Submitted:
14 May 2025
Posted:
15 May 2025
You are already at the latest version
Abstract
Protein Disulfide Isomerases (PDIs) are emerging targets in anticancer therapy, with several PDI inhibitors demonstrating anticancer efficacy in preclinical models. Research has largely focused on “canonical” PDIs, such as PDIA1, which contain CXXC active site motifs where C represents Cysteine. Canonical PDIs have well-studied, critical roles in forming, breaking, and exchanging/scrambling disulfide bonds during protein folding. In contrast, non-canonical PDIs, which harbor CXXS active site motifs, remain less well-studied despite their role as sensors or effectors of protein folding quality control during protein trafficking in the secretory pathway. Here, we provide a review of the literature relating to the non-canonical PDIs ERp44, AGR2, and AGR3, which have been identified as strong dependencies in specific cancer subtypes according to the DepMap database. The biological and biochemical functions of ERp44, AGR2, and AGR3 are discussed, highlighting the role of ERp44 in two mechanisms of protein folding quality control, AGR2 as a selective sensor of mucin protein misfolding, and a unique role for AGR3 in cilia. Finally, we discuss recent efforts to develop small molecule inhibitors of ERp44, AGR2, and AGR3 as tool compounds and experimental therapeutics.
Keywords:
protein disulfide isomerases
; PDIA1
; AGR2
; AGR3
; ERp44
1. Introduction
Protein synthesis homeostasis, or proteostasis, represents an important therapeutic target in cancer, since tumors are characterized by high rates of protein synthesis driven in part by activation of oncogenes and inactivation of tumor suppressor genes [1,2,3]. This elevated rate of protein synthesis requires high levels of amino acids and frequently exceeds the ability of cancer cells to facilitate native protein folding, resulting in cellular stress responses. Promising anticancer strategies include limiting amino acid levels and exacerbating protein misfolding. Consistent with this approach, depleting asparagine levels using asparaginase is a clinically established treatment for Acute Lymphoblastic Leukemia (ALL) [4,5]. Additional emerging strategies include the use of arginase [6] and inhibitors of amino acid transporters [7,8,9,10,11]. Protein misfolding is implicated in various disease pathologies, including diabetes [12,13,14,15,16] and neurodegenerative disease [17,18,19,20] and is an established characteristic of certain malignancies such as breast cancer [21,22,23]. With respect to breast cancer, endoplasmic reticulum (ER) stress has been implicated as a therapeutic vulnerability in HER2+ breast cancer [24,25], and more recently in HER2+ breast cancer metastasis [26].
The folding of transmembrane and secretory proteins occurs in the ER. Eukaryotic cells have evolved response mechanisms to protein misfolding in the ER referred to collectively as the Unfolded Protein Response (UPR) or “ER stress” response. The three primary sensors of ER stress are the transmembrane nuclease Inositol Requiring Enzyme 1 (IRE1) [27,28], the transcription factor ATF6 [29], and the transmembrane kinase Protein Kinase R-like ER Kinase (PERK) [30,31,32]. These sensors initiate signaling pathways that either restore protein homeostasis, or if ER stress is severe and unresolvable, trigger apoptosis, as reviewed previously [33,34,35,36]. ER stress causes PERK-mediated phosphorylation of the translation factor eIF2, resulting in the suppression of cap-dependent mRNA translation (reviewed in [37]). Similarly, the kinases GCN2, PKR, and HRI are activated and phosphorylate eIF2 in response to amino acid deprivation, double-stranded RNA/viral infection, or heme deprivation, respectively. These pathways collectively form the integrated stress response (ISR), as they converge on eIF2 phosphorylation to regulate protein synthesis [38]. Multiple modes of posttranslational modification promote and maintain native protein folding within the ER including N-glycosylation, cis-trans proline isomerization, and disulfide bonding between cysteine residues. In studies of purified proteins, disulfide bonding and proline isomerization represent rate-limiting steps in protein folding [39,40,41,42,43,44,45], with similar findings observed in studies of protein folding in cultured cells [46,47].
2. Protein Disulfide Isomerases as a Druggable Vulnerability in a Subset of Human Cancers
Based on sequence similarity, the human genome encodes 22 members of the protein disulfide isomerase (PDI) family [48] (Table 1). Many of these proteins contain either a catalytic CXXC thioredoxin repeat or a non-canonical CXXS thioredoxin repeat, where C, S, and X represent Cysteine, Serine, or any amino acid residue, respectively. A classic study [49] showed that mammalian PDIs containing CXXC or CXXS complemented the loss of yeast PDI, but that in biochemical studies PDI(CXXC) was capable of disulfide bond formation (oxidation), reduction, or isomerization/”scrambling”, while CXXS catalyzed disulfide isomerization, but not oxidation or reduction. The observation that the CXXS mutant of human PDI complemented yeast PDI inactivation indicated that for yeast viability, disulfide isomerization is the essential PDI function. These distinct PDI catalytic functions are highlighted in Figure 1.
Recent studies indicated that the first cysteine (Cys) of the motif initiates nucleophilic attack of client disulfide-bonded Cys residues, while the second Cys is “resolving” and mediates substrate release [50]. Consistent with this mechanism, mutation of the second Cys, or in some cases the intervening (XX) residues, generates trapping mutants with prolonged client binding half-lives [51,52]. Interestingly, several human PDIs, including ERp44, AGR2, AGR3, and TMX5, possess CXXS motifs (Table 1), suggesting that these non-canonical PDIs play distinct roles in protein folding compared to PDIs with CXXC active site sequences.
In addition to the thioredoxin repeat, many PDIs contain motifs that anchor them in the ER, preventing their secretion, including transmembrane sequences in the case of TMX1-5, or variations of the C-terminal KDEL sequence. These KDEL and KDEL-like sequences bind to the KDEL receptors 1-3 (KDELR1-3) in the Golgi apparatus and facilitate retrograde trafficking back to the ER [53,54]. The precise functions of KDELR1-3 in the secretory pathway and their degree of redundancy are incompletely understood, making this an area of active investigation.
Given the critical role of protein folding in both cancer cells and in normal tissues, and the mystery of why humans have 22 different PDIs rather than one as in some yeast strains, it is important to determine whether a subset of PDIs is indispensable for cancer cells, but dispensable in normal adult tissues. The Dependency Map (DepMap), developed by the Broad Institute and accessible at depmap.org, employs gene knockout/knockdown technology to classify genes as “common essential” if their inactivation is broadly lethal. In contrast, “strongly selective” genes are those whose inactivation is without effect on most cancer lines, but essential for the survival of a subset of cancer lines. Targeting gene products that are strongly selective for a given cancer may sidestep the systemic toxicities associated with many anticancer medicines. As of 12/22/2024, DepMap data indicate that among the 22 PDIs, only four, ERp44, AGR2, AGR3, and TMX1 are strongly selective based on CRISPR knockout screens. Notably, three of these, ERp44, AGR2, and AGR3 are non-canonical PDIs. Why ERp44, AGR2, or AGR3 are essential for specific cancer cell lines is unclear but may relate to their unique biological and biochemical functions as outlined below.
3. Roles of Strongly Selective Disulfide Isomerases in Protein Folding and Cellular Signaling
ERp44 in Protein Folding Quality Control:ERp44 was identified by Sitia and colleagues as a novel ER protein containing a CRFS thioredoxin repeat [55]. Shortly thereafter, ERp44 was shown to retain a subset of clients termed “partner proteins” in the ER by forming a disulfide bond with these clients, thereby preventing their secretion. Examples of ERp44-retained ER partner proteins include ERO1, which generates oxidizing equivalents to drive disulfide bonding in the secretory pathway [56], ER Aminopeptidase 1 (ERAP1), which plays a key role in preparing peptide antigens for presentation to T cells [57], and Peroxiredoxin 4, which reduces hydrogen peroxide to water [58].
ERp44 plays a role in several physiological processes. Through its ability to regulate the secretion or retention of ERAP1, ERp44 may influence both blood pressure and antigen processing for T-cell mediated immunity [59]. ERAP1 controls processing of peptide antigens presented to T cells [60,61,62,63,64] suggesting that the ERp44/ERAP1 cassette may control T cell-mediated immunity in a redox-dependent manner. Additionally, secreted ERAP1 degrades angiotensin II (Ang II), a key regulator of blood pressure [59], indicating that the ERp44/ERAP1 cassette may also modulate blood pressure in response to changes in ER redox status. Depleting ERp44 in db/db mice exacerbates diabetic nephropathy [65], and knockout studies have demonstrated its role in glucose and lipid metabolism [66,67]. Collectively, these findings highlight the involvement of ERp44 in multiple physiological systems.
In addition to its role of retaining partner proteins in the ER, ERp44 has emerged as part of two key checkpoints in protein folding and client trafficking. First, ERp44 was shown to ensure the quality control of disulfide bonded homo-oligomers, such as immunoglobin M (IgM) [68,69] and adiponectin [70,71] by recycling misfolded proteins from the Golgi back to the ER via retrograde trafficking. More recently, Tirosh and colleagues discovered selective ER retention (sERr) of specific client receptor proteins whereby mis-disulfide bonded tyrosine kinases, such as EGFR, MET, and RET, become aggregated in high molecular mass, disulfide bonded complexes that incorporate ERp44 [72]. sERr is potentiated by PERK inhibitors or ISRIB, which override the integrated stress response (ISR), and permit continued cap-dependent translation in the presence of ER stress.
ERp44 Regulation and Cellular Functions: The localization, conformation, and activity of ERp44, as well as its binding and release of client proteins are controlled in a complex, integrated manner by the pH [73], Zinc [74], and redox gradients that exist between various ER and Golgi sub-compartments. Since these external factors affect the binding/release of ERp44 to/from its clients and partners, these factors have been exploited to some extent in therapeutic or diagnostic experiments. For example, abnormal levels of ERAP1 were attributed to the perturbation of the pH gradient in the ER/Golgi that negatively impacted the interaction between ERp44 and ERAP1.
The role of ERp44 in disulfide-bonding quality control is clear. The cysteine residue C29 in its non-canonical active site (CRFS) is essential for ERp44 to form disulfide bonds with clients, while the cysteine residue, C63, is speculated to participate in the release of clients trapped in a mixed disulfide intermediate state [75,76]. As such, targeting the cysteine residues of ERp44, especially C29, presents an attractive strategy for its inhibition. A related approach is the disruption of ERp44-client complexes. In 2017, Hampe et al. pursued this tactic to improve obesity-related metabolic disorders by modulating the interactions between ERp44 and adiponectin [77]. They synthesized two sets of cell-penetrating peptides (CPPs) modeled after two ERp44 partners, namely, IgM and adiponectin. The synthetic peptides successfully competed with endogenous adiponectin for the ERp44 active site with the goal of releasing adiponectin from ERp44, thus counteracting impaired adiponectin oligomerization and restoring adiponectin secretion. Modulation of the ERp44-adiponectin interaction was more pronounced for the adiponectin-derived CPPs than for the IgM-derived CPPs, a response attributed to their respective binding affinities.
To further investigate the interactions between ERp44 and its clients or partners, the same research group synthesized a brominated variant of the adiponectin-derived CPP, WT36-44-Br [78]. The variant peptide produced an ERp44-peptide mimetic complex by selectively forming an irreversible covalent thioether bond to the C29 residue in the active site of ERp44 as opposed to its free C63 residue. Moreover, the complex was generated quickly and efficiently with a 95% substitution yield after a 12-hour incubation, was chemically stable under various pH conditions, and deemed amenable to more extensive structural elucidation by the authors [78]. This approach outlined by Hampe and colleagues could be extended to more recently discovered ERp44 clients and partners such as ATP citrate lyase (ACLY) [79], protein tyrosine phosphatase receptor type-O (PTPR-O) [80], TMX5/TXNDC15 [81], and vascular endothelial growth factor-A (VEGF-A) [82], to study their interactions in greater detail.
Strategies to target ERp44 extend beyond synthetic peptides and include miR-101, a microRNA that targets ERp44 [83]. A negative correlation between miR-101 and ERp44 levels in HTR-8/SVneo trophoblast cells and in human placental tissues with preeclampsia (PE) was consistently observed. Further analysis revealed that miR-101 interacts with the 3’- untranslated region (UTR) of ERp44, and overexpression of miR-101 in trophoblast cells was associated with the downregulation of important ER stress sensors and effectors such as ATF-6, CHOP, caspase-12, and PERK. As a result, the authors posited that miR-101 influences the pathogenesis of PE by modulating trophoblast cell apoptosis through an ERp44-mediated mechanism [83]. Additionally, the small molecule polyphenol, honokiol, was found to alter ERp44 levels [84]. Treatment of oral squamous cancer cell carcinoma (OSCC) cell lines, HN-22 and HSC-4, with honokiol caused degradation of ERp44 in a dose- and time-dependent manner without affecting ERp44 mRNA expression. Honokiol significantly induced apoptosis of tumors in nude mice, as reflected by increased levels of cleaved PARP, with no adverse side effects on the body weight. The binding of honokiol to ERp44 was confirmed by pull-down assay experiments. Moreover, the ERp44-honokiol complex was modeled in silico. Docking studies suggested that honokiol interacts with ERp44 via non-covalent interactions, namely through hydrogen bonds with Glu 365 and Trp 28, and through pi-pi stacking with Arg 372. Collectively, the authors concluded that honokiol can potentially serve as an effective anti-ERp44 agent to treat OSCC [84].
AGR2 and AGR3 are non-canonical disulfide isomerases with intracellular and extracellular functions: Human AGR2 was first discovered as the ortholog of the Xenopus laevis cement gland gene Xenopus Anterior Gradient-2 (XAG-2) [85,86]. Both AGR2 and AGR3 contain a single non-canonical (CXXS) thioredoxin repeat and are relatively small compared to other PDIs. The biological importance of AGR2 is perhaps best illustrated by its diverse functions across species. XAG-2 specifies dorsoanterior ectodermal fate [86,87] and contributes to tissue regeneration in amphibians and fish [88,89,90]. In humans, AGR2 deficiency causes inflammatory bowel disease in part because AGR2 is required for production of the mucus proteins that form the colonic barrier that limits intestinal inflammation [91,92]. This observation is recapitulated in AGR2 knockout mice [93]. Additionally, biallelic AGR2 mutations cause a clinical disorder that mimics cystic fibrosis, characterized by decreased production of components of the mucociliary machinery [94].
Several observations suggest that AGR2 plays a role in cancer progression, including its necessity for EGFR presentation at the cell surface [95], its upregulation in Estrogen Receptor-positive breast cancers [96,97,98], and its ability to transform colonic epithelial cells [99]. Some of the documented pro-cancer effects of AGR2 relate to its ER-localized disulfide isomerase activity, which depends on its the active site thioredoxin Cys residue and the KTEL C-terminal ER retention motif [100]. AGR2 forms disulfide bonds with EGFR [95], suggesting a role in the folding of EGFR and potentially other HER-family members. Notably, HER1-4 are unique among receptor tyrosine kinases due to their two extracellular Cys-rich repeats per receptor molecule (reviewed in [101]).
Multiple studies show that secreted AGR2 contributes to tumor growth and progression, particularly through its role in angiogenesis [102,103,104] and as a ligand for cell surface receptors including C4.4A [105] and LYPD3 [106,107]. Moreover, extracellular AGR2 promotes the conversion of non-tumor organoids to tumor organoids, enhancing growth of the organoids approximately ten-fold [108]. Proteins that either promote or inhibit AGR2 dimerization have been identified, indicating a complicated, regulated role for AGR2 in carrying out functions in the ER, cell signaling, and in the tumor microenvironment [109].
Recent studies have identified yet another important AGR2 function specific to mucus-producing cells [110,111,112]. AGR2 is one of the top dependencies of multiple colon and pancreatic cancer cell lines, as indicated by the DepMap (depmap.org) (Table 2). Several of these AGR2-dependent cell lines also list components of the Wnt/-catenin pathway as top dependencies (Table 3). Interestingly, the strongest predictor of cancer cell dependence on AGR2 in the DepMap is the high expression of IRE1(depmap.org) (Table 4)a paralog of IRE1that is selectively expressed in mucus-producing cells of the lungs and intestines [111,112]. AGR2 inhibits IRE1 by binding to its luminal domain, thus preventing the dimerization-dependent activation of its nuclease function [111,112]. Ectopic IRE1expression triggers cell death [32], an effect that is overcome by AGR2 co-expression [111,112]. Although AGR2 disulfide bonding to IRE1 was not detected [111,112], its catalytic Cys 81 is required for binding, suggesting a role for the AGR2 active site in IRE1 binding. Together, these results reveal a Mucin-AGR2-IRE1axis whereby misfolded mucin sequesters AGR2, de-inhibiting IRE1which mediates the response to mucin misfolding. If confirmed, this mechanism would establish AGR2 as a sensor for specific types of protein misfolding.
In contrast to the broader expression pattern of AGR2, AGR3 is expressed in ciliated cells of the epithelium [113], where it regulates ciliary beat frequency. Decreased ciliary beat frequency is associated with impaired mucociliary clearance [113]. Furthermore, AGR3 is overexpressed in several cancers, including colorectal [114], breast [115], prostate [116] and ovarian cancers [117]. While the expression of AGR3 in the serous type of ovarian cancer predicts prolonged patient survival [117], AGR3 expression in colorectal cancer (CRC) predicts poor survival [114], which may be attributed to the ability of AGR3 to promote Wnt/b-catenin signaling and stemness in CRC cells [114].
AGR2-Targeted Therapeutic Strategies: As a validated oncogene, AGR2 remains the most thoroughly investigated “strongly selective” PDI in the literature. However, therapeutic targeting of AGR2 is complicated by the fact that it carries out divergent functions within the ER and extracellular compartments. Various macromolecules, including antibodies, peptides and microRNAs, have been developed to target AGR2. The mouse monoclonal antibody (mAb) 18A4 is the first AGR2-directed agent capable of selectively binding both recombinant and native AGR2 in cell extracts [118]. mAb also inhibits the growth of MCF-7 breast cancer cells in vitro by inhibiting extracellular AGR2. Its therapeutic properties were exploited by Negi et al. who reported that treatment of a preclinical mouse model with mAb 18A4 substantially reduced tumor size in A549 and H460 xenograft models [119]. Additionally, 18A4 suppressed tumor metastasis in the B16-F10 pulmonary melanoma metastasis mouse model, improving survival with no adverse side effects [120]. This study also found that mAb 18A4 activates the tumor suppressor p53, while deactivating ERK1/2-MAPK, a signaling pathway important for cellular proliferation. These findings highlight the involvement of extracellular AGR2 as a regulator in those two pathways and further confirm its role as an oncogene that facilitates cancer cell survival.
To mitigate the risks of immunogenicity associated with the murine mAb 18A4, a humanized variant, mAb 18A4Hu 1, was developed and optimized [120]. This variant preserves its affinity for AGR2 and retains its anti-tumor efficacy against SKOV-3 ovarian cancer xenografts in nude mice. Notably, the study identified two key binding sites, E60-H76 and A86-E153, through which mAb 18A4Hu1 may inhibit AGR2 function [120]. The same group that produced the humanized variant 18A4Hu 1 combined it with an anti-PD1 sequence to produce a bispecific antibody (BsAb) called AGR2xPD1 [121]. The latter exhibited greater anti-tumor activity than its individual components by redirecting lymphocytes to lung cancer cells, reducing the migration of H460 cells, and modulating the interaction between PD1 and its ligand [121]. AGR2xPD1 is currently being investigated in vivo [121].
Anti-AGR2 peptides with therapeutic properties have also been reported. Using an advanced mRNA display technique, Garri et al. screened a vast library of 1011 peptides and identified H10 as the strongest AGR2 binding partner with an affinity of 6.4 nM [122]. The binding interface between H10 and AGR2 was carefully studied. Since H10 lacked appreciable affinity toward AGR3, regions with the greatest structural diversity between AGR2 and AGR3 were selected for site-directed mutagenesis. Two amino acids, P41 and E60, that stabilize AGR2 dimerization, and amino acid E96 were revealed as essential participants in H10 binding. These results were supported by in silico docking simulations between H10 and AGR2. Combined data revealed the binding of H10 to AGR2 in its dimeric form. Additionally, attempts to block AGR2 homodimerization such as decreasing the concentrations of AGR2 to favor its monomeric form or mutating the E60 residue decreased the binding affinity between H10 and AGR2 [122]. Inhibition of AGR2 homodimer formation is a potential therapeutic strategy pursued by Ullah et al. in a theoretical study in which they successfully disrupted the AGR2 homodimer with five repurposed FDA-approved small molecule drugs that were selected via structure-based screening [123]. The drugs were found to interact with amino acid residues E60-K64 from the AGR2 dimerization domain. Whether the drugs can bind the AGR2 homodimer in vitro and whether this approach holds therapeutic value have yet to be assessed.
Zhang and colleagues designed an anti-AGR2 hexapeptide, NTAIYY, to target pancreatic ductal adenocarcinoma (PDAC) by competitively disrupting the AGR2-RNA polymerase II (RNAPII) complex [124]. NTAIYY was encapsulated in liposomes and tested in vitro and in vivo. NTAIYY consistently produced the desired therapeutic effects on a series of cancer cell lines harboring wild-type p53, including MCF7 and MDA-MB-231 (breast), HCT116 (colon), HT29 (colorectal), A549 and H2087 (lung) and Capan-2 and HPAC (PDAC). Disruption of the AGR2-RNAPII complex In the KC pancreatic ductal adenocarcinoma mouse model, lead to activation of p53, upregulation of p-H2axS139, and suppression of tumor growth with no adverse side effects on healthy tissues. Additionally, NTAIYY enhanced the sensitivity of wild-type p53-harboring cancers to various therapeutic agents, improving their selectivity and overall cytotoxicity. For comparison, a previously reported hexapeptide, PTTIYY [125], was evaluated alongside NTAIYY, with both exhibiting identical biological activity in vitro and in vivo. Moreover, NTAIYY was mutated at the sixth position to generate NTAIYA, revealing that this tyrosine is essential to effectively bind AGR2 and disrupt the AGR2-RNAPII complex [124]. Any additional interactions between AGR2 and NTAIYY were not investigated.
The third subset of biologics utilized to inhibit AGR2 are microRNAs. In 2018, Pan et al. observed a negative correlation between the expression levels of miR-217 and AGR2 in the tyrosine-kinase inhibitor (TKI)-resistant chronic myeloid leukemia cell line K562DR [126]. This inverse relationship between miR-217 and AGR2 was confirmed as the product of miR-217 binding to the 3’-UTR of AGR2 mRNA which impacted AGR2 translation and thus protein expression. Consequently, miR-217 overexpression induced therapeutic effects such as reduced tumor burden in the liver and prolonged lifespan of mice transplanted with K562DR through a mechanism involving simultaneous downregulation of AGR2 and sensitization to Dasatinib [126]. Similarly, expression of the miRNA, miR-199a-3p, negatively correlated with AGR2 expression in lung adenocarcinoma tissues in a study performed by Liu et al [127]. miR-199a-3p was also found to bind directly to the 3’-UTR of the AGR2 gene to repress the proliferation of non-small cell lung carcinoma PC-9 and lung adenocarcinoma Calu-3 cells and to induce cancer cell apoptosis.
Other AGR2 inhibition strategies include: (1) 125I seeds, which upregulated p-p38 MAPK and p-p53, induced apoptosis in cholangiocarcinoma cells (CCA) and reduced AGR2 levels in AGR2-overexpressing CCA xenograft tumors [128]; (2) repurposing etravirine, a non-nucleoside reverse transcriptase inhibitor, to downregulate AGR2 via autophagy-mediated lysosomal degradation in both non-resistant (A2780) and resistant (A2780ADR) ovarian cancer cells, where it demonstrated therapeutic effects alone and in synergy with paclitaxel [129]; and (3) co-administration of allicin, a sulfhydryl-reactive natural product, and lovastatin, a cholesterol lowering drug, to bypass AGR2-mediated resistance in lovastatin treatment [130].
Inhibition of AGR3: Fewer AGR3 inhibitors are reported compared to AGR2 inhibitors since AGR2 is the more extensively studied PDI of the two. The disparity largely stems from the limited expression pattern of AGR3 in disease states and the resulting uncertainty regarding its pro-oncogenic potential in different cancers. Because AGR2 and AGR3 share structural similarities, owing to a single non-canonical CXXS core thioredoxin motif and their relatively small size, strategies to inhibit AGR2 and AGR3 often overlap. For instance, Gray et al. developed MAGR3-1, a monoclonal antibody with high affinity and selectivity for AGR3 over AGR2, to investigate whether extracellular AGR3 is pro- or anti-oncogenic [131]. The more specific roles of AGR3 in breast cancer and tumorigenesis were further investigated in a study by Obacz and colleagues [132]. The authors found that extracellular AGR3 (eAGR3) promotes breast cancer cell adhesion and migration by participating in the c-Src signaling pathway and by inducing the phosphorylation of tyrosine kinases. Dasatinib, a tyrosine kinase inhibitor mentioned previously, was able to control cell migration and to counteract the effects of eAGR3 in T47D breast cancer cells [132]. The relationship between AGR3 and estrogen receptor expression in cancers is intriguing. While AGR3 expression appears to be independent of estrogen receptor status in ovarian cancer [131], a positive correlation was observed in certain breast cancers by Jian et al [133]. Their study identified AGR3 as an estrogen-responsive gene through Gene Set Enrichment Analysis (GSEA) and demonstrated that AGR3 knockdown reduced cell viability, suggesting a role for AGR3 in promoting the viability of estrogen receptor positive breast cancer cells. The selective estrogen receptor modulator 4-hydroxytamoxifen also downregulated AGR3 in T47D breast cancer cells [133]. Xu and colleagues examined the role of AGR3 in invasive ductal carcinoma (IDC) and found that AGR3 was predominantly expressed in luminal subtypes, with AGR3 expression associated with various outcomes, including a higher risk of recurrence and metastasis, increased cancer cell invasion and proliferation, and enhanced sensitivity to chemotherapeutic 5-fluoropyrimidines [134]. Although emerging evidence suggests that AGR3 may contribute to the progression of ovarian and breast cancer, its precise role as an oncogene remains unclear.
The development of PDI inhibitors for therapeutic purposes is gaining momentum with most efforts focused on targeting “common essential” PDIs. In contrast, strategies to inhibit “strongly selective” PDIs remain less common and typically involve large molecules and biologics targeting extracellular PDIs. As highlighted in this review, there are two cases where small molecules have been employed to target “strongly selective” PDIs and inhibit their function [84,130]. Only one of these cases may involve covalent binding of a natural small molecule, allicin, to the active site of AGR2 [130].
4. Therapeutic Strategies Targeting Strongly Selective and Non-Canonical Disulfide Isomerases
A series of bicyclic thiosulfonate compounds termed Disulfide bond Disrupting Agents (DDAs) were reported to exhibit activity against breast cancer cells in vitro and breast tumors in vivo [135,136,137,138,139], as well as to induce ER stress and UPR [36]. Recent work identified PDIA1, AGR2, AGR3, and ERp44 as direct cellular targets of DDAs [140]. AGR2 and ERp44 loss-of-function experiments partially mimicked the effects of DDAs on cancer cells, and DDAs were found to disrupt disulfide-mediated AGR2 dimerization, as well as disulfide bonding between ERp44 or PDIA1 to their client proteins. These findings confirm AGR2, ERp44 and PDIA1 as bona fide cellular DDA target proteins. Additionally, DDA treatment causes secretion of ERAP1, a known ERp44 partner protein, from breast cancer cells [140]. Given the role of secreted ERAP1 in lowering blood pressure by mediating Ang II degradation [59] and altering the immunopeptidome [57,64,141] as discussed above, DDAs may hold therapeutic potential not only as anticancer agents but also as antihypertensive or immunomodulatory compounds.
5. DDAs: Disulfide Isomerase Inhibitors with a Unique Selectivity Profile
To improve the selectivity of DDAs toward their target PDIs, their chemical structures were partially optimized through a structure-activity relationship (SAR) campaign. The pharmacophore, which consists of a disulfide-bond separated from a sulfinate anion by a four-carbon linker (highlighted in blue in Figure 2 was first identified in 2015 after screening a series of linear sulfur-containing compounds against various human cancer cell lines [142]. Compounds with the pharmacophore, such as NSC624205 and RBF3, displayed stronger effects toward EGFR+ (MDA-MB-468) and HER2+ (BT-474) breast cancers, compared with the pancreatic cancer cell line (BxPC-3). Any structural deviation from this motif resulted in biological inactivity. Field et al., who studied some of these compounds as radioprotective agents [143], pertinently noted that a cyclic thiosulfonate, 1,2-dithiane-1,1-dioxide or DTDO, can be released by an intramolecular cyclization reaction involving the pharmacophore [144]. Field’s observation combined with our recent discovery that certain cyclic DDAs are capable of covalently engaging the active sites of select ER-resident PDIs, strongly support an intramolecular mechanism of action. Accordingly, the required compound permeability for both the plasma and ER membranes likely results from the uncharged nature/lipophilicity of the cyclic form of the pharmacophore, DTDO. As expected, both the 5-membered ring (D5DO) and 7-membered ring (D7DO) showed no biological activity in EGFR- and HER2-overexpressing breast cancer cells since they deviate structurally from the pharmacophore by having a three- and a five-carbon linker respectively [145]. This difference in biological activity was investigated in silico by Ghilardi et al. and was attributed to a higher chemical reactivity of D5DO and D7DO toward free thiols and subsequently lower selectivity toward PDIs [145]. This chemical reactivity-selectivity dichotomy, whereby D5DO and D7DO favor their ring-opened forms and off-target binding, is supported by various kinetic and thermodynamic factors detailed in the same work [145]. Hence, DTDO was selected as the most potent first-generation DDA.
Our initial efforts to generate more potent cyclic DDAs started with the functionalization of DTDO at the 4 and 5 positions with hydroxy (dHDTDO) and acetoxy (dAcDTDO) substituents [146]. Despite the feasibility of these chemical modifications, the resulting DDAs exhibited no biological activity. It is hypothesized that dAcDTDO is hydrolyzed by esterases into dHDTDO which is inactive, likely due to a decrease in lipophilicity and membrane permeability. This likely hampers the access of dHDTDO to the target PDIs, since PDI inhibitors must traverse both the plasma and ER membranes to reach their protein targets. Subsequently, a series of bicyclic thiosulfonates were prepared [145]. The fused cycloalkane rings were expected to not only increase the lipophilicity of the DDAs but also favor their ring closing reaction thus improving their selectivity by minimizing off-target binding. Theoretical modeling of cyclic thiosulfonate reactivity and DFT simulations indicated that the opened form of bicyclic DDAs is less thermodynamically stable than their monocyclic counterparts, supporting their enhanced self-excision capacity following disulfide-bond formation unless appropriately “trapped” [145]. Overall, bicyclic DDAs exhibit increased potencies and more pronounced biological responses in EGFR+ and HER2+ breast cancer cells [145]. These improvements are attributed to the pre-organization of the reactive end groups, which presumably facilitates ring closure by a pseudo gem-dialkyl effect after possible off target interactions; an effect consistent with our computational studies as well as the independent findings of Bruice and Pandit [147] and Houk and Whitesides [148]. From the library of bicyclic DDAs, the cyclohexane-fused candidate, tcyDTDO, was identified as the most-potent second generation DDA and was nominated as the lead compound in the design of third-generation DDAs [145].
At this stage, the results gathered from our SAR campaign rationally prompted the design of lipophilic DDAs. This was achieved through the addition of non-polar substituents to the cyclohexane ring of the tcyDTDO scaffold at the more distal 5 and 6 positions to yield dMtcyDTDO [128]. This first derivative, bearing two methoxy groups, displayed improved cytotoxicity as indicated by lower IC50 and IC90 values compared to the parent tcyDTDO [145]. Conversely, dHtcyDTDO with two polar hydroxyl groups, was biologically inactive [145]. Guided by the experimental observation that lipophilicity plays a crucial role in DDA potency, we introduced fluorine to the tcyDTDO scaffold to generate mono-fluoro (FMtcyDTDO1 and FMtcyDTDO2) and di-fluoro (dFtcyDTDO) derivatives [145]. All fluorinated DDA bioisosteres exhibited improved cytotoxicity and more pronounced biological responses in EGFR+ breast cancer cells compared to tcyDTDO [145]. Of the three fluorinated-DDAs, dFtcyDTDO was the most potent [145]. Considering all DDAs prepared in our laboratory, dMtcyDTDO stands out as the most potent DDA thus far likely due to its elevated lipophilicity and the size of its substituents.
DDAs are the first small molecule active site covalent inhibitors of AGR2, AGR3, and ERp44. Mechanistically, we hypothesize that PDI inhibition by the DDAs is initiated by nucleophilic attack on the thiosulfonate group of DDAs by the thiolate nucleophile from the CXXS motif of “strongly selective” PDIs. This results in a covalently linked DDA-PDI complex containing a ring-opened DDA in a mixed disulfide state. The rate of DDA ring closure and self-excision is, we suppose, reduced after stabilization of the opened DDA by non-covalent interactions with neighboring residues inside the PDI active site pocket, the precise nature of which is unknown but currently under investigation by our group via molecular docking and structure elucidation studies. The reason behind PDI selectivity is somewhat unclear; for instance, why DDAs bind to only one “common essential” PDI, namely PDIA1, but not to ERp57 or ERp5/PDIA6 which also contain two identical CGHC repeats is not known. Possible reasons for the observed DDA selectivity may relate to different pKa values and/or redox potentials among PDI active site Cys residues, variables currently under investigation. Nonetheless, the ability of DDAs to act as “tempered electrophiles” that can discriminate between thiolates and can selectively bind certain therapeutically relevant PDIs is strongly supported. A promising future direction is the design and synthesis of trifluoromethyl and/or trifluoromethoxy DDAs since these groups combine the numerous pharmacokinetic benefits of fluorine with molecular sizes that are more comparable to methoxy groups for biologically enhanced fourth-generation DDAs.
With respect to future drug discovery, the development of “tempered electrophiles” holds significant potential to expand the druggable genome [149]. Compounds that utilize both non-covalent interactions and thiol-reactive mechanisms for target recognition have led to innovative approaches in the development of tyrosine kinase inhibitors for cancer therapy, targeting proteins such as JAK3 [150], BTK, [151,152], and EGFR [153,154]. Covalent inhibitors targeting the cancer-specific K-Ras mutant G12C marked a major breakthrough, resulting in the first FDA-approved inhibitors of this previously “undruggable” target (reviewed in [155]). Given the role of cysteine residues in mediating substrate recognition [156], allosteric regulation [157], and catalytic activity [158] of PDIs, tempered or rapidly reversible compounds such as DDAs may be well-suited for developing clinical PDI inhibitors. Additionally, DDA-like compounds featuring cyclic thiosulfonates or cyclic disulfides are being developed for other purposes, including thiol-mediated uptake, as demonstrated in the work of Matile and colleagues [159,160,161,162], and bioreductive probes or bifunctional reagents for thiol redox biology, as explored by Oliver Thorn-Seshold and colleagues [163,164].
6. Unanswered Questions and Future Directions
The anticancer activity of DDAs may stem from their ability to inhibit PDIA1, AGR2, AGR3, and ERp44 in parallel (Figure 3A). However, agents that are selective for AGR2, AGR3, or ERp44 would be valuable tools for investigating the specific functions of these proteins and could reduce adverse effects that might be associated with less selective inhibitors. In addition, the mechanisms that determine the selectivity of PDIs for different client proteins are not well understood, and it remains unclear whether the folding of some proteins requires the involvement of multiple PDIs. With respect to ERp44, it will be important to ascertain how its roles in two protein folding checkpoints, sERr and Golgi-ER recycling, are integrated. It is also necessary to investigate whether ERp44 functions only in regulating the trafficking of client and partner proteins or if it also catalyzes disulfide isomerization during protein folding.
Perhaps the least is known regarding why AGR3 is strongly selective against subsets of cancer cell lines. Given its sequence similarity to AGR2, AGR3 may perform similar biochemical functions. It will be important to determine whether double knockout mice lacking both AGR2 and AGR3 display a more severe phenotype than single knockouts, which would suggest some degree of functional redundancy. Since AGR2 is required for proper formation of the mucociliary machinery [91,92,110,112], and AGR3 is required for calcium-mediated regulation of ciliary function [113], double AGR2/AGR3 knockout may exhibit severe lung phenotypes resembling those seen in cystic fibrosis. Interestingly, despite its normally restricted expression, elevated AGR3 levels have been observed in estrogen receptor-positive breast cancers, both within cells and in secreted forms [131,165,166,167], raising the possibility that AGR3 acts as an endoplasmic reticulum foldase for cell surface receptors, analogous to AGR2. AGR3 may also be secreted and regulate cell surface receptors in a manner like AGR2.
Most cancer therapies, including targeted cancer treatments, involve the use of drug combinations. The approval rate of targeted combination regimens is increasing faster than that of targeted monotherapies [168]. Future studies should focus on identifying inhibitor-based combination regimens that produce strong synergistic effects against cancer cells without elevating toxicity profiles. It is likely that the relevant PDI client proteins will differ across tumor types, and that the mechanisms underlying sensitivity or resistance to PDI inhibitor therapy will vary among different malignancies. There is a need for new PDI inhibitor-based affinity probes designed to monitor how these drugs affect interactions between PDIs and their client proteins. Similarly, PDI inhibitor-based imaging probes are required to track drug distribution within cells, tumors and throughout the body. Under the appropriate conditions, PDI inhibitors such as DDAs may covalently “trap” or “freeze” binding events.
While most published studies have focused on the roles of PDIs individually, many of these proteins form physical complexes with one another, which are likely to have important biological consequences. For example, the DDA targets PDIA1, AGR2, and ERp44 were found to form non-covalent complexes with each other [140]. This and other observations have led to the concept that PDIs may function as a network to maintain protein folding homeostasis, as proposed by Tirosh and colleagues [72]. Data compiled from the BioGrid website shows that, except for AGR3, all DDA-target PDIs have been observed to associate with at least one other PDI (Figure 3B). AGR2 lies at the opposite end of the spectrum from AGR3, having been detected in associations with six other PDIs. This added layer of complexity, contributed by protein-protein interactions among PDIs, must be considered when analyzing the effects of genetically or pharmacologically ablating the activity of individual PDIs. Another unresolved question is why different PDIs harbor distinct C-terminal ER retention anchors (-KDEL, -RDEL, KTEL, and QSEL in the cases of PDIA1, ERp44, AGR2, and AGR3, respectively). The DepMap collection of CRISPR co-dependency data in cancer may offer insights into this question, particularly if KDELR1-3 each have distinct sequences preferences for these retention motifs. Notably, ERp44 and AGR3 are co-dependent, and KDELR1 and KDELR2 are co-dependent (Figure 3C). In addition, ERp44 is a KDELR1 co-dependency, while KDELR3 is an AGR2 co-dependency. A more complete understanding of how ERp44, AGR2, and AGR3 facilitate the maintenance of proteostasis will require detailed profiling of their associations and colocalizations with one another and with other PDIs, along with analysis of how this information correlates with the pattern of KDELR1-3 usage by each PDI.
In summary, non-canonical disulfide isomerases appear to mediate distinct biological and biochemical functions. According to current DepMap data, three non-canonical PDIs AGR2, AGR3, and ERp44 show selective dependencies, suggesting that they may be promising anticancer therapeutic targets. Although the development of PDI inhibitors as novel therapeutic agents is progressing, efforts remain primarily geared toward the inhibition of common essential PDIs such as PDIA1. A great deal more work is required to fully understand the roles of strongly selective PDIs in normal health and disease and to fully realize their potential as targets for anticancer therapy.
Author Contributions
Wrote and edited the manuscript: MEL, ZMD, BH, AK, RC, MRM, GA, SN, RKC, BKL.
Acknowledgments
This work was funded in part by the following grants to BL and RC: NIH/NCI R21 CA252400, NIH/NCI R21 CA277485, Florida Department of Health, James & Esther King Cancer Research Program grants 23K06 and 22K04, Florida Department of Health, Bankhead-Coley Research Program grant 23B03, and a grant from the Florida Breast Cancer Foundation.
Conflicting Interests
The authors declare no potential conflicts of interest.
References
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A 2013, 110, 11988–11993. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res 2009, 69, 8839–8843. [Google Scholar] [CrossRef] [PubMed]
- Smit, W.L.; Spaan, C.N.; Johannes de Boer, R.; Ramesh, P.; Martins Garcia, T.; Meijer, B.J.; et al. Driver mutations of the adenoma-carcinoma sequence govern the intestinal epithelial global translational capacity. Proc Natl Acad Sci U S A 2020, 117, 25560–25570. [Google Scholar] [CrossRef] [PubMed]
- Stams, W.A.; den Boer, M.L.; Holleman, A.; Appel, I.M.; Beverloo, H.B.; van Wering, E.R.; et al. Asparagine synthetase expression is linked with L-asparaginase resistance in TEL-AML1-negative but not TEL-AML1-positive pediatric acute lymphoblastic leukemia. Blood 2005, 105, 4223–4225. [Google Scholar] [CrossRef]
- Wells, R.J.; Woods, W.G.; Lampkin, B.C.; Nesbit, M.E.; Lee, J.W.; Buckley, J.D.; et al. Impact of high-dose cytarabine and asparaginase intensification on childhood acute myeloid leukemia: a report from the Childrens Cancer Group. J Clin Oncol 1993, 11, 538–545. [Google Scholar] [CrossRef]
- Chow, A.K.; Ng, L.; Sing Li, H.; Cheng, C.W.; Lam, C.S.; Yau, T.C.; et al. Anti-tumor efficacy of a recombinant human arginase in human hepatocellular carcinoma. Curr Cancer Drug Targets 2012, 12, 1233–1243. [Google Scholar]
- Bo, T.; Kobayashi, S.; Inanami, O.; Fujii, J.; Nakajima, O.; Ito, T.; et al. LAT1 inhibitor JPH203 sensitizes cancer cells to radiation by enhancing radiation-induced cellular senescence. Transl Oncol 2021, 14, 101212. [Google Scholar] [CrossRef]
- Gauthier-Coles, G.; Broer, A.; McLeod, M.D.; George, A.J.; Hannan, R.D.; Broer, S. Identification and characterization of a novel SNAT2 (SLC38A2) inhibitor reveals synergy with glucose transport inhibition in cancer cells. Front Pharmacol 2022, 13, 963066. [Google Scholar] [CrossRef]
- Nishikubo, K.; Ohgaki, R.; Liu, X.; Okanishi, H.; Xu, M.; Endou, H.; et al. Combination effects of amino acid transporter LAT1 inhibitor nanvuranlat and cytotoxic anticancer drug gemcitabine on pancreatic and biliary tract cancer cells. Cancer Cell Int 2023, 23, 116. [Google Scholar] [CrossRef]
- Jakobsen, S.; Nielsen, C.U. Exploring Amino Acid Transporters as Therapeutic Targets for Cancer: An Examination of Inhibitor Structures, Selectivity Issues, and Discovery Approaches. Pharmaceutics 2024, 16. [Google Scholar] [CrossRef]
- Rii, J.; Sakamoto, S.; Mizokami, A.; Xu, M.; Fujimoto, A.; Saito, S.; et al. L-type amino acid transporter 1 inhibitor JPH203 prevents the growth of cabazitaxel-resistant prostate cancer by inhibiting cyclin-dependent kinase activity. Cancer Sci 2024, 115, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.E.; Dalton, L.; Malzer, E.; Marciniak, S.J. Unravelling the story of protein misfolding in diabetes mellitus. World J Diabetes 2011, 2, 114–118. [Google Scholar] [CrossRef]
- Ashraf, G.M.; Greig, N.H.; Khan, T.A.; Hassan, I.; Tabrez, S.; Shakil, S.; et al. Protein misfolding and aggregation in Alzheimer's disease and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.C.; Soto, C. Type 2 diabetes as a protein misfolding disease. Trends Mol Med 2015, 21, 439–449. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Edwards Iii, G.; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer's disease through cross-seeding of protein misfolding. Mol Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef]
- Costes, S. Targeting protein misfolding to protect pancreatic beta-cells in type 2 diabetes. Curr Opin Pharmacol 2018, 43, 104–110. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Agorogiannis, E.I.; Agorogiannis, G.I.; Papadimitriou, A.; Hadjigeorgiou, G.M. Protein misfolding in neurodegenerative diseases. Neuropathol Appl Neurobiol 2004, 30, 215–224. [Google Scholar] [CrossRef]
- Kwong, L.K.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M. TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals 2008, 16, 41–51. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Cell death: protein misfolding and neurodegenerative diseases. Apoptosis 2009, 14, 455–468. [Google Scholar] [CrossRef]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 2009, 69, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Sharma, N.; Golden, E.B.; Cho, H.; Agarwal, P.; Gaffney, K.J.; et al. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett 2012, 325, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, H.; Jiang, C.C.; Fang, L.; Chen, C.; Zhang, X.D.; et al. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int J Mol Med 2014, 34, 772–781. [Google Scholar] [CrossRef]
- Arora, S.; Golemis, E.A. A new strategy to ERADicate HER2-positive breast tumors? Sci Signal 2015, 8, fs11. [Google Scholar] [CrossRef]
- Singh, N.; Joshi, R.; Komurov, K. HER2-mTOR signaling-driven breast cancer cells require ER-associated degradation to survive. Sci Signal 2015, 8, ra52. [Google Scholar] [CrossRef]
- Calvo, V.; Zheng, W.; Adam-Artigues, A.; Staschke, K.A.; Huang, X.; Cheung, J.F.; et al. A PERK-Specific Inhibitor Blocks Metastatic Progression by Limiting Integrated Stress Response-Dependent Survival of Quiescent Cancer Cells. Clin Cancer Res 2023, 29, 5155–5172. [Google Scholar] [CrossRef]
- Urano, F.; Bertolotti, A.; Ron, D. IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci 2000, 113 Pt 21, 3697–3702. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular Biology of the Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Brewer, J.W.; Diehl, J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A 2000, 97, 12625–12630. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.H.; Zeng, H.Q.; Wek, R.; Schapira, M.; et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; et al. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat Cell Biol 2001, 3, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K.; Murakami, S. Role of the unfolded protein response in cell death. Apoptosis 2006, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Hiss, D.C.; Gabriels, G.A. Implications of endoplasmic reticulum stress, the unfolded protein response and apoptosis for molecular cancer therapy. Part I: targeting p53, Mdm2, GADD153/CHOP, GRP78/BiP and heat shock proteins. Expert Opin Drug Discov 2009, 4, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Hazari, Y.M.; Bashir, A.; Haq, E.U.; Fazili, K.M. Emerging tale of UPR and cancer: an essentiality for malignancy. Tumour Biol 2016, 37, 14381–14390. [Google Scholar] [CrossRef]
- Wang, M.; Law, M.E.; Castellano, R.K.; Law, B.K. The unfolded protein response as a target for anticancer therapeutics. Critical Reviews in Oncology / Hematology 2018, 127, 66–79. [Google Scholar] [CrossRef]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of eukaryotic translation initiation factor 2alpha coordinates rRNA transcription and translation inhibition during endoplasmic reticulum stress. Mol Cell Biol 2009, 29, 4295–4307. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Shin, H.C.; Scheraga, H.A. Catalysis of the oxidative folding of bovine pancreatic ribonuclease A by protein disulfide isomerase. J Mol Biol 2000, 300, 995–1003. [Google Scholar] [CrossRef]
- Scheraga, H.A.; Konishi, Y.; Ooi, T. Multiple pathways for regenerating ribonuclease A. Adv Biophys 1984, 18, 21–41. [Google Scholar] [CrossRef]
- Konishi, Y.; Ooi, T.; Scheraga, H.A. Regeneration of RNase A from the reduced protein: models of regeneration pathways. Proc Natl Acad Sci U S A 1982, 79, 5734–5738. [Google Scholar] [CrossRef]
- Isenman, D.E.; Lancet, D.; Pecht, I. Folding pathways of immunoglobulin domains. The folding kinetics of the Cgamma3 domain of human IgG1. Biochemistry 1979, 18, 3327–3336. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.X.; Baldwin, R.L. Acid catalysis of the formation of the slow-folding species of RNase A: evidence that the reaction is proline isomerization. Proc Natl Acad Sci U S A 1978, 75, 4764–4768. [Google Scholar] [CrossRef] [PubMed]
- McPhie, P. Kinetic studies on the unfolding and refolding of pepsinogen in urea. The nature of the rate-limiting step. J Biol Chem 1980, 255, 4048–4052. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.E.; Ruan, B.; Alexander, P.A.; Wang, L.; Bryan, P.N. Mechanism of the kinetically-controlled folding reaction of subtilisin. Biochemistry 2007, 46, 640–651. [Google Scholar] [CrossRef]
- Nagradova, N. Enzymes catalyzing protein folding and their cellular functions. Curr Protein Pept Sci 2007, 8, 273–282. [Google Scholar] [CrossRef]
- Page, A.P. Cyclophilin and protein disulfide isomerase genes are co-transcribed in a functionally related manner in Caenorhabditis elegans. DNA Cell Biol 1997, 16, 1335–1343. [Google Scholar] [CrossRef]
- Okumura, M.; Kadokura, H.; Inaba, K. Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic Biol Med 2015, 83, 314–322. [Google Scholar] [CrossRef]
- Laboissiere, M.C.; Sturley, S.L.; Raines, R.T. The essential function of protein-disulfide isomerase is to unscramble non-native disulfide bonds. J Biol Chem 1995, 270, 28006–28009. [Google Scholar] [CrossRef]
- Bulleid, N.J. Disulfide bond formation in the mammalian endoplasmic reticulum. Cold Spring Harb Perspect Biol 2012, 4. [Google Scholar] [CrossRef]
- Walker, K.W.; Gilbert, H.F. Scanning and escape during protein-disulfide isomerase-assisted protein folding. J Biol Chem 1997, 272, 8845–8848. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Flaumenhaft, R. Thiol isomerases in thrombus formation. Circ Res 2014, 114, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Johannes, L.; Mallard, F.; Girod, A.; Grill, S.; Reinsch, S.; et al. Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J Cell Biol 1999, 147, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Murshid, A.; Presley, J.F. ER-to-Golgi transport and cytoskeletal interactions in animal cells. Cell Mol Life Sci 2004, 61, 133–145. [Google Scholar] [CrossRef]
- Anelli, T.; Alessio, M.; Mezghrani, A.; Simmen, T.; Talamo, F.; Bachi, A.; et al. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J 2002, 21, 835–844. [Google Scholar] [CrossRef]
- Otsu, M.; Bertoli, G.; Fagioli, C.; Guerini-Rocco, E.; Nerini-Molteni, S.; Ruffato, E.; et al. Dynamic retention of Ero1alpha and Ero1beta in the endoplasmic reticulum by interactions with PDI and ERp44. Antioxid Redox Signal 2006, 8, 274–282. [Google Scholar] [CrossRef]
- Yan, J.; Parekh, V.V.; Mendez-Fernandez, Y.; Olivares-Villagomez, D.; Dragovic, S.; Hill, T.; et al. In vivo role of ER-associated peptidase activity in tailoring peptides for presentation by MHC class Ia and class Ib molecules. J Exp Med 2006, 203, 647–659. [Google Scholar] [CrossRef]
- Kakihana, T.; Araki, K.; Vavassori, S.; Iemura, S.; Cortini, M.; Fagioli, C.; et al. Dynamic regulation of Ero1alpha and peroxiredoxin 4 localization in the secretory pathway. J Biol Chem 2013, 288, 29586–29594. [Google Scholar] [CrossRef]
- Hisatsune, C.; Ebisui, E.; Usui, M.; Ogawa, N.; Suzuki, A.; Mataga, N.; et al. ERp44 Exerts Redox-Dependent Control of Blood Pressure at the ER. Molecular cell 2015, 58, 1015–1027. [Google Scholar] [CrossRef]
- Firat, E.; Saveanu, L.; Aichele, P.; Staeheli, P.; Huai, J.; Gaedicke, S.; et al. The role of endoplasmic reticulum-associated aminopeptidase 1 in immunity to infection and in cross-presentation. J Immunol 2007, 178, 2241–2248. [Google Scholar] [CrossRef]
- Li, L.; Batliwala, M.; Bouvier, M. ERAP1 enzyme-mediated trimming and structural analyses of MHC I-bound precursor peptides yield novel insights into antigen processing and presentation. J Biol Chem 2019, 294, 18534–18544. [Google Scholar] [CrossRef]
- Rastall, D.P.; Aldhamen, Y.A.; Seregin, S.S.; Godbehere, S.; Amalfitano, A. ERAP1 functions override the intrinsic selection of specific antigens as immunodominant peptides, thereby altering the potency of antigen-specific cytolytic and effector memory T-cell responses. Int Immunol 2014, 26, 685–695. [Google Scholar] [CrossRef] [PubMed]
- York, I.A.; Brehm, M.A.; Zendzian, S.; Towne, C.F.; Rock, K.L. Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims MHC class I-presented peptides in vivo and plays an important role in immunodominance. Proc Natl Acad Sci U S A 2006, 103, 9202–9207. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Momburg, F.; Bhutani, N.; Goldberg, A.L. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a "molecular ruler" mechanism. Proc Natl Acad Sci U S A 2005, 102, 17107–17112. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Zhang, Y.; Shi, X.; Li, D.; Han, J. ERp44 depletion exacerbates ER stress and aggravates diabetic nephropathy in db/db mice. Biochem Biophys Res Commun 2018, 504, 921–926. [Google Scholar] [CrossRef]
- Nyirimigabo, E.; Jin, M.; Yang, Z.; Wang, J.; Zhai, K.; Mao, Y.; et al. The role of ERp44 in glucose and lipid metabolism. Arch Biochem Biophys 2019, 671, 175–184. [Google Scholar] [CrossRef]
- Bi, Y.; Chang, Y.; Liu, Q.; Mao, Y.; Zhai, K.; Zhou, Y.; et al. ERp44/CG9911 promotes fat storage in Drosophila adipocytes by regulating ER Ca(2+) homeostasis. Aging (Albany NY) 2021, 13, 15013–15031. [Google Scholar] [CrossRef]
- Anelli, T.; Ceppi, S.; Bergamelli, L.; Cortini, M.; Masciarelli, S.; Valetti, C.; et al. Sequential steps and checkpoints in the early exocytic compartment during secretory IgM biogenesis. EMBO J 2007, 26, 4177–4188. [Google Scholar] [CrossRef]
- Cortini, M.; Sitia, R. ERp44 and ERGIC-53 synergize in coupling efficiency and fidelity of IgM polymerization and secretion. Traffic 2010, 11, 651–659. [Google Scholar] [CrossRef]
- Wang, Z.V.; Schraw, T.D.; Kim, J.Y.; Khan, T.; Rajala, M.W.; Follenzi, A.; et al. Secretion of the adipocyte-specific secretory protein adiponectin critically depends on thiol-mediated protein retention. Mol Cell Biol 2007, 27, 3716–3731. [Google Scholar] [CrossRef]
- Wolf, G. New insights into thiol-mediated regulation of adiponectin secretion. Nutr Rev 2008, 66, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Mahameed, M.; Boukeileh, S.; Obiedat, A.; Darawshi, O.; Dipta, P.; Rimon, A.; et al. Pharmacological induction of selective endoplasmic reticulum retention as a strategy for cancer therapy. Nat Commun 2020, 11, 1304. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Harayama, M.; Kanemura, S.; Sitia, R.; Inaba, K. Structural basis of pH-dependent client binding by ERp44, a key regulator of protein secretion at the ER-Golgi interface. Proc Natl Acad Sci U S A 2017, 114, E3224–E3232. [Google Scholar] [CrossRef]
- Watanabe, S.; Amagai, Y.; Sannino, S.; Tempio, T.; Anelli, T.; Harayama, M.; et al. Zinc regulates ERp44-dependent protein quality control in the early secretory pathway. Nat Commun 2019, 10, 603. [Google Scholar] [CrossRef]
- Anelli, T.; Alessio, M.; Bachi, A.; Bergamelli, L.; Bertoli, G.; Camerini, S.; et al. Thiol-mediated protein retention in the endoplasmic reticulum: the role of ERp44. EMBO J 2003, 22, 5015–5022. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Vavassori, S.; Li, S.; Ke, H.; Anelli, T.; et al. Crystal structure of human ERp44 shows a dynamic functional modulation by its carboxy-terminal tail. EMBO Rep 2008, 9, 642–647. [Google Scholar] [CrossRef]
- Hampe, L.; Xu, C.; Harris, P.W.R.; Chen, J.; Liu, M.; Middleditch, M.; et al. Synthetic peptides designed to modulate adiponectin assembly improve obesity-related metabolic disorders. Br J Pharmacol 2017, 174, 4478–4492. [Google Scholar] [CrossRef]
- Hampe, L.; Harris, P.W.R.; Rushton, B.; Radjainia, M.; Brimble, M.A.; Mitra, A.K. Engineering a stable complex of ERp44 with a designed peptide ligand for analyzing the mode of interaction of ERp44 with its clients. Peptide Science 2021, 113, e24230. [Google Scholar] [CrossRef]
- Tian, H.; Shi, S.; You, B.; Zhang, Q.; Gu, M.; You, Y. ER resident protein 44 promotes malignant phenotype in nasopharyngeal carcinoma through the interaction with ATP citrate lyase. J Transl Med 2021, 19, 77. [Google Scholar] [CrossRef]
- Yang, Y.; Qiu, X.; Wang, F. Protein tyrosine phosphatase receptor type O (PTPRO) knockdown enhances the proliferative, invasive and angiogenic activities of trophoblast cells by suppressing ER resident protein 44 (ERp44) expression in preeclampsia. Bioengineered 2021, 12, 9561–9574. [Google Scholar] [CrossRef]
- Solda, T.; Galli, C.; Guerra, C.; Hoefner, C.; Molinari, M. TMX5/TXNDC15, a natural trapping mutant of the PDI family is a client of the proteostatic factor ERp44. Life Sci Alliance 2024, 7. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Yang, Z.; Jin, M.; Zhai, K.; Wang, J.; Mao, Y.; et al. ERp44 is required for endocardial cushion development by regulating VEGFA secretion in myocardium. Cell Prolif 2022, 55, e13179. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Jiang, Z.; Yu, X.; Zhang, Y.; Sun, M.; Wang, W.; et al. MiR-101 regulates apoptosis of trophoblast HTR-8/SVneo cells by targeting endoplasmic reticulum (ER) protein 44 during preeclampsia. J Hum Hypertens 2014, 28, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Jeon, Y.J.; Park, S.M.; Shin, J.C.; Lee, T.H.; Jung, S.; et al. Multifunctional effects of honokiol as an anti-inflammatory and anti-cancer drug in human oral squamous cancer cells and xenograft. Biomaterials 2015, 53, 274–284. [Google Scholar] [CrossRef]
- Thompson, D.A.; Weigel, R.J. hAG-2, the human homologue of the Xenopus laevis cement gland gene XAG-2, is coexpressed with estrogen receptor in breast cancer cell lines. Biochem Biophys Res Commun 1998, 251, 111–116. [Google Scholar] [CrossRef]
- Aberger, F.; Weidinger, G.; Grunz, H.; Richter, K. Anterior specification of embryonic ectoderm: the role of the Xenopus cement gland-specific gene XAG-2. Mech Dev 1998, 72, 115–130. [Google Scholar] [CrossRef]
- Shih, L.J.; Lu, Y.F.; Chen, Y.H.; Lin, C.C.; Chen, J.A.; Hwang, S.P. Characterization of the agr2 gene, a homologue of X. laevis anterior gradient 2, from the zebrafish, Danio rerio. Gene Expr Patterns 2007, 7, 452–460. [Google Scholar] [CrossRef]
- Ivanova, A.S.; Shandarin, I.N.; Ermakova, G.V.; Minin, A.A.; Tereshina, M.B.; Zaraisky, A.G. The secreted factor Ag1 missing in higher vertebrates regulates fins regeneration in Danio rerio. Sci Rep 2015, 5, 8123. [Google Scholar] [CrossRef]
- Ivanova, A.S.; Tereshina, M.B.; Ermakova, G.V.; Belousov, V.V.; Zaraisky, A.G. Agr genes, missing in amniotes, are involved in the body appendages regeneration in frog tadpoles. Sci Rep 2013, 3, 1279. [Google Scholar] [CrossRef]
- Li, S.; Wang, Y.; Zhang, Y.; Lu, M.M.; DeMayo, F.J.; Dekker, J.D.; et al. Foxp1/4 control epithelial cell fate during lung development and regeneration through regulation of anterior gradient 2. Development 2012, 139, 2500–2509. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, M.; Liu, X.; Ji, X.; Li, S.; Jin, E. New insights into the unfolded protein response (UPR)-anterior gradient 2 (AGR2) pathway in the regulation of intestinal barrier function in weaned piglets. Anim Nutr 2023, 15, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Al-Shaibi, A.A.; Abdel-Motal, U.M.; Hubrack, S.Z.; Bullock, A.N.; Al-Marri, A.A.; Agrebi, N.; et al. Human AGR2 Deficiency Causes Mucus Barrier Dysfunction and Infantile Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol 2021, 12, 1809–1830. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Zhen, G.; Verhaeghe, C.; Nakagami, Y.; Nguyenvu, L.T.; Barczak, A.J.; et al. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U S A 2009, 106, 6950–6955. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.; Hotakainen, R.; Al Shehhi, M.; Urzi, A.; Pareira, C.; Marais, A.; et al. A disorder clinically resembling cystic fibrosis caused by biallelic variants in the AGR2 gene. J Med Genet 2022, 59, 993–1001. [Google Scholar] [CrossRef]
- Dong, A.; Wodziak, D.; Lowe, A.W. Epidermal growth factor receptor (EGFR) signaling requires a specific endoplasmic reticulum thioredoxin for the post-translational control of receptor presentation to the cell surface. J Biol Chem 2015, 290, 8016–8027. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, Q.; Chen, H.; Hu, L.; Negi, H.; Zheng, Y.; et al. Binding of anterior gradient 2 and estrogen receptor-alpha: Dual critical roles in enhancing fulvestrant resistance and IGF-1-induced tumorigenesis of breast cancer. Cancer Lett 2016, 377, 32–43. [Google Scholar] [CrossRef]
- Salmans, M.L.; Zhao, F.; Andersen, B. The estrogen-regulated anterior gradient 2 (AGR2) protein in breast cancer: a potential drug target and biomarker. Breast Cancer Res 2013, 15, 204. [Google Scholar] [CrossRef]
- Liu, D.; Rudland, P.S.; Sibson, D.R.; Platt-Higgins, A.; Barraclough, R. Human homologue of cement gland protein, a novel metastasis inducer associated with breast carcinomas. Cancer Res 2005, 65, 3796–3805. [Google Scholar] [CrossRef]
- Wang, Z.; Hao, Y.; Lowe, A.W. The adenocarcinoma-associated antigen, AGR2, promotes tumor growth, cell migration, and cellular transformation. Cancer Res 2008, 68, 492–497. [Google Scholar] [CrossRef]
- Gupta, A.; Dong, A.; Lowe, A.W. AGR2 gene function requires a unique endoplasmic reticulum localization motif. J Biol Chem 2012, 287, 4773–4782. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Guo, Y.; Zhu, D.; Zhang, N.; Li, L.; Jiang, J.; et al. Pro-metastatic activity of AGR2 interrupts angiogenesis target bevacizumab efficiency via direct interaction with VEGFA and activation of NF-kappaB pathway. Biochim Biophys Acta Mol Basis Dis 2018, 1864, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, Q.; Yu, X.; Merugu, S.B.; Mangukiya, H.B.; Smith, N.; et al. Tumor-secreted anterior gradient-2 binds to VEGF and FGF2 and enhances their activities by promoting their homodimerization. Oncogene 2017, 36, 5098–5109. [Google Scholar] [CrossRef]
- Hong, X.Y.; Wang, J.; Li, Z. AGR2 expression is regulated by HIF-1 and contributes to growth and angiogenesis of glioblastoma. Cell Biochem Biophys 2013, 67, 1487–1495. [Google Scholar] [CrossRef]
- Arumugam, T.; Deng, D.; Bover, L.; Wang, H.; Logsdon, C.D.; Ramachandran, V. New Blocking Antibodies against Novel AGR2-C4.4A Pathway Reduce Growth and Metastasis of Pancreatic Tumors and Increase Survival in Mice. Mol Cancer Ther 2015, 14, 941–951. [Google Scholar] [CrossRef]
- Hu, Y.D.; Wu, K.; Liu, Y.J.; Zhang, Q.; Shen, H.; Ji, J.; et al. LY6/PLAUR domain containing 3 (LYPD3) maintains melanoma cell stemness and mediates an immunosuppressive microenvironment. Biol Direct 2023, 18, 72. [Google Scholar] [CrossRef]
- Cocce, K.J.; Jasper, J.S.; Desautels, T.K.; Everett, L.; Wardell, S.; Westerling, T.; et al. The Lineage Determining Factor GRHL2 Collaborates with FOXA1 to Establish a Targetable Pathway in Endocrine Therapy-Resistant Breast Cancer. Cell Rep 2019, 29, 889–903 e810. [Google Scholar] [CrossRef]
- Fessart, D.; Domblides, C.; Avril, T.; Eriksson, L.A.; Begueret, H.; Pineau, R.; et al. Secretion of protein disulphide isomerase AGR2 confers tumorigenic properties. Elife 2016, 5. [Google Scholar] [CrossRef]
- Maurel M, Obacz J, Avril T, Ding YP, Papadodima O, Treton X et al. Control of anterior GRadient 2 (AGR2) dimerization links endoplasmic reticulum proteostasis to inflammation. EMBO Mol Med, 2019; 11.
- Tonelli, C.; Yordanov, G.N.; Hao, Y.; Deschenes, A.; Hinds, J.; Belleau, P.; et al. A mucus production programme promotes classical pancreatic ductal adenocarcinoma. Gut 2024. [CrossRef]
- Neidhardt, L.; Cloots, E.; Friemel, N.; Weiss, C.A.M.; Harding, H.P.; McLaughlin, S.H.; et al. The IRE1beta-mediated unfolded protein response is repressed by the chaperone AGR2 in mucin producing cells. EMBO J 2023.
- Cloots, E.; Guilbert, P.; Provost, M.; Neidhardt, L.; Van de Velde, E.; Fayazpour, F.; et al. Activation of goblet-cell stress sensor IRE1β is controlled by the mucin chaperone AGR2. The EMBO Journal, 2023; 1-24-24. [Google Scholar]
- Bonser, L.R.; Schroeder, B.W.; Ostrin, L.A.; Baumlin, N.; Olson, J.L.; Salathe, M.; et al. The Endoplasmic Reticulum Resident Protein AGR3. Required for Regulation of Ciliary Beat Frequency in the Airway. Am J Respir Cell Mol Biol 2015, 53, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Zhang, H.; Hu, J.; Song, Y.; Li, J.; Wang, L.; et al. AGR3 promotes the stemness of colorectal cancer via modulating Wnt/β-catenin signalling. Cellular Signalling 2020, 65, 109419. [Google Scholar] [CrossRef]
- Adam, P.J.; Boyd, R.; Tyson, K.L.; Fletcher, G.C.; Stamps, A.; Hudson, L.; et al. Comprehensive Proteomic Analysis of Breast Cancer Cell Membranes Reveals Unique Proteins with Potential Roles in Clinical Cancer*. Journal of Biological Chemistry 2003, 278, 6482–6489. [Google Scholar] [CrossRef] [PubMed]
- Maresh, E.L.; Mah, V.; Alavi, M.; Horvath, S.; Bagryanova, L.; Liebeskind, E.S.; et al. Differential expression of anterior gradient gene AGR2 in prostate cancer. BMC Cancer 2010, 10, 680. [Google Scholar] [CrossRef]
- Armes, J.E.; Davies, C.M.; Wallace, S.; Taheri, T.; Perrin, L.C.; Autelitano, D.J. AGR2 expression in ovarian tumours: a potential biomarker for endometrioid and mucinous differentiation. Pathology 2013, 45, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Zhu, Q.; Gao, G.W.; Zhou, C.C.; Li, D.W. [Preparation, characterization and potential application of monoclonal antibody 18A4 against AGR2]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2010, 26, 49–51. [Google Scholar]
- Negi, H.; Merugu, S.B.; Mangukiya, H.B.; Li, Z.; Zhou, B.; Sehar, Q.; et al. Anterior Gradient-2 monoclonal antibody inhibits lung cancer growth and metastasis by upregulating p53 pathway and without exerting any toxicological effects: A preclinical study. Cancer Letters 2019, 449, 125–134. [Google Scholar] [CrossRef]
- Guo, H.; Chen, H.; Zhu, Q.; Yu, X.; Rong, R.; Merugu, S.B.; et al. A humanized monoclonal antibody targeting secreted anterior gradient 2 effectively inhibits the xenograft tumor growth. Biochem Biophys Res Commun 2016, 475, 57–63. [Google Scholar] [CrossRef]
- Roy, D.; Liu, G.-S.; Zeling Wang, A.; Zhou, B.; Yunus, F.-U.-N.; Raza, G.; et al. Construction and stable gene expression of AGR2xPD1 bi-specific antibody that enhances attachment between T-Cells and lung tumor cells, suppress tumor cell migration and promoting CD8 expression in cytotoxic T-cells. Saudi Pharmaceutical Journal 2023, 31, 85–95. [Google Scholar] [CrossRef]
- Garri, C.; Howell, S.; Tiemann, K.; Tiffany, A.; Jalali-Yazdi, F.; Alba, M.M.; et al. Identification, characterization and application of a new peptide against anterior gradient homolog 2 (AGR2). Oncotarget 2018, 9, 27363–27379. [Google Scholar] [CrossRef]
- Ullah, S.; Khan, S.U.; Khan, A.; Junaid, M.; Rafiq, H.; Htar, T.T.; et al. Prospect of Anterior Gradient 2 homodimer inhibition via repurposing FDA-approved drugs using structure-based virtual screening. Molecular Diversity 2022, 26, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, H.; Deng, Y.; Schuck, K.; Raulefs, S.; Maeritz, N.; et al. AGR2-Dependent Nuclear Import of RNA Polymerase II Constitutes a Specific Target of Pancreatic Ductal Adenocarcinoma in the Context of Wild-Type p53. Gastroenterology 2021, 161, 1601–1614e1623. [Google Scholar] [CrossRef] [PubMed]
- Mohtar, M.A.; Hernychova, L.; O'Neill, J.R.; Lawrence, M.L.; Murray, E.; Vojtesek, B.; et al. The Sequence-specific Peptide-binding Activity of the Protein Sulfide Isomerase AGR2 Directs Its Stable Binding to the Oncogenic Receptor EpCAM. Mol Cell Proteomics 2018, 17, 737–763. [Google Scholar] [CrossRef]
- Pan, B.; Yang, J.; Wang, X.; Xu, K.; Ikezoe, T. miR-217 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitors by targeting pro-oncogenic anterior gradient 2. Experimental Hematology 2018, 68, 80–88e82. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Y.; Wang, Y.; Wu, D.; Zhang, H. miR-199a-3p plays an anti-tumorigenic role in lung adenocarcinoma by suppressing anterior gradient 2. Bioengineered 2021, 12, 7859–7871. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, W.; Dou, M.; Li, Z.; Liu, Z.; Li, J.; et al. 125I seeds inhibit proliferation and promote apoptosis in cholangiocarcinoma cells by regulating the AGR2-mediated p38 MAPK pathway. Cancer Letters 2022, 524, 29–41. [Google Scholar] [CrossRef]
- Ly, T.T.G.; Yun, J.; Ha, J.-S.; Kim, Y.-J.; Jang, W.-B.; Van Le, T.H.; et al. Inhibitory Effect of Etravirine, a Non-Nucleoside Reverse Transcriptase Inhibitor, via Anterior Gradient Protein 2 Homolog Degradation against Ovarian Cancer Metastasis. International Journal of Molecular Sciences 2022, 23, 944. [Google Scholar] [CrossRef]
- Sheng, N.; Wang Y-q Wang C-f Jia M-q Niu H-m Lu, Q.-q.; et al. AGR2-induced cholesterol synthesis drives lovastatin resistance that is overcome by combination therapy with allicin. Acta Pharmacologica Sinica 2022, 43, 2905–2916. [Google Scholar] [CrossRef]
- Gray, T.A.; MacLaine, N.J.; Michie, C.O.; Bouchalova, P.; Murray, E.; Howie, J.; et al. Anterior Gradient-3, A novel biomarker for ovarian cancer that mediates cisplatin resistance in xenograft models. Journal of Immunological Methods 2012, 378, 20–32. [Google Scholar] [CrossRef]
- Obacz, J.; Sommerova, L.; Sicari, D.; Durech, M.; Avril, T.; Iuliano, F.; et al. Extracellular AGR3 regulates breast cancer cells migration via Src signaling. Oncol Lett 2019, 18, 4449–4456. [Google Scholar] [CrossRef]
- Jian, L.; Xie, J.; Guo, S.; Yu, H.; Chen, R.; Tao, K.; et al. AGR3 promotes estrogen receptor-positive breast cancer cell proliferation in an estrogen-dependent manner. Oncol Lett 2020, 20, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Shao, Y.; Zhang, J.; Zhang, H.; Zhao, Y.; Liu, X.; et al. Anterior Gradient 3 Promotes Breast Cancer Development and Chemotherapy Response. Cancer Res Treat 2020, 52, 218–245. [Google Scholar] [CrossRef] [PubMed]
- Law, M.E.; Davis, B.J.; Ghilardi, A.F.; Yaaghubi, E.; Dulloo, Z.M.; Wang, M.; et al. Repurposing Tranexamic Acid as an Anticancer Agent. Frontiers in Pharmacology (Original Research), 2022; 12. [Google Scholar]
- Wang, M.; Law, M.E.; Davis, B.J.; Yaaghubi, E.; Ghilardi, A.F.; Ferreira, R.B.; et al. Disulfide bond-disrupting agents activate the tumor necrosis family-related apoptosis-inducing ligand/death receptor 5 pathway. Cell Death Discov 2019, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ferreira, R.B.; Law, M.E.; Davis, B.J.; Yaaghubi, E.; Ghilardi, A.F.; et al. A novel proteotoxic combination therapy for EGFR+ and HER2+ cancers. Oncogene 2019, 38, 4264–4282. [Google Scholar] [CrossRef]
- Besse, L.; Besse, A.; Mendez-Lopez, M.; Vasickova, K.; Sedlackova, M.; Vanhara, P.; et al. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mitochondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica 2019, 104, e415–e419. [Google Scholar] [CrossRef]
- Law, M.E.; Dulloo, Z.M.; Eggleston, S.R.; Takacs, G.P.; Alexandrow, G.M.; Lee, Y.i.; et al. DR5 disulfide bonding functions as a sensor and effector of protein folding stress. Molecular Cancer Research 2024. [CrossRef]
- Law, M.E.; Yaaghubi, E.; Ghilardi, A.F.; Davis, B.J.; Ferreira, R.B.; Koh, J.; et al. Inhibitors of ERp44, PDIA1, and AGR2 induce disulfide-mediated oligomerization of Death Receptors 4 and 5 and cancer cell death. Cancer Lett 2022, 534, 215604. [Google Scholar] [CrossRef]
- Koumantou, D.; Barnea, E.; Martin-Esteban, A.; Maben, Z.; Papakyriakou, A.; Mpakali, A.; et al. Editing the immunopeptidome of melanoma cells using a potent inhibitor of endoplasmic reticulum aminopeptidase 1 (ERAP1). Cancer Immunol Immunother 2019, 68, 1245–1261. [Google Scholar] [CrossRef]
- Ferreira, R.B.; Law, M.E.; Jahn, S.C.; Davis, B.J.; Heldermon, C.D.; Reinhard, M.; et al. Novel agents that downregulate EGFR, HER2, and HER3 in parallel. Oncotarget 2015, 6, 10445–10459. [Google Scholar] [CrossRef]
- Field, L.; Khim, Y.H. Organic disulfides and related substances. 33. Sodium 4-(2-acetamidoethyldithio)butanesulfinate and related compounds as antiradiation drugs. J Med Chem 1972, 15, 312–315. [Google Scholar] [CrossRef]
- Srivastava, P.K.; Field, L. Organic Disulfides and Related Substances.45. Synthesis and Properties of Some Disulfide Sulfinate Salts Containing No Nitrogen. J Chem Eng Data 1986, 31, 252–254. [Google Scholar] [CrossRef]
- Ghilardi, A.F.; Yaaghubi, E.; Ferreira, R.B.; Law, M.E.; Yang, Y.; Davis, B.J.; et al. Anticancer Agents Derived from Cyclic Thiosulfonates: Structure-Reactivity and Structure-Activity Relationships. ChemMedChem 2022, 17, e202200165. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.B.; Wang, M.; Law, M.E.; Davis, B.J.; Bartley, A.N.; Higgins, P.J.; et al. Disulfide bond disrupting agents activate the unfolded protein response in EGFR- and HER2-positive breast tumor cells. Oncotarget 2017, 8, 28971–28989. [Google Scholar] [CrossRef]
- Bruice, T.C.; Pandit, U.K. Intramolecular Models Depicting the Kinetic Importance of "Fit" in Enzymatic Catalysis. Proc Natl Acad Sci U S A 1960, 46, 402–404. [Google Scholar] [CrossRef]
- Houk, J.; Whitesides, G.M. Characterization and Stability of Cyclic Disulfides and Cyclic Dimeric Bis(Disulfides). Tetrahedron 1989, 45, 91–102. [Google Scholar] [CrossRef]
- Kavanagh, M.E.; Horning, B.D.; Khattri, R.; Roy, N.; Lu, J.P.; Whitby, L.R.; et al. Selective inhibitors of JAK1 targeting an isoform-restricted allosteric cysteine. Nat Chem Biol 2022, 18, 1388–1398. [Google Scholar] [CrossRef]
- Shi, L.; Zhong, Z.; Li, X.; Zhou, Y.; Pan, Z. Discovery of an Orally Available Janus Kinase 3 Selective Covalent Inhibitor. J Med Chem 2019, 62, 1054–1066. [Google Scholar] [CrossRef]
- Barragan, A.M.; Ghaby, K.; Pond, M.P.; Roux, B. Computational Investigation of the Covalent Inhibition Mechanism of Bruton's Tyrosine Kinase by Ibrutinib. J Chem Inf Model 2024, 64, 3488–3502. [Google Scholar] [CrossRef]
- Awoonor-Williams, E.; Abu-Saleh, A.A.A. Molecular Insights into the Impact of Mutations on the Binding Affinity of Targeted Covalent Inhibitors of BTK. J Phys Chem B 2024, 128, 2874–2884. [Google Scholar] [CrossRef]
- Shi, S.; Du, Y.; Huang, L.; Cui, J.; Niu, J.; Xu, Y.; et al. Discovery of novel potent covalent inhibitor-based EGFR degrader with excellent in vivo efficacy. Bioorg Chem 2022, 120, 105605. [Google Scholar] [CrossRef] [PubMed]
- Bouffard, E.; Zaro, B.W.; Dix, M.M.; Cravatt, B.; Wong, C.H. Refinement of Covalent EGFR Inhibitor AZD9291 to Eliminate Off-target Activity. Tetrahedron Lett 2021, 74. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, C.F.; Zhang, W.; Rao, G.W. A New Dawn for Targeted Cancer Therapy: Small Molecule Covalent Binding Inhibitor Targeting K-Ras (G12C). Curr Med Chem 2023. [CrossRef] [PubMed]
- Selles, B.; Zannini, F.; Couturier, J.; Jacquot, J.P.; Rouhier, N. Atypical protein disulfide isomerases (PDI): Comparison of the molecular and catalytic properties of poplar PDI-A and PDI-M with PDI-L1A. PLoS One 2017, 12, e0174753. [Google Scholar] [CrossRef]
- Lu, X.; Gilbert, H.F.; Harper, J.W. Conserved residues flanking the thiol/disulfide centers of protein disulfide isomerase are not essential for catalysis of thiol/disulfide exchange. Biochemistry 1992, 31, 4205–4210. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.; Otto, A.; Maskos, K.; Mucke, M.; Seckler, R.; Glockshuber, R. Efficient catalysis of disulfide formation during protein folding with a single active-site cysteine. J Mol Biol 1995, 247, 28–33. [Google Scholar] [CrossRef]
- Coelho, F.; Saidjalolov, S.; Moreau, D.; Thorn-Seshold, O.; Matile, S. Inhibition of Cell Motility by Cell-Penetrating Dynamic Covalent Cascade Exchangers: Integrins Participate in Thiol-Mediated Uptake. JACS Au 2023, 3, 1010–1016. [Google Scholar] [CrossRef]
- Shybeka, I.; Maynard, J.R.J.; Saidjalolov, S.; Moreau, D.; Sakai, N.; Matile, S. Dynamic Covalent Michael Acceptors to Penetrate Cells: Thiol-Mediated Uptake with Tetrel-Centered Exchange Cascades, Assisted by Halogen-Bonding Switches. Angew Chem Int Ed Engl 2022, 61, e202213433. [Google Scholar] [CrossRef]
- Kato, T.; Lim, B.; Cheng, Y.; Pham, A.T.; Maynard, J.; Moreau, D.; et al. Cyclic Thiosulfonates for Thiol-Mediated Uptake: Cascade Exchangers, Transporters, Inhibitors. JACS Au 2022, 2, 839–852. [Google Scholar] [CrossRef]
- Laurent, Q.; Martinent, R.; Lim, B.; Pham, A.T.; Kato, T.; Lopez-Andarias, J.; et al. Thiol-Mediated Uptake. JACS Au 2021, 1, 710–728. [Google Scholar] [CrossRef]
- Zeisel, L.; Felber, J.G.; Scholzen, K.C.; Schmitt, C.; Wiegand, A.J.; Komissarov, L.; et al. Piperazine-Fused Cyclic Disulfides Unlock High-Performance Bioreductive Probes of Thioredoxins and Bifunctional Reagents for Thiol Redox Biology. J Am Chem Soc 2024. [CrossRef]
- Felber, J.G.; Kitowski, A.; Zeisel, L.; Maier, M.S.; Heise, C.; Thorn-Seshold, J.; et al. Cyclic Dichalcogenides Extend the Reach of Bioreductive Prodrugs to Harness Thiol/Disulfide Oxidoreductases: Applications to seco-Duocarmycins Targeting the Thioredoxin System. ACS Cent Sci 2023, 9, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Jian, L.; Xie, J.; Guo, S.; Yu, H.; Chen, R.; Tao, K.; et al. AGR3 promotes estrogen receptor-positive breast cancer cell proliferation in an estrogen-dependent manner. Oncol Lett 2020, 20, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Obacz, J.; Brychtova, V.; Podhorec, J.; Fabian, P.; Dobes, P.; Vojtesek, B.; et al. Anterior gradient protein 3 is associated with less aggressive tumors and better outcome of breast cancer patients. Onco Targets Ther 2015, 8, 1523–1532. [Google Scholar] [PubMed]
- Garczyk, S.; von Stillfried, S.; Antonopoulos, W.; Hartmann, A.; Schrauder, M.G.; Fasching, P.A.; et al. AGR3 in breast cancer: prognostic impact and suitable serum-based biomarker for early cancer detection. PLoS One 2015, 10, e0122106. [Google Scholar] [CrossRef]
- Yang, J.; Kang, H.; Lyu, L.; Xiong, W.; Hu, Y. A target map of clinical combination therapies in oncology: an analysis of clinicaltrials.gov. Discov Oncol 2023, 14, 151. [Google Scholar] [CrossRef]
Figure 1.
Model for PDIs catalyzing disulfide bond formation (oxidation), cleavage (reduction), or scrambling/exchange with no net.change in redox state.
Figure 1.
Model for PDIs catalyzing disulfide bond formation (oxidation), cleavage (reduction), or scrambling/exchange with no net.change in redox state.
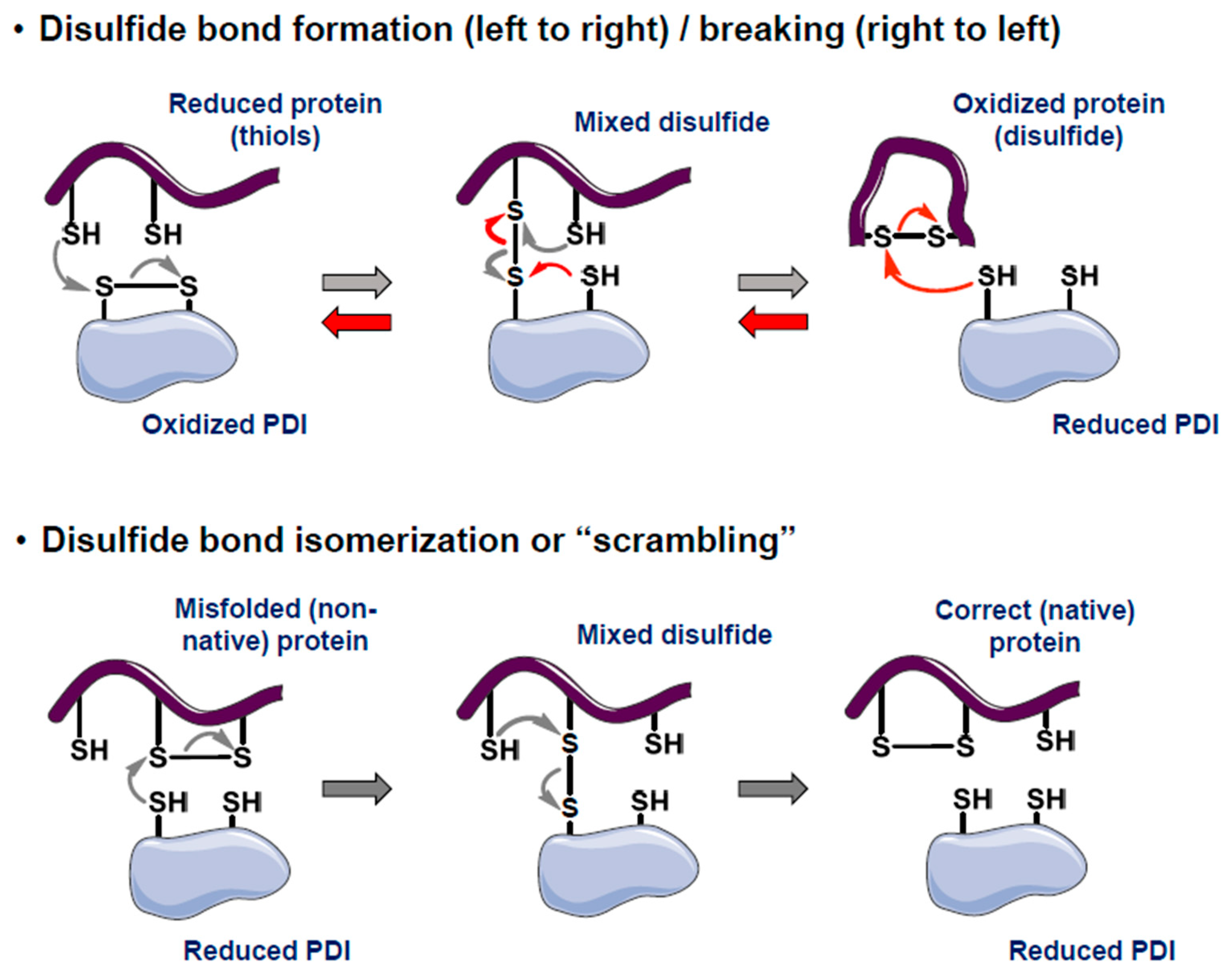
Figure 2.
A timeline showing the rational optimization of DDA structures guided by experimental and theoretical structureactivity and structure-property relationships. The most potent DDA for each generation is highlighted in dotted-line boxes. Proposed structures of fourth-generation DDAs bearing more complicated fluorinated motifs such as CF3 and OCF3 are also presented. Some of these DDAs are newly synthesized and currently undergoing biological evaluation.
Figure 2.
A timeline showing the rational optimization of DDA structures guided by experimental and theoretical structureactivity and structure-property relationships. The most potent DDA for each generation is highlighted in dotted-line boxes. Proposed structures of fourth-generation DDAs bearing more complicated fluorinated motifs such as CF3 and OCF3 are also presented. Some of these DDAs are newly synthesized and currently undergoing biological evaluation.
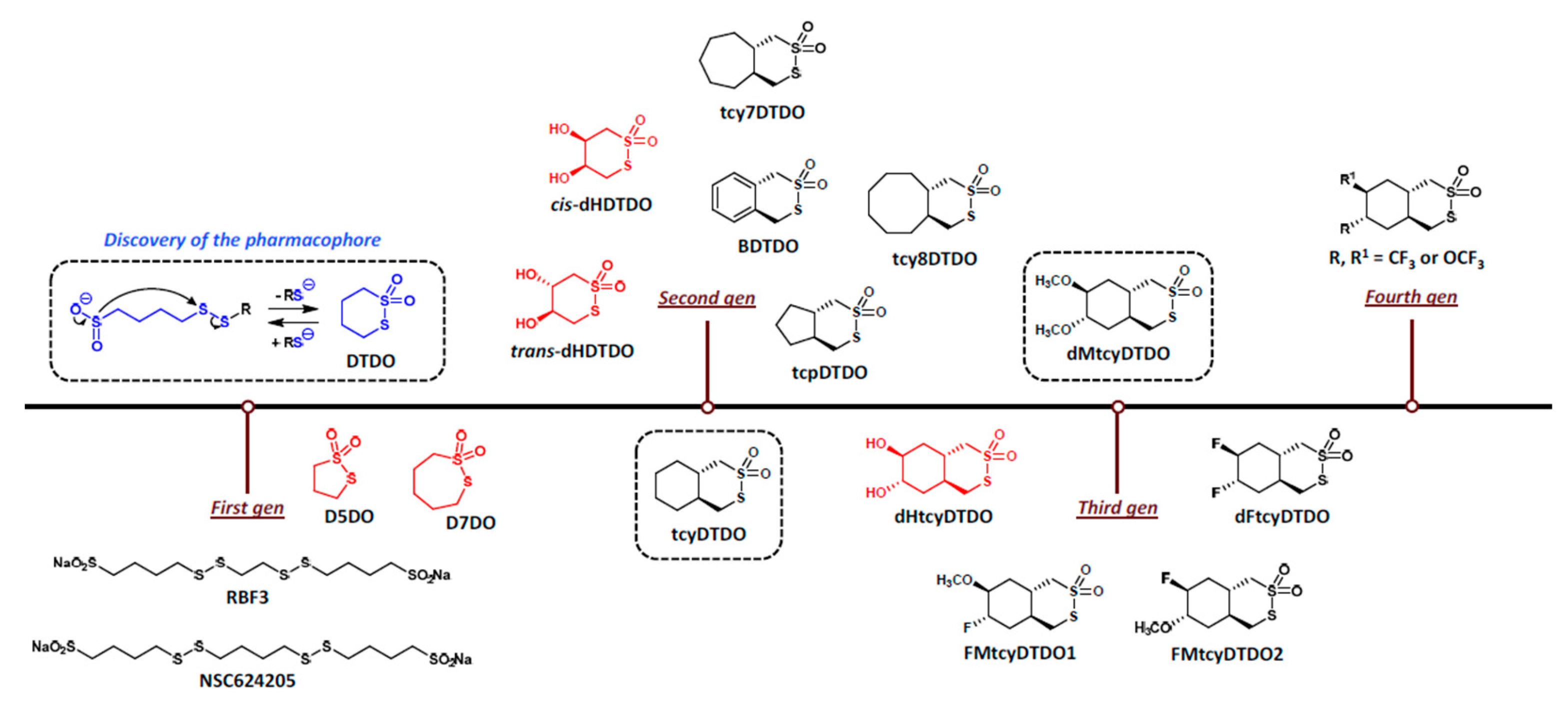
Figure 3.
DDA targets and their cancer dependencies, physical complexes with other PDIs, and co-dependencies on each other and KDEL receptors. A. DDA targets identified to date include the PDIs PDIA1, AGR2, AGR3, and ERp44. AGR2, AGR3, and ERp44 are “strongly selective” PDIs in the Broad Institute’s Map of Cancer Dependencies (DepMap at depmap.org). B. Summary of protein-protein interactions among AGR2, AGR3, ERp44, and other PDIs from BioGrid 4.4 (https://thebiogrid.org/). C. Co-dependencies among the strongly selective DDA targets with each other and the KDEL receptors that mediate ER retention. Arrows denote the direction of co-dependency. This information was obtained from the DepMap.
Figure 3.
DDA targets and their cancer dependencies, physical complexes with other PDIs, and co-dependencies on each other and KDEL receptors. A. DDA targets identified to date include the PDIs PDIA1, AGR2, AGR3, and ERp44. AGR2, AGR3, and ERp44 are “strongly selective” PDIs in the Broad Institute’s Map of Cancer Dependencies (DepMap at depmap.org). B. Summary of protein-protein interactions among AGR2, AGR3, ERp44, and other PDIs from BioGrid 4.4 (https://thebiogrid.org/). C. Co-dependencies among the strongly selective DDA targets with each other and the KDEL receptors that mediate ER retention. Arrows denote the direction of co-dependency. This information was obtained from the DepMap.
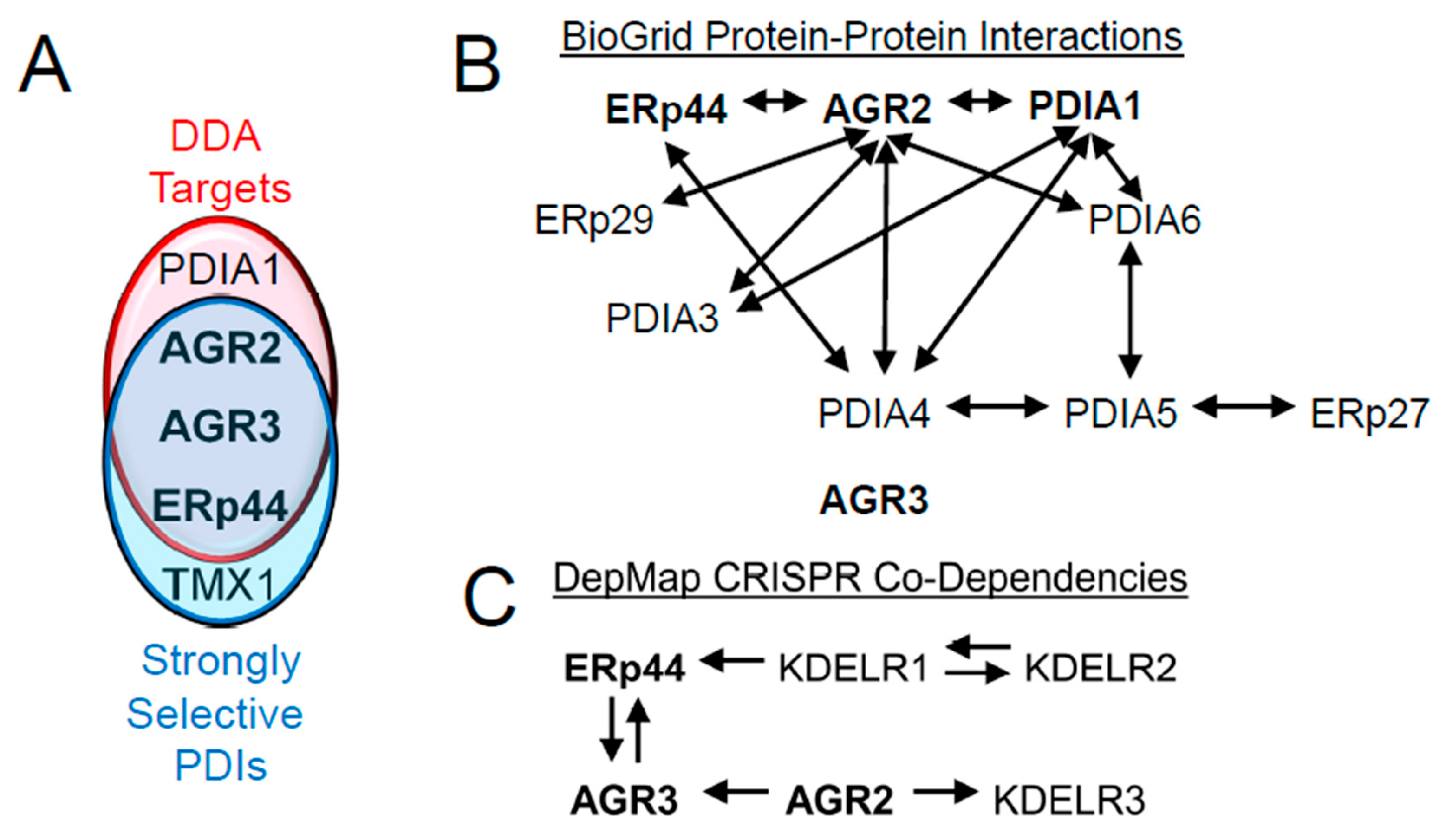

Table 1.
Human Protein Disulfide Isomerases.
| PDIA# | Alternate Names | Thioredoxin Repeat(s) | Enzymatic Motif | ER Retention | Strongly Selective in Broad DepMap (CRISPR; 12/24/24) |
Confirmed as DDA Targets | ||
| 1 | P4HB | CGHC, CGHC | + | -KDEL | - | + | ||
| 2 | PDA2, PDIp | CGHC, CTHC | + | -KEEL | - | - | ||
| 3 | ERp57 | CGHC, CGHC | + | -QEDL | - | - | ||
| 4 | ERp72 | CGHC, CGHC, CGHC | + | -KEEL | - | - | ||
| 5 | PDIR | CSMC, CGHC, CPHC | + | -KEEL | - | - | ||
| 6 | ERp5 | CGHC, CGHC | + | -KDEL | - | - | ||
| 7 | PDILT | SKQS, SKKC | - | -KEEL | - | - | ||
| 8 | ERp27 | - | - | -KTEL | - | - | ||
| 9 | ERp29 | - | - | -KEEL | - | - | ||
| 10 | ERp44, TXNDC4 | CRFS | + | -RDEL | + | + | ||
| 11 | TMX1 | CPAC | + | Transmembrane | + | - | ||
| 12 | TMX2 | SNDC | - | Transmembrane | - | - | ||
| 13 | TMX3 | CGHC | + | Transmembrane | - | - | ||
| 14 | TMX4 | CPSC | + | Transmembrane | - | - | ||
| 15 | TXNDC5, ERp46 | CGHC, CGHC, CGHC | + | -KDEL | - | - | ||
| 16 | TXNDC12, AGR1 | CGAC | + | -KTEL | - | - | ||
| 17 | AGR2 | CPHS | + | -KTEL | + | + | ||
| 18 | AGR3 | CQYS | + | -QSEL | + | + | ||
| 19 | DnaJ10, DnaJ | CSHC, CPPC, CHPC, CGPC | + | -KDEL | - | - | ||
| PDIB1 | Calsequestrin 1 | - | - | - | - | - | ||
| PDIB2 | Calsequestrin 2 | - | - | - | - | - | ||
| TMX5, TXNDC15 | CRFS | + | Transmembrane | - | - | |||
Table 2.
Strongly AGR2-Dependent Human Cancer Lines (DepMap; CRISPR).
| AGR2 Selectively Dependent Cancer Line |
AGR2 Essentiality Rank (out of 17,347 genes) |
Tumor Type | Notes on top 10 essential genes: Gene essentiality rank is in parentheses. Fractions denote the number of top-10 most essential genes involved in Wnt signaling. |
|---|---|---|---|
| C80 | 1 | Colorectal | CDX2 (2) |
| COLO205 | 4 | FOXP4 (2), FOXA2 (10) | |
| LS513 | 3 | CDX2 (1), CDX1 (6) | |
| SNUC4 | 5 | 2/10 Wnt signaling | |
| MAPACHS77 | 2 | Pancreatic | 7/10 Wnt signaling |
| HSC39 | 7 | Esophagogastric | |
| 2313287 | 9 | Gastric | 3/10 Wnt signaling |
Table 3.
Top AGR2 Co-Dependencies (DepMap; CRISPR).
| Rank | Gene |
|---|---|
| 1 | SOX9 |
| 2 | TTC7A |
| 3 | RAB10 |
| 4 | CTNNB1 (b-catenin) |
| 5 | AGR3 |
Table 4.
Top Predictors of AGR2 Dependency (DepMap; CRISPR).
| Rank | Gene | Importance | Corr. Type |
|---|---|---|---|
| 1 | ERN2 (IRE1b) | 23.1% | Expression |
| 2 | SH3BGRL2 | 2.1% | Expression |
| 3 | LRRC19 | 2.1% | Expression |
| 4 | RAB31 | 1.2% | Expression |
| 5 | NOX1 | 1.1% | Expression |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



