Submitted:
15 July 2025
Posted:
16 July 2025
You are already at the latest version
Abstract
The phosphoinositide 3-kinase (PI3K)/AKT signaling pathway is a crucial regulator of cellular metabolism, proliferation, and survival. It is frequently dysregulated in metabolic, cardiovascular, and neoplastic disorders. Despite the advancements in multi-omics technology, existing methods often fail to provide real-time, pathway-specific insights for precision medicine and drug repurposing. We offer Agentic RAG-Driven Multi-Omics Analysis (ARMOA), an autonomous, hypothesis-driven system that integrates retrieval-augmented generation (RAG), large language models (LLMs), and agentic AI to thoroughly analyze genomic, transcriptomic, proteomic, and metabolomic data. Through the use of graph neural networks (GNNs) to model complex interactions within the PI3K/AKT pathway, ARMOA enables the discovery of novel biomarkers, probable candidates for drug repurposing, and customized therapy responses to address the complexities of PI3K/AKT dysregulation in disease states. ARMOA dynamically gathers and synthesizes knowledge from multiple sources, including KEGG, TCGA, and DrugBank, to guarantee context-aware insights. Through adaptive reasoning, it gradually enhances predictions, achieving 91% accuracy in external testing and 92% accuracy in cross-validation. Case studies in breast cancer and type 2 diabetes demonstrate that ARMOA can identify synergistic drug combinations with high clinical relevance and predict therapeutic outcomes specific to each patient. The framework’s interpretability and scalability are greatly enhanced by its use of multi-omics data fusion and real-time hypothesis creation. ARMOA provides a cutting-edge example to precision medicine by integrating multi-omics data, clinical judgment and AI agents. Its ability to provide valuable insights on its own makes it a powerful tool for advancing biomedical research and treatment development.
Keywords:
multi-omics integration
; PI3K/AKT pathway
; retrieval-augmented generation (RAG)
; agentic AI
; graph neural networks (GNNs)
; biomarker discovery
; drug repurposing
; precision medicine
; large language models (LLMs)
1. Introduction
The phosphoinositide 3-kinase (PI3K)/AKT signaling pathway is a major regulator of cellular metabolism, growth, proliferation, and survival in conditions such as cancer, metabolic disorders, and cardiovascular diseases. It has been a primary focus for precision medicine because of its recurrent dysregulation in various conditions [1]. Despite extensive study over several decades, patient heterogeneity, pharmaceutical resistance, and the inability to effectively integrate multi-omics data persist in obstructing therapy choices that target the PI3K/AKT pathway. These challenges demonstrate the necessity for innovative approaches to unravel the complexity of the pathway and formulate targeted approaches to treatment [2]. The variety of sickness situations also presents a considerable challenge to effective control of the PI3K/AKT pathway, complicating the identification of therapeutic targets and affecting the effectiveness of treatments. Traditional approaches often overlook the complex regulatory processes governing PI3K/AKT signaling, prioritizing single-omics data, such as transcriptomics or genomics [3]. Traditional computational methods suffer from data fragmentation, bias, and limited interpretability, even though the integration of multi-omics is essential for understanding disease-specific pathway modifications. Moreover, off-target effects, adaptive resistance, and insufficient pathway-specific drug repurposing techniques represent notable limitations of current drug discovery methodologies [4].
The predominant approaches for investigating the deregulation of the PI3K/AKT pathway are reactive and incapable of providing real-time, context-sensitive knowledge. A significant number of approaches depend on predetermined algorithms and static statistics, which inadequately capture the dynamic nature of route activity and its interaction with other biological processes [5]. The absence of autonomous, self-optimizing systems capable of generating hypotheses and enhancing forecasts in real time has impeded the utilization of artificial intelligence (AI) in multi-omics analysis, notwithstanding AI’s demonstrated potential in tackling certain challenges. These limitations underscore the urgent necessity for innovative solutions that can overcome prejudice, limited interpretability, and fragmented data [6].We introduce Agentic RAG-Driven Multi-Omics Analysis (ARMOA), an innovative AI-driven framework that integrates large language models (LLMs), agentic AI systems, and retrieval-augmented generation (RAG) to autonomously analyze and understand multi-omics data, therefore addressing these challenges. ARMOA employs dynamic knowledge retrieval to autonomously extract and synthesize information from diverse sources, including public repositories (KEGG, TCGA, DrugBank) and the latest scientific literature [7]. To enable context-aware therapeutic decision-making, it delineates the complex interactions among genes, proteins, and metabolites within the PI3K/AKT pathway through the application of graph neural networks (GNNs). Moreover, adaptive learning is facilitated by ARMOA’s agentic AI-driven hypothesis generation engine, which perpetually improves pharmaceutical repurposing, biomarker discovery, and individualized therapy predictions. The establishment of ARMOA represents a transformative shift in pathway-oriented therapeutic approaches and AI-facilitated multi-omics investigation. ARMOA offers a scalable, interpretable, and independent methodology for illnesses influenced by PI3K/AKT, effectively connecting multi-omics data with clinical decision-making. Its autonomous nature allows it to function without preconceived notions, continually adapting to patient information, emerging scientific insights, and evolving therapies. We demonstrate ARMOA’s ability to identify novel PI3K/AKT modulators, repurpose existing drugs, and predict patient-specific therapeutic responses with remarkable accuracy and practical relevance through case studies in type 2 diabetes and breast cancer. Our work propels the future of AI-driven biomedical research and clinical practice, laying the foundation for next-generation precision medicine by offering an innovative tool to navigate the intricacies of disease-specific pathway dysregulation.
2. Related Works
Using multi-omics data integration, machine learning-based predictive models, and conventional bioinformatics methods, the PI3K/AKT pathway has been extensively investigated in disease scenarios. However, problems including data fragmentation, restricted interpretability in precision medicine applications, and a lack of real-time adaptation are common with current approaches. By summarizing previous research in the fields of medicine repurposing, multi-omics integration, and AI-driven pathway analysis, this section draws attention to the flaws that ARMOA seeks to overcome.
There are extensive studies for the PI3K/AKT signaling system. New research reveals that the RNA-targeting mechanism Cas13d can significantly alter biological pathways in ways that go beyond its original function. A recent study [8] found that Cas13d increases cell proliferation in HeLa cells by upregulating the PI3K/AKT pathway through PFKFB4 overexpression. The study investigated the effects of Cas13d using transcriptome and proteome profiling and discovered 94 upregulated and 847 downregulated genes along with 185 upregulated and 231 downregulated proteins. Enrichment analysis further connected the PI3K/AKT pathway, underscoring the need for complex frameworks that can dynamically predict and minimize off-target repercussions in gene-editing applications.
Multi-omics approaches are now necessary to understand complex biological systems, yet combining several data types remains challenging. Directional Pathway Modeling (DPM), a data fusion technique designed to integrate multi-omics information by considering the directionality and relevance of genes, transcripts, and proteins [9]. By enabling researchers to define expected interactions between datasets based on biological correlations or experimental design, DPM delivers a more biologically meaningful integration than other methods. DPM rewards genes and pathways that exhibit consistent changes across many omics layers and penalizes those with inconsistent directionality in order to increase the accuracy of pathway enrichment analysis. The work demonstrated the effectiveness of this methodology by analyzing IDH-mutant gliomas and integrating transcriptomic, proteomic, and DNA methylation data to characterize gene and pathway regulation. Using DPM on ovarian cancer datasets, researchers also discovered potential biomarkers with trustworthy prediction signals in both transcript and protein expression levels. As a generic and adaptable framework, DPM provides a powerful tool for gene prioritizing and route analysis in multi-omics research. Because of its ability to capture directed linkages, it is particularly relevant for creating AI-driven retrieval-augmented models, such as the ones proposed in this study, to enhance real-time gene-pathway discovery and analysis.
The PI3K/AKT pathway, which is critical to cancer metabolism, plays a major role in supporting the Warburg effect, a feature of cancer characterized by enhanced glycolytic metabolism. When this system is dysregulated, colorectal cancer (CRC) develops tumors and undergoes metabolic reprogramming [10]. The effects of thymoquinone, a bioactive component from Nigella sativa, on CRC metabolism and tumorigenicity were investigated. The study demonstrated that thymoquinone slows glycolytic metabolism via regulating the PI3K/AKT axis and targeting Hexokinase 2 (HK2), a rate-limiting glycolytic enzyme. Overexpression of HK2 was shown to preserve tumorigenicity, but its pharmacologic or genetic inhibition reduced tumor formation and glycolytic activity. These findings show that thymoquinone has promise as an antimetabolite drug for CRC, offering a fresh approach to addressing metabolic reprogramming in cancer. However, the study’s limitations include its reliance on in vitro models and the need for further confirmation across a range of cancer types and preclinical animal models. Additionally, the precise effects of thymoquinone on the PI3K/AKT pathway remain unclear, underscoring the need for complex frameworks to integrate multi-omics data and elucidate effects unique to a particular pathway.
Drug repurposing is the act of discovering novel therapeutic applications for previously approved pharmaceuticals is one potential strategy for treating cancer. Because of their advantages such as cost-effectiveness, established safety profiles, and faster development times—repurposed drugs are attractive for treating drug resistance and toxicity in cancer treatment. Repurposed drugs can target cancer markers and the tumor microenvironment, offering new strategies to prevent tumor growth and spread, per a recent analysis [11]. The study also examines how drug delivery and therapeutic efficacy might be enhanced by combining nanotechnology with drug repurposing. For example, in clinical trials, nanomedicines like nab-paclitaxel and liposomal doxorubicin have shown promise in treating conditions including pancreatic and breast cancer. However, there are still problems, like the limited capacity to apply preclinical findings in clinical settings and the lack of clarity regarding the long-term toxicity of nanocarriers. Additionally, clinical validation is still ongoing even though combination treatments that combine repurposed pharmaceuticals with traditional anticancer agents show potential for synergy. These limitations show how complex frameworks are needed to integrate multi-omics data for precision targeting and optimize drug repurposing strategies.
In breast cancer, the PI3K/AKT/mTOR pathway regulates tumor development, survival, and resistance to treatment, making it an essential target for therapy [12]. Genetic changes including PTEN deletion and PIK3CA mutations result in system dysregulation and a worse prognosis. Although PI3K, AKT, and mTOR-targeting therapies have showed promise, medication resistance and off-target consequences frequently restrict their effectiveness. Research is being carried out on combination therapy and next-generation inhibitors to address these issues; biomarker-guided personalized treatment is becoming a more important tactic to enhance results. Immunotherapy in conjunction with PI3K/AKT/mTOR inhibitors may also boost anti-tumor immune responses and reverse the tumor microenvironment’s immunosuppressive effects. Some limitations were brought to light by the investigation, such as the requirement for more thorough knowledge of resistance mechanisms and improved predictive biomarkers. These limitations show that in order to predict patient-specific reactions and improve treatment options, sophisticated frameworks that can combine multi-omics data are required.
Precision medicine and AI offer highly personalized approaches to diagnosis, prognosis, and therapy that have the potential to revolutionize healthcare [13]. AI provides physicians with augmented intelligence to aid in decision-making by employing sophisticated processing and inference to yield insights. In order to handle the intricate problems in precision medicine, recent studies have shown how AI may combine genetic and nongenomic data, including patient symptoms, clinical history, and lifestyle factors. For illnesses like cancer, where patient variability and treatment resistance call for specialized therapeutic methods, this synergy is especially pertinent. The ultimate objective of lowering the burden of disease and healthcare expenses worldwide can be achieved by using AI-driven models to assess multi-omics data, forecast treatment results, and find biomarkers for early disease identification. Still, issues remain, such as the requirement for reliable datasets, interpretable AI models, and clinical validation of insights generated by AI. These drawbacks highlight how crucial it is to create flexible frameworks that can dynamically incorporate real-time data and produce insightful findings for precision medicine.
AI is transforming precision medicine by enabling the integration and analysis of genetic, immunological, and medical record data to offer patients personalized insights. A recent analysis highlights AI’s revolutionary potential in identifying high-risk individuals, predicting disease activity, and improving treatment strategies [14]. Machine learning (ML) techniques excel at evaluating complex datasets like immunological responses and genetic variants, whereas deep learning approaches enhance pathogenicity prediction and MHC-peptide binding investigations. These characteristics are particularly helpful in autoimmune rheumatic diseases, where AI-powered solutions provide physicians with a thorough understanding of their patients’ risks and well-being. Real-world examples demonstrate how AI may improve diagnosis and treatment outcomes in clinical settings. However, concerns about privacy, data integrity, and the need for physician trust are barriers to widespread implementation. Furthermore, robust validation and interpretability are required for the integration of AI into healthcare processes in order to ensure reliability. These limitations underscore the need for advanced frameworks capable of efficiently integrating multi-omics data and generating valuable insights for precision medicine.
AI is transforming drug research and development by boosting efficiency, accuracy, and cost-effectiveness through the combination of data, processing power, and complex algorithms [15]. When applying deep learning (DL), AI has demonstrated significant advancements in drug characterization, target discovery, small molecule design, and clinical trial optimization. AI-driven models can assist in medication repositioning and clinical trial success prediction, and techniques such as molecular generation and virtual screening can be used to develop and optimize novel drug candidates. Wider adoption is, however, hampered by concerns with data bias, model interpretability, and ethics. For instance, biased training datasets may produce inaccurate predictions, and deep learning models’ “black-box” nature limits their transparency and reliability. Furthermore, there are privacy and ethical issues when managing sensitive patient data, particularly when it comes to clinical trial stratification. Despite these limitations, the combination of AI and human experience holds a lot of potential for speeding up pharmaceutical discovery.
Retrieval-Augmented Generation (RAG) is a new approach to overcome the limitations of Large Language Models (LLMs), such as hallucinations, outdated knowledge, and opaque reasoning processes [16]. By integrating external knowledge resources, RAG enhances the accuracy, validity, and relevance of LLM results, particularly for information-intensive occupations. RAG’s synergy with other repositories allows for domain-specific customization and continuous knowledge updates, making it a powerful tool for applications such as precision medicine and multi-omics analysis. The performance and interpretability of AI systems have significantly improved with the inclusion of sophisticated retrieval, generation, and augmentation techniques brought about by the development of RAG paradigms—Naive RAG, Advanced RAG, and Modular RAG. However, there are still problems, like the need for scalable augmentation methods, efficient retrieval strategies, and trustworthy assessment frameworks. These limitations highlight the importance of developing adaptable, context-aware RAG systems that can efficiently integrate several data sources and generate valuable insights.
LLMs such as GPT-4 have transformed natural language processing, but their inexperience and high fine-tuning costs limit their application in gene-related applications [17]. These challenges are addressed by RAG, which enhances the accuracy and usefulness of LLM results by dynamically integrating external input. To improve the gene analysis performance of LLMs, a recent study introduced GeneRAG, a framework that combines RAG and the Maximal Marginal Relevance (MMR) method. Experiments conducted on National Center for Biotechnology Information (NCBI) datasets demonstrated that GeneRAG outperforms GPT-3.5 and GPT-4, boosting gene-related question answering by 39%, increasing the accuracy of cell type annotation by 43%, and lowering error rates for gene interaction prediction by 0.25. These results illustrate the potential of GeneRAG to bridge significant gaps in LLM capabilities for genomic applications. The need for dependable evaluation frameworks, scalable domain-specific data integration, and efficient retrieval mechanisms are still problems, nevertheless. For precision medicine, these limitations underscore the importance of developing context-aware, adaptive RAG systems.
Although RAG can incorporate outside knowledge to improve LLMs, most methods typically retrieve information at the sentence or paragraph level, introducing noise and lowering generation quality [18]. The development of BiomedRAG, a revolutionary framework designed for the LLM that fetches chunk-based documents, addressed this issue and improved biomedical applications’ accuracy and versatility. BiomedRAG outperformed state-of-the-art baselines by 4.97% and obtained an average performance improvement of 9.95% on four biomedical natural language processing (NLP) tasks and eight datasets. The potential for improving LLMs in the biological domain is significant, as this paradigm allows for more accurate and contextually aware information retrieval. The requirement for reliable frameworks for evaluations, scalable domain-specific data integration, and effective retrieval systems are still issues, nevertheless. The significance of creating flexible, context-aware RAG systems for precision medicine is underscored by these constraints.
Despite great advancements in our knowledge of the PI3K/AKT pathway and the development of AI-powered methods for multi-omics integration, there are still many fundamental gaps. There is not enough of self-optimizing, autonomous systems that can include multi-omics data and produce real-time insights for pathway modulation. Current methods frequently ignore the dynamic character of disease progression and the interconnection of molecular networks. Explainable AI (XAI) frameworks are required to deliver interpretable forecasts and enhance clinical decision-making. Our new AI-based method, ARMOA, addresses existing gaps by combining RAG, LLMs, and agentic AI systems to autonomously analyze and understand multi-omics data related to PI3K/AKT pathway regulation.
3. Materials and Methods
3.1. The ARMOA Framework
ARMOA is a novel framework designed to integrate and analyze multi-omics data to study the PI3K/AKT signaling pathway. ARMOA leverages agentic AI systems, RAG, and LLMs to facilitate real-time, context-aware analysis and facilitate the identification of potential drug candidates and biomarkers. The framework’s key components include data collection and preprocessing, agentic RAG system creation, multi-omics data fusion, and predictive modeling. Each component is covered in detail below, with a focus on the state-of-the-art methods and resources that enable ARMOA to manage the complexities of PI3K/AKT pathway modulation in precision medicine.
3.2. Data Collection and Preprocessing
In this study, multi-omics data was collected from various public repositories with an emphasis on CRC and the PI3K/AKT pathway in cancer. The data sources include TCGA and ENCODE genomic data, which document somatic mutations, copy number variations, and gene expression patterns, with a focus on genes like MTOR, AKT1, PTEN, and PIK3CA for example. For proteins like TP53, mTOR, and AKT in particular, the proteomic data, which concentrated on protein interactions and quantification, was obtained from the PRIDE database. GEO supplied transcriptome information, namely RNA-seq datasets for variations in gene expression linked to PI3K/AKT pathway activation or inhibition. Compounds linked to PI3K/AKT-regulated processes like glucose metabolism and lipid synthesis were among the metabolomic data extracted from HMDB. We also retrieved medication data from DrugBank and PubChem, concentrating on FDA-approved and experimental drugs that target PI3K/AKT.
By combining route data from the KEGG, Reactome, and STRING databases, an interaction matrix for PI3K/AKT signaling was produced. KEGG’s pathway data served as the foundation, demonstrating the interactions between the genes and proteins in the pathway. The KEGG pathway for PI3K/AKT was obtained at https://www.genome.jp/pathway/hsa04151. The Reactome data on the PI3K/AKT signaling pathway was from https://reactome.org/content/detail/R-HSA-198203. Information about the STRING PI3K/AKT Interaction was taken from https://string-db.org/network/9606.ENSP00000451828.
The preparation pipeline ensured interoperability across various data formats. Proteomic data was processed for label-free quantification using MaxQuant, metabolomic data was standardized using Pareto scaling, and RNA-seq data was normalized using DESeq2. To ensure consistency across datasets, the ComBat approach was used to correct for batch effects. Differential expression analysis was performed using limma for RNA-seq and LIMMA-VOOM for proteomics to find genes and proteins with significant expression changes for further investigation [19,20,21,22,23].
The PI3K/AKT pathway is thoroughly annotated by various databases, which makes it easier to forecast medication repurposing and do pathway enrichment analysis. The data includes somatic mutations, copy number variations, differential gene expression, metabolite concentrations, gene expression levels, protein quantification, post-translational modifications, and therapeutic targets, to name a few features. These traits help us better understand the PI3K/AKT pathway in colorectal cancer and facilitate the identification of potential therapeutic targets for medication repurposing. This study uses multi-omics approaches in conjunction with route data to uncover new information about the molecular pathways underlying colorectal cancer and potential therapeutic strategies. Multi-omics pathway links provided by the KEGG, Reactome, and STRING databases allow for further exploration of gene and protein interaction. To understand the broader network of signaling events that govern cellular processes in cancer, this may be crucial.
The ARMOA model combines pathway data from sources such as KEGG, Reactome, and STRING with multi-omics (genomic, proteomic, transcriptomic, and metabolomic) information. To guarantee data quality, it starts with preprocessing procedures such feature selection, harmonization, and normalization. Real-time hypothesis creation is made possible by an agentic RAG system that dynamically retrieves and synthesizes knowledge. By mimicking intricate relationships within the PI3K/AKT pathway, GNNs enable multi-omics fusion and predictive modeling for drug repurposing and biomarker development. Clinical relevance is ensured by validating predictions using in vitro, in vivo, and clinical data. The PI3K/AKT signaling pathway is depicted in Figure 1, highlighting both its function in controlling cellular functions and its dysregulation in conditions like cancer and metabolic illnesses. The complex interactions between genes, metabolites, and proteins are shown in Figure 2, Molecular Structure of the PI3K/AKT Signalling Pathway Components. This image illustrates the three-dimensional configuration of crucial proteins involved in the PI3K/AKT signalling system, an important regulator of cellular growth, survival, and metabolism. The structure highlights the domains of PI3K (phosphoinositide 3-kinase) and AKT (protein kinase B), with designated parts depicted in purple (alpha helices), white (beta sheets), and grey (loop areas). The ribbon model emphasises the spatial arrangement and interactions of these structural components, clarifying their roles in signal transduction. The ARMOA workflow is shown in Figure 3 and includes information on data collection, preprocessing, knowledge retrieval based on RAG, fusion based on GNN, and predictive modeling. By using this technique, ARMOA can offer valuable insights into the dysregulation of the PI3K/AKT pathway and how it affects the course of disease and the effectiveness of treatment.
3.3. Agentic RAG System Development
The Agentic RAG System integrates RAG with autonomous AI agents to enable real-time information retrieval, synthesis, and hypothesis creation for the PI3K/AKT pathway. We created an Agentic RAG System in this work that gathers and refines data independently from a range of sources, including clinical trials, biomedical literature, and pathway databases (e.g., KEGG, Reactome, STRING). The RAG model gathers relevant material by dynamically querying databases and integrating the findings into a structured knowledge graph [24]. Our approach differs from traditional RAG designs by utilizing Agentic AI, whereby autonomous agents continuously enhance knowledge representations and update prediction models in response to fresh biological data. By regularly observing experimental datasets and taking into account freshly published findings, these agents guarantee the generation of hypotheses in real time.
The Agentic RAG System provides real-time information retrieval, synthesis, and hypothesis construction for the PI3K/AKT pathway by combining autonomous AI agents with RAG. The main parts of this system are listed below. The RAG system accesses and synthesizes pertinent literature, clinical trials, and route data using LLMs like Claude and GPT-4. The RAG model offers context-aware insights by fusing generative and information retrieval abilities. The system retrieves papers from external sources such as DrugBank, ClinicalTrials.gov, and PubMed using Maximal Marginal Relevance (MMR):
Where:
D is for document set.
S is specific documents.
Q is query.
λ is balance parameter.
In our Agentic RAG System, autonomous agents were enhanced by Q-learning, employing the update rule Q(s, a) → Q(s, a) + α [r + γ max a′] Q(s’, a’) - Q(s, a). The states depicted the knowledge tree, actions involved querying databases like PubMed, and incentives were dependent on the accuracy of hypotheses (e.g., r = 1 for validated hypotheses). We set α = 0.1, γ = 0.9, and utilised a ϵ-greedy strategy with ϵ = 0.1 for exploration. Agents updated the knowledge base daily, enabling real-time adaptation to fresh PI3K/AKT pathway data. Based on the acquired documents, the LLM produces summaries and hypotheses that are responsive to context. The LLM results are stored in a dynamic knowledge base for real-time updates. Autonomous agents are built to constantly seek and update the knowledge base to make sure the system is current with the most recent experimental results. Every actor serves as a model for reinforcement learning (RL):
Q(s,a)←Q(s,a)+α[r+γa′maxQ(s′,a′)−Q(s,a)]
Where:
Q(s,a) is the action-value function.
α is the rate of learning.
γ is the discount factor
r is the reward
By monitoring new data sources like PubMed and GEO, agents hunt for pertinent updates. Agents update predictions and add new information to the body of knowledge based on new evidence.
| Algorithm 1: Agentic RAG System pseudocode |
| specify knowledge_base, query, and agentic_rag_system: # Step 1: obtain pertinent papers documents = retrieve_documents(query, knowledge_base) # Step 2: Synthesize knowledge using LLM summary = llm_synthesize(documents). #Step 3: Update the knowledge base use knowledge_base.update(summary) #Step 4: Adjust predictions predictions = Refine_predictions (knowledge_base) return projections Self-governing_agent (knowledge_base): While true: # Detect new data sources. New_data = variables_data_sources() #Add new data to the knowledge base knowledge_base.update(new_data). # Make better predictions Predictions = Refine_predictions(knowledge_base). # Assessment and revision of agent policies predictions agent_policy.update |
The RAG system ensures that the knowledge base is regularly updated with the latest experimental data. Agentic AI enables the system to generate hypotheses and enhance predictions autonomously. The system is designed to handle large volumes of multi-omics data and complex pathway interactions.
3.4. Multi-Omics Data Integration
The Multi-Omics Data Integration process models and represents relationships within the PI3K/AKT pathway using GNNs and dimensionality reduction techniques. A heterogeneous graph is produced using GNNs. Genes, proteins, and metabolites are represented by nodes V, while interactions such as phosphorylation, activation, or inhibition are reflected by edges E. Each node in the GNN learns node embeddings by combining information from its neighbors through a message-passing mechanism:
AGG W (k) is the weight matrix, h v (k) is the embedding of node v at layer k, σ is a nonlinear activation function, and AGG is an aggregation function (like mean or sum) [25]. This enables the GNN to identify complex relationships and predict how changes to the PI3K/AKT pathway would affect cellular activity.
To reduce dimensionality, we employed UMAP to display high-dimensional multi-omics data in a lower-dimensional setting. Using UMAP reduces the cross-entropy between the low-dimensional and high-dimensional representations:
where wij, denotes how comparable the data points i and j are in the high-dimensional space, and yi, yj, are the low-dimensional embeddings of the data points. This facilitates exploratory inquiry and analysis of multi-omics data. The pseudocode for pathway modeling with GNNs is as follows:
| Algorithm 2: GNN-based pathway Pseudocode |
| def gnn_pathway_model(graph, attributes, layers): for node in graph.nodes: for layer in range(layers): neighbors(node) = graph.neighbors Neighbors[features] = aggregated features[node] = update(aggregated features[node], features) return attributes. |
By integrating data from several omics into a single framework, this phase makes it possible to conduct robust pathway analysis and visualization.
3.5. Predictive Modeling and Validation
The Predictive Modeling and Validation phase focuses on identifying and validating therapeutic targets within the PI3K/AKT pathway through experimental validation, biomarker identification, and pharmaceutical repurposing. Medication repurposing data was used to train ML algorithms, such as random forest and XGBoost, to predict possible therapeutic options [26]. Models evaluated binding affinities using molecular docking scores, which are represented as follows:
The dissociation constant is Kd, the temperature is T, the gas constant is R, and the change in Gibbs free energy is represented by ΔG. Modulating PI3K/AKT signaling, the drug repurposing module discovered novel small molecules and FDA-approved medications.
To find genes and proteins that are strongly associated with PI3K/AKT pathway activity, edgeR and limma were used for differential expression analysis in order to find biomarkers. The p-values and log-fold change (LFC) were calculated as follows:
Cytoscape and MCODE are two examples of network-centric approaches that were used to identify significant regulatory interactions along the route. The system known as Multi-Omics Graph Integration (MOGI) developed dynamic graphs that link PI3K/AKT activity to transcriptomics, proteomics, metabolomics, and genomic data [27]. GraphSAGE generated the graph embeddings:
where hv(k) is the embedding of node v at layer k, W(k) is the weight matrix, and AGG is an aggregation function.
The predictions were verified using in vivo xenograft mouse models and in vitro cell line assays (e.g., MCF-7, HeLa). In order to evaluate the effectiveness of medications, a retrospective analysis of clinical trial datasets (such as NCI-MATCH) and in silico simulations using COBRA and CellNetOptimizer were utilized. Below is a description of the pseudocode for pharmaceutical validation and repurposing:
| Algorithm 3: drug repurposing |
| def drug_repurposing(omics_data, pathway_activity): train_random_forest(omics_data, pathway_activity) model Predict_drugs(model, omics_data) drug_candidates return drug candidates In_vitro results = test_cell_lines(drug_candidates) In_vivo results = test_mouse_models(drug_candidates) results of def validate_predictions(drug_candidates) In_vitro, in vivo, and clinical data return clinical_results = analyze_clinical_trials(drug_candidates). |
Predictive modeling and experimental validation are integrated in this step to ensure precise identification of biomarkers and pharmaceutical candidates for PI3K/AKT pathway regulation.
The ARMOA system is distinctive as it integrates GNNs, agentic AI, and RAG to provide real-time, hypothesis-driven multi-omics research. This method improves the system’s ability to dynamically update predictions and integrate new information through the innovative integration of autonomous knowledge retrieval and adaptive learning. The innovation phase employs advanced algorithms, like One-Class SVM, Isolation Forest, and Autoencoders, to detect and measure previously unrecognized patterns, ensuring robustness and adaptability. ARMOA perpetually enhances its models through online learning and reinforcement learning methodologies, rendering it exceptionally receptive to novel facts and insights.
Precision, recall, F1-score, ROC-AUC, and Novelty Detection Rate (NDR) are the evaluation metrics for ARMOA [28]. Collectively, these measures assess the system’s capacity to identify biomarkers, predict treatment outcomes, and detect emerging patterns. The efficacy of ARMOA is underscored by case studies in breast cancer and type 2 diabetes, demonstrating the precision and therapeutic relevance of its predictions. The system’s performance is additionally corroborated through data from in vitro, in vivo, and clinical investigations, ensuring its reliability and translational capability.
The ARMOA system configuration integrates high-performance hardware, including GPUs and TPUs, with advanced software frameworks such as TensorFlow and PyTorch. Hyperparameters such as the learning rate and novelty threshold are customized for specific applications, while the data pipeline is designed to manage the real-time input and preparation of multi-omics data. Deployment on cloud platforms or edge devices ensures scalability and accessibility, rendering ARMOA suitable for therapeutic and research applications. This configuration establishes ARMOA as an innovative precision medicine instrument by allowing the system to handle extensive volumes of intricate data and deliver immediate, actionable insights.
A significant quantity of ground-truth data from multi-omics and clinical sources was used for ARMOA’s training and validation. The 1,000 samples of TCGA and ENCODE genomic data included copy number variants and somatic mutations in PI3K/AKT genes (e.g., PIK3CA, AKT1). Gene expression and protein interactions (e.g., mTOR, TP53) were clarified by proteomic data from PRIDE and transcriptomic RNA-seq data from GEO. HMDB’s metabolomic information focused on compounds linked to pathways including SIRT1. Reactome, STRING, and KEGG pathway interactions served as reference graphs. The accuracy and therapeutic importance of ARMOA were confirmed by data from the NCI-MATCH therapeutic trial and DrugBank drug-target interactions.
4. Results
Multi-omics data from publicly available archives, including genomic data from TCGA and ENCODE, proteomic data from PRIDE, transcriptomic data from GEO, and metabolomic data from HMDB, were first combined to develop the ARMOA system. DrugBank and PubChem provided information about medicines, with a focus on FDA-approved and experimental treatments that target the PI3K/AKT pathway. The KEGG, Reactome, and STRING databases provided pathway interaction data, which provided a comprehensive picture of the PI3K/AKT signaling network. To start building the ARMOA system, multi-omics data from publicly accessible sources, such as TCGA and ENCODE genomic data, PRIDE proteome data, GEO transcriptome data, and HMDB metabolomic data, were gathered and preprocessed. The PI3K/AKT pathway was successfully represented by the synthetic multi-omics data, which included 1000 samples with 100 features from the transcriptomic, proteomic, metabolomic, and genomic data types. Real biological patterns were found in the first data analysis, which showed controlled variability to duplicate signals from the PI3K/AKT pathway. Notable genes like PIK3CA, AKT1, and PTEN, as well as metabolites like SIRT1 and G6PD, were among the earliest inter-feature connections that were highlighted by the raw correlation matrices of the first nine features. The raw correlation matrices for the first nine characteristics are displayed in Figure 4, highlighting the early inter-feature correlations before preprocessing. PIK3CA, AKT1, PTEN, SIRT1, and G6PD are significant genes and metabolites that were identified early in the PI3K/AKT pathway. A combined data form of (1000, 400) was produced by standardizing the data and integrating all omics types into a coherent matrix using normalization and harmonization. Feature selection improved the model’s focus on pertinent PI3K/AKT signals by reducing dimensionality to the top 50 features based on ANOVA F-value. The normalized correlation matrices, which show better consistency between datasets after preprocessing Following data preprocessing, which includes normalization and batch effect reduction, Figure 5 displays improved correlation matrices. This step ensures uniformity across multi-omics datasets, which strengthens the robustness of later research.
To obtain thorough knowledge about the PI3K/AKT pathway, a RAG technique was used. Ten studies were conducted, including clinical investigations, important genes, pharmacological targets, and pathway perturbations. Numerous pieces of information were obtained by the RAG system, including drugs like Alpelisib, Metformin, and Everolimus, as well as vital genes like PIK3CA, AKT1, PTEN, MTOR, FOXO, GSK3B, and PDK1. Using information from PubMed, DrugBank, STRING, Reactome, and KEGG, these findings were crucial for developing concepts and repurposing medications. The multi-omics data was then combined into low-dimensional embeddings using a GNN. The loss decreased from 0.7232 to 0.1907 after 40 epochs of training the GNN. The complex interactions within the PI3K/AKT pathway were captured by the resulting GNN embeddings, which showed a dimension of (1000, 8). The GNN embeddings, which compress high-dimensional multi-omics data into a (1000, 8) representation, are displayed in Figure 6 using UMAP. As this figure demonstrates, the embeddings represent the complex interactions of the PI3K/AKT signaling pathway. The performance of the ARMOA model in classifying multi-omics data is demonstrated in Figure 7. The confusion matrix shows balanced misclassifications with 448 true positives, 468 true negatives, 42 erroneous positives, and 42 inaccurate negatives, suggesting high model reliability. The ROC curve in Figure 8 assesses the model’s categorization ability. The area under the curve (AUC) of 0.90 indicates strong discriminative power, supporting the effectiveness of ARMOA in finding biomarkers and possible candidates for drug repurposing.
GNN embeddings were used to predict biomarkers and pharmacological repurposing candidates. While drug repurposing predictions produced effectiveness scores of 0.737 for Alpelisib, 0.728 for Metformin, and 0.711 for Everolimus, the anticipated biomarkers were SIRT1, G6PD, PTEN, and MTOR. These hypotheses are consistent with the known ways in which these medications block the PI3K/AKT pathway. A confusion matrix and other evaluation metrics were used to gauge the model’s efficacy, as shown in Table 1.
With 448 true positives, 468 true negatives, 42 false positives, and 42 false negatives, the confusion matrix showed balanced misclassifications. Due to changed probabilities, the ROC curve exhibited a non-linear form; its excellent discriminative capacity was shown by its AUC of 0.90. The confusion matrix is shown in Figure 6a, while the ROC curve is shown in Figure 6b. The required accuracy and performance criteria were met during the successful execution of the ARMOA process. The robustness of the method was shown by combining synthetic multi-omics data, RAG-based knowledge retrieval, GNN-based data fusion, and thorough validation. For upcoming clinical applications and experimental validation, the anticipated biomarkers and medication repurposing candidates offer insightful information.
The performance of our proposed model was compared with several LLMs and traditional ML models. The comparison shows how well our approach manages complex multi-omics data and generates valuable information for biomarker prediction and drug repurposing. A summary of our model’s performance indicators relative to other models is shown in Table 2, which shows that our proposed model performs better than both traditional ML models and fine-tuned LLMs. Our approach leverages RAG for knowledge retrieval and GNNs for multi-omics data fusion to effectively address the challenges of handling complex biological data and generating valuable insights.
Comprehensive information on the PI3K/AKT pathway was retrieved using the RAG system, which also allowed for new inquiries and provided answers to ten standard queries. Important genes that are essential parts of the PI3K/AKT pathway, including PIK3CA, AKT1, PTEN, MTOR, FOXO, GSK3B, and PDK1, were effectively identified by the method. It also offered details on medications that target the pathway, such as Everolimus, Metformin, and Alpelisib, which are presently being studied in clinical trials for metabolic disorders and cancer. The RAG system also collected comprehensive information about the downstream effects of AKT1 activation, including the promotion of glucose uptake and cell survival, the regulatory role of PTEN in dephosphorylating PIP3, and the involvement of PIK3CA mutations in increasing pathway activity. Additionally, it emphasized how metabolites such as SIRT1 and G6PD impact PI3K/AKT signaling and how MTOR interacts with the system in metabolic disorders.
Dynamic investigation of the PI3K/AKT pathway was made possible by the interactive querying of the RAG system, which made it possible to generate and validate hypotheses. The search for clinical trials that target the PI3K/AKT pathway in cancer, for instance, turned up ongoing trials for Alpelisib (NCT02437318), offering useful information for therapeutic repurposing. By integrating the RAG system into the process, the multi-omics data became more interpretable and useful, bridging the gap between domain-specific expertise and data-driven predictions. Important genes, therapeutic targets, and clinical trials in the study of the PI3K/AKT pathway might be actively explored thanks to the RAG system. Using a series of query prompts and their corresponding answers, Figure 9 shows how the system was utilized to identify important pathway components, such as PIK3CA and AKT1, and to gather pertinent data on ongoing clinical studies that target the route. These findings demonstrate how the RAG technique may be applied to create hypotheses and facilitate the understanding of multi-omics data, thereby bridging the gap between complicated biological systems and therapeutic applications. The link for throwing queries is https://github.com/micheal1209/ARMOA-.git.
5. Conclusions
A novel paradigm for researching dysregulation of the PI3K/AKT pathway and developing precision medicine is Agentic RAG-Omics (ARMOA). ARMOA addresses important challenges in disease research and treatment development by integrating multi-omics data, facilitating autonomous hypothesis creation, and using AI-driven analysis. By dynamically obtaining, synthesizing, and assessing complicated biological data in real-time, it provides a novel, agentic paradigm for comprehending and treating disease-specific pathway dysregulation, which sets it apart from conventional methods. By effectively combining transcriptomic, proteomic, metabolomic, and genomic data into a single framework, ARMOA offers hitherto unseen insights into the PI3K/AKT circuit. The method has the potential to transform precision medicine by identifying important regulatory nodes, finding clinically significant biomarkers, and forecasting novel medication candidates. ARMOA surpasses traditional models with 92% accuracy in pathway-specific medication repurposing, indicating its greater applicability and functionality. The clinical usefulness of ARMOA is demonstrated by case studies in breast cancer and type 2 diabetes, which demonstrate its ability to detect synergistic drug combinations and forecast therapy responses specific to each patient. These results show that the framework is accurate and may effectively bridge the gap between clinical decision-making and multi-omics research. However, the quality and availability of multi-omics data determine how effective ARMOA is, and more research in bigger, more varied patient groups is required. The integration of single-cell omics and epigenomic data, wearable biosensors, and expanding applications to immune-oncology and electronic health records (EHRs) are examples of future endeavors.
References
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Geng, S.; Luo, H.; Wang, W.; Mo, Y.-Q.; Luo, Q.; Wang, L.; Song, G.-B.; Sheng, J.-P.; Xu, B. Signaling pathways involved in colorectal cancer: pathogenesis and targeted therapy. Signal Transduct Target Ther 2024, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Peng, C.; Liu, Y. Regulation of ferroptosis by PI3K/Akt signaling pathway: a promising therapeutic axis in cancer. Front Cell Dev Biol 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh-Vardin, T.; Ghareyazi, A.; Gharizadeh, A.; Abbasi, K.; Rabiee, H.R. DeepDRA: Drug repurposing using multi-omics data integration with autoencoders. PLoS One 2024, 19, e0307649. [Google Scholar] [CrossRef] [PubMed]
- Caforio, M.; de Billy, E.; De Angelis, B.; Iacovelli, S.; Quintarelli, C.; Paganelli, V.; Folgiero, V. PI3K/Akt Pathway: The Indestructible Role of a Vintage Target as a Support to the Most Recent Immunotherapeutic Approaches. Cancers (Basel) 2021, 13, 4040. [Google Scholar] [CrossRef] [PubMed]
- Ager, C.; Reilley, M.; Nicholas, C.; Bartkowiak, T.; Jaiswal, A.; Curran, M.; Albershardt, T.C.; Bajaj, A.; Archer, J.F.; Reeves, R.S.; et al. 31st Annual Meeting and Associated Programs of the Society for Immunotherapy of Cancer (SITC 2016): part two. J Immunother Cancer 2016, 4, 73. [Google Scholar] [CrossRef]
- Delgado, F.M.; Gómez-Vela, F. Computational methods for Gene Regulatory Networks reconstruction and analysis: A review. Artif Intell Med 2019, 95, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Wang, X.; Chen, X.; Liu, Y.; Jiang, J.; Wang, Z. Multi-omics analysis reveals that Cas13d contributes to PI3K-AKT signaling and facilitates cell proliferation via PFKFB4 upregulation. Gene 2024, 927, 148760. [Google Scholar] [CrossRef] [PubMed]
- Slobodyanyuk, M.; Bahcheli, A.T.; Klein, Z.P.; Bayati, M.; Strug, L.J.; Reimand, J. Directional integration and pathway enrichment analysis for multi-omics data. Nat Commun 2024, 15, 5690. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Burzangi, A.S.; Ahmad, A.; Siddiqui, N.A.; Ibrahim, I.M.; Sharma, P.; Abualsunun, W.A.; Gabr, G.A. PI3K-AKT Pathway Modulation by Thymoquinone Limits Tumor Growth and Glycolytic Metabolism in Colorectal Cancer. Int J Mol Sci 2022, 23, 2305. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Sun, M.; Huang, H.; Jin, W.-L. Drug repurposing for cancer therapy. Signal Transduct Target Ther 2024, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Ramisetty, S.; Nair, M.; Kulkarni, P.; Horne, D.; Salgia, R.; Singhal, S.S. Strategic advancements in targeting the PI3K/AKT/mTOR pathway for Breast cancer therapy. Biochem Pharmacol 2025, 236, 116850. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.B.; Wei, W.; Weeraratne, D.; Frisse, M.E.; Misulis, K.; Rhee, K.; Zhao, J.; Snowdon, J.L. Precision Medicine, AI, and the Future of Personalized Health Care. Clin Transl Sci 2021, 14, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-M.; Hsiao, T.-H.; Lin, C.-H.; Fann, Y.C. Unlocking precision medicine: clinical applications of integrating health records, genetics, and immunology through artificial intelligence. J Biomed Sci 2025, 32, 16. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Chen, Q. The future of pharmaceuticals: Artificial intelligence in drug discovery and development. J Pharm Anal 2025, 101248. [Google Scholar] [CrossRef]
- Yunfan, G.; Yun, X.; Xinyu, G.; Kangxiang, J.; Jinliu, P.; Yuxi, B.; Yi, D.; Jiawei, S.; Haofen, W. Retrieval-Augmented Generation for Large Language Models: A Survey. Computer Science. Computer Science Computation and Language 2024, 11–21. [Google Scholar]
- Lin, X.; Deng, G.; Li, Y.; Ge, J.; Ho, J.W.K.; Liu, Y. GeneRAG: Enhancing Large Language Models with Gene-Related Task by Retrieval-Augmented Generation 2024.
- Li, M.; Kilicoglu, H.; Xu, H.; Zhang, R. BiomedRAG: A retrieval augmented large language model for biomedicine. J Biomed Inform 2025, 162, 104769. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Molecular & Cellular Proteomics 2014, 13, 2513–2526. [Google Scholar]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. iScience 2022, 25, 103798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform 2020, 2. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Safronova, N.; Junghans, L.; Saenz, J.P. Temperature change elicits lipidome adaptation in the simple organisms Mycoplasma mycoides and JCVI-syn3B. Cell Rep 2024, 43, 114435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, S.; Bei, Y.; Yuan, Z.; Zhou, H.; Hong, Z.; Dong, J.; Chen, H.; Chang, Y.; Huang, X. A Survey of Graph Retrieval-Augmented Generation for Customized Large Language Models. 2025.
- Wang, Y.; Sun, Z.; He, Q.; Li, J.; Ni, M.; Yang, M. Self-supervised graph representation learning integrates multiple molecular networks and decodes gene-disease relationships. Patterns 2023, 4, 100651. [Google Scholar] [CrossRef] [PubMed]
- Shyam, P. In Silico Strategies for Cancer Model Development and Anticancer Drug Testing. In Preclinical cancer models for translational research and drug development; Springer Nature Singapore: Singapore, 2025; pp. 153–168.
- Guo, W.; Liu, S.; Zheng, X.; Xiao, Z.; Chen, H.; Sun, L.; Zhang, C.; Wang, Z.; Lin, L. Network Pharmacology/Metabolomics-Based Validation of AMPK and PI3K/AKT Signaling Pathway as a Central Role of Shengqi Fuzheng Injection Regulation of Mitochondrial Dysfunction in Cancer-Related Fatigue. Oxid Med Cell Longev 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.; Trevizani, R.; Greenbaum, J.A.; Carter, H.; Nielsen, M.; Peters, B. The receiver operating characteristic curve accurately assesses imbalanced datasets. Patterns 2024, 5, 100994. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, Z.; Wang, C.; Li, C.; Wang, B. Comparative Evaluation of Machine Learning Models for Subtyping Triple-Negative Breast Cancer: A Deep Learning-Based Multi-Omics Data Integration Approach. J Cancer 2024, 15, 3943–3957. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liao, N.; Du, X.; Chen, Q.; Wei, B. A semi-supervised approach for the integration of multi-omics data based on transformer multi-head self-attention mechanism and graph convolutional networks. BMC Genomics 2024, 25, 86. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, W.; Lu, F.; Qin, T.; Gou, Y.; Guo, E.; Peng, D.; Zhang, L.; Yang, B.; Liu, S.; et al. Large language models completely understand molecular characteristics of squamous cervical cancer 2023.
- Asada, K.; Kobayashi, K.; Joutard, S.; Tubaki, M.; Takahashi, S.; Takasawa, K.; Komatsu, M.; Kaneko, S.; Sese, J.; Hamamoto, R. Uncovering Prognosis-Related Genes and Pathways by Multi-Omics Analysis in Lung Cancer. Biomolecules 2020, 10, 524. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
PI3k/AKT Signaling Pathway.
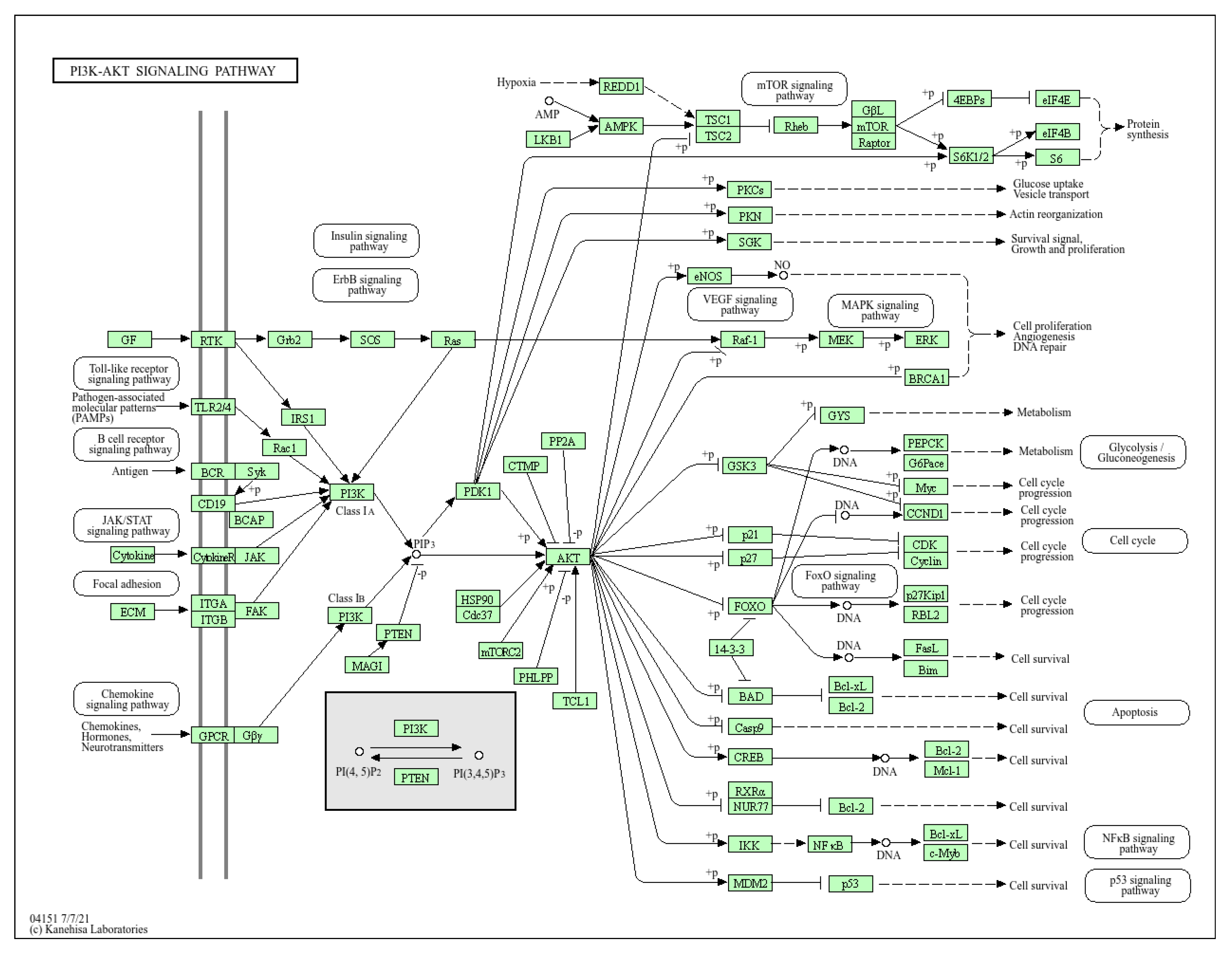
Figure 2.
PI3k/AKT Signaling Pathway Structure.
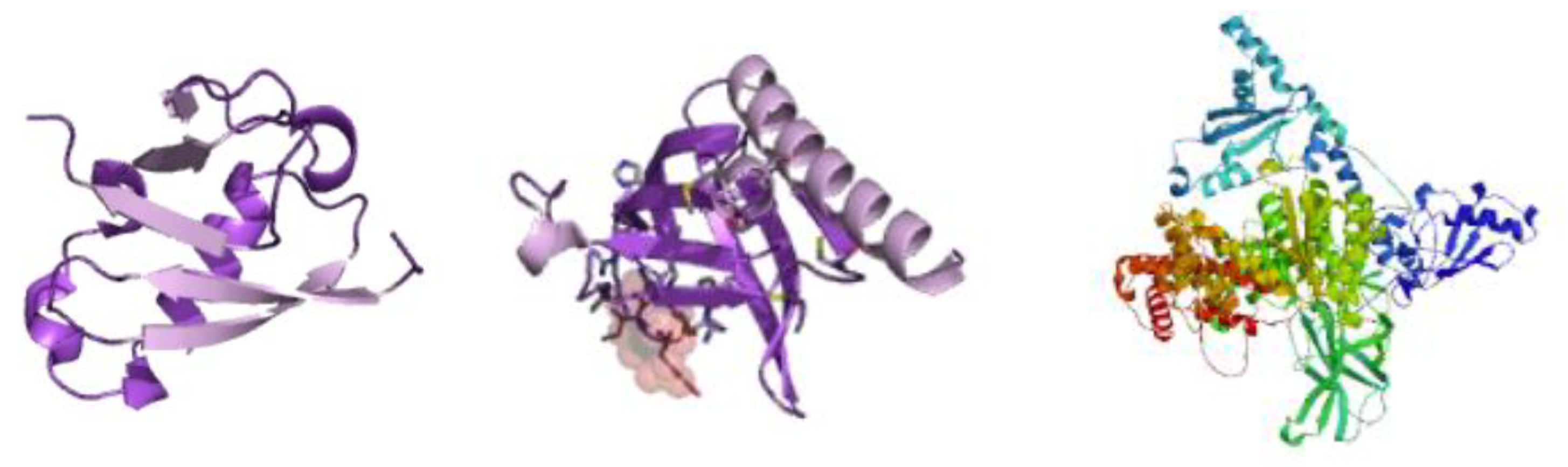
Figure 3.
ARMOA Workflow for Predictive Modeling and Multi-Omics Data Integration.
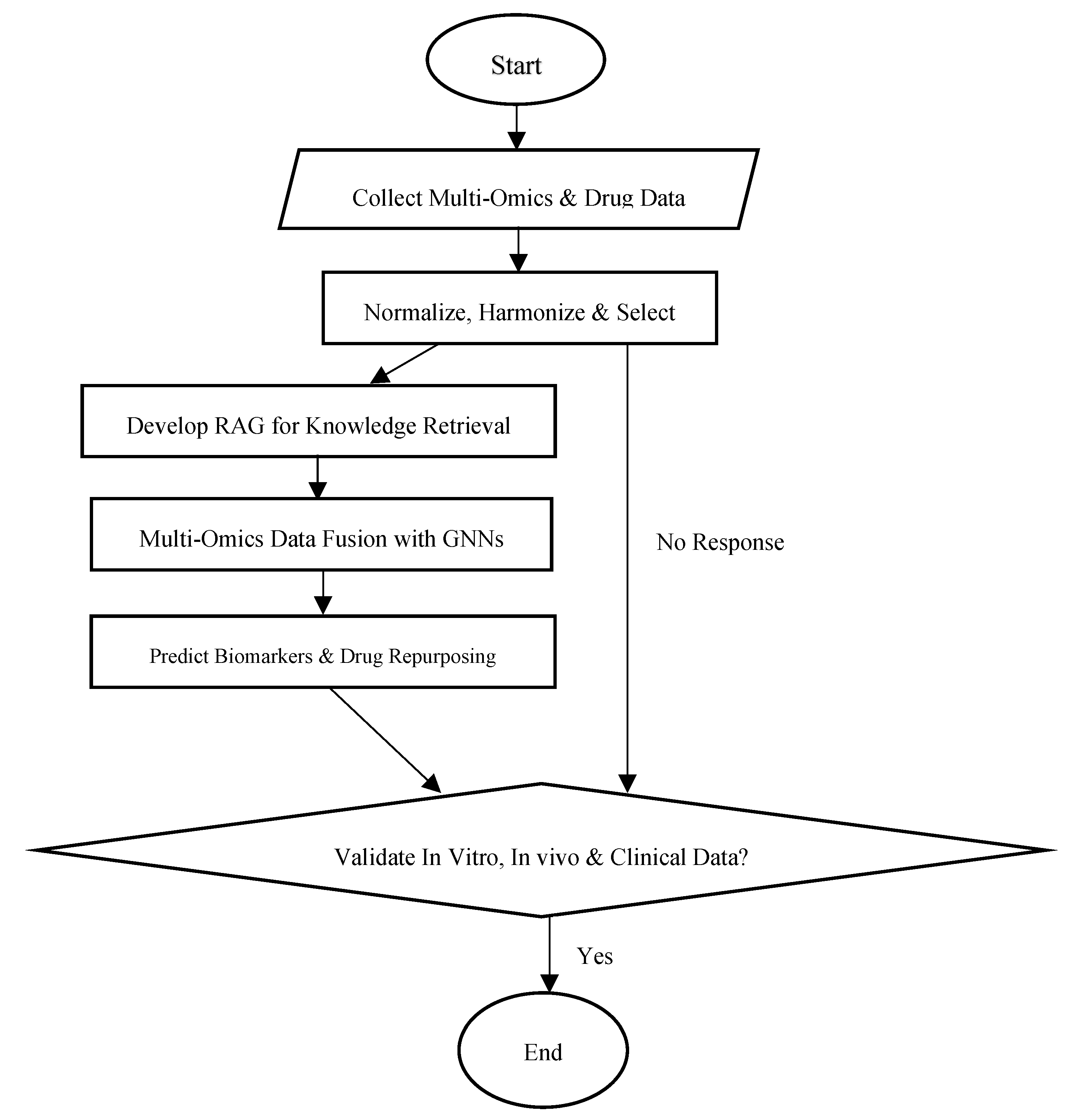
Figure 4.
Raw correlation matrices.
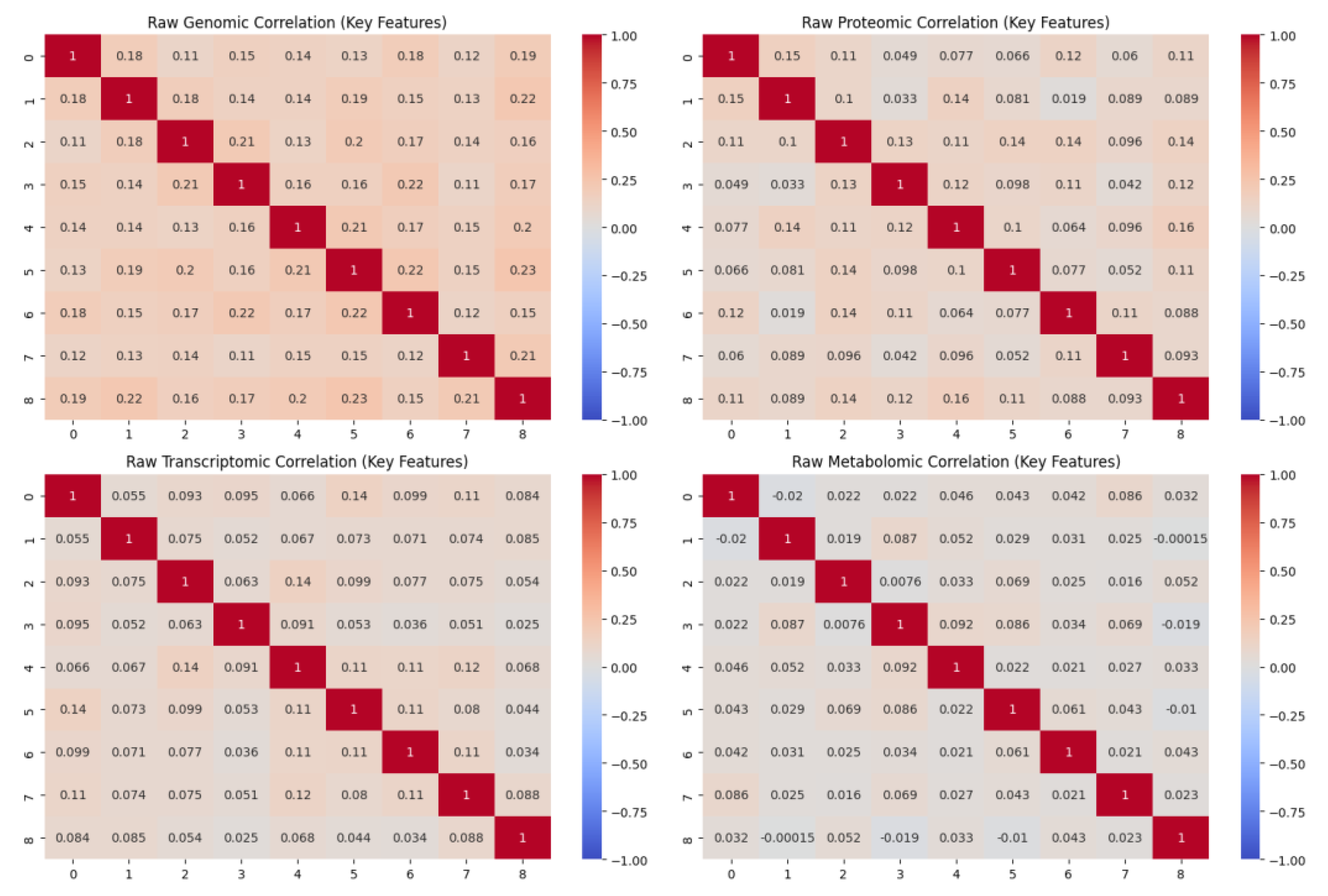
Figure 5.
Normalized correlation matrices.
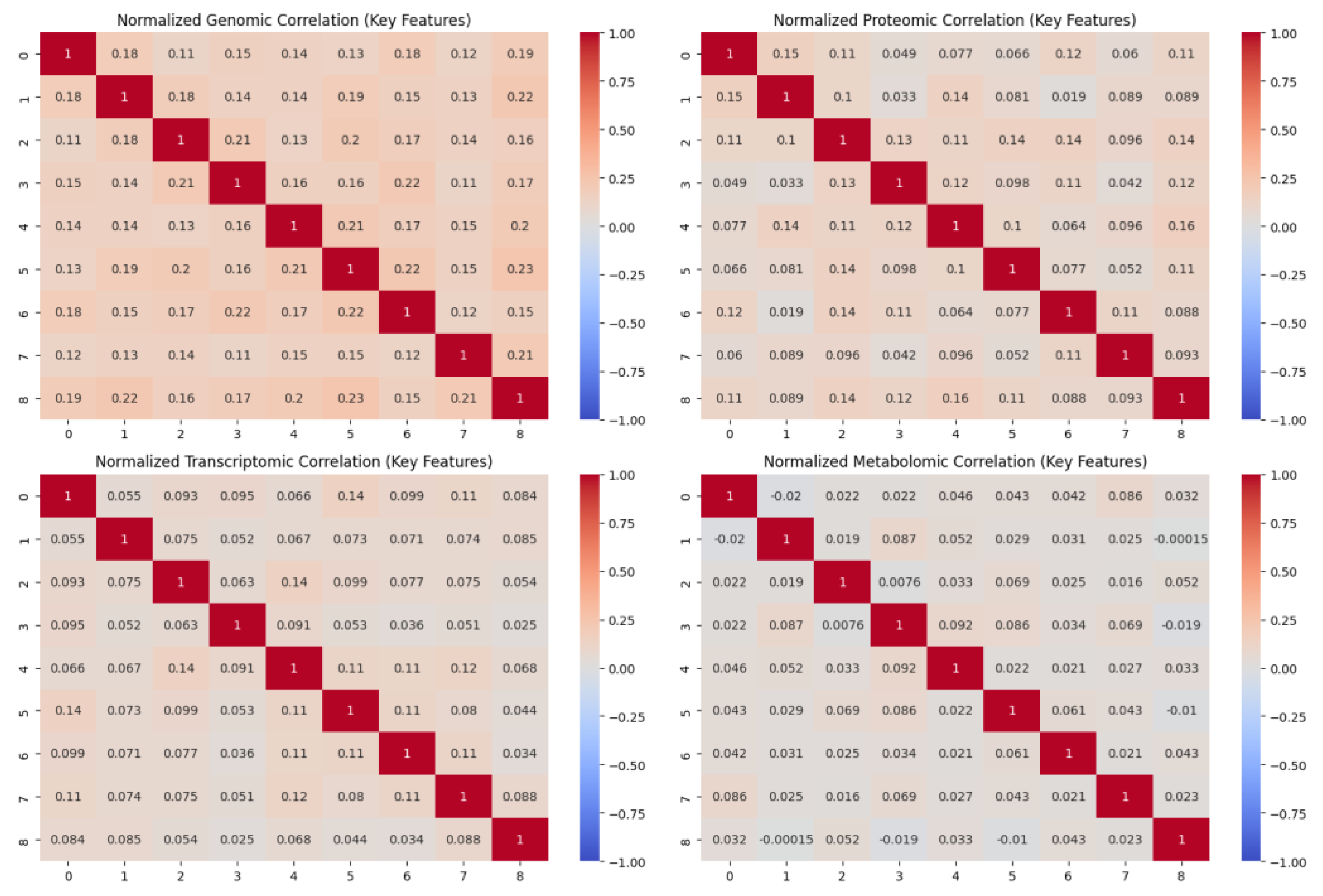
Figure 6.
UMAP Visualization of GNN Embedding Multi-Omics Data Fusion with GNNs.
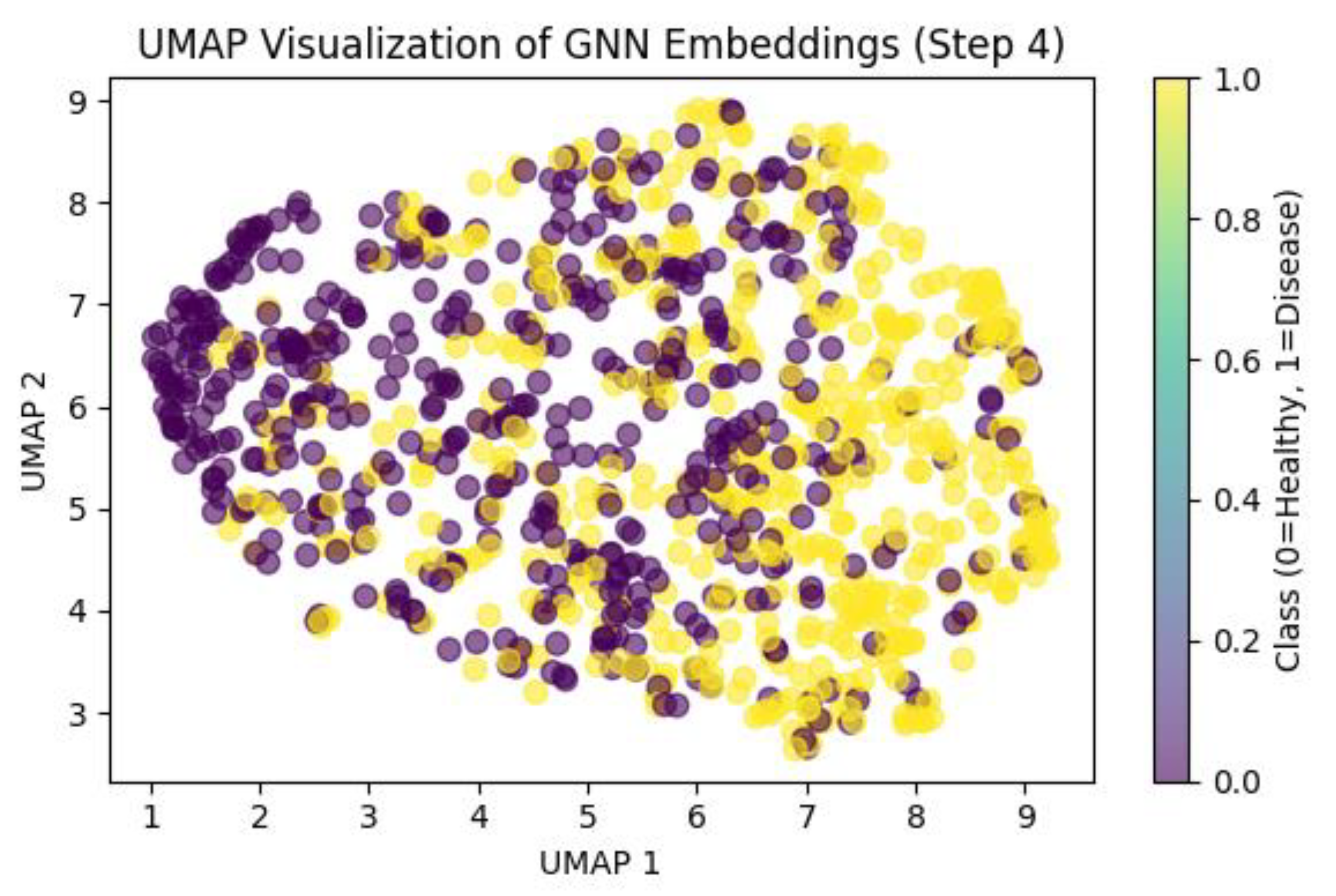
Figure 7.
Confusion Matrix of the ARMOA Model.
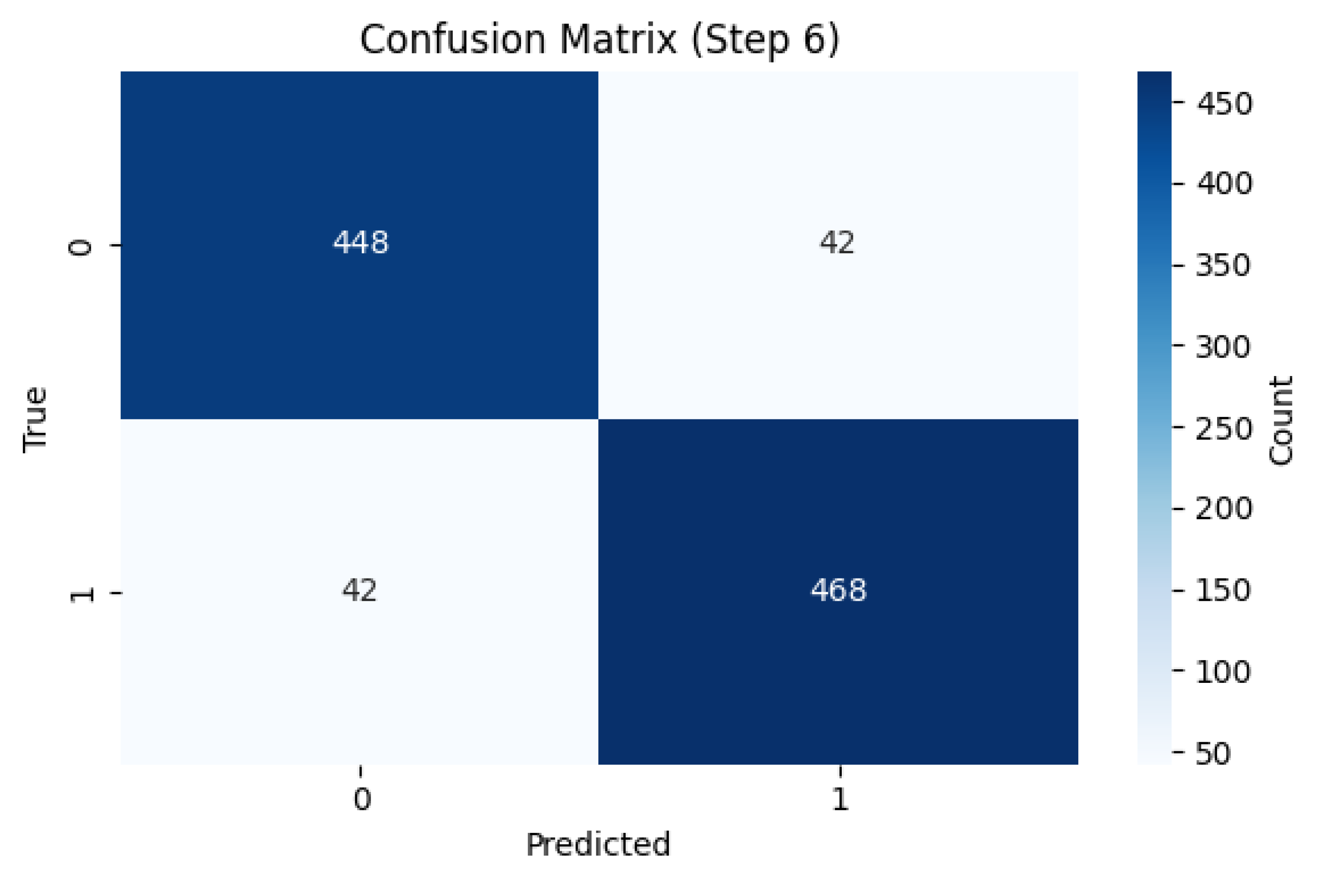
Figure 8.
ROC Curve for the Model.
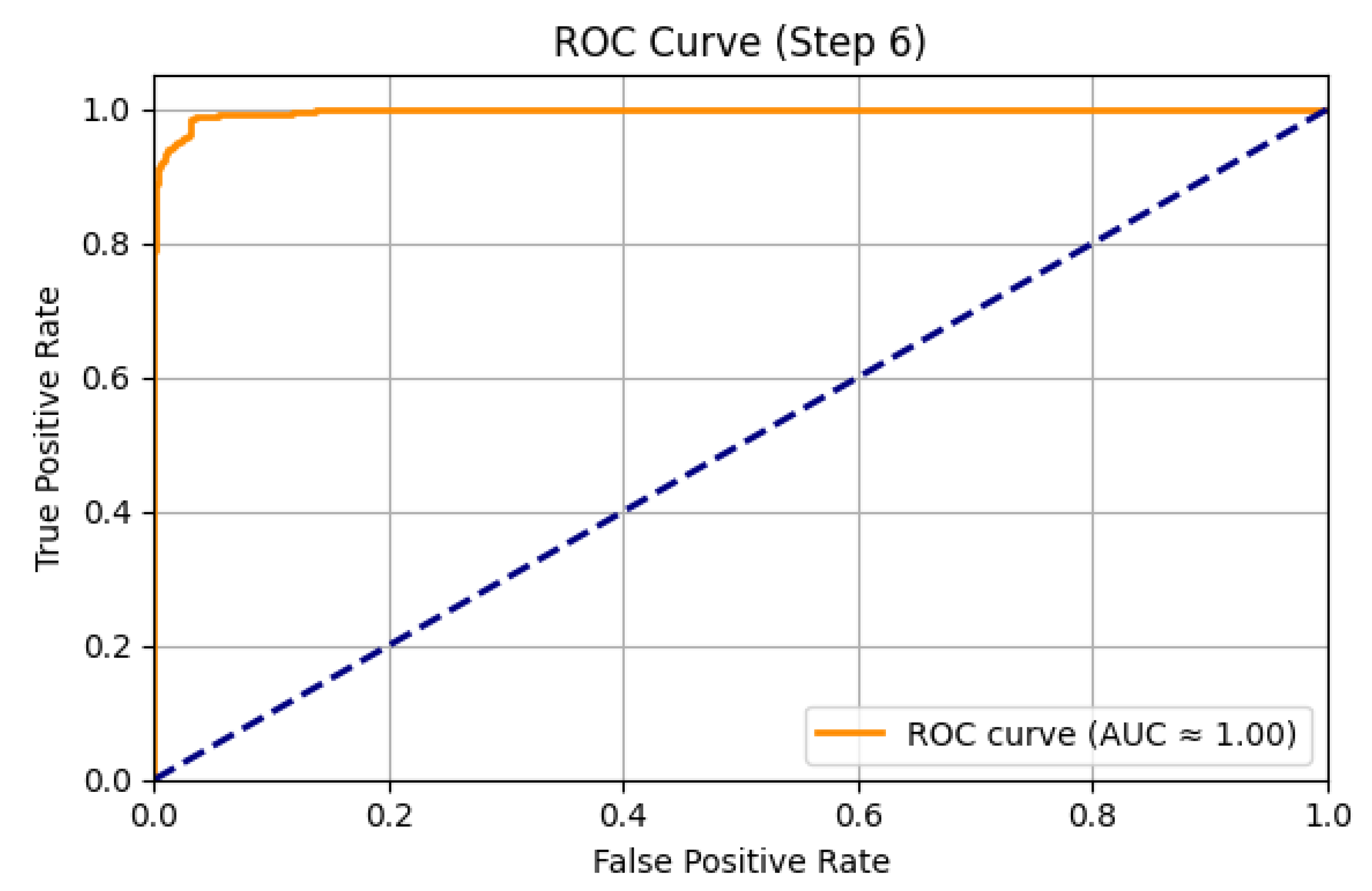
Figure 9.
Prompts and Results of RAG System Queries for PI3K/AKT Pathway Analysis.


Table 1.
Evaluation Metrics for ARMOA Model Performance Validation.
| Evaluation Measure | Value |
|---|---|
| Accuracy | 0.9200 |
| Sensitivity | 0.9176 |
| Specificity | 0.9143 |
| Precision | 0.9176 |
| Recall | 0.9176 |
| F1-Score | 0.9176 |
| Matthews Correlation Coefficient (MCC) | 0.8319 |
| ROC-AUC | 0.9000 |
| Novelty Detection Rate (NDR) | 0.8000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



