Submitted:
11 July 2025
Posted:
15 July 2025
You are already at the latest version
Abstract
Cancer remains a leading global health challenge, with recent artificial intelligence (AI) and machine learning (ML) technologies enabling more accurate predictions of its future impact. Conventional treatments like surgery, radiotherapy, and chemotherapy have advanced with the emergence of targeted and immunotherapy approaches. However, therapeutic resistance and relapse remain major barriers to long-term success in cancer treatment, often driven by cancer stem cells (CSCs). These rare, resilient cells can survive therapy and drive tumour regrowth, urging deeper investigation into the mechanisms underlying their persistence. CSCs express ion channels typical of excitable tissues, which, beyond electrophysiology, critically regulate CSC fate. However, the underlying regulatory mechanisms of these channels in CSCs remain largely unexplored and poorly understood. Nevertheless, the therapeutic potential of targeting CSC ion channels is immense, as it offers a powerful strategy to disrupt vital signalling pathways involved in numerous pathological conditions. In this review, we explore the diverse repertoire of ion channels expressed in CSCs and highlight recent mechanistic insights into how these channels modulate CSC behaviours, dynamics, and functions. We present a concise overview of ion channel-mediated CSC regulation, emphasizing their potential as novel diagnostic markers and therapeutic targets, and identifying key areas for future research.
Keywords:
cancer stem cell
; ion channels
; cellular signalling
; tumour microenvironment
; ion channel-targeted therapy
; cancer therapeutics
1. Introduction
Recent advancements in AI and ML have opened new avenues for predicting global cancer trends with unprecedented accuracy. Alarmingly, AI/ML-based predictive models estimate that by 2050, cancer will account for nearly thousand million new cases and approximately four hundred million deaths worldwide, positioning it as the leading cause of mortality [1]. While ongoing innovations have significantly improved cancer treatment, urgent attention is needed to address the rising burden. Fundamental research remains critical, especially in understanding cancer biology at its root. Among the emerging areas, CSCs have garnered significant attention for their role in therapy resistance, tumour initiation, and relapse [2,3,4,5,6].
Normal stem cells are undifferentiated or partially differentiated cells capable of self-renewal and specialization [7,8]. In mammals, key types include embryonic stem cells (ESCs) from the blastocyst and adult stem cells (ASCs) found in tissues like bone marrow and liver, which remain mostly dormant but activate for tissue repair [7]. Induced pluripotent stem cells (iPSCs), generated by reprogramming adult cells with factors such as Oct4 and Sox2, mimic ESC behaviour [7]. A distinct population, CSCs, has been identified in various tumours [7,8]. CSCs, also known as tumour-initiating cells, are a small but aggressive subpopulation within tumours [8]. CSCs can arise either from normal tissue-resident stem cells or from differentiated cancer cells that acquire stem-like traits, often influenced by signals from the tumour microenvironment [8]. The concept of CSCs, first proposed in the late 1990s, has since become a focal point of intense investigation [9,10]. Over the past decades, extensive research has sought to reveal their roles in cancer biology [11]. CSCs have been identified and extensively characterized across a wide spectrum of tumour types [11]. These cells are identified by specific surface and intracellular markers such as CD44, CD24, CD133, ALDH1, and EpCAM [8]. CSCs have been identified across numerous cancer types such as breast, colon, lung, prostate, liver, melanoma, leukaemia, head and neck, ovarian, pancreatic, and brain tumours [12]. Several key developmental pathways, including Notch, Hedgehog, and Wnt/β-catenin, regulate CSC behavior [8]. These cells possess hallmark properties such as self-renewal, multilineage differentiation, and the unique ability to initiate and sustain tumour growth [13]. Importantly, CSCs are also endowed with a high proliferative potential, enabling them to drive tumour progression and heterogeneity. Due to these intrinsic capabilities, CSCs are increasingly recognized as the root cause of tumour recurrence and metastasis [11,14]. Even after aggressive clinical interventions such as surgical resection, radiotherapy and chemotherapy, a small population of CSCs often survives. These residual cells can evade conventional therapies, regenerate the tumour microenvironment, and ultimately lead to disease relapse [11]. Consequently, many patients experience tumour recurrence and metastasis within a few years post-treatment, emphasizing the critical role of CSCs in cancer persistence and poor long-term prognosis [5]. While CSCs share several characteristics with normal stem cells, such as the capacity for self-renewal and differentiation, they are distinguished by aberrant signalling cascades and a pronounced resistance to conventional therapies [15]. Among the diverse molecular regulators implicated in the maintenance and plasticity of CSCs, ion channels have recently emerged as fundamental modulators [16].
Ion channels, traditionally studied in the context of excitable tissues, are transmembrane proteins that regulate the selective movement of vital ions-such as calcium (Ca²⁺), potassium (K⁺), sodium (Na⁺), and chloride (Cl⁻)-across cellular membranes [17]. Beyond their classical roles in maintaining membrane potential and cellular excitability, ion channels profoundly influence crucial biological processes including cell proliferation, migration, apoptosis, and stemness-related signalling pathways [18]. Recent findings suggest that the dysregulation of specific ion channel families play a crucial role in maintaining the CSC phenotype, supporting their survival, invasiveness, and resistance to therapy [16]. Therefore, it is essential to gain a clear understanding of the ion channel expression profiles in CSCs and their regulatory roles in CSC fate and function, as this knowledge could pave the way for future research aimed at effectively combating cancer. Although a few reviews have discussed ion channels in CSCs, most focus on individual channel type. To the best of our knowledge, this is the first comprehensive review that encompasses a broad spectrum of ion channels expressed across various CSC types and their associated functions. We explore the wide range of ion channels found in CSCs and shed light on recent discoveries regarding their roles in controlling CSC behaviour, functional properties, and cellular dynamics. Special emphasis is placed on acid-sensing ion channels (ASICs) and aquaporin (AQP) channels, which, despite their significant roles in regulating CSC fate and function, have not been comprehensively reviewed until now.
2. AI-Based Generative Prediction of Near-Future Global Cancer Cases
As we deepen our understanding of cancer biology through cellular and molecular insights, particularly the role of CSCs and their regulation by ion channels, it becomes equally important to complement this knowledge with predictive tools that can forecast the future problem of disease. Recent breakthroughs in AI and ML offer powerful methods to model complex datasets and anticipate global cancer trends. This chapter integrates such computational approaches to reinforce the urgency of developing targeted cancer therapies, including those focused on CSC-specific molecular features such as ion channels.
We employed AI and ML to predict global cancer trends. Data were sourced from https://ourworldindata.org/grapher/number-of-people-with-cancer. Using Generative Adversarial Networks (GANs) and Long Short-Term Memory (LSTM) networks, we enhanced datasets for forecasting future global cancer cases. GANs simulated patient data over 30 years, with real and generated data refined via a discriminator network. LSTM predicted mutation trends using both historical and AI-generated data. The dataset was split 80/20 for training and testing, respectively, to evaluate the LSTM model’s forecasting accuracy. In generative adversarial networks (GANs) for generating time series data, the generator and comparator (or discriminator) units work in a dynamic, adversarial process [19]. The generator and comparators units are described as follows
The input and output of the generator are described by Equation 1 and Equation 2 respectively.
z ∼ pz [z]
xˆ = G[z; θg]
Where pz and θgare the distribution of the latent noise and trainable parameter of the generator. The comparator unit evaluates whether an input time series ’x’ is real x ∼ p data [x] or synthetic x = G [z]. The input to the comparator is the output of the generator which is described in the equation. The output of the comparator is described as follows:
D [x; θd] ∈ [0, 1]
Using data from published literature (Table 1) from the past thirty years, along with the above-mentioned method, we predicted global cancer trends for the next 20 years (Table 1 and Figure 1). The sharp projected increase in cancer cases indicating the urgent need for advanced therapeutic strategies and improved management. These AI-generated projections serve as a clarion call for more precise and targeted interventions. Given the rising global cancer burden, it becomes increasingly important to identify molecular vulnerabilities unique to CSCs, such as ion channels. In the following section, we explore into the expression patterns and functional roles of various ion channels in CSCs, aiming to uncover critical insights that could guide future research and pave the way for innovative therapeutic strategies.
3. Ion Channel Expression Profiles and Their Functions in CSCs
A diverse array of functional ion channels has been identified across various types of CSCs, exhibiting significant heterogeneity. CSCs exhibit a unique ion channel expression profile that distinguishes them from bulk tumour cells and normal stem cells. These ion channels span various classes, each contributing to distinct aspects of CSC biology. Through an extensive PubMed-based literature search focusing on ion channels in CSCs, we have compiled a comprehensive list of these channels, summarized in Table 1, deciphering their functional relevance and potential as therapeutic targets (Table 2 and Figure 2). The following section details specific ion channels and their involvement in CSC function.
The figure illustrates key membrane-bound and intracellular ion channels in CSCs, highlighting their downstream signalling pathways and collective roles in regulating ion homeostasis and CSC stemness. Several ion channels contribute to CSC maintenance and proliferation: Na⁺ and K+ channels: Activation of these channels initiates downstream signalling cascades that converge on the β-catenin pathway, ultimately upregulating the CSC-specific transcription factor Oct4 and the oncogene c-Myc, both of which are essential for CSC proliferation. Ca²⁺ channels: Ca²⁺ influx activates the MAPK signalling cascade, leading to ERK phosphorylation and subsequent activation of β-catenin-mediated transcriptional programs driving CSC proliferation [11]. Acid-sensing ion channels (ASICs): ASIC expression has been reported in various tumours and is implicated in tumour progression and metastasis [11,105]. Specifically, Ca²⁺ influx via ASIC1 activates PP2A-dependent apoptotic pathways [105], while Na⁺ entry via ASICs may activate β-catenin signalling and support CSC proliferation (indicated by green and purple dashed lines in the figure). AQPs: These water channels contribute to CSC migration and metastasis through activation of the RhoA/Ras-JNK signalling axis, facilitating disease progression [126]. CLIC1: This membrane protein is crucial for CSC proliferation by regulating the cell cycle, especially by promoting the G1-to-S phase transition [52]. TRP channels: Ca²⁺-impermeable TRPM4 and TRPM5 channels are implicated in maintaining cancer stemness. However, the molecular mechanisms underlying their roles remain largely undefined and warrant further investigation [38]. In addition to plasma membrane channels, the figure also depicts essential organelle membrane channels involved in CSC function: Mitochondrial outer membrane voltage-dependent anion channels (VDACs): These channels supply the elevated energy demand of CSCs during proliferation and metastasis. VDACs are strategically localized near the endoplasmic reticulum (ER) to acquire Ca²⁺ released via IP₃ receptors. The imported Ca²⁺ is subsequently transferred to the mitochondrial matrix through the mitochondrial calcium uniporter (MCU) complex, thus enhancing mitochondrial bioenergetics [3,92]. A portion of the IP₃-released Ca²⁺ also directly promotes CSC stemness and proliferation [3]. All ion channels and their mechanistic roles are described in detail in the main text.
4. Calcium Ion Channels and Their Functions
Intracellular calcium dynamics play a key role in CSCs functions with multiple calcium channels-including store operated calcium entry channels (SOCE), voltage gated calcium channels (VGCC), transient receptor potential (TRP) channels, calcium release channels (CRCs) and mechanosensitive channels [3,20,21,22,23,24,25,26,27,28,29,30].
4.1. SOCE Channels and Their Functions
SOCE is a key mechanism for calcium influx, activated by ER calcium depletion via PLC and IP3 signalling [8]. It involves STIM proteins (ER Ca²⁺ sensors) and Orai channels in the plasma membrane. Ca2+-release activated Ca2+-channels or CRAC channels, mainly formed by Orai proteins, mediate highly selective Ca²⁺ entry to bombard a specific downstream target [8,21,22]. Variants like Orai1α and Orai1β show distinct activation patterns. Dysregulation of SOCE components can disrupt calcium homeostasis, promoting tumour growth, poor prognosis, and drug resistance [8]. Beyond SOCE, Orai1 also supports alternative calcium entry pathways through interactions with secretory pathway Ca2+-ATPase 2 (SPCA2), small conductance calcium-activated potassium channel (SK3 or KCa2.3), and Kv10.1 channels. These interactions can sustain calcium entry and promote CSC survival and migration [8]. Using knockdown and rescue experiments in MDA-MB-231 cells, Jardin et al [22] showed that Orai1 is crucial for spheroid formation and self-renewal in human epidermal growth factor receptor 2 positive (HER2+) and triple-negative breast cancer stem cells (BCSCs), but not in estrogen receptor positive (ER+) BCSCs or BSCs [22]. Both Orai1α and Orai1β equally support SOCE, COX activation, and mammosphere formation in BCSCs [22]. Orai1 interacts with SPCA2 to drive glycolysis-dependent growth in BCSC-like cells via store-independent calcium entry (SICE), while Orai3 and STIM1 promote glycolysis-independent SOCE [20]. In Acute myeloid leukaemia (AML), Orai1-SOCE correlates with leukemic stem cell (LSC) proportion and drug resistance, indicating its utility as a biomarker [21]. Orai3 is enriched in CSCs and promotes stemness via the Orai3-ID1 (Orai3-inhibitor of DNA binding 1) axis in oral/oropharyngeal squamous cell carcinoma (OSCC) [23]. Terrié et al [24] demonstrated that SOC entry supports glioblastoma stem cell (GSC) maintenance, likely through CaMKII and NFAT activation, highlighting calcium’s central role in stemness and tumour progression [24]. However, the precise contribution of SOC in CSCs remains to be fully defined.
4.2. TRP Channels and Their Functions
TRP ion channels, well-known for their roles in sensing physical and chemical stimuli, are increasingly recognized as important regulators of stem cell behaviour, including CSCs [26]. Several TRP family members including TRPC1/3/6, TRPV1/2/4/6, and TRPM7/8 are dysregulated during cancer progression, contributing to diverse oncogenic processes [33,34].
TRPA1 and TRPV1 are modulated by ERK signalling and transcription factor Runx1/Aml1 isoforms, influencing glioblastoma stem cell (GSC) differentiation and survival [27]. TRPC3 is frequently overexpressed in triple-negative breast cancer (TNBC), where it drives proliferation and chemoresistance via the TRPC3–RASA4–MAPK (TRP3-Ras GTPase-activating protein 4-MAPK) axis. Additionally, TRPC3 promotes NF-κB activation in cancer-associated fibroblasts, thereby enhancing tumour growth [35]. In breast cancer, TRPV4 and TRPV6 are upregulated and facilitate calcium influx, which supports cell proliferation, angiogenesis, and tumour progression [36].
TRPM7 plays a particularly versatile role in cancer biology. It promotes metastasis in non-small cell lung cancer (NSCLC) by enhancing O-GlcNAcylation and stabilizing key oncogenic proteins such as c-Myc and caveolin-1 [33]. In lung CSCs, elevated TRPM7 expression is associated with increased levels of stemness and metastatic markers including SOX2, KLF4, CD133, Hsp90α, uPA, and MMP2 [34]. Therapeutically, TRPM7 has emerged as a promising target [33,34]. Oridonin, a bioactive compound from Rabdosia rubescens, suppresses bladder cancer growth by targeting TRPM7 and inhibiting the ERK/AKT signalling pathway [37]. Interestingly, TRPM7 is unique among TRP channels in containing both an ion channel and an α-kinase domain. While its kinase domain regulates sensitivity to Mg²⁺ and cAMP, it is dispensable for the ion channel function. In glioblastoma, TRPM7 activates STAT3 signalling to upregulate ALDH1, thus driving CSC proliferation and invasion [38]. However, further investigation is needed to delineate the specific contributions of its kinase versus channel activities in modulating STAT3, ALDH1, and Notch pathways. Moreover, TRPM7 has been reported to be critical for neuroblastoma cell migration and SNAI2-driven metastasis [39]. In head and neck squamous cell carcinoma (HNSCC), high TRPM7 expression correlates with invasiveness, cisplatin resistance, and poor prognosis. Silencing TRPM7 not only impairs tumorsphere formation but also increases chemosensitivity [40].
Tumour-initiating cells (TICs), which include CSCs, exhibit elevated calcium sensitivity compared to their differentiated counterparts, likely due to enriched expression of ion channels. In glioma-initiating cells (GICs), this sensitivity correlates with neural stem cell markers such as GRIA1, NES, and FABP7 and diminishes upon differentiation. These findings highlight calcium homeostasis as a potential vulnerability in immature, stem-like cancer cells [41].
4.3. VGCCs and Their Functions
VGCCs, composed of α, β, δ, and γ subunits, regulate calcium entry and play critical roles in cellular functions [25]. Among them, CACNG4, a γ-subunit of VGCCs has been implicated in several cancers [25]. Shiozaki et al [42] investigated ion channel expression in gastric cancer stem cells (GCSCs) and found elevated levels of VGCC genes, especially CACNA2D1 and CACNB4, in CD44-high cells isolated from MKN74 gastric cancer cell line [42]. These GCSCs were more resistant to cisplatin but showed increased sensitivity to VGCC inhibitors, amlodipine and verapamil, which reduced tumorsphere formation and tumour growth when combined with cisplatin [42]. They also explored ion channel expression in hepatocellular carcinoma (HCC) CSCs (HepG2 line), revealing upregulation of several channel-related genes, including CACNG4 [25]. Lee et al [43] reported that ovarian cancer’s five-year survival rate remains low due to recurrence and drug resistance, largely driven by CSCs [43]. They investigated the effects of trimebutine maleate (TM) on ovarian CSCs using A2780-SP cells derived from A2780 ovarian cancer cells. They proved that TM showed selective toxicity to CSCs (GI₅₀ ~0.4 µM) compared to non-CSCs (>100 µM), inducing cell cycle arrest and cell death. In mouse models, TM significantly reduced tumour growth [43]. TM targets overexpressed VGCC and calcium-activated potassium channels (BKCa) channels in CSCs, and dual inhibition of these channels further reduced cell viability and stemness markers. Although they explained that TM effectively and selectively reduces stemness and proliferation in ovarian CSCs, potentially involving Wnt/β-catenin signalling [43], but other pathways may also play a role. Additional signalling pathways should be investigated to fully clarify TM’s underlying mechanism of action.
4.4. Ca2+ Release Channels (CRCs)
CRCs like ryanodine receptors (RyRs) and inositol trisphosphate receptors (IP3Rs) regulate calcium flux from the endoplasmic reticulum and are increasingly linked to CSC functions [3]. RyR1 expression is elevated in chemotherapy-resistant BCSCs, where it promotes calcium release via the glutathione S-transferase omega 1 (GSTO1) pathway; blocking this channel reduces CSC markers like Nanog and tumour volume in mouse models [3]. Similarly, downregulation of RyR3 by miR-367 in medulloblastoma enhances stemness traits, including Oct4 and CD133 expression [3]. IP3Rs, though better known for roles in oncogenesis and stem cell differentiation, have also been implicated in CSC maintenance; loss of Selenoprotein K (SELENOK) impairs IP3R function in melanoma, reducing intracellular calcium and stemness gene expression such as PROM1 [3]. These findings suggest both RyRs and IP3Rs contribute to CSC maintenance, though isoform-specific effects require further study.
5. Mechanosensitive Channels and Their Functions
Recently, mechanosensitive channels have gained significant attention for their emerging role in regulating CSCs. Piezo1, a mechanosensitive, calcium-permeable ion channel, regulates focal adhesion dynamics in normal cells by localizing to adhesions in a force-dependent manner [44]. It promotes adhesion maturation and disassembly via localized calcium influx, likely through calpain activation, in an integrin-dependent manner. However, in cancer cells, Piezo1 fails to localize to adhesions and has little effect on adhesion turnover, suggesting a tumour-associated functional shift [44]. Piezo1 supports colon cancer stem cell (CCSC) stemness via calcium/NFAT1 signalling [30]. Piezo1 knockdown reduced Yoda1-induced Ca2+ influx and nuclear NFAT1 protein, without affecting its mRNA. It also destabilized NFAT1 and suppressed stem-like traits, while Piezo1 overexpression increased nuclear NFAT1 and stemness. NFAT1 downregulation blocked Piezo1-driven sphere formation and gene expression [30]. In AML, PIEZO1 is upregulated and associated with better survival [29]. While activation has minimal effect, silencing PIEZO1 impairs proliferation, induces cell cycle arrest, and promotes apoptosis by disrupting DNA repair pathways [29]. In glioblastoma, exosomal circZNF800 from glioma stem-like cells promotes tumorigenesis by activating the PIEZO1/Akt pathway via miR-139-5p sponging. Silencing circZNF800 reduces GBM growth and improves survival in vivo. Exosomes from glioma stem-like cells (GSLCs) contribute to glioblastoma (GBM) aggressiveness by transferring specific circular RNAs (circRNAs) [28]. Zhang et al [28] identified circZNF800 as highly enriched in GSLC-derived exosomes and associated with poor prognosis in GBM patients. CircZNF800 enhances GBM cell proliferation, migration, and survival by sponging miR-139-5p, thereby activating the PIEZO1/Akt signalling pathway. Silencing circZNF800 reduced tumour growth and improved survival in a GBM mouse model [28]. PIEZO1 links cell morphology and cancer progression. In breast cancer, PIEZO1 activation via YODA1 triggers epithelial-mesenchymal plasticity (EMP), a key step in metastasis and therapy resistance, by remodelling calcium signalling in response to shape changes [45].
6. Chloride Channels and Their Functions
Chloride ions, beyond their classical functions in maintaining membrane potential, cell volume, and intracellular pH, are increasingly recognized for their crucial roles in cancer biology, particularly in tumour cell proliferation, migration, and metastasis [46]. Chloride channels including calcium-activated chloride channels (CaCCs), voltage-gated chloride channels (ClCs), acid-sensitive chloride channels, and intracellular chloride channels (CLICs) are deeply involved in tumorigenesis, particularly through their support of CSC proliferation, survival, invasion, and adaptation to the tumour microenvironment [46,47,48,49,50,51,52].
CaCCs such as TMEM16A (also known as ANO1) are overexpressed in various cancers, including lung, breast, colorectal, and gliomas. ANO1 promotes tumour growth through activation of EGFR, PI3K/AKT, and MAPK signalling pathways. In colorectal cancer (CRC), its high expression is associated with poor prognosis, and pharmacological inhibition of ANO1 suppresses tumour growth by downregulating Wnt/β-catenin signalling [46,47]. Another CaCC, Bestrophin-1, has also been implicated in cancer progression, particularly in CRC and oral squamous carcinoma, potentially through modulation of calcium signalling and functional interaction with ANO1 [46].
ClCs such as ClC-2 and ClC-3 are upregulated in gliomas and contribute to enhanced proliferation and motility. Functional assays using antisense oligonucleotides and patch-clamp studies have demonstrated their role in chloride-mediated cell volume regulation and invasive behaviour within the dense extracellular matrix of the brain [48]. ClC-3 is also overexpressed in hepatocellular carcinoma (HCC), CRC, nasopharyngeal, and breast cancers, where it facilitates tumour progression via Wnt/β-catenin and IGF/ERK signalling. Silencing ClC-3 leads to reduced proliferation, suppressed IGF-1 signalling, and decreased tumour growth in breast cancer models [46]. ClC-4, another voltage-gated channel, supports CRC cell invasion and migration by maintaining pH homeostasis in acidic tumour microenvironments [46].
Acid-sensitive chloride channels, particularly TMEM206, are upregulated in CRC, breast, and liver cancers. TMEM206 enables tumour cells to adapt to extracellular acidosis and promotes CRC cell growth and invasion through the AKT/ERK pathway [46].
CLICs, mainly CLIC1 and CLIC4, are significantly overexpressed in several malignancies, including glioblastoma, prostate, colon, and HCC [46,49,51]. In glioblastoma, which is driven by CSCs, CLIC1 has been shown to be selectively active in CSCs but not in mesenchymal stem cells (MSCs) and is crucial for their proliferation and self-renewal [11]. It localizes to the plasma membrane during the cell cycle, where it mediates chloride flux in response to oxidative stress and pH shifts. Blocking CLIC1 disrupts these signals, effectively halting CSC growth without impacting normal stem cells [11]. Additionally, CLIC1 expression in glioblastoma stem cells (GSCs) correlates with poor prognosis, and its inhibition via RNA silencing or antibody targeting reduces proliferation, clonogenicity, and tumorigenicity both in vitro and in vivo [50,51,52].
7. Potassium (K⁺) Channels and Their Functions
Potassium (K⁺) channels are crucial regulators of membrane potential and cell signalling, classified into voltage-gated (Kv), inwardly rectifying (Kir), and two-pore (K2P) families [53]. In CSCs, specific K⁺ channels such as Kv1.3, KCa3.1, and voltage-sensitive human ERG (hERG, Kv11.1) regulate stemness, survival, and proliferation. Inhibition of these channels induces cell cycle arrest and reduces CSC proliferation, as observed in leukaemia models [11,54]. Similarly, K+ channel tetramerization domain containing 12 (KCTD12), a K⁺ channel-associated protein, serves as a prognostic marker in gastrointestinal cancer. In colorectal CSC-like cells, its expression is reduced [11]. Silencing KCTD12 boosts self-renewal and drug resistance, while its overexpression suppresses these traits, highlighting its role in regulating CSC functions and chemoresistance. [11].
Various K⁺ channels including Kv, KCa, hEag, KATP, and K2P are overexpressed in CSCs from prostate, colon, lung, and breast cancers [53]. Blocking these channels suppresses tumour growth or enhances chemotherapy response. Kv (e.g., Eag1, HERG, Kv1.3), Kir3.1, and KCa (KCa1.1, KCa3.1) are especially linked to tumour progression [53]. In glioblastoma CSCs, KCa3.1 is upregulated and exhibits stronger currents in CD133⁺ cells [54].
In prostate cancer, overexpression of IKCa1 (KCa3.1) supports proliferation. Its activity hyperpolarizes the membrane and enhances calcium influx via TRPV6, potentially driving tumour growth [55]. In glioblastoma, M2 muscarinic receptor (M2 mAChR) activation suppresses CSC proliferation and migration partly by inhibiting IKCa currents. Direct IKCa inhibition with TRAM-34 reduces CSC motility and invadopodia formation [56]. In pancreatic CSCs, Shiozaki et al [57] found upregulation of Kv channel genes in ALDH1A1⁺ cells from the PK59 line. Treatment with 4-aminopyridine (4-AP) selectively impaired CSC viability and reduced tumor growth in vivo. In breast cancer, multiple K⁺ channels are linked to CSC maintenance and tumor progression [58]. Kv1.3 is silenced in aggressive tumours via promoter methylation and associated with poor prognosis. Kv10.1 is elevated in invasive and TNBC, responding to astemizole. Kv11.1 (hERG1) is linked to better survival in ER-negative patients, whereas Kir3.1 correlates with metastasis in ER-positive tumours. KCNK9 (K2P) promotes migration in TNBC, while KCa1.1 and KCa3.1 are associated with higher tumour grade and metastasis [58].
8. Sodium (Na⁺) Channels and Their Functions
Na⁺ channels, particularly voltage-gated sodium channels (VGSCs), are membrane proteins that regulate Na⁺ influx and initiate action potentials [59]. In cancer, aberrant VGSC expression is increasingly linked to enhanced invasion, metastasis, and maintenance of CSC traits [59]. Although compounds like eicosapentaenoic acid and tamoxifen show anti-tumour effects by targeting VGSCs, their clinical use is limited due to poor subtype selectivity. However, recent efforts are focused on developing VGSC subtype-specific small molecules and peptides as promising therapeutic tools [59].
In glioblastoma stem cells (GSCs), Giammello et al [60] reported that VGSCs help maintain the depolarized resting membrane potential (RMP), particularly during the G0 phase, supporting stemness. Blocking VGSCs induced GSC differentiation, reduced self-renewal, increased temozolomide sensitivity, and reactivated ERK signalling, suggesting a new approach for glioblastoma therapy [60]. TNBC, characterized by high plasticity and drug resistance, exhibits elevated activity of VGSCs [61]. Nav1.5 (SCN5A) enhances tumour cell invasion and drives epithelial-to-mesenchymal transition (EMT) [61]. Targeting VGSCs in TNBC has been linked to reduced stemness and tumour aggression [61].
9. ASICs and Their Functions
ASICs are voltage-insensitive sodium channels that detect extracellular acidity or proton levels [62]. Several ASIC subtypes, including ASIC1, ASIC1a, and ASIC3, have recently been identified in CSCs [62,63,64]. In glioblastoma stem cells (GSCs), which are key contributors to therapy resistance in malignant glioma, functional ASIC1a and ASIC3 channels are expressed [62]. In contrast, ASIC2, known for its anti-oncogenic role, is absent. Electrophysiological studies confirmed that ACCN2 and ACCN3 transcripts are translated into active ASIC1a- and ASIC3-containing channels. ASIC1b was present at low levels and largely non-functional [62]. Clusmann et al [63] found that under prolonged acidosis, ASIC1a activation induces necrotic cell death, partially through necroptosis-like pathways. They proved that inhibiting ASIC1a or knocking it out protects GSCs from acid-induced death, while activating it reduces tumorsphere formation even at neutral pH [63]. Although their findings suggest that targeting ASIC1a could be a novel therapeutic approach by exploiting the acidic tumour microenvironment to eliminate CSC, but no clear underlying mechanism has been described [63].
10. AQPs or Water Channels and Their Functions
AQP, known primarily for their roles in water and solute transport, are now recognized for their broader involvement in key CSC-related processes and their regulations [65,66,67,68,69,70,71,72,73,74]. Recent research describes the crucial role of aquaporin channels in cancer, particularly in CSCs. Several isoforms, including AQP1, AQP3, AQP4, AQP5, AQP8 and AQP9 are expressed in CSC populations [65,66,67,68,69,70,71,72,73,74]. AQP1 overexpression in rat C6 glioma CSCs enhanced cell viability and migration, potentially promoting glioma progression through transcriptional networks involving Foxo4, Maz, and E2F factors [65]. AQP3 has emerged as a key oncogenic factor across multiple cancers [66,67,68,69]. In breast cancer, AQP3 is upregulated in ER-positive invasive ductal carcinoma, where it enhances migration, invasion, and EMT. Its expression is estrogen-inducible via an estrogen response element (ERE) in the promoter. Clinically, elevated AQP3 correlates with poor differentiation, lymph node metastasis, and premenopausal status [66]. Similarly, AQP3 is highly expressed in hepatocellular carcinoma (HCC), where it correlates with advanced stage, metastasis, and poor prognosis [67]. Mechanistically, AQP3 promotes stemness by enhancing CD133 expression through activation of the JAK/STAT3 pathway, which facilitates STAT3 phosphorylation, nuclear translocation, and histone H3 acetylation at the CD133 promoter [67,68]. Additionally, Liu et al [69] reported that AQP3 inhibits differentiation and apoptosis in liver cancer stem cells (LCSCs) by suppressing components of the Wnt/GSK-3β/β-catenin pathway, including β-catenin, GSK-3β, and STAT3. Collectively, these findings position AQP3 as a key driver of cancer progression and stemness through distinct signalling mechanisms [66,67,68,69]. In glioblastoma (GBM), AQP4-loaded extracellular vesicles (EVs) influence tumour behaviour [70]. EVs containing AQP4-tetramers promote cell migration, while those with AQP4-orthogonal arrays of particles (OAPs) induce apoptosis, highlighting a novel mechanism by which AQP4 modulates the tumour microenvironment [70]. AQP5 is highly overexpressed in breast cancer, but not in normal tissue [71]. Its expression is linked to loss of polarity, increased proliferation, and poor prognosis, particularly in ER/PR-negative and HER2-positive cases. Gene amplification may drive this overexpression [72]. In gastric cancer, AQP5 serves as a surface marker of cancer stem cells (GC-CSCs), co-localizing with LGR5 [72]. It enhances growth, invasion, and tumour formation by activating autophagy and promoting ULK1 ubiquitination [72]. AQP8 is overexpressed in cervical cancer and promotes viability, migration, and EMT, while suppressing apoptosis [73]. It has been reported that AQP9 is expressed and significantly downregulated in LCSCs, and its restoration suppresses stemness by promoting reactive oxygen species (ROS) accumulation. This leads to reduced β-catenin–TCF4 interaction and enhanced β-catenin-FOXO3a association, ultimately inhibiting LCSC self-renewal [74].
10.1. Intracellular Organelles Ion Channels and Their Functions
Intracellular organelle ion channels play essential roles in the survival and function of CSCs by regulating ion homeostasis, apoptosis, and metabolic signalling. Mitochondrial ion channels such as the mitochondrial calcium uniporter (MCU) are critical for tumour growth, influencing cell cycle progression and energy metabolism essential for CSC maintenance [75]. MCU-driven mitochondrial calcium uptake enhances breast cancer cell migration via store-operated Ca²⁺ entry; inhibition of MCU suppresses metastasis in vivo and slows tumour growth through impaired mitochondrial redox signaling [76]. Additionally, ER-resident channels like TMCO1 modulate calcium dynamics and resistance to apoptosis, contributing to CSC survival under therapeutic stress [77]. Synthetic ion transporters targeting mitochondrial and lysosomal membranes have shown potential in selectively inducing apoptosis in CSCs, revealing novel avenues for anti-CSC therapies [78]. Recently, Zheng et al [79] proved that circular RNA hsa_circ_0007905 is upregulated in cervical cancer (CC) and enriched in cancer stem-like cells [79]. Its knockdown impairs proliferation, invasion, self-renewal, and promotes apoptosis. Mechanistically, it sponges miR-330-5p, relieving repression of VDAC1. Restoring VDAC1 or inhibiting miR-330-5p reverses these effects. They suggested that this axis may serve as a therapeutic target in CC [79]. These findings highlight the functional significance of organellar ion channels in CSC biology and emphasize their therapeutic potential in targeting resistant cancer populations.
11. Interplay Between Ion Channels and Epigenetic Control in CSCs
Understanding ion channel expression and function in CSCs reveals their role in regulating key cellular processes. Epigenetic mechanisms further modulate these channels, affecting their expression, localization, and activity. The next section explores how this interplay supports CSC maintenance, plasticity, and therapy resistance.
CSCs, recognized as subpopulations within tumours, contribute significantly to tumour heterogeneity through both genetic and epigenetic mechanisms [80]. This heterogeneity enhances the adaptability of cancer cells, facilitating invasion and resistance to therapy [81]. Epigenetic regulation-comprising DNA methylation, histone modifications, and non-coding RNAs-maintains CSC plasticity and stemness without altering the DNA sequence [82,83]. Emerging evidence reveals a dynamic crosstalk between ion channel signalling and epigenetic machinery in CSCs [84]. Ion channels, especially those mediating calcium influx like the store-operated Ca²⁺ channel Orai1, activate intracellular cascades such as CaMKII-AKT/PI3K and MAPK pathways, which in turn upregulate EMT drivers like TWIST (TWIST related protein1 ir Twist1), thereby promoting CSC-like states [85,86]. TWIST is not only central to EMT but also facilitates tumour initiation and metastasis. Epigenetic mechanisms reciprocally modulate ion channel expression; for example, DNA methylation silences Kv1.3 in colorectal cancer, impacting cellular excitability and tumour behaviour [86]. Additionally, post-translational modifications of histones-such as methylation and acetylation-govern transcription of key epithelial markers like CDH1, often repressed by the polycomb protein EZH2 [87]. In BCSCs, hyperactivation of the nuclear respiratory factor 2 (NRF2) pathway is mediated by the epigenetic reader Zinc Finger MYND-Type Containing 8 (ZMYND8), which enhances antioxidative capacity and promotes resistance to oxidative damage and ferroptosis [88]. Emerging evidence suggests that this NRF2-ZMYND8 axis modulate the expression and activity of redox-sensitive ion channels, such as TRP channels and calcium-permeable channels, thereby contributing to CSC survival, metabolic flexibility, and therapeutic resistance under oxidative stress conditions [89]. TRPM2, a member of the TRP melastatin subfamily, is highly expressed in variety of tumours and CSCs and plays a key role in protecting cell viability by modulating oxidative stress [89,90]. Interestingly, the inhibition of TRPM2 channels results in mitochondrial dysfunction, elevated ROS, and reduced levels of antioxidant cofactors such as GSH, NADPH, and NADH-effects linked to suppressed NRF2 signalling and decreased expression of its antioxidant gene targets. Importantly, TRPM2-dependent stabilization of NRF2 involves IQGAP1 and calcium signalling, pointing toward a regulatory loop where epigenetic factors like ZMYND8 may indirectly influence TRPM2 expression and function, further coordinating redox balance and stemness in CSCs [89,90]. Recent research demonstrated that mitoepigenetic dysregulation in CSCs affects mitochondrial function and redox balance, contributing to tumour progression. Recent findings show that AIF-induced mitochondrial Ca2+ overload, due to impaired mitochondrial Ca2+ uniporter (MCU) regulation, activates calpain and retrograde signalling, enhancing oncogenic gene expression in CSCs. This ROS-Ca2+-calpain axis, integrated with epigenetic modulation of mitochondrial DNA, links mitochondrial dynamics to CSC survival and aggressiveness in numerous tumours [91,92]. In glioblastoma stem-like cells (GSCs), VDAC1 depletion has been shown to trigger extensive epigenetic reprogramming, leading to changes in chromatin organization and transcriptional activity. Specifically, silencing of VDAC1 in GSCs results in reduced mitochondrial metabolism and NAD⁺ levels, which in turn downregulates the activity of sirtuin deacetylases SIRT1 and SIRT6-key epigenetic regulators [93,94]. This inhibition leads to hyperacetylation of histone H3 (at Lys9 and Lys27) and histone H4, altering the expression of genes involved in stemness and oncogenic signalling. Notably, this also affects the expression of calcium and potassium ion channels such as CACNA1C (L-type calcium channel) and KCNMA1 (BK channel), both of which are epigenetically regulated and critical for CSC maintenance and invasiveness [94]. These bidirectional interactions between ion channels and epigenetic modifiers represent a complex regulatory axis, critical to CSC maintenance, tumour progression, and therapeutic resistance.
12. Ion Channel-Targeted Therapies in CSCs
Targeting ion channels presents a promising therapeutic approach, especially in cancer, where their dysregulation contributes to tumour growth, invasion, and treatment resistance. By modulating these channels, it is possible to disrupt critical signalling pathways and impair CSC functions, contribution a novel avenue for more effective treatments [95]. CSC ion channel blockers have shown significant potential in cancer therapy, influencing tumour cell proliferation, differentiation, apoptosis, and metastasis [96]. This chapter explores the emerging strategies aimed at modulating ion channel activity to selectively target CSCs, offering novel avenues for more effective and durable cancer treatments (Table 3 and Figure 3).
The figure illustrates the pharmacological targeting of ion channels in CSCs impairs stemness and promotes apoptosis. Blockade of voltage-gated Na⁺ channels using temozolomide (TMZ) and tetrodotoxin (TTX) reduces SOX2 and NANOG expression, suppressing glioma stem cell (GSC) self-renewal and shifting cell cycle progression toward the G1 phase [63,127]. L-type Ca²⁺ channel blockers (manidipine, lomerizine HCl, and benidipine HCl) inhibit stemness by suppressing AKT/ERK signalling, triggering apoptosis [98]. Aquaporin-mediated ROS elevation impairs Wnt/β-catenin signalling, disrupting proliferation and inducing apoptosis, while ROS scavenging via NAC restores stemness [74]. Activation of ASIC3 by 2-Guanidine-4-methylquinazoline (GMQ) selectively depolarizes GBM CSCs, causing cell damage [105,128]. In ovarian CSCs, inhibition of K⁺ channels (VGCC and KCa) by trimebutine maleate (TM) downregulates stemness markers and Wnt/β-catenin signalling, sensitizing cells to therapy. Chloride channel modulators such as chlorotoxin and bufalin reduce migration and survival by modulating MMP-2 and the PI3K/Akt/mTOR pathway, respectively. TRPM4 inhibition via 9-phenanthrol impairs breast CSC stemness and induces apoptosis [38].
The therapeutic potential of targeting various calcium channels in CSCs is increasingly recognized for its ability to disrupt tumour progression and overcome treatment resistance. FDA-approved manidipine and lacidipine inhibit L- and T-type voltage-gated calcium channels, leading to reduced sphere formation, viability, and proliferation of ovarian CSCs [97]. They also induce apoptosis and suppress stemness by inhibiting the AKT and ERK signalling pathways, which are crucial for maintaining CSC properties [97]. Mibefradil inhibits the T-type calcium channel Cav3.2, which is overexpressed in glioblastoma stem-like cells (GSCs) [98]. This inhibition leads to decreased proliferation, survival, and stemness of GSCs, and sensitizes them to temozolomide chemotherapy. This effect is mediated through suppression of the pro-survival AKT/mTOR pathway and activation of apoptotic regulators such as survivin and BAX [98]. SOCE channels are essential for calcium influx in glioblastoma CSCs [26]. Pharmacological inhibition of SOCE reduces proliferation, impairs self-renewal, and decreases the expression of stem cell markers like SOX2, thereby targeting the CSC population responsible for tumour recurrence [26]. Verapamil, a Ca²⁺ channel blocker, targets MDR-related proteins, reducing proliferation and inducing apoptosis in gemcitabine-resistant pancreatic CSCs [99]. In GBM, combining the KCa3.1 inhibitor TRAM-34 with temozolomide (TMZ) significantly decreases DNA synthesis and CSC survival compared to TMZ alone, also reducing tumour cell infiltration [100,101].
TRPC6 supports breast cancer stem cell traits and chemoresistance by regulating integrin α6 mRNA splicing [102]. It suppresses epithelial splicing regulatory protein 1 (ESRP1), promoting the α6B splice variant, which activates transcriptional co-activator with pDZ-binding motif (TAZ) and reduces Myc expression. Blocking TRPC6 disrupts this pathway, sensitizing TNBC cells to chemotherapy [102]. Several compounds have been reported to irreversibly suppresses the TRPM7 channels, such as waixenicin A, an organic extract of the soft coral S. edmondsoni 14, but they lack specificity and therefore are of limited use [38]. The activation of STAT3 and Notch signalling pathway that leads to the regulation of ALDH1 pathway genes in GSC identify potential therapeutic targets for the treatment of glioma [38].
In hepatocellular carcinoma CSCs, the VGCC subunit CACNG4 is implicated in maintaining stemness, and its inhibition by amlodipine suppresses CSC characteristics [25].
Potassium (K⁺) channel inhibition promotes apoptosis and may reverse multidrug resistance (MDR). Kv1.3 inhibitors like margatoxin (MgTX) and non-selective blockers such as 4-aminopyridine (4-AP) suppress metastasis in prostate cancer and reduce lung cancer cell growth, with MgTX also promoting apoptosis by inducing G1-S phase arrest [103].
Glioblastoma CSCs show strong resistance to BCNU (bis-chloroethylnitrosourea) chemotherapy; however, combining BCNU with the chloride channel inhibitor DIDS (4,4’-diisothiocyanostilbene-2,2’-disulfonic acid) promotes apoptosis and reduces proliferation [104]. CLIC1 contributes to this resistance and may act as a drug efflux channel. Inhibiting CLIC1, either pharmacologically (e.g., with metformin) or via RNA interference, enhances CSC sensitivity to treatment. Similarly, blocking the CLCN3 channel also suppresses CSC proliferation. These findings highlight chloride and calcium channel inhibitors as potential tools to overcome multidrug resistance in cancer therapy [104]. New biguanide compounds Q48 and Q54 block CLIC1 more effectively than metformin, reducing GSC proliferation, invasion, and self-renewal with minimal toxicity, showing promise as targeted glioblastoma therapies [51].
Recent studies have highlighted the therapeutic potential of targeting ASICs in CSCs, particularly within glioblastoma, where acidic tumour microenvironments activate these channels. ASIC1a has been shown to induce necroptosis in GSCs through a RIPK1-dependent pathway [62]. This cell death mechanism can be inhibited by the ASIC1a antagonist PcTx1, indicating that pharmacological modulation of ASIC1a could be a viable therapeutic strategy [62]. Additionally, ASIC3 has been identified as a contributor to GSC proliferation and migration, with its inhibition leading to reduced tumour growth, suggesting that ASIC3 is another promising target for therapy [105]. Beyond glioblastoma, ASIC1a’s role extends to other cancers; for instance, in breast and prostate cancers, its activation leads to reactive oxygen species (ROS) production and activation of survival pathways like AKT and NF-κB, while in pancreatic cancer, it promotes epithelial-mesenchymal transition via RhoA signalling [106]. These findings describe the significance of ASICs in cancer progression and the potential of ASIC-targeted therapies across various tumour types.
AQP channels also regulate CSC functions, offering novel targets for cancer therapy. In gastric cancer, AQP3 is overexpressed and correlates with the CSC marker CD44. This overexpression enhances stem-like traits in gastric cancer cells by activating the Wnt/GSK-3β/β-catenin signalling pathway. Experimental manipulation of AQP3 levels directly influences tumorigenic potential and self-renewal capacities, emphasizing its role in maintaining CSC characteristics. Targeting AQP3 could, therefore, disrupt these pathways and diminish CSC-driven tumour growth [107]. Similarly, AQP5 is upregulated in gastric cancer stem cells and contributes to their self-renewal by modulating autophagy. Inhibiting AQP5 expression has been shown to reduce the stemness of these cells, suggesting that AQP5 is integral to the maintenance of CSC properties and represents a potential therapeutic target [108]. In hepatocellular carcinoma, AQP9 expression is typically reduced in liver CSCs. Restoring AQP9 levels suppresses CSC characteristics by increasing reactive oxygen species (ROS) accumulation, which in turn inhibits β-catenin activity and promotes its interaction with FOXO3a. This mechanism leads to the attenuation of stemness features in liver CSCs, highlighting AQP9 as a promising target for therapeutic intervention [78]. Collectively, these findings emphasize the diverse roles of AQPs in sustaining CSC properties across different cancer types. Therapeutic strategies aimed at modulating AQP function or expression could, therefore, be effective in targeting CSCs and impeding cancer progression.
Many ion channels enriched in GSCs belong to broader families with overlapping functions, making it challenging to target specific channels therapeutically [109]. To address this, Pollak et al [109] analysed enrichment patterns at the family level, identifying 12 ion channel families (including epithelial Na⁺ channels, GABA_A receptors, and ionotropic glutamate receptors) with significant GSC-specific expression. Pharmacological blockade of these enriched ion channels, such as with TTX, TEA, 4-AP, CPP, CNQX, ω-Conotoxin MVIIC, and CdCl₂, significantly reduced GSC viability in vitro, confirming their functional relevance and therapeutic potential [109].
Taken together, the expanding landscape of ion channel-targeted therapies offers a promising avenue for eradicating CSCs and overcoming treatment resistance. However, to fully realize their clinical potential, these strategies must be integrated with precise diagnostic tools, molecular profiling, and advanced drug delivery technologies.
13. Challenges and Strategies in Ion Channel Research for CSCs
Ion channels are key regulators of CSC functions, but despite their therapeutic potential, significant challenges persist in the research and development of ion channel-targeted drugs for CSCs [95,110]. One of the primary obstacles is the remarkable heterogeneity and plasticity of CSCs [95,111,112,113]. CSCs display remarkable intra- and inter-tumoral heterogeneity. Their phenotypic plasticity allows them to shift dynamically between stem-like and differentiated states, which is often accompanied by changes in ion channel expression and function [95,111,112,113]. This variability complicates the development of universal ion channel-targeted strategies [95,111,112,113]. Ion channels in CSCs often exhibit functional redundancy. Upregulated expression of several voltage-gated potassium (Kv) channels, including KCNB1, KCNC1, and KCND1 have been reported in pancreatic CSCs (PK59 CSCs) [114]. Inhibition of KCNB1 with 4-aminopyridine (4-AP) reduced CSC proliferation, suggesting that while multiple Kv channels are overexpressed, targeting specific ones like KCNB1 can impact CSC viability. This indicates potential redundancy among Kv channels in supporting CSC functions [114]. Many ion channels involved in CSC maintenance are also expressed in normal stem cells or vital organs such as the heart and brain. This raises safety concerns and necessitates the development of CSC-specific delivery systems or modulators with high selectivity [95]. Conventional cancer models poorly mimic the CSC niche and ion channel expression patterns [115]. Sphere-forming assays and xenografts are useful but have limitations in throughput and physiological relevance. Additionally, ion channel function is difficult to assess in rare and non-adherent CSCs using traditional electrophysiological tools. High-throughput, label-free technologies suitable for CSCs are still in development [116]. The CSC microenvironment, which are characterized by hypoxia, acidosis, and stromal interaction, profoundly influences ion channel activity [95,117]. Hypoxic conditions can modulate the expression and gating properties of specific ion channels, thereby altering drug sensitivity. Ion channel-targeted therapies must therefore be evaluated in microenvironmentally relevant models to ensure clinical translatability. Identifying CSCs remains challenging, relying on surface markers, functional assays, and gene expression [95]. In cancers like AML and colorectal cancer, CSCs resemble normal stem cells from their tissue of origin. Markers like CD47 have emerged as promising therapeutic targets, with anti-CD47 antibodies showing success in reducing CSCs in AML. Advances also highlight roles for microRNAs in CSC differentiation and metastasis, offering new avenues for treatment [95].
Overcoming the challenges in targeting ion channels in CSCs must prioritize a multidisciplinary and precision medicine approach. To begin with, the development of CSC-specific ion channel biomarkers using single-cell transcriptomics and spatial proteomics can help distinguish tumour-specific isoforms from those expressed in normal tissues, thus minimizing off-target effects [110,118,119]. Integration of functional electrophysiological profiling with gene expression data will also be crucial to confirm ion channel activity in situ and during different CSC states, including quiescence and EMT [120]. Next-generation screening platforms using organotypic 3D tumour models and patient-derived organoids will be essential for assessing the efficacy and toxicity of ion channel modulators in a physiologically relevant context [121]. Moreover, structure-based drug design using cryo-EM and computational modelling of ion channels can lead to highly selective inhibitors that distinguish CSC-related channel conformations [122]. Targeting ion channel-regulated signalling pathways, such as Ca²⁺-dependent Wnt/β-catenin, Notch, or NFAT pathways, in combination with conventional therapies, may suppress CSC resistance and plasticity [18]. The application of nanoparticle-based delivery systems to selectively transport ion channel inhibitors to CSC niches holds promise for enhancing bioavailability while reducing systemic toxicity [12]. Personalized medicine frameworks that incorporate ion channel mutation profiles, CSC surface markers, and drug response phenotypes could guide individualized therapy regimens. These strategic directions, coupled with robust translational pipelines and regulatory support, could unlock the full therapeutic potential of ion channels in combating cancer at its root-through the eradication of CSCs.
14. Conclusions and Future Perspectives
Although substantial advancements in cancer therapies have extended patient survival, drug resistance-largely driven by CSCs-remains a significant clinical hurdle. A growing body of evidence highlights the pivotal role of ion channels in CSC biology, including their contributions to tumour initiation, progression, and resistance to therapy, hence earning the designation of “oncogenic channels” [113]. Aberrant expression and dysregulation of these channels enable CSCs to evade conventional treatments through diverse mechanisms. Targeting ion channels in CSCs approaches a promising strategy to overcome therapeutic resistance. Ideal ion channel targets should exhibit minimal expression in normal tissues, high expression in tumour tissues, selective ligand binding, and low toxicity profiles [18,113]. However, few candidates have progressed into early-phase clinical trials, indicating the pressing need for focused translational research. Existing ion channel inhibitors-already approved for cardiac or neurological disorders-are now being repurposed in oncology, showing encouraging preclinical and early clinical results [17]. Emerging findings emphasize that CSC-specific ion channels, such as Na⁺, K⁺, Ca²⁺, and Cl⁻ channels, modulate key processes including proliferation, invasion, and apoptosis. For instance, inhibition of IKCa and BKCa channels has shown therapeutic promise in glioblastoma models [113], where glioma stem cells exhibit high malignancy and resistance. These results emphasize the translational value of targeting CSC-associated ion channels, particularly in highly aggressive and heterogeneous cancers such as glioblastoma, where standard therapy still relies on maximal resection, radiotherapy, and chemotherapy [123]. Therefore, understanding of ion channel expression and regulation in CSCs is crucial. Herein, we explore the diverse range of ion channels present in CSCs, highlighting recent insights into their roles in regulating CSC behaviour, functional characteristics, and cellular dynamics. Particular attention is given to ASICs and AQPs, as their crucial involvement in controlling CSC behaviour and dynamics has remained underexplored in prior reviews. ASIC1 and ASIC3 promote CSC survival and invasiveness under acidic tumour microenvironments by activating PI3K/Akt and ERK/MAPK pathways [124]. Inhibiting these channels has reduced tumour growth in preclinical models, making them compelling pH-responsive therapeutic targets. Similarly, overexpression of AQP1, AQP3, AQP4, AQP5, and AQP8 has been reported in CSC-enriched tumours, where they regulate cell motility, redox signalling, and chemoresistance [65,66,67,68,69,70,71,72,73]. Among them, AQP3 is particularly implicated in promoting EMT [66]. Small molecule inhibitors and siRNAs targeting AQPs have demonstrated success in sensitizing tumours to standard therapies [17]. Moreover, advances in drug delivery and natural product pharmacology have shown promise. Natural compound-based modulators like limonin and silibinin, which inhibit TMEM16A, exhibit anticancer potential in lung and prostate cancers [113]. Additionally, existing ion channel blockers such as verapamil, mibefradil, and riluzole have shown clinical benefit when repurposed for cancers like metastatic breast cancer, glioblastoma, and melanoma brain metastases, respectively [113,118]. Despite these advances, a major bottleneck remains: the absence of robust biomarkers to predict therapeutic response to ion channel modulators. Therefore, future studies should emphasize the discovery and validation of such biomarkers through genomics, proteomics, and metabolomics. Progress in biomarker development, rational drug design, and precision delivery systems will be crucial to realizing the full potential of ion channel-targeted therapies [113,125].
Intracellular ion channels such as VDAC in the outer mitochondrial membrane and MCU in the inner mitochondrial membrane have emerged as key regulators of CSC maintenance. However, their potential as therapeutic targets remain largely unexplored. Similarly, ER-resident Ca²⁺ channels like the ryanodine receptor (RyR) and IP3 receptor play crucial roles in cancer stemness, yet their modulation for CSC-targeted therapy has not been extensively investigated. Future research focusing on these ion channels may reveal novel strategies for cancer treatment. Exploring these underexplored areas holds promise for advancing targeted therapies against CSCs [3,93].
While CSCs have been identified across various cancer types, data on ion channel expression in these cells remain incomplete. However, more research is needed to map ion channel profiles across different CSC types. Moreover, future research must explore how ion channels interact with epigenetic and metabolic pathways that drive CSC plasticity and therapeutic evasion. Understanding these interactions will help refine combination therapies that integrate ion channel inhibitors with chemotherapy, immunotherapy, or radiotherapy. These combinations should ideally be guided by biomarker-based patient stratification to maximize efficacy while minimizing off-target effects.
In summary, targeting ion channels in CSCs offers a novel and promising strategy to overcome drug resistance and enhance cancer therapy. While challenges remain, particularly in achieving CSC specificity and minimizing side effects, the integration of ion channel modulators into combination therapies-guided by biomarker-based patient stratification-holds great promise. We hope future research will advance these strategies toward clinical application, ultimately offering more durable and personalized treatment options for cancer patients.
Author Contributions
Conceptualization: KS & PK; Writing original draft: KS & PK; Writing review, data analyses & editing: GSVSRR, NKS, KS and PK.
Acknowledgments
Financial support from SRM University-AP (Grant No: SRMAP/URG/SEED/2024-25/042) for our research is gratefully acknowledged. G.S.R.R thanks SRM University-AP for providing PhD fellowships. We also extend our sincere thanks to Prof Ivan Ahel and Dr. Osamu Suyari from the Dunn School of Pathology, Oxford University, for their help and support in our work.
Competing interests
Authors declare that they have no competing interests.
References
- Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007; 23:675-99. [CrossRef]
- Fuchs E. The tortoise and the hair: slow-cycling cells in the stem cell race. Cell. 2009 May 29;137(5):811-9. [CrossRef]
- O’Reilly D, Buchanan P. Calcium channels and cancer stem cells. Cell Calcium. 2019 Jul; 81:21-28. [CrossRef]
- Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013 Jun;34(6):732-40. [CrossRef]
- Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017 Oct 6;23(10):1124-1134. [CrossRef]
- Clevers H. The cancer stem cell premises, promises and challenges. Nat Med. 2011 Mar;17(3):313-9. [CrossRef]
- Li GR, Deng XL. Functional ion channels in stem cells. World J Stem Cells. 2011 Mar 26;3(3):19-24. [CrossRef]
- Jardin I, Lopez JJ, Sanchez-Collado J, Gomez LJ, Salido GM, Rosado JA. Store-operated Calcium entry and its implications in cancer stem cells. Cells. 2022 Apr 13;11(8):1332. [CrossRef]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, CaceresCortes J, Minden M, Paterson B, Caligiuri MA and Dick JE: A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367: 645-648, 1994.
- Bonnet D and Dick JE: Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730-737, 1997.
- Cheng Q, Chen A, Du Q, Liao Q, Shuai Z, Chen C, Yang X, Hu Y, Zhao J, Liu S, Wen GR, An J, Jing H, Tuo B, Xie R, Xu J. Novel insights into ion channels in cancer stem cells (Review). Int J Oncol. 2018 Oct;53(4):1435-1441.
- Ertas YN, Abedi Dorcheh K, Akbari A, Jabbari E. Nanoparticles for Targeted Drug Delivery to Cancer Stem Cells: A Review of Recent Advances. Nanomaterials (Basel). 2021 Jul 5;11(7):1755. [CrossRef]
- Dalerba P, Cho RW and Clarke MF: Cancer stem cells: models and concepts. Annu Rev Med 58: 267-284, 2007.
- Shiozawa Y, Nie B, Pienta KJ, Morgan TM and Taichman RS:Cancer stem cells and their role in metastasis. Pharmacol Ther 138: 285-293, 2013.
- Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX, Ivy SP. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015 Aug;12(8):445-64. [CrossRef]
- Zhao J, Li M, Xu J, Cheng W. The modulation of ion channels in cancer chemo-resistance. Front Oncol. 2022 Aug 10; 12:945896. [CrossRef]
- Prevarskaya N, Skryma R, Shuba Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol Rev (2018) 98(2):559–621. [CrossRef]
- Prevarskaya N, Skryma R, Shuba Y. Ion channels and the hallmarks of cancer. Trends Mol Med. 2010 Mar;16(3):107-21. [CrossRef]
- Lu CK, Reddy P, Wang D, Nie and Ning Y Multi-Label Clinical Time-Series generation via conditional GAN, in IEEE transactions on knowledge and data engineering. (2024) 36 (4):1728-1740. [CrossRef]
- Zhuo D, Lei Z, Dong L, Chan AML, Lin J, Jiang L, Qiu B, Jiang X, Tan Y, Yao X. Orai1 and Orai3 act through distinct signalling axes to promote stemness and tumorigenicity of breast cancer stem cells. Stem Cell Res Ther. 2024; 13;15(1):256. [CrossRef]
- Lewuillon C, Guillemette A, Titah S, Shaik FA, Jouy N, Labiad O, Farfariello V, Laguillaumie MO, Idziorek T, Barthélémy A, Peyrouze P, Berthon C, Tarhan MC, Cheok M, Quesnel B, Lemonnier L, Touil Y. Involvement of ORAI1/SOCE in human AML cell lines and primary cells according to ABCB1 Activity, LSC Compartment and Potential Resistance to Ara-C Exposure. Int J Mol Sci. 2022 May 16;23(10):5555. [CrossRef]
- Jardin I, Alvarado S, Jimenez-Velarde V, Nieto-Felipe J, Lopez JJ, Salido GM, Smani T, Rosado JA. Orai1α and Orai1β support calcium entry and mammosphere formation in breast cancer stem cells. Sci Rep. 2023 Nov 9;13(1):19471. [CrossRef]
- Nguyen A, Sung Y, Lee SH, Martin CE, Srikanth S, Chen W, Kang MK, Kim RH, Park NH, Gwack Y, Kim Y, Shin KH. Orai3 Calcium channel contributes to oral/oropharyngeal cancer stemness through the elevation of ID1 expression. Cells. 2023 Sep 7;12(18):2225. [CrossRef]
- Terrié E, Déliot N, Benzidane Y, Harnois T, Cousin L, Bois P, Oliver L, Arnault P, Vallette F, Constantin B, Coronas V. Store-operated Calcium channels control proliferation and self-renewal of cancer stem cells from glioblastoma. Cancers (Basel). 2021 Jul 8;13(14):3428. [CrossRef]
- Shiozaki A, Kurashima K, Kudou M, Shimizu H, Kosuga T, Takemoto K, Arita T, Konishi H, Yamamoto Y, Morimura R, Komatsu S, Ikoma H, Kubota T, Fujiwara H, Otsuji E. Cancer stem cells of hepatocellular carcinoma are suppressed by the voltage-gated Calcium channel inhibitor Amlodipine. Anticancer Res. 2023 Nov;43(11):4855-4864. [CrossRef]
- Guo J, Shan C, Xu J, Li M, Zhao J, Cheng W. New Insights into TRP Ion Channels in Stem Cells. Int J Mol Sci. 2022 Jul 14;23(14):7766. [CrossRef]
- Santoni G, Nabissi M, Amantini C, Santoni M, Ricci-Vitiani L, Pallini R, Maggi F, Morelli MB. ERK phosphorylation regulates the Aml1/Runx1 splice variants and the TRP channels expression during the differentiation of glioma stem cell lines. Cells. 2021 Aug 10;10(8):2052. [CrossRef]
- Zhang N, Wu P, Mu M, Niu C, Hu S. Exosomal circZNF800 derived from glioma stem-like cells regulates glioblastoma tumorigenicity via the PIEZO1/Akt Axis. Mol Neurobiol. 2024 Sep;61(9):6556-6571. [CrossRef]
- Lebon D, Collet L, Djordjevic S, Gomila C, Ouled-Haddou H, Platon J, Demont Y, Marolleau JP, Caulier A, Garçon L. PIEZO1 is essential for the survival and proliferation of acute myeloid leukemia cells. Cancer Med. 2024 Jan;13(2): e6984. [CrossRef]
- Li R, Wang D, Li H, Lei X, Liao W, Liu XY. Identification of Piezo1 as a potential target for therapy of colon cancer stem-like cells. Discov Oncol. 2023 Jun 12;14(1):95. [CrossRef]
- Kar P, Nelson C, Parekh AB. Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem. 2011 Apr 29;286(17):14795-803. [CrossRef]
- Samanta K, Kar P, Mirams GR, Parekh AB. Cachannel re-localization to plasma-membrane microdomains strengthens activation of Ca2+-dependent nuclear gene expression. Cell Rep. 2015 Jul 14;12(2):203-16. [CrossRef]
- Luanpitpong S, Rodboon N, Samart P, Vinayanuwattikun C, Klamkhlai S, Chanvorachote P, Rojanasakul Y, Issaragrisil S. A novel TRPM7/O-GlcNAc axis mediates tumour cell motility and metastasis by stabilising c-Myc and caveolin-1 in lung carcinoma. Br J Cancer. 2020 Oct;123(8):1289-1301. [CrossRef]
- Liu K, Xu SH, Chen Z, Zeng QX, Li ZJ, Chen ZM. TRPM7 overexpression enhances the cancer stem cell-like and metastatic phenotypes of lung cancer through modulation of the Hsp90α/uPA/MMP2 signaling pathway. BMC Cancer. 2018 Nov 26;18(1):1167. [CrossRef]
- Xue C, Gao Y, Li X, Zhang M, Yang Y, Han Q, Sun Z, Bai C, Zhao RC. Mesenchymal stem cells derived from adipose accelerate the progression of colon cancer by inducing a MT-CAFs phenotype via TRPC3/NF-KB axis. Stem Cell Res Ther. 2022 Jul 23;13(1):335. [CrossRef]
- So CL, Milevskiy MJG, Monteith GR. Transient receptor potential cation channel subfamily V and breast cancer. Lab Invest. 2020 Feb;100(2):199-206. [CrossRef]
- Che X, Zhan J, Zhao F, Zhong Z, Chen M, Han R, Wang Y. Oridonin Promotes Apoptosis and Restrains the Viability and Migration of Bladder Cancer by Impeding TRPM7 Expression via the ERK and AKT Signaling Pathways. Biomed Res Int. 2021 Jul 5; 2021:4340950. [CrossRef]
- Liu M, Inoue K, Leng T, Guo S, Xiong ZG. TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways. Cell Signal. 2014 Dec;26(12):2773-81. [CrossRef]
- Middelbeek J, Visser D, Henneman L, Kamermans A, Kuipers AJ, Hoogerbrugge PM, Jalink K, van Leeuwen FN. TRPM7 maintains progenitor-like features of neuroblastoma cells: implications for metastasis formation. Oncotarget. 2015 Apr 20;6(11):8760-76. [CrossRef]
- Chen TM, Huang CM, Hsieh MS, Lin CS, Lee WH, Yeh CT, Liu SC. TRPM7 via calcineurin/NFAT pathway mediates metastasis and chemotherapeutic resistance in head and neck squamous cell carcinoma. Aging (Albany NY). 2022 Jun 29;14(12):5250-5270. [CrossRef]
- Wee S, Niklasson M, Marinescu VD, Segerman A, Schmidt L, Hermansson A, Dirks P, Forsberg-Nilsson K, Westermark B, Uhrbom L, Linnarsson S, Nelander S, Andäng M. Selective calcium sensitivity in immature glioma cancer stem cells. PLoS One. 2014 Dec 22;9(12): e115698. [CrossRef]
- Shiozaki A, Katsurahara K, Kudou M, Shimizu H, Kosuga T, Ito H, Arita T, Konishi H, Komatsu S, Kubota T, Fujiwara H, Okamoto K, Otsuji E. Amlodipine and Verapamil, voltage-gated Ca2+ channel inhibitors, suppressed the growth of gastric cancer stem cells. Ann Surg Oncol. 2021 Sep;28(9):5400-5411. [CrossRef]
- Lee H, Kwon OB, Lee JE, Jeon YH, Lee DS, Min SH, Kim JW. Repositioning trimebutine maleate as a cancer treatment targeting ovarian cancer stem cells. Cells. 2021 Apr 16;10(4):918. [CrossRef]
- Yao M, Tijore A, Cheng D, Li JV, Hariharan A, Martinac B, Tran Van Nhieu G, Cox CD, Sheetz M. Force- and cell state-dependent recruitment of Piezo1 drives focal adhesion dynamics and calcium entry. Sci Adv. 2022 Nov 11;8(45): eabo1461. [CrossRef]
- So CL, Robitaille M, Sadras F, McCullough MH, Milevskiy MJG, Goodhill GJ, Roberts-Thomson SJ, Monteith GR. Cellular geometry and epithelial-mesenchymal plasticity intersect with PIEZO1 in breast cancer cells. Commun Biol. 2024 Apr 17;7(1):467. [CrossRef]
- Kim HJ, Lee PC, Hong JH. Chloride channels and transporters: roles beyond classical cellular homeostatic pH or ion balance in cancers. Cancers (Basel). 2022 Feb 9;14(4):856. [CrossRef]
- Yan Y, Ding X, Han C, Gao J, Liu Z, Liu Y, Wang K. Involvement of TMEM16A/ANO1 upregulation in the oncogenesis of colorectal cancer. Biochim Biophys Acta Mol Basis Dis. 2022 Jun 1;1868(6):166370. [CrossRef]
- Olsen ML, Schade S, Lyons SA, Amaral MD, Sontheimer H. Expression of voltage-gated chloride channels in human glioma cells. J Neurosci. 2003 Jul 2;23(13):5572-82. [CrossRef]
- Zhou FM, Huang YY, Tian T, Li XY, Tang YB. Knockdown of Chloride channel-3 inhibits breast cancer growth In Vitro and In Vivo. J Breast Cancer. 2018 Jun;21(2):103-111. [CrossRef]
- Peretti M, Raciti FM, Carlini V, Verduci I, Sertic S, Barozzi S, Garré M, Pattarozzi A, Daga A, Barbieri F, Costa A, Florio T, Mazzanti M. Mutual influence of ROS, pH, and CLIC1 membrane protein in the regulation of G1-S phase progression in human glioblastoma stem cells. Mol Cancer Ther. 2018 Nov;17(11):2451-2461. [CrossRef]
- Barbieri F, Bosio AG, Pattarozzi A, Tonelli M, Bajetto A, Verduci I, Cianci F, Cannavale G, Palloni LMG, Francesconi V, Thellung S, Fiaschi P, Mazzetti S, Schenone S, Balboni B, Girotto S, Malatesta P, Daga A, Zona G, Mazzanti M, Florio T. Chloride intracellular channel 1 activity is not required for glioblastoma development but its inhibition dictates glioma stem cell responsivity to novel biguanide derivatives. J Exp Clin Cancer Res. 2022 Feb 8;41(1):53. [CrossRef]
- Setti M, Savalli N, Osti D, Richichi C, Angelini M, Brescia P, Fornasari L, Carro MS, Mazzanti M, Pelicci G. Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J Natl Cancer Inst. 2013 Nov 6;105(21):1644-55. [CrossRef]
- Zúñiga L, Cayo A, González W, Vilos C, Zúñiga R. Potassium Channels as a Target for Cancer Therapy: Current Perspectives. Onco Targets Ther. 2022 Jul 20; 15:783-797. [CrossRef]
- Ruggieri P, Mangino G, Fioretti B, Catacuzzeno L, Puca R, Ponti D, Miscusi M, Franciolini F, Ragona G, Calogero A. The inhibition of KCa3.1 channels activity reduces cell motility in glioblastoma derived cancer stem cells. PLoS One. 2012;7(10):e47825. [CrossRef]
- Lallet-Daher H, Roudbaraki M, Bavencoffe A, Mariot P, Gackière F, Bidaux G, Urbain R, Gosset P, Delcourt P, Fleurisse L, Slomianny C, Dewailly E, Mauroy B, Bonnal JL, Skryma R, Prevarskaya N. Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene. 2009 Apr 16;28(15):1792-806. [CrossRef]
- Guerriero C, Fanfarillo R, Mancini P, Sterbini V, Guarguaglini G, Sforna L, Michelucci A, Catacuzzeno L, Tata AM. M2 muscarinic receptors negatively modulate cell migration in human glioblastoma cells. Neurochem Int. 2024 Mar; 174:105673. [CrossRef]
- Shiozaki A, Konishi T, Kosuga T, Kudou M, Kurashima K, Inoue H, Shoda K, Arita T, Konishi H, Morimura R, Komatsu S, Ikoma H, Toma A, Kubota T, Fujiwara H, Okamoto K, Otsuji E. Roles of voltage-gated potassium channels in the maintenance of pancreatic cancer stem cells. Int J Oncol. 2021 Oct;59(4):76. [CrossRef]
- Lastraioli E. Focus on triple-negative breast cancer: Potassium channel expression and clinical correlates. Front Pharmacol. 2020 May 20; 11:725. [CrossRef]
- Bian Y, Tuo J, He L, Li W, Li S, Chu H, Zhao Y. Voltage-gated sodium channels in cancer and their specific inhibitors. Pathol Res Pract. 2023 Nov; 251:154909. [CrossRef]
- Giammello F, Biella C, Priori EC, Filippo MADS, Leone R, D’Ambrosio F, Paterno’ M, Cassioli G, Minetti A, Macchi F, Spalletti C, Morella I, Ruberti C, Tremonti B, Barbieri F, Lombardi G, Brambilla R, Florio T, Galli R, Rossi P, Brandalise F. Modulating voltage-gated sodium channels to enhance differentiation and sensitize glioblastoma cells to chemotherapy. Cell Commun Signal. 2024 Sep 9;22(1):434. [CrossRef]
- Djamgoz MBA. Stemness of Cancer: A study of triple-negative breast cancer from a neuroscience perspective. Stem Cell Rev Rep. 2025 Feb;21(2):337-350. [CrossRef]
- Tian Y, Bresenitz P, Reska A, El Moussaoui L, Beier CP, Gründer S. Glioblastoma cancer stem cell lines express functional acid sensing ion channels ASIC1a and ASIC3. Sci Rep. 2017 Oct 20;7(1):13674. [CrossRef]
- Clusmann J, Franco KC, Suárez DAC, Katona I, Minguez MG, Boersch N, Pissas KP, Vanek J, Tian Y, Gründer S. Acidosis induces RIPK1-dependent death of glioblastoma stem cells via acid-sensing ion channel 1a. Cell Death Dis. 2022 Aug 12;13(8):702. [CrossRef]
- King P, Wan J, Guo AA, Guo S, Jiang Y, Liu M. Regulation of gliomagenesis and stemness through acid sensor ASIC1a. Int J Oncol. 2021 Oct;59(4):82. [CrossRef]
- Guan Y, Han J, Chen D, Zhan Y, Chen J. Aquaporin 1 overexpression may enhance glioma tumorigenesis by interacting with the transcriptional regulation networks of Foxo4, Maz, and E2F families. Chin Neurosurg J. 2023 Dec 6;9(1):34. [CrossRef]
- Huang YT, Zhou J, Shi S, Xu HY, Qu F, Zhang D, Chen YD, Yang J, Huang HF, Sheng JZ. Identification of estrogen response element in Aquaporin-3 gene that mediates estrogen-induced cell migration and invasion in estrogen receptor-positive breast cancer. Sci Rep. 2015 Jul 29; 5:12484. [CrossRef]
- Wang Y, Wu G, Fu X, Xu S, Wang T, Zhang Q, Yang Y. Aquaporin 3 maintains the stemness of CD133+ hepatocellular carcinoma cells by activating STAT3. Cell Death Dis. 2019 Jun 13;10(6):465. [CrossRef]
- Liu YC, Yeh CT, Lin KH. Cancer stem cell functions in hepatocellular carcinoma and comprehensive therapeutic strategies. Cells. 2020 May 26;9(6):1331. [CrossRef]
- Liu C, Liu L, Zhang Y, Jing H. Molecular mechanism of AQP3 in regulating differentiation and apoptosis of lung cancer stem cells through Wnt/GSK-3β/β-Catenin pathway. J BUON. 2020 Jul-Aug;25(4):1714-1720.
- Simone L, Pisani F, Binda E, Frigeri A, Vescovi AL, Svelto M, Nicchia GP. AQP4-dependent glioma cell features affect the phenotype of surrounding cells via extracellular vesicles. Cell Biosci. 2022 Sep 7;12(1):150. [CrossRef]
- Jang SJ, Moon C. Aquaporin 5 (AQP5) expression in breast cancer and its clinicopathological characteristics. PLoS One. 2023 Jan 27;18(1): e0270752. [CrossRef]
- Zhao R, He B, Bie Q, Cao J, Lu H, Zhang Z, Liang J, Wei L, Xiong H, Zhang B. AQP5 complements LGR5 to determine the fates of gastric cancer stem cells through regulating ULK1 ubiquitination. J Exp Clin Cancer Res. 2022 Nov 14;41(1):322. Erratum in: J Exp Clin Cancer Res. 2022 Dec 13;41(1):342. doi: 10.1186/s13046-022-02557-1. [CrossRef]
- Li W, Song Y, Pan C, Yu J, Zhang J, Zhu X. Aquaporin-8 is a novel marker for progression of human cervical cancer cells. Cancer Biomark. 2021;32(3):391-400. [CrossRef]
- Zheng X, Li C, Yu K, Shi S, Chen H, Qian Y, Mei Z. Aquaporin-9, mediated by IGF2, suppresses liver cancer stem cell properties via augmenting ROS/β-Catenin/FOXO3a signaling. Mol Cancer Res. 2020 Jul;18(7):992-1003. [CrossRef]
- Fernandez Garcia E, Paudel U, Noji MC, Bowman CE, Rustgi AK, Pitarresi JR, Wellen KE, Arany Z, Weissenrieder JS, Foskett JK. The mitochondrial Ca2+ channel MCU is critical for tumor growth by supporting cell cycle progression and proliferation. Front Cell Dev Biol. 2023 Jun 8; 11:1082213. [CrossRef]
- Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F. Mitochondrial Ca²⁺ uniporter is critical for store-operated Ca²⁺ entry-dependent breast cancer cell migration. Biochem Biophys Res Commun. 2015 Feb 27;458(1):186-93. [CrossRef]
- Bong AHL, Robitaille M, Lin S, McCart-Reed A, Milevskiy M, Angers S, Roberts-Thomson SJ, Monteith GR. TMCO1 is upregulated in breast cancer and regulates the response to pro-apoptotic agents in breast cancer cells. Cell Death Discov. 2024 Oct 1;10(1):421. [CrossRef]
- Shen FF, Dai SY, Wong NK, Deng S, Wong AS, Yang D. Mediating K+/H+ transport on organelle membranes to selectively eradicate cancer stem cells with a small molecule. J Am Chem Soc. 2020 Jun 17;142(24):10769-10779. [CrossRef]
- Zheng J, Feng Y, Yuan C. Hsa_circ_0007905 as a modulator of miR-330-5p and VDAC1: Enhancing stemness and reducing apoptosis in cervical cancer stem cells. Gen Physiol Biophys. 2025 May;44(3):213-226. [CrossRef]
- Seoane J. Cancer: Division hierarchy leads to cell heterogeneity. Nature. 2017 Sep 14;549(7671):164-166. [CrossRef]
- O’Brien-Ball C, Biddle A. Reprogramming to developmental plasticity in cancer stem cells. Dev Biol. 2017 Oct 15;430(2):266-274. [CrossRef]
- Sun XX, Yu Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. 2015 Oct;36(10):1219-27. [CrossRef]
- Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010 Jan;1805(1):105-17. [CrossRef]
- Kang Q, Peng X, Li X, Hu D, Wen G, Wei Z, Yuan B. Calcium Channel Protein ORAI1 Mediates TGF-β Induced Epithelial-to-Mesenchymal Transition in Colorectal Cancer Cells. Front Oncol. 2021 May 12; 11:649476. [CrossRef]
- Beck B, Lapouge G, Rorive S, Drogat B, Desaedelaere K, Delafaille S, Dubois C, Salmon I, Willekens K, Marine JC, Blanpain C. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell. 2015 Jan 8;16(1):67-79. [CrossRef]
- He T, Wang C, Zhang M, Zhang X, Zheng S, Linghu E, Guo M. Epigenetic regulation of voltage-gated potassium ion channel molecule Kv1.3 in mechanisms of colorectal cancer. Discov Med. 2017 Mar;23(126):155-162.
- Herranz N, Pasini D, Díaz VM, Francí C, Gutierrez A, Dave N, Escrivà M, Hernandez-Muñoz I, Di Croce L, Helin K, García de Herreros A, Peiró S. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol. 2008 Aug;28(15):4772-81. [CrossRef]
- Luo M, Bao L, Xue Y, Zhu M, Kumar A, Xing C, Wang JE, Wang Y, Luo W. ZMYND8 protects breast cancer stem cells against oxidative stress and ferroptosis through activation of NRF2. J Clin Invest. 2024 Jan 23;134(6): e171166. [CrossRef]
- Bao L, Festa F, Freet CS, Lee JP, Hirschler-Laszkiewicz IM, Chen SJ, Keefer KA, Wang HG, Patterson AD, Cheung JY, Miller BA. The Human Transient Receptor Potential Melastatin 2 Ion Channel Modulates ROS Through Nrf2. Sci Rep. 2019 Oct 1;9(1):14132. [CrossRef]
- Cheng, D., Wu, R., Guo, Y. & Kong, A. N. regulation of Keap1-Nrf2 signaling: The role of epigenetics. Curr Opin Toxicol 1, 134–138, doi.org/10.1016/j.cotox.2016.10.008 (2016).
- Samanta K, Ahel I, Kar p; Deciphering of the reactive oxygen species (ROS) induced calpain activation in cancer progression and its therapeutic potential, Advances in Redox Research, 2025, 10024.
- K. Chen, P. Lu, N.M. Beeraka, O.A. Sukocheva, S.V. Madhunapantula, J. Liu, M. Y. Sinelnikov, V.N. Nikolenko, K.V. Bulygin, L.M. Mikhaleva, I.V. Reshetov, Y. Gu, J. Zhang, Y. Cao, S.G. Somasundaram, C.E. Kirkland, R. Fan, G. Aliev, Mitochondrial mutations and mitoepigenetics: focus on regulation of oxidative stress-induced responses in breast cancers, Semin. Cancer Biol. (2020) 30203–30210. S1044-579X.
- Arif T, Shteinfer-Kuzmine A, Shoshan-Barmatz V. Decoding cancer through silencing the mitochondrial gatekeeper VDAC1. Biomolecules. 2024 Oct 15;14(10):1304. [CrossRef]
- Shoshan-Barmatz V, Krelin Y, Shteinfer-Kuzmine A, Arif T. Voltage-dependent anion channel 1 as an emerging drug target for novel anti-cancer therapeutics. Front Oncol. 2017 Jul 31; 7:154. [CrossRef]
- Alnasser SM. Advances and Challenges in Cancer Stem Cells for Onco-Therapeutics. Stem Cells Int. 2023 Dec 6;2023:8722803. [CrossRef]
- Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002 Apr;82(2):503-68. [CrossRef]
- Lee H, Kim JW, Kim DK, Choi DK, Lee S, Yu JH, Kwon OB, Lee J, Lee DS, Kim JH, Min SH. Calcium channels as novel therapeutic targets for ovarian cancer stem cells. Int J Mol Sci. 2020 Mar 27;21(7):2327. [CrossRef]
- Zhang Y, Cruickshanks N, Yuan F, Wang B, Pahuski M, Wulfkuhle J, Gallagher I, Koeppel AF, Hatef S, Papanicolas C, Lee J, Bar EE, Schiff D, Turner SD, Petricoin EF, Gray LS, Abounader R. Targetable T-type Calcium Channels Drive Glioblastoma. Cancer Res. 2017 Jul 1;77(13):3479-3490. [CrossRef]
- Zhao L, Zhao Y, Schwarz B, Mysliwietz J, Hartig R, Camaj P, Bao Q, Jauch KW, Guba M, Ellwart JW, Nelson PJ, Bruns CJ. Verapamil inhibits tumor progression of chemotherapy-resistant pancreatic cancer side population cells. Int J Oncol. 2016 Jul;49(1):99-110. [CrossRef]
- Fraser SP, Grimes JA, Djamgoz MB. Effects of voltage-gated ion channel modulators on rat prostatic cancer cell proliferation: comparison of strongly and weakly metastatic cell lines. Prostate. 2000 Jun 15;44(1):61-76. [CrossRef]
- D’Alessandro G, Grimaldi A, Chece G, Porzia A, Esposito V, Santoro A, Salvati M, Mainiero F, Ragozzino D, Di Angelantonio S, Wulff H, Catalano M, Limatola C. KCa3.1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget. 2016 May 24;7(21):30781-96. [CrossRef]
- Mukhopadhyay D, Goel HL, Xiong C, Goel S, Kumar A, Li R, Zhu LJ, Clark JL, Brehm MA, Mercurio AM. The calcium channel TRPC6 promotes chemotherapy-induced persistence by regulating integrin α6 mRNA splicing. Cell Rep. 2023 Nov 28;42(11):113347. [CrossRef]
- Comes N, Bielanska J, Vallejo-Gracia A, Serrano-Albarrás A, Marruecos L, Gómez D, Soler C, Condom E, Ramón Y Cajal S, Hernández-Losa J, Ferreres JC, Felipe A. The voltage-dependent K (+) channels Kv1.3 and Kv1.5 in human cancer. Front Physiol. 2013 Oct 10; 4:283. [CrossRef]
- Kang MK, Kang SK. Pharmacologic blockade of chloride channel synergistically enhances apoptosis of chemotherapeutic drug-resistant cancer stem cells. Biochem Biophys Res Commun. 2008 Sep 5;373(4):539-44. [CrossRef]
- Balboni A, D’Angelo C, Collura N, Brusco S, Di Berardino C, Targa A, Massoti B, Mastrangelo E, Milani M, Seneci P, Broccoli V, Muzio L, Galli R, Menegon A. Acid-sensing ion channel 3 is a new potential therapeutic target for the control of glioblastoma cancer stem cells growth. Sci Rep. 2024 Sep 3;14(1):20421. [CrossRef]
- Gründer S, Vanek J, Pissas KP. Acid-sensing ion channels and downstream signalling in cancer cells: is there a mechanistic link? Pflugers Arch. 2024 Apr;476(4):659-672. [CrossRef]
- Zhou Y, Wang Y, Wen J, Zhao H, Dong X, Zhang Z, Wang S, Shen L. Aquaporin 3 promotes the stem-like properties of gastric cancer cells via Wnt/GSK-3β/β-catenin pathway. Oncotarget. 2016 Mar 29;7(13):16529-41. [CrossRef]
- Gao P, Chen A, Tian H, Wang F, Wang N, Ge K, Lian C, Wang F, Zhang Q. Investigating the mechanism and the effect of aquaporin 5 (AQP5) on the self-renewal capacity of gastric cancer stem cells. J Cancer. 2024 Jun 11;15(13):4313-4327. [CrossRef]
- Pollak J, Rai KG, Funk CC, Arora S, Lee E, Zhu J, Price ND, Paddison PJ, Ramirez JM, Rostomily RC. Ion channel expression patterns in glioblastoma stem cells with functional and therapeutic implications for malignancy. PLoS One. 2017 Mar 6;12(3): e0172884. [CrossRef]
- Shi Q, Yang Z, Yang H, Xu L, Xia J, Gu J, Chen M, Wang Y, Zhao X, Liao Z, Mou Y, Gu X, Xie T, Sui X. Targeting ion channels: innovative approaches to combat cancer drug resistance. Theranostics. 2025 Jan 1;15(2):521-545. [CrossRef]
- Zheng X, Yu C, Xu M. Linking Tumor Microenvironment to Plasticity of Cancer Stem Cells: Mechanisms and Application in Cancer Therapy. Front Oncol. 2021 Jun 28; 11:678333. [CrossRef]
- Tan KK, Giam CS, Leow MY, Chan CW, Yim EK. Differential cell adhesion of breast cancer stem cells on biomaterial substrate with nanotopographical cues. J Funct Biomater. 2015 Apr 21;6(2):241-58. [CrossRef]
- Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013 Jan;59(1):168-79. [CrossRef]
- Rao VR, Perez-Neut M, Kaja S, Gentile S. Voltage-gated ion channels in cancer cell proliferation. Cancers (Basel). 2015 May 22;7(2):849-75. [CrossRef]
- Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015 Mar 5;16(3):225-38. [CrossRef]
- Dallas ML, Bell D. Advances in ion channel high throughput screening: where are we in 2023? Expert Opin Drug Discov. 2024 ;19(3):331-337. [CrossRef]
- Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, McLendon R, Lindner D, Sloan A, Rich JN. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011 May;18(5):829-40. [CrossRef]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 Jun 20;344(6190):1396-401. [CrossRef]
- Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015 May;33(5):495-502. [CrossRef]
- Liu J, Gustafson P, Huo D. Bayesian adjustment for the misclassification in both dependent and independent variables with application to a breast cancer study. Stat Med. 2016 Oct 15;35(23):4252-63. [CrossRef]
- Clevers H. Modeling Development and Disease with Organoids. Cell. 2016 Jun 16;165(7):1586-1597. [CrossRef]
- Sun J, MacKinnon R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell. 2017 Jun 1;169(6):1042-1050.e9. [CrossRef]
- Rodríguez-Camacho A, Flores-Vázquez JG, Moscardini-Martelli J, Torres-Ríos JA, Olmos-Guzmán A, Ortiz-Arce CS, Cid-Sánchez DR, Pérez SR, Macías-González MDS, Hernández-Sánchez LC, Heredia-Gutiérrez JC, Contreras-Palafox GA, Suárez-Campos JJE, Celis-López MÁ, Gutiérrez-Aceves GA, Moreno-Jiménez S. Glioblastoma Treatment: State-of-the-Art and Future Perspectives. Int J Mol Sci. 2022 Jun 29;23(13):7207. [CrossRef]
- Gründer S, Vanek J, Pissas KP. Acid-sensing ion channels and downstream signalling in cancer cells: is there a mechanistic link? Pflugers Arch. 2024 Apr;476(4):659-672. [CrossRef]
- Lastraioli E, Iorio J, Arcangeli A. Ion channel expression as promising cancer biomarker. Biochim Biophys Acta. 2015;1848(10 Pt B):2685-702. [CrossRef]
- Bhattacharjee A, Jana A, Bhattacharjee S, Mitra S, De S, Alghamdi BS, Alam MZ, Mahmoud AB, Al Shareef Z, Abdel-Rahman WM, Woon-Khiong C, Alexiou A, Papadakis M, Ashraf GM. The role of Aquaporins in tumorigenesis: implications for therapeutic development. Cell Commun Signal. 2024; 9;22(1):106. [CrossRef]
- Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol. 2020; 21(10):607-632. [CrossRef]
- Bortner CD, Gomez-Angelats M, Cidlowski JA. Plasma membrane depolarization without repolarization is an early molecular event in anti-Fas-induced apoptosis. J Biol Chem. 2001 Feb 9;276(6):4304-14. [CrossRef]
Figure 1.
Forecasting global cancer incidence with an AI Model. (a) Flowchart illustrating the use of GAN and LSTM networks for forecasting cancer cases. (b) An AI model predicting the future global burden of cancer, showing projected trends over the next 20 years. The fine-tuned model parameters for predicting region-wise prostate cancer cases are as follows: number of epochs = 200, look-back value = 3, number of LSTM units = 50, optimizer = Adam, loss function = MSE, and final loss = 0.002.
Figure 1.
Forecasting global cancer incidence with an AI Model. (a) Flowchart illustrating the use of GAN and LSTM networks for forecasting cancer cases. (b) An AI model predicting the future global burden of cancer, showing projected trends over the next 20 years. The fine-tuned model parameters for predicting region-wise prostate cancer cases are as follows: number of epochs = 200, look-back value = 3, number of LSTM units = 50, optimizer = Adam, loss function = MSE, and final loss = 0.002.
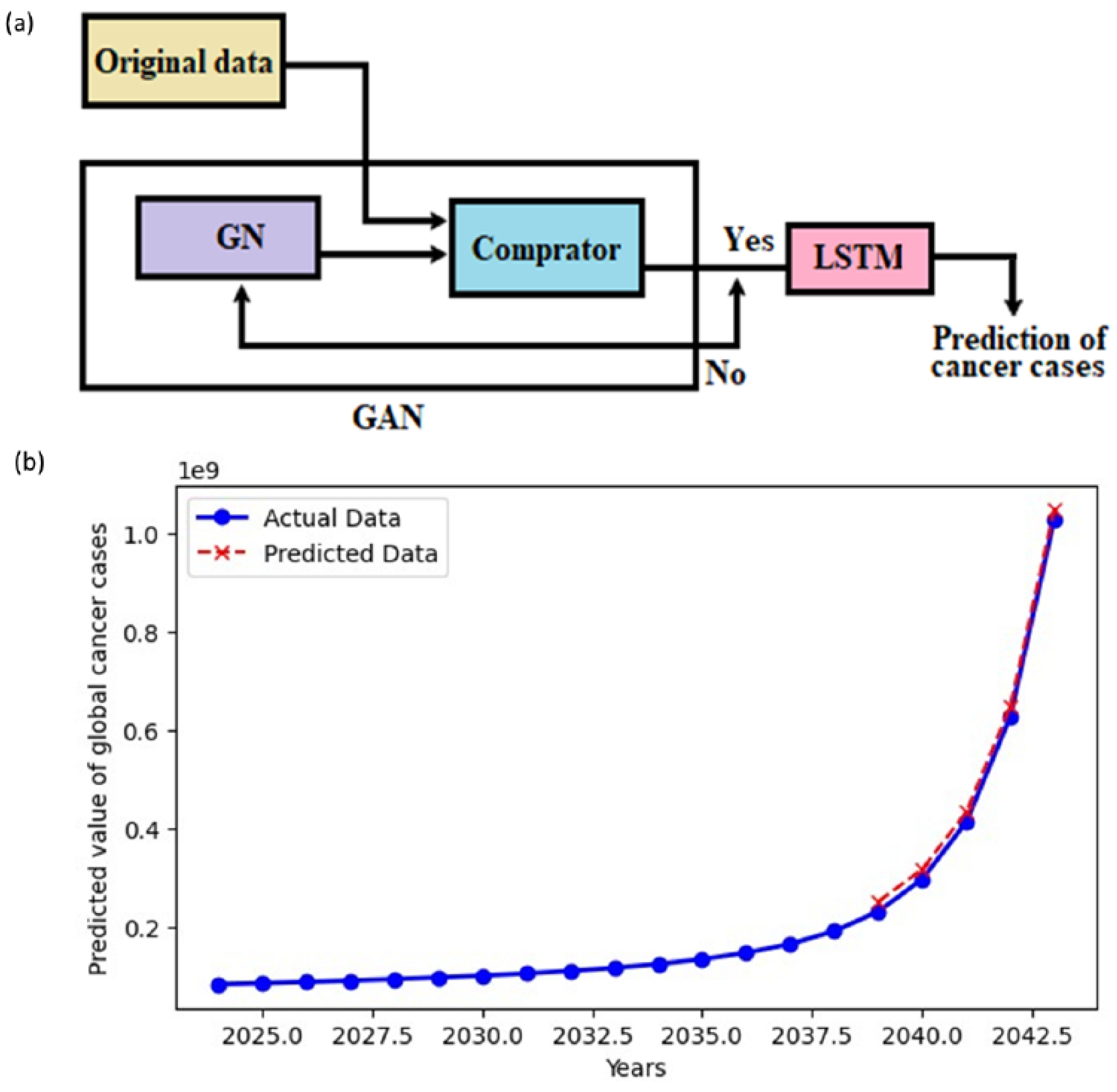
Figure 2.
Fundamental ion channels in CSCs and their functional roles in maintaining stemness.
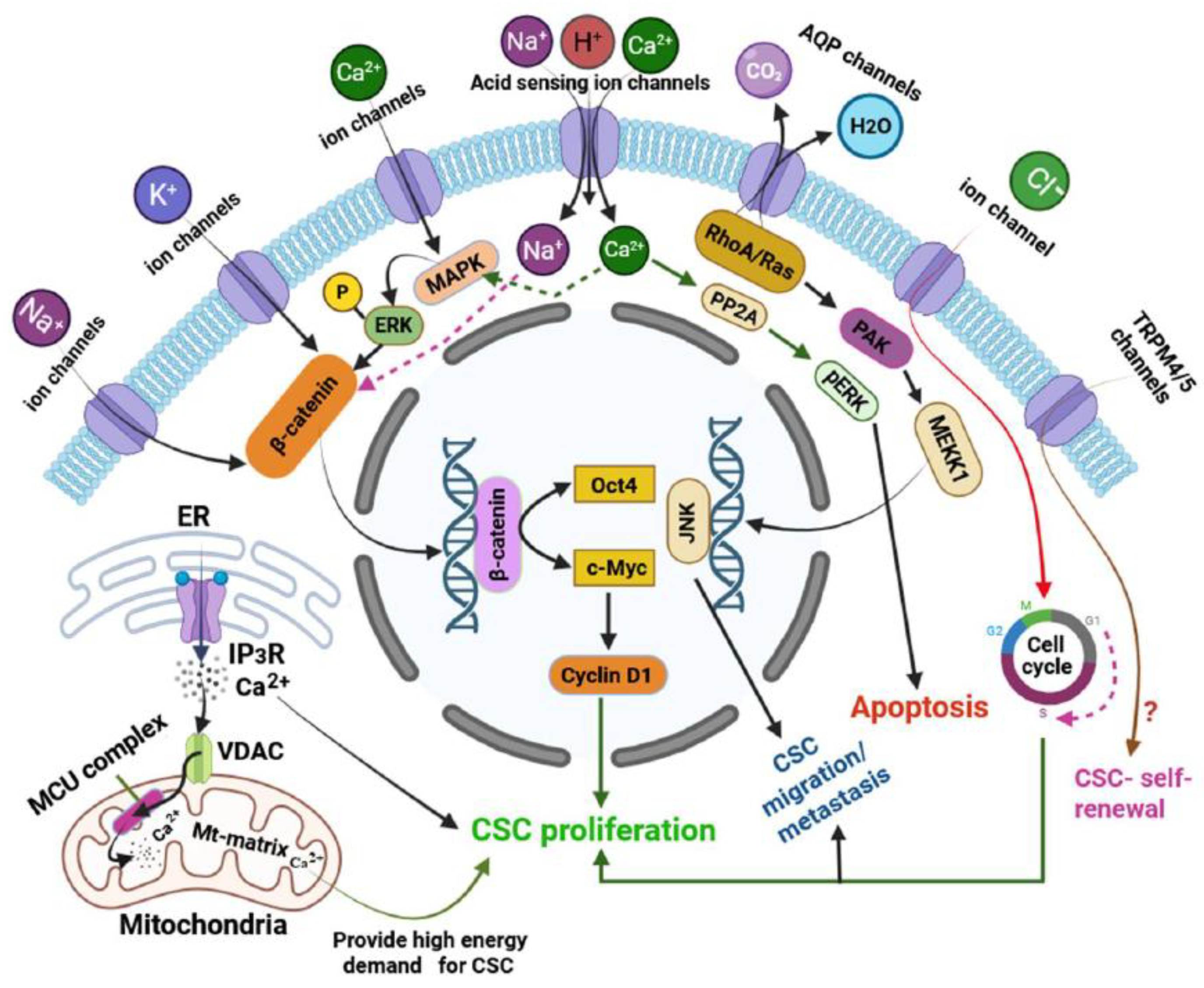
Figure 3.
Targeting ion channels in CSCs as a promising therapeutic strategy.
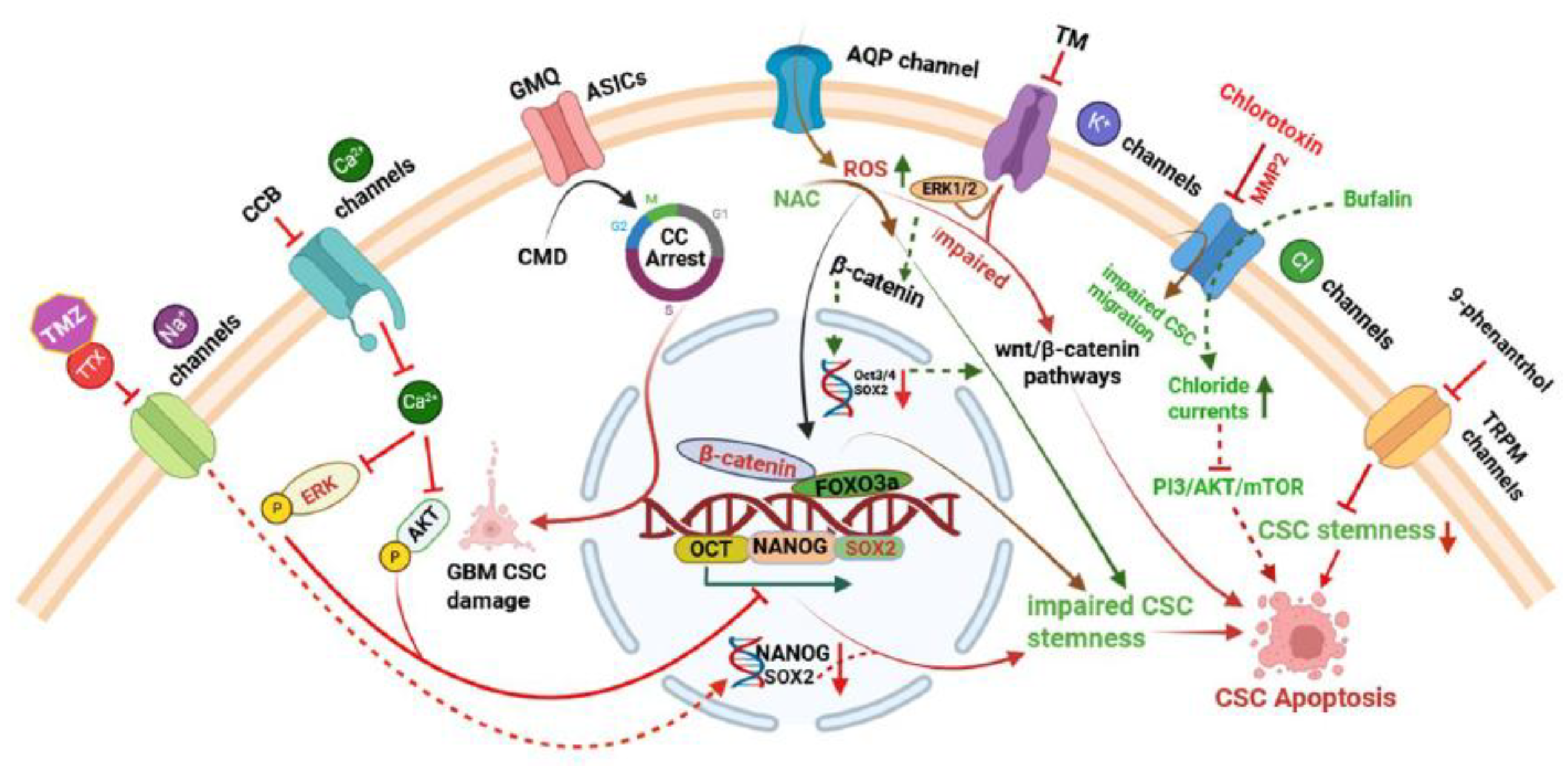

Table 1.
Global cancer case (1990–2022).
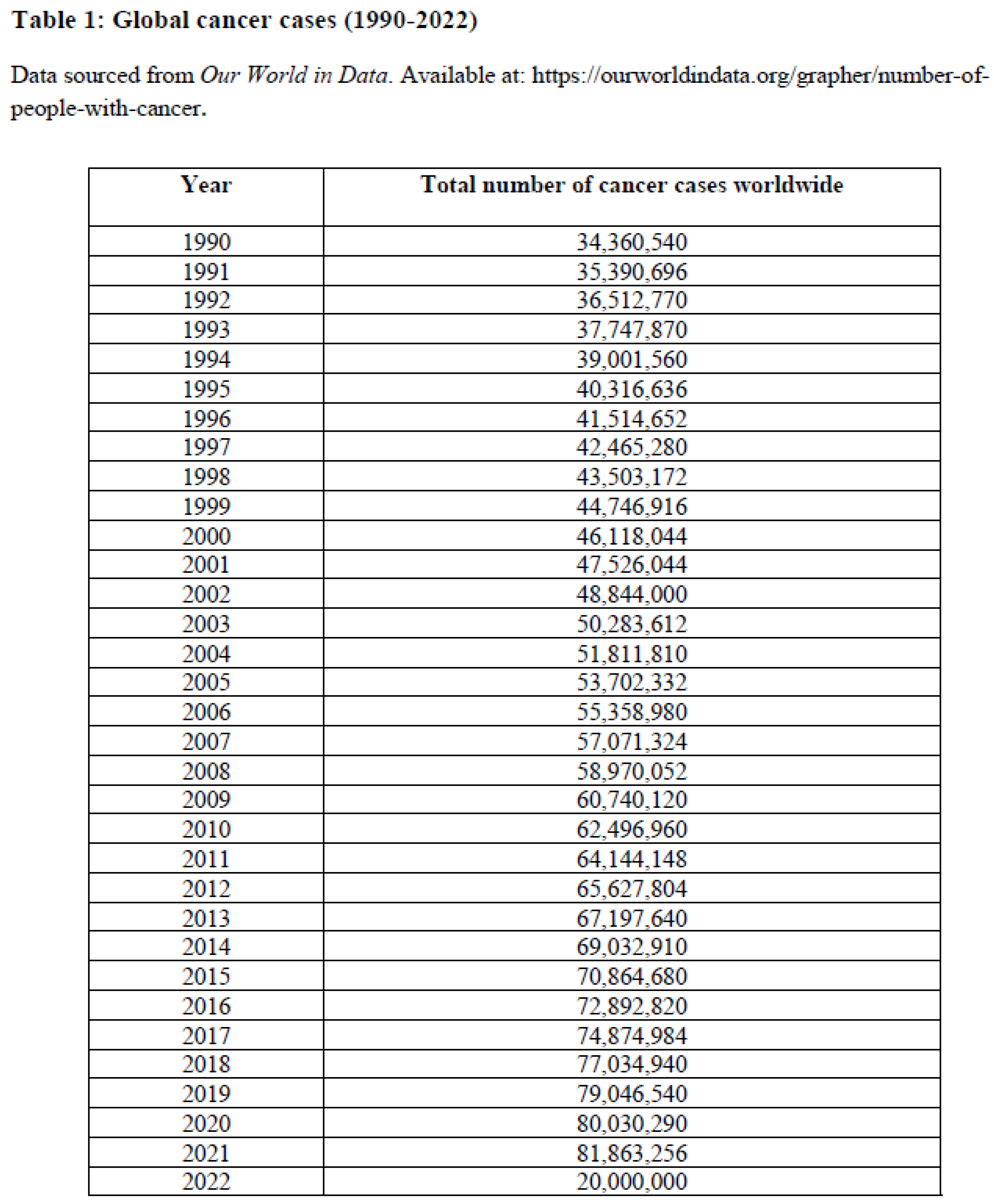
Table 2.
Ion channel expression and functions in CDCs.
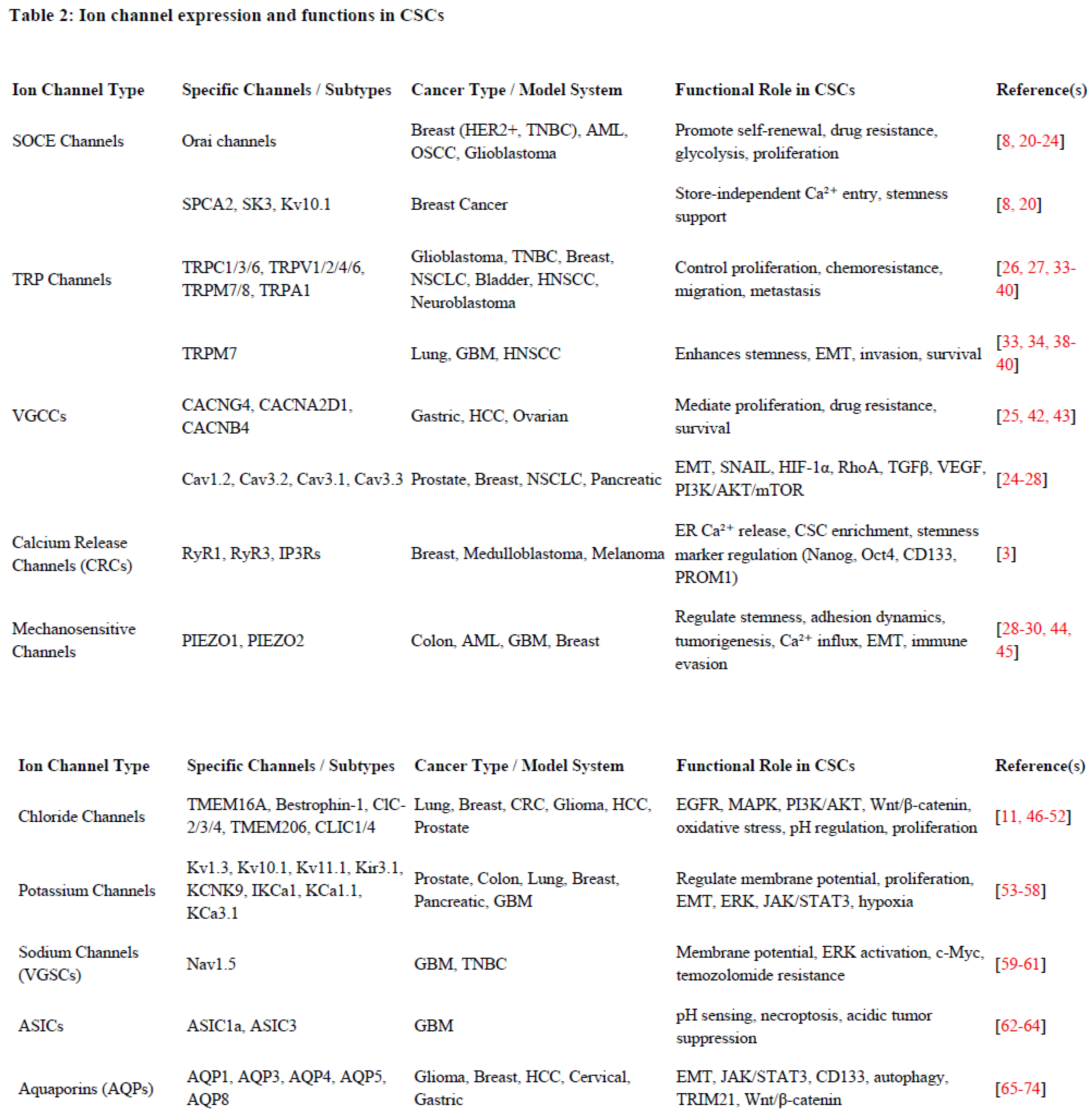
Table 3.
Therapies targeting ion channels in CSCs.
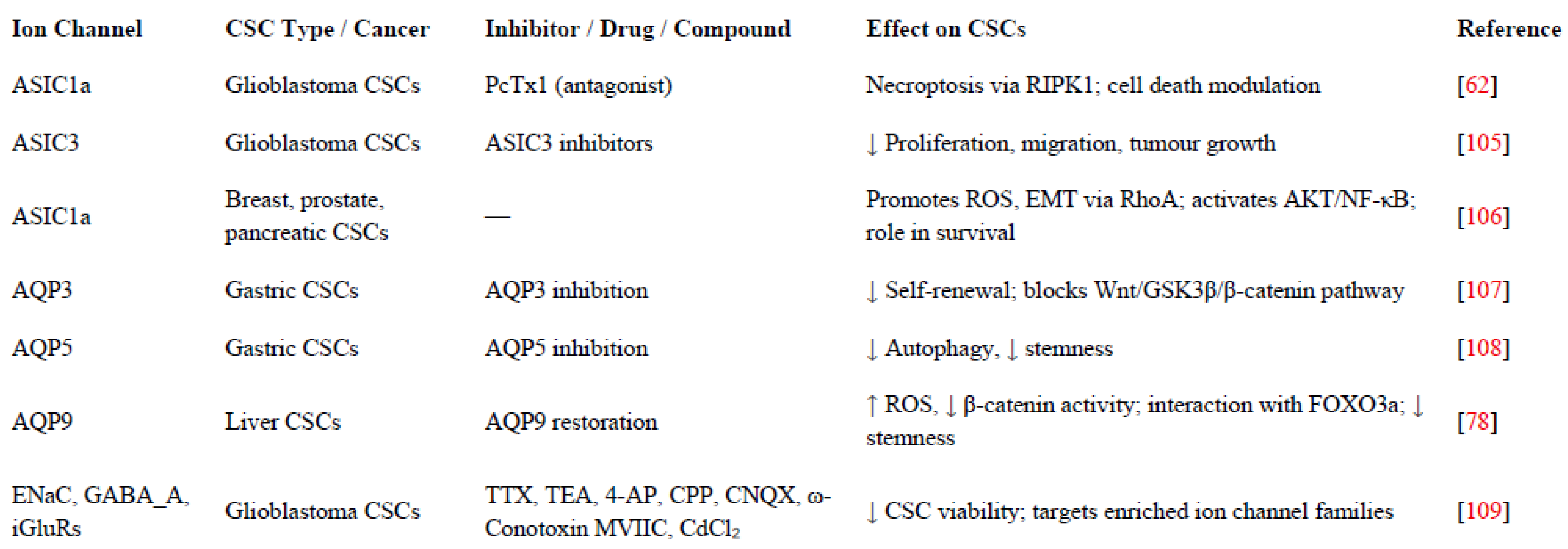
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



