Submitted:
12 June 2025
Posted:
16 June 2025
You are already at the latest version
Abstract
Dual-specificity protein phosphatases (DUSPs) are a family of proteins that dephosphorylate both phospho-serine/threonine and phospho-tyrosine residues of Mitogen-Activated Protein Kinases (MAPKs). MAPKs are involved in a large number of cellular processes, including pro-liferation, differentiation, apoptosis and stress response. Therefore, dysregulation, or improper functioning of the MAPK signalling is involved in the onset and progression of several diseas-es, including cancer. Likewise, dysregulation of DUSPs markedly affects cancer biology. The importance of MAPKs in the modulation of tumour development has been known for a long time, and MAPKs are consistently used as molecular targets for cancer therapy. However, in the last decade, DUSPs have acquired a greater interest as possible therapeutic targets to regulate MAPK activity, and to prevent resistance mechanisms to MAPK-targeting therapies. Moreover, the possibility of exploiting DUSPs as biomarkers for the diagnosis and prognosis of specific types of cancer is also emerging. In this review, we report what is known in the literature on the role of DUSPs in cancer onset and progression, focusing on those active on the extracellular signal-regulated kinases (ERK), in particular on ERK1/2 and ERK5 conventional MAPKs. The specific role of each ERK-targeting DUSP in supporting or hampering cancer progression in the context of different types of cancer is also discussed.
Keywords:
dual-specificity
; protein phosphatase
; mitogen-activated protein kinase
; cancer
; targeted therapy
; ERK1/2/5
1. Introduction
Mitogen-activated protein kinases (MAPKs) are involved in a number of physiological processes, and are activated by a large variety of stimuli. There are four conventional MAPKs that include the extracellular signal-regulated kinase 1 and 2 (ERK1/2), c-Jun N-terminal kinase (JNK) 1-3, p38 (α, β, γ, and δ) and ERK5. Atypical MAPKs are ERK3, ERK4, ERK8 (also known as ERK7) and the Nemo-like kinase. MAPKs phosphorylate specific serines and threonines of target proteins, to turn on or off the activity of the substrate protein, and regulate cellular activities ranging from gene expression, survival and proliferation to migration, metabolism and apoptosis. Because of the many important cellular functions controlled by MAPKs, they have been studied extensively to define their roles in cellular physiology and human diseases. [1,2].
Dual-specificity protein phosphatases (DUSPs, also referred to as mitogen-activated protein kinase phosphatases, MKPs) are a subfamily of protein tyrosine phosphatases (PTPs) implicated in the regulation of MAPKs in mammalian cells. They act by dephosphorylating both tyrosine and threonine residues at the T-X-Y motif located within the activation loop of the substrate kinases [3]. Structure-wise, all DUSPs perform their function thanks to the presence of a conserved phosphatase domain containing aspartic acid, cysteine and arginine residues in the catalytic site. A subset of DUSPs (namely DUSP1, DUSP2, DUSP4, DUSP5, DUSP6, DUSP7, DUSP8, DUSP9, DUSP10, DUSP16 and DUSP24) contains an N-terminal region composed by two CDC25 homology 2 domains. Additionally, based on the presence or the absence of the kinase-interacting motif (KIM), a cluster of basic amino acids that mediates enzyme-substrate interaction by binding to the common domain (CD) of MAPKs, DUSPs are generally divided into two groups. The KIM-containing DUSPs, also referred to as “typical” DUSPs, includes DUSP1, DUSP2, DUSP4, DUSP5, DUSP6, DUSP7, DUSP8, DUSP9, DUSP10 and DUSP16, while those (DUSP3, DUSP11, DUSP12, DUSP13, DUSP14, DUSP15, DUSP18, DUSP19, DUSP21, DUSP22, DUSP23, DUSP24, DUSP26, DUSP27 and DUSP28) that do not contain the KIM domain, are considered “atypical” DUSPs [4,5]. DUSPs belonging to the typical group can be further divided into three subgroups based on sequence homology, intracellular localization and substrate specificity. The first group includes DUSP1, DUSP2, DUSP4 and DUSP5 that are inducible nuclear proteins, while the second is formed by the cytoplasmic ERK-selective DUSP6, DUSP7 and DUSP9. The last group includes DUSP8, DUSP10 and DUSP16 that preferentially inactivate JNK and p38 MAPKs, and are located in both the cytoplasm and the nucleus [6].
MAPKs are often deregulated in various types of cancer, and, since DUSPs are closely involved in their regulation, several studies have been carried out to shed light on the possible role of the latter in tumour development. We review here the contribution of DUSPs in MAPK regulation in cancer, focusing on ERK-targeting DUSPs (Figure 1).
2. Role of ERK-Targeting DUSPs in Cancer
2.1. DUSP1
DUSP1, also referred to as MKP-1, was the first DUSP to be discovered, and participates in the inactivation of several MAPKs, including JNK, ERK1/2, ERK5 and p38 [7,8]. DUSP1 is widely expressed in various tissues, and its expression is regulated by a wide variety of stimuli, such as cellular stress, cytokines, lipopolysaccharide and glucocorticoids [9,10,11]. Together with DUSP2, DUSP4 and DUSP5, DUSP1 belongs to the family of inducible nuclear DUSPs.
Several lines of evidence seem to indicate that DUSP1 may support cancer onset and development. As for breast cancer (BC) in particular, An early report showed that DUSP1 is expressed in poorly differentiated or in late stages BC [12]. Along this line, DUSP1 was later found to be expressed at low levels in normal breast and in ductal hyperplasia, while at higher levels in in situ carcinoma, and to be overexpressed in approximately 50% of infiltrating BC [13]. Among the mechanisms that may support the increased expression of DUSP1 in BC, KLF5, the high expression of which has been associated with shorter survival in BC patients [14], is a possible candidate. Indeed, KLF5 overexpression increased DUSP1 protein levels in oestrogen receptor positive (ER-positive) MCF7 and triple-negative (i.e. lacking ER, progesterone receptor and HER2 overexpression) BC (TNBC) Hs578T cells. At the same time, KLF5 ablation determined the down-regulation of DUSP1 as well the increase of apoptosis in immortalized mammary epithelial MCF10A and in the TNBC BT20 cell lines. Mechanistically, DUSP1 protein levels were regulated by KLF5 via a MEK1/2-ERK1/2-dependent phosphorylation, and proved to be necessary and sufficient for KLF5-supported cell survival [15]. The upregulation of DUSP1 by tumour-supporting genes has also been described in a study performed in TNBC MDA-MB-231 and MDA-MB-468 cells, showing that Β2-adrenergic receptor, which may act as a pro-tumoral gene [16], determines an increase of DUSP1 and a reduction of ERK1/2 phosphorylation. The latter was DUSP1-dependent, since it was prevented by either genetic or pharmacological (with the DUSP1/6 inhibitor BCI, 17) DUSP1 inhibition [18]. Finally, using a BCI-derivative, it was later shown that BCI-215 elicits apoptosis in MDA-MB-231 cells and sensitizes the latter to immune cell killing. In these cells, treatment with BCI-215 activated ERK1/2 signalling without generating reactive oxygen species (ROS) DNA damage, pointing to good potential for its translation to the clinics. Along this line, BCI-215 did not induce toxicity in zebrafish embryos or epithelial cells [19].
As far as colon cancer (CC) is concerned, despite an early report that DUSP1 is overexpressed in the early phases of carcinogenesis and progressively lost in the course of progression [12], several lines of evidence point to a pro-tumoral role of DUSP1 in CC. In a study performed using HCT116 cells, glucose deprivation resulted in ERK1/2-dependent apoptosis. However, AMPK-dependent increased levels of DUSP1/2 protected HCT116 cells from glucose deprivation by suppressing the pro-apoptotic effect of ERK1/2 [20]. In another study, Lee and colleagues focused on a possible relationship between the topoisomerase I inhibitor camptothecin (CPT; [21]) and DUSP1 and ERK1/2 phosphorylation in CC cells. Treatment with CPT induced apoptosis in HCT116 cells, and determined a marked decrease of DUSP1 expression with a concurrent increase of ERK1/2 phosphorylation and nuclear localization. Suppression of ERK1/2 activity with MEK inhibitors (U0126 and PD98059; [22]) decreases CPT-induced apoptosis. Taken together, these findings seem to indicate that DUSP1 inhibition, and the related ERK1/2 activation, could be of potential interest for the treatment of CC [23]. Accordingly, NSC 95397, a quinone-based inhibitor of DUSP1 and DUSP6 [24], reduces the viability and induces apoptosis of CC cells (SW480, SW620 and DLD-1), by inducing p21 expression and caspase-3 activity via enhanced expression of ERK1/2 [25]. Using a model of BALB/c-foxn1nu nude mice and two different cell lines (namely HCT116 and SW480), it was shown that DUSP1 promotes colorectal cancer (CRC) growth in vivo (Table 1). Overexpression of DUSP1 determined the inactivation of the ERK1/2 and p38 signaling pathways. Conversely, the long non-coding RNA CDKN2B-AS1, shown to target DUSP1 in vitro and in vivo, lead to the suppression of CRC growth by activating the ERK1/2 pathway [26]. Finally, as for malignant peripheral nerve sheath tumors (MPNSTs), knock-down (KD) of DUSP1 and/or DUSP6, which are highly expressed in these tumours as compared to normal tissue, leads to reduced MPNST cell growth and to increased ERK1/2 phosphorylation. In vivo, administration of the DUSP1/DUSP6 inhibitor BCI increased ERK1/2 and JNK activation, caused tumour necrosis and fibrosis, and reduced tumor volume in MPNST xenografts established with S462.TY cells or patient-derived cells [27].
Concerning a possible role of DUSP1 in the support of cancer aggressiveness, it has also been reported that this DUSP supports drug resistance. Indeed, the overexpression of DUSP1 rendered non-small cell lung carcinoma (NSCLC) H460 cells resistant to cisplatin, while its down-regulation sensitized these cells to cisplatin-induced cell death. Interestingly, in both NSCLC H460 and ovarian cancer (OC) OVCAR3 cell lines, cisplatin induced DUSP1 expression through a post-transcriptional mechanism regulated by ERK1/2. Another study conducted in NSCLC cell lines (PC-9 and HCC827) demonstrated that the overexpression of DUSP1 is responsible for the resistance to the EGFR tyrosine kinase inhibitor Osimertinib, by inhibiting ERK1/2 phosphorylation. The KD of DUSP1 reverted this effect, and restored the sensitivity to the Osimertinib [28]. In line with the protective role of DUSP1 to cisplatin-induced toxicity, apoptosis induced by the latter was marked in DUSP1(-/-), while being minimal in DUSP1(+/+) mouse embryonic fibroblasts. Mechanistically, induction of DUSP1 by cisplatin correlated with inactivation of JNK but not of ERK1/2 or p38 [29]. In order to clarify the role of DUSP1 in cisplatin-resistance, in a subsequent work the same authors showed that in OC cell lines (OVCA432, TOV112D, CAOV3, OVCA420, OV433, and RMG-1) cisplatin induces DUSP1 through ERK2-mediated phosphorylation. Indeed, impairment of ERK2 activation (by the MEK1/2 inhibitor U0126) or expression (by siRNA) prevented cisplatin-induced DUSP1 increased expression, and enhanced cisplatin-induced cell death. The latter effect was also achieved upon KD of either ERK2 or DUSP1, likely by decreasing the level of Bcl-2 protein. Collectively, these results suggested that targeting ERK1/2-DUSP1 signaling could overcome cisplatin resistance in OC [30]. Finally, in another study, the same authors demonstrated that DUSP1 KD by shRNA in CAOV3 cells increased both basal and rapamycin-induced autophagic flux, while DUSP1 overexpression had the opposite effect. Interestingly, cisplatin-resistant CAOV3 (CAOV3-CR) cells exhibited elevated DUSP1 expression levels, and were refractory to rapamycin-induced autophagy and cytostatic effects. DUSP1 KD in CAOV3-CR cells restored sensitivity to rapamycin. Collectively, this work identifies a previously unrecognized role for DUSP1 in regulating autophagy, and suggests that suppression of DUSP1 may enhance the therapeutic activity of rapamycin [31]. Besides providing evidence of the role of DUSP1 in supporting cisplatin resistance, the above studies indicate that DUSP1 supports the malignant phenotype of OC cells. A more recent study supports this role of DUSP1 in OC. Indeed, Chondroitin sulfate N-acetylgalactosami-nyltransferase-2 (CSGALNACT2), a Golgi transferase, downregulated the levels of DUSP1 and the phosphorylation of ERK1/2 while inhibit inhibiting cell migration and invasiveness [32]. DUSP1 has also been associated with AKT-targeting agents. In a study performed using cell lines from urothelial bladder cancer (BLCA), a tumour in which 50–70% of cases show activation of the PI3K/AKT/mTOR signalling [33], the pan-AKT inhibitor MK-2206 decreased cell viability in a number of cell lines. However, MK-2206-sensitive and resistant cell lines showed opposite responses as for the levels of ERK1/2 phosphorylation, with a reduction of the latter in the sensitive ones and an increase in those that were resistant. The possibility that this effect was due to DUSP1 was supported by the occurrence of an inverse relationship between DUSP1 expression and ERK1/2 phosphorylation in all the analysed cell lines, and by the fact that KD of DUSP1 reverted the effect of MK-2206 [34].
One of the first lines of evidence of a possible role of DUSP1 as a tumour suppressor reported that DUSP1 expression and the subsequent ERK1/2 dephosphorylation might support apoptosis in BC. Indeed, exposure to nitric oxide (NO) determined an increase of DUSP1 transcripts and a decrease of ERK1/2 phosphorylation, and induced apoptosis in ER-positive ZR7530 cells and in TNBC MDA-MB-468 cells. On the contrary, NO failed to increase DUSP1 expression, ERK1/2 inactivation and apoptosis in TNBC MDA-MB-231 cells [35].
Some studies provided evidence of the role of DUSP1 as an oncosuppressor in NSCLC. In an early paper, the authors assessed, by immunohistochemistry and RT-PCR analysis respectively, DUSP1 expression and ERK1/2 phosphorylation in samples obtained from twenty patients with lung squamous cell carcinoma (SCC) and five normal lungs. Expression of DUSP1 negatively correlated with tumour differentiation, and, accordingly, both the phosphorylation and expression of ERK1/2, positively correlated with tumour differentiation. This study suggested that DUSP1 may be a candidate as a marker for the prognosis of lung SCC [36]. In further support of the negative role of DUSP1 in supporting lung cancer cell proliferation, treatment of mice with a γ-secretase inhibitor (LY-411575) reduced K-Ras(G12V)-driven NSCLC growth, while upregulating DUSP1 protein level and decreasing ERK1/2 phosphorylation. Carcinomas from treated mice presented reduced levels of the transcriptional repressor HES1, which was shown, in vitro, to directly bind to and repress DUSP1 promoter [37]. DUSP1 expression has also been associated with gefitinib sensitivity. Indeed, in NSCLC PC9 cells, DUSP1 overexpression abrogated long non-coding RNA CASC9-induced resistance to gefitinib and reduced ERK1/2 phosphorylation [38].
Concerning hepatocellular carcinoma (HCC), several lines of evidence indicate that DUSP1 may counteract tumorigenesis, making it a possible target in this neoplasm. In a first report, Liu and colleagues showed that hypoxia enhances DUSP1 transcription in HepG2 cells. DUSP1-KD enhanced HIF-1α phosphorylation and transcriptional activity, as witnessed by the increased expression of the HIF-regulated gene erythropoietin [39]. In a later work, the same authors provided evidence of the underlying mechanisms, showing that suppression of DUSP1 facilitates the interaction between the HIF-1α subunit and the coactivator p300, likely by increasing ERK1/2 phosphorylation. Indeed, treatment with the MEK1/2 inhibitor PD98059 prevented the increase of HIF-1 activity caused by DUSP1 suppression. Taken together, the above results suggest that hypoxia-induced DUSP1 protects overactivation of HIF-1 through the inhibition of ERK1/2 kinase activity [40]. In line with an anti-tumoral role of DUSP1, HCC patients with poorer prognosis showed low DUSP1 protein levels. DUSP1 inactivation resulted to be due to either ERK1/2-SKP2-CKS1-dependent ubiquitination and subsequent DUSP1 degradation or promoter hypermethylation associated with loss of heterozygosity at the DUSP1 locus. Functional studies revealed that DUSP1 reactivation led to the suppression of ERK1/2-SKP2-CKS1 activity, inhibition of proliferation and induction of apoptosis in human hepatoma cell lines 7703, HuH7, SNU-182, and SNU-387 [41]. Interestingly, in a transgenic rat model expressing a dominant-negative mutant of Cx32 (major hepatocyte gap junction protein) in the hepatocytes, the downregulation of DUSP1 with consequent upregulation of ERK1/2 led to an increase of ethanol-related hepatocarcinogenesis. In particular, oral ethanol administration promoted hepatocarcinogenesis with a higher incidence in transgenic compared with wt rats, pointing to an important anti-tumoral role of DUSP1 in vivo [42]. Another study performed using HepG2 and HBV-genome-transfected HepG2 (HepG2.2.15) cells suggested that Hepatitis B virus X (HBx) protein may support HCC development by inhibiting DUSP1 [43]. Thus, in HCC, DUSP1 may represent a valuable prognostic marker as well as a potential therapeutic target.
Additional lines of evidence have been collected in support of an antitumoral role of DUSP1 in other types of cancer. With regard to endometrioid adenocarcinoma (EEA), a type of endometrial cancer (EC), IHC analysis on tumour and normal tissue provided evidence that DUSP1 downregulation correlates with advanced stages. Accordingly, it was found that the Ishikawa cell line was the only one with high levels of DUSP1 mRNA and protein among a number of EEA cell lines (Hec1A, Hec1B, RL952 and Ishikawa). In the former, DUSP1 silencing promoted cell proliferation and migration along with an increase of ERK1/2 phosphorylation [44]. DUSP1 is also crucial in prostate cancer (PC), since its expression is downregulated in advanced and metastatic carcinomas [45]. This is consistent with another study in which it was shown that most of apparently normal glands, benign prostatic hyperplasia, and low-grade prostatic intraepithelial neoplasia (PIN) samples show high DUSP1 expression, while DUSP1 expression levels are low or even absent in high-grade PIN and PC IHC samples [46]. These data are at variance with an earlier report, performed using in situ hybridization in 50 cases, showing that DUSP1 mRNA is overexpressed in high-grade prostatic intraepithelial neoplasia (PIN) compared with normal tissue. [47]. A correlation between DUSP1 and the expression of SNAIL, ERK1/2, JNK and P38 in either cell lines (DU145 and PC3) and tissue samples from patients diagnosed with benign prostatic hypertrophy or PC has also been reported. DUSP1 k/o caused an increase of SNAIL and ERK1/2 phosphorylation, and lead to increased tumour growth and invasion, while DUSP1 overexpression decreased tumour growth [48]. More recently, in line with a possible antitumoral activity of DUSP1, Doronzo and colleagues found that in D4M BRAFV600E murine melanoma cells, TFEB silencing prevents DUSP-1 inactivation, reduces ERK1/2 activity and tumour growth, highlighting its critical role in melanoma cell proliferation and metabolic adaptation [49]. Concerning the role of DUSP1 in esophageal squamous cell carcinoma (ESCC), it was demonstrated that the circadian transcription factor ARNTL (also known as BMAL1) overexpression upregulates the expression of DUSP1 by inducing its transcription. This overexpression of DUSP1 blocks the phosphorylation of ERK1/2 and mediates anti-cancer activity in both in vitro and in vivo models [50]. Finally, Cheng and colleagues found that the administration of baculovirus-derived recombinant DUSP1 reduced the level of ERK1/2 phosphorylation in cell lines from different types of cancer (GC-7901, MCF-7, HeLa, A49, PC-2 and HepG2), and decreased the proliferation in HeLa cells [51].
DUSP1 is one of the first discovered and most studied DUSPs, and its role in cancer onset and progression is well established. However, DUSP1 exhibits both pro-tumoral and anti-tumoral activities depending on the type of cancer, as it has, for example, a clear anti-tumoral role in NSCLC and HCC and a pro-tumoral role in OC and BC, while its function in melanoma and endometrial cancer needs further clarification to understand its possible therapeutic value. The modulation of DUSP1 activity may be thus included among the possible therapeutic strategies in a subset of cancers but not in others (Figure 2).
2.2. DUSP2
DUSP2 is a nuclear protein widely expressed in various tissues, and its expression is regulated by a wide variety of different stimuli, including cellular stress, cytokines, lipopolysaccharide and glucocorticoids. DUSP2 participates in the inactivation of MAPK signaling components, including ERK1/2, p38, ERK3 and ERK4 [52,53,54].
The first evidence in support of an anti-tumoral role of DUSP2, was reported by Lin and colleagues, who demonstrated that DUSP2 expression is markedly reduced or completely absent in many human cancers, and that its level of expression inversely correlates with that of HIF-1α and with cancer malignancy. Experiments performed with cancer cell lines (HeLa, HCT116, Hep3B) indicated that HIF-1α inhibited DUSP2 transcription and translation, thus resulting in prolonged phosphorylation of ERK1/2 and increased chemoresistance. Consistently, DUSP2 KD increased drug resistance under normoxia, while the forced expression of DUSP2 abolished hypoxia-induced chemoresistance to Paclitaxel, Cisplatin and Oxaliplatin in vitro and in vivo. These results demonstrate that DUSP2 is a key downstream regulator of HIF-1-mediated tumor progression and chemoresistance, so that, among the possible beneficial outcomes of HIF-targeting, we may add the re-expression of DUSP2 [55]. Along this line, a later study confirmed that HIF-1α negatively regulates the expression of DUSP2 in BC cells, while increasing ERK1/2 phosphorylation. Genetic inhibition of DUSP2 led to lapatinib resistance, while its overexpression increased cell sensitivity to lapatinib. Interestingly, low expression of DUSP2 and high expression of HIF-1α were associated with poor prognosis in ER-negative BC patients, further supporting the relevance of a HIF1α/DUSP2/ERK axis in cancer [56]. An anti-tumoral role of DUSP2 has also been reported in pancreatic ductal adenocarcinoma (PDAC). Indeed, overexpression of Mir-361-3p in PDAC cells led to increased EMT and invasion, but not proliferation, along with downregulation of DUSP2 and increased ERK1/2 phosphorylation. DUSP2 overexpression led to inactivation of the ERK1/2 pathway and to a reduction of Mir-361-3p-induced EMT [57]. A recent paper reporting IHC analysis of 164 gastric cancer (GC) cases revealed that high expression of SKA3 (spindle and kinetochore-related complex subunit 3), implicated in the regulation of mitosis, negatively correlates with DUSP2 expression and is related to N stage, peritoneal metastasis, and poor prognosis. Mechanistically, SKA3 negatively regulates the tumor suppressor DUSP2 and activates the MAPK/ERK1/2 pathway to promote GC in vitro (in the MGC-803 and HGC-27GC cell lines), and in vivo. These results indicate that the SKA3-DUSP2-ERK1/2 axis is involved in GC progression, and could be a potential therapeutic target for this cancer [58]. The antitumoral role of DUSP2 has been also confirmed in the context of BLCA by Zou and colleagues. Indeed, they demonstrated both in vitro and in vivo that the overexpression of DUSP2 suppress cancer progression through the inhibition of ERK1/2 phosphorylation and M2 macrophages chemotaxis [59].
Collectively, all the above studies point to a protective role of DUSP2 against tumor growth.
2.3. DUSP3
DUSP3, also referred to as Human Vaccinia H1-related (VHR) phosphatase, is one of the members of the atypical DUSP group. This constitutively expressed nuclear DUSP is active on JNKs and ERK1/2, and is involved in various cellular functions ranging from cell cycle regulation to immune response [60,61].
The very first article reporting a possible role of DUSP3 in cancer showed that, in HeLa cells, DUSP3 KD determines a block of cell cycle at the G1-S and G2-M transitions, and induces the initial signs of cellular senescence. These effects were JNK- and ERK1/2- dependent, because cell cycle arrest did not occur upon JNK and ERK1/2 inhibition [62].
However, accumulating evidence point to a possible role of DUSP3 as a tumor suppressor. In NSCLC, it has been reported that DUSP3 expression is epigenetically repressed by the histone H3 lysine 36 demethylase KDM2A (also called FBXL11 and JHDM1A), which is frequently overexpressed in NSCLC tumors and cell lines, and the high level of expression of which correlates with poor prognosis. KDM2A overexpression in NSCLC cells with low KDM2A levels increased cell proliferation and invasiveness (H460 and H2122 cells), while KDM2A KD abrogated tumor growth and invasiveness in NSCLC (H1792 cells) xenografts. These effects were likely supported by ERK1/2 activation following KDM2A-induced DUSP3 repression [63]. Another study showed that DUSP3 may play a role in miR-1915-3p-driven development of BC. Indeed, overexpression of miR-1915-3p, whose level is increased in BC patients compared with healthy volunteers, determined a reduction of the target gene DUSP3 while increasing the phosphorylation of ERK1/2. Functional in vitro experiments showed that miR-1915-3p enhanced cell proliferation and migration, in keeping with the evidence that patients with infiltrating carcinoma or lymph node metastasis have a higher serum level of miR-1915-3p than patients with in situ carcinoma or without lymph node metastasis [64]. A tumor soppressing role of DUSP3 has also been demonstrated in the most recent study of this phosphatases in which the author showed that L-Methionine epigenetically induces the expression of DUSP3 leading to a decreased activation of the ERK1/2 pathway and to increased sensitivity to Sorafeninb of both HepG2 cells and in a model of sorafenib-resistant HCC in rats [65].
Although initially reported as a possible tumor-supporting signal in cervical cancer cells, DUSP3, mainly through the regulation of ERK1/2, emerges as an interesting signaling molecule in other types of cancer, such as BC, NSCLC and HCC.
2.4. DUSP4
DUSP4/MKP-2 is a nuclear DUSP that is responsible for the dephosphorylation and inactivation of ERK1/2, JNK and p38, and is encoded by a highly inducible gene, which is rapidly up-regulated in response to both mitogenic and stress stimuli [66]. DUSP4 is one of the most closely related enzymes of the well characterized DUSP1, and its role in cancer progression gained importance in the last decade.
One of the first pieces of evidence of DUSP4 implication in cancer comes from a work of Yip and colleagues which provided evidence in support of the fact that pancreatic tumor cells expressing oncogenic K-RAS compensate the activation of the ERK1/2 pathway by up-regulating the expression of DUSP4 [67]. Along this line, a later study demonstrated that DUSP4 has an anti-tumoral role in the progression to invasive pancreatic cancer. Expression profile studies in pancreatic cancer cell lines (PANC-1, KP4-1 and MIApaca2) identified DUSP4 as one of the most downregulated gene upon 8p11.22-ter loss. Restoration of DUSP4 expression did not affect the proliferation but impaired invasiveness in vitro. Xenografts with restored expression of DUSP4 in PANC-1 cells with 8p loss, showed longer survival and reduced tumor growth and metastasis compared with mice transplanted with WT PANC-1. The authors also demonstrated that the activation of ERK1/2 following the decrease of DUSP4 is responsible for enhanced resistance to anoikis and invasiveness of pancreatic carcinomas [68]. A possible oncosuppressor role of DUSP4 has also been identified in head and neck SCC (HNSCC), in which the expression of G9A/EHMT2 histone methyltransferase was found to be associated with tumor growth and poor prognosis. Orthotopic xenografts using FaDu HNSCC cells with inducible-KD of G9a showed reduced growth, and increased autophagy. In vitro, KD of G9A led to increased expression of DUSP4 and to decreased phosphorylation of ERK1/2 as well as to reduced cell growth (FaDu and SAS HNSCC cell lines) [69]. Likewise, DUSP4 showed antitumor effects in GC, where IHC analysis of tumor specimens showed that DUSP4 downregulation is associated with enhanced tumor size, depth of invasion and distant metastasis. Furthermore, DUSP4 overexpression in GC cell lines (SGC-7901 and HGC-27) reduced cell viability and invasive potential, and induced apoptosis and cycle arrest. Interestingly, in the same study, it was shown that sanguinarine, a natural anti-tumoral benzophenanthridine alkaloid extracted from plants of the Papaveraceae family, inhibits growth and invasion of GC cells through the DUSP4/ERK1/2 pathway [70]. In BC, low expression of DUSP4 positively correlates with high Ki-67 score following neoadjuvant chemotherapy, and with basal-like BC subtypes. In BC cell lines (BT-549, MDA-MB-231 and MDA-MB-436, T47D, SUM159PT), low DUSP4 expression correlated with increased ERK1/2 phosphorylation. DUSP4 overexpression inhibited ERK1/2 phosphorylation and reduced cell viability, while DUSP4 KD attenuated docetaxel toxicity. Additionally, MEK1/2i (Selumetinib, U0126, or CI1040) improved chemotherapy-induced apoptosis [71]. A later study directed to deepen this latter effect showed that low DUSP4 expression is associated with high MEK1/2 phosphorylation in BC specimens and cell lines (MDA-MB-231, ZR751, MDA-MB-468, SUM159PT, BT549, MFM223, HCC1143) endowed with cancer stem cell characteristics, and that DUSP4 KD enhances mammosphere formation [72]. Another study analyzing 22 different BC cell lines showed that DUSP4 is downregulated in TNBC cell lines compared to other BC subtypes. Moreover, TNBC cells were found to be more sensitive, in vitro, to the anti-proliferative effects of simvastatin, a statin used in clinics to reduce hyperlipidaemia [73]. Importantly, simvastatin determined an increase of DUSP4 in TNBC compared with non-TNBC cell lines. Accordingly, DUSP4 overexpression, like simvastatin, reduced cell viability. Conversely, DUSP4 KD determined opposite effects. Expression of DUSP4 was accompanied by the reduction of ERK1/2 phosphorylation, while DUSP4 inhibition resulted in increased ERK1/2 phosphorylation, suggesting the involvement of ERK1/2 in the above effects [74]. Along this line, another study reported that DUSP4 KD stimulated ERK1/2 and p38 MAPK pathways, stem-like properties and metastatic capacity both in vitro in ER-positive MCF-7 cells and in vivo in xenografts of mice injected with DUSP4-KD cells derived from ER-positive and TNBC patients [75]. In another work it was demonstrated that DUSP4 is upregulated in MCF-7 cells after treatment with tamoxifen and in tamoxifen-resistant MCF-7 cells, along with increased ERK1/2 phosphorylation. However, DUSP4 overexpression did not affect tamoxifen sensitivity of MCF-7 cells, while suppressed E2-induced (17-beta-estradiol) proliferation, and abrogated ERK1/2 phosphorylation with respect to control cells. These data suggest that ERK1/2 and DUSP4 are involved in tamoxifen sensitivity. Tamoxifen may initially activate ERK1/2, inducing DUSP4 expression to reduce phospho-ERK1/2 levels. However, if phospho-ERK1/2 levels are too high, DUSP4 may be unable to fully dephosphorylate them, potentially contributing to tamoxifen resistance. [76]. A recent study showed that overexpression of oncogenic NRAS (G12V) alone is enough to induce hepatocellular carcinoma (HCC) in mice. During tumor progression, the RAS/RAF/MEK/ERK1/2 pathway was consistently activated, along with upregulation of DUSP4, DUSP6, and the cancer stem cell marker CD133. DUSP4 and DUSP6 act as negative feedback regulators, limiting MAPK signalling to prevent oncogene-induced senescence. Their silencing increased cell proliferation and ERK1/2 activation in liver cancer cells, confirming their role. The study also found a correlation between RAS pathway activation and DUSP4/6 expression in both HCC and cholangiocarcinoma (CCA), suggesting broader relevance. These findings identify DUSP4 and DUSP6 as biomarkers of RAS activity and potential therapeutic targets in RAS-driven liver cancers [77].
At variance with the above studies, a study from Cagnol and colleagues suggests that the negative feedback exerted by DUSP4 on duration and magnitude of nuclear ERK1/2 activation may support intestinal tumorigenesis. In particular, the expression of either K-RAS(G12V) or BRAF(V600E) in normal intestinal epithelial crypt cells (IECs, IEC-6) stimulated ERK1/2 activity and morphological transformation. ERK1/2 phosphorylation was restricted to the cytoplasm, while nuclear ERK1/2 dephosphorylation was found to be tightly correlated with the rapid MEK1/2-dependent expression of DUSP4. Likewise, in human CRC cells (SW480), ERK1/2 phosphorylation was confined within the cytoplasm, while treatment with pervanadate, a tyrosine phosphatase inhibitor effective on DUSP, was necessary to activate ERK1/2 in the nucleus. Accordingly, DUSP4 mRNA was found to be highly expressed, in an MEK-dependent manner, in all CRC cells analysed [78].
On the whole, DUSP4 plays an important role in cancer onset and progression, possibly with the exception of CRC, so that DUSP4-modulating agents aimed to increase its expression may provide new therapeutic options for the treatment of different types of cancer including PDAC, GC, HNSCC and BC.
2.5. DUSP5
The expression of DUSP5, also referred to as VH1-like phosphatase-3 (VH3), is induced by either heat shock or growth factor stimulation in mammalian cells [79,80]. At variance with other inducible DUSPs, DUSP5 is highly selective in its ability to bind and inactivate ERK1 and ERK2 in vitro and in vivo, and is localized in the nucleus [81]. One of the first studies linking DUSP5 to cancer reported that its promoter is hypermethylated in GC tissues but not in healthy gastric mucosa. Importantly, GC patients with hypermethylated DUSP5 promoter exhibit significantly shorter survival than those without hypermethylation. Overexpression of DUSP5 in DUSP5-low expressing GC (MKN-74 and SNU-638) cells decreased growth and colony-formation ability, and resulted in nuclear ERK1/2 dephosphorylation. This study therefore provides evidence that DUSP5 suppression by promoter hypermethylation results in increased ERK1/2 phosphorylation that supports cell proliferation, possibly driving gastric carcinogenesis. The authors also proposed that DUSP5 promoter methylation may serve as a prognostic marker for GC [82]. Another paper, supporting the antiproliferative role of DUSP5, showed that DUSP5 k/o mice showed increased sensitivity to mutant H-Ras (HRasQ61L)-driven papilloma formation in the 7,12-Dimethylbenz[a]anthracene/12-O-tetradecanoylphorbol-13-acetate (DMBA/TPA) model of skin carcinogenesis, and an increased phosphorylation and nuclear ERK1/2 that was associated with the support of tumour growth [83]. Along this line, a IHC study showed that DUSP5 is downregulated in PC with respect to normal tissues, and that DUSP5 lower expression is associated with progression and poor prognosis, pointing to the ability of DUSP5 to act as an onco-suppressor in PC [84]. Another IHC study showed that DUSP5 is also downregulated in CRC specimens with respect to normal tumour-surrounding tissue. In the same study, it was obtained in vitro evidence that DUSP5 is a negative feedback regulator of the ERK1/2 pathway upon EGF stimulation in the LIM1215 CRC cell line. Interestingly, the overexpression of DUSP5 in mouse intestinal epithelium did not affect ERK1/2 signalling, so that the authors suggested that DUSP5 does not regulate ERK1/2 signalling in the normal intestinal epithelium, while showing limited tumour suppressive activity in CRC [85]. In a more recent study, quercetin was shown to increase the expression of DUSP5 following binding of Serum Response Factor (SRF) transcription factor at DUSP5 promoter, to reduce ERK1/2 activity in the nucleus and cell proliferation, resulting in a tumour-suppressive effect [86]. In another study, DUSP5 emerged as a possible oncosuppressor in glioma by counteracting the pro-tumoral role of the p68 RNA helicase [87], that had been previously demonstrated to be overexpressed in glioma and to be correlated with poor overall survival [88]. Indeed, genetic inhibition of p68 led to increased DUSP5 expression, and to decreased ERK1/2 phosphorylation. Moreover, both p68 inhibition and DUSP5 overexpression lead to a decrease of ERK1/2 phosphorylation and of the proliferation, invasion, and migration of the glioma cell line U87 [87]. DUSP5 has also been proven to reduce proliferation and invasiveness of TNBC MDA-MB-231 and MDA-MB-468 cell lines [89]. In contrast to all the studies above, a recent work showed that DUSP5 is overexpressed in BRAF-mutant thyroid cancer cells, and that DUSP5 KD reduces proliferation and migration. It was also demonstrated that DUSP5 KD inhibits ERK1/2 phosphorylation, and that its KD enhances the anti-tumor effect of sorafenib [90].
On the whole, the almost all the above studies indicate that DUSP5 counteracts cancer onset and development in a number of different types of tumour, so that agents, including epigenetic drugs, that are able to increase/restore its expression should be sought after.
2.6. DUSP6
DUSP6/MKP-3 is an inducible cytosolic DUSP active on ERK1/2 and ERK5 [91,92,93,94,95], and is involved in many physiological functions, including DNA damage repair and stress responses, as well as cancer onset and progression [6,96]. Similarly to what observed for other inducible DUSPs, ERK1/2 activation supports both transcriptional and post-transcriptional modifications that collectively increase DUSP6 protein levels, resulting in a negative feedback in both normal and neoplastic cells that avoids excessive activation of ERKs-dependent signals, including cell proliferation [97,98,99]. Conversely, DUSP6 may also function as a tumour-supporting protein by helping tumour cells to adapt to an abnormally enhanced ERK signalling [100].
A number of studies have demonstrated a clear role of DUSP6 as an oncosuppressor in different types of cancer, lung cancer being among the most studied. In silico analysis using Gene Expression Omnibus (GEO) and Cancer Genome Atlas (TCGA) databases revealed an association between a low expression of DUSP6 and a poor prognosis in NSCLC patients. In agreement with these clinical data, DUSP6 emerged as a key player in cell migration and tumour growth, as well as in the down-regulation of several genes involved in epithelial-to-mesenchymal transition (EMT). Referring to the latter, RNA-seq identified EGFR, TGF-β and WNT signalling pathways among the genes upregulated in DUSP6 KD cells. In these cells, activation of the EGFR signalling was proven to occur via ERK5, and not via ERK1/2 [101]. Interestingly, a later IHC study performed using human specimens from 66 human lung adenocarcinoma (LUAD) showed that DUSP6 expression positively correlates with a lower expression of the proliferation marker KI67, and lower histological grade, likely acting as a negative feedback regulator of ERKs in LUAD progression [102]. In support of this hypothesis, a later study reported that DUSP6 is overexpressed in human LUAD cell lines, and impairs ERK1/2 activation [103]. Notably, aberrant ERK1/2 activation is a frequent event in NSCLC as a consequence of constitutive activating mutations of either EGFR or KRAS [104]. On the other hand, synthetic lethality has been described when mutant KRAS and EGFR proteins are co-expressed in LUAD cells (PC9, H358 and H1975), revealing the biological basis for the observed mutual exclusivity of KRAS and EGFR mutations [105]. Interestingly, DUSP6 might be involved in the toxicity elicited as above. By combining pharmacological and genetic approaches to inhibit KRAS or EGFR, it was shown that downregulation of DUSP6 (with siRNA or BCI) or overexpression of ERK1/2 can lead to RAS-induced toxicity in LUAD cell lines. This study provides evidence that tumours with mutant RAS must restrain the activity of ERK1/2 to avoid excessive toxicities while enabling tumour growth, and identifies a pro-tumoral role of DUSP6 in LUAD cancer, showing that DUSP6 may protect from the occurrence of synthetic lethality resulting when mutant KRAS and EGFR proteins are co-expressed [103]. In another work it has been reported that DUSP6 expression tracks in tandem with low ERK1/2 phosphorylation in both NSCLC patient-derived samples and cell lines. Mechanistically, DUSP6 KD in NSCLC cells expressing high levels of DUSP6 (H441 cells) significantly increased ERK1/2 phosphorylation and cell proliferation, whereas overexpression of DUSP6 in low DUSP6-expressing cells (H1975) significantly reduced ERK1/2 phosphorylation and cellular proliferation, and promoted apoptosis. These results indicate that DUSP6 expression is regulated by ERK1/2 signalling, and that DUSP6 exerts antitumor effects via negative feedback regulation of ERK1/2 in NSCLC [106]. In another study performed in LUAD cells, the repression of DUSP6 induced the overactivation (i.e. phosphorylation) of Traslation Initiation Factor 2(eIF2) and ERK1/2. Using xenografts of LUAD cells (KRAS G12D eIF2αS/S and eIF2αA/A), the authors established a connection between ERK1/2 phosphorylation and the increased phosphorylation of eIF2. In particular, they demonstrated that the repression of DUSP6 is caused by activation of eIF2 that in turn leads to the activation of ERK1/2. Consistently, either genetic or pharmacological inhibition of eIF2 determined an upregulation of DUSP6 that impairs the formation of LUAD both in vitro and in vivo [107]. In line with the above observed tumor suppressor roles of DUSP6, in A549 and H1975 NSCLC cells, inhibition of GPx3, a ROS scavenger, determined ROS-induced inactivation of DUSP6 as well as the increase of ERK1/2 phosphorylation and cell proliferation [108]. Regarding the possible mechanisms involved in ERK1/2-mediated DUSP6 expression in NSCLC, it has been reported that ETS1, a well-known nuclear target of activated ERK1/2, positively modulates DUSP6 expression by binding to its promoter region [106]. Furthermore, a recent study reported that the transcription factor ZNF251 inhibits the expression of DUSP6, by directly binding to its promoter region, and activates ERK1/2 signalling in NSCLC cell lines (A549, H520, H23, H460, and SPC-A-1) and in a mouse model with KRAS mutation (G12D) [109].
As for melanoma, DUSP6 overexpression in human melanoma cells (A375) reduced tumour take and growth of xenografts in mice. In the same study, DUSP6 levels were not found to0 be correlated with a better survival of patients with thick melanomas, Furthermore, DUSP6 overexpression in immortalized murine melanocytes enhanced anchorage-independent growth and invasive ability pointing to a protumoral role of DUSP6, at least in normal murine cells [110]. However, a recent work performed using the B16-F10 melanoma cell line demonstrated that hinokitiol, a plant-derived metal chelator, induces an increase of DUSP6 along with a DUSP6-dependent decrease of ERK1/2 phosphorylation and of cell proliferation [111]. In another study, the same authors identified a relevant role of DUSP6 in cisplatin resistance. Indeed, cisplatin increased the expression of the DNA repair protein ERCC1 and XPF, along with a reduction of DUSP6 protein. These effects were dependent on ERK1/2, because a MEK1/2 inhibitor (PD0325901) prevented their occurrence. DUSP6 overexpression prevented cisplatin-dependent induction of both ERCC1 and XPF, and restored sensitivity to cisplatin, confirming that DUSP6 has an anti-tumoral role in melanoma [112]. In a later work based on IHC of melanoma specimens, it was found that DUSP6 protein, as well as STAG2 and STAG3, two cohesin complex components, are decreased following treatment with BRAF or MEK (dabrafenib, trametinib, vemurafenib) inhibitors. Consistently, melanoma cell lines (A375, Mel1617, WM902, WM983 and M14) resistant to BRAFi or MEKi showed reduced expression of STAG2 and STAG3, and their KD led to a further decrease of DUSP6 as well as of BRAFi sensitivity, along with an increase of ERK1/2 phosphorylation. The same effects were observed using A375 and WM902 xenografts in mice, further confirming the protective role of DUSP6 in melanoma [113]. Finally, overexpression of DUSP6 was shown to counteract the pro-tumoral role of Mir-211in melanoma. Indeed, A375 cells overexpressing Mir-211 showed enhanced ability to form xenografts into SCID mice. Mir-211 overexpression led also to the increase of ERK5 phosphorylation and to the downregulation of DUSP6, which was found to be responsible for ERK5 dephosphorylation. Overexpression of DUSP6 in A375/211 cells led to the reduction of tumour growth of xenografts in mice. In the same in vivo model, they also demonstrated that Mir-211 overexpression confers resistance to vemurafenib (BRAFi) and cobimetinib (MEKi) through the activation of ERK5, a MAPK supporting melanoma growth [114] suggesting that DUSP6 may potentially prevent this undesirable effect [115].
Loss of DUSP6 expression, at either mRNA or protein level, has been proven to correlate with high ERK1/2 phosphorylation in primary human OC cells (OV2008, C13*, A2780s, A2780cp, DOV13-5, OVCAR3, SK/OV3, OV420, OV429 and OV433). Loss of DUSP6 protein resulted to be proteasome-dependent, as a consequence of high intracellular ROS. DUSP6 KD resulted in increased ERK1/2 activity, cell proliferation, anchorage-independent growth and resistance to cisplatin. Conversely, overexpression of DUSP6 in DUSP6-deficient OC cells significantly reduced ERK1/2 activity and inhibited cell prolife
ration, anchorage-independent growth and tumour development in nude mice. Furthermore, DUSP6 overexpression sensitized OC cells in vitro and xenografts in mice (A2780 cell line) to cisplatin-induced apoptosis. On the whole, accumulation of ROS during OC progression may cause the degradation of DUSP6, which in turn leads to aberrant ERK1/2 activation, thus contributing to tumorigenicity and chemoresistance of human OC cells [116]. In support of a relevant role of ubiquitination in DUSP6 downregulation in OC cells, it has been reported that gene amplification of the oncoprotein tripartite motif containing 59 (TRIM59), an E3 ubiquitin ligase, is associated with decreased expression of DUSP6 and a shorter overall survival in OC. Genetic inhibition of TRIM59 in different OC cell lines (SK/OV3 and OVCAR3) leds to a decrease of ERK1/2 phosphorylation and to an increase of DUSP6 protein level but not mRNA [117]. Along this line, a study dealing with pancreatic cancer investigated the effects of the loss of the DUSP6-dependent negative feedback on ERK1/2. By IHC analysis, the authors found a down-regulation of DUSP6 in invasive carcinoma. Overexpression of DUSP6 into invasive pancreatic cancer cells (PCI-35 and PK-8) determined a reduction of phosphorylated ERK1/2 and resulted in cell growth suppression and apoptosis [118]. An oncosuppressive role for DUSP6 has also been described in CRC. Depletion of DUSP6 in colonic epithelium in mice caused an increase of the levels of ERK1/2 phosphorylation and of KI67. It also induced colonoid (i.e. colon-derived organoid) development and intestinal tumorigenesis in Apc Min/+ mice. Additionally, genetic silencing of DUSP6 in CRC cell lines (HT29 and HCT116) determined an increase of ERK1/2 phosphorylation and of cell migration and invasion ability in vitro [119]. In line with the above work, another recent study demonstrated that the interaction of the protein Gasdermin B (GSDMB) with insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) leads to an increase of DUSP6 protein levels and subsequent inhibition of ERK1/2 phosphorylation. In both cell lines (HT-29) and mice it emerged that the increased expression of GSDMB and subsequently that of DUSP6 reduced cell proliferation and tumour growth [120]. In non-solid tumors, Xu and colleagues recently showed that in acute myeloid leukemia (AML), the Wilms tumor 1 (WT1) protein can activate DUSP6 transcription, but only in the presence of the RUNX1::RUNX1T1 fusion (also known as t(8;21)). This leads to inhibition of the ERK1/2 pathway. In contrast, when the fusion is absent, WT1 fails to induce DUSP6 expression, resulting in increased ERK1/2 phosphorylation and promoting leukemogenesis [121]. The role of DUSP6 in tumorigenesis has also been investigated in ESCC and nasopharyngeal carcinoma (NPC). Significant loss of DUSP6 was observed in 100% and 71% of ESCC and NPC cell lines, respectively. DUSP6 expression was down-regulated in 40% and 75% of tumor specimens of ESCC and NPC, respectively, compared to normal counterparts. Notably, tissue microarray analysis revealed a clinical association of DUSP6 expression with better patient survival. Overexpression of DUSP6 in ESCC (SLMT-1, KYSE70 and KYSE450) and NPC cell lines (HONE1, HNE1, CNE2 and SUNE1) reduced tumour growth and invasiveness in vitro and in xenografts in mice. This study thus provides evidence of the functional impact of DUSP6 in tumorigenesis and metastasis of ESCC and NPC [122]. Along this line, another study performed in endometrial cancer cells, showed that DUSP6 overexpression in the Ishikawa cell line led to a decrease of ERK1/2 phosphorylation and of E-cadherin expression. Additionally, DUSP6 inhibited cell growth, invasion and migration abilities [123]. Despite this study providing evidence that DUSP6 may counteract endometrial cancer progression, it has not been reported whether DUSP6 expression is reduced or increased in this type of tumor. However, hypermethylation of DUSP6 promoter, that may determine a reduction of DUSP6 expression, has been reported to be a rare event in endometrial cancer [124]. Furthermore, another recent work supported the tumour suppressor role of DUSP6 in cervical cancer, demonstrating that the protein Ribosomal L22-like 1 (RPL22L1) is overexpressed in this type of tumour and is capable of binding and sequestering DUSP6. This led to an increased phosphorylation of ERK1/2 and conferred resistance to Sorafenib as demonstrated with both in vitro and in vivo assays [125]. DUSP6 seems to have an oncosuppressor role also in Renal cell carcinoma (RCC) as it was demonstrated by Liu and colleagues. They showed that DUSP6 downregulation correlates with advanced disease stage and poor clinical outcome. They also demonstrated that Hepatocyte Nuclear Factor 4 alpha (HNF-4α) regulates DUSP6 expression and that the treatment with Calcium saccharate (CAS) can upregulates DUSP6 expression. Moreover, CAS treatment sensitized both parental and sunitinib resistant RCC cells (786-O) to sunitinib both in vitro and in vivo [126].
Several lines of evidence indicate a pro-tumoral role for DUSP6 in other types of cancer. DUSP6 is overexpressed and is linked to a poorer overall survival in GC. In GC cell lines (BGC823, SGC7901 and cisplatin-resistant SGC7901), DUSP6 KD inhibited proliferation, migration, invasion and induced apoptosis. Consistently, the DUSP inhibitor BCI reduced invasion, migration and proliferation, and enhanced cisplatin cytotoxicity in GC cells in vitro. Despite increasing ERK1/2 phosphorylation, BCI treatment led to the reduction of the mRNA of some ERK1/2-regulated genes. More importantly, BCI enhanced the antitumor effects of cisplatin in GC cell-line-based and patient-derived xenograft models [127]. Another study performed in HCC showed that DUSP6 expression and ERK1/2 phosphorylation were higher in tumour samples compared to peritumoral and normal tissue, as determined by IHC. Additionally, higher expression of DUSP6 in tumour tissue correlated with HCC recurrence after resection [128]. Another retrospective IHC study showed that the expression of ERK1/2, DUSP6, c-Fos, c-Myc, cyclin D1, and PCNA is increased in papillary thyroid carcinoma (PTC) compared to benign neoplasms. Moreover, high DUSP6 expression was associated with the level of ERK1/2 expression and with high-risk biological features, including tumour size. Consistent with the pro-tumoral role of DUSP6 in PTC, DUSP6 silencing decreased cell viability and migration of anaplastic thyroid carcinoma FRO cells [129]. In another work performed in PTC, expression of RET/PTC3, HRASV12 and BRAFV600E in PCCL3 cells determined an increase of DUSP5 and DUSP6 mRNA as well as of MEK1/2 and ERK1/2 phosphorylation. Inhibition of MEK, but not of PI3K/AKT, partially prevented the above increase of DUSP5 and DUSP6. Using two BRAFV600E thyroid carcinoma cell lines (BCPAP and 8505c), the same authors showed that genetic inhibition of DUSP5 and DUSP6 did not affect cell proliferation or ERK1/2 phosphorylation, while reducing cell migration and invasion [130]. Upregulation of DUSP6 has been reported to show a tumour-promoting role in human glioblastomas. Indeed, DUSP6 mRNA and protein levels are upregulated in primary and long-term cultures of human glioblastoma cells (U251, T98G and U87MG) compared to normal human astrocytes. DUSP6 overexpression led to reduced ERK1/2 phosphorylation as well as to the increase of cisplatin-resistance of U87MG cells in vitro and in vivo, and was associated with increased xenografts growth [131]. Finally, a study conducted on sarcoma cells (NCC_CDS_X1) revealed that the oncoprotein CIC-DUX binds to DUSP6 causing a decrease of ERK1/2 phosphorylation and a consequent overexpression of the oncoprotein. Pharmacological (BCI) or genetic (shRNA or siRNA) inhibition of DUSP6 leads to an increase of ERK1/2 activation and a decrease in the level of CIC-DUX, resulting in increased apoptosis. This may represent an innovative therapeutic approach to targeting CIC-fused sarcoma [132].
As far as BC is concerned, the role of DUSP6 has been more controversial. Gene expression profile analysis provided evidence that high DUSP6 mRNA levels are associated with resistance to the anti-oestrogen tamoxifen in both patient samples and cell lines. In line with this fact, overexpression of DUSP6 rendered ERα-positive BC cells (MCF7) resistant to tamoxifen. DUSP6 overexpression was also associated with lower levels of ERK1/2 phosphorylation. The MEK inhibitor PD98059 blocked the proliferation of tamoxifen-resistant cells, pointing to a role of ERK1/2 activation in the above process. These results suggested that tamoxifen increases ERK1/2 activity via the loss of DUSP6, and that this activity remains sensitive to MEK inhibition. Therefore, patients with tamoxifen-resistant disease and elevated DUSP6 may be markedly sensitive to MEK inhibitors [133]. A possible pro-tumoral role of DUSP6 has also been reported in TNCB. In particular, DUSP6 expression resulted higher in the MDA-MB-231 TNBC cell line than in patient-derived ER-positive BC cells. In the former, DUSP6 KD (siRNA) reduced cell growth, migration and invasion, and the same result was obtained using mir-145 to inhibit DUSP6. Whether DUSP6 expression was correlated to ERK1/2 phosphorylation was not addressed [134]. The above results are at variance with those of the work carried out by Bergholz and colleagues with MDA-MB-231 cells, where DUSP6 emerged as an inhibitor of tumour invasiveness. Indeed, the authors showed that overexpression of the p63 isoform ΔNp63α inhibits cell invasion by eradication of ERK1/2 signalling via DUSP6 in vitro and in vivo [135]. Importantly, the BCI derivative BCI-215 has been reported to induce apoptosis and to sensitize MDA-MB-231 cells to immune cell killing [19]. Another study identified the increased expression of DUSP6 as a key factor in the progression of secondary acute myeloid leukemia (sAML) by promoting JAK-STAT and ERK signaling, inflammatory cytokine production, and resistance to JAK2 inhibitors. Targeting DUSP6 inhibited these pathways, reduced disease severity, and suppressed sAML development in preclinical models, including Jak2V617F and MPLW515L mouse models, without affecting healthy cells, indicating a protumoral role of DUSP6 also in non-solid tumors [136].
On the whole, DUSP6, one of the most studied DUSPs in cancer, has been shown to have both protumoral and antitumoral properties depending on the type of cancer analyzed. In particular, it exerts antitumoral effects in OC, melanoma and NSCLC, while showing a protumoral role in GC, sarcoma and glioblastoma. Furthermore, it emerged that DUSP6 could be used as a marker for the detection of some tumors and it can also be used as a target for the treatment of others. It also emerged the importance of continuing the study of this DUSP and its signalling, trying to better understand its role in other types of cancer, especially in those tumours, like BC, where it has been shown to have a controversial behaviour.
2.7. DUSP7
DUSP7/MKP-X, also known as PYST2, is a cytoplasmic phosphatase active on ERK1/2 [137,138]. Like other DUSPs, DUSP7 binds to its targets through a conserved arginine-rich kinase interaction motif, which is located amino-terminally of the phosphatase domain [139]. Although there are few reports linking DUSP7 to cancer, it seems that this phosphatase plays mostly an antitumor role. In the MCF-7 BC cell line, oestrogen deprivation induces the expression of the long non-coding RNA LincRNA regulator of reprogramming (Linc-RoR) and activates ERK1/2. This activation is Linc-RoR-dependent because it is abolished upon Linc-RoR KD. Importantly, Linc-RoR KD caused an increase of DUSP7 protein level as well as a decrease of ERK1/2 phosphorylation and of cell proliferation. In keeping with a possible protective role of DUSP7 in ER-positive BC, in silico data provided evidence that reduced DUSP7 expression correlates with a worse prognosis in ER-positive BC patients and with a shorter relapse-free survival [140]. Another study performed using GC cells addressed the hypothesis that asporin (ASPN), a member of the small leucine-rich repeat proteoglycan (SLRP) family of proteins [141], regulates the proliferation of GC cells through the downstream target proteasome 26S subunit non-ATPase 2 (PSMD2). PSMD2 participates in the down-regulation of DUSP7, WIP1 and PTEN. For this reason, genetic inhibition of ASPN determined an increase of DUSP7, WIP1 and PTEN, and the inactivation of the MAPK signaling pathway along with a reduction of cell proliferation [142]. In line with an antitumoral action of DUSP7, a recent work demonstrated that in CRC with SMAD4 deletion, SETD2, a histone methyltransferase, promotes the transcription of DUSP7, leading to an inactivation of the RAS/ERK pathway and reduced tumor growth [143]. In contrast with the above studies, a recent work performed in CRC identified the microRNA let-7c-5p as a tumor suppressor in HCT8 cells. Its overexpression reduced the levels of DUSP7 and also the phosphorylation of ERK1/2, leading to the increased expression of the pro-apoptotic marker BAX, shedding light for the first time on a protumoral role of DUSP7 [144].
Thus, DUSP7 seems to exert an antitumoral role in both BC and GC malignancies. Instead, it has been demonstrated that DUSP7 may have a controversial role in the context of colorectal cancer, but considering the paucity of studies focused on the role of DUSP7 in cancer , it could be interesting to further explore its role in other types of cancer.
2.8. DUSP9
DUSP9/MKP-4 is a cytosolic DUSP active on ERK1/2 but also on JNK and p38, and belongs to a subfamily of three closely related cytoplasmic dual-specificity MAPK phosphatases, which includes the ERK1/2-specific enzymes DUSP6 and DUSP7. As for its possible antitumoral effect, in a clonal model of epidermal carcinogenesis, DUSP9 was found to be down-regulated at initiation, and lost at malignant conversion. Reconstitution of DUSP9 expression in H1299 SCC cells led to cell death and tumour growth suppression, as a consequence of G(2)-M associated cell death and microtubule disruption [145]. In line with the antitumoral role of DUSP9 in this cancer, another study reported that DUSP9 was significantly downregulated in CRC tissues compared with peritumoral ones. Additionally, transcriptomic profiling performed in CRC SW480 cells revealed that DUSP9 KD was associated with activation of ERK1/2 signalling [146]. Other studies reported that DUSP9 KD may be associated with reduced cell proliferation. In particular, it was found that the expression of DUSP9 is upregulated upon retinoic acid receptor (RAR) activation. Indeed, RAR, which promotes DUSP9 expression through direct binding to its promoter region, increases spontaneous differentiation of CRC cells, while ERK1/2 activation suppresses it, favouring proliferation. DUSP9 KD significantly delayed the differentiation of CACO-2 cell line, pointing to a role of RAR/DUSP9 signalling in supporting CRC differentiation. Thus, the authors provided evidence that RAS/ERK1/2 and RAR signaling pathways antagonistically interact with each other to regulate CRC cell fate [147].
Regarding a possible protumoral role of DUSP9, it has been reported that hypoxia-inducible factor (HIF)-1-dependent regulation of MAPK via DUSP9 contributes to the enrichment of chemotherapy-induced BC stem cells in MDA-MB-231 and SUM-159 TNBC cells. In particular, chemotherapy increased DUSP9 expression while decreasing that of DUSP16, in a HIF1-dependent manner, leading to inhibition of ERK1/2 and activation of p38 signaling pathways. Inhibition of ERK1/2 caused transcriptional induction of the pluripotency factor Nanog, while activation of p38 stabilized Nanog and Klf4 mRNA, collectively resulting in the support of the BC stem cell phenotype. Inhibition of HIF-1 or p38 signaling blocked chemotherapy-induced pluripotency factor expression and BCSC enrichment [148].
The above literature is limited and quite discordant, providing evidence of both a protumoral and antitumoral role regarding DUSP9, even in the same type of cancer. For this reason, DUSP9 is considered an ambiguous phosphatase in the context of cancer onset and development, so that further studies are needed to dissect, possibly in a context-specific manner, its possible exploitation as a biomarker and/or a target for cancer therapy.
2.9. DUSP10
DUSP10/MKP-5, located in both cytoplasm and nucleus, selectively dephosphorylates p38 and JNK, showing minor activity towards ERK1/2. It is involved in cell proliferation, differentiation and migration [66,149]. Interestingly, DUSP10 has been proven to interact with and to retain ERK1/2 in the cytoplasm, thus preventing ERK1/2-dependent transcription. These results identified a novel DUSP10 function as a scaffold protein for the ERK1/2 pathway. In the same study, it was reported that DUSP10 mRNA is frequently upregulated in CRC but not in lung carcinoma, suggesting a possible association with the malignant phenotype of the former [150].
With regard to the possible antitumoral role of DUSP10, it has been reported a decreased DUSP10 protein level along with an increased ERK1/2 phosphorylation in HCC metastasis. KD of the stress-induced tumour suppressor gene TP53INP1, which is downregulated in HCC, led to an increase of the invasion capability of immortalized normal liver (MIHA) and HCC (MHCC97L) cell lines, and of the metastasis in nude mice injected with MHCC97L-siTP53INP1 cell with respect to control MHCC97L cells. Overexpression of DUSP10 in MHCC97L-siTP53INP1 led to ERK1/2 inactivation and to a decreased capability of invasion in vitro and metastasis in vivo. Finally, the authors provided evidence that p73 is involved in TP53INP1-dependent regulation of DUSP10 [151].
The limited number of available studies regarding the role of DUSP10 in the modulation of ERKs is in line with the marginal role of DUSP10 in the regulation of ERK1/2. DUSP10 inactivated ERK1/2 and showed antitumor properties in HCC, while having opposite effects in CC.
3. Conclusions
Evidence about the correlation of altered expression of DUSPs to cancer onset and progression is accumulating over the years, pointing to the exploitation of their targeting as an intriguing strategy for cancer treatment. Among the MAPKs that are DUSP substrates, ERK1/2/5 in particular are implicated in important biological events including cell proliferation, survival, and migration, and are often deregulated in cancer, so that they are frequent targets for molecularly-tailored therapies [152]. In addition, activation of these MAPKs is often responsible for the development of resistance to anti-cancer treatments [153,154]. Based on that, DUSPs may thus represent an additional target to modulate the activation of MAPKs. A critical aspect of targeting DUSPs, relies on the fact that these phosphatases can act as either tumour promoters or suppressors, according to the cancer type and to the stage of disease. In this regard, DUSP small molecule inhibitors have been proven to be a promising approach in the contexts in which DUSPs support cancer growth and progression (Figure 2). However, due to the high number of different DUSPs with a relatively well conserved catalytic mechanism, the possibility to obtain highly specific inhibitors seems to be hampered, at least at the moment. Among the DUSPs inhibitors that have demonstrated a good antitumor efficacy, BCI, a benzoil-derivative small-molecule inhibitor of DUSP1 and DUSP6, has been reported to reduce BC, MPNSTs, LUAD, GC and sarcoma growth [17,18,19]. Moreover, NSC 95397, a quinone-based small molecule inhibitor of DUSP1 and DUSP6, reverted the pro-tumoral role of DUSP1 in CC [24,25]. Another interesting approach to exploit the antitumoral effect of DUSPs is the one described by Cheng et colleagues, that administered baculovirus-derived DUSP1 in the medium of Hela cells obtaining a reduction of their proliferation [51].
Concerning the possibility to boost the antitumoral effect of DUSPs by increasing their expression and/or activation, sanguinarine, a natural antitumoral benzophenanthridine alkaloid, has been reported to upregulate the expression of DUSP4, thus counteracting GC progression [70]. On the other hand, Lee and colleagues have reported that the increased expression of mir211 impairs the antitumoral role of DUSP6 in melanoma [115], pointing to the use of non-coding RNAs as an additional strategy to modulate the activity of DUSPs. Another interesting strategy for the modulation of DUSPs is related to the possibility of using small molecules to inhibit or enhance the expression of different transcription factors. Along this line, it has been reported that the transcription factor ETS1 positively modulates DUSP6 expression by binding to its promoter region, leading to a decrease of NSCLC progression [106]. Another study demonstrated that the transcription factor ZNF251 inhibits the expression of DUSP6, activating ERK1/2 signaling and promoting NSCLC growth [109]. On the whole, the available data established the basis for the use of DUSPs as both tumor markers and therapeutic targets in many types of cancer. Despite the fact that DUSP1 and DUSP6 have been studied in detail and that there are inhibitors available, further extensive work is needed to better understand the role of all ERK-targeting DUSPs within the various types of tumours, and to develop DUSP modulators, both inhibitors and activators, for their utilization in the clinics.
Author Contributions
A.T., D.P. and E.R. reviewed all the literature and wrote the manuscript; I.T provided valuable critical revision of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The work in Rovida's lab is supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC, IG-15282 and IG-21349), Ente Fondazione Cassa di Risparmio di Firenze (ECRF), and Università degli Studi di Firenze (Fondo di Ateneo ex-60%). A. Tubita is supported by a “Carlo Zanotti” Fondazione Italiana per la Ricerca sul Cancro (FIRC)-AIRC fellowship (ID-23847).
Acknowledgments
The authors are grateful to Persio Dello Sbarba for the critical review of the manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2012, 76, 496. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and p38 Protein Kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Camps, M.; Nichols, A.; Arkinstall, S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000, 14, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Tan, T.-H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef]
- Chen, H.-F.; Chuang, H.-C.; Tan, T.-H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef]
- Liao, Q.; Guo, J.; Kleeff, J.; Zimmermann, A.; Büchler, M.W.; Korc, M.; Friess, H. Down-regulation of the dual-specificity phosphatase MKP-1 suppresses tumorigenicity of pancreatic cancer cells. Gastroenterology 2003, 124, 1830–1845. [Google Scholar] [CrossRef]
- Teng, F.; Xu, Z.; Chen, J.; Zheng, G.; Zheng, G.; Lv, H.; Wang, Y.; Wang, L.; Cheng, X. DUSP1 induces apatinib resistance by activating the MAPK pathway in gastric cancer. Oncol. Rep. 2018, 40, 1203–1222. [Google Scholar] [CrossRef]
- Chi, H.; Barry, S.P.; Roth, R.J.; Wu, J.J.; Jones, E.A.; Bennett, A.M.; Flavell, R.A. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. 2006, 103, 2274–2279. [Google Scholar] [CrossRef]
- Liu, Y.; Shepherd, E.G.; Nelin, L.D. MAPK phosphatases--regulating the immune response. Nat Rev Immunol. 2007, 7, 202–12. [Google Scholar] [CrossRef]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-Activated Protein (MAP) Kinase/MAP Kinase Phosphatase Regulation: Roles in Cell Growth, Death, and Cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef] [PubMed]
- Loda, M.; Capodieci, P.; Mishra, R.; Yao, H.; Corless, C.; Grigioni, W.; Wang, Y.; Magi-Galluzzi, C.; Stork, P.J. Expression of mitogen-activated protein kinase phosphatase-1 in the early phases of human epithelial carcinogenesis. . 1996, 149, 1553–64. [Google Scholar] [PubMed]
- Rojo, F.; González-Navarrete, I.; Bragado, R.; Dalmases, A.; Menéndez, S.; Cortes-Sempere, M.; Suárez, C.; Oliva, C.; Servitja, S.; Rodriguez-Fanjul, V.; et al. Mitogen-Activated Protein Kinase Phosphatase-1 in Human Breast Cancer Independently Predicts Prognosis and Is Repressed by Doxorubicin. Clin. Cancer Res. 2009, 15, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Czerwenka, K.; Heinze, G.; Ryffel, M.; Schuster, E.; Witt, A.; Leodolter, S.; Zeillinger, R. Expression of KLF5 is a Prognostic Factor for Disease-Free Survival and Overall Survival in Patients with Breast Cancer. Clin. Cancer Res. 2006, 12, 2442–2448. [Google Scholar] [CrossRef]
- Liu, R.; Zheng, H.-Q.; Zhou, Z.; Dong, J.-T.; Chen, C. KLF5 Promotes Breast Cell Survival Partially through Fibroblast Growth Factor-binding Protein 1-pERK-mediated Dual Specificity MKP-1 Protein Phosphorylation and Stabilization. J. Biol. Chem. 2009, 284, 16791–16798. [Google Scholar] [CrossRef]
- Liu, D.; Deng, Q.; Sun, L.; Wang, T.; Yang, Z.; Chen, H.; Guo, L.; Liu, Y.; Ma, Y.; Guo, N.; et al. A Her2-let-7-β2-AR circuit affects prognosis in patients with Her2-positive breast cancer. BMC Cancer 2015, 15, 832. [Google Scholar] [CrossRef]
- Molina, G.; Vogt, A.; Bakan, A.; Dai, W.; de Oliveira, P.Q.; Znosko, W.; E Smithgall, T.; Bahar, I.; Lazo, J.S.; Day, B.W.; et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 2009, 5, 680–687. [Google Scholar] [CrossRef]
- Tuglu, M.M.; Bostanabad, S.Y.; Ozyon, G.; Dalkiliç, B.; Gurdal, H. The role of dual-specificity phosphatase 1 and protein phosphatase 1 in β2-adrenergic receptor-mediated inhibition of extracellular signal regulated kinase 1/2 in triple negative breast cancer cell lines. Mol Med Rep. 2018, 17, 2033–2043. [Google Scholar] [CrossRef]
- Kaltenmeier, C.T.; Vollmer, L.L.; Vernetti, L.A.; Caprio, L.; Davis, K.; Korotchenko, V.N.; Day, B.W.; Tsang, M.; Hulkower, K.I.; Lotze, M.T.; et al. A Tumor Cell-Selective Inhibitor of Mitogen-Activated Protein Kinase Phosphatases Sensitizes Breast Cancer Cells to Lymphokine-Activated Killer Cell Activity. J. Pharmacol. Exp. Ther. 2017, 361, 39–50. [Google Scholar] [CrossRef]
- Kim, M.-J.; Park, I.-J.; Yun, H.; Kang, I.; Choe, W.; Kim, S.-S.; Ha, J. AMP-activated Protein Kinase Antagonizes Pro-apoptotic Extracellular Signal-regulated Kinase Activation by Inducing Dual-specificity Protein Phosphatases in Response to Glucose Deprivation in HCT116 Carcinoma. 2010, 285, 14617–14627. [CrossRef]
- Venditto, V.J.; Simanek, E.E. Cancer Therapies Utilizing the Camptothecins: A Review of thein VivoLiterature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef]
- Rovida, E.; Stecca, B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin. Cancer Biol. 2015, 35, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, M. ; Young, Kim, S. ; Kim, J.; Kim, H.S.; Kim, S.M.; Kim, E.J. Mitogen-activated protein kinase phosphatase-1 inhibition and sustained extracellular signal-regulated kinase 1/2 activation in camptothecin-induced human colon cancer cell death. Cancer Biol Ther. 2013, 14, 1007–15. [Google Scholar]
- Vogt, A.; McDonald, P.R.; Tamewitz, A.; Sikorski, R.P.; Wipf, P.; Skoko, J.J., 3rd; Lazo, J.S. A cell-active inhibitor of mitogen-activated protein kinase phosphatases restores paclitaxel-induced apoptosis in dexamethasone-protected cancer cells. Mol. Cancer Ther. 2008, 7, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Dubey, N.K.; Peng, B.Y.; Lin, C.M.; Wang, P.D.; Wang, J.R.; Chan, C.H.; Wei, H.J.; Deng, W.P. NSC 95397 Suppresses Proliferation and Induces Apoptosis in Colon Cancer Cells through MKP-1 and the ERK1/2 Pathway. Int J Mol Sci. 2018, 19, 1625. [Google Scholar] [CrossRef]
- Pan, J.; Lin, M.; Xu, Z.; Xu, M.; Zhang, J.; Weng, Z.; Lin, B.; Lin, X. CDKN2B antisense RNA 1 suppresses tumor growth in human colorectal cancer by targeting MAPK inactivator dual-specificity phosphatase 1. Carcinog. 2021, 42, 1399–1409. [Google Scholar] [CrossRef]
- Ramkissoon, A.; Chaney, K.E.; Milewski, D.; Williams, K.B.; Williams, R.L.; Choi, K.; Miller, A.; Kalin, T.V.; Pressey, J.G.; Szabo, S.; et al. Targeted Inhibition of the Dual Specificity Phosphatases DUSP1 and DUSP6 Suppress MPNST Growth via JNK. Clin. Cancer Res. 2019, 25, 4117–4127. [Google Scholar] [CrossRef]
- He, W.; Liu, P.; Lei, Q.; Xu, J.; Liu, L. DUSP1 Promotes Osimertinib Drug-Tolerant Persistence by Inhibiting MAPK/ERK Signaling in Non-small Cell Lung Cancer. Mol. Biotechnol. 2024, 67, 1256–1268. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, J.; Zhou, J.-Y.; Liu, Y.; Wu, G.S. Mitogen-Activated Protein Kinase Phosphatase-1 Is Required for Cisplatin Resistance. Cancer Res. 2006, 66, 8870–8877. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.-Y.; Wu, G.S. ERK-Dependent MKP-1–Mediated Cisplatin Resistance in Human Ovarian Cancer Cells. Cancer Res. 2007, 67, 11933–11941. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.Y.; Kho, D.; Reiners, J.J. Jr.; Wu, G.S. Role for DUSP1 (dual-specificity protein phosphatase 1) in the regulation of autophagy. Autophagy. 2016, 12, 1791–1803. [Google Scholar] [CrossRef]
- Ma, M.; Wang, C.; Wu, M.; Gu, S.; Yang, J.; Zhang, Y.; Cheng, S.; Xu, S.; Zhang, M.; Wu, Y.; et al. CSGALNACT2 restricts ovarian cancer migration and invasion by modulating MAPK/ERK pathway through DUSP1. Cell. Oncol. 2023, 47, 897–915. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Platt, F.M.; Ross, R.L.; Hurst, C.D. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Sathe, A.; Guerth, F.; Cronauer, M.V.; Heck, M.M.; Thalgott, M.; Gschwend, J.E.; Retz, M.; Nawroth, R. Mutant PIK3CA controls DUSP1-dependent ERK 1/2 activity to confer response to AKT target therapy. Br J Cancer. 2014, 111, 2103–13. [Google Scholar] [CrossRef] [PubMed]
- Pervin, S.; Singh, R.; A Freije, W.; Chaudhuri, G. MKP-1-induced dephosphorylation of extracellular signal-regulated kinase is essential for triggering nitric oxide-induced apoptosis in human breast cancer cell lines: implications in breast cancer. . 2003, 63, 8853–60. [Google Scholar]
- Wang, K.; Zhang, M.; Qian, Y.-Y.; Ding, Z.-Y.; Lv, J.-H.; Shen, H.-H. Imbalanced expression of mitogen-activated protein kinase phosphatase-1 and phosphorylated extracellular signal-regulated kinases in lung squamous cell carcinoma. J. Zhejiang Univ. B 2011, 12, 828–834. [Google Scholar] [CrossRef]
- Maraver, A.; Fernandez-Marcos, P.J.; Herranz, D.; Cañamero, M.; Muñoz-Martin, M.; Gómez-López, G.; Mulero, F.; Megías, D.; Sanchez-Carbayo, M.; Shen, J.; et al. Therapeutic Effect of γ-Secretase Inhibition in KrasG12V-Driven Non-Small Cell Lung Carcinoma by Derepression of DUSP1 and Inhibition of ERK. Cancer Cell 2012, 22, 222–234. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, Q.; Cheng, Z.; Gu, J.; Feng, W.; Lei, T.; Huang, J.; Pu, J.; Chen, X.; Wang, Z. Long non-coding RNA CASC9 promotes gefitinib resistance in NSCLC by epigenetic repression of DUSP1. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Liu, C.; Shi, Y.; Han, Z.; Pan, Y.; Liu, N.; Han, S.; Chen, Y.; Lan, M.; Qiao, T.; Fan, D. Suppression of the dual-specificity phosphatase MKP-1 enhances HIF-1 trans-activation and increases expression of EPO. Biochem. Biophys. Res. Commun. 2003, 312, 780–786. [Google Scholar] [CrossRef]
- Liu, C.; Shi, Y.; Du, Y.; Ning, X.; Liu, N.; Huang, D.; Liang, J.; Xue, Y.; Fan, D. Dual-specificity phosphatase DUSP1 protects overactivation of hypoxia-inducible factor 1 through inactivating ERK MAPK. Exp. Cell Res. 2005, 309, 410–418. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Pinna, F.; Meloni, F.; Ladu, S.; Pellegrino, R.; Sini, M.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Virdis, P.; Frau, M.; Tomasi, M.L.; Seddaiu, M.A.; Muroni, M.R.; Feo, F.; Pascale, R.M. Dual-specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res. 2008, 68, 4192–200. [Google Scholar] [CrossRef]
- Kato, H.; Naiki-Ito, A.; Naiki, T.; Suzuki, S.; Yamashita, Y.; Sato, S.; Sagawa, H.; Kato, A.; Kuno, T.; Takahashi, S. Connexin 32 dysfunction promotes ethanol-related hepatocarcinogenesis via activation of Dusp1-Erk axis. Oncotarget 2015, 7, 2009–2021. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Zhou, H.; Liang, H.; Li, C. Regulation of ERK and AKT pathways by hepatitis B virus X protein via the Notch1 pathway in hepatocellular carcinoma. Int. J. Oncol. 2017, 51, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, J.-Y.; Zhao, L.-J.; Gao, B.-R.; Wan, X.-P.; Wang, J.-L. Dual-specificity Phosphatase 1 Deficiency Induces Endometrioid Adenocarcinoma Progression via Activation of Mitogen-activated Protein Kinase/Extracellular Signal-regulated Kinase Pathway. Chin. Med J. 2016, 129, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Rauhala, H.E.; Porkka, K.P.; Tolonen, T.T.; Martikainen, P.M.; Tammela, T.L.; Visakorpi, T. Dual-specificity phosphatase 1 and serum/glucocorticoid-regulated kinase are downregulated in prostate cancer. Int. J. Cancer 2005, 117, 738–745. [Google Scholar] [CrossRef]
- Gil-Araujo, B.; Lobo, M.-V.T.; Gutiérrez-Salmerón, M.; Gutiérrez-Pitalúa, J.; Ropero, S.; Angulo, J.C.; Chiloeches, A.; Lasa, M. Dual specificity phosphatase 1 expression inversely correlates with NF-κB activity and expression in prostate cancer and promotes apoptosis through a p38 MAPK dependent mechanism. Mol. Oncol. 2013, 8, 27–38. [Google Scholar] [CrossRef]
- Magi-Galluzzi, C.; Montironi, R.; Cangi, M.G.; Wishnow, K.; Loda, M. Mitogen-activated protein kinases and apoptosis in PIN. Virchows Arch. 1998, 432, 407–413. [Google Scholar] [CrossRef]
- Martínez-Martínez, D.; Toledo Lobo, M.-V.; Baquero, P.; Ropero, S.; Angulo, J.C.; Chiloeches, A.; Lasa, M. Downregulation of Snail by DUSP1 Impairs Cell Migration and Invasion through the Inactivation of JNK and ERK and Is Useful as a Predictive Factor in the Prognosis of Prostate Cancer. Cancers 2021, 13, 1158. [Google Scholar] [CrossRef]
- Ariano, C.; Costanza, F.; Akman, M.; Riganti, C.; Corà, D.; Casanova, E.; Astanina, E.; Comunanza, V.; Bussolino, F.; Doronzo, G. TFEB inhibition induces melanoma shut-down by blocking the cell cycle and rewiring metabolism. Cell Death Dis. 2023, 14, 1–21. [Google Scholar] [CrossRef]
- Wang, J.; Jia, Q.; Sun, J.; Wu, S.; Wei, L.; Yao, W. Arntl-induced upregulation of DUSP1 inhibits tumor progression in esophageal squamous cell carcinoma by inactivating ERK signaling. Cancer Biol. Ther. 2024, 25, 2408042. [Google Scholar] [CrossRef]
- Cheng, P.; Zhu, S.; Jun, L.; Huang, L.; Hong, Y. Production of DUSP1 protein using the baculovirus insect cell expression system and its in vitro effects on cancer cells. Int. J. Mol. Med. 2015, 35, 1715–1719. [Google Scholar] [CrossRef]
- Rohan, P.J.; Davis, P.; Moskaluk, C.A.; Kearns, M.; Krutzsch, H.; Siebenlist, U.; Kelly, K. PAC-1: a Mitogen-Induced Nuclear Protein Tyrosine Phosphatase. Science 1993, 259, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Hammer, M.; Mages, J. DUSP Meet Immunology: Dual Specificity MAPK Phosphatases in Control of the Inflammatory Response. J. Immunol. 2006, 177, 7497–7504. [Google Scholar] [CrossRef] [PubMed]
- Perander, M.; Al-Mahdi, R.; Jensen, T.C.; Nunn, J.A.L.; Kildalsen, H.; Johansen, B.; Gabrielsen, M.; Keyse, S.M.; Seternes, O.-M. Regulation of atypical MAP kinases ERK3 and ERK4 by the phosphatase DUSP2. Sci. Rep. 2017, 7, srep43471. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.-W.; Lee, J.-C.; Yeh, Y.-C.; Hsu, K.-F.; Lai, Y.-Y.; Lin, S.-C.; Tsai, S.-J. Suppression of dual-specificity phosphatase–2 by hypoxia increases chemoresistance and malignancy in human cancer cells. J. Clin. Investig. 2011, 121, 1905–1916. [Google Scholar] [CrossRef]
- Karakashev, S.V.; Reginato, M.J. Hypoxia/HIF1α induces lapatinib resistance in ERBB2-positive breast cancer cells via regulation of DUSP2. Oncotarget 2015, 6, 1967–1980. [Google Scholar] [CrossRef]
- Hu, J.; Li, L.; Chen, H.; Zhang, G.; Liu, H.; Kong, R.; Chen, H.; Wang, Y.; Li, Y.; Tian, F.; et al. MiR-361-3p regulates ERK1/2-induced EMT via DUSP2 mRNA degradation in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, S.; Tan, Y.; Pan, S.; An, W.; Chen, Q.; Wang, X.; Xu, H. The SKA3-DUSP2 Axis Promotes Gastric Cancer Tumorigenesis and Epithelial-Mesenchymal Transition by Activating the MAPK/ERK Pathway. Front. Pharmacol. 2022, 13, 777612. [Google Scholar] [CrossRef]
- Zou, F.; Rao, T.; Chen, W.; Song, T.; Li, T.; Hu, W.; Li, L.; Yu, W.; Cheng, F. DUSP2 affects bladder cancer prognosis by down-regulating MEK/ERK and P38 MAPK signaling pathways through PTPN7. Cell. Signal. 2023, 112, 110893. [Google Scholar] [CrossRef]
- Todd, J.L.; Tanner, K.G.; Denu, J.M. Extracellular Regulated Kinases (ERK) 1 and ERK2 Are Authentic Substrates for the Dual-specificity Protein-tyrosine Phosphatase VHR. J. Biol. Chem. 1999, 274, 13271–13280. [Google Scholar] [CrossRef]
- Alonso, A.; Saxena, M.; Williams, S.; Mustelin, T. Inhibitory Role for Dual Specificity Phosphatase VHR in T Cell Antigen Receptor and CD28-induced Erk and Jnk Activation. J. Biol. Chem. 2001, 276, 4766–4771. [Google Scholar] [CrossRef]
- Rahmouni, S.; Cerignoli, F.; Alonso, A.; Tsutji, T.; Henkens, R.; Zhu, C.; Louis-dit-Sully, C.; Moutschen, M.; Jiang, W.; Mustelin, T. Loss of the VHR dual-specific phosphatase causes cell-cycle arrest and senescence. Nat Cell Biol. 2006, 8, 524–31. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.W.; Alam, H.; Dhar, S.S.; Giri, U.; Li, N.; Wei, Y.; Giri, D.; Cascone, T.; Kim, J.H.; Ye, Y.; Multani, A.S.; Chan, C.H.; Erez, B.; Saigal, B.; Chung, J.; Lin, H.K.; Wu, X.; Hung, M.C.; Heymach, J.V.; Lee, M.G. KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J Clin Invest. 2013, 123, 5231–46. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, C.; Wang, W.; Liu, Y.; He, H.; Chen, C.; Xiang, R.; Luo, Y.; Coleman, W.B. Identification of serum miR-1915-3p and miR-455-3p as biomarkers for breast cancer. PLOS ONE 2018, 13, e0200716. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kabeer, S.W.; Sharma, S.; Tikoo, K. l-Methionine potentiates anticancer activity of Sorafenib by epigenetically altering DUSP3/ERK pathway in hepatocellular carcinoma. J Biochem Mol Toxicol. 2024, 38, e23663. [Google Scholar] [CrossRef]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Lin, A.; Marshall, M.S. Pancreatic Tumor Cells with Mutant K-ras Suppress ERK Activity by MEK-Dependent Induction of MAP Kinase Phosphatase-2. Biochem. Biophys. Res. Commun. 2001, 280, 992–997. [Google Scholar] [CrossRef]
- Hijiya, N.; Tsukamoto, Y.; Nakada, C.; Tung, Nguyen, L. ; Kai, T. ; Matsuura, K.; Shibata, K.; Inomata, M.; Uchida, T.; Tokunaga, A.; Amada, K.; Shirao, K.; Yamada, Y.; Mori, H.; Takeuchi, I.; Seto, M.; Aoki, M.; Takekawa, M.; Moriyama, M. Genomic Loss of DUSP4 Contributes to the Progression of Intraepithelial Neoplasm of Pancreas to Invasive Carcinoma. Cancer Res. 2016, 76, 2612–25. [Google Scholar]
- Li, K.-C.; Hua, K.-T.; Lin, Y.-S.; Su, C.-Y.; Ko, J.-Y.; Hsiao, M.; Kuo, M.-L.; Tan, C.-T. Inhibition of G9a induces DUSP4-dependent autophagic cell death in head and neck squamous cell carcinoma. Mol. Cancer 2014, 13, 172–172. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, G.; Zhang, P.F.; Zhang, J.; Huang, Y.X.; Lu, Y.M.; Da, W. ; Sun,Q. ; Zhu, J.S. Sanguinarine inhibits growth and invasion of gastric cancer cells via regulation of the DUSP4/ERK pathway. J Cell Mol Med. 2017, 21, 1117–1127. [Google Scholar]
- Balko, J.M.; Cook, R.S.; Vaught, D.B.; Kuba, M.G.; Miller, T.W.; E Bhola, N.; E Sanders, M.; Granja-Ingram, N.M.; Smith, J.J.; Meszoely, I.M.; et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat. Med. 2012, 18, 1052–1059. [Google Scholar] [CrossRef]
- Balko, J.M.; Schwarz, L.J.; Bhola, N.E.; Kurupi, R.; Owens, P.; Miller, T.W.; Gómez, H.; Cook, R.S.; Arteaga, C.L. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 2013, 73, :6346–58. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.A.; de Barros, A.L.B.; Leite, E.A. The potential use of simvastatin for cancer treatment: A review. Biomed. Pharmacother. 2021, 141, 111858. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.H.; Lee, S.-H.; Kim, J.-Y.; Ahn, J.S.; Park, Y.H.; Im, Y.-H. Statins affect ETS1-overexpressing triple-negative breast cancer cells by restoring DUSP4 deficiency. Sci. Rep. 2016, 6, 33035. [Google Scholar] [CrossRef] [PubMed]
- Gaggianesi, M.; Turdo, A.; Chinnici, A.; Lipari, E.; Apuzzo, T.; Benfante, A.; Sperduti, I.; Di Franco, S.; Meraviglia, S.; Presti, E.L.; et al. IL4 Primes the Dynamics of Breast Cancer Progression via DUSP4 Inhibition. Cancer Res. 2017, 77, 3268–3279. [Google Scholar] [CrossRef]
- Haagenson, K.K.; Zhang, J.W.; Xu, Z.; Shekhar, M.P.; Wu, G.S. Erratum: Functional analysis of MKP-1 and MKP-2 in breast cancer tamoxifen sensitivity. Oncotarget 2018, 9, 35286–35286. [Google Scholar] [CrossRef]
- Klemm, S.; Evert, K.; Utpatel, K.; Muggli, A.; Simile, M.M.; Chen, X.; Evert, M.; Calvisi, D.F.; Scheiter, A. Identification of DUSP4/6 overexpression as a potential rheostat to NRAS-induced hepatocarcinogenesis. BMC Cancer. 2023, 23, 1086. [Google Scholar] [CrossRef]
- Cagnol, S.; Rivard, N. Oncogenic KRAS and BRAF activation of the MEK/ERK signaling pathway promotes expression of dual-specificity phosphatase 4 (DUSP4/MKP2) resulting in nuclear ERK1/2 inhibition. Oncogene 2012, 32, 564–576. [Google Scholar] [CrossRef]
- Ishibashi, T.; Bottaro, D.; Michieli, P.; A Kelley, C.; A Aaronson, S. A novel dual specificity phosphatase induced by serum stimulation and heat shock. J. Biol. Chem. 1994, 269, 29897–29902. [Google Scholar] [CrossRef]
- Patterson, K.I.; Brummer, T.; O'Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef]
- Mandl, M.; Slack, D.N.; Keyse, S.M. Specific Inactivation and Nuclear Anchoring of Extracellular Signal-Regulated Kinase 2 by the Inducible Dual-Specificity Protein Phosphatase DUSP5. Mol. Cell. Biol. 2005, 25, 1830–1845. [Google Scholar] [CrossRef]
- Shin, S.-H.; Park, S.-Y.; Kang, G.H. Down-Regulation of Dual-Specificity Phosphatase 5 in Gastric Cancer by Promoter CpG Island Hypermethylation and Its Potential Role in Carcinogenesis. Am. J. Pathol. 2013, 182, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, L.K.; Kidger, A.M.; Delavaine, L.; Stewart, G.; van Schelven, S.; Davidson, J.; Bryant, C.J.; Caddye, E.; East, P.; Caunt, C.J.; et al. Dual-specificity phosphatase 5 regulates nuclear ERK activity and suppresses skin cancer by inhibiting mutant Harvey-Ras (HRas Q61L )-driven SerpinB2 expression. Proc. Natl. Acad. Sci. 2014, 111, 18267–18272. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, J.-Y.; Han, Z.-D.; He, H.-C.; Chen, J.-H.; Chen, Y.-R.; Yang, S.-B.; Wu, Y.-D.; Zeng, Y.-R.; Zou, J.; et al. Down-regulation of dual-specificity phosphatase 5 predicts poor prognosis of patients with prostate cancer. Int J Clin Exp Med. 2015, 8, 4186–94. [Google Scholar] [PubMed]
- Tögel, L.; Nightingale, R.; Wu, R.; Chüeh, A.C.; Al-Obaidi, S.; Luk, I.; Dávalos-Salas, M.; Chionh, F.; Murone, C.; Buchanan, D.D.; et al. DUSP5 is methylated in CIMP-high colorectal cancer but is not a major regulator of intestinal cell proliferation and tumorigenesis. Sci. Rep. 2018, 8, 1767. [Google Scholar] [CrossRef]
- Boonruang, K.; Kim, I.; Kwag, C.; Ryu, J.; Baek, S.J. Quercetin induces dual specificity phosphatase 5 via serum response factor. BMB Rep. 2023, 56, 508–513. [Google Scholar] [CrossRef]
- Wang, R.; Bao, H.; Du, W.; Chen, X.; Liu, H.; Han, D.; Wang, L.; Wu, J.; Wang, C.; Yang, M.; et al. P68 RNA helicase promotes invasion of glioma cells through negatively regulating DUSP5. Cancer Sci. 2019, 110, 107–117. [Google Scholar] [CrossRef]
- Wang, R.; Jiao, Z.; Li, R.; Yue, H.; Chen, L. p68 RNA helicase promotes glioma cell proliferation in vitro and in vivo via direct regulation of NF- B transcription factor p50. Neuro-Oncology 2012, 14, 1116–1124. [Google Scholar] [CrossRef]
- Nokin, M.-J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; van Laere, S.; et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. 2019, 21, 1–19. [Google Scholar] [CrossRef]
- Chen, P.; Wang, J.; Yao, Y.; Qu, Y.; Ji, M.; Hou, P. Targeting DUSP5 suppresses malignant phenotypes of BRAF-mutant thyroid cancer cells and improves their response to sorafenib. Endocrine 2024, 85, 1268–1277. [Google Scholar] [CrossRef]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef]
- Keyse, S.M.; Ginsburg, M. Amino acid sequence similarity between CL100, a dual-specificity MAP kinase phosphatase and cdc25. Trends Biochem. Sci. 1993, 18, 377–378. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Rovida, E. Impact of ERK5 on the Hallmarks of Cancer. Int. J. Mol. Sci. 2019, 20, 1426. [Google Scholar] [CrossRef]
- Ahmad, M.K.; Abdollah, N.A.; Shafie, N.H.; Yusof, N.M.; Razak, S.R.A. Dual-specificity phosphatase 6 (DUSP6): a review of its molecular characteristics and clinical relevance in cancer. Cancer Biol Med. 2018, 15, 14–28. [Google Scholar]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008, 412, 287–98. [Google Scholar] [CrossRef]
- Bermudez, O.; Jouandin, P.; Rottier, J.; Bourcier, C.; Pagès, G.; Gimond, C. Post-transcriptional regulation of the DUSP6/MKP-3 phosphatase by MEK/ERK signaling and hypoxia. J. Cell. Physiol. 2010, 226, 276–284. [Google Scholar] [CrossRef]
- Zeliadt, N.A.; Mauro, L.J.; Wattenberg, E.V. Reciprocal regulation of extracellular signal regulated kinase 1/2 and mitogen activated protein kinase phosphatase-3. Toxicol Appl Pharmacol. 2008, 232, 408–17. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. V600E BRAF is associated with disabled feedback inhibition of RAF–MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. 2009, 106, 4519–4524. [Google Scholar] [CrossRef]
- Moncho-Amor, V.; Pintado-Berninches, L.; de Cáceres, I.I.; Martín-Villar, E.; Quintanilla, M.; Chakravarty, P.; Cortes-Sempere, M.; Fernández-Varas, B.; Rodriguez-Antolín, C.; de Castro, J.; et al. Role of Dusp6 Phosphatase as a Tumor Suppressor in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2019, 20, 2036. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.M.; Huang, S.-M.; Park, S.-K.; Kim, D.-H.; Kim, D.H.; Lee, C.S.; Suh, K.S.; Yi, E.S.; Kim, K.-H. Differential expression of DUSP6 with expression of ERK and Ki-67 in non-small cell lung carcinoma. Pathol. - Res. Pr. 2011, 207, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Unni, A.M.; Harbourne, B.; Oh, M.H.; Wild, S.; Ferrarone, J.R.; Lockwood, W.W.; Varmus, H. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. eLife 2018, 7. e33718. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Unni, A.M.; Lockwood, W.W.; Zejnullahu, K.; Lee-Lin, S.-Q.; Varmus, H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. eLife 2015, 4, e06907. [Google Scholar] [CrossRef]
- Zhang, Z.; Kobayashi, S.; Borczuk, A.C.; Leidner, R.S.; LaFramboise, T.; Levine, A.D.; Halmos, B. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinog. 2010, 31, 577–586. [Google Scholar] [CrossRef]
- Ghaddar, N.; Wang, S.; Woodvine, B.; Krishnamoorthy, J.; van Hoef, V.; Darini, C.; Kazimierczak, U.; Ah-Son, N.; Popper, H.; Johnson, M.; Officer, L.; Teodósio, A.; Broggini, M.; Mann, K.K.; Hatzoglou, M.; Topisirovic, I.; Larsson, O.; Le Quesne, J.; Koromilas, A.E. The integrated stress response is tumorigenic and constitutes a therapeutic liability in KRAS-driven lung cancer. Nat Commun. 2021, 12, 4651. [Google Scholar] [CrossRef]
- An, B.C. ; Choi, Y,D. ; Oh, I.J.; Kim, J.H.; Park, J.I.; Lee, S.W. GPx3-mediated redox signaling arrests the cell cycle and acts as a tumor suppressor in lung cancer cell lines. PLoS One. 2018, 13, e0204170. [Google Scholar]
- Zhong, C.; Chen, C.; Yao, F.; Fang, W. ZNF251 promotes the progression of lung cancer by activating ERK signaling. Cancer Sci. 2020, 111, 3236–3244. [Google Scholar] [CrossRef]
- Li, W.; Song, L.; Ritchie, A.; Melton, D.W. Increased levels of DUSP6 phosphatase stimulate tumourigenesis in a molecularly distinct melanoma subtype. Pigment. Cell Melanoma Res. 2012, 25, 188–199. [Google Scholar] [CrossRef]
- Wei, K.C.; Chen, R.F.; Chen, Y.F.; Lin, C.H. Hinokitiol suppresses growth of B16 melanoma by activating ERK/MKP3/proteosome pathway to downregulate survivin expression. Toxicol Appl Pharmacol. 2019, 366, 35–45. [Google Scholar] [CrossRef]
- Li, W.; Melton, D.W. Cisplatin regulates the MAPK kinase pathway to induce increased expression of DNA repair gene ERCC1 and increase melanoma chemoresistance. Oncogene 2012, 31, 2412–2422. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H.; Kim, S.H.; Trousil, S.; Frederick, D.T.; Piris, A.; Yuan, P.; Cai, L.; Gu, L.; Li, M.; Lee, J.H.; et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat. Med. 2016, 22, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Urso, C.; Borgognoni, L.; Wang, J.; Deng, X.; Gray, N.S.; Stecca, B.; et al. ERK5 is activated by oncogenic BRAF and promotes melanoma growth. Oncogene 2018, 37, 2601–2614. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Sahoo, A.; Sawada, J.; Marchica, J.; Sahoo, S.; Layng, F.I.; Finlay, D.; Mazar, J.; Joshi, P.; Komatsu, M.; et al. MicroRNA-211 Modulates the DUSP6-ERK5 Signaling Axis to Promote BRAFV600E-Driven Melanoma Growth In Vivo and BRAF/MEK Inhibitor Resistance. J. Investig. Dermatol. 2020, 141, 385–394. [Google Scholar] [CrossRef]
- Chan, D.W.; Liu, V.W.; Tsao, G.S.; Yao, K.-M.; Furukawa, T.; Chan, K.K.; Ngan, H.Y. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinog. 2008, 29, 1742–1750. [Google Scholar] [CrossRef]
- Tong, X.; Mu, P.; Zhang, Y.; Zhao, J.; Wang, X. TRIM59, amplified in ovarian cancer, promotes tumorigenesis through the MKP3/ERK pathway. J Cell Physiol. 2020, 235, 8236–8245. [Google Scholar] [CrossRef]
- Furukawa, T.; Sunamura, M.; Motoi, F.; Matsuno, S.; Horii, A. Potential Tumor Suppressive Pathway Involving DUSP6/MKP-3 in Pancreatic Cancer. Am. J. Pathol. 2003, 162, 1807–1815. [Google Scholar] [CrossRef]
- Beaudry, K.; Langlois, M.; Montagne, A.; Cagnol, S.; Carrier, J.C.; Rivard, N. Dual-specificity phosphatase 6 deletion protects the colonic epithelium against inflammation and promotes both proliferation and tumorigenesis. J. Cell. Physiol. 2019, 234, 6731–6745. [Google Scholar] [CrossRef]
- Jiang, H.; Deng, L.; Lin, Z.; Yang, K.; Yang, J.; Zhao, W.; Gong, W. GSDMB interacts with IGF2BP1 to suppress colorectal cancer progression by modulating DUSP6-ERK pathway. Int. Immunopharmacol. 2024, 143, 113280. [Google Scholar] [CrossRef]
- Xu, N.; Dao, F.; Shi, Z.; Sun, K.; Qin, Y. WT1 together with RUNX1::RUNX1T1 targets DUSP6 to dampen ERK activity in acute myeloid leukaemia. Br. J. Haematol. 2024, 205, 1848–1859. [Google Scholar] [CrossRef]
- Wong, V.C.L.; Chen, H.; Ko, J.M.Y.; Chan, K.W.; Chan, Y.P.; Law, S.; Chua, D.; Kwong, D.L.; Lung, H.L.; Srivastava, G.; et al. Tumor suppressor dual-specificity phosphatase 6 (DUSP6) impairs cell invasion and epithelial-mesenchymal transition (EMT)-associated phenotype. Int. J. Cancer 2011, 130, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.-J.; Liang, S.-M.; He, P.-J.; Zhao, X.-B.; Li, M.-J.; Geng, F. Dusp6 inhibits epithelial-mesenchymal transition in endometrial adenocarcinoma via ERK signaling pathway. Radiol. Oncol. 2019, 53, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Rimel, B.; Massad, L.S.; Goodfellow, P.J. Infrequent methylation of the DUSP6 phosphatase in endometrial cancer. Gynecol. Oncol. 2010, 119, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, M.; Jiang, P.; Zhou, Y.; Yan, X.; Zhou, C.; Mu, Y.; Xiao, S.; Ji, G.; Wu, N.; et al. RPL22L1 fosters malignant features of cervical cancer via the modulation of DUSP6-ERK axis. J. Transl. Med. 2025, 23, 1–14. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Shi, X.; Yin, L.; Zhai, W.; Gao, S.; Chen, Y.; Zhang, T. Calcium saccharate/DUSP6 suppresses renal cell carcinoma glycolytic metabolism and boosts sunitinib efficacy via the ERK-AKT pathway. Biochem. Pharmacol. 2024, 224, 116247. [Google Scholar] [CrossRef]
- Wu, Q.-N.; Liao, Y.-F.; Lu, Y.-X.; Wang, Y.; Lu, J.-H.; Zeng, Z.-L.; Huang, Q.-T.; Sheng, H.; Yun, J.-P.; Xie, D.; et al. Pharmacological inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Lett. 2018, 412, 243–255. [Google Scholar] [CrossRef]
- Yang, B.; Tan, Y.; Sun, H.; Fan, J.; Tang, Z.; Ji, Y. Higher intratumor than peritumor expression of DUSP6/MKP-3 is associated with recurrence after curative resection of hepatocellular carcinoma. Chin Med J (Engl). 2014, 127, 1211–7. [Google Scholar]
- Lee, J.U.; Huang, S.; Lee, M.H.; Lee, S.E.; Ryu, M.J.; Kim, S.J.; Kim, Y.K.; Kim, S.Y.; Joung, K.H.; Kim, J.M.; et al. Dual specificity phosphatase 6 as a predictor of invasiveness in papillary thyroid cancer. Eur. J. Endocrinol. 2012, 167, 93–101. [Google Scholar] [CrossRef]
- Buffet, C.; Hecale-Perlemoine, K.; Bricaire, L.; Dumont, F.; Baudry, C.; Tissier, F.; Bertherat, J.; Cochand-Priollet, B.; Raffin-Sanson, M.-L.; Cormier, F.; et al. DUSP5 and DUSP6, two ERK specific phosphatases, are markers of a higher MAPK signaling activation in BRAF mutated thyroid cancers. PLOS ONE 2017, 12, e0184861. [Google Scholar] [CrossRef]
- Messina, S.; Frati, L.; Leonetti, C.; Zuchegna, C.; Di Zazzo, E.; Calogero, A.; Porcellini, A. Dual-specificity phosphatase DUSP6 has tumor-promoting properties in human glioblastomas. Oncogene 2011, 30, 3813–3820. [Google Scholar] [CrossRef]
- Lin, Y.K.; Wu, W.; Ponce, R.K.; Kim, J.W.; Okimoto, R.A. Negative MAPK-ERK regulation sustains CIC-DUX4 oncoprotein expression in undifferentiated sarcoma. Proc. Natl. Acad. Sci. 2020, 117, 20776–20784. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Parra, I.; Zhang, M.; Hilsenbeck, S.G.; Tsimelzon, A.; Furukawa, T.; Horii, A.; Zhang, Z.-Y.; Nicholson, R.I.; Fuqua, S.A. Elevated Expression of Mitogen-Activated Protein Kinase Phosphatase 3 in Breast Tumors: A Mechanism of Tamoxifen Resistance. Cancer Res. 2006, 66, 5950–5959. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wu, C.; Wei, C.; Li, D.; Hua, K.; Song, J.; Xu, H.; Chen, L.; Fang, L. Silencing of DUSP6 gene by RNAi-mediation inhibits proliferation and growth in MDA-MB-231 breast cancer cells: an in vitro study. . 2015, 8, 10481–90. [Google Scholar] [PubMed]
- Bergholz, J.; Zhang, Y.; Wu, J.; Meng, L.; Walsh, E.M.; Rai, A.; Sherman, M.Y.; Xiao, Z.X. ΔNp63α regulates Erk signaling via MKP3 to inhibit cancer metastasis. Oncogene. 2014, 33, 212–24. [Google Scholar] [CrossRef]
- Kong, T.; Laranjeira, A.B.A.; Yang, K.; Fisher, D.A.C.; Yu, L.; De La Frégonnière, L.P.; Wang, A.Z.; Ruzinova, M.B.; Fowles, J.S.; Fulbright, M.C.; et al. DUSP6 mediates resistance to JAK2 inhibition and drives leukemic progression. Nat. Cancer 2023, 4, 108–127. [Google Scholar] [CrossRef]
- Tischer, T.; Schuh, M. The Phosphatase Dusp7 Drives Meiotic Resumption and Chromosome Alignment in Mouse Oocytes. Cell Rep. 2016, 17, 1426–1437. [Google Scholar] [CrossRef]
- Castro-Sánchez, P.; Ramirez-Munoz, R.; Lamana, A.; Ortiz, A.; González-Álvaro, I.; Roda-Navarro, P. mRNA profilin identifies low levels of phosphatases dual-specific phosphatase-7 (DUSP7) and cell division cycle-25B (CDC25B) in patients with early arthritis. Clin. Exp. Immunol. 2017, 189, 113–119. [Google Scholar] [CrossRef]
- Chuang, H.-C.; Tan, T.-H. MAP4K Family Kinases and DUSP Family Phosphatases in T-Cell Signaling and Systemic Lupus Erythematosus. Cells 2019, 8, 1433. [Google Scholar] [CrossRef]
- Peng, W.-X.; Huang, J.-G.; Yang, L.; Gong, A.-H.; Mo, Y.-Y. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol. Cancer 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Ikegawa, S. Expression, Regulation and Function of Asporin, A Susceptibility Gene in Common Bone and Joint Diseases. Curr. Med. Chem. 2008, 15, 724–728. [Google Scholar] [CrossRef]
- Li, P.; Zhang, Z.; Li, H.; Zhao, Y.; Guo, Q.; Yu, Y.; Zhu, S.; Zhang, S.; Min, L. Asporin promotes cell proliferation via interacting with PSMD2 in gastric cancer. Front. Biosci. 2019, 24, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Liu, M.; Feng, W.; Rao, H.; Zhang, W.; Liu, C.; Xu, Y.; Wang, Z.; Teng, Y.; Yang, X.; et al. Loss of SETD2 aggravates colorectal cancer progression caused by SMAD4 deletion through the RAS/ERK signalling pathway. Clin. Transl. Med. 2023, 13, e1475. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhou, L.; Li, S.; He, W.; Zheng, J.; Hou, Z.; He, P. Let-7c-5p Down Regulates the Proliferation of Colorectal Cancer Through the MAPK-ERK-Signaling Pathway. Biochem. Genet. 2023, 62, 3231–3243. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lagowski, J.; Sundholm, A.; Sundberg, A.; Kulesz-Martin, M. Microtubule Disruption and Tumor Suppression by Mitogen-Activated Protein Kinase Phosphatase 4. Cancer Res. 2007, 67, 10711–10719. [Google Scholar] [CrossRef]
- Qiu, Z.; Liang, N.; Huang, Q.; Sun, T.; Xue, H.; Xie, T.; Wang, X.; Wang, Q. Downregulation of DUSP9 Promotes Tumor Progression and Contributes to Poor Prognosis in Human Colorectal Cancer. Front. Oncol. 2020, 10. 547011. [Google Scholar] [CrossRef]
- Imajo, M.; Kondoh, K.; Yamamoto, T.; Nakayama, K.; Nakajima-Koyama, M.; Nishida, E. Antagonistic Interactions between Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase and Retinoic Acid Receptor Signaling in Colorectal Cancer Cells. Mol. Cell. Biol. 2017, 37, e00012–17. [Google Scholar] [CrossRef]
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and p38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202. [Google Scholar] [CrossRef]
- Mishra, A.; Oulès, B.; Pisco, A.O.; Ly, T.; Liakath-Ali, K.; Walko, G.; Viswanathan, P.; Tihy, M.; Nijjher, J.; Dunn, S.-J.; et al. A protein phosphatase network controls the temporal and spatial dynamics of differentiation commitment in human epidermis. eLife 2017, 6. e27356. [Google Scholar] [CrossRef]
- Nomura, M.; Shiiba, K.; Katagiri, C.; Kasugai, I.; Masuda, K.; Sato, I.; Sato, M.; Kakugawa, Y.; Nomura, E.; Hayashi, K.; Nakamura, Y.; Nagata, T.; Otsuka, T.; Katakura, R.; Yamashita, Y.; Sato, M.; Tanuma, N.; Shima, H. Novel function of MKP-5/DUSP10, a phosphatase of stress-activated kinases, on ERK-dependent gene expression, and upregulation of its gene expression in colon carcinomas. Oncol Rep. 2012, 28, 931–6. [Google Scholar]
- Ng, K.Y.; Chan, L.H.; Chai, S.; Tong, M.; Guan, X.Y.; Lee, N.P. , Yuan, Y. ; Xie, D.; Lee, T.K.; Dusetti, N.J., Carrier, A.; Ma, S. TP53INP1 Downregulation Activates a p73-Dependent DUSP10/ERK Signaling Pathway to Promote Metastasis of Hepatocellular Carcinoma. Cancer Res. 2017, 77, 4602–4612. [Google Scholar]
- Hou, J.; He, Z.; Liu, T.; Chen, D.; Wang, B.; Wen, Q.; Zheng, X. Evolution of Molecular Targeted Cancer Therapy: Mechanisms of Drug Resistance and Novel Opportunities Identified by CRISPR-Cas9 Screening. Front. Oncol. 2022, 12, 755053. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Brown, J.S. The Evolution and Ecology of Resistance in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2020, 10, a040972. [Google Scholar] [CrossRef] [PubMed]
- Tubita, A.; Tusa, I.; Rovida, E. Playing the Whack-A-Mole Game: ERK5 Activation Emerges Among the Resistance Mechanisms to RAF-MEK1/2-ERK1/2- Targeted Therapy. Front. Cell Dev. Biol. 2021, 9. 647311. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of ERK-targeting DUSPs involved in cancer biology. https://biorender.com/ny85ho7.
Figure 1.
Schematic representation of ERK-targeting DUSPs involved in cancer biology. https://biorender.com/ny85ho7.
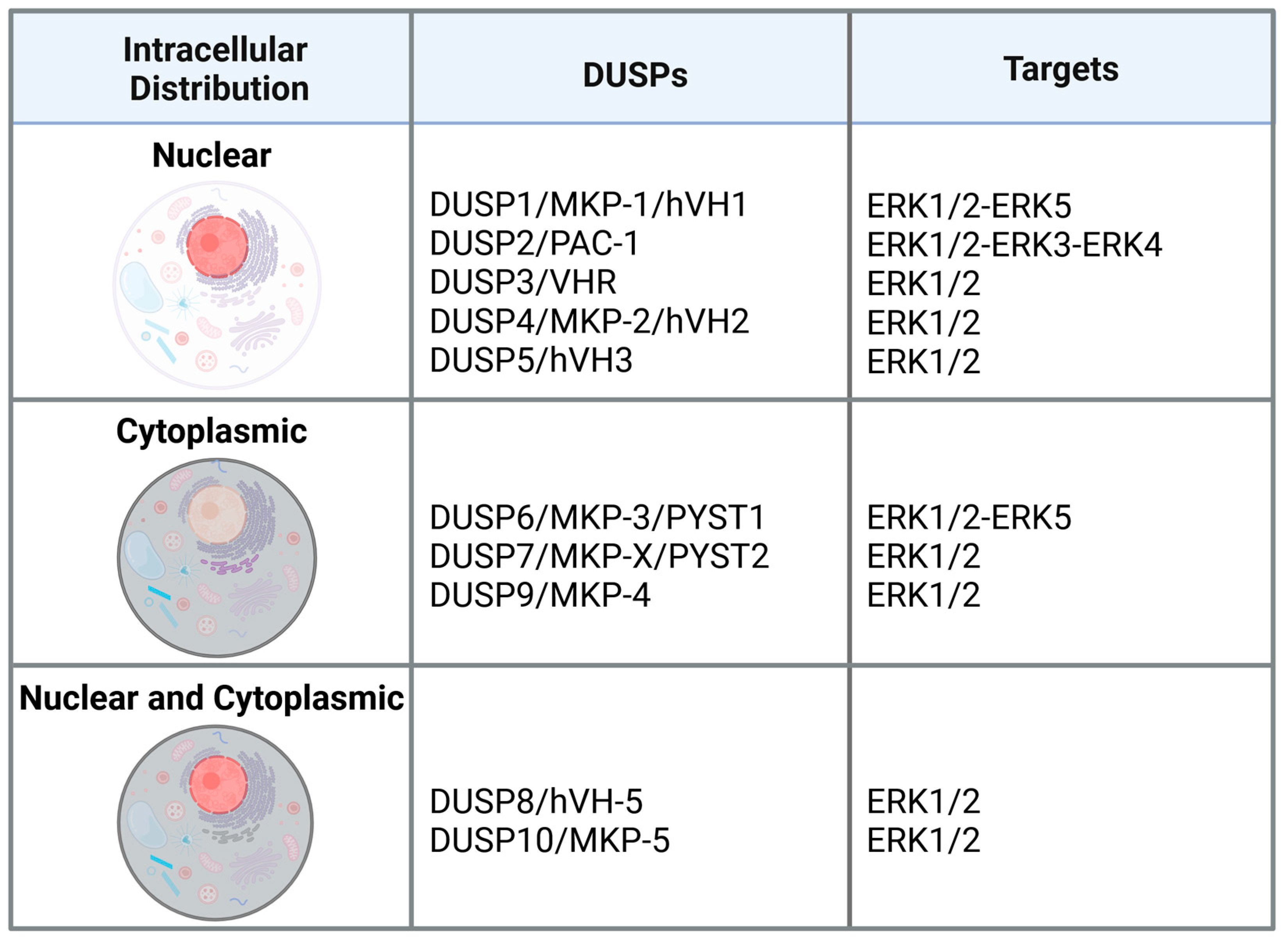
Figure 2.
Role of DUSPs in the hallmarks of cancer. https://biorender.com/n0m03f4.
Figure 2.
Role of DUSPs in the hallmarks of cancer. https://biorender.com/n0m03f4.
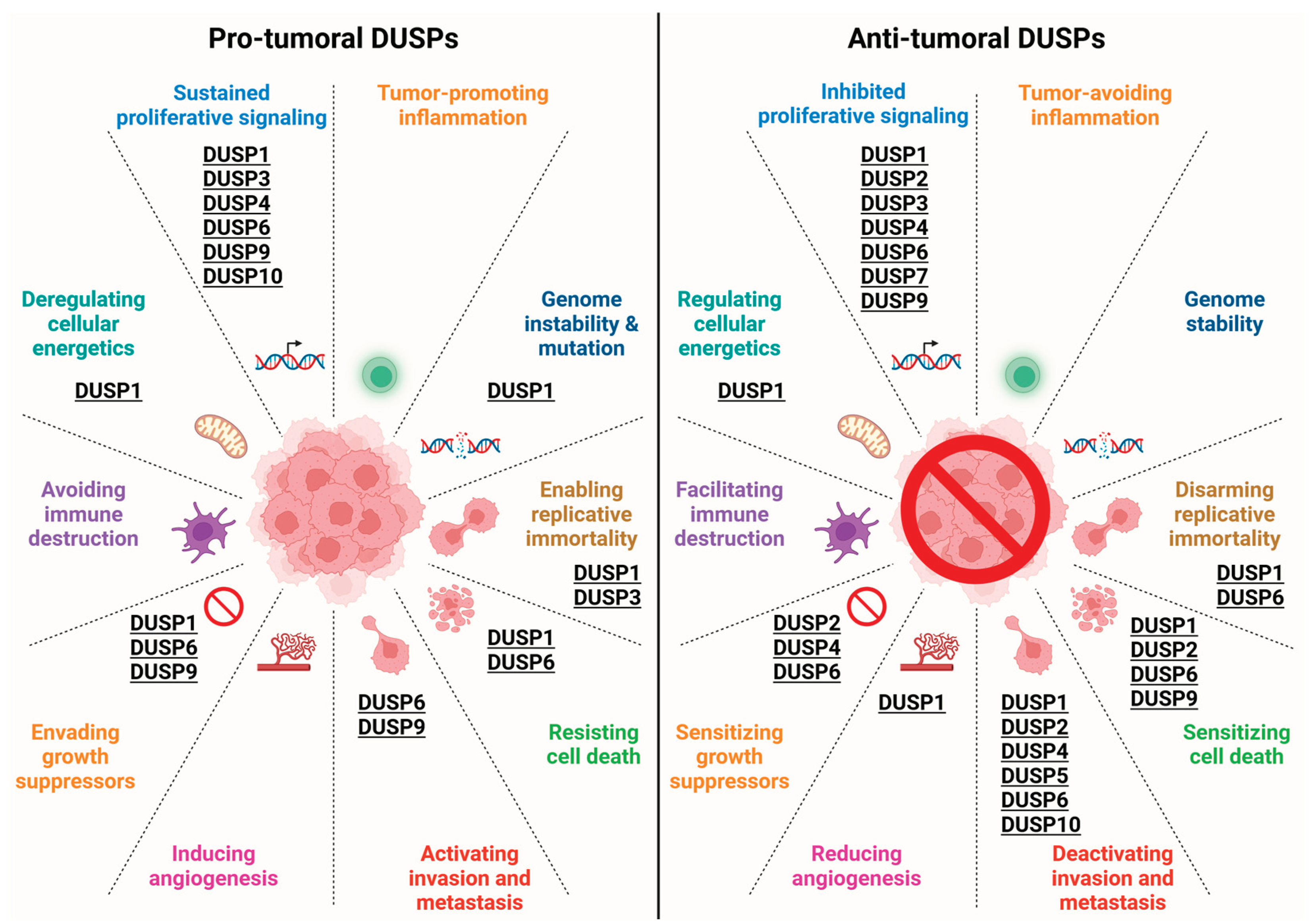

Table 1.
In vivo evidence of the role of ERK-targeting DUSPs in cancer.
| Gene/MKP | Tumour type (xenografted cell line/animal model) | Treatment/genetic manipulation (observed secondary effect on DUSPs) | Biological outcome | Effect on ERKs | Ref |
|---|---|---|---|---|---|
| DUSP1 | HCC | Cx32 k/o (Reduced DUSP1) |
Increased ethanol-induced HCC | Increased pERK1/2 | [42] |
| DUSP1 | CRC (HCT116 and SW480) |
DUSP1 overexpression | Increased tumour growth | Reduced pERK1/2 | [26] |
| DUSP1 | BLCA (HT1197 and RT112 |
AKTi (Increased DUSP1) |
Reduced tumour growth | Reduced pERK1/2 | [34] |
| DUSP1 | ESCC (TE-1) |
ARNTL overexpression (Increased DUSP1) |
Reduced tumour growth | Reduced pERK1/2 | [50] |
| DUSP2 | PDAC (PANC-1, SW1990, BxPC-3, CFPAC-1) |
Mir-361-3p overexpression (Reduced DUSP2) |
Increased metastasis | Increased pERK1/2 | [57] |
| DUSP2 | GC (MGC-803, HGC-27) |
SKA3 KD (Increased DUSP2) |
Reduced tumour growth | Reduced pERK1/2 | [58] |
| DUSP2 | BLCA | DUSP2 overexpression | Reduced tumor growth | Reduced pERK1/2 | [59] |
| DUSP3 | NSCLC (H1792) |
KDM2A KD (Increased DUSP3) |
Reduced tumour growth | Reduced pERK1/2 |
[63] |
| DUSP3 | HCC (diethylnitrosamine) |
DUSP3 overexpression | Reduced tumour growth/Increased sorafenib sensitivity | Reduced pERK1/2 | [65] |
| DUSP4 | BC (MCF7) |
DUSP4 KD | Increased tumour growth | Increased pERK1/2 | [75] |
| DUSP4 | PDAC (PANC-1) |
DUP4 overexpression | Reduced tumour growth | Reduced pERK1/2 | [68] |
| DUSP5 | Skin cancer (HRasQ61L mice) |
DUSP5 KO | Increased papilloma formation | Increased pERK1/2 | [83] |
| DUSP5 | Thyroid Cancer (K1) |
DUSP5 KO | Reduced tumour growth | Reduced pERK1/2 | [90] |
| DUSP6 | Ovarian Cancer (A2780) |
DUSP6 overexpression | Reduced tumour growth/increased cisplatin sensitivity | Reduced pERK1/2 | [116] |
| DUSP6 | Melanoma (A375) |
DUSP6 overexpression | Reduced tumour growth | Reduced pERK5 | [115] |
| DUSP6 | CRC (Apc Min/+ mice) |
DUSP6 KO |
Increased intestinal tumorigenesis | Increased pERK1/2 | [119] |
| DUSP6 | GC (BGC823, SGC7901, SGC7901/DDP on PDX and xenografts) |
DUSP6 KO BCI |
Reduced tumour growth | Increased pERK1/2 | [127] |
| DUSP6 | Melanoma (A375) |
STAG2 or STAG3 KD (Reduced DUSP6) |
Reduced BRAFi sensitivity | Increased pERK1/2 | [113] |
| DUSP6 | Cervical Cancer (HeLa) |
RPL22L1 KD (Increased DUSP6) |
Reduced tumour growth | Reduced pERK1/2 | [125] |
| DUSP6 | CRC (GSDMB transgenic mice) |
GSDMB overexpression (Increased DUSP6) |
Reduced tumor formation | Reduced pERK1/2 | [120] |
| DUSP6 | AML (Kasumi-1 and HL60) |
WT1 overexpression/KD (Increased/reduced DUSP6) |
Reduced/increased tumour growth | Reduced/increased pERK1/2 | [121] |
| DUSP6 | Renal Cell Carcinoma (786-O) | Sunitinib and Calcium saccharate (Increased DUSP6) |
Reduced tumour growth | Reduced pERK1/2 | [126] |
| DUSP6 | ESCC (SLMT-1) NPC (HONE1) |
DUSP6 overexpression | Reduced tumour growth | Reduced pERK1/2 | [122] |
| DUSP6 | NSCLC (H460) |
DUSP6 KD | Increased tumour growth | Increased pERK5 | [101] |
| DUSP6 | AML (CD45.1+ C57BL/6J mice) |
BCI | Reduced tumour progression | Reduced pERK1/2 | [136] |
| DUSP7 | CRC | SETD2/SMAD4 KO (Reduced DUSP7) | Increased tumour growth | Reduced pERK1/2 | [143] |
| DUSP1 and DUSP6 | MPNSTs (S462.TY PDX and xenografts) |
DUSP1 and/or DUSP6 KD BCI |
Reduced tumour growth | Increased pERK1/2 | [27] |
| DUSP10 | HCC (MHCC97L) |
TP53INP1 KD (Reduced DUSP10) |
Increased metastasis | Increased pERK1/2 | [151] |
PDX, patient derived xenograft; pERK1/2, phosphorylated ERK1/2; KO, knock out; KD, knock down; GC, gastric cancer; HCC, hepatocellular carcinoma; MPNST, malignant peripheral nerve sheath tumor; AML, acute myeloid leukemia; NSCLC, non-small cell lung cancer; PDAC, pancreatic ductal adenocarcinoma; ESCC, esophageal squamous cell carcinoma; NPC, nasopharyngeal carcinoma; CRC, colorectal cancer; BC, breast cancer; BLCA, urothelial bladder carcinoma.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



