Submitted:
15 May 2025
Posted:
16 May 2025
You are already at the latest version
Abstract
Natural products have been crucial in cancer treatment due to their ability to selectively target cancer cells. Andrographolide, a major bioactive compound from the plant Andrographis paniculata, and its derivatives exhibited potential anticancer properties. As nitrogen-containing heteroaromatic compounds, particularly pyrazole derivatives, are key building blocks in the design of anticancer drugs, the primary focus of this study was to blend the pyrazole pharmacophore and andrographolide. In this work, twenty novel 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide and isoandrographolide were synthesized and characterized using UV-Vis, FT-IR, NMR, and HRMS. Following initial screening for anticancer activity against 60 human cancer cell lines at NCI, USA, compound 1f (R=4-F) was selected for detailed in vitro analysis on the breast cancer cell line MDA-MB-231. MTT assay results demonstrated that compound 1f exhibited strong, dose-dependent anti-proliferative effects. Apoptotic assay revealed a progressive increase in apoptotic cells upon treatment with 1f. Additionally, cell cycle analysis indicated that 1f caused G1/M phase arrest in MDA-MB-231 cells. The synthesized compounds were also tested for antioxidant activity using the DPPH assay, and compounds 1b (R=3-NO₂, 60.68%) and 2b (R=3-NO₂, 61.17%) showed the strongest radical scavenging activity. Finally, the most promising compounds were subjected to in-silico Lipinski’s assessments to evaluate their potential as lead candidates.
Keywords:
pyrazole acetals
; andrographolide
; isoandrographolide
; anticancer
; antioxidant activity
1. Introduction
Cancer is a group of diseases that occur when cells divide uncontrollably and consistently[1]. Despite the achievements of oncology, cancer continues to be one of the most fatal diseases and the second leading cause of mortality in the world[2]. As stated by the GLOBOCAN 2020 global cancer statistical report, ~19.3 million new cancer cases were recorded and accounted for ~10 million deaths worldwide (barring non-melanoma skin cancer)[3]. Breast, lung, colon, prostate, skin, and stomach are the most common cancers, and their fatality rate is considerably high[4]. The most prevalent cancer diagnosed now is breast cancer, with an estimated 2 million new cases annually, and remains the 5th highest cause of cancer death, with over half a million deaths[5,6]. Approximately 28 million new cases of cancer are anticipated worldwide over the next twenty years, a 47% increase from 2020[3,7]. Current anticancer drugs used in treatment often cause adverse effects such as weakened immune function, hair loss, cardiotoxicity, and infertility in women with active ovaries[8,9,10]. Consequently, alternative treatments involving natural products, which offer greater potential with fewer side effects for cancer patients, should be prioritized[11,12,13,14].
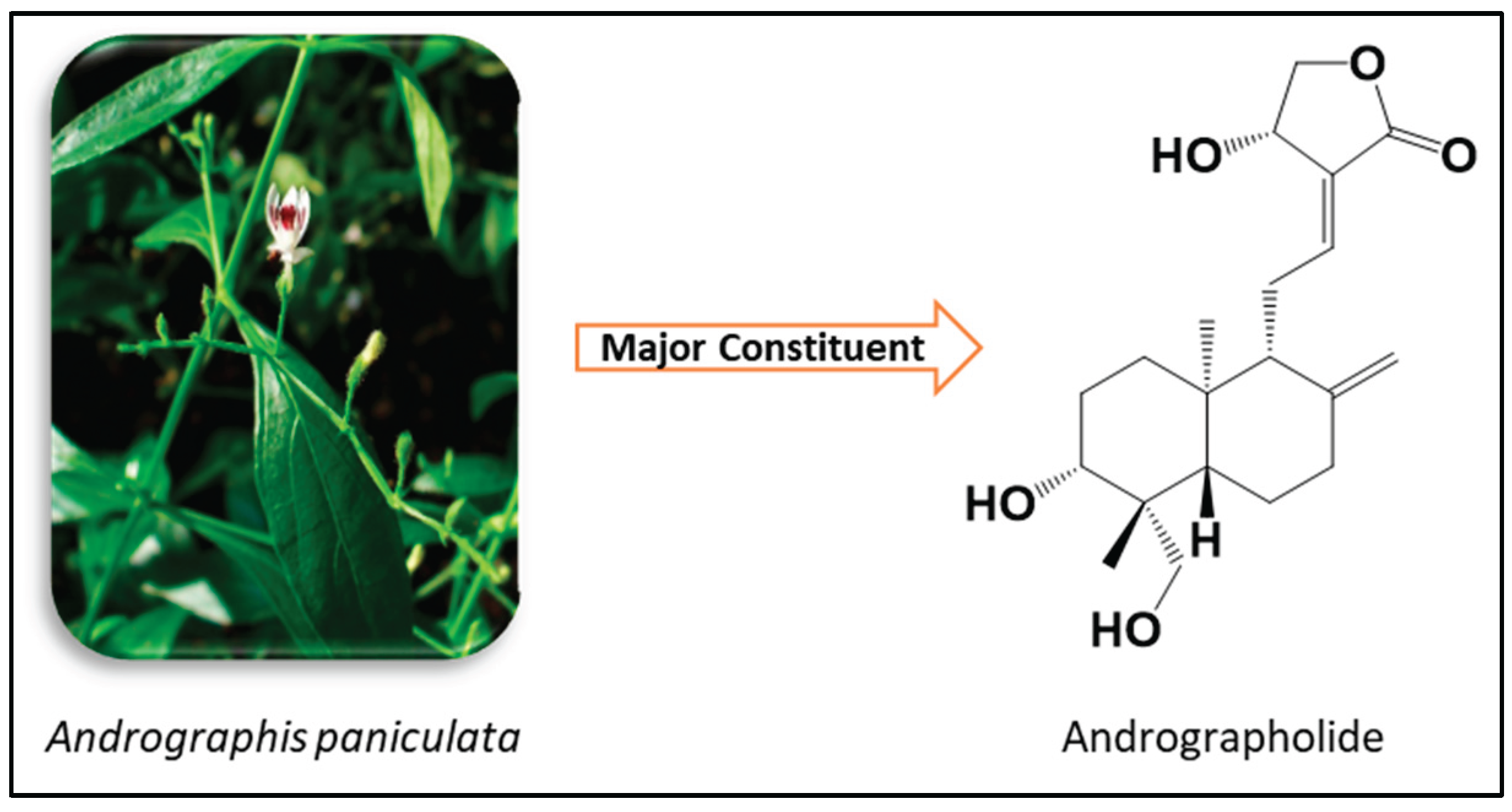
Natural products have been an excellent primary source of bioactive molecules. These bioactive molecules have acquired great value in cancer drug discovery due to their structural features and wide range of medicinal properties[15,16,17,18]. One such bioactive molecule is Andrographolide (ADG). It is an active diterpenoidal constituent of Andrographis paniculata Nees (Figure 1) which belongs to the family Acanthaceae[19,20]. It is primarily accumulated in the leaves, while modest amounts can also be found in the flowering tops, stems, and roots[21]. It is considered an easily isolable compound with a high yield. Andrographis paniculata, also commonly known as the ‘King of Bitter’, is widely cultivated in tropical and subtropical Southeast Asia, China, and India[22]. In traditional Chinese and Indian medicine, the leaves and roots of this plant were used as a remedy for various ailments such as fever, common cold, mouth ulcers, skin infections, urinary tract infections, gastrointestinal disorders, to dispel the toxins from the body, venomous snakebites, etc. [23,24,25,26]. Several research groups investigated the extracts and compounds isolated from A. paniculata and reported that they displayed a wide range of pharmacological activities, including anticancer, antiviral, anti-inflammatory, antimalarial, antioxidant, immunostimulatory activities, etc. In addition, these investigations revealed that the observed anticancer properties of the methanolic extracts of the plant are mainly attributed to andrographolide[26,27,28,29,30,31,32,33,34,35].
In various human cancer cell lines, andrographolide was evaluated for its anticancer potential both in vitro and in vivo[36]. The studies disclosed that andrographolide targets several signalling pathways and has a significant promise for chemoprevention in various types of cancers. By interacting with a variety of targets, andrographolide affects several cancer-related characteristics, such as increase in the expression of pro-apoptotic genes which lead to apoptosis by either intrinsic mitochondrial death or extrinsic TRAIL-related death, cell cycle arrest at different stages with enhancement of pro-apoptotic gene and protein activities, anti-angiogenic properties, inhibition of the VEGF pathway, NF- κβ inhibitor, ubiquitin-mediated proteasomal degradation of proteins, and inhibition of IL-mediated signals[36,37,38,39,40,41]. Though it exhibited excellent therapeutic potential, it has limited oral bioavailability due to its quick elimination and high plasma protein binding capacity[42]. This makes creating formulations for clinical treatment extremely difficult. However, owing to its unique structural features, several structural changes have been made to various parts of andrographolide to enhance anticancer activity and oral bioavailability. Interestingly, these modifications resulted in a significant increment in the anticancer potential and bioavailability. Like andrographolide, pyrazole derivatives also exhibit remarkable anticancer effects via the inhibition of various kinds of enzymes, proteins, and receptors, such as CDKs, kinases, EGFR, VEGF, TGF-β, etc.[43,44,45,46,47].
In this context, we have synthesized twenty novel 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide and isoandrographolide. Our main intention to link the pyrazole scaffold to andrographolide is due to its widespread pharmacological effects in medicinal chemistry, mainly as anticancer and anti-inflammatory pharmacophores. As expected, the synthesized derivatives have displayed excellent anticancer activity, several-fold increased activity compared to andrographolide, against the breast cancer cell line MDA-MB-231.
2. Results and Discussion
2.1. Chemistry
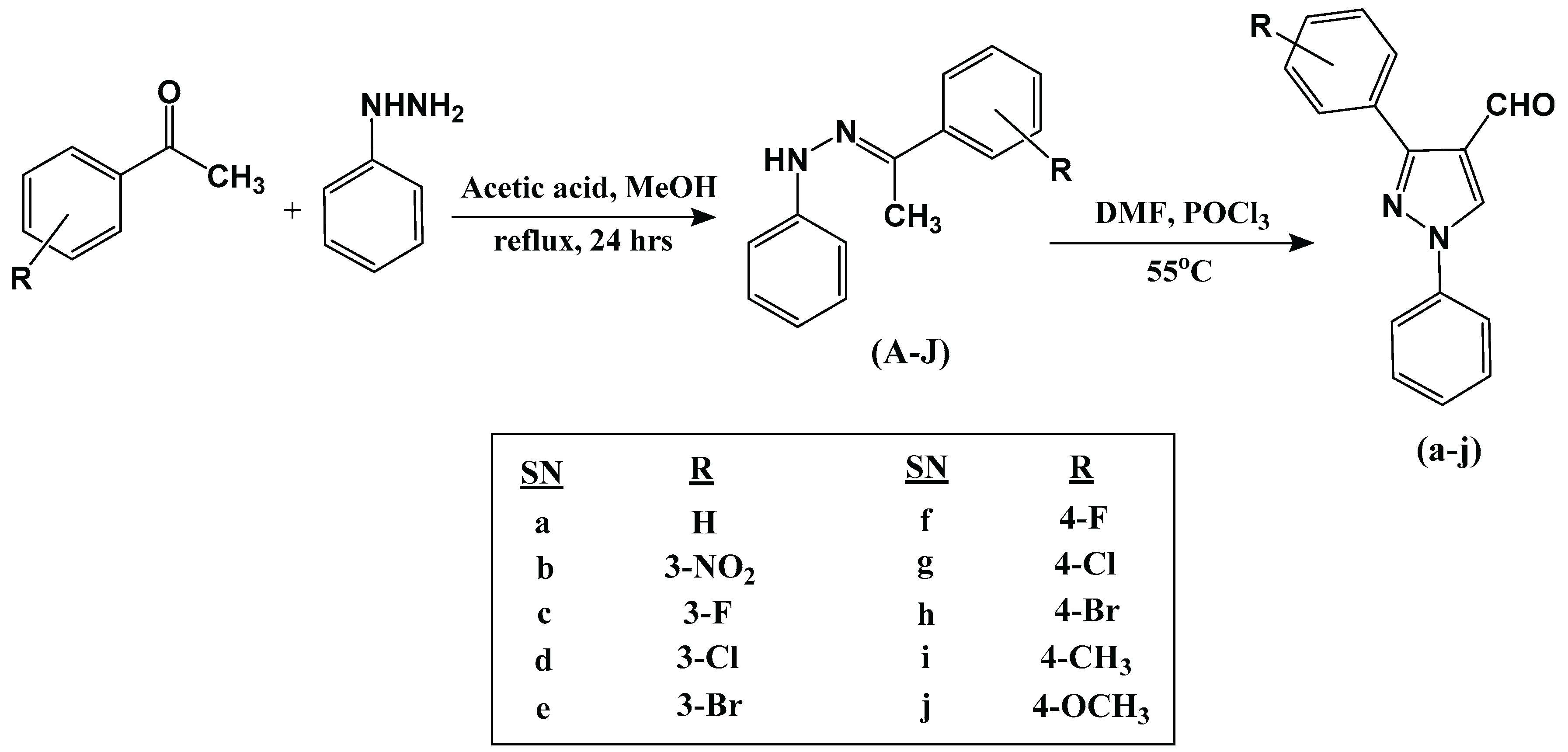
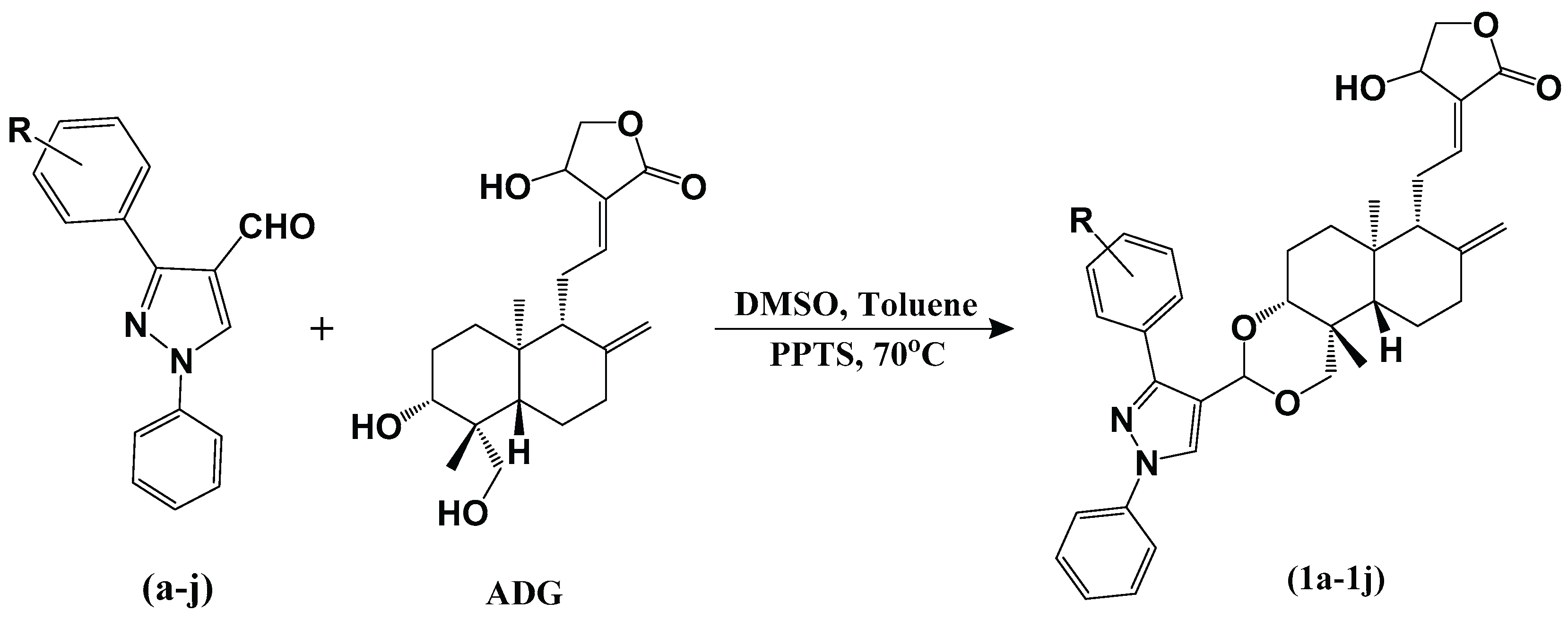
The synthesis of 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide and isoandrographolide entails a series of consecutive steps. This synthesis encompasses three distinct steps, and the schematics detailing the adopted synthetic strategies are presented in Scheme 1, Scheme 2 and Scheme 3[48]. In the initial step, a total of ten 1-phenyl-2-(1-arylethylidene) hydrazines (A-J) were synthesized. Then, in the second step, these compounds were transformed into the corresponding N-aryl-pyrazole-4-carboxaldehydes (a-j). Lastly, in the third step, these aldehydes were subjected to reactions with andrographolide (ADG) and isoandrographolide (ISOADG), resulting in the final attainment of 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide and isoandrographolide (1a-1j and 2a-2j).
2.2. Characterization
The UV-Vis spectra exhibited bands between 275 to 280 nm for all the synthesized compounds (1a-1j and 2a-2j). In the IR spectra of 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide (1a-1j), peaks at ~3400 cm-1 and ~1672 cm-1 were observed for –OH and exocyclic C=C groups. These peaks were absent for 3,19-(N-phenyl-3-aryl-pyrazole) acetals of isoandrographolide (2a-2j). The presence of the C-H stretching band within the 3089-3070 cm-1 range and the aromatic skeletal bands spanning from 1600 to 1450 cm-1, confirm the existence of the aromatic group. The band situated at 2948-2920 cm-1 corresponds to the sp3 C-H stretch originating from the diterpene moiety. The peak between the range 1732-1759 cm-1 corresponds to the carbonyl group (C=O) of the butyrolactone ring. Distinct absorptions at approximately 1220 and 1100 cm-1 can be attributed to the C-O stretching. Compounds with aryl chloride have a band at 1071-1060 cm-1 (Ar-Cl stretch). Compounds with aryl fluorides have a band around 1245 and 1220 cm-1 (Ar-F stretch), and compounds with aryl bromides have a band between 1020-1011 cm-1(Ar-Br stretch).
The attachment of substituted pyrazole moiety is discernible in 1H NMR and 13C NMR spectra of all the acetals. In 1H NMR, the peak at ~ δ 8.18 is due to the presence of only hydrogen in the pyrazole ring, whereas peaks around δ 8.76 to 6.99 correspond to the protons on the aromatic ring attached to the pyrazole ring. In 13C NMR, the carbons in the pyrazole ring and aromatic ring were identified in the ~ δ 163 to 115 range. The bridged carbon, which was bonded to two oxygen atoms, provides strong evidence for the formation of the acetals, and its peak was observed at δ 90 in the 13C NMR spectrum, and the proton attached to the acetal carbon is observed at ~ δ 5.87 in the 1H NMR. Whereas the carbonyl group (C=O) signals in the acetals appeared approximately within the range of δ 173-170. In 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide, peaks at ~148 and 110 in the 13C NMR spectra correspond to an exocyclic C=C, which is absent in 3,19-(N-phenyl-3-aryl-pyrazole) acetals of isoandrographolide[49,50]. More detailed information on the characterization data is provided in the supplementary information.
2.3. In Vitro Anticancer Studies
2.3.1. NCI Screening
All the synthesized compounds (1a-1j and 2a-2j) were initially assessed for their anticancer potential at the National Cancer Institute (NCI, USA) against a diverse panel of 60 human cancer cell lines, representing nine distinct cancer types including leukemia, non-small lung cancer, colon, CNS, melanoma, ovarian, renal, prostate, and breast. The testing protocol employed by NCI encompasses two stages: initial assessment at a single dose and subsequent evaluation at five doses. Compounds that demonstrated growth inhibition exceeding 32% at the single dose stage were identified as active candidates.
2.3.2. Single Dose Study
Initially, a comprehensive anti-cancer screening of all twenty compounds (1a-1j and 2a-2j) was conducted using a 10 µM dose across all the 60 cell lines. In this assessment, a subset of ten compounds (1a-1j) demonstrated impressive percent growth inhibition in most of the cell lines. As a result, these ten compounds were chosen for five dose studies. Of particular interest is the observation that all ten compounds exhibiting notable activity across the majority of cell lines are 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide (1a-1j). Conversely, 3,19-(N-phenyl-3-aryl-pyrazole) acetals of isoandrographolide (2a-2j) displayed a lower % growth inhibition, suggesting that these derivatives are less potent across the majority of cell lines. Consequently, compounds 2a-2j did not meet the criteria for further advancement in the study.
2.3.3. Five Dose Study
In the five-dose study, 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide (1a-1j) were tested at concentrations 0.01 µM, 0.1 µM, 1 µM, 10 µM, and 100 µM against 60 cancer cell line panels. The GI50, TGI, and LC50 of the ten potent compounds on the cancer cell lines are given in Table 1.
It is interesting to observe that all ten compounds have exhibited excellent GI50 values across all the cell lines. However, among these compounds, 1f demonstrated the best activity, followed by 1j and 1h. Compounds 1i and 1b were slightly less active as compared to other compounds. In addition, we note that para-substituted derivatives (1f, 1g, 1h, 1i, and 1j) exhibited better GI50 values than meta-substituted derivatives (1b, 1c, 1d, and 1e). The observed anticancer activity order of these compounds is as follows 1f > 1j > 1h > 1g > 1d > 1c > 1e > 1a > 1b > 1i.
2.3.4. GI50 Values of the Compounds on Individual Cancer Types
Leukemia Cancer: The compound 1f displayed the most potent activity on most of the leukemia cancer cell lines. However, the best GI50 (0.40 µM) shown by 1f was against the cell line CCRF-CEM, followed by the compounds 1h (GI50: 0.48 µM), 1a (GI50: 0.59 µM), 1j (GI50: 0.64 µM), 1e (GI50: 0.70 µM), and 1b (GI50: 1.14 µM). Whereas the compounds 1c, 1d, 1g, and 1i were better against the cell line MOLT-4.
Non-Small Cell Lung Cancer: The compound 1g was observed to have the best GI50 (1.02 µM) against the NCI-H322M cell line. While the compounds 1a, 1f, 1i, and 1j were active against the HOP-92 cell line, and 1b, 1c, 1d, and 1e were active against the NCI-H522 cell line.
Colon Cancer: All the compounds except 1b, 1c, and 1i were found to be active against the HCT-116 cell line. Compound 1f demonstrated the best activity with GI50: 0.39 µM. Compounds 1b, 1c, and 1i showed the best GI50 against HCT-15 and HCC-2998 cell lines.
CNS Cancer: Among the CNS cancer cell lines, these compounds showed promising effects against three cell lines. All the para-substituted derivatives (1f, 1g, 1h, and 1i) except 1j displayed excellent growth inhibition against the SNB-19 cell line, whereas 1j, 1a, and 1d are more active against SNB-75 and 1d (GI50: 1.12 µM) being the best among all. On the other hand, meta-substituted derivatives (1b, 1c, and 1e) showed the best GI50s against SF-295.
Melanoma Cancer: As observed in CNS cancer, against the melanoma cancer cell line LOX IMVI, the para-substituted derivatives (1f, 1g, 1h, and 1i) demonstrated outstanding growth inhibition with GI50s: 1.32, 1.58, 1.55, and 1.43 µM, respectively. All other compounds displayed better activity on various melanoma cancer cell lines.
Ovarian Cancer: These compounds were observed to have very good activity against different ovarian cancer cell lines. However, among all, compound 1j (GI50: 1.35 µM) was found to be the most potent against OVCAR-8 followed by 1d (GI50: 1.39 µM) against the IGROV1 cell line.
Renal Cancer: As in the case of CNS and melanoma cancer, para-substituted derivatives (1f, 1g, 1h, and 1i), except 1j exhibited the best activity with GI50s: 1.27, 1.33, 1.19, and 1.33 µM against TK-10. However, compound 1j (GI50: 0.89 µM) was the most active against RXF 393 amongst all renal cancer cell lines. While the meta-substituted derivatives (1b, 1d, and 1e), except 1c (active against TK-10, GI50: 1.41 µM) displayed the best activity against RXF 393 with GI50s: 1.62, 1.05 and 1.18 µM.
Prostate Cancer: Between the two types of cancer cell lines (PC-3 and DU-145) against which the compounds were tested, four compounds 1a, 1b, 1f, and 1i showed the best GI50s against PC-3, while remaining six compounds 1c, 1d, 1e, 1g, 1h, and 1j showed the best GI50s against DU-145.
Breast Cancer: Including 1a and 1b (GI50: 1.42 and 1.76 µM), the para-substituted derivatives (1f (GI50: 1.19 µM), 1g (GI50: 1.39 µM), 1h (GI50: 1.18 µM) and 1i (GI50: 1.14 µM)) also demonstrated the highest growth inhibition against the T-47D cell line. While the compounds 1j and 1d (GI50: 1.46 and 1.48 µM) against MCF-7 and 1c and 1e (GI50: 1.30 and 1.46 µM) against BT-549 were more active.
2.3.5. NCI Compare Analysis
The NCI compare analysis assesses and arranges compounds based on their similarity in response to the NCI 60 cell lines compared to marketed drugs[51]. This similarity is quantified using the Pearson Correlation Coefficient (PCC). When a marketed drug ranks high in this comparison alongside the compound being studied, it suggests that the studied compound might share a similar mechanism of action. The most promising compounds 1f, 1h, and 1j from the five-dose study were chosen for NCI compare analysis, and their GI50 values were compared to those of marketed drugs (Table 2).
The NCI compare analysis revealed that the three compounds exhibited correlations with various marketed drugs that are recognized for their ability to inhibit DNA synthesis and promote apoptosis. This observation strongly suggests that these compounds could potentially impede cancer growth through similar mechanisms.
2.3.6. Effect of 1f on the Proliferation of MCF-10a and MDA-MB-231
As compound 1f exhibited the best anticancer activity among all the screened compounds in the NCI study, its cytotoxicity on breast cells MCF-10a (normal breast cells) was examined. Compound 1f was found to have a low inhibitory concentration with CC50: 50.01 µM against the breast cells MCF-10a. Further, an MTT assay was performed for compound 1f against the MDA-MB-231 cell line to check its cytotoxicity. These MDA-MB-231 breast cancer cells were treated with 1f at different concentrations: 0, 1.56, 3.125, 6.25, 12.5, 25, 50, 100, and 200 µM for 72 h. As shown in Figure 2, the absorbance decreased with the increasing concentration of the drug. The results indicate that 1f has strong dose-dependent anti-proliferation activity against the MDA-MB-231 breast cancer cell line.
2.3.7. Effect of 1f on Cell Apoptosis Inducement
Apoptosis is considered a preventive mechanism against cancer dissemination, as it plays a crucial role in eliminating excessively proliferating and mutated cancer cells from the body[52,53]. Following the administration of cytotoxic drugs as treatment for various types of cancer, apoptosis is a significant contributor to cellular death[54]. Hence, an enhanced understanding of the processes and mechanisms underlying apoptosis can aid in the development of novel therapeutic drugs for cancer[55]. We proceeded to investigate whether compound 1f triggers apoptosis or not. MDA-MB-231 breast cancer cell line was treated with 1f for 24 h, 48 h, and 72 h. A quantitative evaluation of apoptotic activity was done using FACS.
As depicted in Figure 3, a notable rise in apoptotic cells was noticed in a time-dependent manner. We observed 17.55% apoptotic cells in the control (6.25% in early apoptosis and 11.3 % in late apoptosis). Upon treatment with 1f, we observed a gradual increase of apoptotic cells by 24 % at 24 h (9.38 % in early apoptosis and 14.6 % at late apoptosis), 44 % at 48 h (10.3 % in early apoptosis and 33.0 % at late apoptosis), and 55 % at 72 h (14.7% in early apoptosis and 40.6% at late apoptosis) respectively compared to the control at 0 h. Thus confirming that compound 1f promoted apoptosis in MDA-MB-231 breast cancer cells.
2.3.8. Effects of 1f on MDA-MB-231 Cell Cycle Distribution
Cell proliferation is well associated with the regulation of cell-cycle progression. Therefore, the effect of 1f on cell-cycle distribution was evaluated by flow cytometry in PI-stained cells. As shown in Figure 4, after treatment for 24 h with 1f at concentrations: 3.33, 5.0, and 6.66 µM, cells in G2/M distributions decreased from 22.41 % to 8.91 % compared to control cells (at 0 hours), whereas the percentage of cells in G1-phase increased from 74.96 % to 82.43 % and cell in S phase increased gradually from 2.62% to 3.26%, 8.29% and 8.64% respectively (Table 3). The result indicates that 1f could induce cell cycle arrest in the S Phase.
2.4. Antioxidant Activity
The onset and advancement of cancer have been associated with oxidative stress due to its role in escalating DNA mutations, triggering DNA impairment, fostering genome instability, and promoting cellular proliferation[56,57,58]. Therefore, to find out the antioxidant properties of the compounds, DPPH radical scavenging activity was performed. As shown in Figure 5, all the synthesized compounds showed the DPPH radical scavenging activity between 56.06 % to 61.17 % as compared to ADG (58.74 %), ISOADG (57.94 %), and the positive control ascorbic acid (AA, 69.26 %). All the compounds demonstrated more or less similar antioxidant potential compared to ADG and ISOADG. However, the compounds with the strongest electron withdrawing groups that are 1b (R= 3-NO2, 60.68 %) and 2b (R= 3-NO2, 61.17 %) are the most active, whereas the compounds 1i (R= 4-CH3, 56.31 %) and 2i (R= 4-CH3, 56.06 %) are the least active. Among the halogen-substituted derivatives, chloro-substituted compounds in both the series (1d, 59.32 % and 2d, 59.81 %) have shown superior activity. Another interesting observation was that all the meta-substituted derivatives displayed slightly better radical scavenging activity than all the para-substituted derivatives in both series, except in the case of 2c.
2.5. Drug-Likeness Studies for the Active Compounds
Using the pkCSM tool, a computer-based assessment was conducted on the active compounds (1a-1j) to assess their suitability as drug candidates by examining their Lipinski parameters[59].
Analysis of the results as given in Table 4 revealed that their in silico molecular properties align with Lipinski's rule of five (RO5), except for their molecular weights, which fall within the range of 580-658 Daltons, slightly surpassing the recommended limit (<500 Dalton). Nevertheless, it is worth noting that natural products often deviate from Lipinski's rules regarding molecular weights[60]. The compounds exhibited a TPSA ranging from 78 to 126, whereas the Log P value slightly exceeded the acceptable range for some compounds. Additionally, they have less than five N-H and O-H hydrogen bond donors and less than ten nitrogen and oxygen-hydrogen bond acceptors (HBA). Overall, despite the slight deviation in molecular weights, the compounds largely adhere to Lipinski's parameters, suggesting their suitability as potential anticancer agents.
3. Experimental
3.1. Synthesis
All the chemicals used in the experiments were procured from Merck and S.D. Fine chemicals. Andrographolide (98 %) was obtained from Maysar Herbals, Faridabad, Haryana, and purified by recrystallization. Solvents such as methanol and dichloromethane were distilled before use.
3.1.1. Synthesis of Aryl Hydrazones (A-J)
To a solution of acetophenone (25 mmol) in methanol (80 mL), phenylhydrazine (25 mmol) was added and stirred at room temperature. To this, 2 mL acetic acid was added, and the mixture was refluxed for 24 hours. The progress of the reaction was checked using TLC. After completion of the reaction, crushed ice was added, and the solid formed was filtered and dried. The pure product obtained was then subjected to the next reaction[48].
3.1.2. Synthesis of 3-aryl-4-Pyrazole Carboxaldehydes (a-j)
To a cold solution of N, N-dimethylformamide (40 mL), aliquot amounts of arylhydrazone (20 mmol) were added and stirred over 30 minutes. Then, POCl3 (100 mmol) was added dropwise to the solution while maintaining the temperature at 0-5°C. After the addition, the reaction mixture was heated at 55°C for 4 hours. After completion of the reaction, the resulting mixture was cooled to room temperature and poured into crushed ice and stirred for 20 min. The white solid formed was filtered, dried, and purified using column chromatography (hexane: ethyl acetate, 75:25). In case the solid was not formed after being poured into crushed ice, the solution was neutralized with saturated Na2CO3 to obtain the solid[48].
3.1.3. Synthesis of 3,19-(N-phenyl-3-aryl-pyrazole) Acetals of Andrographolide (1a-1j)
Andrographolide (1 mmol) in DMSO (2 mL) was added to 3-aryl-4-pyrazole carboxaldehydes (4 mmol) in toluene (3 mL) at room temperature. To this catalytic amount (0.08 mmol) of pyridinium para-toluene sulfonate (PPTs) were added and heated at 70°C for 24 hours. After completion of the reaction, the mixture was cooled to room temperature and then neutralized with a freshly prepared, cold saturated sodium bicarbonate solution. The resulting solution was then extracted three times (15 mL each) with DCM, and the obtained organic layer was washed thoroughly with brine solution and dried over anhydrous sodium sulphate. DCM was removed using a rota evaporator. The crude product obtained was then purified using column chromatography (silica gel: 230-400 mesh, hexane: acetone, 80:20)[48].
3.1.4. Synthesis of Isoandrographolide and 3,19-(N-phenyl-3-aryl-pyrazole) Acetals of Isoandrographolide (2a-2j)
To synthesize isoandrographolide, andrographolide (8 g) was dissolved in concentrated hydrochloric acid (HCl, 150 mL) and stirred at room temperature for 24 hours. After completion of the reaction (monitored by TLC), the reaction mixture was poured into crushed ice and extracted with DCM. Then, the organic layer was thoroughly washed with brine and freshly prepared saturated sodium bicarbonate solution. The crude solid was then dried over anhydrous sodium sulphate and recrystallized from ethyl acetate to get a pure product[48].
3,19-(N-phenyl-3-aryl-pyrazole) acetals of isoandrographolide were synthesized as described for the synthesis of 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide (in this case, isoandrographolide was used as a starting material instead of andrographolide).
3.2. Instrumentation
The 1H NMR and 13C NMR spectra were recorded using a BRUKER ASCEND NMR spectrometer at 400 MHz and 100 MHz, respectively. CDCl3 was used as the solvent to dissolve the compounds. TMS was an internal standard. AGILENT 6550 Q-TOF LC/MS (HRMS) was employed for the mass spectra. UV-vis spectra were recorded in acetonitrile between the range 200-600 nm using a Shimadzu 2450 spectrophotometer. An AGILENT Cary 630 spectrophotometer was used to record FT-IR spectra between 400 and 4000 cm-1 using KBr pellets. For HPLC, the Agilent 1260 Infinity high-performance liquid chromatography system, equipped with a quaternary solvent delivery system, inline degasser, autosampler, and photodiode array detector, was used. Chromatographic separation was carried out using an RP-HPLC Zorbax Extend-C18 (4.6 mm × 250 mm, 5 µm) column. The mobile phase consisted of acetonitrile (25%) and water (75%) and was maintained at a flow rate of 1 mL/min. The temperature of the column was controlled at 25°C with an injection volume of 10 µl. Detection wavelengths were set at the respective λmax for each compound, and a diode array detector was employed to detect the eluted peaks between 220-250 nm.
3.3. In Vitro Anticancer Studies
3.3.1. Cell Proliferation Assay
Briefly, breast cancer cells MDA-MB-231 and normal breast cells MCF-10a were seeded in 96-well plates at 4×103 density per well, and cultured at 37°C overnight, then treated with 1f at different concentrations (0, 1.56, 3.125, 6.25, 12.5, 25, 50, 100 and 200 µM) and incubated for 72h. Untreated cells were utilized as controls. This was followed by the addition of 10 μL of 5 mg/mL MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide), which is an indicator of cell viability, to the media in each well, and was further kept for incubation for 3 hours. Then, the media was removed, and absorbance at 570 nm was taken using a spectrophotometer (Promega GloMax, USA).
3.3.2. Apoptotic Assay
The effect of 1f on the apoptosis inducement of cells was further determined by Annexin V-FITC/PI staining. In a 12-well plate, 1 × 105 cells/well were seeded, and after incubating with the drug for 24 h, 48 h, and 72 h, the cells were harvested using 1X trypsin. The cells and media were then collected and centrifuged at 4000 rpm for 5 minutes. The resulting cell pellet was resuspended in 100 μL of 1X Annexin V binding buffer and stained with Annexin V–FITC, followed by a 15-minute incubation in the dark at 4°C. The cells were then washed and resuspended in a fresh 1X Annexin V binding buffer. Propidium iodide staining was performed, and the cells were immediately analyzed using a flow cytometer.
3.3.3. Cell Cycle Assay
Cell cycle analysis was performed using a flow cytometer after cellular staining with PI. 3 × 105 cells/well were seeded in a 6-well plate. After 24 hours, the medium was replaced with a serum-free medium to synchronize the cells. Following an 8-hour synchronization period, the cells were treated with compound 1f at different concentrations for 24 hours. The cells were then harvested using 1X trypsin and washed with cold 1X PBS. They were fixed with chilled 70% ethanol at 4°C for 2 hours. After washing twice with PBS, the cells were resuspended and incubated overnight in a staining buffer containing ribonuclease A (1 μL, 10 mg/mL) and propidium iodide (5 μL, 1 mg/mL) in 1X PBS. The cells were subsequently acquired and analyzed using a flow cytometer.
3.4. DPPH Assay
A stable free radical DPPH (2,2-Diphenyl-1-picrylhydrazyl) assay was employed to test the antioxidant activity of all the synthesized compounds. Concisely, 25 µM of the test compounds were dissolved in acetonitrile and combined with 0.1 mM of DPPH (in acetonitrile) in a 1:2 ratio (80 µl and 160 µl). Then, the mixture was left undisturbed in the dark for 30 minutes at room temperature. After 30 minutes, the absorbance of the solutions was instantly measured at 517 nm using a Varioskan Multiplate reader (Thermo Scientific)[61]. Ascorbic acid (in acetonitrile) was used as the positive control, andrographolide and isoandrogapholide (in acetonitrile) were used as the standards. The following equation was used to determine the DPPH radical scavenging activity:
where, Ac = Absorbance of the blank (DPPH solution), As = Absorbance of the test samples
% Radical scavenging activity = (Ac-As) / Ac × 100
4. Conclusion
In conclusion, twenty novel 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide and isoandrographolide (1a-1j and 2a-2j) were synthesized and characterized. All the synthesized compounds were initially screened for their anticancer activity against 60 human cancer cell lines at the NCI, USA. Single-dose study results revealed that all 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide (1a-1j) displayed superior anticancer activity compared to 3,19-(N-phenyl-3-aryl-pyrazole) acetals of isoandrographolide (2a-2j). Therefore, only compounds 1a-1j were selected by NCI for five dose studies to find out GI50s, TGI, and LC50s. Among the compounds tested, compound 1f exhibited the best GI50s, TGI, and LC50s on most cell lines, followed by 1h and 1j, respectively. Therefore, compound 1f was further selected for detailed in vitro analysis on normal breast cells MCF-10a and the breast cancer cell line MDA-MB-231. The cytotoxicity study of compound 1f on MCF-10a was found to have a low inhibitory concentration. The MTT assay results demonstrated that compound 1f exhibited strong, dose-dependent anti-proliferative activity on the cells. In addition, the apoptotic analysis revealed that compound 1f induced apoptosis in the MDA-MB-231 cells and increased the apoptotic cells from 10% to 55% over 72 hours. Furthermore, cell cycle analysis indicated that 1f caused S-phase arrest in MDA-MB-231 cells. The antioxidant activity of the synthesized compounds was evaluated using the DPPH assay. Of all the compounds tested, 1b (R=3-NO2, 60.68%) and 2b (R=3-NO2, 61.17%) demonstrated the highest radical scavenging activity. Moreover, the in-silico Lipinski’s analyses of the most active compounds indicate their potential to be developed as promising therapeutic leads for cancer treatment.
Supplementary Information
Supplementary data contains spectral characterization, 1H-NMR, 13C-NMR, HRMS spectra, and NCI in vitro results. .
Author Contributions
Siva Kumar Rokkam conducted the synthesis, characterization, in silico studies, data analysis, and drafted the manuscript. Shahjalal Chowdhury and Yashwanth Inabathina performed and analyzed the in vitro anticancer studies. Balasubramanyam Karanam supervised the anticancer results and drafted the manuscript. Nageswara Rao Golakoti, Sripada Lakshminath, and Nanduri Srinivas conceived the study, designed it, analyzed the results, and contributed to the manuscript's drafting. All authors read and approved the final manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability
The data supporting this article have been included as part of the Supplementary Information.
Acknowledgments
The authors dedicate this work to the Founder-Chancellor Bhagawan Sri Sathya Sai Baba. We thank the Central Research Instruments Facility-SSSIHL for providing the characterization facilities. The authors also thank Mr. Manohar Bhujel for his valuable inputs. GNR and NS are grateful to the Council of Scientific & Industrial Research (CSIR) for funding this project (No. 02(0304)/17/EMR-II).
Conflicts of Interest
There are no conflicts to declare.
References
- Majérus, M.-A. The Cause of Cancer: The Unifying Theory. Adv. Cancer Biol. - Metastasis 2022, 4, 100034. [Google Scholar] [CrossRef]
- Cao, W.; Chen, H.-D.; Yu, Y.-W.; Li, N.; Chen, W.-Q. Changing Profiles of Cancer Burden Worldwide and in China: A Secondary Analysis of the Global Cancer Statistics 2020. Chin. Med. J. (Engl). 2021, 134, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.V.S.; Sharma, J.; Kumar, S. GLOBOCAN 2020 Report on Global Cancer Burden: Challenges and Opportunities for Surgical Oncologists. Ann. Surg. Oncol. 2022, 29, 6497–6500. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Houghton, S.C.; Hankinson, S.E. Cancer Progress and Priorities: Breast Cancer. Cancer Epidemiol. biomarkers Prev. 2021, 30, 822–844. [Google Scholar] [CrossRef]
- Hong, R.; Xu, B. Breast Cancer: An Up-to-date Review and Future Perspectives. Cancer Commun. 2022, 42, 913–936. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Kroschinsky, F.; Stölzel, F.; von Bonin, S.; Beutel, G.; Kochanek, M.; Kiehl, M.; Schellongowski, P. New Drugs, New Toxicities: Severe Side Effects of Modern Targeted and Immunotherapy of Cancer and Their Management. Crit. Care 2017, 21, 1–11. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalt. Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Iwamoto, T. Clinical Application of Drug Delivery Systems in Cancer Chemotherapy: Review of the Efficacy and Side Effects of Approved Drugs. Biol. Pharm. Bull. 2013, 36, 715–718. [Google Scholar] [CrossRef]
- Huang, M.; Lu, J.-J.; Ding, J. Natural Products in Cancer Therapy: Past, Present and Future. Nat. Products Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Qu, J.; Sun, Y.; Zhou, W. Role of Natural Products in Tumor Therapy from Basic Research and Clinical Perspectives. Acta Mater. Medica 2024, 3, 163–206. [Google Scholar] [CrossRef]
- Ouyang, L.; Luo, Y.; Tian, M.; Zhang, S.; Lu, R.; Wang, J.; Kasimu, R.; Li, X. Plant Natural Products: From Traditional Compounds to New Emerging Drugs in Cancer Therapy. Cell Prolif. 2014, 47, 506–515. [Google Scholar] [CrossRef]
- Mukherjee, A.K.; Basu, S.; Sarkar, N.; Ghosh, A.C. Advances in Cancer Therapy with Plant Based Natural Products. Curr. Med. Chem. 2001, 8, 1467–1486. [Google Scholar] [CrossRef]
- Ali Abdalla, Y.O.; Subramaniam, B.; Nyamathulla, S.; Shamsuddin, N.; Arshad, N.M.; Mun, K.S.; Awang, K.; Nagoor, N.H. Natural Products for Cancer Therapy: A Review of Their Mechanism of Actions and Toxicity in the Past Decade. J. Trop. Med. 2022, 2022, 5794350. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, J.; Bai, J.; Tong, R.; An, F.; Jiao, P.; He, L.; Zeng, D.; Long, E.; Yan, J. The Application of Natural Products in Cancer Therapy by Targeting Apoptosis Pathways. Curr. Drug Metab. 2018, 19, 739–749. [Google Scholar] [CrossRef]
- Huang, M.; Lu, J.-J.; Huang, M.-Q.; Bao, J.-L.; Chen, X.-P.; Wang, Y.-T. Terpenoids: Natural Products for Cancer Therapy. Expert Opin. Investig. Drugs 2012, 21, 1801–1818. [Google Scholar] [CrossRef]
- Demain, A.L.; Vaishnav, P. Natural Products for Cancer Chemotherapy. Microb. Biotechnol. 2011, 4, 687–699. [Google Scholar] [CrossRef]
- Hossain, M.S.; Urbi, Z.; Sule, A.; Rahman, K.M.H. Andrographis Paniculata (Burm. f.) Wall. Ex Nees: A Review of Ethnobotany, Phytochemistry, and Pharmacology. Sci. World J. 2014, 2014, 1–28. [Google Scholar] [CrossRef]
- Jiang, M.; Sheng, F.; Zhang, Z.; Ma, X.; Gao, T.; Fu, C.; Li, P. Andrographis Paniculata (Burm.f.) Nees and Its Major Constituent Andrographolide as Potential Antiviral Agents. J. Ethnopharmacol. 2021, 272, 113954. [Google Scholar] [CrossRef]
- Kandanur, S.G.S.; Tamang, N.; Golakoti, N.R.; Nanduri, S. Andrographolide: A Natural Product Template for the Generation of Structurally and Biologically Diverse Diterpenes. Eur. J. Med. Chem. 2019, 176, 513–533. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, T.; Hsieh, C.-Y.; Lee, J.-J.; Sheu, J.-R. Experimental and Clinical Pharmacology of Andrographis Paniculata and Its Major Bioactive Phytoconstituent Andrographolide. Evidence-Based Complement. Altern. Med. 2013, 2013, 846740. [Google Scholar] [CrossRef] [PubMed]
- Bharati, B.D.; Sharma, P.K.; Kumar, N.; Dudhe, R.; Bansal, V. Pharmacological Activity of Andrographis Paniculata: A Brief Review. Pharmacologyonline 2011, 2, 10. [Google Scholar]
- Okhuarobo, A.; Ehizogie Falodun, J.; Erharuyi, O.; Imieje, V.; Falodun, A.; Langer, P. Harnessing the Medicinal Properties of Andrographis Paniculata for Diseases and beyond: A Review of Its Phytochemistry and Pharmacology. Asian Pacific J. Trop. Dis. 2014, 4, 213–222. [Google Scholar] [CrossRef]
- Jadhav, A.K.; Karuppayil, S.M. Andrographis Paniculata (Burm. F) Wall Ex Nees: Antiviral Properties. Phyther. Res. 2021, 35, 5365–5373. [Google Scholar] [CrossRef]
- Lim, J.C.W.; Chan, T.K.; Ng, D.S.; Sagineedu, S.R.; Stanslas, J.; Wong, W.F. Andrographolide and Its Analogues: Versatile Bioactive Molecules for Combating Inflammation and Cancer. Clin. Exp. Pharmacol. Physiol. 2012, 39, 300–310. [Google Scholar] [CrossRef]
- Wei, S.; Tang, Y.-B.; Hua, H.; Ohkoshi, E.; Goto, M.; Wang, L.-T.; Lee, K.-H.; Xiao, Z. Discovery of Novel Andrographolide Derivatives as Cytotoxic Agents. Bioorg. Med. Chem. Lett. 2013, 23, 4056–4060. [Google Scholar] [CrossRef]
- Suebsasana, S.; Pongnaratorn, P.; Sattayasai, J.; Arkaravichien, T.; Tiamkao, S.; Aromdee, C. Analgesic, Antipyretic, Anti-Inflammatory and Toxic Effects of Andrographolide Derivatives in Experimental Animals. Arch. Pharm. Res. 2009, 32, 1191–1200. [Google Scholar] [CrossRef]
- Liu, G.; Chu, H. Andrographolide Inhibits Proliferation and Induces Cell Cycle Arrest and Apoptosis in Human Melanoma Cells. Oncol. Lett. 2018, 15, 5301–5305. [Google Scholar] [CrossRef]
- Aromdee, C.; Sriubolmas, N.; Wiyakrutta, S.; Suebsasna, S.; Khunkitti, W. Effect of the Derivatives of Andrographolide on the Morphology of Bacillus Subtilis. Arch. Pharm. Res. 2011, 34, 71–77. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kumar, R.A.; Deevi, D.S.; Satyanarayana, C.; Rajagopalan, R. Andrographolide, a Potential Cancer Therapeutic Agent Isolated from Andrographis Paniculata. J. Exp. Ther. Oncol. 2003, 3, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Berman, S.H.; Babish, J.G.; Ma, X.; Shinto, L.; Dorr, M.; Wells, K.; Wenner, C.A.; Standish, L.J. A Phase I Trial of Andrographolide in HIV Positive Patients and Normal Volunteers. Phyther. Res. 2000, 14, 333–338. [Google Scholar] [CrossRef]
- Chen, J.X.; Xue, H.J.; Ye, W.C.; Fang, B.H.; Liu, Y.H.; Yuan, S.H.; Yu, P.; Wang, Y.Q. Activity of Andrographolide and Its Derivatives against Influenza Virus in Vivo and in Vitro. Biol. Pharm. Bull. 2009, 32, 1385–1391. [Google Scholar] [CrossRef]
- Yu, B.C.; Chen, W.C.; Cheng, J.T. Antihyperglycemic Effect of Andrographolide in Streptozotocin-Induced Diabetic Rats. Planta Med. 2003, 69, 1075–1079. [Google Scholar]
- Wang, Z.; Yu, P.; Zhang, G.; Xu, L.; Wang, D.; Wang, L.; Zeng, X.; Wang, Y. Design, Synthesis and Antibacterial Activity of Novel Andrographolide Derivatives. Bioorg. Med. Chem. 2010, 18, 4269–4274. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Xie, X.; Song, Y.; Yan, W.; Luo, Y.; Jiang, Y. The Therapeutic Potential of Andrographolide in Cancer Treatment. Biomed. Pharmacother. 2024, 180, 117438. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Attar, R.; Sabitaliyevich, U.Y.; Alaaeddine, N.; de Sousa, D.P.; Xu, B.; C. Cho, W. The Prowess of Andrographolide as a Natural Weapon in the War against Cancer. Cancers (Basel). 2020, 12. [Google Scholar] [CrossRef]
- Lim, J.C.W.; Chan, T.K.; Ng, D.S.W.; Sagineedu, S.R.; Stanslas, J.; Wong, W.S.F. Andrographolide and Its Analogues: Versatile Bioactive Molecules for Combating Inflammation and Cancer. Clin. Exp. Pharmacol. Physiol. 2012, 39, 300–310. [Google Scholar] [CrossRef]
- Mishra, S.K.; Tripathi, S.; Shukla, A.; Oh, S.H.; Kim, H.M. Andrographolide and Analogues in Cancer Prevention. Front Biosci (Elite Ed) 2015, 7, 255–266. [Google Scholar] [CrossRef]
- Malik, Z.; Parveen, R.; Parveen, B.; Zahiruddin, S.; Khan, M.A.; Khan, A.; Massey, S.; Ahmad, S.; Husain, S.A. Anticancer Potential of Andrographolide from Andrographis Paniculata (Burm. f.) Nees and Its Mechanisms of Action. J. Ethnopharmacol. 2021, 272, 113936. [Google Scholar] [CrossRef]
- Kandanur, S.G.S.; Kundu, S.; Cadena, C.; Juan, H.S.; Bajaj, A.; Guzman, J.D.; Nanduri, S.; Golakoti, N.R. Design, Synthesis, and Biological Evaluation of New 12-Substituted-14-Deoxy-Andrographolide Derivatives as Apoptosis Inducers. Chem. Pap. 2019, 73, 1669–1675. [Google Scholar] [CrossRef]
- Elsheikh, M.A.; Rizk, S.A.; Elnaggar, Y.S.R.; Abdallah, O.Y. Nanoemulsomes for Enhanced Oral Bioavailability of the Anticancer Phytochemical Andrographolide: Characterization and Pharmacokinetics. AAPS PharmSciTech 2021, 22, 1–12. [Google Scholar] [CrossRef]
- Ebenezer, O.; Shapi, M.; Tuszynski, J.A. A Review of the Recent Development in the Synthesis and Biological Evaluations of Pyrazole Derivatives. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. Biologically Active Pyrazole Derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Bennani, F.E.; Doudach, L.; Cherrah, Y.; Ramli, Y.; Karrouchi, K.; Ansar, M.; Faouzi, M.E.A. Overview of Recent Developments of Pyrazole Derivatives as Anticancer Agents in Different Cell Lines. Bioorg. Chem. 2020, 97, 103470. [Google Scholar] [CrossRef]
- Kumar, H.; Saini, D.; Jain, S.; Jain, N. Pyrazole Scaffold: A Remarkable Tool in the Development of Anticancer Agents. Eur. J. Med. Chem. 2013, 70, 248–258. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Guruvayoorappan, C. Anti-Inflammatory and Anti-Tumor Activity of the Marine Mangrove Rhizophora Apiculata. J. Immunotoxicol. 2012, 9, 341–352. [Google Scholar] [CrossRef]
- Rokkam, S.K.; Bhujel, M.; Jain, D.; Sripada, L.; Nanduri, S.; Bajaj, A.; Golakoti, N.R. Synthesis of Novel Pyrazole Acetals of Andrographolide and Isoandrographolide as Potent Anticancer Agents. RSC Adv. 2024, 14, 26625–26636. [Google Scholar] [CrossRef]
- Robert, M. Silverstein, Francis X. Webster, David J. Kiemle, D.L.B. Spectrometric Identification of Organic Compounds, 8th Edition, 2014. [Google Scholar]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S. Introduction to Spectroscopy-a Guide for Students of Organic Chemistry; Thomson Learning, Inc., 2001.
- Naasani, I. COMPARE Analysis, a Bioinformatic Approach to Accelerate Drug Repurposing against Covid-19 and Other Emerging Epidemics. SLAS Discov. Adv. Sci. Drug Discov. 2020, 26, 345–351. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and Anti-Death: Tumour Resistance to Apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Hickman, J.A. Apoptosis Induced by Anticancer Drugs. Cancer Metastasis Rev. 1992, 11, 121–139. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The Biochemistry of Apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative Stress and Cancer: An Overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive Oxygen Species in Cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Ganesan, A. The Impact of Natural Products upon Modern Drug Discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Kumar Rokkam, S.; Mas-Rosario, J.A.; Joshi, B.P.; Joshi, M.; Choudhury, A.R.; Kar, S.; Golakoti, N.R.; Farkas, M.E. Diarylidene-N-Methyl-4-Piperidones and Spirobibenzopyrans as Antioxidant and Anti-Inflammatory Agents. Chem. Biodivers. 2023, 20. [Google Scholar] [CrossRef]
Figure 1.
Andrographis paniculata and the chemical structure of andrographolide.
Scheme 1.
Synthesis of 1-phenyl-2-(1-arylethylidene) hydrazines (A-J) and N-aryl-pyrazole-4-carboxaldehydes (a-j).
Scheme 1.
Synthesis of 1-phenyl-2-(1-arylethylidene) hydrazines (A-J) and N-aryl-pyrazole-4-carboxaldehydes (a-j).
Scheme 2.
Synthesis of 3,19-(N-phenyl-3-aryl-pyrazole) acetals of andrographolide (1a-1j).
Scheme 3.
Synthesis of 3,19-(N-phenyl-3-aryl-pyrazole) acetals of isoandrographolide (2a-2j).
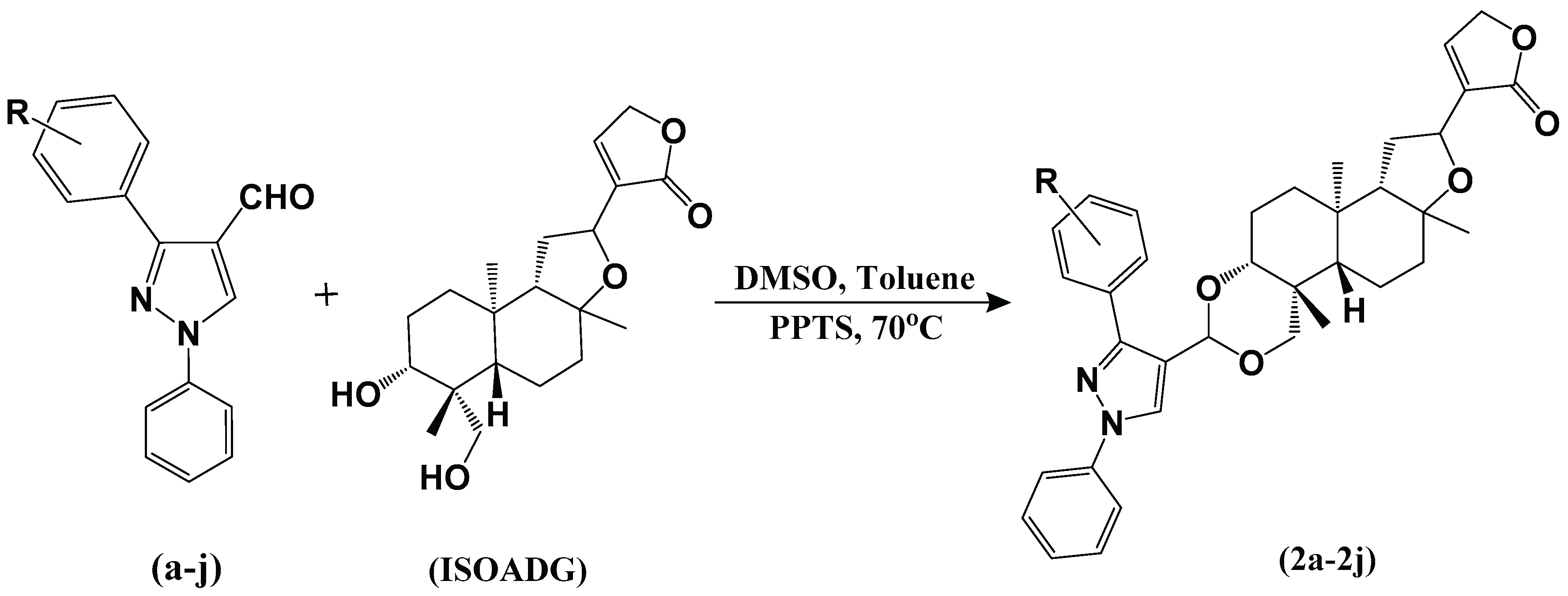
Figure 2.
Dose-dependent cytotoxicity of compound 1f against the breast cancer cell line MDA-MB-231.
Figure 2.
Dose-dependent cytotoxicity of compound 1f against the breast cancer cell line MDA-MB-231.
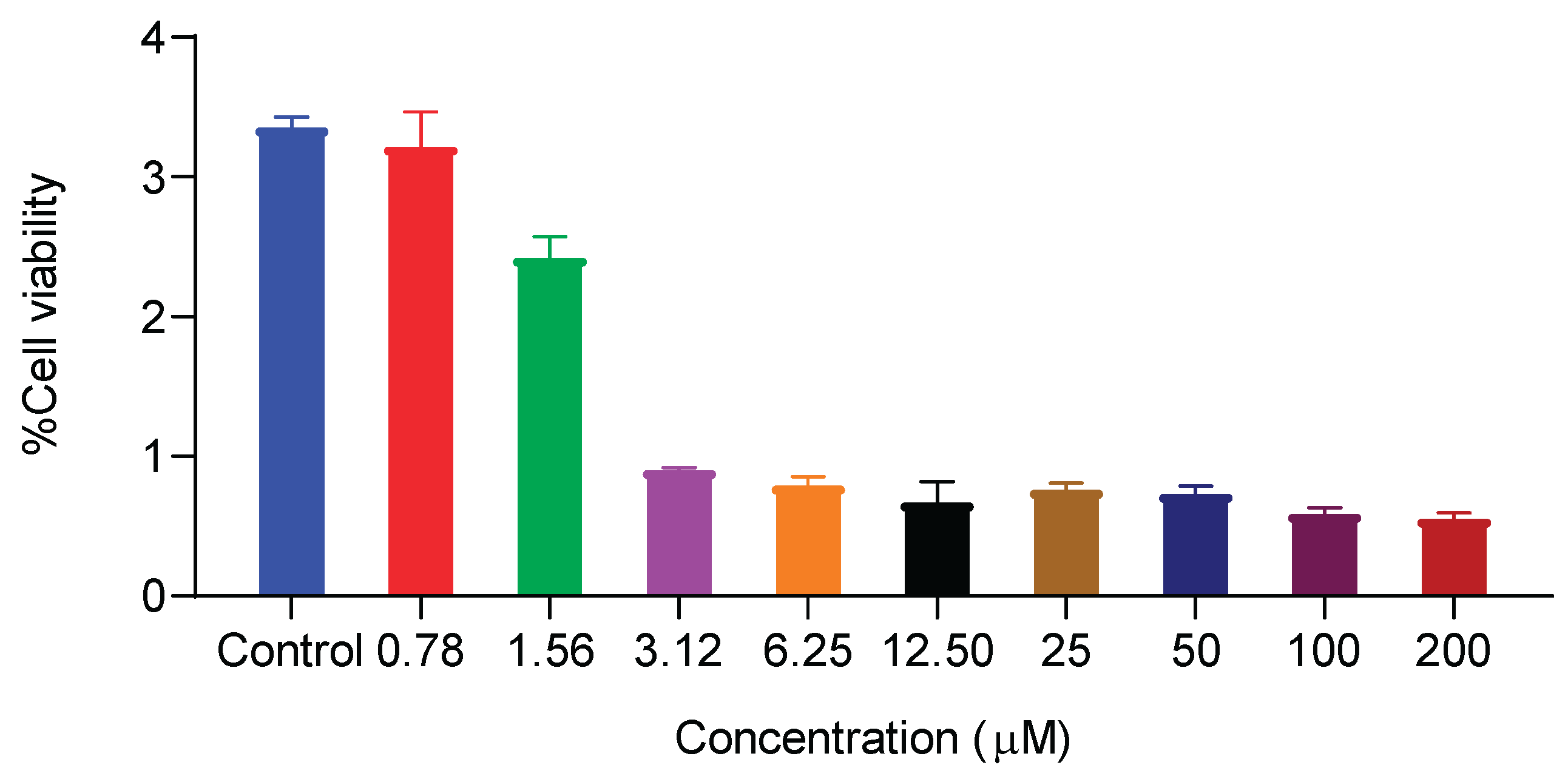
Figure 3.
Total apoptotic cells at 0 h (A). Total apoptotic cells induced by 1f: at 24 h (B), 48 h (C), 72 h (D).
Figure 3.
Total apoptotic cells at 0 h (A). Total apoptotic cells induced by 1f: at 24 h (B), 48 h (C), 72 h (D).
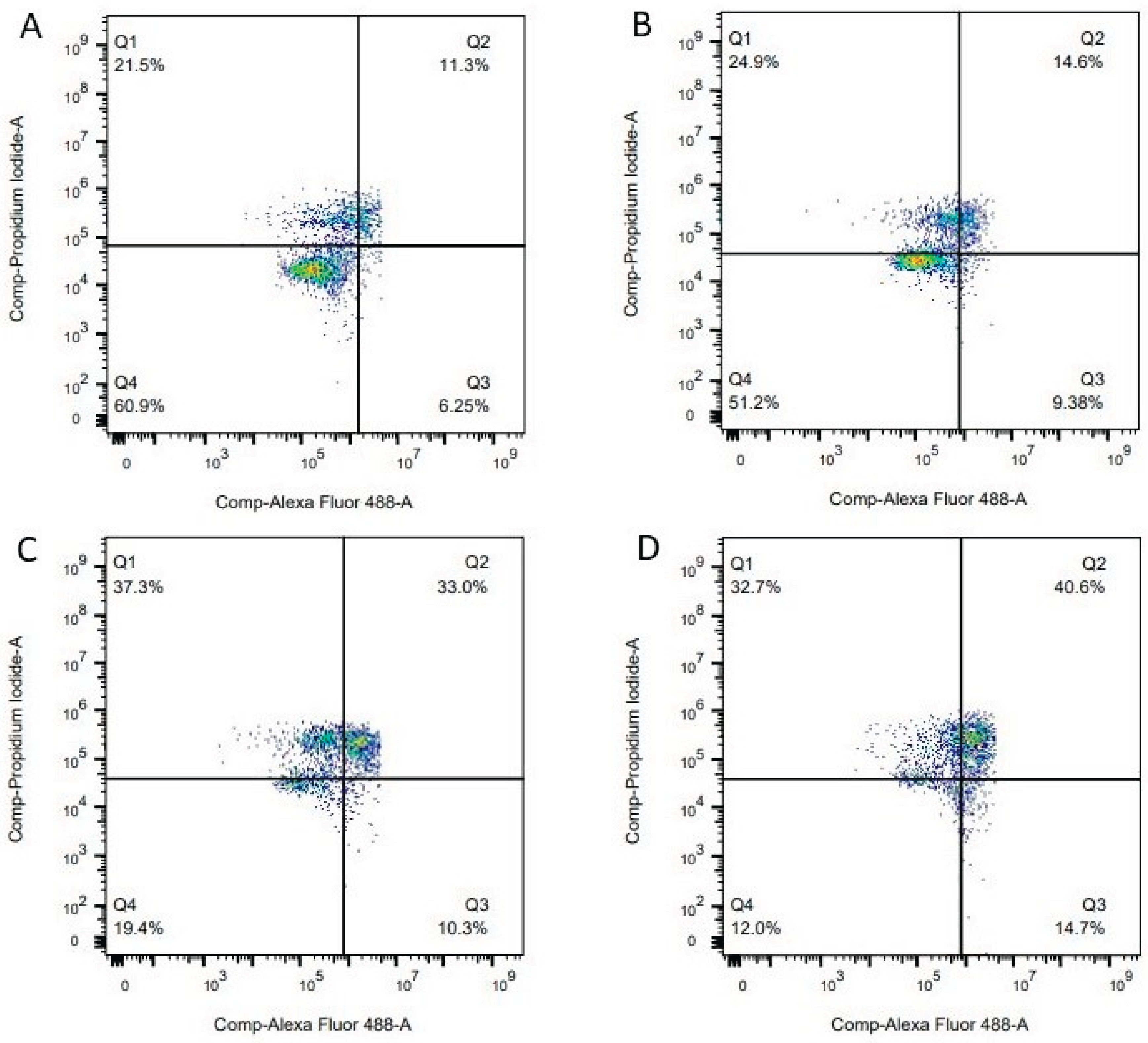
Figure 4.
Analysis of compound 1f on MDA-MB-231 cell cycle distribution: A) control at 0 h; B) after treatment for 24 h with 1f at concentration 3.33 µM; C) after treatment for 24 h with 1f at concentration 5.0 µM; D) after treatment for 24 h with 1f at concentration 6.66 µM.
Figure 4.
Analysis of compound 1f on MDA-MB-231 cell cycle distribution: A) control at 0 h; B) after treatment for 24 h with 1f at concentration 3.33 µM; C) after treatment for 24 h with 1f at concentration 5.0 µM; D) after treatment for 24 h with 1f at concentration 6.66 µM.
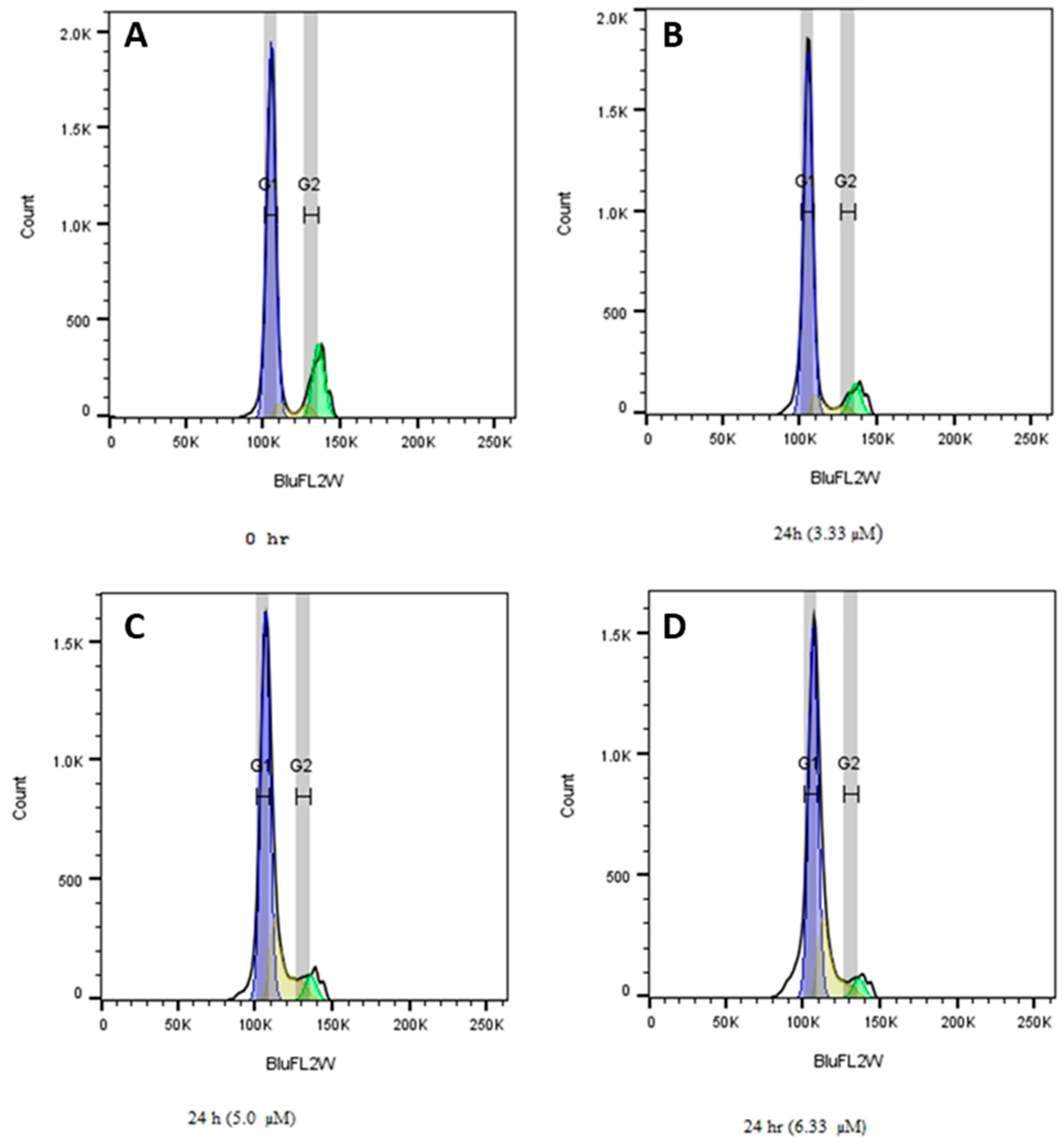
Figure 5.
% DPPH radical scavenging activity of the compounds 1a-1j and 2a-2j.
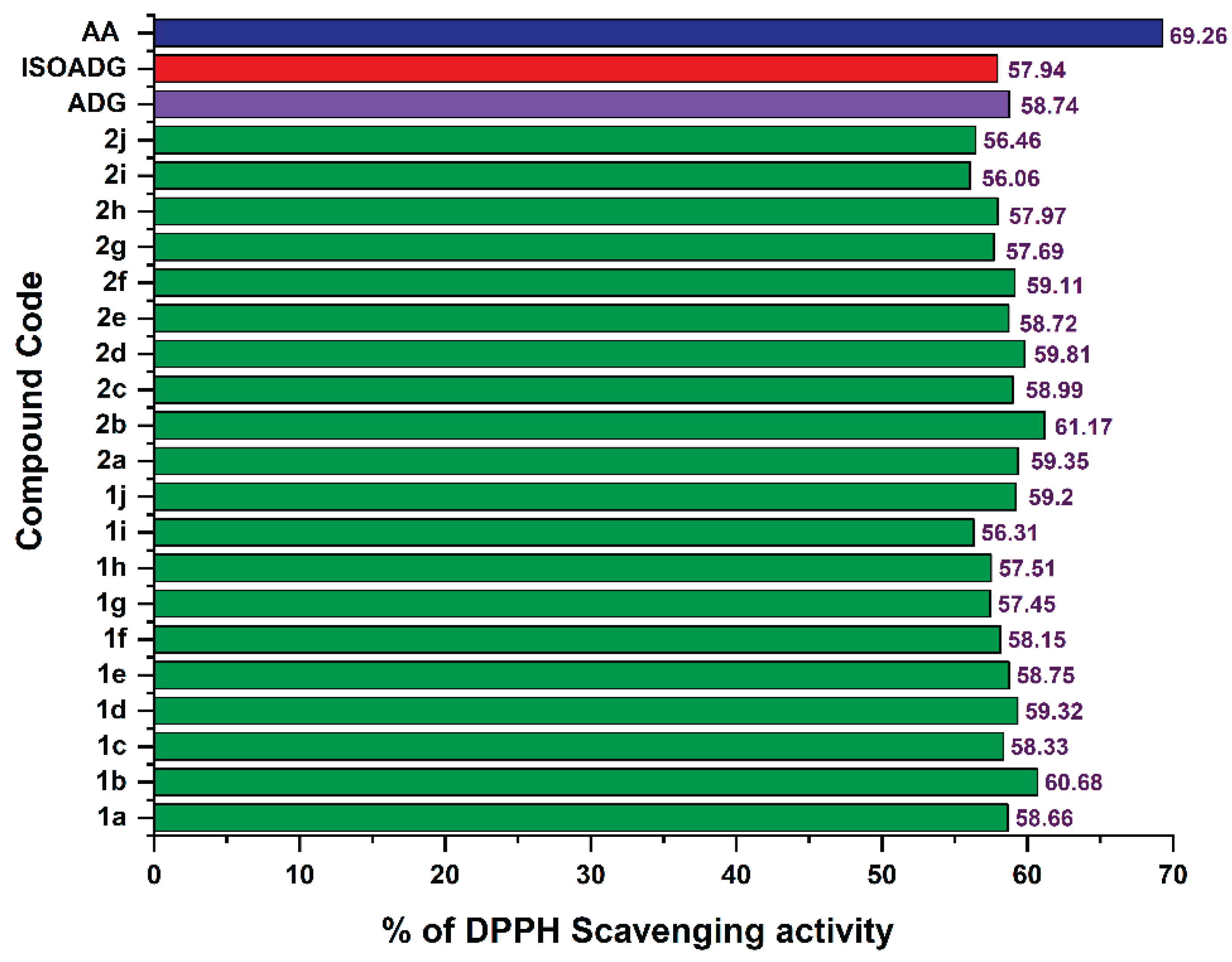

Table 1.
GI50s of the compounds 1a-1j against NCI human cancer cell line panel.
| Cancer | Sub Panel | GI50 (µM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | 1e | 1f | 1g | 1h | 1i | 1j | ||
| Leukemia | CCRF-CEM | 0.59 | 1.14 | 2.06 | 1.32 | 0.70 | 0.40 | 0.92 | 0.48 | 8.40 | 0.64 |
| HL-60(TB) | 2.32 | 1.73 | 0.76 | 1.38 | 1.17 | 0.64 | 2.67 | 0.67 | 2.17 | 1.08 | |
| K-562 | 2.97 | 2.16 | 2.14 | 2.41 | 2.89 | 2.34 | 3.67 | 2.07 | 3.82 | 1.90 | |
| MOLT-4 | 1.97 | 2.87 | 0.69 | 1.14 | 0.91 | 0.60 | 0.88 | 0.61 | 2.11 | 1.71 | |
| RPMI-8226 | 2.02 | 2.45 | 2.63 | 1.38 | 2.75 | 0.49 | 2.46 | 1.28 | 8.60 | 0.92 | |
| SR | 3.79 | 3.46 | 2.20 | 1.29 | 1.77 | 1.70 | 2.53 | 1.69 | 2.74 | 1.74 | |
| Non-Small Cell Lung | A549/ATCC | 2.49 | 3.62 | 3.88 | 2.82 | 3.07 | 2.14 | 3.57 | 3.10 | 3.09 | 2.39 |
| EKVX | 2.07 | 2.75 | 2.21 | 2.21 | 2.10 | 2.06 | 2.24 | 2.21 | 2.13 | 1.96 | |
| HOP-62 | 1.87 | 3.15 | 1.74 | 1.78 | 2.08 | 1.62 | 2.36 | 1.80 | 2.12 | 2.65 | |
| HOP-92 | 1.53 | 1.86 | 1.53 | 1.56 | 1.75 | 1.29 | 1.50 | 1.38 | 1.43 | 1.47 | |
| NCI-H226 | 1.95 | 2.38 | 1.67 | 1.76 | 1.75 | 1.51 | 1.55 | 1.36 | 1.47 | 1.72 | |
| NCI-H23 | 1.69 | 1.83 | 1.79 | 1.75 | 1.90 | 1.70 | 1.73 | 1.71 | 1.76 | 1.71 | |
| NCI-H322M | 3.35 | 1.76 | 4.22 | 3.26 | 6.06 | 3.34 | 1.02 | 4.20 | 4.92 | 2.94 | |
| NCI-H460 | 1.66 | 2.27 | 1.81 | 1.68 | 1.78 | 1.69 | 1.79 | 1.59 | 1.83 | 1.74 | |
| NCI-H522 | 1.90 | 1.75 | 1.52 | 1.54 | 1.50 | 1.95 | 1.58 | 1.69 | 1.75 | 1.73 | |
| Colon | COLO 205 | 2.01 | 2.41 | 2.06 | 1.98 | 2.09 | 1.48 | 2.30 | 1.70 | 1.97 | 1.95 |
| HCC-2998 | 1.68 | 1.92 | 1.73 | 1.81 | 1.72 | 1.76 | 1.88 | 1.83 | 1.63 | 1.70 | |
| HCT-116 | 1.61 | 1.99 | 3.48 | 1.04 | 1.24 | 0.39 | 1.20 | 0.94 | 3.91 | 1.40 | |
| HCT-15 | 1.72 | 1.74 | 1.57 | 1.58 | 1.60 | 1.61 | 1.59 | 1.60 | 1.59 | 1.58 | |
| HT29 | 2.37 | 1.82 | 1.70 | 1.49 | 1.66 | 2.38 | 1.69 | 2.16 | 1.90 | 1.70 | |
| KM12 | 2.06 | 1.99 | 2.26 | 1.67 | 1.83 | 1.84 | 2.29 | 1.68 | 3.01 | 1.66 | |
| SW-620 | 1.73 | 2.21 | 5.71 | 1.53 | 1.72 | 4.49 | NT | NT | 5.42 | 1.69 | |
| CNS | SF-268 | 3.20 | 7.65 | 2.27 | 1.60 | 2.02 | 2.36 | 2.39 | 2.11 | 2.61 | 1.67 |
| SF-295 | 1.64 | 1.68 | 1.52 | 1.42 | 1.36 | 1.55 | 1.50 | 1.54 | 1.58 | 1.43 | |
| SF-539 | 1.84 | 1.74 | 1.77 | 1.63 | 1.78 | 1.77 | 1.72 | 1.76 | 1.79 | 1.69 | |
| SNB-19 | 1.85 | 3.64 | NT | 2.24 | 2.17 | 1.44 | 1.40 | 1.47 | 1.36 | 2.46 | |
| SNB-75 | 1.36 | 7.34 | NT | 1.12 | 1.57 | 1.66 | 1.69 | 1.51 | 1.71 | 1.15 | |
| U251 | 1.84 | 1.74 | 1.81 | 1.59 | 1.69 | 2.36 | 2.39 | 2.11 | 2.61 | 1.66 | |
| Melanoma | LOX IMVI | 1.54 | 1.72 | 1.44 | 1.61 | 1.64 | 1.32 | 1.58 | 1.55 | 1.43 | 1.57 |
| MALME-3M | 1.73 | 1.78 | 1.87 | 1.47 | 1.87 | 1.58 | 1.82 | 1.60 | 2.46 | 1.54 | |
| M14 | 1.81 | 2.08 | 1.75 | 1.69 | 1.86 | 1.50 | 1.84 | 1.71 | 1.76 | 1.72 | |
| MDA-MB-435 | 1.86 | 3.03 | 1.50 | 1.56 | 1.72 | 1.68 | 1.65 | 1.67 | 1.61 | 1.54 | |
| SK-MEL-2 | 1.74 | 1.78 | 1.79 | 1.57 | 1.78 | 1.69 | 1.77 | 1.76 | 1.79 | 1.57 | |
| SK-MEL-28 | 1.77 | 2.04 | 1.77 | 1.80 | 1.73 | 1.66 | 1.78 | 1.73 | 1.74 | 1.94 | |
| SK-MEL-5 | 1.40 | 1.63 | 1.65 | 1.63 | 1.54 | 1.62 | 1.70 | 1.60 | 1.59 | 1.54 | |
| UACC-257 | 1.90 | 1.80 | 1.93 | 1.77 | 1.77 | 1.65 | 1.80 | 1.70 | 1.74 | 1.64 | |
| UACC-62 | 1.65 | 1.69 | 1.65 | 1.71 | 1.68 | 1.58 | 1.68 | 1.58 | 1.64 | 1.91 | |
| Ovarian | IGROV1 | 1.77 | 2.08 | 2.07 | 1.39 | 2.20 | 1.84 | 4.29 | 1.67 | 3.95 | 1.37 |
| OVCAR-3 | 1.82 | 1.87 | 1.95 | 1.60 | 1.69 | 2.02 | 1.78 | 1.52 | 1.95 | 1.50 | |
| OVCAR-4 | 2.31 | 4.05 | 1.74 | 2.09 | 2.49 | 1.75 | 2.03 | 1.89 | 1.92 | 1.72 | |
| OVCAR-5 | 1.86 | 2.06 | 1.92 | 1.78 | 1.85 | 1.74 | 1.98 | 1.93 | 1.93 | 1.86 | |
| OVCAR-8 | 1.81 | 1.90 | 1.77 | 1.53 | 1.76 | 1.48 | 1.95 | 1.55 | 1.67 | 1.35 | |
| NCI/ADR-RES | 4.75 | 2.98 | 1.77 | 1.88 | 2.15 | 1.54 | 1.83 | 1.83 | 1.65 | 1.94 | |
| SK-OV-3 | 1.77 | 2.08 | 2.70 | 5.86 | 4.16 | 1.88 | 3.04 | 2.03 | 3.47 | 3.44 | |
| Renal | 786-0 | 2.03 | 3.04 | 1.78 | 2.31 | 2.59 | 1.60 | 1.99 | 2.07 | 1.77 | 2.30 |
| A498 | 2.34 | 1.90 | 4.13 | 2.90 | 4.33 | 3.31 | 5.20 | 4.35 | 4.03 | 2.63 | |
| ACHN | 1.64 | 1.89 | 1.80 | 1.64 | 1.61 | 1.66 | 1.89 | 2.07 | 1.83 | 1.52 | |
| CAKI-1 | 1.86 | 2.81 | 1.56 | 1.32 | 1.42 | 1.58 | 1.63 | 1.59 | 1.71 | 1.26 | |
| RXF 393 | 1.40 | 1.62 | 1.64 | 1.05 | 1.18 | 1.60 | 1.79 | 1.71 | 1.49 | 0.89 | |
| SN12C | 1.87 | 2.93 | 1.59 | 1.65 | 1.57 | 1.55 | 1.67 | 1.51 | 1.66 | 1.98 | |
| TK-10 | 1.78 | 2.22 | 1.41 | 2.14 | 2.12 | 1.27 | 1.33 | 1.19 | 1.33 | 1.95 | |
| UO-31 | 1.35 | 1.82 | 1.78 | 1.30 | 1.45 | 1.60 | 1.99 | 2.07 | 1.77 | 1.22 | |
| Prostate | PC-3 | 1.95 | 2.75 | 1.97 | 2.07 | 2.29 | 1.55 | 2.07 | 2.03 | 1.92 | 2.18 |
| DU-145 | 2.05 | 4.11 | 1.97 | 1.88 | 2.12 | 1.82 | 2.05 | 1.77 | 2.05 | 2.04 | |
| Breast | MCF7 | 2.00 | 2.64 | 1.63 | 1.48 | 1.77 | 1.43 | 1.51 | 1.49 | 1.57 | 1.46 |
| MDA-MB-231 | 1.72 | 1.84 | 1.72 | 1.49 | 1.68 | 1.68 | 1.68 | 1.59 | 1.70 | 1.47 | |
| HS 578T | 2.31 | 2.69 | 1.66 | 1.91 | 1.94 | 1.51 | 1.75 | 1.52 | 1.85 | 1.86 | |
| BT-549 | 1.64 | 1.94 | 1.30 | 1.57 | 1.46 | 1.31 | 1.49 | 1.47 | 1.20 | 1.65 | |
| T-47D | 1.42 | 1.76 | 1.39 | 1.63 | 2.29 | 1.19 | 1.31 | 1.18 | 1.14 | 1.80 | |
| MDA-MB-468 | 1.69 | 1.76 | 1.63 | 1.74 | 1.82 | 1.53 | 2.00 | 1.93 | 1.85 | 1.71 | |
Table 2.
NCI compare analysis of the compounds 1f, 1h, and 1j.
| Compound | Pearson Correlation Coefficient (pcc) | Marketed drug | Mechanism of action | |
|---|---|---|---|---|
| 1f | GI50 | 0.51 | Nelarabine | Inhibits DNA elongation, apoptosis, and cellular destruction. |
| 1h | GI50 | 0.60 | Melphalan | Inhibits DNA synthesis or transcription. |
| 1j |
GI50 |
0.50 |
Trisenox 3 |
DNA fragmentation characteristic of apoptosis in NB4 human promyelocytic leukemia cells. |
Table 3.
Percentage of the MDA-MB-231 cells in G1, S, and G2 phases before and after treatment with compound 1f.
Table 3.
Percentage of the MDA-MB-231 cells in G1, S, and G2 phases before and after treatment with compound 1f.
| Time | G1 (%) | S (%) | G2 (%) |
|---|---|---|---|
| 0 hr | 74.96 | 2.62 | 22.41 |
| 24 hr (3.33 µM) | 83.53 | 3.26 | 13.19 |
| 24 hr (5.00 µM) | 80.50 | 8.29 | 11.2 |
| 24 hr (6.66 µM) | 82.43 | 8.64 | 8.91 |
Table 4.
Lipinski parameters for the active compounds.
| Compound | *MW | LogP | HBD | HBA | TPSA |
|---|---|---|---|---|---|
| Ia | 580.29 | 5.69 | 1 | 7 | 82.81 |
| Ib | 625.28 | 5.55 | 1 | 10 | 125.95 |
| Ic | 598.28 | 5.87 | 1 | 7 | 82.81 |
| Id | 614.25 | 6.28 | 1 | 7 | 82.81 |
| Ie | 658.20 | 6.38 | 1 | 7 | 82.81 |
| If | 598.28 | 6.35 | 1 | 7 | 78.88 |
| Ig | 614.25 | 6.27 | 1 | 7 | 82.81 |
| Ih | 658.20 | 6.38 | 1 | 7 | 82.81 |
| Ii | 594.31 | 6.09 | 1 | 7 | 82.81 |
| Ij | 610.30 | 5.71 | 1 | 8 | 92.04 |
*MW – Molecular Weight, Log P – Partition coefficient, HBD – No. of. hydrogen bond donors, HBA – No. of. hydrogen bond acceptors, TPSA – Topological Polar Surface Area.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



