Submitted:
25 April 2025
Posted:
28 April 2025
You are already at the latest version
Abstract
Due to the high analytical sensitivity of current routine high-sensitivity cardiac troponin assays, unexpectedly elevated test results without an obvious clinical correlate are increasingly frequent in daily clinical practice. In these patients, myocardial injury may sometimes go undetected by imaging, including cardiac magnetic resonance imaging. This has led to an increased interest in the pathophysiology of cardiac troponin release, in particular on the fact, whether troponin can be released from reversibly injured myocardium and thereby resulting in an increase of cardiac troponin in the systemic circulation. On the other hand, a variety of circulating cardiac troponin forms were described in human blood samples using different analytical methods, raising the question whether the cause of myocardial injury can be reliably determined by measurement of specific circulating cardiac troponin forms. This review aims to provide an up-to-date overview of this current cardiac troponin research topics.
Keywords:
cardiac troponin
; myocardial injury
; pathophysiology
; release
; circulating forms
; myocardial infarction
1. Background
Alongside the electrocardiogram (ECG), laboratory parameters for the detection of myocardial injury have been a cornerstone in the clinical assessment of patients with suspected acute myocardial infarction (AMI) since the 1950s [1,2,3]. The landscape of cardiac biomarkers has evolved dramatically since then. For the routine laboratory diagnosis of myocardial injury, cardiac troponin I (cTnI) and cardiac troponin T (cTnT) emerged as the laboratory parameters of first choice, because they are currently the most sensitive and cardiac-specific laboratory parameters available for routine laboratories [1,2,3,4,5,6]. However, due to the high analytical sensitivity of current high-sensitivity (hs) cardiac troponin (cTn) assays, the positive predictive value for AMI of hs-cTn is markedly lower compared with previous cTn assay generations, and even unexpectedly elevated cTn test results without an obvious clinical correlate are increasing in daily clinical practice [4,5,6]. In the vast majority of these patients, these cTn elevations are caused by acute or chronic myocardial injury due to a variety of cardiac or primarily non-cardiac pathologies with cardiac involvement unrelated to an AMI [3,4,5,6]. In some of these patients, myocardial injury may even go undetected by imaging because of the higher sensitivity of hs-cTn assays, which can detect cTn even in the femtomolar range in blood samples [3,4,5,6]. The huge variety of possible mechanisms underlying myocardial injury has been extensively reviewed [1,2,3,4,5,6,7]. The most frequent causes are summarised in Table 1. They include, myocardial ischaemia caused by an acute coronary syndrome (ACS; i.e. coronary plaque rupture or erosion with intracoronary thrombus formation; type 1 AMI), myocardial ischaemia or hypoxia unrelated to an ACS (type 2 AMI), and myocardial injury unrelated to myocardial ischaemia or hypoxia, such as inflammation (e.g. in myocarditis), increased myocardial wall stress (e.g. in heart failure), toxic myocardial injury and trauma (e.g. cardiac contusion) [4,5]. Analytically false positive test results are very rare [6,8,9].
Table 1. Differential diagnosis of cardiac troponin increases in peripheral blood samples - cardiac diseases and primarily non-cardiac diseases leading to myocardial injury.
However, many aspects of intramyocardial cTn degradation, tissue release, and degradation within and clearance from the human circulation are still incompletely understood. The aim of this review is to provide an update on what is known about cTn degradation and circulating forms of human cTnI and cTnT. Gaps in current knowledge and potential future applications of the measurement of different circulating cTn forms for the differential diagnosis of underlying diseases leading to myocardial injury are highlightened.
2. Pathophysiology of Cardiac Troponin Release from Injured Myocardium
The cTn complex is part of the thin filaments of the myocardium, and its biochemistry and pathophysiology have been extensively reviewed previously [10,11,12]. In summary, the cTn complex plays a central role in calcium dependent regulation of actin thin filament function, it is essential for control of striated muscle contraction. Only troponin I (TnI), the actomyosin adenosine triphosphatase inhibitory subunit, which confers calcium-sensitivity to striated muscle contraction (about 24 kDa), and troponin T (TnT), the tropomyosin binding subunit (about 35-36 kDa depending on the addional cTnT isoform variation, which is a result of alternative splicing), but not troponin C (TnC), the calcium binding subunit, which initiates the sequence of conformational changes on the thin filament, (about 18kDa), are encoded as cardiac-specific isoforms (cTnI and cTnT) in humans (see Table 2) [13,14,15,16,17]. cTnT and cTnI have a cardiac specific N-terminal extension.
Initially, small (approximately 5% of the total content) "cytosolic" pools of cTnI and cTnT were reported in human myocardium [18,19,20]. However, considering the preparation protocols used by the investigators and the poor solubility of cTnI and cTnT in the hydrophilic sarcoplasma, a more appropriate term may be "rapidly degraded, loosely bound, early releasable pool". When not incorporated into myofilaments, e.g. cTnT is rapidly degraded (e.g. by caspase or µ-calpain) within cardiomyocytes to avoid toxic effects [21,22,23,24]. However, later data suggest that the fraction of cTnT that can rapidly dissociate from myofibrils in vivo is substantially higher than the 5-10% previously reported [25]. Thus, all cTn released is likely to be of myofibrillar origin.
A variety of factors (e.g. molecular mass, concentration gradient from cardiomyocytes to interstitial space, local blood and lymphatic flow) influence the onset of release of cardiac markers from injured myocardium and the subsequent increase in the systemic circulation. The most important factor appears to be intracellular compartmentation [20,26,27]. Myocardial necrosis is preceded by a substantial reversible pre-lethal phase [28]. In contrast to cytosolic proteins, the release of structurally bound molecules, such as cTns, requires both a leaky plasma membrane and dissociation from or degradation of cellular structures, such as the contractile filaments, and this is a slower process. In addition, the susceptibility of a structural protein to early intracellular degradation by cytosolic proteases, such as the cytoplasmic calpains or caspases, which are rapidly activated during myocardial injury [21,22,23,24], is of great importance. In contrast, lysosomes are stable within the first 3-4 hours after the onset of cell injury, and therefore, lysosomal enzymes are not involved in the early degradation of structurally bound proteins.
Both cTnI and cTnT are substrates of the cytoplasmic enzyme µ-calpain [21,22,23,24], which is activated by increased cytoplasmic calcium, an important and early feature of cell injury. This occurs through increased influx and redistribution of calcium from intracellular compartments (e.g. endoplasmic reticulum, mitochondria). Increased cytoplasmic calcium also activates phospholipases and endonucleases [29]. pH-dependent dissociation of the troponin complex may be another important factor in early cTn release from injured myocardium (see Figure 1).
The pathophysiological background of the limited N-terminal intramyocardial proteolysis of cTn by caspase and calpain may be that cTns are involved in the sensitivity of the myocardium to decreases in intracellular pH, and thus, this may be an early specific functional adaptive response to myocyte injury rather than a simple destructive degradation [21,22,23,24]. cTnI is also a substrate of caspase-3, which has a key role in apoptosis, it targets the N-terminal region of cTnT as well [24]. Additional proteolysis of troponins may occur in the interstitial space e.g. by matrix metalloproteinase-2 [30].
Once the integrity of the cardiomyocyte plasmalemma is disrupted, cardiomyocytes rapidly release intracellular macromolecules into the interstitial space. In addition, cytoplasmic blebbing may already occur during the reversible phase of cell injury [28]. These blebs contain intracellular macromolecules, such as degraded cTn, and detach with resealing of the plasma membrane without cell death in cell culture experiments and may be responsible for limited cTn release from cardiomyocytes [28]. In contrast, sarcolemmal disruption with subsequent massive release of intracellular molecules into the interstitial space is thought to morphologically indicate the "point of no return" of cell injury, i.e. the occurrence of necrosis.
In addition, intramyocardial posttranslational modifications of cTn occur, such as oxidation or phosphorylization, which may enhance or inhibit the susceptibility to proteolysis [21,23,24,31,32,33]. Thrombin-mediated cTnT degradation at the N-terminal end has been describd as well [34], which could be involved in the degradation of circulating cTnT in blood and has to be considered when testing serum samples. The currently commercially available routine hs-cTnT assay, however, utilizes antibodies which target to the amino acid 125-147 fragment is not affected by thrombin-mediated cTnT degradation [35].
In summary, cTn degradation in response to myocardial injury starts within cardiomyocytes. cTnI and cTnT show a rapid release after myocardial injury, comparably fast to cytosolic proteins, such as myoglobin or creatine kinase isoenzyme MB [19,20,26,36,37,38]. The sustained increase in cTn after myocardial infarction is probably a combination of slow washout and prolonged local tissue degradation. Unrestricted blood flow, e.g. from invasive early reperfusion of the infarct-related coronary artery, results in more rapid extraction and clearance of cTn from damaged myocardium [27,39,40]. The only generally accepted mechanism of cTn release into the systemic circulation in humans currently is myocardial necrosis.
Experimental data on cTn release from reversibly injured myocardium
There is evidence from experimental in-vitro models, that macromolecules including cTn are released without histological evidence for cardiomyocyte necrosis [28,41,42,43,44,45]. These observations could be confirmed by more recent thoroughly conducted large animal studies in pigs by Weil et al. [46,47], which confirm previous in-vitro and in-vivo reports. After applying only 10 minutes of left anterior descending artery (LAD) occusion cTnI increases were found in a porcine heart model, which was associated with reversible myocardial dysfunction but without evidence of myocardial necrosis in tissue analysis [46]. Apart from ischemia or hypoxia, mechanical stretch in response to pressure or volume overload may trigger the activation of proteases associated with intracellular troponin degradation and release of troponin fragments from injured cardiomyocytes without evidence of necrosis despite reversible ventricular dysfunction [47]. In this model, intravenous phenylepinephrine infusion lead to preload-induced myocardial injury by acutely increased wall tension with apoptosis but without signs of necrosis [47]. In addition, it was previously reported in cell culture experiments, that tachycardia may stimulate integrins, triggering release of cTnI in the absence of necrosis [42], and additionally the cTn content of cardiomyocytes already decreases before necrosis occurs [48].
Thus, reversible myocardial injury could be also an option of small, limited cTn increases in humans using hs-cTn assay for testing. However, there is often a contiuum of apoptosis and necrosis in response to myocardial stress, and the lack of histological data and the limited sensitivity of imaging compared with hs-cTn testing will make it very difficult or even impossible to prove cTn release from reversibly injured myocardium in humans [49,50]. It has been postulated that for every 1 µg of human myocardium injured, the cTnI and cTnT concentrations increase by approximately 4 ng/L in peripheral blood samples [51].
Clinical data suggesting possible cTn release from reversibly injured myocardium
From the publication of the first research radioimmunoassay for the detection of cTnI [52] the analytical sensitivity of present hs-cTn assays improved dramatically from 500 ng/L to approximately 1-3 ng/L [35]. This advancement in the analytical performance of cTn assays with maintained cardiospecificity lead to a dramatic improvement in the clinical sensitivity for the detection of myocardial injury, which could not be imagined at the beginning of clinical cTn research. Originally, cTn could not be detected in healthy individuals, but with hs-cTn testing cTn can be detected in >50% of healthy individuals [51]. As analytical interferences have been ruled out [53,54], detectable hs-cTn concentrations in humans with normal hearts suggest a constant limited turnover of cTn and/or cardiomyocytes with cTn release into the systemic circulation. In a functioning sarcomere, protein synthesis, processing, and degradation occur continuously as a part of physiological turnover [55]. cTnT undergoes rapid turnover with a half-life of approximately 3.5 days within cardiomyocytes [56]. If not incorporated into myofilaments, cTnT is rapidly degraded to avoid toxic effects [55,56].
As mentioned previously, it is probably impossible to prove cTn release from reversibly injured myocardium in humans. However, there are increasing reports in the literature on cTn release in clinical scenarios in which myocardial necrosis is very unlikely [57,58,59,60,61]. For example, cTnI release measured by a very sensitive assay has been demonstrated in vivo in humans following nuclear perfusion scintigraphy with peak concentrations associated with the extent of myocardial ischemia during stress testing [57]. In addition, it has been reported that even normal individuals have elevated circulating cTn levels in response to dobutamine stress or exercise testing [58,59]. Small, but significant cTn increases in coronary sinus blood samples within 30 minutes were found after brief episodes of incremnental rapid atrial pacing using a protocol with comparable myocardial stress to moderate brief physical exercise [60]. This cTnT release in coronary sinus blood was mirrowed subsequently in a delayed, small but significant hs-cTnT concentration increase in peripheral blood samples after 3 hours (doubling to tripling of baseline values) even in the subgroups without significant coronary artery disease (CAD) or without net myocardial lactate production as a proof of myocardial ischemia. In patients without significant CAD and no net myocardial lactate production hs-cTnT concentrations stayed within the upper reference limit (URL) in all patients.
Challenges in the interpretation of hs-cTn after elective percutaneous coronary interventions in clinical practice
cTn when measured with hs-cTn assays increases significantly in individuals with angiographically normal coronary arteries even after LAD occlusion of only 30 seconds duration in peripheral venous blood samples, a setting in which necrosis is highly unlikely [61]. cTn increases started as early as 15-30 minutes after balloon occlussion. The highest cTn values were found at the end of the blood sampling period at 4 hours after LAD occlussion with the highest relative increases found after 90 seconds lasting LAD occlussion (all participants developed angina, assay dependent increase ranging from about 3-8 fold of baseline values). This observation explains, why cTn increases are found in almost all and even completely uneventful patients after elective percutaneous coronary interventions (PCI) (see Figure 2), which makes the clinical interpretation of cTn increases in the individual symptomatic patient without knowing baseline values difficult.
Baseline values correlate with the severity and complexity of CAD and may be already increased [7,60,62]. In general, the cTn increase after elective PCI is related to CAD severity and the complexicity of the performed interventions (see supplemental figure 1).
Apart from the obvious cases of peri-interventionel AMI with complications seen in coronary angiography (see supplemental figure 2) its diagnosis is not always straightforward in clinical practice. The clinical application of the criteria of the 4th Universal Definition of Myocardial in Infarction is sometimes demanding as it requires the careful consideration of cTn increase together with clinical symptoms, angiographic, ECG or imaging evidence for acute myocardial ischemia [3,7]. However, it has been shown that the diagnosis of peri-interventional type 4a AMI complicating elective PCI has prognostic implications [63], it was associated with an about 2-fold higher cardiovascular 1-year event rate and an about 3-fold higher, but still low 1-year mortality (3%) in patients with normal cTn baseline concentrations [63,64]. Other societies (see Table 3) preferred a simpler approach being based on cTn release with markedly higher cTn decision limits in combination with gross ECG evidence for occurrence of AMI (e.g. development of new pathological Q waves) [65,66]. A major limitation of all suggested cTn decision limits is that the time dependence of cTn release after myocardial injury is not considered (see figure 2). No clear criteria on blood sampling regimens and the point in time after PCI at which these decision limits are to be used are given in all current recommendations [3,65,66]. Therefore, all recommendations have to be reworked considering these issues, although still limited data on the optimal blood sampling regimen after elective PCI is available so far. From a practical point of view, it is suggested to test cTn at baseline, which is also useful for risk stratiufication [7,62,63,64], and, if clinically indicated, 4-6 hours after PCI, and in the morning of the next day, if the patient stays overnight in the hospital.

Table 3.
Criteria suggested for the diagnosis of peri-procedural AMI in elective PCI.
| cTn baseline ≤URL |
cTn baseline >URL | Clinical criteria (≥1 needed) | |
| UDMI Type 4a AMI [3] | >5x URL plus ECG or imaging criteria | Increase >20% + >5x URL plus ECG or imaging criteria | New signs of myocardial ischemia as evidenced by ECG, imaging, or coronary flow-limiting complications |
| SCAI clinically relevant AMI [65] | ≥70x URL or ≥35x URL plus ECG criteria | ≥70x URL or ≥35x URL plus ECG criteria | New Q waves in ≥ 2 contiguous leads, New persistent LBBB |
| ARC-2 peri-procedural AMI [66] | ≥35x URL plus ECG or imaging criteria |
≥35x URL plus ECG or imaging criteria |
New Q waves or equivalents, evidence in imaging, coronary flow-limiting complications |
Abbreviations: percutaneous coronary intervention (PCI), Universal Definition of Myocardial Infarction (UDMI), Society for Cardiovascular Angiography and Interventions (SCAI), Academic Research Consortium (ARC), acute myocardial infarction (AMI), upper reference limit (URL), left bundle branch block (LBBB), cardiac troponin (cTn), electrocardiogram (ECG).
Table 4.
Summary of current knowledge on circulating cardiac troponin complexes and cardiac troponin forms in relation to the cause of myocardial injury .
Table 4.
Summary of current knowledge on circulating cardiac troponin complexes and cardiac troponin forms in relation to the cause of myocardial injury .
| Intact and truncated cTnTIC complexes | Free partially truncated (HMM) cTnT | Intact and truncated cTnIC complexes | Free heavily truncated (LMM) cTnT | |
| AMI | x | x# | x* | x |
| ESRD | x | x | ||
| Post heavy endurance exercise (e.g. marathon) | x | x |
*Main form of circulating cTnI after AMI (acute phase). # Main form of circulating cTnT after AMI (acute phase)cTnI is mainly found in blood as a partially degraded cTnIC complex in response to all. etiologies of myocardial damage, some is mainly partially degraded cTnTIC complex (e.g. early after AMI), and little is free degraded cTnI. By contrast, according to preliminary clinical results, HMM cTnT is mainly only found in blood in AMI patients during the acute phase. Abbreviations: acute myocardial infarction (AMI), end-stage renal disease (ESRD), low-molecular mass (LMM), high molecular mass (HMM), cardiac troponin I (cTnI), cardiac troponin T (cTnT).
In summary, the distinction between type 4a AMI, PCI-related acute myocardial injury indicated by cTn release not fulfilling these criteria, and chronic myocardial injury indicated already by increased baseline cTn concentrations before PCI without a significant increase thereafter can be challenging in individual patients undergoing elective PCI (see supplemental figures 1 and 2), particularly, if baseline concentrations are not available. This distinction is clinically important, and careful integration of all available clinical data is essential for correct classification. Current clinical practice usually is to test cTn after PCI only when clinically indicated by symptoms or complications during PCI.
Challenges in the clinical interpretation of cTn in symptomatic athletes after competitions or training sessions involving heavy endurance exercise
Of even greater debate on a potential clinical significance are cTn increases in asymptomatic endurance athletes (e.g. marathon runners) after competitions or heavy training sessions with hs-cTn concentrations above the URL and a rising and/or falling pattern [59,67,68,69,70,71,72]. Peak concentrations usually do not increase above 3-times the URL and occur seceral hours after finishing, and concentrations return into the URL within 48-72 hours after exercise [69]. This frequent cTn release in asymptomatic athletes makes the interpretation of cTn very challenging in athletes who develop symptoms during or after competitions or training sessions (see Figure 3).
The symptomatic athlete should be assessed as any other high-risk patient presenting to the emergency department. However, the usual post-exercise increase in cTn makes its interpretation more difficult. Serial cTn testing with considering the magnitudes of cTn increase and the rate of change, and, in the absence of unequivocal ECG findings of acute myocardial ischemia, the combination with imaging, in particular coronary computed tomography angiography (CCTA), are essential to rule out acute myocardial ischemia as the cause of symptoms particularly in older (>35 years) male recreational athletes.
Most healthy individuals have elevated cTn after extreme endurance exercise [59,68,69,70,71,72]. After prolonged endurance events (e.g. marathon, triathlon, ultramarathon) post-race typically mild and transient acute reversible systolic dysfunction of the right ventricle but not the left ventricle has been described [69,73,74]. There was no close correlation between post-race cTnI concentrations and right ventricular ejection fraction decreases [73]. In marathon runners the extent of cTn release did not correlate with myocardial injury [69,73] in imaging as well as the time course of cTn concentrations differed from AMI [69,73]. In athletes there is no association of post-exercise cTn release with worse prognosis [69,73,74,75]. This appears to be different in elderly physical active individuals from the general population [76]. There is experimental data supporting the possibility of cTn release from reversibly injured myocardium in athletes. In a rat animal model of an endurance exercise challenge cTnT release was only associated with reversible changes in cardiomyocyte structure [67]. This is confirmed by a reported small cTnT release during and after a marathon in healthy completely asymptomatic humans on a motorized treadmill [68]. cTnT increased in all 9 participants during running, at completion or within 1 hour cTnT returned to baseline values. All but one participant showed a further small increase within a 24 hour recovery period starting several hours after finishing, 5 participants still had increased cTnT concentrations 24 hours after exercise. The release of cTnT within the first 60 minutes of running indicates that exercise-induced cTn release is not necessarily restricted to prolonged endurance exercise. It appears very unlikely that these minor cTn increases reflect myocardial necrosis in this healthy, extremely fit individuals. It may be part of an adaptive process [22]. A high training volume and a long duration in training appear to be protective for post-exercise cTn release [72]. Exercise-induced cTn release itself, however, appears to be related to the overall cardiac workload (duration and in particular intensity) of the competition or training session [69]. Exercise-induced cTn release did not differ significantly in patients with rule-out of CAD (calcium score 0, no plaques in CCTA) and patients with signs of coronary arteriosclerosis in CCTA but without evidence of exercise-induced myocardial ischemia and, therefore, clearance to perform exercise [70]. However, despite being asymptomatic clinically, the exercise-induced cTnI release in individuals with hemodynamically significant CAD was reported to be higher [77].
Although cardiac magnetic resonance imaging (MRI) studies in participants of marathons found no myocardial edema or late gadolinium enhancement despite increased cTn concentrations after exercise [69,73,74], its limited sensitivity does not rule out necrosis as the cause of cTn release from the heart. It has been speculated, that long-term exercise training could produce myocardial damage from repetetive high-intensity exercise sessions, which could explain, why lifelong endurance athletes have more late gadolinium enhancement compared with their physically less active peers [69,78]. There was a relation between the amount of late gadolinium enhancement with the number of race competitions and years of training as well [78].
In summary, brief periods of myocardial ischemia, without obvious myocardial infarction or imaging evidence of necrosis, may cause low-level hs-cTn increases in peripheral blood, and cTn is released after intensive myocardial workload, even among individuals without objective evidence of myocardial ischemia or CAD. Whether these limited cTn increases are due to reversible myocardial injury remains speculative. Although release of cTn could be demonstrated in cell culture experiments from reversibly injured cardiomyocytes by the formation of blebs that bud off from the plasma membrane [28,45], the clinical implication of this observation remains unclear. Experimental studies demonstrated [46,47] that cTnI was released from porcine myocardium in the absence of detectable tissue necrosis, but not in the absence of other forms of cell death, as apoptosis was detected in myocardium.
3. Clearance of cTnI and cTnT from the Circulation in Humans
There are three possible mechanisms of elimination of circulating proteins in blood, i.e. enzymatic degradation within the circulation, metabolism in organs with a high metabolic rate (e.g. liver, pancreas, kidneys, skeletal muscles, endocytosis by the reticuloendothelial system), and elimination via glomerular filtration and excretion in urine. Thus, in cases of impaired clearance from blood (e.g. renal failure, hypothyroidism) prolonged biomarker increases may be found or higher baseline values may be seen then usual.
Most proteins released from injured myocardium, including cTn, appear to be catabolized in tissues with a high metabolic rate, in particular in the liver probably by scavenger receptor mediated endocytosis and subsequent degradation in lysosomes [79]. In this respect, the reports of cTnT degradation by thrombin [34], a rather indiscriminate serine protease, which cleaves cTnT between R68 and S69, add interesting information and may explain the heparin interference of a previous generation of cTnT assays [34,80]. However, the significance of thrombin for cTnT elimination remains to be shown. Small molecules, such as myoglobin, pass through the glomerular filtration membranes of the kidneys and can be found in the urine [81]. These proteins are mainly reabsorbed and subsequently metabolised in the tubular epithelial cells. Thus, in the presence of impaired renal clearance from blood prolonged increases in such biomarkers are found. Intact free cTnT is too large for glomerular filtration, but smaller cTnT fragments would be small enough for renal clearance. By contrast, cTnI appears to be mainly found in blood as part of complexes (see below) which are too large for glomerular filtration. At high cTnT concentrations (e.g. after AMI) extra-renal cTnT clearance dominates [79], but at low concentrations (<100 ng/L, e.g. in patients with chronic cardiac diseases) renal clearance appears to dominate based on experiments where renal perfusion was reduced [82]. As cTn drop to lower concentrations such as those often observed in patients with stable cTn elevations recent data suggest that cTnT clearance becomes slower and more renal dependent [82]. Smaller, circulating, degraded cTn fragments which are predominantly found in blood after chronic myocardial injury (see below) would be small enough for glomerular filtration [82]. However, local degradation and tubular reabsorption could preclude the detection of cTnT and cTnI in urine. At these low steady-state concentrations, cTnT levels are roughly twice as high if kidney function is reduced by 50% [82]. However, other mechanisms such as increased myocardial stress and wall tension related to a variety of different causes may also contribute to the cTnT elevations seen in patients with chronic renal failure [6,8].
When cTn enters the bloodstream, if follows an exponential 2-phase model with a distribution and elimination phase. Initially, cTn half life times of about 1-2 hours similar to that of myoglobin or CKMB were estimated [14,40,83]. Once cTnT and cTnI reach the circulation, they are initially cleared with a short half-life of about 0.5 hours in dogs and rats [83]. In a recent, carefully designed clinical study [84] with autologous re-transfusion of plasma, which was obtained by plasmapheresis during the subacute phase of AMI, several weeks thereafter the median half-life of cTnT calculated in humans was about 2 hours with a clearance of about 80 ml/min, the half life of cTnI was longer with a median of about 4 hours with a clearance of about 40 ml/min. By this study design the problem of ongoing release of cTn from damaged myocardium is avoided. However, different cTn fragments may have different half-lifes [82,85]. Recently, the half-life of high molecular mass (long-form) cTnT was reported to be about 9 hours, which was calculated from concentration time course analyis of AMI patients [85]. This approach, however, may overestimate the true half-life because of ongoing cTn release from the damaged myocardium.
4. Circulating Forms of Cardiac Troponins in Human Blood
The biochemical and pathophysiological background with post-translational modifications (e.g. phosphorylation, oxidation), intramyocardial N-terminal proteolysis of cTnI and cTnT by caspase and calpain and dissociation of cTn complexes and free cTn forms from the thin filament in response to myocardial injury with the potential ability of rebinding to neighboring filaments, as well as further degradation in the interstitial space and the blood after release from cardiomyocytes is very complex and challenging (see above [10,11,12,30,31,32,33,34,45,46,47,48]). Serum or plasma is a complex matrix with many abundant proteins, such as albumin, and comparably low concentrations of cTn. This is a major analytical challenge for the characterization of circulating cTn forms using standard techniques of proteomics, such as mass spectrometry [86,87,88], the criterion method of proteomics, or gel filtration, because highly sensitive methods are needed. There are in principal two primary approaches, i.e. antibody based techniques and physical seperation techniques, such as western blotting, gel filtration chromatography, and mass spectrometry, which all have strengths and limitations (e.g. denaturing conditions, analytical sensitivity). For example, specific sample preparation and purification procedures are needed to eliminate this background noise of abundant blood proteins and to enrich cTn in the sample to be able to be detected by mass spectrometry [86,87]. Nonetheless its analytical sensitivity is still limited and very high cTn concentrations in samples are needed [87]. A denaturing technique is not suitable for distinguishing the different forms and sizes of the ternary cTnTIC complex or cTnIC complex as a whole.
An obviously easier approach appears to be the development of specific and sensitive immunoassays for the detection of particular circulating cTn forms [85] after characterization of the relevant circulating cTn forms released in response to different causes of myocardial injury by more sophisticated analytical techniques. Thus, for practical reasons, sensitive and specific sandwich immunoassays are the methods of choice for research and potential future routine use [85,89,90]. They have the potential to be adopted for routine use on automated, high-throughput platforms.
The currently available data on circulating cTn forms is summarized in table 3. It is based on different analytical approaches (mainly Western blotting and gel filtration chromatography, sandwich immunoassays) with their method-specific limitations, and it is also mainly based on stored samples. Testing of stored samples has limitations compared to testing of freshly drawn blood samples as storage conditions and freeze thaw cycles may affect in-vitro stability and consequently circulating degradation forms within samples. In addition, in serum samples thrombin generation may affect in particular cTnT forms, and in EDTA plasma samples the anticoagulant may have effects on cTn complexes [91]. Ideally the reported results should be confirmed by using fresh heparinized plasma samples for testing.
Published results [92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109] can be summarized as follows: Intact cTn forms disappear rapidly from the circulation in the first hours after AMI. After AMI, mainly binary truncated cTnIC with a varying quantity of ternary mostly truncated cTnICT complexes are found [92,93,94,96,97]. The so called “large size” c TnIC and cTnTIC complexes are only found early after AMI. 75-100% of the total cTnI content in serum of post-AMI patients is part of a complex [92,93,94,100,102,106]. By contrast, cTnT also circulates in a significant amount as a free unbound form [101,103,104]. Intact cTnT is rapidly degraded at the N-terminal end, and early after AMI it has been described mainly as a high molecular mass (HMM, ≥29kDa), “long” form including complex bound cTnT [97,104,105]. Over time, heavily truncated cTnT forms (14-16 kDa) increase probably by degradation at both the C-terminal and N-terminal ends [101,103,104]. In AMI patients, cTnI primarily exists as truncated cTnIC and truncated cTnTIC complexes. For cTnI degradation the data is less consistent, but degradation at both ends has been reported as well [102,106,107]. By contrast to the N- and C-terminal regions, the central portions of cTnI and cTnT appear to be stable [94,97,102,104,109]. These epitopes appear to the best targets for the development of routine cTn immunoassay with high analytical sensitivity and high in-vitro stability [35].
Interestingly, the cause of myocardial injury may impact circulating cTn forms (see table 2). HMM, long cTnT forms predominate in the acute phase of AMI, whereas in chronic end-stage renal diseases or in individuals after strenous endurance exercise (e.g. marathon running or triathlon) only LMM cTnT forms could be detected [101,104,105]. These observations show potential for future routine use of cTn composition detection in blood samples to define the cause of myocardial injury and to increase the specificity of cTn testing for AMI.
Commercially available routine hs-cTn assays are targeting measurement of total cTnI or cTnT
The practical relevance of complex formation, post-translational modifications of cTn and degradation of cTn is that they may lead to changes in the availability of specific epitopes and thus to different recoveries of cTn variants in different cTn assays. The antibodies of currently commercially available hs-cTn assays target epitopes within stable central regions of the cTnI and cTnT molecules [35]. Therefore, most currently commercially available assays detect total cTnI or cTnT including intact cTn (free or complex-bound) and a comprehensive but varying mixture of cTn degradation products [35].
Research cTn assays detecting specific circulating cTn forms:
Recently immunoassays to quantify intact or only moderately degraded cTnT (so called “long-form”) have been developed [85,89,90], that do not detect heavily degraded so called “small-form” cTnT in blood samples. In pilot studies, the ratio of “long-form” to total cTnT demonstrated discrimimation potential of acute from chronic myocardial injury and of different aetiologies of myocardial injury [90,105,110,111,112,113,114]. Similar findings have not been reported for cTnI yet [106]. However, a hs-cTnI/hs-cTnT ratio in blood samples could have similar discriminatory power for seperating type 1 AMI from other causes of myocardial injury [115,116]. These promising results of pilot studies of cTn composition testing, however, have to be confirmed in large-scaled clinical studies testing fresh blood samples.
5. Are There Clinically Relevant Differences Between cTnI and cTnT?
When critically looking on the published literature on this topic, it has to be considered, that cTnI and cTnT assays are neither standardized nor harmonized, and absolute values are, therefore, hard to compare. The only way to make meaningful comparisons is to transform absolute values into multiplies of the URL of each assay. However, even by doing this, there may be still a bias, if one cTn assay has a markedly higher analytical sensitivity then the other assay, and, therefore, URLs should be used, which were calculated from the same reference population, such as the American Association of Clinical Chemistry and Laboratory Medicine reference population biobank, using the same statistical method for URL calculation. In addition, comparisons were mainly made on stored samples, and differences in the in vitro stabilities of cTns must be excluded as the simplest reason for observed discrepancies. In many studies, samples were stored for months or even years before measurement. Thus, more information is frequently needed, including immediate cTn measurements without sample storage and studies with cardiac MRI imaging as the most sensitive imaging modality currently available to assess myocardial injury to confirm reported differences in clinical sensitivities and specificities of cTnI and cTnT in specific clinical scenarios, e.g. in patients with chronic skeletal muscle myopathies or patients with severe chronic renal failure.
cTnT and cTnI concentration time courses after an AMI
Although cTn show different release kinetics with cTnT having a broader diagnostic window after AMI [27,39,40,117], cTnT and cTnI are considered equally effective in terms of diagnostic performance during the acute phase of AMI [26,118,119]. No clinically relevant differences in their early diagnostic sensitivities for AMI have been published so far. Both cTns generally detect myocardial damage equally well [120].
After AMI with occluded coronary arteries, cTn time courses are influenced by the fact, whether early tissue reperfusion of the infarcted myocardium occurred [27,39,40]. With early reperfusion of the infarct-related coronary artery, cTnT and cTnI peaks are usually found about 12 hours after symptom onset (see supplemental figure 3). Both troponins show a biphasic release pattern with second peak values several days after AMI. However, this feature is much more pronounced in cTnT concentration time courses (see supplemental figure 3), and cTnT increases usually last a couple of days longer than cTnI increases [27]. These second cTnI peaks are much lower than the first peaks (see supplemental figure 4) and may be missed with infrequent (e.g. only daily) blood sampling [27]. In AMI patients with ST-elevations (STEMI) and successfulI primary PCI they were even absent [117]. In case of failed early tissue reperfusion of the infarcted myocardium cTnI peaks occur about 24 hours after symptom onset and the maximum cTnT concentrations are found several days after AMI (see supplemental figure 5) [27,39,40]. The concentration time courses of AMI patients without ST-segment elevations (non-STEMI) resemble those of STEMI with successful primary PCI [121]. cTn peaks and release correlate with infarct size [38,122,123,124]. The early cTn concentration time courses within the first few hours after symptom onset do not differ significantly on admission between STEMI and non-STEMI patients, and the rate in increase within the first 1-2 hours – although usually higher in STEMI – does not allow a reliable discrimination as well [120,121]. Similarly, they do not allow an accurate discrimination between AMI patients and patients with myocardial injury unrelated to myocardial ischemia as well [120].
Despite the lack of differences of early diagnostic sensitivities for AMI [26,118,119] it has been postulated that cTnI is released faster after myocardial damage [125]. However, as mentioned above, although absolute concentrations of hs-cTnI may be markedly higher, this conclusion is not possible, and differences in absolute values in in-vitro and animal experiments may be just related to differences of cTn assay crossreactivities with cTn of animals in cTn assays which were designed to detect human cTns. The freeze-thawed human myocardial tissue damage model used in one study [125] is very unphysiological. From a pathophysiological point of view it is likely that cTnI and cTnT are released equimolarly after myocardial damage from the thin filaments. However, theoretically an intramyocardial rebindung of cTnT to insoluble structures of cardiomyocytes would be possible.
cTn discrepancies in patients with chronic skeletal muscle diseases:
A potential pathophysiological background of reported discrepancies between cTnI and cTnT concentrations (see Figure 4) with much more frequently increased cTnT concentrations in patients with chronically stressed skeletal muscle [126,127,128,129,130,131,132,133] is that during fetal development, cTnT is expressed in cardiac and embryonic and neonatal skeletal muscles [134,135,136,137].
cTnT is downregulated in skeletal muscle during development, and after birth, cTnT gradually disappears from skeletal muscle. Healthy adult human skeletal muscle does not contain cTnT. By contrast, cTnI has not been reported to be expressed outside the heart so far [10,15]. When human skeletal muscle is chronically injured, such as in patients with chronic skeletal muscle myopathies, it recapitulates embryonic myogenesis with re-expression of fetal proteins, potentially including cTnT isoforms [134]. Re-expression is likely dependent on disease severity, and re-expressed proteins can be released into the circulation from damaged skeletal muscles, such as in patients with chronic myositis, myopathies or storage diseases.
Messenger ribonucleid acid (mRNA) coding cTnT in human biopsy samples from patients with chronic skeletal muscle diseases was repeatedly reported in the literature [128,130,132,133,138,139,140,141]. Data on re-expression of cTnT in humans on the protein level is mainly based on immunohistochemistry and western-blotting [128,133,138,139]. Given the inherent problems of specificity of these technologies, these observations are not a definitive proof of re-expression. More recently, however, cTnT expression in skeletal muscle biopsy specimens of patients with Pompe disease was demonstrated by detection of cTnT fragments using nanoflow LC-MS mass spectrometry and by simultaneous detection of cTnT mRNA [130]. By contrast, cTnT was not detected in skeletal muscle of healthy controls, and Wens et al. [130] detected cTnT isoform 6, which is the cTnT isoform expressed in healthy hearts, in skeletal muscle of some patients with Pompe disease. This identifies skeletal muscle of a potential source of circulating cTnT in these patients in case of missing cardiac involvement. In contrast, Schmid et al [131] reported no evidence of cTnT re-expression in skeletal muscle biopsies from patients with myopathies and myositis by mass spectrometry, although significantly more patients had elevated peripheral blood cTnT concentrations compared with cTnI. These conflicting results, may be explained by disease dependent differences in cTnT concentrations within skeletal muscle and/or on differences in the analytical sesnitivities of mass spectrmetry based analytical methods used in both studies. cTnT mRNA expression in skeletal muscle specimens was not tested in the study of Schmid et al. [131].
In summary, there is accumulating evidence of cTnT expression in several human chronic skeletal muscle diseases, and cTnI is the most cardiac-specific marker in these rare patient populations. It is easier to interpret when AMI is suspected in these patients although serial testing is required anyway to confirm acute myocardial injury by a significant change in cTnI concentrations. In addition, with increasing age of patients with skeletal muscle myopathies the frequency of cardiac involvement increases as well, which may lead to chronic increases in both cTnT and cTnI.
cTnI and cTnT in chronic renal failure patients
As already discussed in detail above, discrepancies of cTnI and cTnT concentrations are frequent in patients with advanced renal failure [142,143,144], which may be at least in part due to differences in renal clearances of cTn fragments from blood [79,82]. In hemodialysis patients the changes in cTns during hemodialysis depend on the used membrane and modality of dialysis [145,146], which both influence the permeabilities and clearances of proteins including cTn fragments. In addition, the adherences of cTn complexes and cTn fragments to the dialyzer membrane may be different and membrane dependent, and the recovery of cTn fragments is assay depent as well. In addition, myocardial stress due to a variety of causes (e.g. CAD, hypertension, left ventricular hypertrophy, heart failure) in this patient population may lead to a stable chronic increase in both cTnI and cTnT. cTnI increases, however, are less frequent [142,143,144]. Ricchiuti et al. [140] reported cTnT mRNA expression and cTnT protein expression by Western blotting analysis without cTnI expression in about 50% of skeletal muscle specimens of hemodialysis patients in whom abdominal wall, back muscles and arm muscles were tested. It was not reported which muscle specimens tested positive. By contrast, Haller et al. [147] reported the absence of cTnT expression in abdominal wall or back skeletal muscle biopsy specimen of 5 patients with end-stage renal failure. However, truncal skeletal muscles are typically not involved in uremic skeletal myopathy. Despite more frequently seen cTnT increases and irrespective of the mechanisms of increase, both hs-cTnT and hs-cTnI maintain their prognostic value in patients with chronic renal failure [142]. Similar to chronic skeletal muscle diseases, the cardiacspecificity of cTnI for the detection of acute myocardial injury appears to be higher in this patient population as well, although additionally serial testing of cTnI is also required for AMI diagnosis to document a significant change.
6. Summary and Conclusions
Currently, cTnI and cTnT remain the most accurate laboratory paramaters for routine laboratory diagnosis of myocardial injury, which are hard to be replaced in the future as well. In general, there are no clinically relevant differences in the diagnostic performances of cTnI and cTnT. However, after AMI cTnT tends to stay increased longer than cTnI, and in specific, but rare patient populations with chronic skeletal muscle diseases or chronic renal failure, the clinical specificity of cTnI for the detection of acute myocardial injury is higher. Even mild forms of myocardial injury can be reliably detected by using hs-cTn assays for measurement. Unexpectedly elevated cTn test result should not be neglected, because analytical interferences with false positive test results are very rare. The extreme analytical sensitivity of hs-cTn assays substantially improved the negative predicitive value of cTn in patients admitted with suspected acute coronary syndromes. However, this comes at the cost of clinical specificity for AMI. A promising approach to improve the clinical specificity for type 1 AMI is studying circulating specific cTn degradation forms by the development of sensitive and specific immunoassays for the detection of particular cTn forms, e.g. the so called “long-form” of cTnT or the calculation of a cTnI/ cTnT ratio in blood samples. However, these results of promising pilot studies have to be confirmed in large clinical studies with testing fresh blood samples.
Supplementary Materials
Figure S1: Peri-interventional cardiac troponin T interpretation in a 60 year-old male with known coronary artery disease with succsessful, uncomplicated chronic total occlussion (CTO) percutaneous coronary intervention (PCI) of the right coronary artery for stable angina, Figure S2: A patient with angiographically documented peri-interventional type 4a acute myocardial infarction, Figure S3: Cardiac troponin T concentration time course in a patient with successful primary percutaneous coronary intervention of a dominant right coronary artery, Figure S4: Cardiac biomarker time courses in a 45-year old male patient with ST-segment elevation acute myocardial infarction, Figure S5: Cardiac troponin T concentration time course in a patient with inferior wall ST-segment elevation acute myocardial infarction with no reflow after primary percutaneous coronary intervention.
Funding
none.
Institutional Review Board Statement
not applicable.
Informed Consent Statement
not applicable.
Data Availability Statement
data are contained within the article.
Conflicts of Interest
The author reports research collaboration on cardiac biomarker point-of-care diagnostics with Siemens Healthineers, The Netherlands.
References
- Thygesen, K.; Mair, J.; Katus, H.; Plebani, M.; Venge, P.; Collinson, P.; et al. Study Group on Biomarkers in Cardiology of the ESC Working Group on Acute Cardiac Care. Recommendations for the use of cardiac troponin measurement in acute cardiac care. Eur. Heart J. 2010, 31, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Mair, J.; Giannitsis, E.; Mueller, Ch.; Lindahl, B.; Blankenberg, S.; et al. the Study group on Biomarkers in Cardiology of the ESC Working group on Acute Cardiac Care. How to use high-sensitivity cardiac troponins in acute cardiac care. Eur. Heart J. 2012, 33, 2252–2257. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.B.; Morrow, D.A.; et al. Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth universal definition of myocardial infarction (2018). Eur. Heart J. 2019, 40, 237–269. [Google Scholar]
- Mair, J.; Cullen, L.; Giannitsis, E.; Hammarsten, O.; Huber, K.; Jaffe, A.; et al. Application of the fourth universal definition of myocardial infarction in clinical practice. Biomarkers 2020, 25, 322–330. [Google Scholar] [CrossRef]
- Lindahl, B.; Baron, T.; Albertucci, M.; Prati, F. Myocardial infarction with non-obstructive coronary artery disease. Eurointervention 2021, 17, e875–e887. [Google Scholar] [CrossRef]
- Mair, J.; Lindahl, B.; Müller, Ch.; Giannitsis, E.; Huber, K.; Möckel, M.; et al. What to do when you question cardiac troponin values. Eur. Heart J. Acute Cardiovasc. Care.
- Mair, J.; Jaffe, A.; Lindahl, B.; Mills, N.; Möckel, M.; Cullen, L.; et al. The clinical approach to diagnosing peri-procedural myocardial infarction after percutaneous coronary interventions according to the fourth universal definition of myocardial infarction - from the study group on biomarkers of the European Society of Cardiology (ESC) Association for Acute CardioVascular Care (ACVC). Biomarkers 2022, 27, 407–417. [Google Scholar]
- Mair, J.; Giannitsis, E.; Mills, N.L.; Mueller, C. Study Group on Biomarkers of the European Society of Cardiology Association for Acute CardioVascular Care. How to deal with unexpected cardiac troponin results. Eur. Heart J. Acute Cardiovasc. Care, 11.
- Mair, J.; Hammarsten, O. Potential analytical interferences in cardiac troponin immunoassays. J. Lab. Precis. Med.
- Parmacek, M.S.; Solaro, R.J. Biology of troponin complex in cardiac myocytes. Prog. Cardiovasc. Dis. 2004, 47, 159–176. [Google Scholar] [CrossRef]
- Katrukha, I.A. Human cardiac troponin complex. Structure and functions. Biochemistry (Moscow) 2013, 73, 1447–1465. [Google Scholar] [CrossRef]
- Wei, B.; Jin, J.P. Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch. Biochem. Biophys. 2011, 505, 144–54. [Google Scholar] [CrossRef]
- Takeda, S.; Yamashita, A.; Maeda, K.; Maeda, Y. Structure of the core domain of human cardiac troponin in the Ca2+ saturated form. Nature 2003, 424, 35–41. [Google Scholar] [CrossRef]
- Schreier, T.; Kedes, L.; Gahlmann, R. Cloning, structural analysis, and expression of the human slow twitch skeletal muscle/cardiac troponin C gene. J. Biol. Chem. 1990, 265, 21247–21253. [Google Scholar] [CrossRef]
- Vallins, W.J.; Brand, N.J.; Dabhade, N.; Butler-Browne, G.; Yacoub, M.H.; Barton, P.J. Molecular cloning of human cardiac troponin I using polymerase chain reaction. FEBS Lett. 1990, 270, 57–61. [Google Scholar] [CrossRef]
- Mesnard, L.; Samson, F.; Espinasse, I.; Durand, J.; Neveux, J.Y.; Mercadier, J.J. Molecular cloning and developmental expression of human cardiac troponin T. FEBS Lett. 1993, 328, 139–144. [Google Scholar] [CrossRef]
- Anderson, P.A.W; Greig, A.; Mark, T.M.; Malouf, N.N.; Oakeley, A.E.; Ungerleider, R.M.; et al. Molecular basis of human cardiac troponin T isoforms expressed in developing, adult, anf failing heart. Circ. Res. 1995, 76, 681–686. [Google Scholar] [CrossRef]
- Martin, A.F. Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin I. J. Biol. Chem.
- Remppis, A.; Scheffold, T.; Greten, J.; Haas, M.; Greten, T.; Kübler, W.; et al. Intracellular compartmentation of troponin T: release kinetics after global ischemia and calcium paradox in the isolated perfused rat heart. J. Mol. Cell. Cardiol.
- Bleier, J.; Vorderwinkler, K.P.; Falkensammer, J.; Mair, P.; Dapunt, O.; Puschendorf, B.; et al. Different intracellular compartmentations of cardiac troponins and myosin heavy chains: a causal connenction to their different early release after myocardial damage. Clin. Chem. 1998, 44, 1912–1918. [Google Scholar] [CrossRef]
- De Lisa, F.; De Tullio, R.; Salamino, F.; Barbato, R.; Melloni, E.; Siliprandi, N.; et al. Specific degradation of troponin T and I by µ-calpain and its modulation by substrate phosphorylation. Biochem. J. 1995, 308, 57–61. [Google Scholar] [CrossRef]
- Communal, C.; Sumandea, M.; de Tombe, P.; Narula, J.; Solaro, R.J.; Hajjar, R.J. Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 2002 99, 6252–6256. [Google Scholar] [CrossRef]
- Martin-Garrido, A.; Biesiadecki, B.J.; Sakhi, H.E.; Shaifta, Y.; G Dos Remiedios, C.; Ayaz-Guner, S.; et al. Monophosphorylation of cardiac troponin-I at Ser-23/24 is sufficient to regulate cardiac myofibrillar Ca2+ sensitivity and calpain-induced proteolysis. J. Biol. Chem. 2018, 293, 8588–8599. [Google Scholar] [CrossRef]
- Ke, L.; Qi, X.Y.; Dijkhuis, A.J.; Chartier, D.; Nattel, S.; Henning, R.H.; et al. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J. Mol. Cell. Cardiol. 2008, 45, 685–693. [Google Scholar] [CrossRef]
- Starnberg, K.; Jeppsson, A.; Lindahl, B.; Hammarsten, O. Revision of the troponin T release mechanism from damaged human myocardium. Clin. Chem. 2014, 60, 1098–1104. [Google Scholar] [CrossRef]
- Mair, J.; Morandell, D.; Genser, N.; Lechleitner, P.; Dienstl, F.; Puschendorf, B. Equivalent early sensitivitties of myoglobin, creatine kinase MB mass, creatine kinase isoform ratios and cardiac troponin I and T after acute myocardial infarction. Clin. Chem. 1995, 41, 1266–1272. [Google Scholar] [CrossRef]
- Mair, J.; Thome-Kromer, B.; Wagner, I.; Lechleitner, P.; Dienstl, F.; Puschendorf, B.; et al. Concentration time courses of troponin and myosin subunits after acute myocardial infarction. Coron. Artery Dis. 1994, 5, 865–872. [Google Scholar]
- Piper, H.M.; Schwartz, P.; Spahr, R.; Hütter, J.F.; Spieckermann, G. Early enzyme release from myocardial cells is not due to irreversible cell damage. J. Mol. Cell. Cardiol. 1984, 16, 385–388. [Google Scholar] [CrossRef]
- Trump, B.F.; Berezesky, I.K. The role of cytosolic Ca2+ in cell injury, necrosis and apoptosis. Curr Opin Cell Biol 1992, 2, 227–232. [Google Scholar] [CrossRef]
- Lin, N.N.; Cheng, C.C.; Lee, Y. F; Chen, J-S.; Ho, S-P.; et al. Early activation of myocardial matrix metalloproteinases and degradation of cardiac troponin I after experimental subarachnoid haemorrhage. J. Surg. Res. 2013, 179, e41–e48. [Google Scholar] [CrossRef]
- Burkart, E.M.; Sumandea, M.P.; Kobayashi, T.; Nili, M.; Martin, A.F.; Homsher, E.; et al. Phosphorylation or Glutamic Acid Substitution at Protein Kinase C Sites on Cardiac Troponin I Differentially Depress Myofilament Tension and Shortening Velocity. J. Biol. Chem. 2003, 278, 11265–11272. [Google Scholar] [CrossRef]
- Sumandea, M.P.; Vahebi, S.; Sumandea, C.A.; Garcia-Cazarin,M. L.; Staidle, J.; Homsher, E. Impact of Cardiac Troponin T N-Terminal Deletion and Phosphorylation on Myofilament Function. Biochemistry 2009, 48, 7722–7731. [Google Scholar] [CrossRef]
- Mahmud, Z.; Zahran, S.; Liu, P.B.; Reiz, B.; Chan, B.Y.H.; Roczkowsky, A.; et al. Structure and proteolytic susceptibility of the inhibitory C-terminal tail of cardiac troponin I. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 661–671. [Google Scholar] [CrossRef]
- Katrukha, I.A.; Kogan, A.E.; Vylegzhanina, A.V.; Serebryakova, M.V.; Koshkina, E.V.; Bereznikova, A.V.; et al. Thrombin-mediated degradation of human cardiac troponin T. Clin. Chem. 2017, 63, 1094–1100. [Google Scholar] [CrossRef]
- Committee on Clinical Applications of Cardiac Biomarkers (C-CB). High-Sensitivity Cardiac Troponin I and T Assay Analytical Characteristics Designated by Manufacturer v062024. High-Sensitivity-Cardiac-Troponin-I-and-T-Assay-Analytical-Characteristics-Designated-By-Manufacturer-v062024.pdf (assessed on th 2025). 25 April.
- Asayama, J.; Yamahara, Y. , Ohta; B., Miyazaki; H., Tatsumi; T., Matsumoto; T., et al. Release kinetics of cardiac troponin T in coronary effluent from isolated rat hearts during hypoxia and reoxygenation. Bas. Res. Cardiol., 1992, 87, 428–436. [Google Scholar] [CrossRef]
- Bertinchant, J.P.; Polge, A.; Robert, E.; Sabbah, N.; Fabbo-Paray, P.; Poirey, S.; et al. Time-course of cardiac troponin I release from isolated perfused rat hearts during hypoxia/reoxygenation and ischemia/reperfusion, Clin. Chim. Acta 1999, 283, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Metzler, B.; Hammerer-Lercher, A.; Jehle, J.; Dietrich, H.; Pachinger, O.; Xu, Q.; et al. Plasma cardiac troponin T closely correlates with infarct size in a mouse model of acute myocardial infarction. Clin. Chim. Acta. 2002, 87–90. [Google Scholar] [CrossRef]
- Zabel, M.; Hohnloser, S.H.; Köster, W.; Prinz, M.; Kasper, W.; Just, H. Analysis of creatine kinase; CK-MB, myoglobin, and troponin T time-activity curves for early assessment of coronary artery reperfusion after intravenous thrombolysis. Circulation 1993, 87, 1542–1550. [Google Scholar] [CrossRef]
- Katus, H.A.; Remppis, A.; Scheffold, T.; Diederich, K.W.; Kuebler, W. Intracellular compartmentation of cardiac troponin T and its release kinetics in patients with reperfused and nonreperfused myocardial infarction. Am. J. Cardiol. 1991, 67, 1360–1367. [Google Scholar] [CrossRef]
- Cooper, S.T; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1440. [Google Scholar] [CrossRef]
- Hessel, M.H.; Michielsen, E.C.; Atsma, D.E.; Schalij, M.J.; van der Valk, E.J.; Bax, W.H.; et al. Release kinetics of intact and degraded troponin I and T after irreversible cell damage. Exp. Mol. Pathol. 2008, 85, 90–95. [Google Scholar] [CrossRef]
- Piper, H.M.; Schwartz, P.; Hutter, J.F.; Spieckermann, P.G. Energy metabolism and enzyme release of cultured adult rat heart muscle cells during anoxia, J. Mol. Cell. Cardiol. 1984, 16, 995–1007. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Saffitz, J.E.; Mealman, T.L.; Grace, A.M.; Roberts, R. Reversible myocardial ischemic injury is not associated with increased creatine kinase activity in plasma. Clin. Chem. 1997, 43, 467–475. [Google Scholar] [CrossRef]
- Hammersten, O.; Mair, J.; Möckel, M.; Lindahl, B.; Jaffe, A.S. Possible mechanisms behind cardiac troponin elevations. Biomarkers 2018, 23, 725–734. [Google Scholar] [CrossRef]
- Weil, B.R.; Young, R.F.; Shen, X.; Suzuki, G.; Qu, J. Malhotra S; Canty JM; Jr. Brief Myocardial Ischemia Produces Cardiac Troponin I Release and Focal Myocyte Apoptosis in the Absence of Pathological Infarction in Swine. J. Am. Coll. Cardiol. Basic Transl. Sci. 2017, 2, 105–114. [Google Scholar]
- Weil, B.R.; Suzuki, G.; Young, R.F.; Iyer, V; Canty, J. M.Jr. Troponin Release and Reversible Left Ventricular Dysfunction After Transient Pressure Overload. J. Am. Coll. Cardiol. 2018, 71, 2906–2916. [Google Scholar] [CrossRef] [PubMed]
- Streng, A.S.; Jacobs, L.H.; Schwenk, R.W.; Cardinaels, E.P.; Meex, S.J.; Glatz, J.F.; et al. Cardiac troponin in ischemic cardiomyocytes: intracellular decrease before onset of cell death. Exp. Mol. Pathol. 2014, 96, 339–345. [Google Scholar] [CrossRef]
- Nassenstein, K.; Breuckmann, F.; Bucher, C.; Kaiser, K.; Konzora, T.; Schäfer, L.; et al. How much myocardial damage is necessary to enable detection of focal gadolinium enhancement at cardiac MR imaging? Radiology 2008, 249, 829–835. [Google Scholar] [CrossRef]
- Salatzki, J.; Giannitsis, E.; Hegenbarth, A.; Mueller-Hennessen, M.; André, F.; Frey, N.; et al. Absence of visible infarction on cardiac magnetic resonance imaging despite the established diagnosis of myocardial infarction by 4th Universal Definition of Myocardial Infarction. Eur. Heart J. Acute Cardiovasc. Care 2024, 13, 24–35. [Google Scholar] [CrossRef]
- Marjot, J.; Kaier, T.E.; Martin, E.D.; Reji, S.S.; Copeland, O.; Iqbal, M.; et al. Quantifying the Release of Biomarkers of Myocardial Necrosis from Cardiac Myocytes and Intact Myocardium. Clin. Chem. 2017, 63, 990–996. [Google Scholar] [CrossRef]
- Cummins, B.; Auckland, M.L.; Cummins, P. Cardiac-specific troponin-I radioimmunoassay in the diagnosis of acute myocardial infarction. Am. Heart J. 1987, 113, 1333–1344. [Google Scholar] [CrossRef]
- Giannitsis, E.; Kurz, K.; Hallermayer, K.; Jarausch, J.; Jaffe, A.S.; Katus, H.A. Analytical validation of a high-sensitivity cardiac troponin T assay. Clin Chem 2010, 56, 254–261. [Google Scholar] [CrossRef]
- Krintus, M.; Kozinski, M.; Boudry, P.; Capell, N.E.; Köller, U.; Lackner, K.; et al. European multicenter analytical evaluation of the Abbott Architect STAT high sensitivity troponin I assay. Clin. Chem. Lab. Med. 2014, 52, 1657–1665. [Google Scholar]
- Portbury, A.L.; Willis, M.S.; Patterson, C. Tearin’ Up My Heart: Proteolysis in the Cardiac Sarcomere. J. Biol. Chem. 2011, 286, 9929–9934. [Google Scholar] [CrossRef] [PubMed]
- Martin, AF. Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin I. J. Biol. Chem. 1981, 256, 964–968. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Morrow, D.A.; de Lermos, J.A.; Jarolim, P.; Braunwald, E. Detection of acute changes in circulating troponin in the setting of transient stress test-induced myocardial ischemia using an ultrasensitive assay: results from TIMI 35. Eur. Heart. J 2009, 30, 162–169. [Google Scholar] [CrossRef]
- Siriwardena, M.; Campbell, V.; Richards, A.M.; Pemberton, C.J. Cardiac Biomarker Responses to Dobutamine Stress Echocardiography in Healthy Volunteers and Patients with Coronary Artery Disease. Clin. Chem. 2012, 58, 1492–1494. [Google Scholar] [CrossRef]
- Jannssen, S.L.J.E.; de Fries, F.; Mingels, A.M.A.; Kleinnibbelink, G.; Hopman, M.T.E.; Mosterd, A.; et al. Exercise-induced cardiac troponin release in athletes with versus without coronary atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2024, 326, H1045–H1052. [Google Scholar] [CrossRef]
- Turer, A.T.; Addo, T.A.; Martin, J.L.; Sabatine, M.S.; Lewis, G.D.; Gerszten, R.E.; et al. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. J. Am. Coll. Cardiol. 2011, 57, 2398–405. [Google Scholar] [CrossRef]
- Arnadottir, A.; Pedersen, S.; Bo Hasselbalch, R.; Goetze, J.P.; Friis-Hansen, L.J; . Bloch-Munster, A.M.; et al. Temporal Release of High-Sensitivity Cardiac Troponin T and I and Copeptin After Brief Induced Coronary Artery Balloon Occlusion in Humans. Circulation 2021, 143, 1095–104. [Google Scholar] [CrossRef]
- Oemrawsingh, R.M.; Cheng, J.M.; Garcia-Garcia, H.M.; van Schaik, R.H.N.; Regar, E.; van Geus, R-J. ; et al. High-sensitivity troponin T in relation to coronary plaque characteristics in patients with stable coronary artery disease; results of the ATHEROREMO-IVUS study. Atherosclerosis 2016, 247, 135–141. [Google Scholar] [CrossRef]
- Silvain, J.; Zeitouni, M.; Paradies, V.; Zheng, H.L.; Ndrepepa, G.; Cavallini, C.; et al. Cardiac procedural myocardial injury, infarction, and mortality in patients undergoing elective percutaneous coronary intervention: a pooled analysis of patient-level data. Eur. Heart J. 2021, 42, 323–334. [Google Scholar] [CrossRef]
- Zeitouni, M.; Silvain, J.; Guedeney, P.; Kerneis, M.; Yan, Y.; Overtchouk, P.; et al.; ACTION Study Group Periprocedural myocardial infarction and injury in elective coronary stenting. Eur. Heart J. 2018, 39, 1100–1109. [Google Scholar] [CrossRef]
- Moussa, I.D.; Klein, L.W.; Shah, B.; Mehran, R.; Mack, M.J.; Brilakis, E.S.; et al. Consideration of a new definition of clinically relevant myocardial infarction after coronary revascularization: an expert consensus document from the Society for Cardiovascular Angiography and Interventions (SCAI). J. Am. Coll. Cardiol. 2013, 62, 1563–1570. [Google Scholar] [CrossRef]
- Garcia-Garcia, H.M.; McFadden, E.P.; Farb, A.; Mehran, R.; Stone, G.W.; Spertus, J.; et al.; Academic Research Consortium Standardized End Point Definitions for Coronary Intervention Trials: The Academic Research Consortium-2 Consensus Document. Eur. Heart J. 2018, 39, 2192–2207. [Google Scholar] [CrossRef]
- Nie, J.; George, K.; Duan, F.; Tong, T.K.; Tian, Y. Histological evidence for reversible cardiomyocyte changes and serum cardiac troponin T elevations after exercise in rats. Physiol. Rep. 2016, 4, e13083. [Google Scholar] [CrossRef] [PubMed]
- Middleton, N.; George, K.; Whyte, G.; Gaze, D.; Collinson, P.; Shave, R. Cardiac troponin T release is stimulated by endurance exercise in healthy humans. J. Am. Coll. Cardiol. 2008, 52, 1813–1814. [Google Scholar] [CrossRef]
- Aengevaeren, V.L.; Baggish, A.L.; Chung, E.H.; George, K.; KLeiven, O.; Mingels, A.M.A; et al. Exercise-induced cardiac troponin elevation: from underlying mechanisms to clinical relevance. Circulation 2021, 144, 1955–1972. [Google Scholar] [CrossRef]
- Marshall, L.; Lee, K.K.; Stewart, S.D.; Wild, A.; Fujisawa, T.; Ferry, A.V.; et al. Effects of exercise intensity and duration on cardiac troponin release. Circulation 2020, 141, 83–85. [Google Scholar] [CrossRef]
- Scharhag, J.; Urhausen, A. , Schneider, G.; Hermann, M.; Schuhmacher, K.; Haschke, M.; et al. Reproducibility and clinical significance of exercise-induced increases in cardiac troponins and N-terminal N-termional pro brain natriuretic peptide in endurance athletes. Eur. J. Cardiovasc. Prev. Rehabil. 2006, 13, 388–397. [Google Scholar] [CrossRef]
- Mehta, R.; Gaze, D.; Mohan, S.; Williams, K.L.; Sprung, V.; George, K.; et al. Post-exercise cardiac troponin release is related to exercise training history. Int. J. Sports Med. 2012, 33, 333–337. [Google Scholar] [CrossRef]
- La Gerche, A.; Burns, A.T.; Mooney, D.J.; Inder, W.J.; Taylor, A.J.; Bogaert, J.; et al. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur. Heart J. 2012, 33, 998–1006. [Google Scholar] [CrossRef]
- Shave, R.; Oxborough, D. Exercise-induced cardiac injury: evidence from novel imaging techniques and highly sensitive cardiac troponin assays. Prog. in Cardiovasc. Disease 2012, 54, 407–415. [Google Scholar] [CrossRef]
- Möhlenkamp, S.; Leineweber, K.; Lehman, N.; et al. Coronary atherosclerosis burden, but not transient troponin elevation, predicts long-term outcome in recreational marathon runners. Basic Res. Card. 2014, 109, 391. [Google Scholar] [CrossRef]
- Aengevaeren, V.L.; Hopman, M.T.E.; Thompson, P.D.; Bakker, E.A.; George, K.P.; et al. Exercise-induced cardiac troponin I increase and incident mortality and cardiovascular events. Circulation 2019, 140, 804–814. [Google Scholar] [CrossRef]
- Skadberg, O.; Kleiven, O.; Bjorkavoll-Bergeth, M.; Melberg, T.; Bergseth, R.; Selvag, J.; et al. Highly increased troponin I levels following high-intensity endurance cycling may detect subclinical coronary artery disease in presumably healthy leisure sport cyclists: The North Sea Race Endurance Exercise Study (NEEDED) 2013. Eur. J. Prev. Cardiol. 2017, 24, 885–894. [Google Scholar] [CrossRef]
- van de Schoor, F.R.; Aengevaeren, V.L.; Hopman, M.T.; Oxborough, D.L.; George, K.P.; Thompson, P.D.; et al. Myocardial fibrosis in athletes. Mayo Clin. Proc. 2016, 91, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Muslimovic, A.; Friden, V.; Tenstad, O.; Starnberg, K.; Nystrom, S.; Wesen, E.; et al. The liver and kidneys mediate clearance of cardiac troponin in the rat. Sci. Rep. 6791. [Google Scholar]
- Streng, A.S.; de Boer, D.; van Doorn, W.P.; Kocken, J.M.; Bekers, O.; Wodzig, W.K. Cardiac troponin T degradation in serum is catalysed by human thrombin. Biochem. Biophys. Res. Commun. 2016, 481, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Klocke, F.J.; Copley, D.P.; Krawczyk, J.A.; Reichlin, M. Rapid renal clearance of immunoreactive canine plasma myoglobin. Circulation 1982, 65, 1522–1528. [Google Scholar] [CrossRef]
- Friden, V.; Starnberg, K.; Muslimovic, A.; Ricksten, S-E. ; Bjurman, C.; Forsgard, N.; et al. Clearance of cardiac troponin T with and without kidney function. Clin. Biochem. 2017, 50, 468–474. [Google Scholar] [CrossRef]
- Dunn, M.E.; Coluccio, G.; Hirkaler, I.; Mikaelian, R.; Nicklaus, R.; Lipschutz, S.E.; et al. The complete pharmacokinetic profile of serum cardiac troponin I in the rat and dog. Toxicol. Sci. 2011, 123, 368–373. [Google Scholar] [CrossRef]
- Kristensen, J.H.; Hasselbalch, R.B.; Strandkjaer, N.; Jorgensen, N.; Ostergaard, M.; Moller-Sorensen, P.H.; et al. Half-Life and Clearance of Cardiac Troponin I and Troponin T in Humans. Circulation 2024, 150, 1187–1198. [Google Scholar] [CrossRef]
- Salonen, S.M.; Tuominen, T.J.K.; Raiko, K.I.S; Vasankari, T.; Aalto, R.; Hellman, T.A.; et al. Highly sensitive immunoassay for long form of cardiac troponin T using upconversion luminescence. Clin. Chem. 2024, 70, 1037–1045. [Google Scholar] [CrossRef]
- Hammerer-Lercher, A.; Halfinger, B.; Sarg, B.; Mair. J.; Puschendorf, B.; Griesmacher, A.; et al. Analysis of circulating forms of proBNP and NT-proBNP in patients with severe heart failure. Clin. Chem. 2008, 54, 858–865. [Google Scholar] [CrossRef]
- Amplatz, B.; Sarg, B.; Faserl, K.; Hammerer-Lercher, A.; Mair, J.; Lindner, H.H. Exposing the High Heterogeneity of Circulating Pro B-Type Natriuretic Peptide Fragments in Healthy Individuals and Heart Failure Patients. Clin. Chem. 2020, 66, 1200–1209. [Google Scholar] [CrossRef]
- Peronnet, E.; Becquart, L.; Poirier, F.; Cubizolles, M.; Choquet-Kastylevsky, G.; Jolivet-Reynaud, C. SELDI-TOF MS analysis of the cardiac troponin I forms present in plasma from patients with myocardial infarction. Proteomics 2006, 6, 6288–6299. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.; Katrukha, I.A.; Zhang, L.; Shu, X.; Xu, A.; et al. Design and analytical evaluation of novel cardiac troponin assays targeting multiple forms of the cardiac troponin I–cardiac troponin T–troponin C complex and fragmentation forms. Clin. Chem. 2025, 71, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, K.E.J.; Aalto, R.; Hellman, T.; Vasankari, T.; Lahtinen, A.; Wittfooth, S. Novel Troponin Fragmentation Assay to Discriminate Between Troponin Elevations in Acute Myocardial Infarction and End-Stage Renal Disease. Circulation 2022, 146, 1408–1410. [Google Scholar] [CrossRef]
- Riabkova, N.S.; Kogan, A.E.; Katrukha, I.A.; Vylegzhanina, A.V.; Bogomolova, A.P.; Alieva, A.K.; et al. Influence of Anticoagulants on the Dissociation of Cardiac Troponin Complex in Blood Samples. Int. J. Mol. Sci.
- Katrukha, A.G.; Bereznikova, A.V.; Esakova, T.V.; Petterson, K.; Lövgren, T.; Severina, M.E.; et al. Troponin I is released in blood stream of patients with acute myocardial infarction not in free form but as complex. Clin. Chem. 1997, 43, 1379–1385. [Google Scholar] [CrossRef]
- Wu, A.H.B.; Feng, Y.J.; Moore, R.; Apple, F.S.; McPherson, P.H.; Buechler, K.F.; Bodor, G. Characterization of cardiac troponin subunit release into serum after acute myocardial infarction and comparison of assays for troponin T and I. Clin. Chem. 1998, 44, 1198–1208. [Google Scholar] [CrossRef]
- McDonough, J.L.; Arrell, D.K.; Van Eyk, J.E. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ. Res.
- McDonough, J. .L; Labugger, R.; Pickett, W.; Tse, M.Y.; MacKenzie, S.; Pang, S.C.; et al. Cardiac troponin I is modified in the myocardium of bypass patients. Circulation 2001, 103, 58–64. [Google Scholar] [CrossRef]
- Giuliani, I.; Bertinchant, J.P.; Granier, C.; Laprade, M.; Chocron, S.; Toubin, G.; et al. Determination of cardiac troponin I forms in the blood of patients with acute myocardial infarction and patients receiving crystalloid or cold blood cardioplegia. Clin. Chem. 1999, 45, 213–222. [Google Scholar] [CrossRef]
- Labugger, R.; Organ, L.; Collier, C.; Atar, D.; van Eyk, J.E. Extensive troponin I and troponin T modification detected in serum from patients with acute myocardial infarction. Circulation 2000, 102, 1221–1226. [Google Scholar] [CrossRef]
- Giuliani, I.; Bertinchant, J.P.; Lopez, M.; Coquelin, H.; Granier, C.; Laprade, M.; et al. Determination of cardiac troponin I forms in the blood of patients with unstable angina pectoris. Clin. Biochem. 2002, 35, 111–117. [Google Scholar] [CrossRef]
- Fahie-Wilson, M.N.; Carmichael, D.J.; Delaney, M.P.; Stevens, P.E.; Hall, E.M.; Lamb, E.J. Cardiac troponin T circulates in the free, intact form in patients with kidney failure. Clin. Chem. 2006, 52, 414–420. [Google Scholar] [CrossRef]
- Madsen, L.H.; Christensen, G.; Lund, T.; Serebruany, V.L.; Granger, C.B.; Hoen, I.; et al. Time course of degradation of cardiac troponin I in patients with acute ST-elevation myocardial infarction: the ASSENT-2 troponin substudy. Circ. Res. 2006, 99, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Michielsen, E.C.; Diris, J.H.; Kleijnen, V.W.; Wodzig, W.K.; Van Dieijen-Visser, M.P. Investigation of release and degradation of cardiac troponin T in patients with acute myocardial infarction. Clin. Biochem. 2007, 40, 851–855. [Google Scholar] [CrossRef]
- Bates, K.J.; Hall, E.M.; Fahie-Wilson, M.N.; Kindler, H.; Bailey, C.; Lythall, D.; Lamb, E.J. Circulating immunoreactive cardiac troponin forms determined by gel filtration chromatography after acute myocardial infarction. Clin. Chem. 2010, 56, 952–958. [Google Scholar] [CrossRef]
- Cardinaels, E.P.; Mingels, A.M.; van Rooij, T.; Collinson, P.O.; Prinzen, F.W.; van Dieijen-Visser, M.P. Time-dependent degradation pattern of cardiac troponin T following myocardial infarction. Clin Chem. 2013, 59, 1083–1090. [Google Scholar] [CrossRef]
- Streng, A.S.; de Boer, D.; van Doorn, W.P.T.M.; Bouwman, F.G.; Mariman, E.C.M.; Bekers, O.; et al. Identification and characterization of cardiac troponin T fragments in serum of patients suffering from acute myocardial infarction. Clin. Chem. 2017, 63, 563–572. [Google Scholar] [CrossRef]
- Mingels, A.M.; Cardinaels, E.P.; Broers, N.J.; van Sleeuwen, A.; Streng, A.S.; van Dieijen-Visser, M.P.; et al. Cardiac Troponin T: Smaller Molecules in Patients with End-Stage Renal Disease than after Onset of Acute Myocardial Infarction. Clin. Chem. 2017, 63, 683–690. [Google Scholar] [CrossRef]
- van Wijk, X.M.R.; Claassen, S.; Enea, N.S.; Li, P.; Yang, S.; Brouwer, M.A.; et al. Cardiac troponin I is present in plasma of type 1 myocardial infarction patients and patients with troponin I elevations due to other etiologies as complex with little free I. Clin. Biochem. 2019, 73, 3543. [Google Scholar] [CrossRef]
- Vylegzhanina, A.V.; Kogan, A.E.; Katrukha, I.A.; Koshkina, E.V.; Bereznikova, A.V.; Filatov, V.L.; et al. Full-size and partially truncated cardiac troponin complexes in the blood of patients with acute myocardial infarction. Clin. Chem. 2019, 65, 882–892. [Google Scholar] [CrossRef]
- Damen, S.A.J.; Cramer, G.E.; Dieker, H-J. ; Gehlmann, H.; Ophuis, T.J.M.O.; Aengevaeren, W.R.M., et al. Cardiac troponin composition characterization after non ST-elevation myocardial infarction: relation with culprit artery, ischemic time window, and severity of injury. Clin. Chem. 2021, 67, 227–236. [Google Scholar] [CrossRef]
- Katrukha, I.A.; Riabkova, N.S.; Kogan, A.E.; Vylegzhanina, A.V.; Mukharyamova, K.S.; Bogomolova, A.P.; et al. Fragmentation of human cardiac troponin T after acute myocardial infarction. Clin. Chim. Acta 2023, 542, 117281. [Google Scholar] [CrossRef]
- Vroemen, W.H.M.; Mezger, S.T.P.; Masotti, S.; Clerico, A.; Bekers, O.; de Boer, D.; et al. Cardiac troponin T: only small molecules in recreational runners after marathon completion. J. Appl. Lab. Med. 2019, 3, 909–911. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, K.E.J.; Paana, T.; Vasankari, T.; Salonen, S.; Tuominen, T.; Linko-Parvinen, A.; et al. Composition of cardiac troponin release differs after marathon running and myocardial infarction. Open Heart, 0029. [Google Scholar]
- Airaksinen, K.E.J.; Tuominen, T.; Paana, T.; Hellman, T.; Vasankari, T.; Salonen, S.; et al. Novel troponin fragmentation assay to discriminate between Takotsubo syndrome and acute myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care 2024, 13, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, Y.; Katrukha, I.A.; Zhang, L.; Shu, X.; Xu, A.; et al. Characterization of cardiac troponin fragment composition reveals potential for differentiating etiologies of myocardial injury. Clin. Chem. 2025, 71, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Damen, S.A.J.; Vroemen, W.H.M.; Brouwer, M.A.; Mezger, S.T.P.; Suryapranata, H.; van Royen, N.; et al. Multi-Site Coronary Vein Sampling Study on Cardiac Troponin T Degradation in Non-ST segment-Elevation Myocardial Infarction: Toward a More Specific Cardiac Troponin T Assay. J. Am. Heart. Assoc. 2019, 8, e012602. [Google Scholar] [CrossRef]
- Eggers, K.M.; Hammarsten, O.; Lindahl, B. Differences between high-sensitivity cardiac troponin T and I in stable populations: underlying causes and clinical implications. Clin. Chem. Lab. Med. 2023, 61, 380–387. [Google Scholar] [CrossRef]
- Eggers, K.M.; Hammarsten, O.; Aldous, S.J.; Cullen, L.; Greenslade, J.H.; Lindahl, B.; et al. Diagnostic and prognostic performance of the ratio between high-sensitivity cardiac troponin I and troponin T in patients with chest pain. PLoS One 2022, 17, e0276645. [Google Scholar] [CrossRef]
- Laugaudin, G.; Kuster, N.; Petiton, A.; et al. Kinetics of high-sensitivity cardiac troponin T and I differ in patients with ST-segment elevation myocardial infarction treated by primary coronary intervention. Eur. Heart. J. Acute Cardiovasc. Care 2016, 5, 354–363. [Google Scholar] [CrossRef]
- Cullen, L.; Aldous, S.; Than, M.; Greenslade, J.H.; Tate, J.R.; George, P.M.; et al. Comparison of high sensitivity troponin T and I assays in the diagnosis of non-ST elevation acute myocardial infarction in emergency patients with chest pain. Clin. Biochem. 2014, 47, 321–326. [Google Scholar] [CrossRef]
- Rubini Gimenez, M.; Twerenbold, R.; Reichlin, T.; Wildi, K.; Haaf, P.; Schaefer, M.; et al. Direct comparison of high-sensitivity-cardiac troponin I vs. T for the early diagnosis of acute myocardial infarction. Eur. Heart J. 2014, 35, 2303–2311. [Google Scholar] [CrossRef]
- Wereski, R.; Kimenai, D.M.; Taggart, C.; Doudesis, D.; Lee, K.K.; Lowry, M.T.H.; et al. Cardiac Troponin Thresholds and Kinetics to Differentiate Myocardial Injury and Myocardial Infarction. Circulation 2021, 144, 528–538. [Google Scholar] [CrossRef]
- van Doorn, W.; Vroemen, W.H.M.; Smulders, M.W.; van Suijlen, J.D.; van Cauteren, Y.J.M.; Bekkers, S.; et al. High-Sensitivity Cardiac Troponin I and T Kinetics after Non-ST-Segment Elevation Myocardial Infarction. J. Appl. Lab. Med. 2020, 5, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Reinstadler, S.J.; Feistritzer, H.J.; Klug, G.; Mair, J.; Minh-Duc Tu, A.; Kofler, M.; et al. High-sensitivity troponin T for prediction of left ventricular function and infarct size one year following ST-elevation myocardial infarction. Int. J. Cardiol. 2016, 202, 188–93. [Google Scholar] [CrossRef] [PubMed]
- Mair, J.; Wagner, I.; Morass, B.; Fridrich, L.; Lechleitner, P.; Dienstl, F.; et al. Cardiac troponin I release correlates with myocardial infarction size. Eur. J. Clin. Chem. Clin. Biochem. 1995, 33, 869–872. [Google Scholar] [PubMed]
- Wagner, I.; Mair, J.; Fridrich, L.; Artner-Dworzak, E.; Lechleitner, P.; Morass, B.; et al. Cardiac troponin T release in acute myocardial infarction is associated with scintigraphic estimates of myocardial scar. Coron. Artery Dis. 1993, 4, 537–544. [Google Scholar] [CrossRef]
- Starnberg, K.; Friden, V.; Muslimovic, A.; Ricksten, S-V. ; Nystrom, S.; Forsgard, N.; et al. A possible mechanism behind faster clearance and higher peak concentrations of cardiac troponin I compared with troponin T in acute myocardial infarction. Clin. Chem. 2020, 66, 333–341. [Google Scholar] [CrossRef]
- Hammerer-Lercher, A.; Erlacher, P.; Bittner, R.; Korinthenberg, R.; Skladal, D.; Sorichter, S.; et al. Clinical and experimental results on cardiac troponin expression in Duchenne muscular dystrophy. Clin. Chem. 2001, 47, 451–458. [Google Scholar] [CrossRef]
- Erlacher, P.; Lercher, A.; Falkensammer, J.; Nassanov, M.I. , Shtutman, V.Z., Puschendorf, B.; et al. Cardiac troponin and beta-type myosin heavy chain concentrations in patients with polymyositis or dermatomyositis. Clin. Chim. Acta 2001, 306, 27–33. [Google Scholar] [CrossRef]
- Jaffe, A.S.; Vasile, V.C.; Milone, M.; Saenger, A.K.; Olson, K.N.; Apple, F.S. Diseased skeletal muscle. A noncardiac source of increased circulating concentrations of cardiac troponin T. J. Am. Coll. Cardiol. 2011, 58, 1819–1824. [Google Scholar] [CrossRef]
- Rittoo, D.; Jones, A.; Lecky, B.; Neithercut, D. Elevation of cardiac troponin T but not cardiac troponin I in patients with neuromuscular diseases: implications for the diagnosis of myocardial infarction. J. Am. Coll. Cardiol. 2014, 63, 2411–2420. [Google Scholar] [CrossRef]
- Wens, S.C.A.; Schaaf, G.J.; Michels, M.; Kruijshaar, M.E.; van Gestel, T.J.M.; In`t Groen, S.; et al. Elevated plasma cardiac troponin T levels caused by skeletal muscle damage in Pompe disease. Circ. Cardiovasc. Genet. 2016, 9, 6–13. [Google Scholar] [CrossRef]
- Schmid, J.; Liesinger, L.; Birner-Gruenberger, R.; Stojakovic, T.; Scharnagl, H.; Dieplinger, B.; et al. Elevated cardiac troponin T in patients with skeletal myopathies. J. Am. Coll. Cardiol. 2018, 71, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Du Fay de Lavallaz, J.; Prepoudis, A.; Wendebourg, M.J.; Kesenheimer, E.; Kyburz, D.; Daikeler, T.; et al. Skeletal muscle disorders: a non-cardiac source of cardiac troponin T. Circulation 2022, 145, 1764–1779. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.H.; Heckmann, M.B.; Bailly, G.; Finke, D.; Procureur, A.; Power, J.R.; et al. Cardiomuscular Biomarkers in the Diagnosis and Prognostication of Immune Checkpoint Inhibitor Myocarditis. Circulation 2023, 148, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Saggin, L.; Gorza, L.; Ausoni, S.; Schiaffino, S. Cardiac troponin T in developing, regenerating and denervated rat skeletal muscle. Development 1990, 110, 547–554. [Google Scholar] [CrossRef]
- Anderson, P.A.W.; Malouf, N.N.; Oakeley, A.E.; Pagani, E.D.; Allen, P.D. Troponin T isoform expression in humans: a comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ. Res. 1991, 69, 1226–1233. [Google Scholar] [CrossRef]
- Sabry, M.A.; Dhoot, G.K. Identification of and pattern of transitions of cardiac, adult slow and slow skeletal musle-like embryonic isoforms of troponin T in developing rat and human skeletal muscles. J. Muscle Res. Cell Motil. 1991, 12, 262–270. [Google Scholar] [CrossRef]
- Mesnard, L.; Samson, F.; Espinasse, I.; Durand, J.; Neveux, J.Y.; Mercadier, J.J. Molecular cloning and developmental expression of human cardiac troponin T. FEBS Letters 1993, 328, 139–144. [Google Scholar] [CrossRef]
- McLaurin, M.D.; Apple, F.S.; Voss, E.M.; Herzog, C.A.; Sharkey, S.W. Cardiac troponin I, cardiac troponin T, and creatine kinase MB in dialysis patients without ischemic heart disease: evidence of cardiac troponin T reexpression in skeletal muscle. Clin. Chem. 1997, 43, 976–982. [Google Scholar] [CrossRef]
- Bodor, G.S.; Survant, L.; Voss, E.M.; Smith, S.; Porterfield, D.; Apple, F.S. Cardiac troponin T composition in normal and regenerating human skeletal muscle. Clin. Chem. 1997, 43, 476–484. [Google Scholar] [CrossRef]
- Ricchiuti, V.; Apple, F.S. RNA expression of cardiac troponin T isoforms in diseased human skeletal muscle. Clin. Chem. 1999, 45, 2129–2135. [Google Scholar] [CrossRef]
- Messner, B.; Baum, H.; Fischer, P.; Quasthoff, S.; Neumeier, D. Expression of messenger RNA of cardiac isoforms of troponin T and I in myopathic skeletal muscle. Am. J. Clin. Pathol. 2000, 114, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Hickman, P.E.; McGill, D.; Potter, J.M.; Koerbin, G.; Apple, F.S.; Talaulikar, G. Multiple biomarkers including cardiac troponin I and T measured by high-sensitivity assays, as predictors of long-term mortality in patients with chronic renal failure who underwend dialysis. Am. J. Cardiol. 2015, 115, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Buiten, M.S.; de Bie, M.K.; Rotmans, J.I.; et al. Serum cardiac troponin I is superior to troponin T as a marker for left ventricular dysfunction in clinically stable patients with end-stage renal disease. PloS One 2015, 10, e0134245. [Google Scholar] [CrossRef] [PubMed]
- Twerenbold, R.; Wildi, K.; Jaeger, C.; Rubini Gimenez, M.; Reiter, M.; Reichlin, M.; et al. Optimal cutoff levels of more sensitive cardiac troponin assays for early diagnosis of myocardial infarction in patients with renal dysfunction. Circulation 2015, 131, 2041–2050. [Google Scholar] [CrossRef]
- Kolland, M.; Amenitsch, J.; Schreiber, N.; Ginthör, N.; Schuller, M.; Riedl, R.; et al. Changes in cardiac troponin during hemodialysis depend on hemodialysis membrane and modality: a randomized crossover trial. Clin. Kidney J. 2023, 17, sfad297. [Google Scholar] [CrossRef]
- Gaze, D.C.; Collinson, P.O. Cardiac troponin I but not cardiac troponin T adheres to polysulfone dialyser membranes in an in vitro hemodialysis model: explanation for lower cTnI concentrations following dialysis. Open Heart, 0001. [Google Scholar]
- Haller, C.; Zehelein, J.; Remppis, A.; Müller-Bardorff, M.; Katus, H.A. Cardiac troponin T in patients with end stage renal disease: absence of expression in truncal skeletal muscle. Clin. Chem. 1998, 44, 930–938. [Google Scholar] [CrossRef]
Figure 1.
Potential cardiac troponin release mechanisms from injured human myocardium. Three possible mechanisms of cardiac troponin increase in the systemic circulation are summarized, i.e. cardiomyocyte necrosis, which is the only generally accepted cause, cardiomyocyte apoptosis, and reversible myocardial injury by cTn leakage via a temporarilly leaky pllasma membrane (via cell wounds, cytoplasmatic blebbing, extracellular vesicle formation).Abbreviations: cardiac troponin (cTn).
Figure 1.
Potential cardiac troponin release mechanisms from injured human myocardium. Three possible mechanisms of cardiac troponin increase in the systemic circulation are summarized, i.e. cardiomyocyte necrosis, which is the only generally accepted cause, cardiomyocyte apoptosis, and reversible myocardial injury by cTn leakage via a temporarilly leaky pllasma membrane (via cell wounds, cytoplasmatic blebbing, extracellular vesicle formation).Abbreviations: cardiac troponin (cTn).
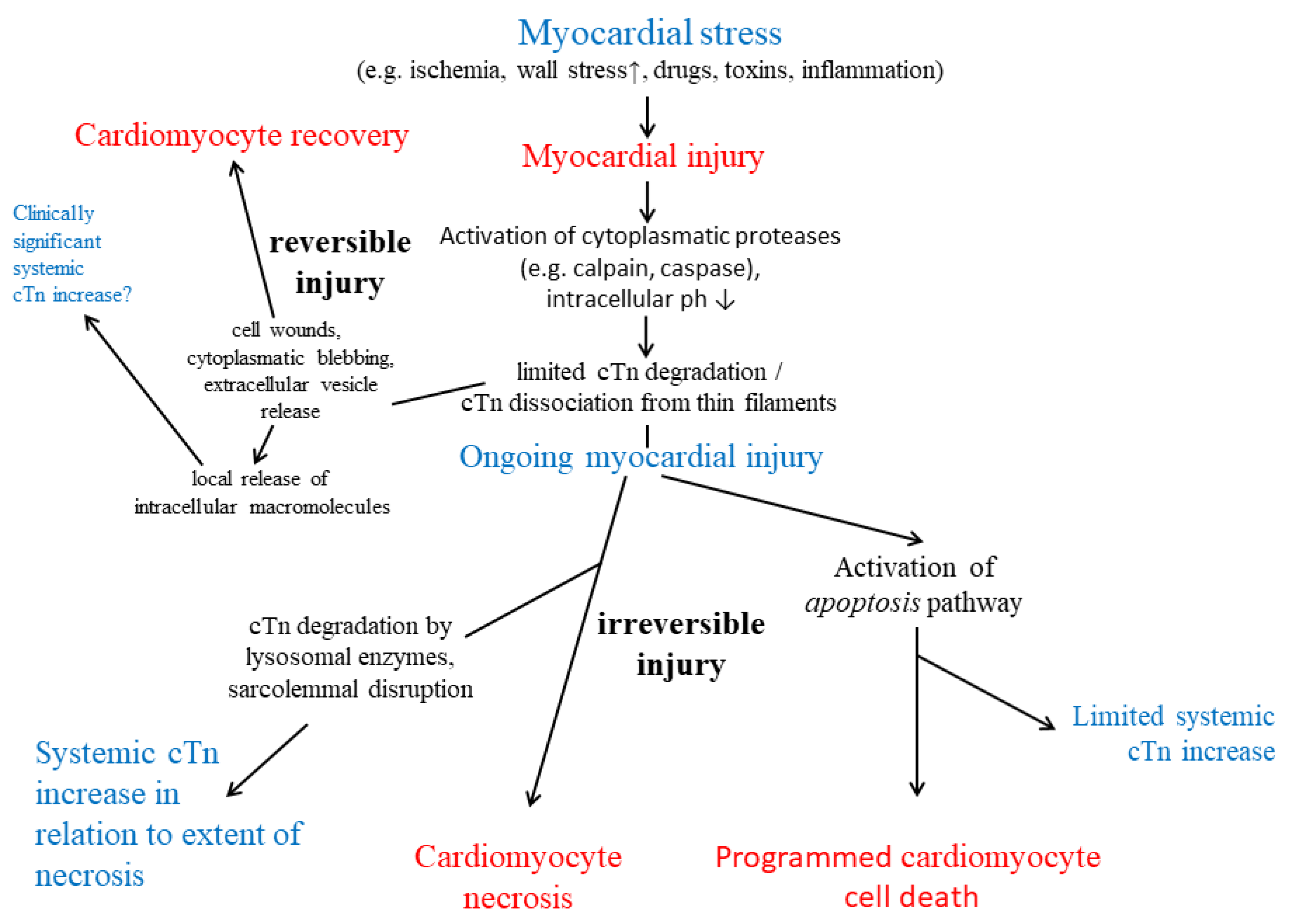
Figure 2.
Cardiac biomarker release in a patient with an uncomplicated bifurcation percutaneous tranluminal coronary intevention for unstable angina. This 55-year old male with known CAD was admitted for symptoms of unstable angina. He underwent PCI of the left circimflex coronary artery 11 years ago in another hospital. He showed no signs of acute myocardial ischemia in serial ECGs before and after PCI. Serial hs-cTnT testing in the emergency department before PCI revealed concentrations within the normal range without a significant change. After treatment in the emergency department the patient was asymptomatic, the criteria of an AMI were not fulfilled. Urgent coronary angiography on the next day revealed a subtotal in-stent restenosis at the distal end of the stent involving the distal circumflex artery bifurcation (Medina 1/1/1, marked with a circle, the angiography of the left coronary artery is shown in A). All other coronary arteries showed no significant stenoses. PCI was performed by dilatation and stent implantation in the marginal branch and dilatation of the smaller distal RCX with subsequent treatment with a drug eluting balloon after succsessful predilatation with an excellent primary result without any complications (B, marked with circle). The biomarker time courses are listed in C. Interestingly, hs-cTnT showed no significant increase within 4 hours after PCI, but it increased above the upper reference limit of 14 ng/L on the morning of the next day. This case nicely demonstrates the importance of a clear specification on the point in time of a proposed cTn decision limit for diagnosing peri-interventional AMI after elective PCI. Notably creatine kinase activities were within the reference limit throughout even showing a constant fall of activities (the patient was a construction worker of tunnel building with heavy physical activities at work). Abbreviations: high-sensitivity troponin T (hs-cTnT), percutaneous coronary intervention (PCI), circumflex artery (RCX), coronary artery disease (CAD), acute myocardial infarction (AMI).
Figure 2.
Cardiac biomarker release in a patient with an uncomplicated bifurcation percutaneous tranluminal coronary intevention for unstable angina. This 55-year old male with known CAD was admitted for symptoms of unstable angina. He underwent PCI of the left circimflex coronary artery 11 years ago in another hospital. He showed no signs of acute myocardial ischemia in serial ECGs before and after PCI. Serial hs-cTnT testing in the emergency department before PCI revealed concentrations within the normal range without a significant change. After treatment in the emergency department the patient was asymptomatic, the criteria of an AMI were not fulfilled. Urgent coronary angiography on the next day revealed a subtotal in-stent restenosis at the distal end of the stent involving the distal circumflex artery bifurcation (Medina 1/1/1, marked with a circle, the angiography of the left coronary artery is shown in A). All other coronary arteries showed no significant stenoses. PCI was performed by dilatation and stent implantation in the marginal branch and dilatation of the smaller distal RCX with subsequent treatment with a drug eluting balloon after succsessful predilatation with an excellent primary result without any complications (B, marked with circle). The biomarker time courses are listed in C. Interestingly, hs-cTnT showed no significant increase within 4 hours after PCI, but it increased above the upper reference limit of 14 ng/L on the morning of the next day. This case nicely demonstrates the importance of a clear specification on the point in time of a proposed cTn decision limit for diagnosing peri-interventional AMI after elective PCI. Notably creatine kinase activities were within the reference limit throughout even showing a constant fall of activities (the patient was a construction worker of tunnel building with heavy physical activities at work). Abbreviations: high-sensitivity troponin T (hs-cTnT), percutaneous coronary intervention (PCI), circumflex artery (RCX), coronary artery disease (CAD), acute myocardial infarction (AMI).
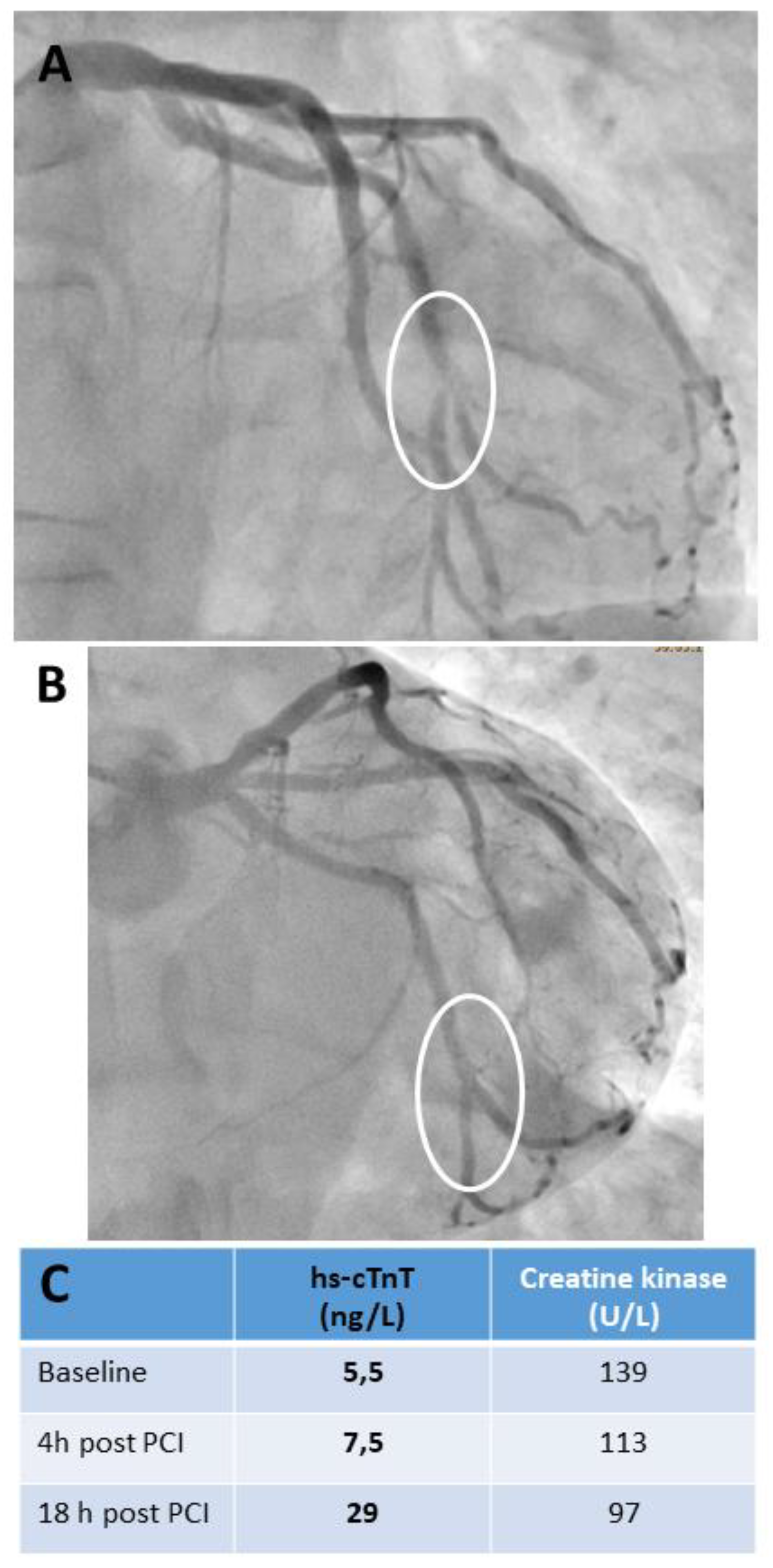
Figure 3.
Cardiac troponin T interpretation in a 39-year old male amateur cyclist, who had to abort a marathon cycling event after approximately 20 hours. The patient was admitted to the emergency department with the key symptoms dyspnoe worsened on exertion and body position and breathing-related angina. His ECG showed signs of early repolarisation (A) without change in serial recordings. The chest X-ray performed on admission showed no pathologies, in particular no signs of pneumonia. His key laboratory parameters are listed in B. A coronary computed tomography angiography revealed a calcium score of 0 with normal coronary arteries without arteriosclerotic plaques and coronary anomalies. As typical for long-term endurance sportsman the left ventricular stroke volume was increased and the left ventricle slightly dilated, the left ventricular ejection fraction and right ventricular function were normal. These findings were confirmed by echocardiography. Before start of the competition he was fit without any signs of infection, on admission he was reporting cough, the physical examination including oxygenation and body temperature were normal. hs-cTnT was slightly increased on admission with a constant fall, and concentrations were within the URL before discharge. The maximum concentration was found on admission and was <3-fold URL consistent with an exercise-induced cTn release. The high creatine kinase activities are to be expected with this high skeletal muscle workload. However, because of his advanced age coronary computed tomography angiography was performed to rule-out coronary artery disease. C-reactive protein was slightly increased on admission, which is consistent with a heavy endurance exercise workload, it returned to normal limits until discharge. Leucocytes were within reference limits, but the differential blood count in the morning after admission revealed a relative lymphocytosis of 71%. The patient wanted to be discharged as he was from abroad. His discharge diagnosis was suspicion of a viral respiratory infection. Acute myocarditis was unlikely but cardiac magnetic resonance imaging for ruling it out definitively was not possible before discharge, this imaging modality was recommended to be performed at home in case of persistent symptoms. Abbreviations: high-sensitivity troponin T (hs-cTnT), creatine kinase (CK), C-reactive protein (CRP), upper reference limit (URL), electrocardiogram (ECG)
Figure 3.
Cardiac troponin T interpretation in a 39-year old male amateur cyclist, who had to abort a marathon cycling event after approximately 20 hours. The patient was admitted to the emergency department with the key symptoms dyspnoe worsened on exertion and body position and breathing-related angina. His ECG showed signs of early repolarisation (A) without change in serial recordings. The chest X-ray performed on admission showed no pathologies, in particular no signs of pneumonia. His key laboratory parameters are listed in B. A coronary computed tomography angiography revealed a calcium score of 0 with normal coronary arteries without arteriosclerotic plaques and coronary anomalies. As typical for long-term endurance sportsman the left ventricular stroke volume was increased and the left ventricle slightly dilated, the left ventricular ejection fraction and right ventricular function were normal. These findings were confirmed by echocardiography. Before start of the competition he was fit without any signs of infection, on admission he was reporting cough, the physical examination including oxygenation and body temperature were normal. hs-cTnT was slightly increased on admission with a constant fall, and concentrations were within the URL before discharge. The maximum concentration was found on admission and was <3-fold URL consistent with an exercise-induced cTn release. The high creatine kinase activities are to be expected with this high skeletal muscle workload. However, because of his advanced age coronary computed tomography angiography was performed to rule-out coronary artery disease. C-reactive protein was slightly increased on admission, which is consistent with a heavy endurance exercise workload, it returned to normal limits until discharge. Leucocytes were within reference limits, but the differential blood count in the morning after admission revealed a relative lymphocytosis of 71%. The patient wanted to be discharged as he was from abroad. His discharge diagnosis was suspicion of a viral respiratory infection. Acute myocarditis was unlikely but cardiac magnetic resonance imaging for ruling it out definitively was not possible before discharge, this imaging modality was recommended to be performed at home in case of persistent symptoms. Abbreviations: high-sensitivity troponin T (hs-cTnT), creatine kinase (CK), C-reactive protein (CRP), upper reference limit (URL), electrocardiogram (ECG)
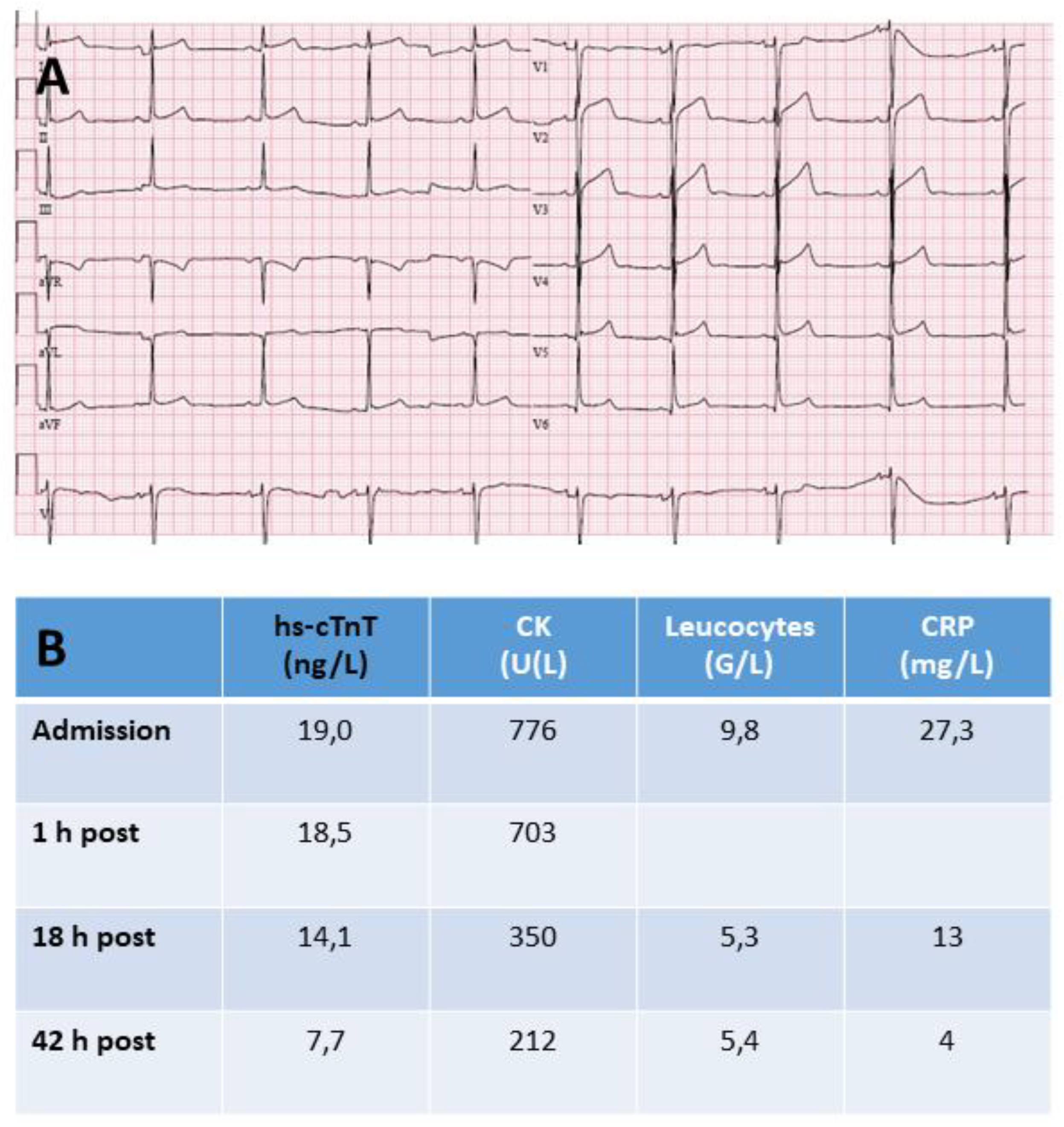
Figure 4.
Cardiac biomarker testing results of 2 recent visits of a 19-year old male with skeletal muscle dystrophy Becker. His father also suffered from this disease. The diagnosis was additionally confirmed by genetic testing with demonstration of a mutation of the dystrophin gene (deletion of exons 45-47). This patient had no cardiac symptoms throughout his whole life with a normal exercise capacity. The ECGs and echocardiograms performed at both visits were normal. NT-proBNP was normal as well. In addition, cardiac magnetic resonance imaging performed at the last visit was normal as well. Disordant hs-cTnT and hs-cTnI results are frequently seen in patients with chronic skeletal muscle dystrophies, which is consistent with re-expression of the cTnT gene in chronically injured skeletal muscle. It has to be stressed that cardiomyopathy in patients with Becker disease, although frequent, usually does not start to develop before the 3rd decade of life. Abbreviations: high-sensitivity troponin T (hs-cTnT), high-sensitivity cardiac troponin I (hs-cTnI), N-terminal B-type natriuretic peptide (NT-proBNP), upper reference limt (URL), electrocardiogram (ECG), B-type natriuretic peptide (BNP).
Figure 4.
Cardiac biomarker testing results of 2 recent visits of a 19-year old male with skeletal muscle dystrophy Becker. His father also suffered from this disease. The diagnosis was additionally confirmed by genetic testing with demonstration of a mutation of the dystrophin gene (deletion of exons 45-47). This patient had no cardiac symptoms throughout his whole life with a normal exercise capacity. The ECGs and echocardiograms performed at both visits were normal. NT-proBNP was normal as well. In addition, cardiac magnetic resonance imaging performed at the last visit was normal as well. Disordant hs-cTnT and hs-cTnI results are frequently seen in patients with chronic skeletal muscle dystrophies, which is consistent with re-expression of the cTnT gene in chronically injured skeletal muscle. It has to be stressed that cardiomyopathy in patients with Becker disease, although frequent, usually does not start to develop before the 3rd decade of life. Abbreviations: high-sensitivity troponin T (hs-cTnT), high-sensitivity cardiac troponin I (hs-cTnI), N-terminal B-type natriuretic peptide (NT-proBNP), upper reference limt (URL), electrocardiogram (ECG), B-type natriuretic peptide (BNP).
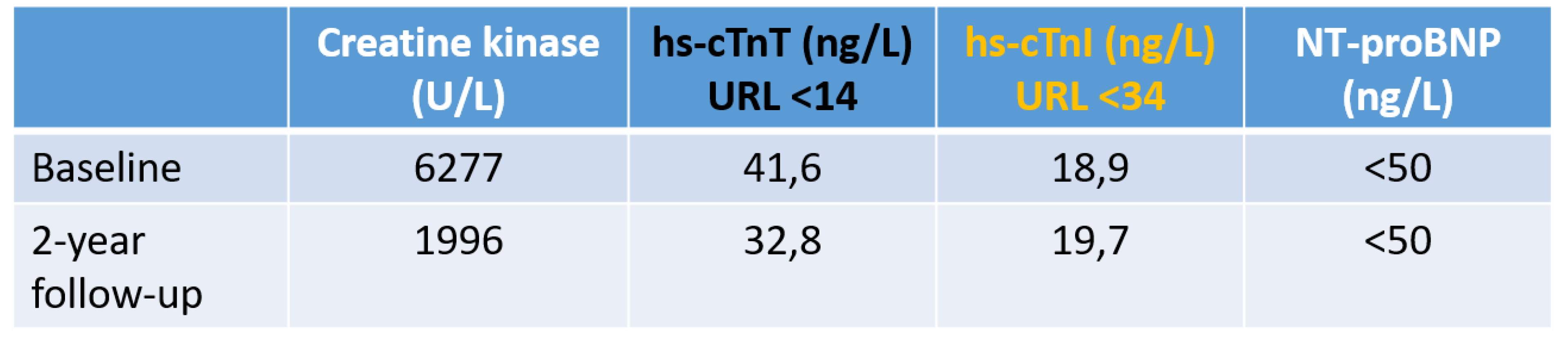
Table 2.
Biochemistry of cardiac troponin isoforms in humans.
| Isoform | Gene, location |
Expression | Aminoacids | Molecular mass |
|---|---|---|---|---|
| cTnC | TNNC1, 3p21.1 |
Heart, slow-twitch skeletal muslce |
161 | 18.4 kDa |
| cTnI | TNNI3, 19q13.42 |
Heart | 210 | 24 kDa |
| cTnT | TNNT2, 1q32.1 |
Heart, skeletal muscle (fetal period, chronic injury) | 287-298* | about 34-36 kDa |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



