Submitted:
09 June 2025
Posted:
10 June 2025
You are already at the latest version
Abstract
The gut microbiota includes a diverse community of microorganisms, including both beneficial and pathogenic species that play a crucial role in human health and disease. Emerging evidence highlights its involvement in colorectal cancer (CRC) pathogenesis through multiple mechanisms, including chronic inflammation, bacterial virulence factors, genotoxic compounds, metabolic alterations, and biofilm formation. CRC remains a major global health burden and is among the leading causes of cancer-related mortality. Recent studies have implicated Fusobacterium nucleatum as a key contributor to CRC progression, facilitating tumor cell proliferation through var-ious virulence-associated mechanisms. Furthermore, the molecular mutations, which have a direct impact on proliferation of CRC, especially KRAS mutations that approximately shape around 60% of CRC cases. This re-view aims to elucidate the role of gut microbiota, molecular mutations in cancer, with a particular focus on CRC, detailing the molecular pathways involved in tumor initiation and progression. Additionally, we discussed the mechanisms by which F. nucleatum promotes CRC as well as how genotypic biomarkers are being utilized to en-hance our understanding of cancer biology, establish prognostic markers, and forecast therapeutic success.
Keywords:
gut microbiota
; Fusobacterium nucleatum(Fn)
; colorectal cancer (CRC)
; molecular mutations
; genotypic biomarkers
; KRAS
1. Introduction
Colorectal cancer (CRC) refers to cancer of the colon or rectum. It is considered more common in women than in men and is defined as the third most common cancer diagnosed in men and the second most common cancer in women[1,2]. In 2010, a study focused on the prevalence and impact of CRC on public health. The study reported more than one million new cases of CRC worldwide [3]. CRC has been identified as the third most prevalent malignancy by the International Agency for Research on Cancer. In 2018, the World Health Organization (WHO) classified CRC as an aggressive cancer and as the second leading cause of cancer-related mortality worldwide [4]. In 2030, the death rate due to CRC is expected to increase by 25%, whereas in 2018, it was estimated to be approximately 0.88 million deaths [5]. Recently, in China, the infection rate was 74.6 per 100,000 men and 58.3 per 100,000 women. A recent estimate reported approximately 36,000 new CRC cases and approximately 191,000 deaths related to CRC occurring annually [6]. According to certain studies, those who consume red meat have a greater prevalence of CRC. Anemia, weight loss, abdominal tenderness, and rectal bleeding are all considered common symptoms of CRC [7].
The gut microbiota includes approximately one billion microorganisms, such as bacteria, viruses, archaea, fungi, and protozoa [8]. It plays a crucial role in the body by promoting immunity to pathogens, influencing the immune system, and affecting digestion and metabolism [9], regulating epithelial cell proliferation and differentiation, moderating insulin resistance, influencing insulin secretion, and affecting the host’s behavioral and neurological functions [10]. One of the most significant factors contributing to the rise in CRC is lifestyle, particularly the increase in obesity due to decreasing physical activity [11].
More than 90% of the gastrointestinal tract has been linked to dietary choices, according to research evaluating the relationship between CRC and diet [12]. Currently, the precise mechanisms for developing CRC remain unclear in most patients, while some risk factors have been identified as genetic; others are linked to environmental and epigenetic factors [13]. There are several treatment methods for CRC, such as chemotherapy, radiotherapy, and surgery [14]; however, all these methods have side effects that prompt researchers to explore alternative treatments. Such an approach involves using bacteria for CRC treatment. For instance, using probiotics or genetically modified bacteria, which have the ability to accelerate cancer detection [15]. Although bacteria are associated with many cancers, they also possess antifungal and antimicrobial properties, which may allow them to be applied as cancer therapies [16,17]. In animal models, a study by Yang et al [18], investigated the significant role of Bifidobacteria sp., Salmonellae sp. and Clostridia sp, which act as a vector to deliver a massive range of genes such as apoptosis genes, anti-angiogenic genes, tumor-linked antigens, and tumor suppressor genes.
Fusobacterium nucleatum (Fn) does not require oxygen for growth, cannot form spores, and is a gram-negative, spindle-shaped bacterium [19]. It plays a key role in human health and illness, acting as a bridge [20]. Fn is commonly found in the oral cavity [21], and is also associated with diseases such as abscesses in the brain and liver [22]. Furthermore, it is linked to intrauterine inflammation related to pregnancy complications [23]. Additionally, Fn affects colorectal cancer, as it may inhibit the immune system and promote tumor growth [24]. Understanding the pathogenic pathways of Fn is valuable for developing targeted interventions and improving human health [25].
Through the analysis of the bacteria’s 16S rDNA sequencing and quantitative PCR, which included tumor tissues and feces, higher densities of Fusobacterium nucleatum (Fn) were discovered in CRC patients than in healthy individuals [26]. Due to changes in gut composition, the percentage of Fn in CRC gradually increases from colon cancer to rectal cancer [27]. The exact mechanism by which Fn impacts the development and drug resistance of CRC is still inadequately understood [28], despite the abundance of data [29]. Fn is a virulence factor that strongly contributes to CRC through various mechanisms [30]. FadA and lipopolysaccharides (LPS), which play significant roles in Wnt/-catenin signaling stimulation, lead to CRC cell generation [31]. Fibroblast activation protein 2 (Fap2) binds with D-galactose-(1–3)-N-acetyl-D-galactosamine (Gal-GalNAc). Fap2 and RaD proteins interact, ultimately leading to apoptosis and suppression of CRC[32].
The main objective of this paper is to review several factors related to CRC in addition to the key mutations, which participate closely in CRC disease. Moreover, we discuss the vital role of genotyping biomarkers in understanding the cancer biology, developing prognostic markers, and predict therapeutic success. First, we will discuss the dangers associated with gut microbiota, confirming its importance as a health concern and its role in various types of cancer, including CRC. Second, the associations between gut microbiota and CRC will be examined, focusing on the probable role of specific microorganisms in CRC development and progression. Fusobacterium nucleatum (Fn) is involved in CRC, and it is believed that some of its virulence factors contribute to increasing the risk of the disease. The review will provide in-depth information on the specific virulence factors produced by Fn and how they contribute to the proliferation of CRC cells that can adhere to host cells, modulate immune responses, and promote inflammation within the gut microenvironment. Additionally, different mechanisms by which Fn affects CRC will be discussed. Finally, we elaborated the KRAS biomarker and its role to inform the targeted therapy for CRC.
2. Gut Microbiota
Gut microbiota refers to the collection of microorganisms in the human intestine [33], while the gut microbiome represents the microbial genome [34]. The gut microbiome participates in many important processes, such as large-scale digestion and vitamin synthesis [35]. The composition of microbes in the microbiota varies throughout the digestive tract, and small intestine [36], whereas the colon harbors a greater diversity of microbes. Most intestinal bacteria are anaerobic, and microorganisms are found in high numbers [37]. For example, the cecum contains a variety of microorganisms, including Firmicutes [38], Bacteroidetes [39], Actinobacteria [40], Proteobacteria [41], Bacteroides [42], Clostridium [43], Bifidobacterium [44], and Ruminococcus [45].
Gut microbiota can help maintain health by defending against pathogens, producing various antimicrobial compounds, and enhancing the immune system [46]. It also plays a role in food absorption and metabolism [47], controls cell proliferation in epithelial cells [48], modifies insulin resistance and secretion [49], and affects brain and nerve functions by influencing communication between the brain and gut[50].
There are many factors affecting gut microbiota and its composition, such as diet, which are crucial for influencing the gut microbiota, potentially in both positive and negative ways. These changes can occur by enhancing or reducing the abundance of certain microorganisms in the gut of infants, diet is a significant factor in determining gut microbiota [51]. After birth, the gut microbiota is enriched with genes that help digest oligosaccharides obtained from breast milk. Over time, genes associated with the metabolism of vitamins and polysaccharides become more prevalent in the metagenome [51]. Additionally, the microbial composition of infant microbiota is influenced by the method of feeding [52]. As mentioned earlier, gut microbiota formation includes some pathogenic phyla, such as Actinobacteria, Firmicutes, and Proteobacteria. Breastfed infants may show a decrease in Firmicutes and Proteobacteria in addition to an increase in Actinobacteria [53]. In contrast, formula-fed infants experience a decrease in Bacteroides, Streptococci, Clostridia, and Enter bacteria. Furthermore, breast milk contains oligosaccharides that these bacterial species can effectively metabolize, leading to the production of short-chain fatty acids (SCFAs) that enhance the expression of immunoglobulin G [54].
The extent of microbiota colonization becomes apparent immediately after birth. Aerobic bacterial strains, such as Proteobacteria, are considered the first bacteria detected in the intestine. These bacteria lead to a deficiency of oxygen, allowing anaerobic bacterial strains like Firmicutes, Bacteroides, and Actinobacteria to colonize [55]. Host genetics also influence species diversity, individual variations, and pathogen sensitivity, linking the microbiome and genes to the host’s innate immune system. Pattern recognition receptors detect bacteria in the intestines and thus regulate the configuration of the microbiome and microbiome-related illnesses [56].
The diversity of the microbiota increases due to exercise. Compared to non-athletes, athletes have higher proportions of Firmicutes and lower levels of Bacteroidetes.
Lactobacillus, Bifid bacterium, and Akkermansia are among the bacterial taxa that respond to exercise, while Proteobacteria, Turicibacter, and Rikenellaceae show a reduction [57]. SCFAs (Short- chain fatty acids) are a measure of gut health. Additionally, it has been demonstrated that the production of butyrate-producing taxa, such as Clostridiales, Roseburia, Lachnospiraceae, and Erysipelotrichaceae, increases in response to exercise and plays a crucial role in moderating the effects of stress on the host gut microbiota [58].
Although antibiotics are used to kill harmful bacteria, they can be detrimental because they affect beneficial microorganisms, leading to dysbiosis, a disruption of the gut microbiota [59]. Antibiotics interfere with the competitive exclusion process that the microbiota uses to prevent pathogen growth [60]. Depending on the type of antibiotics used and the duration of treatment, different antibiotics will have varying impacts on gut flora [61]. For instance, two years of clindamycin therapy can alter the gut microbiota without restoring the Bacteroides population [62]. Additionally, the use of ciprofloxacin has been reported to reduce Ruminococcus, which had not recovered six months after treatment, whereas clarithromycin, used to treat Helicobacter pylori, causes a decrease in the number of Actinobacteria [63]. Vancomycin therapy reduces Bacteroidetes, Fuminococcus, and Faecalibacterium species but increases Proteobacteria species [64].
Another factor affecting gut microbiota is smoking [65]. The fecal microbiota of healthy individuals who quit smoking showed significant changes, including an increase in the relative abundance of Firmicutes and Actinobacteria and a reduction in Bacteroidetes and Proteobacteria. Additionally, research has demonstrated that the oral microbiota of smokers and non-smokers differs significantly [66]. Smokers’ oral cavities are particularly affected by an increase in Porphyromonas and Neisseria species and a reduction in Gemella species. However, there is little difference between smokers and non-smokers in their lung microbiota [67]. According to geographical location, the composition of phyla in the human gut microbiota varies [68]. These geographical differences are influenced by various lifestyle factors, including genetics, nutrition, and environmental characteristics [69]. Studies have shown that individuals in non-industrialized countries have higher Firmicutes counts, while in Westernized countries; the ratio of Bacteroidetes to Firmicutes appears to be higher [70].
3. Gut Microbiota and Cancer
The gut microbiota can influence several types of cancer [71]. Studies have demonstrated that the gut microbiota plays a significant role in cancer development [72]. Anaerobic bacteria are widely present in the gut microbiota [73]. They can ferment mucin once undigested food reaches the large intestine, ultimately producing metabolites, some of which may be beneficial while others can be harmful [74].
3.1. Gut Microbiota and Colorectal Cancer (CRC)
Although there are differences in gut microbiota, many bacterial species are relevant to CRC. Streptococcus bovis is considered a dangerous aspect of CRC; it is a gram-positive cocci bacterium [75]. Diarrhea and inflammatory bowel disease (IBD) are associated with the toxin produced by entero-toxigenic Bacteroides fragilis [76]. Fusobacterium nucleatum is regarded as a critical factor in CRC that may contribute to the progression of the disease from adenoma to cancer due to its role in human colorectal function[77]. In this review, we will illustrate the role of F. nucleatum as a member of the gut microbiota, which has been extensively studied in patients at the early stages of CRC [78]. According to certain studies, CRC patients have higher levels of Enterococcus faecalis than healthy individuals [79]. Superoxide, which damages DNA in epithelial cells, is produced in response to Enterococcus faecalis [80]. Additionally, research has shown that CRC patients exhibit greater levels of Escherichia coli than healthy individuals [81]. Samples from the fecal and mucosal microbiotas of CRC patients have been found to be enriched with Peptostreptococcus anaerobius [82].
3.2. Gut Microbiota and Colorectal Cancer (CRC)
Carcinogenesis of colorectal is extremely complicated Figure 1. It involves environmental, mental, and genetic factors. Recent studies suggest that various mechanisms are involved, including inflammation, virulence factors from pathogenic bacteria, biofilm formation, genotoxins, the production of oxidative stress elements, and numerous bacterial metabolites [83].
3.2.1. Inflammation
Over the past ten years, substantial evidence has been gathered demonstrating that inflammation plays a critical role in tumorigenesis [84]. The gastrointestinal microbiota interacts closely with the host immune system. Inflammation has been evaluated in the context of colorectal cancer (CRC). Bacterial stimulation of the immune response can accelerate chronic low-grade inflammation [85]. Typically, the gastrointestinal microbiota is separated from immune cells by the intestinal mucosal barrier, which is composed of layers of intestinal epithelial cells (IECs) connected by tight junctions (TJ) [86]. Disruption of the gastrointestinal mucosal barrier using dextran sodium sulfate-induced colitis leads to increased susceptibility to CRC [87].
The distinct removal of matriptase, a membrane-associated serine protease that enhances the intestinal epithelial barrier by promoting tight junction formation, contributes to the development of CRC [88]. This enzyme alters the decline of intestinal epithelial cells (IECs) to establish an effective barrier, allowing commensal microorganisms and their metabolites to interact with the tumor microenvironment. The microbiota is recognized by various pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), which regulate the inflammatory response to microbe-associated molecular patterns, including lipopolysaccharides (LPS) [89]. The activation of commensal bacteria and their components through TLRs on cancer-infiltrating myeloid cells triggers the MyD88-mediated production of inflammatory cytokines, particularly interleukin (IL)-23, which subsequently stimulates the production of IL-17A, IL-6, and IL-22 [90].
Finally, the activation of the nuclear factor kappa B (NF-B) and STAT3 signaling pathways initiates the proliferation of malignant growth cells [91]. Furthermore, IL-17C in modified intestinal epithelial cells (IECs) upregulates commensal bacteria and their metabolites through TLR/MyD88-dependent signaling. B-cell lymphoma 2 (Bcl-2) and Bcl-xL are activated by IL-17C, and they are expressed in IECs in an autocrine manner to promote cancer cell survival and tumorigenesis [92]. Peptostreptococcus anaerobius is capable of creating a favorable, inflammatory, and immune microenvironment that facilitates tumorigenesis. In Apc Min/+ mice, Peptostreptococcus anaerobius significantly induces the expression of pro-inflammatory cytokines, which subsequently recruits a range of tumor-infiltrating immune cells, particularly immunosuppressive myeloid-derived suppressor cells, tumor-associated macrophages, and granulocytic tumor-associated neutrophils, to promote tumor growth [82].
3.2.2. Virulence Factors
Many pathogenic bacteria play a crucial role in colorectal cancer (CRC), with Streptococcus bovis being commonly found in the human gastrointestinal tract [93]. In the context of CRC progression, IL-1, cyclooxygenase-2 (COX-2), and IL-8 are among the key mediators of inflammation-driven carcinogenesis [94].
3.2.3. Genotoxins
Bacteria produce genotoxins that are associated with colonic carcinogenesis due to their DNA-damaging effects. A genomic island in Escherichia coli (E. coli) contributes to the production of polyketide synthase (pks), which encodes the synthesis of the polyketide-peptide genotoxic colibactin [83]. Studies have shown that colibactin toxin induces cellular changes that lead to tumor cell proliferation in liver cells [95]. Additionally, Campylobacter jejuni produces genotoxins that cause DNA damage, facilitating the development of CRC [96]. Furthermore, Salmonella produces genotoxins and typhoid toxin, which induce DNA damage through the PI3K pathway in the epithelial cells of the colon [97].
3.2.4. Oxidative Stress
Oxidative stress is an imbalance between the production of pro-oxidative molecules, such as reactive oxygen species (ROS), and the body’s ability to eliminate them. It is well-known for contributing to chronic inflammation caused by gut microorganisms. In the context of chronic inflammation, various ROS are produced by inflammatory cells, which can enhance DNA damage and further promote the progression of colorectal cancer (CRC) by activating oncogenes. The gut microbiota can generate ROS directly. For instance, infection with Enterococcus faecalis activates superoxide production, leading to DNA damage in epithelial cells through a bystander effect [98]. Additionally, cholesterol synthesis is stimulated by the action of TLR2/TLR4, and Propionibacterium anaerobius can promote cell proliferation [99]. Moreover, ROS production is increased by Escherichia coli in colonic epithelial cells [100].
3.2.5. Metabolism
A new study found that 38.3% of CRC cases are associated with poor dietary habits, characterized by a high intake of red and processed meats, and a low consumption of grains and dairy products [101]. The risk of CRC increases by 19% due to obesity, and being overweight has been recognized as a significant risk factor for CRC [102]. Diet influences CRC, particularly when an imbalance in food intake alters the composition of the gut microbiome. A diet rich in animal fat and protein promotes the dominance of Bacteroides enterotypes [103]. The gut microbiota serves as an intermediary factor, playing a critical role in food digestion. Intestinal microbes ferment undigested food components, such as fructo-oligosaccharides, and bile acids, which are produced by the host. Short-chain fatty acids (SCFAs), including acetate, butyrate, and propionate, are organic acids formed through fermentation in individuals who maintain a balanced diet [104]. Conversely, pro-carcinogenic substances, such as secondary bile acids and N-nitroso compounds (NOCs), are produced through microbial metabolism when dietary intake is unbalanced [105].
Butyrate is the primary energy source for colonocytes and a key regulator of epithelial proliferation. Firmicutes primarily produce it through the fermentation of resistant starches and dietary fiber. Butyrate can suppress the activity of histone deacetylases in immune cells and colonocytes, which in turn reduces pro-inflammatory cytokines and induces apoptosis in CRC cells [106].
3.2.6. Biofilms
The concept of biofilm is still being researched in relation to colorectal cancer (CRC) and gut microbiota. Biofilms are aggregates of microbial groups encased in a polymeric matrix that have a direct connection with epithelial cells by invading the colonic mucosal layer. The formation of biofilms increases colonic epithelial permeability, facilitating bacterial antigen conversion and promoting pro-carcinogenic inflammation [107]. Previous researchers have found that the gut microbiota plays a key role in various physiological processes. It assists in the digestion and metabolism of nutrients, helps maintain the health and integrity of the intestinal lining, and participates in immune system functions [108]. Although the gut microbiota is essential for human health, disruptions in its composition and function can lead to dysbiosis, which has been linked to various health conditions, including CRC [109]. Researchers have investigated the complex relationship between gut microbiota and CRC development, seeking to understand how changes in microbiota composition or metabolic activities may influence CRC progression [110]. Imbalances in gut microbiota, particularly with the presence of pathogenic bacteria, can initiate the development of CRC [111]. Previous studies have shown higher levels of certain bacteria associated with CRC, such as Escherichia coli, Fusobacterium nucleatum, and Streptococcus bovis. Among these bacteria, Fusobacterium nucleatum has been identified as one of the causative agents of CRC in humans [112].
4. Fusobacterium nucleatum Mechanisms of Action
In the investigation of colorectal cancer (CRC) tissue structure and normal tissue in relation to the gut microbiota, Fusobacterium nucleatum (Fn) was discovered in CRC tissue, establishing it as a causative agent of CRC[113]. Through the application of 16S rRNA and metagenomic analysis, a significant increase in Fusobacterium species sequences associated with CRC has been observed (Figure 2) [114]. Fusobacterium species are known to act as pathogens in oral infections, appendicitis, and inflammatory bowel disease [115].
Recent research has provided experimental evidence supporting the role of Fusobacterium nucleatum in the initiation of colonic tumorigenesis. Apc (Min/+) mice continuously exposed to F. nucleatum show a significant increase in colon and small bowel tumors[116]. The data indicate that F. nucleatum can modulate the colonic tumor microenvironment, leading to an increase in myeloid-derived immune cells and the upregulation of inflammation-associated genes. Additionally, a higher abundance of F. nucleatum was detected in fecal samples from healthy individuals compared to those with tumors. These findings suggest that the detection of Fusobacterium may not be an adequate biological marker for identifying individuals at high risk of developing CRC. Various factors associated with Fusobacterium nucleatum aid in the initiation and progression of CRC (Figure 3( [111].
4.1. Fusobacterium nucleatum Virulence Factors
FadA and lipopolysaccharides (LPS) are virulence factors demonstrated by Fusobacterium nucleatum. The β-catenin signaling pathway is activated by the action of FadA through its connection with E-cadherin [28]. LPS binds to Toll-like receptor 4 (TLR4), leading to the activation of PAK1 (P21-activated kinase), which results in the phosphorylation of -catenin at Ser675. Phosphorylated -catenin enhances transcriptional activity and promotes the expression of CCND and MYC [117].
Fibroblast activation protein 2 (Fap-2), which binds to D-galactose-(1–3)-N-acetyl- D-galactosamine (Gal-GalNAc), is overexpressed in CRC [118]. Fap-2 interacts with TIGIT, inhibiting the activity of natural killer (NK) cells and lymphocytes. This interaction supports CRC growth. Endothelial and epithelial cells are the targets of FadA interaction, while only Gal-GalNAc is bound by Fap-2 [119].
4.1.1. FadA and LPS Virulence Factors Mechanisms in Colorectal Cancer
FadA, an adhesion protein, has been identified as the most notable virulence factor of Fn , as it can fully attach to host cells, facilitate bacterial adhesion, and disrupt epithelial cells[120]. Several studies have shown that FadA positive F. nucleatum was detected in biopsy specimens from CRC patients. Patients with CRC have detectable levels of FadA, in contrast to patients without CRC. It has been found that FadA can bind to E-cadherin on the surface of CRC epithelial cells. This binding leads to the expression of various transcription factors, oncogenes, and Wnt-regulated genes. The activation of the β-catenin signaling pathway due to Fad A and E-cadherin binding results in inflammation that promotes CRC cell growth [121].
Fad A enhances the stimulation of E-cadherin/β-catenin proteins through the phosphorylation and internalization of E-cadherin Figure 4. This interaction causes β-catenin to undergo nuclear translocation and cytoplasmic accumulation [122]. Downstream genes, such as Cyclin D1, are activated by nuclear β-catenin. Furthermore, FadA modifies annex in A1 via E-cadherin to activate -catenin signaling, accelerating colorectal cancer growth [88]. LPS-containing F. nucleatum activates P21-activated kinase 1 (PAK1). Tumor cell proliferation is driven by the phosphorylation of β-catenin at Ser675 [117]. LPS also triggers TLR4 signaling, which increases the expression of microRNA- 21. Numerous microRNAs play significant roles in the development of colorectal cancer (CRC). Some, like microRNA-21, lower the level of the RAS GTPase RASA1, stimulating tumor growth and development in mouse CRC cells, and have also been utilized to diagnose the disease[88]. TLR4 may accelerate tumor growth by stimulating the Wnt/β-catenin pathway [28].
4.1.2. Fap2 and Radiation Gene (RadD) Virulence Factors Mechanisms in Colorectal Cancer
Fap2 plays a significant role in Fusobacterium nucleatum infection [60]. It mediates the enhancement of Fn in colorectal cancer (CRC) by interacting with the host factor (Gal-GalNAc) expressed by tumors, contributing to the colonization and adherence of Fn to CRC tissue. Additionally, Fn inhibits immune system cells, such as natural killer (NK) cells, through binding with TIGIT [123]. It also induces lymphocytic apoptosis by binding Fap2 to RadD Figure 5 [124].
In summary, we can outline the Fn components in colorectal disease through the following pathways: (1) The binding and activity of FadA from Fn to human epithelial and endothelial cells, while pro-inflammatory cytokine levels such as IL-6, IL-8, IL-10, and NF-B increase in a pro-inflammatory microenvironment, thereby promoting the progression of colorectal tumors. (2) FadA’s interaction with E-cadherin in the signaling pathway. (3) Fn-infected cells increase miRNA expression by activating costimulatory receptors, leading to the release of miRNAs. Human lymphocyte activity is reduced by Fusobacterium nucleatum in a tumor-associated microenvironment. (4) Fap2 from F. nucleatum interacts with the human inhibitory receptor TIGIT, which leads to the death of human lymphocyte cells, creating an inflammatory environment that accelerates the progression of CRC [125].
4.2. Outer Membrane of Fusobacterium Nucleatum
It has been stated that outer membrane vesicles (OMVs) can be secreted by gram-negative bacteria, serving as vehicles for virulence factors [126]. OMVs can communicate with host epithelial cells due to their surface proteins and binding particles [127]. E-cadherin, a tight junction protein, is degraded by intra-OMV proteases from Fusobacterium nucleatum, facilitating bacterial invasion and triggering inflammatory reactions. Furthermore, OMVs promote epithelial-mesenchymal transition (EMT), a significant pathway in cancer metastasis. F. nucleatum and OMVs can upregulate EMT markers, which include N-cadherin, integrin-5, bone morphogenetic proteins (BMPs), Snail, Bend, ZEB-1, and fibronectin-1 [128].
4.3. MicroRNA
Several microRNAs (miRNAs) are involved in colorectal cancer (CRC) tumorigenesis; a few of them are used as diagnostic tools and to track the progress of CRC in patients [129]. MiR-21 is crucial in the development of colitis related to CRC and chronic intestinal inflammation [130]. The potential impact and underlying mechanism of Fusobacterium nucleatum on colorectal cancer (CRC) in both laboratory settings and living organisms have been suggested. When CRC cells were treated with F. nucleatum, an increase in cell proliferation and invasion was observed. Similarly, APC Min/+ mice infected with F. nucleatum showed an increase in tumor burden and tumor size. Microarray analysis conducted after treating CRC cell lines with F. nucleatum revealed that out of the 657 miRNAs examined, 50 miRNAs were upregulated and 52 miRNAs were downregulated [131]. Among these, miR-21 showed the highest expression rate of all the miRNAs. It was proposed that miR-21 is a direct target of the bacteria. Subsequent analysis demonstrated that miR-21 directly targets RASA1, and RASA1 expression is upregulated when miR-21 is inhibited. The RAS oncoprotein can bind to RASA1 and become inactive . The TLR4/MYD88/NF-B axis can be activated by F. nucleatum in CRC cells, and miR-21 expression is regulated by this signaling pathway[131].
4.4. Bacterial Metabolism
Amino acids and peptides are used as nutritional sources by Fusobacterium nucleatum in the cancer microenvironment. Myeloid cell chemoattractant includes short chain fatty acids (SCFAs) and formyl methionyl-leucyl-phenylalanine, which are products of Fusobacterium’s amino acid metabolism. Furthermore, Fusobacterium’s unique electron transport chain allows it to proliferate in the hypoxic tumor microenvironment [20].
5. Molecular Mutations of Colorectal Cancer (CRC) and Targeted Therapy
A molecular mutation is a change in the nuclear material of an organism’s genome. These mutations may occur due to errors during DNA replication, such as the substitution, insertion, or deletion of nucleotides in the DNA sequence. Additionally, defects in DNA repair mechanisms, along with exposure to chemical or radiation mutagens, can be responsible for these mutations [132]. Mutations in a collection of tumor suppressor genes or oncogenes participate in the proliferation and development of colorectal cancer (CRC) worldwide. Crucial genes associated with these mutations are listed in Table 1, indicating their role in the presence of CRC[133].
5.1. Genotypic Biomarkers in Colorectal Cancer (CRC): Understanding Cancer Biology, Prognosis, and Therapeutic Success
Genotypic biomarkers include mutations, polymorphisms, and epigenetic changes, providing insights into the field of cancer biology. As prognostic indicators, they inform therapeutic strategies. The percentages of prevalence of biomarkers in colorectal cancer (CRC) are illustrated in Figure 6 [134,135]. In the context of CRC, these biomarkers are crucial for therapeutic purposes. We summarize in Table 2 the key genotypic biomarkers used in CRC, their roles in cancer biology, and their significance as prognostic and therapeutic indicators, with a focus on mutations.
5.2. Therapeutic Achievements
KRAS G12C mutations carry out in around 4% of CRC disease. Recently achievements in aimed treatment, have led to develop small- molecules, covalent KRAS G12C inhibitors, such as sotorasib and adagrasib[136,137] . One of the most apparently proto-oncogenes known to date is Kirsten Rat Sarcoma gene (KRAS), where it accounts for around 60% of all colorectal cancer mutations[138]. These mutations promote tumor growth by activating continuous significant signaling pathways, such as RAS-RAF-Make-made (MAPK) and PI3K actor, to promote the development of tumors, cause treatment resistance and diagnose adverse diseases [139]. Lately, progress in targeted therapy has resulted in the creation of small-molecule, covalent KRAS G12C inhibitors like sotorasib and adagrasib. These drugs specifically and permanently attach to the mutant KRAS G12C protein, keeping it in an inactive GDP-bound form and shutting down its cancer-causing signals[139].
A study involved 129 patients with advanced stage tumor because of KRAS G12C mutation. 57% participants experienced with random grade of treatment-related adverse events (TRAEs), where 12% with grade 3 or 4. Another study involved 62 patients with advanced stages of CRC with KRAS G12C-mutant, to dose of 960 mg from sotorasib monotherapy once a day. A partial response has been recorded in 6 patients[140].
A combination therapy strategy is being recommended, where KRYSTAL-1 and CodeBreaK 100 have displayed moderate response rates[141]. Panitumumab and sotorasib (960 mg once daily) have been combined and evaluated in 40 patients with chemotherapy-refractory KRAS-G12C-mutant mCRC, leading to a 30% objective response rate (ORR)[142]. Furthermore, a total of 94 patients were treated with a combination of cetuximab and 600 mg twice daily dose of adagrasib, leading to 40 % ORR[143]. A 42% ORR was recorded in among of 38 patients received a combination treatment of cetuximab and around 150 mg twice daily dose[144].
6. Conclusions
This review highlights the important role of gut microbiota and genotype biomarkers in CRC pathogenesis and progression. Integration of microbioma analysis with genotype profiles promises avenues for individual therapy in CRC, facilitates the development of targeted treatments and improves patient results through better development. Ongoing research is necessary to highlight the columns in these interactions and translate conclusions into clinical practice.

Table 1.
Key Molecular Mutations in Colorectal Cancer.
| Gene/Mutation location | Prevalence in CRC (%) | Biological Implication | Reference |
| TP53 (Exons 5–8) | ~50% | Loss of DNA-binding ability, impairs cell cycle arrest and apoptosis; late event in adenoma-to-carcinoma transition | [145,146] |
| KRAS (Codons 12/13) | 25–60% | Constitutively active Ras protein, drives proliferation via MAPK pathway | [147,148] |
| KRAS (Codon 61) | ~5% | Similar to codons 12/13; promotes uncontrolled cell growth | [149] |
| BRAF (V600E) | 5–10% | Constitutively active kinase, activates MAPK pathway in a RAS-independent manner | [150] |
| APC (Truncations, Hypermethylation) | 20–48% (hypermethylation); ~70% (mutations) | Disrupts Wnt signaling, increases β-catenin activity, promotes proliferation; early event in CRC | [151,152] |
| β-Catenin (Point mutations, Deletions) | Up to 10% | Stabilizes β-catenin, activates Wnt signaling; mutually exclusive with APC mutations | [153] |
| SMAD4 (MH2 Region) | ~10–20% | Disrupts TGF-β signaling, impairs growth regulation | [154,155] |
| AXIN1/AXIN2 (Point mutations, Deletions) | ~5–10% | Disrupts β-catenin destruction complex, activates Wnt signaling | [156,157] |
Table 2.
Genotypic Biomarkers in Colorectal Cancer.
| Biomarker | Role in Cancer Biology | Prognostic Significance | Therapeutic Prediction | Reference |
| KRAS | Activates MAPK pathway, drives proliferation | Worse survival in metastatic CRC, higher recurrence | Resistance to anti-EGFR therapies; use VEGF inhibitors or chemotherapy | [158] |
| TP53 | Impairs cell cycle arrest/apoptosis, increases genomic instability | Poorer prognosis in advanced CRC | May influence chemotherapy response; p53-targeted therapies in trials | [159,160] |
| BRAF | Activates MAPK pathway, RAS-independent | Poor prognosis, shorter survival | Resistance to anti-EGFR; BRAF/MEK inhibitors | [161,162] |
| APC | Activates Wnt pathway, promotes proliferation | Linked to FAP and sporadic CRC progression | Wnt inhibitors in trials | [163] |
| β-Catenin | Activates Wnt pathway, mutually exclusive with APC mutations | Variable; may indicate aggressive disease | Wnt inhibitors in trials | [164] |
| SMAD4 | Disrupts TGF-β signaling, impairs growth regulation | Worse prognosis in metastatic CRC | TGF-β modulators in trials | [165] |
| AXIN1/AXIN2 | Activates Wnt pathway via β-catenin dysregulation | May indicate tumor aggressiveness | Wnt inhibitors in trials | [166] |
| NRAS | Activates MAPK pathway, similar to KRAS | Poorer prognosis in metastatic CRC | Resistance to anti-EGFR therapies | [167,168] |
| PIK3CA | Activates PI3K/AKT pathway | Variable; exon 20 mutations linked to worse outcomes | Partial anti-EGFR resistance; PI3K inhibitors in trials | [169,170,171] |
| MSI-High | MMR defects (e.g., MLH1, MSH2) cause genomic instability | Favorable prognosis, better survival | Predicts response to immunotherapy (e.g., pembrolizumab) | [172,173] |
| CIMP | Promoter hypermethylation silences tumor-suppressor genes | Poor prognosis in some subtypes, often with BRAF mutations | May influence chemotherapy response | [174,175] |
Author Contributions
Conceptualization, A.D.; Methodology, A.D.; Software , A.D.; Validation, A.D.; Formal Analysis, A.D.; Investigation, A.D and M.M.; Resources, A.D.; Data Curation, A.D.; Writing – Original Draft Preparation, A.D. and M.M; Writing – Review & Editing, A.D and M.M ; Visualization, A.D.; Supervision, M.M.; Project Administration, M.M.; Funding Acquisition, M.M.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Lewandowska A, Rudzki G, Lewandowski T, Stryjkowska-Góra A, Rudzki S. Risk Factors for the Diagnosis of Colorectal Cancer. Cancer Control [Internet]. 2022 Jan 9;29:10732748211056692. Available from. [CrossRef]
- Hashiguchi Y, Muro K, Saito Y, Ito Y, Ajioka Y, Hamaguchi T, et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2019 for the treatment of colorectal cancer. Int J Clin Oncol. 2020;25:1–42.
- Schliemann D, Matovu N, Ramanathan K, Muñoz-Aguirre P, O’Neill C, Kee F, et al. Implementation of colorectal cancer screening interventions in low-income and middle-income countries: a scoping review protocol. BMJ Open. 2020;10(6):e037520. [CrossRef]
- Mahmood B, Muhammad B, Abbas R, Rabia A, Kumar A, Hamed H, et al. Nanodiagnosis and nanotreatment of colorectal cancer: an overview. J Nanoparticle Res. 2021;23(1).
- Mattiuzzi C, Sanchis-Gomar F, Lippi G. Concise update on colorectal cancer epidemiology. Ann Transl Med. 2019;7(21):609. [CrossRef]
- Gao R, Gao Z, Huang L, Qin H. Gut microbiota and colorectal cancer. Eur J Clin Microbiol Infect Dis. 2017;36:757–69.
- McCulloch SM, Aziz I, Polster A V, Pischel AB, Stålsmeden H, Shafazand M, et al. The diagnostic value of a change in bowel habit for colorectal cancer within different age groups. United Eur Gastroenterol J. 2020;8(2):211–9. [CrossRef]
- Fong W, Li Q, Yu J. Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer. Oncogene. 2020;39(26):4925–43. [CrossRef]
- Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature [Internet]. 2018;555(7695):210–5. Available from. [CrossRef]
- Hasan N, Yang H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ. 2019;7:e7502. [CrossRef]
- Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019;16(12):713–32. [CrossRef]
- Yang J, Yu J. The association of diet, gut microbiota and colorectal cancer: what we eat may imply what we get. Protein Cell. 2018 May;9(5):474–87.
- Soofiyani SR, Ahangari H, Soleimanian A, Babaei G, Ghasemnejad T, Safavi SE, et al. The role of circadian genes in the pathogenesis of colorectal cancer. Gene. 2021;804:145894.
- Hossain MS, Karuniawati H, Jairoun AA, Urbi Z, Ooi DJ, John A, et al. Colorectal cancer: a review of carcinogenesis, global epidemiology, current challenges, risk factors, preventive and treatment strategies. Cancers (Basel). 2022;14(7):1732.
- Ebrahimzadeh S, Ahangari H, Soleimanian A, Hosseini K, Ebrahimi V, Ghasemnejad T, et al. Colorectal cancer treatment using bacteria: focus on molecular mechanisms. BMC Microbiol. 2021;21:1–12. [CrossRef]
- Seyfi R, Kahaki FA, Ebrahimi T, Montazersaheb S, Eyvazi S, Babaeipour V, et al. Antimicrobial peptides (AMPs): roles, functions and mechanism of action. Int J Pept Res Ther. 2020;26:1451–63. [CrossRef]
- Tarhriz V, Eyvazi S, Shakeri E, Hejazi MS, Dilmaghani A. Antibacterial and antifungal activity of novel freshwater bacterium Tabrizicola aquatica as a prominent natural antibiotic available in Qurugol Lake. Pharm Sci. 2020;26(1):88–92. [CrossRef]
- Yang J, Wu Z, Chen Y, Hu C, Li D, Liao Y, et al. A novel radio-sensitization method for lung cancer therapy: enhanced radiosensitization induced by antigens/antibodies reaction after targeting tumor hypoxia using Bifidobacterium. 2020.
- Kang MS, Park GY. In vitro evaluation of the effect of oral probiotic weissella cibaria on the formation of multi-species oral biofilms on dental implant surfaces. Microorganisms. 2021;9(12):2482. [CrossRef]
- Alon-Maimon T, Mandelboim O, Bachrach G. Fusobacterium nucleatum and cancer. Periodontol 2000. 2022;89(1):166–80.
- Abed J, Maalouf N, Manson AL, Earl AM, Parhi L, Emgård JEM, et al. Colon cancer-associated Fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front Cell Infect Microbiol. 2020;10:400. [CrossRef]
- Pignatelli P, Nuccio F, Piattelli A, Curia MC. The role of Fusobacterium nucleatum in oral and colorectal carcinogenesis. Microorganisms. 2023;11(9):2358. [CrossRef]
- Vander Haar EL, So J, Gyamfi-Bannerman C, Han YW. Fusobacterium nucleatum and adverse pregnancy outcomes: epidemiological and mechanistic evidence. Anaerobe. 2018;50:55–9.
- Lee JA, Yoo SY, Oh HJ, Jeong S, Cho NY, Kang GH, et al. Differential immune microenvironmental features of microsatellite-unstable colorectal cancers according to Fusobacterium nucleatum status. Cancer Immunol Immunother. 2021;70:47–59. [CrossRef]
- Bundgaard-Nielsen C, Baandrup UT, Nielsen LP, Sørensen S. The presence of bacteria varies between colorectal adenocarcinomas, precursor lesions and non-malignant tissue. BMC Cancer. 2019;19:1–13. [CrossRef]
- Zhang FF, Cudhea F, Shan Z, Michaud DS, Imamura F, Eom H, et al. Preventable cancer burden associated with poor diet in the United States. JNCI Cancer Spectr. 2019;3(2):pkz034. [CrossRef]
- Park CH, Eun CS, Han DS. Intestinal microbiota, chronic inflammation, and colorectal cancer. Intest Res. 2018;16(3):338.
- Wu Z, Ma Q, Guo Y, You F. The role of Fusobacterium nucleatum in colorectal cancer cell proliferation and migration. Cancers (Basel). 2022;14(21):5350. [CrossRef]
- Liu Y, Baba Y, Ishimoto T, Iwatsuki M, Hiyoshi Y, Miyamoto Y, et al. Progress in characterizing the linkage between Fusobacterium nucleatum and gastrointestinal cancer. J Gastroenterol. 2019;54:33–41. [CrossRef]
- Tran HNH, Thu TNH, Nguyen PH, Vo CN, Doan K Van, Nguyen Ngoc Minh C, et al. Tumour microbiomes and Fusobacterium genomics in Vietnamese colorectal cancer patients. npj Biofilms Microbiomes. 2022;8(1):87. [CrossRef]
- Shao Y, Zeng X. Molecular mechanisms of gut microbiota-associated colorectal carcinogenesis. Infect Microbes Dis. 2020;2(3):96–106. [CrossRef]
- Altmann A, Haberkorn U, Siveke J. The latest developments in imaging of fibroblast activation protein. J Nucl Med. 2021;62(2):160–7. [CrossRef]
- Ragonnaud E, Biragyn A. Gut microbiota as the key controllers of “healthy” aging of elderly people. Immun Ageing. 2021;18:1–11.
- Zou Y, Xue W, Luo G, Deng Z, Qin P, Guo R, et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat Biotechnol. 2019;37(2):179–85. [CrossRef]
- Rudzki L, Stone TW, Maes M, Misiak B, Samochowiec J, Szulc A. Gut microbiota-derived vitamins–underrated powers of a multipotent ally in psychiatric health and disease. Prog Neuro-Psychopharmacology Biol Psychiatry. 2021;107:110240. [CrossRef]
- Crespo-Piazuelo D, Estellé J, Revilla M, Criado-Mesas L, Ramayo-Caldas Y, Óvilo C, et al. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci Rep. 2018;8(1):12727. [CrossRef]
- Sundin OH, Mendoza-Ladd A, Zeng M, Diaz-Arévalo D, Morales E, Fagan BM, et al. The human jejunum has an endogenous microbiota that differs from those in the oral cavity and colon. BMC Microbiol. 2017;17:1–17. [CrossRef]
- Mohamadkhani, A. Gut microbiota and fecal metabolome perturbation in children with autism spectrum disorder. Middle East J Dig Dis. 2018;10(4):205. [CrossRef]
- Faniyi TO, Adegbeye MJ, Elghandour M, Pilego AB, Salem AZM, Olaniyi TA, et al. Role of diverse fermentative factors towards microbial community shift in ruminants. J Appl Microbiol. 2019;127(1):2–11. [CrossRef]
- Popov I V, Algburi A, Prazdnova E V, Mazanko MS, Elisashvili V, Bren AB, et al. A review of the effects and production of spore-forming probiotics for poultry. Animals. 2021;11(7):1941. [CrossRef]
- Grabowicz, M. Lipoprotein Transport: Greasing the Machines of Outer Membrane Biogenesis: Re-Examining Lipoprotein Transport Mechanisms Among Diverse Gram-Negative Bacteria While Exploring New Discoveries and Questions. BioEssays. 2018;40(4):1700187.
- Shin Y, Park SJ, Paek J, Kim JS, Rhee MS, Kim H, et al. Bacteroides koreensis sp. nov. and Bacteroides kribbi sp. nov., two new members of the genus Bacteroides. Int J Syst Evol Microbiol. 2017;67(11):4352–7. [CrossRef]
- Burgess SA, Palevich FP, Gardner A, Mills J, Brightwell G, Palevich N. Occurrence of genes encoding spore germination in Clostridium species that cause meat spoilage. Microb Genomics. 2022;8(2):767. [CrossRef]
- Olofsson TC, Modesto M, Pascarelli S, Scarafile D, Mattarelli P, Vasquez A. Bifidobacterium mellis sp. nov., isolated from the honey stomach of the honey bee Apis mellifera. Int J Syst Evol Microbiol. 2023;73(3):5766. [CrossRef]
- Togo AH, Diop A, Bittar F, Maraninchi M, Valero R, Armstrong N, et al. Description of Mediterraneibacter massiliensis, gen. nov., sp. nov., a new genus isolated from the gut microbiota of an obese patient and reclassification of Ruminococcus faecis, Ruminococcus lactaris, Ruminococcus torques, Ruminococcus gnavus and Clostri. Antonie Van Leeuwenhoek. 2018;111:2107–28.
- Hou K, Wu ZX, Chen XY, Wang JQ, Zhang D, Xiao C, et al. Microbiota in health and diseases. Signal Transduct Target Ther. 2022;7(1):135.
- Jing TZ, Qi FH, Wang ZY. Most dominant roles of insect gut bacteria: digestion, detoxification, or essential nutrient provision? Microbiome. 2020;8(1):1–20. [CrossRef]
- Michaudel C, Sokol H. The gut microbiota at the service of immunometabolism. Cell Metab. 2020;32(4):514–23. [CrossRef]
- Lee CJ, Sears CL, Maruthur N. Gut microbiome and its role in obesity and insulin resistance. Ann N Y Acad Sci. 2020;1461(1):37–52. [CrossRef]
- Zheng P, Zeng B, Liu M, Chen J, Pan J, Han Y, et al. The gut microbiome from patients with schizophrenia modulates the glutamate-glutamine-GABA cycle and schizophrenia-relevant behaviors in mice. Sci Adv. 2019;5(2):eaau8317. [CrossRef]
- Hills RD, Pontefract BA, Mishcon HR, Black CA, Sutton SC, Theberge CR. Gut microbiome: profound implications for diet and disease. Nutrients. 2019;11(7):1613.
- Vandenplas Y, Carnielli VP, Ksiazyk J, Luna MS, Migacheva N, Mosselmans JM, et al. Factors affecting early-life intestinal microbiota development. Nutrition. 2020;78:110812. [CrossRef]
- Młynarska E, Gadzinowska J, Tokarek J, Forycka J, Szuman A, Franczyk B, et al. The role of the microbiome-brain-gut axis in the pathogenesis of depressive disorder. Nutrients. 2022;14(9):1921. [CrossRef]
- Zuurveld M, Van Witzenburg NP, Garssen J, Folkerts G, Stahl B, Van’t Land B, et al. Immunomodulation by human milk oligosaccharides: the potential role in prevention of allergic diseases. Front Immunol. 2020;11:801. [CrossRef]
- Del Chierico F, Vernocchi P, Petrucca A, Paci P, Fuentes S, Pratico G, et al. Phylogenetic and metabolic tracking of gut microbiota during perinatal development. PLoS One. 2015;10(9):e0137347. [CrossRef]
- Iebba V, Totino V, Gagliardi A, Santangelo F, Cacciotti F, Trancassini M, et al. Eubiosis and dysbiosis: the two sides of the microbiota. New Microbiol. 2016;39(1):1–12.
- Hughes, RL. A review of the role of the gut microbiome in personalized sports nutrition. Front Nutr. 2020;6:504337. [CrossRef]
- Allen JM, Mailing LJ, Niemiro GM, Moore R, Cook MD, White BA, et al. Exercise alters gut microbiota composition and function in lean and obese humans. Med Sci Sport Exerc. 2018;50(4):747–57. [CrossRef]
- Kandpal M, Indari O, Baral B, Jakhmola S, Tiwari D, Bhandari V, et al. Dysbiosis of gut microbiota from the perspective of the gut–brain axis: role in the provocation of neurological disorders. Metabolites. 2022;12(11):1064. [CrossRef]
- Shah T, Baloch Z, Shah Z, Cui X, Xia X. The intestinal microbiota: impacts of antibiotics therapy, colonization resistance, and diseases. Int J Mol Sci. 2021;22(12):6597. [CrossRef]
- Ianiro G, Tilg H, Gasbarrini A. Antibiotics as deep modulators of gut microbiota: between good and evil. Gut. 2016;65(11):1906–15. [CrossRef]
- Park JY, Dunbar KB, Mitui M, Arnold CA, Lam-Himlin DM, Valasek MA, et al. Helicobacter pylori clarithromycin resistance and treatment failure are common in the USA. Dig Dis Sci. 2016;61:2373–80. [CrossRef]
- Avcı A, İnci İ, Baylan N. Adsorption of ciprofloxacin hydrochloride on multiwall carbon nanotube. J Mol Struct. 2020;1206:127711. [CrossRef]
- Isaac S, Scher JU, Djukovic A, Jiménez N, Littman DR, Abramson SB, et al. Short-and long-term effects of oral vancomycin on the human intestinal microbiota. J Antimicrob Chemother. 2016;72(1):128–36. [CrossRef]
- Savin Z, Kivity S, Yonath H, Yehuda S. Smoking and the intestinal microbiome. Arch Microbiol. 2018;200(5):677–84.
- Biedermann L, Zeitz J, Mwinyi J, Sutter-Minder E, Rehman A, Ott SJ, et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS One. 2013;8(3):e59260. [CrossRef]
- Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187(10):1067–75. [CrossRef]
- Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, et al. Gut microbiome of the Hadza hunter-gatherers. Nat Commun. 2014;5(1):3654. [CrossRef]
- Martínez I, Stegen JC, Maldonado-Gómez MX, Eren AM, Siba PM, Greenhill AR, et al. The gut microbiota of rural papua new guineans: composition, diversity patterns, and ecological processes. Cell Rep. 2015;11(4):527–38. [CrossRef]
- Zhu A, Sunagawa S, Mende DR, Bork P. Inter-individual differences in the gene content of human gut bacterial species. Genome Biol. 2015;16:1–13. [CrossRef]
- Zhao Y, Liu Y, Li S, Peng Z, Liu X, Chen J, et al. Role of lung and gut microbiota on lung cancer pathogenesis. J Cancer Res Clin Oncol. 2021;147(8):2177–86. [CrossRef]
- Fujita K, Matsushita M, Banno E, De Velasco MA, Hatano K, Nonomura N, et al. Gut microbiome and prostate cancer. Int J Urol. 2022;29(8):793–8. [CrossRef]
- Pascal Andreu V, Fischbach MA, Medema MH. Computational genomic discovery of diverse gene clusters harbouring Fe-S flavoenzymes in anaerobic gut microbiota. Microb Genomics. 2020;6(5):e000373. [CrossRef]
- Piccioni A, Covino M, Candelli M, Ojetti V, Capacci A, Gasbarrini A, et al. How do diet patterns, single foods, prebiotics and probiotics impact gut microbiota? Microbiol Res (Pavia). 2023;14(1):390–408.
- Pasquereau-Kotula E, Martins M, Aymeric L, Dramsi S. Significance of Streptococcus gallolyticus subsp. gallolyticus association with colorectal cancer. Front Microbiol. 2018;9:614. [CrossRef]
- Chung L, Orberg ET, Geis AL, Chan JL, Fu K, Shields CED, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018;23(2):203–14.
- Lo CH, Wu DC, Jao SW, Wu CC, Lin CY, Chuang CH, et al. Enrichment of Prevotella intermedia in human colorectal cancer and its additive effects with Fusobacterium nucleatum on the malignant transformation of colorectal adenomas. J Biomed Sci. 2022;29(1):88. [CrossRef]
- Liu X, Cheng Y, Shao L, Ling Z. Alterations of the predominant fecal microbiota and disruption of the gut mucosal barrier in patients with early-stage colorectal cancer. Biomed Res Int. 2020;2020(1):2948282. [CrossRef]
- De Almeida CV, Lulli M, di Pilato V, Schiavone N, Russo E, Nannini G, et al. Differential responses of colorectal cancer cell lines to Enterococcus faecalis’ strains isolated from healthy donors and colorectal cancer patients. J Clin Med. 2019;8(3):388. [CrossRef]
- Zhang Z, Li T, Xu L, Wang Q, Li H, Wang X. Extracellular superoxide produced by Enterococcus faecalis reduces endometrial receptivity via inflammatory injury. Am J Reprod Immunol. 2021;86(4):e13453. [CrossRef]
- Chervy M, Barnich N, Denizot J. Adherent-Invasive E. coli: Update on the Lifestyle of a Troublemaker in Crohn’s Disease. Int J Mol Sci. 2020;21(10):3734.
- Long X, Wong CC, Tong L, Chu ESH, Ho Szeto C, Go MYY, et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat Microbiol. 2019;4(12):2319–30. [CrossRef]
- Cheng Y, Ling Z, Li L. The intestinal microbiota and colorectal cancer. Front Immunol. 2020;11:615056. [CrossRef]
- Brennan CA, Garrett WS. Gut microbiota, inflammation, and colorectal cancer. Annu Rev Microbiol. 2016;70(1):395–411.
- Yu L, Zhang MM, Hou JG. Molecular and cellular pathways in colorectal cancer: apoptosis, autophagy and inflammation as key players. Scand J Gastroenterol. 2022;57(11):1279–90. [CrossRef]
- Lee T, Huang Y, Lu Y, Yeh Y, Yu LC. Hypoxia-induced intestinal barrier changes in balloon-assisted enteroscopy. J Physiol. 2018;596(15):3411–24.
- Chi H, Wang D, Chen M, Lin J, Zhang S, Yu F, et al. Shaoyao decoction inhibits inflammation and improves intestinal barrier function in mice with dextran sulfate sodium-induced colitis. Front Pharmacol. 2021;12:524287. [CrossRef]
- Chen J, Pitmon E, Wang K. Microbiome, inflammation and colorectal cancer. In: Seminars in immunology. Elsevier; 2017. p. 43–53.
- Goodman B, Gardner H. The microbiome and cancer. J Pathol. 2018;244(5):667–76.
- Cremonesi E, Governa V, Garzon JFG, Mele V, Amicarella F, Muraro MG, et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut. 2018;67(11):1984–94. [CrossRef]
- Bai Y, Li H, Lv R. Interleukin-17 activates JAK2/STAT3, PI3K/Akt and nuclear factor-κB signaling pathway to promote the tumorigenesis of cervical cancer. Exp Ther Med. 2021;22(5):1291.
- Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity. 2014;40(1):140–52. [CrossRef]
- Şahin T, Kiliç Ö, Acar AG, Severoğlu Z. A review the role of Streptococcus bovis in colorectal cancer. Arts Humanit Open Access J. 2023;5:165–73. [CrossRef]
- Kim J, Lee HK. Potential role of the gut microbiome in colorectal cancer progression. Front Immunol. 2022;12:807648. [CrossRef]
- Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014;63(12):1932–42. [CrossRef]
- Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol. 2016;17(3):230–40. [CrossRef]
- Martin OCB, Bergonzini A, d’Amico F, Chen P, Shay JW, Dupuy J, et al. Infection with genotoxin-producing Salmonella enterica synergises with loss of the tumour suppressor APC in promoting genomic instability via the PI3K pathway in colonic epithelial cells. Cell Microbiol. 2019;21(12):e13099.
- de Almeida CV, Taddei A, Amedei A. The controversial role of Enterococcus faecalis in colorectal cancer. Therap Adv Gastroenterol. 2018;11:1756284818783606. [CrossRef]
- Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, et al. Peptostreptococcus anaerobius induces intracellular cholesterol biosynthesis in colon cells to induce proliferation and causes dysplasia in mice. Gastroenterology. 2017;152(6):1419–33.
- Elatrech I, Marzaioli V, Boukemara H, Bournier O, Neut C, Darfeuille-Michaud A, et al. Escherichia coli LF82 differentially regulates ROS production and mucin expression in intestinal epithelial T84 cells: implication of NOX1. Inflamm Bowel Dis. 2015;21(5):1018–26. [CrossRef]
- Baena R, Salinas P. Diet and colorectal cancer. Maturitas. 2015;80(3):258–64.
- Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body fatness and cancer—viewpoint of the IARC Working Group. N Engl J Med. 2016;375(8):794–8. [CrossRef]
- Siddiqui R, Boghossian A, Alharbi AM, Alfahemi H, Khan NA. The pivotal role of the gut microbiome in colorectal cancer. Biology (Basel). 2022;11(11):1642. [CrossRef]
- Zhang N, Jin M, Wang K, Zhang Z, Shah NP, Wei H. Functional oligosaccharide fermentation in the gut: Improving intestinal health and its determinant factors-A review. Carbohydr Polym. 2022;284:119043. [CrossRef]
- Zhou CB, Fang JY. The regulation of host cellular and gut microbial metabolism in the development and prevention of colorectal cancer. Crit Rev Microbiol. 2018;44(4):436–54. [CrossRef]
- Martin-Gallausiaux C, Marinelli L, Blottière HM, Larraufie P, Lapaque N. SCFA: mechanisms and functional importance in the gut. Proc Nutr Soc. 2021;80(1):37–49. [CrossRef]
- Mirzaei R, Mirzaei H, Alikhani MY, Sholeh M, Arabestani MR, Saidijam M, et al. Bacterial biofilm in colorectal cancer: What is the real mechanism of action? Microb Pathog. 2020;142:104052.
- Cummings JH, Antoine JM, Azpiroz F, Bourdet-Sicard R, Brandtzaeg P, Calder PC, et al. PASSCLAIM 1—gut health and immunity. Eur J Nutr. 2004;43:ii118–73. [CrossRef]
- Illiano P, Brambilla R, Parolini C. The mutual interplay of gut microbiota, diet and human disease. FEBS J. 2020;287(5):833–55. [CrossRef]
- Li J, Zhang A hua, Wu F fang, Wang X jun. Alterations in the gut microbiota and their metabolites in colorectal cancer: recent progress and future prospects. Front Oncol. 2022;12:841552.
- Vacante M, Ciuni R, Basile F, Biondi A. Gut microbiota and colorectal cancer development: a closer look to the adenoma-carcinoma sequence. Biomedicines. 2020;8(11):489. [CrossRef]
- Khodaverdi N, Zeighami H, Jalilvand A, Haghi F, Hesami N. High frequency of enterotoxigenic Bacteroides fragilis and Enterococcus faecalis in the paraffin-embedded tissues of Iranian colorectal cancer patients. BMC Cancer. 2021;21(1):1353. [CrossRef]
- Sun CH, Li BB, Wang B, Zhao J, Zhang XY, Li TT, et al. The role of Fusobacterium nucleatum in colorectal cancer: from carcinogenesis to clinical management. Chronic Dis Transl Med. 2019;5(03):178–87. [CrossRef]
- Drewes JL, White JR, Dejea CM, Fathi P, Iyadorai T, Vadivelu J, et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ biofilms microbiomes. 2017;3(1):34.
- Chen Y, Shi T, Li Y, Huang L, Yin D. Fusobacterium nucleatum: the opportunistic pathogen of periodontal and peri-implant diseases. Front Microbiol. 2022;13:860149. [CrossRef]
- Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14(2):207–15. [CrossRef]
- Ou S, Wang H, Tao Y, Luo K, Ye J, Ran S, et al. Fusobacterium nucleatum and colorectal cancer: From phenomenon to mechanism. Front Cell Infect Microbiol. 2022;12:1020583. [CrossRef]
- Ganesan K, Guo S, Fayyaz S, Zhang G, Xu B. Targeting programmed Fusobacterium nucleatum Fap2 for colorectal cancer therapy. Cancers (Basel). 2019;11(10):1592. [CrossRef]
- Padma S, Patra R, Sen Gupta PS, Panda SK, Rana MK, Mukherjee S. Cell surface fibroblast activation protein-2 (Fap2) of fusobacterium nucleatum as a vaccine candidate for therapeutic intervention of human colorectal cancer: an immunoinformatics approach. Vaccines. 2023;11(3):525. [CrossRef]
- Zeng XY, Li M. Looking into key bacterial proteins involved in gut dysbiosis. World J Methodol. 2021;11(4):130. [CrossRef]
- Kasprzak, A. Angiogenesis-related functions of Wnt signaling in colorectal carcinogenesis. Cancers (Basel). 2020;12(12):3601. [CrossRef]
- Rubinstein MR, Baik JE, Lagana SM, Han RP, Raab WJ, Sahoo D, et al. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019;20(4):e47638.
- Abed J, Emgård JEM, Zamir G, Faroja M, Almogy G, Grenov A, et al. Fap2 mediates Fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed Gal-GalNAc. Cell Host Microbe. 2016;20(2):215–25. [CrossRef]
- Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod-Levi T, et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun. 2020;11(1):3259.
- Ranjbar M, Salehi R, Haghjooy Javanmard S, Rafiee L, Faraji H, Jafarpor S, et al. The dysbiosis signature of Fusobacterium nucleatum in colorectal cancer-cause or consequences? A systematic review. Cancer Cell Int. 2021;21:1–24. [CrossRef]
- Wang S, Gao J, Wang Z. Outer membrane vesicles for vaccination and targeted drug delivery. Wiley Interdiscip Rev Nanomedicine Nanobiotechnology. 2019;11(2):e1523. [CrossRef]
- Chatterjee D, Chaudhuri K. Vibrio cholerae O395 outer membrane vesicles modulate intestinal epithelial cells in a NOD1 protein-dependent manner and induce dendritic cell-mediated Th2/Th17 cell responses. J Biol Chem. 2013;288(6):4299–309. [CrossRef]
- Munshi, RM. The association between Fusobacterium nucleatum outer membrane vesicles and colonic cancer. Trinity College Dublin; 2017.
- Hur K, Toiyama Y, Okugawa Y, Ide S, Imaoka H, Boland CR, et al. Circulating microRNA-203 predicts prognosis and metastasis in human colorectal cancer. Gut. 2017;66(4):654–65. [CrossRef]
- Shi C, Yang Y, Xia Y, Okugawa Y, Yang J, Liang Y, et al. Novel evidence for an oncogenic role of microRNA-21 in colitis-associated colorectal cancer. Gut. 2016;65(9):1470–81. [CrossRef]
- Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor− κB, and up-regulating expression of microRNA-21. Gastroenterology. 2017;152(4):851–66.
- Stefl S, Nishi H, Petukh M, Panchenko AR, Alexov E. Molecular mechanisms of disease-causing missense mutations. J Mol Biol. 2013;425(21):3919–36. [CrossRef]
- Sameer, AS. Colorectal cancer: molecular mutations and polymorphisms. Front Oncol. 2013;3:114. [CrossRef]
- Coppedè F, Lopomo A, Spisni R, Migliore L. Genetic and epigenetic biomarkers for diagnosis, prognosis and treatment of colorectal cancer. World J Gastroenterol WJG. 2014;20(4):943.
- de Assis JV, Coutinho LA, Oyeyemi IT, Oyeyemi OT. Diagnostic and therapeutic biomarkers in colorectal cancer: a review. Am J Cancer Res. 2022;12(2):661.
- Strickler JH, Yoshino T, Stevinson K, Eichinger CS, Giannopoulou C, Rehn M, et al. Prevalence of KRAS G12C mutation and co-mutations and associated clinical outcomes in patients with colorectal cancer: a systematic literature review. Oncologist. 2023;28(11):e981–94. [CrossRef]
- Salem ME, El-Refai SM, Sha W, Puccini A, Grothey A, George TJ, et al. Landscape of KRAS G12C, associated genomic alterations, and interrelation with immuno-oncology biomarkers in KRAS-mutated cancers. JCO Precis Oncol. 2022;6:e2100245.
- Teo MYM, Fong JY, Lim WM, In LLA. Current advances and trends in KRAS targeted therapies for colorectal cancer. Mol Cancer Res. 2022;20(1):30–44.
- Miao R, Yu J, Kim RD. Targeting the KRAS Oncogene for Patients with Metastatic Colorectal Cancer. Cancers (Basel). 2025;17(9):1512. [CrossRef]
- Fakih MG, Kopetz S, Kuboki Y, Kim TW, Munster PN, Krauss JC, et al. Sotorasib for previously treated colorectal cancers with KRASG12C mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022;23(1):115–24. [CrossRef]
- Ryan MB, Coker O, Sorokin A, Fella K, Barnes H, Wong E, et al. KRASG12C-independent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy. Cell Rep. 2022;39(12). [CrossRef]
- Kuboki Y, Fakih M, Strickler J, Yaeger R, Masuishi T, Kim EJ, et al. Sotorasib with panitumumab in chemotherapy-refractory KRAS G12C-mutated colorectal cancer: a phase 1b trial. Nat Med. 2024;30(1):265–70. [CrossRef]
- Desai J, Alonso G, Kim SH, Cervantes A, Karasic T, Medina L, et al. Divarasib plus cetuximab in KRAS G12C-positive colorectal cancer: a phase 1b trial. Nat Med. 2024;30(1):271–8. [CrossRef]
- Hollebecque A, Kuboki Y, Murciano-Goroff YR, Yaeger R, Cassier PA, Heist RS, et al. Efficacy and safety of LY3537982, a potent and highly selective KRAS G12C inhibitor in KRAS G12C-mutant GI cancers: Results from a phase 1 study. American Society of Clinical Oncology; 2024. [CrossRef]
- Pool, R. Library to deacidify books. Nature. 1991;351(6326). [CrossRef]
- Antonarakis SE, Group NW. Recommendations for a nomenclature system for human gene mutations. Hum Mutat. 1998;11(1):1–3.
- Watzinger F, Lion T. K-RAS (Kristen rat sarcoma 2 viral oncogene homolog). Atlas Genet Cytogenet Oncol Haematol. 1999.
- Donovan S, Shannon KM, Bollag G. GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta (BBA)-Reviews Cancer. 2002;1602(1):23–45. [CrossRef]
- Bazan V, Migliavacca M, Zanna I, Tubiolo C, Grassi N, Latteri MA, et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann Oncol. 2002;13(9):1438–46. [CrossRef]
- Domingo E, Schwartz Jr S. BRAF. Atlas Genet Cytogenet Oncol Haematol. 2004.
- Fearnhead NS, Britton MP, Bodmer WF. The abc of apc. Hum Mol Genet. 2001;10(7):721–33.
- Tirnauer, J. APC (adenomatous polyposis coli). Atlas Genet Cytogenet Oncol Haematol URL http//AtlasGeneticsOncology org/Genes/APC118 html. 2005. [Google Scholar]
- Debuire B, Lemoine A, Saffroy R. CTNNB1 (Catenin, beta-1). Atlas Genet Cytogenet Oncol Haematol http//AtlasGeneticsOncology org/Genes/CTNNB1ID71 html. 2002.
- Saffroy R, Lemoine A, Debuire B. SMAD4 (mothers against decapentaplegic homolog 4 (Drosophila)). http//AtlasGeneticsOncology org. 2004;294. [CrossRef]
- Shi, Y. Structural insights on Smad function in TGFβ signaling. Bioessays. 2001;23(3):223–32.
- Lammi L, Arte S, Somer M, Järvinen H, Lahermo P, Thesleff I, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74(5):1043–50. [CrossRef]
- Xu HT, Wei Q, Liu Y, Yang LH, Dai SD, Han Y, et al. Overexpression of axin downregulates TCF-4 and inhibits the development of lung cancer. Ann Surg Oncol. 2007;14:3251–9. [CrossRef]
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–34. [CrossRef]
- Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hosteller R, Cleary K, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342(6250):705–8. [CrossRef]
- Harris CC, Hollstein M. Clinical implications of the p53 tumor-suppressor gene. N Engl J Med. 1993;329(18):1318–27. [CrossRef]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. [CrossRef]
- Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–12. [CrossRef]
- Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000;60(16):4366–71.
- Diederich, M. Signal transduction pathways, chromatin structures, and gene expression mechanisms as therapeutic targets: Preface. Ann N Y Acad Sci. 2004;1030:xiii.
- Feilchenfeldt J, Tötsch M, Sheu SY, Robert J, Spiliopoulos A, Frilling A, et al. Expression of galectin-3 in normal and malignant thyroid tissue by quantitative PCR and immunohistochemistry. Mod Pathol. 2003;16(11):1117–23. [CrossRef]
- Mazzoni SM, Petty EM, Stoffel EM, Fearon ER. An AXIN2 mutant allele associated with predisposition to colorectal neoplasia has context-dependent effects on AXIN2 protein function. Neoplasia. 2015;17(5):463–72. [CrossRef]
- Summers MG, Smith CG, Maughan TS, Kaplan R, Escott-Price V, Cheadle JP. BRAF and NRAS locus-specific variants have different outcomes on survival to colorectal cancer. Clin Cancer Res. 2017;23(11):2742–9.
- Cercek A, Braghiroli MI, Chou JF, Hechtman JF, Kemeny N, Saltz L, et al. Clinical features and outcomes of patients with colorectal cancers harboring NRAS mutations. Clin cancer Res. 2017;23(16):4753–60.
- Jin J, Shi Y, Zhang S, Yang S. PIK3CA mutation and clinicopathological features of colorectal cancer: a systematic review and Meta-Analysis. Acta Oncol (Madr). 2020;59(1):66–74. [CrossRef]
- Stec R, Semeniuk-Wojtaś A, Charkiewicz R, Bodnar L, Korniluk J, Smoter M, et al. Mutation of the PIK3CA gene as a prognostic factor in patients with colorectal cancer. Oncol Lett. 2015;10(3):1423–9. [CrossRef]
- Mei ZB, Duan CY, Li CB, Cui L, Ogino S. Prognostic role of tumor PIK3CA mutation in colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2016;27(10):1836–48. [CrossRef]
- Whiffin N, Broderick P, Lubbe SJ, Pittman AM, Penegar S, Chandler I, et al. MLH1-93G> A is a risk factor for MSI colorectal cancer. Carcinogenesis. 2011;32(8):1157–61. [CrossRef]
- Gatalica Z, Vranic S, Xiu J, Swensen J, Reddy S. High microsatellite instability (MSI-H) colorectal carcinoma: a brief review of predictive biomarkers in the era of personalized medicine. Fam Cancer. 2016;15(3):405–12. [CrossRef]
- Curtin K, Slattery ML, Ulrich CM, Bigler J, Levin TR, Wolff RK, et al. Genetic polymorphisms in one-carbon metabolism: associations with CpG island methylator phenotype (CIMP) in colon cancer and the modifying effects of diet. Carcinogenesis. 2007;28(8):1672–9. [CrossRef]
- Chang SC, Li AFY, Lin PC, Lin CC, Lin HH, Huang SC, et al. Clinicopathological and molecular profiles of sporadic microsatellite unstable colorectal cancer with or without the CpG island methylator phenotype (CIMP). Cancers (Basel). 2020;12(11):3487. [CrossRef]
Figure 1.
Gut microbiota mechanisms in colorectal carcinogenesis. 1. Myeloid cells are tumor-related cells that can be activated by products of pathogenic bacteria. 2. The adhesion of pathogenic bacterial virulence factors to intestinal epithelial cells (IECs) promotes tumorigenesis. 3. By damaging DNA in IECs and accelerating the production of genotoxins, this process is considered the first step in CRC development. 4. DNA damage is activated by reactive oxygen species (ROS) and reactive nitrogen species (RNS), which are produced by inflammatory cells. 5. Many bacterial metabolites, including NOCs and secondary bile acids, contribute to DNA damage, resulting in the promotion of CRC carcinogenesis. 6. Biofilm formation by different pathogenic bacteria enhances carcinogenesis through IL-6 and by activating the STAT3.
Figure 1.
Gut microbiota mechanisms in colorectal carcinogenesis. 1. Myeloid cells are tumor-related cells that can be activated by products of pathogenic bacteria. 2. The adhesion of pathogenic bacterial virulence factors to intestinal epithelial cells (IECs) promotes tumorigenesis. 3. By damaging DNA in IECs and accelerating the production of genotoxins, this process is considered the first step in CRC development. 4. DNA damage is activated by reactive oxygen species (ROS) and reactive nitrogen species (RNS), which are produced by inflammatory cells. 5. Many bacterial metabolites, including NOCs and secondary bile acids, contribute to DNA damage, resulting in the promotion of CRC carcinogenesis. 6. Biofilm formation by different pathogenic bacteria enhances carcinogenesis through IL-6 and by activating the STAT3.
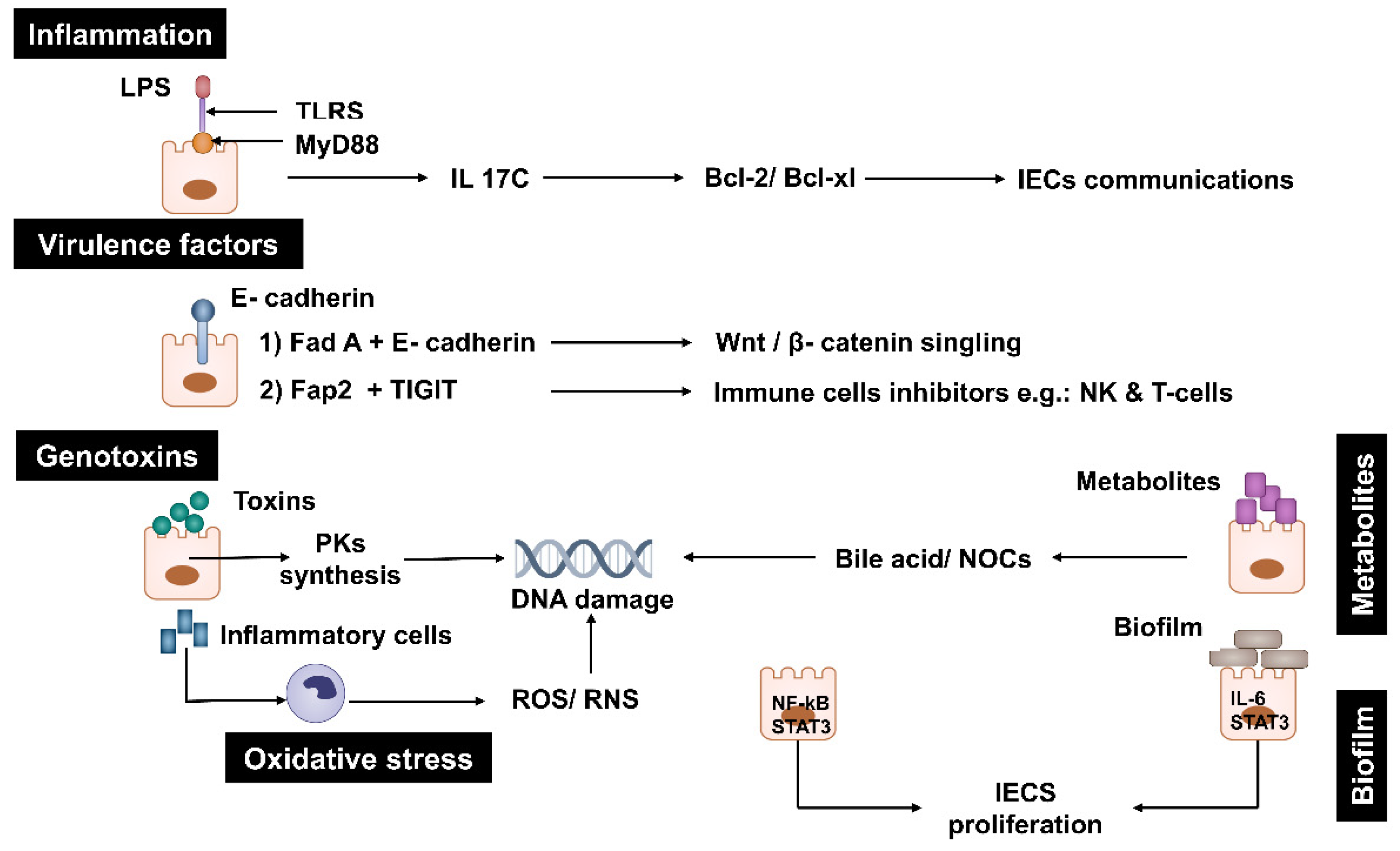
Figure 2.
Relative Abundance of Fusobacterium nucleatum and Other Microbial Groups in CRC vs. Normal Tissue.F. nucleatum abundance in CRC tissues is estimated at 15% to reflect its significant enrichment (noted as a dominant fusobacterial species in 94.1% of biofilm-positive tumors). Paired normal tissues have minimal F. nucleatum (2%). Healthy biopsies have even lower abundance (1%), based on Figure 5a showing significant differences from CRC tissues. Other Fusobacterium species (F. necrophorum, F. periodonticum, L. trevisanii) are less abundant, estimated at 5% in CRC tissues. “Other Bacteria” (e.g., Bacteroidetes, Lachnospiraceae, Proteobacteria) dominate the remaining composition.
Figure 2.
Relative Abundance of Fusobacterium nucleatum and Other Microbial Groups in CRC vs. Normal Tissue.F. nucleatum abundance in CRC tissues is estimated at 15% to reflect its significant enrichment (noted as a dominant fusobacterial species in 94.1% of biofilm-positive tumors). Paired normal tissues have minimal F. nucleatum (2%). Healthy biopsies have even lower abundance (1%), based on Figure 5a showing significant differences from CRC tissues. Other Fusobacterium species (F. necrophorum, F. periodonticum, L. trevisanii) are less abundant, estimated at 5% in CRC tissues. “Other Bacteria” (e.g., Bacteroidetes, Lachnospiraceae, Proteobacteria) dominate the remaining composition.
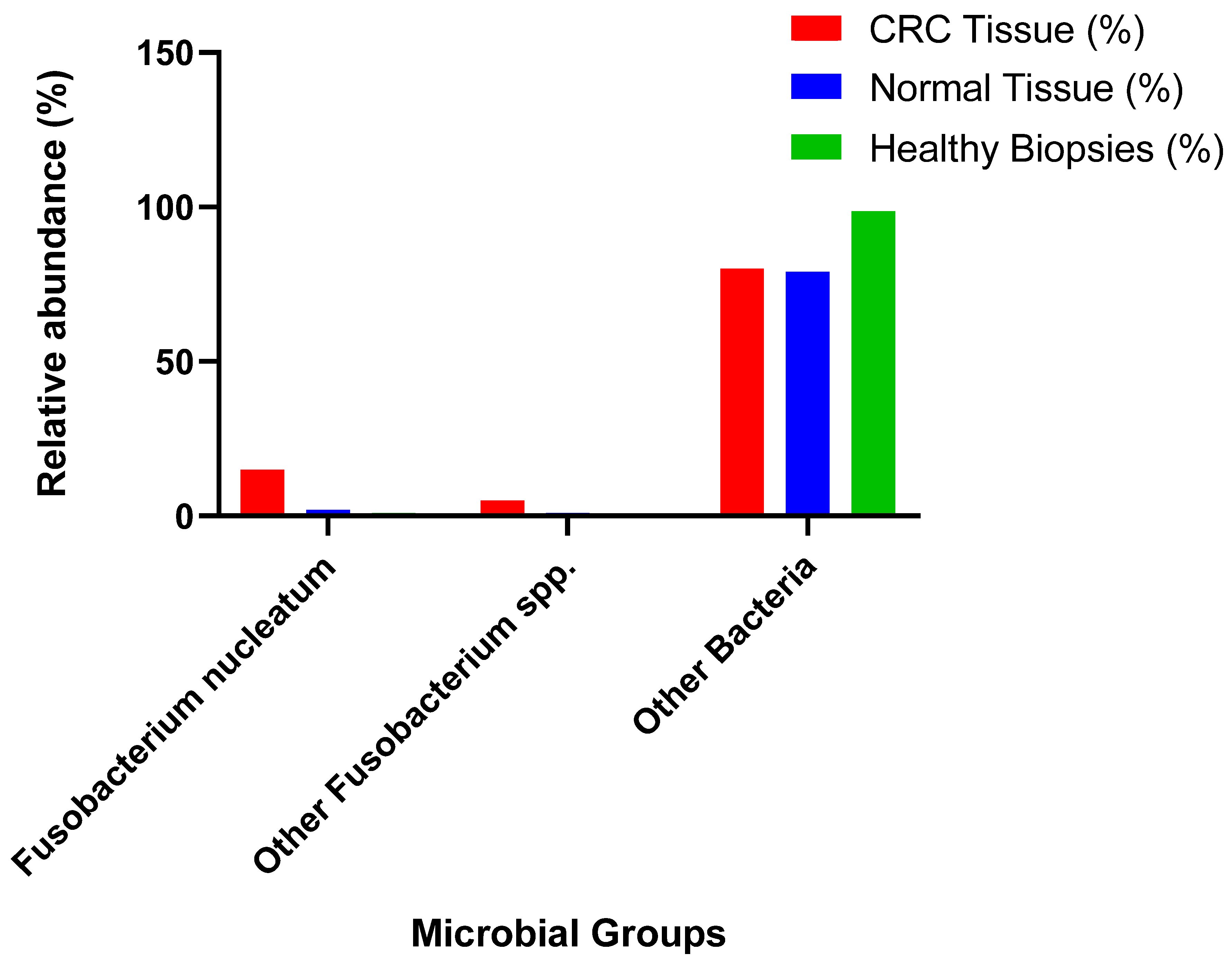
Figure 3.
The role of Fusobacterium nucleatum in the initiation and progression of colorectal cancer. There are various factors, which play a significant role in the initiation and progression of colorectal cancer as described above. Abbreviations; EMT: epithelial-mesenchymal transition; Fap2: fibroblast activation protein 2; Gal-GalNAc: D-galactose-(1–3)-N-acetyl-D-galactosamine; NK: natural killer; OMV: outer membrane vesicle.
Figure 3.
The role of Fusobacterium nucleatum in the initiation and progression of colorectal cancer. There are various factors, which play a significant role in the initiation and progression of colorectal cancer as described above. Abbreviations; EMT: epithelial-mesenchymal transition; Fap2: fibroblast activation protein 2; Gal-GalNAc: D-galactose-(1–3)-N-acetyl-D-galactosamine; NK: natural killer; OMV: outer membrane vesicle.
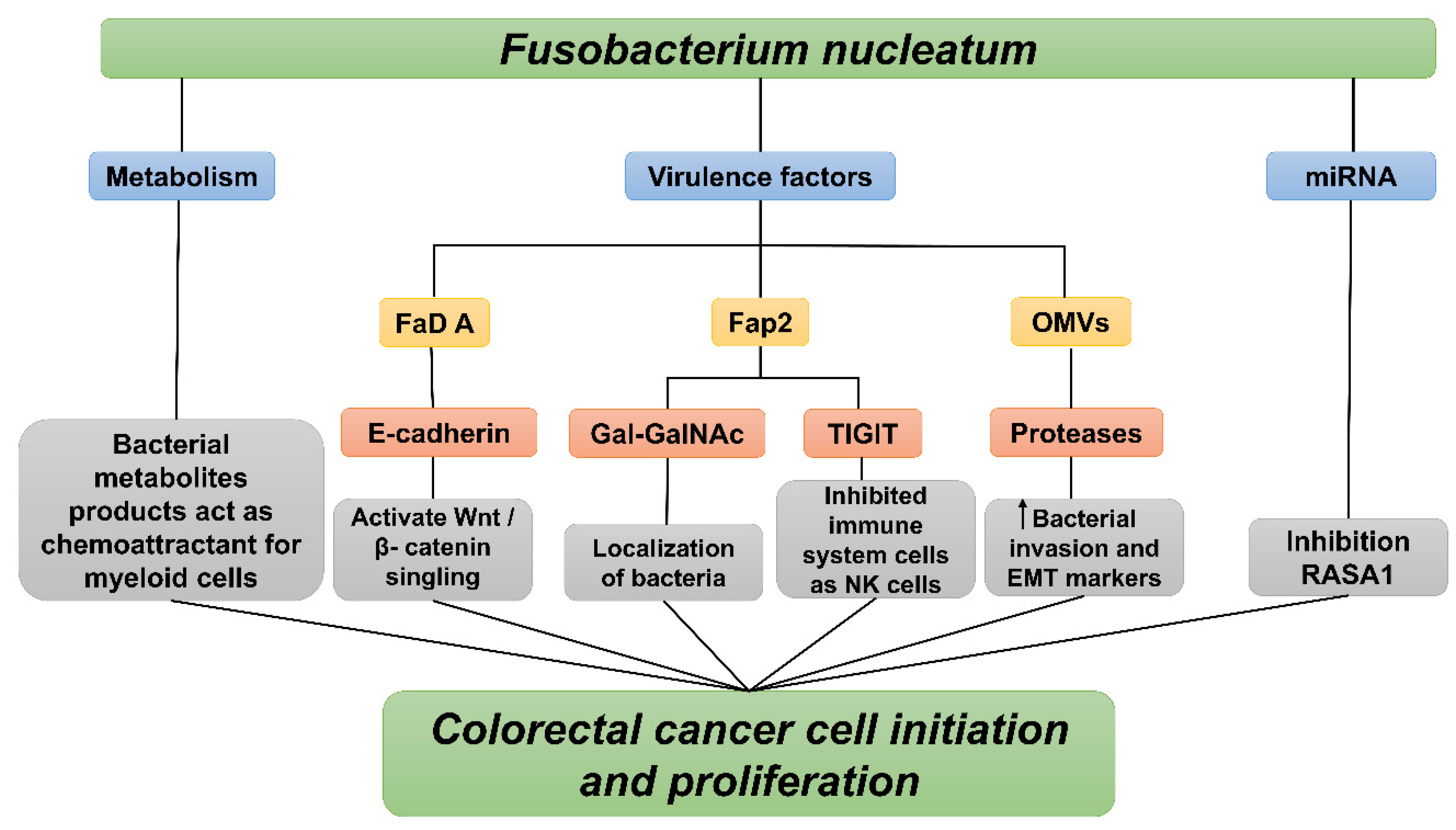
Figure 4.
The role of Fusobacterium nucleatum virulence factors in enhancing CRC cell proliferation. 1.FadA protein binds to E-cadherin on epithelial cells. At the same time, Annexin A (ANXA1) is regulated by FadA in conjunction with E-cadherin, leading to the activation of the Wnt/β-catenin pathway. 2. PAK1 is activated by LPS binding through TLR4. PAK1 phosphorylates -catenin at ser675. 3. This phosphorylation increases the nuclear accumulation of -catenin. 4. The activation of the Cyclin D1 (CCND1) gene by nuclear -catenin leads to CRC promotion. 5. Transcription of miR- 21 occurs due to the activation signaling of TLR4, MYD88, and NF-B. 6. MAPK is activated by miR-21 through RASA1 inhibition, leading to accelerated cancer cell proliferation.
Figure 4.
The role of Fusobacterium nucleatum virulence factors in enhancing CRC cell proliferation. 1.FadA protein binds to E-cadherin on epithelial cells. At the same time, Annexin A (ANXA1) is regulated by FadA in conjunction with E-cadherin, leading to the activation of the Wnt/β-catenin pathway. 2. PAK1 is activated by LPS binding through TLR4. PAK1 phosphorylates -catenin at ser675. 3. This phosphorylation increases the nuclear accumulation of -catenin. 4. The activation of the Cyclin D1 (CCND1) gene by nuclear -catenin leads to CRC promotion. 5. Transcription of miR- 21 occurs due to the activation signaling of TLR4, MYD88, and NF-B. 6. MAPK is activated by miR-21 through RASA1 inhibition, leading to accelerated cancer cell proliferation.
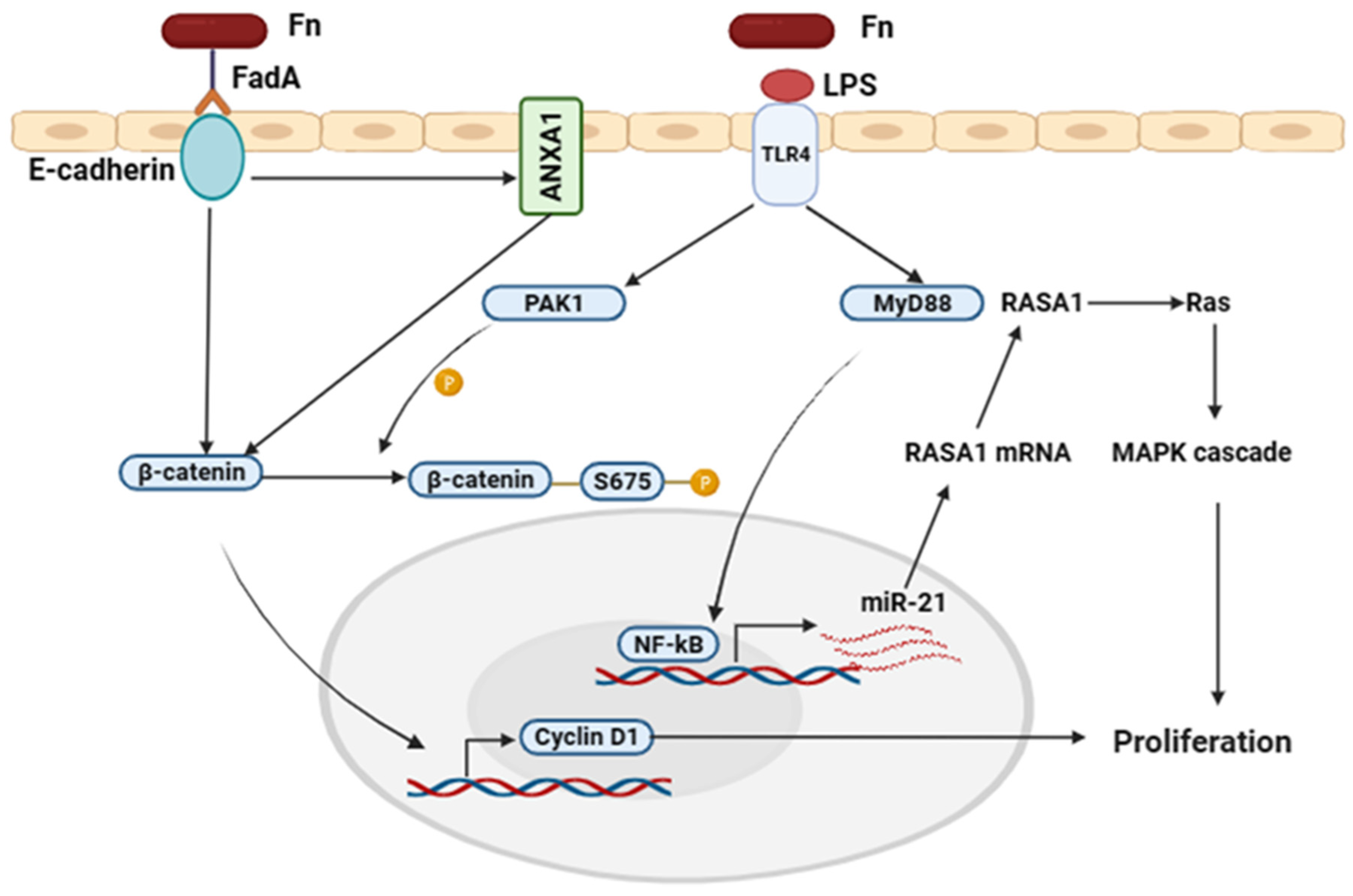
Figure 5.
Fap2 and Rad D mechanisms in colorectal cancer: (1) Fap2 binding to Gal-GalNAc leads to colonization of F.nucletum in CRC tissue (2) Function of the immune system has been inhibited because of Fap2– TIGIT binding (3) Fap2 and Rad D connection leads to lymphocytic apoptosis induction. (4) Additionally, a kynurenine-enriched toxic environment could be created by F. nucleatum inside the immune cells' tryptophan metabolism.
Figure 5.
Fap2 and Rad D mechanisms in colorectal cancer: (1) Fap2 binding to Gal-GalNAc leads to colonization of F.nucletum in CRC tissue (2) Function of the immune system has been inhibited because of Fap2– TIGIT binding (3) Fap2 and Rad D connection leads to lymphocytic apoptosis induction. (4) Additionally, a kynurenine-enriched toxic environment could be created by F. nucleatum inside the immune cells' tryptophan metabolism.
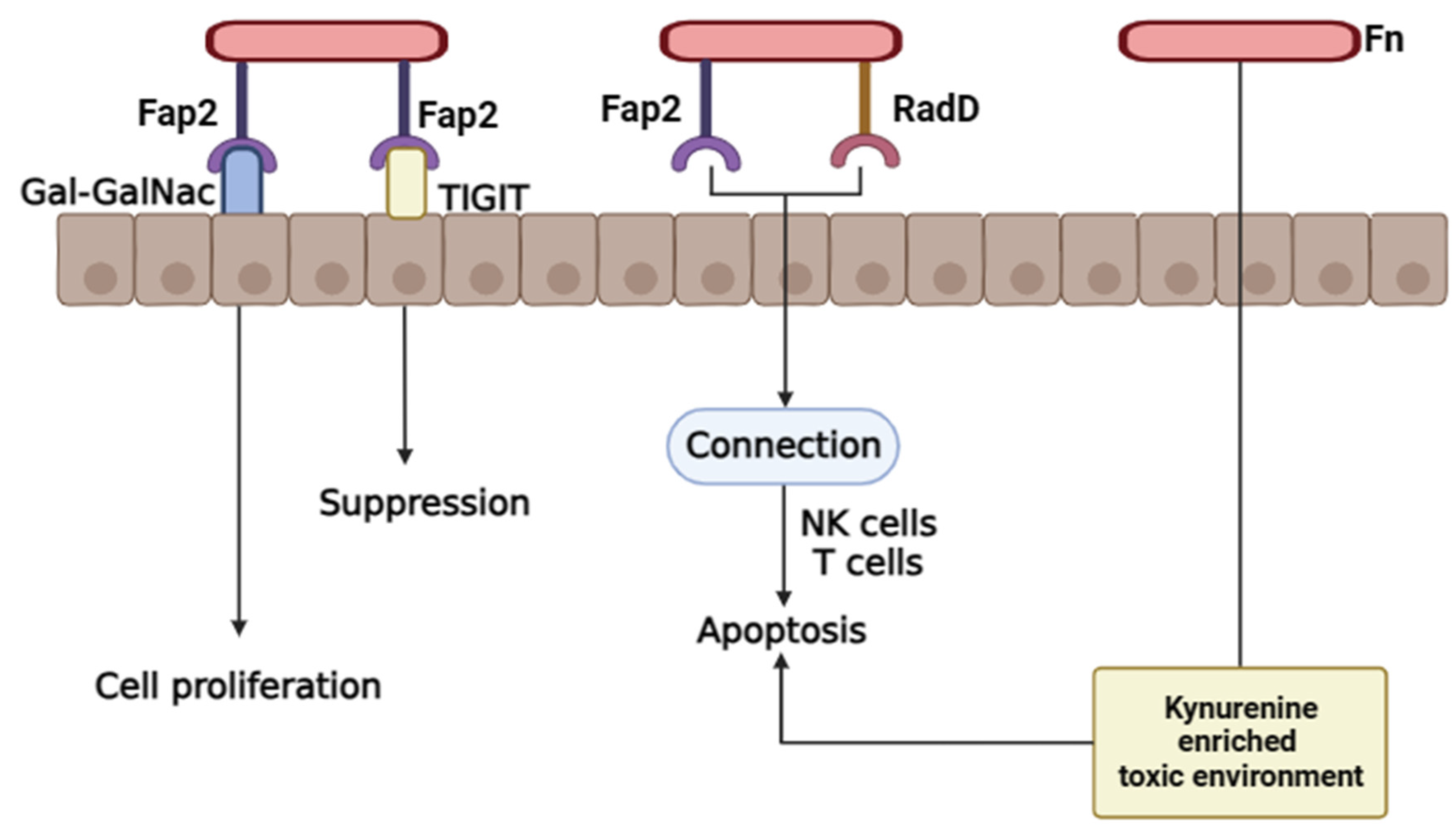
Figure 6.
The prevalence of Genotypic Biomarkers in CRC. In particular, MYC shows a striking incidence of 100%, indicating that this mutation exists among universally studied samples. The PEN mutations are observed in 30% of cases, while CIMP is present in 55% of patients, suggesting significant participation in the CRC pathology. KRAS mutation, a well -known oncogenic driver, appears in 35% of the samples, while TP53 mutations are found in 55%, highlighting their importance in the progression of the tumor. In addition, PIK3CA mutations and BRAF mutations show the spread of 17.5% and 12.5%, respectively, which indicates their roles in the disease. Finally, the presence of microsatellite instability (MSI) observed in 15% cases, as well as Lynch syndrome genes of 3%, reflects their contribution to hereditary cancer syndrome. Overall, this data forms the varying occurrence of the important major biomarkers of CRC's diverse genetic landscape and individual treatment strategies. Abbreviations: MSI: Microsatellite Instability, CIMP: CpG Island Methylator Phenotype, KRAS: Kirsten Rat Sarcoma Viral Oncogene Homolog, BRAF: B-Raf Proto-Oncogene, PIK3CA: Phosphoinositide-3-Kinase Catalytic Subunit Alpha, TP53: Tumor Protein p53, PTEN: Phosphatase and Tensin Homolog , MYC: MYC Proto-Oncogene.
Figure 6.
The prevalence of Genotypic Biomarkers in CRC. In particular, MYC shows a striking incidence of 100%, indicating that this mutation exists among universally studied samples. The PEN mutations are observed in 30% of cases, while CIMP is present in 55% of patients, suggesting significant participation in the CRC pathology. KRAS mutation, a well -known oncogenic driver, appears in 35% of the samples, while TP53 mutations are found in 55%, highlighting their importance in the progression of the tumor. In addition, PIK3CA mutations and BRAF mutations show the spread of 17.5% and 12.5%, respectively, which indicates their roles in the disease. Finally, the presence of microsatellite instability (MSI) observed in 15% cases, as well as Lynch syndrome genes of 3%, reflects their contribution to hereditary cancer syndrome. Overall, this data forms the varying occurrence of the important major biomarkers of CRC's diverse genetic landscape and individual treatment strategies. Abbreviations: MSI: Microsatellite Instability, CIMP: CpG Island Methylator Phenotype, KRAS: Kirsten Rat Sarcoma Viral Oncogene Homolog, BRAF: B-Raf Proto-Oncogene, PIK3CA: Phosphoinositide-3-Kinase Catalytic Subunit Alpha, TP53: Tumor Protein p53, PTEN: Phosphatase and Tensin Homolog , MYC: MYC Proto-Oncogene.
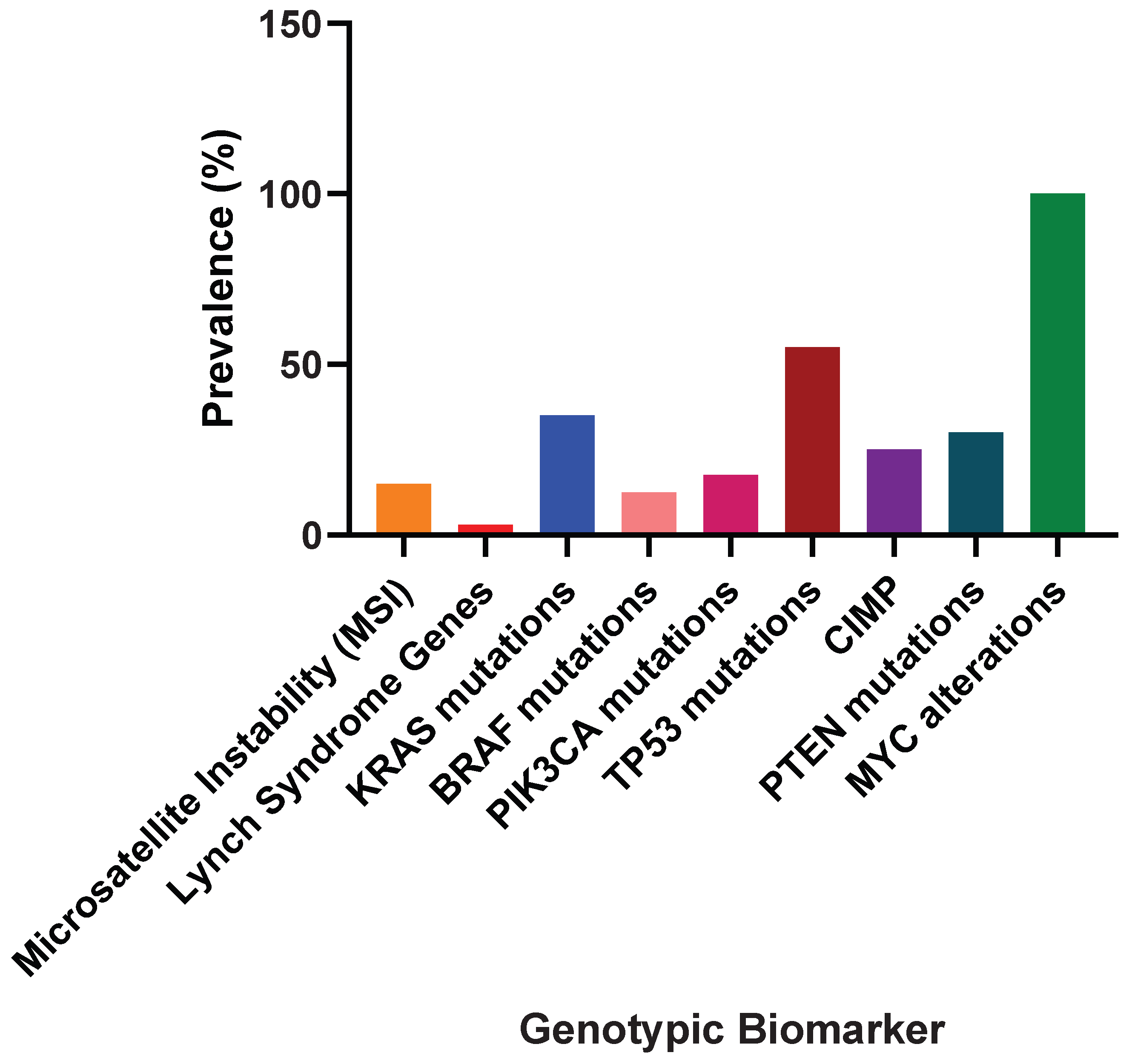
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



