Submitted:
26 May 2025
Posted:
27 May 2025
You are already at the latest version
Abstract
In vitro results and murine experiments consistently demonstrate the cytotoxic effect of vitamin C (AA) on cancer cells, but current clinical evidence for high-dose i.v. AA’s (HDVC) therapeutic effect is ambivalent. The discrepancy may be due to the lack of knowledge regarding the AA's mode of action.
Recently, we introduced a novel hypothesis suggesting an alternative synthesis mechanism of adenosine triphosphate (ATP) involving specific cellular structures: Structure for Energy Transformation. Fe-S clusters, AA, ATP, D-glucose, uric acid, NO, and H2PO4- molecules are involved in the glucose – ATP energy transformation process.
We suppose that AA is responsible for the continuous electron transfer in cells. AA initiates the energy transformation in the multiplex electron transfer chains, where O2- radicals are formed for carbon oxidation. These radicals destroy cancer cells when glucose is unavailable in the reaction. Using supportive high-dose vitamin C therapy might increase the effectiveness of immunotherapy for cancer.
Keywords:
cancer
; immunotherapy
; ascorbic acid
; ATP synthesis
; Fe-S clusters
; cell energetics
; electron transfer chain
1. Development of Cancer
The cells of the living kingdom provide continuous oxygenation. In humans and vertebrates, this is achieved through respiration and blood flow. Cells die if they are deprived of oxygen. However, it is usually possible to restore the original state if the damage does not affect the whole body, but only part of it is lost due to injury/illness. The hypoxia-induced system controls the regeneration. It switches cell metabolism to the ancient energy system using aerobic glycolysis as described by Wartburg. Normally this system works very well. The oxygen-sensitive Hypoxia-Induced Factor (HIF) system converts the tissues to aerobic glycolysis metabolism, which develops new blood vessels, cleanses dead tissue, and restores the original state. In the case of hypoxia, the HIF system leads to a modulation of about 200 genes. The most important changes are: the induction of neovascularisation, induction of pluripotent cells, increased adhesion molecule expression and reduced sensitivity to apoptotic signals.
Cancer is a disease of tissue growth regulation. The genes that regulate cell growth and differentiation must be altered for a normal cell to become a cancer cell. Typically, changes in several genes are required to turn a normal cell into a cancer cell. Several mutations cause cancer.
Once cancer has started, this ongoing process, called clonal evolution, drives progression to more invasive stages. During tumour formation, the rapidly growing tumour cells lack sufficient blood vessels, and the limited capillary support often leads to hypoxia within the tumour cells. At this point, the failure to hydrolyse HIF-1α triggers malignant cells to become more malignant. A new type of tumour cell has been developed that adapts to the lack of oxygen. HIF-1α enables neovascularisation and tumour growth. By inducing adhesion molecules (which allow the destruction of dead tissue in wound healing), it is possible to develop the metastatic property of tumour cells. HIF-1α will lead to a specific Darwinian selection and will become the conductor of suicide evolution. Tumour progression is also favoured by the induction of pluripotency and reduced sensitivity to apoptosis.
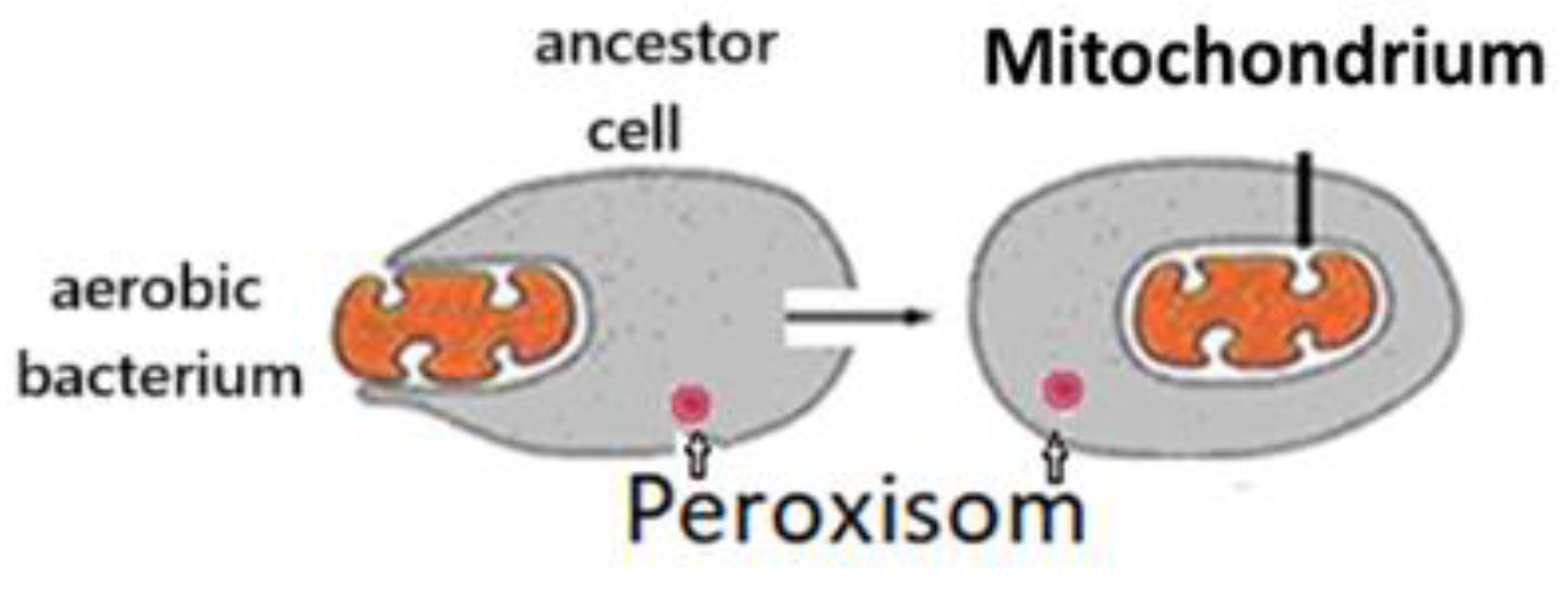
During the evolution of the living world, the development of the structure for energy conversion was one of the most critical determinants. Combining a cell living without O2 with a cell using O2 resulted in a qualitative advance. Lynn Margulis (1938-2011) suggested that the ancestor of eukaryotic cells avoided being destroyed by oxygen by entering into a symbiotic relationship with aerobic bacterium. This gave rise to the mitochondria of eukaryotes (Figure 1) [1]. Thus, eukaryotes have genetic material from both cells and can live in hypoxic and normoxic environments. All eukaryotic cells have mitochondria. Mitochondrial Pyruvate dehydrogenase results in an increased energy supply and effective protection against Reactive Oxygen Species (ROS).
During energy conversion, adenosine triphosphate (ATP) is formed from glucose. According to our hypothesis, the energy conversion is mediated by unique structures, the Structure of Energy Transformation for Aerobic Glycolysis (SET-AG) of the ancestor cell and the Structure of Energy Transformation for Oxidative Phosphorylation (SET-OP) of the mitochondrion. SET-AG is located in the peroxisomes. Understanding their function could be an important step towards optimising tumour therapy by glucose deprivation or intravenous vitamin C therapy.
Figure 1.
Symbiosis of ancestor cell with an aerobic cell.
Cancer is a group of diseases in which abnormal cell growth, invades and spreads to other parts of the body. There are more than 100 types of cancer, usually named after the organs or tissues in which the tumours form [2].
Approximately 90-95% of cancers are caused by genetic mutations due to environmental and lifestyle factors. The remaining 5-10% are inherited [3]. Common environmental factors contributing to cancer include diet and obesity (30-35%), tobacco (25-30%), infections (15-20%), radiation (both ionising and non-ionising, up to 10%), stress, physical inactivity and pollution [3,4]. Cancer is a disease of tissue growth regulation. Therefore, the genes that regulate cell growth and differentiation must be altered [5]. The genes involved are oncogenes (genes that promote cell growth and reproduction) or tumour suppressor genes (genes that inhibit cell division and survival). Malignant transformation can occur through the formation of new oncogenes, the inappropriate overexpression of normal oncogenes, or the underexpression or silencing of tumour suppressor genes. Typically, changes in several genes are required to transform a normal cell into a cancer cell. Thus, a series of mutations causes cancer. In addition, each mutation slightly alters the behaviour of the cell [6].
Replication of the information contained in the DNA of living cells can lead to mutations. Normal metabolic activity and environmental factors such as radiation can cause DNA damage in cells, resulting in up to 1 million individual molecular lesions per cell per day [7]. Complex error correction and prevention are built into the cell’s defences against cancer. If a significant error occurs, the damaged cell can self-destruct through programmed cell death (apoptosis). If the error control processes fail, the mutated cell will survive and be passed on to daughter cells.
Some environments make it more likely that errors will occur and cancer will develop. Such environments may include disruptive substances called carcinogens, repeated physical injury, heat, ionising radiation or hypoxia. In this way, the errors that cause cancer are self-reinforcing and cumulative.
1.1. Protective options against malignant tumours
Living organisms are constantly defending themselves against harmful environmental factors. Specialised systems control both invaders and abnormally functioning cells. In animals (and humans), immune cells can usually recognise and destroy abnormal cells. Unfortunately, there are many situations in which malignant cells escape the control of the immune system and begin to multiply uncontrollably, leading to death.
Treatment options for malignant tumours are becoming more widespread. While surgery is the definitive treatment for early-stage tumours, the current therapies against advanced cancers do not yet provide effective, long-lasting control of the lesions and a satisfactory impact on patient survival. Thus, research is also focused on novel treatments that could potentiate the current therapies.
1.2. Controlling apoptosis
Apoptosis is the process of programmed cell death that can occur in multicellular organisms. Between 50 and 70 billion cells die by apoptosis every day in the average human adult. Binding of nuclear receptors by glucocorticoids, heat, nutrient deprivation, radiation, viral infection and hypoxia can trigger the release of intracellular apoptotic signals. Many cellular components, such as poly-ADP-ribose polymerase, can also facilitate the regulation of apoptosis [8].
1.3. The chain reaction of cancer development
The transformation of a normal cell into cancer is a chain reaction caused by initial mistakes that compound into more serious mistakes, each of which allows the cell to escape more and more of the controls that regulate the growth of healthy tissue. This rebellious scenario, suicidal evolution, is an undesirable survival of the fittest tumour cell in which the driving forces of evolution work against the body’s design and enforcement of order. Once cancer has begun to develop, this ongoing process, known as clonal evolution, can lead to more invasive stages that result in death.
Clonal evolution leads to intra-tumour heterogeneity. The genetic properties and metabolism (oxidative phosphorylation or aerobic glycolysis) of tumour cells are different. This heterogeneity makes it difficult to develop effective treatment strategies.
The first step in the development of a malignant tumour is the appearance of a mutant cell. The second problem is that the error control mechanism is altered. The mutant cell is not eliminated. The multiplying mutant cells are the precursor cells of the tumour (T1), using SET OP (Figure 2). When the distance between the T1 containing growing tumour cells and the supplying blood vessel exceeds 70 nm [9], the tissue partial pressure of O2 (pO2) is not sufficient for SET OP to function properly. At the same time, HIF-1 α will not be hydrolysed, resulting in the further development of the tumour, cells capable of living in an anoxic environment (tumour stem cells) will be created (T2). The system of HIF will lead to the further development of the tumour, creating tumour cells with new behaviours (T3, T4, Tn). There is competition between these cells. The majority of cancers are the most malignant cells.
Malignant tumours regularly send cells into the body through lymph and blood vessels. A cell’s ability to metastasise depends on its surface properties, which are influenced by unique adhesion molecules. As the tumour evolves, it may develop cells capable of forming metastases.
The HIF system initiates the induction of pluripotent cells, the expression of adhesion molecules and the installation of neovascularisation and reduced sensitivity to apoptotic signals. Thus, the HIF system is the conductor of tumour cells on the road to death (Figure 2).
1.4. Vitamin C and Cancer
There is a large body of literature on the treatment of cancer with sugar withdrawal or vitamin C treatment. Despite several decades of research, their clinical efficacy has not been proven. The importance of this topic is reflected in the large number of clinical trials, both completed and ongoing. According to the literature, sugar deprivation and pharmacological doses of vitamin C kill tumour cells through reactive oxygen species (ROS). Both treatments, in vitro and animal experiments reproducibly destroy many tumour cells and tumours. However, the results of clinical trials are contradictory [10,11,12,13,14,15,16].
2. The Supposed Way of Action Regarding Vitamin C Therapy
The earliest experience of using high-dose vitamin C (intravenous [i.v.] and oral) for cancer treatment was published by Cameron, and Campbell, in the 1970s [17]. Later, Cameron and Pauling detected prolonged survival times in terminal human cancer by supplemental ascorbate in the supportive treatment of cancer [18,19]. In contrast, in the late 1970s and early 1980s, Creagan, Moertel, and O’Fallon, et al. published the failure of High-dose Ascorbic Acid Therapy (HAAT) to benefit patients with advanced cancer [20,21,22].
The serum level of AA is significantly lower after a high oral AA dose than after a high dose of i.v. AA [23]. This difference explains the different results observed by Creagan and Cameron, as Creagan/Moertel used oral [20,21], while Cameron/Pauling used oral + parenteral AA [17,18,19]. Although Cameron’s treatment was mainly oral therapy, they used i.v. AA as an initiation to the use of oral AA.
Pharmacologic ascorbate sensitizes cancer cells to damage by increasing intracellular L-ascorbate engagement through sodium-dependent vitamin C transporter 2 (SVCT-2) acting as a pro-drug [24]. HAAT is an aggressive adjunctive cancer treatment, widely used in naturopathic and integrative oncology settings, although it is not accepted as an effective drug for cancer patients by health authorities (FDA, EMA).
Until today, many publications have been available regarding the HAAT of malignant tumours. However, in vitro and animal experiments have proved AA’s pharmacological cytotoxic effect on cancer cells, while human clinical observations and studies show contradicting data. Thus, the Pauling–Moertel debate continues to this day.
2.1. The Biochemical Motor for Energy Transformation
Previously, we described a hypothetical structure, the SET, which might be responsible for the proper energy transformation, leading to the continuous membrane potential, production of H+, and ATP in living cells [25]. We suppose that the SET of aerobe glycolysis (SET-AG) is responsible for managing aerobic glycolysis, while the SET of oxidative phosphorylation (SET-OP) for the process of oxidative phosphorylation by Pyruvate dehydrogenase. The multiplex electron transfer chain (METC) is the basic structure of both SETs. The METC consists of 2 electron transfer chains (ETC)s, containing 2 X ten Fe-S clusters and 2 X four L-AA [25].
2.2. Fe-S clusters, Cysteine
2.2.1. Fe-S clusters
Many Fe-S clusters are known in organometallic chemistry as precursors to synthetic analogues of the biological clusters. In the simplest polymetallic system, the [Fe2S2] cluster comprises two iron ions bridged by two sulfide ions and coordinated by four cysteine ligands (in Fe2S2 ferredoxins).
The most abundant Fe-S clusters are of the rhombic [Fe2-S2] and cubic [Fe4-S4] types, but [Fe3–S4], [Mo-Fe3–S4], [Fe4–S3] and [Fe8-S7] clusters of nitrogenase have also been described.
2.2.2. Cystein
Cysteine (Cys) is a semi-essential proteinogenic amino acid with the formula HOOC−CH(−NH2)−CH2−SH (Figure 3). Cys is a deterministic part of all Fe-S clusters, as it provides the continuity of the electron transfer in the METC, (Figure 3). We suppose the 2 x 10 Fe-S clusters create a continuous electron chain by connecting through their cys parts. Two cys of two Fe-S clusters can create continuity between clusters as presented in Figure 3. The Nitrogen-Oxygen connection results in N-C bound + H2O while one C-NH2 and one C=O will remain.
3. The Continuous Electron Transfer
3.1. The Structure of Energy Transformation
The condition for life is a continuous flow of electrons ensured by a complex electron transfer chain.
The Fe-S clusters are connected by cys-S bounds, forming one METC, where the energy transformation is completed. We suppose that the METC contains two ETCs. One ETC has four [Fe8-S7], four [Fe2-S2], one [Fe4-S4], and one molybdenum iron sulfide [MoFe3-S4] cluster (Figure 4).
The connection of the two ETCs results in electron transfer mediated by 8 AA. (Figure 5).
Each ETC produces 32 PO33-s, 64 H+, 24 CO2, and 4 ATP (Table I).

Table I. Results of structures of energy transformations in the electron transfer chain
| Fe-S clusters | Results of the structures
|
|||||||||
| NH2-UA | adenine | Pi | H+ | CO2 | ribose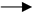
|
acetic acid | Pyru-vate | new ADP | ATP | |
| 4 Fe8-S7 | 4 | 4 | 8 | 16 | 12 | 4 | 4 | 4 | 4 | new 4 |
| 4 Fe2-S2 | 16 | 32 | 8 | ADP-ATP 20 |
||||||
| 1 Fe4-S4 | 4 | 8 | 2 | |||||||
| 1 MoFe3-S4 | 4 | 8 | 2 | |||||||
| ETC | 4 | 32 | 64 | 24 | 4 | 4 | 4 | 4 | 24 | |
Abbreviation: NH2-UA: nitrified uric acid; Pi: PO33-
3.2. Oxygen binding points of the multiplex electron transfer chain.
The [Fe8S7(C3H7NO2S)6]2- clusters obtain six cys parts. It thus offers places for six oxygen-containing molecules. The cluster bounds one UA by the C6-positioned oxygen, two aminated UA by the C2 or C8-positioned oxygen, two H2PO4- and one NO molecule by the double-bonded oxygen of the molecules (Figure 6).
Oxygen-containing molecules will replace the sulphur atoms after the ETC is initiated. The ETC has 48 cys structures. The four Fe8-S7 clusters contain 6x4=24, the four [Fe2-S2], the [MoFe3-S4], and the [Fe4-S4] clusters contain 6x4=24 cys, providing 48 oxygen binding points (Table II).
Four aminated uric acid molecules connected to nicotinamide and Flavine molecules link the four [Fe8S7 (SCH2CH3)6]e2- clusters of nitrogenase (Illustration 7). The four NO, four UA, eight D-glucose four F2S2, the F3S4, the Fe4S4 clusters and the 32 H2PO4- molecules are not presented.
 |
3.3. Continuous electron flow and ATP production, the two steps of ATP production
The energy conversion process runs in the ETC. The first step produces aminated uric acid, PO33-, H+, CO2, and ADP molecules. Finally, ATP synthase produces ATP from ADP + PO33-[24].
3.3.1. ATP-uric acid-ATP cycle
The view that UA is the end-product of ATP metabolism is widely accepted. Gounaris et al. Described that the hypoxanthine molecule could evolve as an effective capture of inorganic nitrogen species. These authors have also reported that hypoxanthine, the biochemical precursor of adenine and guanine, trapped nitrite ions and was reductively converted to adenine [25]
We predict that UA is not an end-product but one element of an ATP – UA – NH2-UA– adenosine – ADP – ATP - UA cycle.
3.4. The synchronised activity of the neighbour ETCs.
The neighbour ETCs (ETC1 - ETC2) work synchronised. They mutually activate each other, resulting in continuous electron transfer (Table III). First, 20 ATP, and 3 AA initiates the ETC1 resulting in 4 UA, 4 aminated UA, 4 NO, 36 H2PO4 and 8 Glucose arrival. The electron flow consequences the energy transformation, resulting in 4 ADP, 24 PO33-, 4 aminated UA, 4 Pyruvate and 4 acetic acid molecules. At the same time, the neighbour ETC2 will be activated, where the same reaction takes place, followed by the activation of ETC1 resulting in continuous electron flow (Table III; Figure 8).
 |
3.5. The three states of the electron transfer chains.
The energy transfer involves the active site of the two ETCs, cycling between three states.
Energy investment and initialisation.
First, AA and ATP molecules initiate the ETC1, wherein the “loose” state, UA, NH-UA, Glucose, and H2PO4, enter the active site; in Figure 8 this is shown as an empty ring.
At the same time, the energy of the initiating ATP molecule will help the transformation in the ETC2. The ETC1 then undergoes a change and the transformation of these molecules, with the active site (shown in red) preparing the newly produced ADP molecule.
Finally, the active site cycles back to the open state (brown), releasing ADP and being ready for the next cycle (Figure 9). The ADP and phosphate, are forming ATP in the ATP synthase.
3.6. Structure for Energy Transformation of Aerobe Glycolysis
Two ETCs and the ATP synthase enzymes might form the METC, the SET-AG, which produces 2 X [four ATP, 64 H+ and 24 CO2 (Table I). The 48 O2- reacts with 24 carbons, producing 24 CO2. The carbon atom of the CO2 originates from the eight carbon atoms of the eight glucose and eight acetic acid molecules. ATP synthetase and many other specific enzymes are responsible for the proper function of the METC.
ATP and AA are determinants of the continuity of electron transfer. The energy investment results in 4 new ATP + 20 ADP molecules. The ADP molecules will be transformed into ATP in the ATP synthase using the energy obtained from the oxidation of 24 carbon atoms before the transformation ends.
3.7. The Structure of Energy Transformation for Oxidative Phosphorylation
The SET-OP consists of one SET-AG and Pyruvate dehydrogenase enzyme, which is responsible for oxidative phosphorylation (Figure 9).
3.8. The energy source for new ATP production.
The ETC converts 8 D-glucose to 4 new ATP, 4 acetic acid and 4 Pyruvate molecules. Building new ATP requires 5 ATP’s energy (Figure 10). Twenty ATP are responsible for the 4 new ATP production, resulting in 20 ADP.
In the ETC, 20 ATPs start the reaction, producing 20 ADPs. During the transformation, 2 X 24 (SET-AG) or 2 X 36 (SET-OP) carbon atoms are oxidised, producing energy. This energy, together with the energy of the 20 ATP—ADP conversion, creates four new ATP molecules and regenerates the 20 ADP molecules.
3.9. Vitamin C and ATP are the activators and initiators of energy transformation.
Korth et al. published that vitamin C molecules are located in the pocket of NADPH, presumably at the adenine binding site of the inner mitochondrial membrane [27].
Kinga Linowiecka et al. stated that ascorbic acid (AA) is an oxidative stress sensor and a gene expression regulator. In addition, they pointed out that the change of AA to dehydroascorbic acid (DHA) regulates the modulation of the iron’s electron state in 2-oxoglutarate and Fe2+ dependent dioxygenases (2-OGDD) (Figure 11). Two H+ are liberated during the AA – DHA transformation. AA crucially increases the rate of the reaction of 2-OGGD by targeting its catalytic domain and regenerating iron ions from Fe3+ to Fe2+ [28].
This change might also be valid for the Fe atoms in the Fe-S clusters. The reaction results in a sulphur-oxygen exchange, creating four O2- in the Fe2-S2 cluster.
A similar reaction might occur when the two OHs of the ribose part of the ATP activate the Fe-S clusters.
Partially supported by this observation, we developed our hypothesis regarding the SETs. The four AAs and the 20 ATP in the ETC allow the conversion of Fe3+ to Fe2+, initiating the process leading to the production of ADP, PO33-, aminated uric acid and finally ATP.
Based on the publication by Kinga Linowiecka et al. [28], we assume that a continuous AA- dehydro-AA - AA conversion could cause a change of Fe ions (Fe3+ - Fe2+ - Fe3+) in the Fe-S clusters of ETCs, resulting in constant energy and ATP production and permanent maintenance of the membrane potential. Figure 12 illustrates the Fe3+ - FE2+ change in a Fe4-S4 cluster.
Table IV Activation molecules of the Fe-S clusters
| Fe-S Cluster | ATP | AA |
| [Fe8-S7] 1 | 3 | 1 |
| [Fe8-S7] 2 | 4 | |
| [Fe8-S7] 3 | 3 | 1 |
| [Fe8-S7] 4 | 4 | |
| [Fe2-S2] 1 | 1 | |
| [Fe2-S2] 2 | 1 | |
| [Fe2-S2] 3 | 1 | |
| [Fe2-S2] 4 | 1 | |
| [MoFe3-S4] | 1 | 1 |
| [Fe4-S4] | 1 | 1 |
| all | 20 | 4 |
Twenty ATP molecules activate the ETC, while four AAs initiate the reaction. Two OH of the ribose on the ATP bound to two Fe atoms of the Fe-S clusters, while the NH2 part of the ATP’s adenine will be bound to the C=O part of the connecting point. In that way, the 20 ATP molecules will bind 20 C=O of the connected cys. The C-NH parts of the connecting points bound 4 L-AA, 8 D-glucose and 8 acetic acid molecules.
3.10. The Proposed Way of Action by Parenteral AA on Sensitive Cancer Cells
Vitamin C enters the cell through SVCT-2 [29] and changes the intracellular glucose/AA ratio. If the ratio of glucose to vitamin C changes and the ratio of glucose in the cell decreases, this can lead to cell death. O2- is produced by sulphur-iron clusters at the start of the energy conversion process. If glucose is not available during the conversion, free radicals, which should produce CO2 (glucose + 2 O2- = ribose + CO2), multiply in the cell. The catalase system neutralizes them. When the catalase system is exhausted, the free radicals kill the cell [30]. Accordingly, several reports recommend the use of molecules that inhibit sugar transporters on the cell membrane [31]. In addition, the effect of intravenous (IV) vitamin C gives better results with a ketogenic diet [32].
The in vitro cytotoxic effect of ascorbic acid’s pharmacological concentration on cancer cells is mediated through a chemical reaction that generates H2O2 [33]. In addition, ascorbic acid induces reactive oxygen species and impaired mitochondrial membrane potential [34].
Our novel hypothesis regarding ATP synthesis via the Structure for Energy Transformation (SET) integrates known biochemical components. It remains a theoretical model that has not yet been empirically validated. The following limitations should be considered:
- The SET hypothesis has not been tested through direct biochemical assays or in vivo experiments.
- The role of Fe-S clusters, uric acid, and vitamin C in ATP synthesis within this framework is speculative and requires further experimental confirmation.
- Although the hypothesis suggests a potential link between vitamin C metabolism and cancer treatment, no clinical studies have directly tested this mechanism.
Future research should aim to experimentally validate the SET model and assess its physiological relevance before clinical applications are considered.
3.11. Increasing the efficacy of high ascorbic acid cancer treatment
Caspase inhibitors, sodium-dependent vitamin C transporters (SVCT), glucose transporters (GLUT), ribose, and 2-deoxy-d-glucose (2DG) might increase the efficacy of HAAT.
3.12. Caspase inhibitors
Caspases are an evolutionary conserved family of cysteine-dependent proteases that are actively involved in the execution of apoptosis. To date, fourteen mammalian caspases have been identified.
Multiple caspase inhibitors have been designed and synthesized as a potential therapeutic tool for the treatment of cell death-related pathologies. Their effect is based on the fact, that caspase helps the survival of sugar-deficient tumour cells [30].
3.12.1. Sodium-Dependent Vitamin C Transporter
Lv, H et al. demonstrated that L-ascorbate has a selective killing effect influenced by SVCT-2 in human hepatocellular cancer cells [29]. Furthermore, SVCT-2 expression was absent or weak in healthy tissues but strongly detected in tumour samples obtained from breast cancer patients, indicating that tumour cells contain elevated levels of AA [35].
3.13. Glucose Transporters
Numerous observations suggest that starvation, ketogenic diet, and glucose transporter inhibition have antitumor activity, but clinical studies to support this are lacking.
The glucose entrance inside the cell occurs by facilitated diffusion and mainly depends on GLUTs. There are three different classes of GLUTs with tissue-specific distribution and distinct affinity for glucose and other carbohydrates. Class 1 comprises four members, GLUT1–LUT4, whose preferential substrate is glucose, and ribose, while the other two classes, class 2 (GLUT5) and 3 (GLUT6, 8, 10), are more selective for other sugars [36].
GLUTs are expressed 10–12-fold higher in cancer cells than in healthy tissues, especially in highly proliferative and malignant tumours, contributing to the high glycolytic flux observed in this kind of tissue [33]. GLUT1 and GLUT3, whose expression is regulated by HIF-1α, can be considered the main over-expressed isoforms in a wide range of human cancers [37]. Moreover, they are correlated with poor prognosis and the radio-resistance of several types of human tumours. Hence, the activation of their expression can be considered a typical feature of the malignant phenotype. GLUT has a fundamental role in tumour cells of aerobic glycolysis. Thus, GLUT inhibition may represent an appealing way of attacking cancer by blocking its direct nutrient uptake, thus reducing the glycolytic flux and causing cell death by starvation and H2O2.
3.14. Treatment of Tumors by Influencing Glucose Metabolism
In most cancer cells, especially the most aggressive phenotypes, there is a substantial uncoupling of glycolysis from oxidative phosphorylation with the consequent production of high lactate levels (Warburg effect). This metabolic modification gives the tumour an evolutionary advantage, adapting to the local, more-or-less transient hypoxic conditions occurring during the disease’s progress. Furthermore, the remarkably higher glucose uptake also shows this metabolic preference by cancer cells through transmembrane glucose transporters, compensating for the higher energy demand of rapidly growing cells. Due to this feature, any enzyme or transporter promoting the glycolytic flux may be considered a potential target to block tumour progression [ 32, 33, 41].
Recently, human cancer cells were shown to be more susceptible to glucose deprivation-induced cytotoxicity and oxidative stress relative to non-transformed human cell types. These results indicated that some biochemical processes provided a mechanistic link between glucose metabolism and the expression of phenotypic characteristics associated with malignancy [42].
The most direct way to target exaggerated aerobic glycolysis in tumours is to reduce glucose availability to cancer cells. It can be achieved through either dietary or pharmacological interventions. The pharmacologic influence might be realized either by controlling glucose uptake of the tumour cell by inhibiting glucose transporters or by inhibiting glycolytic enzymes [ 33, 41].
3.14.1. Short-Term Fasting
Short-term fasting (STF) 48 to 72 hours before chemotherapy appears to be more effective than intermittent fasting. Preliminary data show that STF is safe but challenging in cancer patients receiving chemotherapy.
Clinical trials should explore whether STF can reduce toxicity and increase the efficacy of chemotherapy regimens in daily practice [ 41].
3.14.2. Ketogenic Diet
The ketogenic diet consists of high fat, moderate to low protein, and extremely low carbohydrates. The goal is to develop ketosis in the body. It occurs when the body is forced to use fat for energy without glucose [41].
3.14.3. Clinical Trials of Fasting
3.15. Ribose - on melanoma cell line
The killing effect of potassium ascorbate with ribose (PAR) treatment on the human melanoma cell line, A375, in 2D and 3D models was published by Cavicchio et al. [44]. In the 2D model, in line with the current literature, the pharmacological treatment with PAR decreased cell proliferation and viability. In addition, an increase in Connexin 43 mRNA and protein was observed. This novel finding was confirmed in PAR-treated melanoma cells cultured in 3D, where an increase in functional gap junctions and higher spheroid compactness was observed. Moreover, in the 3D model, a remarkable decrease in the size and volume of spheroids was observed, further supporting the treatment efficacy observed in the 2D model. In conclusion, these results suggest that PAR could be used as a safe adjuvant approach in support of conventional therapies for the treatment of melanoma.
3.15.1. Deoxy-D-Glucose
Increased pro-oxidant production and profound perturbations in thiol metabolism have been observed during glucose deprivation, suggesting that metabolic oxidative stress is induced [43]. In addition, co-treatment with a thiol antioxidant could suppress glucose-deprivation-induced cytotoxicity [45]. Lin et al. stated that 2DG is a potent inhibitor of glucose metabolism by mimicking glucose deprivation in vivo. When HeLa cells were exposed to 2DG (4-10 mM) for 4-72 h, cell survival decreased (20-90%) in a dose - and time-dependent fashion. When HeLa cells were treated with 6 mM 2DG for 16 h before ionizing radiation exposure, radio-sensitization was observed with a sensitiser enhancement ratio of 1.4 at 10% survival. Treatment with 2DG was also found to cause decreases in intracellular total glutathione content (50%) [43].
Simultaneous treatment with the thiol antioxidant N-acetylcysteine (NAC; 30 mM) protected HeLa cells against the cytotoxicity and radio-sensitizing effects of 2DG without altering radiosensitivity in the absence of 2DG. Furthermore, treatment with NAC partially reversed the 2DG-induced decreases in total glutathione content and augmented intracellular cysteine content.
These results support the hypothesis that exposure to 2DG causes cytotoxicity and radio-sensitization via a mechanism involving perturbations in thiol metabolism.
Lee et al. observed that glucose deprivation induces cell death in multidrug-resistant human breast carcinoma cells (MCF-7/ADR). Their results suggest that glucose deprivation-induced cytotoxicity and alterations in MAPK signal transduction are mediated by oxidative stress in MCF-7/ADR. These results also support the speculation that a common mechanism of glucose deprivation-induced cytotoxicity in mammalian cells may involve metabolic oxidative stress. [43].
3.15.2. Flavonoids
Some natural products belonging to the family of flavonoids, which are polyphenolic compounds widely found in plants, exert inhibitory effects on glucose transporters. For example, naringenin, a flavanone found mainly in grapefruit and shown to bind selectively to the estrogen receptor beta [46], has been reported to inhibit insulin-stimulated glucose uptake in breast cancer cells by interfering with insulin-induced GLUT4 translocation from intracellular compartments to the plasma membrane. A concentration of 100 μM naringenin inhibited glucose uptake by about 50% and cell proliferation by about 20% [47]. Other flavonoids such as myricetin, fisetin, quercetin and its glycoside analogue isoquercitrin have been shown to inhibit GLUT2, an isoform primarily found in the intestine, with potential antidiabetic/antiobesity effects [48]; however, no data are currently available on their effects on GLUT1 and GLUT3, which are the most exciting isoforms as anti-cancer targets.
3.16. Complex treatment of malignant tumours
Several mutations cause cancer. Normally, the immune system eliminates infected and cancerous cells. However, during this process, malignant cells stimulate the immune system. Tumour-derived peptides or proteins, together with costimulatory molecules and cytokines, are critical factors in the process of immune defence [49,50].
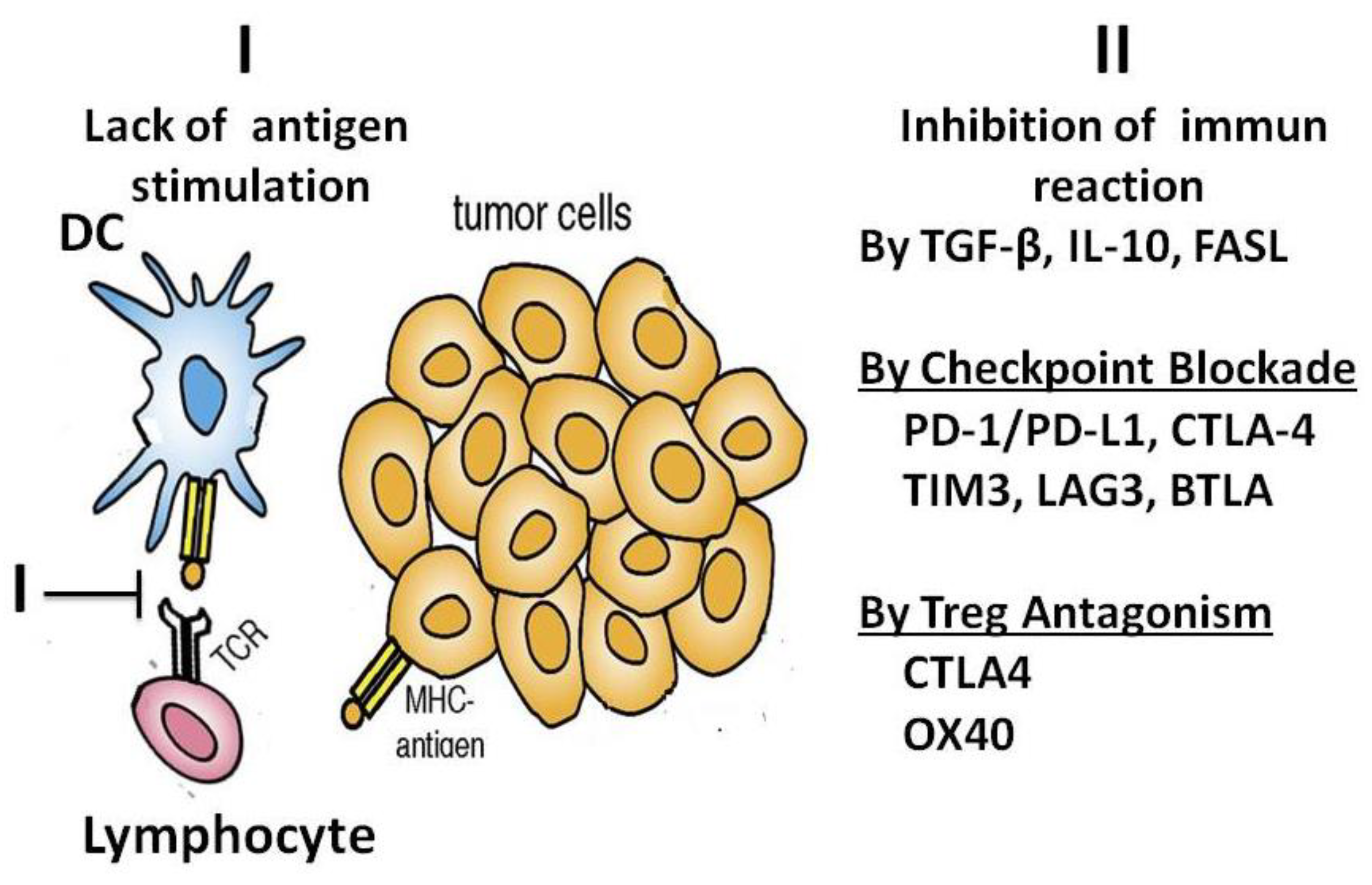
When immune defence is triggered, dendritic cells (DCs) take up and process tumour-derived peptides or proteins. After maturation, they present the peptide antigens on major histocompatibility complex (MHC) molecules and activate T cells specific for these antigenic epitopes. As a result of these processes, the T cells expand into short-lived effector and long-lived memory populations. Essentially, these T cells are ready to respond rapidly to subsequent antigenic encounters [49,51,52], Figure 13).
We investigated the dendritic cell-based active immunotherapies in patients with colorectal cancer patients using adoptively transferred DC-based immunotherapies. Our results indicate that vaccination by autologous DCs loaded with autologous oncolysates containing various tumour antigens represents a well-tolerated therapeutic modality in patients with colorectal cancer without any detectable adverse effects [52].
Tumour destruction is only possible if the number of newly formed tumour cells grows more slowly than the number of destroyed cells. As this is a cell-to-cell reaction, the ratio of cancer cells to immune cells is crucial.
If cell-mediated immune stimulation fails, malignancy develops. Inhibition of the immune defence is another way in which cancer develops (Figure 14).
Immune protection against cancer can be impaired for several reasons, such as
I: Failure of antigen processing;
II: Inhibition of immunity by TGF-β, IL-10 and fatty acid synthase ligand (FasL);
Checkpoint blockade by programmed death ligand 1 (PD-L-1), programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), mucin domain containing-3 (TIM-3) protein, lymphocyte activation gene 3, as well as B- and T-lymphocyte-attenuator protein; Tumour-associated macrophages (TAMs) secreting immunosuppressive factors and cytokines to block the activity of natural killer (NK) cells and CTLs.
Regulatory T cell (Treg) antagonism via CTLA-4 and OX40 (a secondary costimulatory immune checkpoint molecule). Tumour cells and host stromal cells (including fibroblasts and lymphatic endothelium) can also express TGF-β, checkpoint ligands and FasL to directly induce T-cell apoptosis).
Immunotherapy has excellent potential to treat cancer and prevent future recurrence by activating the immune system to recognise and kill cancer cells. Various strategies are being developed in the laboratory and the clinic, including therapeutic non-cellular (vector- or subunit-based) cancer vaccines and dendritic cell vaccines [51,52]. There is also evidence that many other factors, including age, sex, metabolic state, immune history and even gut microbiota, can influence the immune system [53].
Engineered T cells [54,55,56,57,58,59,60,61,62,63,64] and immune checkpoint blockade therapies have led to exciting recent clinical advances in cancer immunotherapy [61,62,63,64,65,66,67,68].
Despite their promise, much more research is needed to understand how and why certain cancer patients fail to respond to immunotherapy and to predict which therapeutic strategies are best for a cancer patient. Many successful tumour therapies are based on this knowledge, but we are far from the final solution.
We believe the way to success is to treat patients as a whole. The aim is to reduce the tumour mass, stimulate the immune system and reverse the inhibition of the immune defence. In addition, the ratio of tumour cells to protective immune cells can influence the success of immunotherapy. It is therefore important to reduce the tumour mass. In addition to surgery, this can be achieved with X-rays and pharmacological vitamin C treatment.
Pharmacological vitamin C treatment kills tumour cells using Warburg’s aerobic glycolysis, mitotic cells are the primer targets, while cells with oxidative phosphorylation survive.
Inhibition of checkpoint blockade, complemented by dendritic cell immune stimulation and pharmacological vitamin C treatment, could be synergistic.
References
- Lynn Margulis: Origin of eukaryotic cells New Haven: Yale University Press, 1970.
- Anand P, Kunnumakkara A, Sundaram C, Harikumar K, Tharakan S, Lai O, Sung B, Aggarwal B: Cancer is a preventable disease that requires major lifestyle changes. Pharmaceutical Research 2008, 25 (9):2097–2116.
- Islami F, Sauer A, Miller K, Siegel R, Fedewa S, Jacobs E, McCullough M: Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA CANCER J CLIN 2018, 68(1):31-54. [CrossRef]
- Croce C: Oncogenes and cancer. The New England Journal of Medicine 2008, 358 (5):502-511.
- Knudson A: Two genetic hits (more or less) to cancer. Nature Reviews Cancer 2001, 1(2):157-162.
- Lodish H, Berk A, Matsudaira P, Kaiser C, Krieger M, Scott M, Zipursky, Darnell J: Molecular Biology of the Cell, 5th edn. New York; 2004.
- Nelson D, Tan T, Rabson A, Anderson D, Degenhardt K, White E: Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes & Development 2004, 18(17):2095–2107.
- Merlo L, Pepper J, Reid B, Maley C: Cancer as an evolutionary and ecological process. Nature Reviews Cancer 2006, 6(12):924-935.
- Padhani AR, Krohn KA, Lewis JS, Alber M. Imaging oxygenation of human tumors. Eur Radiol 2007;17:861-72.
- Hunyady, J: The Result of Vitamin C Treatment of Patients with Cancer: Conditions Influencing the Effectiveness. Int J Mol Sci. 2022 Apr 15;23(8):4380. [CrossRef]
- Fritz, H.; Flower, G.; Weeks, L.; Cooley, K.; Callachan, M.; McGowan, J.; Skidmore, B.; Kirchner, L.; Seely, D. Intravenous Vitamin C and Cancer: A systematic review. Integr. Cancer Ther. 2014, 13, 280–300. [CrossRef].
- Andrés, S., Pesántez, V., Salinas, MAF et al.:Vitamin C and its Action on Cancer Cells International Journal of Medical, Pharmacy and Drug Research (IJMPD) [Vol-6, Issue-5, Sep-Oct 2022] ISSN: 2456-8015. [CrossRef]
- Nauman, G.; Gray, J.C.; Parkinson, R.; Levine, M.; Paller, C. Systematic review of intravenous ascorbate in cancer clinical trials. Antioxidants 2018, 7, 89. [CrossRef] [PubMed]. [CrossRef]
- Van Gorkom, G.N.Y.; Lookermans, E.L.; Van Essen, C.H.; Bos, G.M.J. The effect of Vitamin C (ascorbic acid) in the treatment of patients with cancer: A systematic review. Plasma Nutr. 2019, 11, 977. [CrossRef] [PubMed].
- Bazzan, A.J.; Zabrecky, G.; Wintering, N.; Newberg, A.B.; Monti, D.A. Retrospective evaluation of clinical experience with intravenous ascorbic acid in patients with cancer. Integr. Cancer Ther. 2018, 17, 912–920. [CrossRef].
- High-Dose Vitamin C (PDQ®)–Health Professional Version—National Cancer Institute. Available online: https://www.cancer. gov/about-cancer/treatment/cam/hp/vitamin-c-pdq (accessed on 12 February 2021).
- Cameron, E.; Campbell, A. The orthomolecular treatment of cancer II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem. Biol. Interact. 1974, 9, 285–315.
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689.
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4538–4542.
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of high-dose Vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N. Engl. J. Med. 1979, 301, 687–690.
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-dose Vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized, double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141. [CrossRef]
- Parrow NL, Leshin JA, Levine M. Parenteral ascorbate as a cancer therapeutic: a reassessment based on pharmacokinetics. Antioxid Redox Signal. 2013 Dec 10;19(17):2141-56. Epub 2013 Jun 19.PMID: 23621620. [CrossRef]
- Verrax, J.; Calderon, P. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic. Biol. Med. 2009, 47, 32–40. [CrossRef]
- Levine, M.; Padayatty, S.; Espey, M. Vitamin C. A concentration-function approach yields pharmacology and therapeutic discoveries. Adv. Nutr. 2011, 2, 78–88.
- Hunyady, J. The role of Vitamin C in the energy supply of cells Hypothetical structure for energy transformation 2021. JSRR 2021, 7, 30–44.
- Gounaris Y, Litinas C, Evgenidou E, Petrotos C. “A hypothesis on the possible contribution of free hypoxanthine and adenine bases in prebiotic amino acid synthesis.” Hypothesis. 2015;13.
- Korth H, Meier A, Auferkamp O, Sicking W, de-Groot H, Sustmann R, Kirsch M: Ascorbic acid reduction of compound I of mammalian catalases proceeds via specific binding to the NADPH binding pocket. Biochemistry 2012, 51(23):4693-4703.
- Linowiecka, K., Foksinski, M., & Brożyna, A. A. (2020). Vitamin C Transporters and Their Implications in Carcinogenesis. Nutrients, 12, 3869. [CrossRef]
- Lv, H.; Wang, C.; Fang, T.; Li, T.; Lv, G.; Han, Q.; Yang, W.; Wang, H. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis. Oncol. 2018, 2, 1.
- Shanel Dhani, Yun Zhao and Boris Zhivotovsky: A long way to go: caspase inhibitors in clinical use. Cell Death and Disease (2021) 12:949. [CrossRef]
- Macheda M, Rogers S, Best J: Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005 202(3):654-662.
- Abdelwahab M, Fenton K, Preul M, Rho J, Lynch A, Stafford P, Scheck A: The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLoS One 2012, 7(5):e36197.
- Riester, M.; Xu, Q.; Moreira, A.; Zheng, J.; Michor, F.; Downey, R.J. The Warburg effect: Persistence of stem-cell metabolism incancers as a failure of differentiation Annalsof. Oncology 2018, 29, 264–270.
- Takemura, Y.; Satoh, M.; Satoh, K.; Hamada, H.; Sekido, Y.; Kubota, S. High dose of ascorbic acid induces cell death in mesothelioma cells. Biochem. Biophys. Res. Commun. 2010, 394, 249–253.
- Hong, S.; Lee, S.; Moon, J.; Hwang, J.; Kim, D.; Ko, E.; Kim, H.; Cho, I.; Kang, J.; Kim, D.; et al. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene 2013, 32, 1508–1517.
- Zhao F, Keating A: Functional properties and genomics of glucose transporters. Curr Genomics 2007 22:113-128.
- Caccialanza R, Cereda E, De-Lorenzo F, Farina G, Pedrazzoli P: To fast, or not to fast before chemotherapy, that is the question. BMC Cancer 2018 18(1):337.
- Younes M, Brown R, Stephenson M, Gondo M, Cagle P: Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer Cell 1997 80(6):1046-1051.
- Rastogi S, Banerjee S, Chellappan S, Simon G: Glut-1 antibodies induce growth arrest and apoptosis in human cancer cell lines. Cancer Lett 2007 257(2):244-251.
- Chan K, Chan J, Chung K, Fung K: Inhibition of cell proliferation in human breast tumor cells by antisense oligonucleotides against facilitative glucose transporter 5. J Cell Biochem 2004 93(6):1134-1142.
- Spitz, D.; Sim, J.; Ridnour, L.; Galoforo, S.; Lee, Y. Glucose deprivation-induced oxidative stress in human tumour cells: A fundamental defect in metabolism? Ann. N. Y. Acad. Sci. 2000, 899, 349–362.
- Galoforo, S.; Berns, C.; Erdos, G.; Corry, P.; Lee, Y. Hypoglycemia induced AP-1 transcription factor and basic fibroblast growth factor gene expression in multidrug-resistant human breast carcinoma MCF-7/ADR cells. Mol. Cell. Biochem. 1996, 155, 163–171. [CrossRef]
- Lin X, Zhang F, Bradbury C, Kaushal A, Li L, Spitz D, Aft R, Gius D: 2-Deoxy-D-Glucose-induced Cytotoxicity and Radiosensitization in Tumor Cells Is Mediated via Disruptions in Thiol Metabolism. CANCER RESEARCH 2003, 63:3413-3417.
- Cavicchio C, et al., Potassium Ascorbate with Ribose: Promising Therapeutic Approach for Melanoma Treatment. Oxidative Medicine and Cellular Longevity Volume 2017, Article ID 4256519, 12 pages. [CrossRef]
- Lee Y, Galoforo S, Berns C, Chen J, Davis B, Sim J, Corry P, Spitz D: Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J Biol Chem 1998, 273:5294-5299.
- Minutolo F, Macchia M, Katzenellenbogen B, Katzenellenbogen J: Estrogen receptor β ligands: recent advances and biomedical applications. Med Res Rev 2011 31(3):364-442.
- Harmon A, Patel Y: Naringenin inhibits glucose uptake in MCF-7 breast cancer cells: a mechanism for impaired cellular proliferation. Breast Cancer Res Treat 2004 85(2):103-110.
- Harmon O, Eck P, Chen S, Corpe C, Lee J, Kruhlak M, Levine M: Inhibition of the intestinal glucose transporter GLUT2 by flavonoids. FASEB J 2007 21(2):366-377.
- Jeanbart L, Swartz MA Engineering opportunities in cancer immunotherapy. PNAS November 24, 2015 112 (47) 14467-14472.
- Sharma P, Allison JP The future of immune checkpoint therapy. Science 2015 348 (6230):56–61.
- Chiang CLL, Balint K, Coukos G, Kandalaft LE Potential approaches for more successful dendritic cell-based immunotherapy. Expert Opin Biol Ther 2015 15(4):569–582.
- Hunyadi J, András C, Szabó I, Szántó J, Szluha K, Sipka S, Kovács P, Kiss A, Szegedi G, Altorjay I, Sápy P, Antal-Szalmás P, Tóth L, Fazekas G, Rajnavölgyi É. Autologous dendritic cell based adoptive immunotherapy of patients with colorectal cancer-A phase I-II study. Pathol Oncol Res. 2014 Apr;20(2):357-65.
- Pulendran B Systems vaccinology: Probing humanity’s diverse immune systems with vaccines. Proc Natl Acad Sci USA 2014 111(34):12300–12306.
- Pedersen SR, Sørensen MR, Buus S, Christensen JP, Thomsen AR Comparison of vaccine-induced effector CD8 T cell responses directed against self- and non-self-tumor antigens: Implications for cancer immunotherapy. J Immunol 2013 191(7):3955–3967.
- Linnemann C et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 2015 21(1):81–85.
- Matsushita H et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012 482(7385):400–404.
- Kershaw MH, Westwood JA, Darcy PK Gene-engineered T cells for cancer therapy. Nat Rev Cancer 2013 13(8):525–541.
- Gill S, June CH Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 2015 263(1):68–89.
- Stephan MT, Stephan SB, Bak P, Chen J, Irvine DJ Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials 2012 33(23):5776–5787.
- Huang B, et al. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci Transl Med 2015 7(291):291ra94.
- Gubin MM, et al. Checkpoint blockade cancer immunotherapy targets tumor-specific mutant antigens. Nature 2014 515(7528):577–581.
- Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013 369(2):122–133.
- Larkin J, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med. 2019 Oct 17;381(16):1535-154.
- Pentcheva-Hoang T, Simpson TR, Montalvo-Ortiz W, Allison JP Cytotoxic T lymphocyte antigen-4 blockade enhances antitumor immunity by stimulating melanoma-specific T-cell motility. Cancer Immunol Res 2014 2(10):970–980.
- Phan GQ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA 2003 100(14):8372–8377.
- Attia P et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol 2005 23(25):6043–6053.
- Topalian SL, Drake CG, Pardoll DM Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015 27(4):450–461.
- Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012 366(26):2443–2454.
Figure 2.
Evolution of malignant tumour cells.
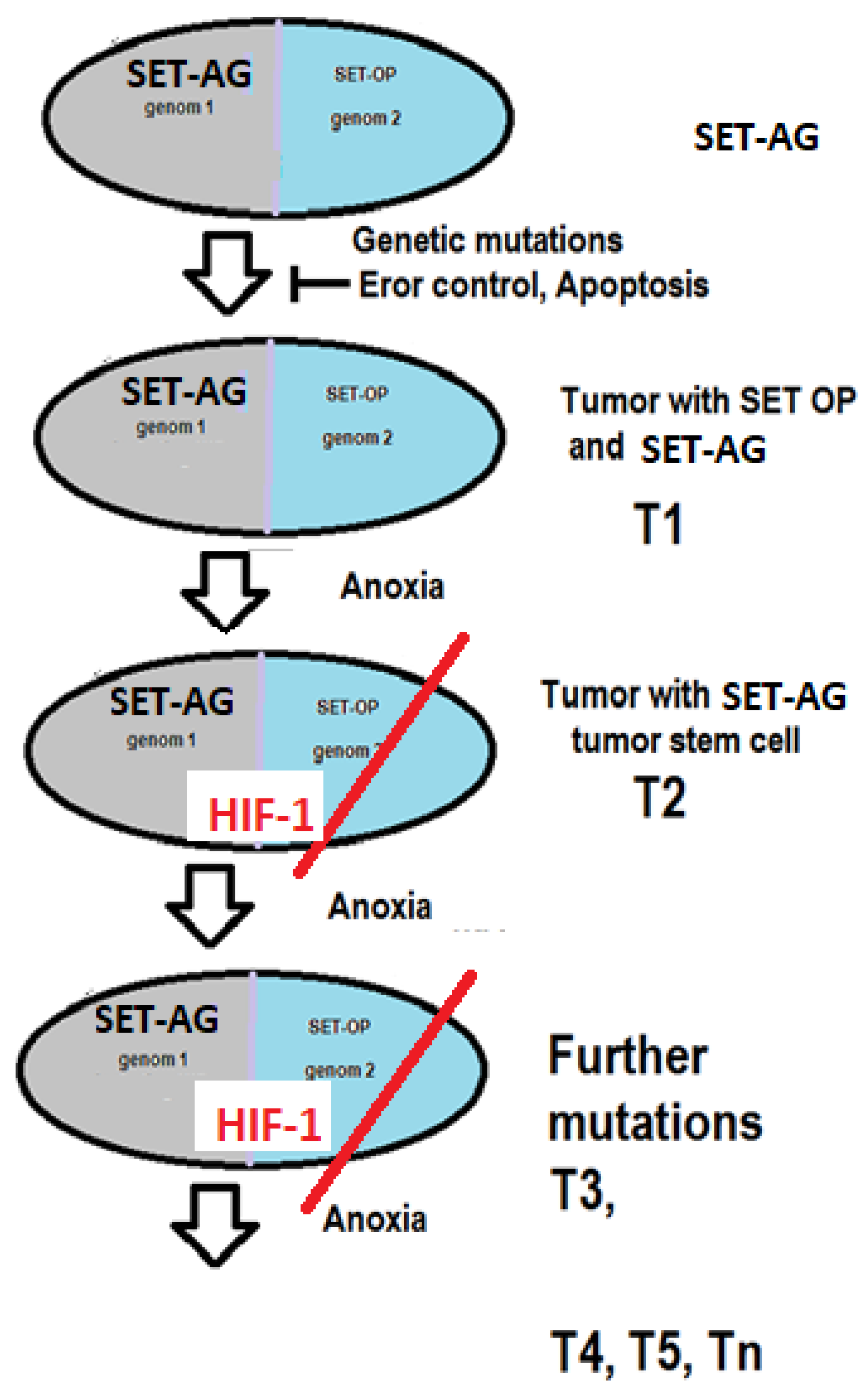
Figure 3.
Connection of two L-cysteines.
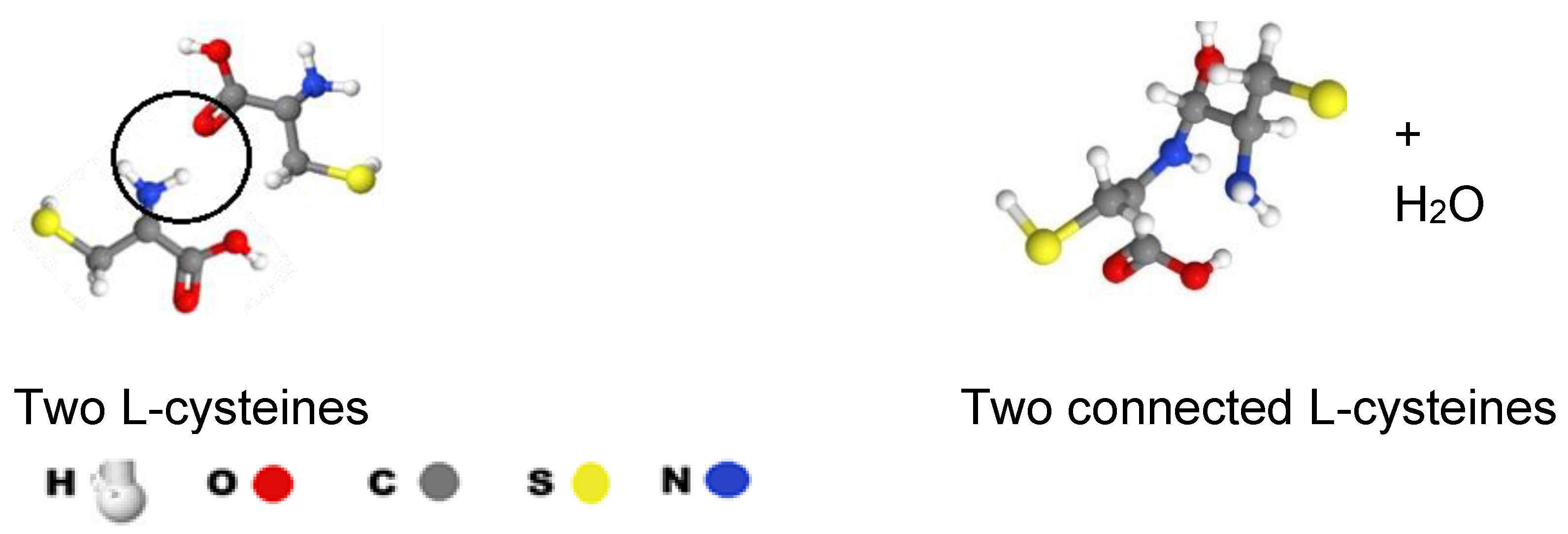
Figure 4.
Electron Transfer Chain.
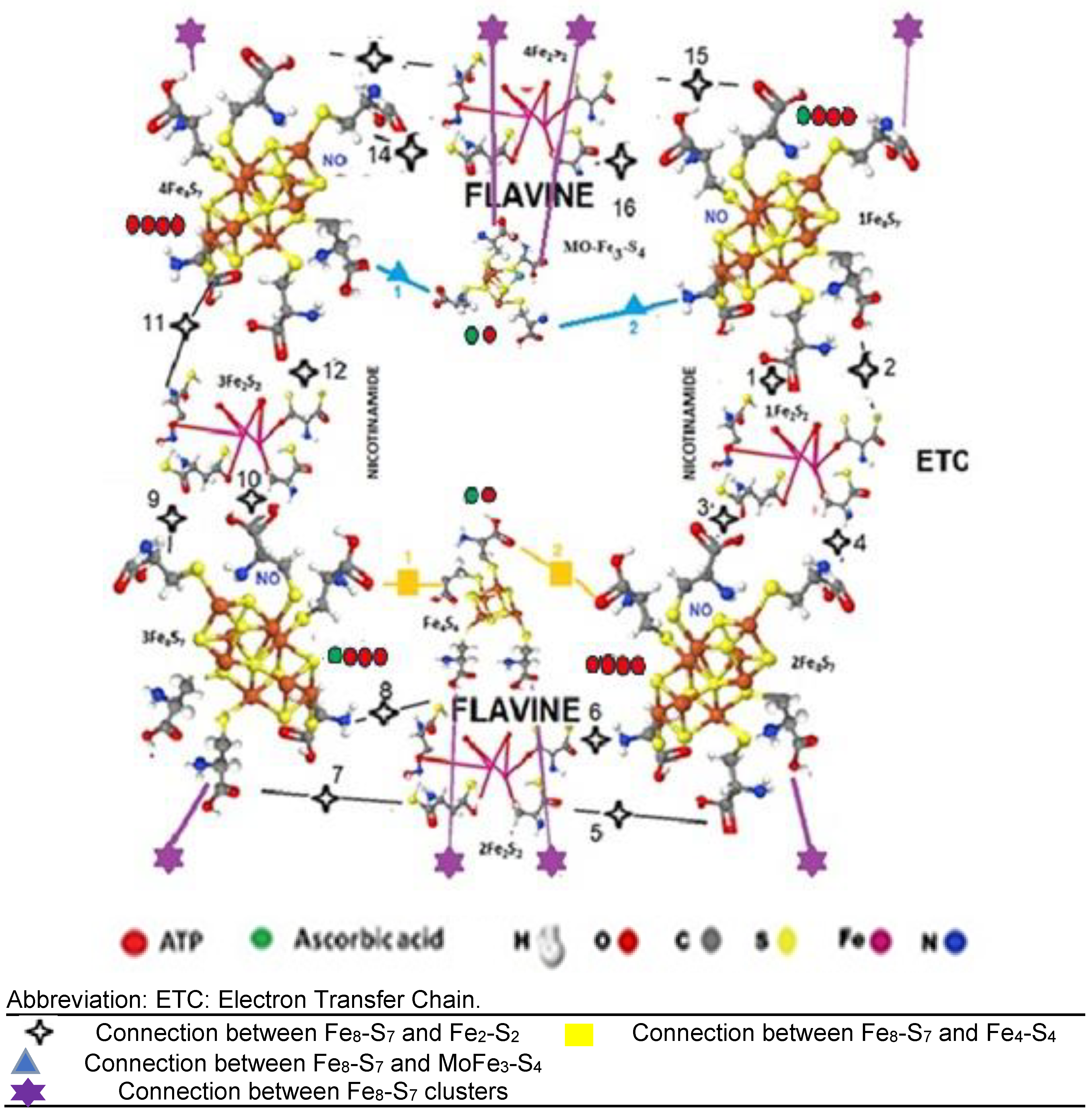
Figure 5.
Multiple Electron Transfer Chain Abbreviation: ETC: Electron Transfer Chain, METC: Multiple ETC.
Figure 5.
Multiple Electron Transfer Chain Abbreviation: ETC: Electron Transfer Chain, METC: Multiple ETC.
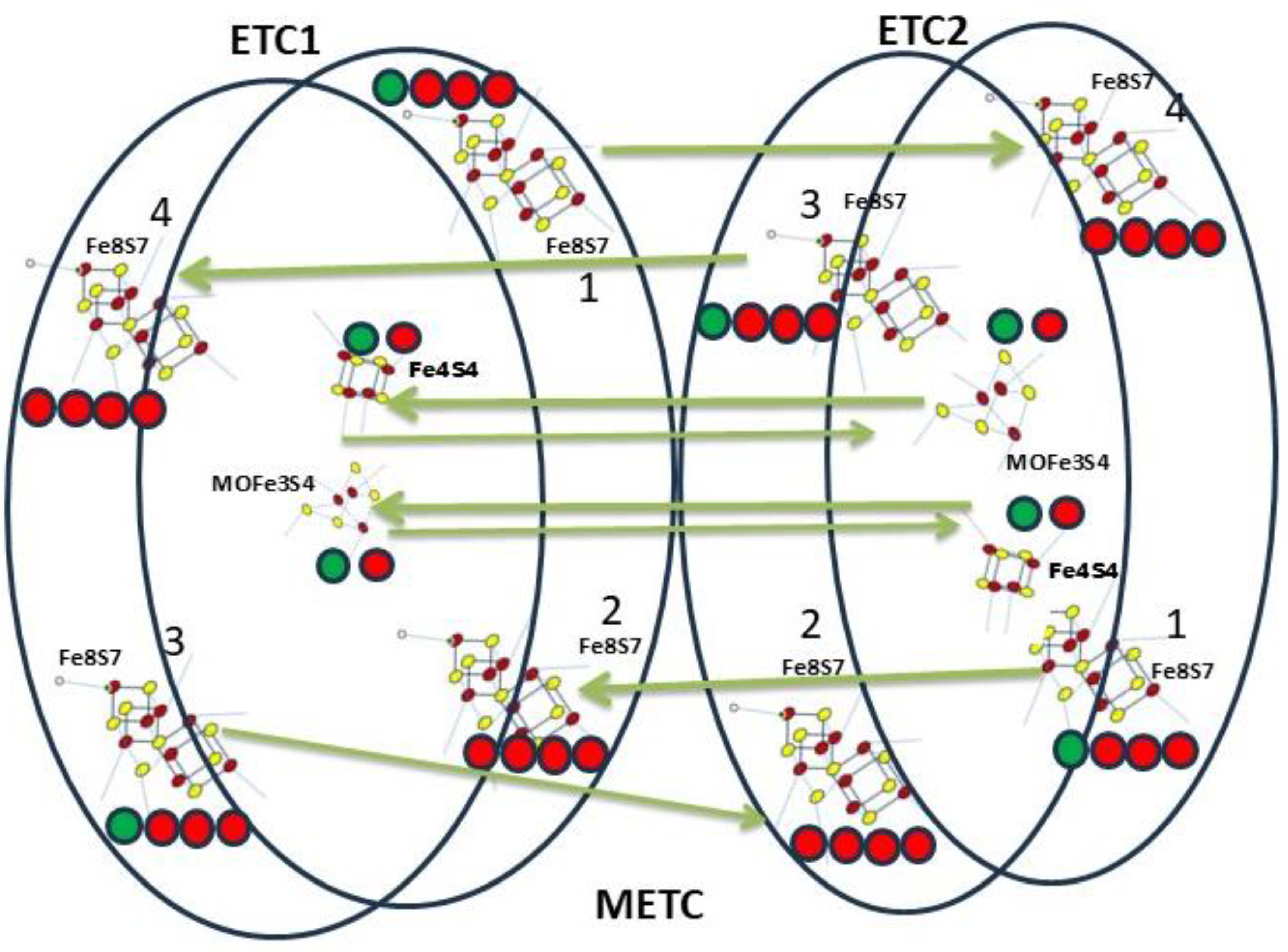
Figure 6.
The connecting points of the Fe8S7 cluster.
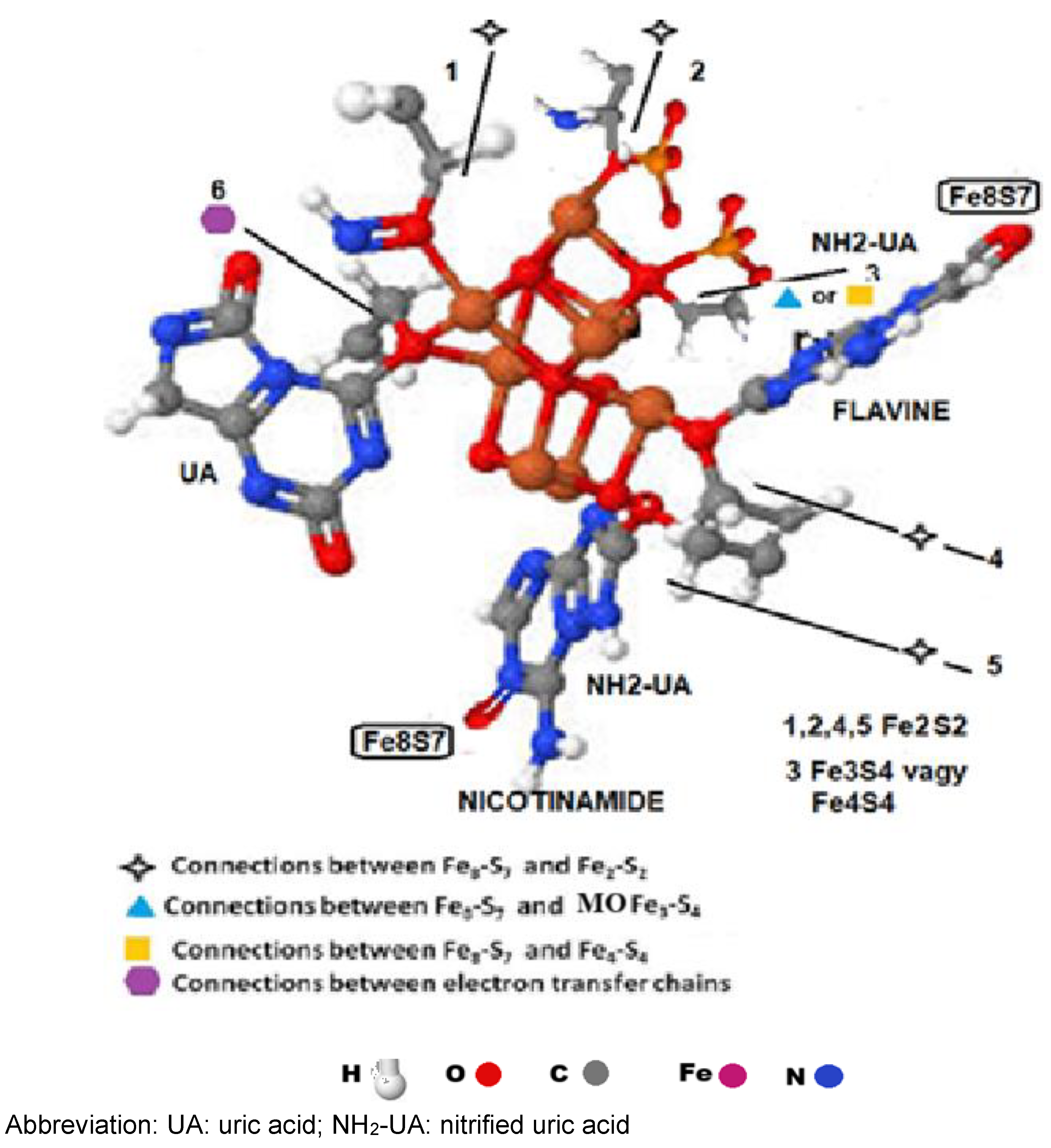
Illustration 7. Four aminated uric acid molecules connected to nicotinamide and Flavine molecules link the four[Fe8S7 (SCH2CH3)6]e2− clusters of nitrogenase.
Illustration 7. Four aminated uric acid molecules connected to nicotinamide and Flavine molecules link the four[Fe8S7 (SCH2CH3)6]e2− clusters of nitrogenase.
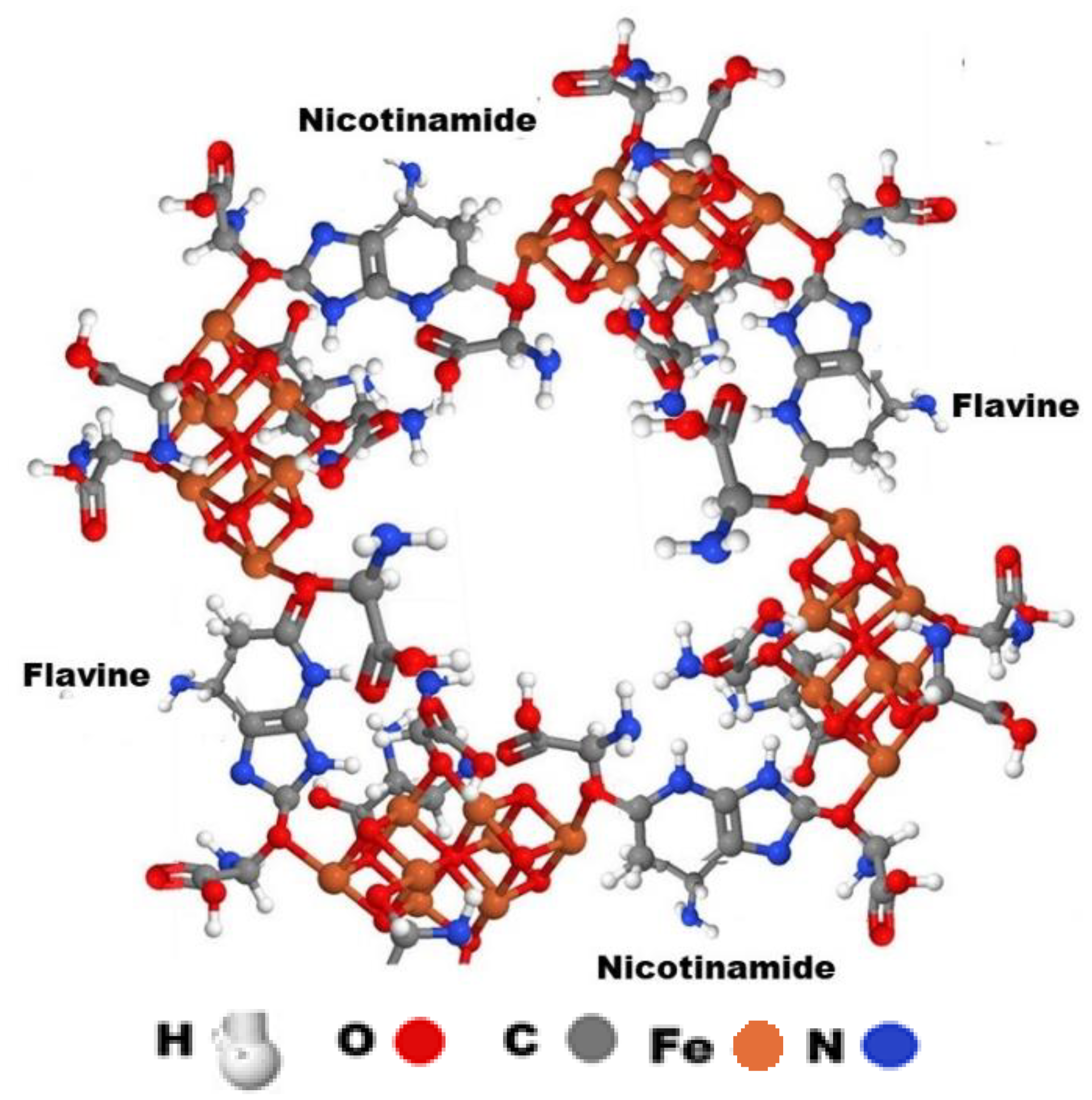
Figure 8.
Synchronized function of the two electron transfer chains.
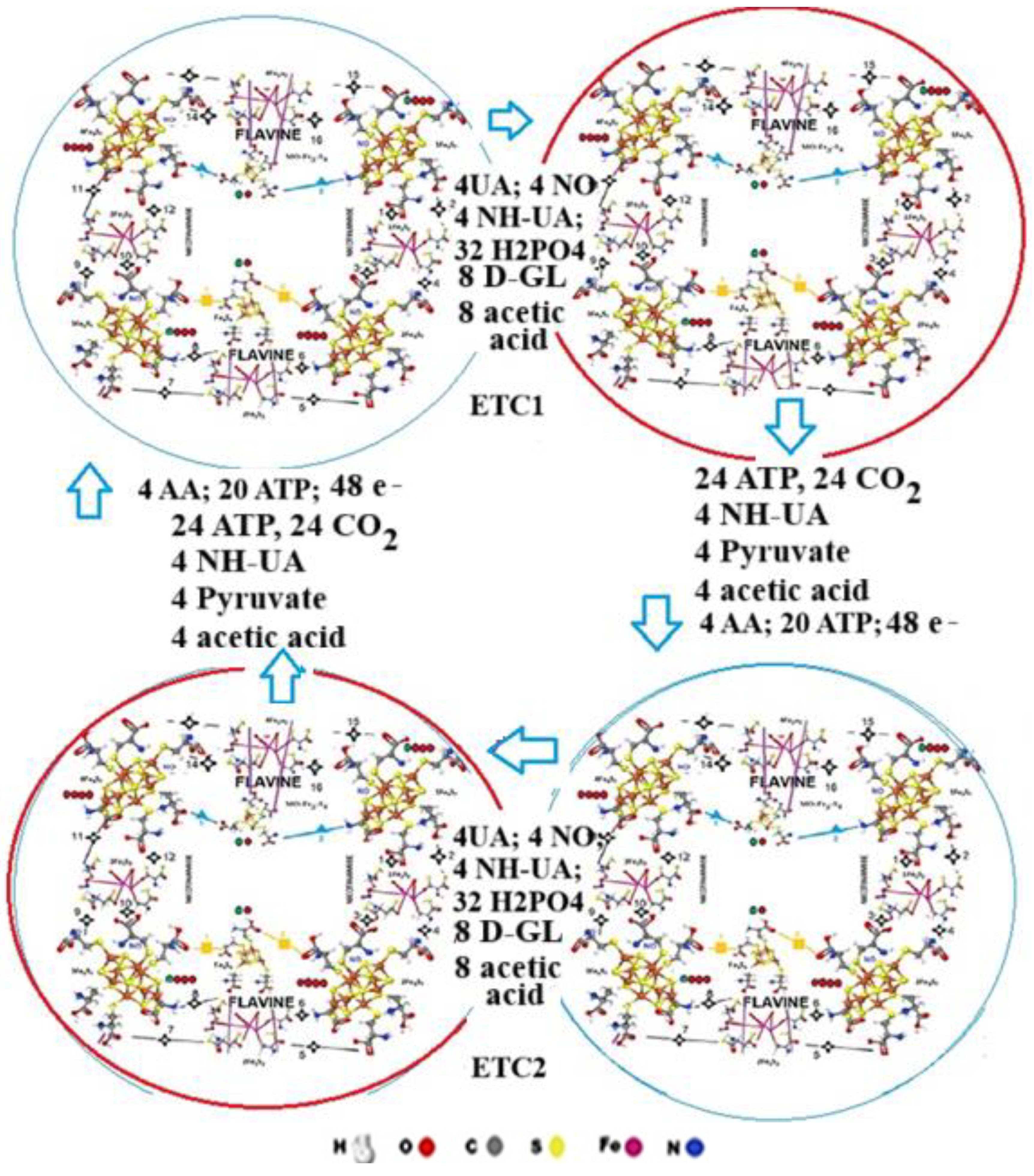
Figure 9.
The three states of the electron transfer chains.
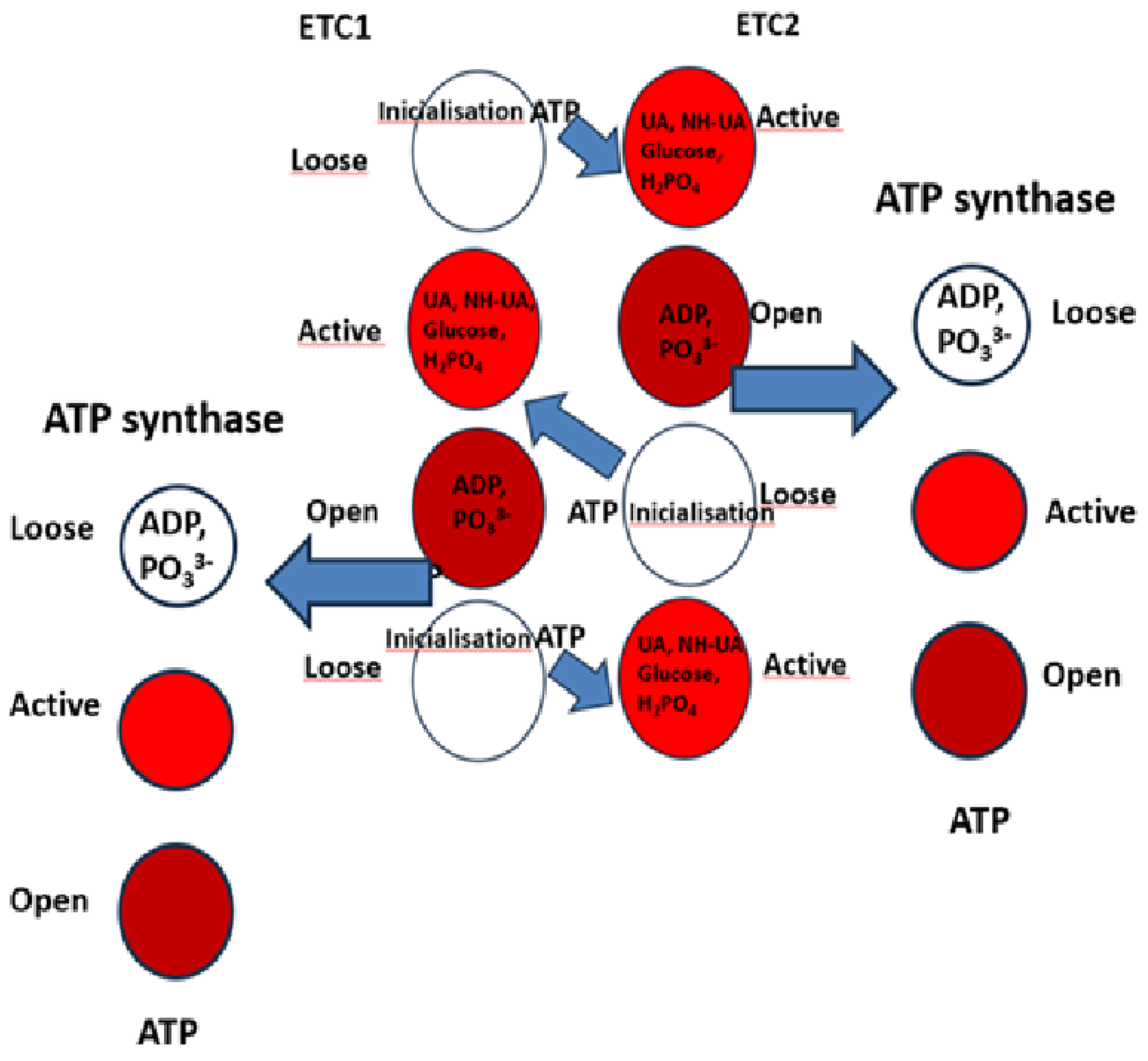
Figure 9.
The Structure of Energy Transformation for Oxidative Phosphorylation.
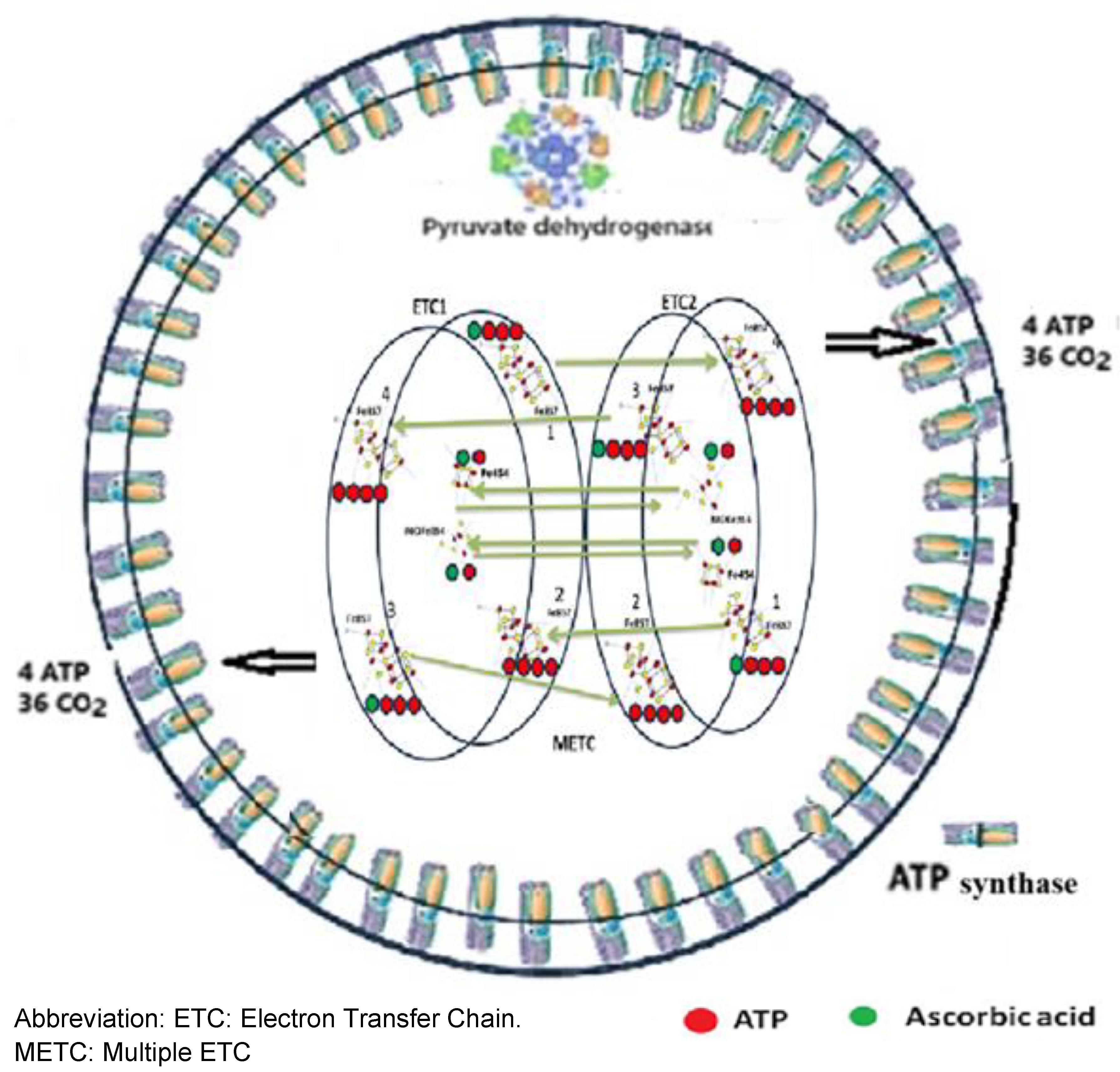
Figure 10.
Energy investment is required to develop a new ATP.
Figure 11.
Vitamin C’s role in Fe3+ - Fe2+ transformation [28]. Abbreviations: AA: ascorbic acid; DHA: dehydroascorbic acid.
Figure 11.
Vitamin C’s role in Fe3+ - Fe2+ transformation [28]. Abbreviations: AA: ascorbic acid; DHA: dehydroascorbic acid.
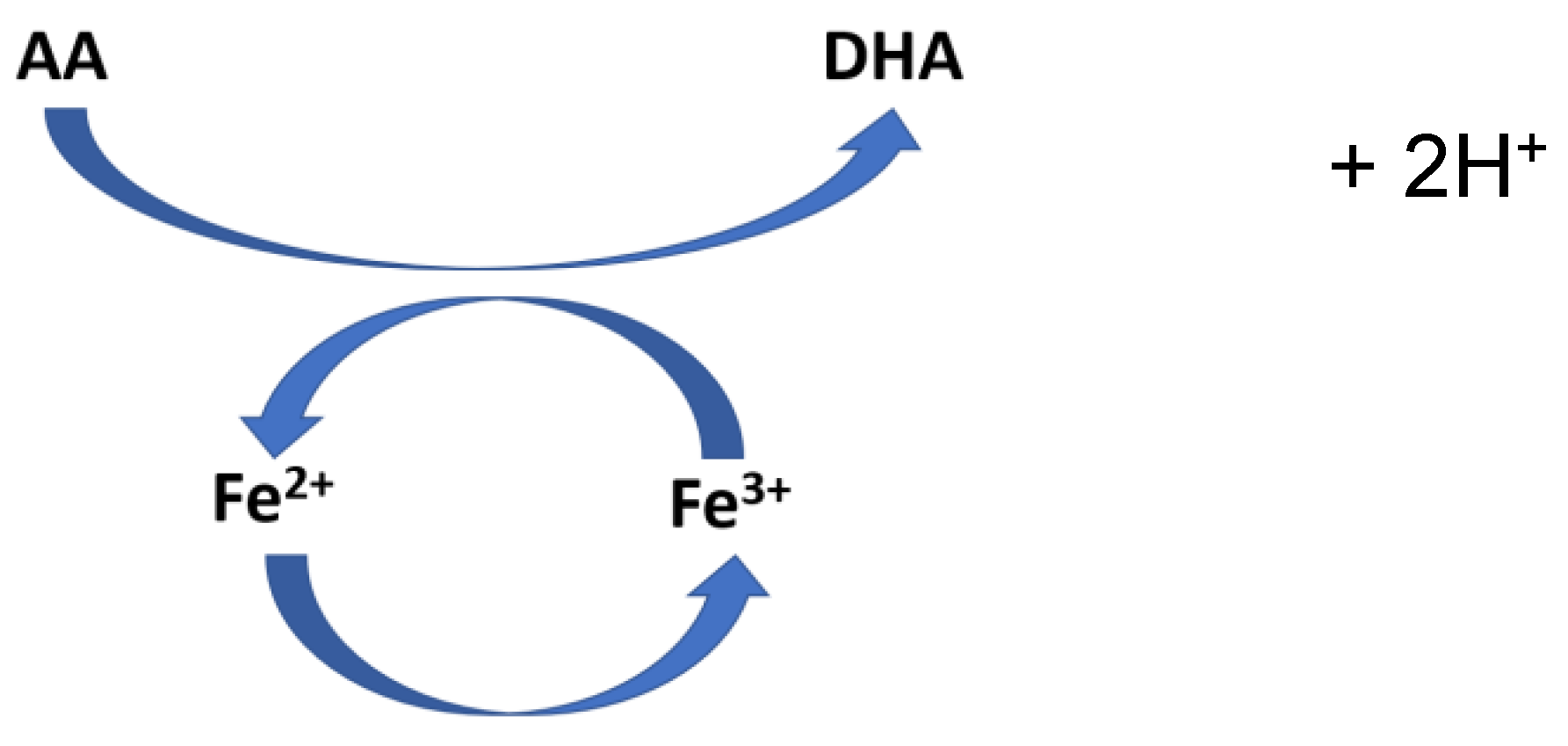
Figure 12.
Ascorbic acid leads to a sulphur-oxygen change in Fe4-S4 clusters, Each [Fe8-S7] cluster has four places for activation, each [Fe2-S2] cluster and the [MoFe3-S4] cluster have one, while the [Fe4-S4] cluster has two activation points. In the [Fe8-S7]1, [Fe8-S7] 2 and [Fe4-S4] clusters one AA is responsible for the activation. (Table IV).
Figure 12.
Ascorbic acid leads to a sulphur-oxygen change in Fe4-S4 clusters, Each [Fe8-S7] cluster has four places for activation, each [Fe2-S2] cluster and the [MoFe3-S4] cluster have one, while the [Fe4-S4] cluster has two activation points. In the [Fe8-S7]1, [Fe8-S7] 2 and [Fe4-S4] clusters one AA is responsible for the activation. (Table IV).
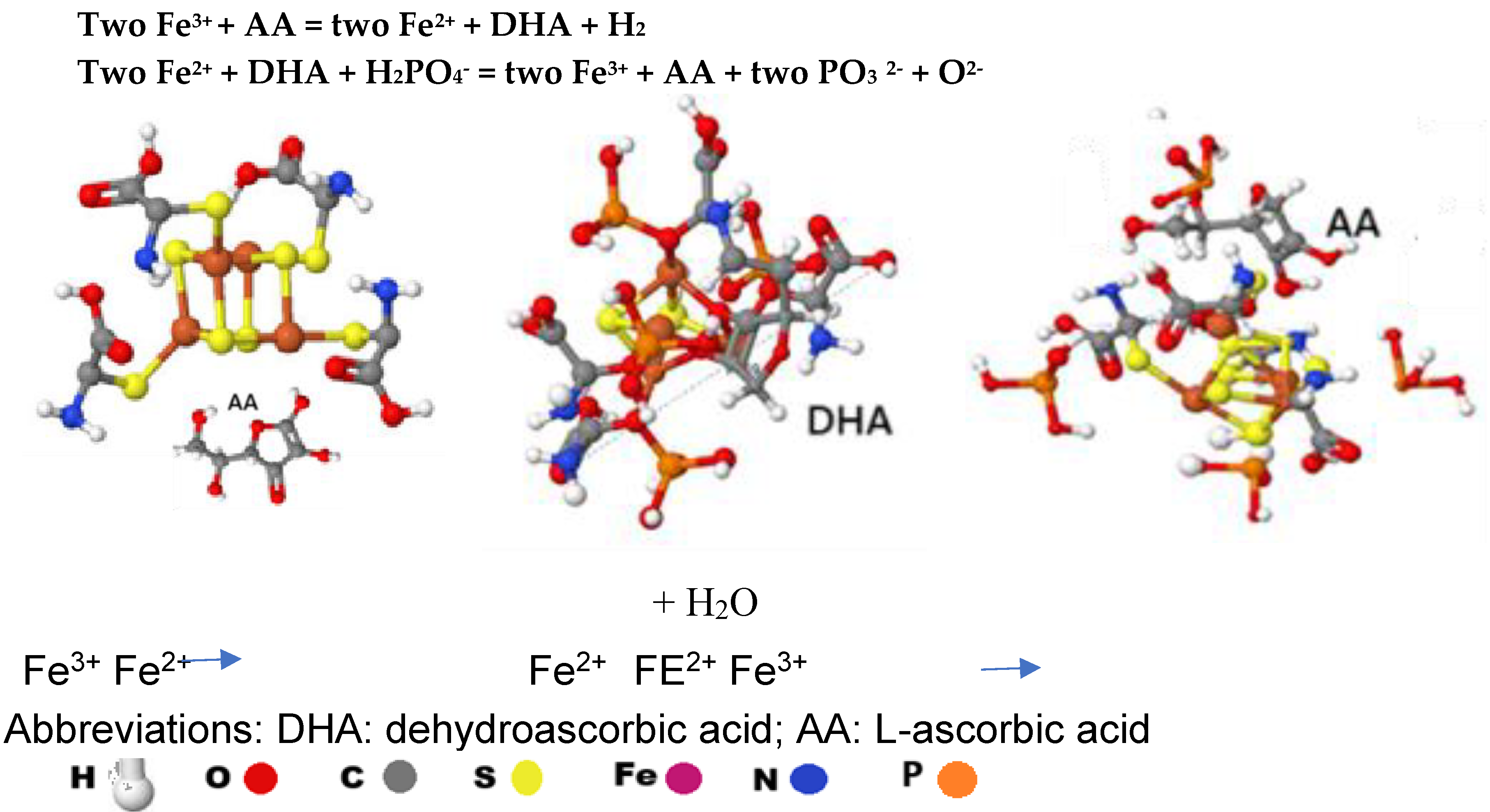
Figure 13.
Immune stimulation against cancer. A: Dendritic cells (DCs) take up and process tumour-derived peptides or proteins. After maturation, they present the peptide antigens on MHC molecules and activate T cells specific for these antigenic epitopes. B: Effector lymphocytes and natural killer (NK) cells kill the tumour cells.
Figure 13.
Immune stimulation against cancer. A: Dendritic cells (DCs) take up and process tumour-derived peptides or proteins. After maturation, they present the peptide antigens on MHC molecules and activate T cells specific for these antigenic epitopes. B: Effector lymphocytes and natural killer (NK) cells kill the tumour cells.

Figure 14.
Reasons for the lack of immune protection against cancer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



