Submitted:
09 July 2025
Posted:
10 July 2025
You are already at the latest version
Abstract
This review examines ALN-APP, a small interfering RNA (siRNA) formulated for intrathecal injection, intended to reduce the formation of beta-amyloid precursor protein (APP), a critical factor in the pathology of Alzheimer’s disease (AD). ALN-APP incorporates a 16-carbon chain (C16-siRNA) to enhance its delivery to the central nervous system (CNS) while integrating advancements in specificity and duration of action based on previous FDA-approved drugs. The development of ALN-APP involved a thorough analysis of the optimal carbon chain length and its conjugation position to the siRNA. Preclinical studies conducted in male Sprague Dawley rats, mice, and non-human primates demonstrated the efficacy of ALN-APP. In rats, intrathecal (IT) injection of C16-siRNAs at a concentration of 30 mg/mL, delivering a dose of 0.9 mg, resulted in cranial distribution via cerebrospinal fluid and led to a 75% reduction in copper-zinc superoxide dismutase 1 (SOD1) mRNA levels. These effects were dose-dependent and persisted for over three months across multiple brain regions. Furthermore, studies in non-human primates indicated reductions in soluble APP levels (sAPPα and sAPPβ) to below 25% sustained for two months. In the CVN mouse model of AD, administration of 120 µg of siRNA via the intracerebroventricular route produced reductions in APP expression, with mRNA levels remaining suppressed for 60 days in the ventral cortex. Following these promising results in animal models, ALN-APP advanced to a Phase 1 trial, designated ALN-APP-001, assessing its safety and efficacy in 12 participants with early-onset Alzheimer's disease (EOAD). Initial findings revealed a 55% reduction in soluble APPα and a 69% reduction in APPβ by day 15. In a subsequent cohort of 36 patients, the administration of the 75 mg dose via IT injection produced mean reductions of 61.3% in sAPPα and 73.5% in sAPPβ after one month. These silencing effects persisted over a six-month period and were associated with important decreases in Aβ42 and Aβ40 levels. These results highlight ALN-APP's potential to address Alzheimer’s pathology while maintaining a favorable safety profile. Collectively, advancements in ALN-APP represent a promising strategy to reduce beta-amyloid formation in AD, with significant biomarker reductions suggesting potential disease-modifying effects. Continued development may pave the way for innovative treatments for neurodegenerative diseases.
Keywords:
gene therapy
; RNA interference
; β-amyloid
; Alzheimer’s disease
; C16-siRNA
1. Introduction
Dementia has increasingly become a focal point in scientific research and public health initiatives. This attention is driven by the condition’s rising prevalence among the aging global population, as well as its profound impact on both affected individuals and their families [1]. Currently, approximately 50 million individuals worldwide are diagnosed with dementia, a figure projected to triple by 2050 [2]. Alzheimer’s disease (AD) is the most common form of dementia globally. Despite considerable progress in understanding the brain pathology associated with AD, treating this complex disorder remains challenging [1,3].
The incidence of AD is influenced by non-modifiable factors such as advancing age, female sex, lower educational attainment, and genetic predisposition, particularly the presence of the APOE (Apolipoprotein E) ε4 allele. Modifiable factors include cardiovascular conditions (hypertension, diabetes, hypercholesterolemia), lifestyle elements (Mediterranean diet, physical activity, cognitive engagement), and environmental exposures (e.g., air pollution). Depression, obesity, subjective cognitive decline, neurodegenerative markers on imaging, and co-pathologies also contribute to AD risk [4,5,6,7,8,9,10,11,12].
The pathological features of AD encompass the accumulation of β-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs) composed of tau protein within the brain. Additional changes of note include an increase in reactive oxygen species, the proliferation of glial cells, impaired insulin sensitivity, and modifications in the microbiome [13]. These characteristics have been extensively studied due to their critical importance for both diagnostic and potential therapeutic applications [14]. Amyloid plaques are extracellular deposits of Aβ peptides that follow a predictable regional progression, starting in the neocortex and advancing to other brain areas [15,16,17,18,19].
Under normal physiological conditions, Aβ exists in a balance between its production and clearance. Several mechanisms aid in clearing Aβ from the brain, including non-enzymatic pathways, such as its transport across blood vessel walls into the bloodstream, and enzymatic pathways involving neprilysin and insulin-degrading enzymes [20,21]. Cerebral Aβ is transported across the blood-brain barrier through scavenger receptors such as lipoprotein receptor-related protein 1 (LRP1) and very low-density lipoprotein receptor (VLDLR), which facilitate its clearance into the bloodstream in the human brain. Sequester proteins have been shown to enhance the binding affinity of Aβ to these scavenger receptors [22], stabilizing monomeric Aβ and inhibiting its aggregation [23]. Aβ is primarily present in two forms: Aβ40 and Aβ42. The main difference between them lies in the two extra residues at the C-terminus in the Aβ42, which alter its metabolism, physiological functions, toxicities and aggregation mechanism [24,25]. Among these forms, Aβ42 is more toxic and more strongly associated with the development of AD-related pathologies compared to Aβ40 [22]. Growing evidence establishes that Aβ42 begin to accumulate in the brain many years, or even decades, before the clinical symptoms of AD appear [26].
Currently, the treatment landscape for AD is limited, with only two major classes of drugs available: cholinesterase inhibitors and N-methyl D-aspartate (NMDA) antagonists. These medications primarily provide symptomatic relief and do not cure or prevent the disease. Furthermore, they often come with undesirable side effects, highlighting the significant limitations of current therapies [27,28,29]. Monoclonal antibodies (mAbs) have recently gained attention as potential disease-modifying therapies for AD. The Food and Drug Administration (FDA) has approved mAbs like aducanumab and lecanemab, which show promise in their ability to potentially modify disease progression. However, the long-term efficacy and safety of these treatments remain uncertainties that require further investigation [30,31,32].
Given the existing limitations of current therapies, including their focus on symptom management and associated side effects, there is a pressing need to explore new treatment strategies and targets. This ongoing search for safer and more effective interventions is critical to improving the outcomes for individuals affected by AD.
2. siRNAs – Concept and Mechanism of Action
Small interfering RNAs (siRNAs) are short, double-stranded RNA molecules that play a crucial role in the regulation of gene expression through a process known as RNA interference (RNAi). These molecules, composed of 19 to 23 ribonucleotides, exhibit specificity in binding to messenger RNA (mRNA) via Watson-Crick base pairing [33].
The discovery of siRNAs and the elucidation of the RNAi process can be traced back to the groundbreaking research conducted by Andrew Fire and Craig Mello, who were awarded the Nobel Prize for their work in this area. They characterized the gene-silencing effects of short RNAs and introduced the term “RNA interference” in their seminal publication [34]. The mechanisms governing RNAi-mediated gene silencing are illustrated in Figure 1 and have been reviewed extensively in the literature [35,36,37,38,39,40].
Inside the cell, siRNA duplexes are first processed by enzymes that remove the sense strand, after which they are incorporated into a protein complex known as the RNA-induced silencing complex (RISC). Upon entry, RISC first removes one strand of the siRNA duplex, referred to as the passenger strand, thereby retaining the guide strand. This guide strand subsequently searches for complementary nucleotides in target mRNAs through a trial-and-error mechanism, eventually forming stable hydrogen bonds with matching sequences [41,42]. Once bound, the RISC complex, assisted by the guide strand, executes cleavage of the targeted mRNA [43,44].
siRNAs have successfully transitioned into clinical settings and are now recognized as U.S. Food and Drug Administration (FDA)-approved therapeutics. Patisiran, for instance, was the first siRNA therapeutic to complete all phases of clinical development successfully and receive regulatory approval for market entry. This drug operates as a short RNA molecule that selectively binds to and induces the cleavage of target mRNA through the RNAi pathway (Figure 2), a process facilitated by RNAi-associated proteins [45,46,47].
Patisiran is indicated for the treatment of hereditary transthyretin-mediated (hATTR) amyloidosis [47,48], and it represents a leading example among ten RNAi-based therapeutics that entered Phase II–III clinical trials beginning in 2017, ultimately receiving approval from the FDA in the following year [35,49]. This innovative class of therapeutics represents a substantial advancement in pharmacology, introducing a unique mechanism of action based on sequence-specific gene silencing at the post-transcriptional level [35].
3. Reaching the Liver: The First Two FDA-Approved siRNAs, Patisiran and Givosiran
The therapeutic efficacy of patisiran heavily relies on its lipid nanoparticle (LNP) delivery system, which facilitates targeted delivery to hepatocytes (Figure 3). The LNPs used for patisiran are coated with ApoE, which mediates opsonization of the nanoparticles. This process allows the particles to traverse the fenestrated hepatic endothelium and bind specifically to ApoE receptors on the surface of hepatocytes. After receptor-mediated endocytosis, the LNPs are internalized, and the ionizable lipids within the particles are protonated in the acidic endosomal environment. This protonation induces fusion of the LNP membrane with the endosomal membrane, destabilizing the lipid structure and leading to the release of free siRNAs into the cytoplasm [52,53].
Once in the cytoplasm, the siRNAs are incorporated into the RISC, where the antisense (guide) strand directs the complex to its complementary sequence on the TTR mRNA. This interaction triggers RNAi-mediated degradation of the TTR transcript (Figure 3F). The specificity of this process is determined by the base-pairing between the siRNA guide strand and its target mRNA. For instance, the guide strand of patisiran, with the sequence 3′-CAUUGGUUCUCAUAAGGUA-5′, exhibits perfect complementarity to the TTR mRNA sequence 5′-GUAACCAAGAGUAUUCCAU-3′, as shown in Figure 2C. Notably, the 3′ terminal triplet "CAU" of the siRNA pairs precisely with the 5′ triplet "GUA" of the TTR mRNA, underscoring the critical importance of sequence complementarity for efficient RNAi-based gene silencing [54,55,56].
Givosiran was the second FDA-approved siRNA and brought two significant improvements compared to patisiran: the absence of a nanoparticle for delivery and a different route of administration [51]. First, the company identified a ligand capable of vectorizing siRNAs to hepatocytes without the LNP previously used in patisiran, illustrating their commitment to site-specific delivery [57]. Consequently, a novel hepatic-targeting strategy was developed by conjugating N-acetylgalactosamine (GalNAc) to the siRNA passenger strand (Figure 4). GalNAc specifically binds to the asialoglycoprotein receptor (ASGPR) on hepatocytes, facilitating efficient siRNA uptake and gene silencing without the need for nanoparticle-based delivery systems (Figure 4C). Gene knockdown was observed in a dose-dependent manner [57,58,59].
Upon internalization by hepatocytes, the siRNA duplex engages the Dicer/TRBP complex, which processes the duplex and loads the antisense strand into the RISC. Within RISC, the guide strand directs the complex to the complementary ALAS1 mRNA sequence, leading to its degradation through the RNAi pathway. This reduction in ALAS1 enzyme levels results in decreased synthesis of the neurotoxic heme intermediates δ-aminolevulinic acid (ALA) and porphobilinogen (PBG), addressing a central pathological mechanism in acute hepatic porphyria.
Second, a key advancement of the GalNAc-conjugated givosiran formulation is the transition from intravenous administration, which previously required medical supervision, to subcutaneous injection. This modification significantly enhances patient convenience and allows for a more user-friendly administration regimen at home [58,59].
4. C16-siRNAs
The therapy for chronic-degenerative diseases of the central nervous system (CNS), whether stemming from genetic dysfunction or other origins, remains a significant challenge in the field of medicine [28,60]. Many of these diseases are characterized by the accumulation of pathological proteins that lead to neuronal death, as observed in conditions such as Parkinson's and Alzheimer's disease, with alpha-synuclein and beta-amyloid peptides being notable examples, respectively [27,29,61]. In Parkinson's disease, despite the majority of cases being idiopathic, a percentage can be attributed to genetic mutations, including the A53T mutation, which involves the substitution of the amino acid alanine with threonine at position 53 [62]. This mutation promotes the formation of aggregates of the protein, resulting in neuronal damage. In genetic disorders that cause neurological impairments, such as de novo DEAF1 (deformed epidermal autoregulatory factor-1), specific mutations compromise the function of this protein, which serves as a zinc-finger to regulate the activity of promoter regions, thereby adversely affecting the expression of various cerebral genes [63,64,65].
Given these examples, the strategy of silencing pathogenic proteins through RNAi has emerged as a promising alternative. This approach became more tangible in 2018 when the first siRNA, patisiran, was approved by the FDA, as mentioned earlier. However, the utilization of siRNAs for the treatment of diseases posed significant challenges, particularly regarding the delivery to the brain, due to the blood-brain barrier and the inherent difficulty of transfecting neuronal cells. While siRNA therapies have proven effective in treating hepatic diseases using vectorization molecules, as seen in patisiran and givosiran, the brain poses a considerably greater challenge [66,67]. This led to skepticism regarding the progress of gene therapy in the CNS, where many diseases manifest [68]. Thus, the development of an effective strategy to overcome these obstacles is commendable [69,70,71].
The seminal study conducted by Brown et al., published in 2022, provided compelling evidence that the attachment of a hexadecyl lipid at the 2’ position of the ribose in siRNA molecules significantly enhances their biodistribution across various regions of the brain [72]. These meticulously engineered oligonucleotides, referred to as 2'-O-Hexadecyl (C16)-siRNA Conjugates, are administered through the intrathecal route, paving the way for siRNA-based therapeutic interventions for neurological disorders. Furthermore, C16-siRNAs demonstrate notable biodistribution in ocular and pulmonary tissues, thereby expanding the potential for addressing diseases affecting these important organs.
The research began over a decade before the publication of the article. In 2019, Alnylam Pharmaceuticals, Inc. was granted a patent titled "Extrahepatic Delivery" (WO 2019/217459 A1). The abstract of the patent states: ‘The invention relates to a method of gene silencing, comprising administering to a cell or a subject in need thereof a therapeutically effective amount of lipophilic moieties-conjugated double-stranded iRNAs at one or more internal positions on at least one strand, optionally via a linker or carrier.’ The term “therapeutically effective amount” holds particular importance in pharmacology, as it aligns with the widely recognized notion that a drug will produce the desired therapeutic effect when it reaches an adequate concentration in the target organ or biophase. This concept is thoroughly discussed in our pharmacology textbook, Goodman & Gilman’s The Pharmacological Basis of Therapeutics.
Vasant Jadhav and Martin Maier were the corresponding authors of the cited article that warrants consideration for the cover of Nature Biotechnology journal [72]. The proposed design features a detailed 3D model that illustrates the interaction between the C16-conjugated siRNA (depicted in orange), the RISC (shown in white), and the mRNA (represented in green) involved in the process of gene silencing. This model was constructed by Erin Dewalt from Alnylam Pharmaceuticals. At the top of the cover, the phrase "Extrahepatic Delivery of RNA Therapeutics" would prominently headline the visual representation (https://www.nature.com/nbt/volumes/40/issues/10).
The complete history of C16 development was told by the authors that measures in two decades the time needed for scientific basic studies until testing the clinical potential of a siRNA-based drug [73]. The previous experiences with other siRNAs make a significant contribution. The development of siRNAs and their efficient in vivo delivery to target cells are of paramount importance. For liver-targeted delivery, this was achieved either by encapsulating siRNAs within LNPs or by directly conjugating stable siRNAs to a targeting ligand that includes GalNAc, recognized by the ASGPR present on liver hepatocytes, as previously discussed [51,56].
The authors referenced several studies that have investigated lipophilic compounds for conjugation with siRNAs, including cholesterol, aiming to achieve robust activity in biological systems, such as cell culture and animal model assays [74,75,76]. In this context, researchers have explored alternative molecules that could enhance the ability of siRNAs to cross the lipid bilayer of cells, thereby improving their lipophilicity and overall delivery efficiency. Importantly, these compounds are designed to be non-toxic to the organism. Vasant Jadhav and Martin Maier noted that the first successful outcome of an siRNA conjugated with a lipophilic compound was achieved in 2018 after intrathecal injection in rats. Following this breakthrough, a series of assays were conducted to optimize the performance of this siRNA, with the objective of developing a product suitable for clinical application. This section will first provide an overview of siRNA itself, followed by a discussion of the experimental results that demonstrate its enhanced distribution, particularly within the brain [72].
4.1. Innovations in the Design of Brain-Delivered C16-siRNAs: A Functional Perspective
C16 conjugates are characterized by the 2’-O-hexadecyl (C16) modification, which involves the covalent attachment of a short fatty acid chain to siRNA molecule. This modification is illustrated in Figure 5 as a blue tail, identified as number 1 within the yellow-filled circle. The hydrophobic nature of this modification significantly enhances the lipophilicity of the siRNA, thereby improving its interaction with cellular membranes and associated membrane proteins. This interaction is illustrated by the blue C16 tail, which is connected to schematic representations of membrane proteins (number 2). Such interactions are crucial for the effective uptake of siRNA across diverse cell types, including those found in the CNS, as well as in lung and ocular tissues. The diagram also illustrates the mechanism of cellular uptake, which involves the internalization of siRNA into endosomes, followed by the critical process of endosomal escape (numbers 3 and 4). This endosomal escape is essential for allowing siRNA to exit the endosome and freely access the cytosol (number 5), where it can initiate its silencing effects, as previously described.
In Figure 6, we depict the interaction between C16 and siRNA from both a chemical perspective and through spatial modeling, as outlined by Vasant Jadhav and Martin Maier [73]. The schematic representation illustrates the interaction between the oxygen atom located at the 2′ position of the adenine ribose in the sense strand of siRNA and the 16-carbon chain (2′-O-C16). This interaction is highlighted on the left by a dashed red circle and is labeled as number 1 within a yellow-filled circle. In the middle, the C16 chain, which consists of 16 carbon atoms, is identified as number 2. On the right, the figure presents a spatial representation of the siRNA, where the C16 molecule is depicted as a protruding gray structure (labelled as number 3). It is crucial to acknowledge that the lipophilic C16 molecule resembles a tail, functioning effectively as an anchor with significant spatial implications. While its size is relatively modest compared to the overall siRNA molecule, Brown et al. (2022) undertook a thorough investigation of the potential effects of C16 incorporation and determined the optimal positioning for its integration, as detailed below. The spatial conformation of siRNA is pivotal for its recognition by the RISC and the subsequent initiation of RNAi-mediated gene silencing [72].
The authors conducted a series of experimental tests to optimize the design of C16-siRNAs, beginning with the most critical component: the lipophilic moiety. The C16, a relatively large molecule consisting of 16 carbon atoms, was conjugated to a small double-stranded RNA with 21 nucleotides (siRNA moiety) to enhance biodistribution across CNS regions. This modification was meticulously considered to ensure that it did not interfere with silencing efficacy, which depends on the incorporation and activation of the RISC, as previously outlined.
To achieve a carbon chain length of 16, this study investigated the in vivo efficacy of siRNAs targeting the enzyme copper-zinc superoxide dismutase 1 (SOD1), composed of 10, 12, 14, 16, and 18 carbon atoms [72]. The effects of RNAi were assessed in the brain and spinal cord tissues of rats two weeks after intrathecal administration of 0.9 mg of siRNA (n=4 per group). Furthermore, the research explored the optimal positioning for the attachment of the C16 tail by evaluating the silencing efficiency of siRNA attachments at various nucleotide positions on the sense (1-21) and antisense (2-23) strands in cell culture. In rat tissues, the evaluated sense strands included nucleotides 1, 2, 5, 6, 7, 10, 11, 16, 17, and 21, while the antisense strands comprised nucleotides 5, 15, and 16, with assessments conducted on day 28. The most effective RNAi silencing was observed at nucleotide number 6 of the sense strand. At this point, the research successfully identified the optimal lipophilic chain length of 16 carbon atoms, as well as the advantageous attachment position at the 6th nucleotide of sense strand of the siRNAs.
The double-stranded C16-siRNA structure, composed of 21 nucleotides, was designated as compound number XVIII, as outlined in Supplementary Table S1 of the original article [72]. Figure 7 illustrates the key components, highlighting the central feature: the sixteen carbon atoms of the lipophilic moiety, referred to as C16, which are represented by 16 blue filled circles. A double-headed orange arrow indicates the conjugation of the 2′-O-C16 ligand to the sixth nucleotide from the 5’ end of the sense strand, N6, which is shown as the adenine nucleotide (also depicted in blue). Upper-case and lower-case letters in the diagram signify the modifications of the ribosugar: upper-case for 2′-deoxy-2′-fluoro (2′-F) and lower-case for 2′-O-methyl (2′-OMe). Additionally, the underlined uppercase letters, such as the letter "A" found in the antisense strand, signify a specific modification of glycol nucleic acid (GNA). The symbol (•) denotes phosphorothioate (PS) linkages, while "VP" indicates the presence of 5'-(E)-vinylphosphonate [72].
The assessment of these additional enhancements aimed to enhance siRNA efficacy and included a series of chemical modifications that were gradually integrated into the original siRNAs developed by the manufacturer, such as patisiran and givosiran [51,56]. Notably, one of the primary modifications involved substituting certain phosphodiester linkages with phosphorothioate (PS) linkages. This alteration was specifically aimed at decreasing the susceptibility of siRNA to enzymatic degradation, particularly by ribonucleases, thereby improving the overall stability of the siRNA molecules. These structural changes are critical for increasing the therapeutic potential of siRNAs by ensuring longer-lasting effects in biological systems [78].
The incorporation of 2′-deoxy-2′-fluoro and 2′-O-methyl modifications in C16-siRNAs is a feature previously utilized in other siRNAs developed by Alnylam, resulting in enhanced potency, as reported in the literature [79,80,81,82,83,84]. Additionally, the C16-siRNA contains glycol nucleic acid (GNA) in the antisense seed region, which contributes to increased specificity in vivo [85,86]. Furthermore, the authors incorporated a vinylphosphonate at the 5' end of the antisense strand. This stable phosphate mimic enhances the efficacy of siRNAs in vivo by improving loading onto the RISC, ultimately increasing the potency of RNA interference (RNAi) in cells [87,88]. SiRNAs engineered with both C16 and the 5'-vinylphosphonate modifications demonstrated increased RNAi activity compared to the individual components, achieving 75% and 95% silencing of SOD1 rat mRNA in the brain and spinal cord on day 28, respectively, following intrathecal administration of 0.9 mg.
An elegantly detailed image of the work was produced using immunohistochemistry (IHC) with an in-house anti-siRNA rabbit polyclonal antibody, which was subsequently detected with an anti-rabbit HRP secondary antibody. This analysis revealed a notable penetration of siRNAs conjugated with C16 on the right, in contrast to the unconjugated on the left (Figure 8). C6-siRNAs demonstrated a notable biodistribution throughout the rat brain following intrathecal injection, effectively reaching the cortex and hippocampus − regions critically involved in cognition and memory, which are negatively impacted in Alzheimer’s disease, the target condition for the current C16-siRNA therapeutics. Additionally, the signal was observed in the olfactory bulb and brainstem. Interestingly, C16-siRNAs exhibited limited penetration into the striatum, an area of considerable relevance in other neurodegenerative disorders, such as Parkinson's disease.
During the progression of neurodegenerative disorders such as AD, various cell types are involved, including neurons and glial cells associated with neuroinflammation, particularly astrocytes and microglia [89,90]. Thus, the cellular uptake of siRNAs across these different cell types is crucial for the therapeutic outcome. To investigate this aspect, the authors conducted immunohistochemistry assays using siRNAs targeted to mRNAs uniquely expressed in the studied cell types: neurons, astrocytes, microglia, oligodendrocytes, and perivascular/endothelial macrophages. The target proteins included MAP2, GFAP, Iba1, MBP, and PECAM1, respectively. The results demonstrated successful uptake of the siRNAs in neurons, astrocytes, and microglia. In contrast, no signals were detected in oligodendrocytes.
It is important to clarify the route of administration for this innovative siRNA delivered to CNS: intrathecal (IT) injection. IT injection was performed in male Sprague Dawley rats. The C16-siRNAs were initially diluted in artificial cerebrospinal fluid (aCSF) at a concentration of 30 mg/mL, and a volume of 30 μL was administered. Consequently, each animal received an administered dose of 0.9 mg. The IT injection was executed by puncturing the lumbar region of the spinal cord between the L3 and L5 vertebrae (Figure 9). Notably, there was cranial distribution of the siRNAs, presumably via the CSF, from the lumbar injection site to other spinal regions as well as to brain areas, as evidenced by qPCR results. Three regions of the spinal cord were assessed: lumbar, thoracic, and cervical, in addition to the cerebellum and brainstem. Gene expression assays via RT-qPCR were consistent with the biodistribution results obtained from immunohistochemistry in the aforementioned areas. A marked silencing of the targets under investigation was observed following IT injection of 0.9 mg siRNA in neurons (MAP2 C16-siRNA at 28 days), astrocytes (GFAP C16-siRNA at 14 days), and microglia (Iba1 C16-siRNA at 32 days) (n=3-6 animals per group).
Visual analysis of the bar graph indicates a greater intensity of gene silencing in the spinal cord segments, particularly in regions closer to the intrathecal injection site, compared to the cerebellum and brainstem, specifically for GFAP, Iba1, and MAP2. The graphs demonstrate the effects of gene silencing, with GFAP, Iba1, and MAP2 levels reduced to less than 50% in all spinal cord segments. In the cerebellum, reductions below 50% were observed for GFAP and MAP2, indicating effects on astrocytes and neurons, but not on microglia. In the brainstem, similar reductions were noted for GFAP and Iba1, though not for MAP2. As statistical analyses of the experiments were not provided, it is prudent to consider that the observed differences may not have reached statistical significance. However, it can be inferred that the efficacy of gene silencing following IT injection in the L3-L5 lumbar region was influenced by the distance from the injection site and the specific neuronal or glial cell type present in the spinal cord, cerebellum, and brainstem. As anticipated, silencing effects were minimal in oligodendrocytes and vascular/perivascular macrophages, suggesting that other neuropathologies involving these specific cell types may not benefit from treatment with C16-siRNAs.
This anatomical aspect represents a limitation of the IT administration route in the lumbar region, particularly considering the necessity for siRNA to reach more cranial areas of the CNS, such as the cerebral cortex and hippocampus, which are affected in AD. However, the targets examined (GFAP, Iba1, and MAP2) were utilized more to characterize the penetration of siRNAs into different cell types rather than to present a pattern of physiological gene expression within the brain. In this context, the authors aim to conduct further tests utilizing the mRNA of the enzyme SOD1 as the target. SOD1 is an intracellular enzyme with antioxidant properties, thereby regulating baseline levels of oxidative stress in healthy individuals, while also playing a role in mechanisms underlying various neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and Parkinson's disease [91]. The genetic structure of SOD1 and its gene expression regulation are well characterized, making it a widely expressed gene within the nervous system and suitable for assessing the efficacy of silencing systems such as those described in this study [92,93].
The study conducted two significant pharmacological analyses: a dose-response curve and a temporal analysis regarding the duration of siRNA silencing, initiated after a single IT administration of 0.9 mg [72]. The siRNAs reduced the expression of SOD1 mRNA to below 50% for all three tested doses (0.07 mg, 0.3 mg, and 0.9 mg) at day 28, across the thoracic spinal cord, cerebellum, and frontal cortex. Notably, only the experimental group that received the lowest dose of 0.07 mg exhibited a minimal reduction in the frontal cortex.
The silencing effect was found to be dose-dependent, with the highest dose of 0.9 mg reducing SOD1 mRNA to 25% of the control value in all three regions assessed. This dose also produced a durable effect across various brain regions, with a silencing effect exceeding 50% that persisted for over three months in most regions analyzed, including all spinal cord segments. The silencing effects followed an expected gradient, being most pronounced near the injection site (lumbar), followed by thoracic and cervical regions. Additionally, effects were observed in the cerebellum, frontal cortex, temporal cortex, and hippocampus.
In both the spinal cord and frontal cortex, the RNAi effect remained above 50% after five months post-IT injection. The authors also tested the administration of a reduced dose of one-third of the original concentration (0.3 mg), given monthly for five months, which yielded positive results.
Assays conducted with non-human primates (NHPs) confirmed the findings obtained from rat studies. In this phase of the research, cynomolgus monkeys were administered 60 mg of IT siRNAs. The results indicated that the efficacy of siRNAs featuring both the C16 tail and VP modifications was higher than that of the isolated use of these modifications in most brain regions. Similar to the rat studies, NHPs also exhibited limited penetration of siRNAs in the striatum. An important finding in rats and NHP was the absence of RNAi effects on organs outside the CNS, liver and kidney. This indicates that the C16-siRNAs administered via the IT route maintain a target-specific distribution. Notably, the siRNAs reduced the CSF levels of soluble APPα (sAPPα) and soluble APPβ (sAPPβ) to below 25% for up to two months, and subsequently to below 50% for five months.
The authors subsequently conducted studies using the CVN mouse model of Alzheimer's disease, administering siRNA via the intracerebroventricular (ICV) route at a dose of 60 μg. The siRNA treatment resulted in a significant reduction of amyloid precursor protein (APP) expression at 30 days post-administration, affecting both mRNA and peptide levels. Notably, the effect on APP mRNA in animals treated with 120 μg persisted for 60 days in the ventral cortex and was also observed in other brain regions over extended time points. Additionally, behavioral assessments indicated changes in total distance traveled and rearing frequency during the open-field test in the siRNA-treated animals.
Based on the positive pharmacological results from preclinical testing of C16-siRNAs in rodents and non-human primates, which demonstrated their potential for durably lowering amyloid precursor mRNAs and protein levels, the compound has progressed to a Phase 1 clinical trial involving patients with early-onset Alzheimer’s disease (EOAD).
5. Phase 1 Clinical Trial – NCT05231785: Experimental Design and Preliminary Results
The Phase 1 clinical study (ALN-APP-001; NCT05231785) is a multi-center, randomized, double-blind, placebo-controlled trial designed to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of intrathecally administered ALN-APP in patients with EOAD. The trial is structured in two parts: Part A: Single ascending dose (SAD); Part B: Multiple ascending dose (MAD) [94].
ALN-APP utilizes Alnylam’s proprietary C16-siRNA conjugate technology to facilitate enhanced delivery into CNS cells via intrathecal administration. The prolonged PD effects suggest sustained CNS exposure and long tissue half-life, although full PK profiles are still under analysis. The intrathecal route allows direct access to the CSF, maximizing CNS bioavailability while minimizing systemic exposure.
Eligible participants were adults with symptom onset before the age of 65 and a Mini-Mental State Examination (MMSE) score greater than 20 [95]. All participants had confirmed amyloid pathology, either through CSF analysis or positron emission tomography (PET) imaging. They were at the stage of mild cognitive impairment (MCI) or mild dementia, representing early-stage AD in a young-onset population. Unlike traditional Phase 1 trials conducted in healthy individuals, this study was designed to evaluate safety and preliminary efficacy in patients who may potentially benefit from the reduction or suppression of amyloid production [96].
In the initial patient cohort, 12 participants were enrolled and randomized in a 2:1 ratio to receive either ALN-APP or placebo within the 25 mg and 75 mg dose groups. ALN-APP demonstrated robust target engagement following a single IT administration. Dose-dependent reductions in CS levels of soluble APPα and APPβ (sAPPα and sAPPβ) were observed on day 15 post-administration. Mean reductions from baseline were 55% for sAPPα and 69% for sAPPβ, with maximal reductions of 71% and 83%, respectively, observed in the 75 mg cohort (n = 4) [95].
ALN-APP also demonstrated a favorable safety profile in this initial cohort. All reported adverse events (AEs) were mild to moderate in severity. The most commonly observed AEs included back pain, headache, post-lumbar puncture syndrome, and syncope, each occurring in 2 participants (17%). Additional AEs were reported in 1 participant each (8%) and included constipation, hemorrhoids, injection site pain, procedural pain, elevated alanine aminotransferase (ALT), amyloid-related imaging abnormalities with microhemorrhages and hemosiderin deposits (ARIA-H), presyncope, and vomiting. No serious AEs or dose-limiting toxicities were observed. Moreover, no safety signals were identified based on routine CSF biomarkers or exploratory markers such as neurofilament light chain (NfL) [95].
In a new cohort study involving 36 patients with EOAD (mean age 60.9 years; mean MMSE score 24.6), participants were assigned to escalating dose cohorts (25, 35, 50, and 75 mg) and evaluated over a minimum follow-up period of six months. Extended monitoring continued for up to 12 months to assess the durability of treatment effects [97]. CSF samples were collected on Day 15 and at Months 1, 2, 3, 4, 5, and 6 for the analysis of APP and Aβ biomarkers [96].
AEs were common; however, the majority were mild to moderate in severity and not classified as serious. The most frequently reported AE was post–lumbar puncture headache, observed in 41.7% of participants. This event was more prevalent in the lower dose groups, occurring in 75% of participants in the 50 mg cohort, 50% in both the 25 mg and 35 mg cohorts, and only 14.3% in the 75 mg cohort [97].
Only three participants experienced AEs that were considered possibly related to the investigational drug. One individual in the 50 mg group reported mild headache and nausea, both deemed related to IT administration as well as to the study drug. Two participants in the 75 mg group experienced moderate AEs, including headache, neck pain, vomiting, and lymphocytopenia. The first three events were also attributed to the lumbar puncture procedure. A serious and fatal AE of acute pancreatitis was reported in the 75 mg group on day 277 following administration of a single dose; however, this event was assessed as unrelated to mivelsiran [97].
Regarding pharmacodynamics, a dose-dependent and sustained reduction in the levels of sAPPα and sAPPβ was observed in the CSF. In the 25 mg cohort, reductions were modest and transient. In contrast, doses of 35 mg and above resulted in marked and sustained decreases. The 75 mg dose elicited the most pronounced effect, with mean reductions of 61.3% in sAPPα and 73.5% in sAPPβ observed at one month post-administration. These reductions were maintained through six months in all cohorts receiving ≥35 mg, and persisted for up to 12 months in the 50 mg and 75 mg groups [97].
In addition, the effects of mivelsiran on Aβ peptides were assessed. Significant reductions in CSF Aβ42 and Aβ40 levels were noted in cohorts receiving 35 mg or higher, with the magnitude of reduction increasing with dose escalation. Once again, the 75 mg dose was the most effective, achieving mean reductions of 49.3% for Aβ42 and 66.5% for Aβ40 at one month. These effects were sustained for at least six months in the 35 mg, 50 mg, and 75 mg groups, suggesting effective inhibition of the amyloidogenic cascade in response to APP gene silencing [97].
These findings support the hypothesis that ALN-APP may suppress the production of all APP-derived cleavage products, both intracellular and extracellular, thereby potentially addressing multiple pathological pathways involved in AD and cerebral amyloid angiopathy (CAA).
The consistency of the pharmacodynamic effects, coupled with a favorable safety profile, supports the continued clinical development of ALN-APP as a potential disease-modifying therapy for AD and other conditions associated with Aβ deposition, such as cerebral amyloid angiopathy [97].
6. Conclusions
Advancements in therapeutic siRNAs, particularly ALN-APP, represent a significant step forward in the effort to address neurodegenerative diseases, notably AD. The approval of patisiran in 2018 set an important precedent, while the development of ALN-APP showcases innovative strategies to overcome the challenges associated with delivering siRNAs to the CNS. The IT is particularly critical as it allows direct access to the cerebrospinal fluid, facilitating effective delivery to target neurons that are otherwise difficult to transfect with nucleic acids. Historically, anatomical barriers have limited the effectiveness of available treatments; however, ALN-APP presents a promising alternative.
This specific siRNA formulation targets the reduction of beta-amyloid protein production, which is a key contributor to the pathogenesis of AD. As a C16-siRNA, ALN-APP incorporates a 16-carbon chain to enhance specificity and extend its therapeutic effects, building on the successes of earlier FDA-approved therapies. Preclinical studies conducted in various animal models, including rats, mice, and non-human primates, have demonstrated its efficacy by showing significant reductions in critical biomarkers related to amyloid production.
The positive outcomes from the Phase 1 trial, ALN-APP-001, further highlight this potential, revealing reductions in soluble APP levels that indicate meaningful impacts on the pathology of the disease. Importantly, reductions of 55% in soluble APPα and 69% in APPβ by day 15, along with sustained effects across patient cohorts for up to six months, suggest that ALN-APP may represent a disease-modifying therapy capable of influencing the progression of AD.
Moreover, the favorable safety profile of ALN-APP enhances its suitability as a therapeutic option, suggesting its potential for long-term use in patients with EOAD. As research continues to progress, the insights gained from studies involving ALN-APP will be crucial for refining siRNA delivery methods and developing effective therapeutic strategies for neurodegenerative diseases.
In summary, the ongoing development and testing of ALN-APP not only expand our understanding of siRNA technology but also hold promise for innovative treatments for Alzheimer's disease and related neurodegenerative disorders, marking an important progression in the field of neurological therapeutics.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, R.T.A.; writing—R.T.A., G.M.O.G.; T.C.S.; S.S.T.A.; original draft preparation, R.T.A.; writing—review and editing, R.T.A., G.M.O.G.; T.C.S.; S.S.T.A.; supervision, R.T.A. and S.S.T.A. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| aCSF | Artificial cerebrospinal fluid |
| AD | Alzheimer’s disease |
| AEs | Adverse events |
| AGO2 Argonaute 2 enzyme | Argonaute 2 enzyme |
| AHP | Acute hepatic porphyria |
| ALA | δ-aminolevulinic acid |
| ALS | Amyotrophic lateral sclerosis |
| ALT | Alanine aminotransferase |
| APOE | Apolipoprotein E |
| ApoE | Apolipoprotein E |
| ARIA-H | Amyloid-related imaging abnormalities-haemosiderin |
| ASGPR | Asialoglycoprotein receptor |
| Aβ | β-amyloid |
| CAA | Cerebral amyloid angiopathy |
| CNS | Central nervous system |
| DEAF1 | Deformed epidermal autoregulatory factor-1 |
| ds-siRNA | Double-stranded siRNA |
| EOAD | Early-onset Alzheimer’s disease |
| FDA | Food and Drug Administration |
| GalNAc | N-acetylgalactosamine |
| GNA | Glycol nucleic acid |
| hATTR | Hereditary transthyretin amyloidosis |
| IT | Intrathecal |
| IV | Intravenous |
| LNP | Lipid nanoparticle |
| LRP1 | Lipoprotein receptor-related protein |
| MAD | Multiple ascending dose |
| MCI | Mild cognitive impairment |
| MMSE | Mini-Mental State Examination |
| mRNA | Messenger RNA |
| NfL | Neurofilament light chain |
| NFT’s | Neurofibrillary tangles |
| NHPs | Non-human primates |
| PACT | Protein activator of the interferon-induced protein kinase |
| PBG | Porphobilinogen |
| PD | Pharmacodynamics |
| PET | Positron emission tomography |
| PK | Pharmacokinetics |
| PS | Phosphorothioate |
| RISC | RNA-induced silencing complex |
| RNAi | RNA interference |
| ROS | Reative oxygen species |
| SAD | Single ascending dose |
| sAPPα | Soluble APPα |
| sAPPβ | Soluble APPβ |
| siRNA | Small interfering RNA |
| Sod1 | Superoxide dismutase 1 |
| TRBP | Transactivation response RNA-binding protein |
| TTR | Transthyretin |
| VLDLR | Very low-density lipoprotein receptor |
| VP | Vinylphosphonate |
| Wt | Wild type |
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat Rev Dis Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the Global Prevalence of Dementia in 2019 and Forecasted Prevalence in 2050: An Analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Madnani, R.S. Alzheimer’s Disease: A Mini-Review for the Clinician. Front Neurol 2023, 14. [Google Scholar] [CrossRef]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical Diagnosis of Alzheimer’s Disease: Recommendations of the International Working Group. Lancet Neurol 2021, 20, 484–496. [Google Scholar] [CrossRef]
- Zhao, N.; Ren, Y.; Yamazaki, Y.; Qiao, W.; Li, F.; Felton, L.M.; Mahmoudiandehkordi, S.; Kueider-Paisley, A.; Sonoustoun, B.; Arnold, M.; et al. Alzheimer’s Risk Factors Age, APOE Genotype, and Sex Drive Distinct Molecular Pathways. Neuron 2020, 106, 727–742.e6. [Google Scholar] [CrossRef]
- Grant, W.B. A Brief History of the Progress in Our Understanding of Genetics and Lifestyle, Especially Diet, in the Risk of Alzheimer’s Disease. Journal of Alzheimer’s Disease 2024, 100, S165–S178. [Google Scholar] [CrossRef]
- Nordestgaard, L.T.; Christoffersen, M.; Frikke-Schmidt, R. Shared Risk Factors between Dementia and Atherosclerotic Cardiovascular Disease. Int J Mol Sci 2022, 23, 9777. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Wang, H.-F.; Jiang, T.; Tan, M.-S.; Tan, L.; Zhao, Q.-F.; Li, J.-Q.; Wang, J.; Yu, J.-T. Meta-Analysis of Modifiable Risk Factors for Alzheimer’s Disease. J Neurol Neurosurg Psychiatry 2015, jnnp-2015-310548. [Google Scholar] [CrossRef]
- Li, J.-Q.; Tan, L.; Wang, H.-F.; Tan, M.-S.; Tan, L.; Xu, W.; Zhao, Q.-F.; Wang, J.; Jiang, T.; Yu, J.-T. Risk Factors for Predicting Progression from Mild Cognitive Impairment to Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Cohort Studies. J Neurol Neurosurg Psychiatry 2016, 87, 476–484. [Google Scholar] [CrossRef]
- Arenaza-Urquijo, E.M.; Vemuri, P. Resistance vs Resilience to Alzheimer Disease. Neurology 2018, 90, 695–703. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat Genet 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Monsell, S.E.; Mock, C.; Fardo, D.W.; Bertelsen, S.; Cairns, N.J.; Roe, C.M.; Ellingson, S.R.; Morris, J.C.; Goate, A.M.; Kukull, W.A. Genetic Comparison of Symptomatic and Asymptomatic Persons With Alzheimer Disease Neuropathology. Alzheimer Dis Assoc Disord 2017, 31, 232–238. [Google Scholar] [CrossRef]
- Prpar Mihevc, S.; Majdič, G. Canine Cognitive Dysfunction and Alzheimer’s Disease – Two Facets of the Same Disease? Front Neurosci 2019, 13. [Google Scholar] [CrossRef]
- Panek, W.K.; Murdoch, D.M.; Gruen, M.E.; Mowat, F.M.; Marek, R.D.; Olby, N.J. Plasma Amyloid Beta Concentrations in Aged and Cognitively Impaired Pet Dogs. Mol Neurobiol 2021, 58, 483–489. [Google Scholar] [CrossRef]
- Thal, D.R.; Capetillo-Zarate, E.; Del Tredici, K.; Braak, H. The Development of Amyloid β Protein Deposits in the Aged Brain. Science of Aging Knowledge Environment 2006, 2006. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging–Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease. Alzheimer’s & Dementia 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol 2012, 123, 1–11. [Google Scholar] [CrossRef]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 173–185. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol Neurodegener 2019, 14, 32. [Google Scholar] [CrossRef]
- Cummings, B.J.; Su, J.H.; Cotman, C.W.; White, R.; Russell, M.J. β-Amyloid Accumulation in Aged Canine Brain: A Model of Early Plaque Formation in Alzheimer’s Disease. Neurobiol Aging 1993, 14. [Google Scholar] [CrossRef]
- Czasch, S.; Paul, S.; Baumgärtner, W. A Comparison of Immunohistochemical and Silver Staining Methods for the Detection of Diffuse Plaques in the Aged Canine Brain. Neurobiol Aging 2006, 27. [Google Scholar] [CrossRef]
- Bates, K.A.; Sohrabi, H.R.; Rodrigues, M.; Beilby, J.; Dhaliwal, S.S.; Taddei, K.; Criddle, A.; Wraith, M.; Howard, M.; Martins, G.; et al. Association of Cardiovascular Factors and Alzheimer’s Disease Plasma Amyloid-β Protein in Subjective Memory Complainers. Journal of Alzheimer’s Disease 2009, 17, 305–318. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE Influences Amyloid-β (Aβ) Clearance despite Minimal ApoE/Aβ Association in Physiological Conditions. Proc Natl Acad Sci U S A 2013, 110. [Google Scholar] [CrossRef]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 Peptides Form Interlaced Amyloid Fibrils. J Neurochem 2013, 126, 305–311. [Google Scholar] [CrossRef]
- Qiu, T.; Liu, Q.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Aβ42 and Aβ40: Similarities and Differences. Journal of Peptide Science 2015, 21, 522–529. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-Wide Meta-Analysis Identifies New Loci and Functional Pathways Influencing Alzheimer’s Disease Risk. Nat Genet 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent Advances in Alzheimer’s Disease: Mechanisms, Clinical Trials and New Drug Development Strategies. Signal Transduct Target Ther 2024, 9, 211. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J Cent Nerv Syst Dis 2020, 12. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Lacorte, E.; Ancidoni, A.; Zaccaria, V.; Remoli, G.; Tariciotti, L.; Bellomo, G.; Sciancalepore, F.; Corbo, M.; Lombardo, F.L.; Bacigalupo, I.; et al. Safety and Efficacy of Monoclonal Antibodies for Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Published and Unpublished Clinical Trials. Journal of Alzheimer’s Disease 2022, 87. [Google Scholar] [CrossRef]
- Ramanan, V.K.; Armstrong, M.J.; Choudhury, P.; Coerver, K.A.; Hamilton, R.H.; Klein, B.C.; Wolk, D.A.; Wessels, S.R.; Jones, L.K. Antiamyloid Monoclonal Antibody Therapy for Alzheimer Disease: Emerging Issues in Neurology. Neurology 2023, 101. [Google Scholar] [CrossRef]
- Cummings, J.; Osse, A.M.L.; Cammann, D.; Powell, J.; Chen, J. Anti-Amyloid Monoclonal Antibodies for the Treatment of Alzheimer’s Disease. BioDrugs 2024, 38. [Google Scholar] [CrossRef]
- Meister, G.; Tuschl, T. Mechanisms of Gene Silencing by Double-Stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Titze-de-Almeida, R.; David, C.; Titze-de-Almeida, S.S. The Race of 10 Synthetic RNAi-Based Drugs to the Pharmaceutical Market. Pharm Res 2017, 34, 1339–1363. [Google Scholar] [CrossRef]
- Wilson, R.C.; Doudna, J.A. Molecular Mechanisms of RNA Interference. Annu Rev Biophys 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of MiRNAs and SiRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Bushati, N.; Cohen, S.M. MicroRNA Functions. Annu Rev Cell Dev Biol 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Svoboda, P. Renaissance of Mammalian Endogenous RNAi. FEBS Lett 2014, 588, 2550–2556. [Google Scholar] [CrossRef]
- Ketting, R.F. The Many Faces of RNAi. Dev Cell 2011, 20, 148–161. [Google Scholar] [CrossRef]
- Pratt, A.J.; MacRae, I.J. The RNA-Induced Silencing Complex: A Versatile Gene-Silencing Machine. J Biol Chem 2009, 284, 17897–17901. [Google Scholar] [CrossRef]
- Jinek, M.; Doudna, J.A. A Three-Dimensional View of the Molecular Machinery of RNA Interference. Nature 2009, 457, 405–412. [Google Scholar] [CrossRef]
- Zamore, P.D. RNA Interference: Big Applause for Silencing in Stockholm (Reprinted from Cell, Vol 127, Pg 1083-1086, 2006). Cell 2007. [Google Scholar] [CrossRef]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-Stranded RNA Directs the ATP-Dependent Cleavage of MRNA at 21 to 23 Nucleotide Intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Meister, G.; Tuschl, T. Mechanisms of Gene Silencing by Double-Stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu Rev Pharmacol Toxicol 2019, 59, 605–630. [Google Scholar] [CrossRef]
- Adams, D.; Suhr, O.B.; Hund, E.; Obici, L.; Tournev, I.; Campistol, J.M.; Slama, M.S.; Hazenberg, B.P.; Coelho, T. First European Consensus for Diagnosis, Management, and Treatment of Transthyretin Familial Amyloid Polyneuropathy. Curr Opin Neurol 2016. [CrossRef]
- Ledford, H. Gene-Silencing Technology Gets First Drug Approval after 20-Year Wait. Nature 2018, 560, 291–292. [Google Scholar] [CrossRef]
- FDA - Food and Drug Administration. FDA Approves New Treatments for Heart Disease Caused by a Serious Rare Disease, Transthyretin Mediated Amyloidosis.
- Nikam, R.R.; Gore, K.R. Journey of SiRNA: Clinical Developments and Targeted Delivery. Nucleic Acid Ther 2018, 28, 209–224. [Google Scholar] [CrossRef]
- de Paula Brandão, P.R.; Titze-de-Almeida, S.S.; Titze-de-Almeida, R. Leading RNA Interference Therapeutics Part 2: Silencing Delta-Aminolevulinic Acid Synthase 1, with a Focus on Givosiran. Mol Diagn Ther 2020, 24. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Molecular Therapy 2017. [CrossRef]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther 2018. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A. V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi Therapeutic for the Treatment of Hereditary Transthyretin-Mediated Amyloidosis. Neurodegener Dis Manag 2019. [Google Scholar] [CrossRef]
- Titze-de-Almeida, S.S.; Brandão, P.R. de P.; Faber, I.; Titze-de-Almeida, R. Leading RNA Interference Therapeutics Part 1: Silencing Hereditary Transthyretin Amyloidosis, with a Focus on Patisiran. Mol Diagn Ther 2020, 24.
- An, G. Pharmacokinetics and Pharmacodynamics of GalNAc-Conjugated SiRNAs. J Clin Pharmacol 2024, 64. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kelin, A.V.; Milstein, S.; et al. Multivalent N -Acetylgalactosamine-Conjugated SiRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J Am Chem Soc 2014, 136. [Google Scholar] [CrossRef]
- Willoughby, J.L.S.; Chan, A.; Sehgal, A.; Butler, J.S.; Nair, J.K.; Racie, T.; Shulga-Morskaya, S.; Nguyen, T.; Qian, K.; Yucius, K.; et al. Evaluation of GalNAc-SiRNA Conjugate Activity in Pre-Clinical Animal Models with Reduced Asialoglycoprotein Receptor Expression. Molecular Therapy 2018, 26. [Google Scholar] [CrossRef]
- Gadhave, D.G.; Sugandhi, V.V.; Jha, S.K.; Nangare, S.N.; Gupta, G.; Singh, S.K.; Dua, K.; Cho, H.; Hansbro, P.M.; Paudel, K.R. Neurodegenerative Disorders: Mechanisms of Degeneration and Therapeutic Approaches with Their Clinical Relevance. Ageing Res Rev 2024, 99, 102357. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson Disease. Nat Rev Dis Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science (1979) 1997, 276. [Google Scholar] [CrossRef]
- Nabais Sá, M.J.; Jensik, P.J.; McGee, S.R.; Parker, M.J.; Lahiri, N.; McNeil, E.P.; Kroes, H.Y.; Hagerman, R.J.; Harrison, R.E.; Montgomery, T.; et al. De Novo and Biallelic DEAF1 Variants Cause a Phenotypic Spectrum. Genetics in Medicine 2019, 21. [Google Scholar] [CrossRef]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-Finger Proteins in Health and Disease. Cell Death Discov 2017, 3. [Google Scholar] [CrossRef]
- Krishna, S.S. Structural Classification of Zinc Fingers: SURVEY AND SUMMARY. Nucleic Acids Res 2003, 31. [Google Scholar] [CrossRef]
- Germain, N.D.; Chung, W.K.; Sarmiere, P.D. RNA Interference (RNAi)-Based Therapeutics for Treatment of Rare Neurologic Diseases. Mol Aspects Med 2023, 91. [Google Scholar] [CrossRef]
- Li, V.; Huang, Y. Oligonucleotide Therapeutics for Neurodegenerative Diseases. NeuroImmune Pharmacology and Therapeutics 2025, 4, 1–11. [Google Scholar] [CrossRef]
- Sheridan, C. Billion-Dollar Deal Propels RNAi to CNS Frontier. Nat Biotechnol 2019, 37. [Google Scholar] [CrossRef]
- Won Lee, J.; Kyu Shim, M.; Kim, H.; Jang, H.; Lee, Y.; Hwa Kim, S. RNAi Therapies: Expanding Applications for Extrahepatic Diseases and Overcoming Delivery Challenges. Adv Drug Deliv Rev 2023, 201. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down Barriers: Advances in SiRNA Delivery. Nat Rev Drug Discov 2009, 8, 129–138. [Google Scholar] [CrossRef]
- Wittrup, A.; Lieberman, J. Knocking down Disease: A Progress Report on SiRNA Therapeutics. Nat Rev Genet 2015, 16, 543–552. [Google Scholar] [CrossRef]
- Brown, K.M.; Nair, J.K.; Janas, M.M.; Anglero-Rodriguez, Y.I.; Dang, L.T.H.; Peng, H.; Theile, C.S.; Castellanos-Rizaldos, E.; Brown, C.; Foster, D.; et al. Expanding RNAi Therapeutics to Extrahepatic Tissues with Lipophilic Conjugates. Nat Biotechnol 2022, 40. [Google Scholar] [CrossRef]
- Jadhav, V.; Maier, M. Behind the Paper - Expanding the Reach of RNAi Therapeutics with Next Generation Lipophilic SiRNA Conjugates Available online:. Available online: https://communities.springernature.com/posts/expanding-the-reach-of-rnai-therapeutics-with-next-generation-lipophilic-sirna-conjugates (accessed on 5 July 2025).
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Gelck, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic Silencing of an Endogenous Gene by Systemic Administration of Modified SiRNAs. Nature 2004, 432. [Google Scholar] [CrossRef]
- Alterman, J.F.; Hall, L.M.; Coles, A.H.; Hassler, M.R.; Didiot, M.C.; Chase, K.; Abraham, J.; Sottosanti, E.; Johnson, E.; Sapp, E.; et al. Hydrophobically Modified SiRNAs Silence Huntingtin MRNA in Primary Neurons and Mouse Brain. Mol Ther Nucleic Acids 2015, 4. [Google Scholar] [CrossRef]
- Tai, W. Chemical Modulation of SiRNA Lipophilicity for Efficient Delivery. Journal of Controlled Release 2019, 307. [Google Scholar] [CrossRef]
- Alnylam Pharmaceutics, Inc. Alnylam. Delivery Platforms - C16 Conjugates Available online:. Available online: https://www.alnylam.com/our-science/sirna-delivery-platforms (accessed on 5 July 2025).
- Titze-de-Almeida, R.; David, C.; Titze-de-Almeida, S.S. The Race of 10 Synthetic RNAi-Based Drugs to the Pharmaceutical Market. Pharm Res 2017, 34, 1339–1363. [Google Scholar] [CrossRef]
- Cummins, L.L.; Owens, S.R.; Risen, L.M.; Lesnik, E.A.; Freier, S.M.; Mc Gee, D.; Cook, C.J.; Cook, P.D. Characterization of Fully 2’-Modified Oligoribonucleotide Hetero-and Homoduplex Hybridization Andnuclease Sensitivity. Nucleic Acids Res 1995, 23. [Google Scholar] [CrossRef]
- Layzer, J.M.; McCaffrey, A.P.; Tanner, A.K.; Huang, Z.; Kay, M.A.; Sullenger, B.A. In Vivo Activity of Nuclease-Resistant SiRNAs. RNA 2004, 10. [Google Scholar] [CrossRef]
- Takahashi, M.; Minakawa, N.; Matsuda, A. Synthesis and Characterization of 2′-Modified-4′-ThioRNA: A Comprehensive Comparison of Nuclease Stability. Nucleic Acids Res 2009, 37. [Google Scholar] [CrossRef]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2′-Modified Oligonucleotide Duplexes with Improved in Vitro Potency and Stability Compared to Unmodified Small Interfering RNA. J Med Chem 2005, 48. [Google Scholar] [CrossRef]
- Prakash, T.P.; Kinberger, G.A.; Murray, H.M.; Chappell, A.; Riney, S.; Graham, M.J.; Lima, W.F.; Swayze, E.E.; Seth, P.P. Synergistic Effect of Phosphorothioate, 5′-Vinylphosphonate and GalNAc Modifications for Enhancing Activity of Synthetic SiRNA. Bioorg Med Chem Lett 2016, 26. [Google Scholar] [CrossRef]
- Foster, D.J.; Brown, C.R.; Shaikh, S.; Trapp, C.; Schlegel, M.K.; Qian, K.; Sehgal, A.; Rajeev, K.G.; Jadhav, V.; Manoharan, M.; et al. Advanced SiRNA Designs Further Improve In Vivo Performance of GalNAc-SiRNA Conjugates. Molecular Therapy 2018, 26. [Google Scholar] [CrossRef]
- Janas, M.M.; Schlegel, M.K.; Harbison, C.E.; Yilmaz, V.O.; Jiang, Y.; Parmar, R.; Zlatev, I.; Castoreno, A.; Xu, H.; Shulga-Morskaya, S.; et al. Selection of GalNAc-Conjugated SiRNAs with Limited off-Target-Driven Rat Hepatotoxicity. Nat Commun 2018, 9. [Google Scholar] [CrossRef]
- Schlegel, M.K.; Foster, D.J.; Kel’In, A.V.; Zlatev, I.; Bisbe, A.; Jayaraman, M.; Lackey, J.G.; Rajeev, K.G.; Charissé, K.; Harp, J.; et al. Chirality Dependent Potency Enhancement and Structural Impact of Glycol Nucleic Acid Modification on SiRNA. J Am Chem Soc 2017, 139. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Roux, L.; Coles, A.H.; Turanov, A.A.; Alterman, J.F.; Echeverria, D.; Godinho, B.M.D.C.; Aronin, N.; Khvorova, A. 5’-Vinylphosphonate Improves Tissue Accumulation and Efficacy of Conjugated SiRNAs in Vivo. Nucleic Acids Res 2017, 45. [Google Scholar] [CrossRef]
- Parmar, R.; Willoughby, J.L.S.; Liu, J.; Foster, D.J.; Brigham, B.; Theile, C.S.; Charisse, K.; Akinc, A.; Guidry, E.; Pei, Y.; et al. 5′-(E)-Vinylphosphonate: A Stable Phosphate Mimic Can Improve the RNAi Activity of SiRNA-GalNAc Conjugates. ChemBioChem 2016, 17. [Google Scholar] [CrossRef]
- Heneka, M.T.; van der Flier, W.M.; Jessen, F.; Hoozemanns, J.; Thal, D.R.; Boche, D.; Brosseron, F.; Teunissen, C.; Zetterberg, H.; Jacobs, A.H.; et al. Neuroinflammation in Alzheimer Disease. Nat Rev Immunol 2025, 25, 321–352. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat Rev Neurol 2021, 17. [Google Scholar] [CrossRef]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angewandte Chemie - International Edition 2021, 60. [Google Scholar] [CrossRef]
- Milani, P.; Gagliardi, S.; Cova, E.; Cereda, C. SOD1 Transcriptional and Posttranscriptional Regulation and Its Potential Implications in ALS. Neurol Res Int 2011, 2011. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.A.; Saccon, R.A.; Fratta, P.; Fisher, E.M.C. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21. [Google Scholar] [CrossRef]
- Alnylan Pharmaceuticals, Inc. Alnylam Reports Additional Positive Interim Phase 1 Results for ALN-APP, in Development for Alzheimer’s Disease and Cerebral Amyloid Angiopathy Available online:. Available online: https://investors.alnylam.com/press-release?id=27761 (accessed on 5 July 2025).
- Cohen, S.; Ducharme, S.; Brosch, J.R.; Vijverberg, E.G.B.; Apostolova, L.G.; Sostelly, A.; Goteti, S.; Makarova, N.; Avbersek, A.; Guo, W.; et al. Interim Phase 1 Part A Results for ALN-APP, the First Investigational RNAi Therapeutic in Development for Alzheimer’s Disease. Alzheimer’s & Dementia 2023, 19. [Google Scholar] [CrossRef]
- Cohen, S.; Meglio, M. Overviewing Encouraging Early-Stage Data on Mivelsiran, RNA Therapeutic for Alzheimer Disease: Sharon Cohen, MD, FRCPC.
- Cohen, S.; Ducharme, S.; Brosch, J.R. Single Ascending Dose Results from an Ongoing Phase 1 Study of Mivelsiran (ALN-APP), the First Investigational RNA Interference Therapeutic Targeting Amyloid Precursor Protein for Alzheimer’s Disease.; Philadelphia, PA, 2024;
Figure 1.
The siRNA pathway mediating post-transcriptional gene silencing. RNA interference (RNAi) is a highly conserved cellular pathway that allows for targeted gene silencing at the post-transcriptional level. Synthetic siRNA duplexes, typically delivered to the cytoplasm via nanoparticle carriers, consist of a guide (green) and a sense (red) strand. Once inside the cytoplasm, the duplex is recognized by a protein complex that includes the ribonuclease Dicer, the transactivation response RNA-binding protein (TRBP), and the protein activator of the interferon-induced protein kinase (PACT). This complex facilitates the removal and degradation of the sense strand, while the guide strand is retained and incorporated into the RNA-induced silencing complex (RISC). The mature RISC, which includes Argonaute 2 (AGO2) and the guide strand, is directed by the guide RNA to bind complementary sequences on target messenger RNAs (mRNAs). Upon binding, AGO2 catalyzes the cleavage of the target mRNA, resulting in sequence-specific degradation and subsequent gene silencing. Abbreviations: AGO2, Argonaute 2 enzyme; mRNA, messenger RNA; PACT, protein activator of the interferon-induced protein kinase; RISC, RNA-induced silencing complex; RNAi, RNA interference; siRNA, small interfering RNA; TRBP, HIV-1 transactivation response (TAR) RNA-binding protein.
Figure 1.
The siRNA pathway mediating post-transcriptional gene silencing. RNA interference (RNAi) is a highly conserved cellular pathway that allows for targeted gene silencing at the post-transcriptional level. Synthetic siRNA duplexes, typically delivered to the cytoplasm via nanoparticle carriers, consist of a guide (green) and a sense (red) strand. Once inside the cytoplasm, the duplex is recognized by a protein complex that includes the ribonuclease Dicer, the transactivation response RNA-binding protein (TRBP), and the protein activator of the interferon-induced protein kinase (PACT). This complex facilitates the removal and degradation of the sense strand, while the guide strand is retained and incorporated into the RNA-induced silencing complex (RISC). The mature RISC, which includes Argonaute 2 (AGO2) and the guide strand, is directed by the guide RNA to bind complementary sequences on target messenger RNAs (mRNAs). Upon binding, AGO2 catalyzes the cleavage of the target mRNA, resulting in sequence-specific degradation and subsequent gene silencing. Abbreviations: AGO2, Argonaute 2 enzyme; mRNA, messenger RNA; PACT, protein activator of the interferon-induced protein kinase; RISC, RNA-induced silencing complex; RNAi, RNA interference; siRNA, small interfering RNA; TRBP, HIV-1 transactivation response (TAR) RNA-binding protein.
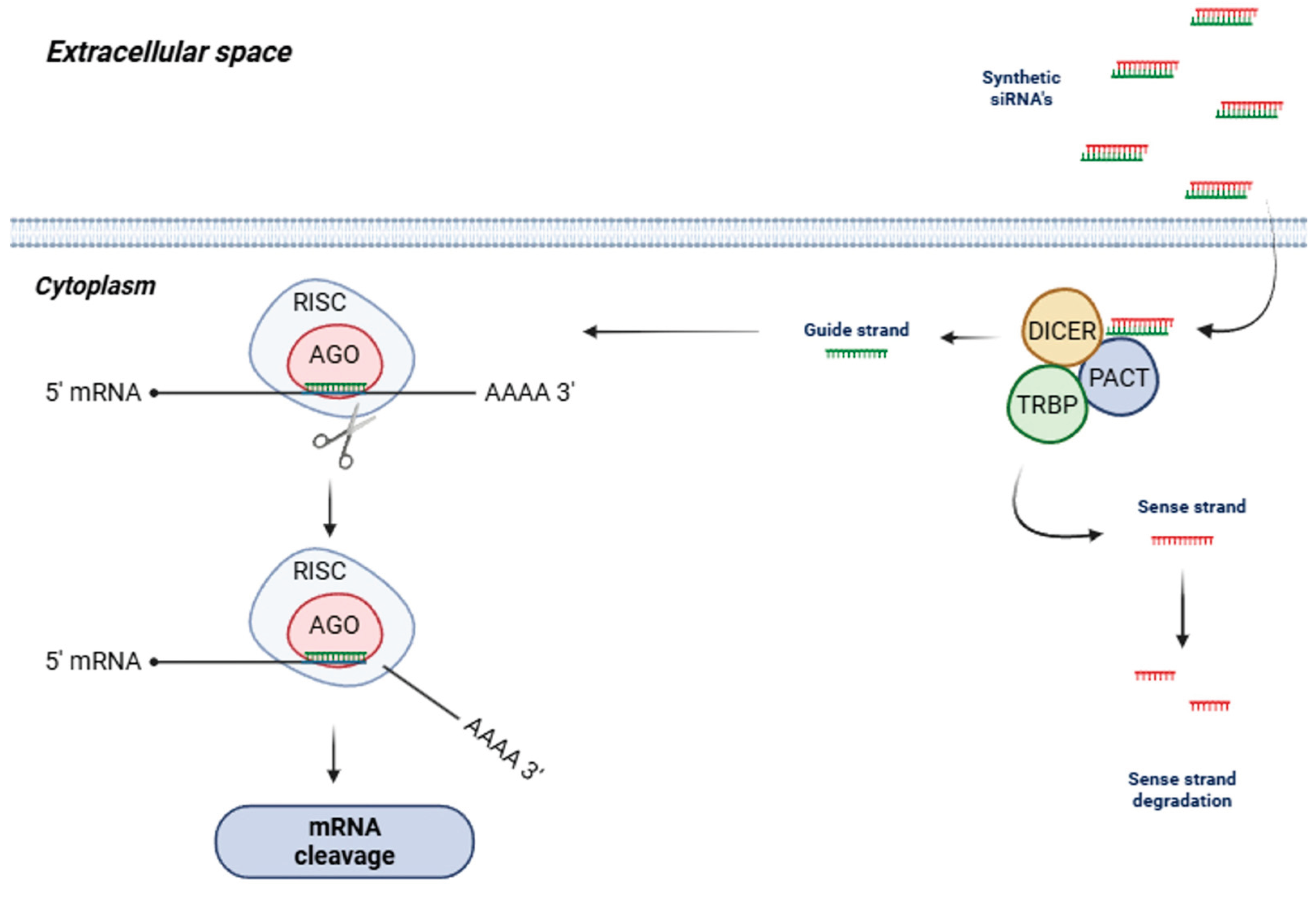
Figure 2.
RNAi-mediated gene silencing by the mechanism of action and cell effects of patisiran. (A) The TTR gene expresses the TTR mRNA that in the 3′-UTR region contains a sequence conserved across wild-type (wt) and all TTR variant forms. (B) Patisiran is a ds-siRNA composed by a passenger and guide strand with overhangs at the 3′ ends. (C) The passenger strand is eliminated and the guide strand (without overhangs) finds the complementary nucleotides in the 3′ UTR region of TTR mRNA (mutated or wt) for base pairing. (D) The binding of patisiran to TTR mRNA induces the RISC complex to execute RNAi-mediated gene silencing by cleavage of this 3′ UTR region and synthesis of the TTR protein is suppressed. siRNA- double-strand small-interfering RNA, mRNA- messenger RNA, iRNA- RNA interference, TTR transthyretin, UTR- untranslated region, wt- wild-type.
Figure 2.
RNAi-mediated gene silencing by the mechanism of action and cell effects of patisiran. (A) The TTR gene expresses the TTR mRNA that in the 3′-UTR region contains a sequence conserved across wild-type (wt) and all TTR variant forms. (B) Patisiran is a ds-siRNA composed by a passenger and guide strand with overhangs at the 3′ ends. (C) The passenger strand is eliminated and the guide strand (without overhangs) finds the complementary nucleotides in the 3′ UTR region of TTR mRNA (mutated or wt) for base pairing. (D) The binding of patisiran to TTR mRNA induces the RISC complex to execute RNAi-mediated gene silencing by cleavage of this 3′ UTR region and synthesis of the TTR protein is suppressed. siRNA- double-strand small-interfering RNA, mRNA- messenger RNA, iRNA- RNA interference, TTR transthyretin, UTR- untranslated region, wt- wild-type.
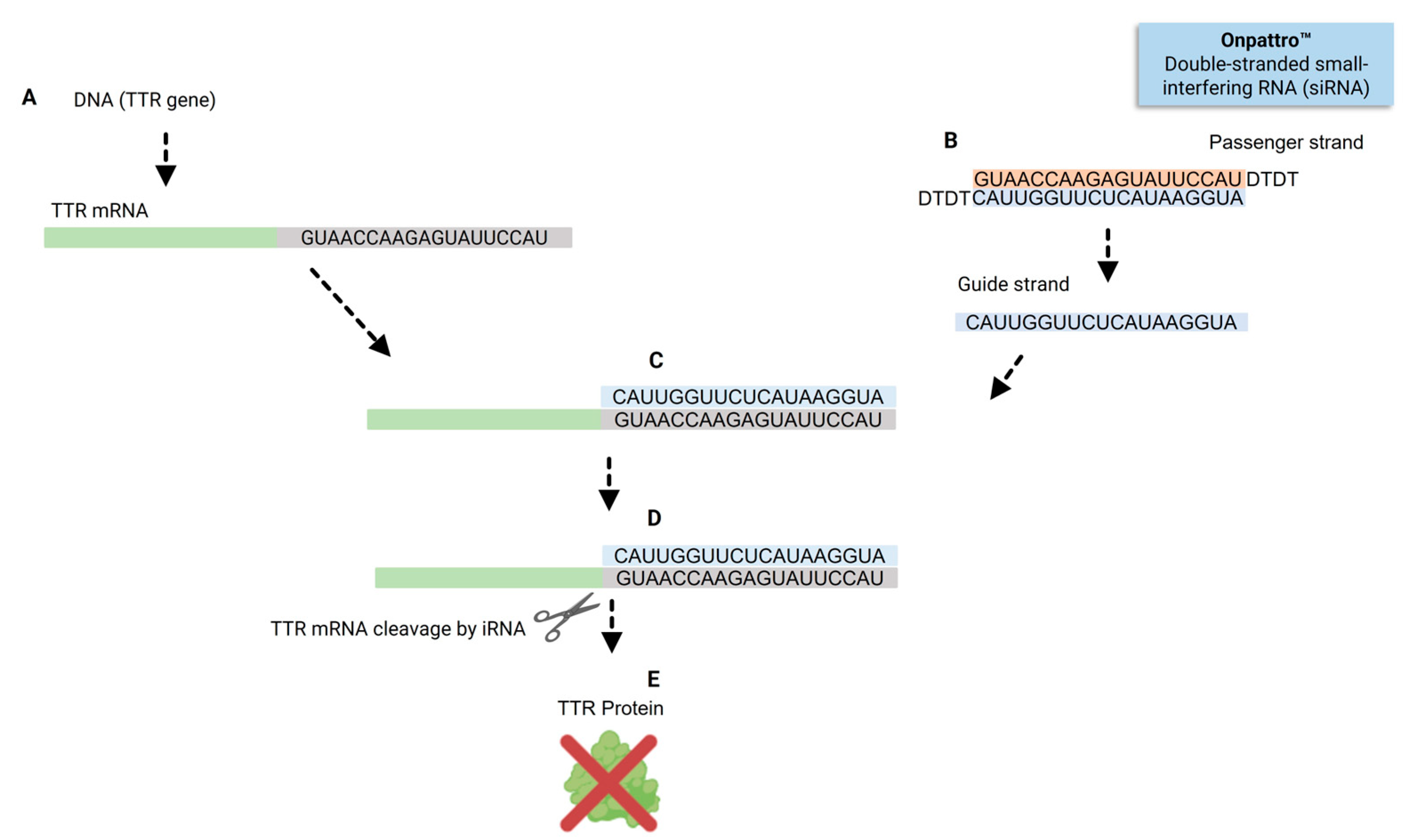
Figure 3.
Mechanism of patisiran therapy for the treatment of hereditary transthyretin amyloidosis (hATTR amyloidosis). Patisiran, the first FDA-approved siRNA therapeutic for hATTR amyloidosis, reduces transthyretin (TTR) expression through RNA interference (RNAi). (A) Patients receive patisiran via intravenous (IV) infusion. (B) Patisiran is encapsulated in lipid nanoparticles (LNPs), which are naturally opsonized by apolipoprotein E (ApoE) in the circulatory system. (C) The ApoE-coated LNPs bind to ApoE receptors on hepatocytes and are internalized via receptor-mediated endocytosis. (D) Within the acidic environment of endocytic vesicles, the ionizable lipids in the LNPs become protonated, acquiring a positive charge. This promotes electrostatic interaction with the negatively charged endosomal membrane, leading to membrane fusion and cytoplasmic release of the double-stranded siRNA (ds-siRNA). (E) In the cytoplasm, the ds-siRNA is processed by a protein complex consisting of Dicer (a ribonuclease), TRBP (HIV-1 trans-activation response RNA-binding protein), and PACT (protein activator of the interferon-induced protein kinase). This complex removes the passenger (sense) strand, while the guide (antisense) strand is loaded into the RNA-induced silencing complex (RISC). (F) The Argonaute 2 (AGO2) enzyme within RISC directs the guide strand to bind complementary sequences in TTR mRNA, resulting in targeted cleavage and degradation of the transcript. (G) Suppression of TTR mRNA translation reduces the production of unstable TTR tetramers, thereby preventing their misfolding and aggregation into amyloid fibrils. This reduction in amyloid burden stabilizes or improves clinical manifestations such as neuropathy and cardiomyopathy. Abbreviations: AGO2, Argonaute 2 enzyme; ApoE, apolipoprotein E; ds-siRNA, double-stranded small interfering RNA; hATTR amyloidosis, hereditary transthyretin amyloidosis; IV, intravenous; LNPs, lipid nanoparticles; mRNA, messenger RNA; PACT, protein activator of the interferon-induced protein kinase; RISC, RNA-induced silencing complex; RNAi, RNA interference; siRNA, small interfering RNA; TRBP, HIV-1 trans-activation response (TAR) RNA-binding protein; TTR, transthyretin.
Figure 3.
Mechanism of patisiran therapy for the treatment of hereditary transthyretin amyloidosis (hATTR amyloidosis). Patisiran, the first FDA-approved siRNA therapeutic for hATTR amyloidosis, reduces transthyretin (TTR) expression through RNA interference (RNAi). (A) Patients receive patisiran via intravenous (IV) infusion. (B) Patisiran is encapsulated in lipid nanoparticles (LNPs), which are naturally opsonized by apolipoprotein E (ApoE) in the circulatory system. (C) The ApoE-coated LNPs bind to ApoE receptors on hepatocytes and are internalized via receptor-mediated endocytosis. (D) Within the acidic environment of endocytic vesicles, the ionizable lipids in the LNPs become protonated, acquiring a positive charge. This promotes electrostatic interaction with the negatively charged endosomal membrane, leading to membrane fusion and cytoplasmic release of the double-stranded siRNA (ds-siRNA). (E) In the cytoplasm, the ds-siRNA is processed by a protein complex consisting of Dicer (a ribonuclease), TRBP (HIV-1 trans-activation response RNA-binding protein), and PACT (protein activator of the interferon-induced protein kinase). This complex removes the passenger (sense) strand, while the guide (antisense) strand is loaded into the RNA-induced silencing complex (RISC). (F) The Argonaute 2 (AGO2) enzyme within RISC directs the guide strand to bind complementary sequences in TTR mRNA, resulting in targeted cleavage and degradation of the transcript. (G) Suppression of TTR mRNA translation reduces the production of unstable TTR tetramers, thereby preventing their misfolding and aggregation into amyloid fibrils. This reduction in amyloid burden stabilizes or improves clinical manifestations such as neuropathy and cardiomyopathy. Abbreviations: AGO2, Argonaute 2 enzyme; ApoE, apolipoprotein E; ds-siRNA, double-stranded small interfering RNA; hATTR amyloidosis, hereditary transthyretin amyloidosis; IV, intravenous; LNPs, lipid nanoparticles; mRNA, messenger RNA; PACT, protein activator of the interferon-induced protein kinase; RISC, RNA-induced silencing complex; RNAi, RNA interference; siRNA, small interfering RNA; TRBP, HIV-1 trans-activation response (TAR) RNA-binding protein; TTR, transthyretin.
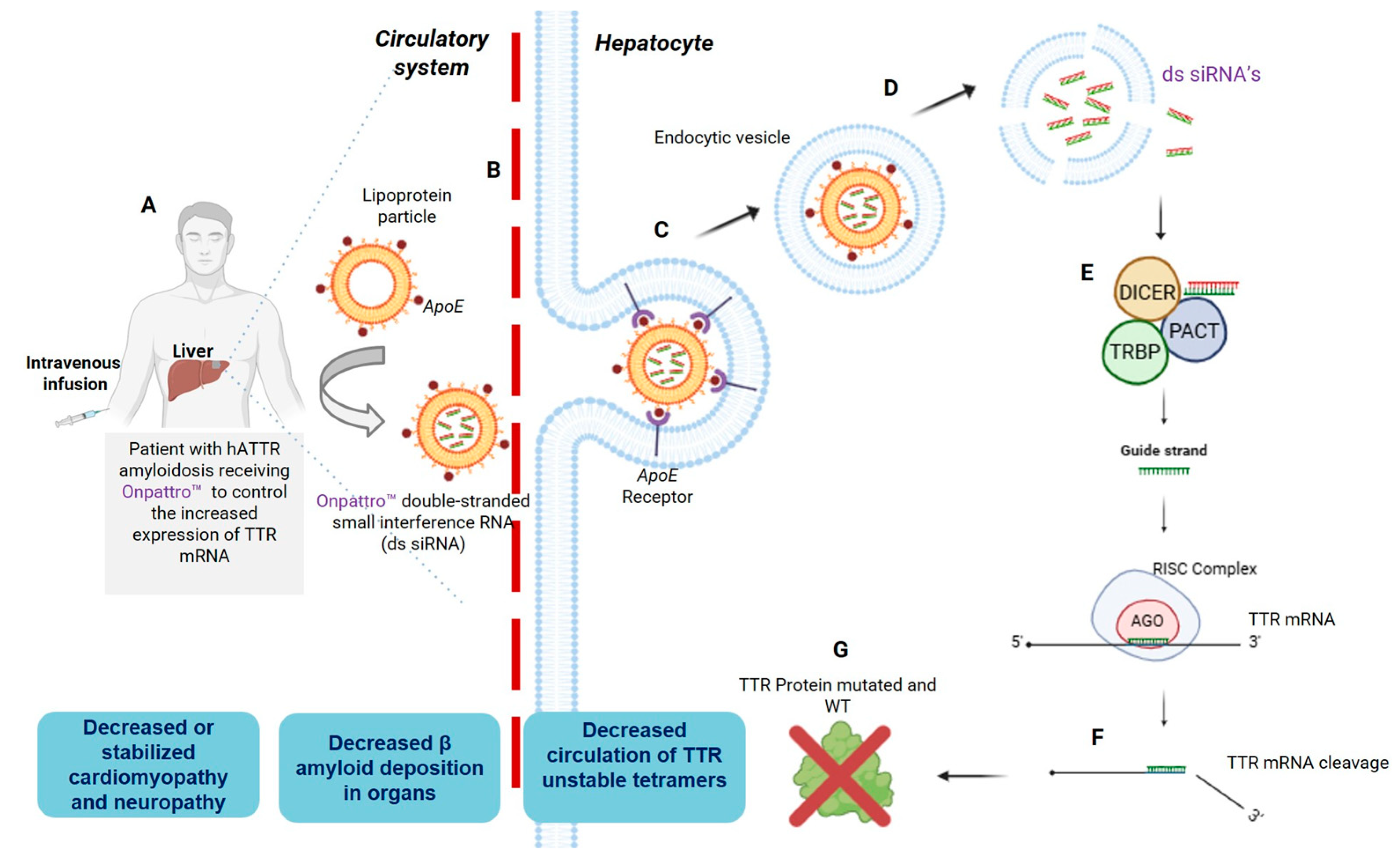
Figure 4.
Givosiran therapy for reducing acute hepatic porphyria attacks (AHP). (A) Patients with AHP receive givosiran by subcutaneous injection. (B) Anti-ALAS1 siRNA contains an N-acetyl galactosamine (GalNac) molecule conjugated at the 3’-end of the passenger strand. (C) GalNac moiety binds to asialoglycoprotein receptor (ASGPR) on hepatocytes. (D) An endocytic vesicle is formed and internalize the siRNA molecule into the cell cytoplasm. (E) siRNA GalNac conjugated is delivered in the cytoplasm. (F) The siRNA molecule forms a complex with Dicer, TRBP, and PACT proteins, that will eliminate the passenger strand. (G) The remaining siRNA guide strand loaded in RISC complex binds to complementary sequences in ALAS1 mRNA, allowing AGO enzyme to execute the cleavage of ALAS1 mRNA. (H) Knock-down of ALAS1 expression prevents further formation of toxic intermediates (ALA and PBG), reducing clinical signs of porphyria. AGO, Argonaute 2; Dicer, ribonuclease enzyme; PACT, Protein activator of the interferon-induced protein kinase (PKR); RISC, RNA-induced silencing complex; TRBP, HIV-1 Trans-activation response (TAR) RNA-binding protein.
Figure 4.
Givosiran therapy for reducing acute hepatic porphyria attacks (AHP). (A) Patients with AHP receive givosiran by subcutaneous injection. (B) Anti-ALAS1 siRNA contains an N-acetyl galactosamine (GalNac) molecule conjugated at the 3’-end of the passenger strand. (C) GalNac moiety binds to asialoglycoprotein receptor (ASGPR) on hepatocytes. (D) An endocytic vesicle is formed and internalize the siRNA molecule into the cell cytoplasm. (E) siRNA GalNac conjugated is delivered in the cytoplasm. (F) The siRNA molecule forms a complex with Dicer, TRBP, and PACT proteins, that will eliminate the passenger strand. (G) The remaining siRNA guide strand loaded in RISC complex binds to complementary sequences in ALAS1 mRNA, allowing AGO enzyme to execute the cleavage of ALAS1 mRNA. (H) Knock-down of ALAS1 expression prevents further formation of toxic intermediates (ALA and PBG), reducing clinical signs of porphyria. AGO, Argonaute 2; Dicer, ribonuclease enzyme; PACT, Protein activator of the interferon-induced protein kinase (PKR); RISC, RNA-induced silencing complex; TRBP, HIV-1 Trans-activation response (TAR) RNA-binding protein.
Figure 5.
The role of C16 molecules in siRNA internalization across lipidic cell membranes. This illustration depicts the mechanism of internalization for C16-modified siRNA molecules into cells. (1) The 2’-O-hexadecyl (C16) modification is represented as a blue tail, signifying a short fatty acid chain that enhances the siRNA's lipophilicity. (2) The C16 tail interacts with cellular membranes and membrane proteins, facilitating initial contact. (3, 4) Subsequently, siRNA is internalized into endosomes. (5) Endosomal escape enables the siRNA to exit the endosomal compartment and subsequently initiate RNAi-mediated gene silencing. Modified from Anlylam, Delivery Platforms – C16 Conjugates (2023) [77].
Figure 5.
The role of C16 molecules in siRNA internalization across lipidic cell membranes. This illustration depicts the mechanism of internalization for C16-modified siRNA molecules into cells. (1) The 2’-O-hexadecyl (C16) modification is represented as a blue tail, signifying a short fatty acid chain that enhances the siRNA's lipophilicity. (2) The C16 tail interacts with cellular membranes and membrane proteins, facilitating initial contact. (3, 4) Subsequently, siRNA is internalized into endosomes. (5) Endosomal escape enables the siRNA to exit the endosomal compartment and subsequently initiate RNAi-mediated gene silencing. Modified from Anlylam, Delivery Platforms – C16 Conjugates (2023) [77].
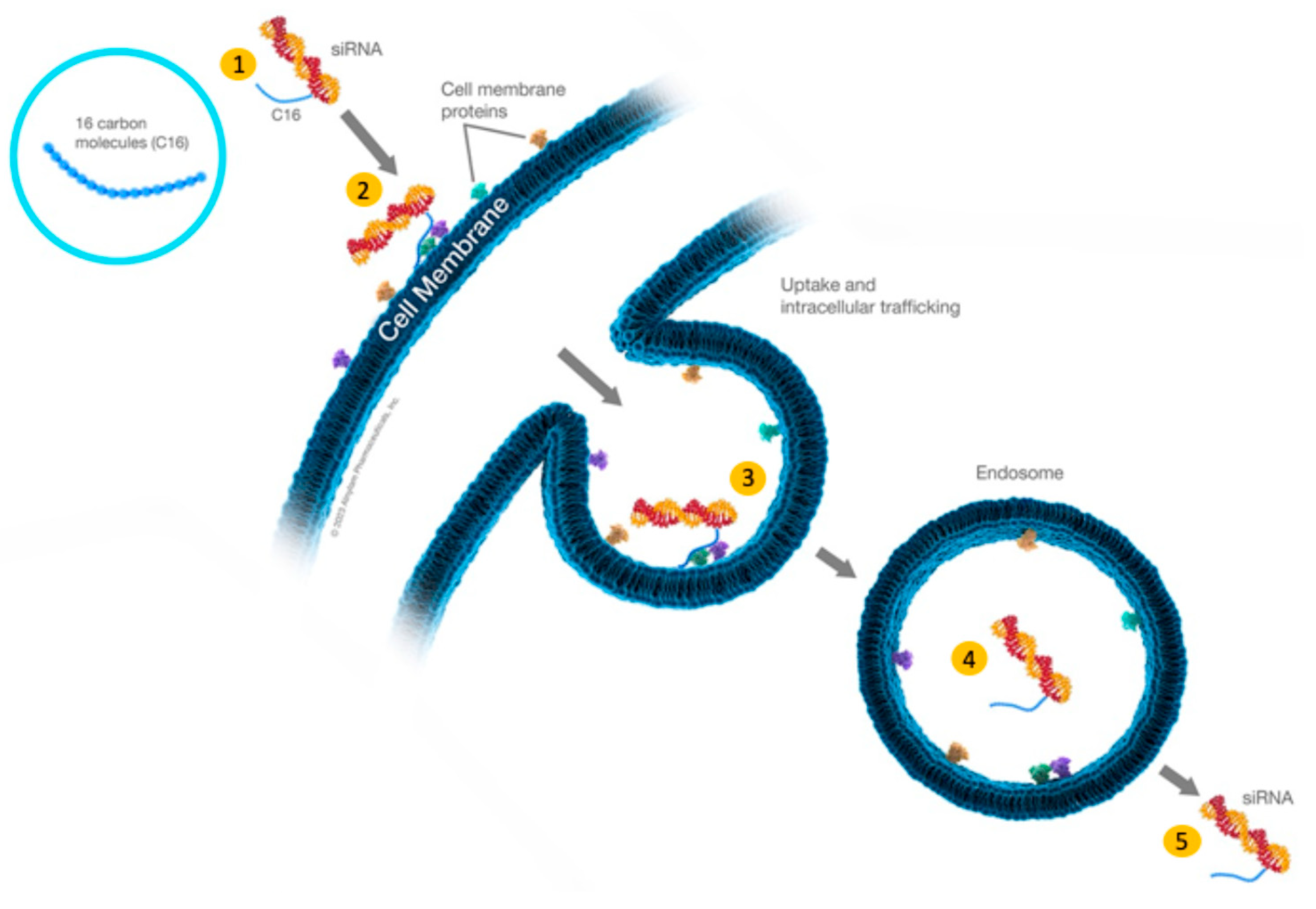
Figure 6.
Schematic Representation of a Lipophilic-siRNA Conjugate. This figure presents a chemical and spatial modeling approach to illustrate the interaction between C16 and siRNA. (1) On the left, the oxygen atom at the 2′ position of the adenine ribose in the sense strand of siRNA interacts with the 16-carbon chain (2′-O-C16), indicated within a dashed red circle. (2) The central section displays the C16 chain, which consists of 16 carbon atoms. (3) On the right, a spatial representation of siRNA is shown, with the C16 molecule depicted as a protruding gray structure. Modified from Vasant Jadhav and Martin Maier (2022) [73].
Figure 6.
Schematic Representation of a Lipophilic-siRNA Conjugate. This figure presents a chemical and spatial modeling approach to illustrate the interaction between C16 and siRNA. (1) On the left, the oxygen atom at the 2′ position of the adenine ribose in the sense strand of siRNA interacts with the 16-carbon chain (2′-O-C16), indicated within a dashed red circle. (2) The central section displays the C16 chain, which consists of 16 carbon atoms. (3) On the right, a spatial representation of siRNA is shown, with the C16 molecule depicted as a protruding gray structure. Modified from Vasant Jadhav and Martin Maier (2022) [73].
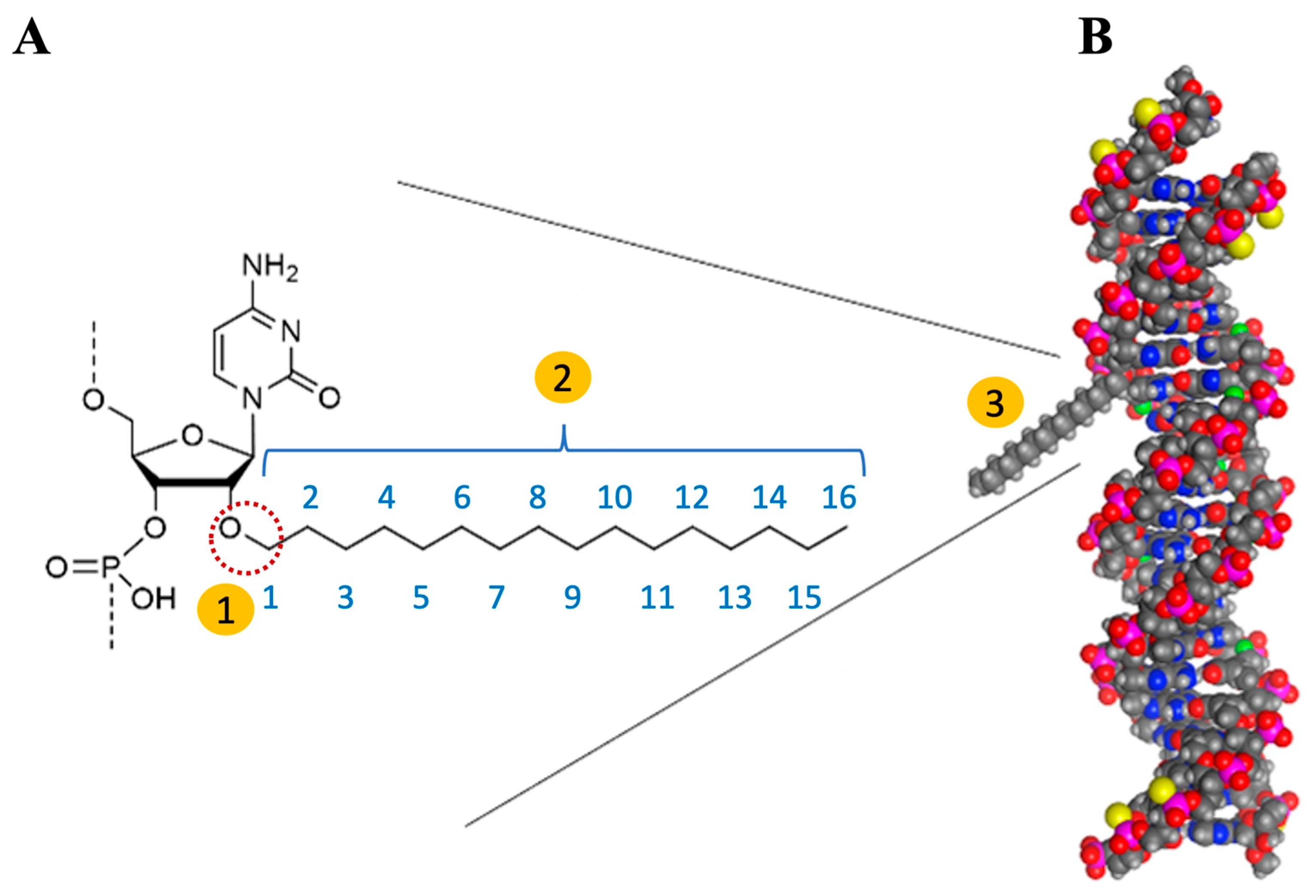
Figure 7.
Structure of Human APP-Targeting siRNA XVIII. This illustration depicts the C16-siRNA structure, as referenced in Brown et al. (2022) [72]. The key components include: (i) the sixteen carbon atoms of the lipophilic moiety (C16), represented by blue-filled circles; (ii) a double-headed orange arrow indicating the conjugation of the C16 ligand to the 2' position of the pentose in the adenine nucleotide (colored blue); (iii) upper-case and lower-case letters denoting the 2′-deoxy-2′-fluoro (2′-F) and 2′-O-methyl (2′-OMe) ribosugar modifications, respectively; (iv) vertical orange lines connecting the sense and antisense strands, illustrating Watson and Crick hydrogen bonds between complementary nucleotides; (v) underlined uppercase letter A in the antisense strand indicates a modification of glycol nucleic acid (GNA); (vi) the symbol (•) indicating phosphorothioate (PS) linkages; and (vii) "VP," which refers to the incorporation of 5'-(E)-vinylphosphonate.
Figure 7.
Structure of Human APP-Targeting siRNA XVIII. This illustration depicts the C16-siRNA structure, as referenced in Brown et al. (2022) [72]. The key components include: (i) the sixteen carbon atoms of the lipophilic moiety (C16), represented by blue-filled circles; (ii) a double-headed orange arrow indicating the conjugation of the C16 ligand to the 2' position of the pentose in the adenine nucleotide (colored blue); (iii) upper-case and lower-case letters denoting the 2′-deoxy-2′-fluoro (2′-F) and 2′-O-methyl (2′-OMe) ribosugar modifications, respectively; (iv) vertical orange lines connecting the sense and antisense strands, illustrating Watson and Crick hydrogen bonds between complementary nucleotides; (v) underlined uppercase letter A in the antisense strand indicates a modification of glycol nucleic acid (GNA); (vi) the symbol (•) indicating phosphorothioate (PS) linkages; and (vii) "VP," which refers to the incorporation of 5'-(E)-vinylphosphonate.
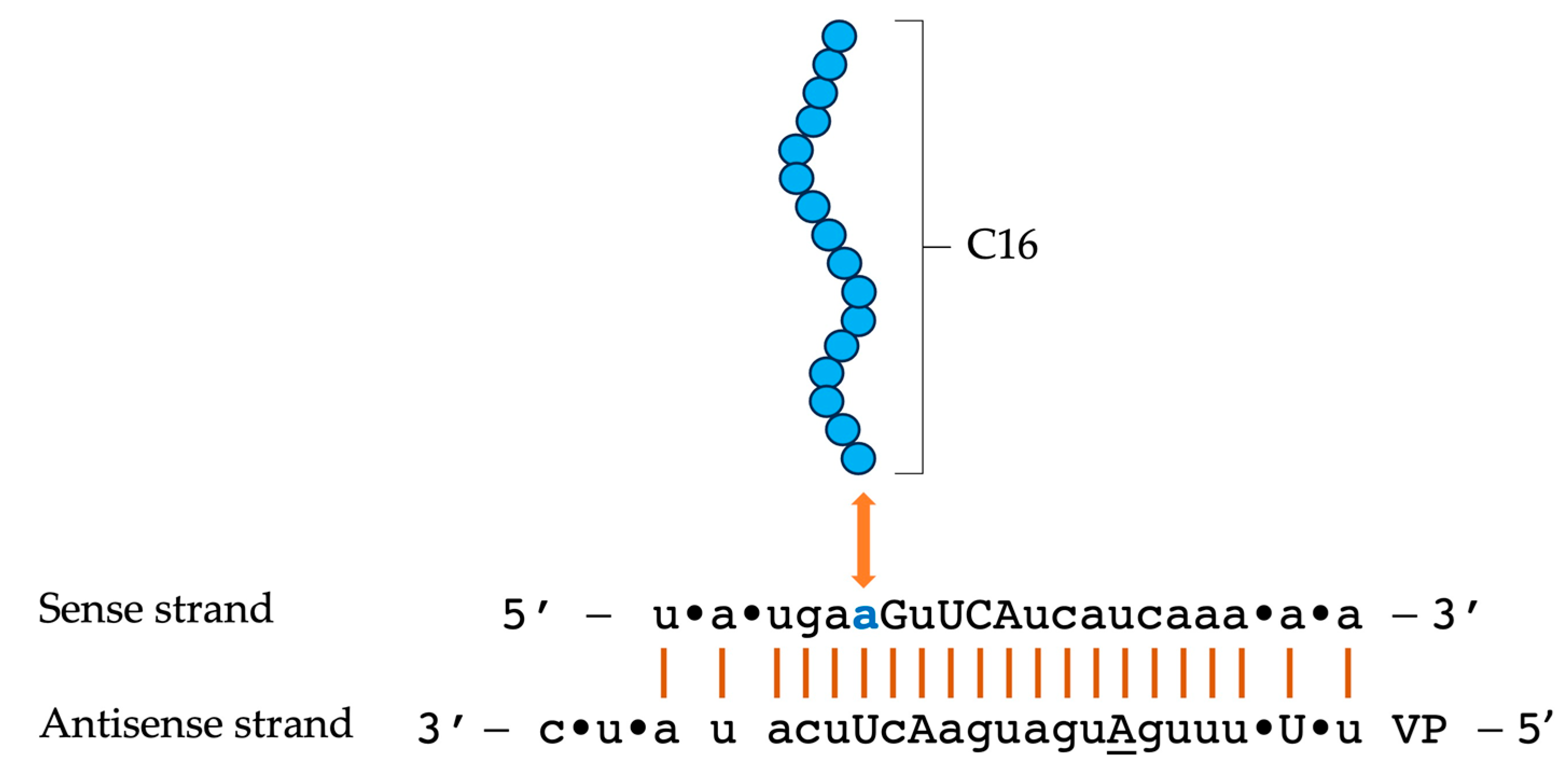
Figure 8.
Biodistribution of C16-siRNAs across various brain regions after intrathecal injection in rats. Immunohistochemistry (IHC) was performed using an anti-siRNA rabbit polyclonal antibody, which was detected with an anti-rabbit HRP secondary antibody. C16-siRNAs (right) demonstrated substantial penetration compared to unconjugated siRNAs (left), effectively reaching the cortex and hippocampus. Additionally, signals were detected in the olfactory bulb and brainstem.
Figure 8.
Biodistribution of C16-siRNAs across various brain regions after intrathecal injection in rats. Immunohistochemistry (IHC) was performed using an anti-siRNA rabbit polyclonal antibody, which was detected with an anti-rabbit HRP secondary antibody. C16-siRNAs (right) demonstrated substantial penetration compared to unconjugated siRNAs (left), effectively reaching the cortex and hippocampus. Additionally, signals were detected in the olfactory bulb and brainstem.
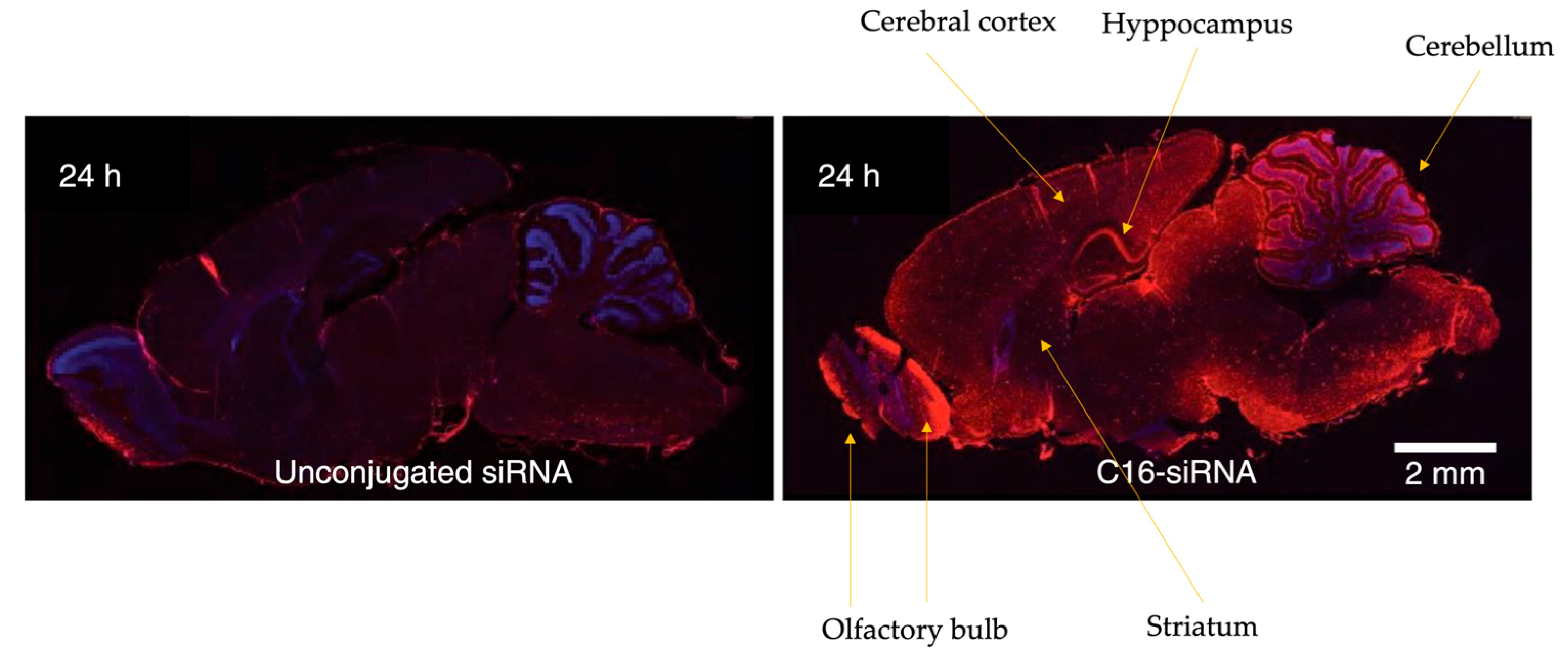
Figure 9.
Intrathecal injection of C16-siRNAs. Intrathecal (IT) injections were performed in male Sprague Dawley rats using C16-siRNAs diluted in artificial cerebrospinal fluid at a concentration of 30 mg/mL. A total volume of 30 μL was administered, resulting in a dose of 0.9 mg per animal. The IT injection was executed by puncturing the lumbar region of the spinal cord between the L3 and L5 vertebrae.
Figure 9.
Intrathecal injection of C16-siRNAs. Intrathecal (IT) injections were performed in male Sprague Dawley rats using C16-siRNAs diluted in artificial cerebrospinal fluid at a concentration of 30 mg/mL. A total volume of 30 μL was administered, resulting in a dose of 0.9 mg per animal. The IT injection was executed by puncturing the lumbar region of the spinal cord between the L3 and L5 vertebrae.
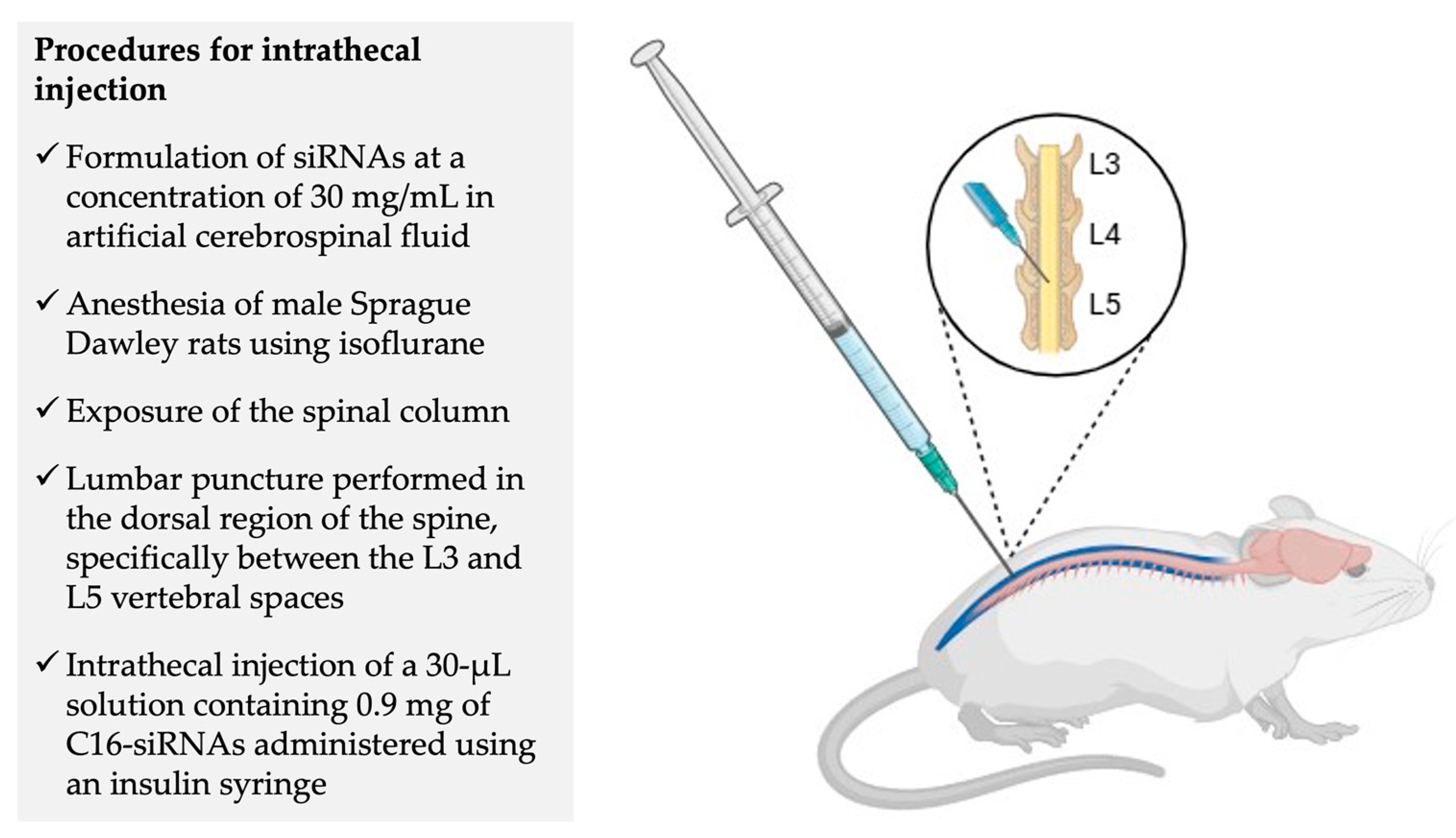
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



