Submitted:
23 May 2025
Posted:
26 May 2025
You are already at the latest version
Abstract
The development of orally bioavailable non-peptidomimetic glucagon-like peptide-1 receptor agonists (GLP-1RAs) offers a promising therapeutic avenue for the treatment of type 2 diabetes mellitus (T2DM) and obesity. An extensive in silico approach combining structure-based drug design, and ligand-based strategies together with pharmacokinetic properties and drug-likeness predictions, is implemented to identify novel non-peptidic GLP-1RAs from the COCONUT and Marine Natural Products (CMNPD) libraries. More than 700,000 compounds were screened by shape-based similarity filtering in combination with precision docking against the orthosteric site of the GLP-1 receptor (PDB ID: 6X1A). The docked candidates were further assessed with the molecular mechanics MM-GBSA tool to check the binding affinities; the final list of candidates was validated by running a 500 ns long MD simulation. Twenty final hits were identified, ten from each database. The hits contained compounds with reported antidiabetic effect but with no evidence of GLP-1 agonist activity including hits 1, 6, 7, 10. These findings proposed a novel mechanism for these hits through GLP-1 activity and positioned the other hits as potential promising scaffolds. Among the studied compounds—especially hits 1, 5, and 9, possessed strong and stable interactions with critical amino acid residues such as TRP-203, PHE-381, and GLN-221 at the active site of the 6X1A-substrate along with favorable pharmacokinetic profiles. Moreover, the RMSF and RMSD plots further suggested the possibility of stable interactions. Specifically, hit 9 possessed the best docking score with a ΔG_bind value of −102.78 kcal/mol, surpassing even the control compound in binding affinity. The ADMET profiling also showed desirable drug-likeness and pharmacokinetic characteristics for hit 9. The pipeline of computational integration underscores the potential of non-peptidic alternatives in natural product libraries to pursue GLP-1-mediated metabolic therapy into advanced preclinical validation.
Keywords:
GLP-1R
; virtual screening
; molecular dynamics
; oral therapy
; repurposing
; chemical scaffolds
1. Introduction
Type 2 diabetes (T2D) and obesity are among the most critical challenges faced by global public health. The International Diabetes Federation (IDF) has projected that the number of individuals suffering from diabetes worldwide may reach 783.2 million in 2045 [1]. Moreover, diabetes will be positioned as the seventh leading cause of mortality across the globe by the year 2030 [2]. The worldwide rise in diabetes mellitus (DM) is predominantly fueled by increasing obesity levels. Recent research demonstrates that global obesity rates have nearly tripled over the past 40 years, a trend that has been closely linked to a significant uptick in the occurrence of T2D [3].
In the management of T2D and obesity, glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1RAs) have evolved into cornerstone therapy [4]. These agents augment insulin secretion, inhibit glucagon release, slow gastric emptying, and increase satiety; thus, they change the face of treatment for diabetes (Figure 1) [5]. As important as the discovery of the GLP-1 receptor (GLP-1R), which was more than 30 years ago, therapeutic agents directed toward this receptor bring astonishing effects in aspects such as glycemic control and weight reduction. The first success of peptide-based GLP-1RAs, exenatide, increased interest in other injectable formulations whenever further improvements were made to the diabetes treatment [6]. Since then, limited number of GLP-1 RAs have received approval from the U.S. Food and Drug Administration (FDA) for the treatment of T2DM and, in certain cases, for the management of obesity. Among these, liraglutide, semaglutide, and dulaglutide stand out as some of the most recognized agents [7,8,9]. All these are synthetic peptide-based analogs of GLP-1 [10]. For both weight management and diabetes control, only semaglutide and liraglutide have received the FDA approval [11,12,13].
However, peptide-based therapies have major drawbacks, the most notable being their administration by injection, which affects compliance and convenience for patients [14,15]. This is where the research focus has shifted toward developing orally bioavailable GLP-1RAs. The oral administration route is expected to significantly improve patient compliance due to the reduced burden associated with injectable therapies. Transitioning from peptide-based therapies to non-peptide agonists (NPAs) is a promising avenue; however, developing small molecules replicating endogenous GLP-1 pharmacokinetics remains challenging [14]. Recent advancements in cryogenic electron microscopy (cryo-EM) promise insights into the structural activation of GLP-1R have also led the way in discovering new classes of non-peptide GLP-1R agonists [15,16]. Compounds like orforglipron, danuglipron, and TT-OAD2 have shown promising pharmacokinetic profiles, and early clinical results suggest that they would be an efficient and practical alternative to existing peptide oral GLP-1RAs (Figure 2) [17,18,19,20,21,22,23]. The opening up of oral delivery was semaglutide, the first oral GLP-1RA approved in 2019 by the FDA for the management of T2DM, has made a huge milestone in this area [24]. It has been shown that oral semaglutide is effective in lowering hemoglobin A1c (HbA1c) levels and in weight loss, like the results of other routes [24]. This demonstration gave extra credence to the therapeutic potency of oral GLP-1RAs and further motivated research of small molecules that would mimic the effects of peptide-based GLP-1RAs. One among those is cinchonine, which has been presented as a promising contender with anti-hyperglycemic effects and benefits in obesity-affiliated conditions like non-alcoholic steatohepatitis NASH [25]. Indeed, the small molecules derived from various discoveries, yet with similar pharmacological profiles to peptide-based GLP-1RAs, but open to oral administration, ushering in new hope for treating T2D and obesity. Important pharmacokinetic modifications targeting specific carriers and surface coatings will ultimately improve the bioavailability and stability of GLP-1RAs when administered orally. Such designs overarchingly counteract challenges usually associated with peptide medications, including poor intestinal absorption and enzymatic degradation within the gastrointestinal tract [26]. However, apart from the hyperglycemia control, GLP-1RAs have cardiovascular benefits and protection for renal and hepatic organs, which would further build on the therapeutic interest of patients suffering from co-morbid conditions such as chronic kidney disease and non-alcoholic fatty liver diseases (NAFLD) [27,28]. Yet despite these advances, an apparent void exists in identifying novel non-peptide GLP-1-RAs featuring optimized pharmacokinetic profiles regarding effectiveness and safety for long-term T2DM and obesity treatment. Although many small-molecule agonists have shown some promise, a need for intense evaluation of alternative sources for new GLP-1RAs exists.
Natural products (NPs) are accessible and cost-effective sources that can help solve the challenge of identifying an oral GLP-1 therapeutic agent. COCONUT and Comprehensive Marine Natural Products (CMNPD) databases, with their vast chemical space and diverse bioactivity, offer a vast, understudied ground for discovering novel non-peptide GLP-1RAs. Such compounds have demonstrated therapeutic promise in disease areas such as metabolic disturbances and may be particularly well-suited to offer distinct advantages in structural diversity and pharmacological activity [29,30,31,32]. In this study, we employed an integrative molecular modeling strategy to predict candidate hits capable of binding to the orthosteric sites of GLP-1R. By screening a large and diverse library of natural and marine compounds, our approach demonstrates a potentially unbiased and powerful strategy for the discovery of novel non-peptide GLP-1 receptor agonists (GLP-1RAs).
2. Results and Discussion
Using integrated structure-based and ligand-based methods, two different natural product databases, including COCONUT and CMNPD, were screened for GLP-1R activation. To identify potential candidates as well as structural scaffolds for GLP-1R modulation, a combined virtual screening and drug repurposing protocol was subsequently carried out. Marine and natural compounds with known antibiotic activity that might activate GLP-1 were also evaluated in the results.
2.1. Database Preparation for Virtual Screening
2.1.1. Physicochemical Parameters and Drug-Likeness
Finding GLP-1 agonists in both natural and marine sources is the primary goal of this endeavor. Therefore, a filtering technique is developed, emphasizing the calculation of drug-like properties. Several rules and principles that describe the physicochemical properties of a target drug, such as the rule of 3, the rule of 5, the Zinc, CNS, and respiratory drug-likeness principles, are combined with a statistical analysis of approved drugs to create the quantitative estimation of the drug-likeness (QED) model developed in FAFDrugs4 [33,34]. The filter thresholds were validated when it was discovered that the obtained chemical space could retain up to 90% of the oral medications. The COCONUT database is an extensive library of natural products (NPs) gathered from public databases [35]. It includes 63 NP databases, such as Zinc NPs, NPAtlas, PubChem NPS, NCI developing medicines, and ChemSpider NPs. As for its most recent version, this massive database contains 695,133 NPs. Additionally, the CMNPD provides a plethora of compound scaffolds covering a vast chemical space with intriguing possibilities for medicinal chemistry. Specifically, the database comprises approximately 47,000 compounds isolated from various marine organisms [36]. The databases were successfully downloaded from the COCONUT and the CMNPD websites and then further filtered using Schrödinger’s Canvas by removing duplicate structures followed by physicochemical descriptors originally included in the database, such as MW 100-600, logP −3-6, number of rings < 6, total number of heavy atoms < 50, total charge −4-4, number of H-donors < 7, number of H-acceptors < 12, number of rotatable bonds < 11, and another total charge condition of −4-4 [37]. The QED incorporates eight physicochemical characteristics—including molecular weight, LogP, H-bond donors, H-bond acceptors, charge, aromaticity, stereochemistry, and solubility—into a score ranging from 0 to 1. Therefore, a molecule will appear more drug-like the closer its QED score is to 1 [34,38].
2.1.2. Shape Screening
The GLP-1 co-crystallized ligand was screened against the filtered COCONUT database, which had 40,332 natural products, and the CMNPD, which contained over 12,000 marine compounds, using the shape-screening feature of Schrödinger’s software. A cutoff for shape similarity indexes ≥ 0.3 was used to refine the shape screening results [39]. Following this procedure, 33,227 natural products and 1,306 marine compounds were chosen for further virtual screening against the GLP-1R binding pocket.
2.2. Virtual Screening
The resultant databases underwent three steps of virtual screening in Schrödinger’s Glide: high throughput virtual screening (HTVS), standard precision (SP), and extra precision (XP) docking [40]. Ten percent of the top-scoring compounds were retained in each phase, while the scoring hierarchy was maintained due to the protocol’s flexible docking approach, which included post-docking energy minimization. The final output following the XP docking stage included 276 and 36 compounds from the COCONUT and Marine databases, respectively. The XP scores of the top 10 hits were correlated with their binding energies (MM-GBSA) (Figure 3). The docking methodology was first verified by calculating the root mean square deviation (RMSD) of the crystal and docked poses of the co-crystallized ligand, resulting in 2.03 Å, indicating that Glide can predict binding to the GLP-1R binding site with high accuracy.
The results of the top hits from the COCONUT and Marine databases, as shown in Table 1 and Table 2, provide valuable insights into how well each compound binds to the GLP-1 receptor (PDB ID: 6X1A) through the descriptors of XP Score, MM-BBSA dG_Bind, and Shape Similarity. In the COCONUT database, the top hits displayed XP scores ranging from -13.835 to -12.532, indicating strong binding affinities to the GLP-1 receptor, with 1 exhibiting the highest XP score of -13.835. The MM-BBSA dG Bind values, indicative of binding affinity and measured in kcal/mol for these hits, ranged from -84.69 to -66.07 showing a significant correlation to the XP scores and further implying strong interactions between the ligands and the receptor. The Shape Similarity value reflects the geometric similarity of the hits compared to a reference ligand, with hits falling in the intermediate similarity range of 0.305-0.346 range compared to the control compound (the co-crystallized agonist PF-06882961) (Shape Similarity = 1.000). In the Marine database, the mean XP scores for top 10 hit compounds were between -12.80 and -10.190 kcal/mol. The best among them is 11, which got the highest binding affinity (-12.82 kcal/mol). The binding energies--MM-BBSA dG_Bind values for those compounds are between -100.43 and -52.04 kcal/mol, from which compound 16 exhibited the most favorable chemical affinity of -100.43 kcal/mol. As for the comparisons of Shape Similarity of Marine hits from 0.302 to 0.375, different compounds fit moderate to high degrees of similarity to the control compound. It needs to be noted that only one compound from the Marine database, 15, obtained a Shape Similarity score of 0.375, which further underpins its potential as a promising hit. The results confirm the existence of compounds within the COCONUT and Marine databases that possess strong binding affinities and some promising shape characteristics to the reference ligand, making them suitable for further examination as non-peptide GLP-1RAs. Favorable binding-based 2D and 3D docking interactions with key amino acid residues involved LYS-197, TRP-203, and PHE-381, which proved essential for the high-affinity interactions observed with the GLP-1R binding pocket (Figure 3). These results imply that the screened compounds are promising hits for targeting the GLP-1 receptor, enabling a more potent and selective interaction.
Analysis of the binding interactions of the identified ligands was based on spotting their interactions with the key residues identified in the crystal structure as crucial for binding the GLP-1 peptide, namely, LYS-197, TRP-203, and PHE-381. Common additional interactions were also searched to identify potential binding poses and important residues that might drive future drug design and development.
Strong π-cation interactions were noted for hit 1 with the pyridine nitrogen and TRP-203, and PHE-381. The indole ring further anchored the compound through a π- π interaction with TRP-33. An additional H bond was established between the acetate sidechain and GLN-221.
Hit 5 was found to bind the active site with a similar binding pose to hit 1. The pyridine nitrogen interacted through π-cation interactions with TRP-203, and PHE-381. The polycyclic aromatic system established several π- π interactions with TRP-33 and the dioxolane oxygen interacted with H bond as acceptor with GLN-221. The same interactions were completely spotted between hit 9 and the same previously mentioned residues. Hit 11, however, lacked the interaction with PHE-381 while keeping the same interaction profile with TRP-203, and GLN-221. The missed interaction was compensated for by additional H bonds with SER-31 and LEU-32. On the other hand, hit 13 lacked the interaction with PHE-381 without additional bonding (Figure 5).
In summary, the binding profile of the top hits exhibited consistent binding with the key residues TRP-203, and GLN-221 through π-cation or π- π interactions and H bond as acceptor respectively. The binding analysis also identified TRP-33 as an important binding residue that would act as a gatekeeper at the entrance of the binding site and that is preferably bound through a π- π interaction with a planar aromatic system. Moreover, the high values of the docking scores (-13) and binding energy calculations (-85) validated the significance of the drawn binding pattern and residues.
2.3. MD Simulations and Binding Free Energy Calculations for MD Frames
In mutagenesis studies, the activity of the non-peptide control compound was found to be reduced by alanine mutations of many residues, including ARG-380, GLU-373, PHE-385, LYS-197, TRP-33, PHE-230, LEU-141, TRP-203, PHE-381, LEU-384, and ARG-190 [9]. With this knowledge, we have used MD simulations over a period of 100 and 500 nano seconds to evaluate the stability of ligand-target complexes, the binding poses from the docking calculations, and the interaction pattern between each ligand with key GLP-1R amino acids. The studies were performed for top select candidates from virtual screening, namely compounds 1, 5, and 9 and for the control compound as a reference. To assess the stability of the protein, we evaluated the RMSD values of Cα atoms (Figure 4).
The proteins in the complexes of the control and compounds 1 and 5 stabilized at around 200 ns, while the protein in the 9-GLP-1R complex stabilized at around 250 ns. The proteins in all complexes remained stable for the rest of the simulation time with no significant structural deviations, indicating that they had reached equilibrium. Fluctuations from average Cα-atom RMSD values were within an acceptable order of 1–3.5 Å, which is expected since the GLP-1R protein is a large, multi-domain, G-protein coupled structure. The average protein Cα atoms RMSD values were in the range of 4.7–6.5 Å. RMSF analysis shows that the ligand binding site in the TM region remains stable throughout the simulation for all the complexes (Figure 5).
Figure 5.
RMSF analysis for Cα atoms in different complexes. The TM region is indicated in the yellow box. Residues that interact with each ligand are shown as green vertical bars. A) control, B) compound 1, C) compound 5, and D) compound 9.
Figure 5.
RMSF analysis for Cα atoms in different complexes. The TM region is indicated in the yellow box. Residues that interact with each ligand are shown as green vertical bars. A) control, B) compound 1, C) compound 5, and D) compound 9.
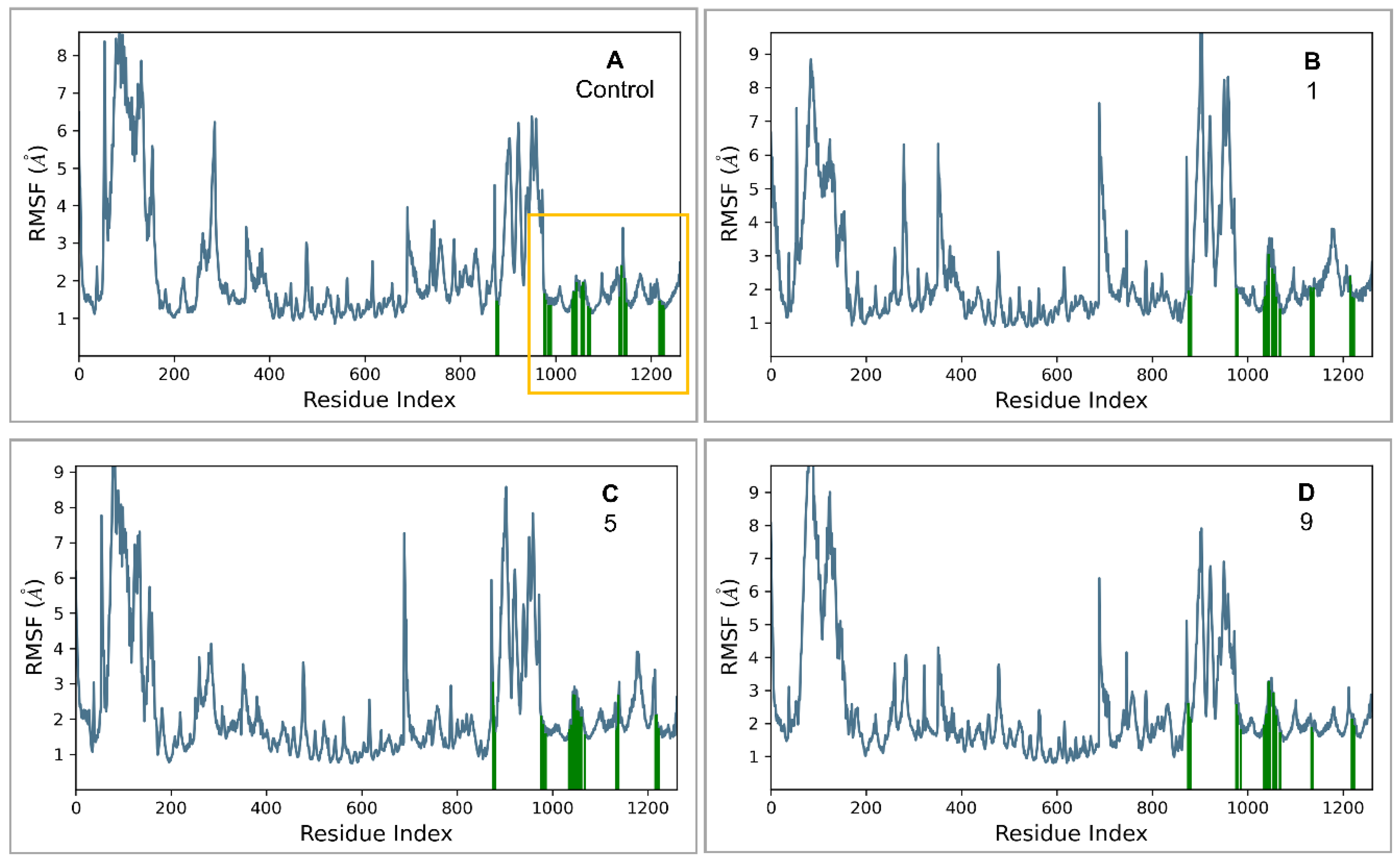
To assess ligand stability, RMSD values for ligand heavy atoms were measured with respect to the protein (Figure 6). None of the average ligand heavy-atom RMSD values is significantly larger than the average protein Cα atom RMSD value. This finding indicates that compounds 1, 5, 9, and the control were all bound efficiently within the binding site, which was also confirmed visually (supplementary files S1−S4, residues that interact for 45% or more of the simulation time are shown). Based on RMSD analysis and visual inspection, compound 5 seems to have changed its orientation after 200 ns has passed, but it remains stable. Similar to the control, compound 9 remains stable throughout the simulation time. Compound 1 shows some fluctuations within the binding side until the pyridoindole group is in an optimum position to form H-bonds with GLN-221, and the alcohol group is in an optimum position to form H-bonds with THR-298 and ARG-299. Compound 1 then binds stably to these residues for the rest of the simulation time. Except for compound 1, all ligands had no major structural deviations from their starting docking conformations. Average RMSD values for ligand heavy atoms ranged from 3.2–5 Å. Compounds 9 and 1 had the closest average RMSD values to the control, while compound 5 had the highest RMSD value.
- Control-GLP-1R complex
The control compound forms a hydrogen bond with GLN-221. More importantly, it forms strong and stable π-cation interactions with three key amino acids, TRP-33, TRP-203, and PHE-381, for 87%, 80%, and 49% of the simulation time, respectively (Figure 7). These key interactions stabilize the complex and contribute to the known agonist activity of the control compound. The average binding free energy for the control-GLP-1R complex was −97.30 kcal/mol.
- Compound 1-GLP-1R complex
Compound 1 forms H-bonds with GLN-221, THR-298, and ARG-299 (Figure OB), and the former two can stabilize the complex since they last for more than half the simulation time. It also forms a π–π stacking interaction with TRP-214 and a π–cation interaction with TRP-203. Although TRP-203 is important for the activity of non-peptide GLP-1R agonists, the interaction of this residue with compound 1 is not strong. The average binding free energy for the compound 1-GLP-1R complex was −97.87 kcal/mol, similar to the control ligand. This could indicate that compound 1 is a good binder.
- Compound 5-GLP-1R complex
Compound 5 forms a H-bond with GLU-138 (Figure OC). It also forms π–π stacking interactions and π–cation interactions with two key amino acids, TRP-33 and TRP-203, with the latter lasting for more than half the simulation time. Such strong interactions with TRP-203 can stabilize the complex and contribute to compound activity, potentially as a GLP-1R agonist. The average binding free energy for the compound 5-GLP-1R complex was −92.50 kcal/mol, a value close to that of the control ligand. These findings indicate that compound 5 may be a potential GLP-1R agonist.
- Compound 9-GLP-1R complex
Compound 9 forms a H-bond with ASP-198 through a water bridge (Figure OD). It also forms π–π stacking interactions with important amino acids TRP-203 and PHE-381 for more than half the simulation time. Stable π–cation interactions are also observed between compound 9 and the key amino acids TRP-33 and TRP-203, lasting for 89% and 93% of the simulation time, respectively. The average binding free energy for the compound 9-GLP-1R complex was −102.78 kcal/mol, the lowest value among all four complexes. Compound 9 seems the most promising as a GLP-1R agonist.
The average MM-GBSA ΔGbind values obtained from the MD simulation frames of the control and the three compounds were low, comparable, and indicative of strong binding. The affinity ranking of compounds based on MM-GBSA ΔGbind values often correlates with the ranking observed in experimental settings. Accordingly, the binding affinity of the ligands can be predictively ranked as 9 (−102.78 kcal/mol) > 1 (−97.87 kcal/mol) > control (−97.30 kcal/mol) > 5 (−92.50 kcal/mol). Compound 9 outperformed compounds 1, 5, and the control in terms of predicted binding affinity.
Results from the MD simulations and the binding free energy calculations indicate that compound 9 is the most promising since it demonstrates the highest predicted affinity. For most of the simulation time, this compound maintains stable interactions with three key amino acids that are important for non-peptide agonist activity. Its binding profile in terms of energy, interactions, and stability within the binding site is similar to, and sometimes better than, that of the control, which further supports its potential as a promising non-peptide GLP-1R agonist. On the other hand, compound 1 has favorable energetics and seems to strongly bind to GLP-1R through different amino acids than the other three compounds. This warrants further investigation into its activity to determine whether such binding behavior will also result in GLP-1R activation. Compounds 1 and 9 have comparable ligand heavy-atom average RMSD values to the control and remain stable in the binding site. Compound 5 maintains interactions with one important amino acid for more than half the simulation time and has a favorable binding free energy. All three hits interact with at least one residue for more than 50% of the simulation time, show protein stability, and exhibit minimum structural fluctuations for at least half of the simulation time.
Despite not having direct experimental or clinical evidence concerning the exact molecules, a complete review of specific compounds from the COCONUT and CMNPD (marine) databases reveals a diverse array of structural classes with potential anti-diabetic properties (Table 3 and Table 4). Among the compounds originating from coconut, CNP0593098.1 (3,4,5,6-tetradehydroyohimbine) stands out. No research has directly addressed this derivative, but anti-diabetic activity has been demonstrated for its parent compound, yohimbine, in animal models via α2-adrenoceptor antagonism, resulting in increased insulin secretion and improved glucose tolerance. Neither this derivative nor other derivatives of yohimbine have, however, been able to show GLP-1 agonist activity.
Other compounds, as found from COCONUT, include CNP0402650.0 and CNP0311770.0 (chromen derivatives). These belong to structural classes with an established reputation for antioxidant activities, inhibitory effects on carbohydrate-hydrolyzing enzymes (like α-glucosidase), and insulin-sensitizing effects. Despite this, no studies have researched these specific molecules for these functions. Other compounds from coconut, such as CNP0542406.3, CNP0510864.0, and CNP0294111.0, are also characterized by their structural cores of indole, pyridine, and benzofuran, which are recurrent in anti-diabetic studies, mainly for their oxidative stress reduction, insulin sensitizing, and pancreatic β-cell protecting effects; once again, no literature records exist for these particular compounds.
Continuing in the same vein, marine-derived compounds cataloged under CMNPD appear to harbor equally enticing avenues. Some of the quinazoline alkaloids such as CMNPD5314 (fumiquinazoline B) and CMNPD27343 (versiquinazoline B) resemble structurally known compounds that inhibit α-glucosidase and protein tyrosine phosphatase 1B (PTP1B), two well-studied anti-diabetic targets. Other marine substances, comprising CMNPD2041 (waixenicin A), CMNPD24592 and CMNPD24593 (speradines D and E), and CMNPD26002 (a gliotoxin derivative), are diterpenes and alkaloids that are known, at least in structurally related forms, for anti-inflammatory and metabolic regulating actions, including PTP1B inhibition and AMPK activation.
Marine-derived epipolythiodioxopiperazines, in addition, activate the Nrf2/HO-1 pathway and thus would confer cytoprotective and antioxidant effects that preserve β-cell function. Even though there are no direct studies regarding anti-diabetic or GLP-1 activities for specific compounds such as CMNPD27661, CMNPD4014, CMNPD9270, and CMNPD22795, the structural classes to which they belong, including polyketides, prostaglandin derivatives, and benzofurans, are increasingly associated with redox modulation, enzyme inhibition, and generalized metabolic benefits.
In summary, compounds from both coconut and marine natural product databases present strong potential as anti-diabetic agents from the standpoints of structural features, mechanistic plausibility, and class-based pharmacological evidence. Compounds from coconut are notably important for insulinotropic and β-cell-protective effects, while those marine ones would provide additional scaffolds for enzyme and redox pathways involved in glucose homeostasis. Although bioactivity data on these particular molecules are absent, the confluence of evidence available for their structurally related counterparts renders them promising agents in multi-target anti-diabetic drug discovery and clearly worthy of intensive evaluation.
The literature-supported findings affirm the pharmacophore hypotheses and in silico prediction proposed in the current investigation. They promote the contention that natural product scaffolds, selected via rational filters combined with docking and shape-similarity methods, constitute a rich source of non-peptidic GLP-1 receptor modulators.
2.8. ADMET and drug-likeness
Water solubility in all compounds ranges from –3.851 to –2.791 log mol/L, compound 3 having the highest solubility and compound 6 the lowest solubility. Despite this difference, most compounds have moderate to low solubility, with compounds 5 below average. With respect to the Caco-2 permeability scale, 1, 2, 5, 6, 8, and 9 can be stated to have good membrane permeability (>0.5 log Papp), allowing easy passage across the intestinal epithelial barriers; while 3, 4, 7, and 10, get below this threshold, showing less permeability. Intestinal absorption from the human GI tract is generally high, above 95% for compounds 1, 2, 5, 6, 8, and 9, implying a good oral bioavailability. On the contrary, 3 is poorly absorbed (3.62% absorption), with subpar values for 4 and 10 (48%). Nearly all the candidates are P-glycoprotein substrates, except for 3 and 7, which probably increase retention inside the cell.
On the permeability of the blood-brain barrier (BBB), all compounds show a log BB value under the +0.3 threshold, thereby implying low or just moderate penetration into the CNS. Compounds 1, 2, and 8, albeit close to this threshold, are considered moderate. CNS Permeability (log PS) values are uniformly low, as all compounds apart from compound 8 (-1.894) are below the –2.0 mark, confirming little CNS exposure and so little risk of CNS-associated side effects.
Compounds 1, 2, 6, and 8 are substrates of CYP3A4. Compounds 3 and 4 are substrates of CYP2D6, which may impede their metabolic stability. Regarding CYP450 inhibitory activity, the scenario changes since compounds 1, 2, 4, 5, 6, and 8 inhibit at least one isoform, interfering with the metabolism of some co-administered drugs. Compound 4 inhibits three CYP enzymes (i.e., CYP1A2, CYP2C9, and CYP3A4) and can, therefore, be considered at greater risk for metabolic interaction. Compounds 3, 7, and 10 show little or no CYP inhibition and therefore seem to possess a clean metabolic profile.

Table 5.
ADMET profiling of the 10 hits from the CMNPD database.
| ADMET Parameters | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Control |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Absorption | |||||||||||
| Water solubility (log mol/L) | -3.506 | -3.489 | -2.791 | -2.998 | -3.011 | -.851 | -3.745 | -3.583 | -3.32 | -3.005 | -2.96 |
| Caco2 permeability (log Papp in 10-6 cm/s) | 1.135 | 1.198 | -0.546 | 0.151 | 0.994 | 1.23 | -0.003 | 0.829 | 1.158 | -0.305 | 0.986 |
| Intestinal absorption (human) (% Absorbed) | 97.322 | 96.618 | 3.62 | 48 | 95 | 100 | 54 | 96 | 100 | 48 | 63.055 |
| P-glycoprotein substrate (Yes/No) | Yes | Yes | No | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes |
| Distribution | |||||||||||
| BBB permeability (log BB) | -0.043 | 0.228 | -1.479 | -1.378 | -0.133 | -0.398 | -1.031 | 0.058 | -0.485 | -1.047 | -1.635 |
| CNS permeability (log PS) | -2.209 | -2.002 | -3.698 | -2.508 | -2.361 | -3.778 | -3.464 | -1.894 | -2.565 | -2.895 | -3.746 |
| Metabolism | |||||||||||
| CYP2D6 substrate (Yes/No) | No | No | Yes | Yes | No | No | No | No | No | Yes | No |
| CYP3A4 substrate (Yes/No) | Yes | Yes | No | No | No | Yes | No | Yes | Yes | No | No |
| CYP1A2 inhibitior (Yes/No) | Yes | Yes | No | Yes | Yes | No | No | Yes | Yes | No | No |
| CYP2C19 inhibitior (Yes/No) | No | No | No | No | Yes | Yes | No | No | No | No | No |
| CYP2C9 inhibitior (Yes/No) | No | No | No | Yes | No | Yes | No | No | No | No | No |
| CYP2D6 inhibitior (Yes/No) | No | Yes | No | No | Yes | No | No | No | No | No | No |
| CYP3A4 inhibitior (Yes/No) | No | No | No | No | Yes | Yes | No | No | Yes | No | No |
| Excretion | |||||||||||
| Total Clearance (log ml/min/kg) | 1.003 | 1.189 | 1.021 | 0.497 | 1.11 | 0.763 | 0.099 | 1.1 | 0.928 | 0.844 | 0.664 |
| Renal OCT2 substrate (Yes/No) | No | No | No | No | Yes | No | No | Yes | No | No | No |
| Toxicity | |||||||||||
| AMES toxicity (Yes/No) | Yes | Yes | No | No | No | No | No | Yes | No | No | No |
| Max. tolerated dose (human) (log mg/kg/day) | -0.349 | -0.779 | 0.31 | 0.742 | -0.154 | 0.528 | 0.595 | 0.122 | -0.647 | 0.62 | 0.664 |
| hERG I inhibitor (Yes/No) | No | No | No | No | No | No | No | No | No | No | No |
| Hepatotoxicity (Yes/No) | Yes | Yes | Yes | Yes | No | Yes | No | Yes | Yes | Yes | Yes |
Table 6.
Physicochemical properties, drug-likeness, and medicinal chemistry prediction of 10 hits from the CMNPD database. .
Table 6.
Physicochemical properties, drug-likeness, and medicinal chemistry prediction of 10 hits from the CMNPD database. .
| Molecule properties | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Control |
| Physicochemical properties | |||||||||||
| Molecular Weight | 351.42 | 349.40 | 397.38 | 404.42 | 336.32 | 417.41 | 354.31 | 344.36 | 389.43 | 333.34 | 543.59 |
| LogP | 1.98 | 2.08 | 0.68 | 2.04 | 2.11 | 3.22 | 1.63 | 2.65 | 2.07 | 1.69 | 1.96 |
| #Acceptors | 3 | 3 | 7 | 6 | 5 | 7 | 7 | 3 | 4 | 6 | 8 |
| #Donors | 2 | 1 | 5 | 4 | 2 | 2 | 3 | 1 | 1 | 3 | 1 |
| #Heavy atoms | 26 | 26 | 29 | 30 | 25 | 31 | 26 | 26 | 29 | 24 | 40 |
| #Arom. heavy atoms | 13 | 13 | 9 | 20 | 18 | 22 | 12 | 19 | 20 | 10 | 21 |
| Fraction Csp3 | 0.43 | 0.33 | 0.20 | 0.13 | 0.11 | 0.12 | 0.21 | 0.14 | 0.18 | 0.35 | 0.33 |
| #Rotatable bonds | 2 | 2 | 8 | 8 | 1 | 6 | 2 | 2 | 6 | 7 | 10 |
| Molar refractivity | 101.04 | 100.49 | 108.21 | 111.41 | 93.35 | 114.92 | 88.51 | 99.10 | 112.34 | 88.21 | 149.31 |
| TPSA (Ų) | 66.20 | 55.20 | 152.08 | 115.56 | 72.25 | 109.86 | 121.13 | 69.03 | 90.57 | 116.84 | 117.53 |
| Drug-likeness | |||||||||||
| Lipinski alert | yes | yes | no | yes | yes | yes | yes | yes | yes | Yes | yes |
| Ghose | yes | yes | no | yes | yes | yes | yes | yes | yes | Yes | no |
| Veber | yes | yes | no | yes | yes | yes | yes | yes | yes | Yes | Yes |
| Egan | yes | yes | no | yes | yes | yes | yes | yes | yes | Yes | Yes |
| Muegge | yes | yes | no | yes | yes | yes | yes | yes | yes | Yes | Yes |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.56 | 0.55 | 0.55 | 0.56 | 0.55 |
| Medicinal chemistry | |||||||||||
| PAINS | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Brenk | 1 | 1 | 0 | 0 | 3 | 0 | 0 | 1 | 2 | 1 | 0 |
| Synthetic accessibility | 3.99 | 4.36 | 4.61 | 3.70 | 2.68 | 4.06 | 3.88 | 3.00 | 2.94 | 3.18 | 4.75 |
Total clearance is from 0.099 to 1.189 log ml/min/kg. A greater clearance above 1 is for compounds 1, 2, 5, and 8, which means that they are quickly eliminated systemically. Compound 7 has a very low clearance value and therefore remains exposed longer. Only compounds 5 and 8 are substrates of renal OCT2, suggesting that renal excretion may occur, a factor that should be kept in mind when considering dosage for patients with renal impairment.
Still, toxicological profiling is suspected to be a risk for several candidates. Compounds 1, 2, and 8 are AMES-positive, which points toward mutagenicity. The maximum tolerated dose varies among compounds 3, 4, 6, 7, and 10, and is generally high. Hepatotoxicity seems to be the leading cause for concern, observed in all except compounds 5 and 7, thus supporting the presumption that liver safety is a top priority. It is interesting to note that no compounds are considered to inhibit hERG I channel, thus leaving cardiotoxicity out of consideration.
Most of the compounds (1, 2, 5, 6, and 8) exhibit appreciable absorption; however, compound 3 arises as poorly soluble and absorbed and in need of structural modification. None of the compounds are presumed to cross the BBB or to promote significant CNS penetration. Compounds 3, 7, and 10 contain minimal CYP inhibition, while compounds 4 and 6 represent high metabolic liability. Most compounds were cleared efficiently, with compound 7 being an outlier. In terms of toxicity, compound 5 has a better profile, free from AMES toxicity and hepatotoxicity. These profiles are taken to lay the groundwork for prioritization for experimental validation.
3. Materials and Methods
3.1. Materials and Software
For our computational studies, we utilized the Schrödinger’s Maestro molecular modeling software (Versions 2024-4 and 2025-1) [32,67,68]. Our computational work was conducted on a desktop workstation equipped with an Intel® Core™ i7-10700F Processor, running the Linux Ubuntu 22.10 Operating System, and featuring an RTX 5000 graphics card (RDIA 12990-iau-2023-iau-R-3-1-HW: P.O. 6947 License key: 03cb87b8-723c-4fec-9b8c-8a58137d7a76).
3.2. Database Preparation
We obtained a remarkable comprehensive natural products (NPs) database that contains 695,133 NPs from the Coconut website https://coconut.naturalproducts.net/;” URL (accessed on 15 November 2024) [35]. In addition, we downloaded over 46,000 compounds from the Comprehensive Marine Natural Products database (CMNPD), which was retrieved from the online server (https://www.cmnpd.org/);” URL (accessed on 18 November 2024)” [36].
The collected structures were filtered using Schrödinger’s Canvas based on their physicochemical descriptors, including MW, logP, number of rings, total heavy atoms count, overall charge, number of H-donors, number of H-acceptors, number of rotatable bonds, total charge, and QED score. The final database filtration ensured compliance with the rule of five [69] and maintained a natural product-like (NPL) score <2 [70].
3.3. Shape Screening
Our investigation was carried out using Schrödinger's Maestro 's form screening tool. We started by adding a default RMSD of 0.30 Å, specifically for non-hydrogen atoms, to the filtered database in order to decrease its energy using the Optimized Potentials for Liquid Simulations 3 (OPLS3) force field. Following this optimization, shape screening was carried out with the crystal ligand structure serving as a guide. We used a number of different pharmacophore types in conjunction with a volume-scoring algorithm to precisely evaluate the compounds. We ranked the compounds according to their shape similarity score, setting a limit of 0.3 to further refine our virtual screening database. The evaluation was based on pharmacophore features [71].
3.4. Crystal Structures
3.5. Protein Preparation
This protein structure was prepared for docking using Maestro’s Protein Preparation Workflow. To fine-tune the ionization states, the preparation and minimization process were carried out at a pH 7.4. We added polar hydrogens and eliminated extraneous water molecules from the structures. Using the OPLS3 force field to optimize the receptors was the last stage. Under the OPLS3 force field, the ligand-protein complexes were optimized and minimized using a preset RMSD value of 0.30 for non-hydrogen atoms [73]. Then, receptor grids with a radius of 1.00 A van der waals (vdW) were made using the center of the bound ligand. In this procedure, a vdW radius was applied and the threshold for partial charges was set at 0.25 [74]. The binding sites were contained within a grid box of 20 Å using default parameters and without any constraints
3.6. Ligand Library Preparation
The LigPrep tool, integrated into the virtual screening workflow of Maestro, was used to prepare the filtered ligands. The three-dimensional structures of the ligands were created by the addition of hydrogen atoms, followed by the generation of the most probable ionization states at pH 7±2 using Epik. The geometry of the ligands was then optimized with the OPLS3 force field, which included tautomer generation, desalting, and producing a maximum of 32 isomers per ligand [67,75]. The resulting conformations served as the starting input structures for the virtual screening workflow.
3.7. Validation of Molecular Docking
The molecular docking protocols were evaluated in order to confirm Maestro Glide’s accuracy in predicting the ligand’s docking poses for the protein under study [76,77,78]. The library screening criteria were used to dock the cognate ligand into the GLP1 receptor. The Maestro structure superimposition function has been used to further align the docking poses with the lowest binding energy with the crystal structure conformation, and the aligned RMSD has been calculated.
3.8. Virtual Screening Workflow
Three distinct methods were employed to structure the virtual screening workflow: Standard Precision Docking, Extra Precision Docking, and High-Throughput Virtual Screening. Therefore, flexible docking was utilized in conjunction with post-docking minimization for each approach, where each compound generated three poses and retained up to 10% of the highest-scoring compounds. During the docking procedure, no additional filters or constraints were applied. The actual parameters included a vdw radius scaling factor of 0.80 and a partial charge cut-off of 0.15. The ligands and hit molecules were ranked using the XP score. For binding free energy calculations in molecular mechanics-generalized Born surface area (MM-GBSA), the virtual screening pipeline estimated the binding affinity of top-scoring hits using the Prime MM-GBSA module (Figure 9).
3.9. Molecular Dynamics (MD) Simulations
Molecular dynamics (MD) simulations were conducted for compounds 1, 5, 9, and the control compound, using the Desmond Module in the Schrödinger suite [79,80,81]. Desmond’s system builder tool was used to prepare the protein-ligand complexes for MD simulations. Since GLP-1R is a transmembrane (TM) protein and our ligands mainly bind in the TM region, a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayer membrane was added at 300 K [82]. The simple point charge (SPC) water model was used for an orthorhombic box with dimensions of 10 Å × 10 Å × 10 Å [83]. A 20 Å distance from the ligand covered the region for exclusion of ion and salt placement. The number of required counter ions was calculated and subsequently added to neutralize the system. Sodium and chloride ions were used to maintain a 0.15 M salt concentration, and the OPLS5 force field was selected for running the simulations [84]. The environmental conditions included a temperature of 300 K, a pressure of 1 bar, and the NP γT ensemble were set [85,86]. The membrane model system was relaxed before running 100-500 ns simulations. Coordinates were recorded every 500 ps, and 1000 frames were generated for the trajectory. The temperature was controlled using the Nose-Hoover chain coupling scheme with a 1 ps coupling constant. The pressure was controlled using the Martyna-Tobias-Klein coupling scheme with a 2 ps coupling constant. A time step of 2 fs was selected for RESPA (Reference System Propagator Algorithm), and a 9 Å cutoff radius was set for Coulombic short-range interactions [86]. Desmond’s Simulation Interaction Diagram tool was used to analyze data generated from the MD simulations [87]. The RMSD values were evaluated to determine how stable the ligand-protein complexes are, and root-mean square fluctuation (RMSF) was used to assess individual residue fluctuations [88].
3.10. Binding Free Energy Calculations for MD Frames
The binding free energy was calculated for frames generated from MD simulations for compounds 1, 5, 9, and the control. Schrödinger’s MM-GBSA continuum solvation method and the thermal_mmgbsa script were utilized [89]. A step size of five was used, and average ΔGbind values were reported. These calculations provide more thorough and accurate insights into the binding energetics for these compounds and can help rank them based on predicted affinities [90,91].
3.11. ADMET Profiling
To predict the ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties as well as drug-likeness characteristics for the selected putative inhibitors used as modeling input, we utilized the pkCSM web server (http://biosig.unimelb.edu.au/pkcsm/prediction accessed on 13 December 2024) [91]. Eight molecular descriptors were generated based on the ADMET descriptors identified in the potential hits. Additionally, Swiss ADME (www.swissadme.ch/ accessed 13 December 2024) was employed for the in silico calculations of physicochemical parameters, medicinal chemistry-like properties, and drug-likeness attributes [92,93].
4. Conclusions
More than 700,000 compounds from natural sources obtained from the COCONUT and Marine Natural Products (CMNPD) libraries were screened for agonist activity on the orthosteric site of GLP-1R to identify novel non-peptide compounds that would have better clinical usefulness. A filtration protocol combining both receptor-based and ligand-based approaches was utilized including shape screening, precision docking, binding energy (MM-GBSA) calculations and 500 ns molecular dynamic simulations. Pharmacokinetics prediction was also performed on the hit compounds to establish their drug-likeness potential and clinical applicability. Twenty final hits were identified from both databases with some hits including 1, 6, 7, 10 having previously reported antidiabetic activity but no evidence of a GLP-1 activity. The most active hits 1, 5, 9 showed excellent binding profile with key residues and binding stability as indicated by the binding scores and the RMSF and RMSD values respectively. These results opened the floor for further experimental studies to confirm the GLP-1 mediated antidiabetic activity of those hits. These results also substantiated the primary identification of other hits and positioned them as novel non-peptide structural scaffolds for GLP-1 orthosteric agonist activity with subsequent antidiabetic effect. The predicted ADMET profiling was acceptable especially in terms of drug-likeness and bioavailability. The current study identified new lead compounds for GLP-1R orthosteric agonist activity that on one hand represents novel non-peptide scaffolds for optimization and preclinical development and on the other hand proposed a GLP-1R mediated activity for those hits with known antidiabetic activity for further experimental testing.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.S.A., and M.S.G.; methodology, M.S.A., R.A.A., M.S.G., D.H., A.H.A.K., and T.A.R.; software, M.S.A., R.A.A., M.S.G., D.H., A.H.A.K., and T.A.R.; validation, M.S.A., R.A.A., M.S.G., D.H., N.T., A.H.A.K., M.F.A., and T.A.R.; writing—original draft preparation, M.S.A., R.A.A., M.S.G., D.H.,N.T., A.H.A.K., M.F.A., S.A.A., and T.A.R.; writing—review and editing, M.S.A., R.A.A., M.S.G., D.H.,N.T., A.H.A.K., M.F.A., S.A.A., and T.A.R.; supervision, M.S.A.; funding acquisition, M.S.A., R.A.A., M.S.G., D.H., A.H.A.K., M.F.A., and T.A.R.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Acknowledgments
We would like to acknowledge the RDIA and the integrative and translation research lab and for the use of the Schrodinger software suite. We would also like to thank the STU and IAU for their support and guidance. The authors would also like to express their thanks to the University of Utah, United States of America, and the Council for Scientific and Industrial Research—Center for High-Performance Computing (CSIR/CHPC) in Cape Town, South Africa.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; Pavkov, M.E.; Ramachandaran, A.; Wild, S.H.; James, S.; Herman, W.H.; Zhang, P.; Bommer, C.; Kuo, S.; Boyko, E.J.; Magliano, D.J. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006, 3, e442. [Google Scholar] [CrossRef]
- Sarzani, R.; Landolfo, M.; Di Pentima, C.; Ortensi, B.; Falcioni, P.; Sabbatini, L.; Massacesi, A.; Rampino, I.; Spannella, F.; Giulietti, F. Adipocentric origin of the common cardiometabolic complications of obesity in the young up to the very old: pathophysiology and new therapeutic opportunities. Front Med (Lausanne) 2024, 11, 1365183. [Google Scholar] [CrossRef]
- Smith, N.K.; Hackett, T.A.; Galli, A.; Flynn, C.R. GLP-1: Molecular mechanisms and outcomes of a complex signaling system. Neurochem Int 2019, 128, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Abiola, J.O.; Oluyemi, A.A.; Idowu, O.T.; Oyinloye, O.M.; Ubah, C.S.; Owolabi, O.V.; Somade, O.T.; Onikanni, S.A.; Ajiboye, B.O.; Osunsanmi, F.O.; et al. Potential Role of Phytochemicals as Glucagon-like Peptide 1 Receptor (GLP-1R) Agonists in the Treatment of Diabetes Mellitus. Pharmaceuticals 2024, 17, 736. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.; Kleinman, W.A.; Singh, L.; Singh, G.; Raufman, J.P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem 1992, 267, 7402–7405. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schäffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; Strauss, H.M.; Gram, D.X.; Knudsen, S.M.; Nielsen, F.S.; Thygesen, P.; Reedtz-Runge, S.; Kruse, T. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J Med Chem 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Jimenez-Solem, E.; Rasmussen, M.H.; Christensen, M.; Knop, F.K. Dulaglutide, a long-acting GLP-1 analog fused with an Fc antibody fragment for the potential treatment of type 2 diabetes. Curr Opin Mol Ther 2010, 12, 790–797 . [Google Scholar] [CrossRef]
- Knudsen, L.B.; Agerso, H.; Bjenning, C.; Bregenholt, S.; Carr, R.D.; Godtfredsen, C.; Holst, J.J.; Huusfeldt, P.O.; Larsen, M.O.; Larsen, P.J. GLP-1 derivatives as novel compounds for the treatment of type 2 diabetes: selection of NN2211 for clinical development. Drugs of the Future 2001, 26. [Google Scholar] [CrossRef]
- Zheng, Z.; Zong, Y.; Ma, Y.; Tian, Y.; Pang, Y.; Zhang, C.; Gao, J. Glucagon-like peptide-1 receptor: mechanisms and advances in therapy. Signal Transduction and Targeted Therapy 2024, 9, 234. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.D.; Wadden, T.A. Once-weekly semaglutide in adults with overweight or obesity. New England Journal of Medicine 2021, 384, 989–1002. [Google Scholar] [CrossRef]
- Xie, Z.; Yang, S.; Deng, W.; Li, J.; Chen, J. Efficacy and Safety of Liraglutide and Semaglutide on Weight Loss in People with Obesity or Overweight: A Systematic Review. Clin Epidemiol 2022, 14, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, X.; Astrup, A.; Fujioka, K.; Greenway, F.; Halpern, A.; Krempf, M.; Lau, D.C.; le Roux, C.W.; Violante Ortiz, R.; Jensen, C.B.; Wilding, J.P. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N Engl J Med 2015, 373, 11–22. [Google Scholar] [CrossRef]
- Gogineni, P.; Melson, E.; Papamargaritis, D.; Davies, M. Oral glucagon-like peptide-1 receptor agonists and combinations of entero-pancreatic hormones as treatments for adults with type 2 diabetes: where are we now? Expert Opin Pharmacother 2024, 25, 801–818. [Google Scholar] [CrossRef]
- Drucker, D.J. Advances in oral peptide therapeutics. Nature Reviews Drug Discovery 2020, 19, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, B.; Feng, D.; Hu, H.; Chu, M.; Qu, Q.; Tarrasch, J.T.; Li, S.; Sun Kobilka, T.; Kobilka, B.K.; Skiniotis, G. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 2017, 546, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sun, B.; Yoshino, H.; Feng, D.; Suzuki, Y.; Fukazawa, M.; Nagao, S.; Wainscott, D.B.; Showalter, A.D.; Droz, B.A.; Kobilka, T.S.; Coghlan, M.P.; Willard, F.S.; Kawabe, Y.; Kobilka, B.K.; Sloop, K.W. Structural basis for GLP-1 receptor activation by LY3502970, an orally active nonpeptide agonist. Proc Natl Acad Sci U S A 2020, 117, 29959–29967. [Google Scholar] [CrossRef]
- Griffith, D.A.; Edmonds, D.J.; Fortin, J.-P.; Kalgutkar, A.S.; Kuzmiski, J.B.; Loria, P.M.; Saxena, A.R.; Bagley, S.W.; Buckeridge, C.; Curto, J.M.; Derksen, D.R.; Dias, J.M.; Griffor, M.C.; Han, S.; Jackson, V.M.; Landis, M.S.; Lettiere, D.; Limberakis, C.; Liu, Y.; Mathiowetz, A.M.; Patel, J.C.; Piotrowski, D.W.; Price, D.A.; Ruggeri, R.B.; Tess, D.A. A Small-Molecule Oral Agonist of the Human Glucagon-Like Peptide-1 Receptor. Journal of Medicinal Chemistry 2022, 65, 8208–8226. [Google Scholar] [CrossRef]
- Zhao, P.; Liang, Y.-L.; Belousoff, M.J.; Deganutti, G.; Fletcher, M.M.; Willard, F.S.; Bell, M.G.; Christe, M.E.; Sloop, K.W.; Inoue, A.; Truong, T.T.; Clydesdale, L.; Furness, S.G.B.; Christopoulos, A.; Wang, M.-W.; Miller, L.J.; Reynolds, C.A.; Danev, R.; Sexton, P.M.; Wootten, D. Activation of the GLP-1 receptor by a non-peptidic agonist. Nature 2020, 577, 432–436. [Google Scholar] [CrossRef]
- Wharton, S.; Blevins, T.; Connery, L.; Rosenstock, J.; Raha, S.; Liu, R.; Ma, X.; Mather Kieren, J.; Haupt, A.; Robins, D.; Pratt, E.; Kazda, C.; Konig, M. Daily Oral GLP-1 Receptor Agonist Orforglipron for Adults with Obesity. New England Journal of Medicine 2023, 389, 877–888. [Google Scholar] [CrossRef]
- Pratt, E.; Ma, X.; Liu, R.; Robins, D.; Haupt, A.; Coskun, T.; Sloop, K.W.; Benson, C. Orforglipron (LY3502970), a novel, oral non-peptide glucagon-like peptide-1 receptor agonist: A Phase 1a, blinded, placebo-controlled, randomized, single- and multiple-ascending-dose study in healthy participants. Diabetes Obes Metab 2023, 25, 2634–2641. [Google Scholar] [CrossRef] [PubMed]
- Pratt, E.; Ma, X.; Liu, R.; Robins, D.; Haupt, A.; Coskun, T.; Sloop, K.W.; Benson, C. Orforglipron (LY3502970), a novel, oral non-peptide glucagon-like peptide-1 receptor agonist: a Phase 1a, blinded, placebo-controlled, randomized, single-and multiple-ascending-dose study in healthy participants. Diabetes, Obesity and Metabolism 2023, 25, 2634–2641, (Duplicate of #21; same DOI). [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Dvergsten, C.; Dunn, I.; Valcarce, C. In TTP273, Oral (nonpeptide) GLP-1R agonist: improved glycemic control without nausea and vomiting in phase 2, 2017, AMER DIABETES ASSOC 1701 N BEAUREGARD ST, ALEXANDRIA, VA 22311-1717 USA: pp. A326-A326. (Conference abstract; no DOI).
- Davies, M.; Pieber, T.R.; Hartoft-Nielsen, M.L.; Hansen, O.K.H.; Jabbour, S.; Rosenstock, J. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial. Jama 2017, 318, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Xing, H.J.; Wang, B.; Fu, C.; Zhang, Y.S.; Qiao, X.; Guo, C.; Zhang, X.L.; Hu, B.; Zhao, X.; Deng, L.J.; Zhu, X.C.; Zhang, Y.; Liu, Y.F. Cinchonine, a Potential Oral Small-Molecule Glucagon-Like Peptide-1 Receptor Agonist, Lowers Blood Glucose and Ameliorates Non-Alcoholic Steatohepatitis. Drug Des Devel Ther 2023, 17, 1417–1432. [Google Scholar] [CrossRef]
- Deacon, C.F.; Nauck, M.A.; Meier, J.; Hücking, K.; Holst, J.J. Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab 2000, 85, 3575–3581. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; Steinberg, W.M.; Stockner, M.; Zinman, B.; Bergenstal, R.M.; Buse, J.B. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Berndt, J.; Ooi, S.L.; Pak, S.C. What Is the Mechanism Driving the Reduction of Cardiovascular Events from Glucagon-Like Peptide-1 Receptor Agonists?—A Mini Review. Molecules 2021, 26. [Google Scholar] [CrossRef]
- Grover, J.K.; Yadav, S.; Vats, V. Medicinal plants of India with anti-diabetic potential. Journal of Ethnopharmacology 2002, 81, 81–100. [Google Scholar] [CrossRef]
- Akawa, A.B.; Oyinloye, B.E.; Ajiboye, B.O. Computer-aided Identification of Bioactive Compounds from Brachystegia eurycoma with Therapeutic Potential against Drug Targets of Type 2 Diabetes mellitus. Biointerface Res. Appl. Chem 2022, 13, 454. [Google Scholar] [CrossRef]
- Ajiboye, B.O.; Iwaloye, O.; Owolabi, O.V.; Ejeje, J.N.; Okerewa, A.; Johnson, O.O.; Udebor, A.E.; Oyinloye, B.E. Screening of potential antidiabetic phytochemicals from Gongronema latifolium leaf against therapeutic targets of type 2 diabetes mellitus: multi-targets drug design. SN Applied Sciences 2021, 4, 14. [Google Scholar] [CrossRef]
- Gomaa, M.S.; Alturki, M.S.; Tawfeeq, N.; Hussein, D.A.; Pottoo, F.H.; Al Khzem, A.H.; Sarafroz, M.; Abubshait, S. Discovery of Non-Peptide GLP-1 Positive Allosteric Modulators from Natural Products: Virtual Screening, Molecular Dynamics, ADMET Profiling, Repurposing, and Chemical Scaffolds Identification. Pharmaceutics 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nature Chemistry 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. Journal of Cheminformatics 2021, 13, 2. [Google Scholar] [CrossRef]
- Lyu, C.; Chen, T.; Qiang, B.; Liu, N.; Wang, H.; Zhang, L.; Liu, Z. CMNPD: a comprehensive marine natural products database towards facilitating drug discovery from the ocean. Nucleic Acids Research 2021, 49, D509–D515. [Google Scholar] [CrossRef]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: insights into database screening performance using eight fingerprint methods. J Mol Graph Model 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Alzyoud, L.; Bryce, R.A.; Al Sorkhy, M.; Atatreh, N.; Ghattas, M.A. Structure-based assessment and druggability classification of protein–protein interaction sites. Scientific Reports 2022, 12, 7975. [Google Scholar] [CrossRef]
- Veeramachaneni, G.K.; Raj, K.K.; Chalasani, L.M.; Annamraju, S.K.; Js, B.; Talluri, V.R. Shape based virtual screening and molecular docking towards designing novel pancreatic lipase inhibitors. Bioinformation 2015, 11, 535–542. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; Shaw, D.E.; Francis, P.; Shenkin, P.S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. Journal of Medicinal Chemistry 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Tsuneki, H.; Maeda, T.; Takatsuki, M.; Sekine, T.; Masui, S.; Onishi, K.; Takeda, R.; Sugiyama, M.; Sakurai, T.; Yanagisawa, M.; Wada, T.; Sasaoka, T. Bromocriptine Improves Glucose Tolerance in Obese Mice via Central Dopamine D2 Receptor-Independent Mechanism. PLoS ONE 2025, 20(3), e0320157. [Google Scholar] [CrossRef]
- Tamaddonfard, E.; Naderi, S.; Nafisi, S.; Soltanalinejad, F. Effect of Thymoquinone on Acetic Acid-Induced Visceral Nociception in Rats: Role of Central Cannabinoid and A2-Adrenergic Receptors. Vet Res Forum 2024, No. Online First. [CrossRef]
- Romeo, I.; Vallarino, G.; Turrini, F.; Roggeri, A.; Olivero, G.; Boggia, R.; Alcaro, S.; Costa, G.; Pittaluga, A. Presynaptic Release-Regulating Alpha2 Autoreceptors: Potential Molecular Target for Ellagic Acid Nutraceutical Properties. Antioxidants 2021, 10(11), 1759. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A. C.; Steyn, L. V.; Kitzmann, J. P.; Anderson, M. J.; Mueller, K. R.; Hart, N. J.; Lynch, R. M.; Papas, K. K.; Limesand, S. W. Function and Expression of Sulfonylurea, Adrenergic, and Glucagon-like Peptide 1 Receptors in Isolated Porcine Islets. Xenotransplantation 2014, 21(4), 385–391. [Google Scholar] [CrossRef]
- Li, S.; Xu, H.; Cui, S.; Wu, F.; Zhang, Y.; Su, M.; Gong, Y.; Qiu, S.; Jiao, Q.; Qin, C.; Shan, J.; Zhang, M.; Wang, J.; Yin, Q.; Xu, M.; Liu, X.; Wang, R.; Zhu, L.; Li, J.; Xu, Y.; Jiang, H.; Zhao, Z.; Li, J.; Li, H. Discovery and Rational Design of Natural-Product-Derived 2-Phenyl-3,4-Dihydro-2H-Benzo[f]Chromen-3-Amine Analogs as Novel and Potent Dipeptidyl Peptidase 4 (DPP-4) Inhibitors for the Treatment of Type 2 Diabetes. J. Med. Chem. 2016, 59(14), 6772–6790. [Google Scholar] [CrossRef] [PubMed]
- Menteşe, E.; Baltaş, N.; Bekircan, O. Synthesis and Kinetics Studies of N′-(2-(3,5-disubstituted-4H-1,2,4-triazol-4-yl)Acetyl)-6/7/8-substituted-2-oxo-2H-chromen-3-carbohydrazide Derivatives as Potent Antidiabetic Agents. Archiv der Pharmazie 2019, 352 (12). [CrossRef]
- Zhu, G.; Luo, Y.; Xu, X.; Zhang, H.; Zhu, M. Anti-Diabetic Compounds from the Seeds of Psoralea Corylifolia. Fitoterapia 2019, 139, 104373. [Google Scholar] [CrossRef]
- Seong, S. H.; Ha, M. T.; Min, B. S.; Jung, H. A.; Choi, J. S. Moracin Derivatives from Morus Radix as Dual BACE1 and Cholinesterase Inhibitors with Antioxidant and Anti-Glycation Capacities. Life Sciences 2018, 210, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Thabet, H. K.; Abusaif, M. S.; Imran, M.; Helal, M. H.; Alaqel, S. I.; Alshehri, A.; Mohd, A. A.; Ammar, Y. A.; Ragab, A. Discovery of Novel 6-(Piperidin-1-Ylsulfonyl)-2H-Chromenes Targeting α-Glucosidase, α-Amylase, and PPAR-γ: Design, Synthesis, Virtual Screening, and Anti-Diabetic Activity for Type 2 Diabetes Mellitus. Computational Biology and Chemistry 2024, 111, 108097. [Google Scholar] [CrossRef]
- Hosseini Ghazvini, S. M. B.; Safari, P.; Mobinikhaledi, A.; Moghanian, H.; Rasouli, H. Synthesis, Characterization, Anti-Diabetic Potential and DFT Studies of 7-Hydroxy-4-Methyl-2-Oxo-2H-Chromene-8-Carbaldehyde Oxime. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2018, 205, 111–131. [Google Scholar] [CrossRef]
- Parmar, K.; Aggarwal, G.; Sharma, U. α-Glucosidase Inhibiting Chromene Derivatives from Ageratum Conyzoides L. Essential Oil Extracted via NADES-Assisted Hydrodistillation. Chemistry & Biodiversity 2025, 22 (1). [CrossRef]
- Chen, Y.; Zhang, Y.; Li, X.; Wang, Y. The Role of Natural Products in Drug Discovery: Current Status and Future Perspectives. Front. Pharmacol. 2022, *13*, 830103. [CrossRef]
- Smith, A. B.; Jones, C. D. Fungal Secondary Metabolites: Biosynthesis and Applications. Front. Fungal Biol. 2023, *4*, 1088966. [CrossRef]
- Brown, E. F.; Green, G. H. Phytochemical Analysis of Medicinal Plants: Recent Advances. Phytochemistry 2024, *215*, 114206. [CrossRef]
- Wilson, D.; Taylor, R. Natural Compounds as Potential Therapeutic Agents: A Pharmacological Review. Biomed. Pharmacother. 2017, *88*, 1234–1242. [CrossRef]
- Lee, S.; Kim, H.; Park, J. Antioxidant and Anti-Inflammatory Effects of Plant-Derived Compounds. Oxid. Med. Cell. Longev. 2021, *2021*, 2691525. [CrossRef]
- Martinez, L.; Rodriguez, M. Advances in the Pharmacology of Natural Bioactive Molecules. Biomed. Pharmacother. 2018, *98*, 456–465. [CrossRef]
- Harris, P.; Robinson, K. Omega-3 Fatty Acids and Their Health Benefits. Prostaglandins Leukot. Essent. Fatty Acids 2005, *73* (5), 327–335. [CrossRef]
- Thompson, R.; Clark, S. Recent Advances in the Isolation and Characterization of Natural Products. Nat. Prod. Res. 2024, *38* (15), 1–15. [CrossRef]
- Anderson, K.; White, L. Bioactive Compounds from Traditional Medicinal Plants: Pharmacological Evaluation. Fitoterapia 2019, *137*, 104373. [CrossRef]
- Zhang, Y.; Wang, L.; Chen, X.; Li, J. Advances in Natural Product Drug Discovery: Mechanisms and Applications. J. Nat. Prod. 2020, *83* (6), 1895–1908. [CrossRef]
- Kumar, S.; Patel, R.; Sharma, M. Therapeutic Potential of Flavonoids in Neurodegenerative Diseases. Eur. J. Med. Chem. 2023, *245*, 114892. [CrossRef]
- Brown, A. D.; Smith, E. F. Antimicrobial Properties of Plant-Derived Alkaloids. Phytochemistry 2014, *97*, 112–120. [CrossRef]
- Wilson, K. L.; Garcia, M. P. Recent Advances in Cancer Chemoprevention Using Natural Compounds. Bioorg. Med. Chem. 2023, *88-89*, 117306. [CrossRef]
- Lee, H.; Park, S. Y. Anti-Inflammatory Effects of Terpenoids: Mechanisms and Clinical Implications. J. Ethnopharmacol. 2019, *239*, 111920. [CrossRef]
- Johnson, R. B.; Miller, T. K. Bioactive Compounds from Marine Organisms: A Review of Their Pharmacological Properties. Mar. Drugs 2014, *12* (12), 5986–6003. [CrossRef]
- Rants'o, T.A.; Johan van der Westhuizen, C.; van Zyl, R.L. Optimization of covalent docking for organophosphates interaction with Anopheles acetylcholinesterase. Journal of Molecular Graphics and Modelling 2022, 110, 108054. [Google Scholar] [CrossRef]
- Sithole, M.; Gabriels, G.; Rants’o, T.A. In silico analysis of the melamine structural analogues interaction with calcium-sensing receptor: A potential for nephrotoxicity. Computational Toxicology 2024, 32, 100333. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural Product-likeness Score and Its Application for Prioritization of Compound Libraries. Journal of Chemical Information and Modeling 2008, 48, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Advances in the Development of Shape Similarity Methods and Their Application in Drug Discovery. Front Chem 2018, 6, 315. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; Ganesan, S.; Goodsell, D.S.; Ghosh, S.; Green, R.K.; Guranović, V.; Guzenko, D.; Hudson, B.P.; Lawson, C.L.; Liang, Y.; Lowe, R.; Namkoong, H.; Peisach, E.; Persikova, I.; Randle, C.; Rose, A.; Rose, Y.; Sali, A.; Segura, J.; Sekharan, M.; Shao, C.; Tao, Y.P.; Voigt, M.; Westbrook, J.D.; Young, J.Y.; Zardecki, C.; Zhuravleva, M. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res 2021, 49, D437–d451. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; Kaus, J.W.; Cerutti, D.S.; Krilov, G.; Jorgensen, W.L.; Abel, R.; Friesner, R.A. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. Journal of Chemical Theory and Computation 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Ban, T.; Ohue, M.; Akiyama, Y. Multiple grid arrangement improves ligand docking with unknown binding sites: Application to the inverse docking problem. Computational Biology and Chemistry 2018, 73, 139–146. [Google Scholar] [CrossRef]
- Rants'o, T.A.; van Greunen, D.G.; van der Westhuizen, C.J.; Riley, D.L.; Panayides, J.L.; Koekemoer, L.L.; van Zyl, R.L. The in silico and in vitro analysis of donepezil derivatives for Anopheles acetylcholinesterase inhibition. PLoS One 2022, 17, e0277363. [Google Scholar] [CrossRef]
- Jaundoo, R.; Bohmann, J.; Gutierrez, G.E.; Klimas, N.; Broderick, G.; Craddock, T.J.A. Using a Consensus Docking Approach to Predict Adverse Drug Reactions in Combination Drug Therapies for Gulf War Illness. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Alturki, M.S.; Gomaa, M.S.; Tawfeeq, N.; Al Khzem, A.H.; Shaik, M.B.; Alshaikh Jafar, M.; Alsamen, M.; Al Nahab, H.; Al-Eid, M.; Almutawah, A.; et al. A Multifaceted Computational Approach to Identify PAD4 Inhibitors for the Treatment of Rheumatoid Arthritis (RA). Metabolites 2025, 15, 156. [Google Scholar] [CrossRef]
- Hussein, D. In Silico Investigation of the Human GTP Cyclohydrolase 1 Enzyme Reveals the Potential of Drug Repurposing Approaches towards the Discovery of Effective BH4 Therapeutics. International Journal of Molecular Sciences 2023, 24(2), 1210. [Google Scholar] [CrossRef] [PubMed]
- Alturki, M.S. Exploring Marine-Derived Compounds: In Silico Discovery of Selective Ketohexokinase (KHK) Inhibitors for Metabolic Disease Therapy. Mar. Drugs 2024, 22, 455. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. In Scalable algorithms for molecular dynamics simulations on commodity clusters, 2006, pp. 84-es. (Conference proceedings; no DOI).
- Al Khzem, A.H.; Shoaib, T.H.; Mukhtar, R.M.; Alturki, M.S.; Gomaa, M.S.; Hussein, D.; Tawfeeq, N.; Bano, M.; Sarafroz, M.; Alzahrani, R.; et al. Repurposing FDA-Approved Agents to Develop a Prototype Helicobacter pylori Shikimate Kinase (HPSK) Inhibitor: A Computational Approach Using Virtual Screening, MM-GBSA Calculations, MD Simulations, and DFT Analysis. Pharmaceuticals 2025, 18, 174. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhao, F.; Zhang, Y.; Yang, D.; Wang, M.-W. Structural pharmacology and mechanisms of GLP-1R signaling. Trends in Pharmacological Sciences 2025, 46, 422–436. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, Israel, April 13–16, 1981; Pullman, B., Ed.; Springer Netherlands: Dordrecht, 1981; pp. 331–342. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. Journal of the American Chemical Society 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Ikeguchi, M. Partial rigid-body dynamics in NPT, NPAT and NPγT ensembles for proteins and membranes. Journal of Computational Chemistry 2004, 25, 529–541. [Google Scholar] [CrossRef]
- Lippert, R.A.; Predescu, C.; Ierardi, D.J.; Mackenzie, K.M.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. The Journal of Chemical Physics 2013, 139, 164106. [Google Scholar] [CrossRef]
- Braga, L.; Manione, R.; Renditore, P. A Formal Description Language for the Modelling and Simulation of Timed Interaction Diagrams. In Formal Description Techniques IX: Theory, application and tools; Gotzhein, R., Bredereke, J., Eds.; Springer US: Boston, MA, 1996; pp. 245–260. [Google Scholar] [CrossRef]
- Alturki, M.S.; Tawfeeq, N.; Alissa, A.; Ahbail, Z.; S. Gomaa, M.; Al Khzem, A.H.; Rants'o, T.A.; Akbar, M.J.; Alharbi, W.S.; Alshehri, B.Y.; Alotaibi, A.N.; Almughem, F.A.; Alshehri, A.A. Targeting KRAS G12C and G12S mutations in lung cancer: In silico drug repurposing and antiproliferative assessment on A549 cells. Informatics in Medicine Unlocked 2025, 52, 101612. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; Donini, O.; Cieplak, P.; Srinivasan, J.; Case, D.A.; Cheatham, T.E. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Accounts of Chemical Research 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The comprehensive antidiabetic effects of GLP-1 signaling.
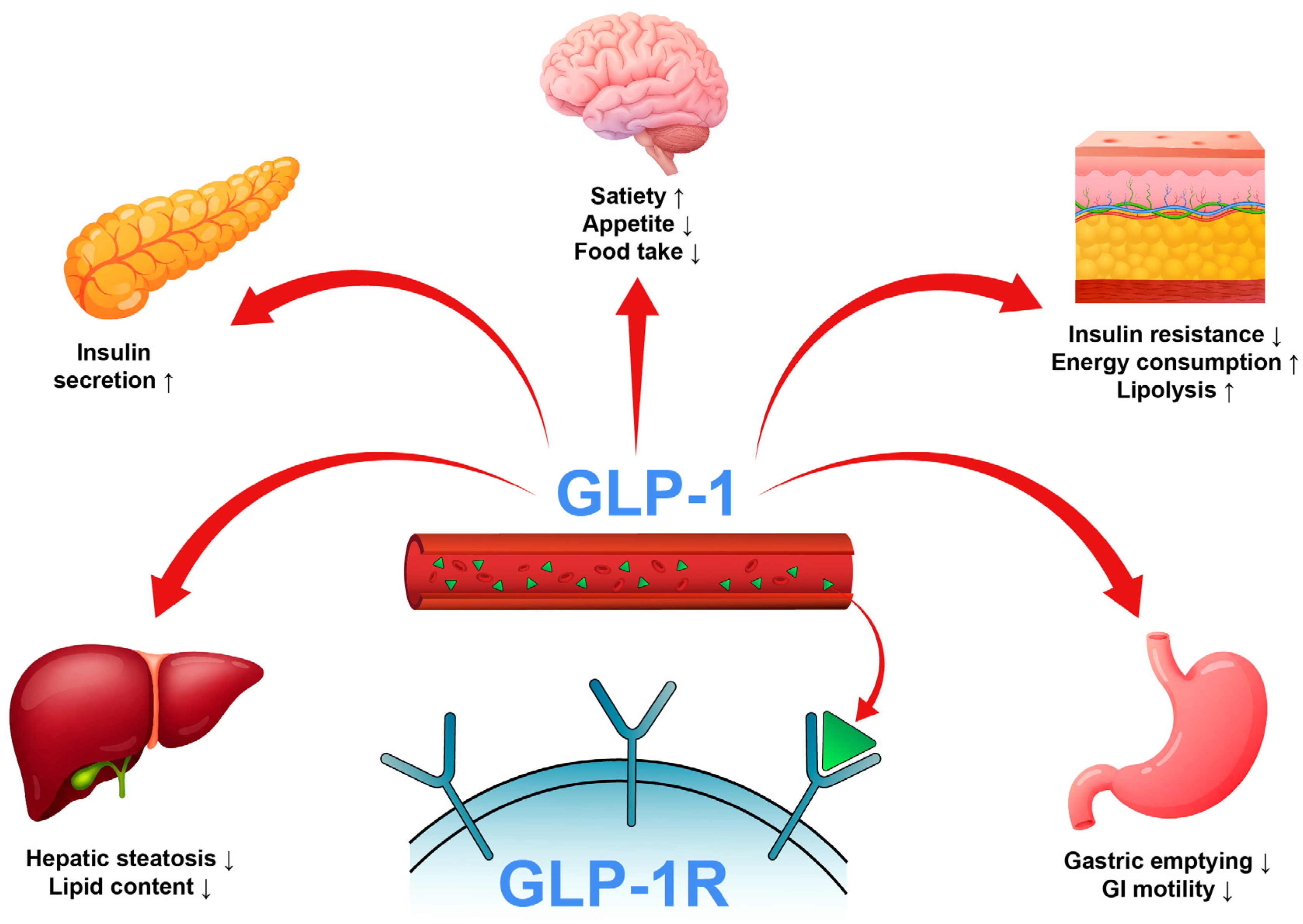
Figure 2.
Examples of non-peptide GLP-1R agonists.
Figure 3.
(a and b) 3D and 2D representation of the binding interactions between 1 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in red), and the key residues are shown as sticks (carbon atoms colored in green). (c and d) 3D and 2D representation of the binding interactions between 5 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in salmon), and the key residues are shown as sticks (carbon atoms colored in green). (e and f) 3D and 2D representation of the binding interactions between 9 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in magenta), and the key residues are shown as sticks (carbon atoms colored in green). (g and h) 3D and 2D representation of the binding interactions between 11 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in plum), and the key residues are shown as sticks (carbon atoms colored in green). (i and j) 3D and 2D representation of the binding interactions between hit 13 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in blue), and the key residues are shown as sticks (carbon atoms colored in green).
Figure 3.
(a and b) 3D and 2D representation of the binding interactions between 1 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in red), and the key residues are shown as sticks (carbon atoms colored in green). (c and d) 3D and 2D representation of the binding interactions between 5 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in salmon), and the key residues are shown as sticks (carbon atoms colored in green). (e and f) 3D and 2D representation of the binding interactions between 9 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in magenta), and the key residues are shown as sticks (carbon atoms colored in green). (g and h) 3D and 2D representation of the binding interactions between 11 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in plum), and the key residues are shown as sticks (carbon atoms colored in green). (i and j) 3D and 2D representation of the binding interactions between hit 13 and GLP1 (PDB ID: 6X1A). Ligand atoms are shown as sticks (carbon atoms colored in blue), and the key residues are shown as sticks (carbon atoms colored in green).
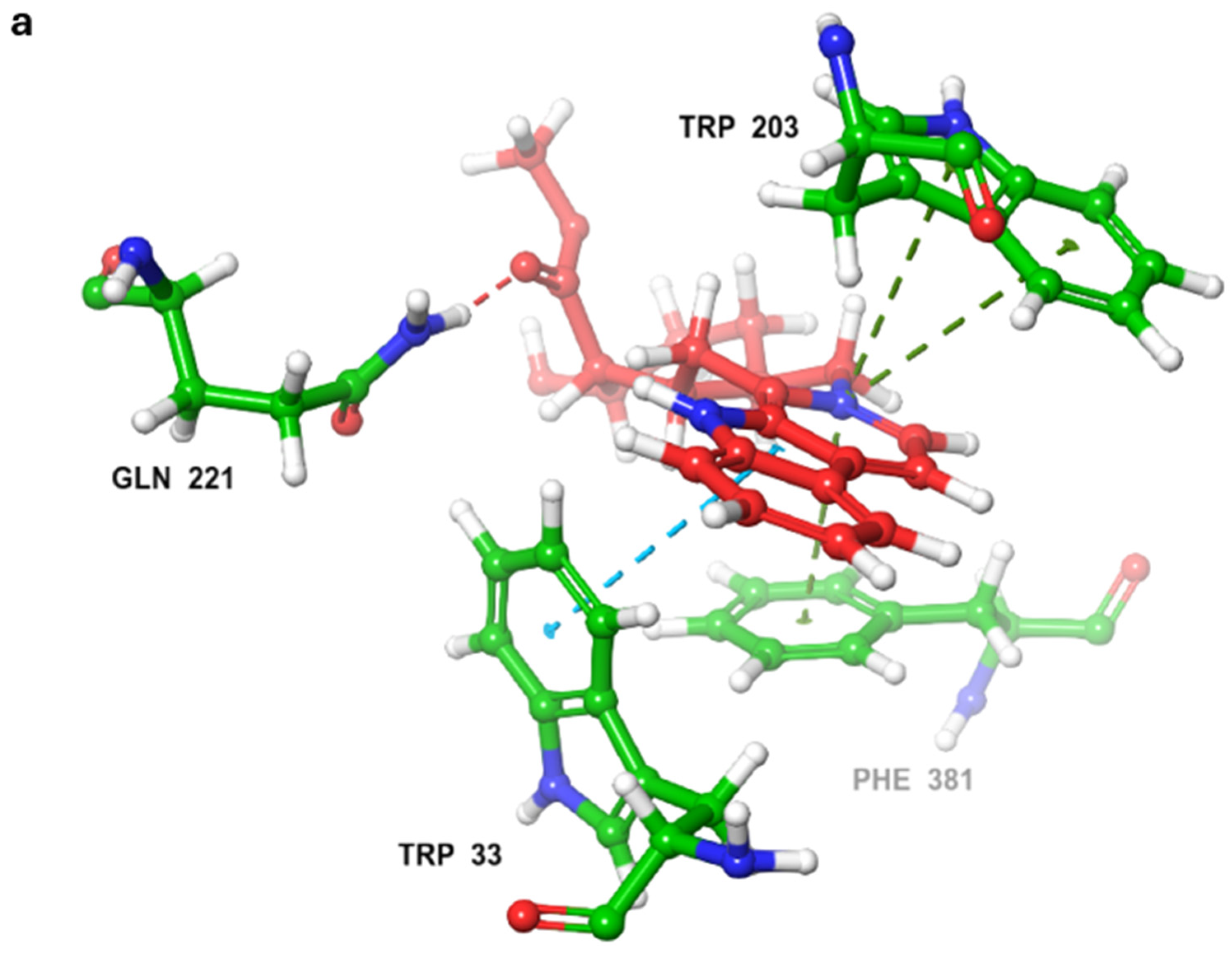
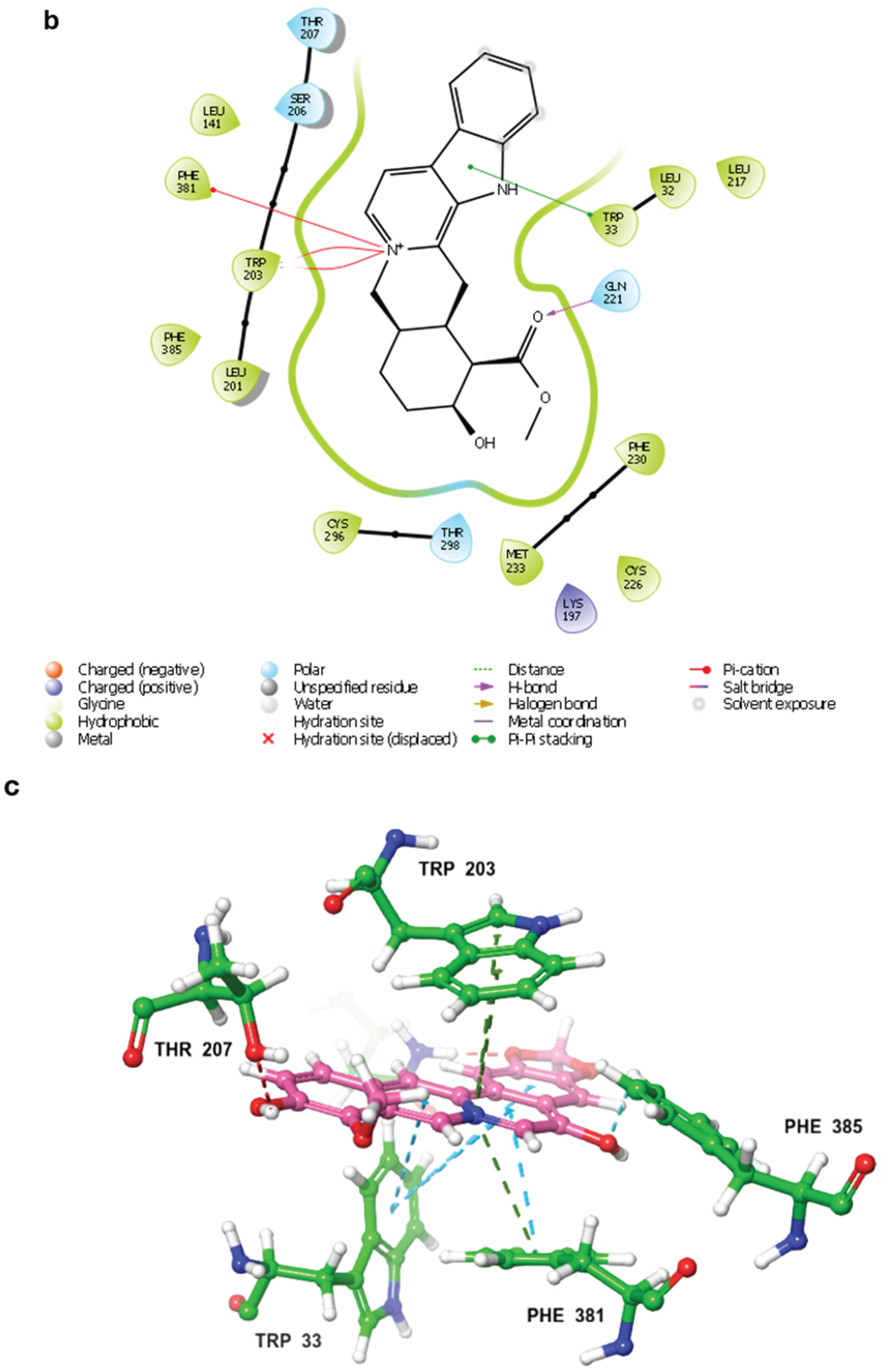
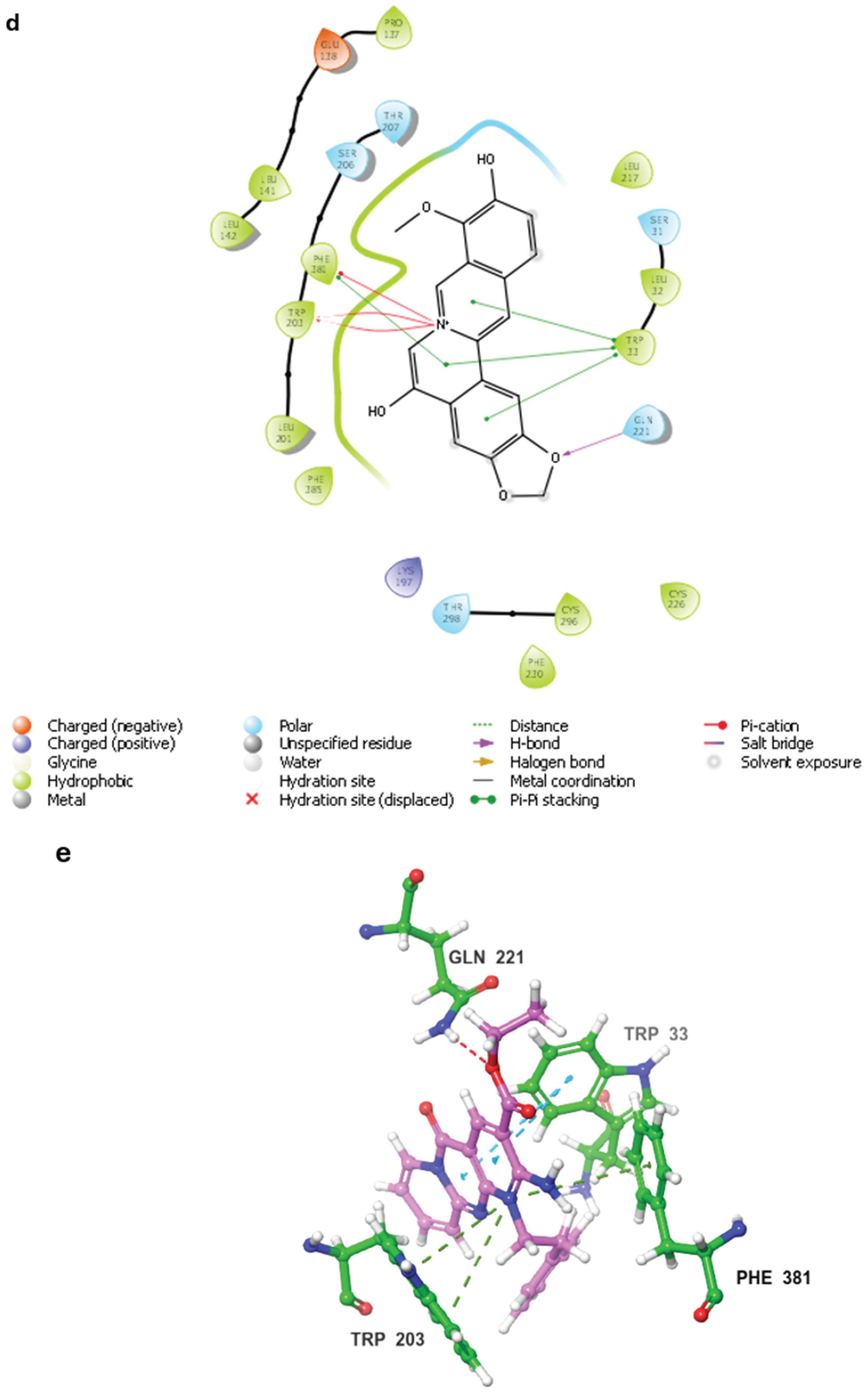
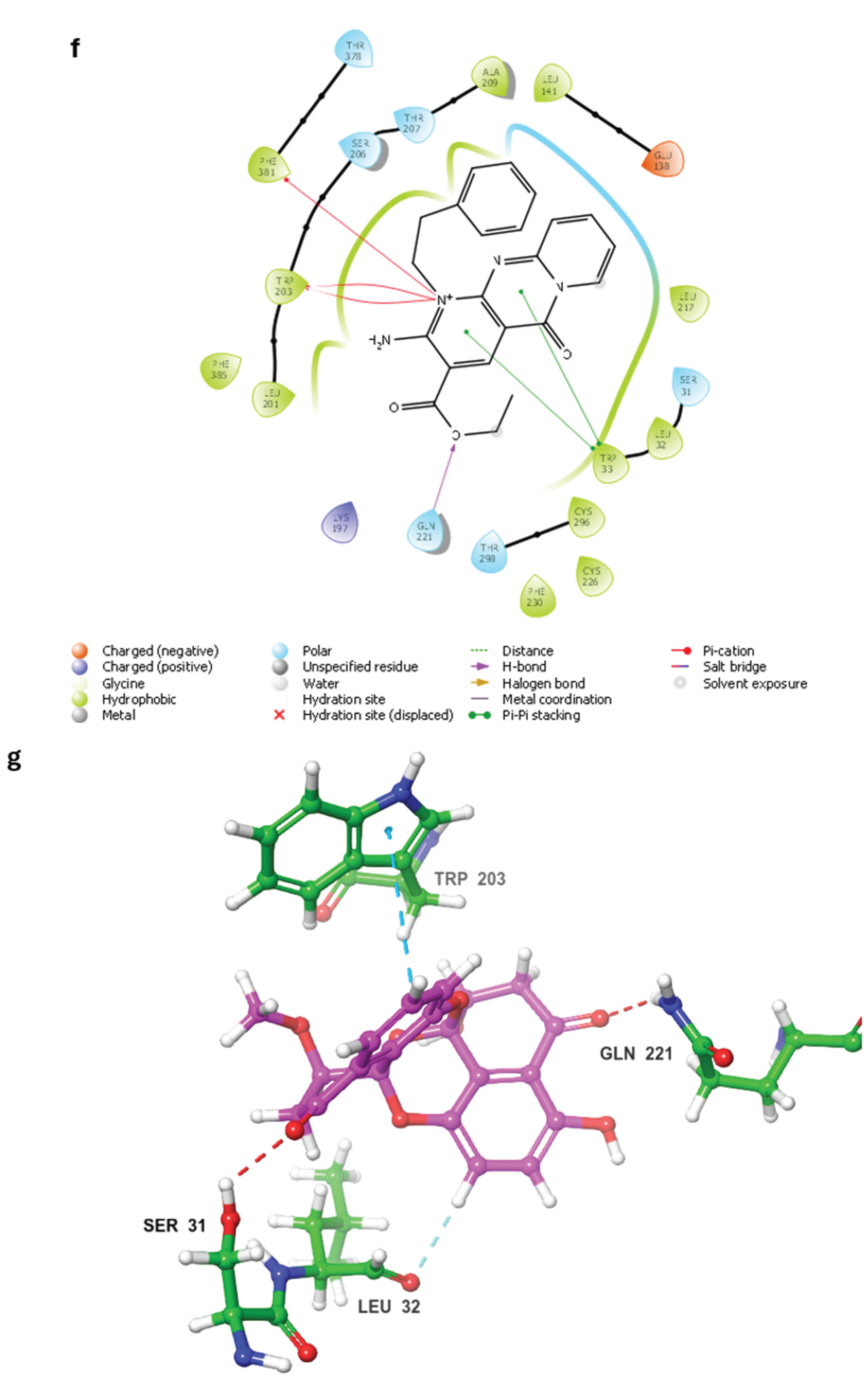
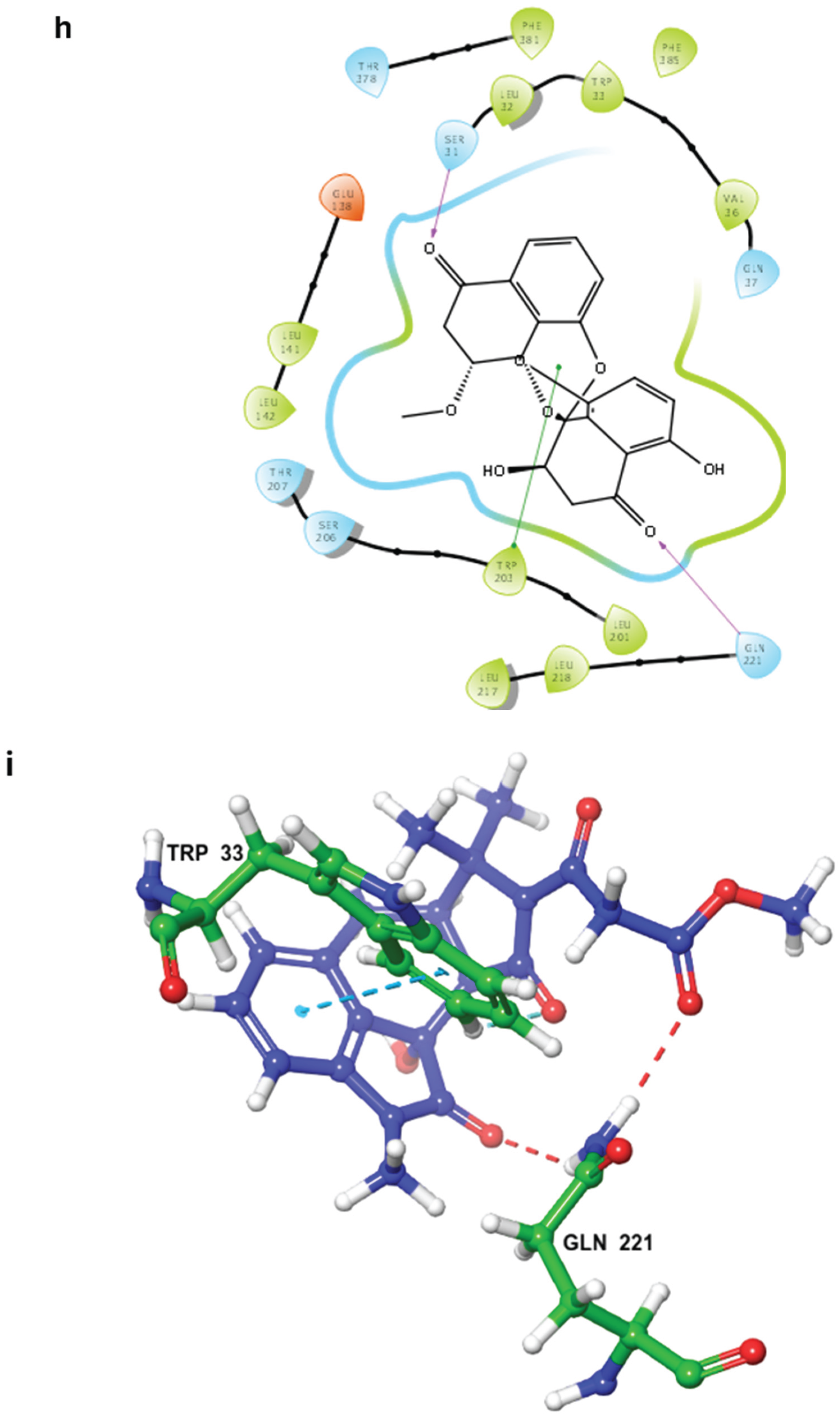
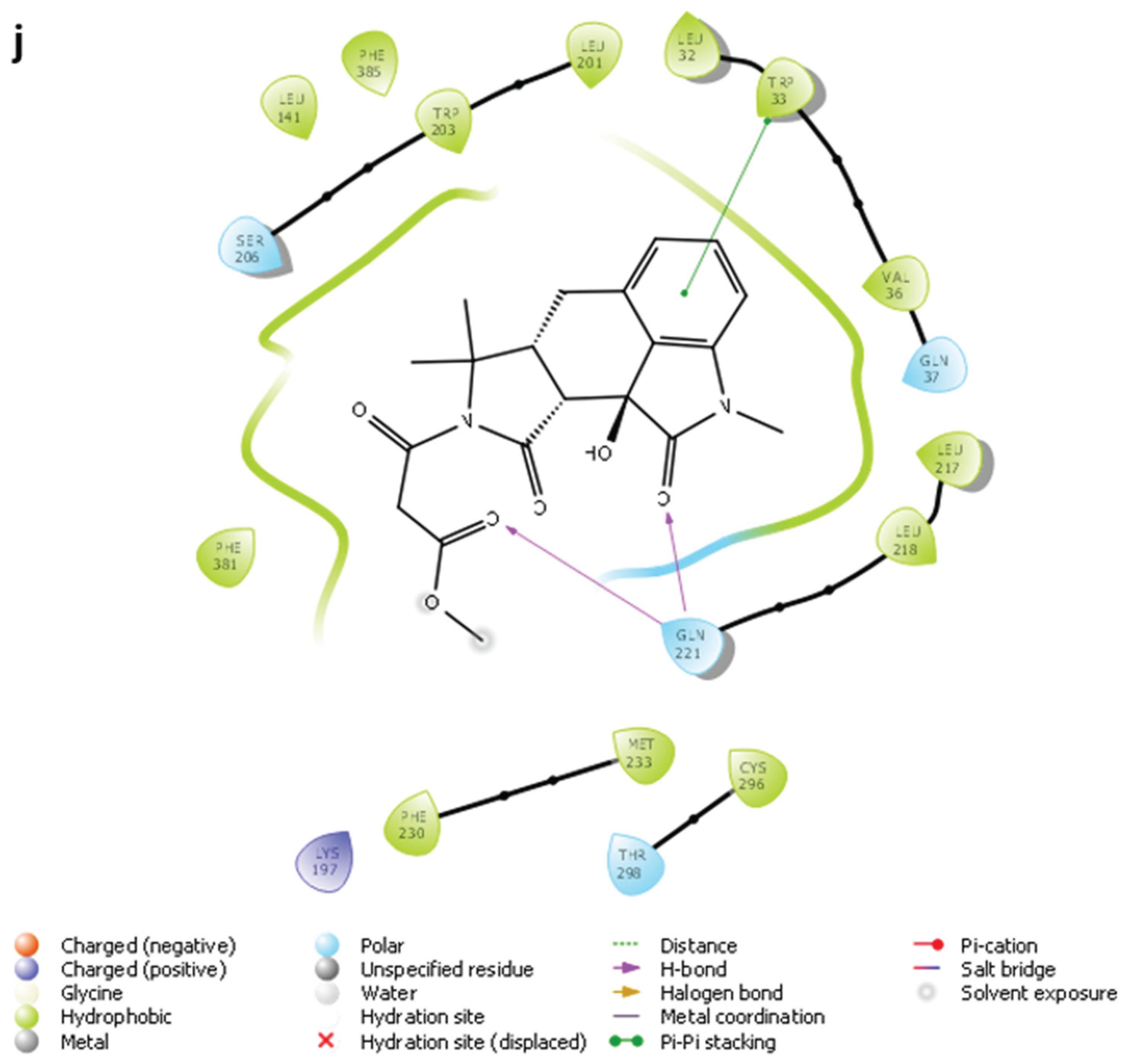
Figure 4.
RMSD analysis for protein Cα atoms in different complexes. Average RMSD values are shown between parentheses.
Figure 4.
RMSD analysis for protein Cα atoms in different complexes. Average RMSD values are shown between parentheses.
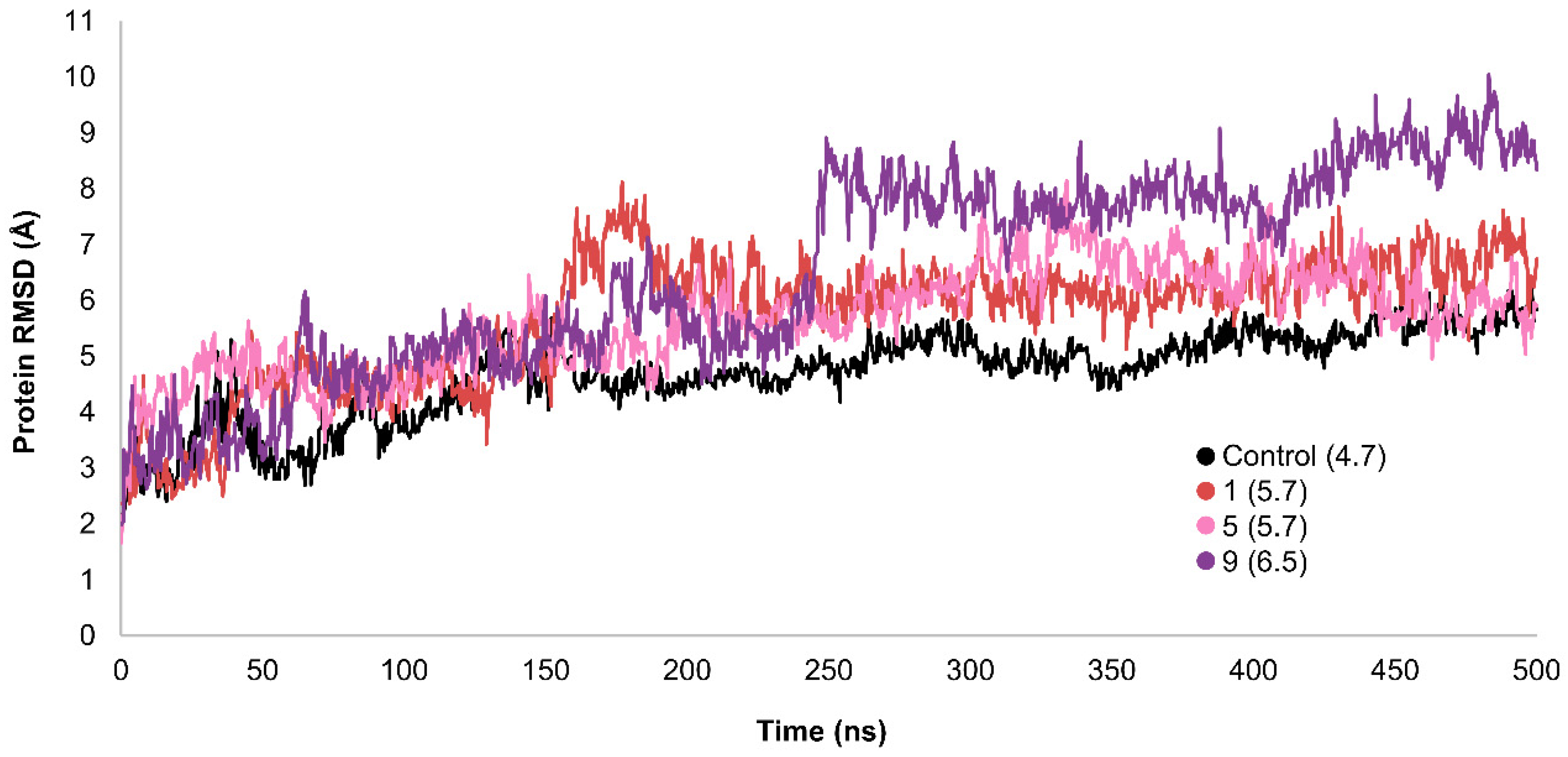
Figure 6.
RMSD analysis for ligand heavy atoms in different complexes. Average RMSD values are shown between parentheses.
Figure 6.
RMSD analysis for ligand heavy atoms in different complexes. Average RMSD values are shown between parentheses.
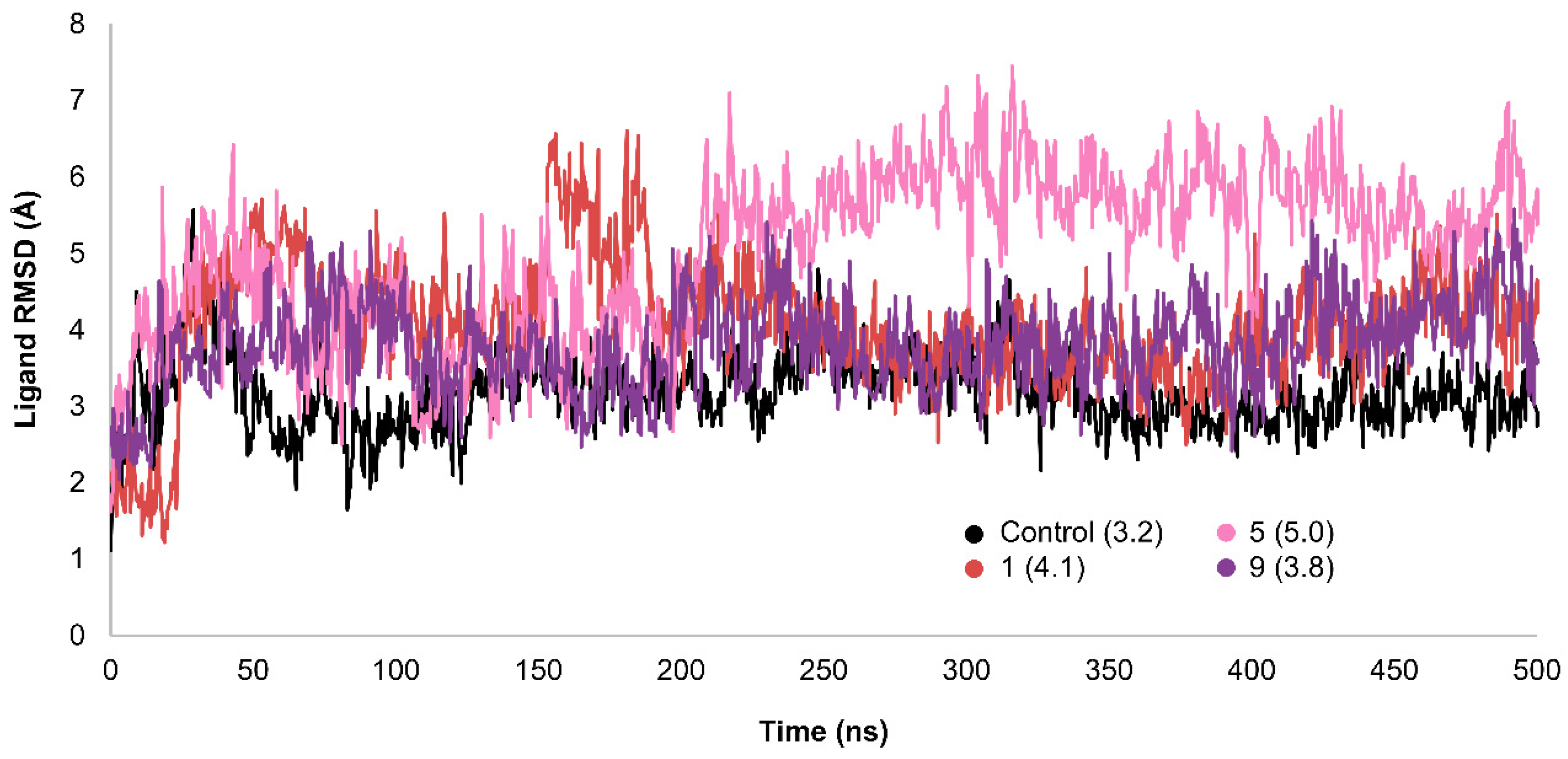
Figure 7.
Interactions between the ligands and residues in the binding site of GLP-1R during MD simulations. A) control, B) compound 1, C) compound 5, and D) compound 9.
Figure 7.
Interactions between the ligands and residues in the binding site of GLP-1R during MD simulations. A) control, B) compound 1, C) compound 5, and D) compound 9.
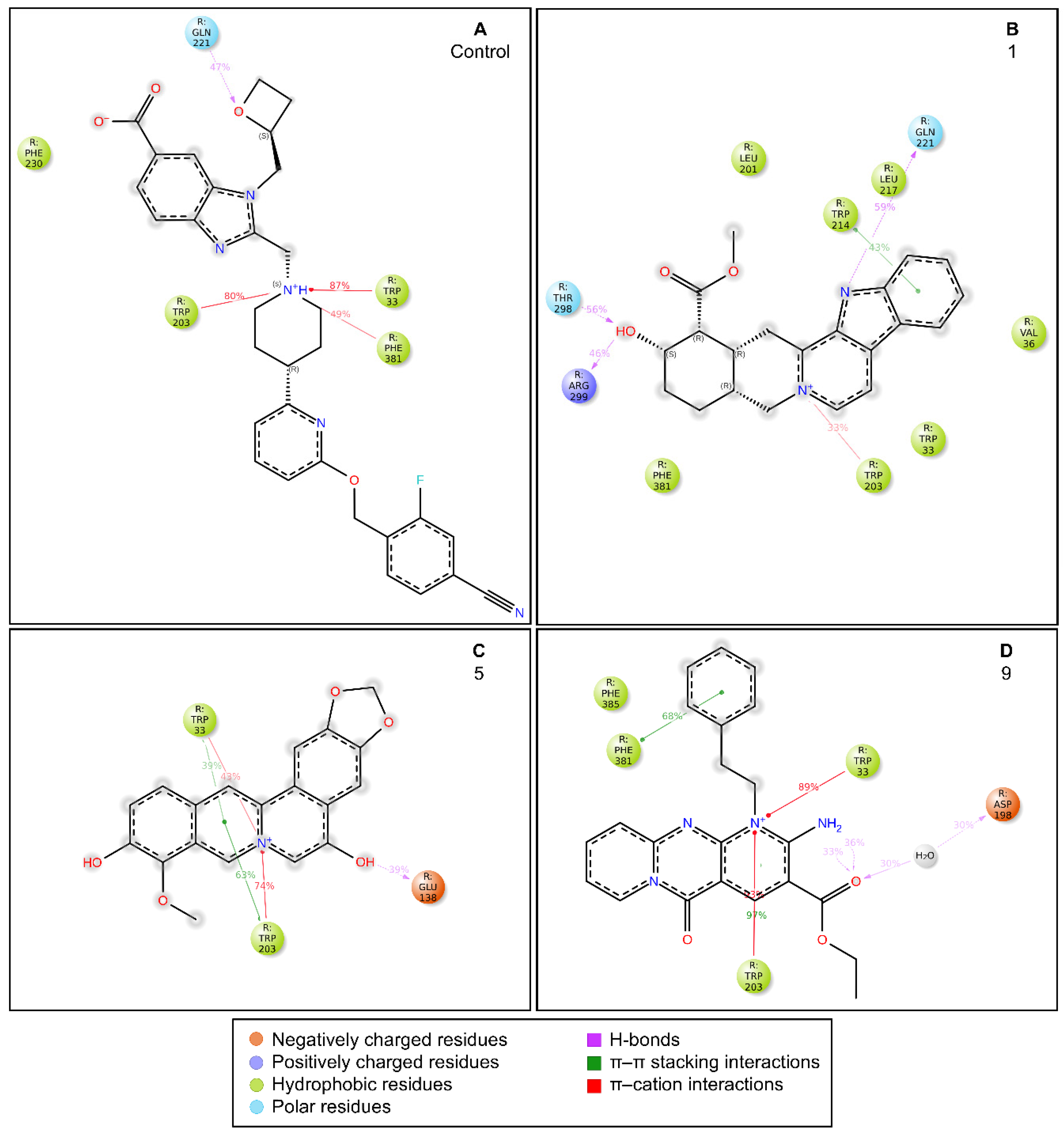
Figure 8.
The crystal structure of the orthosteric GLP-1 receptor (PDB ID: 6X1A) is depicted as a cartoon model, with the non-peptide agonist PF-06882961 bound to the receptor. The PF-06882961 molecule is shown as sticks, with carbon atoms colored in green.
Figure 8.
The crystal structure of the orthosteric GLP-1 receptor (PDB ID: 6X1A) is depicted as a cartoon model, with the non-peptide agonist PF-06882961 bound to the receptor. The PF-06882961 molecule is shown as sticks, with carbon atoms colored in green.
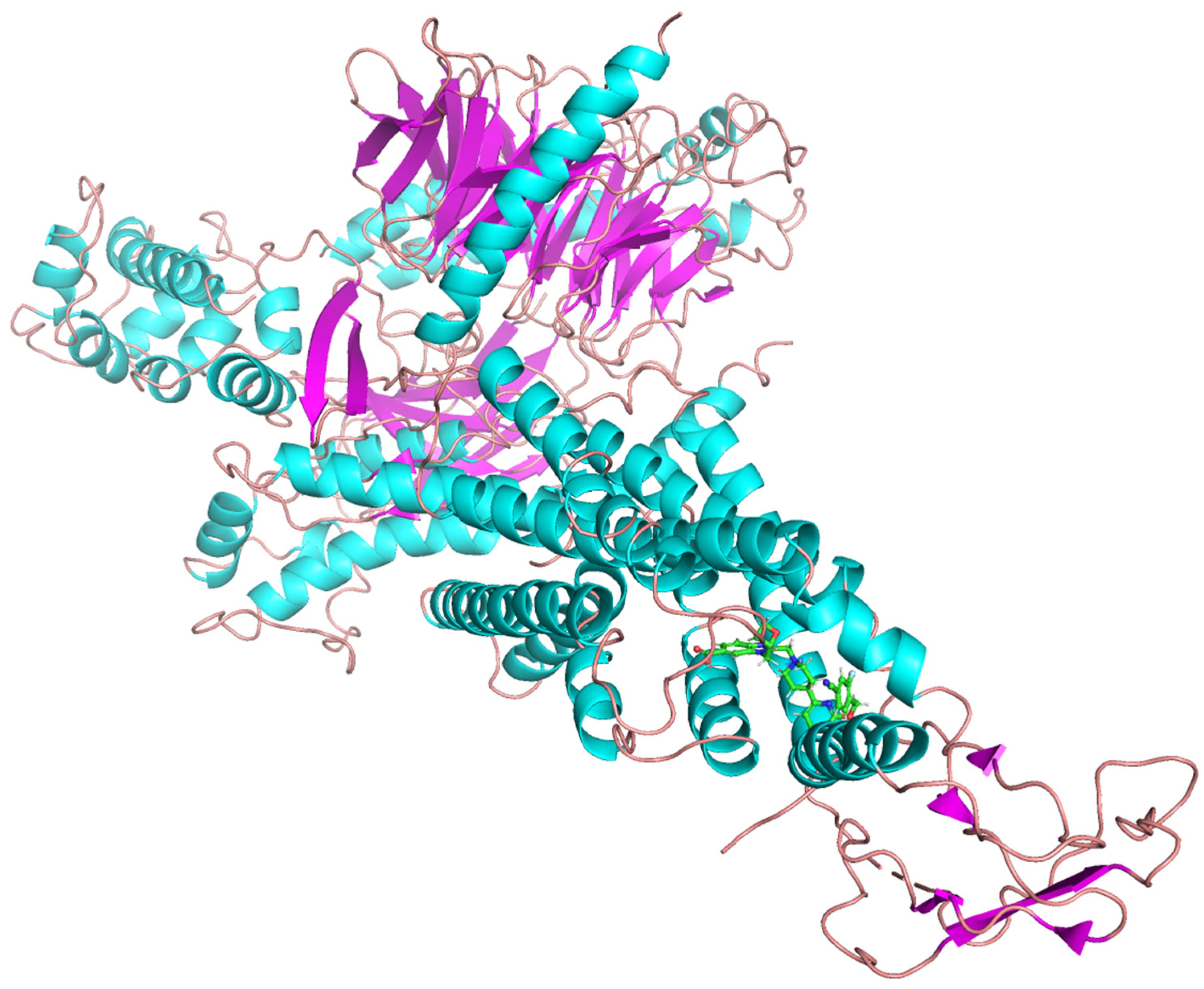
Figure 9.
Overview of the non-peptide GLP-1RA in silico identification workflow used in this study.
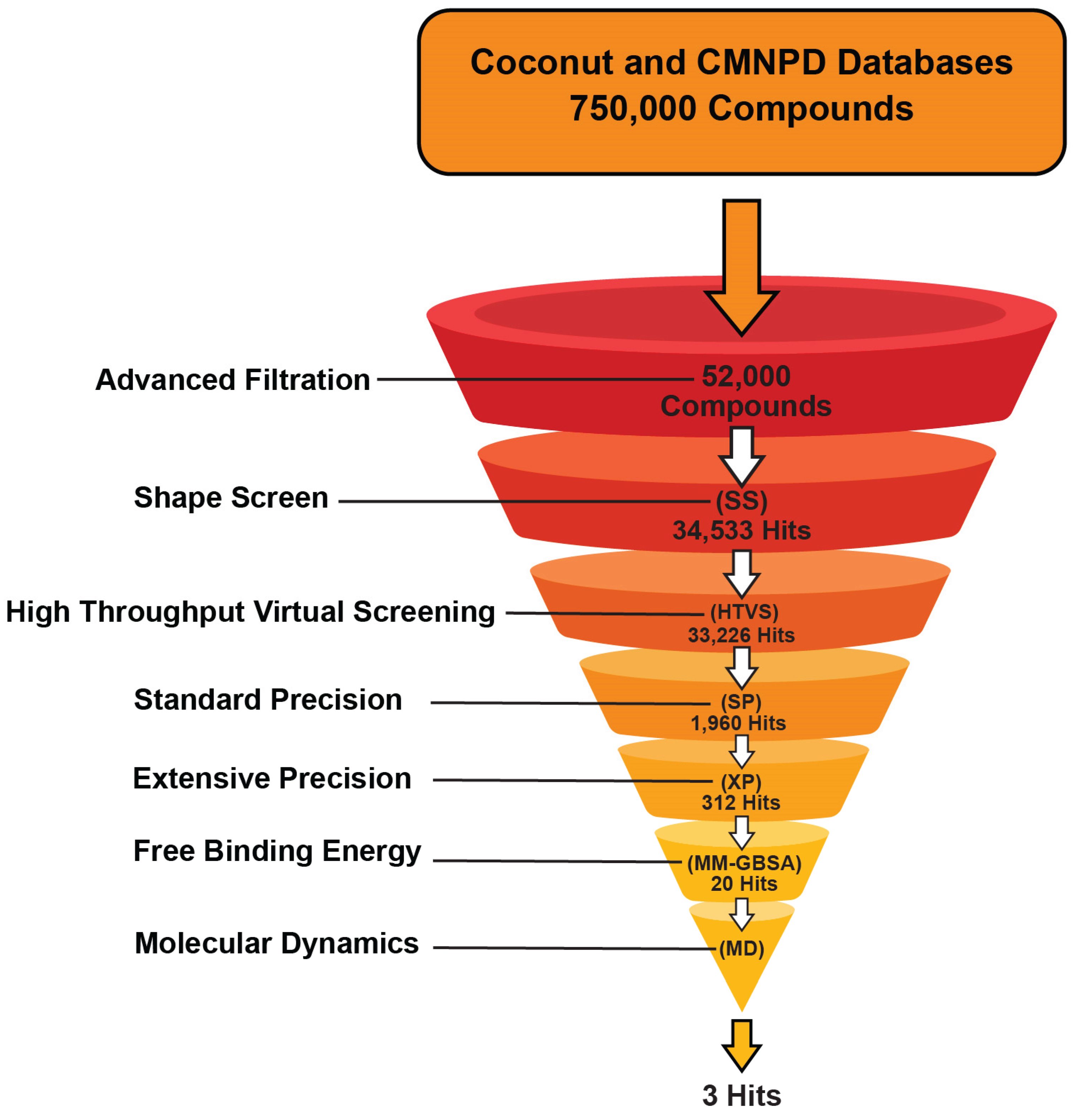
Table 1.
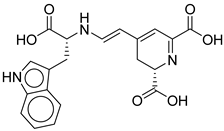
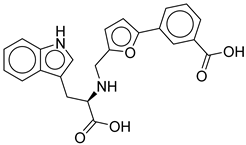
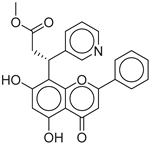
Structure, XP score, MM-GBSA, and shape similarity of the top 10 hits from the Coconut database with GLP-1R binding site (PDB ID: 6X1A).
Table 1.
Structure, XP score, MM-GBSA, and shape similarity of the top 10 hits from the Coconut database with GLP-1R binding site (PDB ID: 6X1A).
| Hit No.* | Structure | ID | XP Score | MM-BBSA dG Bind | Shape Similarity* |
|---|---|---|---|---|---|
|
1 |
 |
CNP0593098.1 |
-13.835 |
-84.69 |
0.346 |
|
2 |
 |
CNP0542406.3 |
-13.657 |
-83.94 |
0.315 |
|
3 |
 |
CNP0510864.0 |
-13.513 |
-82.51 |
0.305 |
|
4 |
 |
CNP0294111.0 |
-13.506 |
-84.61 |
0.344 |
| 5 | 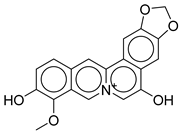 |
CNP0513629.0 |
-13.114 |
-76.49 |
0.308 |
|
6 |
 |
CNP0402650.0 |
-13.003 |
-86.43 |
0.325 |
|
7 |
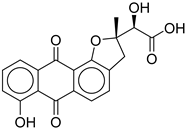 |
CNP0449126.4 |
-12.954 |
-66.07 |
0.320 |
|
8 |
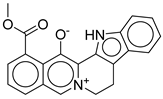 |
CNP0576495.0 |
-12.770 |
-76.87 |
0.312 |
|
9 |
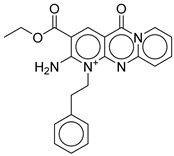 |
CNP0510059.0 |
-12.563 |
-84.80 |
0.339 |
|
10 |
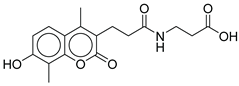 |
CNP0311770.0 | -12.532 | -85.80 | 0.310 |
|
Control |
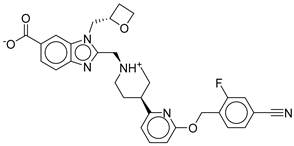 |
-12.040 |
-116.06 |
1.000 |
*Shape similarity of the hits and control. Similarity ranges: 0.5–1 (High), ≥0.3–0.49 (Intermediate), <0.3 (Low). Cutoff score ≥0.3.
Table 2.
Structure, XP score, MM-GBSA, and shape similarity of the top 10 hits from the Marine database with GLP-1R binding site (PDB ID: 6X1A).
Table 2.
Structure, XP score, MM-GBSA, and shape similarity of the top 10 hits from the Marine database with GLP-1R binding site (PDB ID: 6X1A).
| Hit No.* | Structure | ID | XP Score | MM-BBSA dG Bind | Shape Similarity* |
|---|---|---|---|---|---|
| 11 | 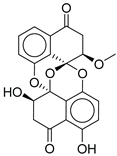 |
CMNPD27661 |
-12.806 |
-71.68 |
0.303 |
| 12 | 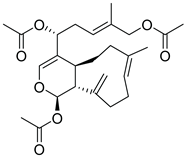 |
CMNPD2041 | -11.609 | -96.80 | 0.327 |
| 13 | 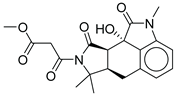 |
CMNPD24592 | -11.575 | -74.09 | 0.315 |
| 14 | 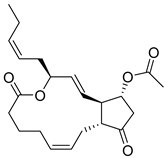 |
CMNPD4014 | -11.495 | -95.02 | 0.302 |
| 15 | 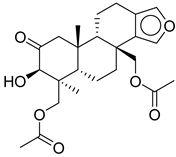 |
CMNPD9270 | -11.427 | -97.33 | 0.375 |
| 16 | 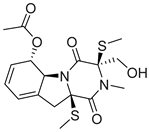 |
CMNPD26002 |
-11.116 |
-100.43 |
0.320 |
| 17 | 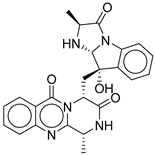 |
CMNPD5314 | -10.805 | -75.01 | 0.343 |
| 18 | 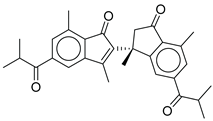 |
CMNPD22795 | -10.798 | -88.67 | 0.363 |
| 19 | 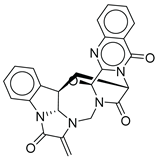 |
CMNPD27343 | -10.372 | -61.54 | 0.311 |
| 20 | 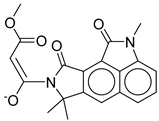 |
CMNPD24593 | -10.190 | -52.04 | 0.352 |
| Control | 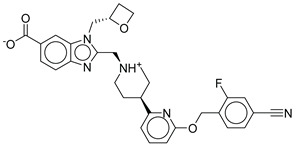 |
-12.040 | -116.06 | 1.000 |
*Shape similarity of the hits and control. Similarity ranges: 0.5–1 (High), ≥0.3–0.49 (Intermediate), <0.3 (Low). Cutoff score ≥0.3.
Table 3.
Potential antidiabetic candidate molecules from the COCONUT database .
| COCONUT ID | Compound Name / Description | Anti-Diabetic/GLP-1 Literature Evidence (2015-2025)[41,42,43,44,45,46,47,48,49,50,51] |
|---|---|---|
| CNP0593098.1 | 3,4,5,6-Tetradehydroyohimbine | Yohimbine (parent compound) has demonstrated antidiabetic effects in STZ-induced diabetic rats. It acts as an alpha-2 adrenoceptor antagonist, improving glucose tolerance, lowering blood glucose, and improving lipid profiles. The mechanism is thought to involve increased insulin secretion by blocking inhibitory catecholaminergic tone in pancreatic islets. No direct evidence for GLP-1 agonism or for the specific dehydro-derivative. |
| CNP0542406.3 | (15S)-19-(methoxycarbonyl)-16-methyl-17-oxa-3,13λ⁵-diazapentacyclo[11.8.0.0²,¹⁰.0⁴,⁹.0¹⁵,²⁰]henicosa-1(13),2(10),4,6,8,11,18-heptaen-13-ylium | No direct literature for this structure |
| CNP0510864.0 | 4-[2-[[1-carboxy-2-(1H-indol-3-yl)ethyl]amino]ethenyl]-2,3-dihydropyridine-2,6-dicarboxylic acid | No direct literature for this structure |
| CNP0294111.0 | 3-[5-[[[1-carboxy-2-(1H-indol-3-yl)ethyl]amino]methyl]furan-2-yl]benzoic acid | No direct studies. |
| CNP0513629.0 | 16-methoxy-5,7-dioxa-13-azoniapentacyclo[11.8.0.0²,¹⁰.0⁴,⁸.0¹⁵,²⁰]henicosa-1,3,8,10,12,14,16,18,20-nonaene-11,17-diol | No direct anti-diabetic or GLP-1related studies for this structure. |
| CNP0402650.0 | methyl 3-(5,7-dihydroxy-4-oxo-2-phenyl-4H-chromen-8-yl)-3-(pyridin-3-yl)propanoate | Chromen derivatives and flavonoids are widely studied for anti-diabetic effects, often through antioxidant and insulin-sensitizing mechanisms, but no direct evidence |
| CNP0449126.4 | (2S)-2-hydroxy-2-[(2S)-7-hydroxy-2-methyl-6,11-dioxo-3H-naphtho[2,3-g]benzofuran-2-yl]acetic acid | Benzofuran derivatives have shown anti-diabetic effects but no direct study for this compound. |
| CNP0576495.0 | 19-methoxycarbonyl-3-aza-13-azoniapentacyclo[11.8.0.0²,¹⁰.0⁴,⁹.0¹⁵,²⁰]henicosa-1(13),2(10),4,6,8,14,16,18,20-nonaen-21-olate | No direct literature for anti-diabetic or GLP-1 effects. |
| CNP0510059.0 | ethyl 6-amino-2-oxo-7-(2-phenylethyl)-1,9-diaza-7-azoniatricyclo[8.4.0.0³,⁸]tetradeca-3(8),4,6,9,11,13-hexaene-5-carboxylate | No direct literature for anti-diabetic or GLP-1 effects. |
| CNP0311770.0 | STL522422; 3-[3-(7-hydroxy-4,8-dimethyl-2-oxo-chromen-3-yl)propanoylamino]propanoic acid | Chromen (coumarin) derivatives show anti-diabetic effects (antioxidant, insulin-sensitizing) |
Table 4.
Antidiabetic Relevance of Selected CMNPD Compounds: Structural Classes, Mechanistic Insights.
Table 4.
Antidiabetic Relevance of Selected CMNPD Compounds: Structural Classes, Mechanistic Insights.
| CMNPD ID | Compound Name | Structural Class / Source | Direct Anti-Diabetic/GLP-1 Evidence [52,53,54,55,56,57,58,59,60,61,62,63,64,65,66] | Mechanistic/Structural Notes |
|---|---|---|---|---|
| CMNPD27661 | preussomerin M | Polyketide (fungal) | No direct anti-diabetic or GLP-1 literature was found for this compound. | Polyketide-type marine metabolites are being explored as anti-diabetic |
| CMNPD2041 | waixenicin A | Diterpene (soft coral) | No direct anti-diabetic or GLP-1 literature found for this compound. | Marine diterpenes show anti-inflammatory and metabolic regulatory effects. |
| CMNPD24592 | speradine D | Alkaloid (marine fungus) | No direct studies for this structure; marine alkaloids are reported to enhance glucose uptake, insulin sensitivity, and AMPK activity. | Alkaloids from marine sources are highlighted as promising anti-diabetic agents via AMPK/GLUT4 pathways. |
| CMNPD4014 | PGE3-1,15-lactone-11-acetate | Prostaglandin derivative | No direct anti-diabetic or GLP-1 evidence; some marine prostaglandins have anti-inflammatory and metabolic effects. | Prostaglandin derivatives may indirectly affect glucose metabolism via inflammation modulation. |
| CMNPD9270 | speradine E | Benzofuran derivative | No direct studies; marine benzofurans are under investigation for antioxidant and enzyme inhibitory effects relevant to diabetes. | Benzofuran derivatives can inhibit α-glucosidase and reduce oxidative stress in β-cells. |
| CMNPD26002 | 6-acetylbisdethiobis(methylthio)gliotoxin | Epipolythiodioxopiperazine | No direct anti-diabetic/GLP-1 studies | N/A |
| CMNPD5314 | fumiquinazoline B | Quinazoline alkaloid | No direct studies for this compound; marine quinazoline alkaloids show anti-diabetic potential via enzyme inhibition and AMPK action. | Quinazoline alkaloids are highlighted for PTP1B inhibition, and AMPK activation. |
| CMNPD22795 | anthogorgiene I | Polycyclic aromatic (coral) | No direct anti-diabetic/GLP-1 evidence; marine polycyclic aromatics may have metabolic effects, but data are limited. | Structural novelty; potential for redox and enzyme modulation. |
| CMNPD27343 | versiquinazoline B | Quinazoline alkaloid | No direct anti-diabetic/GLP-1 studies | N/A |
| CMNPD24593 | speradine E | Alkaloid (marine fungus) | No direct studies | Alkaloids from marine origin reported with anti-diabetic activities |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



