
Submitted:
01 April 2025
Posted:
02 April 2025
You are already at the latest version
Abstract
Donation after circulatory death (DCD) donors serve as an alternative source for heart donors for transplantation. However, the DCD process involves unavoidable ischemia-reperfusion injuries, leading to mitochondrial dysfunction and myocardial damage. Activation of calpain-1 and calpain-2 (CPN1/2) exacerbates mitochondrial damage during ischemia-reperfusion. We investigated the role of CPN1/2 activation in mitochondrial and cardiac injury in DCD hearts. In this study, rat hearts were divided into three groups: (1) control beating-heart donor (CBD) hearts, which were procured without in vivo ischemia and subsequently underwent ex vivo perfusion; (2) DCD hearts, which underwent 25 minutes of in vivo ischemia followed by ex vivo perfusion; and (3) DCD hearts treated with a CPN1/2 inhibitor (MDL-28170, 10 μM) during ex vivo perfusion. MDL treatment significantly reduced infarct size in DCD hearts, supporting the notion that CPN1/2 activation exacerbates cardiac injury. Additionally, MDL treatment reduced mitochondrial permeability transition pore (MPTP) opening in DCD hearts and prevented the degradation of mitofilin, a protein that interacts with cyclophilin D to regulate MPTP opening. Notably, the content of cyclophilin D remained unchanged in DCD hearts. Our study suggests that CPN1/2-mediated mitofilin degradation contributes to increased MPTP opening in DCD hearts. Administering a CPN1/2 inhibitor during early reperfusion is a promising strategy to mitigate cardiac injury in DCD hearts by preserving mitofilin and reducing MPTP opening.
Keywords:
MPTP
; heart transplantation
; calpain inhibitor
; mitochondria
; ischemia-reperfusion
; cyclophilin D
; mitofilin
1. Introduction
Heart transplantation is the definitive treatment for patients with end-stage heart failure. However, due to the limited availability of donor hearts from donation after brain death (DBD), hearts from donation after circulatory death (DCD) donors are also used for clinical transplantation [1,2]. Despite their potential, DCD hearts incur ischemia-reperfusion injury, which cause mitochondrial damage and potentially compromise their suitability for transplantation [3,4]. Ischemia-reperfusion injury impairs the electron transport chain and increases mitochondrial permeability transition pore (MPTP) opening in the DCD hearts [5,6]. While interventions before DCD heart procurement are not permitted, treatment during early reperfusion is a potential strategy to reduce cardiac injury. Reversible blockade of the electron transport chain with amobarbital during early reperfusion has been shown to decrease cardiac injury in DCD hearts [7,8]. Similarly, cyclosporine A treatment at the onset of reperfusion reduces cardiac injury by inhibiting MPTP opening [6,9]. These findings suggest that timely interventions during early reperfusion can effectively mitigate cardiac injury in DCD hearts, improving their potential for successful transplantation [10].
Calpain-1 (CPN1) and calpain-2 (CPN2) are two ubiquitous Ca²⁺-dependent cysteine proteases found in both the cytosol and mitochondria [11,12,13,14,15,16]. Activation of cytosolic CPN1 and CPN2 (cCPN1/2) increases cardiac injury during ischemia-reperfusion by degrading structural proteins, including spectrin [14,17,18,19]. Similarly, activation of mitochondria-localized CPN1 and CPN2 (mCPN1/2) impairs mitochondrial function during ischemia-reperfusion [19,20,21]. Specifically, mCPN1/2 activation contributes to decreased oxidative-phosphorylation and increases susceptibility to mitochondrial permeability transition pore (MPTP) opening in isolated rat hearts following ex vivo ischemia-reperfusion [14,20]. During the DCD process, hearts first undergo in vivo global ischemia before being procured for either ex vivo reperfusion. Thus, ischemia-reperfusion injury is inherent to DCD hearts [3,22,23]. However, it remains to be studied whether the activation of cCPN1/2 and mCPN1/2 contributes to ischemia-reperfusion injury in DCD hearts. In the current study, we investigated:(1) Whether the DCD process leads to the activation of cCPN1/2 and mCPN1/2. (2) Whether administering a CPN1/2 inhibitor at reperfusion would reduce cardiac injury in DCD hearts.
MPTP opening plays a central role in exacerbating cardiac injury in DCD hearts [6,9]. Recent studies indicate that alterations in mitofilin sensitize MPTP opening in hearts following ischemia-reperfusion [24,25,26]. Mitofilin, a key component of the mitochondrial inner membrane organizing system, is essential for maintaining mitochondrial structure and function [27]. It plays a critical role in preserving cristae architecture, which is crucial for sustaining oxidative phosphorylation and ATP production [27,28]. Additionally, mitofilin interacts with other components of the inner mitochondrial membrane organizing system to regulate mitochondrial dynamics, including fusion and fission [24,25,26]. A recent study demonstrated that mitofilin regulates MPTP opening by interacting with cyclophilin D through its C-terminal sequence. Cleaved mitofilin sensitizes MPTP opening due to its dissociation from cyclophilin D [25]. Furthermore, mitofilin content is decreased in rat hearts following ischemia-reperfusion [26]. However, it remains unclear whether the DCD process leads to alterations in mitofilin. Although MPTP opening is increased in DCD heart mitochondria [5], it is unknown whether this increase is due to mitofilin degradation. We hypothesize that mitofilin content is reduced in DCD heart mitochondria. Additionally, we will investigate whether this reduction in mitofilin content in DCD hearts is dependent on CPN1/2 activation.
2. Material and Methods
2.1. Measurement of Cardiac Function and Infarct Size
Male Sprague Dawley rats (3–4 months old) were divided into three groups: CBD group: Rat hearts were procured without in vivo ischemia and then perfused on a Langendorff setup for 90 minutes using modified Krebs-Henseleit (K-H) buffer containing 115 mM NaCl, 4.0 mM KCl, 2.0 mM CaCl₂, 26 mM NaHCO₃, 1.1 mM MgSO₄, 0.9 mM KH₂PO₄, and 5.5 mM glucose. The K-H buffer was oxygenated with 95% O₂/5% CO₂ to maintain a pH of approximately 7.4, and its temperature was regulated at 37°C using a circulatory water bath. Hearts were perfused at a constant pressure of 72 mmHg. Vehicle-treated DCD group: Rat hearts underwent 25 minutes of in vivo global ischemia before being procured and perfused on a Langendorff setup for 90 minutes. MDL-treated DCD group: Rat hearts underwent 25 minutes of in vivo global ischemia before being procured and perfused on a Langendorff setup for 90 minutes with K-H buffer containing MDL (10 µM) (Figure 1A). In DCD hearts following 25 minutes of global ischemia, a latex balloon-tip catheter was inserted into the left ventricle to monitor left ventricular function after 10 minutes of reperfusion. Heart rate and left ventricular developed pressure (LVDP) were monitored and analyzed using LabChart software (ADInstruments Inc., Colorado Springs, CO). The rate-pressure product (RPP = HR (heart rate) × LVDP) was used to account for cardiac function variability associated with heart rate. Infarct size was measured in rat hearts following 90 minutes of reperfusion using triphenyl-tetrazolium chloride (TTC) staining [6,9].
2.2. Isolation of SSM and IFM
Both subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM) were isolated from CBD, untreated DCD, and MDL-treated DCD hearts [21]. In this study, rat hearts were perfused for only 60 minutes on a Langendorff setup, as our previous study showed that this duration of reperfusion causes significant mitochondrial dysfunction in DCD hearts. A single rat heart tissue sample was submerged in cold (4°C) buffer A [100 mM KCl, 50 mM 3-(N-morpholino) propanesulfonic acid (MOPS), 1 mM EGTA, 5 mM MgSO₄·7H₂O, and 1 mM ATP, pH 7.4]. The heart was then blotted dry, weighed, and minced in a beaker on ice. The minced tissue was resuspended in buffer B (buffer A + 0.2% bovine serum albumin) and homogenized using a Polytron tissue processor (Brinkman Instruments, Westbury, NY) for 2.5 seconds at 10,000 rpm. The homogenate was centrifuged at 500 × g for 10 minutes to separate supernatant 1 and pellet 1. Supernatant 1 was further centrifuged at 3,000 × g for 10 minutes to generate supernatant 2 and pellet 2 (crude SSM). Supernatant 2 was then centrifuged at 100,000 × g for 30 minutes to obtain a particle-free cytosol. Pellet 1 from the Polytron homogenate was resuspended in buffer A, homogenized, and incubated with trypsin (5 mg/g wet weight) for 10 minutes at 4°C [15]. The homogenate was then centrifuged at 500 × g for 10 minutes to separate supernatant 3, which contained IFM, from the pellet (cell debris). Supernatant 3 was further centrifuged at 3,000 × g for 10 minutes to isolate crude IFM. The crude SSM and IFM were washed twice with buffer B and then resuspended in KME buffer (100 mM KCl, 50 mM MOPS, and 0.5 mM EGTA). Mitochondrial protein content was measured using the Lowry method, with bovine serum albumin as a standard. Since SSM are more sensitive to ischemic damage in DCD hearts, we focused our study exclusively on SSM in the current investigation [3,5,7].
2.3. Assessing Calcium Retention Capacity (CRC) in the Isolated SSM
CRC was used to reflect the sensitivity of calcium-induced mitochondrial permeability transition pore (MPTP) opening in freshly isolated mitochondria [29]. SSM (400 μg/ml) were incubated in a buffer containing 150 mM sucrose, 50 mM KCl, 2 mM KH₂PO₄, 5 mM succinate, and 20 mM Tris/HCl, pH 7.4. Sequential addition of a known amount of calcium (20 nmol/pulse) was used to induce MPTP opening in the incubated mitochondria [5]. CRC referred to the total amount of calcium additions required to trigger MPTP opening in the isolated mitochondria. The calcium retention capacity is greater in mitochondria oxidizing a complex II substrate compared to a complex I substrate [30]. Thus, succinate was used as the substrate for CRC measurement. Extra-mitochondrial Ca²⁺ concentration was recorded using 0.5 µM Calcium Green-5N, and fluorescence was monitored with excitation and emission wavelengths set at 500 and 530 nm, respectively [29].
2.4. Western Blotting
Cytosol or mitochondrial proteins were separated using non-staining 12% or 4-15% Tris-glycine gels (Bio-Rad, Hercules, CA). The total protein content in the gel was digitally analyzed after UV activation using Image Lab 6.0 software (Bio-Rad, Hercules, CA). Proteins were then transferred from the gel to a PVDF membrane (Fisher Scientific, Hampton, NH) using semi-dry transfer (Bio-Rad) for 60 minutes at 20 V. The blots were incubated for 1 hour at room temperature in 5% (w/v) non-fat dry milk (Bio-Rad) and 1% BSA in TBST (Tris-buffered saline with Tween 20) buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween-20), followed by an overnight incubation at 4°C with primary antibodies (see information in Table 1). The membrane was washed four times (5 minutes each time) with TBST buffer. Then, the membrane was incubated at room temperature for 1 hour in the presence of a 1:10,000 dilution of HRP-conjugated anti-mouse or anti-rabbit IgG F(ab)₂ (GE Healthcare Life Sciences, Piscataway, NJ) secondary antibodies. The membrane was washed three times (10 minutes each time) with TBST buffer. Finally, the blots were developed using ECL Plus Western Blotting Detection Reagents (GE Healthcare Life Sciences, Piscataway, NJ). The protein images on the membranes were digitally recorded and analyzed using Image Lab 6.0 software (Bio-Rad, Hercules, CA) [31].
2.5. Statistical Analysis
Data are expressed as the mean ± standard error [23]. For all analyses, differences between groups (≥ 3 groups) were compared using one-way ANOVA when the data passed normality and equal distribution tests. When a significant F value was obtained, means were compared using the Student-Newman-Keuls test for multiple comparisons. Statistical significance was defined as a p-value of < 0.05.
3. Results
3.1. Inhibition of CPN1/2 Decreases Cardiac Injury in DCD Hearts
The heart rate (HR) and left ventricular developed pressure (LVDP) were decreased in DCD hearts during reperfusion compared to CBD hearts (Figure 1B,C). The rate pressure product (RPP, LVDPxHR) was also decreased in DCD hearts compared to CBD hearts (Figure 1D). These results suggest that cardiac function was impaired in DCD hearts following ischemia and reperfusion. HR, LVDP, and RPP in MDL-treated hearts were still decreased compared to CBD hearts (Figure 1B–D). Although MDL treatment did not improve HR in DCD hearts during reperfusion compared to untreated DCD hearts (Figure 1B), it did slightly improve LVDP and RPP in DCD hearts, especially during late reperfusion, compared to untreated DCD hearts (Figure 1C,D). Our previous study shows that infarct size is increased in DCD hearts compared to CBD hearts [6]. Here we found that MDL treatment led to a decreased infarct size in DCD hearts compared to untreated DCD hearts (Figure 2). These results indicate that inhibition of CPN1/2 reduces cardiac injury in DCD rat hearts following ischemia-reperfusion.
3.2. MDL Treatment Decreased MPTP Opening in DCD Hearts
The CRC was used to assess the sensitivity of MPTP opening in isolated SSM. Figure 3A shows the original tracing for the CRC measurement in the CBD, DCD, and DCD+MDL groups. The CRC in SSM isolated from untreated DCD hearts was decreased compared to the corresponding SSM from CBD hearts (Figure 3A,B), suggesting that the sensitivity of MPTP opening is increased in DCD heart mitochondria. MDL treatment led to an increased CRC in the SSM from treated DCD hearts compared to the SSM from untreated DCD hearts (Figure 3A,B), indicating that activation of CPN1/2 sensitizes MPTP opening in DCD heart mitochondria.
3.3. MDL Treatment Decreased the Activation of cCPN1/2 and mCPN1/2 in DCD Hearts
Spectrin is a substrate of cytosolic CPN1/2 (cCPN1/2) [18], and its activation leads to spectrin cleavage. An increase in cleaved spectrin at 150 kDa serves as a biomarker of cCPN1/2 activation [18,19]. There were no significant differences in spectrin content between the CBD, DCD, and DCD+MDL groups (Figure 4A,B), but the content of cleaved spectrin was significantly higher in DCD hearts compared to CBD hearts (Figure 4A,C), indicating that the DCD process activates cCPN1/2. MDL treatment reduced the content of cleaved spectrin in DCD hearts (Figure 4A,C), supporting its effectiveness in decreasing cCPN1/2 activation in DCD hearts. Total protein was used as a loading control.
Activation of mitochondrial CPN1/2 (mCPN1/2) contributes to mitochondrial damage during ischemia-reperfusion . Apoptosis-inducing factor (AIF) is a known substrate of mCPN1, and mCPN1 activation cleaves AIF, producing truncated AIF (tAIF) [32,33,34,35]. Mitochondrial CPN2 (mCPN2) may also play a role in AIF cleavage [12]. Thus, a decrease in AIF content or an increase in tAIF content serves as an indicator of mCPN1/2 activation. Compared to CBD hearts, AIF content (62 kDa) was reduced in DCD hearts (Figure 4D,E). However, MDL treatment preserved AIF content in DCD hearts (Figure 4D,E). A reduction in AIF should lead to an increase in tAIF (57 kDa) levels. However, no differences in tAIF content were observed among the groups (Figure 4D,F). AIF is anchored to the inner mitochondrial membrane, whereas tAIF is detached from the membrane [12]. Increased MPTP opening can cause tAIF to be released into the cytosol, potentially explaining the absence of elevated tAIF levels in DCD heart mitochondria [12,36]. These results support the activation of both cCPN1/2 and mCPN1/2 in DCD hearts. MDL treatment during reperfusion can still decrease CPN1/2 activation in the DCD hearts.
3.4. MDL Treatment Decreased the Degradation of Mitofilin in DCD Hearts
Recent studies indicate that mitofilin regulates MPTP opening through its interaction with cyclophilin D [24,25,26]. We investigated whether mitofilin content was reduced in DCD hearts subjected to 25 minutes of ischemia followed by 60 minutes of reperfusion. MDL treatment improved CRC in DCD hearts (Figure 3A,B), prompting us to examine whether MDL treatment also preserved mitofilin content in DCD hearts. Mitofilin content in isolated mitochondria was assessed using immunoblotting. Compared to CBD hearts, mitofilin content was significantly reduced in DCD hearts (Figure 5A,B). MDL treatment preserved mitofilin levels in DCD hearts (Figure 5A,B). These results support that the DCD process leads to decreased mitofilin content in a CPN1/2-dependent manner.
Cyclophilin D is a key regulator of MPTP opening [37]. Therefore, the content of cyclophilin D was measured in CBD and DCD heart mitochondria. There were no significant differences in the content of cyclophilin D between the CBD, DCD, and DCD+MDL groups (Figure 5C,D), indicating that the increased MPTP opening in DCD heart mitochondria was not due to a decrease in cyclophilin D content.
4. Discussion
In this study, we found that both cCPN1/2 and mCPN1/2 are activated in the DCD hearts. Reperfusion following ischemia leads to increased MPTP opening and decreased mitofilin content in DCD heart mitochondria. Inhibition of CPN1/2 using MDL reduces cardiac injury in DCD hearts. Additionally, inhibition of CPN1/2 decreases MPTP opening and preserves mitofilin content. These results suggest that activation of CPN1/2 increases MPTP opening in DCD hearts by degrading mitofilin. Mitofilin is a critical target for preserving mitochondrial integrity during ischemia-reperfusion injury. Protecting its function could be a key strategy to minimize cardiac damage and improve outcomes in ischemia-reperfusion injury related heart conditions.
Ischemia-reperfusion leads to activation of cytosolic and mitochondrial calpains in isolated mouse and rat hearts [32,33,38]. In the present study, we found that the formation of cleaved spectrin was increased in the DCD hearts, supporting the idea that the DCD process leads to activation of cytosolic CPN1/2. Mitochondrial CPN1 is identified in the mitochondrial intermembrane space and matrix [11,12], and CPN2 also exists in the matrix of cardiac mitochondria [36]. Activation of mitochondrial CPN1 within the intermembrane space augments cardiac injury during reperfusion by inducing the translocation of truncated AIF from mitochondria to the cytosol and nucleus [11,12]. Activation of mitochondrial CPN2 may also be involved in AIF cleavage [12]. Therefore, a reduced AIF content or an increased truncated AIF content is used as an indicator of mitochondrial CPN1/2 activation, at least in the intermembrane space compartment. AIF content was decreased in DCD hearts compared to CBD hearts, indicating that mitochondrial CPN1/2 was activated in the DCD heart mitochondria. A decrease in AIF content should lead to an increase in truncated AIF content. However, truncated AIF is not increased in the DCD hearts compared to CBD. Although AIF is bound to the inner mitochondrial membrane, truncated AIF is detached from it [12]. Thus, truncated AIF can be released from mitochondria to the cytosol and nucleus when the permeability of the mitochondrial membrane is increased in pathological conditions, including ischemia-reperfusion [39]. MPTP opening is increased in the DCD heart mitochondria, leading to increased permeation of the outer membrane. Consequently, some truncated AIF content can be released into the cytosol in DCD heart mitochondria through increased MPTP opening. This may explain why the DCD process does not lead to an increased content of truncated AIF. MDL treatment decreased spectrin cleavage and maintained AIF content in DCD hearts, supporting that MDL treatment given during reperfusion can effectively prevent both cytosolic and mitochondrial CPN1/2 activation. Thus, inhibition of CPN1/2 is a promising strategy to decrease cardiac injury in DCD hearts.
Opening of the MPTP increases cardiac injury during ischemia-reperfusion [40,41]. Our studies show that MPTP opening is also a key mechanism of cardiac injury in DCD hearts [6]. Recent studies have made significant advances in understanding the structure of the MPTP. The initial components of the MPTP include voltage dependent anion channels, the inorganic phosphate carrier, and the translocator protein. More recent studies suggest that ATP synthase is another key component of the MPTP [42,43]. However, the exact subunits of ATP synthase involved in MPTP formation remain uncertain. An increase in calcium levels within mitochondria leads to calcium binding to ATP synthase, resulting in a conformational change in ATP synthase that favors MPTP opening [42,43]. Cyclophilin D, a soluble protein in the mitochondrial matrix, is a key regulator of the MPTP. The binding of Cyclophilin D to MPTP components, including ATP synthase, markedly decreases the calcium threshold required to trigger MPTP opening [42,43]. Genetic elimination of Cyclophilin D inhibits MPTP opening [37], supporting the key role of Cyclophilin D in the regulation of MPTP opening. Pharmacologic inhibition of Cyclophilin D using cyclosporin A is also well-known to decrease MPTP opening. Treatment with cyclosporin A during early reperfusion leads to decreased cardiac injury in DCD hearts [6,9], supporting that inhibition of MPTP is a promising approach to reduce cardiac injury in DCD hearts.
Recent studies also indicate that mitofilin plays a crucial role in cardiac injury during ischemia-reperfusion by regulating MPTP opening through its interaction with Cyclophilin D via its C-terminal sequence [24,25]. Cleavage of mitofilin dissociates it from Cyclophilin D, which binds to MPTP components and promotes MPTP opening [24,25,26]. Reperfusion, especially following prolonged ischemia, leads to decreased mitofilin content and increased infarct size [26]. In this study, we found that mitofilin content was reduced in DCD hearts compared to CBD hearts following reperfusion. MDL treatment improved mitofilin content and decreased infarct size, supporting the notion that restoring mitofilin content is essential for reducing cardiac injury in DCD hearts.
Inhibition of CPN1/2 leads to decreased MPTP opening during ischemia-reperfusion [33,44]. The mechanisms are related to the protection of complex I, ATP synthase, and other components [20]. In this study, we show that inhibition of CPN1/2 helps preserve mitofilin. MDL treatment preserved mitofilin content, which in turn decreased Cyclophilin D binding to MPTP components, leading to reduced MPTP opening (Graphic abstract). Our results suggest that mitofilin is a substrate of CPN1/2, and activation of CPN1/2 sensitizes MPTP opening by degrading mitofilin in DCD hearts.
There are several limitations to our study. First, only male rats were used in this experiment. Our previous study showed that mitochondrial dysfunction and MPTP opening also occur in female rat DCD hearts [45]. Therefore, we anticipate that CPN1/2 will also be activated in female DCD hearts, and inhibition of CPN1/2 will reduce cardiac injury in these hearts as well. Although MDL is a well-known CPN1/2 inhibitor, it does have some off-target effects [46]. To clarify the relationship between CPN1/2 activation and mitofilin cleavage, future in vitro studies using purified CPN1/2 and mitofilin peptides are needed. Additionally, post-translational modifications of Cyclophilin D may influence MPTP opening [47,48]. While the content of Cyclophilin D did not change in DCD hearts, its post-translational modifications may be altered in these hearts, warranting further investigation.
5. Conclusion
Our study shows that CPN1/2 are activated in DCD hearts, leading to mitofilin degradation, which results in sensitizing the MPTP opening. Administering CPN1/2 inhibitor during early reperfusion is a promising strategy to decrease cardiac injury in the DCD hearts.
Abbreviations
| AIF | Apoptosis-inducing factor |
| CBD | Control beating-heart donor |
| CPN1 | Calpain-1 |
| CPN2 | Calpain-2 |
| CPN1/2 | calpain-1 and calpain-2 |
| cCPN1/2 | cytosolic calpain-1 and calpain-2 |
| CRC | Calcium retention capacity |
| DCD | Donation after circulatory death |
| HR | Heart rate |
| IFM | interfibrillar mitochondria |
| LVDP | left ventricular developed pressure |
| MDL | MDL-28170 |
| mCPN1/2 | Mitochondria-localized calpain-1 and calpain-2 |
| MOPS | 3-(N-morpholino)propanesulfonic acid |
| MPTP | Mitochondrial permeability transition pore |
| RPP | Rate-pressure product |
| SSM | Subsarcolemmal mitochondria |
| tAIF | truncated apoptosis-inducing factor |
| TBST | Tris-buffered saline with Tween 20 |
Author Contributions
Participated in research design: Chen Q., Kienan Z., Lesnefsky E. J., and Quader M. Conducted experiments: Chen Q., Kienan Z., Labate G., and Akande Q. Performed data analysis: Chen Q., Kienan Z., Labate G., and Akande Q. Wrote or contributed to the writing of the manuscript: Chen Q., Kienan Z., Lesnefsky E. J., and Quader M.
Funding
This work was supported by Merit Review Grants awarded to Dr. Mohammed Quader (1I01 BX003859), Dr. Edward J. Lesnefsky (2I01 BX001355), and by funds from the Pauley Heart Center to Drs. Mohammed Quader and Qun Chen.
Institutional Review Board Statement
This study was approved by the Animal Care and Use Committees of the Richmond VA Medical Center and Virginia Commonwealth University (AH00074596). The health and welfare of the animals used in the study were supervised by personnel from both institutions. Every effort was made to minimize animal pain and discomfort throughout the study.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article, and further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors of this manuscript have no financial or other conflicts of interest to disclose.
References
- Quader, M.; Toldo, S.; Chen, Q.; Hundley, G.; Kasirajan, V. Heart transplantation from donation after circulatory death donors: Present and future. J. Card. Surg. 2020, 35, 875–885. [Google Scholar] [CrossRef]
- Al-Tawil, M.; Wang, W.; Chandiramani, A.; Zaqout, F.; Diab, A.H.; Sicouri, S.; Ramlawi, B.; Haneya, A. Survival after heart transplants from circulatory-dead versus brain-dead donors: Meta-analysis of reconstructed time-to-event data. Transplant. Rev. 2025, 39, 100917. [Google Scholar] [CrossRef] [PubMed]
- Quader, M.; Akande, O.; Toldo, S.; Cholyway, R.; Kang, L.; Lesnefsky, E.J.; Chen, Q. The Commonalities and Differences in Mitochondrial Dysfunction Between ex vivo and in vivo Myocardial Global Ischemia Rat Heart Models: Implications for Donation After Circulatory Death Research. Front. Physiol. 2020, 11, 681. [Google Scholar] [CrossRef]
- Siddiqi, H.K.; Trahanas, J.; Xu, M.; Wells, Q.; Farber-Eger, E.; Pasrija, C.; Amancherla, K.; Debose-Scarlett, A.; Brinkley, D.M.; Lindenfeld, J.; et al. Outcomes of Heart Transplant Donation After Circulatory Death. Circ. 2023, 82, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Akande, O.; Chen, Q.; Toldo, S.; Lesnefsky, E.J.; Quader, M. Ischemia and reperfusion injury to mitochondria and cardiac function in donation after circulatory death hearts- an experimental study. PLOS ONE 2020, 15, e0243504. [Google Scholar] [CrossRef]
- Kiernan, Z.; Labate, G.; Chen, Q.; Lesnefsky, E.J.; Quader, M. Infarct Size Reduction with Cyclosporine A in Circulatory Death Rat Hearts: Reducing Effective Ischemia Time with Therapy During Reperfusion. Circ. Hear. Fail. 2024, 17, e011846. [Google Scholar] [CrossRef] [PubMed]
- Akande, O.; Chen, Q.; Cholyway, R.; Toldo, S.; Lesnefsky, E.J.; Quader, M. Modulation of Mitochondrial Respiration During Early Reperfusion Reduces Cardiac Injury in Donation After Circulatory Death Hearts. J. Cardiovasc. Pharmacol. 2022, 80, 148–157. [Google Scholar] [CrossRef]
- Quader, M.; Chen, Q.; Akande, O.; Cholyway, R.; Mezzaroma, E.; Lesnefsky, E.J.; Toldo, S. Electron Transport Chain Inhibition to Decrease Injury in Transplanted Donation After Circulatory Death Rat Hearts. J. Cardiovasc. Pharmacol. 2023, 81, 389–391. [Google Scholar] [CrossRef]
- Quader, M.; Akande, O.; Cholyway, R.; Lesnefsky, E.J.; Toldo, S.; Chen, Q. Infarct Size with Incremental Global Myocardial Ischemia Times: Cyclosporine A in Donation After Circulatory Death Rat Hearts. Transplant. Proc. 2023, 55, 1495–1503. [Google Scholar] [CrossRef]
- Quader, M.; Mezzaroma, E.; Wickramaratne, N.; Toldo, S. Improving circulatory death donor heart function: A novel approach. JTCVS Tech. 2021, 9, 89–92. [Google Scholar] [CrossRef]
- Chen, Q.; Lesnefsky, E.J. Heart mitochondria and calpain 1: Location, function, and targets. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2015, 1852, 2372–2378. [Google Scholar] [CrossRef]
- Ozaki, T.; Yamashita, T.; Ishiguro, S.-I. Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2009, 1793, 1848–1859. [Google Scholar] [CrossRef]
- Wang, J.; Chen, B.; Shi, Q.; Ciampa, G.; Zhao, W.; Zhang, G.; Weiss, R.M.; Peng, T.; Hall, D.D.; Song, L.-S. Preventing Site-Specific Calpain Proteolysis of Junctophilin-2 Protects Against Stress-Induced Excitation-Contraction Uncoupling and Heart Failure Development. Circulation 2025, 151, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Hagihara, H.; Ohga, Y.; Nakajima-Takenaka, C.; Murata, K.-Y.; Taniguchi, S.; Takaki, M. Calpain inhibitor-1 protects the rat heart from ischemia-reperfusion injury: analysis by mechanical work and energetics. Am. J. Physiol. Circ. Physiol. 2005, 288, H1690–H1698. [Google Scholar] [CrossRef]
- Ma, J.; Wei, M.; Wang, Q.; Li, J.; Wang, H.; Liu, W.; Lacefield, J.C.; Greer, P.A.; Karmazyn, M.; Fan, G.-C.; et al. Deficiency of Capn4 Gene Inhibits Nuclear Factor-κB (NF-κB) Protein Signaling/Inflammation and Reduces Remodeling after Myocardial Infarction. J. Biol. Chem. 2012, 287, 27480–27489. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells 2020, 9, 2698. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Hernando, V.; Garcia-Dorado, D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2012, 96, 23–31. [Google Scholar] [CrossRef]
- Hernando, V.; Inserte, J.; Sartório, C.L.; Parra, V.M.; Poncelas-Nozal, M.; Garcia-Dorado, D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J. Mol. Cell. Cardiol. 2010, 49, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Thompson, J.; Hu, Y.; Lesnefsky, E.J.; Willard, B.; Chen, Q. Calpain-mediated protein targets in cardiac mitochondria following ischemia–reperfusion. Sci. Rep. 2022, 12, 1–17. [Google Scholar] [CrossRef]
- Thompson, J.; Hu, Y.; Lesnefsky, E.J.; Chen, Q. Activation of mitochondrial calpain and increased cardiac injury: beyond AIF release. Am. J. Physiol. Circ. Physiol. 2016, 310, H376–H384. [Google Scholar] [CrossRef]
- Zheng, D.; Cao, T.; Zhang, L.-L.; Fan, G.-C.; Qiu, J.; Peng, T.-Q. Targeted inhibition of calpain in mitochondria alleviates oxidative stress-induced myocardial injury. Acta Pharmacol. Sin. 2020, 42, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Quader, M.; Mezzaroma, E.; Kenning, K.; Toldo, S. Targeting the NLRP3 inflammasome to reduce warm ischemic injury in donation after circulatory death heart. Clin. Transplant. 2020, 34, e14044. [Google Scholar] [CrossRef] [PubMed]
- Quader, M.; Mezzaroma, E.; Kenning, K.; Toldo, S. Modulation of Interleukin-1 and -18 Mediated Injury in Donation after Circulatory Death Mouse Hearts. J. Surg. Res. 2020, 257, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Aliagan, A.I.; Tombo, N.; Bopassa, J.C. Mitofilin Heterozygote Mice Display an Increase in Myocardial Injury and Inflammation after Ischemia/Reperfusion. Antioxidants 2023, 12, 921. [Google Scholar] [CrossRef]
- Madungwe, N.B.; Feng, Y.; Lie, M.; Tombo, N.; Liu, L.; Kaya, F.; Bopassa, J.C. Mitochondrial inner membrane protein (mitofilin) knockdown induces cell death by apoptosis via an AIF-PARP-dependent mechanism and cell cycle arrest. Am. J. Physiol. Physiol. 2018, 315, C28–C43. [Google Scholar] [CrossRef]
- Tombo, N.; Aliagan, A.D.I.; Feng, Y.; Singh, H.; Bopassa, J.C. Cardiac ischemia/reperfusion stress reduces inner mitochondrial membrane protein (mitofilin) levels during early reperfusion. Free. Radic. Biol. Med. 2020, 158, 181–194. [Google Scholar] [CrossRef]
- Chaurembo, A.I.; Xing, N.; Chanda, F.; Li, Y.; Zhang, H.-J.; Fu, L.-D.; Huang, J.-Y.; Xu, Y.-J.; Deng, W.-H.; Cui, H.-D.; et al. Mitofilin in cardiovascular diseases: Insights into the pathogenesis and potential pharmacological interventions. Pharmacol. Res. 2024, 203, 107164. [Google Scholar] [CrossRef]
- Thapa, D.; Nichols, C.E.; Lewis, S.E.; Shepherd, D.L.; Jagannathan, R.; Croston, T.L.; Tveter, K.J.; Holden, A.A.; Baseler, W.A.; Hollander, J.M. Transgenic overexpression of mitofilin attenuates diabetes mellitus-associated cardiac and mitochondria dysfunction. J. Mol. Cell. Cardiol. 2014, 79, 212–223. [Google Scholar] [CrossRef]
- Paillard, M.; Gomez, L.; Augeul, L.; Loufouat, J.; Lesnefsky, E.J.; Ovize, M. Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential. J. Mol. Cell. Cardiol. 2009, 46, 902–909. [Google Scholar] [CrossRef]
- Matsuura, T.R.; Bartos, J.A.; Tsangaris, A.; Shekar, K.C.; Olson, M.D.; Riess, M.L.; Bienengraeber, M.; Aufderheide, T.P.; Neumar, R.W.; Rees, J.N.; et al. Early Effects of Prolonged Cardiac Arrest and Ischemic Postconditioning during Cardiopulmonary Resuscitation on Cardiac and Brain Mitochondrial Function in Pigs. Resuscitation 2017, 116, 8–15. [Google Scholar] [CrossRef]
- Chen, Q.; Li, L.; Samidurai, A.; Thompson, J.; Hu, Y.; Willard, B.; Lesnefsky, E.J. Acute endoplasmic reticulum stress-induced mitochondria respiratory chain damage: The role of activated calpains. FASEB J. 2024, 38, e23404. [Google Scholar] [CrossRef]
- Chen, Q.; Paillard, M.; Gomez, L.; Ross, T.; Hu, Y.; Xu, A.; Lesnefsky, E.J. Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia–reperfusion. Biochem. Biophys. Res. Commun. 2011, 415, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Thompson, J.; Hu, Y.; Dean, J.; Lesnefsky, E.J. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. Am. J. Physiol. Physiol. 2019, 317, C910–C921. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Thompson, J.; Hu, Y.; Lesnefsky, E.J. Reversing mitochondrial defects in aged hearts: role of mitochondrial calpain activation. Am. J. Physiol. Physiol. 2022, 322, C296–C310. [Google Scholar] [CrossRef]
- Ozaki, T.; Tomita, H.; Tamai, M.; Ishiguro, S.-I. Characteristics of Mitochondrial Calpains. J. Biochem. 2007, 142, 365–376. [Google Scholar] [CrossRef]
- Shintani-Ishida, K.; Yoshida, K.-I. Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia–reperfusion. Int. J. Cardiol. 2015, 197, 26–32. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W., II.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, G.; Peng, T. Calpain-Mediated Mitochondrial Damage: An Emerging Mechanism Contributing to Cardiac Disease. Cells 2021, 10, 2024. [Google Scholar] [CrossRef]
- Chen, Q.; Thompson, J.; Hu, Y.; Lesnefsky, E.J. The mitochondrial electron transport chain contributes to calpain 1 activation during ischemia-reperfusion. Biochem. Biophys. Res. Commun. 2022, 613, 127–132. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Gerdes, H.J.; Yang, M.; Heisner, J.S.; Camara, A.K.; Stowe, D.F. Modulation of peroxynitrite produced via mitochondrial nitric oxide synthesis during Ca2+ and succinate-induced oxidative stress in cardiac isolated mitochondria. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2020, 1861, 148290–148290. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.-S.; Soukas, A.A. Identity, structure, and function of the mitochondrial permeability transition pore: controversies, consensus, recent advances, and future directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef]
- Endlicher, R.; Drahota, Z.; Štefková, K.; Červinková, Z.; Kučera, O. The Mitochondrial Permeability Transition Pore-Current Knowledge of Its Structure, Function, and Regulation, and Optimized Methods for Evaluating Its Functional State. Cells 2023, 12, 1273. [Google Scholar] [CrossRef] [PubMed]
- Garciadorado, D.; Rodriguezsinovas, A.; Ruizmeana, M.; Inserte, J.; Agulló, L.; Cabestrero, A. The end-effectors of preconditioning protection against myocardial cell death secondary to ischemia–reperfusion. Cardiovasc. Res. 2006, 70, 274–285. [Google Scholar] [CrossRef]
- Chen, Q.; Akande, O.; Lesnefsky, E.J.; Quader, M. Influence of sex on global myocardial ischemia tolerance and mitochondrial function in circulatory death donor hearts. Am. J. Physiol. Circ. Physiol. 2023, 324, H57–H66. [Google Scholar] [CrossRef]
- Samantaray, S.; Ray, S.K.; Banik, N.L. Calpain as a Potential Therapeutic Target in Parkinsons Disease. CNS Neurol. Disord. - Drug Targets 2008, 7, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Baetz, D.; Ovize, M. Cyclophilin D and myocardial ischemia–reperfusion injury: A fresh perspective. J. Mol. Cell. Cardiol. 2015, 78, 80–89. [Google Scholar] [CrossRef]
- Elrod, J.W.; Molkentin, J.D. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef]
Figure 1.
MDL-28170 (MDL) treatment improves cardiac function in DCD hearts. Panel A shows the experimental protocol. MDL was administered throughout the entire reperfusion phase. Compared to CBD hearts, heart rate (HR) was decreased in DCD hearts, and MDL treatment did not improve HR in the DCD hearts (Panel B). Compared to CBD hearts, left ventricular developed pressure (LVDP) was decreased in DCD hearts during the entire reperfusion period (Panel C). MDL treatment slightly improved LVDP during late reperfusion compared to untreated DCD hearts (Panel C). Compared to CBD hearts, rate-pressure product (RPP) was also decreased in DCD hearts during the entire reperfusion period (Panel D). MDL treatment slightly improved RPP during late reperfusion compared to untreated DCD hearts (Panel D). Data are expressed as mean ± SEM; *p < 0.05 vs. CBD; †p < 0.05 vs. untreated DCD hearts. N = 9–10 in each group.
Figure 1.
MDL-28170 (MDL) treatment improves cardiac function in DCD hearts. Panel A shows the experimental protocol. MDL was administered throughout the entire reperfusion phase. Compared to CBD hearts, heart rate (HR) was decreased in DCD hearts, and MDL treatment did not improve HR in the DCD hearts (Panel B). Compared to CBD hearts, left ventricular developed pressure (LVDP) was decreased in DCD hearts during the entire reperfusion period (Panel C). MDL treatment slightly improved LVDP during late reperfusion compared to untreated DCD hearts (Panel C). Compared to CBD hearts, rate-pressure product (RPP) was also decreased in DCD hearts during the entire reperfusion period (Panel D). MDL treatment slightly improved RPP during late reperfusion compared to untreated DCD hearts (Panel D). Data are expressed as mean ± SEM; *p < 0.05 vs. CBD; †p < 0.05 vs. untreated DCD hearts. N = 9–10 in each group.
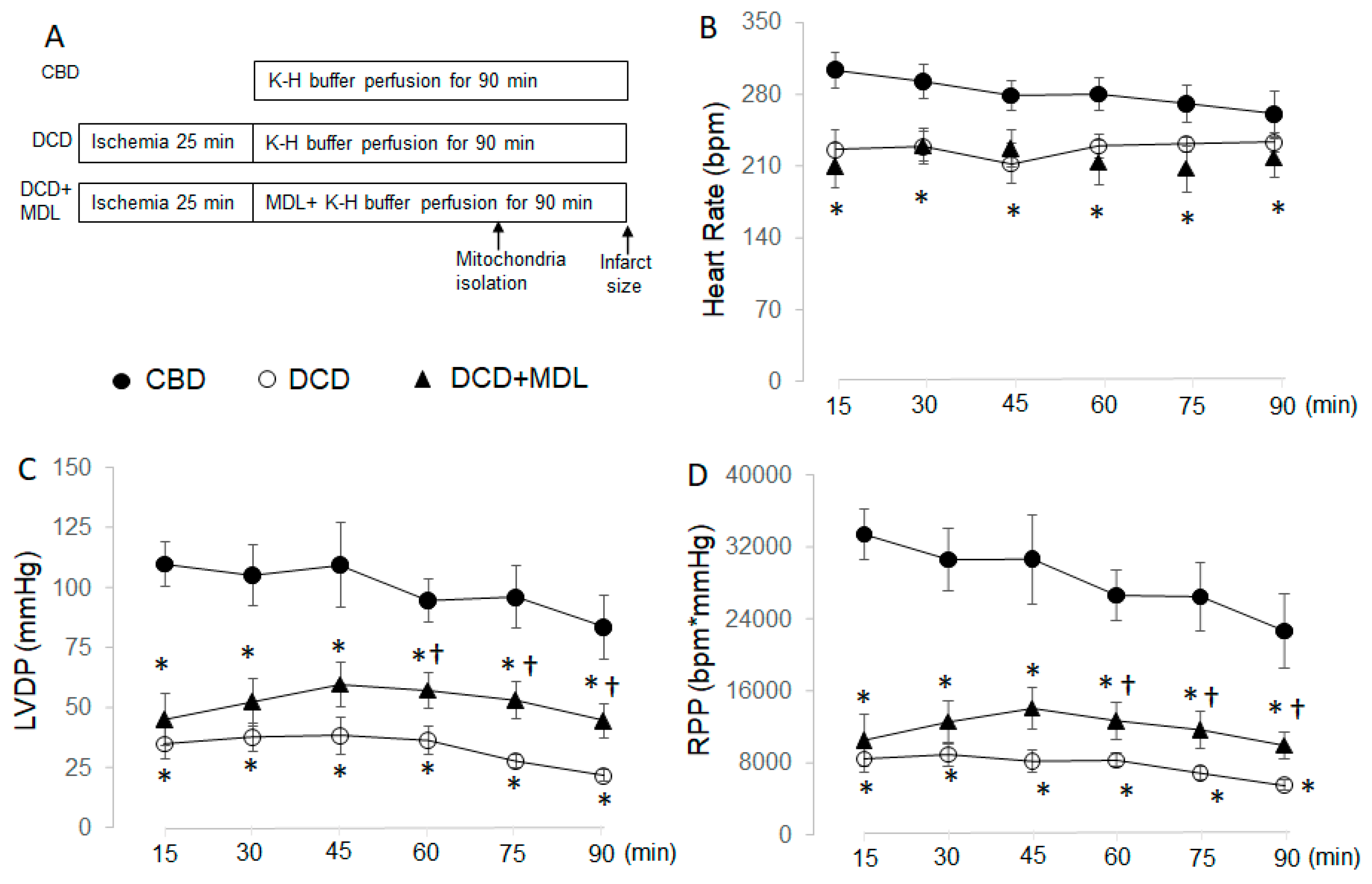
Figure 2.
MDL treatment reduces infarct size in DCD hearts. Administering MDL during reperfusion led to a decrease in infarct size compared to untreated DCD hearts, indicating that inhibition of CPN1/2 reduces cardiac injury in DCD hearts. Data are expressed as mean ± SEM; *p < 0.05 vs. DCD. N = 9–10 in each group.
Figure 2.
MDL treatment reduces infarct size in DCD hearts. Administering MDL during reperfusion led to a decrease in infarct size compared to untreated DCD hearts, indicating that inhibition of CPN1/2 reduces cardiac injury in DCD hearts. Data are expressed as mean ± SEM; *p < 0.05 vs. DCD. N = 9–10 in each group.
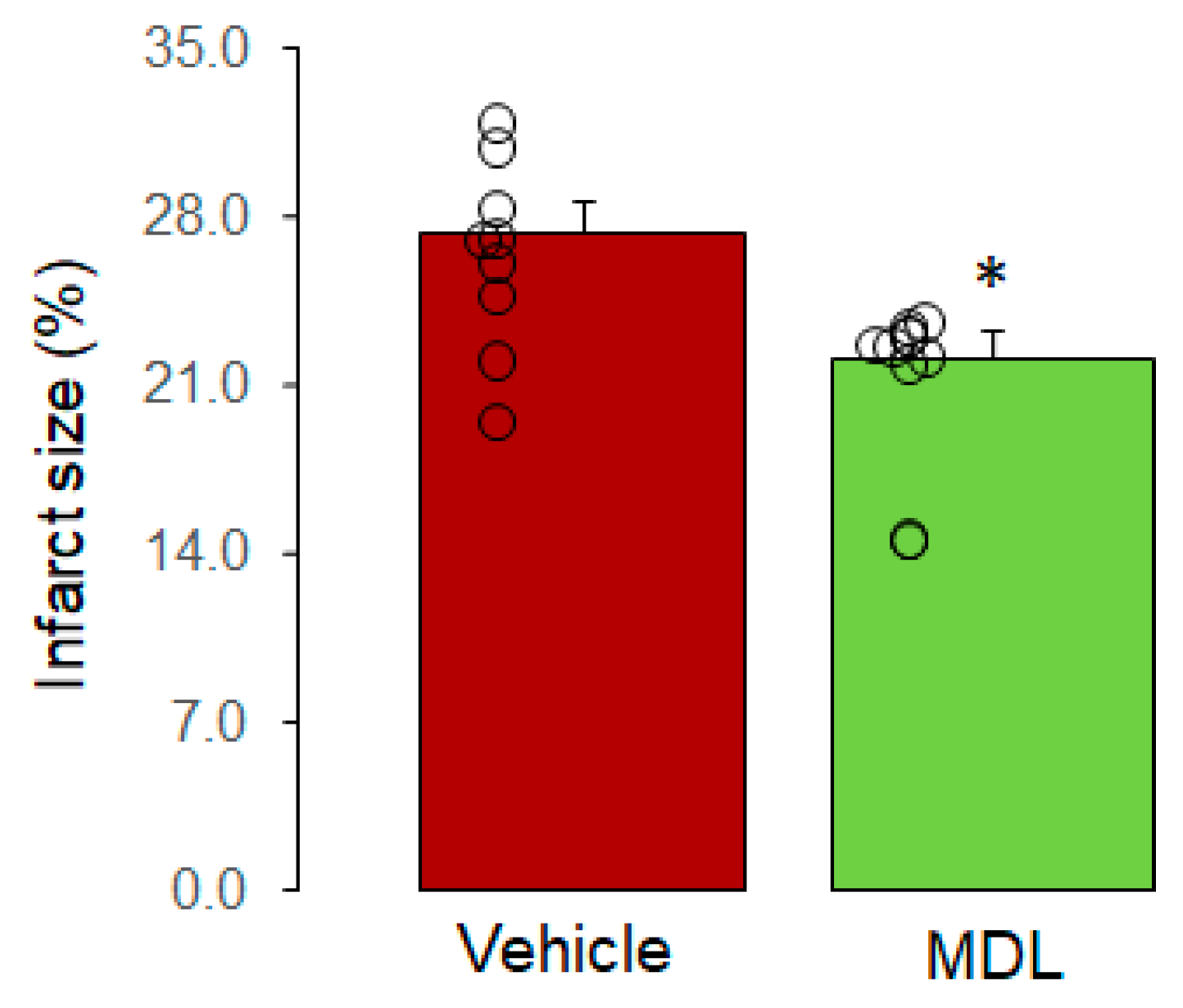
Figure 3.
MDL treatment decreases MPTP opening in DCD hearts. The CRC was used to assess MPTP opening. Panel A shows the original tracing of CRC measurements in CBD, DCD, and DCD+MDL groups. Compared to CBD, the CRC was decreased in SSM from DCD hearts (Panels A and B), indicating increased MPTP opening in DCD hearts. MDL treatment improved the CRC in SSM from DCD hearts, suggesting that inhibition of CPN1/2 decreases MPTP opening in DCD hearts. Data are expressed as mean ± SEM; *p < 0.05 vs. CBD; †p < 0.05 vs. untreated DCD hearts. N = 9–10 in each group.
Figure 3.
MDL treatment decreases MPTP opening in DCD hearts. The CRC was used to assess MPTP opening. Panel A shows the original tracing of CRC measurements in CBD, DCD, and DCD+MDL groups. Compared to CBD, the CRC was decreased in SSM from DCD hearts (Panels A and B), indicating increased MPTP opening in DCD hearts. MDL treatment improved the CRC in SSM from DCD hearts, suggesting that inhibition of CPN1/2 decreases MPTP opening in DCD hearts. Data are expressed as mean ± SEM; *p < 0.05 vs. CBD; †p < 0.05 vs. untreated DCD hearts. N = 9–10 in each group.
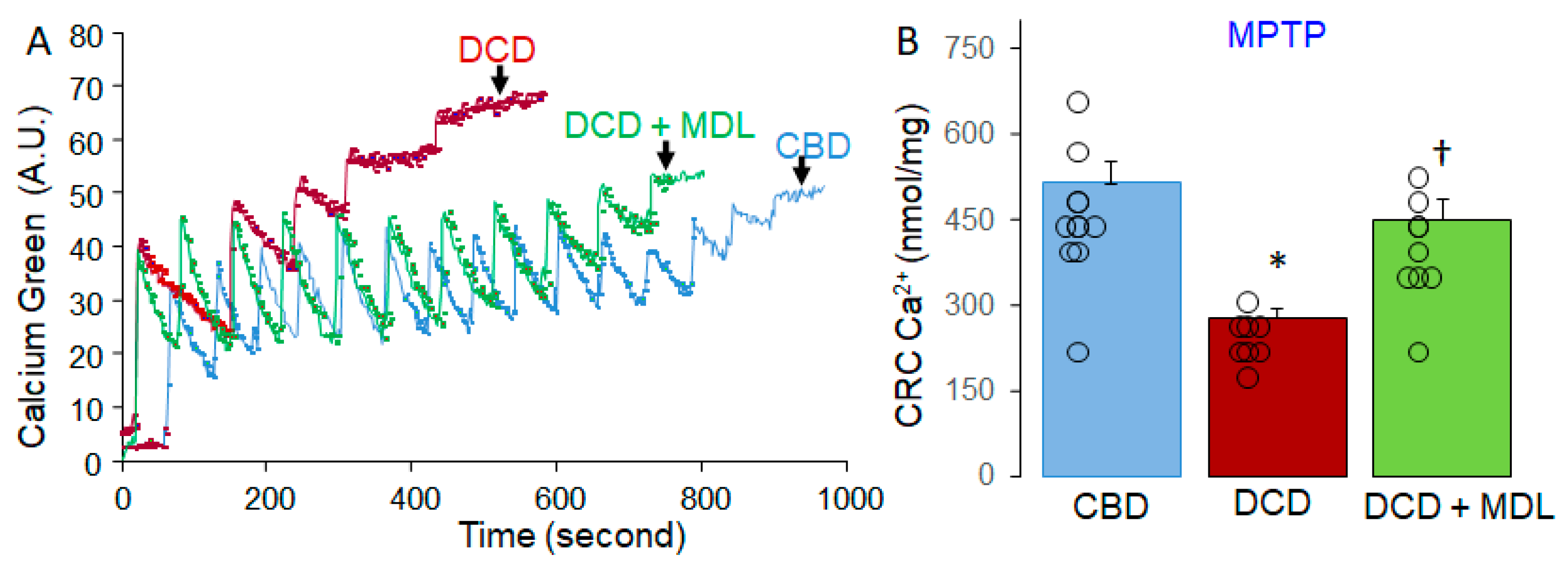
Figure 4.
MDL treatment reduces activation of cytosolic and mitochondrial CPN1/2. Spectrin is a substrate of cytosolic CPN1/2, and an increase in cleaved spectrin content serves as a biomarker of cytosolic CPN1/2 activation. Although there were no significant differences in total spectrin content between CBD, DCD, and DCD+MDL groups (Panels A and B), cleaved spectrin content was significantly higher in DCD hearts compared to CBD (Panel A and C), indicating that the DCD process activates cytosolic CPN1/2. AIF is a substrate of mitochondrial CPN1/2, and a decrease in AIF content or an increase in truncated AIF content is a biomarker of mitochondrial CPN1/2 activation. AIF content was decreased in DCD hearts compared to CBD (Panel D and E), suggesting activation of mitochondrial CPN1/2 in DCD heart mitochondria. MDL treatment preserved AIF content in DCD hearts, supporting that MDL prevents mitochondrial CPN1/2 activation. No significant differences were observed in truncated AIF content between CBD, DCD, and DCD+MDL groups (Panels E and F). Although a decrease in AIF content would typically result in increased truncated AIF, the absence of increased truncated AIF in DCD heart mitochondria likely reflects its release from mitochondria into the cytosol. Total protein was used as the loading control. Data are expressed as mean ± SEM; *p < 0.05 vs. CBD or DCD+MDL. N = 6–8 for each group in mitochondria.
Figure 4.
MDL treatment reduces activation of cytosolic and mitochondrial CPN1/2. Spectrin is a substrate of cytosolic CPN1/2, and an increase in cleaved spectrin content serves as a biomarker of cytosolic CPN1/2 activation. Although there were no significant differences in total spectrin content between CBD, DCD, and DCD+MDL groups (Panels A and B), cleaved spectrin content was significantly higher in DCD hearts compared to CBD (Panel A and C), indicating that the DCD process activates cytosolic CPN1/2. AIF is a substrate of mitochondrial CPN1/2, and a decrease in AIF content or an increase in truncated AIF content is a biomarker of mitochondrial CPN1/2 activation. AIF content was decreased in DCD hearts compared to CBD (Panel D and E), suggesting activation of mitochondrial CPN1/2 in DCD heart mitochondria. MDL treatment preserved AIF content in DCD hearts, supporting that MDL prevents mitochondrial CPN1/2 activation. No significant differences were observed in truncated AIF content between CBD, DCD, and DCD+MDL groups (Panels E and F). Although a decrease in AIF content would typically result in increased truncated AIF, the absence of increased truncated AIF in DCD heart mitochondria likely reflects its release from mitochondria into the cytosol. Total protein was used as the loading control. Data are expressed as mean ± SEM; *p < 0.05 vs. CBD or DCD+MDL. N = 6–8 for each group in mitochondria.
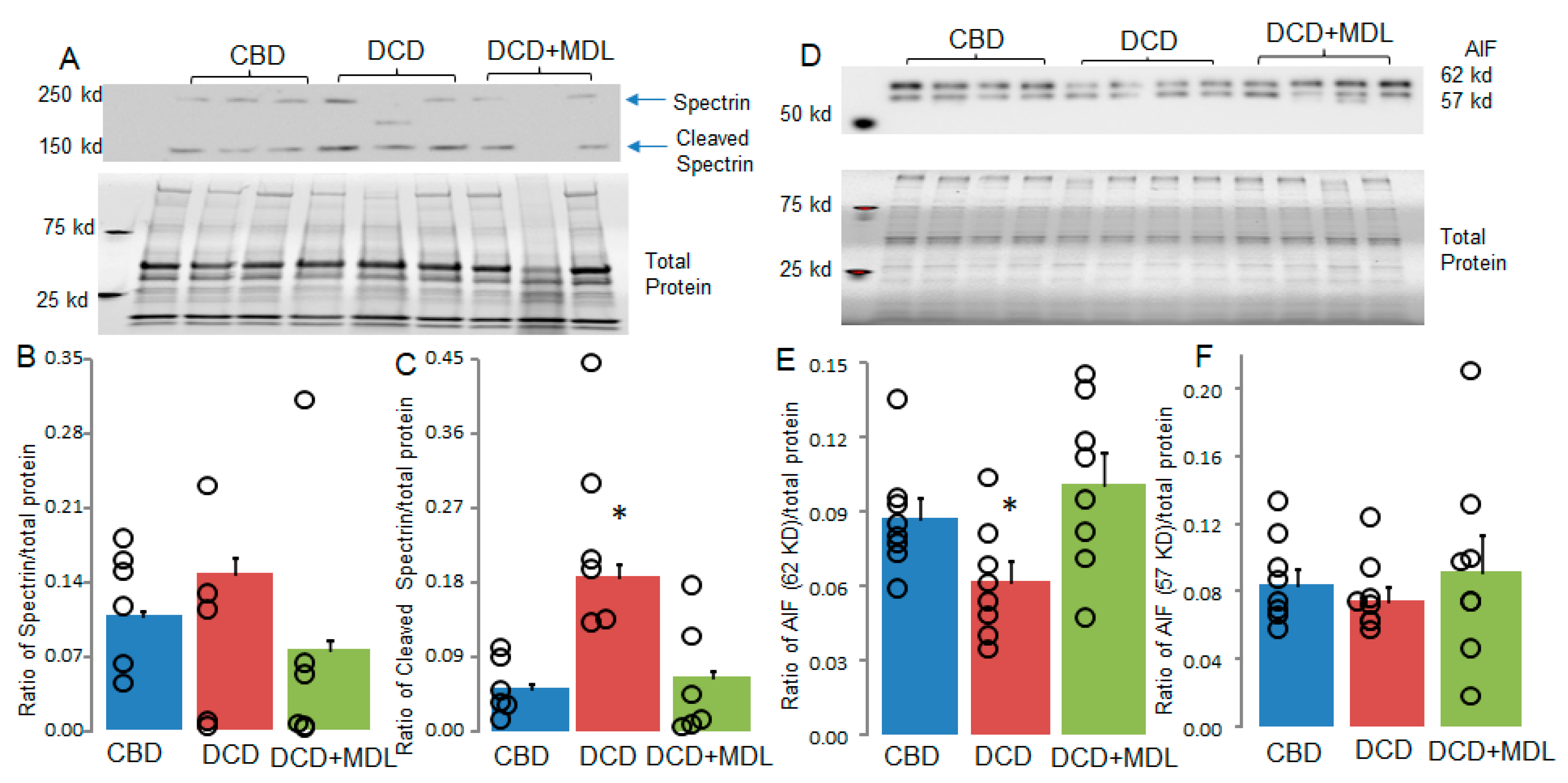
Figure 5.
MDL treatment protects mitofilin but not cyclophilin D in DCD heart mitochondria. Alterations in mitofilin or cyclophilin D content can influence MPTP opening. Mitofilin content was decreased in DCD heart mitochondria compared to CBD (Panels A and B), whereas MDL treatment preserved mitofilin in DCD heart mitochondria (Panels A and B). These results suggest that the decrease in mitofilin content is CPN1/2-dependent. No significant differences in cyclophilin D content were observed between CBD, DCD, and DCD+MDL groups (Panels C and D). Our findings suggest that decreased mitofilin content contributes to increased MPTP opening in DCD heart mitochondria. Data are expressed as mean ± SEM; *p < 0.05 vs. CBD or DCD+MDL. N = 8 in each group.
Figure 5.
MDL treatment protects mitofilin but not cyclophilin D in DCD heart mitochondria. Alterations in mitofilin or cyclophilin D content can influence MPTP opening. Mitofilin content was decreased in DCD heart mitochondria compared to CBD (Panels A and B), whereas MDL treatment preserved mitofilin in DCD heart mitochondria (Panels A and B). These results suggest that the decrease in mitofilin content is CPN1/2-dependent. No significant differences in cyclophilin D content were observed between CBD, DCD, and DCD+MDL groups (Panels C and D). Our findings suggest that decreased mitofilin content contributes to increased MPTP opening in DCD heart mitochondria. Data are expressed as mean ± SEM; *p < 0.05 vs. CBD or DCD+MDL. N = 8 in each group.
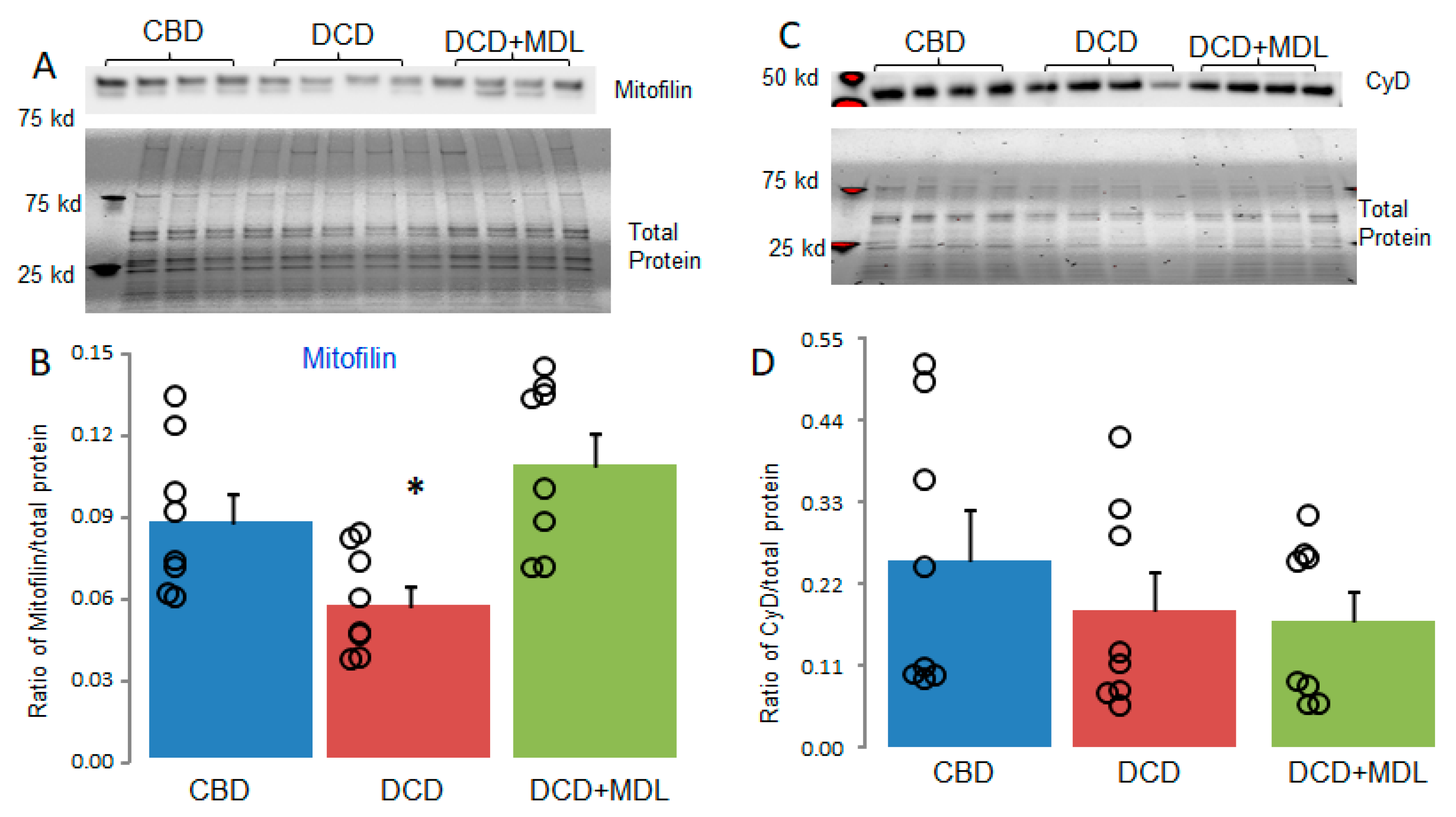

Table 1.
Antibodies used in the current manuscript.
| Antibody name | Company | Catalog number | Concentration |
| AIF | Cell Signaling | 4642 | 1:1000 |
| Mitofilin | ThermoFisher Scientific | MA3-940 | 1:1000 |
| Spectrin | Santa Cruz | csc-46696 | 1:100 |
| VDAC | Abcam | 14715 | 1:2500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



