
Submitted:
09 June 2025
Posted:
10 June 2025
You are already at the latest version
Abstract
Core fucose is one of the most important glycans in HBV infection. In this study, we investigated whether PhoSL, a lectin that specifically binds to core fucose, exerts an inhibitory effect in an HBV infection model of normal human hepatocytes. Similar to previous studies using hepatocellular carcinoma cells (HepG2-C4), coexistence of PhoSL during HBV infection inhibited HBe antigen production and HBV cccDNA in normal human hepatocytes in a PhoSL concentration-dependent manner. Furthermore, this effect of PhoSL was found to be able to suppress HBe antigen production in a treatment period-dependent manner even when PhoSL was administered after HBV infection. The mechanisms of HBV infection inhibition by PhoSL that have been elucidated so far are physical inhibition by binding to the HBV receptor and inhibition of HBV entry into cells by inhibiting phosphorylation of EGFR, a co-receptor for NTCP. Furthermore, this study suggested that PhoSL may also inhibit HBV proliferation in cells through some mechanism. PhoSL is a lectin derived from edible mushrooms that is resistant to acid and heat. In addition, it has a low molecular weight and can be chemically synthesized, so it is expected to be used clinically as a new carbohydrate therapy for HBV in the future.
Keywords:
HBV
; PhoSL
; therapy
; Fut8
; PXB cells
1. Introduction
Hepatitis B virus (HBV) is a major hepatitis virus. This infection is a serious global health problem, with 2 billion people infected worldwide and 350 million suffering from chronic HBV infection, although the incidence rate varies greatly between countries [1]. The main route of infection is mother-to-child transmission of HBV, which is basically established through the bloodstream. As chronic hepatitis caused by HBV progresses, the incidence of liver cirrhosis and hepatocellular carcinoma increases, so aggressive treatment of HBV is recommended [2,3]. Currently, interferon and nucleoside analogues are commonly used for treatment, but the former has a low response rate and side effects, and the latter has the problem that the drug cannot be discontinued [4,5]. Therefore, there is a need for the development of drugs such as direct-acting hepatitis C antivirals (DAAs) that are highly effective in treating hepatitis B virus and do not cause permanent recurrence of the virus even if treatment is discontinued [6,7,8]. In 2012, sodium taurocholate cotransporting polypeptide (NTCP, encoded by SLC10A1) was identified as a functional receptor for HBV by a Chinese group [9]. NTCP is a receptor involved in the enterohepatic circulation of bile acids [10]. To date, several compounds have been reported as HBV entry inhibitors targeting NTCP and are undergoing clinical trials [11,12,13]. Because the HBV receptor is present on the cell surface, possibly as a complex like the hepatitis C virus receptor [14,15,16], such coreceptors may be new targets for HBV treatment.
Many cellular proteins, especially secreted and cell surface proteins, are glycosylated and are known to be involved in the biological functions of cells [17,18,19]. Since many cell surface proteins, including viral receptors and viral envelope proteins, are glycoproteins, glycans have become targets for antiviral therapy [20,21]. Changes in the glycosylation state of proteins may affect the infectivity of virus particles [22]. For example, influenza infection requires the cleavage of sialic acid from certain proteins by sialidases on host cells [23]. Inhibition of sialidases has been a target for anti-influenza therapy [20]. Sialylation of host cells is also required for SARS-CoV-2 infection [24]. Based on these findings, many lectins, i.e., proteins that selectively bind to specific glycans, are considered to be potential candidates for antiviral drugs [25]. For example, the lectin Griffithsin can bind to mannose-rich oligosaccharides and inhibit human immunodeficiency virus (HIV)-1 infection without cytotoxicity [26]. It is also known that human surfactant protein A can inhibit SARS-CoV-2 infection [27].
NTCP is a glycoprotein with four N-linked glycosylation sites [28]. We previously revealed that core fucose is one of the glycosylation modifications most involved in HBV infection using HBV pseudoparticles BNC (bionanocapsules) [29]. Core fucose is fucose added to the innermost N-acetylglucosamine of N-glycans and is specifically synthesized by α1-6 fucosyltransferase Fut8 [30]. Furthermore, we recently reported that PhoSL (Pholiota squarrosa lectin), which specifically recognizes core fucose, inhibits HBV infection and cccDNA synthesis in a concentration-dependent manner in HepG2 cells overexpressing NTCP (HepG2-C4) [31]. Imaging experiments demonstrated that the mechanism by which PhoSL inhibits HBV infection occurs not only by directly binding to NTCP but also by inhibiting EGF receptor phosphorylation. Furthermore, the inhibition of HBV infection by PhoSL was observed even in Fut8 knockout C4 cells, suggesting that PhoSL also binds to HBV itself, is taken up into the cell, and has some effect on the growth and replication of HBV.
PhoSL has been purified from edible mushrooms and has been confirmed to be non-toxic when administered intraperitoneally and orally to animals [32]. In this study, we investigated whether PhoSL also exerts an inhibitory effect on HBV infection in normal human hepatocytes, aiming at future clinical drug discovery. As a result, we found that when PhoSL was administered simultaneously with HBV infection, it could suppress HBe antigen production and cccDNA synthesis in normal human hepatocytes in a concentration-dependent manner. Furthermore, even when PhoSL was administered after HBV infection was established, the inhibitory effect of HBV infection by PhoSL was found to be able to suppress HBe antigen production and cccDNA synthesis in a treatment-period-dependent manner.
2. Materials and Methods
2.1. Cells
PXB-cells were purchased from PhoenixBio (Hiroshima, Japan). PXB-cells are fresh hepatocytes harvested from human hepatocyte chimeric mice by collagenase perfusion. The purity of human hepatocytes in PXB-cells is approximately 93%. Donor cells are derived from a 1-year-old Caucasian boy or a 12-year-old Caucasian girl.
PXB-cells were cultured in DMEM (nacalai-tesque, Kyoto, Japan) containing 10% fetal bovine serum (FBS) (Nichirei, Tokyo, Japan) with 20 mM HEPES (Thermo Fisher, Waltham, MA), 44 mM NaHCO3 (Fujifilm-Wako, Osaka, Japan), 100 U/ml Penicillin G (Thermo Fisher), 100 µg/ml Streptomycin (Thermo Fisher), 15 µg/ml L-proline (Fijifilm-Wako, Osaka, Japan), 0.25 µg/ml Insulin (Sigma-Aldrich, St Louis, MO), 50 nM Dexamethasone (Sigma-Aldrich), 5 ng/ml EGF (Sigma-Aldrich), 0.1 mM L-ascorbic acid 2-phosphate (Fujifilm-Wako), and 2% DMSO (Sigma-Aldrich) in a 37°C incubator under a humidified atmosphere containing 5% CO2.
PhoSL lectin was obtained from Dr. Y. Kobayashi (Mitsubishi Chemical). Myristoylated PreS1 peptide (myrPreS1) was synthesized by Scrum (Tokyo, Japan) [33].
2.2. Preparation of HBV Particles
HepAD38.7 cells are a tetracycline-regulated HBV-producing cell line using the Tet-Off system. To obtain HBV particles for HBV infection experiments, tetracycline was removed from the culture medium and HBV replication was induced in HepAD38.7 cells. The culture medium of confluent HepAD38.7 cells cultured without tetracycline was collected every week for 2 weeks, and HBV particles were precipitated by adding PEG 8000 (final concentration 6%) and stand overnight at 4°C. The precipitate was centrifuged, resuspended in phosphate-buffered saline (PBS), concentrated, and filtered through a 0.45 µm filter (Millipore). HBV DNA was quantified by qPCR and adjusted to 107 HBV/µL.
2.3. Cytotoxicity Assay
PXB cells seeded at 4 x 105 cells/well in 24-well plates were cultured at 37 °C under humidified conditions with 5% CO2. After replacing with fresh medium, cells were co-cultured with HBV with PhoSL (0, 1, 2.5, 5, or 10 μg/mL) for 1 day and then continued to be cultured for an additional 3, 6, 9, or 12 days with medium changes every 3 days. At each time point, culture supernatants were harvested, diluted 25-fold in LDH storage buffer, and stored at -20 °C until analysis. Viability was measured using the LDH-Glo Cytotoxicity Assay (Promega) according to the manufacturer's instructions. Chemiluminescence intensity was measured using the GloMax Discover System (Promega).
2.4. HBV Infection and HBe Antigen Measurement
HBV stocks were prepared as previously described [34]. PXB cells (4 × 105/well) seeded in 24-well plates were replaced with 0.5 ml of fresh medium once upon arrival, and HBV infection experiments were started the next day or later. The cells were infected with HBV (2 × 107 genomes/well [approximately 50 genome infectious equivalents (G.E.I)]) in medium containing 4% polyethylene glycol 8000 (PEG 8000 [Sigma-Aldrich]). After 24 h of incubation, the cells were washed twice with 0.5 ml of PBS and cultured for 12 days in 0.5 ml of the same medium, changing the medium every 3 days. Culture supernatants were collected five times, on the day of medium replacement and at the end of incubation, and hepatitis B e antigen (HBeAg) was quantified by using an HBeAg ELISA kit (Bioneovan, Beijing, China). PhoSL was administered at a final concentration of 10 µg/ml in the following five patterns: 1. Simultaneous administration for 1 day at the time of HBV infection, 2. Administration for 12 days after medium change following completion of HBV infection, 3. Administration for the final 9 days of culture, 4. Administration for the final 6 days of culture, and 5. Administration for the final 3 days of culture.
2.5. Preparation of Extracellular HBV Particle-Associated DNA
Extracellular core particle-associated HBV DNA was prepared as follows: 30% PEG8000 (Sigma-Aldrich) was added to the collected culture supernatant to a final concentration of 6%, and the mixture was mixed by end-over-end at 4°C overnight. After centrifugation at 8000 x g for 30 min at 4°C, the supernatant was removed, and 450 µl of TBS (20 mM Tris-HCl, pH 7.8, 150 mM NaCl) was added to the precipitate, which was then dissolved by end-over-end mixing at 4°C overnight. After centrifugation at 8000 x g for 30 min at 4°C, the supernatant was collected and incubated overnight at 37°C with 12.5 U DNase I (TaKaRa) and 500 ng RNase (Roche) in 1 x DNase buffer (40 mM Tris-HCl, pH 7.9, 10 mM NaCl, 6 mM MgCl2, 10 mM CaCl2, 2 mM DTT) to degrade DNA outside the virions. EDTA (pH 8.0) was added to a final concentration of 10 mM, and the mixture was heated at 80°C for 5 min to inactivate DNase I. 50 µl of this reaction mixture was mixed with 350 µl of Proteinase K reaction buffer (10 mM Tris-HCl, pH 7.8, 10 mM EDTA, 0.5 % SDS) and 4 µl of 600 mU/µl Proteinase K (nacalai-tesque), and digested overnight at 56°C. An equal volume of phenol/chloroform/isoamylalcohol (25:24:1) was added, mixed using a vortex, and centrifuged at 13,500 x g for 30 min at 4°C, and 300 µl of the aqueous layer was collected. 2 µl of 10 mg/ml glycogen (nacalai-tesque), 30 µl of 3M sodium acetate, pH 5.2 (Sigma-Aldrich), and 750 µl of ethanol (nacalai-tesque) were added, mixed using a vortex, centrifuged at 13,500 x g for 30 min at 4°C, and the supernatant was removed. 1,000 µl of 70% ethanol was added, and centrifuged at 13,500 x g for 15 min at 4°C, and the supernatant was removed. The precipitate was air-dried briefly and then dissolved in 20 µl of TE.
2.6. Preparation of Hirt DNA and Quantification of HBV DNA Copies
Hirt DNA was isolated from HBV-infected PXB cells using a standard Hirt extraction method. Briefly, 350 µl of 1% SDS/TE was added to one well of cells, incubated for 10 min, and the lysate was scraped off with a pipette tip. 50 µl of 0.5 M NaCl was then added to the lysate, mixed, and incubated overnight at 4°C. After centrifugation at 18,000 xg for 30 min at 4°C, 200 µl of the supernatant was collected. 1 µl of 500 µg/ml RNase (Roche, Indianapolis, IN) was then added to the supernatant, mixed, and incubated at 65°C for 30 min. An additional 2 µL of 0.2 mg/ml proteinase K (Roche) was added, mixed, and incubated at 56°C overnight. 200 µl of TE-saturated phenol (nacalai-tesque, Kyoto, Japan) was added, mixed by vortexing, and centrifuged at 18,000 xg for 20 min at 4°C, and 200 µl of the supernatant was collected. 200 µl of phenol-chloroform-isoamyl alcohol (PCI)25: 24:1 (nacalai-tesque) was added to the supernatant, mixed by vortexing, and centrifuged at 18,000 xg for 30 min at 4°C, and 150 µl of the supernatant was collected. 2 µl of 10 mg/ml glycogen (Takara Bio, Shiga, Japan), 15 µl of 3M CH3COONa, and 375 µl of ethanol were added to the supernatant, mixed by vortexing, and centrifuged at 20,000 xg for 30 min at 4°C. After removing the supernatant, 550 µl of 70% ethanol was added and centrifuged at 20,000 xg for 15 min at 4°C. The supernatant was removed and the samples were briefly air-dried before dissolving in 20 µl of TE. cccDNA measurements were performed by digesting 10 µl of dissolved Hirt DNA with 0.5 µl of T5 exonuclease (Promega, Madison, WI) for 60 min at 37°C.
Extracellular and intracellular HBV DNA was quantified by real-time PCR. Quantification was calculated by creating a standard curve using a dilution series of HBV DNA from 1.2 x 108 to 1.2 x 103 genome copies. Real-time qPCR was performed using a QuantStudio1 real-time PCR system (Thermo Scientific). The primers used were HBs-qPCR-Fw: 5'-CTTCATCCTGCTGCTATGCCT-3' and HBs-qPCR-Rv: 5'-AAAGCCCAGGATGATGGGAT-3', both at a concentration of 10 µM. After initial denaturation at 95°C for 30 s, PCR reactions consisted of two steps: denaturation at 95°C for 5 s, annealing and extension at 60°C for 10 s, with 40 cycles.
HBV cccDNA was quantified by digital PCR. The amount of RNase P gene was measured using TaqMan RNase P control reagent kit (Thermo) and used as an internal control. Digital PCR was performed using a QuantStudio Absolute Q Digital PCR System (Thermo Fisher, Waltham, MA). The primers used were as follows: Probe (FAM) 5’-CTGTAGGCATAAATTGGT-3’
cccDNA_Fwd: 5’-CGTCTGTGCCTTCTCATCTGC-3’ and cccDNA_Rev: 5’-GCACAGCTTGGAGGCTTGAA-3’
2.7. Statistical Analysis
Data are presented as mean ± SD and significance was tested using Student’s t test with Microsoft Excel software. P < 0.05 was considered to indicate statistical significance.
3. Results and Discussion
3.1. PhoSL Inhibited HBV Infection in Normal Human Hepatocytes in a Concentration-Dependent Manner When Administered Simultaneously with HBV Infection
Figure 1.
A shows an outline of the PhoSL administration experiment.
First, to examine the concentration dependence of PhoSL, PhoSL was allowed to coexist during HBV infection. In our previous study, we found that PhoSL inhibited HBV infection in hepatocellular carcinoma HepG2-C4 cells. Therefore, to clarify whether PhoSL can also inhibit HBV infection in normal hepatocytes, we examined its effect using PXB cells. As shown in Figure 1B, PhoSL did not show significant toxicity to PXB cells up to a concentration of 10 µg/mL.
As shown in Figure 1C, PhoSL was able to suppress HBe antigen production in normal hepatocytes in a concentration-dependent manner, as well as in hepatocellular carcinoma cells. The results in Figure 1C show that the effect of PhoSL coexistence during HBV infection was large, and HBeAg production was dramatically suppressed during the measurement period from the third day after the end of infection. This is thought to be due to PhoSL's strong inhibition of HBV intracellular entry.
However, this is only the result when PhoSL coexists with HBV infection, and to expect the effect of PhoSL during natural HBV infection, it is necessary to take PhoSL continuously as a permanent preventive measure, which is not very realistic. If the infectious disease treatment effect of PhoSL can be confirmed after HBV infection, clinical application is expected. Therefore, using the same normal hepatic PXB cells, we investigated whether a therapeutic effect could be obtained by administering PhoSL for various periods starting one day after HBV infection.
3.2. PhoSL Suppressed HBeAg Production, Intra- and Extracellular HBV DNA and cccDNA Production in a Treatment-Period-Dependent Manner, even When Administered After HBV Infection
Figure 2A outlines an experiment in which PhoSL was administered therapeutically after HBV infection. Cells were infected with HBV for 1 day, washed twice with PBS, and then cultured in the presence of PhoSL for 3, 6, 9, or 12 days. Figure 2B shows the time course of HBe Ag produced in the culture supernatant after HBV infection. When myristoylated PreS1 peptide (myrPreS1) was coexisted during HBV infection, the infection was strongly suppressed, and HBe Ag production in the medium was low even after 12 days.
To verify the feasibility of PhoSL therapy for HBV-infected patients, we used PXB cells to examine whether administration of PhoSL after HBV infection has an infection-suppressing effect, using changes in HBe Ag production and extracellular HBV production as indicators. As shown in the results in Figure 2C, it was found that PhoSL can suppress HBe Ag production in a treatment time-dependent manner, even when administered after HBV infection. This result is almost reflected in the results in Figure 2D, and it was confirmed that the amount of extracellular HBV DNA was also suppressed with increasing PhoSL treatment time.
We found that PhoSL, when administered simultaneously with HBV infection, reduced the efficiency of HBV infection into PXB cells. Based on the results of previous studies, this is thought to be due to inhibition of entry, but in this study, we found that even when treated after HBV infection, the amount of extracellular HBeAg and HBV DNA was suppressed in a treatment-time-dependent manner.
Previous studies have demonstrated that, even in the absence of HBV, PhoSL is internalized into cells, likely through binding to the core fucose of cell surface receptors. [31]
To determine whether this was due to inhibition of extracellular secretion or intracellular replication, we measured the amount of intracellular HBV DNA and cccDNA. The results are shown in Figure 2E,F. The measured intracellular HBV DNA includes HBV DNA in the cytoplasm, relaxed circular DNA (rcDNA) in the nucleus, and covalently closed circular DNA (cccDNA). If the amount of intracellular HBV DNA is unchanged with or without PhoSL administration, and only cccDNA is reduced, it would suggest that PhoSL affects the conversion of rcDNA to cccDNA. The conversion of rcDNA to cccDNA is an important step in the HBV life cycle, and cccDNA is the main template for HBV gene transcription, so it would be groundbreaking if PhoSL could inhibit cccDNA synthesis. However, since PhoSL treatment also reduced the total amount of HBV DNA in the cells, it is possible that the process of rcDNA translocation to the nucleus, denucleation, and reimport of nucleocapsids into the nucleus is inhibited, no conclusion has been reached at this time. Furthermore, core fucosylation may play an important role not only in protein import via endosomes, but also in secretion via the ER-Golgi system, and PhoSL may act on this system to reduce the amount of HBV released outside the cells. However, in a study using HB611 cells [35] established by introducing tandem repeats of the HBV genome into Huh6 cells, PhoSL administration did not reduce the amount of extracellular HBe antigen (data not shown). This cell line is a model for evaluating the release process of formed virus particles outside the cells at the late stage of HBV infection, as they can produce and secrete HBV particles inside the cells. As the results it has been suggested that PhoSL does not affect the final release of HBV particles outside the cells.
3.3. MyrPreS1 Peptide Did Not Affect the Amount of Extracellular HBeAg or the Amount of Intracellular and Extracellular HBV DNA and cccDNA Produced When Administered After HBV Infection
Another possible pathway for the suppressive effect of PhoSL on extracellular HBeAg and HBV DNA levels observed after HBV infection is the suppression of reinfection. Therefore, we investigated the effect of myrPreS1, which is known to be able to suppress the reinfection pathway. Figure 3A shows an outline of an experiment in which myrPreS1 was administered after HBV infection. Cells were infected with HBV for 1 day, washed twice with PBS, and then cultured for 12 days in the presence or absence of myrPreS1.
As shown in Figure 3B, myrPreS1 can strongly suppress HBV infection when it is coexisted during HBV infection, but when it is treated after infection, it cannot suppress the amount of extracellular HBe antigen as shown in Figure 3C, and as shown in Figure 3 (D, E, F), no suppressive effect was observed on the amount of intracellular and extracellular HBV DNA and cccDNA, unlike in the case of PhoSL. In addition, PhoSL recognizes and binds to a glycan structure called core fucose, but since HBV glycosylation is affected by the balance of glycosyltransferases in the host cell, it is thought that HBV produced from PXB cells is not core fucose-modified because normal hepatocytes such as PXB cells hardly express the core fucosyltransferase Fut8. Considering these findings together, it is speculated that the infection-suppressing effect observed when PhoSL was administered after HBV infection is not due to inhibition of reinfection, but rather due to PhoSL that was taken up into the cells by some route and inhibited somewhere in the HBV replication pathway.
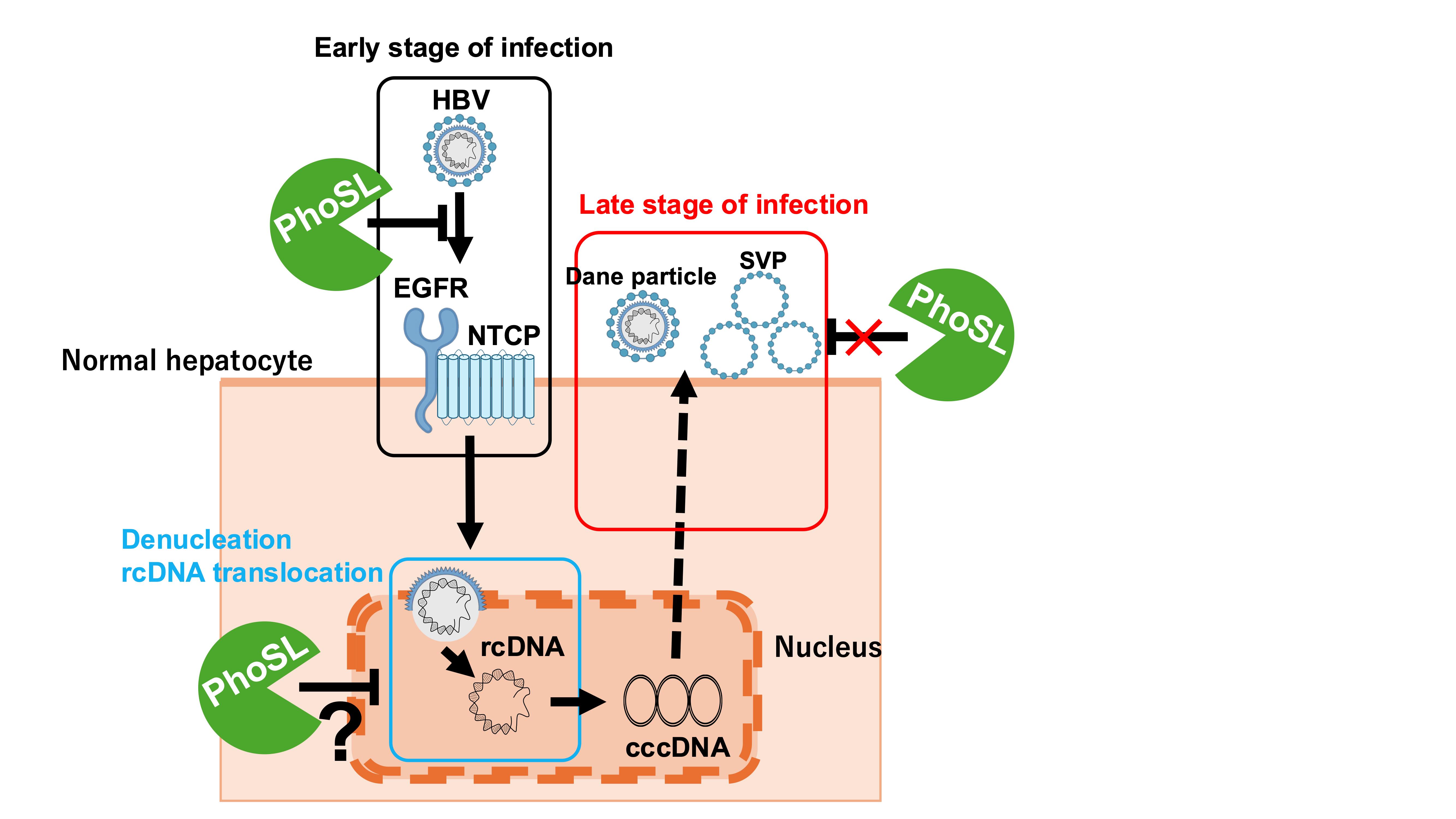
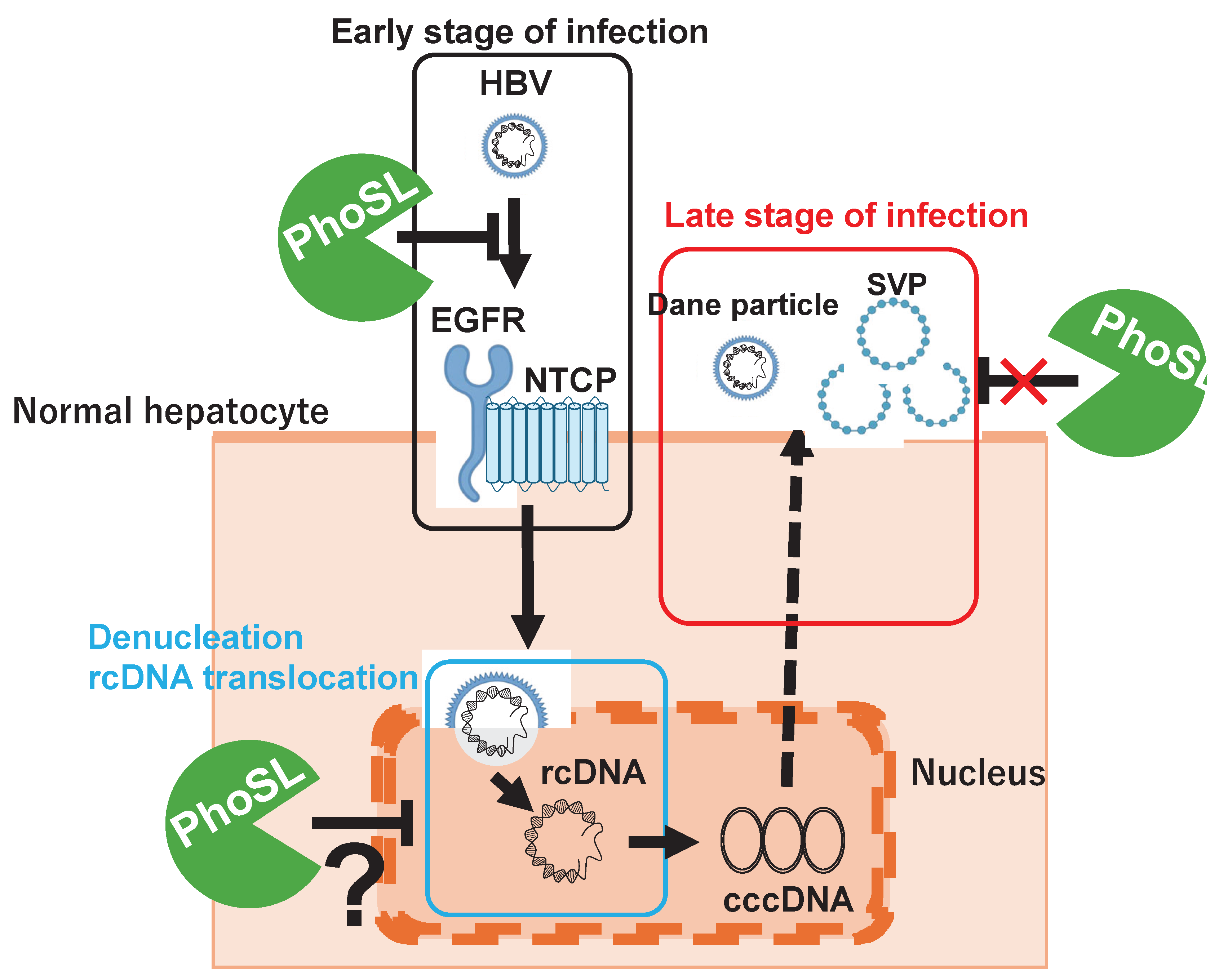
A brief summary of the results of this study is shown in Figure 4. PhoSL binds to the core fucose of NTCP receptor and HBV virus particles, inhibiting HBV entry into cells by steric hindrance. PhoSL taken up into cells by endocytosis together with HBV may also inhibit cccDNA synthesis by some mechanism. However, PhoSL does not appear to affect the secretory pathway of HBV particles after HBV mRNA synthesis at the late stage of infection.
In this study, we demonstrated that PhoSL also has an inhibitory effect on HBV infection in normal hepatocytes. This inhibitory effect can suppress intracellular cccDNA levels in a time-dependent manner even when administered after HBV infection, and is therefore expected to have clinical applications in the future.
Author Contributions
ST, CM, DS, KN, HH performed experiments, ST, CM, JK performed data analysis, DS prepared the graphical abstract, ST wrote a manuscript, KU supervised this study and supported the grant, EM made conception of this study and review a manuscript.
Funding
Supported by a Research Program on Hepatitis grant from the Japan Agency for Medical Research and Development (grant number 23fk0310505h0002).
Abbreviations
| PhoSL | Pholiota squarrosa lectin |
| HBV | Hepatitis B Virus |
| NTCP | Sodium Taurocholate Cotransporting Polypeptide |
| Fut8 | Fucosyltransferase 8 |
| HBe Ag | Hepatitis B e Antigen |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| myrPreS1 | Myristoylated PreS1 peptide |
References
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004, 11, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M. Natural history of chronic hepatitis B virus infection in adults with emphasis on the occurrence of cirrhosis and hepatocellular carcinoma. J Gastroenterol Hepatol 2000, 15 Suppl, E25–30. [Google Scholar] [CrossRef]
- Li, H.; Yan, L.; Shi, Y.; Lv, D.; Shang, J.; Bai, L.; Tang, H. Hepatitis B Virus Infection: Overview. Adv Exp Med Biol 2020, 1179, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jeng, W.J.; Lok, A.S.F. What will it take to cure hepatitis B? Hepatol Commun 2023, 7. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Almajed, M.R.; Fitzmaurice, M.G.; Jafri, S.M. Developments in pharmacotherapeutic agents for hepatitis B - how close are we to a functional cure? Expert Opin Pharmacother 2023, 24, 1001–1011. [Google Scholar] [CrossRef]
- Soriano, V.; Vispo, E.; Poveda, E.; Labarga, P.; Martin-Carbonero, L.; Fernandez-Montero, J.V.; Barreiro, P. Directly acting antivirals against hepatitis C virus. J Antimicrob Chemother 2011, 66, 1673–1686. [Google Scholar] [CrossRef]
- Rosato, V.; Nevola, R.; Dallio, M.; Di Micco, P.; Spinetti, A.; Zeneli, L.; Ciancio, A.; Milella, M.; Colombatto, P.; D'Adamo, G.; et al. Safety of Sofosbuvir-Based Direct-Acting Antivirals for Hepatitis C Virus Infection and Direct Oral Anticoagulant Co-Administration. J Clin Med 2024, 13. [Google Scholar] [CrossRef]
- Zhu, X.; Jia, L.; Yue, M.; Zhang, A.; Xia, X.; Yu, R.; Chen, H.; Huang, P. DAA treatment for HCV reduce risk of hepatocellular carcinoma: a 10-years follow-up study based on Chinese patients with hepatitis C. Sci Rep 2024, 14, 23760. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Döring, B.; Lütteke, T.; Geyer, J.; Petzinger, E. The SLC10 carrier family: transport functions and molecular structure. Curr Top Membr 2012, 70, 105–168. [Google Scholar] [CrossRef]
- Volz, T.; Allweiss, L.; Ben MBarek, M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lütgehetmann, M.; et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol 2013, 58, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Watashi, K.; Kamisuki, S.; Matsunaga, H.; Iwamoto, M.; Kawai, F.; Ohashi, H.; Tsukuda, S.; Shimura, S.; Suzuki, R.; et al. A Novel Tricyclic Polyketide, Vanitaracin A, Specifically Inhibits the Entry of Hepatitis B and D Viruses by Targeting Sodium Taurocholate Cotransporting Polypeptide. J Virol 2015, 89, 11945–11953. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Duan, M.; Wang, X.; Xu, J.; Tian, S.; Xu, X.; Duan, A.; Mahal, A.; Zhu, Y.; Zhu, Q. Synthesis and evaluation of pentacyclic triterpenoids conjugates as novel HBV entry inhibitors targeting NTCP receptor. Bioorg Chem 2024, 147, 107385. [Google Scholar] [CrossRef]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 2002, 21, 5017–5025. [Google Scholar] [CrossRef]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Cocquerel, L.; Voisset, C.; Dubuisson, J. Hepatitis C virus entry: potential receptors and their biological functions. J Gen Virol 2006, 87, 1075–1084. [Google Scholar] [CrossRef]
- Dosaka-Akita, H.; Miyoshi, E.; Suzuki, O.; Itoh, T.; Katoh, H.; Taniguchi, N. Expression of N-acetylglucosaminyltransferase v is associated with prognosis and histology in non-small cell lung cancers. Clin Cancer Res 2004, 10, 1773–1779. [Google Scholar] [CrossRef]
- Isaji, T.; Gu, J.; Nishiuchi, R.; Zhao, Y.; Takahashi, M.; Miyoshi, E.; Honke, K.; Sekiguchi, K.; Taniguchi, N. Introduction of bisecting GlcNAc into integrin alpha5beta1 reduces ligand binding and down-regulates cell adhesion and cell migration. J Biol Chem 2004, 279, 19747–19754. [Google Scholar] [CrossRef]
- Rademacher, T.W.; Parekh, R.B.; Dwek, R.A. Glycobiology. Annu Rev Biochem 1988, 57, 785–838. [Google Scholar] [CrossRef]
- Mawatari, M.; Saito, R.; Hibino, A.; Kondo, H.; Yagami, R.; Odagiri, T.; Tanabe, I.; Shobugawa, Y.; Group, J.I.C.S. Effectiveness of four types of neuraminidase inhibitors approved in Japan for the treatment of influenza. PLoS One 2019, 14, e0224683. [Google Scholar] [CrossRef]
- Lotfi, H.; Sheervalilou, R.; Zarghami, N. An update of the recombinant protein expression systems of Cyanovirin-N and challenges of preclinical development. Bioimpacts 2018, 8, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, D.; Wang, Y.; Su, W.; Liu, G.; Dong, W. The Importance of Glycans of Viral and Host Proteins in Enveloped Virus Infection. Front Immunol 2021, 12, 638573. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Sialobiology of influenza: molecular mechanism of host range variation of influenza viruses. Biol Pharm Bull 2005, 28, 399–408. [Google Scholar] [CrossRef]
- Saso, W.; Yamasaki, M.; Nakakita, S.I.; Fukushi, S.; Tsuchimoto, K.; Watanabe, N.; Sriwilaijaroen, N.; Kanie, O.; Muramatsu, M.; Takahashi, Y.; et al. Significant role of host sialylated glycans in the infection and spread of severe acute respiratory syndrome coronavirus 2. PLoS Pathog 2022, 18, e1010590. [Google Scholar] [CrossRef]
- Mitchell, C.A.; Ramessar, K.; O'Keefe, B.R. Antiviral lectins: Selective inhibitors of viral entry. Antiviral Res 2017, 142, 37–54. [Google Scholar] [CrossRef]
- Mori, T.; O'Keefe, B.R.; Sowder, R.C.; Bringans, S.; Gardella, R.; Berg, S.; Cochran, P.; Turpin, J.A.; Buckheit, R.W.; McMahon, J.B.; et al. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J Biol Chem 2005, 280, 9345–9353. J Biol Chem 2005, 280, 9345–9353. [Google Scholar] [CrossRef]
- Jacob, I.B.; Gemmiti, A.; Xiong, W.; Reynolds, E.; Nicholas, B.; Thangamani, S.; Jia, H.; Wang, G. Human surfactant protein A inhibits SARS-CoV-2 infectivity and alleviates lung injury in a mouse infection model. Front Immunol 2024, 15, 1370511. [Google Scholar] [CrossRef]
- Dennis, J.W.; Granovsky, M.; Warren, C.E. Glycoprotein glycosylation and cancer progression. Biochim Biophys Acta 1999, 1473, 21–34. [Google Scholar] [CrossRef]
- Takamatsu, S.; Shimomura, M.; Kamada, Y.; Maeda, H.; Sobajima, T.; Hikita, H.; Iijima, M.; Okamoto, Y.; Misaki, R.; Fujiyama, K.; et al. Core-fucosylation plays a pivotal role in hepatitis B pseudo virus infection: a possible implication for HBV glycotherapy. Glycobiology 2016, 26, 1180–1189. [Google Scholar] [CrossRef]
- Costache, M.; Apoil, P.A.; Cailleau, A.; Elmgren, A.; Larson, G.; Henry, S.; Blancher, A.; Iordachescu, D.; Oriol, R.; Mollicone, R. Evolution of fucosyltransferase genes in vertebrates. J Biol Chem 1997, 272, 29721–29728. [Google Scholar] [CrossRef]
- Ouchida, T.; Maeda, H.; Akamatsu, Y.; Maeda, M.; Takamatsu, S.; Kondo, J.; Misaki, R.; Kamada, Y.; Ueda, M.; Ueda, K.; et al. The specific core fucose-binding lectin Pholiota squarrosa lectin (PhoSL) inhibits hepatitis B virus infection in vitro. Sci Rep 2023, 13, 6175. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Tateno, H.; Dohra, H.; Moriwaki, K.; Miyoshi, E.; Hirabayashi, J.; Kawagishi, H. A novel core fucose-specific lectin from the mushroom Pholiota squarrosa. J Biol Chem 2012, 287, 33973–33982. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Suwanmanee, Y. ATP5B Is an Essential Factor for Hepatitis B Virus Entry. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Liang, G.; Iwamoto, M.; Marusawa, H.; Uchida, N.; Daito, T.; Kitamura, K.; Muramatsu, M.; Ohashi, H.; Kiyohara, T.; et al. Interleukin-1 and tumor necrosis factor-α trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). J Biol Chem 2013, 288, 31715–31727. [Google Scholar] [CrossRef]
- Tsurimoto, T.; Fujiyama, A.; Matsubara, K. Stable expression and replication of hepatitis B virus genome in an integrated state in a human hepatoma cell line transfected with the cloned viral DNA. Proc Natl Acad Sci U S A 1987, 84, 444–448. [Google Scholar] [CrossRef]
Figure 1.
PhoSL inhibits the infection and replication of HBV in normal hepatocytes (PXB cells). (A) This figure shows an overview of the concentration-dependent experiments of PhoSL cotreatment during HBV infection. The blue arrow indicates the day of medium replacement, and the red arrow indicates the day of HBV infection. PhoSL was co-administered at various concentrations at the time of infection. (B) Cell viability of PXB cells cultured with PhoSL for 3, 6, 9, or 12 days is shown. PhoSL concentrations of 0, 0.5, 1, 2.5, 5, and 10 μg/mL are shown in dark blue, orange, dark green, light blue, and magenta, respectively. Viable cells were measured by LDH-Glo cytotoxicity assay. No statistically significant differences were observed between 1, 2.5, 5, and 10 μg/mL PhoSL and 0 μg/mL PhoSL. Error bars indicate standard deviation; n=3. (C) Time course of HBeAg production after treatment with various concentrations of PhoSL during HBV infection. Each data shows with mean value +/- S.D. *** P < 0.001 (D) Quantification of cccDNA levels by digital PCR in PXB cells treated with various concentrations of PhoSL during HBV infection at day 12 post-infection. Each experiment was performed with N=1.
Figure 1.
PhoSL inhibits the infection and replication of HBV in normal hepatocytes (PXB cells). (A) This figure shows an overview of the concentration-dependent experiments of PhoSL cotreatment during HBV infection. The blue arrow indicates the day of medium replacement, and the red arrow indicates the day of HBV infection. PhoSL was co-administered at various concentrations at the time of infection. (B) Cell viability of PXB cells cultured with PhoSL for 3, 6, 9, or 12 days is shown. PhoSL concentrations of 0, 0.5, 1, 2.5, 5, and 10 μg/mL are shown in dark blue, orange, dark green, light blue, and magenta, respectively. Viable cells were measured by LDH-Glo cytotoxicity assay. No statistically significant differences were observed between 1, 2.5, 5, and 10 μg/mL PhoSL and 0 μg/mL PhoSL. Error bars indicate standard deviation; n=3. (C) Time course of HBeAg production after treatment with various concentrations of PhoSL during HBV infection. Each data shows with mean value +/- S.D. *** P < 0.001 (D) Quantification of cccDNA levels by digital PCR in PXB cells treated with various concentrations of PhoSL during HBV infection at day 12 post-infection. Each experiment was performed with N=1.
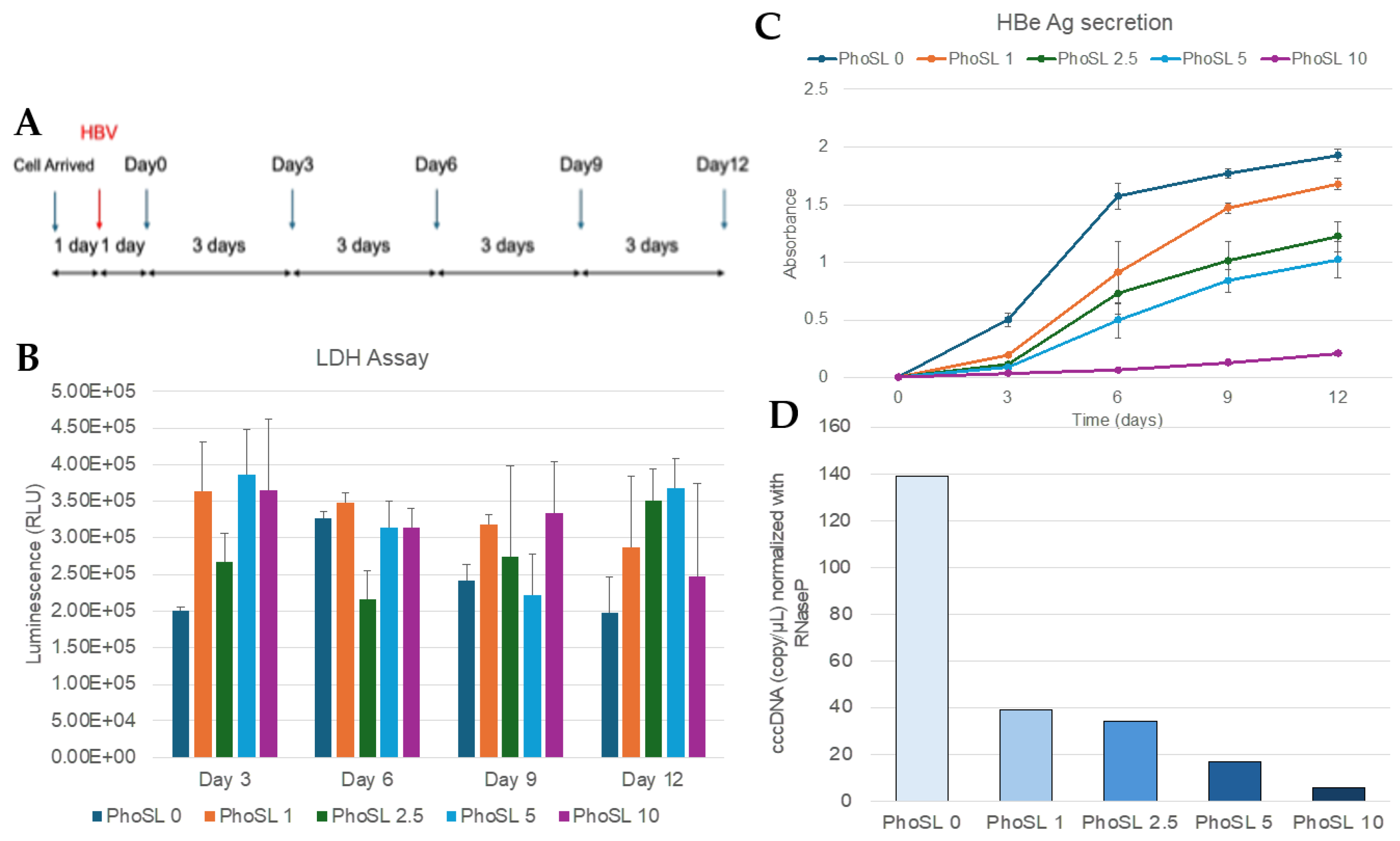
Figure 2.
PhoSL suppressed extracellular HBeAg and HBV DNA production, and also intracellular HBV DNA and cccDNA production in PXB cells in a treatment duration-dependent manner. (A) This figure shows an overview of the experiments measuring the time-dependent inhibitory effect of PhoSL treatment after HBV infection. Arrows indicate the dates when conditioned medium were collected for measuring HBe Ag. The blue arrows indicate the days when the medium was changed, and the red arrows indicate the days when HBV infection was performed. PhoSL was administered at the time of medium change according to the treatment period. (B) Time course of HBeAg levels in the supernatant of PXB cells with or without myristoylated PreS1 peptide treatment during HBV infection. (C) The HBeAg levels secreted by PXB cells treated with 10 µg/mL PhoSL for various periods after HBV infection on day 12 of culture are shown as relative values, with the value for the PhoSL-untreated sample on day 3 set as 1. (D) Inhibitory effects of extracellular HBV DNA production in HBV-infected PXB cells depending on the duration of PhoSL treatment. (E) Quantitative results of intracellular HBV DNA by real-time PCR using HBV genomic DNA as a standard. (F) Quantification of cccDNA levels by digital PCR in PXB cells treated with 10µg/ml PhoSL for various periods after HBV infection. Each experiment was performed with an N of 3 and results are shown as mean +/- standard deviation. * P < 0.05, ** P < 0.01, *** P < 0.005.
Figure 2.
PhoSL suppressed extracellular HBeAg and HBV DNA production, and also intracellular HBV DNA and cccDNA production in PXB cells in a treatment duration-dependent manner. (A) This figure shows an overview of the experiments measuring the time-dependent inhibitory effect of PhoSL treatment after HBV infection. Arrows indicate the dates when conditioned medium were collected for measuring HBe Ag. The blue arrows indicate the days when the medium was changed, and the red arrows indicate the days when HBV infection was performed. PhoSL was administered at the time of medium change according to the treatment period. (B) Time course of HBeAg levels in the supernatant of PXB cells with or without myristoylated PreS1 peptide treatment during HBV infection. (C) The HBeAg levels secreted by PXB cells treated with 10 µg/mL PhoSL for various periods after HBV infection on day 12 of culture are shown as relative values, with the value for the PhoSL-untreated sample on day 3 set as 1. (D) Inhibitory effects of extracellular HBV DNA production in HBV-infected PXB cells depending on the duration of PhoSL treatment. (E) Quantitative results of intracellular HBV DNA by real-time PCR using HBV genomic DNA as a standard. (F) Quantification of cccDNA levels by digital PCR in PXB cells treated with 10µg/ml PhoSL for various periods after HBV infection. Each experiment was performed with an N of 3 and results are shown as mean +/- standard deviation. * P < 0.05, ** P < 0.01, *** P < 0.005.
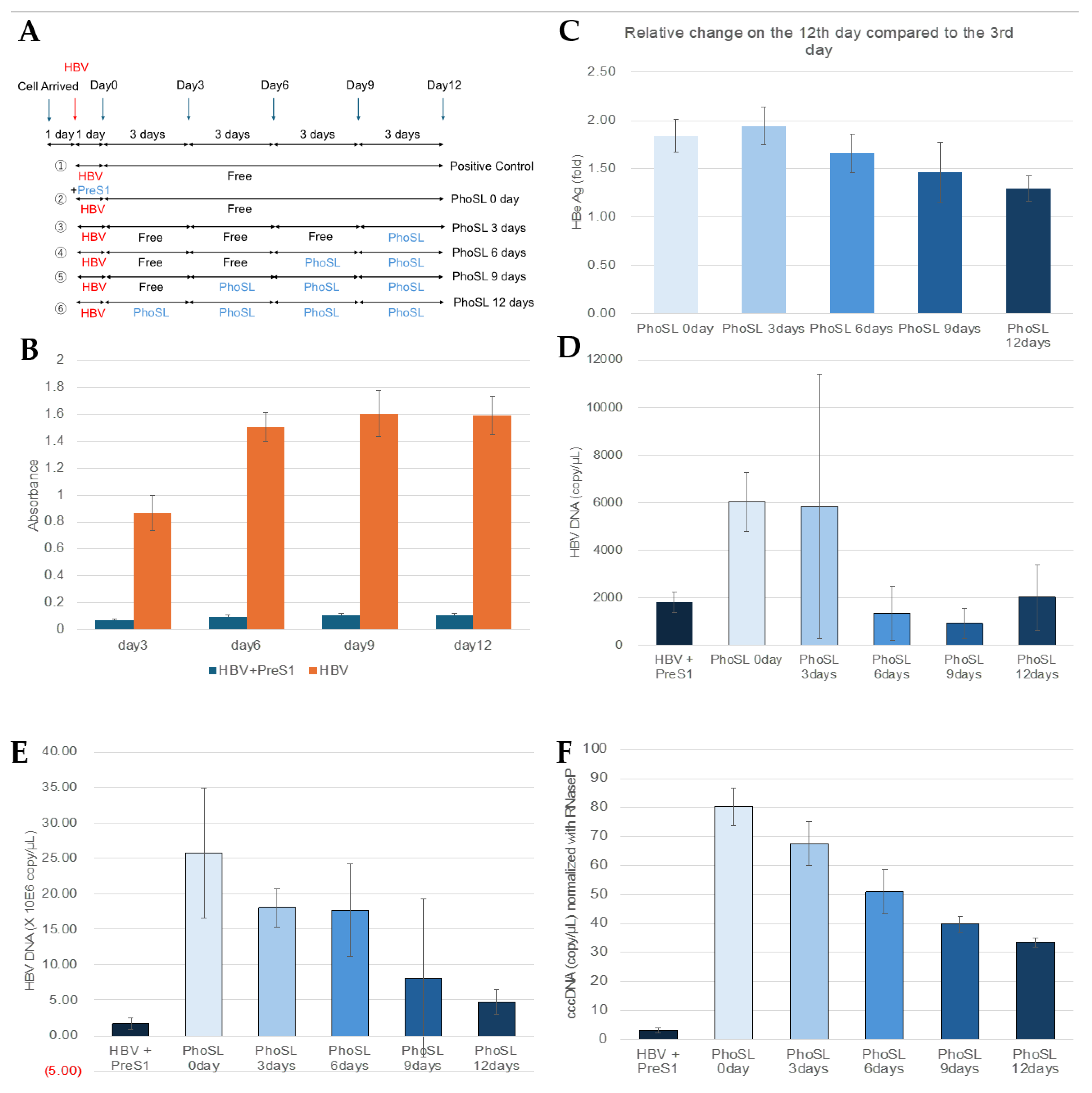
Figure 3.
PreS1 peptide did not affect extracellular HBeAg production and extracellular HBV DNA generation, and also intracellular HBV DNA and cccDNA production in PXB cells when administered after HBV infection. (A) This figure shows an overview of the experiments measuring the inhibitory effect of myrPreS1 treatment after HBV infection. Arrows indicate the dates when conditioned medium were collected for measuring HBe Ag. The blue arrows indicate the days when the medium was changed, and the red arrows indicate the days when HBV infection was performed. MyrPreS1 was administered continuously at medium changes for 12 days after HBV infection. (B) Time course of HBeAg levels in the supernatant of PXB cells with or without myrPreS1 treatment during HBV infection. (C) The HBe antigen levels secreted from PXB cells treated with 1 µM myrPreS1 for 0 and 12 days after HBV infection on day 12 of culture are shown as relative values, with the value for the myrPreS1-untreated sample on day 3 set as 1. (D) Quantification of intracellular HBV DNA by real-time PCR using HBV genomic DNA as a standard. (E) Quantification of extracellular HBV DNA in HBV-infected PXB cells according to the duration of myrPreS1 treatment. Each data shows with mean value +/- S.D. N = 3. (F) Quantification of cccDNA levels by digital PCR in PXB cells treated with 1µM myrPreS1 for various periods after HBV infection. Each experiment was performed with an N of 3 and results are shown as mean +/- standard deviation.
Figure 3.
PreS1 peptide did not affect extracellular HBeAg production and extracellular HBV DNA generation, and also intracellular HBV DNA and cccDNA production in PXB cells when administered after HBV infection. (A) This figure shows an overview of the experiments measuring the inhibitory effect of myrPreS1 treatment after HBV infection. Arrows indicate the dates when conditioned medium were collected for measuring HBe Ag. The blue arrows indicate the days when the medium was changed, and the red arrows indicate the days when HBV infection was performed. MyrPreS1 was administered continuously at medium changes for 12 days after HBV infection. (B) Time course of HBeAg levels in the supernatant of PXB cells with or without myrPreS1 treatment during HBV infection. (C) The HBe antigen levels secreted from PXB cells treated with 1 µM myrPreS1 for 0 and 12 days after HBV infection on day 12 of culture are shown as relative values, with the value for the myrPreS1-untreated sample on day 3 set as 1. (D) Quantification of intracellular HBV DNA by real-time PCR using HBV genomic DNA as a standard. (E) Quantification of extracellular HBV DNA in HBV-infected PXB cells according to the duration of myrPreS1 treatment. Each data shows with mean value +/- S.D. N = 3. (F) Quantification of cccDNA levels by digital PCR in PXB cells treated with 1µM myrPreS1 for various periods after HBV infection. Each experiment was performed with an N of 3 and results are shown as mean +/- standard deviation.
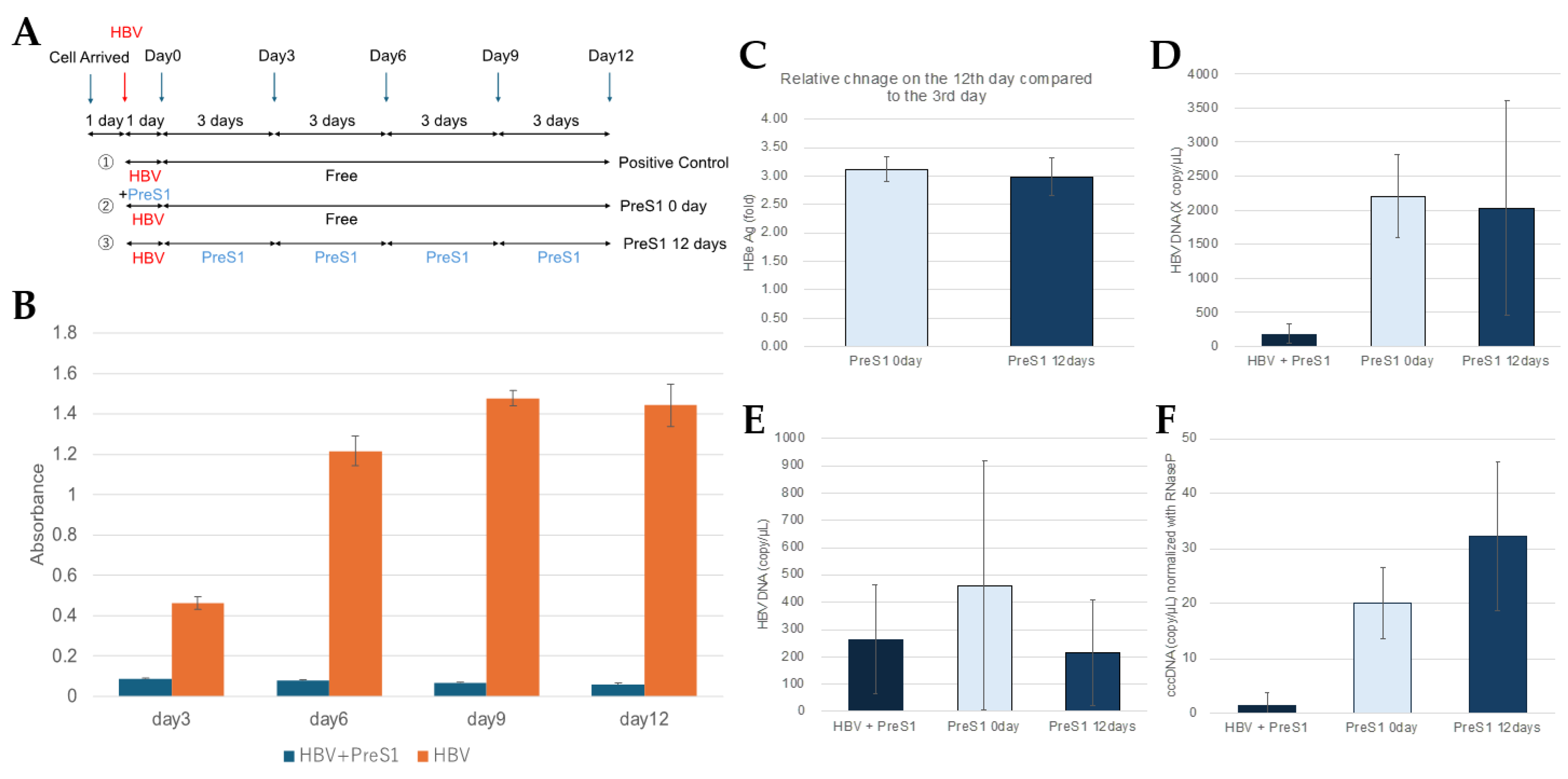
Figure 4.
Graphical Abstract. PhoSL binds to the core fucose of NTCP receptor and HBV virus particles, inhibiting HBV entry into cells by steric hindrance. PhoSL taken up into cells by endocytosis together with HBV may also inhibit cccDNA synthesis by some mechanism. However, PhoSL does not appear to affect the secretory pathway of HBV particles after HBV mRNA synthesis at the late stage of infection.
Figure 4.
Graphical Abstract. PhoSL binds to the core fucose of NTCP receptor and HBV virus particles, inhibiting HBV entry into cells by steric hindrance. PhoSL taken up into cells by endocytosis together with HBV may also inhibit cccDNA synthesis by some mechanism. However, PhoSL does not appear to affect the secretory pathway of HBV particles after HBV mRNA synthesis at the late stage of infection.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



