 and emphasize subtle, yet important, distinctions in band topology with pressure.
Table 2 shows clear trends with increasing pressure for the range 135 GPa to 220 GPa. For example, for calculations with SOC the mid-point of the cosine band, designated by Emid, shifts away from EF to lower energies with increase in pressure to 220 GPa. For calculations in the primitive cell without SOC, Emid is above EF and with increased pressure shifts to energy levels closer to the Fermi level. The intersection of the fold axis (along ΓX/2) with the cosine band, designated as EΓX/2, also shifts to lower energy with increase in pressure to 220 GPa for calculations with SOC.
For calculations without SOC, a similar trend to that for Emid is apparent and with higher estimates for ΔET resulting in a higher value for Tc. However, for calculations with SOC the energy asymmetry of the cosine band, measured as ΔET, generally decreases in value with increased pressure. For completeness, Table S2 (Supplemental Materials) lists derived parameters for other relativistic and non-relativistic calculations. For cubic settings with SOC, estimates for Tc using a cosine curve are substantially higher than with primitive settings; without SOC adjustment estimated Tc values are lower.
2.4. Density of States – van Hove singularities
Using the cubic structure, we calculate the density of states (DOS) for LaH10 at 170 GPa and 200 GPa with and without SOC correction using the LDA functional as shown in Figure 7a. For calculations without SOC, DOS peaks show a shift away from the Fermi level (EF = 0) with higher pressure. This trend for calculations without SOC is also clearly shown for all pressures from 90 GPa to 230 GPa in Figure 7b, noting that the stable pressure range for cubic LaH10 is between 135 GPa and 220 GPa [9,14].
For DFT calculations with SOC, peaks close to the EF overlap at a constant energy value of −0.103(5) eV (for cubic calculation) and −0.122(3) (for primitive calculation) as shown in Figure 7. DOS calculations without SOC correction show peaks that shift to higher energy with increase in applied pressure. The difference in energy between the peaks without SOC and with SOC also changes with pressure. For example, at 170 GPa the delta is 1.08 eV (1.10 eV for primitive) while at 200 GPa the difference is 1.20 eV as shown in Figure 7. These overlapping DOS peaks close to the EF correspond to van Hove singularities [32] that are associated with critical points of the band structure [33].
Over a wider pressure range, the intensity of the DOS peak near EF reduces at higher pressure for calculations with SOC, and to a lesser extent for calculations without SOC. The DFT calculated DOS with SOC using the primitive cell is shown for all pressures from 90 GPa to 250 GPa in Supplemental Figure S8. In this case, the constant energy value is −0.122(3) eV; at a lower value by 20 meV than for the cubic calculation. As anticipated, the total number of states/eV at this van Hove singularity for the cubic structure is ~4x the number for the primitive structure.
The DOS profiles with and without SOC in Figure 7 of this study are unlike the double van Hove singularities calculated by Liu et al. [16] using DFT calculations based on parameters at 300 GPa. We suggest that the Liu et al. [16] DOS profiles with and without SOC (shown as Figure S5 in [16]) partly result from a low cut-off energy (500 eV) used for the plane wave calculation.
3. Discussion
Atomic orbital interactions determine the conditions for bonding in crystals. These interactions include conditions wherein orbital directions must overlap, orbital energies must match approximately, and orbitals must have the correct symmetry [34].
3.1. Atomic Orbitals and Bonding
The LaH10 structure along [h0l], [0kl] and [hk0] orientations features closely spaced hydrogen atoms; Figure 1 shows the [h0l] orientation. In real space, these schemas represent closely linked s-orbitals densely packed in alternating layers interspersed with La+3 ions. Figure 2 also shows that inclusion of SOC further contracts the lattice parameter or invokes an additional pressure compared to calculated results without SOC. This inference appears inconsistent with experimental data [2] as well as Migdal-Eliashberg and SCDFT calculations [14] and first principles calculations [35], that suggest at higher pressures the Tc is lower. We note that DFT calculations are ground state measures (absolute zero temperature) for a particular structure. Inclusion of thermal expansion (thermal energy ~25 meV at 250 K) for cell dimensions calculated with SOC would result in values similar to experimental data. Nevertheless, data in Figure 2 suggest that calculations including SOC have a substantial influence on the crystal structure and hence, the electronic structure, of LaH10 at high pressure.
Calculations without SOC adjustment allocate valence electrons for La to the 6s2 and 5d1 orbitals. Calculations with SOC promote electrons to f-orbitals which are strongly hybridised with H s-orbitals [16]. The valence state for La requires shifting electrons from [core]6s2 5d1 orbitals to [core]4f3 orbitals or to other orbitals hybridized with s-orbitals of surrounding H atoms. Mulliken analysis of orbital populations confirms this outcome for SOC calculations using LDA with K-H treatment for all pressures. In the primitive cell, H8c atoms show the lowest electron population of hydrogen orbitals with the lowest charge while for La, the s, p and f orbitals are occupied. Promoting electron populations onto La f-orbitals accounts for the substantially enhanced overall stability of the LaH10 structure at these pressures.
Calculated atom-atom distances (H–H, La–H and La–La) for LaH10 at a range of pressures are listed in Supplemental Table S3. There is universal acceptance that LaH10 demonstrates metallic bonding based on the formation of high electron-density energy bands at the Fermi level [36]. In general, metallic bonding involves metal nuclei sharing delocalised valence electrons that are free to move in any direction in a solid.
A first principles evaluation of many body corrections to f-orbital systems by Wei et al. [36] showed that f-orbitals are an important characteristic of superconducting rare earth hydrides. Liu et al. [16] show that an unusual bonding network occurs for LaH10 comprising anionic La ions and both anionic and cationic H atoms at H32f sites and H8c sites, respectively. DFT calculations of bond populations (Supplemental Table S3) reveal a similar analysis of electron density or bond population. For example, the two shortest H−H distances shown in Figure 8a show a net positive bond population for 90 GPa < P < 250 GPa. Across these pressures, bond populations range from 0.31 to 0.40 and 0.11 to 0.13, respectively for the H8c−H32f and H32f−H32f distances shown in Table S3.
Three H32f−H32f distances are shown in Supplemental Table S3 and designated with superscripts to identify specific distances. Strong bond populations include the H32f−H32fb and H32f−H32fc distances as shown in Figure 8b (cyan and pastel cyan, respectively). The next-nearest H32f−H32fb distance transects the vertices of the cubic array of H32f atoms surrounding (in six directions) each La atom. Supplemental Table S3 shows a strong electron population along these directions (−0.49 to −0.66 between 90 GPa and 250 GPa) each of which also align with the (h0l), (0kl) and (hk0) planes. This configuration suggests that these cubic sub-units are regions of high electron density that significantly influence electron distribution as well as the inherent vibration states of LaH10.
The H–H interatomic distances between sites in LaH10 suggest that bonding includes metallic hydrogen bonds and perhaps ionic or partially covalent bonds. Orbital configurations for La atom 4f energy levels offer bonding orientations typical for a high symmetry cubic structure. The cubic set of 4f orbitals contains two distinct shapes that have planar and conical lobes with symmetric distribution of bonding and anti-bonding character (i.e. positive and negative values) [37]. Of these, likely orbitals for metal-metal bonding are 4fx3, 4fy3 and 4fz3 with a planar node in the xy plane and two conical nodes along the z direction (see Supplemental Figure S9). The second set, with orbitals 4fx(z2-y2), 4fy(z2-x2), and 4fz(x2-y2), are related to each other by 45° rotations about the x, y, and z-axis, respectively [37]. These orbitals are oriented along the (h0l), (hk0) and (0kl) planes; coincident with the H8c–H32f, H32f–H32fb, and La–H8c distances in the real space cubic structure of LaH10.
The coordination number for La in LaH10 is 32 (i.e. 32 H atoms surround La) although the closest nearest-neighbour coordination of H32f atoms for La is 24. For the H8c next nearest-neighbour atoms, the coordination number for La is 8. Supplemental Table S3 shows that two additional H32f–H32f distances as well as those identified in Figure 8a are less than 2.0 Å. To highlight this abundance of close H–H distances within the LaH10 unit cell, Figure 8b shows the four shortest H–H distances calculated for 180 GPa. These H–H distances delineate the clathrate moiety of a chamfered cube polyhedron [31]. For a convex polyhedron at maximum chamfer (i.e. a symmetric truncation of vertices), the hexagons are equilateral but not regular (i.e. with 120o internal angles).
In an ideal chamfered cube polyhedron, the hexagons are congruent with two internal face vertex angles of ~109.47o and four internal face vertex angles of ~125.26o [38]. Table S3 lists the calculated face vertex angles for the cubic LaH10 structure. The two internal face angles at ~109.47o are consistent with an ideal chamfered cube, but the four internal face angles at ~121.2o (i.e. H32f–H8c–H32f) are substantially different from the ideal angle of ~125.26o. This suggests an implicit structural anisotropy as noted by Harshman and Fiory [31]. We calculate an anisotropy factor for each pressure calculation in Table S3. This factor, f, reduces consistently from 4.1% to 3.8% over the pressure range from 90 GPa to 250 GPa.
The topologies of convex polyhedra, including the chamfered cube, are well known [38,39,40] and have been shown to influence ion diffusion and conduction in solids [26]. The relative change in polyhedral anisotropy between 90 GPa and 250 GPa is gradual for LaH10 (Table S3). We suggest that polyhedral distortion in LaH10 is in response to mixed bonding configurations that gradually shift with change in pressure. Such a gradual change in electron distribution may also affect experimental Tc values for LaH10 resulting in the dome-like trend shown in Figure 6 as pressure changes between 135 GPa and 220 GPa.
The chemistry of bonding is also axiomatic in solid state theory [41] and sometimes can be overlooked in discussions of superconductivity. A recent study of two superconducting hexaborides with the same crystal structure [42] emphasises this point. In a study of YB6 and LaB6, both superconductors at low temperature, Lavroff et al. [42] determine that low-lying, unoccupied 4f atomic orbitals in La are key to understanding the difference between compounds of similar valence electron configuration and phonon modes. Fundamentally different electron-phonon coupling mechanisms are invoked by different chemical bonding that affects lattice coupling and phonon interactions, including phonon softening, for YB6 and LaB6 [42]. By analogy, we posit that further exploration of bonding configurations for LaH10 at a range of pressures, including the critical structural transition(s) identified at 135 GPa [9] and at 220 GPa [14], using the tools identified by Lavroff et al. [42], will validate similar mechanistic insight to the superconducting mechanism(s) of LaH10.
3.2. Spin Orbit Coupling
SOC is ubiquitous in condensed matter systems and describes the interaction between the motion of an electron and its spin. In the reference frame of an electron, the positively charged lattice moves, thus creating a magnetic field which couples to the electron spin [43]. A band that occurs within a narrow energy window near, or centred by, the Fermi energy EF, is referred to as a flat band [44,45]. In a flat section of the EBS, group velocities of electrons approach zero [45]. Figure 3 shows that application of SOC in DFT calculations of LaH10 illuminates a potential flat band in the EBS for either a cubic or primitive lattice.
For unconventional superconductors, an extremely small carrier density, one or two orders of magnitude smaller than in usual metals can be accommodated by an interplay between nonadiabatic effects and a van Hove singularity [32,46]. In particular, the van Hove singularity appears related to the highest values of Tc, to the reduction of the isotope effect and to linear resistivity. In addition, the presence of a van Hove singularity strongly enhances the phase space for small q scattering and leads to a further enhancement of Tc [46,47]
A van Hove singularity is characterised by spikes in the DOS, although this behaviour also requires an association with critical points of the electronic bands, such as saddle points or inflections [16], to qualify as a van Hove singularity . A van Hove singularity is generally a feature of low dimensional systems [33,48] and all two-dimensional band structures have a saddle-point or van Hove singularity at which the DOS diverges logarithmically [49].
The van Hove model only applies to approximately half-filled bands [50] which is consistent with the presence and positioning of the cosine-shaped bands relative to the Fermi level when SOC is included in DFT calculations as shown in Figure 3, Figure 4 and Figure 5. Furthermore, flat bands and van Hove singularities often go together, coexisting near the Fermi energy, such as identified in a two-phase superconductor by Chen et al. [51].
Two specific models,
- (i)
- the flat/steep band and;
- (ii)
- the van Hove,
attribute a flat band, or a resulting high density of electronic states, near the Fermi energy to superconductivity [52]. In the flat/steep band scenario for superconductivity, the band structure reflects the trade-off between pairwise localisation and mobility of electrons [45].
Yang et al.[52] investigated whether flat bands in the calculated EBS are good predictors of superconducting materials. Although the results were not widely conclusive, some statistical validity with hexagonal symmetries seemed to exist for this idea. An important comment arising from this study is that the omission of SOC from existing, widely accessible calculations in databases is a very significant drawback to discovery of new superconductors [52].
In this work, the total DOS calculated with SOC (Figure 7) is consistent with the total DOS determined by Errea et al. [14] using a stochastic self-consistent harmonic approximation for LaH10 between 100 GPa and 300 GPa. We show, over a similar pressure range that the intensity of the DOS peak near EF reduces at higher pressure for calculations with SOC, and to a lesser extent for calculations without SOC. In general, this trend is also consistent with pressure variations shown in earlier publications [14,35].
In Figure 3, the X–R and W–L reciprocal space directions for cubic and primitive unit cells, respectively, display differences in band structures with calculation for SOC. With SOC calculation, an apparent flat section of band aligned with the Fermi level occurs in both the cubic and primitive band structures (circled in Figure 3c and 3d). Enlarged views of the X–R and W–L directions of the EBS for LaH10 with SOC shown in Supplemental Figure S2 show a similar energy “gap” between bands near EF.
However, the band(s) are not flat, but rather curve towards EF with a maximum at reciprocal point L (or R). Perhaps the definition, and recognition, of a “flat” band depends on the resolution, or “granularity”, of a band structure calculation. With this in mind, we refer to the work of Yang et al. [52] wherein evaluations of 980 known superconductors suggest that, in general, there is no link between flat bands and superconductivity. This study also appropriately noted, as have others [53,54], that low resolution DFT calculations hinder the determination of useful aids to predict properties of superconductors [52].
The difference in energy between the bands at reciprocal point L (or R), ΔEL (or ΔER), is 114 meV at 170 GPa whether calculated in the cubic or primitive cell. Relativistic treatments using the primitive cell give ΔEL = 125 meV at 230 GPa (Table 2) This value compares with a value of 125 meV calculated by Liu et al. [16] for LaH10 at 300 GPa.
3.3. Cosine Shaped Bands
In crystalline solids, atomic orbital interactions determine the conditions for bonding in crystals. These interactions include the degree and extent of orbital overlap as well as similarity of orbital energies and correct symmetry [34]. We have shown how orbital character can be inferred from band structures calculated using DFT with a plane wave basis set for MgB2 [18] and for CaC6 [19,20]. In contrast to flat bands for superconductors, cosine-shaped bands near EF seem to be an archetypical characteristic of superconducting materials. In this work, we highlight their presence in DFT calculations of EBS but also show that their topology can be related to bonding-antibonding orbital character and that their asymmetric characteristics display a strong link to superconductivity [18,19].
A similar approach for the primitive cell of LaH10 with SOC calculation suggests that cosine band(s) provide trends consistent with experimental data collected at pressure as shown in Figure 6. Ab initio DFT calculations show that both cubic and primitive lattices for LaH10 allow identification of cosine-shaped bands that cross the Fermi level. Furthermore, these band structure calculations show clear differences in band energy spread and format with and without SOC calculation.
Visual inspection of Figure 3b and 3d, as well as those shown in Figure 4 and Figure 5, show that the cosine band, while it crosses the Fermi level and is “pinned” at reciprocal points, is not evenly distributed above and below EF. We observe these characteristics in published band structures for LaH10, by Liu et al.[16], Wang et al. [35], Quan et al. [12] and Papaconstantopolous et al. [13], particularly along the Γ–X reciprocal space direction. Calculation parameters for these studies are similar to that deployed in this work albeit with variations in initial cell dimensions, pressure and SOC adjustment. We also evaluated the calculated EBS for LaH10 by Krarroubi et al. [55] and identified a cosine-shaped band along the Γ–X direction that crosses the Fermi level at a range of pressures.
3.4. Cosine Inflection in Real and Reciprocal Space
To identify a real space equivalence to the inflection of the cosine curve, keeping in mind the non-bonding character, we examine the P1 asymmetric unit at a range of pressures. For example, Supplemental Figure S10a shows the P1 asymmetric unit for two pressures, 150 GPa and 220 GPa, and the and (101) planes. Each primitive cell is at the same scale and superimposed to show the intersection of the (h0l) planes with atom positions. Using this schematic, we infer that the inflection along the reciprocal Γ–X direction is at the intersection of the (h0l) planes.
This inflection line (plane) – that is, the intersection of the (h0l) planes – shifts incrementally with pressure as does the inflection point in the reciprocal Γ–X direction in band structure calculations (i.e. the values of EΓX/2 in Figure 4 and Figure 5 and listed in Table 2). With four symmetric orientations for these planes (i.e. (h0l), , and ), the multiplicity (or degeneracy) of bands at Γ is unsurprisingly 4. Figure 8 shows the and (101) planes in the cubic cell for LaH10 as viewed along [010]. A key feature of the LaH10 structure is the perfect H–H alignment along (h0l) planes. Only two planes are shown in Figure 8 for simplicity of presentation. All H8c–H32f distances align along (h0l) planes.
With calculation for SOC, these distances range in dimension from 1.13895 Å to 1.09205 Å between 140 GPa and 220 GPa, respectively (Table 2) and are shorter than H32f–H32f distances (blue in Figure 8). In addition, the H32f–H32f distances align with primary crystallographic planes (i.e. (h00), (0k0) and (00l)) of the LaH10 cubic structure. This novel arrangement in a high symmetry structure suggests that H–H interactions play a critical role in the development of superconductivity at specific pressures in LaH10. In comparison, Figure S10b (Supplemental) also shows the La–H8c bonds for the LaH10 structure; notably, these and many, but not all, La-H32f bonds also align with the (h0l), (0kl) or (hk0) planes; suggesting that these bonds may also play an important role for superconductivity. Each face centred La atom is surrounded by 32 hydrogens, of which 24 are on H32f sites as close neighbours and 8 are on further distant H8c sites.
3.5. Lattice Vibrations
Nearest neighbour distances are included in our Monte Carlo simulations to identify key lattice vibrations. Using the cubic unit cell, we calculate the vibration modes for each pressure based on the DFT optimised cell dimensions. Simulations were undertaken to evaluate the influence of:
Figure 9 shows lattice vibration modes with change in pressure in the optical region (< 300 meV) for LaH10. Both cases include the shortest La–La and La–H32f distances noting that the La–H8c distance, not included in these calculations, pertains to the next–nearest La coordination sphere (i.e. CN = 8). A schematic of the LaH10 unit cell that includes the La-H8c distance is shown in Figure S10c (Supplemental).
Supplemental Table S4 lists the lowest energy optical vibration modes for LaH10 at pressures between 90 GPa and 250 GPa. Each mode has equivalent, or nearly equivalent, vibration modes (within 1 meV) along three orthogonal directions of the cubic structure. Higher energy modes are calculated by this method but are not shown in this work.
A signature effect that pertains to superconductors – a softening of vibrations – is clearly shown for the shortest H–H distances for superconducting LaH10 in Figure 9. A substantial reduction in vibration energy occurs at 135 GPa for both H–H bonding scenarios (4 × H–H and 2 × H–H). For Modes 1 and 2, the 2 × H–H model shows a gradual reduction in energy to 220 GPa and, as with Mode 3, dramatically increases (by > 100 meV) at pressures > 220 GPa.
For the 4 × H–H model, the three modes show a significant change in vibration energy (± 100 meV) at 135 GPa and a subsequent gradual increase to 220 GPa (Figure 9). For Modes 2 and 3, the relative difference between the two scenarios (i.e. 2 × H–H vs 4 × H–H) with change in pressure increases towards higher pressure values (Supplemental Table S4). These lattice vibration trends suggest that the two additional H32f bond types (i.e. H32f–H32fb and H32f–H32fc) exert a moderating influence on lattice vibrations with increased pressure.
Subtle shifts in electron distribution, or atom–atom bonding, in response to changes in polyhedral anisotropy caused by applied pressure influences the superconducting behaviour of LaH10 under pressure. The collective behaviour of these lattice vibrations is consistent with our earlier suggestion that the dome-like format for Tc with pressure, reproduced in Figure 6, is influenced by hydrogen bonding in the LaH10 structure. Furthermore, these lattice vibration models for LaH10 are generated from optimised cell dimensions and space group symmetry only.
Visualisation of these LaH10 lattice vibrations, using CrystalMaker V11.5, shows that the dominant direction(s) of atom movement are parallel with the (h0l), (hk0) and (0kl) planes as shown in Figure 8. These primary directions are consistent for all pressures evaluated. At lower pressure (< 135 GPa) and higher pressure (> 220 GPa) minor variations, such as additional modes (e.g. at 110 GPa) or movement parallel to primary axes (i.e. (0k0) etc) also occurs. In all cases, visualisation of La atoms shows minimal motion (< 2 meV) while H atom movements are in articulated groups (e.g. H32f atoms as a cube sub-unit; or H8c atoms linked with 4 × H32f atoms; as shown in Figure 2 and Figure 9) of varying intensity at different frequencies. A comparison of lattice vibrations for an individual clathrate moiety as well as that of next-nearest neighbour bonds are planned for a separate publication.
The frequencies for Modes 1 and 2 identified from Monte Carlo simulations are comparable with optical phonon bands calculated for cubic LaH10 at 200 GPa [9]. Sun et al. [9] show that phonon dispersion (PD) calculations for LaH10 suggest a stable structure at 200 GPa (i.e. no negative frequencies) but not at 180 GPa. Using PDs calculated at 180 GPa and 200 GPa, Sun et al. [9] suggest that low-lying H–H “wagging” vibrations of the transverse acoustic phonon along the Γ–X direction leads to a structural instability and distortion that conforms with a monoclinic C2/m structure.
In addition, Sun et al. [9] observe a trend towards softening of phonons along the Γ–X direction of the cubic LaH10 structure. Wagging motions along Γ–X are assigned to the , (101) and (011) planes [9]. These observations are consistent with lattice vibration trends discussed above and shown in Figure 9. Furthermore, the substantive change in lattice dynamics at pressures previously identified as points of transformation to/from a cubic structure [9,14] suggests a subtle, and flexible dependence on H–H bonding that drives superconductivity in LaH10.
We have attempted PD calculations for LaH10 at a range of pressures (with and without SOC correction) but have been unable to obtain positive values for bands in the acoustic and low energy optical regions. These calculations require high numbers of cores and long time periods to complete full PDs that enable resolution of fine detail. As noted in Supplemental Figure S3, the DFT resolution obtainable using our available computational capacity appears insufficient for convergence of LaH10 PDs. In earlier work on layered superconductors [21,54], we have shown that the k-grid interval is an important determinant for useful PD interpretation. Perhaps phonon calculations using DFT, particularly for high symmetry superconductors, are sensitive to other factors in addition to substantive electron-phonon interactions.
3.3. Tight Binding Potentials and Free Electrons
We have shown in earlier work on layered superconductors, such as MgB2, that electronic bands perpendicular to the layers can be better approximated by revised tight-binding equations [18]. Revised tight-binding equations based on band structure calculations reveal that additional terms are essential in order to accommodate the superconducting gap and to appropriately balance band energies at key symmetry points [18]. For convenience, we outline these revisions below.
Tight-binding equations for layered compounds are normally written as [30]:
where is the transfer coefficient and are the heavy and light effective masses. However, careful inspection of the energy values at key symmetry points indicates that these energies are not part of the same, single cosine function. The energies in the antibonding branch of the Fermi surface are slightly offset by a value 2Δ of energy (for a given ) from the energies of the bonding branch of the Fermi surface. Hence, we adjust Equation (1) as follows:
For high symmetry directions, the equations obtain a singular cosine dependence (e.g. along the [101]* reciprocal direction or when ).
Tight binding equations for s-orbitals and body-centred or face-centred cubic structures include products of cosine functions [56,57]. Again, for high symmetry directions and with some algebra, the equations can be written as a singular cosine dependence. Alternatively, the cosine dependence can be explained as being associated with the corresponding orbital electron density waves, largely driven and reinforced by the acoustic phonons participating in Fermi surface nesting [58]. Fermi surface nesting will be discussed in more detail in a separate publication.
Cosine shaped bands arise when applying the tight-binding (or LCAO) expansion in atomic orbitals to crystalline solids [59]. These types of bands are typically associated with linear chains (or periodic arrays) of identical atoms [59,60]. The cosine band for a chain of s atomic orbitals can be written as:
where d is the inter-atomic distance. The free electron approximation can be combined with the tight-binding approach to generate approximations to the Vsss coefficients as:
ekS = eS + 2 Vsss cos(kd)
Vsss = -(π2/8)ℏ2/(md2)
Approximate interatomic distances can be extracted from the amplitudes of the calculated cosine-shaped bands.
Table 3 compares these approximate interatomic distances based on the amplitude of cosine bands with calculated values determined by DFT for three pressures within the superconducting region. We have estimated the amplitudes of cosine shaped bands for cubic and primitive lattices with and without SOC adjustment (Supplemental Table S2). For DFT calculations with or without SOC, the amplitude increases with pressure resulting in a reduced interatomic distance as expected. The proportionalities (i.e. one-half, one-third) explored in Table 3 give a strong match to experimental values considering that calculations are for absolute zero temperature, while experimental values (which form the basis of this work; see Figure 2) are at near room temperature.
Similar asymmetry can be identified in cosine shaped bands with and without SOC included in our DFT calculations, although centred at different energies from EF. We suggest that the inclusion of SOC provides fine-tuning in energy and amplitude to the dominant electron-phonon coupling for resonance at the Fermi level (via nesting to be discussed in more detail in a separate publication). This electron-phonon coupling is inherently tuned at EF in conventional superconductors and does not require additional interactions to conform to theoretical paradigms.
These attributes may nullify appeals to invoke alternative theories for superconductivity by focusing on finding a separate tuning mechanism for electron-phonon coupling. As noted by Hirsch and colleagues [47,61,62], there is theoretical and practical evidence to suggest that misunderstanding of the role of holes on superconductivity can lead to misinterpretation of subtle yet relevant asymmetry in orbital radii for electron and hole carriers within band structures. In particular, misunderstanding relates to the importance or otherwise, of bands that cross EF into the antibonding states at energies near the non-bonding cross-over, away from the nearly full/empty band maximum/minimum. These studies show that interactions other than electron-ion, such as electron-electron interactions, have a critical role to play in superconductivity mechanisms [61,63] and complement electron-phonon coupling. In this work, with systematic analysis of ab initio DFT models on LaH10, we infer that H–H interactions (which by their nature, involve electron-electron interaction) are worthy of further understanding in the quest for predictable models of superconductivity.
4. Methods
DFT calculations on the EBS and Fermi surface (FS) for LaH10 at pressures from 90 GPa to 300 GPa were undertaken with Materials Studio CASTEP Versions 2023 and 2025 [64]. These pressures encompass the stability region for LaH10 as well as those pressure regimes below and above experimentally determined superconducting cubic phase stability [9,14]. Key parameters such as plane wave cut-off energies, pseudopotentials, and k-point grids are critical enablers of meV resolution for EBS and phonon dispersion (PD) calculations as noted in earlier publications [19,54]. All band structure representations utilise .xlsx files converted from direct .csv outputs of Materials Studio DFT calculations. These data are then re-plotted using SigmaPlot for Windows Version 15 (Grafiti LLC, Palo Alto, CA USA) to enable precise interpretation of band energies and intersections at reciprocal lattice symmetry points.
Crystal structures are re-calculated and visualised with Crystal Maker V 11.5 software (CrystalMaker Software Limited, Begbroke, Oxfordshire, UK) using geometry-optimised cell parameters of experimentally determined values as described below. Modelled crystal vibrations with force field equations use Buckingham and Lennard-Jones atom potentials for solid state inorganic materials with Monte Carlo simulation using Crystal Maker V11.5 software [65].
4.1. Calculation Parameters
All DFT calculations employ cut-off energies of 990 eV, k-point grid of 0.005 Å-1, norm-conserving pseudopotentials and either the Perdew-Zunger Local Density Approximation (LDA) or Generalised Gradient Approximation (GGA) [66,67] for the exchange-correlation functional. Slightly coarser k-grids (e.g. 0.01 Å-1 and 0.007 Å-1) and finer k-grids (e.g. 0.003 Å-1) are used to identify, or exemplify, nuanced electronic band structure changes with k-grid value, particularly at multiple band intersections or crossing points.
Optimisation calculations used the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm employing a total energy/atom convergence tolerance of 0.5 x 10-6 eV, an eigen-energy convergence tolerance of 0.1375 x 10-6 eV, a Fermi energy convergence tolerance of 0.2721 x 10-13 eV, a Gaussian smearing scheme of width 0.1 eV and the Pulay density mixing scheme with charge density mixing g vector of 1.5 Å-1. Both non-relativistic (Schrödinger [68]) and relativistic (Dirac [69,70], Koelling-Harmon [64]) treatments are calculated for the LaH10 structures at a range of pressures. Selected examples are provided in the text and Supplemental section for comparison.
Other pressure-dependent calculations with ultrasoft pseudopotentials [71] within the LDA and GGA approximations were carried out for additional comparisons. For consistency, we report calculations using the LDA functional for ease of comparison across pressure-temperature-structure data for LaH10. Equivalent calculations using the GGA functional show similar trends and closely aligned optimised cell parameters for a cubic unit cell as shown in Supplemental Figure S11. For optimisation calculations, all atoms are allowed to relax along orthogonal directions until residual forces are less than 0.0005 eV/Å. The input lattice constant, a = 5.1019(5) is taken from X-ray diffraction results for LaH10 at 150 GPa from the work of Drozdov et al. [2].
Choice of DFT parameters
We chose the LDA and GGA functionals and the cut-off energy for the plane wave basis set in order to elicit nodal and anti-nodal character of hydrogen atoms in DFT calculations. The published literature includes many DFT calculations that opt to use ultrasoft pseudopotentials (USPs) in order to reduce required cut-off energies for band calculations and often to reduce compute overheads.
Typically, USPs are invoked with a plane-wave basis set for tightly bound orbitals that have a substantial fraction of their weight inside the core region of an atom, violating norm-conservation so as to reduce the need for a high cut-off energy [64]. However, s-orbitals have significant anti-node character with high wave amplitude at the origin of the atom (representing a region of high electron density). A high amplitude weight localized at the origin raises doubts regarding the level of certainty introduced in a calculation by reduced cut-off energies particularly when considering a high proportion of hydrogen atoms (with s-orbitals) in the LaH10 structure.
We explored the effects of reduced cut-off energies, as well as k-grid values, using similar functionals in earlier calculations for MgB2 [54]. We show that for an ab initio calculation:
For some calculations that deploy USPs to describe atoms, the norm-conservation condition is violated since the charge associated with orbitals from the core region is removed during calculation. A generalised ortho-normalization of the charge density applied at the end of a calculation via an augmented core charge density falls short of executing a full wave function normalization during the calculation [64].
To evaluate this maxim, we conducted further tests on LaH10 structures using USPs on plane waves. Calculated results using USPs without SOC appear very similar to those with SOC using norm-conserving pseudopotentials. This outcome is unlike that shown above in Section 2 and so, our reticence to utilise USPs in these calculations. These results indicate that important differences in EBSs may arise when using augmented plane waves compared to full plane waves, particularly when large fractions of s-orbital character are involved.
4.2. Superlattices and Symmetry
A range of calculated band structures for different pressures have been examined using DFT. The calculations follow two fundamental symmetry conditions:
- (i)
- face centred cubic of space group and
- (ii)
- the reduced primitive lattice (space group P1) of the fcc cubic structure.
Supplemental Figures S12 and S13 show the real and reciprocal space projections of the derived primitive cell and Brillouin zone boundaries for LaH10. The transformation(s) between the primitive (P1) asymmetric unit and the cubic unit cell (), or vice versa, involve well established principles as outlined in Supplemental Figure S14.
Using this cubic–primitive transformation, we track key low dimension h0l planes such as (101) and . This enables interpretation of the reciprocal space cosine curve and coeval real space characteristics. Within each space group, we calculate band structures with and without SOC. Using these band structures for cubic or primitive lattices, we show how to estimate the asymmetry of a specific cosine-shaped band by folding the band along key reciprocal space direction(s).
4.3. Previous Experiments and Calculations
Experimental data for LaH10 at high pressures are broadly consistent with replicate experiments undertaken by different laboratories [2,5,8]. Calculations on LaH10 show that it is stable above 135 GPa to 220 GPa [9,14] when quantum effects are considered; consistent with other experimental determinations [3,5,9]. Due to technical difficulties, some reports on high-pressure experiments have not provided either cell dimensions or Tc data for LaH10. Thus, few correlated data points of a, Tc and P for experiments are represented in Figure 2. Drozdov et al. [2] and Sun et al. [9] report all three parameters for LaH10 and are designated by filled crossed diamonds and by solid squares, respectively, in Figure 2 and Figure 6.
For data in Figure 2, other experiments determine either cell dimension or Tc with applied pressure. Somayazulu et al. [5] report cell volume data with a range of values from which we re-calculate cubic cell dimensions for three experiments. For the data point by Geballe et al. [3], we utilize an estimate of error for pressure determination (± 9 GPa) based on the detailed analysis by Sun et al. [9]. Unit cell calculations in this work using DFT with the LDA functional are broadly consistent with bond length trends (but not values) calculated by Wang et al. [35]. A comparison of optimized cell dimensions calculated using the LDA functional across a wide pressure range (Supplemental Table S1) for LaH10 shows that values differ substantially for calculations without SOC and with SOC.
5. Conclusions
We utilise ab initio DFT calculations on LaH10 to compare band structures with and without adjustments for SOC at pressures that include and extend beyond the experimentally determined superconductivity region. In this evaluation, we identify cosine-shaped bands that cross the Fermi level along the Γ–X direction of reciprocal space. These cosine-shaped bands are observable in EBS using either a cubic or a reduced primitive cell with non-relativistic or relativistic treatments. We relate these reciprocal space dimensions to changes in cell dimensions, polyhedral distortions, and hydrogen bonding.
Cosine-shaped bands display asymmetry between the higher energy antibonding region and the lower energy bonding region; an asymmetry clearly noticeable when folding is carried out at the mid-nodal, non-bonding point along Γ–X. This asymmetry carries information on the superconducting gap, and correlates well with experimental data for Tc as a function of pressure. Close examinations of LaH10 band structures show subtle changes in band topology at the Γ and X nodes between models at the non-superconducting pressures (i.e. < 135 GPa and > 220 GPa) and the superconducting regime. Calculation of the cosine band asymmetry enables estimates for Tc based on our earlier evaluations for layered superconductors using the same methodology. Estimates of Tc for the superconducting pressure regime (135 GPa < P < 220 GPa) align closely (within systematic error) with the experimentally determined dome-like format for LaH10.
Calculations with and without SOC show distinctive differences in optimised cell dimensions and the DOS near the Fermi level. DFT calculation with SOC reduces the optimised cubic cell dimension, a, for a particular pressure by an average ~1.2% between 135 GPa and 220 GPa. This change in a is not linear with higher effect as pressure increases. Adjustment for SOC also results in the presence of extra bands near the Fermi level, of which some are often termed “flat bands”. Close examination of LaH10 band structures show that “flat bands” are not flat but curve towards and away from EF at a nodal point. With SOC, a van Hove singularity occurs at the same energy, −0.122(3) eV, between 90 GPa and 250 GPa. Without SOC adjustment, a high DOS occurs at varying positive values above EF and, on average, ~1.1 eV from the value for the respective SOC-generated EBS. These comparative trends are evident for the full range of pressures from 90 GPa to 250 GPa.
Using a reduced cell, we relate the inflection of cosine-shaped curves to the line (point) of intersection of (h0l), (hk0) and (0kl) planes in the real space structure. These planes are parallel to key H–H interatomic distances (e.g. H8c–H32f and H32f–H32f) that define integral surfaces of the clathrate chamfered cube topology. The LaH10 structure for all calculated pressures is a distorted chamfered cube with an inner hexagonal angle (i.e. the angle H32f-H8c-H32f) of 121.2o compared with 125.26o in an ideal truncated rhombic dodecahedron. This distortion reduces with pressure and is in response to mixed H–H bonding configurations (i.e. electron densities) that gradually shift with change in pressure.
Lattice vibration calculations show that four nearest neighbour H–H distances significantly influence atom movement both in intensity and direction. A distinct softening of lattice vibrations for three key modes with energy < 300 meV occurs between 135 GPa and 220 GPa pressure. At pressures < 135 GPa and > 220 GPa, lattice vibrations shift by ~100 meV (i.e. ~806 cm-1) to higher energy. Collective behaviour of these lattice vibrations is also consistent with the dome-like format shown for experimental Tc values with pressure for LaH10. The primary directions of atom movement in the pressure region with vibration softening are parallel with the (h0l), (hk0) and (0kl) planes.
Inclusion of SOC adjustment in DFT calculations results in promotion of electrons to f-orbitals which are strongly hybridized with H s-orbitals. This electronic configuration for LaH10, utilizing 4f orbitals of La atoms, offers bonding orientations conducive to a stable cubic structure and to interatomic distances of a chamfered cube topology. Without adjustment for SOC, DFT calculations allocate La valence electrons to the 6s2 and 5d1 orbitals with limited options for orbital overlap. For LaH10, the crystal structure and consequent atom-atom bonding environments, while not canonical parameters typical of earlier studies, are clearly important for understanding superconductivity mechanisms.
Systematic evaluation of electron distributions in LaH10 offers further insight into the key determinants that enable a specific “superconducting window” of ~85 GPa between 135 GPa and 220 GPa. As noted, the structure is stable within this pressure range and tolerates limited electron density redistributions predominantly centred on H atoms. DFT models that allow for SOC provide definitive detail on band structures that enhance predictive models for ambient pressure superconductors and are encouraged.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Optimised cell dimensions for LaH10 between 140 GPa and 220 GPa. Figure S2: Crystal structure of LaH10; Figure S3: Asymmetric unit with P1 symmetry; Figure S4: Reciprocal space projections for LaH10 primitive cell; Figure S5: EBS for LaH10 with and without SOC; Figure S6: Estimates of systematic error for EBS calculations; Figure S7: Cosine curves at Γ–X without SOC; Figure S8: Cosine curves at Γ–X with SOC; Figure S9: Experimental and calculated values of Tc for LaH10 with applied pressure; Figure S10: EBS for LaH10 showing “flat band” and band topology; Figure S11: Calculated DOS for LaH10 with pressure; Figure S12: Cubic to Primitive transformation; Figure S13: Details of LaH10 lattice planes and bonding; Figure S14: Orbitals of the cubic 4f set; Table S1: Cell dimensions for Cubic LaH10 without and with SOC correction; Table S2: DFT derived parameters for LaH10; Table S3: Inter-atomic distances for LaH10; Table S4: Calculated lattice vibrations for LaH10 with pressure.
Author Contributions
Conceptualization, J.A.A. and I.D.R.M.; methodology, J.A.A. and I.D.R.M.; software, J.A.A. and I.D.R.M.; validation, J.A.A. and I.D.R.M.; formal analysis, J.A.A.; investigation, J.A.A. and I.D.R.M.; data curation, J.A.A. and I.D.R.M.; writing—original draft preparation, J.A.A.; writing—review and editing, I.D.R.M.; visualization, J.A.A. and I.D.R.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Additional data presented in this study are available in Supplemental Information. Raw data files from DFT calculations are available from Open Science Framework [weblink to be provided].
Acknowledgments
The authors acknowledge the e-Research Office at QUT, for access to high-performance computing. During the preparation of this manuscript and study, the authors did not use any artificial intelligence tools. The authors are grateful to Antonio Bianconi who encouraged further investigation of spin orbit coupling.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Drozdov, A.P.; Eremets, M.I.; Troyan, I.A.; Ksenofontov, V.; Shylin, S.I. Conventional superconductivity at 203 kelvin at high pressures in the sulfur hydride system. Nature 2015, 525, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Drozdov, A.P.; Kong, P.P.; Minkov, V.S.; Besedin, S.P.; Kuzovnikov, M.A.; Mozaffari, S.; Balicas, L.; Balakirev, F.F.; Graf, D.E.; Prakapenka, V.B.; et al. , Superconductivity at 250 K in lanthanum hydride under high pressures. Nature 2019, 569, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Geballe, Z.M.; Liu, H.Y.; Mishra, A.K.; Ahart, M.; Somayazulu, M.; Meng, Y.; Baldini, M.; Hemley, R.J. Synthesis and Stability of Lanthanum Superhydrides. Angew Chem Int Ed 2018, 57, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Sun, Y.; Pickard, C.J.; Needs, R.J.; Wu, Q.; Ma, Y.M. Hydrogen Clathrate Structures in Rare Earth Hydrides at High Pressures: Possible Route to Room-Temperature Superconductivity. Phys Rev Lett 2017, 119, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Somayazulu, M.; Ahart, M.; Mishra, A.K.; Geballe, Z.M.; Baldini, M.; Meng, Y.; Struzhkin, V.V.; Hemley, R.J. Evidence for Superconductivity above 260 K in Lanthanum Superhydride at Megabar Pressures. Phys Rev Lett 2019, 122, 1–6. [Google Scholar] [CrossRef]
- Pickard, C.J.; Errea, I.; Eremets, M.I. Superconducting Hydrides Under Pressure, in: Marchetti, M.C. Mackenzie, A.P. (Eds.), Ann Rev Cond Matter Phys 2020, pp. 57-76.
- Minkov, V.S.; Bud’ko, S.L.; Balakirev, F.F.; Prakapenka, V.B.; Chariton, S.; Husband, R.J.; Liermann, H.P.; Eremets, M. Magnetic field screening in hydrogen-rich high-temperature superconductors. Nat Comm 2022, 13, 1–6. [Google Scholar] [CrossRef]
- Hong, F.; Yang, L.; Shan, P.; Yang, P.; Liu, Z.; Sun, J.; Yin, Y.; Yu, X.; Cheng, J.; Zhao, Z. Superconductivity of Lanthanum Superhydride investigated using standard four-probe configuration under high pressures. Chin Phys Lett 2020, 37, 1–5. [Google Scholar] [CrossRef]
- Sun, D.; Minkov, V.S.; Mozaffari, S.; Sun, Y.; Ma, Y.M.; Chariton, S.; Prakapenka, V.B.; Eremets, M.; Balicas, L.; Balakirev, F.F. High-temperature superconductivity on the verge of a structural instability in lanthanum superhydride. Nat Comm 2021, 12, 1–7. [Google Scholar] [CrossRef]
- Eremets, M.I.; Minkov, V.S.; Drozdov, A.P.; Kong, P.P.; Ksenofontov, V.; Shylin, S.I.; Bud’ko, S.L.; Prozorov, R.; Balakirev, F.F.; Sun, D.; et al. , High-Temperature Superconductivity in Hydrides: Experimental Evidence and Details. J Supercond Novel Magnet 2022, 35, 965–977. [Google Scholar] [CrossRef]
- Eremets, M.I.; Minkov, V.S.; Drozdov, A.P.; Kong, P.P. The characterization of superconductivity under high pressure. Nat Mater 2024, 23, 26–27. [Google Scholar] [CrossRef]
- Quan, Y.; Ghosh, S.S.; Pickett, W.E. Compressed hydrides as metallic hydrogen superconductors. Phys Rev B 2019, 100, 1–10. [Google Scholar] [CrossRef]
- Papaconstantopoulos, D.A.; Mehl, M.J.; Chang, P.H. High-temperature superconductivity in LaH10. Phys Rev B 2020, 101, 1–6. [Google Scholar] [CrossRef]
- Errea, I.; Belli, F.; Monacelli, L.; Sanna, A.; Koretsune, T.; Tadano, T.; Bianco, R.; Calandra, M.; Arita, R.; Mauri, F.; et al. , Quantum crystal structure in the 250-kelvin superconducting lanthanum hydride. Nature 2020, 578, 66–72. [Google Scholar] [CrossRef]
- Wang, C.; Yi, S.; Cho, J.H. Multiband nature of room-temperature superconductivity in LaH10 at high pressure. Phys Rev B 2020, 101, 1–5. [Google Scholar] [CrossRef]
- Liu, L.L.; Wang, C.Z.; Yi, S.; Kim, K.W.; Kim, J.; Cho, J.H. Microscopic mechanism of room-temperature superconductivity in compressed LaH10. Phys Rev B 2019, 99, 1–5. [Google Scholar] [CrossRef]
- Liu, H.Y.; Naumov, I.I.; Geballe, Z.M.; Somayazulu, M.; Tse, J.S.; Hemley, R.J. Dynamics and superconductivity in compressed lanthanum superhydride. Phys Rev B 2018, 98, 1–4. [Google Scholar] [CrossRef]
- Alarco, J.A.; Mackinnon, I.D.R. Superlattices, Bonding-Antibonding, Fermi Surface Nesting, and Superconductivity. Cond Matter 2023, 8, 1–13. [Google Scholar] [CrossRef]
- Wang, B.C.; Bianconi, A.; Mackinnon, I.D.R.; Alarco, J.A. Superlattice Symmetries Reveal Electronic Topological Transition in CaC6 with Pressure. Crystals 2024, 14, 1–21. [Google Scholar] [CrossRef]
- Wang, B.C.; Bianconi, A.; Mackinnon, I.D.R.; Alarco, J.A. Superlattice Delineated Fermi Surface Nesting and Electron-Phonon Coupling in CaC6. Crystals 2024, 14, 1–17. [Google Scholar] [CrossRef]
- Alarco, J.A.; Chou, A.; Talbot, P.C.; Mackinnon, I.D.R. Phonon Modes of MgB2: Super-lattice Structures and Spectral Response. Phys Chem Chem Phys 2014, 16, 24443–24456. [Google Scholar] [CrossRef]
- Alarco, J.A.; Shahbazi, M.; Talbot, P.C.; Mackinnon, I.D.R. Spectroscopy of metal hexaborides: Phonon dispersion models. J Raman Spect 2018, 49, 1985–1998. [Google Scholar] [CrossRef]
- Alarco, J.A.; Talbot, P.C.; Mackinnon, I.D.R. Phonon dispersion models for MgB2 with application of pressure. Physica C: Supercond Appl 2017, 536, 11–17. [Google Scholar] [CrossRef]
- Merchant, A.; Batzner, S.; Schoenholz, S.S.; Aykol, M.; Cheon, G.; Cubuk, E.D. Scaling deep learning for materials discovery. Nature 2023, 624, 80–90. [Google Scholar] [CrossRef]
- Riebesell, J.; Surta, T.W.; Goodall, R.E.A.; Gaultois, M.W.; Lee, A.A. Discovery of high-performance dielectric materials with machine-learning-guided search. Cell Reports Phys Sci 2024, 5, 1–21. [Google Scholar] [CrossRef]
- Andersson, S.; Hyde, S.T.; Larsson, K.; Lidin, S. Minimal surfaces and structures - from inorganic and metal crystals to cell-membranes and bio-polymers. Chem Rev 1988, 88, 221–242. [Google Scholar] [CrossRef]
- Perenlei, G.; Talbot, P.C.; Martens, W.N.; Riches, J.; Alarco, J.A. Computational prediction and experimental confirmation of rhombohedral structures in Bi1.5CdM1.5O (M = Nb, Ta) pyrochlores. RSC Advances 2017, 7, 15632–15643. [Google Scholar] [CrossRef]
- Jenkins, T.; Alarco, J.; Cowie, B.C.C.; Mackinnon, I. Validating the Electronic Structure of Vanadium Phosphate Cathode Materials. ACS Appl Mater Interfaces 2021, 13, 45505–45520. [Google Scholar] [CrossRef]
- Jenkins, T.; Alarco, J.; Cowie, B.C.C.; Mackinnon, I. Regulation of surface oxygen activity in Li-rich layered cathodes using band alignment of vanadium phosphate surface coatings. J Mater Chem A 2022, 10, 24487–24509. [Google Scholar] [CrossRef]
- Alarco, J.A.; Gupta, B.; Shahbazi, M.; Appadoo, D.; Mackinnon, I.D.R. THz/Far infrared synchrotron observations of superlattice frequencies in MgB2. Phys Chem Chem Phys 2021, 23, 23922–23932. [Google Scholar] [CrossRef]
- Harshman, D.R.; Fiory, A.T. High-TC Superconductivity in Hydrogen Clathrates Mediated by Coulomb Interactions Between Hydrogen and Central-Atom Electrons. J Supercond Novel Magnet 2020, 33, 2945–2961. [Google Scholar] [CrossRef]
- Cappelluti, E.; Pietronero, L. Van Hove singularities and nonadiabatic effects in superconductivity. Europhys Lett 1996, 36, 619–624. [Google Scholar] [CrossRef]
- Wu, W.B.; Shi, Z.P.; Ozerov, M.; Du, Y.H.; Wang, Y.X.; Ni, X.S.; Meng, X.H.; Jiang, X.Y.; Wang, G.Y.; Hao, C.M.; et al. , The discovery of three-dimensional Van Hove singularity. Nat Comm 2024, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Urch, D.S. Orbitals and Symmetry, Penguin Books Ltd, Harmondsworth, UK, 1970.
- Wang, C.; Yi, S.; Cho, J.-H. Pressure dependence of the superconducting transition temperature of compressed LaH10. Phys Rev B 2019, 100, 1–5. [Google Scholar] [CrossRef]
- Wei, Y.; Chachkarova, E.; Plekhanov, E.; Bonini, N.; Weber, C. Exploring the Effect of the Number of Hydrogen Atoms on the Properties of Lanthanide Hydrides by DMFT. Appl Sci 2022, 12, 1–9. [Google Scholar] [CrossRef]
- The Orbitron, The University of Sheffield https://winter.group.shef.ac.uk/orbitron/atomic_orbitals/4f/index.html (Accessed on 29/05/2025).
- Visual Polyhedra, https://dmccooey.com/polyhedra/index.html (Accessed on 15/05/2025).
- Fogden, A.; Hyde, S.T. Continuous transformations of cubic minimal surfaces. Eur Phys J B 1999, 7, 91–104. [Google Scholar] [CrossRef]
- Hyde, S.T.; Ramsden, S.; Di Matteo, T.; Longdell, J.J. Ab-initio construction of some crystalline 3D Euclidean networks. Solid State Sci 2003, 5, 35–45. [Google Scholar] [CrossRef]
- Miller, G.J.; Zhang, Y.; Wagner, F.R. Chemical Bonding in Solids, Handbook of Solid State Chemistry, pp. 405-489.
- Lavroff, R.H.; Munarriz, J.; Dickerson, C.E.; Munoz, F.; Alexandrova, A.N. Chemical bonding dictates drastic critical temperature difference in two seemingly identical superconductors. Proc Nat Acad Sci 2024, 121 e2316101121.
- Amundsen, M.; Linder, J.; Robinson, J.W.A.; Zutic, I.; Banerjee, N. Colloquium: Spin-orbit effects in superconducting hybrid structures. Rev Modern Phys 2024, 96, 1–33. [Google Scholar] [CrossRef]
- Deng, S.; Simon, A.; Köhler, J. Superconductivity and Chemical Bonding in Mercury. Angew Chem Int Ed 1998, 37, 640–643. [Google Scholar] [CrossRef]
- Deng, S.; Simon, A.; Köhler, J. The origin of a flat band. J Solid State Chem 2003, 176, 412–416. [Google Scholar] [CrossRef]
- Cappelluti, E.; Pietronero, L. Nonadiabatic superconductivity: The role of van Hove singularities. Phys Rev B 1996, 53, 932–944. [Google Scholar] [CrossRef]
- Hutchinson, J.; Hirsch, J.E.; Marsiglio, F. Enhancement of superconducting Tc due to the spin-orbit interaction. Phys Rev B 2018, 97, 1–7. [Google Scholar] [CrossRef]
- Bok, J.; Bouvier, J. Superconductivity and the Van Hove Scenario. J Supercond Novel Magnet 2012, 25, 657–667. [Google Scholar] [CrossRef]
- Markiewicz, R.S. The Van Hove Singularity and High-T Superconductivity: The Role of Nanoscale Disorder and Interlayer Coupling. J Phys Chem Solids 1991, 52, 1363–1370. [Google Scholar] [CrossRef]
- Abd-Shukor, R. Acoustic Debye temperature and the role of phonons in cuprates and related superconductors. Supercond Sci Technol 2002, 15, 435–438. [Google Scholar] [CrossRef]
- Chen, X.Z.; Wang, L.; Ishizuka, J.; Zhang, R.J.; Nogaki, K.; Cheng, Y.W.; Yang, F.Z.; Chen, Z.H.; Zhu, F.Y.; Liu, Z.T.; et al. , Coexistence of near- EF Flat Band and Van Hove Singularity in a Two-Phase Superconductor. Phys Rev X 2024, 14, 1–17. [Google Scholar] [CrossRef]
- Yang, Z.S.; Ferrenti, A.M.; Cava, R.J. Testing whether flat bands in the calculated electronic density of states are good predictors of superconducting materials. J Phys Chem Solids 2021, 151, 1–5. [Google Scholar] [CrossRef]
- Malik, G.P. On the role of fermi energy in determining properties of superconductors: A detailed comparative study of two elemental superconductors (Sn and Pb), a non-cuprate (MgB2) and three cuprates (YBCO, Bi-2212 and Tl-2212). J Supercond Novel Magnet 2016, 29, 2755–2764. [Google Scholar] [CrossRef]
- Mackinnon, I.D.R.; Almutairi, A.; Alarco, J.A. Insights from systematic DFT calculations on superconductors, in: Arcos, J.M.V. (Ed.), Real Perspectives of Fourier Transforms and Current Developments in Superconductivity, IntechOpen Ltd., London UK, 2021, pp. 1-29.
- Krarroubi, C.M.; Benayad, N.; Benosman, F.; Djermouni, M.; Kacimi, S.; Zaoui, A. Exploring superconducting signatures in high-pressure hydride compounds: An electronic-structure analysis. Physica C: Supercond Appl 2025, 628.
- Patterson, J.; Bailey, B. Solid-State Physics: Introduction to the Theory, Second ed., Springer-Verlag Berlin Heidelberg Germany 2010.
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics, Saunders, Philadelphia PA USA, 1976.
- Grüner, G. Density waves in solids, 1st ed., CRC Press, Boca Raton FL USA, 1994.
- Harrison, W.A. Applied Quantum Mechanics, World Scientific, Singapore, 2000.
- Canadell, E.; Doublet, M.-L.; Lung, C. Orbital Approach to the Electronic Structure of Solids, Oxford University Press, Oxford, UK, 2012.
- Hirsch, J.E. Superconductivity begins with H, World Scientific Publishing Co. Pte. Ltd., Singapore, 2020.
- Hirsch, J.E.; Marsiglio, F. Understanding electron-doped cuprate superconductors as hole superconductors. Physica C: Supercond Appl 2019, 564, 29-37.
- Hirsch, J.E. What holes in superconductors reveal about superconductivity. arXiv e-prints, 2025; 1–12. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z Kristallogr 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Palmer, D.; Palmer, S. CrystalMaker: Interactive Crystal and Molecular Modelling, CrystalMaker Software Limited, Oxfordshire, United Kingdom, 2024, pp. 484.
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys Rev Lett 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys Rev B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Suzuki, Y.; Abedi, A.; Maitra, N.T.; Yamashita, K.; Gross, E.K.U. Electronic Schrodinger equation with nonclassical nuclei. Phys Rev A 2014, 89, 1–5. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Wills, J.M. Density functional theory for d- and f-electron materials and compounds. Int J Quantum Chem 2016, 116, 834–846. [Google Scholar] [CrossRef]
- Saue, T.; Bast, R.; Gomes, A.S.P.; Jensen, H.J.A.; Visscher, L.; Aucar, I.A.; Di Remigio, R.; Dyall, K.G.; Eliav, E.; Fasshauer, E.; et al. , The DIRAC code for relativistic molecular calculations. J Chem Phys 2020, 152, 1–18. [Google Scholar] [CrossRef]
- Bartók, A.P.; Yates, J.R. Ultrasoft pseudopotentials with kinetic energy density support: Implementing the Tran-Blaha potential. Phys Rev B 2019, 99, 235103. [Google Scholar] [CrossRef]
Figure 1.
Structure of cubic LaH10 (SG: ) using DFT optimised cell dimensions for 170 GPa with spin orbit coupling as perspective view along [101] (tilted towards the viewer by 3o) showing sequential layers of La atoms and high density of hydrogen atoms between La atoms. A unit cell is outlined in dark green. La–H and La-La bonds have been omitted for clarity. Only the two shortest hydrogen distances (H8c–H32f pink; H32f–H32f blue) are shown.
Figure 1.
Structure of cubic LaH10 (SG: ) using DFT optimised cell dimensions for 170 GPa with spin orbit coupling as perspective view along [101] (tilted towards the viewer by 3o) showing sequential layers of La atoms and high density of hydrogen atoms between La atoms. A unit cell is outlined in dark green. La–H and La-La bonds have been omitted for clarity. Only the two shortest hydrogen distances (H8c–H32f pink; H32f–H32f blue) are shown.
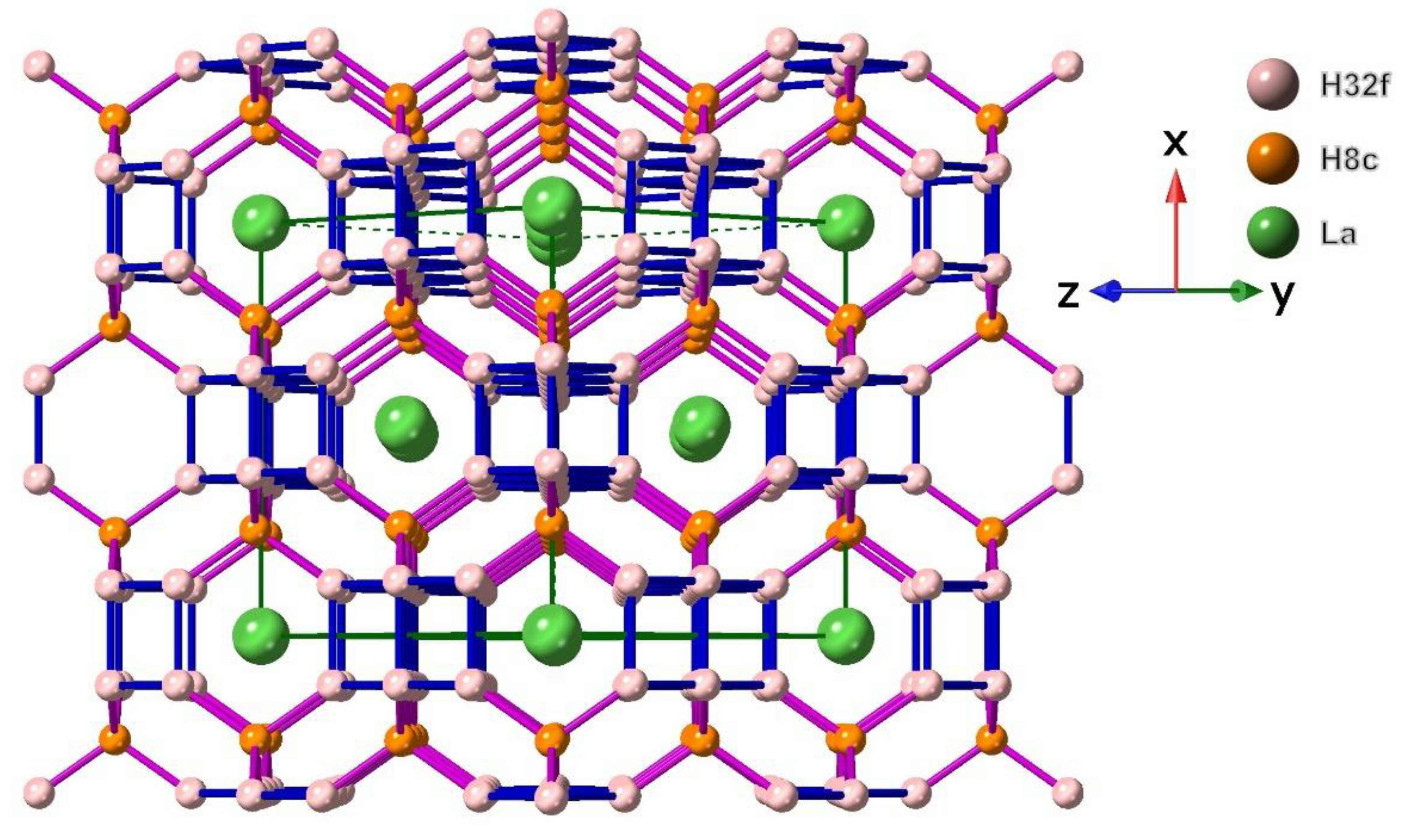
Figure 2.
Plot of cell dimensions for cubic LaH10 (SG: ) determined experimentally (triangles, diamonds, hexagon and square) [2,3,5,7,9] showing estimates of experimental error. Dotted vertical lines delineate the experimentally determined extent of superconductivity in cubic LaH10. Note that data points for ref [2] and [9] overlap at ~136 GPa. Calculated DFT values for optimized cell dimensions at pressure are shown for the LaH10 cubic structure using the LDA functional without spin orbit coupling (red circles) and with spin orbit coupling (half-red filled circle).
Figure 2.
Plot of cell dimensions for cubic LaH10 (SG: ) determined experimentally (triangles, diamonds, hexagon and square) [2,3,5,7,9] showing estimates of experimental error. Dotted vertical lines delineate the experimentally determined extent of superconductivity in cubic LaH10. Note that data points for ref [2] and [9] overlap at ~136 GPa. Calculated DFT values for optimized cell dimensions at pressure are shown for the LaH10 cubic structure using the LDA functional without spin orbit coupling (red circles) and with spin orbit coupling (half-red filled circle).
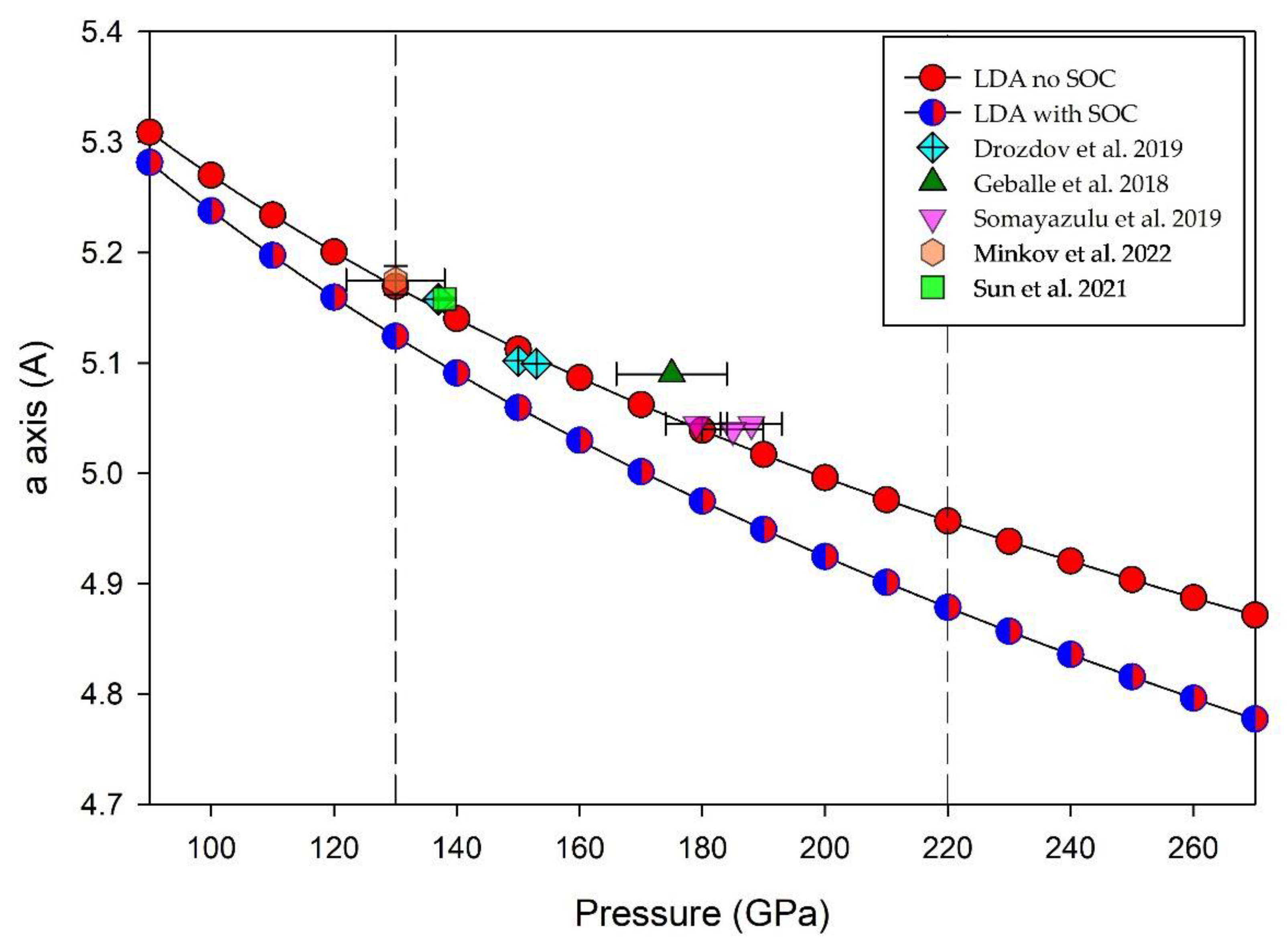
Figure 3.
Calculated band structures for LaH10 at 170 GPa for (a) cubic unit cell without SOC; (b) equivalent primitive unit cell without SOC; (c) cubic unit cell with SOC and (d) equivalent primitive unit cell with SOC. All calculations used k-grid 0.005 Å-1, except for (c) for which k = 0.01 Å-1. Note red highlighted cosine shaped bands along the M–Γ and Γ–X reciprocal directions for cubic and primitive unit cells, respectively. A nearly flat band near EF is circled in (c) and (d).
Figure 3.
Calculated band structures for LaH10 at 170 GPa for (a) cubic unit cell without SOC; (b) equivalent primitive unit cell without SOC; (c) cubic unit cell with SOC and (d) equivalent primitive unit cell with SOC. All calculations used k-grid 0.005 Å-1, except for (c) for which k = 0.01 Å-1. Note red highlighted cosine shaped bands along the M–Γ and Γ–X reciprocal directions for cubic and primitive unit cells, respectively. A nearly flat band near EF is circled in (c) and (d).
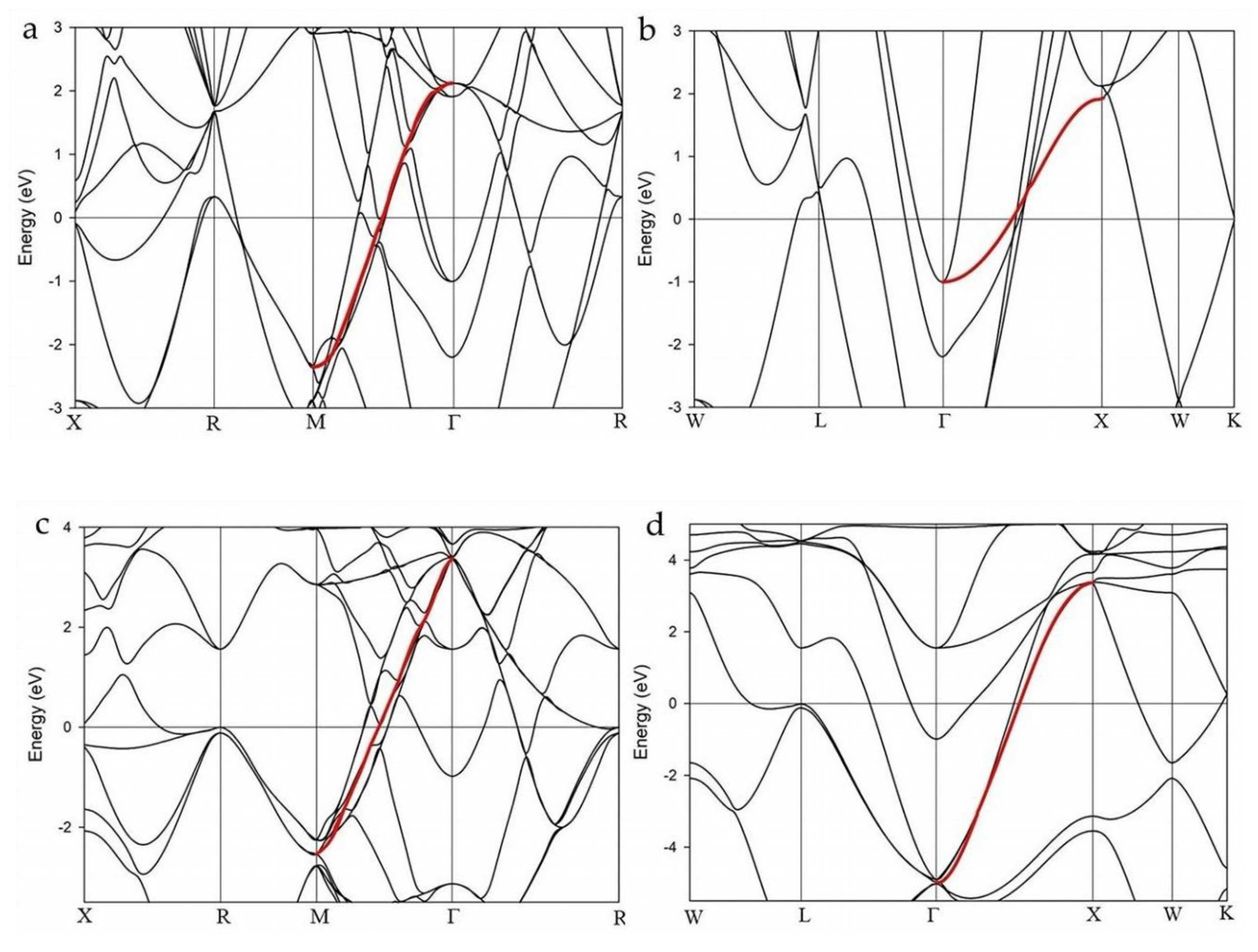
Figure 4.
Enlarged views of the cosine curve for LaH10 along the Γ–X reciprocal direction for DFT calculated EBS for a primitive cell using the LDA functional with k = 0.005 Å-1 and spin orbit coupling at 170 GPa for (a) a Schrödinger, non-relativistic and (b) a K-H relativistic treatment.
Figure 4.
Enlarged views of the cosine curve for LaH10 along the Γ–X reciprocal direction for DFT calculated EBS for a primitive cell using the LDA functional with k = 0.005 Å-1 and spin orbit coupling at 170 GPa for (a) a Schrödinger, non-relativistic and (b) a K-H relativistic treatment.
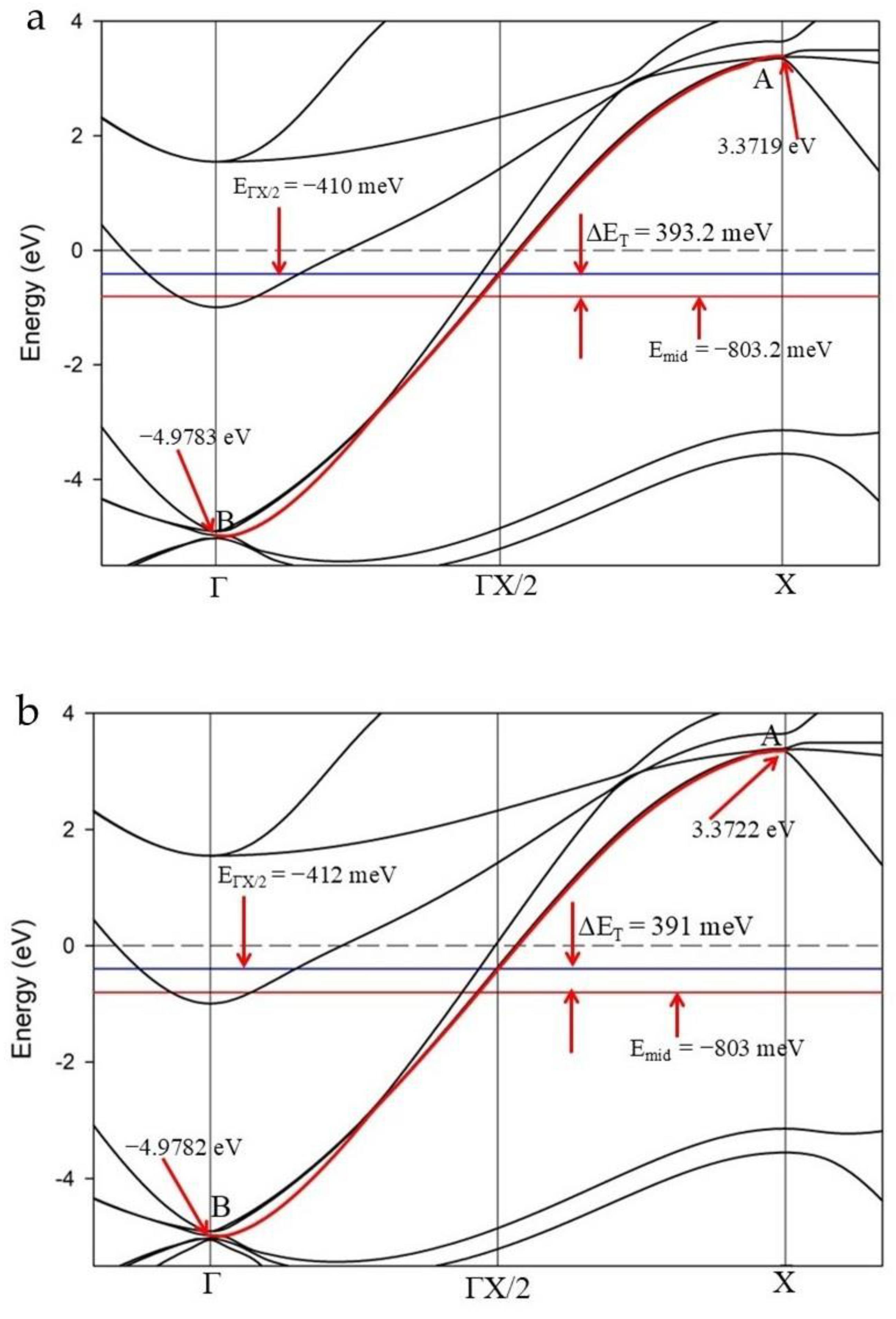
Figure 5.
Enlarged views of the cosine curve for LaH10 along the Γ–X reciprocal direction for DFT calculated EBS of a primitive cell using the LDA functional with k = 0.005 Å-1 and spin orbit coupling at (a) 140 GPa and (b) 220 GPa with K-H relativistic treatments. Note (i) the shift to lower energy with higher pressure for Emid and EΓX/2 and (ii) a wider range of energy for the cosine curve with increase in pressure.
Figure 5.
Enlarged views of the cosine curve for LaH10 along the Γ–X reciprocal direction for DFT calculated EBS of a primitive cell using the LDA functional with k = 0.005 Å-1 and spin orbit coupling at (a) 140 GPa and (b) 220 GPa with K-H relativistic treatments. Note (i) the shift to lower energy with higher pressure for Emid and EΓX/2 and (ii) a wider range of energy for the cosine curve with increase in pressure.
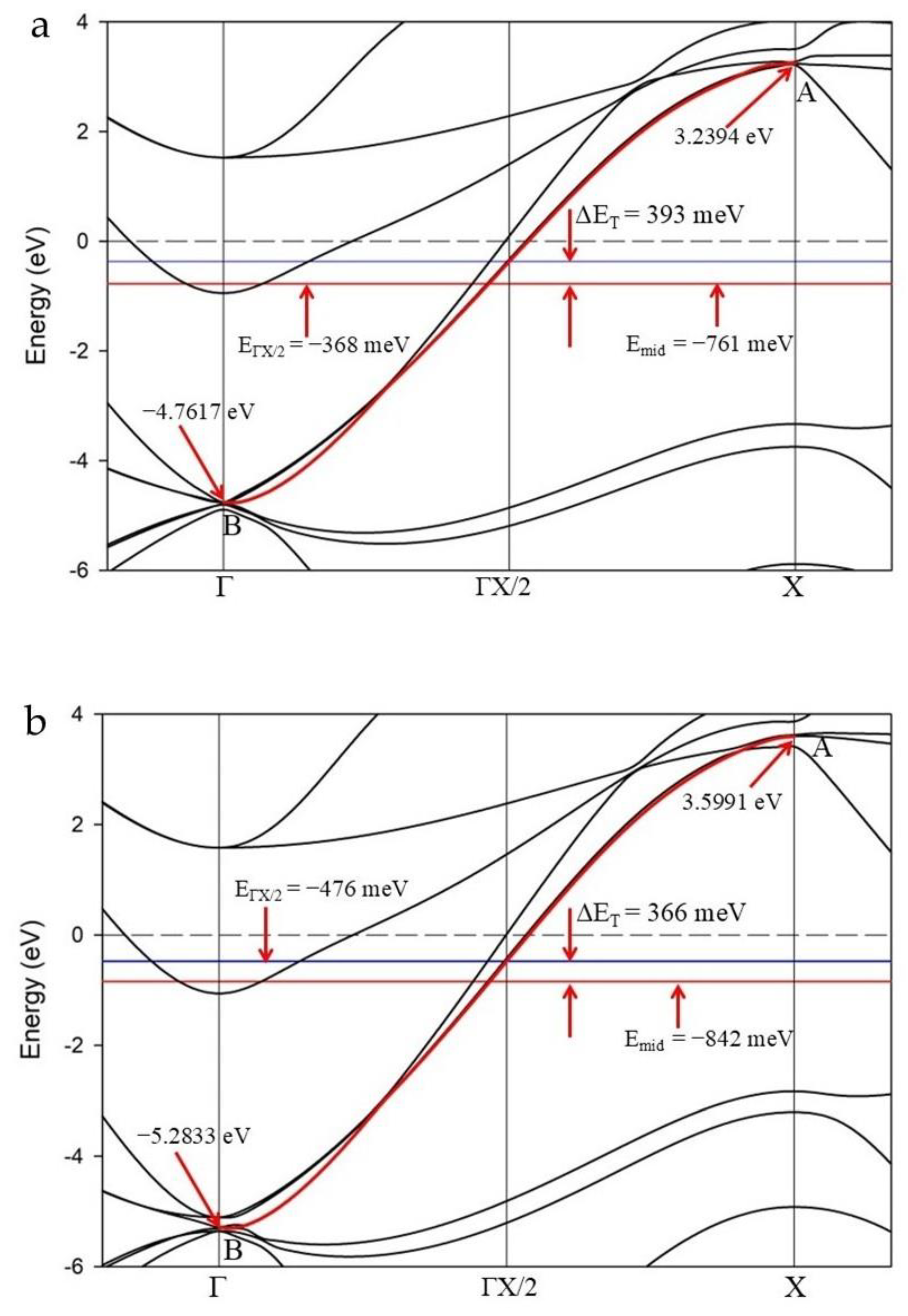
Figure 6.
Plot of (a) experimental Tc values with pressure determined for cubic LaH10 [2,5,7,8,9] (solid diamonds, triangles, hexagon and squares) and (b) ab initio DFT calculated values (circles) from this study using LDA with k = 0.005 Å-1 and SOC. Diamond symbols with an internal cross and filled squares are data points that also include contemporaneous measurement of P, Tc and cell dimension.
Figure 6.
Plot of (a) experimental Tc values with pressure determined for cubic LaH10 [2,5,7,8,9] (solid diamonds, triangles, hexagon and squares) and (b) ab initio DFT calculated values (circles) from this study using LDA with k = 0.005 Å-1 and SOC. Diamond symbols with an internal cross and filled squares are data points that also include contemporaneous measurement of P, Tc and cell dimension.
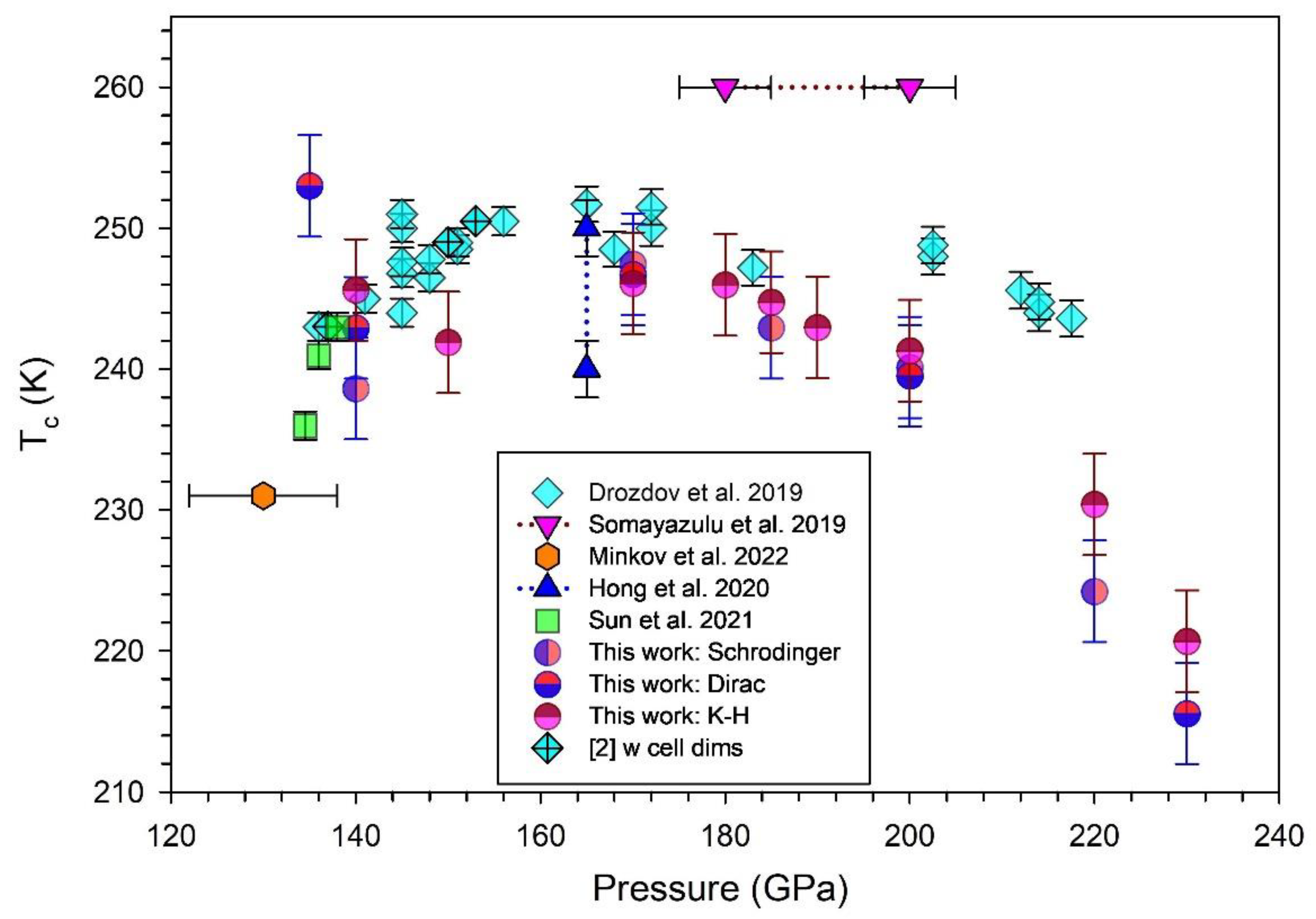
Figure 7.
DFT calculated DOS as a function of pressure for LaH10 based on (a) the cubic unit cell at 170 GPa and 200 GPa without SOC (dotted lines) and with SOC (solid lines) and (b) the primitive unit cell for a range of pressures without SOC (dotted lines) and with SOC (solid lines). Accounting for SOC in these DFT calculations results in the van Hove singularity overlapping for all pressures at −0.122(3) eV closely aligned with the Fermi level (vertical line at EF = 0 eV). Inclusion of SOC induces a shift of ~1.1 eV to key electronic band energies, associated with the van Hove singularity.
Figure 7.
DFT calculated DOS as a function of pressure for LaH10 based on (a) the cubic unit cell at 170 GPa and 200 GPa without SOC (dotted lines) and with SOC (solid lines) and (b) the primitive unit cell for a range of pressures without SOC (dotted lines) and with SOC (solid lines). Accounting for SOC in these DFT calculations results in the van Hove singularity overlapping for all pressures at −0.122(3) eV closely aligned with the Fermi level (vertical line at EF = 0 eV). Inclusion of SOC induces a shift of ~1.1 eV to key electronic band energies, associated with the van Hove singularity.
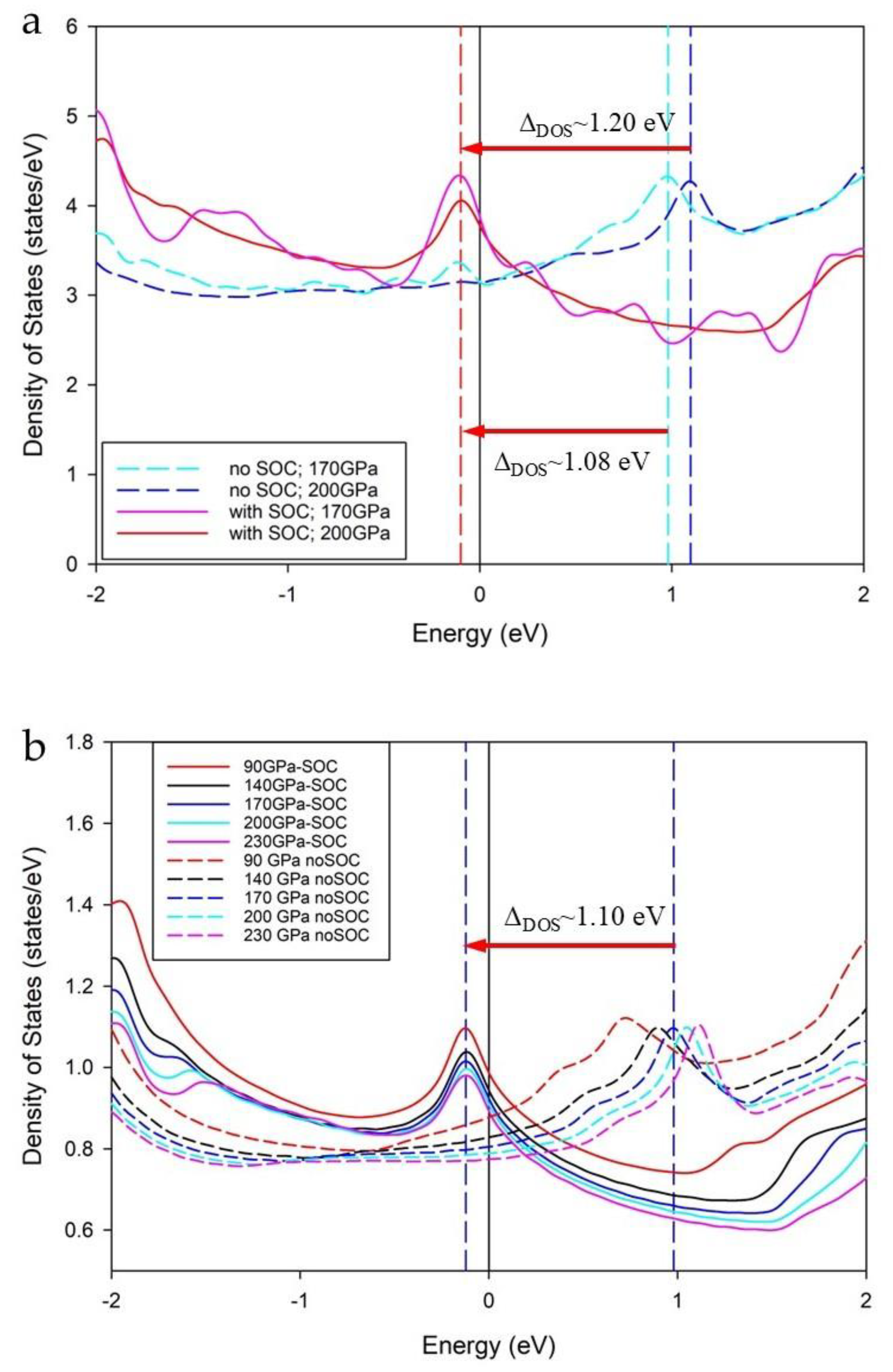
Figure 8.
Calculated LaH10 cubic structure (a) showing the [010] direction and and (101) planes. The structure is rotated 5o about z to highlight the (h0l) planes (red and dark red). These planes – and their symmetric equivalents – align perfectly with the shorter H8c–H32f hydrogen distances (pink). Longer H32f–H32f distances (in blue) also align with the primary (h00), (0k0) and (00l) directions. The two shortest H–H distances only are shown in this figure. (b) Perspective view of the clathrate moiety at 180 GPa along [101] (tilted 4o about y) showing H–H distances less than 2.0 Å and the La-H distance from face centred La sites. The unit cell is outlined in red; other atoms in the unit cell are omitted for clarity.
Figure 8.
Calculated LaH10 cubic structure (a) showing the [010] direction and and (101) planes. The structure is rotated 5o about z to highlight the (h0l) planes (red and dark red). These planes – and their symmetric equivalents – align perfectly with the shorter H8c–H32f hydrogen distances (pink). Longer H32f–H32f distances (in blue) also align with the primary (h00), (0k0) and (00l) directions. The two shortest H–H distances only are shown in this figure. (b) Perspective view of the clathrate moiety at 180 GPa along [101] (tilted 4o about y) showing H–H distances less than 2.0 Å and the La-H distance from face centred La sites. The unit cell is outlined in red; other atoms in the unit cell are omitted for clarity.
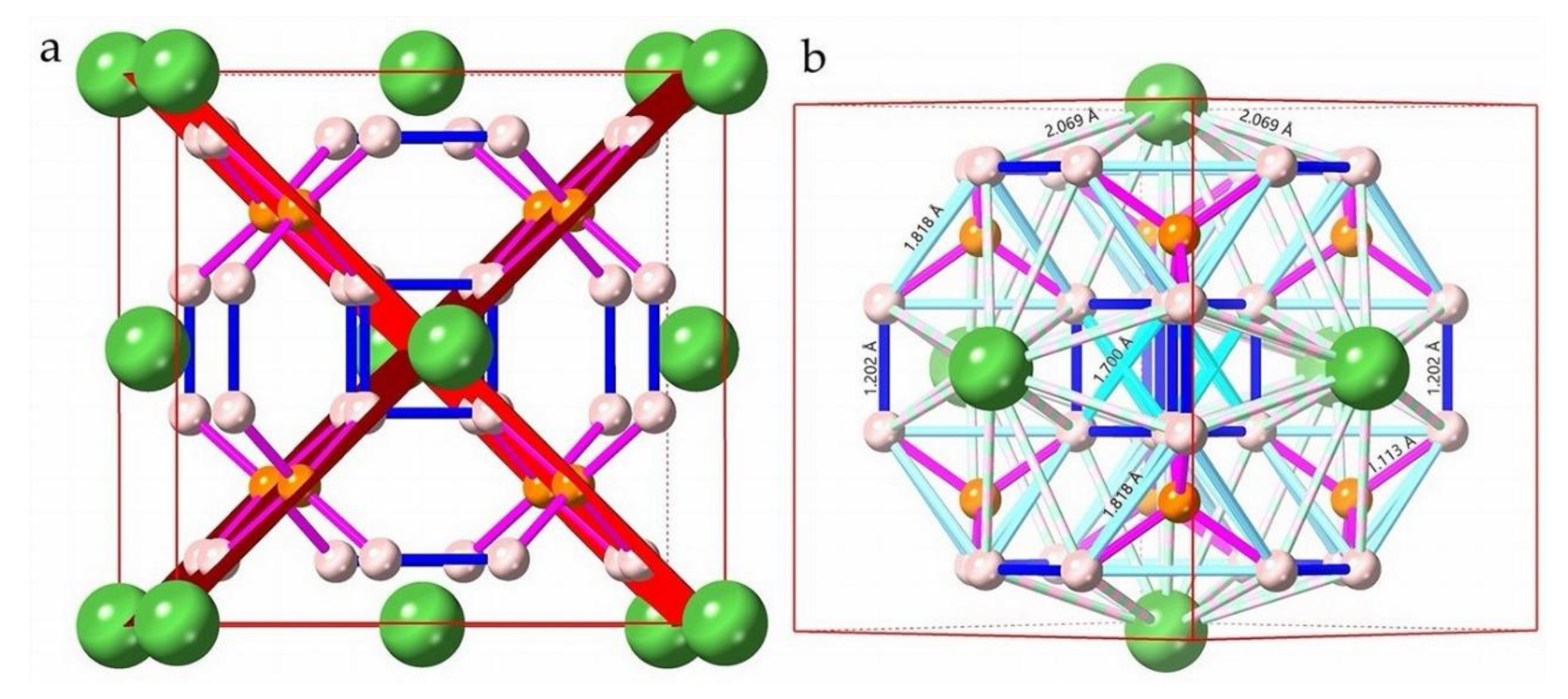
Figure 9.
Plot of lattice vibrations for LaH10 using force field equations based on DFT optimised cell dimensions with SOC. Vertical dashed lines delineate the experimentally determined superconducting region (i.e. 135 GPa < P < 220 GPa) for LaH10. The three lowest frequency optical modes are plotted for two bonding conditions (i) 2 × H–H bonds (i.e. H8c–H32f and H32f–H32fa) and (ii) 4 × H–H bonds (i.e. all four H–H bonds < 2.0).
Figure 9.
Plot of lattice vibrations for LaH10 using force field equations based on DFT optimised cell dimensions with SOC. Vertical dashed lines delineate the experimentally determined superconducting region (i.e. 135 GPa < P < 220 GPa) for LaH10. The three lowest frequency optical modes are plotted for two bonding conditions (i) 2 × H–H bonds (i.e. H8c–H32f and H32f–H32fa) and (ii) 4 × H–H bonds (i.e. all four H–H bonds < 2.0).
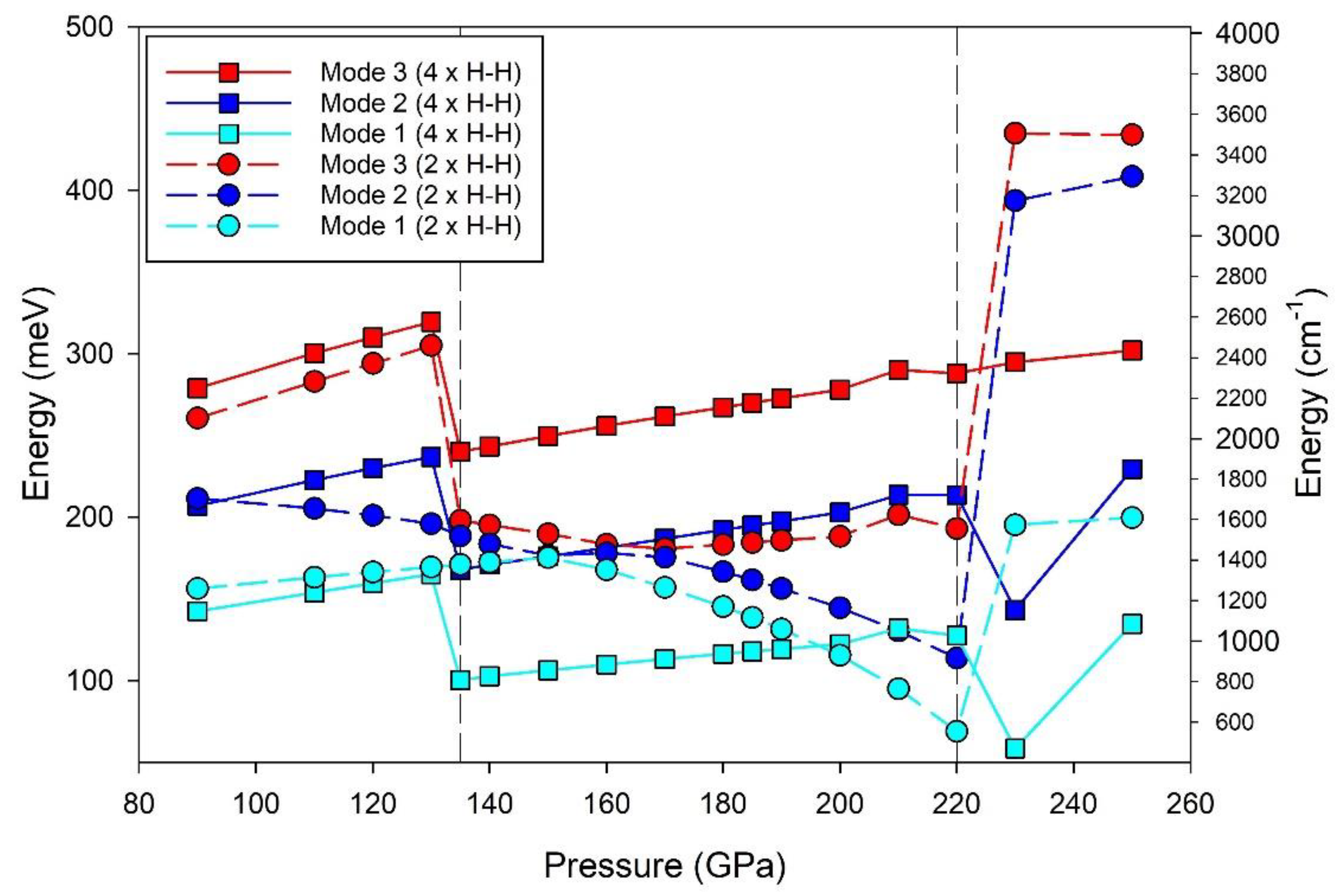

Table 1.
Calculated parameters for optimized unit cells of LaH10 using non-relativistic and relativistic treatments.
Table 1.
Calculated parameters for optimized unit cells of LaH10 using non-relativistic and relativistic treatments.
| Pressure | Symm | Method* | k (Å−1) | SOC | a (Å) | Volume (Å3) | H8c–H32f (Å) | No. of Atoms | Enthalpy (eV x 102) |
|---|---|---|---|---|---|---|---|---|---|
| 140 | Cubic | Schröd | 0.005 | No | 5.14007 | 135.802 | 1.18288 | 44 | −6.3522 |
| 140 | Cubic | Schröd | 0.010 | Yes | 5.09088 | 131.940 | 1.14630 | 44 | −48.2289 |
| 170 | Cubic | Schröd | 0.005 | No | 5.06223 | 129.726 | 1.16964 | 44 | −6.1038 |
| 170 | Cubic | Schröd | 0.010 | Yes | 5.00160 | 125.120 | 1.12619 | 44 | −47.9884 |
| 200 | Cubic | Schröd | 0.005 | No | 4.99600 | 124.700 | 1.15865 | 44 | −5.8657 |
| 200 | Cubic | Schröd | 0.010 | Yes | 4.92460 | 119.430 | 1.14630 | 44 | −47.7596 |
| 220 | Cubic | Schröd | 0.005 | No | 4.95667 | 121.779 | 1.14767 | 44 | −5.7119 |
| 220 | Cubic | Schröd | 0.010 | Yes | 4.87850 | 116.107 | 1.09848 | 44 | −47.6126 |
| 140 | Prim | Schröd | 0.005 | Yes | 3.59986 | 32.987 | 1.13829 | 11 | −12.0572 |
| 170 | Prim | Schröd | 0.005 | No | 3.57967 | 32.435 | 1.16955 | 11 | −1.5260 |
| 170 | Prim | Schröd | 0.005 | Yes | 3.53669 | 31.281 | 1.11894 | 11 | −11.9971 |
| 220 | Prim | Schröd | 0.005 | Yes | 3.44964 | 29.027 | 1.09207 | 11 | −11.9031 |
| 90 | Prim | K-H | 0.005 | No | 3.75407 | 37.410 | 1.21246 | 11 | −1.6990 |
| 90 | Prim | K-H | 0.005 | Yes | 3.73471 | 36.835 | 1.17919 | 11 | −12.1657 |
| 140 | Prim | K-H | 0.005 | No | 3.63462 | 33.952 | 1.18286 | 11 | −1.5881 |
| 140 | Prim | K-H | 0.005 | Yes | 3.59988 | 32.988 | 1.13831 | 11 | −12.0572 |
| 170 | Prim | K-H | 0.005 | No | 3.57960 | 32.433 | 1.16967 | 11 | −1.5260 |
| 170 | Prim | K-H | 0.005 | Yes | 3.53667 | 31.280 | 1.11894 | 11 | −11.9971 |
| 200 | Prim | K-H | 0.005 | No | 3.53272 | 31.176 | 1.15872 | 11 | −1.4664 |
| 200 | Prim | K-H | 0.005 | Yes | 3.48221 | 29.857 | 1.10215 | 11 | −11.9399 |
| 220 | Prim | K-H | 0.005 | Yes | 3.44963 | 29.027 | 1.09206 | 11 | −11.9031 |
| 230 | Prim | K-H | 0.005 | No | 3.49193 | 30.108 | 1.14191 | 11 | −1.4091 |
| 230 | Prim | K-H | 0.005 | Yes | 3.43429 | 28.642 | 1.08730 | 11 | −11.8851 |
| 250 | Prim | K-H | 0.005 | Yes | 3.40526 | 27.921 | 1.07826 | 11 | −11.8498 |
*”Schröd” = Schrödinger method; “K-H” = Koelling-Harmon method.
Table 2.
DFT derived parameters calculated with K-H method and k = 0.005 Å-1 for primitive LaH10*.
| P (GPa) | SOC | ΔEL (eV) | ELmax (eV) | Emid (eV) | EΓX/2 (eV) | ΔET (meV) | ΔEgap (meV) | Tc (K) |
|---|---|---|---|---|---|---|---|---|
| 90 | No | na | na | 0.70728 | 0.5220 | 185.3 | 92.6 | 233.2 |
| 140 | No | na | na | 0.54649 | 0.3420 | 204.5 | 102.2 | 257.4 |
| 170 | No | na | na | 0.45515 | 0.2490 | 206.2 | 103.1 | 259.5 |
| 200 | No | na | na | 0.36578 | 0.1432 | 222.6 | 111.3 | 280.2 |
| 230 | No | na | na | 0.27890 | 0.0613 | 217.6 | 108.8 | 273.9 |
| 100 | Yes | 0.0977 | −0.0405 | −0.74490 | −0.3130 | 431.9 | 108.0 | 271.8 |
| 110 | Yes | 0.1004 | −0.0353 | −0.75782 | −0.3260 | 431.8 | 108.0 | 271.8 |
| 130 | Yes | 0.1053 | −0.0262 | −0.77889 | −0.3540 | 424.9 | 106.2 | 267.4 |
| 135** | Yes | 0.1065 | −0.0243 | −0.76507 | −0.3620 | 403.1 | 100.8 | 253.7 |
| 140 | Yes | 0.1076 | −0.0222 | −0.76113 | −0.3680 | 393.1 | 98.3 | 247.4 |
| 150 | Yes | 0.1098 | −0.0187 | −0.77359 | −0.3850 | 388.6 | 97.1 | 244.6 |
| 170 | Yes | 0.1139 | −0.0124 | −0.80301 | −0.4120 | 391.0 | 97.8 | 246.1 |
| 180 | Yes | 0.1159 | −0.0103 | −0.81384 | −0.4230 | 390.8 | 97.7 | 246.0 |
| 185 | Yes | 0.1169 | −0.0090 | −0.81889 | −0.4300 | 388.9 | 97.2 | 244.8 |
| 190 | Yes | 0.1178 | −0.0080 | −0.82403 | −0.4380 | 386.0 | 96.5 | 243.0 |
| 200 | Yes | 0.1196 | −0.0063 | −0.84157 | −0.4510 | 390.6 | 97.6 | 245.8 |
| 220 | Yes | 0.1231 | −0.0032 | −0.84209 | −0.4760 | 366.1 | 91.5 | 230.4 |
| 230 | Yes | 0.1248 | −0.0022 | −0.84265 | −0.4920 | 350.7 | 87.7 | 220.7 |
| 250 | Yes | 0.1279 | −0.0011 | −0.83064 | −0.5090 | 321.6 | 80.41 | 202.4 |
Table 3.
Comparison of interatomic distances with pressure for LaH10.
| Pressure (GPa) | No SOC | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cosine Band Amplitude | Interatomic Distance | ||||||||
| 4VSSσ (eV) | V (eV) |
d (Å) |
d/3 (Å) | H8c–H32f (Å) |
H32f–H32f (Å) |
||||
| 140 | 2.77338 | 0.6933 | 3.6831 | 1.2277 | 1.1828 | 1.2042 | |||
| 170 | 2.91491 | 0.7287 | 3.5926 | 1.1975 | 1.1696 | 1.1805 | |||
| 200 | 3.04240 | 0.7606 | 3.5165 | 1.1721 | 1.1587 | 1.1600 | |||
| With SOC | |||||||||
| Cosine Band Amplitude | Interatomic Distance | ||||||||
| 4VSSσ (eV) | V (eV) |
d (Å) |
d/2 (Å) | H8c–H32f (Å) |
H32f–H32f (Å) |
||||
| 140 | 8.00114 | 2.0003 | 2.1684 | 1.0842 | 1.1390 | 1.2303 | |||
| 170 | 8.35034 | 2.0876 | 2.1226 | 1.0613 | 1.1190 | 1.2088 | |||
| 200 | 8.70794 | 2.1770 | 2.0785 | 1.0393 | 1.1021 | 1.1896 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



