
Submitted:
04 June 2025
Posted:
05 June 2025
You are already at the latest version
Abstract
Despite advancements around in-field detection of pathogens in aquatic organisms, limited research has evaluated point-of-need DNA extraction methods. This is a critical step in the application of any field deployable molecular assays. This is especially important to applications that use PCR, because carry over of contaminants causes inhibition and false negative results. To address this, we compared the performance of nine rapid DNA extraction methods with a control laboratory method (QIAMP DNA mini kit, QIAGEN). DNA was extracted from fresh, frozen and ethanol-fixed mantle and digestive tissues of Pacific oysters. Extraction performance was assessed by measuring DNA quantity and contaminants using fluorescence and spectrophotometric methods respectively. PCR amplification of the host cytochrome oxidase subunit 1 gene (COX1) was performed to assess the presence of inhibitors. The QuickExtract™ DNA extraction solution (QEDES, Lucigen) yielded the highest DNA quantity and carried over minimal contaminants, resulting in low PCR inhibition. In addition, the suitability of QEDES for detection of oyster pathogens was assessed, using Magallana gigas samples infected with Ostreid herpesvirus 1 and Vibrio aestuarianus and European flat oysters infected with Bonamia ostreae. Conventional and qPCR were used to assess performance. In each instance, the QEDES successfully identified the presence of pathogens tested.

Keywords:
in field
; point of need
; DNA extraction
; Magallana (Crassostrea) gigas
; Vibrio aestuarianus
; Bonamia ostreae
; Ostreid herpesvirus 1
1. Introduction
Aquaculture is one of the fastest growing food sectors globally [1]. The global aquaculture of Pacific oysters Magallana (Crassostrea) gigas is worth £1.29 billion annually and generates on average 620,000 tonnes per year [2]. Disease is considered one of the main limiting factors that impact the production of bivalve molluscs worldwide [3]. The treatment of disease is especially problematic for mollusc aquaculture because the use of drugs to control infection has limited effect and in addition any chemical treatment will be diluted in the open water sites in which the molluscs are grown [4]. Vaccination against infection is not possible as molluscs do not have an adaptive immune response [5,6]. Therefore, control of disease in bivalve molluscs relies mainly on preventive measures to avoid the introduction and spread of pathogens. Two of the main diseases affecting Pacific oysters aquaculture are the Ostreid herpesvirus 1 microvar (OsHV-1 µvar) and the bacterium Vibrio aestuarianus. V. aestuarianus has caused mass mortality of adult M. gigas in France since 2001 and in Ireland, Scotland and Spain since 2011 [2,7,8,9,10]. OsHV-1 has caused considerable worldwide losses [11] and is associated with Pacific oyster mortality syndrome. The production of European flat oysters (Ostrea edulis) in Europe has been adversely affected by diseases caused by the protists Bonamia ostreae and Marteilia refringens [12,13]. The use of rapid onsite screening methods that can be employed in production units or at border control posts are crucial to prevent the spread of disease and help minimise stock losses.
Although numerous manuscripts have outlined the development of in-field disease detection in molluscs [14,15,16,17], few have addressed challenges associated with DNA extraction [16,18,19,20,21,22]. Only a single paper has investigated this in bivalves [23]. However, this step is critical in order to successfully achieve results that are accurate and free from contamination. This requires the transfer of complex laboratory processes, usually completed in controlled conditions into the field. Technological and method developments have allowed this to become achievable. These include, portable laboratory equipment condensed into the size of a laptop bag (e.g. the Bento lab), isothermal amplification technologies such as Loop mediated isothermal amplification (LAMP) and Recombinase Polymerase Amplification (RPA) and portable sequencers with small dimensions for example the MinION by Oxford Nanopore Technologies (ONT). These technological breakthroughs can be combined to form methods that can be utilised at the point-of-need [24]. However, the success of these new technologies depends in part on the ability to extract DNA of suitable quality for downstream applications.
Although there have been a number of reviews on this challenge [25,26,27,28,29,30] the matrix the DNA is extracted from is invariably human or plant derived. Although the methodology and its associated challenges can be applied to tissue from aquatic species such as fish, crustacea and molluscs it is apparent that there is not a ‘one method fits all’ technique. Different sample types specifically host, tissue type, pathogen and quantity of pathogen will all have to be considered and then a range of extraction / sample preparation methods tested and assessed for each scenario. For example, for the amplification of prolific bacteria from fish gills, sample preparation may be as quick as boiling the tissue to inactivate the bacteria and using a small homogenate volume into your chosen point-of-need detection method. However, for the detection of low levels of spore forming pathogen from the hepatopancreas, or a virus from the mantle tissue of molluscs there are many and various known substances that are present in the tissue that can inhibit the detection method resulting in false negatives [31,32]. Furthermore, the tissue matrix itself may require physical disruption, prior to being able to release the DNA from the pathogen itself.
The objective of this work was to evaluate nine different methods for extraction of DNA from oysters and assess their suitability for point-of-need detection of mollusc pathogens by PCR.
2. Materials and Methods
Pacific oysters M. gigas were obtained from a local oyster farm and kept refrigerated at approximately 4 °C while dissections were carried out over a three-day period. Every day a single M. gigas individual was shucked and several pieces of mantle edge (left bottom lobe) and digestive gland were dissected, weighed (between 10 and 20 mg for each tissue type) and placed into separate tubes. Of these, ten pieces of each tissue type (mantle and digestive gland) from each oyster (n=3) were extracted immediately after dissection (fresh tissue). Ten pieces of each tissue type were frozen at -20oC and kept for 7 days prior to extraction and a similar set of samples were placed into 80% ethanol and incubated at room temperature for 1 h prior to extraction. Tissue samples stored in 80% ethanol were removed and blotted on tissue paper prior to DNA extractions.
DNA was extracted from mantle and digestive gland tissues preserved using the different methods (fresh, ethanol and frozen), using a standard DNA extraction method and nine quick extraction methods, namely: (1) QIAmp DNA mini kit (QIAGEN) (2) Buccalyse DNA Release Kit (Isohelix); (3) QIAcard FTA Elute method (QIAGEN), (4) Filter paper method (Whatman); (5) Kapa express extract kit (Roche); (6) Hotshot DNA extraction kit (Bento lab); (7) Dipstick DNA extraction Kit (Bento lab); (8) boiling method; (9) short digestion/boiling method [33] and (10) (QEDES) (Lucigen). DNA extractions were carried out following manufacturer's instructions with minor modifications or as recommend by Batista et al [33] as described below.
For the QIAmp DNA mini kit method, 180 µL of buffer ATL and 20 µL of proteinase K was added to digestive gland and mantle tissues in separate tubes. These were vortexed every 30 min while being incubated at 56 °C for 3 h. Two hundred microlitres of buffer AL was added, tubes vortexed for 15 s and incubated at 70 °C for 10 min. Two hundred microlitres of 100% ethanol was added, tubes vortexed for 15 s and the resulting homogenate pipetted into the QIAmp Mini spin column. The spin column was centrifuged at 8000 rpm for 1 min and the flow through discarded. Five hundred microlitres of buffer AW1 was added and the spin column centrifuged for 8000 rpm for 1 min. The flow through was discarded and 500 µL of buffer AW2 was added and centrifuged for 3 min at 14,000 rpm. The flow through was discarded, a new collection tube added, centrifuged at 14,000 rpm for 1 min to dry. A new collection tube was added along with 100 µL of elution buffer AE which was centrifuged for 8000 rpm for 1 min.
For the Buccalyse DNA Release method, 100 µL of Buccalyse solution was pipetted into both the mantle and digestive gland tubes. The tissues were homogenised using disposable sterile pestles and the resulting homogenates were incubated at 70 °C for 15 min followed by 95 °C for 2 min.
For the QIAcard FTA Elute method, mantle and digestive gland tissues were homogenised in molecular grade water separately using disposable sterile pestles. Approximately 75 µL was smeared onto two separate labelled FTA cards using a sterile pipette tip. The cards were left to dry on the bench for three hours at room temperature prior to storage in the dark for 7 days. Sterile scissors were used to cut off 4 x 3 mm sections of each card which were placed into two 1.5 mL tubes. Five hundred microlitres of TE buffer was pipetted into each tube, vortexed for 5 s and the buffer removed. This was repeated three times. One hundred microlitres of TE buffer was added and tubes were incubated at 95 °C for 30 min with shaking at 1000 rpm. Tubes were centrifuged at 14,000 rpm for one min and the elution solution removed until required.
For the filter paper method, 500 µL of lysis buffer (20 mM Tris [pH 8.0], 25 mM NaCl, 2.5 mM EDTA, and 0.05 % SDS) was pipetted into both the mantle and digestive gland tubes and the tissue homogenised using sterile pestles. A 1 cm diameter circle of Whatman no 1 paper was dipped into the lysate 3 times, and subsequently dipped 3 times into another tube containing 500 µL wash buffer (10 mM Tris, 0.01 % Tween-20). The paper was frozen at -20 °C prior to PCR.
For the Kapa express extract kit method, 88 µL of molecular grade water was pipetted into both the mantle and digestive gland tubes followed by 10 µL of 10X KAPA Express Extract Buffer, homogenised using a pestle and 2 µL of KAPA Express Extract Enzyme was added. The homogenate was transferred into a 0.5 mL PCR tube and incubated at 75 °C for 10 min followed by 95 °C for 5 min. The homogenate was centrifuged briefly and diluted tenfold using TE buffer before use.
For the Hotshot DNA extraction kit, 150 µL of Alkaline Lysis Solution was pipetted into both the mantle and digestive gland tubes and tissue was homogenised using a pestle. The homogenate was transferred into 0.5 mL PCR tubes and incubated at 95 °C for 30 min. Tubes were cooled on ice and 150 µL of neutralising buffer was added to each tube and mixed by pipetting.
For the Dipstick DNA extraction Kit method, 100 µL of extraction buffer was pipetted into both the mantle and digestive gland tubes. The tissue was homogenised using pestles prior to the addition of another 400 μL of extraction buffer. A paper dip stick was incubated in each tube at room temperature for 2 min. The dipstick was rinsed three times in 1 mL of wash buffer and then incubated for 2 min in fresh tubes containing 100 µL TE buffer.
For the boiling method, mantle and digestive gland tissues in separate tubes were incubated at 100 °C for 10 min. Before use, the supernatant was diluted tenfold in molecular grade water. For the short digestion-boiling method [33], 330 µL of extraction buffer (containing 33 µL of Goldstar Taq DNA Polymerase buffer, 297 µL of double distilled water and 0.5 % of Tween-20) was pipetted into both the mantle and digestive gland tubes. Tissue was homogenised using a pestle and 33 µL of Proteinase K (10 mg.mL 1) was added and the tubes incubated at 55 °C for 60 min followed by 100 °C for 20 min. Tubes were spun down briefly using a centrifuge at 1,200 x g for 5 min at 4 °C. Supernatants were diluted tenfold in molecular grade water prior to use.
For the QEDES method, 500 µL of QEDES was added to all tissues. Tubes were vortexed for 15 s, incubated at 65 °C for 10 min, vortexed for another 15 s and heated to 98 °C for 2 min. Each extraction method had an associated negative control (molecular grade water) which was treated identically to the tissues.
DNA samples were quantified using QuantiFluor® ONE dsDNA System (Promega) on a Quantus fluorometer. The NanoDrop One/OneC spectrophotometer was used to measure the 260/280 and 260/230 ratios to determine the purity of the DNA and the presence of contaminants. The DNA extraction yield (in ng/mg) was calculated by multiplying the concentration of DNA (in ng/µl) determined using the fluorometric method by the total elution volume, divided by the tissue weight (mg).
To determine the presence of PCR inhibitors and assess the quality of M.gigas DNA obtained, both neat and 1:10 diluted DNA was used to amplify a 356 bp cytochrome oxidase subunit 1 (COX1) fragment using the primers mlCOIintF 5'- GGWACWGGWTGAACWGTWTAYCCYCC -3' and dgHCO2198 5'- TAAACTTCAGGGTGACCAAARAAYCA -3' [34]. Reactions were set up in a 25 µL volume as follows: 1 x NEBNext Ultra II Q5® Master mix, 0.5 µM of forward primer (mlCOIintF), 0.5 µM of reverse primer (dgHCO2198), 6.5 µL of molecular grade water, 20 µg of BSA and 2 µL of DNA. Thermocycling conditions were 98 °C for 2 min followed by 35 cycles of 98 °C for 40 s, 45 °C for 40 s and 72 °C for 30 s with a final extension of 72 °C for 3 min. Six microlitres of amplification products were run on 2% TBE agarose gels stained with Green Safe dye (NZTECH) and viewed under a UV illuminator. Negative controls were incorporated in all PCR runs containing all the PCR reagents and molecular biology grade water instead of template DNA.
DNA was extracted from three Pacific oysters infected with V. aestuarianus using the QIAcard FTA Elute, digestion/boiling, QEDES and QIAmp DNA Mini kit methods. These three extraction methods plus the control method were selected based on the amplification scores obtained for M. gigas COX1. DNA of V. aestuarianus was quantified using both neat and tenfold diluted DNA by real time PCR using the primers dnaJ-F (5’- GTATGAAATTTTAACTGACCCACAA -3’), dnaJ-R (5’- TCAATTTCTTTCGAACAACCAC -3’), and the probe dnaJ-probe (5’- 6FAM TGGTAGCGCAGACTTCGGCGAC QSY -3’) [35]. In a total reaction volume of 20 µL the following reagents were used: 10 µL of TaqMan™ Fast Advanced Master Mix (2X, no UNG, Applied Biosystems), 0.6 µL of forward primer (dnaJ-F 20 µM), 0.6 µM of reverse primer (dnaJ-R 20 µM), 0.4 µL of probe (dnaJ-probe 10 µM), 3.4 µL of molecular grade water and 5 µL of extracted DNA. Samples were run in duplicate and a standard curve was also run consisting of 10 fold dilutions of V. aestuarianus gDNA from 1 x 106 to 1 x 101 genome copies. The following thermocycling conditions were used: an initial denaturation of 3 min at 95 °C followed by 40 cycles of 1 s at 95 °C and 20 s at 61 °C. Plates were run on a QuantStudio 3 Real Time qPCR system (Applied Biosystems, Thermofisher).
DNA was extracted from mantle edge tissue dissected from three M. gigas individuals infected with OsHV-1 using the QEDES as well as the QIAmp DNA mini kit (Qiagen) as described in 2.2. Each tissue was cut in half and each half extracted separately using the two different methods. The resulting neat and tenfold diluted DNA from both methods was amplified in duplicate following a protocol adapted from Renault and Arzul [36] using the C2: 5’ – CTCTTTACCATGAAGATACCCACC – 3’ and C6: 5’ – GTGCACGGCTTACCATTTTT – 3’ primer pair. Twenty-five microlitre PCR reactions were prepared to achieve the following concentrations of reagents: 1 x GoTaq® Hot Start Polymerase buffer (Promega), 2.5mM MgCl2, 0.5 mM dNTPs, 10 µM C2 forward primer, 10 µM C6 reverse primer, 9.3 µL of molecular grade water and 5 units of GoTaq® Hot Start Polymerase to which was added 2 µL of DNA. Thermocycling conditions used an initial denaturation at 95 °C for 3 min followed by 35 cycles of 95 °C for 30 s, 58 °C for 45 s and 72 °C for 45 s followed by a final extension of 72 °C for 10 min.
DNA was extracted from a pool of gill, digestive gland and mantle tissues preserved in absolute ethanol from twelve European flat oysters Ostrea edulis infected with Bonamia ostreae using the QEDES as described above. From another set of the same tissues and specimens, DNA was extracted using the EZ1 & 2 DNA Tissue Kit. (QIAGEN). Briefly, 100 mg of tissue were grinded in a FastPrep-24 Classic for 1 min at 6.5 m.sec-1 in 1 mL of G2 buffer and 0.2 mg.mL-1 of proteinase K. The tissues were incubated overnight at 56 °C, centrifuged at 7800 x g for 2 min and 50 µL of the supernatant plus 150 µL of G2 buffer was transferred to new tube and subjected to automated DNA isolation on a EZ1 Advanced XL instrument (QIAGEN) using an EZ1 & 2 DNA Tissue Kit cartridge. Using DNA extracted with EZ1 & 2 DNA Tissue Kit, B. ostreae DNA was detected by real time PCR (qPCR) following the SOP available on the EURL for Molluscs Diseases website [37]. The BO2_F primer (5’- AAATGGCCTCTTCCCAATCT -3’), BO2_R primer (5’- CCGATCAAACTAGGCTGGAA -3’), and the BO2 probe (5 ’- TGACGATCGGGAATGAACGC -3’; HEX-BHQ-1) were used. Amplification reactions were performed in duplicate in a total volume of 20 µL with 10 µL of TaqManTM Universal PCR Master Mix (No AmpErase, Applied Biosystems, Thermofisher), 0.6 µL of BO2_F (10 µM), 0.6 µL of BO2_R (10 µM), 0.4 µL of BO2 probe (10 µM), 6.2 µL of molecular biology water and 2.5 µL of DNA elution. Plates were run on a QuantStudio 3 Real Time qPCR system (Applied Biosystems, Thermofisher) with an initial denaturation of 3 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 20 s at 60 °C. Negative controls consisting of 2.5 µL of molecular biology grade water were added to each PCR plate. Moreover, the conventional PCR assay using the primer pair BOSTRE-F/BOSTRE-R developed by Ramilo et al [38] was used to amplify B. ostreae DNA extracted using the QEDES and EZ1 & 2 DNA Tissue Kit.
Statistical analysis to determine differences among DNA extraction methods for yield, 260/280, and 260/230 ratios were carried out using one-way ANOVA. Tukey HSD tests were then used for multiple group comparisons. Analyses were conducted using Python (version 3.11.11) with the following packages: pandas (version 2.2.2), numpy (version 1.26.4) and statsmodels (version 0.14.4). Permutation analyses were used to assess the success of CO1 PCR amplification using the R packages readr (2.1.5), dplyr (1.1.4), stringr (1.5.1) and coin (1.4.3). PCR amplification results were coded as binary variables, with 1 indicating successful amplification and 0 indicating failure. Independent permutation tests were conducted to evaluate the impact of dilution, tissue type, preservation method, and DNA extraction method. All permutation tests utilized 10,000 resampling iterations under a two-sided hypothesis framework.
3. Results
3.1. Spectrophotometric and Fluorometric Measurements
Overall, the highest DNA extraction yields were obtained using the QEDES (Table 1). This method produced results comparable to the reference method (QIAMP DNA mini kit) and even surpassed it when extracting DNA from fresh digestive gland tissue. The Kapa express extract kit also generated high DNA yields, in contrast to the QIAcard FTA Elute method and the Dipstick DNA extraction Kit, which consistently produced low yields.
The purity of DNA, determined by the absorbance ratio at wavelengths 260/280 nm for ten extraction methods tested, is shown in Table S1. The mean 260/280 ratio for DNA extracted using the control method ranged between 1.8 and 2.1 across various tissue types and preservation methods. Except for DNA extracted using the QIAcard FTA Elute and boiling methods from the mantle tissue, most 260/280 values obtained were not significantly different from the control method.
For all extraction methods including the control method the 260/230 values (Table S2) were consistently lower than 2.0 showing that all of the methods tested produce DNA that contain contaminating substances that absorb at 230 nm. The highest 260/230 values were observed in DNA extracted from mantle tissue using the control method (between 1.6 and 1.9). Besides the control method, the boiling also showed slightly higher values across the different tissues and preservation methods test. Most methods showed low variability between replicates as shown by the low coefficient of variation results with exception of the boiling method that showed consistently high coefficient of variation values indicating a larger amount of variability between replicates.
3.2. Amplification of M. gigas DNA
Permutation tests identified significant differences in COX1 PCR amplification success between tissue types (p < 0.001). Mantle tissue exhibited significantly higher success rates than digestive gland, consistent across preservation and extraction methods. Frozen tissues outperformed ethanol-preserved samples (p < 0.001) and no significant differences were observed between the other preservation methods (p > 0.05). Overall, no significant difference was observed between diluted and undiluted samples (p = 0.418), indicating that DNA dilution had no substantial effect on amplification outcomes under the tested conditions. However, for some methods such as the Hotshot DNA extraction kit, diluting 1:10 the DNA considerably improved PCR performance. The QIAMP DNA mini kit consistently achieved high amplification success and was used as the benchmark for post-hoc comparisons. All extraction methods tested performed significantly worse (p < 0.05) than QIAMP DNA mini kit, with exception of the QIAcard FTA Elute and the QEDES methods that showed no significant difference in performance with p-values of 0.497 and 0.054, respectively. Together, these results highlight that DNA extraction method, tissue type, and preservation strategy all strongly influence amplification success.
Overall, besides the control method, the three best methods for extracting DNA from both mantle and digestive gland tissue of M. gigas were the QIAcard FTA Elute method, the digestion/boiling method and the QEDES.

Table 2.
PCR COX1 amplification success out of 3 replicates (total of 360 individual reactions) using different extraction methods from mantle and digestive gland of M. gigas fresh, fixed in ethanol or frozen at –20 °C. Nondiluted and diluted DNA (1:10) were also tested.
Table 2.
PCR COX1 amplification success out of 3 replicates (total of 360 individual reactions) using different extraction methods from mantle and digestive gland of M. gigas fresh, fixed in ethanol or frozen at –20 °C. Nondiluted and diluted DNA (1:10) were also tested.
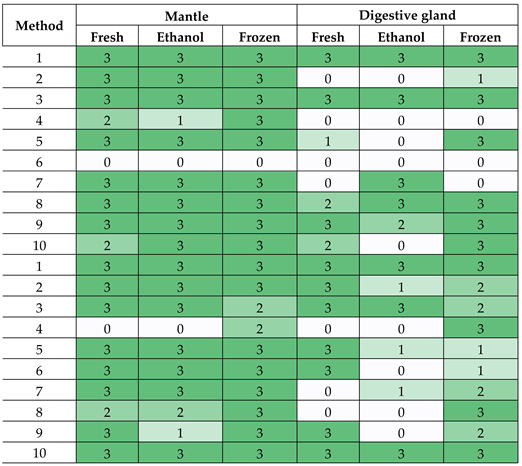 |
1 QIAmp DNA mini kit; (2) Buccalyse DNA Release Kit; (3) QIAcard FTA Elute method, (4) Whatman no 1 Filter paper method; (5) Kapa express extract kit; (6) Hotshot DNA extraction kit; (7) Dipstick DNA extraction Kit; (8) boiling method; (9) short digestion/boiling method; and (10) the QEDES.
3.3. Detection of Oyster Pathogens DNA
3.3.1. Case Study 1: Detection of Vibrio aestuarianus in M. gigas
DNA from M. gigas mantle tissue from three individuals infected with V. aestuarianus (Va) was extracted using the QEDES, the QIAcard FTA Elute method, the Short digestion/Boiling method [33] and the QIAmp DNA mini kit (control), and subsequently amplified using a Va qPCR method. DNA from V. aestuarianus was detected by qPCR in all samples extracted using the QEDES and the QIAmp DNA mini kit. However, a lower number of copies was observed using DNA extracted with QEDES (4454 copies/mg) in comparison with QIAmp DNA mini kit (8424 copies/mg).
Results from the Va qPCR showed that the control method was more efficient at amplifying V. aestuarianus from mantle tissue from all three M. gigas individuals used in the test as determined by the mean number of copies per mg tissue detected. (8424 genome copies/mg). The QEDES was the best in field extraction method tested with a mean value of 4454 copies per mg. The second best method was the QIAcard FTA Elute method obtaining mean copies per mg of 1556. The lowest copy number was obtained using the short digestion / Boiling method was the least good as this method was unable to amplify any V. aestuarianus DNA from sample D13 and extremely low copies from the other two individuals 10 +/- 14. All qPCR and extraction negatives run on the qPCR produced no CT values and all samples were run in duplicate.
3.3.2. Case Study 2: Detection of OsHV-1 in M. gigas
PCR products with the expected size (~ 709 bp) for the primer pair C2/C6 were observed for all samples tested using DNA extracted with both QIAMP DNA mini kit and the QEDES (Figure 1). Similar results were obtained using neat DNA or tenfold diluted DNA extracted using both methods.
3.3.3. Case Study 3: Detection of Bonamia ostreae in O. edulis
DNA from B. ostreae was detected in all twelve O. edulis samples using DNA extracted with both the QEDES and the EZ1 & 2 DNA Tissue Kit. The Cq values ranged from 25.7 to 37 for DNA extracted with the QEDES and from 25.4 to 38.8 for DNA extracted with the EZ1 & 2 DNA Tissue Kit. PCR products with the expected size (~208 bp) for B. ostreae were obtained using the BOSTRE-F/BOSTRE-R primer pair for all samples tested using DNA extracted with both methods (Figure 2).
3.4. Analysis of Throughput Time, Cost and Additional Equipment
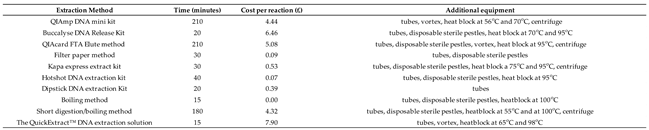
Comparison of the ten extraction methods for time, cost and additional equipment is reported in Table 3. The two fastest methods (taking only 15 minutes) were the QEDES and the boiling method. They also were the most expensive and cheapest options, respectively. The dipstick method and the filter paper method required the least amount of additional equipment. Seven of the methods require the use of a heat block in the field, a portable one such as the MyBlock™ mini dry bath (Merck) and a vehicle power adapter could be used. Two of the methods (the Kapa express extract and short digestion boiling) required the use of centrifuges. A portable option could be the Multi Spin Battery-Powered Mini Centrifuge (TOMY). The QIAcard FTA elute also requires the use of a centrifuge but only during the extraction step which can be done in a lab. The homogenisation step (where the homogenised tissue is fixed onto a paper matrix) is completed in the field.
4. Discussion
This study evaluated the efficacy of nine rapid DNA extraction methods in comparison with a conventional reference method. The performances were assessed on digestive gland and mantle of M. gigas and with different storage processes, specifically fresh, frozen and ethanol fixed. Performance was assessed by assessing yield, purity and efficacy of PCR for both host and pathogen DNA. Although several assays performed well in certain conditions, the QuickExtract™ DNA extraction solution (QEDES) overall was found to be the most reliable method.
This was evidenced by the QEDES providing the consistently high yields and in particular for fresh digestive gland material, even surpassing the reference method. This is particularly relevant as analysis in the field is likely to be performed on fresh material. Furthermore, it generated superior purity DNA extracts, with 260/280 results comparable to the control method and elevated 260/230 ratios, indicating low carry over of contaminants.
PCR amplification of COX1 genes again revealed that QEDES provided good results with the exception of the undiluted digestive gland extract fixed with ethanol. However, this inhibition was completely removed in the diluted sample, highlighting the importance of dilution in onward application. This experiment also highlighted that the mantle provides better DNA extract compared to the digestive gland and frozen tissue better than ethanol-preserved samples. These findings may be explained by the high concentration of inhibitory substances in the digestive gland compared to mantle, residual ethanol inhibiting PCR performance [39] and/or the freeze-thaw cycle providing some cell lysis [40].
Experiments assessing pathogen detection by qPCR showed that QEDES were also notably effective for pathogen detection in oyster tissues, facilitating detection of Vibrio aestuarianus, Ostreid herpesvirus 1 (OsHV-1), and Bonamia ostreae by both conventional and real-time PCR. The method provided comparable copy number recovery when compared to the reference method. Finally although QEDES is the most expensive per reaction it is also very quick to perform taking only 15 minutes.
Previous studies have also highlighted the efficacy of the QEDES being used by Kotov & Taylor [41] and Walthall, Tice & Brown [42] to identify a new Daphinia obtusa lineage and a new species of Amoeboza respectively. Cano et al [14] also found that QEDES was the best method when compared to four other extraction methods for the in-field detection of Neoparamoeba perurans the causative agent of Amoebic gill disease (AGD) in Atlantic salmon gill swabs. Cano et al [15] further applied QEDES for the detection of Cyprinid herpesvirus (CyHV-3) and carp edema virus (CEV), the causative agents of koi herpesvirus disease and koi sleepy disease, respectively. QEDES has also been combined with fluorescence real-time loop-mediated isothermal amplification (LAMP) assays in order to detect these pathogens in the field and determined that the method worked extremely well [15].
In this study the assessment of the COX1 amplification by PCR determined that the Buccalyse DNA Release Kit, the Whatman no 1 filter paper method, the Hotshot DNA extraction kit (Bento lab) and the boiling method [33] extracts caused considerable amounts of inhibition when compared to the control method. It was for this reason these methods were not used in further in our tests, allowing us to focus on the best performing candidates. It is important to mention that the level of inhibition seen on the tests we rejected was not uniform. For example the boiling method [33] showed large amounts of inhibition when digestive gland was used but if the tissue is frozen prior to extraction and used neat rather than diluted then no inhibition was observed. Moreover many of these methods showing a low performance in the present study have previously been proven to work well on different host tissues underlining the fact that the extraction method that may be best in one scenario may not be the best when using a different, host, tissue or pathogen and the type of amplification and detection method also needs to be compatible.
In conclusion, the QuickExtract™ DNA Extraction Solution (QEDES) demonstrated superior performance in terms of DNA yield, purity, and PCR amplification success compared to other rapid DNA extraction methods. Although the most expensive of all the assays tested, its efficacy in detecting pathogens in Pacific oysters and European flat oysters underscores its potential for point-of-need use and justifies the additional cost. Future studies should explore the use of this method for detecting other pathogens in different aquatic species and evaluate its performance under various field conditions.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Mean 260/280 ratio (coefficient of variation) of DNA extracted using in-field methods from mantle and digestive gland of M. gigas fresh, fixed in ethanol or frozen at – 20 °C. Table S2: Mean 260/230 ratio (coefficient of variation) of DNA extracted using in-field methods from mantle and digestive gland of M. gigas fresh, fixed in ethanol or frozen at – 20 °C.
Author Contributions
Conceptualization, F.B. and R.H; formal analysis, F.B and RK; investigation, R.K and M.E; resources, F.B and R.H; data curation, R.K.; writing—original draft preparation, R.K.; writing—review and editing, F.B, R.K and R.H.; visualization, F.B and R.K; supervision, F.B.; funding acquisition, F.B and R.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Seedcorn project DP445 and DEFRA project FC1216.
Institutional Review Board Statement
Not applicable the study used bivalve molluscs.
Data Availability Statement
Data is contained within the article or supplementary material. The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Acknowledgments
The authors would like to thank Adele Cobb for providing laboratory assistance.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| The QuickExtract™ DNA extraction solution | (QEDES) |
| Ostreid herpesvirus 1 microvar | (OsHV-1 µvar) |
| Ostreid herpesvirus 1 | (OsHV-1) |
| cytochrome oxidase subunit 1 gene | (COX1) |
| Recombinase Polymerase Amplification | (RPA) |
| Oxford Nanopore Technologies | (ONT) |
| Vibrio aestuarianus | (Va) |
| Amoebic Gill Disease | (AGD) |
| carp edema virus | (CEV) |
| Cyprinid herpesvirus | (CyHV-3) |
| loop-mediated isothermal amplification | (LAMP) |
References
- FAO. (2018). The state of world fisheries and aquaculture 2018: meeting the sustainable development goals. Food and Agriculture Organization of the United Nations.
- Feil, E.J.; Geary, M.; Waine, A.; Ryder, D.; Coyle, N.M.; Cheslett, D.; Thomas, J.C.L.; Bean, T.P.; Joseph, A.W.; Verner-Jeffreys, D.W.; et al. Vibrio aestuarianus clade A and clade B isolates are associated with Pacific oyster (Magallana gigas) disease outbreaks across Ireland. Microb. Genom. 2023, 9, 001078. [Google Scholar] [CrossRef]
- Bushek, D.; Carnegie, R.B.; Arzul, I. Managing marine mollusc diseases in the context of regional and international commerce: policy issues and emerging concerns. Philos. Trans. R. Soc. B: Biol. Sci. 2016, 371, 20150215. [Google Scholar] [CrossRef]
- Nozal, D.F.; Arzul, I.; Carrasco, N.; Rodgers, C. A literature review as an aid to identify strategies for mitigating ostreid herpesvirus 1 in Crassostrea gigas hatchery and nursery systems. Rev. Aquac. 2018, 11, 565–585. [Google Scholar] [CrossRef]
- Raftos, D.; Allam, B. Immune responses to infectious diseases in bivalves. J. Invertebr. Pathol. 2015, 131, 121–136. [Google Scholar] [CrossRef]
- Novoa, B.; Renault, T.; Roch, P.; Venier, P.; Gestal, C.; Oubella, R.; Figueras, A.; Pallavicini, A.; Paillard, C. Study of Diseases and the Immune System of Bivalves Using Molecular Biology and Genomics. Rev. Fish. Sci. 2008, 16, 133–156. [Google Scholar] [CrossRef]
- Garcia, C., Lupo, C., Travers, M. A., Arzul, I., Tourbiez, D., Haffner, P., & others. (2014). Vibrio aestuarianus and Pacific oyster in France a review of 10 years of surveillance. National Shellfisheries Association 106th Annual Meeting, 33.
- Labreuche, Y.; Garnier, M.; Nicolas, J.-L. Molecular and phenotypic characterization of Vibrio aestuarianus subsp. francensis subsp. nov., a pathogen of the oyster Crassostrea gigas. Syst. Appl. Microbiol. 2008, 31, 358–365. [Google Scholar] [CrossRef]
- Canier, L.; Arzul, I.; Cheslett, D.; Garcia, C.; Geary, M.; Sicard, M.; Orozova, P.; Furones, D.; Tourbiez, D.; Garden, A.; et al. Emergence and clonal expansion of Vibrio aestuarianus lineages pathogenic for oysters in Europe. Mol. Ecol. 2023, 32, 2869–2883. [Google Scholar] [CrossRef]
- Tourbiez, D.; Parizadeh, L.; Haffner, P.; Kozic-Djellouli, A.; Aboubaker, M.; Koken, M.; Dégremont, L.; Travers, M.-A.; Lupo, C. Several strains, one disease: experimental investigation of Vibrio aestuarianus infection parameters in the Pacific oyster, Crassostrea gigas. Veter- Res. 2017, 48, 1–8. [Google Scholar] [CrossRef]
- EFSA Panel on Animal Health and welfare (AHAW) Scientific Opinion on the increased mortality events in Pacific oysters,Crassostrea gigas. EFSA J. 2010, 8. [CrossRef]
- Comps, M., Tige, G., & Grizel, H. (1980). Etude ultrastructurale d’un protiste parasite de l’hutre plate. Ostrea.
- Grizel, H., Comps, M., Bonami, J.-R., Cousserans, F., Duthoit, J.-L., & Le Pennec, M.-A. (1974). Recherche sur l’agent de la maladie de la glande digestive de Ostrea edulis Linné. Science et Pêche, 240, 7–30.
- Paley, R.; Joiner, C.; Mulhearn, B.; Gunning, S.; McCullough, R.; Waine, A.; Cano, I. Non-lethal loop-mediated isothermal amplification assay as a point-of-care diagnostics tool for Neoparamoeba perurans, the causative agent of amoebic gill disease. J. Fish Dis. 2020, 43, 779–790. [Google Scholar] [CrossRef]
- Savage, J.; Wood, G.; Mulhearn, B.; Worswick, J.; Cano, I.; Stone, D.; Paley, R. A Seasonal Study of Koi Herpesvirus and Koi Sleepy Disease Outbreaks in the United Kingdom in 2018 Using a Pond-Side Test. Animals 2021, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Zaczek-Moczydłowska, M.A.; Mohamed-Smith, L.; Furones, M.D.; Bean, T.P.; Hooper, C.; Campàs, M.; Toldrà, A. A Single-Tube HNB-Based Loop-Mediated Isothermal Amplification for the Robust Detection of the Ostreid herpesvirus 1. Int. J. Mol. Sci. 2020, 21, 6605. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Shi, Y.-H.; Li, C.-H.; Zhou, Q.-J.; Yan, X.-J.; Wang, R.-N.; Zhang, D.-M.; Wang, L.; Chen, J. Development and evaluation of a real-time fluorogenic loop-mediated isothermal amplification assay integrated on a microfluidic disc chip (on-chip LAMP) for rapid and simultaneous detection of ten pathogenic bacteria in aquatic animals. J. Microbiol. Methods 2014, 104, 26–35. [Google Scholar] [CrossRef]
- Taengphu, S.; Kawasaki, M.; Gan, H.M.; Kayansamruaj, P.; Senapin, S.; Barnes, A.; Wilkinson, S.; Mohan, C.V.; Delamare-Deboutteville, J.; Debnath, P.P.; et al. Rapid genotyping of tilapia lake virus (TiLV) using Nanopore sequencing. J. Fish Dis. 2021, 44, 1491–1502. [Google Scholar] [CrossRef]
- Falk, K.; Matejusova, I.; Nguyen, L.; Macqueen, D.J.; Gallagher, M.D.; Ruane, N.M. Nanopore sequencing for rapid diagnostics of salmonid RNA viruses. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Prachumwat, A.; Srisala, J.; Suebsing, R.; Kanitchinda, S.; Chaijarasphong, T. CRISPR-Cas fluorescent cleavage assay coupled with recombinase polymerase amplification for sensitive and specific detection of Enterocytozoon hepatopenaei. Biotechnol. Rep. 2020, 27, e00485. [Google Scholar] [CrossRef]
- Li, Y.; Liu, C.; Wang, Q.; Wang, Y.; Liu, X.; Ren, Y.; Zeng, W.; Wang, Y.; Yin, J.; Qu, Y. Development of a real-time recombinase polymerase amplification assay for rapid detection of Aeromonas hydrophila. J. Fish Dis. 2020, 44, 469–477. [Google Scholar] [CrossRef]
- Harold, G.; Adams, A.; Weidmann, M.; Lopez-Jimena, B.; Shahin, K.; Ramirez-Paredes, J.G. Development of a recombinase polymerase amplification assay for rapid detection of Francisella noatunensis subsp. orientalis. PLOS ONE 2018, 13, e0192979. [Google Scholar] [CrossRef]
- Canier, L.; Stone, D.; Arzul, I.; Cano, I.; Wood, G.; Noyer, M. Loop-Mediated Isothermal Amplification for the Fast Detection of Bonamia ostreae and Bonamia exitiosa in Flat Oysters. Pathogens 2024, 13, 132. [Google Scholar] [CrossRef]
- Hatfield, R.G.; Ryder, D.; Tidy, A.M.; Hartnell, D.M.; Dean, K.J.; Batista, F.M. Combining Nanopore Sequencing with Recombinase Polymerase Amplification Enables Identification of Dinoflagellates from the Alexandrium Genus, Providing a Rapid, Field Deployable Tool. Toxins 2023, 15, 372. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M.A.; Ali, N.; Rampazzo, R.d.C.P.; Costa, A.D.T. Current Nucleic Acid Extraction Methods and Their Implications to Point-of-Care Diagnostics. BioMed Res. Int. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Mahilum-Tapay, L.; Dineva, M.A.; Lee, H. Sample preparation: a challenge in the development of point-of-care nucleic acid-based assays for resource-limited settings. Anal. 2007, 132, 1193–1199. [Google Scholar] [CrossRef]
- Lau, H.Y.; Botella, J.R. Advanced DNA-Based Point-of-Care Diagnostic Methods for Plant Diseases Detection. Front. Plant Sci. 2017, 8, 2016. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, Y.; Li, J.; Hoffmann, M.R. Rapid Detection Methods for Bacterial Pathogens in Ambient Waters at the Point of Sample Collection: A Brief Review. Clin. Infect. Dis. 2020, 71, S84–S90. [Google Scholar] [CrossRef]
- Paul, R.; Wei, Q.; Ostermann, E. Advances in point-of-care nucleic acid extraction technologies for rapid diagnosis of human and plant diseases. Biosens. Bioelectron. 2020, 169, 112592–112592. [Google Scholar] [CrossRef]
- Tang, Y.; Yao, J.D. Bridging the divide: Harmonizing polarized clinical laboratory medicine practices. iLABMED 2024. [Google Scholar] [CrossRef]
- Adema, C.M. Sticky problems: extraction of nucleic acids from molluscs. Philos. Trans. R. Soc. B: Biol. Sci. 2021, 376, 20200162. [Google Scholar] [CrossRef]
- Renault, T.; Friedman, C.S.; Ruano, F.; Pepin, J.-F.; Arzul, I.; Boudry, P.; Batista, F.M. Detection of ostreid herpesvirus 1 DNA by PCR in bivalve molluscs: A critical review. J. Virol. Methods 2006, 139, 1–11. [Google Scholar] [CrossRef]
- Renault, T.; Boudry, P.; Batista, F.; Taris, N. Detection of ostreid herpesvirus-1 (OsHV-1) by PCR using a rapid and simple method of DNA extraction from oyster larvae. Dis. Aquat. Org. 2005, 64, 1–4. [Google Scholar] [CrossRef]
- Leray, M.; Ranwez, V.; Agudelo, N.; Yang, J.Y.; Meyer, C.P.; Mills, S.C.; Boehm, J.T.; Machida, R.J. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front. Zoöl. 2013, 10, 34–34. [Google Scholar] [CrossRef] [PubMed]
- Haffner, P.; De Decker, S.; Saulnier, D. Real-time PCR assay for rapid detection and quantification of Vibrio aestuarianus in oyster and seawater: A useful tool for epidemiologic studies. J. Microbiol. Methods 2009, 77, 191–197. [Google Scholar] [CrossRef]
- Renault; Arzul Herpes-like virus infections in hatchery-reared bivalve larvae in Europe: specific viral DNA detection by PCR. J. Fish Dis. 2001, 24, 161–167. [CrossRef]
- EURL for Molluscs Diseases website (EURL for Molluscs Diseases, 2nd edition, February 2023). Avaliable online: https://www.eurlmollusc.eu/content/download/137231/file/B.ostreae%26B.exitiosa%20_TaqmanRealTimePCR_editionN%C2%B02.pdf (Accessed on 20 January 2025).
- Villalba, A.; Abollo, E.; Navas, J.; Ramilo, A. Species-specific diagnostic assays for Bonamia ostreae and B. exitiosa in European flat oyster Ostrea edulis: conventional, real-time and multiplex PCR. Dis. Aquat. Org. 2013, 104, 149–161. [Google Scholar] [CrossRef]
- Shrader, C., Schielke, A., Ellerbroek, L & Johne, R. (2012). PCR inhibitors – occurrence, properties and removal. Journal of Applied Microbiology. Journal of applied microbiology, 113(5), 1014-1026. [CrossRef]
- Jamet, E. An eye-tracking study of cueing effects in multimedia learning. Comput. Human Behav. 2014, 32, 47–53. [Google Scholar] [CrossRef]
- Taylor, D.J.; Kotov, A.A. A new African lineage of the Daphnia obtusa group (Cladocera: Daphniidae) disrupts continental vicariance patterns. J. Plankton Res. 2010, 32, 937–949. [Google Scholar] [CrossRef]
- Walthall, A. C., Tice, A. K., & Brown, M. W. (2016). New species of flamella (Amoebozoa, variosea, gracilipodida) isolated from a freshwater pool in Southern Mississippi, USA. Acta Protozoologica, 55(2), 111–117. [CrossRef]
Figure 1.
Neat and 10 fold dilutions of DNA extracted using the QIAMP DNA mini kit and the QEDES amplified using an OsHV PCR. Six microlitres was run in duplicate on a 2 % TBE agarose gel stained with Greensafe alongside 100bp ladder, eight negative controls and two positive controls. Where lanes 1-6 show OsHV amplification from neat DNA from M. gigas mantle tissue extracted using the QIAmp DNA mini Kit: Lanes 7-12 show OsHV amplification from tenfold diluted DNA from QIAmp DNA mini Kit: Lanes 14-19 show OsHV amplification from neat DNA from the QEDES. Lanes 20-25 show OsHV amplification from DNA from QEDES diluted tenfold: Lane 36 and 37 is OsHV DNA amplified from a positive control sample. Lanes 27- 34 are negative controls. Ladder (100pb:L).
Figure 1.
Neat and 10 fold dilutions of DNA extracted using the QIAMP DNA mini kit and the QEDES amplified using an OsHV PCR. Six microlitres was run in duplicate on a 2 % TBE agarose gel stained with Greensafe alongside 100bp ladder, eight negative controls and two positive controls. Where lanes 1-6 show OsHV amplification from neat DNA from M. gigas mantle tissue extracted using the QIAmp DNA mini Kit: Lanes 7-12 show OsHV amplification from tenfold diluted DNA from QIAmp DNA mini Kit: Lanes 14-19 show OsHV amplification from neat DNA from the QEDES. Lanes 20-25 show OsHV amplification from DNA from QEDES diluted tenfold: Lane 36 and 37 is OsHV DNA amplified from a positive control sample. Lanes 27- 34 are negative controls. Ladder (100pb:L).
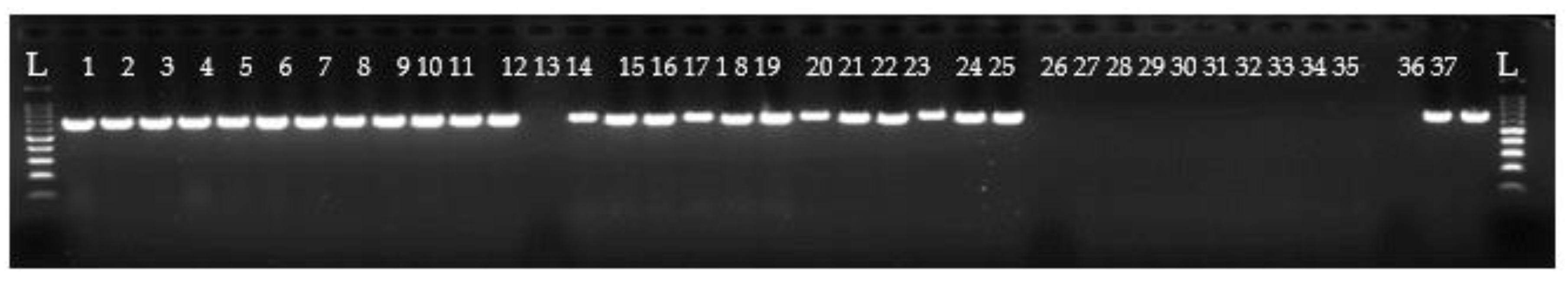
Figure 2.
PCR products obtained with the BOSTRE-F/BOSTRE-R primer pair using DNA extracted with the QEDES from twelve O. edulis infected with B. ostreae (Lanes 1-24 in duplicate). Lane P, positive control; Lane N, negative control; Lanes L, DNA ladder (100 bp ladder, Promega).
Figure 2.
PCR products obtained with the BOSTRE-F/BOSTRE-R primer pair using DNA extracted with the QEDES from twelve O. edulis infected with B. ostreae (Lanes 1-24 in duplicate). Lane P, positive control; Lane N, negative control; Lanes L, DNA ladder (100 bp ladder, Promega).
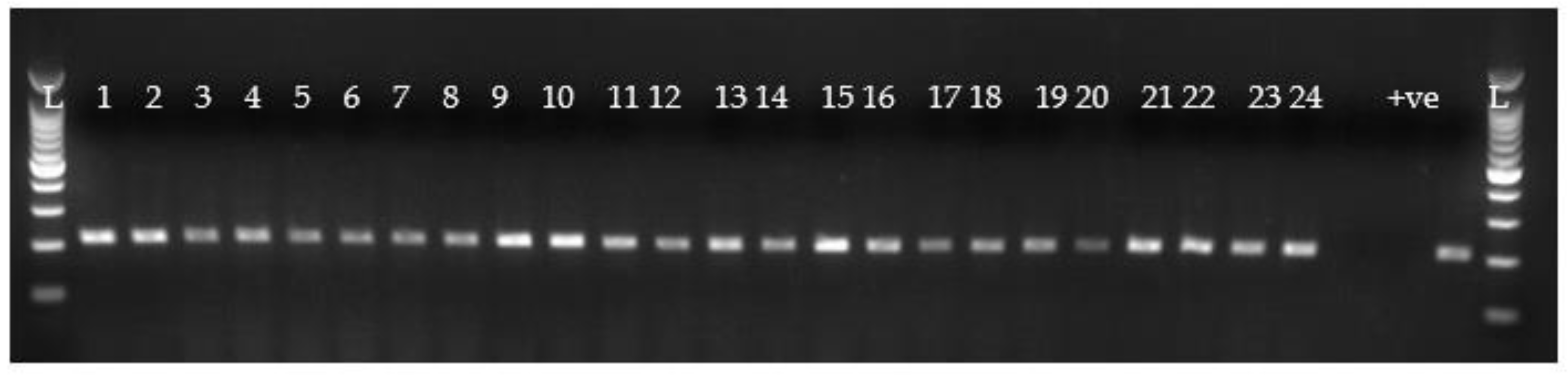
Table 1.
Fluorometer derived mean DNA extraction yields (ng/mg) with coefficient of variation values in parentheses obtained for different DNA extraction methods from mantle and digestive gland of M. gigas fresh, fixed in ethanol or frozen at – 20 °C. Significant statistical differences in each column are shown by different letters for each method.
Table 1.
Fluorometer derived mean DNA extraction yields (ng/mg) with coefficient of variation values in parentheses obtained for different DNA extraction methods from mantle and digestive gland of M. gigas fresh, fixed in ethanol or frozen at – 20 °C. Significant statistical differences in each column are shown by different letters for each method.
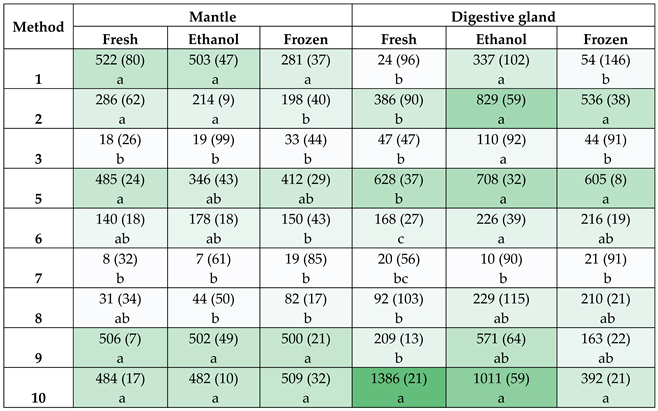 |
1QIAmp DNA mini kit; (2) Buccalyse DNA Release Kit; (3) QIAcard FTA Elute method, (4) Whatman no 1 Filter paper method; (5) Kapa express extract kit; (6) Hotshot DNA extraction kit; (7) Dipstick DNA extraction Kit; (8) boiling method; (9) short digestion/boiling method; and (10) The QuickExtract™ DNA extraction solution (QEDES).
Table 3.
Assessment of consumables, process duration, cost per reaction and additional equipment.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



