
Submitted:
11 July 2025
Posted:
14 July 2025
You are already at the latest version
Abstract
Ischemic stroke triggers a dynamic immune response that influences both acute damage and long-term recovery. This review synthesizes a decade of evidence on immunolog-ical and inflammatory biomarkers in ischemic stroke, emphasizing their prognostic and therapeutic significance. Following ischemic insult, pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, tumour necrosis factor-α (TNF-α), and chemokines like IL-8 rapidly rise, promoting blood-brain barrier disruption, leukocyte infiltration, and neu-ronal death. Conversely, anti-inflammatory mediators such as IL-10 and TGF-β facilitate repair, neurogenesis, and immune regulation in later phases. The balance between these pathways determines outcomes and is reflected in circulating biomarkers. Composite hematological indices including neutrophil-to-lymphocyte ratio (NLR), plate-let-to-lymphocyte ratio (PLR), and systemic immune-inflammation index (SII) offer accessible and cost-effective prognostic tools. Several biomarkers correlate with infarct size, neurological deterioration, and mortality, and may predict complications like hemorrhagic transformation or infection. Therapeutic strategies targeting cytokines, especially IL-1 and IL-6, have shown promise in modulating inflammation and im-proving outcomes. Future directions include personalized immune profiling, real-time cytokine monitoring, and combining immunotherapy with neurorestorative approach-es. By integrating immune biomarkers into stroke care, clinicians may enhance risk stratification, optimize treatment timing, and identify candidates for novel interven-tions. This review underscores inflammation’s dual role and evolving therapeutic and prognostic relevance in ischemic stroke.
Keywords:
ischemic stroke
; inflammation
; cytokines
; biomarkers
; immune response
; prognosis
; prevention
; outcome
1. Introduction
Ischemic stroke remains a global health burden and a leading cause of disability and death. While its acute management has advanced, particularly with the advent of mechanical thrombectomy and thrombolytics, the long-term functional outcome varies widely between patients. Traditional prognostic tools rely heavily on clinical scales and imaging, but in recent years, biomarkers reflecting systemic and neuroinflammation have gained substantial attention [1]. The immune system plays a dual role in stroke. On one hand, inflammation exacerbates blood–brain barrier (BBB) breakdown, oxidative stress, and neuronal death [2,3,4]. On the other, immune-regulated processes also facilitate neurorepair, angiogenesis, and debris clearance in subacute and chronic phases. This duality positions inflammatory mediators not just as mechanistic components, but also as potential biomarkers and therapeutic targets [5,6].
Key cytokines such as interleukin-6 (IL-6), interleukin-1β (IL-1β), tumour necrosis factor-α (TNF-α), and chemokines like IL-8 are rapidly released after stroke onset and are strongly associated with infarct size, neurological deterioration, and poor functional recovery [7,8,9,10]. Meanwhile, anti-inflammatory cytokines such as IL-10 and TGF-β rise in the resolution phase and are linked to tissue repair—but also with stroke-induced immunosuppression and increased infection risk [11,12]. Composite biomarkers such as the neutrophil-to-lymphocyte ratio (NLR), platelet-to-lymphocyte ratio (PLR), and systemic immune-inflammation index (SII = platelet count × neutrophil count / lymphocyte count) provide a cost-effective means of assessing immune balance and have shown predictive value for stroke severity and outcomes in large cohorts [13,14,15]. At the same time, immunogenetic variants and peripheral inflammatory signatures offer further granularity to individual prognosis. Inflammation is not only a consequence but also a modifiable driver of stroke outcomes . Trials with agents like IL-1 receptor antagonists (IL-1Ra), tocilizumab (IL-6R antibody), and colchicine ((NLRP3 (NOD-like Receptor Family Pyrin Domain-Containing 3) inflammasome inhibitor)) suggest that immune-targeted interventions could enhance neuroprotection or prevent recurrence [16,17].
Despite this progress, translation into routine clinical practice remains limited. The evidence is fragmented across cytokine families, outcome timepoints, and stroke subtypes [1,18,19]. Thus, this review synthesizes a decade of literature on immunological and inflammatory biomarkers in ischemic stroke. Our objective is to identify and classify key biomarkers, examine their associations with clinical outcomes, and explore emerging therapeutic strategies grounded in immune modulation.
2. Inflammation and Immune Response in Ischemic Stroke
The brain’s immune privilege is challenged during stroke, as cell injury and necrosis release damage-associated molecular patterns (DAMPs) that activate innate immune pathways [20,21]. Within minutes of an ischemic insult, resident microglia become activated and begin producing pro-inflammatory cytokines such as interleukin-1β (IL-1β) and tumour necrosis factor-α (TNF-α) [22,23]. Microglial activation is an early and dual-edged event, initially beneficial in debris clearance and trophic support, but ultimately harmful due to production of inflammatory mediators and neurotoxic substances [23,24].
As the blood–brain barrier (BBB) becomes compromised, peripheral immune cells infiltrate the brain parenchyma. Neutrophils are among the earliest responders, reaching the infarct zone within hours and peaking around 24 hours post-insult. They release matrix metalloproteinases (MMPs), including MMP-9, and reactive oxygen species (ROS), which exacerbate BBB breakdown and worsen neuronal damage [25,26]. Monocytes and lymphocytes follow in a temporally distinct pattern: monocyte-derived macrophages accumulate over days 3–7, while T lymphocytes infiltrate around day 2–3, secreting additional cytokines that propagate the inflammatory milieu. The sustained invasion of leukocytes contributes to secondary injury and neurological worsening [27,28,29]. IL-8 (CXCL8) and the acute-phase protein C-reactive protein (CRP) rise slightly later, with CRP peaking at approximately 48 hours post-stroke [30,31]. In contrast, IL-10—a key anti-inflammatory cytokine—peaks between 24–72 hours as part of the immune system’s counter-regulatory response [30]. IL-17, produced primarily by infiltrating T-helper 17 (Th17) and γδ T cells, shows a delayed elevation beginning around 48–72 hours, coinciding with later-stage T-cell infiltration and extended inflammation [32]. Figure 1 illustrates the time-course of key inflammatory mediators in the systemic circulation after an ischemic stroke.
Once recruited to the ischemic territory, immune cells engage in complex interactions. DAMPs such as HMGB1 (High-Mobility Group Box 1) and extracellular ATP (Adenosine Triphosphate) released from necrotic cells bind to pattern recognition receptors ((e.g., TLRs (Toll-Like Receptors), RAGE (Receptors for Advanced Glycation End-products), P2X7 (Purinergic Receptor P2X, Ligand-Gated Ion Channel, 7)) on microglia and other immune cells, amplifying inflammatory signaling and transmigration across the BBB [26,33,34]. This facilitates the orchestrated recruitment of immune cells and establishes a dynamic interplay between innate and adaptive immunity. While the infiltration of leukocytes can contribute to debris clearance and subsequent tissue repair, excessive or prolonged invasion aggravates inflammatory signaling and disruption of neurovascular integrity [35]. Pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α aggravate neuronal apoptosis via activation of death receptor pathways, destabilization of mitochondrial function, and exacerbation of excitotoxicity, which further promotes immune cell recruitment [9,22,36]. Figure 2 illustrates the key components of the inflammatory cascade after stroke, where an initial wave of cytokine release triggers a cycle of immune cell recruitment and further cytokine production, leading to expansion of tissue injury.
Figure abbreviations: DAMPs (Damage-Associated Molecular Patterns), HMGB1 (High-Mobility Group Box 1), ATP (Adenosine Triphosphate), IL-1β (Interleukin-1 beta), TNF-α (Tumour Necrosis Factor-alpha), IL-6 (Interleukin-6), CCL2 ((C-C motif chemokine ligand 2, also historically known as MCP-1 (Monocyte Chemoattractant Protein-1)), CXCL8 (C-X-C motif chemokine ligand 8, more commonly known as Interleukin-8), ICAM-1 (Intercellular Adhesion Molecule-1), VCAM-1 (Vascular Cell Adhesion Molecule-1), ROS (Reactive Oxygen Species), NO (Nitric Oxide)
Neurons themselves, particularly in the ischemic penumbra, become an active source of cytokines release, amplifying tissue damage. IL-10, an anti-inflammatory cytokine, is upregulated as a compensatory response to counteract this pro-inflammatory surge but often fails to fully neutralize acute neurotoxicity if the inflammatory response is overwhelming [37,38]. Despite this, inflammation is not inherently deleterious; its role is highly phase dependent. In the subacute and chronic phases, microglia and macrophages can polarize toward an anti-inflammatory phenotype, secreting trophic factors and cytokines that support tissue remodelling and axonal sprouting. IL-10 and TGF-β are central to this reparative shift, promoting immune resolution and neurovascular stabilization. Interestingly, IL-1β, typically associated with acute injury, also contributes to later regenerative processes by stimulating angiogenesis and remodelling of endothelial cells during the repair phase [19,33].
As immune cells continue to accumulate, adhesion processes become central. Endothelial cells upregulate intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and selectins in response to cytokine stimulation and hypoxia. These molecules interact with leukocyte integrins, including LFA-1 (Lymphocyte Function-Associated Antigen 1) ((CD (Cluster of Differentiation) 11a / CD 18), Mac-1 (Macrophage-1 Antigen, also known as CD11b/CD18 or Integrin αMβ2), and VLA-4 (Very Late Antigen-4, also known as integrin α4β1 or CD49d/CD29), to mediate firm adhesion and transmigration into the ischemic tissue. The balance of T cell responses further shapes post-stroke neuroinflammation. Conversely, Th2 and Treg cells produce IL-4, IL-5, IL-13, IL-10, and TGF-β, contributing to immune suppression and repair. CD8+ cytotoxic T cells can directly induce neuronal death through perforin- and granzyme-mediated pathways, exacerbating tissue injury [15,17,35,39,40]. Although adaptive immunity intensifies the inflammatory response initially, it also plays a role in post-stroke immunosuppression, a clinically recognized phenomenon characterized by lymphocyte depletion and increased vulnerability to infections.
Acute ischemic stroke also triggers the release of DAMPs like ATP and HMGB1, which activate inflammasomes such as NLRP3 in microglia via P2X7 receptors. In response, activated microglia secrete pro-inflammatory cytokines including interleukin-1β (IL-1β) and TNF-α, which stimulate endothelial cells of the cerebral vasculature to upregulate adhesion molecules (ICAM-1, VCAM-1) and to release chemokines like interleukin-8 (IL-8/CXCL8) [17,22,33]. This signalling cascade promotes neutrophil adhesion to the endothelium and their extravasation into the brain parenchyma. Once inside, neutrophils release ROS, elastases, and proteolytic enzymes such as MMP-9, leading to degradation of the extracellular matrix and disruption of the BBB [26,35]. Monocytes are subsequently recruited through chemokines such as CCL2 (MCP-1), secreted by activated astrocytes. These monocytes differentiate into macrophages that participate in both propagation of inflammation and phagocytic clearance of necrotic debris [27].
Astrocytes also undergo reactive gliosis, releasing IL-6 and various chemokines that further amplify immune cell infiltration during the acute phase. However, in later stages, astrocytes contribute to scar formation and help restore tissue integrity by secreting structural matrix proteins and regulatory cytokines [11,41]. In the subacute phase (approximately 2–3 days post-stroke), T lymphocytes infiltrate the ischemic brain. CD4+ Th (T helper) cells differentiate into Th1 and Th17 subsets, producing interferon-gamma (IFN-γ) and interleukin-17 (IL-17), respectively, which sustain local inflammation and modulate innate immune cells. CD8+ cytotoxic T cells can directly induce neuronal apoptosis via perforin- and granzyme-mediated pathways, exacerbating tissue injury [18,32,42].
This intricate sequence of events, featuring crosstalk among microglia, astrocytes, neutrophils, macrophages, and lymphocytes, governs the extent of neuroinflammation, BBB compromise, and ultimately neuronal survival in the ischemic penumbra. While early and excessive inflammation promotes irreversible damage, controlled immune activation in the later stages is essential for clearing debris, modulating angiogenesis, and guiding neural repair [2,23,24,33]. Therefore, the timing, magnitude, and cellular context of immune activation are critical determinants of stroke progression and recovery. Specific biomarkers that reflect the evolving phases of immune activity, such as IL-6, IL-10, IL-17, MMP-9, and NLR, are being increasingly recognized for their prognostic value and therapeutic relevance in clinical practice and trials [21,29,30,43]. Interactions between key immune cells in the post-ischemic brain are illustrated in Figure 3.
2.1. Key Inflammatory Biomarkers in Ischemic Stroke
2.1.1. Interleukin-1β (IL-1β)
IL-1β is one of the most pivotal pro-inflammatory cytokines involved in the acute phase of ischemic stroke. It is rapidly produced by activated microglia and infiltrating macrophages in response to necrotic cell debris and DAMPs. IL-1β contributes significantly to BBB disruption, leukocyte adhesion, and infiltration by upregulating adhesion molecules on endothelial cells and activating astrocytes and microglia. Experimental studies showed mice lacking IL-1α and IL-1β exhibit smaller infarct volumes after middle cerebral artery occlusion, underscoring the cytokine’s neurotoxic role in stroke pathophysiology [6,16,22].
Conversely, therapeutic blockade of IL-1 has demonstrated neuroprotective effects in preclinical models. Administration of IL-1 receptor antagonist (IL-1Ra) reduces infarct size, suppresses microglial activation, and attenuates neutrophil infiltration. IL-1β’s pro-inflammatory loop amplifies local damage by inducing IL-6 and TNF-α expression, forming a positive-feedback circuit of neuroinflammation. Clinically, elevated plasma IL-1β levels within the first 24 hours after stroke correlate with stroke severity, larger infarct volumes, and poorer neurological outcomes. Genetic polymorphisms in IL1RN (the gene encoding IL-1Ra) have also been associated with increased stroke susceptibility, although findings have varied across populations [16,44,45].
Therapeutically, recombinant human IL-1Ra (anakinra) has been tested in clinical settings. Phase II studies demonstrated that anakinra administered within 6 hours of stroke onset was safe and led to reductions in circulating neutrophils, CRP, and IL-6 levels, suggesting systemic anti-inflammatory effects. The subsequent SCIL-STROKE trial using subcutaneous IL-1Ra confirmed downregulation of inflammatory biomarkers and indicated possible improvements in functional outcomes, although larger trials are needed for validation. Despite the absence of phase III efficacy data, IL-1β remains a strong candidate for targeted intervention and a valuable biomarker reflecting acute neuroinflammatory activation after stroke [22,46].
2.1.2. Tumour Necrosis Factor-α (TNF-α)
TNF-α is a pivotal early mediator of inflammation following ischemic stroke. It is rapidly released by activated microglia and further amplified as circulating immune cells infiltrate the ischemic brain tissue [23,33,37]. TNF-α exerts diverse effects, including induction of neuronal apoptosis via TNF receptor 1 (TNFR1) signalling, generation of oxidative stress, and disruption of the BBB through upregulation of endothelial adhesion molecules and MMP-9. While these actions contribute to infarct expansion and oedema, TNF-α also exhibits context-dependent neuroprotective functions. For example, it participates in synaptic plasticity, neurogenesis, and cell survival via TNF receptor 2 (TNFR2)-mediated pathways, particularly during the recovery phase. This dual nature has been confirmed in preclinical studies showing that TNF-α inhibition administered acutely may reduce infarct volume and neurological deficits, while delayed blockade can impair recovery by suppressing reparative processes [9,23].
In clinical settings, elevated TNF-α levels in the acute phase have been associated with larger infarcts and worse functional outcomes, although results are not entirely consistent. Some studies highlight that high TNF-α levels independently predict poor prognosis, while others suggest the association weakens after adjusting for confounders. Circulating levels of soluble TNF receptors (sTNFR1 and sTNFR2) have emerged as more stable markers, with elevated sTNFR levels linked to recurrent strokes and early post-stroke seizures. Chronic elevation of TNF-α has also been implicated in stroke risk, particularly in individuals with pro-inflammatory conditions such as diabetes or autoimmune disease. Polymorphisms in the TNF gene and persistently elevated TNF-α are associated with increased susceptibility to ischemic stroke in multiple populations [23,47,48].
Despite its established pathogenic role, TNF-α has not become a clinical target in stroke therapy. Trials involving systemic TNF inhibition (e.g., with etanercept or infliximab) have not shown benefit and may carry risk. Observational data suggest that patients with autoimmune disease on anti-TNF agents might even experience a slight increase in cerebrovascular events, possibly due to impaired vascular repair mechanisms or thrombogenic side effects [1,2,8]. Nevertheless, ongoing research is investigating more selective approaches to modulate TNF-α signalling. For example, targeting TNFR1-mediated necroptosis in cerebral endothelial cells—while preserving TNFR2-dependent protective functions—could offer a refined therapeutic strategy that mitigates inflammation without hindering post-stroke recovery. TNF-α thus remains a critical biomarker reflecting the magnitude of acute inflammation, with complex therapeutic implications [9,23].
2.1.3. Interleukin-6 (IL-6)
IL-6 is a multifunctional cytokine with a central role in the acute phase response to ischemic stroke. It is rapidly upregulated in both brain tissue and systemic circulation within hours of vessel occlusion, and its levels correlate with infarct size, stroke severity, and prognosis [49,50]. IL-6 signals through two major pathways: classical signalling via membrane-bound IL-6 receptors (which may be regenerative or anti-inflammatory) and trans-signalling via soluble IL-6 receptors, which predominantly promote inflammation . In the early phase of stroke, IL-6 is induced by IL-1β and TNF-α and amplifies the inflammatory cascade. It contributes to leukocyte recruitment, upregulation of endothelial adhesion molecules, and stimulation of hepatic acute-phase responses, including CRP synthesis [50]. However, IL-6 also plays a role in tissue repair during later phases by supporting neurogenesis, astrogliosis, and angiogenesis—highlighting its dual role in injury and recovery [51].
Clinically, IL-6 is among the most studied biomarkers in ischemic stroke. Elevated serum levels within the first 24–48 hours post-onset are associated with larger infarcts, higher NIHSS (National Institutes of Health Stroke Scale) scores, and poorer functional outcomes on the modified Rankin Scale (mRS) at 3 months [14,51,52]. Persistently elevated IL-6 levels beyond the acute phase may also predict stroke recurrence and long-term disability. Due to these associations, IL-6 is being explored as a therapeutic target. Tocilizumab, an anti-IL-6 receptor monoclonal antibody, has shown promise in preclinical stroke models, and the ongoing IRIS trial is currently evaluating its utility in patients undergoing mechanical thrombectomy. High IL-6 levels may help identify patients at risk of extensive injury who could benefit from targeted anti-inflammatory strategies [48,51,53,54].
2.1.4. C-Reactive Protein (CRP)
CRP, a well-known acute-phase reactant produced in the liver under the stimulation of IL-6, is widely used in clinical practice as a systemic inflammatory marker. In ischemic stroke, CRP levels rise within 6–24 hours post-onset, often peaking between 48 and 72 hours. High-sensitivity CRP (hs-CRP) assays allow detection of subtle elevations that are clinically meaningful [31]. Meta-analyses and large cohort studies have confirmed that elevated admission CRP is associated with increased risk of early neurological deterioration, mortality, and worse functional outcomes at 3 months. Indeed, patients with CRP levels in the top quartile upon admission have roughly double the risk of 30-day mortality compared to those in the lowest quartile and are also at higher risk for recurrent vascular events [55,56,57].
While CRP may have a direct pathophysiological role in endothelial dysfunction and thrombosis, it is also a reliable surrogate marker for the overall inflammatory burden. CRP levels reflect infarct size, systemic immune activation, and infection status, all of which influence prognosis [31]. Persistently elevated CRP during hospitalization (for example, after thrombolysis) has been linked to poor recovery, even in patients with initially mild strokes. From a preventive standpoint, chronically elevated CRP is a risk factor for ischemic stroke, analogous to its role in coronary artery disease. Therapies such as high-dose statins, anti-IL-1β agents, and colchicine may reduce CRP and improve outcomes in high-risk populations. As such, CRP remains an accessible, cost-effective, and integrative biomarker in stroke care [26,31].
2.1.5. Interleukin-10 (IL-10)
IL-10 is a potent anti-inflammatory cytokine secreted by monocytes, macrophages, and regulatory T cells in response to tissue injury. In stroke, IL-10 modulates the immune response by inhibiting the synthesis of pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α), suppressing antigen presentation, and downregulating adhesion molecules on endothelial cells. It promotes neuronal survival by preventing apoptosis and limiting oxidative damage. Experimental studies have shown that IL-10 overexpression or exogenous administration reduces infarct volumes and improves neurological outcomes in animal models of ischemic stroke. However, its clinical significance is complex and time dependent [47,58,59]. A low IL-10 response in the acute phase (<24 h) is associated with unopposed inflammation, haemorrhagic transformation, and early neurological deterioration. Conversely, elevated IL-10 levels in the subacute phase (days 2–7) may indicate stroke-induced immunosuppression (SIS), increasing the risk of infections such as pneumonia and urinary tract infections [11,21,30].
This duality underscores the importance of context in interpreting IL-10 levels. While protective in moderation, excessive IL-10 may suppress systemic immunity and contribute to adverse outcomes. Some studies propose that IL-10 or IL-10/IL-6 ratios could serve as biomarkers to predict post-stroke complications, including infections or haemorrhagic conversion [6,26]. Although IL-10 is not currently a direct therapeutic target due to concerns about immunosuppression, strategies to modulate endogenous IL-10 or deliver it locally to the brain are being investigated. Ultimately, IL-10 serves as both a marker of immune balance and a potential indicator of whether inflammation is being adequately regulated in the aftermath of stroke [30,39,40].
2.2. Other Important Inflammatory Biomarkers
In addition to major cytokines like IL-1β, IL-6, TNF-α, and IL-10, several other inflammatory mediators play significant roles in ischemic stroke. Two that have received increasing attention are interleukin-8 (IL-8) and interleukin-17 (IL-17), representing innate and adaptive immune responses, respectively [22,32].
2.2.1. Interleukin-8 (CXCL8)
CXCL8 is a neutrophil-attracting chemokine secreted by activated microglia, endothelial cells, and astrocytes in response to ischemia. Blood IL-8 levels rise within hours after stroke onset and correlate with neutrophil infiltration into the brain. Elevated IL-8 levels have been linked to larger infarcts, greater early neurological deterioration, and poor functional outcomes [6]. Mechanistically, IL-8 promotes neutrophil adhesion to endothelium and entry into the brain, where these cells release proteases and reactive oxygen species, worsening BBB disruption. Experimental models show that blocking IL-8 signalling or deleting IL-8 receptors reduces infarct volume and neutrophil infiltration. Moreover, IL-8 may contribute to angiogenesis via stimulation of VEGF, implying a dual role in both injury and repair [42,48,60].
2.2.2. Interleukin-17 (IL-17). IL-17, predominantly produced by Th17 cells and γδ T cells, rises slightly later, typically 24–72 hours post-stroke, as part of the adaptive immune response. IL-17 amplifies inflammation by stimulating the release of IL-1β, TNF-α, and chemokines that recruit neutrophils [20,42,61]. It also directly impairs endothelial function and contributes to BBB disruption. Elevated IL-17 in plasma or CSF has been observed in patients with more severe strokes, and preclinical studies demonstrate that blocking IL-17 or IL-23 reduces infarct size and improves outcomes [6,30].2.2.3. Other relevant biomarkers:
IL-18 and IL-12, both pro-inflammatory cytokines that are elevated after stroke and associated with worse clinical outcomes due to their role in enhancing Th1 responses and innate immune activation;
IL-4 and IL-13, which promote M2 macrophage polarization and may facilitate repair by resolving inflammation and supporting tissue remodelling;
TGF-β and endogenous IL-1 receptor antagonist (IL-1Ra), which act as regulatory cytokines in later stages, limiting immune activation and supporting recovery [6,30,62].
Table 1 Main pro- and anti-inflammatory molecules involved in the pathophysiology of ischemic strokes.
In summary, IL-8, IL-17, and related markers broaden our understanding of the inflammatory response in stroke. Their temporal dynamics, early for IL-8, delayed for IL-17, reflect distinct phases of immune activation. Together with IL-1β, IL-6, and TNF-α, these molecules contribute to acute injury, while IL-10, TGF-β, and IL-1Ra offer counter-regulatory effects. Accurate profiling of these mediators may enhance prognostication and guide immunomodulatory therapies [6,9,32].
Several non-cytokine markers also reflect inflammatory burden and vascular injury. These include soluble ICAM-1, VCAM-1, E-selectin, and MMP-9. Among them, MMP-9 is particularly important for its association with haemorrhagic transformation and BBB degradation due to its proteolytic activity on extracellular matrix proteins [26,37,39,59].
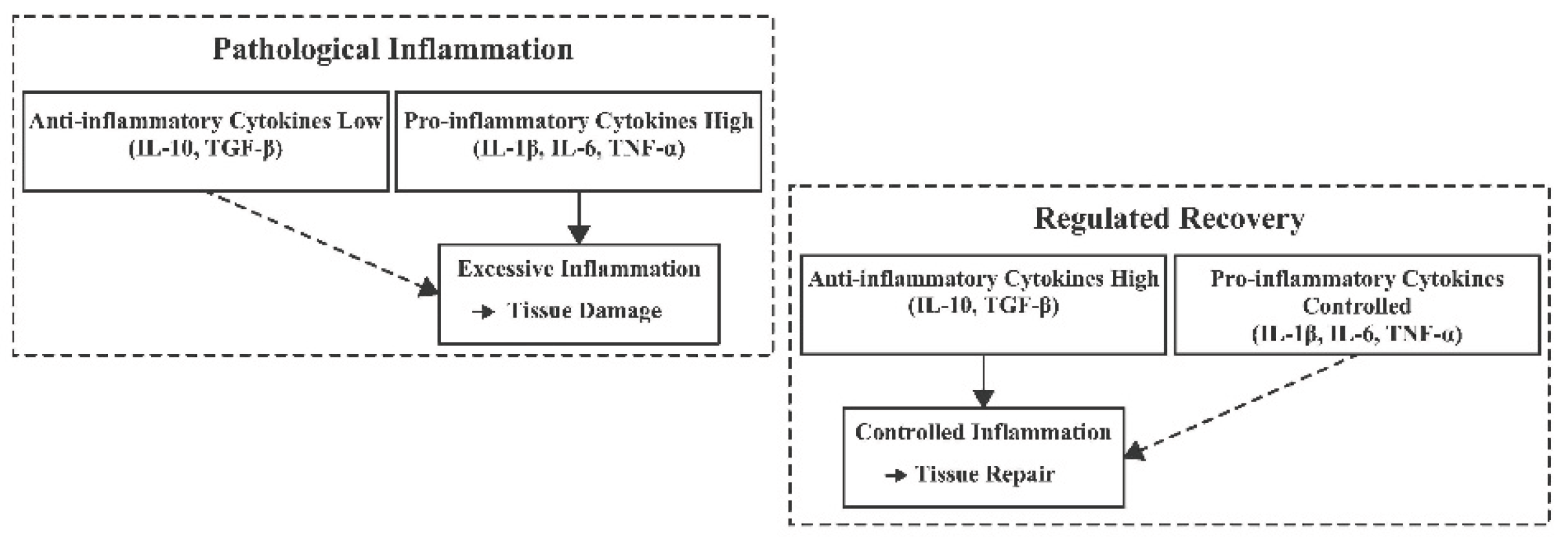
Figure 4.
Comparison of a pathological inflammatory response versus a regulated recovery phase after stroke. In the pathological inflammation scenario (left), there is an excessive production of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) and insufficient anti-inflammatory activity (low IL-10, TGF-β). This imbalance leads to uncontrolled inflammation and exacerbated tissue injury. By contrast, in the regulated recovery phase (right), anti-inflammatory mediators (IL-10, TGF-β) are prevalent and help keep pro-inflammatory cytokines in check, promoting resolution of inflammation and tissue repair. Figure abbreviations: IL-10 (Interleukin-10), TGF-β (Transforming Growth Factor-beta), IL-1β (Interleukin-1 beta), IL-6 (Interleukin-6), TNF-α (Tumour Necrosis Factor-alpha).
Figure 4.
Comparison of a pathological inflammatory response versus a regulated recovery phase after stroke. In the pathological inflammation scenario (left), there is an excessive production of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) and insufficient anti-inflammatory activity (low IL-10, TGF-β). This imbalance leads to uncontrolled inflammation and exacerbated tissue injury. By contrast, in the regulated recovery phase (right), anti-inflammatory mediators (IL-10, TGF-β) are prevalent and help keep pro-inflammatory cytokines in check, promoting resolution of inflammation and tissue repair. Figure abbreviations: IL-10 (Interleukin-10), TGF-β (Transforming Growth Factor-beta), IL-1β (Interleukin-1 beta), IL-6 (Interleukin-6), TNF-α (Tumour Necrosis Factor-alpha).
3. Prognostic Value of Immune Biomarkers in Stroke
Inflammation-related biomarkers offer significant prognostic value beyond traditional clinical predictors in ischemic stroke. Elevated levels of pro-inflammatory cytokines, particularly IL-6, IL-1β, TNF-α, and IL-8, are consistently associated with worse neurological outcomes [1,2,3]. For example, increased IL-6 and TNF-α levels during the acute phase correlate with larger infarct volumes and higher disability scores at three months post-stroke. IL-6 levels measured upon admission have been shown to significantly correlate with both initial stroke severity and long-term functional status. Similarly, higher early concentrations of TNF-α and IL-1β are predictive of more severe neurological deficits and poorer outcomes. These cytokines thus serve as early warning markers of a more aggressive and damaging neuroinflammatory response [7,9].
Among circulating inflammatory markers, C-reactive protein (CRP) has emerged as a particularly robust prognostic biomarker due to its stability, reproducibility, and clinical accessibility. Elevated CRP levels during the subacute phase have been consistently linked with increased mortality, risk of stroke recurrence, and poor functional recovery [31]. Meta-analyses have shown that patients with CRP levels in the top quartile upon admission have approximately double the risk of 30-day mortality compared to those in the lowest quartile and are also at higher risk for recurrent vascular events. As a result, CRP is increasingly used in clinical settings for early risk stratification. A markedly elevated CRP may also prompt clinicians to consider more aggressive secondary prevention or to closely monitor for post-stroke complications such as infection [48,55].
On the other hand, anti-inflammatory cytokines such as IL-10 provide a more nuanced prognostic picture. A blunted IL-10 response in the acute phase suggests unopposed inflammation and has been associated with haemorrhagic transformation and early neurological deterioration [6]. Conversely, excessive IL-10 elevation may indicate compensatory immunosuppression and predispose patients to infectious complications such as pneumonia or urinary tract infection [4,21]. Thus, IL-10 serves as a dual-purpose biomarker: insufficient levels may signal heightened injury risk, while elevated levels may forecast immune suppression and secondary complications.
Beyond individual cytokines, composite inflammatory indices derived from routine blood counts have gained traction as prognostic tools. Indices such as the neutrophil-to-lymphocyte ratio (NLR), platelet-to-lymphocyte ratio (PLR), and systemic immune-inflammation index (SII) provide integrative markers of systemic immune activation and stress [63,64]. These values rise when neutrophils and platelets are elevated and lymphocyte counts are suppressed, patterns typical of acute inflammation and stress-induced immunosuppression. Clinical studies have demonstrated that higher NLR, PLR, and SII levels are independently associated with worse outcomes after stroke [65,66]. In a 2024 cohort study involving over 500 patients, six haematological inflammatory indices, including NLR, PLR, SII, and the systemic inflammation response index (SIRI), were all linked to poor 30-day prognosis, with multivariate models identifying NLR, PLR, and SIRI as independent predictors of early functional decline [14,67,68]. These indices are particularly appealing due to their low cost, ease of use, and their ability to reflect cytokine dynamics—e.g., a high NLR may indirectly indicate IL-8- and TNF-driven neutrophilia along with cortisol-mediated lymphopenia [15,69].
In summary, immune and inflammatory biomarkers offer valuable prognostic insights following ischemic stroke. Elevated pro-inflammatory cytokines (IL-6, IL-1β, TNF-α) are associated with worse clinical trajectories, while the presence of regulatory cytokines like IL-10 and integrative markers such as CRP provide additional layers of prognostic clarity [9,70,71]. Incorporating these biomarkers into clinical prediction models, alongside established factors such as age and NIHSS score, is an active area of research, with the potential to identify high-risk patients who may benefit from closer monitoring, early intervention, or enrolment in trials of targeted immunomodulatory therapies.
4. Therapeutic Modulation of Inflammation: Implications for Treatment and Prevention
The central role of inflammation in stroke pathophysiology raises the possibility that immunomodulation could improve outcomes or reduce the risk of recurrence. This strategy has gained traction particularly considering repeated failures of traditional neuroprotective agents targeting excitotoxicity. Several inflammatory mediators, especially IL-1β, IL-6, and TNF-α, are now under investigation as therapeutic targets [8,22,23].
One of the most advanced approaches involves targeting the IL-1 axis. Given IL-1β’s established role in acute neuroinflammation and BBB disruption, pharmacologic blockade using IL-1 receptor antagonist (IL-1Ra, anakinra) has shown promise in clinical trials [5,22,24]. Phase II studies demonstrated that IL-1Ra administration in acute ischemic stroke was biologically active, reduced inflammatory cytokine levels, and was well tolerated, though sample sizes were insufficient to confirm functional benefit. Further trials are warranted, especially in subgroups with elevated inflammatory markers. Moreover, IL-1 blockade may help limit secondary injury such as haemorrhagic transformation and reperfusion damage, particularly if administered peri-thrombectomy [16]. Ongoing investigations are also exploring IL-1β neutralizing antibodies (e.g., canakinumab) for potential neuroprotection and secondary prevention [38,61].
IL-6 signalling is another compelling target. The IRIS trial is currently evaluating tocilizumab, an IL-6 receptor (IL-6R) blocker, as an adjunct to reperfusion therapy. The rationale stems from evidence that IL-6 mediates reperfusion injury even after successful mechanical thrombectomy, contributing to infarct expansion. Tocilizumab, already in use for cytokine release syndromes, may mitigate this response when administered acutely. However, careful monitoring is essential due to potential side effects, including transient immunosuppression and hepatotoxicity [52,53,54].
Efforts to inhibit TNF-α have produced more ambiguous results. While etanercept (a soluble TNF decoy receptor) has shown benefit in animal models, reducing infarct size and inflammation, it has not yet demonstrated efficacy in clinical trials. Concerns also persist that long-term TNF suppression could increase vascular risk. Consequently, interest has shifted to downstream pathways such as TNF-induced necroptosis, which may be selectively targeted to prevent endothelial injury without broadly impairing TNF function [9].
Findings from cardiovascular trials provide a strong rationale for inflammation-targeted stroke prevention. The CANTOS trial in post-myocardial infarction patients demonstrated that canakinumab (an IL-1β neutralizing antibody) reduced major vascular events, including stroke, independent of lipid levels. These results support the concept that anti-inflammatory therapy alone can reduce vascular risk. While canakinumab remains costly and is associated with neutropenia, other more accessible agents are being investigated. Low-dose colchicine, which suppresses NLRP3 inflammasome activation and neutrophil recruitment, has reduced cardiovascular events in coronary disease and is currently being tested in patients with TIA (transient ischemic attacks) or minor stroke [6,17,22]. Its actions include inhibition of IL-1β production and downstream inflammatory signalling, making it a promising candidate for both primary and secondary prevention.
Beyond pro-inflammatory suppression, attention has also turned to post-stroke immunosuppression, a major contributor to infectious complications such as pneumonia. Trials using prophylactic antibiotics like moxifloxacin have had mixed results, though targeted approaches in patients with evidence of immune exhaustion (e.g., high IL-10 levels or lymphopenia) may prove more effective. An alternative strategy involves using granulocyte colony-stimulating factor (G-CSF) to boost immune cell production. G-CSF not only promotes leukocyte recovery but also has neurotrophic properties; however, clinical trials in stroke have so far shown variable results, and its role in routine practice remains unclear [11,30,37].
Ultimately, a multi-faceted approach to immunomodulation may be necessary. Combining anti-inflammatory therapies (to limit acute injury) with strategies that prevent or reverse post-stroke immunosuppression could yield the best outcomes [60,72]. Personalized medicine approaches, such as tailoring treatments based on a patient’s inflammatory profile or genetic background (e.g., IL-1 or IL-6 polymorphisms), are on the horizon. As our understanding of the immune mechanisms in stroke deepens, it opens the door to innovative therapies that complement existing reperfusion and neuroprotective strategies.
5. Discussion and Future Directions
Over the past decade, mounting evidence has firmly established that immunological and inflammatory biomarkers are integral to predicting and understanding outcomes after ischemic stroke. These biomarkers not only reflect the intensity and duration of the immune response but also offer valuable insight into the potential for injury, recovery, and complications [20,60]. As stroke care increasingly moves toward precision medicine, several future directions are emerging for integrating immune biomarkers into both clinical practice and research frameworks.
5.1. Personalized Inflammatory Profiling
Rather than relying on isolated cytokine measurements, future studies will likely employ multi-biomarker panels, encompassing cytokines, chemokines, acute-phase proteins (e.g., CRP), and leukocyte-derived indices (e.g., NLR, PLR), to construct patient-specific inflammatory signatures. These immune profiles, analysed using machine learning or clustering algorithms, could stratify patients by prognosis or therapeutic responsiveness [14,20,31]. Large-scale initiatives and biobanks are already collecting longitudinal blood samples to correlate inflammatory dynamics with functional outcomes and infarct evolution.
5.2. Real-Time and Point-of-Care Inflammation Monitoring
Technological advances in rapid bioassay platforms may soon enable bedside monitoring of key cytokines such as IL-6 or IL-1β, providing real-time assessment of inflammatory status [22,49]. This would support dynamic treatment decisions, such as the timing of intensive care de-escalation or early rehabilitation initiation. Additionally, serial inflammatory tracking could help identify patients at risk for delayed deterioration, haemorrhagic transformation or post-stroke infection.
5.3. Biomarker-Guided Immunotherapy Trials
Encouraging results from exploratory trials support the need for larger randomized controlled studies evaluating anti-inflammatory agents in stroke. Prominent candidates include IL-1 blockers (e.g., anakinra, canakinumab), IL-6R inhibitors (e.g., tocilizumab), and NLRP3 inflammasome inhibitors [9,16,17]. Another emerging approach involves cell-based immunotherapy, such as regulatory T cell or mesenchymal stem cell infusions, aimed at promoting resolution of inflammation and neurorepair. Future trials may adopt biomarker-enrichment strategies, treating only patients with elevated CRP, IL-6, or SII levels, thereby maximizing therapeutic efficiency and minimizing unnecessary exposure.
5.4. Synergy with Neurorestorative Interventions
Post-stroke inflammation extends beyond the acute phase and modulates repair mechanisms such as neurogenesis, synaptic remodelling, and remyelination. This opens the door for combinatorial approaches that align immunomodulation with neurorestorative therapies. For instance, early anti-IL-17 therapy could reduce lesion volume and secondary damage, while late-phase immune stimulation might enhance plasticity and functional recovery [32,73]. Temporal precision will be crucial, suppressing inflammation during the hyperacute phase and boosting repair-associated immunity during convalescence.
5.5. Inflammation as a Target for Primary Prevention
Growing evidence suggests that chronic low-grade inflammation contributes to stroke risk, independent of traditional vascular factors. Biomarkers such as CRP and IL-6 may soon be incorporated into risk prediction models alongside blood pressure, cholesterol, and smoking status. This raises the possibility of using anti-inflammatory agents (e.g., statins, colchicine) for primary prevention in high-risk populations [34,48,54]. However, the safety of long-term immune modulation must be carefully evaluated, as immunosuppression may predispose to infections or malignancies.
5.6. Translational and Clinical Challenges
Despite clear associations between inflammatory markers and outcomes, causality remains difficult to prove, and therapeutic translation faces several hurdles. Stroke is a heterogeneous condition, and not all patients with elevated inflammatory biomarkers will benefit from immunosuppression. Moreover, precision is essential: excessive suppression may impair recovery or promote infection, while inadequate modulation may be ineffective. Future trials must identify the right patient subsets, timing of intervention, and optimal targets to ensure safety and efficacy [19,58].
6. Conclusions
Immunological and inflammatory biomarkers, including IL-1β, IL-6, TNF-α, IL-10, IL-8, IL-17, and CRP, have become indispensable tools in stroke research, offering insight into how the immune system shapes both the extent of brain injury and the trajectory of recovery [6,8,9,30]. These molecules are not merely reflections of damage but active participants in the pathophysiological cascade, and they hold considerable prognostic value. Elevated pro-inflammatory markers generally correlate with larger infarct volumes, higher rates of disability, and poorer functional outcomes, while anti-inflammatory signals such as IL-10 convey a more regulated immune response and may predict better recovery [74].
These biomarkers are paving the way for a new class of stroke therapies aimed at modulating the immune response. From IL-1 blockade to IL-6 receptor inhibition and inflammasome targeting, clinical trials are now leveraging our growing understanding of neuroinflammation to guide therapeutic strategies. In parallel, haematological indices such as NLR, PLR, and SII are being validated as cost-effective, readily available proxies for immune activity, offering clinicians practical tools for early risk stratification and management decisions [13,61,68,69].
The interaction between the immune and nervous systems in stroke is intricate and dynamic, influenced by time, comorbidities, and genetic background. Yet, untangling these relationships offers a transformative opportunity: to move toward personalized, pathophysiology-guided stroke care [36,47]. As we integrate biomarkers into clinical workflows, their use may extend from prognosis to therapy selection, rehabilitation planning, and secondary prevention.
Ultimately, by monitoring and modulating the inflammatory response, clinicians may be able to reduce secondary brain injury, enhance repair mechanisms, and prevent recurrent events. This strategy has the potential to improve survival, optimize functional recovery, and reduce the overall burden of stroke, a critical goal in the face of aging populations and rising global incidence. The next decade of research will be crucial in translating these insights into standard-of-care tools and treatments, marking a paradigm shift in how stroke is managed at both the individual and population level.
Author Contributions
Conceptualization, M.I., A.B., C.F., C.G., I.C. M.S., D.T., D.B., M.P.; methodology, M.I., A.B., C.F., C.G., I.C. M.S., D.T., D.B., M.P., M.L.P.; software, M.I., A.B., C.G. ,I.C., M.S., D.T., validation, D.B, M.P., C.T., M.L.P.; formal analysis, M.I., A.B., D.T., D.B., M.P., C.T.; investigation, M.I., A.B., C.F., C.G., I.C. M.S., D.T., D.B., M.P.; resources, M.I., A.B., C.F., C.G., I.C. M.S., D.T., D.B., M.P.; data curation, M.I., A.B, D.T., D.B., M.P., C.T., M.L.P.; writing—original draft preparation, M.I., C.F., C.G., I.C. M.S., D.T.; writing—review and editing, M.I., A.B, D.T., D.B., M.P., M.L.P., C.T.; visualization, A.B., M.P., M.L.P., C.T.; supervision, M.P., M.L.P., C.T.; project administration, M.L.P., C.T.; funding acquisition, M.I., A.B., C.F., C.G., I.C. M.S., D.T., D.B., M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ATP | Adenosine Triphosphate |
| BBB | Blood–Brain Barrier |
| BDNF | Brain-Derived Neurotrophic Factor |
| CD | Cluster of Differentiation |
| CRP | C-Reactive Protein |
| CXCL8 | C-X-C Motif Chemokine Ligand 8 (also known as IL-8) |
| DAMPs | Damage-Associated Molecular Patterns |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| HMGB1 | High-Mobility Group Box 1 |
| hs-CRP | High-Sensitivity C-Reactive Protein |
| ICAM-1 | Intercellular Adhesion Molecule-1 |
| IFN-γ | Interferon-Gamma |
| IL | Interleukin |
| IL-1β | Interleukin-1 Beta |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 (CXCL8) |
| IL-10 | Interleukin-10 |
| IL-17 | Interleukin-17 |
| IL-18 | Interleukin-18 |
| IL-21 | Interleukin-21 |
| IL-23 | Interleukin-23 |
| LFA-1 | Lymphocyte Function-Associated Antigen-1 |
| Mac-1 | Macrophage-1 Antigen |
| MMP-9 | Matrix Metalloproteinase-9 |
| mRS | Modified Rankin Scale |
| NLR | Neutrophil-to-Lymphocyte Ratio |
| NLRP3 | NOD-, LRR- and Pyrin Domain-Containing Protein 3 |
| NO | Nitric Oxide |
| PLR | Platelet-to-Lymphocyte Ratio |
| ROS | Reactive Oxygen Species |
| SII | Systemic Immune-Inflammation Index |
| SIRI | Systemic Inflammation Response Index |
| SIS | Stroke-induced Immunosuppression |
| TGF-β | Transforming Growth Factor-Beta |
| Th1 | T Helper Type 1 |
| Th2 | T Helper Type 2 |
| Th17 | T Helper Type 17 |
| TIA | Transient Ischemic Attack |
| TNF-α | Tumour Necrosis Factor-Alpha |
| TNFR1/2 | Tumour Necrosis Factor Receptor 1/2 |
| Treg | Regulatory T Cell |
| VCAM-1 | Vascular Cell Adhesion Molecule-1 |
| VEGF | Vascular Endothelial Growth Factor |
| VLA-4 | Very Late Antigen-4 |

Table 1.
Main roles pro- and anti-inflammatory molecules involved in the pathophysiology of ischemic strokes.
Table 1.
Main roles pro- and anti-inflammatory molecules involved in the pathophysiology of ischemic strokes.
| Cytokine / Interleukin | Classification | Main Functions | 1. Clinical / Prognostic Implications |
2. Timing of Elevation Post-Stroke |
|---|---|---|---|---|
| Interleukin-1β (IL-1β) | Pro-inflammatory | Initiates inflammatory cascade; activates microglia and endothelial cells | Elevated levels associated with larger infarct size and worse neurological outcomes | Peaks within 6–24 hours |
| Interleukin-6 (IL-6) | Pro-inflammatory | Modulates acute phase response; promotes leukocyte recruitment | High levels correlate with increased stroke severity and poor functional outcomes | Rises within hours; remains elevated for days |
| Tumour Necrosis Factor-α (TNF-α) | Pro-inflammatory | Promotes apoptosis; disrupts blood-brain barrier | Elevated levels linked to increased infarct size and neurological deterioration | Peaks within 6–24 hours |
| Interleukin-10 (IL-10) | Anti-inflammatory | Suppresses pro-inflammatory cytokine production; limits immune response | Higher levels associated with reduced infarct size and better outcomes; excessive levels may increase infection risk | Increases within 24–72 hours |
| Transforming Growth Factor-β (TGF-β) | 3. Anti-inflammatory/ Regulatory |
Regulates immune response; promotes tissue repair | Contributes to neuroprotection and recovery; exact prognostic value in stroke is under investigation | Elevation timing varies; involved in later stages |
| Interleukin-17 (IL-17) | Pro-inflammatory | Recruits neutrophils; amplifies inflammatory response | Elevated levels may exacerbate neuroinflammation and tissue damage | Peaks within 24–72 hours |
| Interleukin-8 (IL-8) | Pro-inflammatory | Attracts neutrophils; promotes inflammation | Higher levels associated with increased infarct volume and poorer outcomes | Rises early; peaks within 24 hours |
| Interleukin-1α (IL-1α) | Pro-inflammatory | Acts as an alarmin; initiates sterile inflammation | Early release may contribute to initial inflammatory response; specific prognostic implications in stroke require further study | Released immediately upon cell injury |
References
- Endres, M.; Moro, M.A.; Nolte, C.H.; Dames, C.; Buckwalter, M.S.; Meisel, A. Immune Pathways in Etiology, Acute Phase, and Chronic Sequelae of Ischemic Stroke. Circ. Res. 2022, 130, 1167–1186. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and Foe for Ischemic Stroke. J. Neuroinflammation 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Simion, A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. Int. J. Mol. Sci. 2021, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, S.; He, Q.; Zhang, D.; Chang, J. The Role of Immune Cells in Post-Stroke Angiogenesis and Neuronal Remodeling: The Known and the Unknown. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hu, S.; Li, Y.; Sun, Y.; Xiong, X.; Hu, X.; Chen, J.; Qiu, S. Interleukins and Ischemic Stroke. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Pawluk, H.; Woźniak, A.; Tafelska-Kaczmarek, A.; Kosinska, A.; Pawluk, M.; Sergot, K.; Grochowalska, R.; Kołodziejska, R. The Role of IL-6 in Ischemic Stroke. Biomolecules 2025, 15, 470. [Google Scholar] [CrossRef] [PubMed]
- Bitencourt, A.C.S.; Timóteo, R.P.; Bazan, R.; Silva, M.V.; Da Silveira Filho, L.G.; Ratkevicius, C.M.A.; De Assunção, T.S.F.; De Oliveira, A.P.S.; Luvizutto, G.J. Association of Proinflammatory Cytokine Levels with Stroke Severity, Infarct Size, and Muscle Strength in the Acute Phase of Stroke. J. Stroke Cerebrovasc. Dis. 2022, 31, 106187. [Google Scholar] [CrossRef] [PubMed]
- Băcilă, C.-I.; Vlădoiu, M.-G.; Văleanu, M.; Moga, D.-F.-C.; Pumnea, P.-M. The Role of IL-6 and TNF-Alpha Biomarkers in Predicting Disability Outcomes in Acute Ischemic Stroke Patients. Life 2025, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, M.; Wang, J.; Hu, F. Association between Interleukin-6 Levels and Stroke: A Systematic Review and Meta-Analysis. J. Int. Med. Res. 2024, 52. [Google Scholar] [CrossRef] [PubMed]
- Pawluk, H.; Woźniak, A.; Grześk, G.; Kołodziejska, R.; Kozakiewicz, M.; Kopkowska, E.; Grzechowiak, E.; Kozera, G. <p>The Role of Selected Pro-Inflammatory Cytokines in Pathogenesis of Ischemic Stroke</P>. Clin. Interv. Aging 2020, Volume 15, 469–484. [Google Scholar] [CrossRef]
- Pawluk, H.; Kołodziejska, R.; Grześk, G.; Kozakiewicz, M.; Woźniak, A.; Pawluk, M.; Kosinska, A.; Grześk, M.; Wojtasik, J.; Kozera, G. Selected Mediators of Inflammation in Patients with Acute Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 10614. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gu, L.; Chen, L.; Hu, W.; Feng, X.; Qiu, F.; Fan, Z.; Chen, Q.; Qiu, J.; Shao, B. Neutrophil-to-Lymphocyte Ratio and Platelet-to-Lymphocyte Ratio as Potential Predictors of Prognosis in Acute Ischemic Stroke. Front. Neurol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Liao, J.; Luo, X.; Chen, X. Prognostic Value of the Neutrophil-to-Lymphocyte Ratio in Older Patients with Acute Ischemic Stroke. J. Nippon Med. Sch. 2023, 90, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Rajakumar, I.; Vidya, T.A.; Ramachandran, K.; Hussain, A.; Aarthi, J.; Poovitha, M.; Madhavan, K.; Kumar, J.S. Platelet Indices as Prognostic Markers of Ischemic Stroke and Their Correlation with Lipid Profile. Clin. Neurol. Neurosurg. 2024, 237, 108119. [Google Scholar] [CrossRef] [PubMed]
- Brough, D.; Denes, A. Interleukin-1α and Brain Inflammation. IUBMB Life 2015, 67, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ren, W.; Wu, Q.; Liu, T.; Wei, Y.; Ding, J.; Zhou, C.; Xu, H.; Yang, S. NLRP3 Inflammasome Activation: A Therapeutic Target for Cerebral Ischemia–Reperfusion Injury. Front. Mol. Neurosci. 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Zietz, A.; Gorey, S.; Kelly, P.J.; Katan, M.; McCabe, J.J. Targeting Inflammation to Reduce Recurrent Stroke. Int. J. Stroke 2024, 19, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Sun, H.; Hou, S.; Zhang, W.; Liu, H.; Zhu, L.; Meng, H. Inflammatory Cytokines and Stroke and Its Subtypes: A Genetic Correlation and Two-Sample Mendelian Randomization Study. Front. Mol. Neurosci. 2023, 16. [Google Scholar] [CrossRef] [PubMed]
- Tirandi, A.; Sgura, C.; Carbone, F.; Montecucco, F.; Liberale, L. Inflammatory Biomarkers of Ischemic Stroke. Intern. Emerg. Med. 2023, 18, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liu, Q.; Wang, C.; Zhao, Y.; Jin, C.; Sun, M.; Ge, S. The Interplay between Cytokines and Stroke: A Bi-Directional Mendelian Randomization Study. Sci. Rep. 2024, 14. [Google Scholar] [CrossRef] [PubMed]
- Matys, P.; Mirończuk, A.; Starosz, A.; Grubczak, K.; Kochanowicz, J.; Kułakowska, A.; Kapica-Topczewska, K. Expanding Role of Interleukin-1 Family Cytokines in Acute Ischemic Stroke. Int. J. Mol. Sci. 2024, 25, 10515. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.-Q.; Fang, Z.; Chen, X.-L.; Yang, S.; Zhou, Y.-F.; Mao, L.; Xia, Y.-P.; Jin, H.-J.; Li, Y.-N.; You, M.-F.; et al. Microglia-Derived TNF-α Mediates Endothelial Necroptosis Aggravating Blood Brain–Barrier Disruption after Ischemic Stroke. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Petrovic-Djergovic, D.; Goonewardena, S.N.; Pinsky, D.J. Inflammatory Disequilibrium in Stroke. Circ. Res. 2016, 119, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.A.; Jickling, G.C.; Winship, I.R. Neutrophil Dynamics and Inflammaging in Acute Ischemic Stroke: A Transcriptomic Review. Front. Aging Neurosci. 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- The Importance of Selected Markers of Inflammation and Blood-Brain Barrier Damage for Short-Term Ischemic Stroke Prognosis. J. Physiol. Pharmacol. 2019. [CrossRef]
- Xu, Q.; Liu, Y.; Tian, X.; Xia, X.; Zhang, Y.; Zhang, X.; Wang, Y.; Sun, P.; Meng, X.; Wang, A. Monocyte Chemoattractant Protein-1, Inflammatory Biomarkers, and Prognosis of Patients With Ischemic Stroke or Transient Ischemic Attack: Fndings From a Nationwide Registry Study. J. Am. Heart Assoc. 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Juli, C.; Heryaman, H.; Nazir, A.; Ang, E.-T.; Defi, I.R.; Gamayani, U.; Atik, N. The Lymphocyte Depletion in Patients with Acute Ischemic Stroke Associated with Poor Neurologic Outcome. Int. J. Gen. Med. 2021, Volume 14, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Manoj, H.; Gomes, S.M.; Thimmappa, P.Y.; Nagareddy, P.R.; Jamora, C.; Joshi, M.B. Cytokine Signalling in Formation of Neutrophil Extracellular Traps: Implications for Health and Diseases. Cytokine Growth Factor Rev. 2025, 81, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, M.; Ikram, A.; Suriya, S.; Saleem, S.; Quadri, S.A.; Robinson, M.; Ortega-Gutierrez, S.; Qeadan, F.; Leira, E.; Paul, S.; et al. Cytokine Registry In Stroke Patients (CRISP): Protocol of a Prospective Observational Study. Medicine (Baltimore) 2020, 99, e20921. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pradhan, R. Prognostic Relevance of C-Reactive Protein in Short Term Adverse Outcome in Patients with Acute Ischaemic Stroke. J. Clin. Diagn. Res. 2021. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Yuan, Y.; Wang, H.; Wang, Z.; Zhang, X. Th17 Cells and IL-17A in Ischemic Stroke. Mol. Neurobiol. 2024, 61, 2411–2429. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Ma, L.; Chu, Z.; Xu, H.; Wu, W.; Liu, F. Regulation of Microglial Activation in Stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Tanase, C.; Cruceru, M.L.; Enciu, A.-M.; Popa, A.C.; Albulescu, R.; Neagu, M.; Constantinescu, S.N. Signal Transduction Molecule Patterns Indicating Potential Glioblastoma Therapy Approaches. OncoTargets Ther. 2013, 1737. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Guo, H.; Zhou, Y.; Fang, R.; Zhang, W.; Mei, Z. Neutrophil Extracellular Traps in Cerebral Ischemia/Reperfusion Injury:Friend and Foe. Curr. Neuropharmacol. 2023, 21, 2079–2096. [Google Scholar] [CrossRef] [PubMed]
- Lundin, J.I.; Peters, U.; Hu, Y.; Ammous, F.; Benjamin, E.J.; Bis, J.C.; Brody, J.A.; Cushman, M.; Fuller, H.; Gignoux, C.; et al. Epigenetic Mechanisms Underlying Variation of IL-6, a Well-Established Inflammation Biomarker and Risk Factor for Cardiovascular Disease. Atherosclerosis 2025, 407, 120219. [Google Scholar] [CrossRef] [PubMed]
- Amruta, N.; Rahman, A.A.; Pinteaux, E.; Bix, G. Neuroinflammation and Fibrosis in Stroke: The Good, the Bad and the Ugly. J. Neuroimmunol. 2020, 346, 577318. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.; Cabantan, D.; Monsour, M.; Borlongan, C.V. Neuroinflammation, Stem Cells, and Stroke. Stroke 2022, 53, 1460–1472. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Zhuang, H.; Huang, W.; Sun, J. Neuroinflammation and Energy Metabolism: A Dual Perspective on Ischemic Stroke. J. Transl. Med. 2025, 23. [Google Scholar] [CrossRef] [PubMed]
- Couch, C.; Mallah, K.; Borucki, D.M.; Bonilha, H.S.; Tomlinson, S. State of the Science in Inflammation and Stroke Recovery: A Systematic Review. Ann. Phys. Rehabil. Med. 2022, 65, 101546. [Google Scholar] [CrossRef] [PubMed]
- Monsour, M.; Croci, D.M.; Agazzi, S.; Borlongan, C.V. Contemplating IL-6, a Double-edged Sword Cytokine: Which Side to Use for Stroke Pathology? CNS Neurosci. Ther. 2023, 29, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, T.; Zhai, X.; Ma, Y.; Xie, L.; Lu, B.; Zhang, Y.; Li, Y.; Chen, Z.; Yin, J.; et al. Targeting Neutrophils as a Novel Therapeutic Strategy after Stroke. J. Cereb. Blood Flow Metab. 2021, 41, 2150–2161. [Google Scholar] [CrossRef] [PubMed]
- Prapiadou, S.; Živković, L.; Thorand, B.; George, M.J.; Van Der Laan, S.W.; Malik, R.; Herder, C.; Koenig, W.; Ueland, T.; Kleveland, O.; et al. Proteogenomic Data Integration Reveals CXCL10 as a Potentially Downstream Causal Mediator for IL-6 Signaling on Atherosclerosis. Circulation 2024, 149, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.F.; Wang, D.L.; Zhou, Y.; Xiong, H. Association between the Interleukin-6-174 G/C Polymorphism and Risk of Ischemic Stroke: A Meta-Analysis. Genet. Mol. Res. 2015, 14, 13076–13083. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, L.; Wallén, H.; Aspberg, S.; De Faire, U.; Gigante, B. IL6 Trans-Signaling Associates with Ischemic Stroke but Not with Atrial Fibrillation. BMC Neurol. 2021, 21. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vorst, E.P.C.; Döring, Y.; Weber, C. Chemokines and Their Receptors in Atherosclerosis. J. Mol. Med. 2015, 93, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Gill, D.; Rannikmäe, K.; Traylor, M.; Anderson, C.D.; MEGASTROKE consortium of the International Stroke Genetics Consortium (ISGC); Lee, J. -M.; Kamatani, Y.; Hopewell, J.C.; Worrall, B.B.; et al. Genetically Determined Levels of Circulating Cytokines and Risk of Stroke: Role of Monocyte Chemoattractant Protein-1. Circulation 2019, 139, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.J.; Lemmens, R.; Tsivgoulis, G. Inflammation and Stroke Risk: A New Target for Prevention. Stroke 2021, 52, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Yang, K.; Li, J.; Lin, J.; Jing, J.; Xiong, Y.; Zhao, X.; Wang, Y.; Liu, L.; Meng, X.; et al. Mediation Effect of Stroke Recurrence in the Association between Post-stroke Interleukin-6 and Functional Disability. CNS Neurosci. Ther. 2023, 29, 3579–3587. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, A.; Palaiopanos, K.; Björkbacka, H.; Peters, A.; De Lemos, J.A.; Seshadri, S.; Dichgans, M.; Georgakis, M.K. Circulating Interleukin-6 Levels and Incident Ischemic Stroke: A Systematic Review and Meta-Analysis of Prospective Studies. Neurology 2022, 98. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-H.; Luo, M.-Y.; Liang, N.-; Gong, S.-X.; Chen, W.; Huang, W.-Q.; Tian, Y.; Wang, A.-P. Interleukin-6: A Novel Target for Cardio-Cerebrovascular Diseases. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; deGoma, E.; Shapiro, M.D. IL-6 and Cardiovascular Risk: A Narrative Review. Curr. Atheroscler. Rep. 2024, 27. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, J.; Pan, Y.; Wang, M.; Meng, X.; Li, H.; Wang, Y.; Zhao, X.; Qin, H.; Liu, L.; et al. Interleukin-6 and YKL-40 Predicted Recurrent Stroke after Ischemic Stroke or TIA: Analysis of 6 Inflammation Biomarkers in a Prospective Cohort Study. J. Neuroinflammation 2022, 19. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Buckley, L.; Sun, C.; Al Rifai, M.; Yu, B.; Nambi, V.; Virani, S.S.; Selvin, E.; Matsushita, K.; Hoogeveen, R.C.; et al. Association of Interleukin-6 and Interleukin-18 with Cardiovascular Disease in Older Adults: Atherosclerosis Risk in Communities Study. Eur. J. Prev. Cardiol. 2023, 30, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-I.; Kim, J.-S.; Bae, H.J.; Kim, W.Y. C-Reactive Protein for Stroke Detection in the Emergency Department in Patients With Dizziness Without Neurological Deficits. Front. Neurol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Maida, C.; Arnao, V.; Corte, V.D.; Simonetta, I.; Corpora, F.; Di Bona, D.; Maugeri, R.; et al. Early High-Dosage Atorvastatin Treatment Improved Serum Immune-Inflammatory Markers and Functional Outcome in Acute Ischemic Strokes Classified as Large Artery Atherosclerotic Stroke: A Randomized Trial. Medicine (Baltimore) 2016, 95, e3186. [Google Scholar] [CrossRef] [PubMed]
- Tanase, C.; Enciu, A.M.; Codrici, E.; Popescu, I.D.; Dudau, M.; Dobri, A.M.; Pop, S.; Mihai, S.; Gheorghișan-Gălățeanu, A.-A.; Hinescu, M.E. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? Int. J. Mol. Sci. 2022, 23, 604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, H.; Wang, J.; Xi, X.; Cefalo, P.; Wood, L.J.; Luo, X.; Wang, Q.M. Multiplex Array Analysis of Serum Cytokines Offers Minimal Predictive Value for Cognitive Function in the Subacute Phase after Stroke. Front. Neurol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Meisinger, C.; Freuer, D.; Schmitz, T.; Ertl, M.; Zickler, P.; Naumann, M.; Linseisen, J. Inflammation Biomarkers in Acute Ischemic Stroke According to Different Etiologies. Eur. J. Neurol. 2024, 31. [Google Scholar] [CrossRef] [PubMed]
- Rezaeitalab, F.; Esmaeili, M.; Saberi, A.; Vahidi, Z.; Emadzadeh, M.; Rahimi, H.R.; Ramezani, N.; Mirshabani-Toloti, S.Z. Predictive Value of Inflammatory Markers for Functional Outcomes in Patients with Ischemic Stroke. Curr. J. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; Micieli, G.; Tassorelli, C.; Cuartero, M.I.; Ballesteros, I.; Certo, M.; Moro, M.A.; Lizasoain, I.; Bagetta, G. Rational Modulation of the Innate Immune System for Neuroprotection in Ischemic Stroke. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, C.; Wang, H.; Nan, S. Serum Interleukin-37 Increases in Patients after Ischemic Stroke and Is Associated with Stroke Recurrence. Oxid. Med. Cell. Longev. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhang, X.-G.; Jiang, H.-Y.; Cui, X.-K.; Zhang, D.; Yao, Z.-W.; Yue, Y.-H. An Increase in Neutrophil-to-Lymphocyte Ratio Predicts Poor Functional Outcomes in Older Patients with Acute Ischemic Stroke: A Retrospective Study. J. Integr. Neurosci. 2021, 20. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Cai, L.; Yi, T.; Yi, X.; Hu, Y. Neutrophil-to-lymphocyte Ratio Is Associated with Stroke Progression and Functional Outcome in Patients with Ischemic Stroke. Brain Behav. 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, Y.; Lv, C.; Li, F. The Prognosis of Neutrophil-to-Lymphocyte Ratio and Lymphocyte-to-Monocyte Ratio in Elderly with Acute Ischemic Stroke. Clin. Interv. Aging 2024, Volume 19, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Quan, K.; Wang, A.; Zhang, X.; Meng, X.; Chen, P.; Li, H.; Wang, Y. Neutrophil to Lymphocyte Ratio and Adverse Clinical Outcomes in Patients with Ischemic Stroke. Ann. Transl. Med. 2021, 9, 1047–1047. [Google Scholar] [CrossRef] [PubMed]
- Department of Neurology, Kirikkale University, Faculty of Medicine, Kirikkale, Turkey; Alpua, M.; Say, B.; Yardimci, I.; Ergün, U.; Kisa, U.; Ceylan, O.D. First Admission Neutrophil–Lymphocyte Ratio May Indicate Acute Prognosis of Ischemic Stroke. Rambam Maimonides Med. J. 2021, 12, e0021. [Google Scholar] [CrossRef] [PubMed]
- Memiş, Z.; Gürkaş, E.; Özdemir, A.Ö.; Acar, B.A.; Ögün, M.N.; Aytaç, E.; Akpınar, Ç.K.; Akıl, E.; Çabalar, M.; Özkul, A.; et al. Impact of Neutrophil-to-Lymphocyte Ratio on Stroke Severity and Clinical Outcome in Anterior Circulation Large Vessel Occlusion Stroke. Diagnostics 2024, 14, 2880. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, G.; Liu, F.; Zheng, J.; Jiang, Z.; Hu, S.; Shi, X.; Wang, W.; Xu, L.; Wang, Z. Association of Neutrophil-to-Lymphocyte Ratio with Stroke Morbidity and Mortality: Evidence from the NHANES 1999–2020. Front. Med. 2025, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, S.; Dai, Q.; Li, J. Neutrophil Lymphocyte Ratio Predicts Early Neurological Deterioration in Patients with Anterior Circulation Stroke. Int. J. Gen. Med. 2024, Volume 17, 5325–5331. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Heo, M.Y.; Joo, H.J.; Shim, G.Y.; Chon, J.; Chung, S.J.; Soh, Y.; Yoo, M.C. Neutrophil-to-Lymphocyte Ratio as a Predictor of Short-Term Functional Outcomes in Acute Ischemic Stroke Patients. Int. J. Environ. Res. Public. Health 2023, 20, 898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhu, T.; Li, H.; He, Y.; Zhang, Y.; Huang, N.; Zhang, G.; Li, Y.; Chang, D.; Li, X. <p>Plasma Interleukin-37 Is Elevated in Acute Ischemic Stroke Patients and Probably Associated With 3-Month Functional Prognosis </P>. Clin. Interv. Aging 2020, Volume 15, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Iordache, M.P. Reframing Functional Neurological Disorders in Modern Medicine. Cureus 2025. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Feng, X.; Shao, M.; Liu, W.; Ma, Q.; Wang, E.; Chen, J.; Shao, B. Serum Interleukin-33 as a Novel Marker for Long-term Prognosis and Recurrence in Acute Ischemic Stroke Patients. Brain Behav. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Temporal Profile of Cytokines Post-Stroke. Figure 1. (Interleukin-1 beta), IL-6 (Interleukin-6), TNF-α (Tumour Necrosis Factor-alpha), IL-8 (Interleukin-8), IL-10 (Interleukin-10), IL-17 (Interleukin-17), CRP (C-reactive protein).
Figure 1.
Temporal Profile of Cytokines Post-Stroke. Figure 1. (Interleukin-1 beta), IL-6 (Interleukin-6), TNF-α (Tumour Necrosis Factor-alpha), IL-8 (Interleukin-8), IL-10 (Interleukin-10), IL-17 (Interleukin-17), CRP (C-reactive protein).
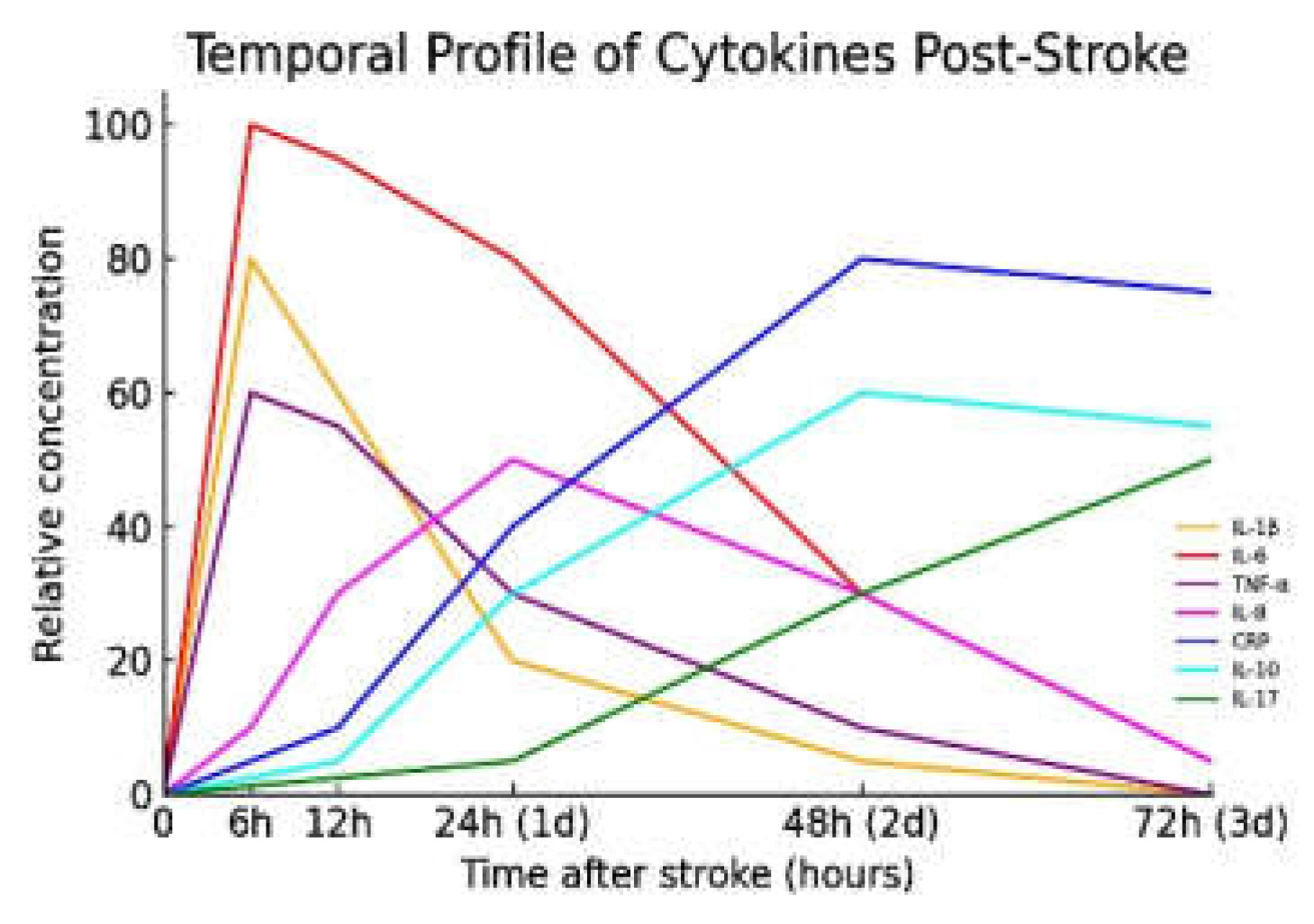
Figure 2.
Schematic illustration of the inflammatory cascade after an ischemic stroke.
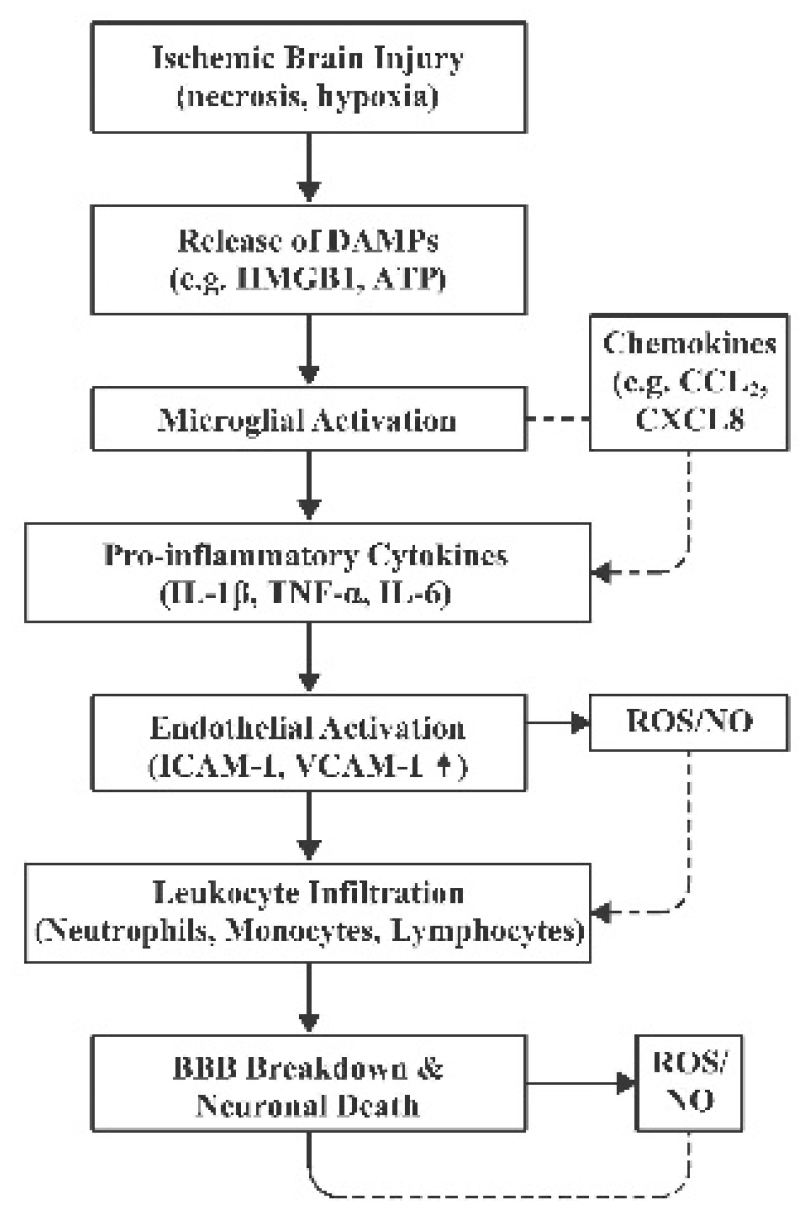
Figure 3.
Key immune cells and their interactions in the post-ischemic brain. Figure abbreviations: DAMPs (Damage-Associated Molecular Patterns), HMGB1 (High-Mobility Group Box 1), ATP (Adenosine Triphosphate), IL-1β (Interleukin-1 beta), TNF-α (Tumour Necrosis Factor-alpha), ICAM-1 (Intercellular Adhesion Molecule 1), IL-8 (Interleukin-8), ROS (Reactive Oxygen Species), MMP-9 (Matrix Metalloproteinase-9), IL-1 (Interleukin-1), IL-6 (Interleukin-6), T-lymphocytes (CD4+ helper and CD8+ cytotoxic T cells), IFN-γ (Interferon gamma), IL-17 (Interleukin-17).
Figure 3.
Key immune cells and their interactions in the post-ischemic brain. Figure abbreviations: DAMPs (Damage-Associated Molecular Patterns), HMGB1 (High-Mobility Group Box 1), ATP (Adenosine Triphosphate), IL-1β (Interleukin-1 beta), TNF-α (Tumour Necrosis Factor-alpha), ICAM-1 (Intercellular Adhesion Molecule 1), IL-8 (Interleukin-8), ROS (Reactive Oxygen Species), MMP-9 (Matrix Metalloproteinase-9), IL-1 (Interleukin-1), IL-6 (Interleukin-6), T-lymphocytes (CD4+ helper and CD8+ cytotoxic T cells), IFN-γ (Interferon gamma), IL-17 (Interleukin-17).
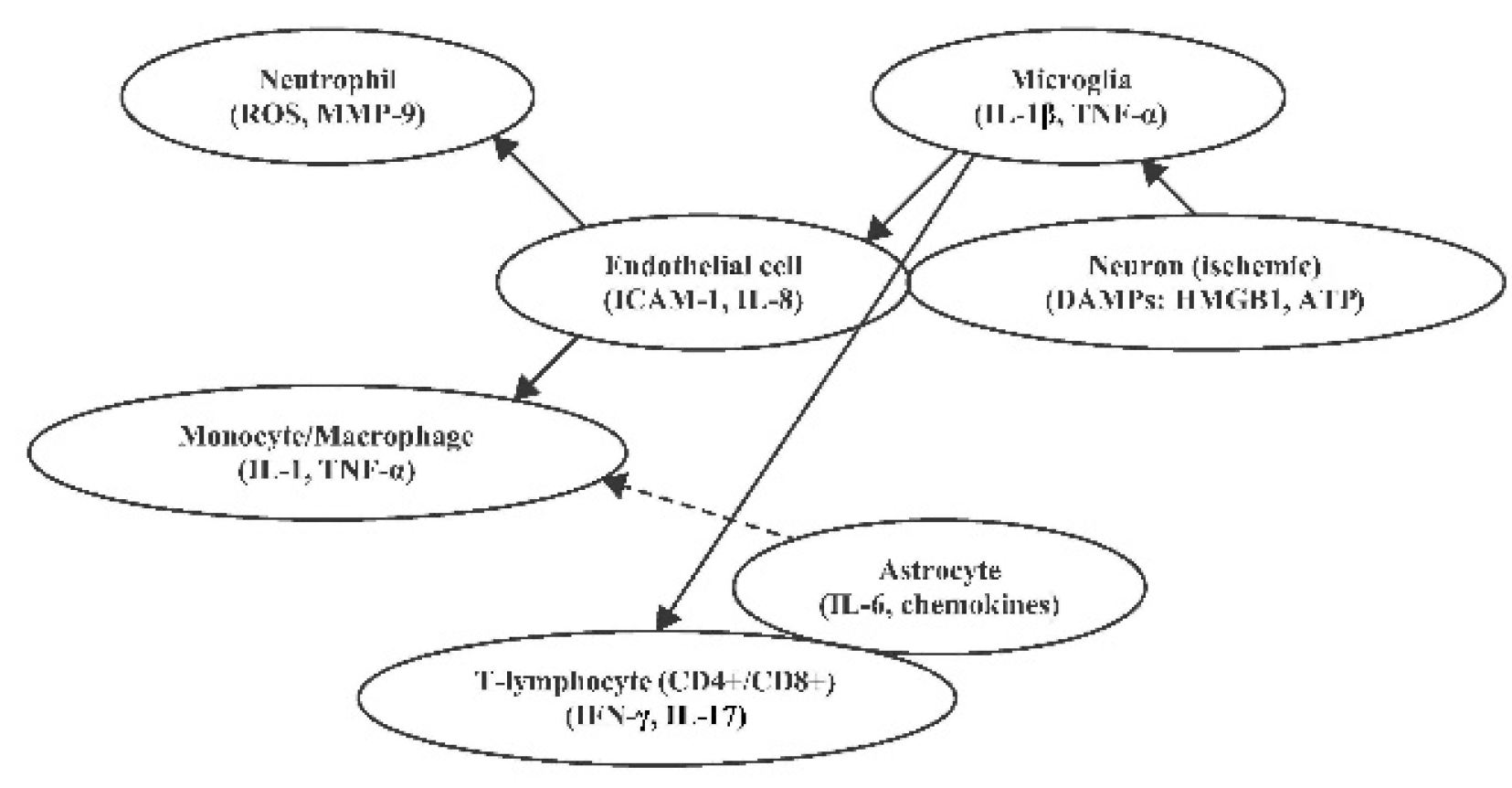
Table 1.
illustrates the main pro- and anti-inflammatory molecules involved in the pathophysiology of ischemic strokes, their main functions, timing and clinical and prognostic implications.
Table 1.
illustrates the main pro- and anti-inflammatory molecules involved in the pathophysiology of ischemic strokes, their main functions, timing and clinical and prognostic implications.
| Cytokine / Interleukin | Classification | Main Functions | Clinical / Prognostic Implications |
Timing of Elevation Post-Stroke |
|---|---|---|---|---|
| Interleukin-1β (IL-1β) | Pro-inflammatory | Initiates inflammatory cascade; activates microglia and endothelial cells | Elevated levels associated with larger infarct size and worse neurological outcomes | Peaks within 6–24 hours |
| Interleukin-6 (IL-6) | Pro-inflammatory | Modulates acute phase response; promotes leukocyte recruitment | High levels correlate with increased stroke severity and poor functional outcomes | Rises within hours; remains elevated for days |
| Tumour Necrosis Factor-α (TNF-α) | Pro-inflammatory | Promotes apoptosis; disrupts blood-brain barrier | Elevated levels linked to increased infarct size and neurological deterioration | Peaks within 6–24 hours |
| Interleukin-10 (IL-10) | Anti-inflammatory | Suppresses pro-inflammatory cytokine production; limits immune response | Higher levels associated with reduced infarct size and better outcomes; excessive levels may increase infection risk | Increases within 24–72 hours |
| Transforming Growth Factor-β (TGF-β) | Anti-inflammatory/ Regulatory |
Regulates immune response; promotes tissue repair | Contributes to neuroprotection and recovery; exact prognostic value in stroke is under investigation | Elevation timing varies; involved in later stages |
| Interleukin-17 (IL-17) | Pro-inflammatory | Recruits neutrophils; amplifies inflammatory response | Elevated levels may exacerbate neuroinflammation and tissue damage | Peaks within 24–72 hours |
| Interleukin-8 (IL-8) | Pro-inflammatory | Attracts neutrophils; promotes inflammation | Higher levels associated with increased infarct volume and poorer outcomes | Rises early; peaks within 24 hours |
| Interleukin-1α (IL-1α) | Pro-inflammatory | Acts as an alarmin; initiates sterile inflammation | Early release may contribute to initial inflammatory response; specific prognostic implications in stroke require further study | Released immediately upon cell injury |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



