Submitted:
24 May 2025
Posted:
26 May 2025
You are already at the latest version
Abstract
Obesity has emerged as a significant global health challenge characterized by excessive fat accumulation and a complex interplay of systemic inflammation and metabolic dysregulation. This condition not only impacts overall health but also affects various organ systems, including the lungs. Recent research highlights how obesity influences lung health through mechanisms involving mechanics and chronic low-grade inflammation, which can alter pulmonary structure and function. Despite increasing evidence that inflammation in adipose tissue plays a crucial role in systemic metabolic disturbances, our understanding of its relationship with pulmonary neutrophilic inflammation remains limited. There is a need to explore how adipose tissue inflammation interacts with pulmonary physiology, particularly in the context of obesity-associated metabolic risk. By investigating the mechanisms underlying pulmonary neutrophilic inflammation, we can identify potential therapeutic targets to mitigate pulmonary dysfunction, as well as obesity-associated metabolic risk. This review emphasizes the importance of adipose tissue-derived inflammatory mediators in pulmonary neutrophilic inflammation in obesity-associated metabolic comorbidities.
Keywords:
obesity
; adipose tissue inflammation
; pulmonary neutrophilic inflammation
; systemic inflammation
; obesity-associated metabolic comorbidity
; insulin resistance
Introduction
Obesity has become one of the main public health concerns worldwide [1]. Obesity is driven by factors such as unhealthy eating and a lack of physical activity, which have increased in recent decades due to lifestyle changes[2]. According to the World Health Organization (2021), more than 1.9 billion adults are overweight, and more than 650 million are obese (World Health Organization, 2021). Obesity is a chronic inflammatory disease resulting from a chronic positive energy balance resulting in increased adipose tissue (AT) [1,3,4]. This condition not only affects people's quality of life but is also linked to an increased risk of chronic diseases, such as type 2 diabetes (T2D) and cardiovascular disease (CVD), as well as pulmonary disease and susceptibility to environmental stressors [5].
Energy accumulates as triglycerides in adipocytes, whereas excess energy remains as free fatty acids (FFAs) [6]. FFAs can activate resident AT macrophages and adipocytes to express a number of genes encoding proinflammatory adipokines and cytokines, enzymes that produce reactive oxygen species (ROS)/nitrogen (RNS) species, chemokines, and adhesion molecules for inflammatory cells [4,7]. This causes, first, the recruitment of neutrophils[8], which then contribute to the recruitment of monocytes from the bloodstream toward the AT [9,10]. Stressed adipocytes and neutrophils help the further recruitment of monocytes. The recruited monocytes then differentiate into macrophages, which can reach 60% of the total cell population in the AT [11]. These macrophages regularly organize in crown-like structures, and macrophages organize as crowns surrounding dead adipocytes. Under the prevalent hypoxic conditions in AT, due to adipocyte hypertrophy and hyperplasia, AT macrophages change gene expression toward a proinflammatory phenotype [12,13]. Inflammation mediators produced by AT inflammation drop into the bloodstream, causing systemic low-grade inflammation or metaflammation. Proinflammatory mediators secreted by inflamed AT, as well as the mechanical effects of growing AT depots, affect pulmonary physiology [14]. AT mediators, such as leptin and proinflammatory cytokines, can increase the expression of adhesion molecules for neutrophils in the pulmonary microvasculature [15]. This causes homing and activation of neutrophils in the lung microvasculature, a process known as pulmonary neutrophilic inflammation [15,16]. Thus, the exposure of obese patients to environmental stressors, such as ash, dust, bacteria, viruses, diesel exhaust, particulate matter and other stressors, can cause the lungs to act as an extra source of inflammatory mediators [16]. These inflammatory mediators contribute to the pool of inflammatory mediators produced by inflamed AT [17]. Consequently, obesity-associated metabolic risk increases [11,18]. Targeting pulmonary neutrophilic inflammation mechanisms can help reduce the burden of obesity-associated metabolic comorbidities in obese patients exposed to environmental stressors (Figure 1).
Herein, we discuss recent evidence showing the mechanism by which obesity sensitizes the lungs to neutrophilic inflammation when patients are exposed to environmental stressors.
Neutrophilia in the Bloodstream of Obese Patients
Neutrophils are the most abundant leukocytes in human blood, the main effector cells of acute inflammation, and the first to respond to infections [19]. Neutrophils are often considered primary leukocytes against infections because of their ability to act as phagocytic cells, degranulate by releasing lytic enzymes, and perform an oxidative burst that produces reactive oxygen species (ROS) and neutrophil extracellular traps (NETs) with antimicrobial potential [20]. Neutrophils are also considered the main effector cells of acute inflammatory reactions, as they are the first leukocytes to be recruited at sites of tissue irritation where they are capable of producing large amounts of cytokines and chemokines, including TNFα, IL-1β, IL-8, and MCP-1 [20]. Consequently, neutrophils induce the second wave of immune cells, such as macrophages and lymphocytes, to become sites of irritation.
During obesity-induced inflammation in animal models, the number of neutrophils in the peripheral circulation increases [21]. From there, neutrophils can infiltrate the AT and endothelium of blood vessels. Therefore, neutrophils play an important role in the early stages of obesity by infiltrating visceral AT[20]. Interestingly, the number of circulating neutrophils is increased in obese individuals, with a clear association between the neutrophil blood count and a higher body mass index (BMI) [22]. Patients who are overweight with neutrophilia mostly have elevated serum C-reactive protein concentrations and larger waist circumferences [23]. In addition, neutrophil counts are significantly greater in people with metabolic syndrome than in lean patients [20]. In addition, neutrophils in obese patients have an activated phenotype, as indicated by elevated plasma concentrations of myeloperoxidase (MPO) and neutrophil elastase (NE), as well as increased expression of CD66b, a marker of neutrophil degranulation [24]. Neutrophil activation in obese individuals was also indicated by stimulation of the NF-κB signaling pathway and by increased ROS generation and cytokine release of proinflammatory mediators [25,26]. Once in adipose tissue, neutrophils establish direct interactions with adipocytes. This occurs through the binding of the integrin αM β2 on neutrophils to the intercellular adhesion molecule 1 (ICAM-1) present on adipocytes. This connection activates neutrophils, causing them to release IL-1β via the NF-κB and NLRP3 inflammasome pathways [27]. Additionally, neutrophils produce TNFα, which further stimulates macrophages. Leptin also plays an important role, as it activates its receptor to promote the NF-κB pathway, inhibiting neutrophil apoptosis and allowing their persistence in adipose tissue [28]. Moreover, FFAs derived from adipocyte lipolysis can attract more neutrophils and stimulate their production of IL-1β [29]. Neutrophils also generate NEL, an enzyme that alters the energy expenditure of AT and directly activates macrophages. Furthermore, the granular protein cathelicidin (LL-37) can trigger the release of more proinflammatory cytokines from macrophages. As neutrophils become activated, they also have the capacity to recruit monocytes that differentiate into macrophages in the AT microenvironment [30].
Notably, weight loss, for example, after gastric band surgery, results in a reduction in both neutrophil blood counts and their proinflammatory activity. These findings suggest that the chronic inflammation associated with obesity contributes to the increase in neutrophils [31,32]. Elevated levels of acute-phase proteins have been reported in obese individuals, and the adipose tissue of these individuals produces increased levels of inflammatory mediators such as TNFα, IL-1β, IL-6, and IL-8 [32]. These mediators stimulate granulopoiesis in the bone marrow, promoting the release of neutrophils into the peripheral circulation and the elimination of these leukocytes from the endothelial wall, leading to neutrophilia. In addition, AT secretes leptin, which has been shown to stimulate hematopoiesis and can promote neutrophil oxidative bursts, induce chemotaxis, and inhibit apoptosis [20]. Taken together, these findings support the idea that AT in obesity plays a crucial role in systemic inflammation, facilitating the generation, increase, and activation of neutrophils, which become the first leukocytes to infiltrate adipose tissues [33].
Activation of Neutrophils in the Bloodstream of Obese Patients
Obesity is associated with a chronic low-grade inflammatory state affecting various tissues, including the lungs. This inflammation is largely due to the expansion of AT, where adipocytes increase in number (hyperplasia) and size (hypertrophy). These adipocytes release proinflammatory cytokines, such as TNF-α and IL-6, which are crucial for the activation of the immune system [34]. The accumulation of these cytokines in adipose tissue and their subsequent release into the bloodstream generate an environment conducive to the activation of neutrophils [25,35].
Activated neutrophils release a variety of inflammatory mediators, including more cytokines, proteases, and reactive oxygen species (ROS), which can aggravate tissue damage and perpetuate inflammation [25]. This inflammatory cascade not only affects adipose tissue but also affects organs such as the liver and lungs, where it can affect their physiology [36].
Furthermore, neutrophil activation in obesity has been associated with an increase in insulin resistance. Systemic inflammation triggered by neutrophils and other immune cells interferes with insulin signaling, hindering glucose uptake by peripheral tissues [24]. This mechanism is a key risk factor for the development of T2D and other obesity-related metabolic complications [37]. Therefore, understanding the relationship between obesity and neutrophil activation is critical for developing therapeutic strategies that address inflammation and its adverse effects on metabolic health [38].
Activated neutrophils in the lungs release more inflammatory cytokines and other chemicals (such ROS). This lung inflammation can be chronic and affect lung function. Pulmonary inflammation is a pathological process that can be triggered by various conditions, including obesity. In this context, neutrophil activation plays a crucial role [37]. When neutrophils are activated in response to inflammatory cytokines, such as TNFα and IL-6, they release a series of inflammatory mediators, including more cytokines, chemokines, and other chemicals. Among the latter, ROS, which are highly reactive molecules that can cause oxidative damage to surrounding cells and tissues, stand out. This damage results in an exacerbation of the inflammatory response and contributes to a vicious cycle that perpetuates inflammation [39].
Recruitment of Monocytes and Further Differentiation into M1 Macrophages Causes Adipose Tissue Inflammation
Adipose tissue is composed of various cell types, including mature adipocytes, preadipocytes, mesenchymal cells, and stromal cells, such as endothelial cells, macrophages, and fibroblasts. Its primary function is lipid storage, thereby regulating the influx of dietary lipids by removing triacylglycerol (TAG) from the circulation and preventing the release of FFAs. In obesity, adipocytes are overloaded with TAG, which reduces their ability to store more FFAs. This leads to an increase in the levels of TAG and FFAs in the blood, promoting ectopic lipid accumulation in organs such as skeletal muscle, liver, and pancreatic islets [40]. FFAs act as ligands for the Toll-2/4-like receptor (TLR2/4) in AT macrophages and contribute to systemic inflammation [41].
During obesity, hypertrophied adipocytes face perfusion problems due to inadequate vasculature, resulting in hypoxia and apoptotic cell death [42]. Dead cell debris induces the production of chemokines such as the chemoattractant protein monocyte-1 (MCP-1, also known as CCL-2), which recruits monocytes and T cells from the peripheral circulation [43]. Monocytes in the tissue differentiate into macrophages. These macrophages, in turn, produce TNFα, IL-6, and other cytokines that inhibit the differentiation of preadipocytes, preventing the formation of new adipocytes that could mitigate the increase in TAG. This perpetuates a cycle of adipocyte hypertrophy, hypoxia, apoptosis, and monocyte recruitment and differentiation into macrophages, resulting in the phenotypic switch of macrophages toward inflammatory macrophages (M1), thus contributing to a state of chronic low-grade systemic inflammation [44]. In obesity, the production of proinflammatory adipocytokines such as leptin, resistin, and visfatin increases, whereas the synthesis of adiponectin, an anti-inflammatory adipocytokine, decreases. Figure 2. There is also an increase in the production of inflammatory mediators such as IL-6, TNFα, acute phase reactants, C-reactive protein, serum amyloid A, complement fragment C3 and other immunomodulators [45].
Adipose Tissue Inflammation Leads to Systemic Inflammation in Obesity
White adipose tissue (WAT) serves two main functions: it is the body's main fat reservoir and acts as the largest endocrine organ, releasing adipokines and cytokines into the bloodstream [34]. Adipokines play crucial roles in various metabolic and physiological pathways, regulating processes such as insulin signaling, glucose uptake, fatty acid oxidation, and other metabolic and energy mechanisms [46]. On the other hand, cytokines modulate inflammation and its resolution, as well as adaptive and reparative angiogenesis [11].
When a patient gains weight or becomes obese, the WAT undergoes a significant phenotypic change. This change manifests itself in the appearance of inflamed and dysfunctional adipocytes, as well as in the infiltration of immune cells in the vascular fraction of the stroma [11]. Inflamed adipocytes release proinflammatory cytokines both locally and into the bloodstream, disrupting the normal function of AT itself and affecting distant organs, such as the lung, pancreas and cardiovascular system. From this perspective, AT can be seen as an organ with immunological and secretory functions, whereas obesity can be considered a chronic inflammatory disease of immunological origin [47].
In obesity, AT acts not only as an energy reservoir but also as an important source of bioactive molecules known as adipokines. These molecules include various cytokines, chemokines, and other proteins that play regulatory roles in the immune system and inflammation. Leptin, which is secreted mainly by adipocytes, belongs to the cytokine family (structurally similar to IL-5, IL-6 and IL-15), and its plasma concentrations correlate with fat mass and respond to changes in energy balance [48]. In this context, the main effect of leptin is to act on the central nervous system and inhibit intake to regulate energy reserves [49]. In addition, it increases basal metabolism, and leptin also stimulates the oxidation of fatty acids and modulates the functionality of pancreatic ß cells [50]. Therefore, a leptin deficiency may be the cause of obesity. However, it has been reported that most obese people have elevated levels of leptin [51]. These individuals do not have a deficiency in leptin production, but in many cases, the leptin transporter from the blood to the interior of the central nervous system, through the blood‒brain barrier, and/or through a peripheral pathway of leptin resistance fails [52]. Therefore, leptin was initially considered an anti-obesity hormone, but experimental evidence has demonstrated the pleiotropic effects of this molecule on reproductive function, hematopoiesis, angiogenesis, lymphoid organ homeostasis, and T-cell functions [53]. In obese patients, high serum leptin concentrations are associated with reduced respiratory drive and impaired hypercapnic responses in men and women, suggesting resistance to the effects of leptin on respiratory function[54]. See Figure 2.
These data indicate that leptin receptors are present not only in the cells of the nervous system but also in many other cells in the body [55]. More specifically, leptin appears to be a link between the Th1 proinflammatory response, nutritional status, and energy balance [56]. Thus, some data indicate that it stimulates the proliferation and activation of peripheral mononuclear cells, stimulates the production of proinflammatory cytokines (e.g., IL-6 and TNFα) by circulating monocytes, and enhances the activation of lymphocytes against mitogens such as phytohemagglutinin or concanavalin A [57]. Leptin also inhibits the production of memory T lymphocytes, increases the production of B lymphocytes and stimulates the production of Th1-type cytokines (IL-2 and IFN-γ) by lymphocytes [52].
Leptin stimulates monocytes to express more leptin receptors and acts on T lymphocytes, causing them to express more leptin receptors and activating them [58]. As a result, monocytes release proinflammatory cytokines; these cytokines stimulate the T lymphocytes that respond by increasing the production of IL-2 and IFN-γ, two cytokines of the Th1 response. The Th1 response is immunoprotective, but it is also proinflammatory; therefore, an exaggerated Th1 response is harmful. On the other hand, IFN-γ acts on monocytes, increasing the production of proinflammatory cytokines; thus, we can consider leptin itself to be a proinflammatory cytokine [55]. Therefore, obesity may be considered an inflammatory disease in which high levels of circulating leptin are frequently observed [58].
IL-6 is primarily secreted by adipocytes and macrophages from AT and contributes significantly to the systemic inflammation associated with obesity. This adipocytokine not only participates in the regulation of the immune response but also has been implicated in the development of metabolic complications such as insulin resistance and endothelial dysfunction. Elevated IL-6 levels may contribute to chronic inflammation and the pathogenesis of metabolic and respiratory diseases [59]. IL-6 is a multifunctional cytokine produced by a wide range of cells, including T lymphocytes, macrophages, fibroblasts, and adipocytes. It acts on numerous cell types to regulate the immune response, inflammation, and hematopoiesis. IL-6 exerts its effects by binding to a specific receptor, the IL-6 receptor (IL-6R), which forms a complex with glycoprotein 130 (gp130) to activate intracellular signaling pathways such as JAK-STAT (Janus kinase - signal transducers and transcription activators), MAPK (mitogen-activated protein kinase) and PI3K (phosphatidylinositol-3 kinase) [59,60].
IL-6 regulates the immune response in both the acute and chronic phases of inflammation, participating in the differentiation and activation of T cells, as well as in the production of antibodies by B lymphocytes. In addition, it plays a role in the transition from acute inflammation to tissue repair [61]. Elevated IL-6 levels are associated with various chronic inflammatory, autoimmune, and metabolic diseases, including rheumatoid arthritis, Crohn's disease, metabolic syndrome, and obesity. In these contexts, IL-6 may contribute to disease progression by modulating inflammation and the immune response [61]. Owing to its central role in inflammation, IL-6 has been investigated as a therapeutic target in the treatment of inflammatory diseases. IL-6 inhibitors such as tocilizumab and sarilumab are used to treat diseases such as rheumatoid arthritis and Castleman disease [60,61].
Another important proinflammatory adipokine secreted primarily by macrophages from adipose tissue is TNFα. TNF-α is involved in regulating the inflammatory response and may contribute to metabolic dysfunction associated with obesity, as well as lung inflammation [62,63]. TNF-α is a key proinflammatory cytokine involved in regulating the immune response and inflammation. In the context of obesity, adipose tissue, especially visceral adipose tissue, serves as an important source of circulating TNF-α [63,64,65]. TNF-α is secreted by several cells, including macrophages, T lymphocytes, and adipocytes, in response to inflammatory stimuli [66]. It regulates both local and systemic inflammation by activating inflammatory signaling pathways such as the NF-κB and MAPK pathways and promotes the production of other proinflammatory cytokines [63].
Elevated levels of TNFα in adipose tissue are associated with insulin resistance, endothelial dysfunction, and increased risk of cardiovascular disease in obese individuals. TNF-α can modulate the secretion of other proinflammatory adipokines, such as IL-6, thereby exacerbating chronic inflammatory states in obese individuals. Obesity induces excessive production of TNFα by AT, contributing to systemic inflammation and the development of metabolic comorbidities, including pulmonary inflammation. TNF-α may interact synergistically with other inflammatory cytokines to potentiate detrimental effects on metabolic and cardiovascular function [64].
Numerous studies in animal and human models have confirmed that increased body fat is associated with AT inflammation due to excessive caloric intake. For example, Lee et al. [67] demonstrated in immunocompromised mice that inflammation plays a key role in the development of insulin resistance induced by a prolonged obesogenic diet. A notable feature of inflammation associated with the growth of WAT is its persistence and low-grade inflammation, known as "metaflammation", which is characterized by chronic inflammation that does not resolve effectively.
Inflammation is an energy-consuming process that can increase energy expenditure by reducing both energy intake and energy storage. Inflammatory cytokines, such as factor necrosis tumor alpha (TNFα), interleukin-1 (IL-1β), and interleukin-6 (IL-6), stimulate energy expenditure by interacting with receptors in the central nervous system and in active metabolic tissues, similar to leptin, which also promotes energy expenditure[68,69]. Leptin, which inhibits appetite and increases energy expenditure [70], is produced in greater amounts in response to inflammation and hypoxia [71,72], and its action is enhanced by TNFα [73], thus facilitating greater energy expenditure.
However, WAT inflammation induced by excess calories does not always translate into a significant increase in energy expenditure, allowing inflammation and weight gain to coexist in obese patients [74]. However, AT inflammation is similar to traditional inflammatory responses, such as the infiltration of immune cells and the release of inflammatory mediators, such as chemokines and cytokines, by both adipocytes and resident immune cells. Inflamed WAT, even in normal-weight patients, can cause widespread systemic inflammation through the release of cytokines into the bloodstream [75].
WAT has diverse inflammatory features, and obesity causes a more complex inflammatory response in visceral WAT (VAT) than in subcutaneous WAT (SAT). VAT contains more macrophages (up to 60% of total cells in the tissue) than SAT does in obese mice, as in humans [76,77]. Obesity and insulin resistance lead to greater adipocyte hypertrophy and hyperplasia in VAT than in SAT [78], and the inflammation of VAT in obese humans is associated with lower expression of lipogenic markers [79]. This occurs because more cells in the VAT adopt an inflammatory phenotype rather than one oriented toward lipid storage, which can lead to metabolic complications, such as lipid accumulation in the muscle and liver [79]. This accumulation interferes with peripheral insulin signaling, causing VAT inflammation to have a significant effect on obesity-related metabolic disorders, such as systemic insulin resistance, T2D, CVD and pulmonary inflammation [80]. However, SAT inflammation has also been shown to play a role in metabolic complications [81].
Contribution of Adipose Tissue Inflammation to Systemic Inflammation
The expansion of AT not only increases the degree of infiltration of macrophages from AT but also causes a change in the polarization of macrophages from the M2 phenotype, with an anti-inflammatory secretory profile effect, toward the M1 phenotype, with a proinflammatory secretory profile [82]. The latter would be responsible for the expression of most cytokines involved in the proinflammatory processes that occur in AT and of the molecules involved in the recruitment of more macrophages to the tissue, establishing a vicious cycle that would amplify the activation of inflammatory pathways [83,84]. This AT inflammation plays a critical role in the pathophysiology of metabolic syndrome, and visceral obesity can be considered the triggering component of metabolic syndrome (MS) [84,85]. Therefore, insulin resistance is clinically defined by the incompetence of a certain concentration of insulin (endogenous or exogenous) to increase cellular glucose utilization. When the amount of insulin in the blood is the same, the withdrawal of circulating glucose is lower, and the cellular performance as metabolic fuel in the target organs is worse, resulting in poor cellular performance (AT, muscle and liver, especially) [84].
MS is a clinical entity characterized by a set of risk factors that include visceral obesity, alterations in lipid metabolism (increased triglycerides and decreased HDL-C) and insulin, hypertension, and the presence of prothrombotic and inflammatory factors [86]. In different studies carried out in both children and adults, a positive correlation has been observed between BMI and the presence of MS, where the higher the BMI is, the greater the presence of MS [87]. In turn, MS is associated with an increased risk of T2DM, ischemic CVD, and premature death [88,89]. In addition to local inflammation at the AT, the inflammatory process triggered by obesity also involves skeletal muscle and the liver in the development of systemic IR and MS [88]. It has been proposed that the skeletal muscle mass index (SMM) could play an important role in the progression of MS. VAT (associated with internal organs) is the most important determinant of MS in obese individuals [90].
In experimental animals, macrophage infiltration into the skeletal muscle of obese mice has been demonstrated, especially intramuscular adipose deposition. These macrophages exhibit a proinflammatory M1 phenotype, accompanied by an increase in the expression of inflammatory factors, which contribute to local IR [90].
Adipose Tissue Inflammation Causes Pulmonary Neutrophilic Inflammation in Obesity
Neutrophilic inflammation in the lungs of obese individuals is an emerging area of research that has gained attention because of the increasing prevalence of obesity and its adverse effects on respiratory health. Obesity is associated with a number of respiratory comorbidities, and excess adipose tissue can negatively influence lung function. The relationship between obesity and pulmonary neutrophilic inflammation is closely linked to the production of proinflammatory cytokines. Adipokines, which are molecules produced by adipose tissue, can exacerbate lung inflammation by activating neutrophils and other inflammatory cells. A study showed that adipokines such as leptin and adiponectin have significant effects on the inflammatory response and can increase neutrophil infiltration into lung tissues [91].
Adipose tissue also responds to proinflammatory stimuli that originate in the lungs and spread through the systemic circulation. Several studies in animal models have shown that exposure to bacteria, ozone (O3), allergens, and particulate matter (PM) activates AT to produce leptin, IL-6, and other adipocytokines that worsen pulmonary neutrophilic inflammation [51,92]. Similarly, systemic administration of bacteria, IL-1β, lipopolysaccharide (LPS), and TNFα has been shown to increase systemic proinflammatory adipocytokine concentrations [93]. These observations suggest that in obese patients exposed to environmental stressors, WAT acts as an endocrine gland that releases adipocytokines, cytokines, and other mediators, intensifying both local and systemic inflammatory responses. Furthermore, impaired neutrophil function in obese lungs can lead to significant tissue damage. The accumulation of neutrophils and the release of their toxic products can induce oxidative stress and damage lung structures, contributing to the development of respiratory diseases such as chronic obstructive pulmonary disease (COPD) and asthma [89].
The impact of neutrophilic inflammation on lung function is also reflected in changes in lung structure. Obesity can induce changes in lung architecture, such as thickening of airway walls and increased mucus production, which can exacerbate respiratory symptoms [94]. Neutrophilic inflammation can induce significant damage to lung tissue. Neutrophils release proteolytic enzymes and ROS that damage epithelial cells and connective tissue. This can lead to alterations in the structure of the airways and lung parenchyma [94].
Pulmonary neutrophilic inflammation can contribute to the pool of inflammatory mediators in the bloodstream, worsening obesity-associated metabolic complications in obese patients exposed to airborne stressors. Chronic lung diseases, such as COPD and asthma, are associated with increased systemic inflammation [95]. Inflammatory mediators released in the lungs can enter the bloodstream and cause systemic inflammation that affects insulin signaling in peripheral tissues [96]. Chronic inflammation in the lungs and the release of inflammatory cytokines can alter adipose and muscle tissue function, exacerbating insulin resistance [95].
Inflamed AT releases mediators that not only impact insulin sensitivity but also may influence lung inflammation [11]. This systemic inflammation manifests in the lung through the secretion of proinflammatory cytokines that affect lung function and metabolic regulation [97]. Visceral adipose tissue, in particular, is associated with increased systemic inflammation and increased risk of respiratory and metabolic diseases [98].
Systemic inflammation resulting from pulmonary neutrophilic inflammation can contribute to metabolic dysfunction, including insulin resistance. Inflammatory mediators present in the bloodstream, originating in the lungs or other inflamed tissues, can interfere with insulin signaling and alter glucose metabolism. Chronic inflammation in multiple body systems amplifies insulin resistance and increases the risk of developing T2D [99]. This bidirectional relationship between pulmonary neutrophilic inflammation and AT inflammation highlights the importance of addressing both respiratory and metabolic inflammation in the management of obesity-associated metabolic comorbidities [100].
Neutrophilic inflammation in the lungs of obese patients may negatively affect lung function through several mechanisms. Chronic inflammation can lead to thickening of the airway walls and increased mucus production, resulting in airway obstruction and difficulty breathing. The presence of neutrophils has also been associated with lower lung elasticity and a greater predisposition to respiratory infections[94,101]. Respiratory dysfunction in obese patients can be especially severe because of the combination of chronic inflammation and mechanical alterations in the lungs [94,101].
The management of pulmonary neutrophilic inflammation in obese lungs is an active area of research. Therapeutic strategies may include reducing inflammation through the use of anti-inflammatory agents and modulating the immune response [102]. For example, drugs that block inflammatory signaling pathways or reduce adipokine production could be beneficial. Furthermore, weight loss interventions may have a positive effect on reducing neutrophilic inflammation and improving lung function [102]
Mechanical Effects of Obesity on Lung Function
The impact of obesity on respiratory function and the increased burden of respiratory disease among obese people are significant [47,94,103]. The impact begins with the critical distribution of fat, which represents a mechanical obstruction of lung function. Android obesity (i.e., fat distributed in the chest, abdomen, and visceral organs) has a more direct effect on lung function than does gynoid obesity (i.e., fat distributed in the subcutaneous tissue of the hips, thighs, arms, and legs) [94]. The greatest impact of android obesity on lung function depends on the increase in abdominal volume and the presence of intrathoracic fat that favors displacement of the diaphragm, reducing lung volume, specifically functional residual capacity (FRC) and expiratory reserve volume (VRE) [94,104]. The reduction in chronic respiratory failure is directly proportional to the severity of obesity: overweight, mildly obese, and severely obese subjects demonstrate reductions of 10%, 22%, and 33%, respectively [94]. In obese subjects, the mechanical load caused by excess adipose tissue reduces the capacity caliber of the airways, resulting in limited expiratory flow and low lung volume[52]. One of the mechanical complications of obesity is increased stress on the respiratory system due to constriction of the airways [94].
Recent studies have revealed a strong association between obesity and pulmonary hypertension, a condition characterized by an increase in mean pulmonary arterial pressure [105]. Alterations in the mechanics of the respiratory system caused by obesity include expiratory flow limitation, atelectasis, and V/Q mismatch (which occurs when ventilation airflow or perfusion blood flow is impaired, limiting primary lung function from supplying oxygen to the blood) with hypoxemia. All of these factors have important implications in the context of critical illness [106]. The tidal volume is also slightly lower in obese people; however, there is no significant effect on residual volume (RV) or total lung capacity [94]. Fat accumulation in the chest and abdomen has been found to increase the elasticity lung rate to 35% in obese individuals, which is exponentially related to body mass index (BMI) [107]. This reduced compliance is a consequence of significant alterations in the mechanical properties of the entire respiratory system in the presence of fat deposits in the mediastinum and abdominal cavities and contributes to breathing difficulties, such as wheezing, difficulty breathing (dyspnea) and shortness of breath during sleep (orthopnea) [94]. However, the mechanical properties of the chest wall in obese individuals indicate a chest mass load rather than stiffness of the chest mass due to elastic loading, which complicates the simplistic association between a constrictive effect and fat volume and may indicate that fat volume increases compliance [107].
Pulmonary Neutrophilic Inflammation in Obesity-Associated Metabolic Risk
Pulmonary inflammation triggered by neutrophil activation, known as pulmonary neutrophilic inflammation, is not limited to an acute response; it can become a chronic process that significantly affects lung function [108]. Under conditions such as chronic obstructive pulmonary disease (COPD) and asthma, sustained accumulation of neutrophils in the lungs is associated with tissue damage, airway remodeling, and decreased respiratory capacity. The continued presence of ROS and proinflammatory cytokines can lead to lung tissue destruction and reduced lung elasticity, contributing to the progression of respiratory diseases [89].
The data in humans are still incipient and do not allow us to determine temporal evolution, as in animal models [109]. When obese populations with or without cardiovascular risk factors are compared, leukocytes in the group with the worst metabolic profile present differences in quantitative and qualitative characteristics [48], suggesting a possible role of immune system activation in the consequences of obesity. Inflammatory markers, including the neutrophil count and myeloperoxidase (MPO) level in the blood, have even been suggested as predictors of obesity-associated cardiovascular morbidity and mortality [104].
Practically all metabolic pathologies associated with excess malnutrition have an inflammatory component within their genesis and/or progression [110]. The activation of the inflammatory pathway in any type of cell (at the level of the transcription factor NF-κB) hinders insulin cell signaling. Insulin resistance is the condition that underlies all entities of metabolic syndrome, but for each cardiovascular risk factor, there is also a direct pathophysiological association with inflammation, such as diabetes, dyslipidemia, endothelial dysfunction, hypertension, and metabolically associated liver disease (MALD) [111].
In obesity, AT- and pulmonary-derived inflammatory mediators contribute to systemic inflammation and thus worsen insulin resistance and other obesity-associated comorbidities. Furthermore, chronic lung inflammation has implications not only for respiratory health but also for systemic health. Persistent inflammation can promote insulin resistance and other metabolic disorders, creating a link between obesity-associated pulmonary neutrophilic inflammation and metabolic health. This is particularly relevant in obese individuals, where systemic inflammation can exacerbate respiratory conditions and contribute to an overall deterioration in quality of life [47,89]. Thus, understanding the role of neutrophils and inflammation in the lungs is critical for developing effective therapeutic strategies to mitigate these adverse effects.
Obesity is a complex metabolic condition that, in addition to pulmonary neutrophilic inflammation, is associated with a number of significant metabolic abnormalities. These abnormalities are crucial to understanding how obesity contributes to the development of chronic diseases such as T2D. Metabolic alterations associated with obesity include insulin resistance, alterations in insulin secretion, and a dysfunctional lipid profile [112]. These alterations not only affect glucose metabolism but also have important implications for cardiovascular health and other aspects of general metabolic well-being [113].
Pulmonary neutrophilic inflammation is a pathological process that can be triggered by various conditions, including obesity. In this context, neutrophil activation plays a crucial role. When neutrophils are activated in response to inflammatory cytokines such as TNF-α and IL-6, they release a series of inflammatory mediators, including more cytokines, chemokines, and chemicals such as reactive oxygen species (ROS) [114]. These highly reactive molecules cause oxidative damage to surrounding cells and tissues, exacerbating the inflammatory response and contributing to a vicious cycle that perpetuates inflammation [115].
Lung inflammation triggered by neutrophil activation is not limited to an acute response; it can become a chronic process that affects not only respiratory function but also systemic health. Sustained accumulation of neutrophils in the lungs is associated with an increase in the release of inflammatory mediators that enter the bloodstream, which can lead to systemic inflammation [89]. This inflammatory state can negatively influence metabolic function, contributing to conditions such as insulin resistance and T2D, as inflammatory cytokines alter insulin signaling in peripheral tissues [88,96]. Furthermore, chronic lung inflammation may have broader repercussions on the overall health of individuals [30]. This is especially relevant in obese individuals, where systemic inflammation can contribute to an overall deterioration in quality of life and an increase in morbidity [116].
Pulmonary neutrophilic inflammation is a key factor in insulin resistance associated with obesity. Excess adipose tissue produces a number of inflammatory mediators, such as proinflammatory cytokines, including TNFα and IL-6 [91]. These cytokines can interfere with insulin signaling in the muscles and liver, exacerbating insulin resistance and contributing to the development of T2D [117]. Chronic inflammation in adipose tissue is also linked to impaired endothelial function and increased predisposition to cardiovascular disease[118].
Chronic inflammation and insulin resistance are interconnected through common inflammatory mechanisms. In obesity, excess adipose tissue releases proinflammatory adipokines such as TNFα, IL-6, and C-reactive protein [119]. These proinflammatory cytokines not only affect metabolic function and insulin sensitivity in peripheral tissues, such as the liver and muscles but also influence lung inflammation [120]. Systemic inflammation and the production of these cytokines contribute to further inflammation in lung tissue [116].
Systemic inflammation is a phenomenon characterized by the presence of proinflammatory cytokines in the bloodstream and is often triggered by obesity [46]. In this state, adipocytes, as they expand, release inflammatory mediators such as TNF-α and IL-6 [121,122]. These cytokines not only activate neutrophils in the lungs and other tissues but also induce a generalized inflammatory response that affects various organs. This chronic inflammation, in particular, has been directly associated with the development of insulin resistance, a metabolic disorder in which cells do not respond adequately to insulin, compromising blood glucose regulation [116].
Insulin resistance originates, in part, from the interference of inflammatory cytokines in insulin signaling pathways. In a state of systemic inflammation, cytokines such as TNFα can inhibit the translocation of glucose transporters to the cell membrane and reduce the effectiveness of insulin in the liver and muscles [123]. This mechanism not only causes an increase in blood glucose levels but also contributes to a vicious cycle in which hyperglycemia and insulin resistance further promote inflammation [80]. Thus, systemic inflammation becomes a central factor in the development of T2D and other metabolic disorders [112].
Furthermore, the interrelationship between systemic inflammation and insulin resistance has broad implications for metabolic health. Chronic inflammation can promote not only insulin resistance but also the risk of cardiovascular disease, dyslipidemia, and other conditions associated with metabolic syndrome[89,111]. In obese individuals, systemic inflammation contributes to an overall deterioration in quality of life and an increase in morbidity. Therefore, addressing inflammation through interventions such as regular exercise, weight loss, and an anti-inflammatory diet is essential to improve metabolic health and prevent complications [86].
Concluding Remarks and Therapeutic Perspectives
Obesity affects the mechanical and biochemical functions of the lungs. AT-derived systemic inflammation is the main contributor to pulmonary neutrophilic inflammation. This, in turn, provides additional inflammatory mediators that can worsen obesity-associated metabolic comorbidities, including insulin resistance. Obesity-associated metabolic risk is greater in obese patients exposed to airborne irritants because it can cause pulmonary neutrophilic inflammation and thus increase systemic inflammation, the main driver of obesity-associated metabolic comorbidities. Therefore, obesity treatments that shrink AT and induce weight loss, such as lifestyle interventions, pharmacotherapy, and bariatric surgery, could represent another approach to treating obesity-associated pulmonary neutrophilic inflammation and reducing obesity-associated metabolic risk. The search for natural and synthetic products to reduce the homing and activation of neutrophils can help reduce the disparities between obese patients and obese patients exposed to airborne stressors.
Acknowledgments
This work was supported by grants from Agencia Nacional para la Promoción de la Ciencia y la Technología, FONCYT, PICT-2018-03435 and PICT-2021-I-A-147. Ministerio de Capital Humano, República Argentina.
References
- Abete, I. et al. Association of lifestyle factors and inflammation with sarcopenic obesity: data from the PREDIMED-Plus trial. J Cachexia Sarcopenia Muscle 10, 974-984. (2019). [CrossRef]
- Swinburn, B. A. et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet (London, England) 378, 804-814. (2011). [CrossRef]
- Nedunchezhiyan, U. et al. Obesity, Inflammation, and Immune System in Osteoarthritis. Front Immunol 13, 907750. (2022). [CrossRef]
- Masenga, S. K., Kabwe, L. S., Chakulya, M. & Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. International Journal of Molecular Sciences 24, 7898 (2023).
- Health Effects of Overweight and Obesity in 195 Countries over 25 Years. New England Journal of Medicine 377, 13-27, (2017). [CrossRef]
- Jaitovich, A. & Hall, J. B. The flux of energy in critical illness and the obesity paradox. Physiol Rev 105, 1487-1552. (2025). [CrossRef]
- Mancuso, P. Obesity and lung inflammation. Journal of Applied Physiology 108, 722-728. (2009). [CrossRef]
- Gummlich, L. Obesity-induced neutrophil reprogramming. Nat Rev Cancer 21, 412. (2021). [CrossRef]
- Kohyama, M. et al. Monocyte infiltration into obese and fibrilized tissues is regulated by PILRalpha. Eur J Immunol 46, 1214-1223. (2016). [CrossRef]
- Shantaram, D. et al. Obesity-associated microbiomes instigate visceral adipose tissue inflammation by recruitment of distinct neutrophils. Nat Commun 15, 5434. (2024). [CrossRef]
- Kawai, T., Autieri, M. V. & Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol 320, C375-C391. (2021). [CrossRef]
- Anderson, E. K., Gutierrez, D. A. & Hasty, A. H. Adipose tissue recruitment of leukocytes. Curr Opin Lipidol 21, 172-177. (2010). [CrossRef]
- Poblete, J. M. S. et al. Macrophage HIF-1alpha mediates obesity-related adipose tissue dysfunction via interleukin-1 receptor-associated kinase M. Am J Physiol Endocrinol Metab 318, E689-E700. (2020). [CrossRef]
- Peris, A. et al. Obesity and inflammatory response in moderate-to-severe acute respiratory distress syndrome: a single center pilot study. Minerva Med 116, 89-93. (2025). [CrossRef]
- Zhou, J., You, L., Zhou, X. & Li, Y. Associations between metabolic score for visceral fat and adult lung functions from NHANES 2007-2012. Front Nutr 11, 1436652. (2024). [CrossRef]
- Gomez Mejiba, S. E. & Ramirez, D. C. Neutrophilic Inflammation and Diseases: Pathophysiology, Biomarkers and Therapeutic Targets. (Eliva Press, 2020).
- Gomez-Mejiba, S. E. et al. Inhalation of environmental stressors & chronic inflammation: autoimmunity and neurodegeneration. Mutat Res 674, 62-72. (2009). [CrossRef]
- Talukdar, S. et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18, 1407-1412. (2012). [CrossRef]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Frontiers in physiology 9, 113. (2018). [CrossRef]
- Uribe-Querol, E. & Rosales, C. Neutrophils Actively Contribute to Obesity-Associated Inflammation and Pathological Complications. Cells 11. (2022). [CrossRef]
- Uribe-Querol, E. & Rosales, C. Neutrophils Actively Contribute to Obesity-Associated Inflammation and Pathological Complications. Cells 11, 1883 (2022).
- Kordonowy, L. L. et al. Obesity is associated with neutrophil dysfunction and attenuation of murine acute lung injury. Am J Respir Cell Mol Biol 47, 120-127. (2012). [CrossRef]
- Li, N. et al. Correlation of White Blood Cell, Neutrophils, and Hemoglobin with Metabolic Syndrome and Its Components. Diabetes, Metabolic Syndrome and Obesity 16, 1347-1355. (2023). [CrossRef]
- Biswas, M. et al. The mechanistic role of neutrophil lymphocyte ratio perturbations in the leading non communicable lifestyle diseases. F1000Research 11, 960. (2022). [CrossRef]
- Baragetti, A. et al. Neutrophil aging exacerbates high fat diet induced metabolic alterations. Metabolism: clinical and experimental 144, 155576. (2023). [CrossRef]
- Suren Garg, S., Kushwaha, K., Dubey, R. & Gupta, J. Association between obesity, inflammation and insulin resistance: Insights into signaling pathways and therapeutic interventions. Diabetes Research and Clinical Practice 200, 110691. (2023). [CrossRef]
- Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature reviews. Immunology 19, 477-489. (2019). [CrossRef]
- Sun, Z., Dragon, S., Becker, A. & Gounni, A. S. Leptin inhibits neutrophil apoptosis in children via ERK/NF-κB-dependent pathways. PloS one 8, e55249. (2013). [CrossRef]
- Ghanim, H. et al. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation 110, 1564-1571. (2004). [CrossRef]
- Herrero-Cervera, A., Soehnlein, O. & Kenne, E. Neutrophils in chronic inflammatory diseases. Cellular & Molecular Immunology 19, 177-191. (2022). [CrossRef]
- Dixon, J. B. & O' Brien, P. E. Obesity and the White Blood Cell Count: Changes with Sustained Weight Loss. Obesity Surgery 16, 251-257. (2006). [CrossRef]
- Mohamed-Ali, V. et al. Subcutaneous Adipose Tissue Releases Interleukin-6, However, Not Tumor Necrosis Factor-α, in Vivo1. The Journal of Clinical Endocrinology & Metabolism 82, 4196-4200. (1997). [CrossRef]
- Bruno, A., Conus, S. b., Schmid, I. s. & Simon, H.-U. Apoptotic Pathways Are Inhibited by Leptin Receptor Activation in Neutrophils1. The Journal of Immunology 174, 8090-8096. (2005). [CrossRef]
- Artemniak-Wojtowicz, D., Kucharska, A. & Pyrżak, B. Obesity and chronic inflammation crosslinking. Central European Journal of Immunology 45, 461-468. (2020). [CrossRef]
- Bahadir, A. et al. Is the neutrophil-to-lymphocyte ratio indicative of inflammatory state in patients with obesity and metabolic syndrome? Anatol J Cardiol 15, 816-822. (2015). [CrossRef]
- Barden, A. et al. Effect of weight loss on neutrophil resolvins in the metabolic syndrome. Prostaglandins Leukot Essent Fatty Acids 148, 25-29. (2019). [CrossRef]
- Della Vedova, M. C. et al. 410 - Neutrophils in the Obese Lung: A Mechanistic Study in a Mouse Model of Metabolic Syndrome. Free Radical Biology and Medicine 100, S171-S172. (2016). [CrossRef]
- Cottam, D. R., Schaefer, P. A., Fahmy, D., Shaftan, G. W. & Angus, L. D. The effect of obesity on neutrophil Fc receptors and adhesion molecules (CD16, CD11b, CD62L). Obes Surg 12, 230-235. (2002). [CrossRef]
- Elgazar-Carmon, V., Rudich, A., Hadad, N. & Levy, R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res 49, 1894-1903. (2008). [CrossRef]
- Goossens, G. H. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiology & behavior 94, 206-218. (2008). [CrossRef]
- Shi, H. et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116, 3015-3025. (2006). [CrossRef]
- Cinti, S. et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of lipid research 46, 2347-2355. (2005). [CrossRef]
- Kim, J. Y. et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117, 2621-2637. (2007). [CrossRef]
- Sethi, J. K. & Vidal-Puig, A. J. Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. Journal of lipid research 48, 1253-1262. (2007). [CrossRef]
- Tilg, H. & Moschen, A. R. Role of adiponectin and PBEF/visfatin as regulators of inflammation: involvement in obesity-associated diseases. Clin Sci (Lond) 114, 275-288. (2008). [CrossRef]
- Antuna-Puente, B., Feve, B., Fellahi, S. & Bastard, J. P. [Obesity, inflammation and insulin resistance: which role for adipokines]. Therapie 62, 285-292. (2007). [CrossRef]
- Brock, J. M., Billeter, A., Müller-Stich, B. P. & Herth, F. Obesity and the Lung: What We Know Today. Respiration; international review of thoracic diseases 99, 856-866. (2020). [CrossRef]
- Ayed, K. et al. Obesity and cancer: focus on leptin. Molecular biology reports 50, 6177-6189. (2023). [CrossRef]
- Münzberg, H., Singh, P., Heymsfield, S. B., Yu, S. & Morrison, C. D. Recent advances in understanding the role of leptin in energy homeostasis. F1000Research 9. (2020). [CrossRef]
- Stern, J. H., Rutkowski, J. M. & Scherer, P. E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell metabolism 23, 770-784. (2016). [CrossRef]
- Hsu, A., Aronoff, D. M., Phipps, J., Goel, D. & Mancuso, P. Leptin improves pulmonary bacterial clearance and survival in ob/ob mice during pneumococcal pneumonia. Clin Exp Immunol 150, 332-339. (2007). [CrossRef]
- Gogiraju, R. et al. Deletion of endothelial leptin receptors in mice promotes diet-induced obesity. Scientific reports 13, 8276. (2023). [CrossRef]
- Matarese, G., Sanna, V., Fontana, S. & Zappacosta, S. Leptin as a novel therapeutic target for immune intervention. Current drug targets. Inflammation and allergy 1, 13-22. (2002). [CrossRef]
- Bassi, M. et al. Control of respiratory and cardiovascular functions by leptin. Life sciences 125, 25-31. (2015). [CrossRef]
- Fan, X., Yuan, W., Huang, W. & Lin, Z. Recent progress in leptin signaling from a structural perspective and its implications for diseases. Biochimie 212, 60-75. (2023). [CrossRef]
- Pérez-Pérez, A., Sánchez-Jiménez, F., Vilariño-García, T. & Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. International journal of molecular sciences 21. (2020). [CrossRef]
- Dixit, V. D. et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. The Journal of clinical investigation 114, 57-66. (2004). [CrossRef]
- Fernández-Riejos, P. et al. Role of leptin in the activation of immune cells. Mediators of inflammation 2010, 568343. (2010). [CrossRef]
- Scheller, J., Chalaris, A., Schmidt-Arras, D. & Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochimica et biophysica acta 1813, 878-888. (2011). [CrossRef]
- Hunter, C. A. & Jones, S. A. IL-6 as a keystone cytokine in health and disease. Nature immunology 16, 448-457. (2015). [CrossRef]
- Tanaka, T., Narazaki, M. & Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harbor perspectives in biology 6, a016295. (2014). [CrossRef]
- Hotamisligil, G. S., Shargill, N. S. & Spiegelman, B. M. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (New York, N.Y.) 259, 87-91. (1993). [CrossRef]
- Mohamed-Ali, V., Pinkney, J. H. & Coppack, S. W. Adipose tissue as an endocrine and paracrine organ. International Journal of Obesity 22, 1145-1158. (1998). [CrossRef]
- Uysal, K. T., Wiesbrock, S. M., Marino, M. W. & Hotamisligil, G. S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389, 610-614. (1997). [CrossRef]
- Kunz, H. E. et al. Adipose tissue macrophage populations and inflammation are associated with systemic inflammation and insulin resistance in obesity. Am J Physiol Endocrinol Metab 321, E105-E121. (2021). [CrossRef]
- Parameswaran, N. & Patial, S. Tumor necrosis factor-α signaling in macrophages. Critical reviews in eukaryotic gene expression 20, 87-103. (2010). [CrossRef]
- Lee, Y. S. et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 60, 2474-2483. (2011). [CrossRef]
- Anforth, H. R. et al. Biological activity and brain actions of recombinant rat interleukin-1alpha and interleukin-1beta. European cytokine network 9, 279-288 (1998).
- Wallenius, V. et al. Interleukin-6-deficient mice develop mature-onset obesity. Nature medicine 8, 75-79. (2002). [CrossRef]
- Myers, M. G., Jr. & Olson, D. P. Central nervous system control of metabolism. Nature 491, 357-363. (2012). [CrossRef]
- Grosfeld, A. et al. Hypoxia-inducible factor 1 transactivates the human leptin gene promoter. The Journal of biological chemistry 277, 42953-42957. (2002). [CrossRef]
- Grunfeld, C. et al. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. The Journal of clinical investigation 97, 2152-2157. (1996). [CrossRef]
- Gan, L. et al. TNF-α upregulates protein level and cell surface expression of the leptin receptor by stimulating its export via a PKC-dependent mechanism. Endocrinology 153, 5821-5833. (2012). [CrossRef]
- Carneiro, I. P. et al. Is Obesity Associated with Altered Energy Expenditure? Advances in nutrition (Bethesda, Md.) 7, 476-487. (2016). [CrossRef]
- Oliveros, E., Somers, V. K., Sochor, O., Goel, K. & Lopez-Jimenez, F. The concept of normal weight obesity. Progress in cardiovascular diseases 56, 426-433. (2014). [CrossRef]
- Altintas, M. M. et al. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. Journal of lipid research 52, 480-488. (2011). [CrossRef]
- Okamoto, Y. et al. Comparison of mitochondrial and macrophage content between subcutaneous and visceral fat in db/db mice. Experimental and molecular pathology 83, 73-83. (2007). [CrossRef]
- Hardy, O. T. et al. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surgery for obesity and related diseases : official journal of the American Society for Bariatric Surgery 7, 60-67. (2011). [CrossRef]
- Poulain-Godefroy, O., Lecoeur, C., Pattou, F., Frühbeck, G. & Froguel, P. Inflammation is associated with a decrease of lipogenic factors in omental fat in women. American journal of physiology. Regulatory, integrative and comparative physiology 295, R1-7. (2008). [CrossRef]
- Klöting, N. et al. Insulin-sensitive obesity. American journal of physiology. Endocrinology and metabolism 299, E506-515. (2010). [CrossRef]
- Ortega Martinez de Victoria, E. et al. Macrophage content in subcutaneous adipose tissue: associations with adiposity, age, inflammatory markers, and whole-body insulin action in healthy Pima Indians. Diabetes 58, 385-393. (2009). [CrossRef]
- Longo, M. et al. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. International journal of molecular sciences 20. (2019). [CrossRef]
- Yunna, C., Mengru, H., Lei, W. & Weidong, C. Macrophage M1/M2 polarization. European journal of pharmacology 877, 173090. (2020). [CrossRef]
- Ros Pérez, M. & Medina-Gómez, G. [Obesity, adipogenesis and insulin resistance]. Endocrinologia y nutricion : organo de la Sociedad Espanola de Endocrinologia y Nutricion 58, 360-369. (2011). [CrossRef]
- Suren Garg, S., Kushwaha, K., Dubey, R. & Gupta, J. Association between obesity, inflammation and insulin resistance: Insights into signaling pathways and therapeutic interventions. Diabetes research and clinical practice 200, 110691. (2023). [CrossRef]
- Fahed, G. et al. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. International journal of molecular sciences 23. (2022). [CrossRef]
- Meshkani, R. & Adeli, K. Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clinical biochemistry 42, 1331-1346. (2009). [CrossRef]
- Iafusco, D. et al. From Metabolic Syndrome to Type 2 Diabetes in Youth. Children (Basel, Switzerland) 10. (2023). [CrossRef]
- Baffi, C. W. et al. Metabolic Syndrome and the Lung. Chest 149, 1525-1534. (2016). [CrossRef]
- Vedova, M. C. D. et al. Diet-Induced Pulmonary Inflammation and Incipient Fibrosis in Mice: a Possible Role of Neutrophilic Inflammation. Inflammation 42, 1886-1900. (2019). [CrossRef]
- Zatterale, F. et al. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front Physiol 10, 1607. (2019). [CrossRef]
- Johnston, R. A., Schwartzman, I. N. & Shore, S. A. Macrophage inflammatory protein-2 levels are associated with changes in serum leptin concentrations following ozone-induced airway inflammation. Chest 123, 369 s-370 s (2003).
- Faggioni, R. et al. IL-1 beta mediates leptin induction during inflammation. The American journal of physiology 274, R204-208. (1998). [CrossRef]
- Dixon, A. E. & Peters, U. The effect of obesity on lung function. Expert review of respiratory medicine 12, 755-767. (2018). [CrossRef]
- Naik, D., Joshi, A., Paul, T. V. & Thomas, N. Chronic obstructive pulmonary disease and the metabolic syndrome: Consequences of a dual threat. Indian journal of endocrinology and metabolism 18, 608-616. (2014). [CrossRef]
- Galicia-Garcia, U. et al. Pathophysiology of Type 2 Diabetes Mellitus. International journal of molecular sciences 21. (2020). [CrossRef]
- Chan, S. M. H., Selemidis, S., Bozinovski, S. & Vlahos, R. Pathobiological mechanisms underlying metabolic syndrome (MetS) in chronic obstructive pulmonary disease (COPD): clinical significance and therapeutic strategies. Pharmacology & therapeutics 198, 160-188. (2019). [CrossRef]
- Wernstedt Asterholm, I. et al. Adipocyte Inflammation Is Essential for Healthy Adipose Tissue Expansion and Remodeling. Cell metabolism 20, 103-118. (2014). [CrossRef]
- Rodriguez-Rada, C. et al. Análisis de la relación entre diabetes mellitus tipo 2 y la obesidad con los factores de riesgo cardiovascular. Journal of Negative and No Positive Results 6, 411-433 (2021).
- Sagun, G. et al. The relation between insulin resistance and lung function: a cross sectional study. BMC pulmonary medicine 15, 139. (2015). [CrossRef]
- Littleton, S. W. Impact of obesity on respiratory function. Respirology (Carlton, Vic.) 17, 43-49. (2012). [CrossRef]
- Jasper, A. E., McIver, W. J., Sapey, E. & Walton, G. M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research 8. (2019). [CrossRef]
- Grassi, L., Kacmarek, R. & Berra, L. Ventilatory Mechanics in the Patient with Obesity. Anesthesiology 132, 1246-1256. (2020). [CrossRef]
- Patel, P. S., Buras, E. D. & Balasubramanyam, A. The role of the immune system in obesity and insulin resistance. Journal of obesity 2013, 616193. (2013). [CrossRef]
- Perrotta, F. et al. Pulmonary Hypertension and Obesity: Focus on Adiponectin. International journal of molecular sciences 20. (2019). [CrossRef]
- Anderson, M. R. & Shashaty, M. G. S. Impact of Obesity in Critical Illness. Chest 160, 2135-2145. (2021). [CrossRef]
- Umbrello, M., Fumagalli, J., Pesenti, A. & Chiumello, D. Pathophysiology and Management of Acute Respiratory Distress Syndrome in Obese Patients. Seminars in respiratory and critical care medicine 40, 40-56. (2019). [CrossRef]
- Ramirez, D. C. & Gomez Mejiba, S. E. Pulmonary Neutrophilic Inflammation and Noncommunicable Diseases: Pathophysiology, Redox Mechanisms, Biomarkers, and Therapeutics. Antioxid Redox Signal 33, 211-227. (2020). [CrossRef]
- Golforoush, P., Yellon, D. M. & Davidson, S. M. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic research in cardiology 115, 73. (2020). [CrossRef]
- Saunders, J. & Smith, T. Malnutrition: causes and consequences. Clinical medicine (London, England) 10, 624-627. (2010). [CrossRef]
- Baker, R. G., Hayden, M. S. & Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell metabolism 13, 11-22. (2011). [CrossRef]
- Klein, S., Gastaldelli, A., Yki-Järvinen, H. & Scherer, P. E. Why does obesity cause diabetes? Cell metabolism 34, 11-20. (2022). [CrossRef]
- Rosen, E. D. & Spiegelman, B. M. What we talk about when we talk about fat. Cell 156, 20-44. (2014). [CrossRef]
- Tilton, S. C. et al. Diet-induced obesity reprograms the inflammatory response of the murine lung to inhaled endotoxin. Toxicol Appl Pharmacol 267, 137-148. (2013). [CrossRef]
- Ubags, N. D. et al. A Comparative Study of Lung Host Defense in Murine Obesity Models. Insights into Neutrophil Function. Am J Respir Cell Mol Biol 55, 188-200. (2016). [CrossRef]
- Zhang, J. M. & An, J. Cytokines, inflammation, and pain. International anesthesiology clinics 45, 27-37. (2007). [CrossRef]
- Hotamisligil, G. S. Inflammation and metabolic disorders. Nature 444, 860-867. (2006). [CrossRef]
- Wang, Z. & Nakayama, T. Inflammation, a link between obesity and cardiovascular disease. Mediators of inflammation 2010, 535918. (2010). [CrossRef]
- Ruck, L., Wiegand, S. & Kühnen, P. Relevance and consequence of chronic inflammation for obesity development. Molecular and cellular pediatrics 10, 16. (2023). [CrossRef]
- Püschel, G. P., Klauder, J. & Henkel, J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. Journal of clinical medicine 11. (2022). [CrossRef]
- Bonamichi, B. & Lee, J. Unusual Suspects in the Development of Obesity-Induced Inflammation and Insulin Resistance: NK cells, iNKT cells, and ILCs. Diabetes Metab J 41, 229-250. (2017). [CrossRef]
- Chmelar, J., Chung, K. J. & Chavakis, T. The role of innate immune cells in obese adipose tissue inflammation and development of insulin resistance. Thromb Hemost 109, 399-406. (2013). [CrossRef]
- de Luca, C. & Olefsky, J. M. Inflammation and insulin resistance. FEBS Lett 582, 97-105. (2008). [CrossRef]
Figure 1.
Obesity-associated insulin resistance is worsened by obesity-induced pulmonary neutrophilic inflammation. Obesity is a metabolic state characterized by excess adipose tissue, which triggers an inflammatory response in the body. This condition is associated with the release of inflammatory cytokines by adipocytes, which activate neutrophils in the lungs and other organs, intensifying pulmonary neutrophilic inflammation. Inflammatory cytokines enter the bloodstream, causing systemic inflammation, a state of chronic inflammation that affects several systems in the body. One of the most serious consequences of this systemic inflammation is insulin resistance, where the signaling of this hormone is compromised, making it difficult to regulate blood glucose. This process is a critical precursor to T2D and other metabolic diseases.
Figure 1.
Obesity-associated insulin resistance is worsened by obesity-induced pulmonary neutrophilic inflammation. Obesity is a metabolic state characterized by excess adipose tissue, which triggers an inflammatory response in the body. This condition is associated with the release of inflammatory cytokines by adipocytes, which activate neutrophils in the lungs and other organs, intensifying pulmonary neutrophilic inflammation. Inflammatory cytokines enter the bloodstream, causing systemic inflammation, a state of chronic inflammation that affects several systems in the body. One of the most serious consequences of this systemic inflammation is insulin resistance, where the signaling of this hormone is compromised, making it difficult to regulate blood glucose. This process is a critical precursor to T2D and other metabolic diseases.
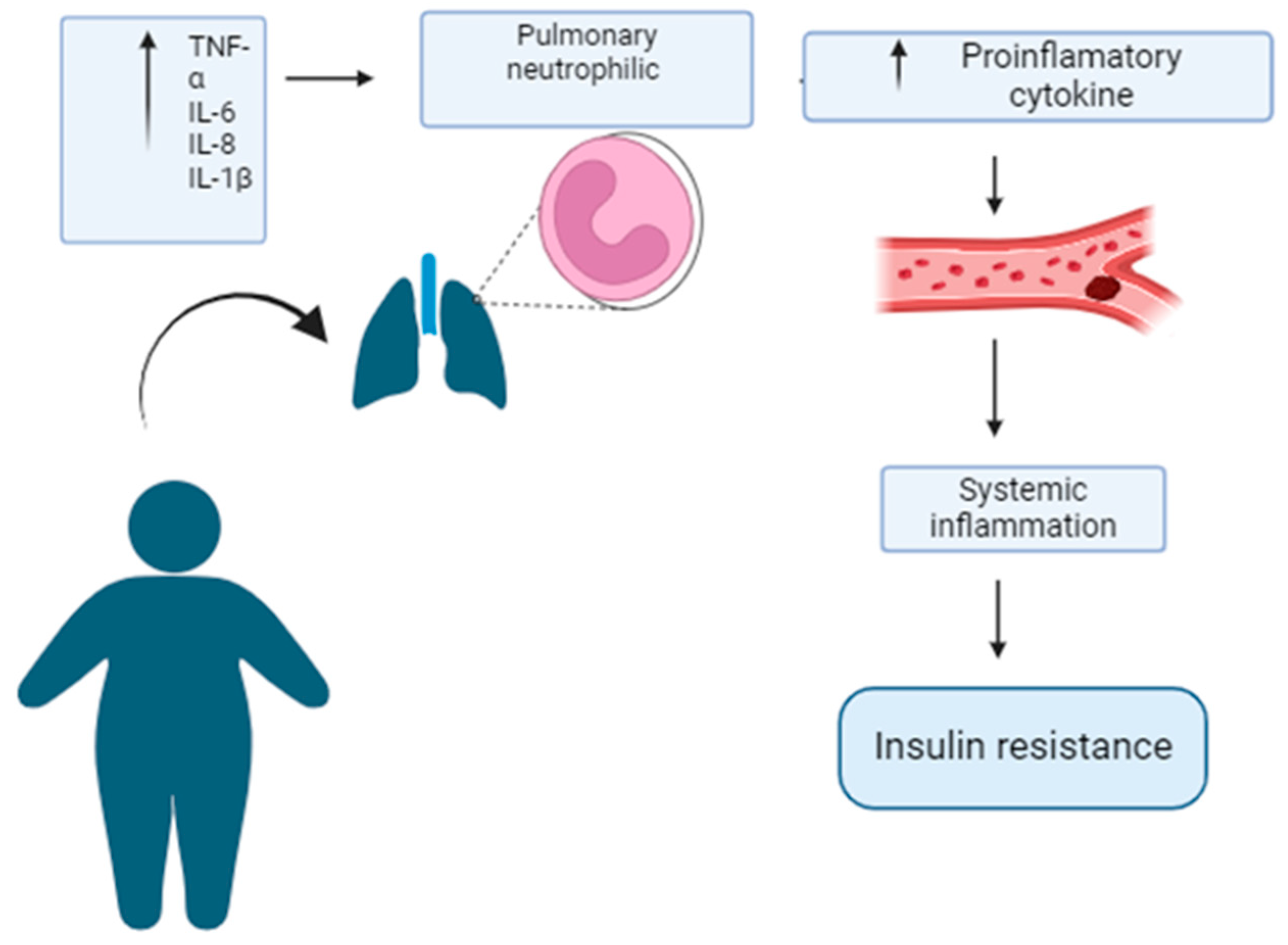
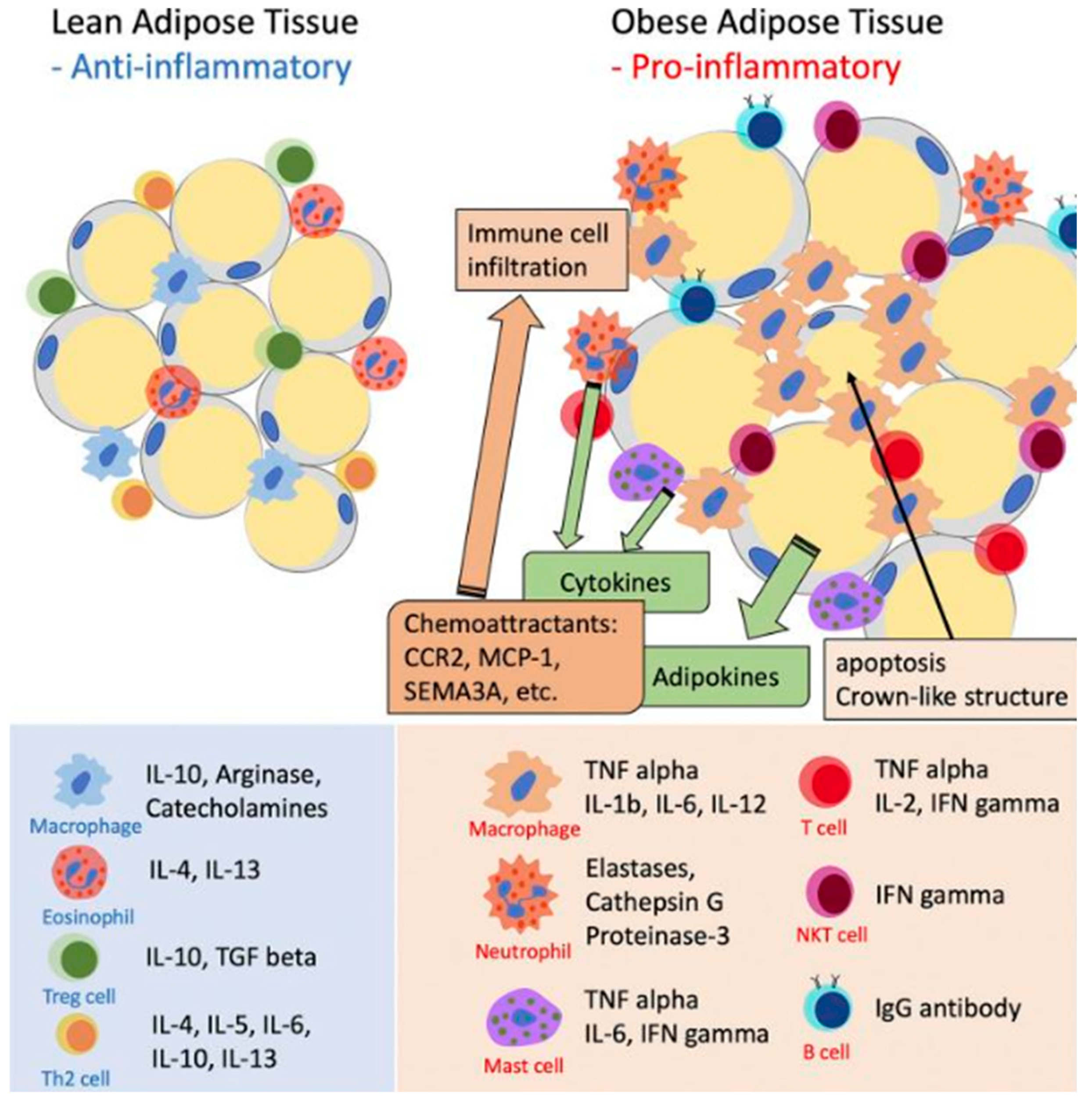
Figure 2.
Role of inflammatory cells in adipose tissue inflammation. The inflammatory phenotype of expanding AT is characterized by a series of changes in adipocytes and resident immune cells. As adipocytes hypertrophy and adipose tissue expands, these adipocytes and immune cells undergo transformations that alter their function. Instead of secreting protective anti-inflammatory cytokines, they begin to release adipokines and proinflammatory cytokines. These substances act both locally and systemically, contributing to the development of insulin resistance in peripheral tissues. Chronic inflammation in adipose tissue is maintained and amplified by the action of adipocyte-derived chemoattractants. These chemoattractants include the chemokine receptor CC type 2 (CCR2), monocyte chemoattractant protein-1 (MCP-1, also known as CCL-2), and traffic lights 3A (SEMA3A). These molecules play crucial roles in attracting and maintaining immune cells in inflamed AT, thereby perpetuating the inflammatory response and contributing to obesity-associated metabolic dysfunctions and low-grade inflammation. Modified from [11].
Figure 2.
Role of inflammatory cells in adipose tissue inflammation. The inflammatory phenotype of expanding AT is characterized by a series of changes in adipocytes and resident immune cells. As adipocytes hypertrophy and adipose tissue expands, these adipocytes and immune cells undergo transformations that alter their function. Instead of secreting protective anti-inflammatory cytokines, they begin to release adipokines and proinflammatory cytokines. These substances act both locally and systemically, contributing to the development of insulin resistance in peripheral tissues. Chronic inflammation in adipose tissue is maintained and amplified by the action of adipocyte-derived chemoattractants. These chemoattractants include the chemokine receptor CC type 2 (CCR2), monocyte chemoattractant protein-1 (MCP-1, also known as CCL-2), and traffic lights 3A (SEMA3A). These molecules play crucial roles in attracting and maintaining immune cells in inflamed AT, thereby perpetuating the inflammatory response and contributing to obesity-associated metabolic dysfunctions and low-grade inflammation. Modified from [11].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



