Submitted:
08 May 2025
Posted:
09 May 2025
You are already at the latest version
Abstract
The increasing market demand and rising prices of raw materials have heightened interest in renewable and sustainable sources. Consequently, the production of building block chemicals from natural products or synthetic consumables has shifted scientific research towards catalytic strategies for the depolymerization of these materials. Polymer chemistry presents significant opportunities for recycling, as the synthesis of polymers typically begins with monomers. Emerging non-destructive techniques enable the recovery of these starting reagents. This review summarizes recent advancements in catalytic methods for the depolymerization of polymers derived from both natural sources, such as cellulose and lignin, and synthetic sources, including conventional plastics. The review is organized into three sections: catalytic depolymerization of cellulose, catalytic depolymerization of lignin, and catalytic depolymerization of plastics. Particular emphasis is placed on recent studies exploring innovative techniques. The raw materials obtained through these strategies can be reintegrated into the production cycle, supporting a fully circular economy.
Keywords:
circular economy
; catalysis
; sustainable chemistry
; polymers
1. Introduction
The overproduction of new materials has led to the exploitation of fossil resources at unprecedented levels, resulting in irreversible effects on ecosystems [1,2]. This shortage of building block chemicals has caused a rise in market prices [3,4]. In recent decades, to address these challenges, both academia and industry have increasingly invested in more sustainable resources and related methodologies [5,6]. Today, sustainability is understood as a concept encompassing not only economic factors but also environmental considerations [7,8]. Polymers, both natural and synthetic, are among the most abundant materials on Earth [9,10]. Biomass, in particular, has gained significant attention over the past few decades as it has been transformed from a natural resource or waste product into a high-potential raw material for the production of chemicals, materials, and energy [11,12,13]. Cellulose, a natural glucose reservoir in plants, has been chemically functionalized over the years to produce materials suitable for various applications [14,15,16]. One of the most extensively studied and intriguing research areas involving this polymer is its depolymerization, a topic that continues to attract significant interest [17,18,19]. Depolymerization breaks the polymer chain into glucose, the fundamental building block, initiating cascade reactions that yield what are now known as platform molecules [20,21,22]. These platform molecules have demonstrated broad application potential and are emerging as viable or even superior alternatives to their fossil-based counterparts [23,24,25]. Lignin, the other primary component of biomass, was historically underutilized but is now regarded as a key resource with promising future potential [26,27,28]. This shift in perspective is due to its status as an underexplored sustainable resource and its abundance as a waste product, particularly from the paper manufacturing industry [29,30,31]. Currently, lignin serves as a renewable source of aromatic compounds, which are of great interest for biofuels and the chemical industry [32,33]. Despite being an aromatic polymer composed of cross-linked phenolic units derived from phenylpropane, with a composition that varies depending on the plant type and other factors [34], lignin can also be used to produce aliphatic molecules such as linear and cyclic alkanes, which were traditionally sourced exclusively from petroleum [35,36]. The biopolymers discussed thus far represent an inexhaustible resource when utilized sustainably, with substantial untapped potential. Despite these advances, fossil-derived raw materials remain widely used due to economic factors, including market demand, as well as familiarity with the properties and applications of the resulting materials. Annually, over 380 million tons of polyethylene terephthalate (PET) are produced globally. PET, commonly used in everyday products, is primarily synthesized from terephthalic acid and ethylene glycol, both historically derived from fossil resources [37]. Other fossil-based plastics that are widely used include polyvinyl chloride (PVC), polyethylene (PE), polystyrene (PS), polypropylene (PP), and many others [38,39,40]. Modern research is increasingly focused on replacing fossil-derived chemicals with sustainable alternatives or developing methods to produce them [41,42]. In particular, chemical depolymerization using eco-friendly catalysts and sustainable strategies is emerging as a promising approach to regenerate the monomers of these plastics, enabling their reintegration into a circular economy, including applications in food packaging [43,44,45]. This review is structured into three sections, each detailing the latest innovations in catalytic methods for the depolymerization of cellulose, lignin, and commonly used plastics. The selected studies highlight strategies that aim to merge the principles of sustainable chemistry with those of green chemistry. The discussion also emphasizes the monomers produced, which not only allow the reproduction of the same materials for a reuse economy but also offer potential for alternative applications.
2. Catalytic Depolymerization of Cellulose
Cellulose is an insoluble aggregate composed of D-glucose units linked by β-1,4-glycosidic bonds, forming a structure with both crystalline and amorphous regions [46,47]. The extensive network of intra- and intermolecular hydrogen bonds results in a fully cross-linked molecular architecture, contributing to its insolubility in most solvents. This unique structural composition also presents significant challenges for the depolymerization of cellulose. The sugars derived from cellulose depolymerization are crucial precursors for various biofuels and valuable chemicals, including ethylene, ethanol, glycerol, lactic acid, glycol, and levulinic acid. Several methods are available for converting cellulose into glucose, with enzymatic hydrolysis and catalytic hydrolysis, using either liquid or solid acid catalysts, being the most widely studied approaches. Enzymatic hydrolysis is particularly promising due to its mild operating conditions and high selectivity, enabling the efficient extraction of fermentable sugars from diverse biomass sources. Notably, enzymatic methods exhibit high selectivity for glucose. However, challenges associated with enzyme recycling, high costs, and low efficiency limit the broader application of enzymatic hydrolysis.
Mineral and organic acids, including H2SO4, HCl, acetic acid, and oxalic acid, have been extensively employed as catalysts for the catalytic hydrolysis of cellulose [47,48]. However, these acids present several challenges: (i) difficulties in separating and recovering the acid from the sugar products, (ii) the need for wastewater treatment, (iii) equipment corrosion, and (iv) reduced sugar yields due to the degradation of sugars into 5-hydroxymethylfurfural (HMF), furfural, and levulinic acid (LA) under high-temperature conditions [48,49]. To address these limitations, solid acid catalysts have emerged as promising alternatives for cellulose hydrolysis. Compared to conventional liquid acids, solid acid catalysts offer easier recovery and reuse, enhancing sustainability and process efficiency.
2.1. Solid Acid Catalyst Depolymerization of Cellulose
Solid acid catalysts play a crucial role in the depolymerization of cellulose and the conversion of biomass into valuable chemicals and biofuels. Compared to homogeneous acids such as H2SO4 and HCl, solid acid catalysts are preferred due to their recyclability, reduced corrosiveness, and enhanced selectivity in product formation. Notable examples include Amberlyst 15, 35, and 70, as well as Nafion and zeolite Y, which are particularly effective for cellulose hydrolysis in ionic liquids. Amberlyst 15 DRY, a sulfonated polystyrene resin, is highly efficient at hydrolyzing cellulose into glucose due to its strong Brønsted acidity and reusability. Additionally, Amberlyst resins exhibit stability in acidic anion ionic liquids, whereas they are prone to rapid degradation in basic anion ionic liquids, such as 1-butyl-3-methylimidazolium acetate. Zeolites, including HZSM-5, provide both Brønsted and Lewis acid sites, making them suitable for isomerization and hydrolysis reactions. Similarly, metal oxides like sulfated zirconia are valuable for high-temperature reactions because of their thermal stability and tunable acidity [50,51,52]. Ionic liquids, such as 1-Allyl-3-methylimidazolium Chloride (AMIMCl), can dissolve cellulose at concentrations of up to 25 wt% [53]. Their unique solvent properties make ionic liquids particularly effective for cellulose depolymerization.
In a notable study, Pang et al. [49] explored the use of ionic liquids and solid acids to process cellulose fibers from various agricultural byproducts, including sugarcane bagasse, sago pith, sawdust, and corn cobs. They prepared a 5% cellulose solution by dissolving these fibers in AMIMCl. Introducing a small amount of Amberlyst catalyst initiated the depolymerization process by facilitating ion exchange with the ionic liquid, generating H3O+ ions. These ions were crucial for cleaving the β-glycosidic bonds in cellulose, leading to the formation of cello-oligomers and byproducts such as reducing sugars and glucose. This innovative approach demonstrates a promising strategy for extracting valuable components from natural biomass (see Scheme 1).
The experiment demonstrated that the degree of polymerization by weight (DPw) of the resulting cello-oligomers was significantly influenced by the initial DPw values of the cellulose fibers subjected to depolymerization. Specifically, cellulose fibers from corn cobs and sawdust, with initial DPw values of approximately 351, produced cello-oligomers with average DPw values of 88 and 72, respectively, after a reaction time of about 40 minutes (Table 1) [49].
In contrast, fibers derived from sugarcane bagasse and sago pith, which had higher initial mean DPw values of 571 and 564, yielded lower DPw values of 37 and 30 for the cello-oligomers after longer reaction times of 60 and 100 minutes, respectively. Notably, sago pith fibers depolymerized at a faster rate than those from sugarcane bagasse. This accelerated depolymerization may be attributed to the more uniform distribution of DPw within the cellulose fibers from sago pith [49].
Although solid acid catalysts offer several advantages, their effectiveness in cellulose hydrolysis is hindered by the high crystallinity of cellulose, which limits the catalyst’s access to glycosidic bonds. This restricted accessibility impedes efficient glucose production, posing a significant challenge for biorefineries that depend on sugar platforms. A promising approach to overcoming this limitation involves the pretreatment of cellulose using molten salt hydrates (MSH) [54]. In a recent study by Paiva et al. [55], cellulose conversion was carried out using a Radleys Carousel Reactor. In a typical experimental setup (Figure 1), 0.05 g of cellulose was combined with either 0.05 g or 0.025 g of catalyst in a solution containing 2.5 g of ZnCl2, maintaining a constant molar ratio of salt to H2O at 3.0.
The mixture was placed in a reaction flask with a magnetic stir bar and heated in the Radleys Carousel at 70 or 90 °C, with continuous stirring at 600 rpm for 2 to 5 hours. After the reaction, the flask was cooled using an ice bath. The catalysts were separated by centrifugation, and 0.3 g of the hydrolysate was diluted tenfold for product analysis [55].
Table 2 summarizes the conversion of cellulose using AMSH (acidic molten salt hydrate) in the presence of various catalytic agents, highlighting the different product yields obtained. Notably, when compared to conventional inorganic acids like H2SO4, the use of lithium bromide (LiBr) acidified MSH as a mesoporous solid hydrolysis agent at 85 °C resulted in a significantly higher glucose yield [56]. Additionally, elevated temperatures in the presence of ZnCl2 promoted substantial production of 5-hydroxymethylfurfural (HMF) [57]. When HZSM-5 (SiO2/Al2O3) was used at 90 °C for 2 hours, the glucose yield was slightly lower than that achieved with the SO42−/TiO2 catalyst [58].
Cellulose conversion using LiBr at elevated temperatures demonstrated variable glucose yields depending on the catalyst and conditions. Specifically, at 120 °C, Beta and ZSM-5 zeolites produced 28% glucose, whereas activated carbon at 110 °C yielded 80% glucose (Liang et al., 2024; Wu et al., 2020). At higher temperatures (175 °C), the use of NbOPO4, Nb2O5, NbOPO4/HZSM-5, and HZSM-5 resulted in 85.1% levulinic acid (LA) and 5% 5-hydroxymethylfurfural (HMF) [59]. Romeo and co-workers [17] developed an interesting green process to convert microcrystalline cellulose into bio-oil and cellulose citrate using an open-air reaction, catalyzed by molten citric acid. The molten carboxylic acid was able to penetrate into the less accessible areas of the cellulose by breaking the network of intra and inter-molecular bonds and promoting the simultaneous hydrolysis into bio-oil and esterification into cellulose citrate.
2.2. Mechanocatalytic Depolymerization of Cellulose
Mechanocatalytic depolymerization of cellulose combines mechanical energy, such as ball milling, with catalytic action to break down cellulose into smaller molecules. This approach disrupts the crystalline structure of cellulose, enhancing its reactivity while lowering energy requirements. Solid acid catalysts, including sulfonated carbon materials (e.g., CMK-3-SO3H) and perfluorinated ionomers (e.g., Aquivion), significantly improve the efficiency of mechanocatalytic depolymerization. This method is environmentally friendly, reducing the need for harsh chemicals and extreme conditions. Key factors influencing its efficiency include milling speed, catalyst properties, and moisture content [60,61]. Zirconia-based catalysts, such as sulfated zirconia, exhibit high acidity, thermal stability, and resistance to deactivation, making them particularly effective for enhancing hydrolysis efficiency. These catalysts can be used alone or in combination with other solid acids to improve cellulose breakdown. In the study by Karam et al. a planetary ball mill (Retsch PM 100) equipped with a zirconia grinding bowl was employed [62]. Depending on the experiment, zirconia, stainless steel, or tungsten carbide grinding balls with diameters of 2 mm, 3 mm, or 4 mm were used. To ensure effective grinding, a 125 mL zirconium oxide bowl was used in combination with twenty 10 mm balls made of the same zirconium oxide material as the bowl (see Figure 2) [62].
Karam et al. subsequently removed the cellulose-milled catalyst mixture to evaluate the solubility of all milled samples. The solubility measurement involved three distinct steps: dispersion, filtration, and drying [62].
As shown in Table 3, the solubility data obtained from the study by Karam et al. demonstrated that less than 5% solubility was achieved for microcrystalline cellulose (MCC) after 24 hours of ball milling under non-catalytic conditions [62]. However, water solubility significantly increased when unmodified MCC, treated in a planetary ball mill, was directly subjected to various solid acid catalysts. This suggests that the inherent acidic properties of solid acid catalysts played a pivotal role in disrupting the β-1,4 glycosidic bonds of cellulose during the mechanical milling process [62].
According to Table 3, high water solubility was achieved for MCC using the Aquivion PW98 catalyst, reaching approximately 90%, followed closely by the CMK-3-SO3H catalyst at about 87%. In contrast, the SBA-SO3H catalyst yielded a lower water solubility of around 60%. Kaolinite was the least effective catalyst, producing a product with only 50% water solubility [62]. Significant differences in performance were observed among the Aquivion catalysts tested. Aquivion PW66 exhibited the highest solubility at approximately 99%, followed by PW98 at 90%, PW87 at 80%, and PW79 at 32%. Further investigations were conducted to explore the effect of ball milling duration and the catalytic efficiency of Aquivion PW66 and PW98, as they were the most effective catalysts in the initial water-soluble synthesis using MCC. The results showed that longer milling times considerably enhanced water solubility, achieving 90% with Aquivion PW98 and an exceptional 99% with PW66 after 24 hours [62].
Table 4 provides detailed information on the grinding balls used in various applications reported in the literature [63,61].
The first report in the scientific literature on mechanocatalytic degradation of cellulose demonstrates that effective depolymerization occurs when solid material is processed in a dry state. In a study utilizing a high-energy shaker mill, the introduction of kaolinite, a clay mineral, yielded high amounts of water-soluble products, including glucose, fructose, and anhydroglucose (levo-glucose). This innovative approach highlights the potential of mechanochemical reactions for synthesizing valuable sugars from cellulose [63]. In their study, Hick et al. proposed that ball milling induces delamination of layered compounds, thereby exposing surface acid sites. Although the authors did not provide detailed product distributions for the resulting monosaccharides and oligomers, they suggested that the layered structure and acidity of the solid additive play crucial roles in the efficient mechanochemical degradation of cellulose [63]. Another promising approach involves the use of layered HNbMoO6, known for its unique layered structure and high acidity, which collectively enhance its catalytic efficiency in various applications [61]. Furusato et al. demonstrated that HNbMoO6 effectively catalyzes the mechanochemical depolymerization of cellulose due to its strong acidity, favorable layered structure, and exceptional catalytic performance in acid-catalyzed reactions such as hydrolysis, esterification, and dehydration. Notably, HNbMoO6 produced water-soluble sugars at a rate three times higher than kaolinite, achieving a high yield of 72% upon complete conversion within 24 hours. Table 5 presents the results reported by Furusato et al. regarding the mechanocatalytic depolymerization of cellulose [61].
All monometallic oxides tested, including NiO, SiO2, TiO2, and Nb2O5, showed no catalytic activity. The two-dimensional material, acid-treated kaolinite, produced 4% water-soluble sugars, while montmorillonite yielded 3%. Among the tested solid catalysts, the layered acid catalyst HNbMoO6 achieved a 14% yield of water-soluble sugars in a solvent-free reaction, outperforming other solid catalysts. Conversely, the Mg-Al hydrotalcite catalyst was ineffective for this reaction. The USY zeolite exhibited moderate activity, yielding 3%, comparable to montmorillonite [61]. The study also revealed that the number of zirconia spheres used significantly influenced the total sugar yield. When one or two zirconia spheres were used, the total sugar yield was below 1%, attributed to inadequate collisions as the spheres primarily moved along the walls of the zirconia vessel. However, increasing the number of spheres to more than four enhanced mechanical energy through more frequent collisions, resulting in sugar yields of 12–14%. The highest water-soluble sugar yield of 72% was achieved after 24 hours of grinding at 800 rpm and room temperature using layered HNbMoO6, with a 99% cellulose conversion based on weight loss. This yield was notably higher than the 1% yield obtained without a catalyst [61]. Yu et al. recently employed an acid-assisted mechanocatalytic depolymerization technique to pretreat rice straw, leading to significant structural changes in cellulose. Compared to untreated straw, the pretreated straw exhibited much smaller particle sizes (from 279 to 11.8 µm), reduced cellulose crystallinity (from 43.05% to 22.71%), a substantially larger surface area (an increase of 177%), and 75% more surface oxygen relative to carbon [64]. Enzymatic hydrolysis of the pretreated straw over 12 hours resulted in pronounced microstructural alterations, yielding a total sugar amount exceeding 95%. This yield was significantly higher than that of untreated straw (36.24%) and also surpassed the yields from acid-impregnated straw (45.20%) and ball-milled straw (73.25%). Consequently, acid-assisted mechanocatalytic depolymerization proved to be a highly effective pretreatment method, significantly enhancing enzymatic hydrolysis and overall conversion efficiency of rice straw [64].
3. Catalytic Depolymerization of Lignin
Lignocellulose is made up of different constituents such as lignin, hemicellulose, cellulose, proteins and oils. Lignin comprises of phenylpropanoid units that is cross-linked via ether and carbon-carbon bonds (see Figure 3). Historically, lignin was considered a low-value byproduct. However, the lignin market, valued at approximately USD 836.8 million in 2023, is projected to grow at a rate of 6% from 2024 to 2032. But only a small fraction (1–2%) of lignin is currently utilized. The complex structure of lignin, rich in polyaromatic compounds, makes it an excellent precursor for the production of high-value chemicals and materials [65]. Lignin adopts a helical configuration and is polymerized from three distinct phenylpropane monomeric units: p-coumaryl alcohol (H), coniferyl alcohol (G), and sinapyl alcohol (S) (see Figure 3). These phenolic monomers are interconnected through ether and carbon-carbon (C–C) linkages. The primary linkages include β-O-4, 4-O-5, 5-5, β-5, β-1, and β-β, with β-O-4 being the most prevalent, constituting approximately 45% to 60% of the total structure. In contrast, β-5 and 5-5 linkages are the next most abundant, comprising 5% to 25% of the structure. The effective depolymerization of lignin into its constituent phenolic monomers is predominantly dependent on the selective cleavage of the β-O-4 linkages [68].
Lignin valorization into value-added products has gained significant attention. However, due to its complex structure, the depolymerization of lignin requires high energy (approximately 500 °C) and high pressure (approximately 200 bar). In comparison, catalytic depolymerization of lignin offers a more promising approach, as it facilitates the breakdown of the polymeric rings into smaller fractions under milder conditions [69,70].
3.1. Challenges and Opportunities
Lignin, a complex and recalcitrant biopolymer, is a major component of plant cell walls and represents a significant source of renewable aromatic compounds. The heterogeneity and irregular structure of lignin, resulting from the random polymerization of phenylpropanoid units, complicates selective bond cleavage [71]. Various C–C and C–O linkages require highly specific catalytic strategies, rendering the process energy-intensive and inefficient [72]. Moreover, structural variations among lignin types, such as: softwood, hardwood, and grass lignin, necessitate tailored depolymerization approaches, further complicating large-scale processing [73]. Despite these challenges, recent advancements offer promising opportunities for efficient lignin depolymerization. Catalytic hydrogenolysis, oxidative depolymerization, and biocatalytic methods are among the most extensively studied techniques [71]. Metal-based catalysts, including those containing Ru, Pd, or Ni, have demonstrated the potential to break down lignin into valuable monomers with high selectivity [74]. Additionally, engineered microbial pathways and enzyme-based strategies are gaining attention due to their potential to enable mild and sustainable lignin valorization [72]. Advances in solvent-based fractionation techniques, such as deep eutectic solvents and ionic liquids, also present promising solutions to enhance lignin solubility and reactivity [75]. The integration of lignin depolymerization processes into existing biorefineries is essential for developing a sustainable bioeconomy. Future research should focus on optimizing catalysts, developing scalable biotechnological methods, and enhancing process economics to make lignin valorization commercially viable. Overcoming these challenges could position lignin as a valuable feedstock for bio-based chemicals, materials, and fuels, thus supporting a circular bioeconomy [71,74].
3.1.1. Homogeneous-Acid Catalysis
Strong acids, such as hydrochloric acid, sulfuric acid, p-toluenesulfonic acid, and phosphoric acid, are commonly employed in homogeneous lignin depolymerization (see Figure 4) [76].
These acids are effective due to their ability to attack ether bonds in lignin, facilitating the cleavage of linkages such as β-O-4 and 4-O-5 through various mechanisms, ultimately leading to the formation of phenolic compounds [77]. The protonation of the ether oxygen also leads to the formation of carbocations, which undergo rearrangements, yielding a complex mixture of products. The efficiency of acid-catalyzed lignin depolymerization is influenced by several factors, including temperature, catalyst concentration, reaction duration, and solvent choice, all of which significantly affect product distribution [70]. Higher acid concentrations enhance the reaction rate by increasing the rate of protonation and nucleophilic attacks. Elevated temperatures reduce activation energy, thereby promoting faster depolymerization rates. However, higher temperatures also increase the likelihood of side reactions, leading to undesired byproducts. Longer reaction durations ensure the comprehensive breakdown of lignin’s complex structure but may also enhance the formation of secondary products. While strong acid catalysis is effective in achieving high depolymerization rates, optimizing the reaction parameters is crucial to maximizing yield and selectivity while minimizing side reactions [78]. Homogeneous acid catalysts are highly active and efficiently depolymerize the complex lignin structure at relatively low cost [79]. However, several limitations hinder their practical application. One major drawback is their low selectivity, which leads to the formation of complex mixtures of byproducts, including phenolic monomers, dimers, oligomers, and various other degradation products. Additionally, the process often requires harsh reaction conditions and involves the use of toxic chemicals, complicating catalyst recovery and increasing purification costs. These factors pose challenges to achieving a sustainable and eco-friendly lignin valorization approach [70]. Moreover, the use of strong acids results in corrosive environments, necessitating specialized equipment and safety measures to mitigate corrosion-related issues. These limitations underscore the challenges associated with homogeneous acid catalysis in lignin depolymerization [70].
3.1.2. Homogeneous- Base Catalysis
Base-catalyzed depolymerization is a commonly used method for extracting phenolic monomers from lignin. This process employs various bases, including calcium hydroxide (Ca(OH)2), sodium hydroxide (NaOH), potassium hydroxide (KOH), and organic bases such as tetramethylammonium hydroxide [80,81]. In this method, the base catalyst provides hydroxyl ions (-OH−), which initiate depolymerization by attacking the phenolic hydroxyl group (-C6H5OH), leading to the removal of a proton (-H+) and the formation of a negatively charged phenoxide ion (-C6H5O−). This phenoxide ion subsequently attacks the β-carbon in the ether linkage, forming a new bond with the phenoxide oxygen. This reaction results in the production of aromatic aldehydes and ketones [82]. Base-catalyzed depolymerization is considered one of the most promising methods due to its high catalytic efficiency at relatively low temperatures. However, it also has certain limitations. Notably, the choice of solvent is critical in base-catalyzed lignin depolymerization, as solvent selectivity significantly influences reaction outcomes. For example, conducting depolymerization in water with a low concentration of base catalyst typically results in minimal depolymerization, occasionally yielding negligible amounts of depolymerized products. This indicates that an aqueous medium is not suitable for facilitating this reaction [83,84]. In a recent work by Bijoy Biswas and co-workers [85] an interesting depolymerization of alkali lignin into phenolic chemicals was developed using a solid base catalysis. Different catalysts were tested, such as: CaO/CeO2, CaO/Al2O3, and CaO/ZrO2. At a process temperature of 180 °C, a screening of different solvents was performed. Ethanol and methanol gave a bio-oil yield of 50%, over CaO/CeO2 and CaO/ZrO2 catalysts. Vanillin was identified as the major component of the bio-oil after the catalytic liquefaction step, and the authors attributed this to the action of the basic catalyst enhancing the β-O-4 cleavage producing both an increase in the bio-oil yield and an increase in the vanillin yield, how schematized in the following Scheme 2.
Combined Acid-Base Catalysis
Interestingly, in the recent years the combination of acid and base was proved to be efficient for lignin depolymerization. Xia Zhang and co-workers [86] recently developed an interesting procedure for the depolymerization of Kraft lignin into liquid fuels over a WO3 modified acid-base coupled hydrogenation catalyst. In this kind of processes, the activation of C-C and C-O bonds together with the stabilization of the produced intermediates is essential for the efficiency of the system. In this study, The Lewis acid sites WOx and TiO2 in combination with the base sites MgO were proved to be efficient for the adsorption, stabilization, polarization and activation of the C-O and C-C lignin bonds. NiO sites are instead essential for the stabilization of reaction intermediates through the activation of molecular H2. Selective hydrodeoxygenation, at 300 °C for 24 hours furnished 63.03% of petroleum ether solubles and 21.56% of monophenol. This study opens new perspectives toward the development of mild, sustainable and efficient conversion of lignin.
3.1.3. Homogeneous- Metal Catalysis
The most commonly used catalysts in lignin depolymerization are metal catalysts due to their diversity, design flexibility, and outstanding catalytic efficiency. Various metals have been utilized, including transition metals, noble metals, and bimetallic catalysts, such as ruthenium (Ru), palladium (Pd), platinum (Pt), copper (Cu), nickel (Ni), and iron (Fe) [87,88,89]. Metal catalysts facilitate the hydrogenolysis reaction and activate hydrogen (H2), leading to the formation of aromatic monomers. They promote the cleavage of C–O and C–C bonds in lignin through multiple mechanisms, including hydrogenolysis, oxidation, and solvolysis:
Hydrogenolysis involves the cleavage of bonds by the addition of hydrogen (H2), resulting in the formation of smaller molecules.
Oxidation entails the removal of electrons from a molecule, leading to the formation of carbonyl groups (C=O) and other oxidized byproducts.
Solvolysis involves the cleavage of bonds through reaction with a solvent, producing smaller molecular fragments.
These mechanisms underscore the versatility and effectiveness of metal catalysts in lignin depolymerization [90]. Ligands attached to transition metal catalysts can significantly modify their catalytic properties by influencing electronic and steric effects. The efficiency of transition metal catalysts in lignin depolymerization varies due to several factors, including temperature, pressure, and solvent choice, all of which can impact reaction rate and product selectivity [91]. Metal catalysts offer several advantages, such as high activity and selectivity for specific products. They can operate under mild reaction conditions and can be supported on various material surfaces, facilitating easy recovery and reuse. However, metal catalysts also have certain limitations. Their primary drawbacks include high cost and the requirement for elevated temperatures and activation pressure to achieve effective depolymerization. Despite these challenges, their high efficiency and selectivity make them valuable for lignin valorization [92]. Florian Walch and co-workers [93] discovered an efficient technology for the oxidative depolymerization of Kraft lignin into aromatics compounds using homogenous vanadium-copper catalyst. The authors used molecular oxygen to produce aromatic compound monomers such as vanillin, vanillic acid and acetovanillone. The overall bio-oil yiled reached 50% under optimized reaction conditions with a yield in aromatic compounds of 27%. The representative scheme of the process is depicted in the following Scheme 3.
In a similar work Omar Y. Abdelaziz and co-workers [94] developed an oxidative kraft linin depolymerization using bimetallic V-Cu/ZrO2 catalysts. This zirconia-supported vanadium–copper catalysts was proved to be efficient for the conversion of softwood LignoBoost Kraft lignin (LB), with a maximum monomer yield of 9% weight and a maximum selectivity in vanillin of 59%. Copper and vanadium were confirmed promising homogeneous catalysts for this kind of applications.
3.1.4. Heterogeneous – Solid acid Catalysis
Heterogeneous solid acid catalysis, including sulfonates, metal oxides, zeolites, heteropoly acids, and phosphates, is widely used for lignin depolymerization. These catalysts promote the cleavage of C–O bonds in lignin, leading to the formation of phenolic compounds [95,96,97]. The solid acid catalyst provides a proton (H+) to facilitate the reaction. The reaction mechanism begins with the protonation of oxygen in the ether bond (Lignin–O–R), increasing the bond’s susceptibility to cleavage. The C–O bond then breaks, transferring the proton to the oxygen and forming a hydroxyl group (-OH). This process yields two products: a phenolic compound or phenolic hydroxyl group (Lignin–OH) and a carbocation (R+). If R is an alkyl group, it forms an alkyl carbocation, whereas if R is aryl, it forms a protonated aromatic compound. These reactive intermediates can undergo further transformations depending on the reaction conditions, potentially leading to alcohol formation (R–OH) when reacting with water molecules. They may also participate in rearrangements or other secondary reactions [98,99]. A key advantage of solid acid catalysts is their regenerability; the H+ ion is released post-reaction, allowing the catalyst to catalyze successive cycles. This enhances their economic and environmental sustainability [100]. A prevalent linkage in lignin is the β-O-4 ether linkage. Its cleavage generates aromatic groups (Ar), and the resulting carbocation intermediates (R+ or CH2=Ar+) are highly reactive, leading to a variety of products. This underscores the potential of solid acid catalysts for efficient lignin depolymerization [101]. One of the most interesting challenges in this area is selective depolymerization. Catalysis plays a key role in this direction. Hongwei Ma et al. described an in-depth investigation of lignin depolymerization catalyzed by nickel supported on zirconium phosphate [102]. Starting from the organosolv lignin via non-noble metallic nickel supported on zirconium phosphate (Ni/ZrP), they achieved about 87% lignin conversion and less than 5% char formation, using 15%Ni/ZrP-2.0 at 260 °C for 4 hours. Furthermore, the yield of phenolic monomers was demonstrated to be about 15% at a hydrogen pressure of 2 MPa. A considerable yield of para ethyl phenol was obtained demonstrating the great potential of this technique also in the future perspective.
In another recent work Zhuang Li and co-workers [103] developed a hydrogen-free strategy using solid acid catalysis to selective depolymerization of lignin into guaiacol. The authors hypothesized, supported by experimental evidence, the breaking of the Car-Cα and Cβ-O bonds. In a 90% aqueous methanol solution, a guaiacol yield of 18.2% was demonstrated. The mechanism was hypothesized by combining experimental evidence with density functional theory. The authors highlighted how there is an interesting action of water, which promotes the decomposition of methanol to produce more reactive hydrogen species, thus favoring the Car-Cα bond cleavage.
3.1.5. Heterogeneous – Metal-supported Catalysis
Metal-supported catalysts are composed of metal nanoparticles deposited on various catalytic supports. This design enhances dispersibility and modifies the physicochemical properties of the catalyst, thereby optimizing its catalytic potential. Several physical and chemical attributes, including high surface area, variable valencies, tunable acidity and basicity, structured porosity, topological arrangement, and exceptional thermal stability, contribute to their effectiveness. Additionally, their sustainability and cost-efficiency make them highly suitable for lignin depolymerization. Common catalytic supports include silica (Si), activated carbon (C), alumina, and zeolites [104]. These metal-supported catalysts exhibit high activity and stability, facilitating the hydrogenolysis of lignin. This process effectively breaks down the complex polymer, leading to the formation of aromatic monomers. The general reaction for lignin hydrogenolysis using metal-supported catalysts is represented as follows:
Lignin + Hydrogen (H2) → Smaller molecules (e.g. aromatic monomers)
The mechanism of lignin hydrogenolysis using metal-supported catalysts involves the adsorption of both lignin and hydrogen molecules on the catalyst’s surface. The nano-sized metal activates the hydrogen molecules, enhancing their reactivity. These activated hydrogen molecules facilitate the cleavage of C–O and C–C bonds within lignin, resulting in the formation of smaller molecular fragments. These smaller molecules subsequently desorb from the catalyst surface, regenerating the catalyst for subsequent reaction cycles [105]. The efficiency of lignin hydrogenolysis using metal-supported catalysts is influenced by several factors. The type of metal is crucial, as different metals exhibit varying catalytic activities and selectivities for specific bond cleavages. The concentration of nano-sized metal on the support directly affects the reaction rate and selectivity, making the decoration and distribution of metal on the support critical. The support material itself significantly impacts the dispersion of metal nanoparticles and the overall catalytic activity. An inert support is essential to avoid unwanted side reactions. Additionally, reaction conditions such as temperature, pressure, and reaction time can significantly influence the efficiency and product distribution of lignin hydrogenolysis [106]. Overall, metal-supported catalysts offer a large surface area for reactions, leading to high catalytic activity. The support material also stabilizes metal nanoparticles, preventing agglomeration and preserving catalytic activity over multiple cycles. This stability, combined with the recoverability and reusability of metal-supported catalysts, enhances their cost-effectiveness for lignin depolymerization [107,108]. Bijoy Biswas and colleagues [109] recently developed an interesting method for the catalytic depolymerization of lignin into phenolic monomers using cobalt-supported calcium catalysis. Different reaction parameters were evaluated such as time, solvent, temperature and catalysts loading. The optimal performance was obtained in methanol with a final yield in bio-oil of 60.2% weight at 160 °C for 60 min and a catalyst loading Co/CaO of 10% weight. Interestingly, this catalysis improves not only the bio-oil yield but also the selectivity in vanillin production with a value of 58.7%. In another work, Yanfang Zhu and co-workers [110] investigated the effect of reaction parameters on lignin depolymerization catalyzed by metal supported hydrotalcite. The catalytic pyrolysis without catalysis was conducted at 500 °C, to obtain a bio-oil yield of 32.4% weight after 60 minutes. When cobalt supported hydrotalcite catalyst (Co/MgAl2O4) was introduced, the yield reached 47.4% in weight. The presence of a selectivity towards G-types phenolic monomer was the demonstration of the catalyst activity towards breakdown of the lignin ether bond (C–O–C).
Lignin-first approach is a known method that solubilize lignin from lignocellulosic biomass through the stabilization of the intermediates that can be accomplished by solvolysis, catalysis or by protecting group chemistry [111]. Shihao Su and co-workers [112] developed an interesting procedure for the lignin-first depolymerization of lignin into monophenols, catalyzed by Carbon Nanotube-Supported Ruthenium. Lignin hydrogenolisis into aromatic monomers was proved to be correlated to lignin source. The yield decrease from hardwoods to softwoods probably due to side condensations. The recycling of the catalyst has given excellent results confirming that this study can open new horizons in the field of lignin conversion into chemical compounds and added value.
3.1.6. Enzyme Catalysis
Enzyme catalysis plays a pivotal role in lignin depolymerization, providing a sustainable and environmentally friendly alternative for breaking down this complex polymer. Unlike conventional chemical methods, enzymatic depolymerization occurs under ambient temperature and neutral pH conditions, utilizing biocatalysts derived from renewable resources. This process frequently employs oxygen or hydrogen peroxide as oxidants, often producing water as a byproduct, thus aligning with the principles of green chemistry. Laccases and peroxidases are the most commonly used enzymes for lignin depolymerization due to their high specificity in targeting specific bonds within the lignin structure [118]. Laccases mainly target phenolic subunits and ether linkages, whereas peroxidases can also break down non-phenolic linkages, expanding the range of lignin structures that can be effectively depolymerized [113].
Due to lignin’s heterogeneous structure, it is challenging to define precise chemical equations for its depolymerization using enzymes. The reaction pathways are influenced by the specific enzyme employed (e.g., laccase, peroxidase) and the presence of mediators. Enzymes can synergistically combine biological and chemical processes to convert lignin into valuable products [114]. Huan Zhang and co-workers [115] described in a recent study a methodology for lignin depolymerization using mediator-enzyme catalysis. They used different ketones as a lignin model and experimented on them the C−C oxidation in analogy to β-O-4 lignin bonds. Novozym 435 was used as the catalys for the C-C oxidation, while hydrogen peroxide was the oxidant in ethyl acetate solvent. Excellent results were obtained using different model compunds and the reaction scheme is depicted in the following Scheme 4.
In another work Justine Dillies and Colleagues reported a strategy for industrial lignin depolymerization (organosolvlignin, Kraft lignin, and sodium lignosulfonate) by laccase in a solvent mixture composed by 1,4-dioxane/water [116]. Laccase from T. versicolor was used, while 1,4 dioxane was used at 25% to improve the solubility of lignin in the reaction medium. Excellent performance was obtained at 50 °C with activity remaining even after several days. Future studies will focus on the type of products obtained through these techniques with in-depth characterizations.
Advantages of Enzyme Catalysis
Mild Reaction Conditions: Enzymatic depolymerization occurs at ambient temperature and neutral pH, reducing energy requirements.
Specificity and Selectivity: High specificity minimizes unwanted side reactions, yielding a more controlled product distribution compared to chemical methods.
Environmental Friendliness: It avoids the use of harsh chemicals and produces minimal waste, supporting sustainable practices [117].
Challenges and Limitations
Despite its advantages, enzyme catalysis faces several challenges:
Sensitivity to Reaction Conditions: Enzymes are sensitive to temperature, pH, and the presence of inhibitors, necessitating careful control of reaction conditions.
Limited Lignin Accessibility: The complex structure of lignin can hinder enzyme accessibility, reducing depolymerization efficiency.
Cost and Production Efficiency: The high cost of enzyme production impacts the economic viability of the process [118].
Strategies for Improvement
To overcome these challenges, research is focused on:
Engineering more efficient enzymes with enhanced stability and activity.
Developing cost-effective production methods.
Implementing pretreatment strategies to improve lignin accessibility [118].
By addressing these limitations, enzyme catalysis holds significant potential for sustainable lignin valorization, paving the way for bio-based chemicals and materials within a circular bioeconomy.
4. Catalytic Depolymerization of Plastics
4.1. Depolymerization of Polyester Plastics
Polyester plastics are among the most widely used plastics due to their ester bonds (-C=O-OR), which provide several advantageous properties, including processability, versatility, ease of production, resistance to chemical and bacterial agents, and a high rate of recyclability. Current research focuses on developing innovative strategies to obtain starting monomers suitable for producing new plastics that retain their original structural properties [119,120,121].
In a recent study, Yu-Ji Luo et al. [122] proposed a closed-loop process for the depolymerization of polyesters into reusable monomers. This method utilizes trifluoromethanesulfonic acid or metal triflates as recyclable catalysts in combination with a carboxylic acid and water (Figure 5).
Before optimizing the reaction, the homogeneous acidolysis of 4-chlorobenzoate using various Lewis and Brønsted acid catalysts with acetic acid was investigated. Water was found to play a crucial role in this reaction. Specifically, the authors reported an initial yield of 49% within 5 minutes using 15 mol/mol of trifluoromethanesulfonic acid (TfOH) at 180 °C, which increased to 78% in 5 minutes upon adding 0.9 wt% of water. The effectiveness of Lewis acids was also positively evaluated. For instance, when 15% of hafnium trifluoromethanesulfonate (Hf(OTf)4) was used, the reaction yield reached 90%. These findings were then applied to the acidolysis depolymerization of PET as a model substrate, yielding recycled terephthalic acid (rTPA) and glycol diacetate (EGDA) as the primary products. The most significant results are summarized in Table 6.
The reaction using Hf(OTf)4 without added water yielded a satisfactory 60%, which increased to 72% with the addition of 0.5 M of water. In contrast, when other Lewis acidic transition metals, such as iron and aluminum triflates, were used under the same conditions, the yield decreased to 64% and 60%, respectively. Excellent results were also obtained with Brønsted acids. Specifically, using trifluoromethanesulfonic acid under the same conditions resulted in a yield of 98%, whereas the yield dropped to 58% when trifluoromethanesulfonimide was used as the catalyst. Figure 6 illustrates the recycling efficiency of the Lewis acid [Fe(OTf)3] and Brønsted acid (TfOH) catalysts used in this study, shown as a function of the yield percentage of recycled terephthalic acid (rTPA).
As shown in Figure 6, the recycling of the catalytic system achieved recycled terephthalic acid yields of 90% after three cycles. These promising results pave the way for future studies in this field. In a study by Shinji Tanaka and co-workers [123], an innovative strategy for the depolymerization of polyester fibers was developed using dimethyl carbonate-aided methanolysis. Global fabric production is estimated at approximately 60 million tons per year, with polyethylene terephthalate being the predominant material used across various industries, including clothing and home furnishings [124,125]. This study focused on recycling polyester fibers primarily composed of polyethylene terephthalate. By employing methanolysis with dimethyl carbonate as a trapping reagent for ethylene glycol, the researchers achieved a low-temperature, efficient, and rapid depolymerization process. This method produced dimethyl terephthalate (DMT) with yields exceeding 90%. DMT, the diester form of terephthalic acid, is widely used in the production of recycled polyethylene terephthalate [126]. Conventionally, the methanolysis reaction is an equilibrium process that is challenging to perform under mild conditions. Scheme 5 illustrates the mechanistic pathways of the dimethyl carbonate-aided methanolysis process.
Methanolysis is a substitution reaction in which the alcoholic portion of an ester is replaced by methanol, as illustrated in Scheme 2 for the reaction of PET (A). This reaction is a reversible equilibrium because the leaving group, ethylene glycol, can react with the newly formed methyl ester. In this process, the addition of dimethyl carbonate enables the trapping of the produced ethylene glycol as ethyl carbonate, thereby shifting the equilibrium to the right, as shown in Scheme 2 (B). Consequently, the depolymerization of PET is driven towards product formation, as depicted in Scheme 1 (C). Table 7 summarizes the key results from the optimization of the reaction conditions, aiming to identify the most critical parameters in the development of this method.
As shown in Table 7, NaOMe and KOMe exhibited higher catalytic activity compared to LiOMe, achieving yields of 95% versus 83%. Notably, NaOMe provided satisfactory yields even with a shorter reaction time of 1 hour. Excellent results were also obtained using lower amounts of DMC and MeOH with a reaction time of 2 hours.
In a recent study, Xiaoshen Bai et al. [127] reported a solvent-free depolymerization of PET into dimethyl terephthalate (DMT) and ethylene glycol (EG) using heterogeneous catalysis, specifically through plastic-catalyst interfacial engineering. During reaction optimization, various metal acetates were screened for the depolymerization of PET microparticles sourced from plastic bottles. The experiments were conducted in closed vials with methanol at 100 °C for two hours. After testing several metal acetates, including MnAc2, CoAc2, NiAc2, CuAc2, ZnAc2, and PbAc2, a volcano-type activity plot was obtained [128]. Zinc acetate showed the most promising results after lead acetate, which was excluded due to toxicity concerns. SEM and TEM morphological studies suggested that the ZnAc2/MeOH interaction induces methanolysis of zinc acetate. This hypothesis was supported by the observation of zinc oxide nanocrystals on the plastic surface, likely acting as catalysts in the PET methanolysis process. The authors proposed that the significant catalytic activity at 140–200 °C is mainly due to ZnO2, while below 140 °C, the catalytic effect is attributed to ZnAc2. Notably, experiments using methanol vapor instead of liquid methanol demonstrated a solid-solid interaction between PET and zinc oxide at the interface, persisting until complete PET degradation due to the absence of interfering liquid solvent molecules. At 160 °C and after two hours, PET conversion was nearly quantitative in both ZnOAc with liquid methanol and in ethanol with ZnO vapors. This study presents new prospects for solvent-free depolymerization techniques for plastics. In another work Tong Chang and co-workers [129], reported the interesting catalytic activity of double metal cyanide complex (DMC), as heterogenous catalyst for polyester depolymerization into monomers and chemicals. Poly(ethylene terephthalate) (PET) was used as the reference plastic, and the most important reaction parameters including the amount of ethylene glycol and the catalyst loading were investigated. In the following Figure 7 was reported a schematic representation of the double metal cyanide (DMC) complex catalyst used in this study.
4.2. Depolymerization of Polyamides
Polyamides, notably Nylon 6,6 and Nylon 6, are widely used in the textile, automotive, construction, and medical industries, with substantial global market demand [130]. Derived from petroleum-based sources, these materials exhibit high resistance to biodegradation and general decomposition [131].
Common disposal methods include incineration and mechanical recycling, although the latter often results in the loss of the original material properties [132]. In contrast, catalytic chemical depolymerization is emerging as an effective strategy to recover the initial monomers. In a recent study, Wei Zhou and co-workers [133] developed a catalytic hydrogenative depolymerization method for polyamide 66 using a Ru pincer complex, as illustrated in Scheme 6.
The authors began optimizing the catalytic system using low molecular weight polyamide 66 (Mw = 8240 g mol−1; amine end groups = 1748 mmol kg−1). The results are summarized in Table 8.
As shown in Table 8, high temperature is crucial for catalyst activation, with the highest yields achieved at 200 °C. Hydrogen pressure is also a key factor, with the best results obtained at 100 bar. Lower pressures led to reduced yields of diamine and diol. After optimizing the reaction conditions, the method was applied to Ultramid® A 27, a commercial high molecular weight polyamide, yielding diamine and diol at 19% and 18%, respectively. Robin Coeck et al. [134] developed a green and sustainable method for Nylon 66 depolymerization using transamidation with a short primary amide, catalyzed by Nb2O5 and assisted by NH3. Acetamide was chosen as the model amide due to its bio-derived and biodegradable properties [135]. In this process, ammonia promotes polymer cleavage through ammonolysis. The reaction scheme is shown in Figure 8.
At 225 °C and a reaction time of 16 hours, Nylon 66 was fully depolymerized into monomers and dimers with yields of 94% and 5%, respectively. At a maximum concentration of 12.5 wt% Nylon 66 in acetamide, valuable monomers such as N,N’-hexamethylene bis(acetamide) and adipamide were obtained, which are suitable for various applications. In another work Kousuke Tsuchiya and co-workers [136] have developed a method both in the direction of polymerization and subsequent depolymerization of semiaromatic polyamide by enzymes. The awareness of the problem of waste has led today to develop not only research aimed only at the production of new high-performance and efficient materials, but also at the eventual disposal process. Polyamides consisting of 4-amino-3-hydroxybenzoic acid and peptide moieties were enzymatically synthesized by papain-catalyzed polymerization in aqueous media, and the same underwent selective degradation into their corresponding monomers when treated with proteinase K, the process is represented in the following Scheme 7.
Another application of a solid acid catalyst for polyamides depolymerization was developed by Yang Liu at al. [137] This was the first report on a sulfonated Fe-MOF catalyst for the polyamide 6 depolymerization into water soluble products. 100% conversion was achieved at 250 °C after only 1 hour. The subsequent characterization step was able to demonstrate by LC-MS the presence of caprolactam and its oligomers as the main products. Even a selectivity of 90% in caprolactam was declared at a process temperature of 250 °C after 5 hours. These studies demonstrate how from widely used materials it is possible to obtain reusable monomers in a sustainable way.
4.3. Depolymerization of Polyurethanes
Polyurethanes are a versatile class of materials known for their unique properties [138]. They are typically synthesized through the reaction of diisocyanates (or polyisocyanates) with diols (or polyols), forming urethane bonds (R-O-C=O-NH-R), with catalysis playing a crucial role in this process [139]. Recent research has focused on replacing fossil-based reagents with sustainable resources to produce bio-based polyurethanes [140]. These materials exhibit high resistance and stability against environmental and biological agents [141]. Aromatic polyurethanes are among the most common due to their high stability, which makes them suitable for various applications [142]. However, their low eco-compatibility poses significant environmental challenges throughout their production and application phases [143].
Viktoriia Zubar and co-workers [144] discovered an innovative hydrogenative depolymerization method for aromatic polyurethanes using a manganese pincer complex (shown in Scheme 8), combined with high temperatures and suitable solvents. This approach enabled the efficient recovery of aromatic diamines and corresponding polyols in excellent yields.
Figure 9 shows three examples of different polyurethanes along with the corresponding optimized reaction conditions.
In the same study, the authors applied the optimized reaction conditions to commercial polyurethanes, in addition to the self-made samples shown in Figure 9 (A, B and C). This was done to assess whether additives like colorants and thickeners would affect the process yield. Surprisingly, depolymerization was achieved with conversion rates ranging from 66% to 100%, demonstrating the robustness of the method.
4.4. Depolymerization of Polyethers
Polyethers are widely used due to their excellent chemical and physical properties, including low-temperature flexibility and resistance to heat and oils [145]. They serve as key reagents in polymer production, driving the continuous pursuit of innovative and eco-friendly synthetic strategies [146]. Significant progress has also been made in polyether depolymerization and recycling, including approaches like ring-closing metathesis reactions [147]. In a study by N. Ansmann et al. [148], the mechanism of silicon-catalyzed C–O bond ring-closing metathesis of polyethers was explored. A second-generation silicon Lewis superacid was identified as an optimal catalyst for this reaction, as shown in Scheme 9.
The mechanism was clarified through DFT computational studies, revealing that the catalyst’s low tendency for chelation and deactivation by polyethers, such as polyethylene glycol, is a key advantage. In contrast, other Lewis acids like Fe(OTf)3 and AlCl3 exhibited mono-, bi-, and tridentate binding with diglyme, adversely affecting product conversion.
Figure 10 illustrates the reaction scheme starting from 1,5-dimethoxypentane (Figure 10A) or polyethylene glycol/polypropylene glycol (Figure 10B), along with the corresponding optimized reaction conditions.
In a recent work, Florian S. Tschernuth et al [149] investigated the interesting catalytic activity of a Lewis superacidic bis(perfluoropinacolato) silane for the degradation of aliphatic ethers. The catalyst is reported in the following Scheme 10.
Ring-closing metathesis is typically affected by the availability of two or three coordination sites on the catalyst, leading to coordination and subsequent deactivation [150]. However, catalysts with blocked or less favorable multiple-coordination sites, such as the acetonitrile-adduct of a Lewis superacidic silane (Si(pinF)2·MeCN), demonstrated enhanced activity. This study revealed that the coordinated acetonitrile is exchanged with the etheric substrate used. Table 9 summarizes the optimized reaction conditions and corresponding products for the degradation of 1,5-dimethoxypentane (1,5-DMP), diglyme, poly(ethylene glycol) dimethyl ether (PEG DME), and 18-crown-6 [149].
5. Summary and Outlook
This review examines innovative catalytic technologies for the depolymerization of polymers of significant interest. In this work, we focus on cellulose, lignin, polyester plastics, polyamides, polyurethanes, and polyethers. Cellulose is highlighted as a primary source of glucose and organic platform molecules for the production of high-value materials, attracting significant interest in the pharmaceutical, food, energy, and various other sectors. Lignin is identified as an important renewable source of aromatic and aliphatic molecules, traditionally derived from petroleum-based resources. In recent years, it has been extensively investigated, particularly in the biofuel and building materials sectors. This review also explores the depolymerization of the most widely used plastics worldwide. Polyester plastics, polyamides, polyurethanes, and polyethers are materials used worldwide, mainly produced from fossil resources. This review demonstrates how through sustainable methodologies it is possible to recycle them into starting reagents that can be used for multiple purposes. The ongoing ecological transition has increased awareness of environmental challenges, underscoring the importance of recycling strategies over conventional disposal methods. In this context, catalysis plays a pivotal role in improving process efficiency and scalability at the industrial level. Developing mild, non-destructive techniques for obtaining monomers—allowing multiple recycling cycles without compromising product properties—represents a new frontier in science.
Future Challenges:
Development of cost-effective and environmentally sustainable catalytic systems. For an innovative process to be successfully scaled up, it must achieve an optimal cost-benefit balance. Many catalysts are expensive materials, and only non-destructive strategies under mild conditions can enable true recycling cycles. Mild reaction conditions are also closely linked to energy efficiency and cost savings. Additionally, solvent-based processes should be minimized or eliminated where possible, as this can significantly reduce costs and environmental impact.
Production of high-purity monomers with minimal by-products and high atom economy. A method is truly sustainable only if it ensures high yield and selectivity. Achieving this requires growing interest and investment in this field of research to drive further advancements.
Design of simple, easily scalable processing plants. Beginning with straightforward laboratory reactors and processes increases the likelihood of successful scale-up beyond the lab.
The circular economy, if implemented sustainably, holds significant potential as a future strategy to minimize the waste of both natural and synthetic resources
Author Contributions
G.O. Software, Writing- Original draft preparation, Writing- Reviewing and Editing; F.O. Methodology, Conceptualization, Supervision; Writing- Original draft preparation, Writing- Reviewing and Editing; A.R. Software, Writing- Original draft preparation, Writing- Reviewing and Editing; M.N. Software, Validation, Writing- Reviewing and Editing; A.P. Software, Validation, Writing- Reviewing and Editing; W.A.A.Q.I.W.-M. Software, Validation, Writing- Reviewing and Editing; P.J. Software, Writing- Original draft preparation, Writing- Reviewing and Editing.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kreps, B.H. The Moral Economy of Debt: A Critical Reappraisal. Am. J. Econ. Sociol. 2020, 79, 695–717. [Google Scholar] [CrossRef]
- Ghazouani, T.; Maktouf, S. Institutions, Natural Resources and Economic Growth in the MENA Region. Nat. Resour. Forum 2024, 48, 211–231. [Google Scholar] [CrossRef]
- Zibunas, C.; Meys, R.; Kätelhön, A.; Bardow, A. Quantifying Climate Benefits of Circular Economy Strategies for Plastics. Comput. Chem. Eng. 2022, 162, 107798. [Google Scholar] [CrossRef]
- Boulamanti, A.; Moya, J.A. Energy Efficiency Trends in the EU. Renew. Sustain. Energy Rev. 2017, 68, 1205–1212. [Google Scholar] [CrossRef]
- Axon, S.; James, D. The Role of Discourse Analysis in Understanding Student Perspectives on Sustainability. Curr. Opin. Green Sustain. Chem. 2018, 13, 140–145. [Google Scholar] [CrossRef]
- Kümmerer, K. Sustainable Chemistry: A Future Guiding Principle. Angew. Chem. Int. Ed. 2017, 56, 16420. [Google Scholar] [CrossRef]
- Zuin, V.G.; Eilks, I.; Elschami, M.; Kümmerer, K. Green and Sustainable Chemistry Education. Green Chem. 2021, 23, 1594–1608. [Google Scholar] [CrossRef]
- Marion, P.; Bernela, B.; Piccirilli, A.; Estrine, B.; Patouillard, N.; Guilbot, J.; Jérôme, C. Sustainable Chemistry: Toward a Cleaner and Greener Future. Green Chem. 2017, 19, 4973–4989. [Google Scholar] [CrossRef]
- Fagnani, D.E.; Tami, J.L.; Copley, G.; Clemons, M.N.; Getzler, Y.D.Y.L.; McNeil, A.J. Chemically Recyclable Polymers: A Circular Economy Solution to Plastic Waste. ACS Macro Lett. 2021, 10, 41–53. [Google Scholar] [CrossRef]
- Guo, X.; Facchetti, A. Polymer Semiconductors for Flexible Electronics. Nat. Mater. 2020, 19, 922–928. [Google Scholar] [CrossRef]
- Tursi, A.; Olivito, F. Lignocellulosic Biomass Conversion. In Biomass Conversion: General Information, Chemistry, and Processes; Rahimpour, M.R., Kamali, R., Makarem, M.A., Manshadi, M.K.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 3–39. [Google Scholar]
- Wang, R.; Feng, Y.; Li, D.; Li, K.; Yan, Y. Catalytic Upgrading of Biomass-Derived Platform Molecules. Green Chem. 2024, 26, 9075–9103. [Google Scholar] [CrossRef]
- Jia, J.; Zang, G.; Paul, M.C. Renewable Energy Applications for Sustainable Development. Int. J. Energy Res. 2021, 45, 15182–15199. [Google Scholar] [CrossRef]
- Thakur, V.; Guleria, A.; Kumar, S.; Sharma, S.; Singh, K. Recent Developments in Biodegradable Polymers. Mater. Adv. 2021, 2, 1872–1895. [Google Scholar] [CrossRef]
- Etale, A.; Onyianta, A.J.; Turner, S.R.; Eichhorn, S.J. Advances in Lignocellulosic Biomass Conversion. Chem. Rev. 2023, 123, 2016–2048. [Google Scholar] [CrossRef]
- Maiuolo, L.; Olivito, F.; Algieri, V.; Costanzo, P.; Jiritano, A.; Tallarida, M.A.; Tursi, A.; Sposato, C.; Feo, A.; De Nino, A. Biobased Polymers from Renewable Feedstocks. Polymers 2021, 13, 2802. [Google Scholar] [CrossRef]
- Romeo, I.; Olivito, F.; Tursi, A.; Algieri, V.; Beneduci, A.; Chidichimo, G.; Maiuolo, L.; Sicilia, E.; De Nino, A. Microwave-Assisted Reactions for Polymer Chemistry. RSC Adv. 2020, 10, 34738–34751. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Shrotri, A.; Fukuoka, A. Catalytic Processes in Biomass Upgrading. Appl. Catal. 2021, 621, 118177. [Google Scholar] [CrossRef]
- Tyufekchiev, M.; Ralph, K.; Duan, P.; Yuan, S.; Schmidt-Rohr, K.; Timko, M.T. Thermochemical Conversion of Biomass. ChemSusChem 2020, 13, 2634. [Google Scholar] [CrossRef]
- Clark, J.H.; Deswarte, F.E.I. Introduction to Chemicals from Biomass; John Wiley & Sons: Chichester, UK, 2008. [Google Scholar]
- Farmer, T.J.; Mascal, M. Overview of Biomass-Derived Chemicals. In Introduction to Chemicals from Biomass; Clark, J., Deswarte, F., Eds.; Wiley: Chichester, UK, 2015. [Google Scholar]
- Yabushita, M.; Kobayashi, H.; Fukuoka, A. Catalytic Conversion of Cellulose. Appl. Catal. B Environ. 2014, 145, 1–9. [Google Scholar] [CrossRef]
- Forsberg, C.W.; Dale, B.E.; Jones, D.S.; Hossain, T.; Morais, A.R.C.; Wendt, L.M. Integration of Renewable Energy Systems. Appl. Energy 2021, 298, 117225. [Google Scholar] [CrossRef]
- Olivito, F.; Jagdale, P.; Oza, G. Sustainable Approaches to Polymer Synthesis. ACS Sustain. Chem. Eng. 2023, 11, 17595–17599. [Google Scholar] [CrossRef]
- Tomishige, K.; Yabushita, M.; Cao, J.; Nakagawa, Y. Green Catalysis for Biomass Utilization. Green Chem. 2022, 24, 5652–5690. [Google Scholar] [CrossRef]
- Mascal, M. Biomass-Derived Platform Molecules. ChemSusChem 2020, 13, 274. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Bai, J.; Innocent, M.T.; Wang, Q.; Xiang, H.; Tang, J.; Zhu, M. Green Energy Technologies for Sustainable Development. Green Energy Environ. 2022, 7, 578–605. [Google Scholar] [CrossRef]
- Sethupathy, S.; Morales, G.M.; Gao, L.; Wang, H.; Yang, B.; Jiang, J.; Sun, J.; Zhu, D.B. Valorization of Agricultural Waste. Bioresour. Technol. 2022, 347, 126696. [Google Scholar]
- Sun, R.C. Lignin Valorization Strategies. ChemSusChem 2020, 13, 4385. [Google Scholar] [CrossRef]
- Haq, I.; Mazumder, P.; Kalamdhad, A.S. Biotechnological Innovations in Waste Management. Bioresour. Technol. 2020, 312, 123636. [Google Scholar]
- Mboowa, D. Sustainable Biomass Technologies for Renewable Fuel Production. Biomass Convers. Bioref. 2024, 14, 1–12. [Google Scholar] [CrossRef]
- Zhang, B.; Meng, Q.; Liu, H.; Han, B. Recent Advances in CO2 Conversion. Acc. Chem. Res. 2023, 56, 3558–3571. [Google Scholar] [CrossRef]
- Karagoz, P.; Khiawjan, S.; Marques, M.P.C.; et al. Lignin-Derived Platform Molecules. Biomass Convers. Bioref. 2024, 14, 26553–26574. [Google Scholar] [CrossRef]
- Salcedo-Puerto, O.; Mendoza-Martinez, C.; Saari, J.; Vakkilainen, E. Renewable Energy from Forest Biomass. Fuel 2024, 373, 132389. [Google Scholar] [CrossRef]
- Sun, P.; Wang, Z.; Li, C.; Tang, B.; Peng, C. Catalytic Fast Pyrolysis of Biomass. Fuel 2024, 361, 130726. [Google Scholar] [CrossRef]
- Diao, X.; Xiong, Y.; Shi, Y.; Ma, L.; Dong, C.; Zhang, S.; Ji, N. Conversion of Cellulose to Biofuels. Green Chem. 2024. Advance Article. [Google Scholar]
- Rosenboom, J.G.; Langer, R.; Traverso, G. Biodegradable Polymers for Sustainable Packaging. Nat. Rev. Mater. 2022, 7, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Awasthi, M.K.; Sheet, N.; Kharde, T.A.; Singh, S.K. Catalysts in Biomass Upgrading. ChemCatChem 2023, 15, e202300574. [Google Scholar] [CrossRef]
- Faust, K.; Denifl, P.; Hapke, M. Biocatalytic Upgrading of Lignin. ChemCatChem 2023, 15, e202300310. [Google Scholar] [CrossRef]
- Yang, S.; Li, Y.; Nie, M.; Liu, X.; Wang, Q.; Chen, N.; Zhang, C. Advanced Materials for Biomass-Derived Electronics. Adv. Mater. 2024, 36, 2404115. [Google Scholar] [CrossRef]
- Kee Wong, M.; Lock, S.S.M.; Chan, Y.H.; Yeoh, S.J.; Tan, I.S. Green Engineering Approaches in Chemical Processes. J. Chem. Eng. 2023, 468, 143699. [Google Scholar]
- Volanti, M.; Cespi, D.; Passarini, F.; Neri, E.; Cavani, F.; Mizsey, P.; Fozer, D. Life Cycle Assessment of Biomass Pathways. Green Chem. 2019, 21, 885–896. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, Q.; Che, Y.; Niu, Z. Photocatalysis in Biomass Valorization. Chem. Sci. 2024, 15, 6200–6217. [Google Scholar] [CrossRef]
- Liu, Y.; Akula, K.C.; Dandamudi, K.P.R.; Liu, Y.; Xu, M.; Sanchez, A.; Zhu, D.; Deng, S. Green Engineering for Polymer Recovery. Chem. Eng. 2022, 446, 137238. [Google Scholar] [CrossRef]
- Cao, J.; Liang, H.; Yang, J.; et al. Photocatalytic Conversion of CO2. Nat. Commun. 2024, 15, 6266. [Google Scholar] [CrossRef]
- Yang, H.; et al. Activated Clays in Biomass Processing. Appl. Clay Sci. 2020, 105512. [Google Scholar] [CrossRef]
- Zhou, L.; et al. Efficient Biomass Conversion Using Green Solvents. Green Chem. 2014, 1519–1524. [Google Scholar]
- Zhou, L.; et al. Lignin-Based Biocomposites. Ind. Crops Prod. 2021, 174, 114179. [Google Scholar] [CrossRef]
- Pang, S.C.; Voon, L.K.; Chin, S.F. Chitosan-Based Hydrogels: Synthesis and Applications. Int. J. Polym. Sci. 2018, 1–11. [Google Scholar] [CrossRef]
- Luo, X.; et al. Advances in Biomass Chemistry. Front. Chem. 2020. [Google Scholar]
- Negi, A.; Kesari, K.K. Green Polymer Composites for Environmental Applications. Polymers 2023, 15, 3671. [Google Scholar] [CrossRef]
- Wu, T.; Li, N.; Pan, X.; Chen, S.L. Nanocellulose Materials: Structure and Function. Cellulose 2020, 27, 9201–9215. [Google Scholar] [CrossRef]
- Brandt, A.; Gräsvik, J.; Hallett, J.P.; Welton, T. Ionic Liquids for Lignin Processing. Green Chem. 2013, 15, 550. [Google Scholar] [CrossRef]
- Liu, Q.; et al. Biomass Pretreatment Using Green Solvents. Green Chem. 2019, 21, 5030–5038. [Google Scholar] [CrossRef]
- Paiva, M.F.; et al. Nanocellulose-Based Films for Packaging. Cellulose 2024, 31, 7953–7972. [Google Scholar] [CrossRef]
- Deng, W.; Kennedy, J.R.; Tsilomelekis, G.; Zheng, W.; Nikolakis, V. Catalytic Conversion of Furfural. Ind. Eng. Chem. Res. 2015, 54, 5226–5236. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Song, Y.N.; Chen, C.Z.; Li, M.F.; Zhang, Z.T.; Fan, Y.M. Hemicellulose-Derived Products from Biomass. BioResources 2017, 12, 7807–7818. [Google Scholar] [CrossRef]
- Wei, W.; Wu, S. Biochar-Based Catalysts in Biofuel Production. Bioresour. Technol. 2017, 241, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; et al. Recent Trends in Biomass Conversion. Biomass Convers. Bioref. 2022. [Google Scholar]
- Wang, J.; et al. Hydrothermal Processing of Biomass. Green Chem. 2020, 22, 4240–4251. [Google Scholar] [CrossRef]
- Furusato, S.; Takagaki, A.; Hayashi, S.; Miyazato, A.; Kikuchi, R.; Oyama, S.T. Catalysis for Biomass-Derived Fuels. ChemSusChem 2018, 11, 888–896. [Google Scholar] [CrossRef]
- Karam, A.; De Oliveira Vigier, K.; Marinkovic, S.; Estrine, B.; Oldani, C.; Jérôme, F. Acid Catalysts in Green Chemistry. ChemSusChem 2017, 10, 3604. [Google Scholar] [CrossRef]
- Hick, S.M.; et al. Lignin Depolymerization Mechanisms. Green Chem. 2010, 12, 468. [Google Scholar]
- Yu, P.; et al. Agronomic Perspectives on Lignocellulosic Feedstocks. Agronomy 2024, 14, 2550. [Google Scholar] [CrossRef]
- Lignin Market – By Raw Material, Product, Application, and Forecast 2024–2032. Market Research Report.
- Fernández-Rodríguez, J.; Erdocia, X.; Sánchez, C.; González Alriols, M.; Labidi, J. Optimization of Lignin Extraction Processes. J. Energy Chem. 2017, 26, 622–631. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Green Chemistry Routes to Biomass-Derived Chemicals. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [PubMed]
- Roy, R.; Jadhav, B.; Rahman, M.S.; Raynie, D.E. Recent Advances in Sustainable Chemical Synthesis. Curr. Res. Green Sustain. Chem. 2021, 4, 100052. [Google Scholar] [CrossRef]
- Rath, S.; Pradhan, D.; Du, H.; Mohapatra, S.; Thatoi, H. Hybrid Materials from Biomass. Adv. Compos. Hybrid Mater. 2024, 7, 27. [Google Scholar] [CrossRef]
- Roy, R.; Rahman, M.S.; Amit, T.A.; Jadhav, B. Biomass as Feedstock for Green Polymers. Biomass 2022, 2, 130–154. [Google Scholar] [CrossRef]
- Patel, R.; Dhar, P.; Babaei-Ghazvini, A.; Nikkhah Dafchahi, M.; Acharya, B.B. Conversion of rice straw into valuable chemicals: Production of levulinic acid. Bioresour. Technol. Rep. 2023, 22, 101463. [Google Scholar]
- Gonçalves, C.C.; Silva, L.F.; Costa, J.A.V.; Bertolini, T.C.; Zepka, L.Q. Biochemical profile and bioactive potential of biomass from Spirulina strains: A comparative analysis. Front. Microbiol. 2020, 11, 123456. [Google Scholar]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef]
- Nghiem, N.P.; Kim, T.H.; Yoo, C.G. (Eds.) Biomass Utilization: Conversion Strategies; Springer International Publishing: Cham, Switzerland, 2022. [Google Scholar]
- Ventura, M.; Domine, M.E.; Chávez-Sifontes, M. Catalytic cracking of waste plastics using natural clays and mesoporous materials. Curr. Catal. 2019, 8, 45–55. [Google Scholar]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C.C. Production of chemicals from lignin: A review. Bioresour. Technol. 2015, 190, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Wanmolee, W.; Daorattanachai, P.; Laosiripojana, N. Effect of temperature on hydrothermal liquefaction of rice straw. Energy Procedia 2016, 100, 173–177. [Google Scholar] [CrossRef]
- Yong, K.J.; Wu, T.Y. Application of deep eutectic solvents in biomass pretreatment: A review. Bioresour. Technol. 2023, 384, 129238. [Google Scholar]
- Wang, Y.; Wei, L.; Hou, Q.; Mo, Z.; Liu, X.; Li, W. Fermentation of lignocellulosic biomass for ethanol production. Fermentation 2023, 9, 386. [Google Scholar] [CrossRef]
- Pazhavelikkakath Purushothaman, R.K.; van Erven, G.; van Es, D.S.; Rohrbach, L.; Frissen, A.E.; van Haveren, J.; Gosselink, R.J.A. Catalytic oxidative depolymerization of lignin in a flow-through system. RSC Adv. 2023, 13, 4898. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Qin, J.; Sun, S.; Gao, D.; Fang, Y.; Chen, G.; Tian, C.; Bao, C.; Zhang, S. Progress in developing methods for lignin depolymerization and elucidating the associated mechanisms. Eur. Polym. J. 2024, 210, 112995. [Google Scholar] [CrossRef]
- Li, C.; Jiang, D.; Cheng, X.; Li, H.; He, S.; Mu, M.; Cao, B.; Esakkimuthu, S.; Wang, S. Elucidating reaction mechanisms in the oxidative depolymerization of sodium lignosulfonate for enhancing vanillin production: A density functional theory study. J. Anal. Appl. Pyrolysis 2024, 179, 106499. [Google Scholar] [CrossRef]
- Paananen, H.; Eronen, E.; Mäkinen, M.; Jänis, J.; Suvanto, M.; Pakkanen, T.T. Base-catalyzed oxidative depolymerization of softwood kraft lignin. Ind. Crops Prod. 2020, 152, 112473. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, Y.I.; Um, B.H. Base-catalyzed depolymerization of organosolv lignin into monoaromatic phenolic compounds. Waste Biomass Valor. 2025, 16, 257–270. [Google Scholar] [CrossRef]
- Biswas, B.; Kumar, A.; Krishna, B.B.; Bhaskar, T. Effects of solid base catalysts on depolymerization of alkali lignin for the production of phenolic monomer compounds. Renew. Energy 2021, 175, 270–280. [Google Scholar] [CrossRef]
- Zhang, X.; Li, W.; Wang, J.; Zhang, B.; Guo, G.; Shen, C.; Jiang, Y. Depolymerization of kraft lignin into liquid fuels over a WO3 modified acid-base coupled hydrogenation catalyst. Fuel 2022, 323, 124428. [Google Scholar] [CrossRef]
- Liu, X.; Bouxin, F.P.; Fan, J.; Budarin, V.L.; Hu, C.; Clark, J.H. Recent developments in lignin valorisation via catalytic processes. ChemSusChem 2020, 13, 4296–4313. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, X.; Ma, R.; Song, G. Catalytic valorization of lignin to functional materials and chemicals. Acc. Chem. Res. 2025, 58, 529–542. [Google Scholar] [CrossRef]
- Kaur, P.; Singh, G.; Arya, S.K. Tandem catalytic approaches for lignin depolymerization: A review. Biomass Convers. Biorefin. 2024, 14, 6143–6154. [Google Scholar] [CrossRef]
- Ji, D.; Wang, Y.; Peng, J.; Yuan, D.; Li, Z.; Ji, D.; Wu, H. Integrated catalytic process for lignin depolymerization: Advances and prospects. Ind. Eng. Chem. Res. 2024, 63, 19916–19935. [Google Scholar] [CrossRef]
- Khan, A.; Evans, L.W.; Martin, D. Tandem catalysis strategies for lignin valorization. Green Chem. 2025, in press. [Google Scholar]
- Song, J.; Zhang, H.; Niu, M.; Guo, Y.; Li, H. Research progress on vanillin synthesis by catalytic oxidation of lignin: A review. Ind. Crops Prod. 2024, 214, 118443. [Google Scholar] [CrossRef]
- Walch, F.; Abdelaziz, O.Y.; Meier, S.; Bjelić, S.; Hulteberg, C.P.; Riisager, A. Catalytic oxidative depolymerization of lignin to aromatic compounds. Catal. Sci. Technol. 2021, 11, 1843–1853. [Google Scholar] [CrossRef]
- Abdelaziz, O.Y.; Clemmensen, I.; Meier, S.; Walch, F.; Bjelić, S.; Riisager, A. Oxidative depolymerization of kraft lignin to aromatics over bimetallic V–Cu/ZrO2 catalysts. Top. Catal. 2023, 66, 1369–1380. [Google Scholar] [CrossRef]
- Martinez, D.V.; Rodriguez, A.; Juarros, M.A.; Martinez, E.J.; Alam, T.M.; Simmons, B.A.; Sale, K.L.; Singer, S.W.; Kent, M.S. Advances in green chemistry for lignin transformation. Green Chem. 2022, 24, 1627–1640. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, L.; Jiang, Y.; Liu, X.; Wang, J.; Tang, Z. Recent advances in the acid-catalyzed conversion of lignin. Biomass Convers. Biorefin. 2023, 13, 519–539. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, C.; Chee, P.L.; Qu, C.; Fok, A.Z.; Yong, F.H.; Ong, Z.L.; Kai, D. Sustainable lignin-derived nanomaterials for energy and environment. Sustain. Energy Fuels 2023, 7, 2953–2964. [Google Scholar] [CrossRef]
- Zhang, B.; Li, W.; Li, X. Selective depolymerization of corn stover lignin via nickel-doped tin phosphate catalyst in the absence of hydrogen. Ind. Crops Prod. 2021, 174, 114211. [Google Scholar] [CrossRef]
- Zhang, C.; Shen, X.; Jin, Y.; Cheng, J.; Cai, C.; Wang, F. Advances in catalytic lignin valorization. Chem. Rev. 2023, 123, 4510–4601. [Google Scholar] [CrossRef]
- Huang, Y.; Duan, Y.; Qiu, S.; Wang, M.; Ju, C.; Cao, H.; Fang, Y.; Tan, T. Efficient catalytic conversion of lignin into valuable biofuels and chemicals. Sustain. Energy Fuels 2018, 2, 637–646. [Google Scholar] [CrossRef]
- Zeng, J.; Mills, M.J.L.; Simmons, B.A.; Kent, M.S.; Sale, K.L. Catalytic conversion of lignin to bioproducts. Green Chem. 2017, 19, 2145–2151. [Google Scholar] [CrossRef]
- Ma, H.; Li, H.; Zhao, W.; Li, L.; Liu, S.; Long, J.; Li, X. Lignin valorization strategies. Green Chem. 2019, 21, 658–667. [Google Scholar] [CrossRef]
- Li, Z.; Bai, X.; Wei, X.Y.; Dilixiati, Y.; Fan, Z.C.; Kong, Q.Q.; Li, L.; Li, J.H.; Lu, K.L.; Zhao, J.; Zhang, X.Q.; Zong, Z.M.; Bai, H.C. Solid acid-catalyzed depolymerization of pine lignin to guaiacol. Fuel Process. Technol. 2023, 249, 107843. [Google Scholar] [CrossRef]
- Knill, C.J.; Kennedy, J.F. Degradation of lignin: A review. Carbohydr. Polym. 2003, 51, 281–300. [Google Scholar] [CrossRef]
- Guan, W.; et al. Biorefining of lignin waste. Bioresour. Technol. 2020, 298, 122432. [Google Scholar]
- Shen, Z.; Shi, C.; Liu, F.; Wang, W.; Ai, M.; Huang, Z.; Zhang, X.; Pan, L.; Zou, J.J. Advances in heterogeneous catalysts for lignin hydrogenolysis. Adv. Sci. 2024, 11, 2306693. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kong, W.; Liang, W.; Guo, Y.; Cui, J.; Hu, Y.; Sun, Z.; Elangovan, S.; Xu, F. Metal-free catalysis in lignin depolymerization. ChemSusChem 2025, 18, e202401399. [Google Scholar] [CrossRef]
- Kumi, D.O.; et al. Catalytic upgrading of lignin. Appl. Catal. B 2018, 232, 492–500. [Google Scholar] [CrossRef]
- Biswas, B.; Kumar, A.; Krishna, B.B.; Baltrusaitis, J.; Adhikari, S.; Bhaskar, T. Catalytic fast pyrolysis of lignin. Energy Fuels 2023, 37, 3813–3824. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, Y.; Song, W.; He, Z.; Yao, R.; Deng, Z. Metal-supported hydrotalcite catalysis for lignin depolymerization. Fuel 2023, 340, 127559. [Google Scholar] [CrossRef]
- Abu-Omar, M.M.; Barta, K.; Beckham, G.T.; Luterbacher, J.S.; Ralph, J.; Rinaldi, R.; Román-Leshkov, Y.; Samec, J.S.M.; Sels, B.F.; Wang, F. Review on catalytic lignin valorization. Energy Environ. Sci. 2021, 14, 262–292. [Google Scholar] [CrossRef]
- Su, S.; Xiao, L.P.; Chen, X.; Wang, S.; Chen, X.H.; Guo, Y.; Zhai, S.R. Catalytic strategies for lignin transformation. ChemSusChem 2022, 15, e202200365. [Google Scholar] [CrossRef]
- Hatakka, A. Lignin-degrading enzymes. FEMS Microbiol. Rev. 1994, 13, 125–135. [Google Scholar] [CrossRef]
- Hofrichter, M. Enzymatic transformation of lignin. Enzyme Microb. Technol. 2002, 30, 454–466. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, R.; Xin, Y.; Li, Y.; Shi, G.; Zhu, R.; Zhang, L. Enzyme-assisted lignin valorization. ACS Sustain. Chem. Eng. 2024, 12, 5842–5849. [Google Scholar] [CrossRef]
- Dillies, J.; Vivien, C.; Chevalier, M.; Rulence, A.; Châtaigné, G.; Flahaut, C.; Senez, V.; Froidevaux, R. Enzymatic depolymerization of industrial lignins by laccase–mediator systems in 1,4-dioxane/water. Biotechnol. Appl. Biochem. 2020, 67, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Qin, J.; Sun, S.; Gao, D.; Fang, Y.; Chen, G.; Tian, C.; Bao, C.; Zhang, S. Progress in developing methods for lignin depolymerization. Eur. Polym. J. 2024, 210, 112995. [Google Scholar] [CrossRef]
- Pajer, N.; Gigli, M.; Crestini, C. Functional lignin derivatives from green processes. ChemSusChem 2024. [Google Scholar]
- Kim, D.H.; Han, D.O.; Shim, K.I.; Kim, J.K.; Pelton, J.G.; Ryu, M.H.; Joo, J.C.; Han, J.W.; Kim, H.T.; Kim, K.H. Molecular insights into lignin depolymerization. ACS Catal. 2021, 11, 3996–4008. [Google Scholar] [CrossRef]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.L.; Texier, H.; Gavalda, S.; et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef]
- Singh, A.; Rorrer, N.A.; Nicholson, S.R.; Erickson, E.; DesVeaux, J.S.; Avelino, A.F.T.; Lamers, P.; Bhatt, A.; Zhang, Y.; Avery, G.; et al. Enzymatic and microbial degradation of plastics. Joule 2021, 5, 2479–2503. [Google Scholar] [CrossRef]
- Luo, Y.; Sun, J.; Li, Z. Catalytic lignin valorization in green chemical engineering. Green Chem. Eng. 2024, 5, 257–265. [Google Scholar] [CrossRef]
- Tanaka, S.; Koga, M.; Kuragano, T.; Ogawa, A.; Ogiwara, H.; Sato, K.; Nakajima, Y. Advanced materials for polymer degradation. ACS Mater. Au 2024, 4, 335–345. [Google Scholar] [CrossRef]
- Li, H.; Aguirre-Villegas, H.A.; Allen, R.D.; Bai, X.; Benson, C.H.; Beckham, G.T.; Bradshaw, S.L.; Brown, J.L.; Brown, R.C.; Cecon, V.S.; et al. Sustainable approaches in lignin valorization. Green Chem. 2022, 24, 8899–9002. [Google Scholar] [CrossRef]
- Majumdar, A.; Shukla, S.; Singh, A.A.; Arora, S. Resource conservation and recycling of polymers. Resour. Conserv. Recycl. 2020, 161, 104915. [Google Scholar] [CrossRef]
- Tanaka, S.; Sato, J.; Nakajima, Y. Catalytic strategies for lignin valorization. Green Chem. 2021, 23, 9412–9416. [Google Scholar] [CrossRef]
- Bai, X.; Aireddy, D.R.; Roy, A.; Ding, K. Catalytic depolymerization of lignin. Angew. Chem. Int. Ed. 2023, 62, e202309949. [Google Scholar] [CrossRef]
- Gordy, W.; Thomas, W.J.O. Nuclear magnetic resonance studies. J. Chem. Phys. 1956, 24, 439–444. [Google Scholar] [CrossRef]
- Chang, T.; Wang, Y.; Li, Z.; Hu, Y.; Shi, N.; Zou, X.; Li, C.; Xu, Y.; Li, N.; Guo, K. Advances in catalytic lignin processing. Catal. Today 2025, 452, 115241. [Google Scholar] [CrossRef]
- Rodríguez, M.P.; Vázquez-Vélez, E.; Galván-Hernández, A.; Martinez, H.; Torres, A. Polymer applications and sustainability. J. Appl. Polym. Sci. 2023, 140, e53763. [Google Scholar] [CrossRef]
- Shakiba, M.; Rezvani Ghomi, E.; Khosravi, F.; et al. Polymer advances and technologies. Polym. Adv. Technol. 2021, 32, 3368–3383. [Google Scholar] [CrossRef]
- Hirschberg, V.; Rodrigue, D. Polymer science insights. J. Polym. Sci. 2023, 61, 1937–1952. [Google Scholar] [CrossRef]
- Zhou, W.; Neumann, P.; Al Batal, M.; Rominger, F.; Hashmi, A.S.K.; Schaub, T. Catalytic lignin conversion. ChemSusChem 2021, 14, 4176–4183. [Google Scholar] [CrossRef]
- Coeck, R.; De Vos, D.E. Catalysis and green chemistry. Chem. Commun. 2024, 60, 1444–1447. [Google Scholar] [CrossRef]
- Kennedy, G.L.; Short, R.D. Toxicology reviews in polymers. CRC Crit. Rev. Toxicol. 1986, 17, 129–182. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Masunaga, H.; Numata, K. Sustainable polymer chemistry. ACS Sustain. Chem. Eng. 2025, 13, 3994–4004. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, G.; Zhu, X.; Zhang, Y.; Liang, C.; Chen, Y. Polymer science innovations. J. Polym. Sci. 2025. [Google Scholar] [CrossRef]
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polymer advances and applications. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef]
- Maiuolo, L.; Olivito, F.; Ponte, F.; Algieri, V.; Tallarida, M.A.; Tursi, A.; Chidichimo, G.; Sicilia, E.; De Nino, A. Reactive polymer chemistry. React. Chem. Eng. 2021, 6, 1238–1245. [Google Scholar] [CrossRef]
- Olivito, F.; Algieri, V.; Jiritano, A.; Tallarida, M.A.; Costanzo, P.; Maiuolo, L.; De Nino, A. Polymer synthesis and characterization. Polymers 2023, 15, 1785. [Google Scholar] [CrossRef]
- Olivito, F.; Jagdale, P.; Oza, G. Toxicology in polymer science. Toxics 2023, 11, 698. [Google Scholar] [CrossRef]
- Mouren, A.; Avérous, L. Polymer environment interactions. Eur. Polym. J. 2023, 197, 112338. [Google Scholar] [CrossRef]
- Corapi, A.; Gallo, L.; Lucadamo, L.; Tursi, A.; Chidichimo, G. Environmental toxicology and polymers. Environ. Toxicol. Chem. 2023, 42, 421–436. [Google Scholar] [CrossRef]
- Zubar, V.; Haedler, A.T.; Schütte, M.; Hashmi, A.S.K.; Schaub, T. Sustainable catalysis for polymers. ChemSusChem 2022, 15, e202101606. [Google Scholar] [CrossRef]
- Shukla, G.; Ferrier, R.C., Jr. Advances in polymer science. J. Polym. Sci. 2021, 59, 2704–2715. [Google Scholar] [CrossRef]
- Li, X.; Yu, Z.; Zhang, L. Applied polymer science. J. Appl. Polym. Sci. 2021, 138, e50154. [Google Scholar] [CrossRef]
- Maeno, Z.; Yamada, S.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K. Green polymer chemistry. Green Chem. 2017, 19, 2612–2619. [Google Scholar] [CrossRef]
- Ansmann, N.; Thorwart, T.; Greb, L. Innovative polymer approaches. Angew. Chem. Int. Ed. 2022, 61, e202210132. [Google Scholar] [CrossRef] [PubMed]
- Tschernuth, F.S.; Bichlmaier, L.; Inoue, S. Catalytic polymer chemistry. ChemCatChem 2023, 15, e202300281. [Google Scholar] [CrossRef]
- Ansmann, N.; Johann, K.; Favresse, P.; Johann, T.; Fiedel, M.; Greb, L. Mechanistic studies in polymer catalysis. ChemCatChem 2024, 16, e202301615. [Google Scholar] [CrossRef]
Scheme 1.
Mechanism of depolymerization of cellulose [49].
Scheme 1.
Mechanism of depolymerization of cellulose [49].
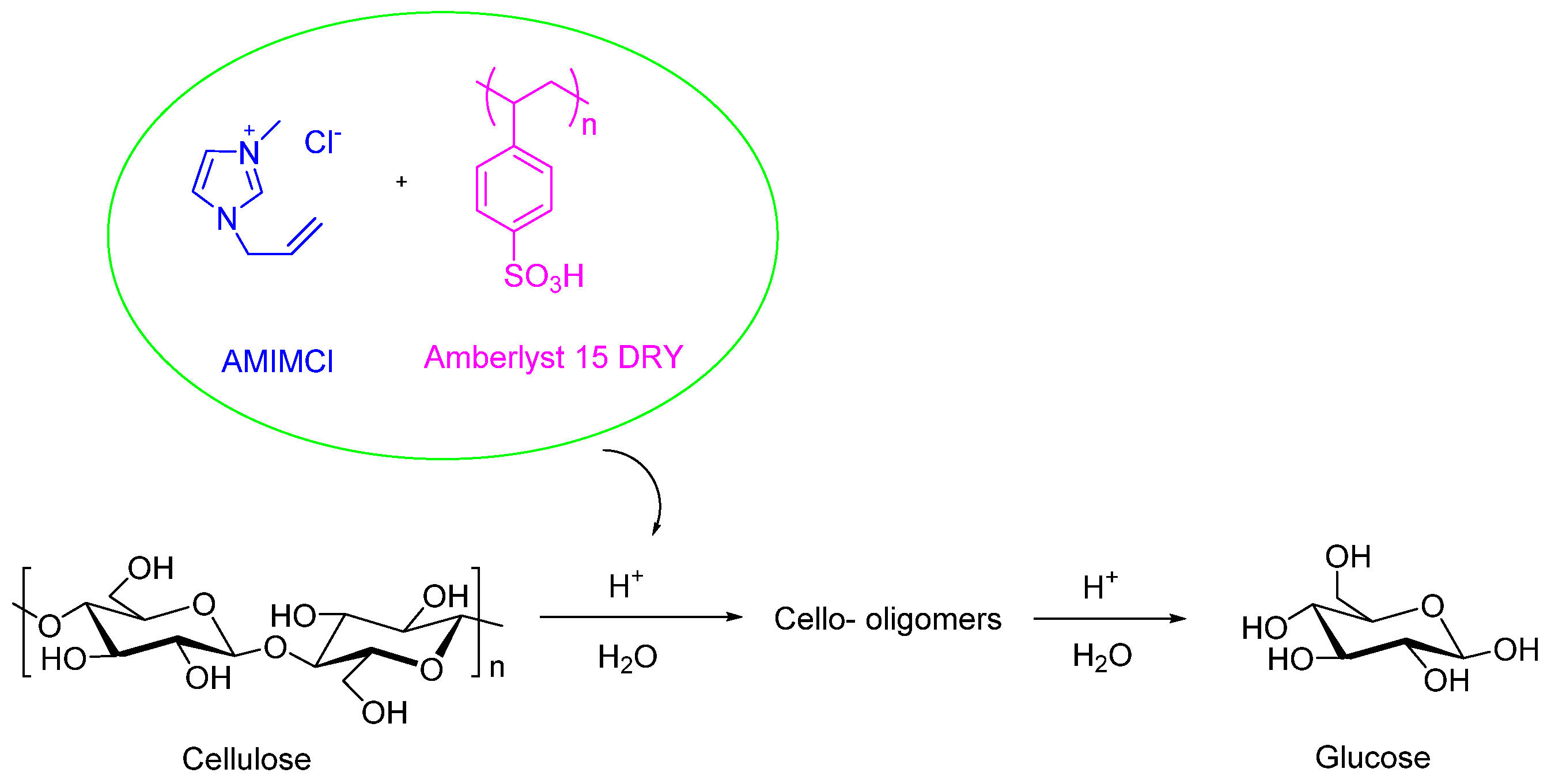
Figure 1.
Methodology for Screening Cellulose Catalytic Conversions [55].
Figure 1.
Methodology for Screening Cellulose Catalytic Conversions [55].
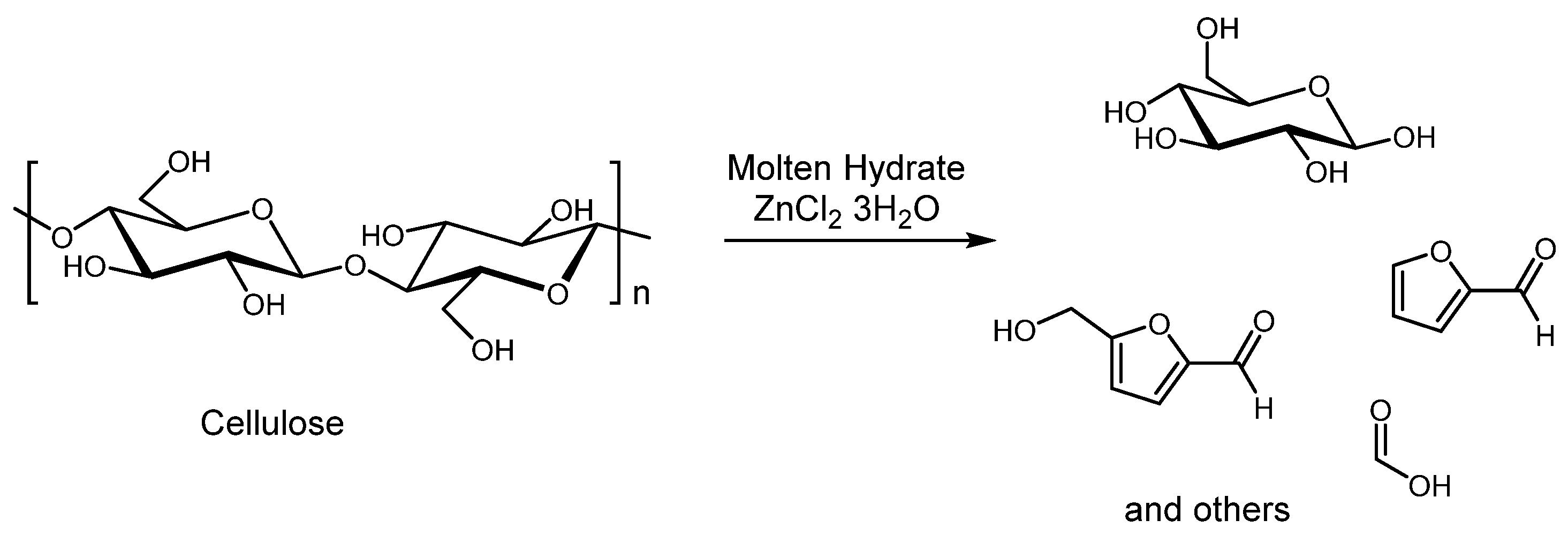
Figure 2.
Mechanocatalytic depolymerization of cellulose results in the formation of water-soluble oligomers [62].
Figure 2.
Mechanocatalytic depolymerization of cellulose results in the formation of water-soluble oligomers [62].
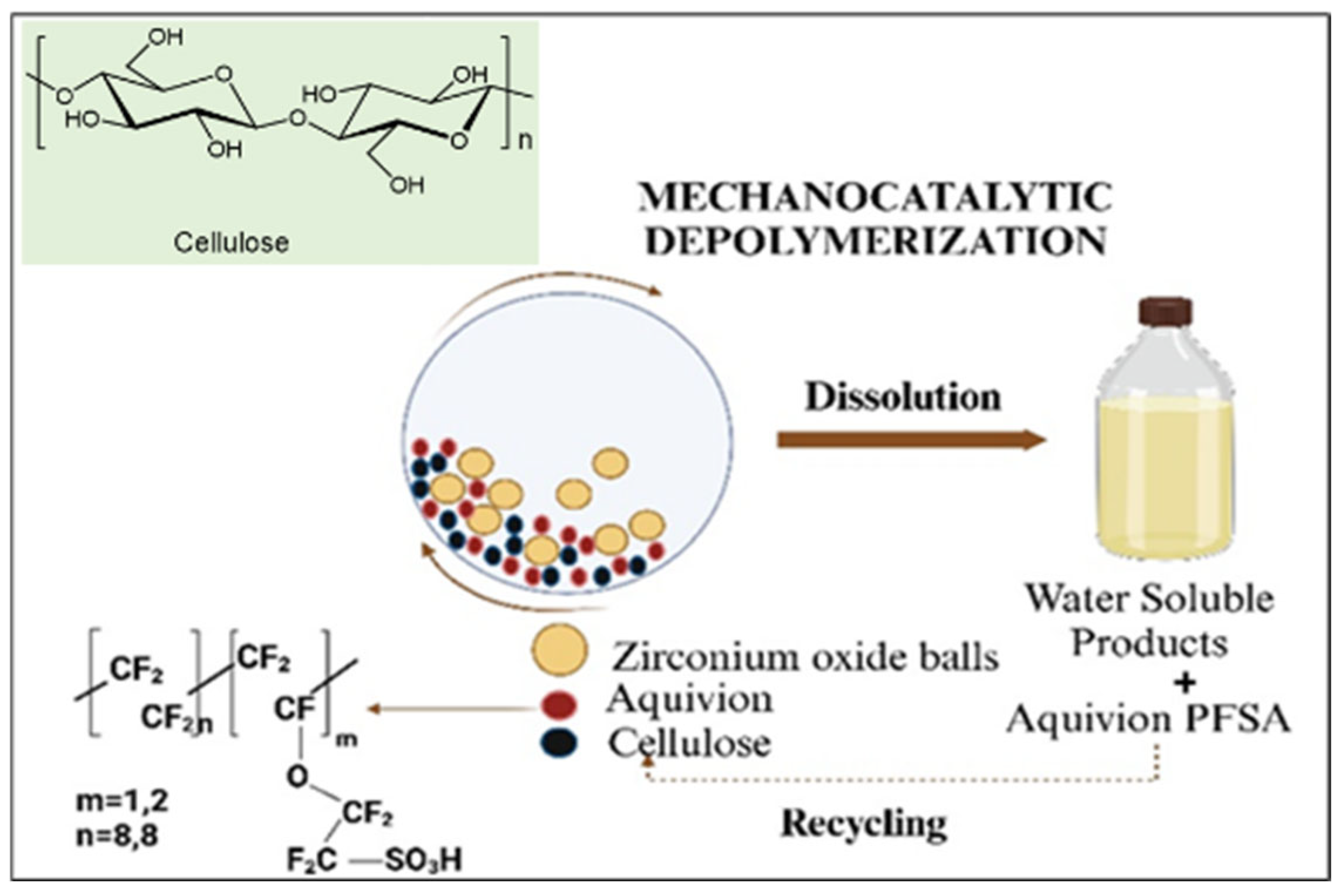
Figure 3.
A segment of macromolecular lignin structure with its H, G, and S moieties. The figure referred from [68].
Figure 3.
A segment of macromolecular lignin structure with its H, G, and S moieties. The figure referred from [68].
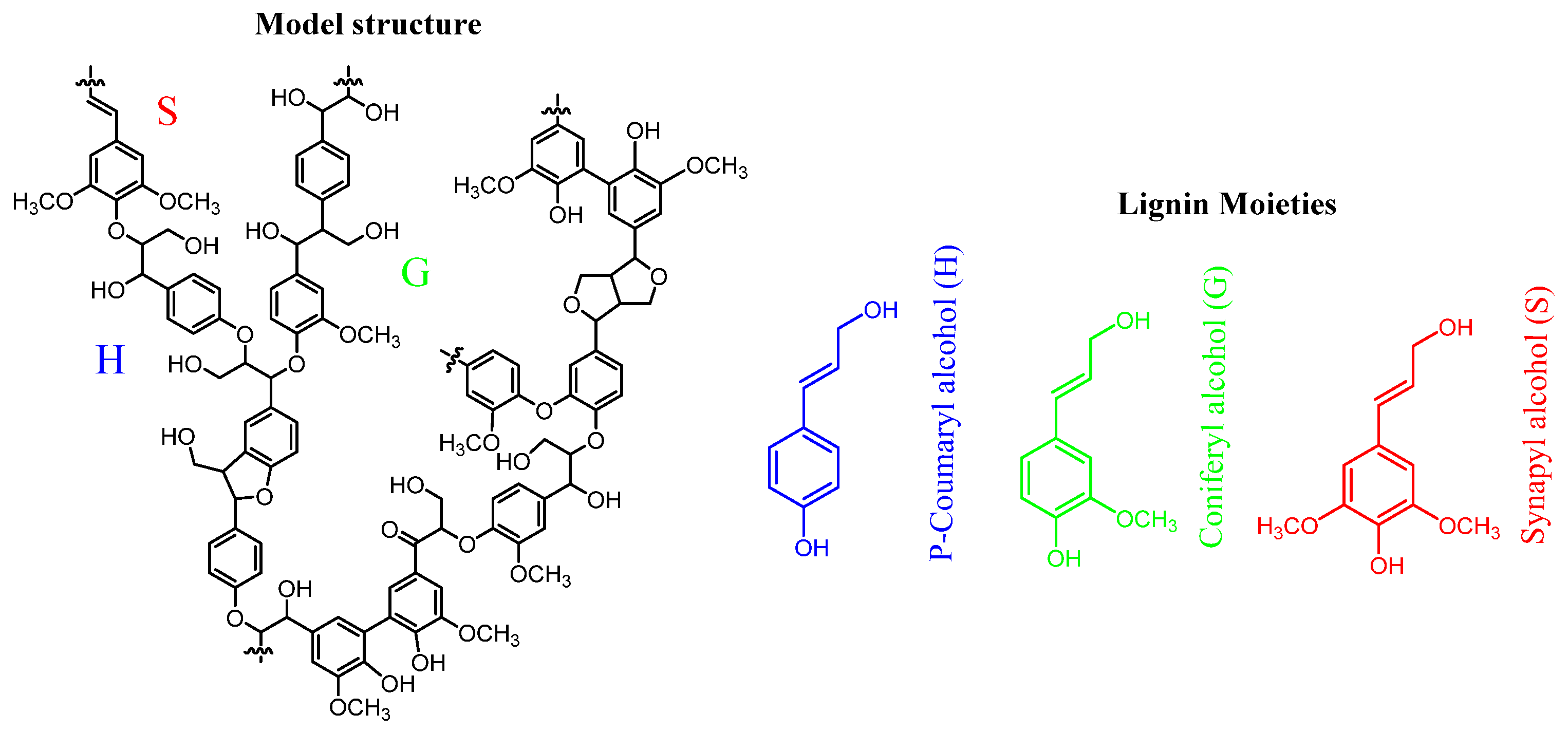
Figure 4.
Typical reaction mechanism of lignin under acidic conditions. (a) Self-condensation between lignin molecules; (b) reaction between lignin and phenol [76].
Figure 4.
Typical reaction mechanism of lignin under acidic conditions. (a) Self-condensation between lignin molecules; (b) reaction between lignin and phenol [76].
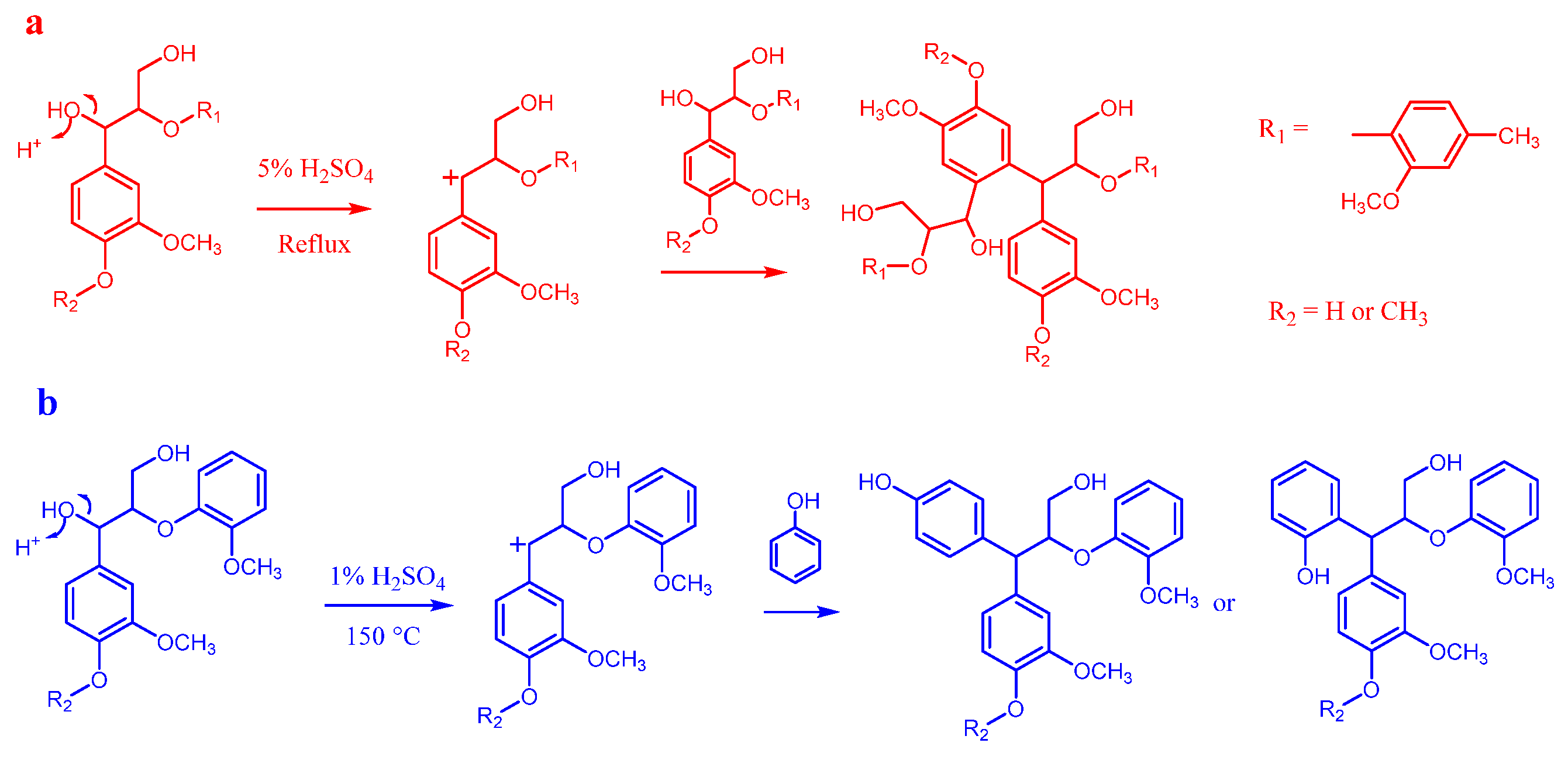
Scheme 2.
Lignin β-O-4 cleavage into vanillin assisted by base catalysis [85].
Scheme 2.
Lignin β-O-4 cleavage into vanillin assisted by base catalysis [85].
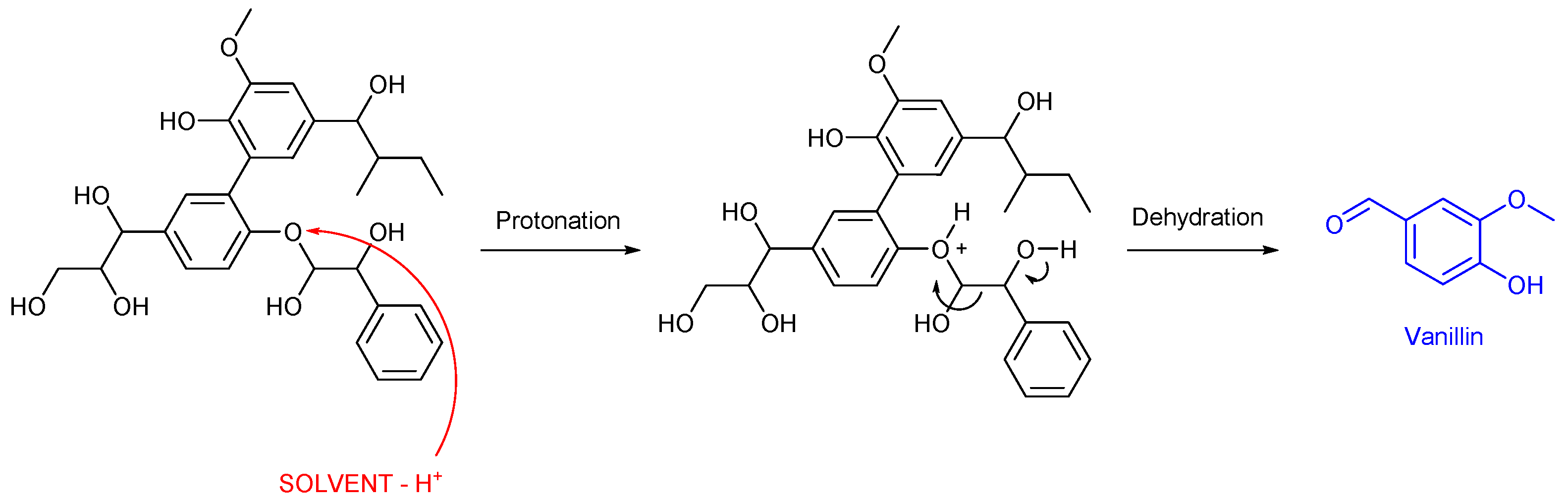
Scheme 3.
Oxidative lignin depolymerization into aromatic compounds [93].
Scheme 3.
Oxidative lignin depolymerization into aromatic compounds [93].
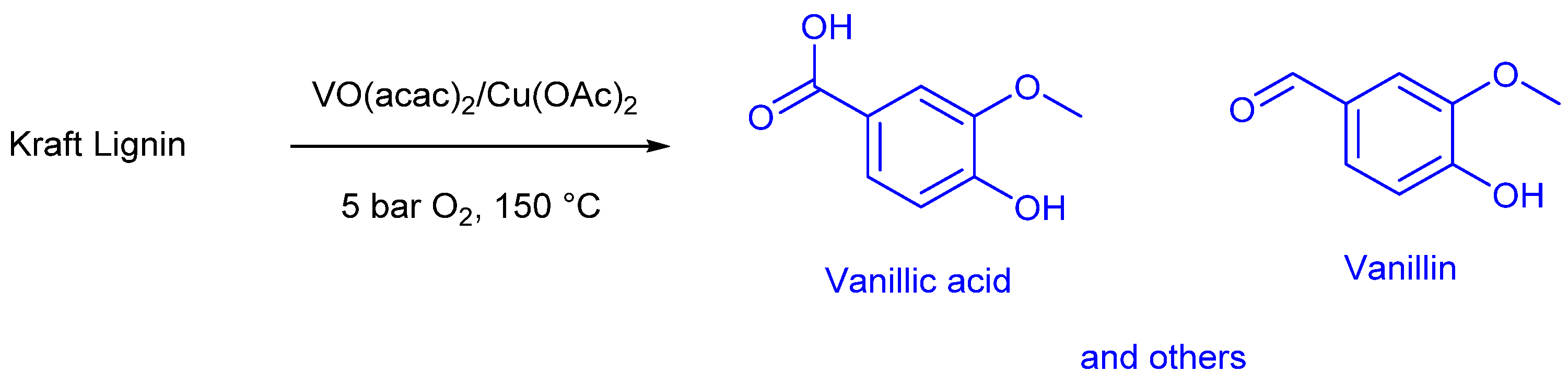
Scheme 4.
Depolymerization of lignin model compounds depolymerized by Mediator-Enzyme System [115].
Scheme 4.
Depolymerization of lignin model compounds depolymerized by Mediator-Enzyme System [115].
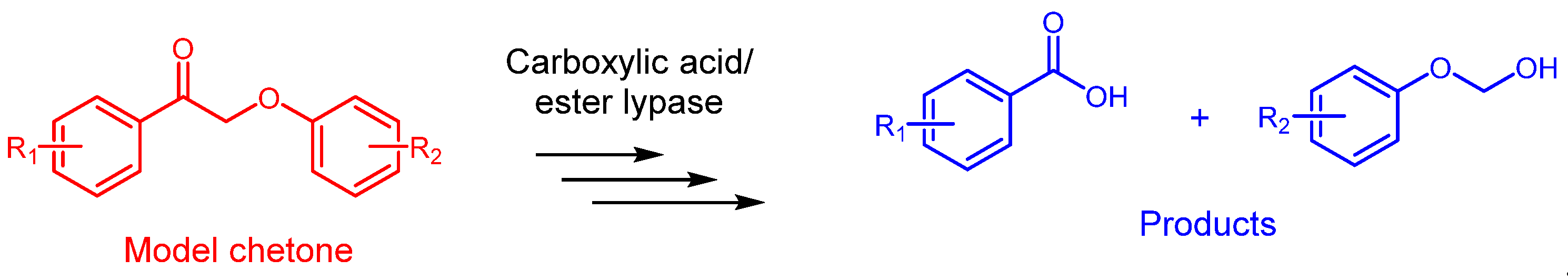
Figure 5.
A schematic representation of the reaction process starting from PET [122].
Figure 5.
A schematic representation of the reaction process starting from PET [122].
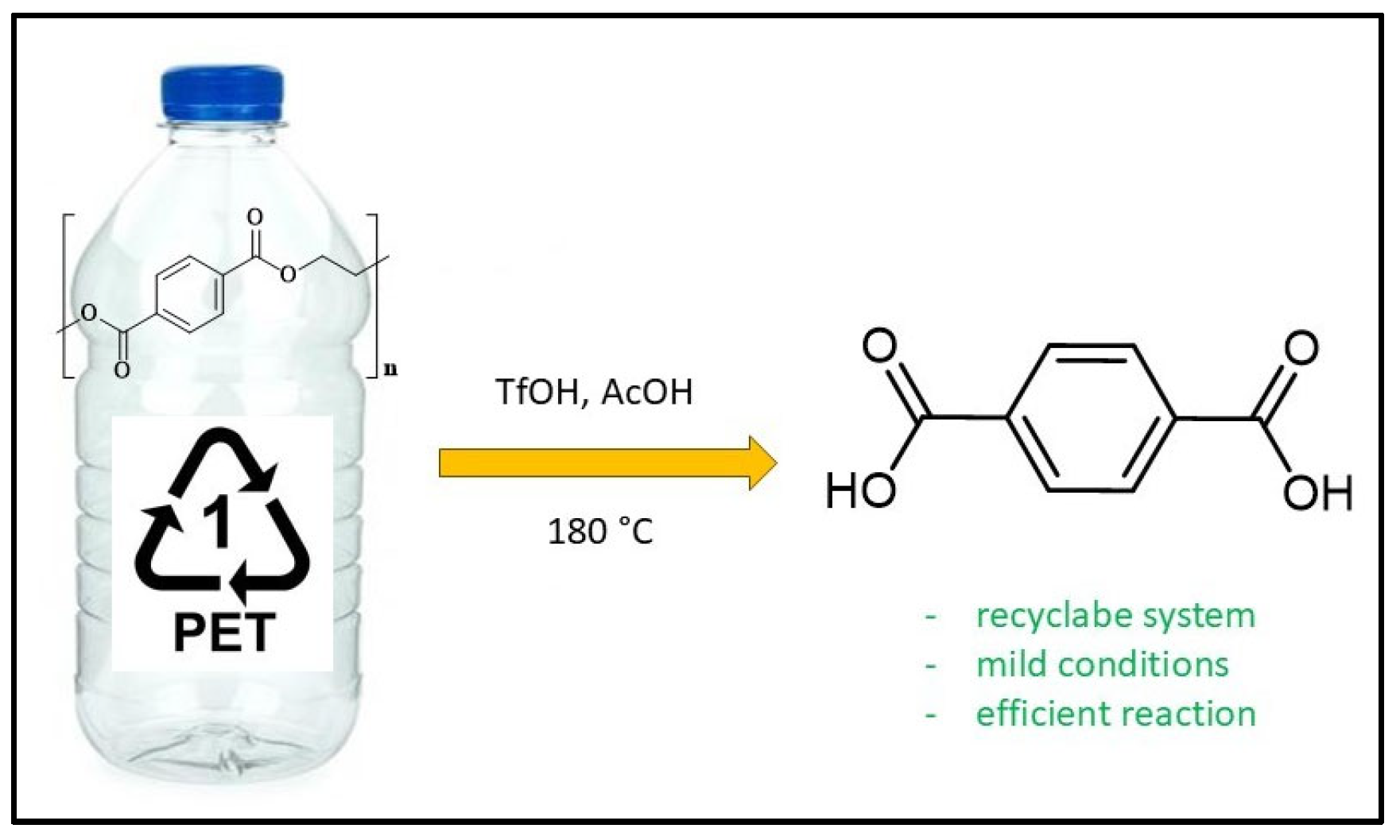
Figure 6.
A schematic representation of the reaction process starting from PET [122].
Figure 6.
A schematic representation of the reaction process starting from PET [122].
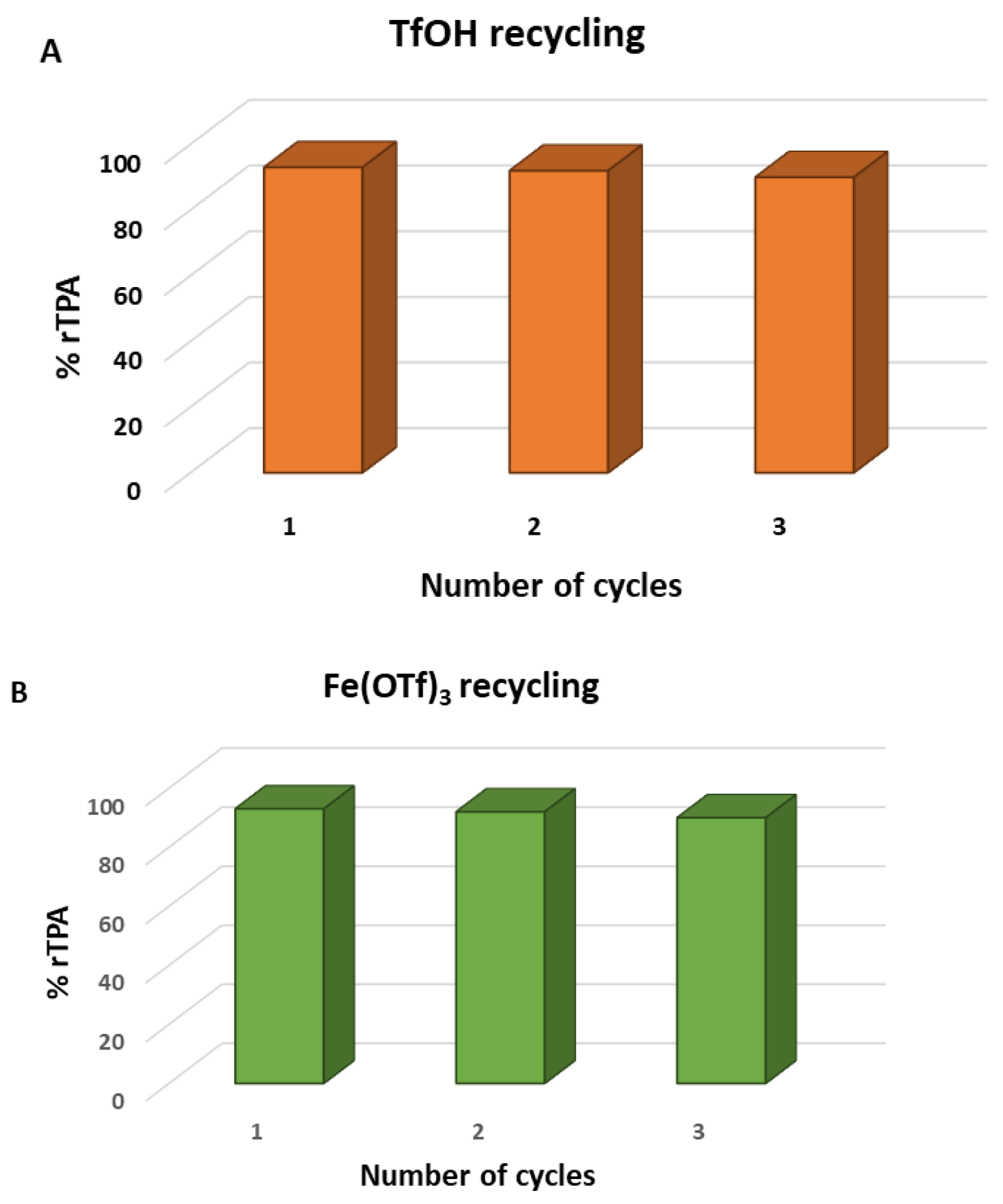
Scheme 5.
Mechanism relative to the overall process divided in 3 reactions (A, B and C) [123].
Scheme 5.
Mechanism relative to the overall process divided in 3 reactions (A, B and C) [123].
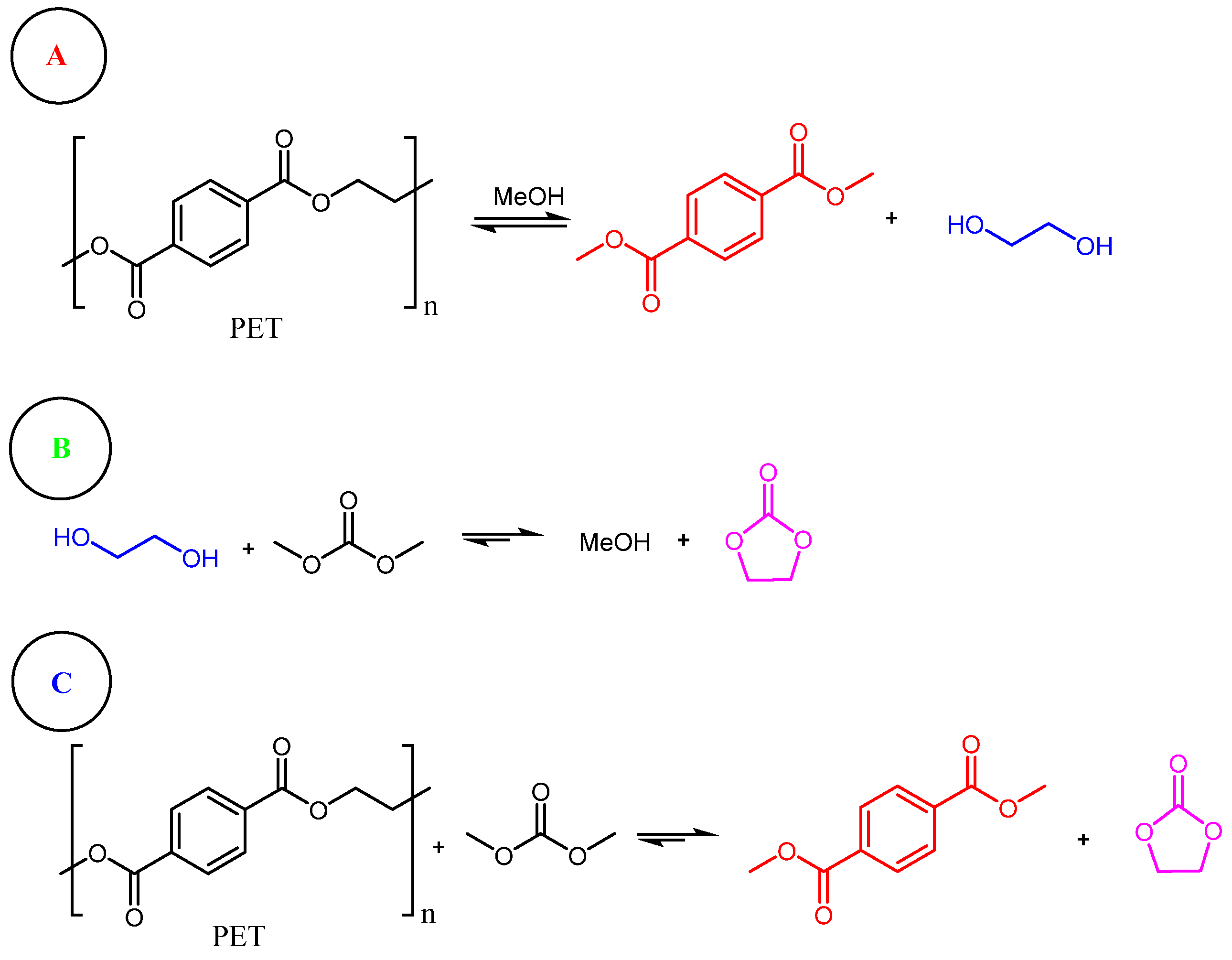
Figure 7.
A schematic representation of the double metal cyanide (DMC) complex catalyst [129].
Figure 7.
A schematic representation of the double metal cyanide (DMC) complex catalyst [129].
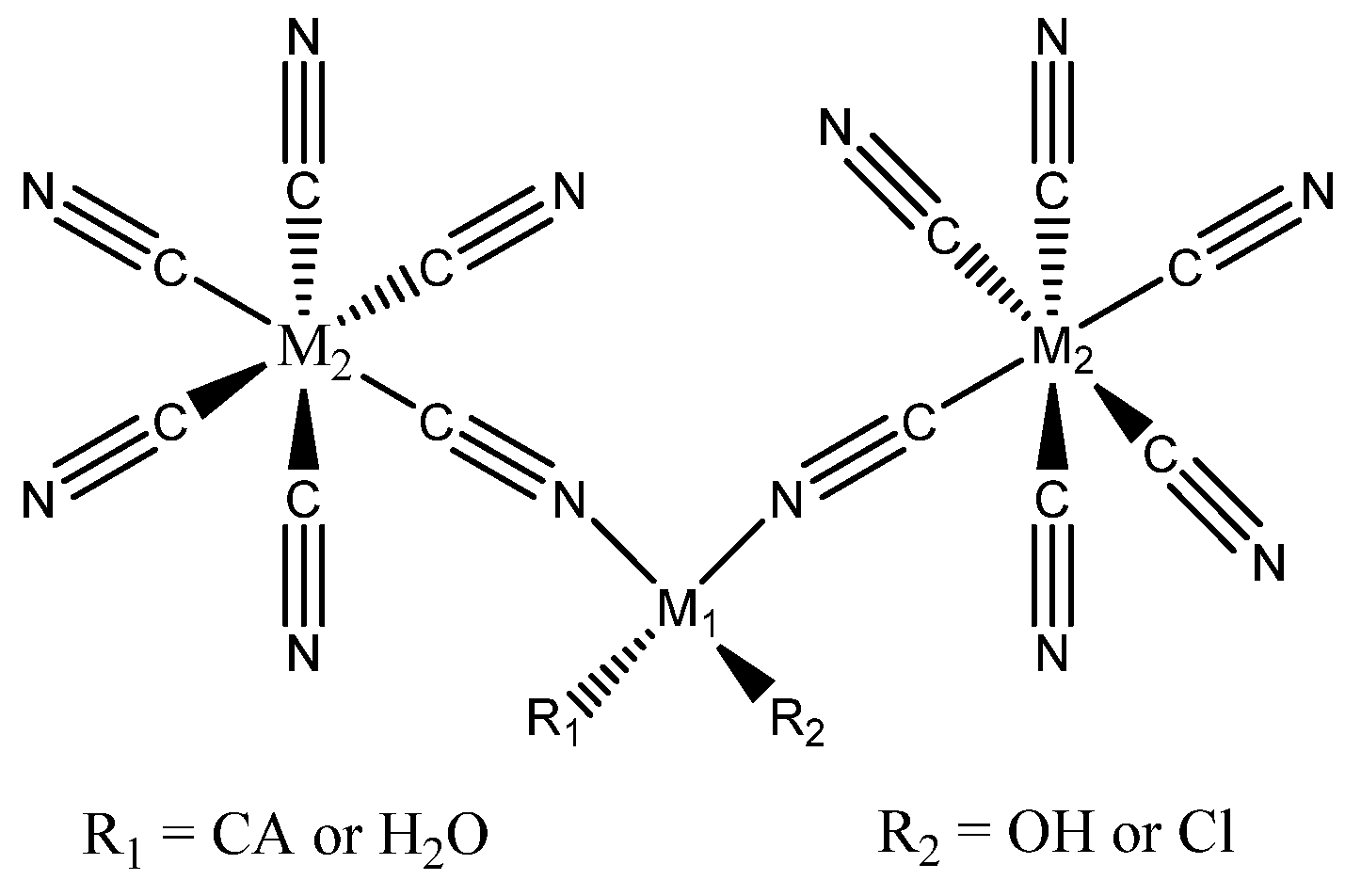
Scheme 6.
Ru pincer complex used to depolymerize low molecular weight polyamide [133].
Scheme 6.
Ru pincer complex used to depolymerize low molecular weight polyamide [133].
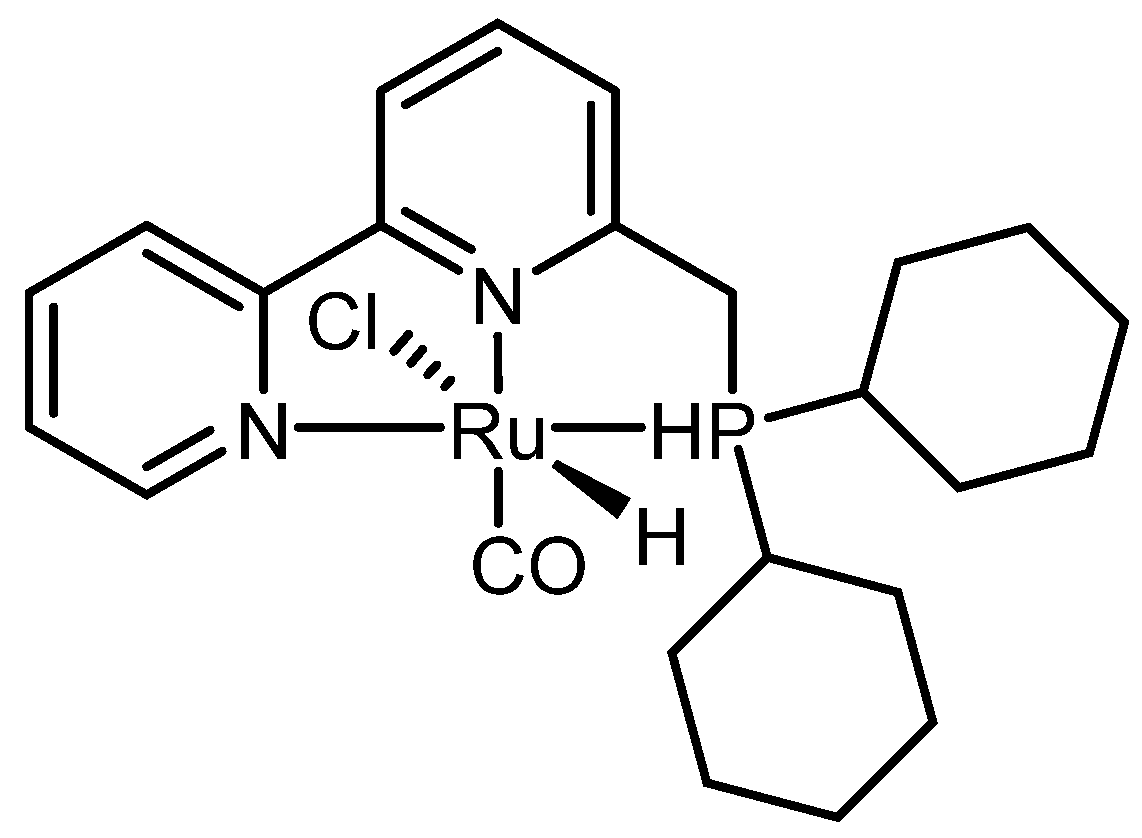
Figure 8.
Reaction scheme for the depolymerization of polyamides (Nylon 66) [134].
Figure 8.
Reaction scheme for the depolymerization of polyamides (Nylon 66) [134].
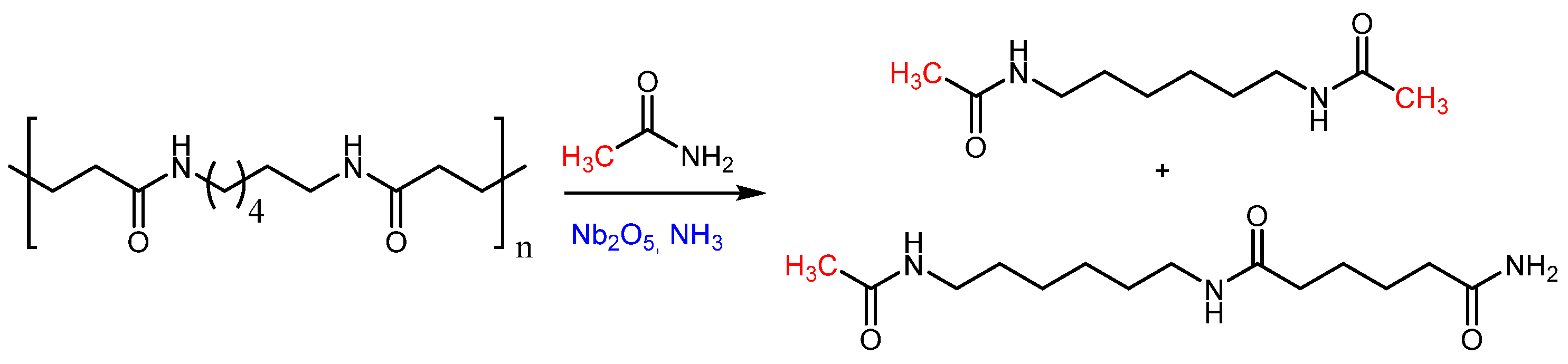
Scheme 7.
Chemoenzymatic Polymerization/Depolymerization of Semiaromatic Polyamides [136].
Scheme 7.
Chemoenzymatic Polymerization/Depolymerization of Semiaromatic Polyamides [136].
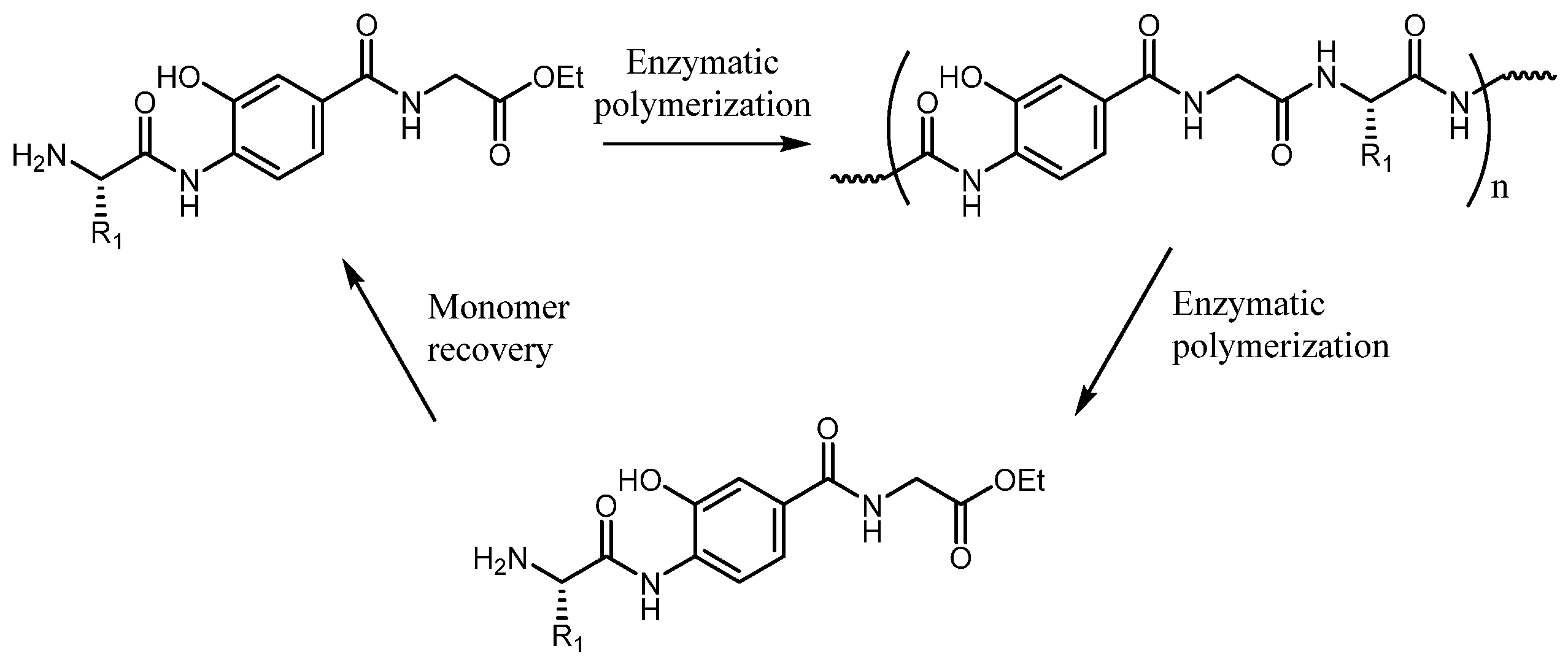
Scheme 8.
Mn pincer complex used to depolymerize polyurethanes [144].
Scheme 8.
Mn pincer complex used to depolymerize polyurethanes [144].
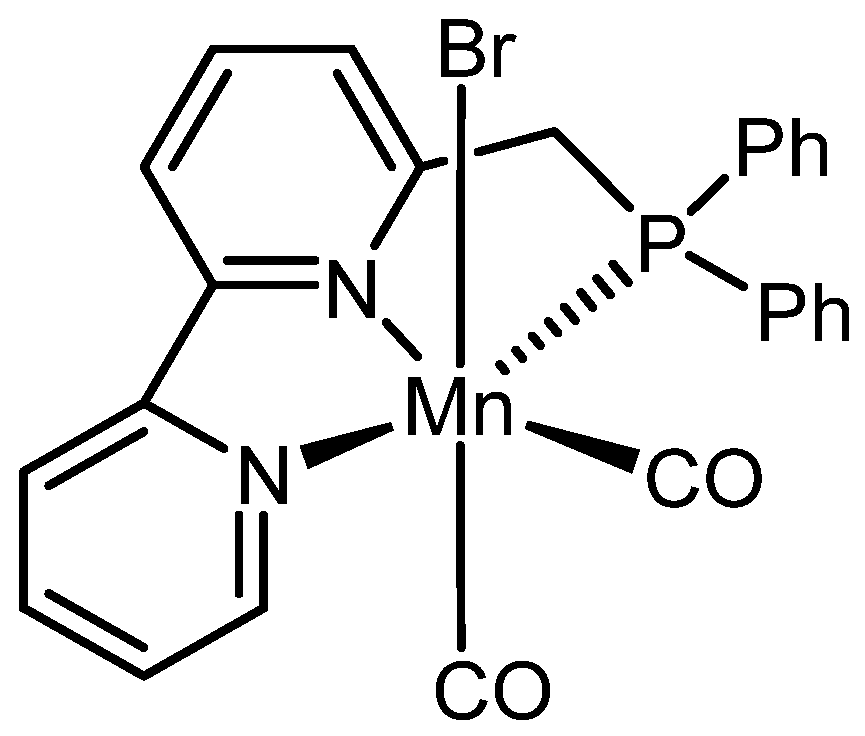
Figure 9.
Catalytic depolymerization of three different polyurethanes (A, B, C) with the relative optimized reaction conditions and isolated yield.
Figure 9.
Catalytic depolymerization of three different polyurethanes (A, B, C) with the relative optimized reaction conditions and isolated yield.
Scheme 9.
Lewis superacid bis(perchlorocatecholato)silane as a catalyst for C-O bond metathesis of polyethers [148].
Scheme 9.
Lewis superacid bis(perchlorocatecholato)silane as a catalyst for C-O bond metathesis of polyethers [148].
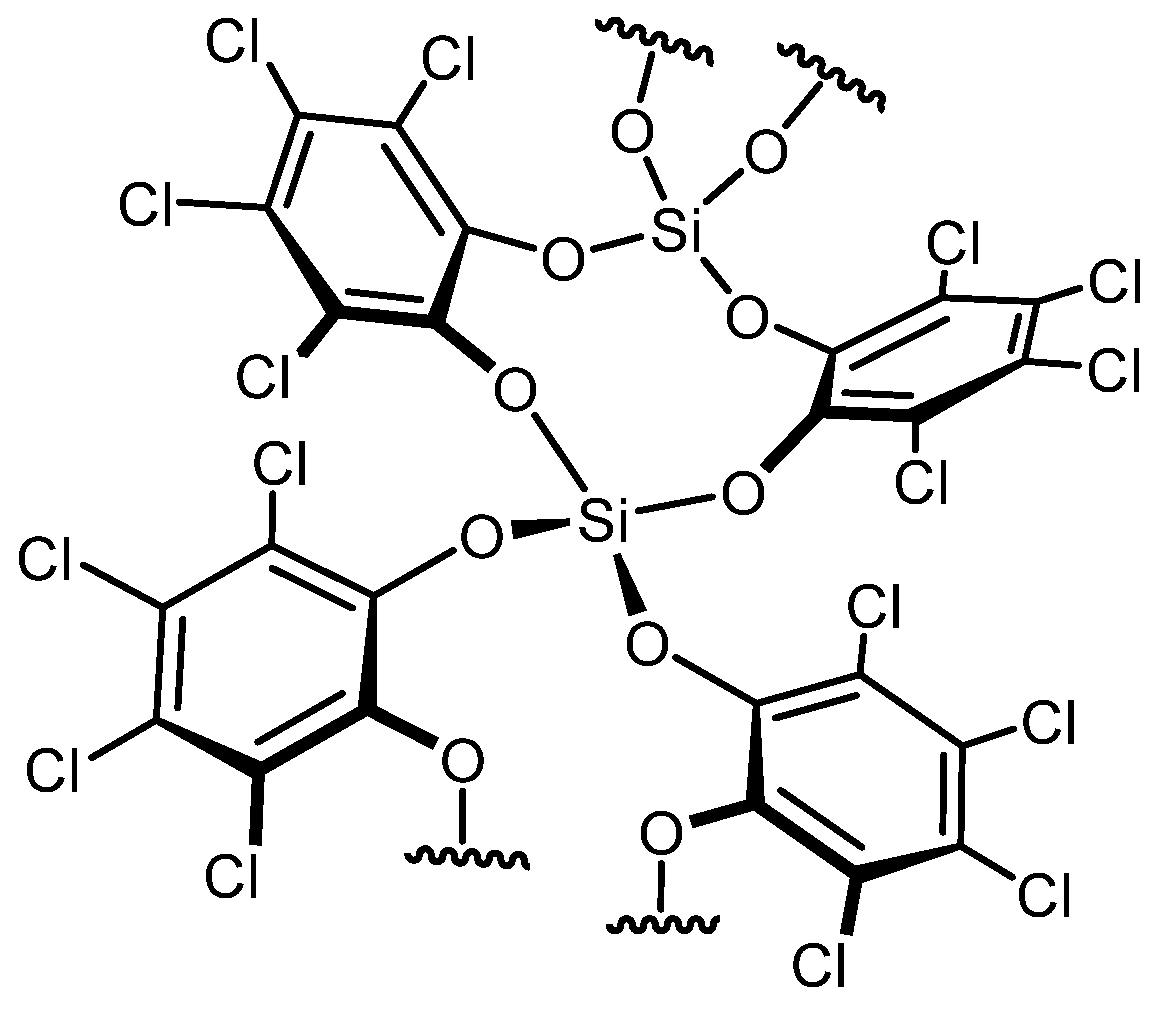
Figure 10.
Reaction scheme of the Lewis acid catalyzed selective degradation of 1,5-dimethoxypentane (A) and selective ring closing C-O bond metathesis of polyethylene glycol and polypropylene glycol (B) [148].
Figure 10.
Reaction scheme of the Lewis acid catalyzed selective degradation of 1,5-dimethoxypentane (A) and selective ring closing C-O bond metathesis of polyethylene glycol and polypropylene glycol (B) [148].
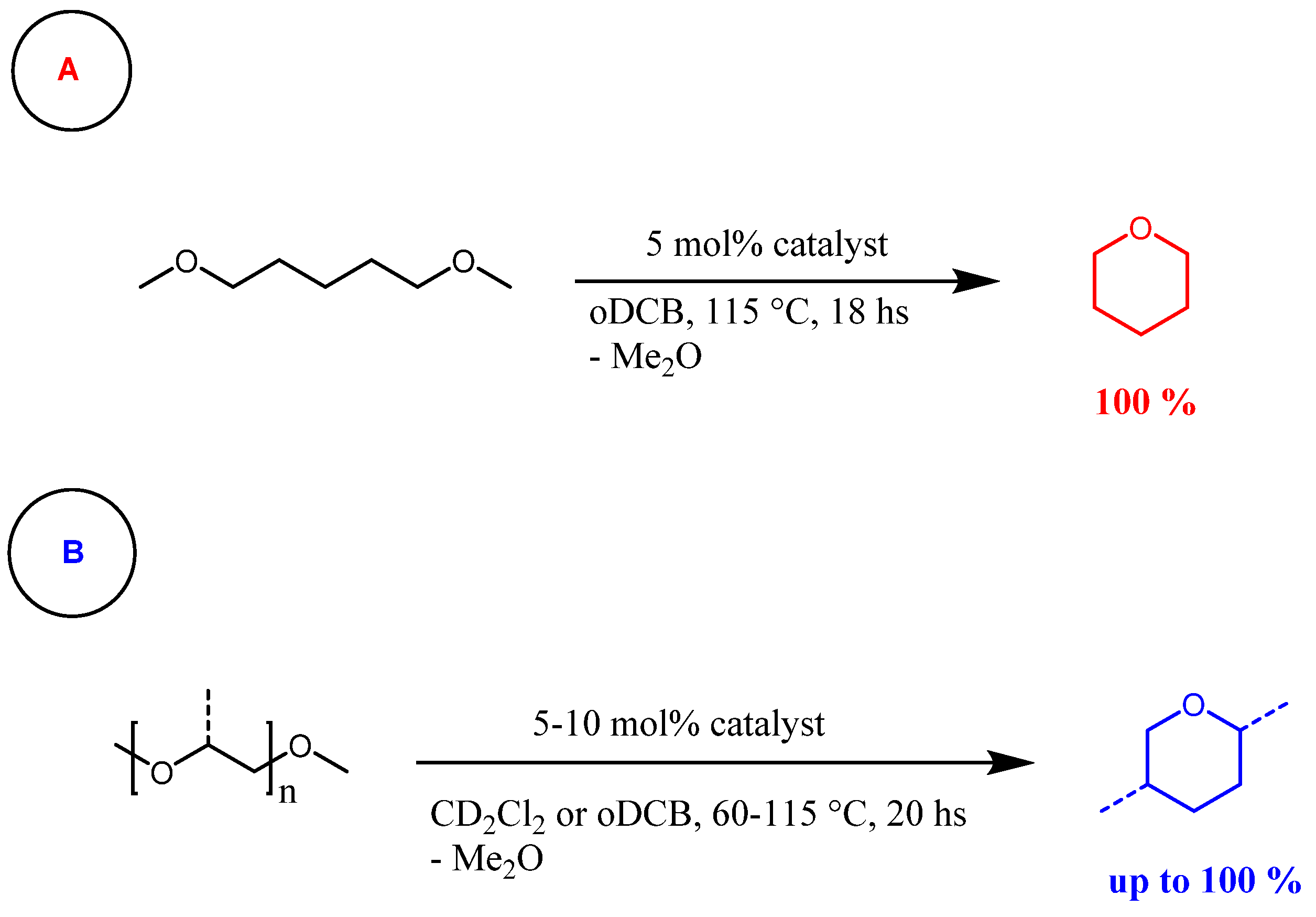
Scheme 10.
Acetonitrile-adduct of a Lewis superacidic silane (Si(pinF)2·MeCN) as a catalyst for C-O bond metathesis of polyethers [149].
Scheme 10.
Acetonitrile-adduct of a Lewis superacidic silane (Si(pinF)2·MeCN) as a catalyst for C-O bond metathesis of polyethers [149].
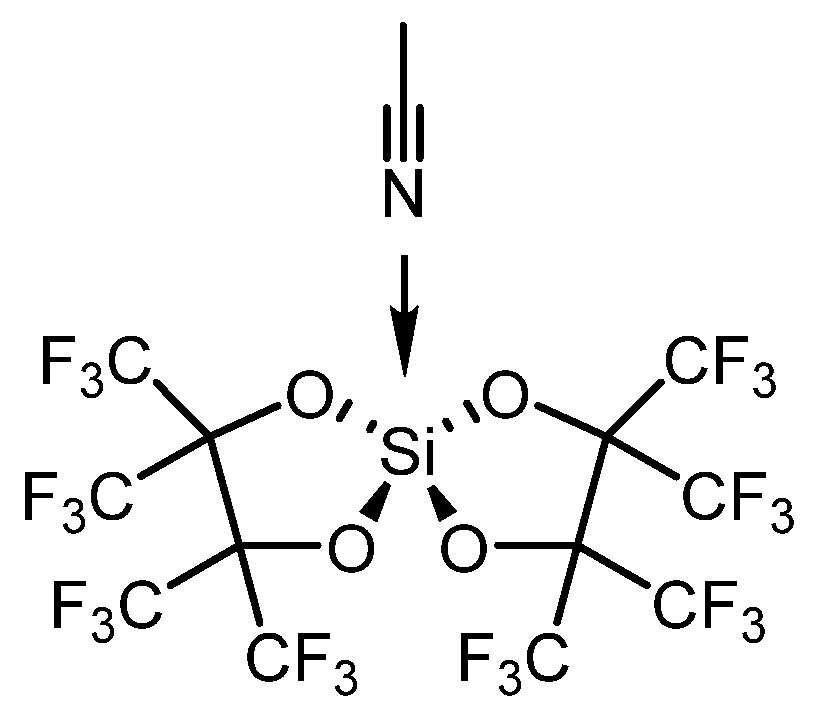

Table 1.
Sources of cellulose with their Mean DPw peak values for cello-oligomers and Percentage change of DPw.
Table 1.
Sources of cellulose with their Mean DPw peak values for cello-oligomers and Percentage change of DPw.
| Source of cellulose | Mean DPw peak 0 min and 120 min |
Percentage change of DPw ΔDPw (%) | |
|---|---|---|---|
| Sawdust | 351 | 72 | 79.5 |
| Sago pith wastes | 564 | 30 | 94.7 |
| Corn Cob | 329 | 88 | 78.1 |
| Sugarcane bagasse | 571 | 37 | 93.5 |
Table 2.
Cellulose conversion in molten salt hydrates solutions with catalysts.
| Substrate | MSH/Catalyst | Main products (%yield) | Ref. |
|---|---|---|---|
| MCC | Beta and ZSM-5 zeolites with SiO2/Al2O3 = 30 and 50 | Gluco oligomers (54.4%), glucose (28.3%) & HMF (0.2%) | [52] |
| MCC | ZnCl2 (72wt%)/HCl (0.2 M) | HMF (69.5%) | [57] |
| Cellulose | ZnCl2·3H2O/SO4/TiO2 | Gluco oligomers (9.4%), glucose (50.5%), HMF (3.4%), fructose (5.9%), & LA (5.1%) | [58] |
| MCC | LiBr (55 wt%) Activated/Activated carbon | Glucose (80%) & LA (4%) | [59] |
Table 3.
Mechanocatalytic depolymerization of cellulose in the presence of different solid acid catalysts.
Table 3.
Mechanocatalytic depolymerization of cellulose in the presence of different solid acid catalysts.
| Catalyst | H+ exchange capacity(mmol/g) | Solubility (%) |
|---|---|---|
| Blank | - | <5 |
| Aquivion PW98 | 1.0 | 90 |
| SBA – SO3H | 0.2 | 60 |
| CMK-3-SO3H | 0.7 | 87 |
| Kaolinite | - | 50 |
| Aquivion PW66 | 1.45 | 99 |
| Aquivion PW79 | 1.26 | 32 |
| Aquivion PW87 | 1.15 | 80 |
Table 4.
Zirconia and other grinding balls with their properties.
| Material | Diameter (mm) | Density (g/cm−3) |
|---|---|---|
| Zirconia | 3 | 5.68 |
| Stainless steel | 2 | 7.8 |
| Stainless steel | 4 | 7.8 |
| Tungsten carbide | 3 | 15.63 |
Table 5.
Catalysts and products yield by mechanocatalytic process. a.
| Catalyst | Products yield (%) |
|---|---|
| HNbMoO6 | 14 |
| kaolinite | 4 |
| NiO | 0.3 |
| SnO2 | 0.6 |
| TiO2 | 0.5 |
| Nb2O5 | 0.9 |
| H-Montmorillonite | 3 |
| USY zeolite | 3 |
| Mg–Al HT | 0 |
a Yields refer to reaction stopped after 4 hours.
Table 6.
Reaction optimization for the acidolysis depolymerization of PET. [a]
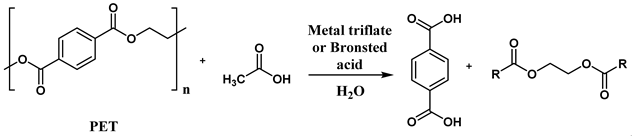 | ||||
|---|---|---|---|---|
| Catalyst | T (°C) | Ccat (M) | CH20 (M) | Yield (%) |
| Hf(OTf)4 | 180 | 0.25 | 0 | 60 |
| Hf(OTf)4 | 180 | 0.25 | 0.5 | 72 |
| Fe(OTf)3 | 180 | 0.25 | 0.5 | 64 |
| Al(OTf)3 | 180 | 0.25 | 0.5 | 60 |
| TfOH | 180 | 0.25 | 0.5 | 98 |
| Tf2NH | 180 | 0.25 | 0.5 | 58 |
[a] Reactions conditions: 0.5 mmol PET was put in 0.2 mL of Cat./H2O/AcOH solution in a capped vial at 180 °C for 40 min.
Table 7.
Reaction optimization for the dimethyl carbonate-aided methanolysis depolymerization of PET. [a]
Table 7.
Reaction optimization for the dimethyl carbonate-aided methanolysis depolymerization of PET. [a]
| Catalyst | DMC (mL) | MeOH (mL) | DMT (%) | EC (%) |
|---|---|---|---|---|
| LiOMe | 1.5 | 0.2 | 83 | 70 |
| KOMe | 1.5 | 0.2 | 95 | 93 |
| NaOMe | 1.5 | 0.2 | 95 | 86 |
| NaOMe [b] | 1.5 | 0.2 | 93 | 92 |
| NaOMe | 1 | 0.13 | 98 | 67 |
| NaOMe | 0.5 | 0.065 | 93 | 60 |
[a] PET textile (100 mg), catalyst (5 mol % based on the PET alternating unit), DMC (1.5 mL), MeOH (0.2 mL), 50 °C, and 2 h. Abbreviations. DMC: dimethyl carbonate, DMT: dimethyl terephthalate, EC: ethylene carbonate. [b] Reaction time: 1 hour.
Table 8.
Reaction optimization for the depolymerization of low molecular weight polyamide 66 using Ru pincer complex. [a].
Table 8.
Reaction optimization for the depolymerization of low molecular weight polyamide 66 using Ru pincer complex. [a].
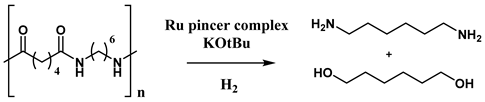 | |||
|---|---|---|---|
| T (°C) | P (H2) Bar | Yield (%) Diamine | Yield (%) Diol |
| 150 | 70 | 12 | < 5 |
| 180 | 100 | 60 | 35 |
| 200 | 100 | 78 | 62 |
| 200 | 80 | 70 | 47 |
[a] Low-molecular weight polyamide (0.3 g, 1.25 mmol according to the repeating unit of polyamide 66), Ru pincer complex as catalyst (0.01 mmol), KOtBu (0.04 mmol), THF (5 mL).
Table 9.
Reaction optimization for the degradation of 1,5-dimethoxypentane (1,5-DMP), diglyme, poly(ethyleneglycol)dimethylether (PEG-DME) and 18-crown-6. [a]
Table 9.
Reaction optimization for the degradation of 1,5-dimethoxypentane (1,5-DMP), diglyme, poly(ethyleneglycol)dimethylether (PEG-DME) and 18-crown-6. [a]
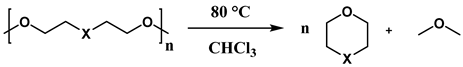 | |||
|---|---|---|---|
| Substrate | Catalyst (%mol) | Time (h) | Yield (%) |
| 1,5 DMP | 1-5 | 1-20 | 96-97 |
| Diglyme | 1-5 | 2.5-96 | 96-99 |
| 18-crown-6 | 30 | 30 | 88 |
| PEG-DME | 225 | 18 | 87 |
[a] %Yield of tetrahydropyran and 1,4-dioxane was conducted by 1H NMR using mesitylene as an internal standard.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



