
Submitted:
29 April 2025
Posted:
30 April 2025
You are already at the latest version
Abstract
Solid-state Nuclear Magnetic Resonance (ssNMR) spectroscopy enables studying complex macromolecules with low solubility. Compared to solution NMR, few tools exist for biomacromolecule-ssNMR data analysis. A key challenge is assigning spin systems due to low peak dispersion. Broad peaks from large quadrupolar couplings and shift anisotropy cause significant overlap and missing peaks. To address this, we introduce ssPINE-POKY, a user-friendly graphical user interface (GUI) integrated into the POKY suite. ssPINE-POKY streamlines the automation of spin system recognition and chemical shift assignment in multidimensional ssNMR spectra by integrating the ssPINE algorithm within an intuitive interface. The platform allows easy and fast job submission, real-time result visualization, and enhanced analysis through additional built-in tools, significantly improving the efficiency of ssNMR data interpretation.
Keywords:
ssPINE
; ssPINE-POKY
; POKY
; ssNMR
; Protein
; Assignment
; Automation
; AI
1. Introduction
Liquid-state nuclear magnetic resonance (NMR) spectroscopy provides detailed information about the structure and dynamics of biomolecules in solution, such as proteins, nucleic acids, and carbohydrates. However, some biomolecules, such as membrane proteins, amyloid fibrils, and large macromolecular assemblies, are challenging to study using liquid-state NMR due to their low solubility and structural complexity. Therefore, magic angle spinning (MAS) solid-state NMR (ssNMR) spectroscopy has become a powerful alternative for studying such systems, as it enables the investigation of complex biomolecules [1,2].
Recent advances in MAS ssNMR have improved the ability to study complex biomolecular systems at atomic resolution. For instance, techniques for solvent suppression, hydrogen/deuterium (H/D) exchange, and deuterium spectroscopy have been developed to improve spectral quality [3]. The use of perdeuterated and labeled proteins combined with ultrafast MAS has demonstrated remarkable improvements in spectral resolution [4]. Additionally, deuterium quadrupolar interactions have enabled site-specific characterization of molecular motions without the need for proton decoupling [5]. Furthermore, the emergence of proton-detected multidimensional experiments under ultrafast MAS conditions has opened new possibilities for studying protein structure and dynamics, even in the absence of complete deuteration [6]. These methodological innovations, together with the application of hybrid structural approaches, have contributed to the high-resolution structure determination of different biomolecule complexes [7,8].
Often, this investigation involves multidimensional NMR techniques, and the pulse sequences have been actively developed to observe different aspects and responses of proteins. The number of pulse sequences is increasing rapidly [2,9,10]; however, the analysis of the resulting multidimensional NMR spectra remains a significant challenge. Once NMR spectra are acquired from a protein sample, most tasks are performed on computers. Therefore, lack of user-friendly software tools for automation would be a huge drawback, which is partly true with biomolecular ssNMR spectroscopy. Considering the potential of ssNMR on challenging and complex biomolecules described above, it is important to develop critical tools analogous to existing in solution NMR.
Tasks in solution NMR are more matured and streamlined than them in ssNMR. For example, the POKY suite provides all-in-one features for routinely conducted analytical activities, which is a great advanced compared to its ancestor, NMRFAM-SPARKY [11,12]. Peak picking is one of the first steps for the processed frequency-domain data from the time-domain data. The APES plugin (two-letter-code ae) of POKY automates peak picking on common twenty solution NMR spectra and cross-validates between them to filter out noise peaks [13]. Subsequently, these identified peaks on spectra are used as inputs to backbone and sidechain chemical shift assignment steps. Combination of the I-PINE webserver and the PINE-SPARKY.2 plugin (two-letter-code ep) provide automated chemical shift assignments for both backbone and sidechain atoms. They support twenty-nine solution NMR experiments, and versions in POKY which has the capability of integrating artificial intelligence (AI)-based three-dimensional (3D) structure and chemical prediction methods in the assignment over versions in NMRFAM-SPARKY. Combined automated peak picking and assignment of NOESY spectra and calculation of 3D structure of a protein is offered in the PONDEROSA plugin (two-letter-code c3). Obtained near complete backbone and sidechain chemical shifts are required to use the plugin. As a matter of fact, automated peak picking, assignment and 3D structure calculation can be fully automated by only running the APES plugin because APES calls PINE-SPARKY.2 that runs the I-PINE webserver if the user agrees and the I-PINE webserver runs PONDEROSA-C/S (client-server) when NOESY data are available. Additionally, the user can intervene any of these steps to improve quality of results, and the existence of these automation tools in the POKY suite provides seamless user experience of the Integrative NMR research platform unlike other automation programs.
This kind of streamlined routine automation tools has focused more on data from solution NMR despite ssNMR’s potential in challenging systems. The large number of peaks and complex coupling patterns in these spectra make it difficult to assign the signals to specific atomic sites and extract meaningful structural information. Still only a limited number of tools [14,15,16,17,18,19] are available for the analysis of complex ssNMR data, and most of these require operation through a command-line interface, requiring coding knowledge to be run. Additionally, users must manually prepare input files, making the process time-consuming and not user-friendly.
To address this issue, we have been implementing new tools in the POKY suite recently. For example, the iPick peak picking program has been made to support both solution NMR and ssNMR. It adopts APES’s cross-validation feature across different experiments. While APES is limited in fixed solution NMR experiments, iPick’s versatile design enables the user to use a wider array of solution and solid-state NMR experiments. Because ssNMR spectra are more difficult for peak picking programs, we have made REDEN to provide a way to investigate clustered regions with multiple peaks on top of each other resulting in a poor resolution. The ssPINE program has been made as an automated algorithm for ssNMR spectra [20]. It automates the process of recognizing, categorizing, and assigning signals from various types of multidimensional ssNMR spectra. This software uses a combination of automated methods to facilitate assignment, and analysis of ssNMR data. With the sequence of the protein and peak lists from fourteen different 2D and 3D ssNMR experiments as inputs, ssPINE generates backbone and side-chain assignments. We have made a web submission form to run ssPINE and receive results via email. Once chemical shifts are assigned, PONDEROSA-C/S in the POKY suite can be used to automate analysis of long-mixing time versions of 2D-(H)CC(H), NCACX and NCOCX spectra for dipolar interaction assignments, restraint generation and automated 3D structure calculation. However, the utilization of ssPINE still represents a laborious web submission process, involving several steps: generation of peak lists, navigating web browsers, file uploads, job submissions, result downloads, extraction of data, and integration with the project.
In this paper, we introduce a new graphical user interface (GUI), ssPINE-POKY, which is now integrated with the popular POKY suite for NMR analysis and biomolecular structure determination. This interface allows users to submit ssPINE jobs easily and supports the visualization, verification, browsing, and downloading of results. With ssPINE-POKY, researchers can quickly obtain automated peak assignments and accelerate 3D structure determination. Furthermore, in contrast to previous alpha version of ssPINE webserver, ssPINE-POKY integrates different analysis tools, including random coil index, hydrophobic core prediction, secondary structure prediction and secondary chemical shift analysis, protein backbone φ/ψ dihedral angles prediction, web results visualization, between others.
Combination of ssPINE algorithm and ssPINE-POKY GUI offers a robust solution for analyzing complex ssNMR data and can significantly facilitate the determination of the structures of large macromolecular assemblies with high structural complexity and low solubility. As such, we recommend ssPINE-POKY as an essential tool for structural biologists studying challenging biomolecules using MAS ssNMR spectroscopy.
2. Materials and Methods
Our ssPINE program for automated ssNMR assignments was previously implemented and made available through a web submission form. While useful, the multiple required steps made the process complex and time-consuming, limiting accessibility. To streamline the workflow, we introduce ssPINE-POKY, which provides a user-friendly graphical user interface (GUI), that eliminates those extra steps and simplifies the use of ssPINE. This GUI enables easy submission, manipulation, and visualization of results, along with a new feature called “web results”, allowing users to have a custom visualization of the results on their web browser. Figure 1 provides a summary of the steps needed to submit a ssPINE-job and view the results.
ssPINE-POKY software was developed using Python, Bash, and HTML, and is integrated with our ServerSide application, which provides a secure and unified web development environment. The client-server architecture of ssPINE and ssPINE-POKY, built around the POKY framework, is illustrated in Figure 2. Users can submit jobs either through ssPINE-POKY or directly via the web page. These requests are handled by the ServerSide application, which forwards the jobs to the MANDU server—where the ssPINE backend is executed. MANDU hosts most of the POKY suite applications.
For structure prediction, ESMFold is run through the ESM Metagenomic Atlas developed by Meta AI. If users do not provide a 3D structure, the predicted structure from ESMFold is used as input for UCBShift and SHIFTX2, both of which are installed within a VirtualEnv on MANDU. The predicted chemical shifts from UCBShift or SHIFTX2, along with data from PACSY, serve as prior probabilities for Bayesian probabilistic assignments. All modules required by ssPINE and ssPINE-POKY are pre-configured to run automatically, requiring no additional user intervention after submission.
To support the web results feature, we developed multiple HTML templates that are dynamically and interactively updated in real time. Each submission is linked to a customized results page, allowing individualized access and visualization of assignment outcomes.
3. Results
3.1. Implementation and Functionalities
To run ssPINE-POKY, users must upload a POKY-project containing the NMR experiments. The minimum set of experiments required to run ssPINE consists of 2D-CC, 2D-NCA, 2D-NCO, 3D-NCACX, 3D-NCOCX, and 3D-CAN(CO)CX, though additional experiments can improve assignment quality (2D-NCACB, 3D-NCACB, 3D-NCOCACB, 3D-NCOCA, NCACO, CANCO, 3D-CAN(CO)CA, 3D-CAN(CO)CACB). Peak picking must be performed before running ssPINE, which can be automated using the integrated iPick program in POKY (two-letter-code iP[21]). Users can access to ssPINE-POKY via the main window under Poky > Automated assignment > Run SSPINE automated assignment (ssPINE-POKY), or by entering the two-letter code EP.
At the top of the ssPINE-POKY automation window, users can optionally enter their name and email address (Figure 3A). If provided, the server will use the email to notify users about job status and send the results upon completion. A zip file containing the results will be attached, along with a URL for accessing the visualized data in a web browser. Providing this information is not required; however, users will need the generated key (Figure 3J) to access their results.
The sequence file input section (Figure 3B) supports three formats: (1) a 3-letter code sequence file with indices (*.seq), (2) a 1-letter code sequence file (*.fasta), or (3) a 3-letter code sequence file without indices (*.txt). Similarly to PINE-SPARKY.2 [22], the three-letter format file will be automatically set if the user has submitted the sequence to POKY using the Sequence Entry window (two-letter code sq).
Users may optionally provide additional data to refine ssPINE results, such as pre-assignment (Figure 3C) and a PDB file (Figure 3H). Additionally, they can choose to run CS-Rosetta [23,24,25,26](Figure 3D) for protein structure prediction based on ssPINE outputs. The "Spectra to run ssPINE/ssPINE-POKY" section (Figure 3E) allows users to specify the spectra ssPINE assignment. This is done by selecting a spectrum and an experiment type (Figure 3F), then clicking the “Add” button in the Button section (Figure 3E).
Additional buttons in Figure 3G provide further functionality: the “Delete” button removes selected spectra, while the “Clear” button empties the spectra box. Once all required inputs are set, users can initiate job submission by clicking the "Submit" button, which sends the job to the ssPINE web server. Additionally, the “ssPINE Web” button redirects users to the ssPINE website (https://poky.clas.ucdenver.edu/ssPINE), where they can also submit jobs by providing manually prepared peak lists and the sequence file. The “User group” button links to the online discussion forum, which includes previously asked questions from other users and provides a platform to start a new conversation in case any issues arise while using the software.
The ssPINE-POKY importer section (Figure 3J) provides tools to visualize the job results. A key number in the Key box is generated automatically when the “Submit” button is clicked. It is a unique identifier that facilitates communication between ssPINE-POKY and the ssPINE web server.
3.2. Output Files
Compared to our previously published ssPINE version with web submission form, ssPINE-POKY introduces several improvements. In addition to providing assignments and a bar graph displaying the assignment probability for each residue, ssPINE-POKY can automatically generate labels in the NMR spectra uploaded to the POKY project, significantly reducing the time required for manual assignments.
The earlier version of ssPINE included two primary tools for providing structural and dynamic parameters: PECAN (Protein Energetic Conformational Analysis from NMR chemical shifts) for protein secondary structure prediction [27] and LACS (Linear Analysis of Chemical Shifts) for chemical shift referencing errors [28]. In contrast, ssPINE-POKY integrates several additional analysis tools. These include RCI-S2 (random coil index order parameter value) for predicting protein flexibility [29], PACSY hydrophobic core prediction based on hydrophobicity scales derived from solvent-accessible surface area [30], and GetSBY secondary structure prediction, which utilizes chemical shifts of N, HN, Cα, Cβ, CO and Hα in the protein backbone [13]. Additionally, PSIPRED [31] predicts secondary structure based on the position specific scoring matrices generated by PSI-BLAST [32]), while TALOS-N applies a chemical shift-based method to predict secondary structure [29] and protein backbone φ/ψ dihedral angles, also visualized as PACSY RAMA plots [33]. For 3D structure prediction, ESMFold uses the primary protein sequence [34], and CS-Rosetta uses the chemical shifts.
3.3. Web Results
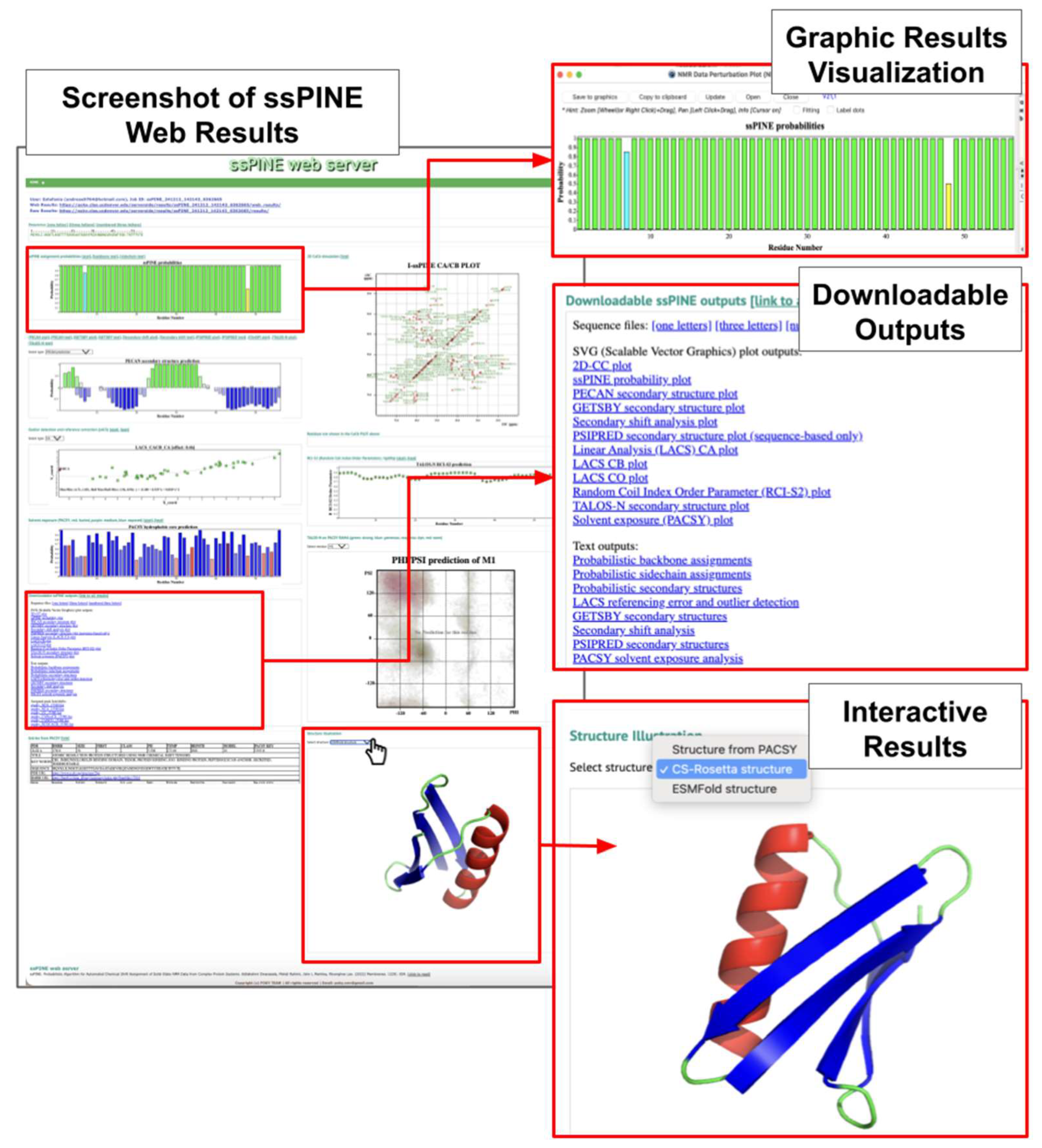
In addition to viewing results within POKY, users can access them via a web browser. Figure 4 presents a screenshot of ssPINE Web Results displayed in the browser. This feature is accessible by clicking the "Web..." button in the ssPINE-POKY interface (Figure 3J).
The web interface provides an interactive graphical representation of the results, allowing users to explore data dynamically. Additionally, raw data can be downloaded directly for further analysis.
3.4. Visual Verification and Resubmission
As illustrated in Figure 1, the “Check” button can be used to import results back into the running POKY project. Like the PINE-SPARKY.2 plugin, ssPINE-POKY communicates with the user interactively when the button is clicked. Standalone visual plots that also exist on the web results will be popped up. Floating color-coded ssPINE labels showing probability will be created. The user will be asked if they want to assign peaks to the best ssPINE candidates with probabilities over 0.5. Once a peak is assigned, the remaining floating color-coded ssPINE candidates will be removed. Similar to its solution NMR counterparts (I-PINE, PINE-SPARKY, and PINE-SPARKY.2), ssPINE-POKY allows the user to use different visual verification tools and resubmission with fixed assignments for incremental assignment iteration. These tools include: probability cutoffs for the assignment (Assign-the-best; two-letter-code ab), graphical peak-by-peak probabilistic assignment (PINE Assigner; two-letter-code pr), graphical residue-by-residue probabilistic assignment (PINE Graph Assigner; two-letter-code pp) and selecting floating labels (two-letter-code se) [35]. Additionally, in cases of highly overlapped areas of the spectra or challenging residues, a semi-automated approach via Versatile Assigner [14] (two-letter-code va) can be used. This can be followed by selecting the “Use pre-assignment” option (Figure 3C) for incremental completion of assignment.
3.5. Tutorial
A YouTube video tutorial for ssPINE-POKY submission can be found at: [https://youtu.be/b6m2AnC7kiM]. Additionally, the user can find POKY team’s help by pressing the buttons “Question?” and “User Forum” located in the POKY main window for general questions regarding using this software.
4. Discussion and Conclusions
In conclusion, ssPINE-POKY offers a valuable solution for analyzing complex solid-state NMR (ssNMR) data in structural biology. The integration of ssPINE into the user-friendly graphical interface of ssPINE-POKY, in conjunction with the widely used POKY software for protein structure determination, significantly enhances software functionality and ease of use. Researchers can quickly submit ssPINE jobs, visualize results, and expedite the determination of 3D structures. This combined tool addresses the challenges of analyzing complex ssNMR data, especially for large macromolecular assemblies with high structural complexity and low solubility. This integration positions ssPINE-POKY as a key tool for structural biologists using ssNMR spectroscopy.
The ssPINE-POKY interface simplifies submission, integrates results into ongoing projects, and allows customizable visualization, empowering users to interpret outcomes according to their preferences. However, there are still notable limitations to be addressed. The quality of ssNMR data, particularly due to overlapping spectral signals, poses an ongoing challenge, leading to time-consuming processes for identifying peaks and analyzing data. Dealing with complex spectra containing multiple peaks and overlapping signals requires further improvement.
Looking forward, we aim to improve ssPINE-POKY by providing more options for users to customize their experience, adding features to make the initial steps of NMR assignment quicker, adding pre-assignment and labeling capabilities to expedite NMR assignment, implementing advanced spectral processing techniques for peak separation, and integrating automatic structural calculation based on NMR assignments. Additionally, considering the inclusion of new experiments to ssPINE's supported repertoire would provide valuable structural information. By addressing these limitations and implementing improvements, ssPINE-POKY can enhance usability, accuracy, and efficiency in structural biology research.
Author Contributions
Conceptualization, W.L.; methodology, A.E.L.G., M.R. and W.L.; software, A.E.L.G., M.R. and W.L.; validation, A.E.L.G., and W.L.; formal analysis, A.E.L.G., and W.L.; investigation, A.E.L.G.; resources, W.L.; data curation, A.E.L.G..; writing—original draft preparation, A.E.L.G., and W.L.; writing—review and editing, A.E.L.G., and W.L.; visualization, A.E.L.G., and W.L.; supervision, W.L.; project administration, W.L.; funding acquisition, W.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Foundation, grant number DBI-2413041, DBI-2051595 and DBI-1902076 to W.L., and University of Colorado Denver.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The ssPINE-POKY program presented in this study are openly available as a plugin in the POKY suite at https://poky.clas.ucdenver.edu. No new data were created or analyzed in this study. Data sharing is not applicable to this article. However, one may simulate datasets as demonstrated in https://youtu.be/HnLubuDoZxM using POKY.
Acknowledgments
We acknowledge useful discussions in the NMR POKY/SPARKY user group (https://groups.google.com/g/nmr-sparky).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shcherbakov, A.A.; Medeiros-Silva, J.; Tran, N.; Gelenter, M.D.; Hong, M. From Angstroms to Nanometers: Measuring Interatomic Distances by Solid-State NMR. Chem. Rev. 2022, 122, 9848–9879. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.Y.; De Paëpe, G.; Hediger, S. Biomolecular and Biological Applications of Solid-State NMR with Dynamic Nuclear Polarization Enhancement. Chem. Rev. 2022, 122, 9795–9847. [Google Scholar] [CrossRef]
- Reif, B. Deuteration for High-Resolution Detection of Protons in Protein Magic Angle Spinning (MAS) Solid-State NMR. Chem. Rev. 2022, 122, 10019–10035. [Google Scholar] [CrossRef] [PubMed]
- Reif, B. Ultra-High Resolution in MAS Solid-State NMR of Perdeuterated Proteins: Implications for Structure and Dynamics. Journal of Magnetic Resonance 2012, 216, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hologne, M.; Faelber, K.; Diehl, A.; Reif, B. Characterization of Dynamics of Perdeuterated Proteins by MAS Solid-State NMR. J. Am. Chem. Soc. 2005, 127, 11208–11209. [Google Scholar] [CrossRef]
- Zhang, R.; Mroue, K.H.; Ramamoorthy, A. Proton-Based Ultrafast Magic Angle Spinning Solid-State NMR Spectroscopy. Acc. Chem. Res. 2017, 50, 1105–1113. [Google Scholar] [CrossRef]
- Mote, K.R.; Gopinath, T.; Veglia, G. Determination of Structural Topology of a Membrane Protein in Lipid Bilayers Using Polarization Optimized Experiments (POE) for Static and MAS Solid State NMR Spectroscopy. Journal of Biomolecular NMR 2013, 57, 91–102. [Google Scholar] [CrossRef]
- Aguion, P.I.; Marchanka, A.; Carlomagno, T. Nucleic Acid–Protein Interfaces Studied by MAS Solid-State NMR Spectroscopy. Journal of Structural Biology: X 2022, 6, 100072. [Google Scholar] [CrossRef]
- Hong, M.; Schmidt-Rohr, K. Magic-Angle-Spinning NMR Techniques for Measuring Long-Range Distances in Biological Macromolecules. Acc. Chem. Res. 2013, 46, 2154–2163. [Google Scholar] [CrossRef]
- Brown, S.P.; Spiess, H.W. Advanced Solid-State NMR Methods for the Elucidation of Structure and Dynamics of Molecular, Macromolecular, and Supramolecular Systems. Chem. Rev. 2001, 101, 4125–4156. [Google Scholar] [CrossRef]
- Lee, W.; Rahimi, M.; Lee, Y.; Chiu, A. POKY: A Software Suite for Multidimensional NMR and 3D Structure Calculation of Biomolecules. Bioinformatics 2021, 37, 3041–3042. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef]
- Shin, J.; Lee, W.; Lee, W. Structural Proteomics by NMR Spectroscopy. Expert Review of Proteomics 2008, 5, 589–601. [Google Scholar] [CrossRef]
- Manthey, I.; Tonelli, M.; II, L.C.; Rahimi, M.; Markley, J.L.; Lee, W. POKY Software Tools Encapsulating Assignment Strategies for Solution and Solid-State Protein NMR Data. Journal of Structural Biology: X 2022, 6, 100073. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R.; Hu, K.-N. A Monte Carlo/Simulated Annealing Algorithm for Sequential Resonance Assignment in Solid State NMR of Uniformly Labeled Proteins with Magic-Angle Spinning. Journal of Magnetic Resonance 2010, 205, 304–314. [Google Scholar] [CrossRef]
- Moseley, H.N.B.; Sperling, L.J.; Rienstra, C.M. Automated Protein Resonance Assignments of Magic Angle Spinning Solid-State NMR Spectra of Β1 Immunoglobulin Binding Domain of Protein G (GB1). Journal of Biomolecular NMR 2010, 48, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.T.; Kulminskaya, N.; Bjerring, M.; Nielsen, N. Chr. Automated Robust and Accurate Assignment of Protein Resonances for Solid State NMR. Journal of Biomolecular NMR 2014, 59, 119–134. [Google Scholar] [CrossRef]
- Schmidt, E.; Gath, J.; Habenstein, B.; Ravotti, F.; Székely, K.; Huber, M.; Buchner, L.; Böckmann, A.; Meier, B.H.; Güntert, P. Automated Solid-State NMR Resonance Assignment of Protein Microcrystals and Amyloids. Journal of Biomolecular NMR 2013, 56, 243–254. [Google Scholar] [CrossRef]
- Weber, D.K.; Wang, S.; Markley, J.L.; Veglia, G.; Lee, W. PISA-SPARKY: An Interactive SPARKY Plugin to Analyze Oriented Solid-State NMR Spectra of Helical Membrane Proteins. Bioinformatics 2020, 36, 2915–2916. [Google Scholar] [CrossRef]
- Dwarasala, A.; Rahimi, M.; Markley, J.L.; Lee, W. ssPINE: Probabilistic Algorithm for Automated Chemical Shift Assignment of Solid-State NMR Data from Complex Protein Systems. Membranes 2022, 12. [Google Scholar] [CrossRef]
- Rahimi, M.; Lee, Y.; Markley, J.L.; Lee, W. iPick: Multiprocessing Software for Integrated NMR Signal Detection and Validation. Journal of Magnetic Resonance 2021, 328, 106995. [Google Scholar] [CrossRef]
- Lee, W.; Markley, J.L. PINE-SPARKY.2 for Automated NMR-Based Protein Structure Research. Bioinformatics 2018, 34, 1586–1588. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lange, O.; Delaglio, F.; Rossi, P.; Aramini, J.M.; Liu, G.; Eletsky, A.; Wu, Y.; Singarapu, K.K.; Lemak, A.; et al. Consistent Blind Protein Structure Generation from NMR Chemical Shift Data. Proceedings of the National Academy of Sciences 2008, 105, 4685–4690. [Google Scholar] [CrossRef]
- Shen, Y.; Vernon, R.; Baker, D.; Bax, A. De Novo Protein Structure Generation from Incomplete Chemical Shift Assignments. Journal of Biomolecular NMR 2009, 43, 63–78. [Google Scholar] [CrossRef]
- Shen, Y.; Bryan, P.N.; He, Y.; Orban, J.; Baker, D.; Bax, A. De Novo Structure Generation Using Chemical Shifts for Proteins with High-Sequence Identity but Different Folds. Protein Science 2010, 19, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Lange, O.F.; Rossi, P.; Sgourakis, N.G.; Song, Y.; Lee, H.-W.; Aramini, J.M.; Ertekin, A.; Xiao, R.; Acton, T.B.; Montelione, G.T.; et al. Determination of Solution Structures of Proteins up to 40 kDa Using CS-Rosetta with Sparse NMR Data from Deuterated Samples. Proceedings of the National Academy of Sciences 2012, 109, 10873–10878. [Google Scholar] [CrossRef]
- Eghbalnia, H.R.; Wang, L.; Bahrami, A.; Assadi, A.; Markley, J.L. Protein Energetic Conformational Analysis from NMR Chemical Shifts (PECAN) and Its Use in Determining Secondary Structural Elements. Journal of Biomolecular NMR 2005, 32, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Eghbalnia, H.R.; Bahrami, A.; Markley, J.L. Linear Analysis of Carbon-13 Chemical Shift Differences and Its Application to the Detection and Correction of Errors in Referencing and Spin System Identifications. Journal of Biomolecular NMR 2005, 32, 13–22. [Google Scholar] [CrossRef]
- Berjanskii, M.V.; Wishart, D.S. A Simple Method To Predict Protein Flexibility Using Secondary Chemical Shifts. J. Am. Chem. Soc. 2005, 127, 14970–14971. [Google Scholar] [CrossRef]
- Lee, W.; Yu, W.; Kim, S.; Chang, I.; Lee, W.; Markley, J.L. PACSY, a Relational Database Management System for Protein Structure and Chemical Shift Analysis. Journal of Biomolecular NMR 2012, 54, 169–179. [Google Scholar] [CrossRef]
- Jones, D.T. Protein Secondary Structure Prediction Based on Position-Specific Scoring matrices11Edited by G. Von Heijne. Journal of Molecular Biology 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Cornilescu, G.; Dashti, H.; Eghbalnia, H.R.; Tonelli, M.; Westler, W.M.; Butcher, S.E.; Henzler-Wildman, K.A.; Markley, J.L. Integrative NMR for Biomolecular Research. J Biomol NMR 2016, 64, 307–332. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-Scale Prediction of Atomic-Level Protein Structure with a Language Model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Westler, W.M.; Bahrami, A.; Eghbalnia, H.R.; Markley, J.L. PINE-SPARKY: Graphical Interface for Evaluating Automated Probabilistic Peak Assignments in Protein NMR Spectroscopy. Bioinformatics 2009, 25, 2085–2087. [Google Scholar] [CrossRef]
Figure 1.
Simplified ssPINE-POKY Workflow. The process begins with opening the POKY project, which includes NMR spectra, peak list, and the protein sequence. The user then opens ssPINE-POKY and provides the required input parameters. Upon submission, multiple analytical programs, and servers, including ssPINE, are executed automatically. Once the process is complete, results can be reviewed directly within POKY using the "Check" button or accessed via the web-based interface using the "Web…" button.
Figure 1.
Simplified ssPINE-POKY Workflow. The process begins with opening the POKY project, which includes NMR spectra, peak list, and the protein sequence. The user then opens ssPINE-POKY and provides the required input parameters. Upon submission, multiple analytical programs, and servers, including ssPINE, are executed automatically. Once the process is complete, results can be reviewed directly within POKY using the "Check" button or accessed via the web-based interface using the "Web…" button.
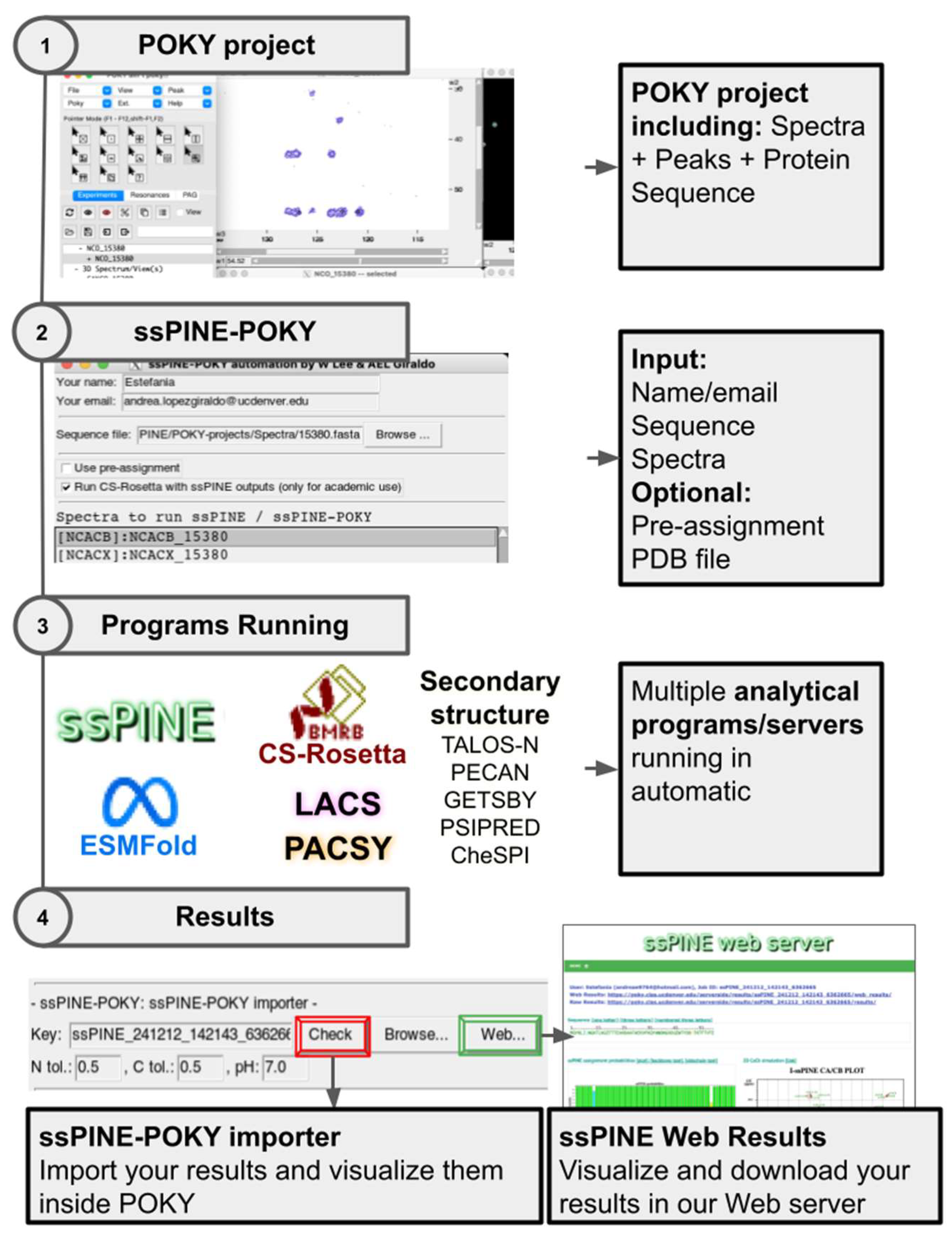
Figure 2.
Client-server architecture of ssPINE and ssPINE-POKY implemented around POKY.
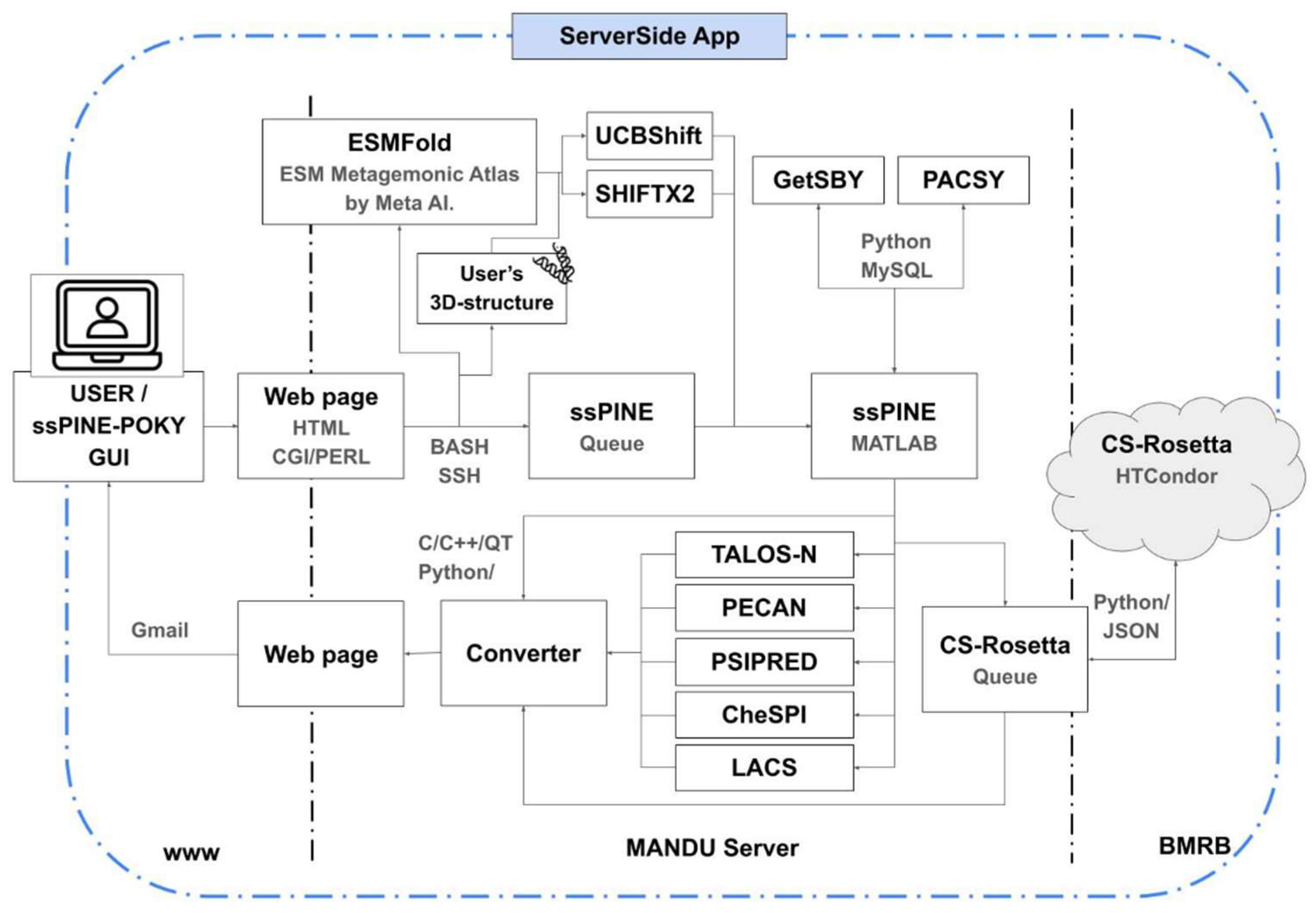
Figure 3.
Screenshot of ssPINE-POKY. The user-friendly graphical user interface (GUI) can be launched as a plugin in POKY using the two-letter-code EP. (A) Name and email fields (optional) for receiving results via email. (B) Sequence file input. (C) Option to use a pre-assignment. (D) Selection of CS-Rosetta for protein structure prediction based on ssPINE outputs. (E) List of user-added spectra. (F) Dropdown menus for spectrum type selection. (G) Row of functional buttons. (H) PDB file upload option to enhance ssPINE results. (I) Threshold values for Chemical Shift prediction. (J) Section for importing ssPINE results; the key is automatically generated upon clicking the “Submit” button. (K) Adjustable 15N and 13C tolerances and sample pH settings.
Figure 3.
Screenshot of ssPINE-POKY. The user-friendly graphical user interface (GUI) can be launched as a plugin in POKY using the two-letter-code EP. (A) Name and email fields (optional) for receiving results via email. (B) Sequence file input. (C) Option to use a pre-assignment. (D) Selection of CS-Rosetta for protein structure prediction based on ssPINE outputs. (E) List of user-added spectra. (F) Dropdown menus for spectrum type selection. (G) Row of functional buttons. (H) PDB file upload option to enhance ssPINE results. (I) Threshold values for Chemical Shift prediction. (J) Section for importing ssPINE results; the key is automatically generated upon clicking the “Submit” button. (K) Adjustable 15N and 13C tolerances and sample pH settings.
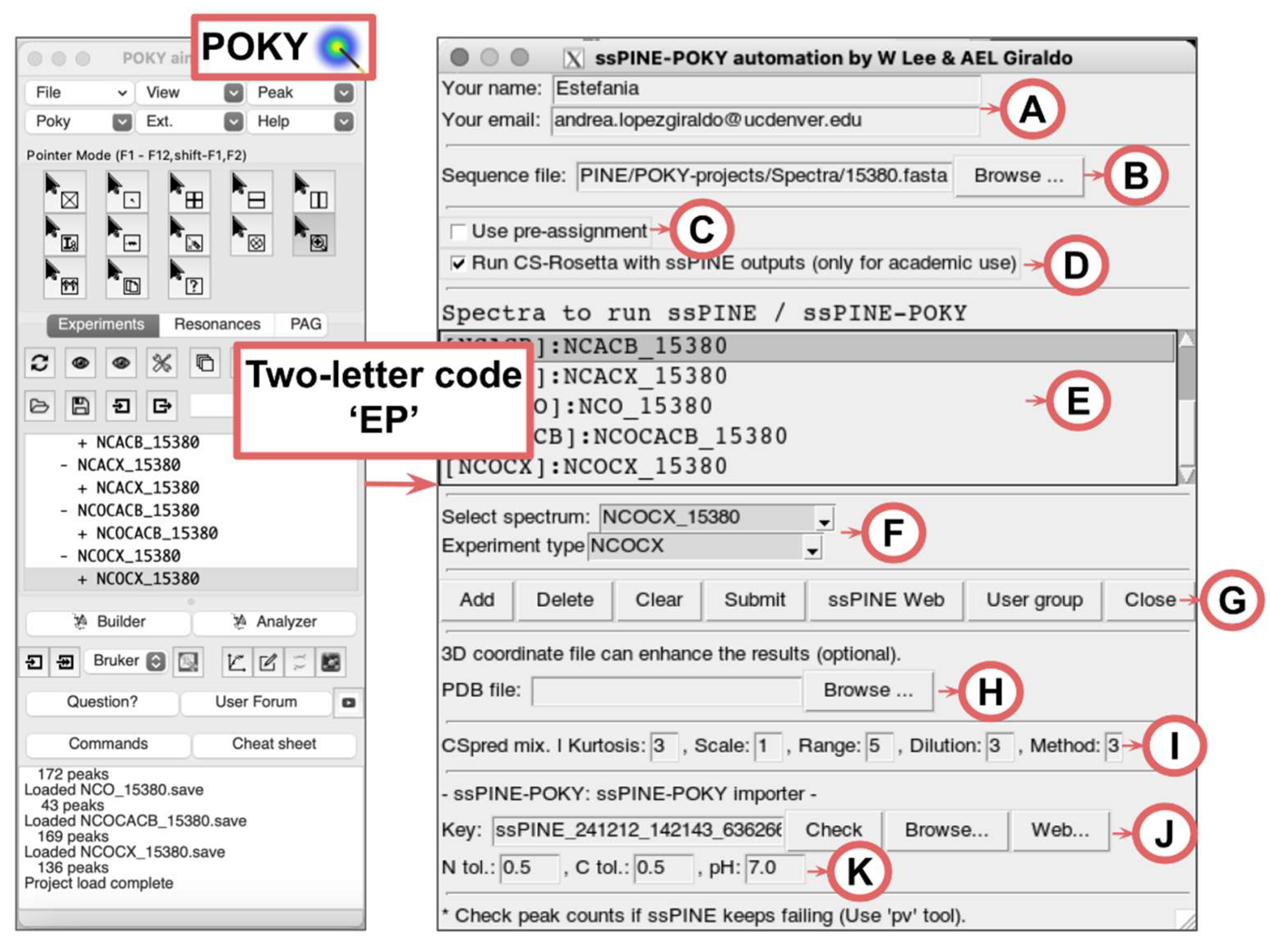
Figure 4.
Screenshot showing the web-based results visualization feature of ssPINE-POKY. This feature enables users to effortlessly view and download all generated results directly from the web browser interface.
Figure 4.
Screenshot showing the web-based results visualization feature of ssPINE-POKY. This feature enables users to effortlessly view and download all generated results directly from the web browser interface.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



