Submitted:
28 March 2025
Posted:
28 March 2025
You are already at the latest version
Abstract
Eliciting DNA damage in tumor cells continues to be one of the most successful strategies against cancer. This is the case for classical chemotherapy drugs and radiotherapy. In the modern era of personalized medicine, this strategy tries to identify specific vulnerabilities identified in each patient’s tumor, to inflict DNA damage in certain cell contexts that end up in massive cancer cell death. Cells rely on multiple DNA repair pathways to fix DNA damage, but cancer cells frequently exhibit defects in these pathways, many times being tolerant to the damage. Key vulnerabilities, such as BRCA1/BRCA2 mutations, have been exploited with PARP inhibitors, leveraging synthetic lethality to selectively kill tumor cells and improving patients’ survival. In the DNA damage response (DDR) network, kinases ATM, ATR, Chk1, and Chk2 coordinate DNA repair, cell cycle arrest, and apoptosis. Inhibiting these proteins enhances tumor sensitivity to DNA-damaging therapies, especially in DDR-deficient cancers. Several small-molecule inhibitors targeting ATM/Chk2 and ATR/Chk1 are currently being tested in preclinical and/or clinical settings, showing promise in cancer models and patients. Additionally, pharmacological blockade of ATM/Chk2 and ATR/Chk1 axes enhances the effects of immunotherapy by increasing tumor immunogenicity, promoting T-cell infiltration and activating immune responses. Combining ATM/Chk2- or ATR/Chk1-targeting drugs with conventional chemotherapy, radiotherapy or immune checkpoint inhibitors offers a compelling strategy to improve treatment efficacy, overcome resistance, and enhance patients’ survival in modern oncology.
Keywords:
ATR
; Chk1
; ATM
; Chk2
; DNA damage
; synthetic lethality
; immunotherapy
1. Introduction
Chemotherapy and radiotherapy are cornerstones for cancer treatment. Many chemotherapy drugs and radiotherapy act through damaging the DNA and causing cell death [1]. Irradiation induces DNA breaks, especially double-strand breaks, which are difficult for cells to repair [2]. Chemotherapy with DNA damaging drugs includes alkylating agents, platinum-based compounds, and topoisomerase inhibitors. These drugs exert their effects by interfering with DNA replication and transcription, ultimately leading to cell death. Cancer cells, which divide more rapidly and frequently than most normal cells, are especially vulnerable to this type of damage, making DNA-damaging chemotherapy an effective therapeutic strategy. Radiotherapy and chemotherapy are many times combined to produce a synergistic anticancer effect in tumors [3]. However, these therapies are also associated with side effects, such as hair loss, bone marrow suppression, and gastrointestinal toxicity. In the era of precision medicine, new approaches to damage the DNA and block DNA repair mechanisms, based on particular alterations of each tumor, are being intensely investigated. It is expected that these therapies, many of which target DNA repair pathways, be highly effective with less toxic consequences. Targeting specific proteins of the DNA repair pathway also offers a promising strategy for sensitizing tumor cells to chemotherapy and radiotherapy [4].
DNA is continuously exposed to various internal and external insults that can lead to mutations, which, if left unrepaired, may contribute to carcinogenesis. To maintain genomic integrity, cells use several DNA repair mechanisms, each specialized in correcting different types of damage. The primary DNA repair pathways include base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), and non-homologous end joining (NHEJ) [5]. BER corrects small, non-helix-distorting base alterations caused by oxidation or alkylation, while NER removes bulky helix-distorting lesions, including those induced by ultraviolet (UV) light [5]. MMR solves replication errors, such as base mismatches and insertion-deletion loops. Double-strand breaks (DSBs), which are particularly harmful for cells, are repaired by HR (an error-free process using sister chromatid as a template) or NHEJ, which ligates broken ends directly and is more error-prone [5].
These repair mechanisms are frequently altered in cancer cells, which are able to adapt by tolerating DNA damage and accumulating mutations, instead of dying. Mutations or deficiencies in proteins of these pathways are common. For instance, BRCA1 and BRCA2 mutations impair HR, increasing susceptibility to DNA damaging drugs in breast and ovarian cancers. This vulnerability has been exploited therapeutically using PARP inhibitors (e.g., olaparib). PARP plays a role in single-strand break repair, and its inhibition in BRCA-deficient cells leads to the so-called synthetic lethality: a synergistic effect produced by blockade of two or more pathways related to a specific cellular process, leading to massive cell death [6]. Exploiting synthetic lethality enables selective targeting of tumor cells while sparing normal tissue. Ongoing research is focused on identifying novel repair pathway dependencies and developing specific inhibitors to enhance the effectiveness and specificity of cancer therapies [7].
The DNA damage response (DDR) pathway plays a crucial role in maintaining genomic stability, and its dysregulation is a hallmark of cancer. Among the central DDR proteins are the kinases ATM (ataxia-telangiectasia mutated), ATR (ATM and Rad3-related), Chk1 (Checkpoint Kinase 1), and Chk2 (Checkpoint Kinase 2), which coordinate cell cycle arrest, DNA repair, and apoptosis in response to DNA damage [5]. ATM is activated primarily by double-strand DNA breaks (DSBs). Upon DNA damage, it activates proteins like p53, Chk2, and BRCA1 to stop the cell cycle and induce the repair mechanisms, or apoptosis if damage is too severe [8,9]. ATM is mainly activated by single-stranded DNA (ssDNA) breaks, often at stalled replication forks, and plays a key role in responding to replication stress. It activates Chk1 and helps stabilize replication forks to maintain genome integrity. Both proteins are crucial for preventing mutations that could promote cancer. Inhibiting the ATM/Chk2 and ATR/Chk1 axes can sensitize tumor cells to DNA-damaging agents, particularly in tumors with DDR deficiencies [8,9,10]. Several small-molecule inhibitors targeting these proteins are in preclinical or clinical development and hold promise for the treatment of cancer patients in a more personalized and effective fashion than regular DNA-damaging chemotherapy agents.
In this study we review the ATM/Chk2 and ATR/Chk1 axes in cancer and the new drugs developed to inhibit these proteins, which are being tested in different phases of preclinical experimentation or clinical trials. We also focus on recent discoveries that involve targeting these proteins in certain tumor genetic backgrounds related to DNA damage, whose inhibition leads to synthetic lethality. Finally, we address the role of ATM/Chk2 and ATR/Chk1 blockade as a way to enhance immunotherapy responses.
2. DNA Damage and Repair Mechanisms
2.1. Main Types of DNA Damage
Cells are continuously exposed to different agents which can damage integrity within DNA strands. Depending on the origin of these agents, DNA damage can be categorized in exogenous or endogenous [11]. Certain environmental hazards, toxic heavy metals, UV and ionizing radiation (IR), among others, have been described as common external agents responsible of causing DNA damage [12]. IR can directly induce a broad range of DNA alterations such as single base lesions or single/double strand breaks (SSB, DSB) [13]. Additionally, the most harmful insult is due to an indirect effect of reactive oxygen species (ROS) caused by IR. [13,14]. UV is another source of exogenous DNA damage. UV has been described to produce bulky lesions, such as cyclobutane-pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs), generation of free radicals and subsequent introduction of strand breaks [15,16].
On the other hand, cells may also suffer internal damage, which can generate multiple mutations or breaks, that can lead to base mismatches and genomic instability, among others. DNA polymerases are able to replicate 6x109 nucleotides in every cell replication cycle [17,18]. Despite having high fidelity and proofreading activity, they can spontaneously make mistakes during the replication process. When cells are damaged during DNA replication (S-phase), replication forks temporally stop their activity (fork stalling). If maintained, this results in “fork collapse” leading to formation of DSBs and triggering DNA damage repair mechanisms [19]. Failure in DNA replication is mainly caused by DNA base damage. This includes abasic sites and alkylation processes [20,21]. Abasic sites (or apurinic/apyrimidinic, AP) can be spontaneously formed by destabilization of N-glycosyl bonds. If AP sites are maintained, they can cause blockade of transcription and DNA replication, leading to genomic instability. Single strand breaks (SSBs) are easily formed, and if not repaired, DSBs lesions can appear [21]. Regarding DNA methylation, it is a reversible epigenetic process essential for maintaining DNA stability [19]. This process is catalyzed by a whole family of DNA Methyltransferases (DNMTs) [19], which transfer a methyl group from S-adenosyl-L-methionine (SAM) to the corresponding nitrogenous base. However, spontaneous methylation of DNA brings about the formation of cytotoxic adducts, such as the 7-methylguanine (7-meG), 3-methyladenine (3-meA) and 3-methylguanine (3-meG) [20].
The diversity of DNA-lesions that may appear in every cell must be localized and repaired. Failures in recognition systems leads to an increased number of mutations, which will increase the risk of cancer development [22].
2.2. Mechanisms of Cellular Damage Response
2.2.1. Single Strand Breaks
Mismatch Repair System (MMR)
During DNA replication, errors occur in base pairing and small loops form due to insertion-deletion events (Figure 1). These base mismatches are repaired through mismatch repair systems [23]. However, a malfunction in this pathway leads to alterations in microsatellites, which are small tandemly repeated DNA sequences. Loops can be formed by repetition of the same sequence, resulting in different sequence lengths between microsatellites, which is known as microsatellite instability (MSI) [24]. The errors produced by DNA polymerases, if not corrected by their proofreading activity, lead to MMR system activation [25]. For strand correction, the intact complementary DNA strand is used as a template [25,26].
In prokaryotes, MutS gene detects mismatches in double strand DNA by recruiting MutL, which allows downstream signaling with effector proteins. MutH belongs to type II family restriction endonucleases and is responsible for strand excision by creating a “nick”, from which the DNA helicase UvrD works to unwind DNA during recombination [26].
There is a high similarity in the DNA repair mechanisms between the described system for E. coli and humans, with key roles of families MSH (MutS Homolog) and MLH/PMS (MutL Homolog/Post-Meiotic Segregation protein) proteins. The MSH complex plays a role in damage recognition, as MSH2-MSH3 and MSH2-MSH6 heterodimers slide on the DNA helix until mispaired bases are detected: larger insertion-deletion loops (MSH2-MSH3) or dinucleotide insertion-deletion distortions and single base mismatches (MSH2-MSH6) [27,28,29]. Then, the proliferating cell nuclear antigen (PCNA), replication factor C (RFC), MLH complex and exonuclease 1 (Exo1) are recruited for DNA repair. The MLH complex is formed by MLH1-PMS2 heterodimer and is responsible for generation of a nick in the unmethylated strand, through its endonuclease activity [30,31,32]. Subsequently, Exo1 degrades the DNA between the error and the nick, and DNA polymerase δ resynthesizes the strand [30,31,33].
Base Excision Repair (BER)
Genomic aberrations that do not distort the helix are mainly caused by oxidative damage [34,35]. Chemical agents and oxygen radicals can promote depurination, depyrimidination, and deamination reactions, affecting DNA strand and dNTP pool. The resulting modified bases are removed by DNA glycosylases through BER mechanisms, generating abasic (AP) sites that can be then replaced with the correct nucleotide [35,36,37]. One single damaged base (short patch repair, SP-BER) or a 2-8 nucleotide synthesized fragment (long patch repair, LP-BER) can be replaced [36]. Generally, oxidation derivatives of purines (8-oxoguanine and formamidopyrimines) and pyrimidines (thymine glycol and 5-OHU) are the most prevalent damage produced [38].
This damage is principally detected by a DNA glycosylase that generates AP sites, which are then recognized by an AP endonuclease 1 (APE1) forming a SSB. This gap is engaged and protected by poly(ADP-ribose) polymerase 1 (PARP1). XRCC1 prevents excessive PARP1 binding. Then, DNA polymerase β along with ligase IIIα (for SP-BER) or DNA polymerases δ/ε along with PCNA/RFC (for LP-BER) cooperate to re-synthesize a corrected insert. The displaced damaged strand is removed by flap endonuclease 1 (FEN1) and the new segment is joint to the adjacent strands by DNA ligase I activity [39,40,41,42].
Nucleotide Excision Repair (NER)
Chemotherapeutic agents, environmental mutagens and UV irradiation induce bulky adducts that distort the structure of the double helix and that can be repaired by NER mechanisms. There are two subtypes within this repair system: the GG-NER (global genome NER), which repairs throughout the genome, and the TC-NER (transcription-coupled NER), a faster process that ensures an efficient transcription process by blocking the elongating type II polymerase complex [43]. Although different proteins are involved in each mechanism, both follow the same pathway. The DNA lesion is recognized, excised and finally restored by polymerization/ligation using the non-damaged strand as a template [41].
In the GG-NER, damage identification is carried out by multiple protein complexes such as UV-damaged DNA-binding protein (UV/DDB) and CETN2-XPC-RAD23B [44,45]. In contrast, in TC-NER, damage recognition occurs through CSA/CSB and RNA polymerase II, which stalls upon detecting damage [46]. These subpathways then converge with the recruitment of transcription factor IIH (TFIIH), which interacts with XPB, to facilitate protein binding [47,48] and XPD, with helicase activity, to unwind DNA and form a bubble. XPA is necessary to verify that the damage has been correctly detected and acts as a scaffold protein [49,50]. Once the strands are separated, RPA binds to the single-stranded DNA to protect it from nuclease attacks during the repair process [51].
Finally, the endonucleases XPF and XPG remove the damaged fragment by performing 5’ and 3’ incisions, respectively [52], and DNA polymerases δ/ε resynthesize the missing DNA, which is ligated to the downstream DNA [53,54]. In this process, PCNA is responsible for the precise incorporation of nucleotides [53].
3. Double Strand Breaks (DSB)
DSBs are directly or indirectly originated by environmental factors such as IR and certain drugs (Figure 1) [55]. These lesions are probably the most harmful among the different DNA injuries described earlier. Thus, organisms must have evolved mechanisms to detect and repair the genomic instability originated by DSBs [55]. Mre11, Rad50 and Nbs1 (MRN complex) are firstly recruited within the DSB site [56,57]. Then, the signaling pathway involving ATM, ATR and the DNA-dependent protein kinase (DNA-PK) are primarily activated [56]. These proteins have a crucial role in the phosphorylation of histone H2AX at Ser139, converting it into H2AX [58,59]. Additionally, apparition of multiple H2AX foci sites ensures the amplification of the signal, recruiting several proteins related with DNA repair, such as 53BP1, MDC1 and BRCA1 [59]. The two main mechanisms to repair DSBs are non-homologous end joining (NHEJ) and homologous recombination (HR).
3.1. Non- Homologous End Joining (NHEJ)
Classical non-homologous end joining is initialized with dimerization of Ku70-Ku80 within the DSB site [60]. This binding will provide a scaffold for recruitment of DNA-PKs, DNA ligase IV, XRCC4, XRCC4-like factor (XLF) and PAXX (an XRCC4-like factor that will stabilize the NHEJ complex on the damaged chromatin) [61]. There is evidence suggesting that the kinase activity of DNA-PKs, together with the XRCC4-XLF complex are crucial in maintaining the two broken ends of DNA close enough to ligate both strands [61,62]. Next, the proteins Artemis and APLF, which have 5’ nuclease activity, are needed to generate a blunt end [63]. For gap filling, DNA pol and DNA pol (members of Pol X family) are required [63] and finally, ligation of both strands is completed by the XRCC4-Ligase IV complex. When ligation process is ended, the NHEJ complex is dissolved.
3.2. Homologous Recombination Repair (HRR)
Homologous recombination represents the second pathway of DSB repair. Unlike NHEJ, in HR a sister chromatid is needed, thus providing a high fidelity, error-free process [60,64]. As HR pathway is limited by the presence of a sister chromatid, this mechanism of repair is constrained to the S/G2 phase of the cell cycle [60].
Endonuclease Mre11 (as part of MRN complex) along with C-terminal interacting protein (CtIP) initiate the digestion on the 5’ strand (direction 5’–3’). A second digestion step is followed by the exonuclease 1 (EXO1) together with the helicase Bloom syndrome protein (BLM) and the endonuclease DNA2. This originates a ‘long range’ resection in 5’ strand of the DNA. Such process generates a ssDNA that is rapidly coated by replication protein A (RPA) [60]. RPA competes with RAD51, which polymerizes and forms stable filaments, protecting the 3’ ssDNA from further uncontrolled resection [65]. The formation of this synaptic complex is a key step and allows the invasion and displacement of the sister chromatid, forming what is known as D-loop. The invasion strand serves as template for DNA synthesis [65]. Finally, the heteroduplex complex is dissociated and after several steps of replication and ligation, the newly synthetized DNA serves as template to restore the complementary strand.
4. The ATR/Chk1 and ATM/Chk2 Axes in DNA Damage Repair
The ATR/Chk1 and ATM/Chk2 axis is crucial in DDR, as regulators of the repair machinery. Downstream signaling of these axes results in either halt of the cell cycle, which allows for DNA repair, or apoptosis and subsequent cell death when the damage is deemed irreparable [8].
4.1. ATR/Chk1 Signaling
The ATR/Chk1 pathway is key in ensuring the integrity of DNA replication; as such, it is triggered by replication stress and a wide range of genotoxic events. In fulfilling its protective function, ATR/Chk1 signaling often culminates with halt of the cell cycle during the replicative or S phase [66]. ATR is a serine/threonine kinase that phosphorylates a wide range of effector molecules in response to replication hindrances or DNA damage. ATR binds to RPA-coated ssDNA overhangs through interaction with ATRIP (ATR-interacting protein) [67]. This complex is characteristic of stressed replication forks, as well as certain intermediaries in DNA repair mechanisms, such as HR or NER [8]. ATR binding promotes stabilization of the stalled replication fork in order to prevent imminent collapse. Moreover, upon allosteric stimulation by TOPBP1 (Topoisomerase Binding Protein 1) or ETAA1 (Ewing’s tumor-associated antigen 1), ATR can phosphorylate and modulate key substrates involved in cell cycle regulation and HR [66,68].
Chk1 is the main downstream effector of ATR. Activated upon its phosphorylation by ATR, Chk1 inhibits Cdc25A and C phosphatases to arrest the cell cycle at intra-S and G2/M checkpoints, respectively [68]. Cdc25C is then ubiquitinated and degraded by the proteasome, followed by decreased CDK (Cyclin-Dependent Kinase) activity, which delays cell cycle progression until the damage has been repaired [61]. In parallel, by means of Wee1 activation, Chk1 maintains CDK and Cyclin suppression and reinforces cell cycle arrest [69].
Beyond the described mechanisms of DNA repair and cell cycle regulation, ATR/Chk1-mediated phosphorylation of p53 and other transcription factors actively contributes to the increased expression of genes related to DNA repair and survival [70]. The ATR/Chk1/Wee1 cascade also contributes to HR by interaction with ssDNA overhangs generated from end-resection in the intermediate steps of repair. In this context, ATR has been proven to recruit Rad51, while Chk1 can phosphorylate both this key substrate and BRCA2. Additionally, Wee1-induced CDK suppression promotes this mechanism of DSBs repair [68]. Other unexpected roles of this axis include reported preventive suppression of chromosome instability and faithful segregation due to mitosis-specific centromeric R-loop formation [71]. Lastly, ATR/Chk1 axis supports and fires normal replication fork origins in unperturbed S phase, ensuring reliable replication and preventing further issues altogether [72,73].
4.2. ATM/Chk2 Signaling
The ATM/Chk2 pathway primarily responds to DSBs in the DNA, which can be induced by IR, oxidative stress and certain forms of replication errors. In this response, ATM is recruited by the MRN complex at the site of DSB, triggering a regulation cascade that initializes with its autophosphorylation [74,75,76]. ATM’s kinase activity is key in the activation of Chk2, p53 and several other downstream targets that orchestrate cell cycle progression [74,77]. Moreover, ATM directly contributes to NHEJ and HR pathways by recruitment of repair proteins, such as DNA-PKcs or BRCA1 to the damage site [75]. In immediate response to DSBs, the Ser139 residue in the C-terminal tail of histone H2AX is rapidly phosphorylated by ATM, serving as a docking site for chromatin remodeling and DNA repair complexes [74].
ATM signaling initiates the p53 checkpoint pathway, leading to cell cycle arrest at the G1/S transition, ultimately triggering senescence or apoptosis in cases of severe and irreparable damage [77]. Furthermore, Chk2 contributes to p53 signaling by phosphorylating specific residues and thus maintaining cellular arrest through stabilization and activation of this tumor suppressor [77]. The induction of the p53 transcriptional target p21 further induces the expression of genes involved in the fate of the affected cell, depending on the extension and severity of the damage [69,77,78]. In addition, the cofactor Strap (stress responsive activator of p300), needed for an effective p53 response, is also targeted by ATM and Chk2. Phosphorylation of distinct residues in Strap by this axis contributes to its stabilization and nuclear accumulation, which, in turn, enhances p53 half-life and signaling [79].
Concurrently, Cdc25A and C kinases are inhibited by Chk2, which results in further cell cycle arrest by depletion and absence of CDKs and cyclin stimulation [76,80]. Interestingly, Chk2-dependent phosphorylation of Cdc25 on Ser 123 upon irradiation has been described to not only contribute to IR-induced DNA damage repair by S-phase delay, but also as a preventive mechanism against accumulation of mutations that confer radioresistance [81].
Aside from intervening in cell cycle arrest, the ATM/Chk2 axis prevents chromatin breakage via the phosphorylation of the scaffold protein INCENP, which promotes its binding to the chromosomal passenger complex (CPC) in cytokinesis, establishing an abscission checkpoint and avoiding DNA damage in the process [82]. In addition to the tumor suppressive character of ATM/Chk2 signaling, recently, CCNDBP1 (cyclin D1 binding protein 1), a protein associated to chemotherapy-induced damage rescuing, has been found as a possible linker between the ATM/Chk2 signaling and chemoresistance [83].
4.3. Cross-Talk Between ATR/Chk1 and ATM/Chk2
The extensive study of Chk1/ATR and Chk2/ATM axes has uncovered the complex network of overlapping and non-redundant interactions that conform DDR (Figure 2). Aside from converging in the established downstream inhibition of Cdc25 phosphatases, the main components of these pathways can exert direct modifications and regulate mutually [74]. This interaction was reported both ways, upon UV-induced DDR in ATR-dependent ATM activation and, vice versa, in IR-induced ATM-induced activation of ATR [84,85]. Further cross-talk reveals connecting points towards the p53 downstream signaling pathway. As an example, the indispensable role of Chk1 in maintaining ATM/p53/p21-triggered G2 cell cycle arrest has been observed in the context of sustained damage [70].
When chemotherapy drugs induce potentially lethal DNA damage in the cancer cells, these may develop resistance by activation of the DDR, which effectively repairs DNA damage [86]. Therefore, inhibition of DDR in this context could result in irreversible damage in cancer cells, making the this combinatory strategy very promising in the clinic [87].
For all the reasons mentioned, the pharmaceutical industry identified the ATR/Chk1 and ATM/Chk2 pathways as highly convenient targets to develop anticancer drugs, particularly in the context of certain mutations related to DDR. Many cancers exhibit defects in DDR, making them more reliant on ATR/Chk1 or ATM/Chk2 for survival. Inhibition of these pathways may selectively kill cancer cells by enhancing genomic instability and preventing repair of DNA damage induced by chemotherapy or radiation. ATR/Chk1 inhibition can be particularly effective in tumors with replication stress, whereas ATM/Chk2 inhibition may sensitize tumors with pre-existing DDR deficiencies, making them more susceptible to DNA-damaging agents. Also, dual inhibition or combination with other therapies, such as PARP inhibitors, can result in synthetic lethality. Moreover, increased DNA damage can lead to generation of neoantigens and enhanced response to immunotherapy. In the next section we describe the main ATR, ATM, Chk1 and Chk2 inhibitors used in preclinical experiments and/or clinical trials and explain how these drugs, alone or in combination, may help in cancer treatment.
4.4. ATR Inhibitors
As previously mentioned, ATR is deeply involved in responses against single-strand DNA damage or replication stress. Therefore, cancer cells that present these deficiencies will more likely depend on the ATR pathway for survival [88]. Among the drugs undergoing clinical testing, berzosertib (M6620, VX-970) has been studied as a single agent or in combination with chemotherapeutic agents (gemcitabine, topotecan, carboplatin, cisplatin) [89]. In preclinical studies of patient-derived lung tumor xenografts, combination of berzosertib with cisplatin led to a substantial tumor regression and delayed regrowth [89]. These results guided different phase I clinical trials, such as the CHARIOT trial (NCT03641547), where berzosertib was evaluated in combination with radiotherapy for palliative treatment of oesophageal cancer, or with cisplatin-capecitabine chemotherapy in solid tumors. Berzosertib was well tolerated and encouraging clinical activity was observed, as 5 patients presented partial responses and 10 showed stable disease [90]. Another phase I trial (NCT02487095) assessed the efficacy of M6620 in combination with topotecan in the second-line treatment of small cell lung cancer (SCLC) patients [91]. The trial demonstrated tolerability of berzosertib combined with topotecan and showed promising clinical activity in refractory SCLC, with 2 confirmed partial responses and 8 cases of prolonged stable disease [91]. In a phase II study where M6620 was evaluated in combination with gemcitabine, in platinum-resistant ovarian cancer (NCT02595892), increased median progression-free survival (mPFS) was observed in the gemcitabine plus berzosertib group (22.9 weeks) compared with the gemcitabine group (14.7 weeks) [92].
AZD6738 (ceralasertib) is an additional ATR inhibitor that has shown antitumor activity in combination with DNA-damaging anticancer agents [8], such as cisplatin, causing rapid regression of ATM-deficient non-small cell lung cancer (NSCLC) xenograft models [93]. A phase I clinical trial studying the effect of combining ceralasertib and paclitaxel (NCT02630199) determined that the treatment was well tolerated and demonstrated promising antitumor activity, especially in patients with advanced melanoma resistant to anti-PD1/L1 treatments. A trial (RP2D) using ceralasertib determined an optimal dose of 240 mg twice daily on days 1–14, combined with paclitaxel at 80 mg/m² on days 1, 8, and 15 of each 28-day cycle. Among the 57 patients treated, the overall response rate (ORR) was 22.6%, with a higher ORR (33.3%) for the 33 patients with melanoma resistant to prior anti-PD1/L1 therapy (mPFS 3.6 months, mOS 7.4 months) [94]. In the CAPRI phase I trial (NCT03462342), patients received olaparib 300 mg twice daily and ceralasertib 160 mg on days 1 to 7 of a 28-day cycle. Six patients presented partial responses, yielding an ORR of 50%. Combination of olaparib and ceralasertib was found to be tolerable and demonstrated activity in HR-deficient, platinum-sensitive recurrent high-grade serous ovarian cancer (HGSOC) patients who had progressed after benefiting from PARP inhibition as their penultimate treatment [95].
BAY 1895344 (elimusertib) is an oral ATR inhibitor that has demonstrated antitumor efficacy as monotherapy in CDX models and has shown synergistic effects when combined with the PARP1 inhibitor olaparib in vivo [96]. In a phase I dose escalation trial (NCT03188965) where elimusertib was administered at doses ranging from 5 to 80 mg, twice daily, to 21 patients with advanced solid tumors, the maximum tolerated dose was determined to be 40 mg twice daily with a 3-day-on/4-day-off schedule. 4 patients presented partial responses and stable disease was noted in 8 patients, with a median duration of response of 315.5 days. In fact, patients that responded had ATM protein loss and/or deleterious mutations of ATM [97]. Currently other trials studying the combination of elimusertib with other agents (pembrolizumab, NCT04095273) are being conducted [97].
4.5. ATM Inhibitors
When DSBs appear, ATM is activated by phosphorylation and repairs the DNA breaks [98]. As a master regulator in damage repair, ATM has been widely studied as a therapeutic target [8]. Preclinical studies of ATM inhibition with AZD0156, which presents high oral bioavailability, have demonstrated that it improves olaparib’s efficacy in two patient-derived triple-negative breast cancer xenograft models. AZD0156 alone was ineffective to inhibit cancer cell growth in colorectal cancer PDX models, but increased antitumor effects were observed when combined with irinotecan [99]. However, a phase I clinical trial (NCT02588105) combining AZD0156 with olaparib or irinotecan did not meet the expected results [99].
AZD1390 is another ATM-targeting DDR inhibitor optimized to present blood-brain barrier permeability. In preclinical models, including syngeneic and glioma PDX, as well as orthotopic lung-brain metastatic models, AZD1390 administered with daily doses of IR significantly resulted in tumor regressions and improved animal survival compared to radiation treatment alone [100]. Currently, there are several Phase I clinical trials actively recruiting participants, such as: the CONCORDE trial (NCT04550104) in patients with NSCLC in combination with conventional radiotherapy [101]; and NCT03423628, to assess the safety and tolerance of AZD1390 in combination with radiation therapies in patients with glioblastoma multiforme and patients with brain metastases [102].
4.6. Chk1/2 Inhibitors
Inhibiting Chk1 and Chk2, highly conserved serine/threonine kinases and key downstream targets of ATR and ATM, respectively, could disrupt the initiation of G2 checkpoint, impair DNA repair mechanisms and promote apoptosis in tumor cells [9,103]. LY-2606368 (prexasertib) has shown promising results in preclinical models. For instance, it was shown that Chk1/2 inhibition in monotherapy or in combination with cisplatin was able to significantly reduce tumor growth in a syngeneic model of SCLC [104]. Moreover, it increased cisplatin and olaparib potential and improved the response in platinum-resistant models [104]. A Phase I trial using prexasertib combined with olaparib showed preliminary clinical activity in BRCA-mutant patients with HGSOC, who had previously progressed on a PARP inhibitor. Notably, 4 of 18 patients with BRCA1-mutant PARP inhibitor–resistant HGSOC, achieved partial responses, suggesting potential for this combination therapy in overcoming resistance [105]. Phase II clinical trials with prexasertib have also shown promising results, particularly in ovarian cancer. In the NCT02203513 trial, prexasertib was particularly effective in patients with platinum-resistant or refractory BRCA-wild type HGSOC, with an 8/24 partial response rate [106]. The NCT03414047 trial reported an ORR of 12.1% in platinum-resistant patients and 6.9% in platinum-refractory patients, with durable activity as a single agent [107]. Phase II studies have also been conducted in other diseases, including triple-negative breast cancer and SCLC, but the efficacy observed in these cases was lower compared to that of ovarian cancer [108,109].
In a preclinical study evaluating the effects of Chk1 inhibition by GDC-0575, in vivo experiments were conducted using soft tissue sarcoma PDX models. The combination of GDC-0575 with gemcitabine demonstrated a synergistic antitumor effect and improved survival in dedifferentiated liposarcoma and leiomyosarcoma xenografts and TP53 mutant PDX models with the combination therapy, compared to either agent alone (no reduction was observed in the TP53 wild type PDX model) [110]. A phase I clinical trial using this drug in combination with gemcitabine (NCT02797964) demonstrated modest tolerability but improved antitumor activity. 4 patients achieved confirmed partial responses, 4 presenting TP53-mutated tumors. Overall, 15% of patients had stable disease for a period over 4 months. While GDC-0575 was in general safe administered both as a monotherapy and in combination with gemcitabine, hematological toxicities were frequent yet manageable [111].
The preclinical evaluation of PHI-101, a novel Chk2 inhibitor, demonstrated its potent anticancer activity against refractory ovarian and breast cancer cells, with improved efficacy both as monotherapy and in combination, in vitro and in vivo [112]. Currently, a phase Ia trial named CREATIVE (NCT04678102), is being conducted with the objective of assessing the dose-limiting toxicity (DLT) and the maximum tolerated dose (MTD) of PHI-101 in recurrent epithelial peritoneal, fallopian or ovarian cancer [113].
5. Synthetic Lethality Approaches to Target the ATR/Chk1 and ATM/Chk2 Axes
As described before, synthetic lethality implies a therapeutic approach by which the simultaneous alteration of two or more genes, or targeting two or more proteins in complementary pathways related to cell survival, results in vast cell death, while alteration of either gene/protein alone has little or modest effect on viability [114]. This type of perturbations are being widely studied along with DNA repair deficiencies in different cancers, receiving the name of “BRCAness”, as it was first described with the discovery of BRCA gene mutations in breast tumors, which were highly sensitive to PARP inhibitors [114,115]. In this section, we summarize some of the most recent synthetic lethality applications to the ATR/Chk1 and ATM/Chk2 regulatory pathways.
Weaknesses in cancer cells that present DNA repair deficiencies or genomic instability can be exploited for synthetic lethality with Chk1/ATR and Chk2/ATM inhibitors [116,117]. ATR/Chk1 signaling, hyperactive in cancer cells as a result from replication stress and uncontrolled proliferation, offers a good opportunity for synthetic lethality [68]. This could be particularly relevant in BRCA-deficient tumors [118,119]. Among the various preclinical and clinical trials conducted so far, ATR/Chk1/Wee1 inhibition has been described to be synergistic with the PARP inhibitor rucaparib [119]. Some studies even showcase successful overcoming of resistance to the PARP inhibitor olaparib in BRCA2-mutant ovarian cancer, by targeted inhibition of ATR/Chk1 [120]. Similarly, TP53 deficiency-induced loss of G1 checkpoint, makes cancer cells particularly vulnerable to ATR/Chk1 inhibition. Sensitivity to elimusertib and other ATR inhibitors is being thoroughly studied, with these drugs having readily entered clinical trials for TP53-deficient triple-negative breast cancer treatment, following on promising preclinical data [121,122].
ATR/Chk1 inhibition has also been used in combination with chemo- and radiotherapy. Potential synergy in cancer cell cytotoxicity has been studied by combining ATR inhibitors AZD6738 and M6620 with a wide arrange of chemotherapeutic agents, from cisplatin and carboplatin to topotecan and gemcitabine. These strategies have shown promising results in preclinical assays, but high toxicities in clinical trials [123]. Interestingly, simultaneous ATR and Chk1 inhibition was synthetic lethal in preclinical studies involving a variety of solid tumor models, with improved effects with hydroxyurea treatment [118]. ATR blockade induced synthetic lethality and overcomed chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells [124]. Using the ATR inhibitor AZD6738, authors showed that cells accumulated unrepaired DNA damage, ultimately leading to cell death by mitotic catastrophe. Inhibition of ATR induced synthetic lethality in mismatch repair-deficient cells and increased the effect of immunotherapy, in the CT26 colorectal cancer model [125]. By using a model of ATR-deficient DLD-1 human colorectal cancer cells, Schneider et al., [126] demonstrated synthetic lethality upon POLA1 inhibition. siRNA-mediated POLA1 depletion sensitized several cancer cell lines to ATR and Chk1 drugs. ATR inhibition was also shown to be synthetic lethal with ERCC1 deficiency [127]. A synthetic lethal screening discovered increased sensitivity to ATR inhibitors in mantle cell lymphoma with ATM loss-of-function [128]. In an orthotopic breast cancer model, co-targeting ATR and Wee1 led to tumor-selective synthetic lethality, with tumor remission and metastasis impairment. Mechanistically, this combination left cells with unrepaired or under-replicated DNA, thus inducing mitotic catastrophe [129].
Synthetic lethality upon combination of Chk1 (SRA737) and Wee1 (AZD1775) inhibitors has also been reported in castration-resistant prostate cancer [130]. In EZH2 deficient T cell acute lymphoblastic leukemia (T-ALL), a synthetic lethal screening identified Chk1 inhibition as an exploitable vulnerability. EZH2 loss was related to a transcriptomic signature in immature T-ALL cells, characterized by upregulation of MYCN and replication stress [131]. ATR-, Chk1- and Wee1-targeting drugs caused HR repair deficiency and induced synthetic lethality with PARP inhibitors [119]. Combination of an HDAC8 inhibitor with the Chk1-targeting drug AZD-7762 produced synthetic lethality in preclinical cancer models. HDAC8 impairment slowed DNA fork progression and induced Chk1/2 activation, allowing the co-targeting strategy that resulted in cell death [132].
Loss of BRCA1 has also been shown to be synthetic lethal with ATM loss [133], suggesting that drugs targeting ATM can be highly effective against tumors in the context of BRCA1 mutation. Synthetic lethality between epigenetic silencing of BEND4, a DNA repair gene, and ATM drugs has been described in pancreatic ductal adenocarcinoma (PDAC). Promoter methylation of BEND4 was found in 58.1% PDAC patients. BEND4 is involved in NHEJ signaling and loss of BEND4 significantly increased the sensitivity of PDAC cells to the ATM inhibitor AZD0156 [134]. Oh et al., [135] described a synthetic lethal strategy using PARP and ATM inhibitors to overcome trastuzumab resistance in HER2-positive cancers. The authors described increased PARP1 levels in trastuzumab-resistant cells. Inhibition of PARP1 with olaparib in the resistant cells restrained proliferation, but activated ATM to maintain genome stability. Dual inhibition of PARP and ATM (with AZD0156) in this context caused synthetic lethality. Ratz et al., [136] showed that combination between an EZH2 inhibitor (GSK126) and an ATM inhibitor (AZD1390) is synthetic lethal in BRCA1-deficient breast cancer. Lethality was manifested in reduced colony formation, increased genotoxic stress, and apoptosis-mediated cell death in vitro, as well as significantly increased anti-tumor activity in vivo.
These and other published results exemplify the opportunity to discover novel cancer vulnerabilities by pharmacological combinations targeting ATM, ATR, Chk1/2 inhibitors, especially in DDR impaired genetic backgrouds.
6. Synergistic Anticancer Effects Elicited by Combining ATM/Chk2 or ATR/Chk1 Inhibition with Immunotherapy
DDR inhibitors have been shown to enhance antitumor immune responses and potentiate immunotherapy. The rationale for this combination is that DDR inhibitors can increase tumor immunogenicity by promoting accumulation of DNA damage, leading to the presence of cytosolic DNA fragments that activate the cGAS-STING pathway. This activation triggers type I interferon responses and upregulation of inflammatory cytokines and chemokines, enhancing recruitment and activation of immune cells, particularly cytotoxic T cells [137]. Examples of this cooperation are copious and summarized below.
The recent link between ATR/Chk1 and modulation of the tumor microenvironment (TME) has sparked interest in the combination of ATR inhibitors with anti-PD-(L)1 therapy [138]. Several ongoing early-phase clinical trials in solid tumors are studying the administration of ATR-targeting drugs (M6620, AZD6738) together with either avelumab or pembrolizumab [139]. The ATR inhibitor ceralasertib potentiated checkpoint-based immunotherapy by up-regulation of type I interferon (IFNI) pathways [140]. Similar findings were reported by Taniguchi et al., in SCLC, using the ATR inhibitor berzosertib [141]. In hepatocellular carcinoma, addition of the ATR targeting drug AZD6738 to radioimmunotherapy boosted the immune cell infiltration and enhanced interferon (IFN)-γ production [142]. ATR inhibition also activated STING and increased tumor expression of MHC-I [143]. Radiation therapy in combination with the ATM-targeting drug AZD0156 increased STING-dependent anticancer responses. A low-dose of this drug plus radiotherapy synergistically increased IFN-β, MHC-I and PD-L1 expression in tumor cells [144]. In lung cancer, combination of the ATR inhibitor berzosertib with ablative radiotherapy remodeled the TME and enhanced immunotherapy responses [145]. In this study, authors showed that ATR inhibition increased radiation-induced damage, with activation of the cGAS/STING pathway. Adding immunotherapy further enhanced antitumor and antimetastatic effects. In a phase 1 clinical trial, the Chk1 inhibitor prexasertib combined to anti-PD-L1 therapy showed evidence of CD8 + T-cell activation in peripheral blood in response to treatment [146]. In preclinical cancer models, combination of the oral Chk1 Inhibitor SRA737 with low-dose gemcitabine enhanced the effect of anti-PD-L1 therapy by modulating the TME. This treatment led to increase in CD8+ cytotoxic T cells, dendritic cells, and M1-like macrophages and IFNβ, CCL5 and CXCL10, as well as decrease in M2-like macrophages and MDSC [147].
Inhibition of the ATM/Chk2 axis also induced cGAS/STING signaling in ARID1A-deficient tumors [148]. ARID1A is a member of the chromatin-remodeling complex SWI/SNF, which is frequently mutated in cancer. Using data from the Cancer Genomic Atlas, authors found augmented expression of Chk2 in ARID1A-mutated/deficient tumors. Inhibition of ATM/Chk2 led to replication stress, accumulation of cytosolic DNA and activation of the STING-mediated innate immune response. This resulted in increased tumor-infiltrating lymphocytes [148].
These examples show the mechanistic and clinical rationale for combining ATR/Chk1 and ATM/Chk2 to enhance the effect of immunotherapy. Clinical trials co-targeting these proteins and other DDR proteins are underway, with great expectation about the potential synergism in anticancer therapy [149].
7. Conclusion and Future Perspectives
Drugs that cause DNA damage are still pillars for cancer treatment. Although DNA damage induced by anticancer drugs can be repaired by different mechanisms to maintain genomic integrity, dysfunctionality of these pathways in cancer cells increases susceptibility to DDR-based treatments. Central to DDR are kinases ATM, ATR, Chk1, and Chk2, which orchestrate repair, cell cycle arrest, or apoptosis upon DNA damage. Inhibiting these kinases can sensitize tumor cells to treatments, including chemotherapy, radiotherapy and DNA repair-targeted therapy, especially in DDR-deficient tumors. Gemomic studies and pharmacological inhibitors targeting ATM/Chk2 and ATR/Chk1 axes have identified novel synthetic lethality approaches, leading to a pronounced antitumor growth. These strategies are revealing novel options for personalized medicine, in some cases being currently tested in clinical trials. Furthermore, ATR/Chk2 and ATM/Chk1 inhibitors enhance immunotherapy by increasing tumor immunogenicity through activation of immune pathways, such as cGAS-STING, increase in mutation load and promotion of T-cell infiltration, particularly when combined with immune checkpoint inhibitors. Therefore, inhibition of ATM, ATR, Chk1, and Chk2 represents a promising strategy in modern oncology. By exploiting these cancer-specific weaknesses, the therapeutic approach based on ATR/Chk2 and ATM/Chk1 inhibition is expected to improve efficacy, overcome resistance and reduce toxicity, potentially increasing patients’ survival.
Acknowledgments
This study has been funded by: This work has been funded by Instituto de Salud Carlos III (ISCIII)-FIS Pl25/01059 (to A.C.), World Wide Cancer Research (Grant# 240390, to A.C.), Gobierno de Navarra-GRANATE (to A.C. and D.S.) and a grant of the International Association for the Study of Lung Cancer (to D.S.). SL was supported by a fellowship from the Asociación de Amigos, University of Navarra; NO was supported by a fellowship from Gobierno de Navarra.
References
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.W. Biological Response of Cancer Cells to Radiation Treatment. Front. Mol. Biosci. 2014, 1. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes (Basel). 2019, 10. [Google Scholar] [CrossRef]
- Boshuizen, J.; Peeper, D.S. Rational Cancer Treatment Combinations: An Urgent Clinical Need. Mol. Cell 2020, 78, 1002–1018. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Drew, Y.; Zenke, F.T.; Curtin, N.J. DNA Damage Response Inhibitors in Cancer Therapy: Lessons from the Past, Current Status and Future Implications. Nat. Rev. Drug Discov. 2025, 24. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kreis, J.; Schelhorn, S.E.; Dahmen, H.; Grombacher, T.; Zühlsdorf, M.; Zenke, F.T.; Guan, Y. Mapping Combinatorial Drug Effects to DNA Damage Response Kinase Inhibitors. Nat. Commun. 2023, 14. [Google Scholar] [CrossRef]
- Choi, W.; Lee, E.S. Therapeutic Targeting of DNA Damage Response in Cancer. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Qian, J.; Liao, G.; Chen, M.; Peng, R.W.; Yan, X.; Du, J.; Huang, R.; Pan, M.; Lin, Y.; Gong, X.; et al. Advancing Cancer Therapy: New Frontiers in Targeting DNA Damage Response. Front. Pharmacol. 2024, 15. [Google Scholar] [CrossRef]
- Zhou, B.B.S.; Elledge, S.J. The DNA Damage Response: Putting Checkpoints in Perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA Damage Repair: Historical Perspectives, Mechanistic Pathways and Clinical Translation for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2021 61 2021, 6, 1–35. [Google Scholar] [CrossRef]
- Maremonti, E.; Brede, D.A.; Olsen, A.K.; Eide, D.M.; Berg, E.S. Ionizing Radiation, Genotoxic Stress, and Mitochondrial DNA Copy-Number Variation in Caenorhabditis Elegans: Droplet Digital PCR Analysis. Mutat. Res. Toxicol. Environ. Mutagen. 2020, 858–860, 503277. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing Radiation-Induced Metabolic Oxidative Stress and Prolonged Cell Injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Kciuk, M.; Marciniak, B.; Mojzych, M.; Kontek, R. Focus on UV-Induced DNA Damage and Repair—Disease Relevance and Protective Strategies. Int. J. Mol. Sci. 2020, 21, 7264. [Google Scholar] [CrossRef]
- Cadet, J.; Richard Wagner, J. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235. [Google Scholar] [CrossRef]
- Sriraman, A.; Debnath, T.K.; Xhemalce, B.; Miller, K.M. Making It or Breaking It: DNA Methylation and Genome Integrity. Essays Biochem. 2020, 64, 687. [Google Scholar] [CrossRef]
- Bordin, D.L.; Lirussi, L.; Nilsen, H. Cellular Response to Endogenous DNA Damage: DNA Base Modifications in Gene Expression Regulation. DNA Repair (Amst). 2021, 99, 103051. [Google Scholar] [CrossRef]
- Thompson, P.S.; Cortez, D. New Insights into Abasic Site Repair and Tolerance. DNA Repair (Amst). 2020, 90, 102866. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nat. 2009 4617267 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2008 181 2007, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.M.; Pearson, C.E. Disease-Associated Repeat Instability and Mismatch Repair. DNA Repair (Amst). 2016, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 535672. [Google Scholar] [CrossRef]
- Sameer, A.S.; Nissar, S.; Fatima, K. Mismatch Repair Pathway: Molecules, Functions, and Role in Colorectal Carcinogenesis. Eur. J. Cancer Prev. 2014, 23, 246–257. [Google Scholar] [CrossRef]
- Brown, M.W.; Kim, Y.; Williams, G.M.; Huck, J.D.; Surtees, J.A.; Finkelstein, I.J. Dynamic DNA Binding Licenses a Repair Factor to Bypass Roadblocks in Search of DNA Lesions. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Jiricny, J. Postreplicative Mismatch Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–23. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA Mismatch Repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Cannavo, E.; Gerrits, B.; Marra, G.; Schlapbach, R.; Jiricny, J. Characterization of the Interactome of the Human MutL Homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 2007, 282, 2976–2986. [Google Scholar] [CrossRef]
- Fishel, R. Mismatch Repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic Function of MutLalpha in Human Mismatch Repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef]
- Prindle, M.J.; Loeb, L.A. DNA Polymerase Delta in DNA Replication and Genome Maintenance. Environ. Mol. Mutagen. 2012, 53, 666–682. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. [CrossRef]
- Hegde, M.L.; Izumi, T.; Mitra, S. Oxidized Base Damage and Single-Strand Break Repair in Mammalian Genomes: Role of Disordered Regions and Posttranslational Modifications in Early Enzymes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153. [Google Scholar] [CrossRef]
- Grundy, G.J.; Parsons, J.L. Base Excision Repair and Its Implications to Cancer Therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef]
- Çaǧlayan, M.; Horton, J.K.; Dai, D.P.; Stefanick, D.F.; Wilson, S.H. Oxidized Nucleotide Insertion by Pol β Confounds Ligation during Base Excision Repair. Nat. Commun. 2017 81 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Base-Excision Repair of Oxidative DNA Damage by DNA Glycosylases. Mutat. Res. 2005, 591, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Frosina, G.; Fortini, P.; Rossi, O.; Carrozzino, F.; Raspaglio, G.; Cox, L.S.; Lane, D.P.; Abbondandolo, A.; Dogliotti, E. Two Pathways for Base Excision Repair in Mammalian Cells. J. Biol. Chem. 1996, 271, 9573–9578. [Google Scholar] [CrossRef]
- Campalans, A.; Kortulewski, T.; Amouroux, R.; Menoni, H.; Vermeulen, W.; Radicella, J.P. Distinct Spatiotemporal Patterns and PARP Dependence of XRCC1 Recruitment to Single-Strand Break and Base Excision Repair. Nucleic Acids Res. 2013, 41, 3115–3129. [Google Scholar] [CrossRef]
- Fousteri, M.; Mullenders, L.H.F. Transcription-Coupled Nucleotide Excision Repair in Mammalian Cells: Molecular Mechanisms and Biological Effects. Cell Res. 2008 181 2008, 18, 73–84. [Google Scholar] [CrossRef]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-Excision Repair of Oxidative DNA Damage. Nat. 2007 4477147 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.J.; Schärer, O.D. Molecular Mechanisms of Mammalian Global Genome Nucleotide Excision Repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Aboussekhra, A.; Biggerstaff, M.; Shivji, M.K.K.; Vilpo, J.A.; Moncollin, V.; Podust, V.N.; Protić, M.; Hübscher, U.; Egly, J.M.; Wood, R.D. Mammalian DNA Nucleotide Excision Repair Reconstituted with Purified Protein Components. Cell 1995, 80, 859–868. [Google Scholar] [CrossRef]
- Sugasawa, K.; Okamoto, T.; Shimizu, Y.; Masutani, C.; Iwai, S.; Hanaoka, F. A Multistep Damage Recognition Mechanism for Global Genomic Nucleotide Excision Repair. Genes Dev. 2001, 15, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, F.; Hennecke, U.; Carell, T.; Cramer, P. CPD Damage Recognition by Transcribing RNA Polymerase II. Science 2007, 315, 859–862. [Google Scholar] [CrossRef]
- Okuda, M.; Kinoshita, M.; Kakumu, E.; Sugasawa, K.; Nishimura, Y. Structural Insight into the Mechanism of TFIIH Recognition by the Acidic String of the Nucleotide Excision Repair Factor XPC. Structure 2015, 23, 1827–1837. [Google Scholar] [CrossRef]
- Volker, M.; Moné, M.J.; Karmakar, P.; Van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.J.; Van Driel, R.; Van Zeeland, A.A.; Mullenders, L.H.F. Sequential Assembly of the Nucleotide Excision Repair Factors in Vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar] [CrossRef]
- Kokic, G.; Chernev, A.; Tegunov, D.; Dienemann, C.; Urlaub, H.; Cramer, P. Structural Basis of TFIIH Activation for Nucleotide Excision Repair. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Sugasawa, K.; Akagi, J. ichi; Nishi, R.; Iwai, S.; Hanaoka, F. Two-Step Recognition of DNA Damage for Mammalian Nucleotide Excision Repair: Directional Binding of the XPC Complex and DNA Strand Scanning. Mol. Cell 2009, 36, 642–653. [Google Scholar] [CrossRef]
- De Laat, W.L.; Appeldoorn, E.; Sugasawa, K.; Weterings, E.; Jaspers, N.G.J.; Hoeijmakers, J.H.J. DNA-Binding Polarity of Human Replication Protein A Positions Nucleases in Nucleotide Excision Repair. Genes Dev. 1998, 12, 2598–2609. [Google Scholar] [CrossRef]
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Schärer, O.D. Coordination of Dual Incision and Repair Synthesis in Human Nucleotide Excision Repair. EMBO J. 2009, 28, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA Polymerases, Recruited by Different Mechanisms, Carry out NER Repair Synthesis in Human Cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Paul-Konietzko, K.; Thomale, J.; Arakawa, H.; Iliakis, G. DNA Ligases I and III Support Nucleotide Excision Repair in DT40 Cells with Similar Efficiency. Photochem. Photobiol. 2015, 91, 1173–1180. [Google Scholar] [CrossRef]
- Aleksandrov, R.; Hristova, R.; Stoynov, S.; Gospodinov, A. The Chromatin Response to Double-Strand DNA Breaks and Their Repair. Cells 2020, 9, 1853. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN Complex in Double-Strand Break Repair and Telomere Maintenance. FEBS Lett. 2010, 584, 3682. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA Double-Strand Break Repair Pathway Choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.D.; Guan, H.; Zhang, S.M.; Tang, Y.; Rang, W.Q.; Zhou, P.K. ΓH2AX Foci Formation in the Absence of DNA Damage: Mitotic H2AX Phosphorylation Is Mediated by the DNA-PKcs/CHK2 Pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef]
- Prabhu, K.S.; Kuttikrishnan, S.; Ahamad, N.; Habeeba, U.; Mariyam, Z.; Suleman, M.; Bhat, A.A.; Uddin, S. H2AX: A Key Player in DNA Damage Response and a Promising Target for Cancer Therapy. Biomed. Pharmacother. 2024, 175, 116663. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019 2011 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Graham, T.G.W.; Walter, J.C.; Loparo, J.J. Two-Stage Synapsis of DNA Ends during Non-Homologous End Joining. Mol. Cell 2016, 61, 850. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA End-Joining for Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2017, 293, 10512. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008 181 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous Recombination and the Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524. [Google Scholar] [CrossRef]
- Simoneau, A.; Zou, L. An Extending ATR-CHK1 Circuitry: The Replication Stress Response and Beyond. Curr. Opin. Genet. Dev. 2021, 71, 92–98. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA Damage Checkpoint Kinases in Cancer. Expert Rev. Mol. Med. 2020, 22. [Google Scholar] [CrossRef]
- Buscemi, G.; Perego, P.; Carenini, N.; Nakanishi, M.; Chessa, L.; Chen, J.; Khanna, K.K.; Delia, D. Activation of ATM and Chk2 Kinases in Relation to the Amount of DNA Strand Breaks. Oncogene 2004, 23, 7691–7700. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Besnard, E.; Fisher, D.; Piette, J.; Dulić, V. Chk1 Is Dispensable for G2 Arrest in Response to Sustained DNA Damage When the ATM/P53/P21 Pathway Is Functional. Oncogene 2011, 30, 4261–4274. [Google Scholar] [CrossRef]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A Mitosis-Specific and R Loop-Driven ATR Pathway Promotes Faithful Chromosome Segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef]
- Petermann, E.; Caldecott, K.W. Evidence That the ATR/Chk1 Pathway Maintains Normal Replication Fork Progression during Unperturbed S Phase. Cell Cycle 2006, 5, 2203–2209. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Yin, Y.; Calderon, M.J.; Qian, C.; Schamus-Haynes, S.; Sugitani, N.; Osmanbeyoglu, H.U.; Rothenberg, E.; Watkins, S.C.; Bakkenist, C.J. An ATR and CHK1 Kinase Signaling Mechanism That Limits Origin Firing during Unperturbed DNA Replication. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 13374–13383. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Smith, J.; Mun Tho, L.; Xu, N.; A. Gillespie, D. The ATM–Chk2 and ATR–Chk1 Pathways in DNA Damage Signaling and Cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef] [PubMed]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs Kinases-the Lessons from the Mouse Models: Inhibition ≠ Deletion. Cell Biosci. 2020, 10. [Google Scholar] [CrossRef]
- Chen, L.; Gilkes, D.M.; Pan, Y.; Lane, W.S.; Chen, J. ATM and Chk2-Dependent Phosphorylation of MDMX Contribute to P53 Activation after DNA Damage. EMBO J. 2005, 24, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Kuk, M.U.; Kim, J.W.; Lee, Y.H.; Lee, Y.S.; Choy, H.E.; Park, S.C.; Park, J.T. ATM Mediated-P53 Signaling Pathway Forms a Novel Axis for Senescence Control. Mitochondrion 2020, 55, 54–63. [Google Scholar] [CrossRef]
- Adams, C.J.; Graham, A.L.; Jansson, M.; Coutts, A.S.; Edelmann, M.; Smith, L.; Kessler, B.; La Thangue, N.B. ATM and Chk2 Kinase Target the P53 Cofactor Strap. EMBO Rep. 2008, 9, 1222. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A Checkpoint Pathway Guards against Radioresistant DNA Synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef]
- Petsalaki, E.; Zachos, G. An ATM-CHK2-INCENP Pathway Prevents Chromatin Breakage by Regulating the Abscission Checkpoint. Mol. Cell. Oncol. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Kamimura, K.; Ogawa, K.; Oda, C.; Tanaka, Y.; Horigome, R.; Ohtsuka, M.; Miura, H.; Fujisawa, K.; Yamamoto, N.; et al. Cyclin D1 Binding Protein 1 Responds to DNA Damage through the ATM-CHK2 Pathway. J. Clin. Med. 2022, 11. [Google Scholar] [CrossRef]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-Dependent Phosphorylation and Activation of ATM in Response to UV Treatment or Replication Fork Stalling. EMBO J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and Cell Cycle-Dependent Regulation of ATR in Response to DNA Double-Strand Breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Jurkovicova, D.; Neophytou, C.M.; Gašparović, A.Č.; Gonçalves, A.C. DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA Damage Response: Next-Generation Anti-Cancer Therapeutic Strategies. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of Tumor Responses to DNA Damaging Therapy by the Selective ATR Inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef]
- Javed, S.R.; Lord, S.; El Badri, S.; Harman, R.; Holmes, J.; Kamzi, F.; Maughan, T.; McIntosh, D.; Mukherjee, S.; Ooms, A.; et al. CHARIOT: A Phase I Study of Berzosertib with Chemoradiotherapy in Oesophageal and Other Solid Cancers Using Time to Event Continual Reassessment Method. Br. J. Cancer 2024, 130, 467–475. [Google Scholar] [CrossRef]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.C.; Wahner Hendrickson, A.E.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus Gemcitabine versus Gemcitabine Alone in Platinum-Resistant High-Grade Serous Ovarian Cancer: A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet. Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembé, A.B.; Cho, H.; HKim, K.M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [CrossRef]
- Wethington, S.L.; Shah, P.D.; Martin, L.; Tanyi, J.L.; Latif, N.; Morgan, M.; Torigian, D.A.; Rodriguez, D.; Smith, S.A.; Dean, E.; et al. Combination ATR (Ceralasertib) and PARP (Olaparib) Inhibitor (CAPRI) Trial in Acquired PARP Inhibitor-Resistant Homologous Recombination-Deficient Ovarian Cancer. Clin. Cancer Res. 2023, 29, 2800–2807. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lucking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bomer, U.; Moosmayer, D.; Eberspacher, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Noor, N.R.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef]
- Hargreaves, C.E.; Salatino, S.; Sasson, S.C.; Charlesworth, J.E.G.; Bateman, E.; Patel, A.M.; Anzilotti, C.; Broxholme, J.; Knight, J.C.; Patel, S.Y. Decreased ATM Function Causes Delayed DNA Repair and Apoptosis in Common Variable Immunodeficiency Disorders. J. Clin. Immunol. 2021, 41, 1315–1330. [Google Scholar] [CrossRef]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The Brain-Penetrant Clinical ATM Inhibitor AZD1390 Radiosensitizes and Improves Survival of Preclinical Brain Tumor Models. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Walls, G.M.; Oughton, J.B.; Chalmers, A.J.; Brown, S.; Collinson, F.; Forster, M.D.; Franks, K.N.; Gilbert, A.; Hanna, G.G.; Hannaway, N.; et al. CONCORDE: A Phase I Platform Study of Novel Agents in Combination with Conventional Radiotherapy in Non-Small-Cell Lung Cancer. Clin. Transl. Radiat. Oncol. 2020, 25, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Jucaite, A.; Stenkrona, P.; Cselényi, Z.; De Vita, S.; Buil-Bruna, N.; Varnäs, K.; Savage, A.; Varrone, A.; Johnström, P.; Schou, M.; et al. Brain Exposure of the ATM Inhibitor AZD1390 in Humans-a Positron Emission Tomography Study. Neuro. Oncol. 2021, 23, 687–696. [Google Scholar] [CrossRef]
- Merry, C.; Fu, K.; Wang, J.; Yeh, I.J.; Zhang, Y. Targeting the Checkpoint Kinase Chk1 in Cancer Therapy. Cell Cycle 2010, 9, 279–283. [Google Scholar] [CrossRef]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.B.; Fan, Y.H.; Li, L.; Glisson, B.S.; et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017, 77, 3870–3884. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; De Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-Grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a Cell Cycle Checkpoint Kinase 1 and 2 Inhibitor, in BRCA Wild-Type Recurrent High-Grade Serous Ovarian Cancer: A First-in-Class Proof-of-Concept Phase 2 Study. Lancet. Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Lee, J. min; Gao, B.; Miller, R.; Lee, J.Y.; Colombo, N.; Vergote, I.; Credille, K.M.; Young, S.R.; McNeely, S.; et al. A Phase 2 Study of Prexasertib (LY2606368) in Platinum Resistant or Refractory Recurrent Ovarian Cancer. Gynecol. Oncol. 2022, 167, 213–225. [Google Scholar] [CrossRef]
- Byers, L.A.; Navarro, A.; Schaefer, E.; Johnson, M.; Özgüroğlu, M.; Han, J.Y.; Bondarenko, I.; Cicin, I.; Dragnev, K.H.; Abel, A.; et al. A Phase II Trial of Prexasertib (LY2606368) in Patients With Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.-J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, 1013–e1824. [Google Scholar] [CrossRef]
- Laroche-Clary, A.; Lucchesi, C.; Rey, C.; Verbeke, S.; Bourdon, A.; Chaire, V.; Algéo, M.P.; Cousin, S.; Toulmonde, M.; Vélasco, V.; et al. CHK1 Inhibition in Soft-Tissue Sarcomas: Biological and Clinical Implications. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1023–1029. [Google Scholar] [CrossRef]
- Italiano, A.; Infante, J.R.; Shapiro, G.I.; Moore, K.N.; LoRusso, P.M.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I Study of the Checkpoint Kinase 1 Inhibitor GDC-0575 in Combination with Gemcitabine in Patients with Refractory Solid Tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef]
- Han, J.H.-J.; Kim, K.-T.; Im, J.; Park, S.; Choi, M.K.; Kim, I.; Nam, K.-Y.; Yoon, J. Abstract 1461: PHI-101, a Potent and Novel Inhibitor of CHK2 in Ovarian and Breast Cancer Cells. Cancer Res. 2021, 81, 1461–1461. [Google Scholar] [CrossRef]
- Park, S.J.; Chang, S.J.; Suh, D.H.; Kong, T.W.; Song, H.; Kim, T.H.; Kim, J.W.; Kim, H.S.; Lee, S.J. A Phase IA Dose-Escalation Study of PHI-101, a New Checkpoint Kinase 2 Inhibitor, for Platinum-Resistant Recurrent Ovarian Cancer. BMC Cancer 2022, 22. [Google Scholar] [CrossRef]
- Murai, J.; Pommier, Y. BRCAness, Homologous Recombination Deficiencies, and Synthetic Lethality. Cancer Res. 2023, 83, 1173–1174. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic Lethality and Cancer. Nat. Rev. Genet. 2017 1810 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Mavroeidi, D.; Georganta, A.; Panagiotou, E.; Syrigos, K.; Souliotis, V.L. Targeting ATR Pathway in Solid Tumors: Evidence of Improving Therapeutic Outcomes. Int. J. Mol. Sci. 2024, Vol. 25, Page 2767 2024, 25, 2767. [Google Scholar] [CrossRef]
- Hopkins, J.L.; Lan, L.; Zou, L. DNA Repair Defects in Cancer and Therapeutic Opportunities. Genes Dev. 2022, 36, 278. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R. V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef]
- Smith, H.L.; Willmore, E.; Prendergast, L.; Curtin, N.J. ATR, CHK1 and WEE1 Inhibitors Cause Homologous Recombination Repair Deficiency to Induce Synthetic Lethality with PARP Inhibitors. Br. J. Cancer 2024, 131, 905–917. [Google Scholar] [CrossRef]
- Biegała, Ł.; Gajek, A.; Szymczak-Pajor, I.; Marczak, A.; Śliwińska, A.; Rogalska, A. Targeted Inhibition of the ATR/CHK1 Pathway Overcomes Resistance to Olaparib and Dysregulates DNA Damage Response Protein Expression in BRCA2MUT Ovarian Cancer Cells. Sci. Rep. 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Haciefendi, A.; Eskiler, G.G. The Suppression of ATR/Chk1 Pathway by Elimusertib ATR Inhibitor in Triple Negative Breast Cancer Cells. Am. J. Transl. Res. 2023, 15, 4902. [Google Scholar] [PubMed]
- Sofianidi, A.; Dumbrava, E.E.; Syrigos, K.N.; Nasrazadani, A. Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets. Cancers (Basel). 2024, 16, 1139. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia Telangiectasia and Rad3-Related Inhibitors and Cancer Therapy: Where We Stand. J. Hematol. Oncol. 2019, 12, 1–8. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR Inhibition Induces Synthetic Lethality and Overcomes Chemoresistance in TP53- or ATM-Defective Chronic Lymphocytic Leukemia Cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef]
- Wang, M.; Ran, X.; Leung, W.; Kawale, A.; Saxena, S.; Ouyang, J.; Patel, P.S.; Dong, Y.; Yin, T.; Shu, J.; et al. ATR Inhibition Induces Synthetic Lethality in Mismatch Repair-Deficient Cells and Augments Immunotherapy. Genes Dev. 2023, 37, 929–943. [Google Scholar] [CrossRef]
- Schneider, H.E.; Schmitt, L.M.; Job, A.; Lankat-Buttgereit, B.; Gress, T.; Buchholz, M.; Gallmeier, E. Synthetic Lethality between ATR and POLA1 Reveals a Potential New Target for Individualized Cancer Therapy. Neoplasia 2024, 57. [Google Scholar] [CrossRef]
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR Pathway Inhibition Is Synthetically Lethal in Cancer Cells with ERCC1 Deficiency. Cancer Res. 2014, 74, 2835–2845. [Google Scholar] [CrossRef]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A Synthetic Lethal Screen Reveals Enhanced Sensitivity to ATR Inhibitor Treatment in Mantle Cell Lymphoma with ATM Loss-of-Function. Mol. Cancer Res. 2015, 13, 120–129. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR Kinases Produces Tumor-Selective Synthetic Lethality and Suppresses Metastasis. J. Clin. Invest. 2019, 129, 1329–1344. [Google Scholar] [CrossRef]
- Chao, Y.; Chen, Y.; Zheng, W.; Demanelis, K.; Liu, Y.; Connelly, J.A.; Wang, H.; Li, S.; Wang, Q.J. Synthetic Lethal Combination of CHK1 and WEE1 Inhibition for Treatment of Castration-Resistant Prostate Cancer. Oncogene 2024, 43, 789–803. [Google Scholar] [CrossRef]
- León, T.E.; Rapoz-D’Silva, T.; Bertoli, C.; Rahman, S.; Magnussen, M.; Philip, B.; Farah, N.; Richardson, S.E.; Ahrabi, S.; Guerra-Assunção, J.A.; et al. EZH2-Deficient T-Cell Acute Lymphoblastic Leukemia Is Sensitized to CHK1 Inhibition through Enhanced Replication Stress. Cancer Discov. 2020, 10, 998–1017. [Google Scholar] [CrossRef]
- Chang, T.Y.; Yan, Y.; Yu, Z.Y.; Rathore, M.; Lee, N.Z.; Tseng, H.J.; Cheng, L.H.; Huang, W.J.; Zhang, W.; Chan, E.R.; et al. Combined HDAC8 and Checkpoint Kinase Inhibition Induces Tumor-Selective Synthetic Lethality in Preclinical Models. J. Clin. Invest. 2024, 134. [Google Scholar] [CrossRef]
- Chen, C.C.; Kass, E.M.; Yen, W.F.; Ludwig, T.; Moynahan, M.E.; Chaudhuri, J.; Jasin, M. ATM Loss Leads to Synthetic Lethality in BRCA1 BRCT Mutant Mice Associated with Exacerbated Defects in Homology-Directed Repair. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7665–7670. [Google Scholar] [CrossRef]
- Yao, Y.; Lv, H.; Zhang, M.; Li, Y.; Herman, J.G.; Brock, M. V.; Gao, A.; Wang, Q.; Fuks, F.; Zhang, L.; et al. Epigenetic Silencing of BEND4, a Novel DNA Damage Repair Gene, Is a Synthetic Lethal Marker for ATM Inhibitor in Pancreatic Cancer. Front. Med. 2024, 18, 721–734. [Google Scholar] [CrossRef]
- Oh, K.S.; Nam, A.R.; Bang, J.H.; Seo, H.R.; Kim, J.M.; Yoon, J.; Kim, T.Y.; Oh, D.Y. A Synthetic Lethal Strategy Using PARP and ATM Inhibition for Overcoming Trastuzumab Resistance in HER2-Positive Cancers. Oncogene 2022, 41, 3939–3952. [Google Scholar] [CrossRef]
- Ratz, L.; Brambillasca, C.; Bartke, L.; Huetzen, M.A.; Goergens, J.; Leidecker, O.; Jachimowicz, R.D.; van de Ven, M.; Proost, N.; Siteur, B.; et al. Combined Inhibition of EZH2 and ATM Is Synthetic Lethal in BRCA1-Deficient Breast Cancer. Breast Cancer Res. 2022, 24. [Google Scholar] [CrossRef]
- Shi, C.; Qin, K.; Lin, A.; Jiang, A.; Cheng, Q.; Liu, Z.; Zhang, J.; Luo, P. The Role of DNA Damage Repair (DDR) System in Response to Immune Checkpoint Inhibitor (ICI) Therapy. J. Exp. Clin. Cancer Res. 2022, 41. [Google Scholar] [CrossRef]
- Murray, C.E.; Kornepati, A.V.R.; Ontiveros, C.; Liao, Y.; de la Peña Avalos, B.; Rogers, C.M.; Liu, Z.; Deng, Y.; Bai, H.; Kari, S.; et al. Tumour-Intrinsic PDL1 Signals Regulate the Chk2 DNA Damage Response in Cancer Cells and Mediate Resistance to Chk1 Inhibitors. Mol. Cancer 2024, 23, 242. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Peng, G.; Yap, T.A. A Tale of Two Checkpoints: ATR Inhibition and PD-(L)1 Blockade. Annu. Rev. Med. 2022, 73, 231–250. [Google Scholar] [CrossRef]
- Hardaker, E.L.; Sanseviero, E.; Karmokar, A.; Taylor, D.; Milo, M.; Michaloglou, C.; Hughes, A.; Mai, M.; King, M.; Solanki, A.; et al. The ATR Inhibitor Ceralasertib Potentiates Cancer Checkpoint Immunotherapy by Regulating the Tumor Microenvironment. Nat. Commun. 2024, 15. [Google Scholar] [CrossRef]
- Taniguchi, H.; Chakraborty, S.; Takahashi, N.; Banerjee, A.; Caeser, R.; Zhan, Y.A.; Tischfield, S.E.; Chow, A.; Nguyen, E.M.; Villalonga, Á.Q.; et al. ATR Inhibition Activates Cancer Cell CGAS/STING-Interferon Signaling and Promotes Antitumor Immunity in Small-Cell Lung Cancer. Sci. Adv. 2024, 10, eado4618. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Huang, Y.; Xiao, Y.; Zhu, Z.; Shen, M.; Zhou, P.; Guo, Z.; Wang, J.; Wang, H.; Dai, W.; et al. ATR Inhibitor AZD6738 Enhances the Antitumor Activity of Radiotherapy and Immune Checkpoint Inhibitors by Potentiating the Tumor Immune Microenvironment in Hepatocellular Carcinoma. J. Immunother. cancer 2020, 8. [Google Scholar] [CrossRef]
- Li, C.; Wang, B.; Tu, J.; Liu, C.; Wang, Y.; Chen, J.; Huang, Y.; Liu, B.; Yuan, X. ATM Inhibition Enhance Immunotherapy by Activating STING Signaling and Augmenting MHC Class I. Cell Death Dis. 2024, 15. [Google Scholar] [CrossRef]
- Jin, W.J.; Zangl, L.M.; Hyun, M.; Massoud, E.; Schroeder, K.; Alexandridis, R.A.; Morris, Z.S. ATM Inhibition Augments Type I Interferon Response and Antitumor T-Cell Immunity When Combined with Radiation Therapy in Murine Tumor Models. J. Immunother. cancer 2023, 11. [Google Scholar] [CrossRef]
- Chen, J.L.Y.; Pan, C.K.; Lin, L.C.; Huang, Y. Sen; Huang, T.H.; Yang, S.J.; Kuo, S.H.; Lin, Y.L. Combination of Ataxia Telangiectasia and Rad3-Related Inhibition with Ablative Radiotherapy Remodels the Tumor Microenvironment and Enhances Immunotherapy Response in Lung Cancer. Cancer Immunol. Immunother. 2024, 74, 8. [Google Scholar] [CrossRef]
- Do, K.T.; Manuszak, C.; Thrash, E.; Giobbie-Hurder, A.; Hu, J.; Kelland, S.; Powers, A.; de Jonge, A.; Shapiro, G.I.; Severgnini, M. Immune Modulating Activity of the CHK1 Inhibitor Prexasertib and Anti-PD-L1 Antibody LY3300054 in Patients with High-Grade Serous Ovarian Cancer and Other Solid Tumors. Cancer Immunol. Immunother. 2021, 70, 2991–3000. [Google Scholar] [CrossRef]
- Sen, T.; Della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 Axis Promotes CGAS/STING Signaling in ARID1A-Deficient Tumors. J. Clin. Invest. 2020, 130, 5951–5966. [Google Scholar] [CrossRef]
- Concannon, K.; Morris, B.B.; Gay, C.M.; Byers, L.A. Combining Targeted DNA Repair Inhibition and Immune-Oncology Approaches for Enhanced Tumor Control. Mol. Cell 2023, 83, 660–680. [Google Scholar] [CrossRef]
Figure 1.
Scheme showing the correlation between different causes and types of DNA damage with its corresponding DNA damage response mechanism. Agents causing single strand breaks (SSBs) induce base mismatch, base damage or bulky lesions which are repaired by mismatch mediated repair (MMR), base excision repair (BER) or nucleotide excision repair (NER) respectively. On the contrary, ionizing radiation and chemotherapeutics can induce double-strand breaks (DSBs) or interstrand crosslinks which are finally repaired by non-homologous end joining (NHEJ) or homologous recombination (HR).
Figure 1.
Scheme showing the correlation between different causes and types of DNA damage with its corresponding DNA damage response mechanism. Agents causing single strand breaks (SSBs) induce base mismatch, base damage or bulky lesions which are repaired by mismatch mediated repair (MMR), base excision repair (BER) or nucleotide excision repair (NER) respectively. On the contrary, ionizing radiation and chemotherapeutics can induce double-strand breaks (DSBs) or interstrand crosslinks which are finally repaired by non-homologous end joining (NHEJ) or homologous recombination (HR).
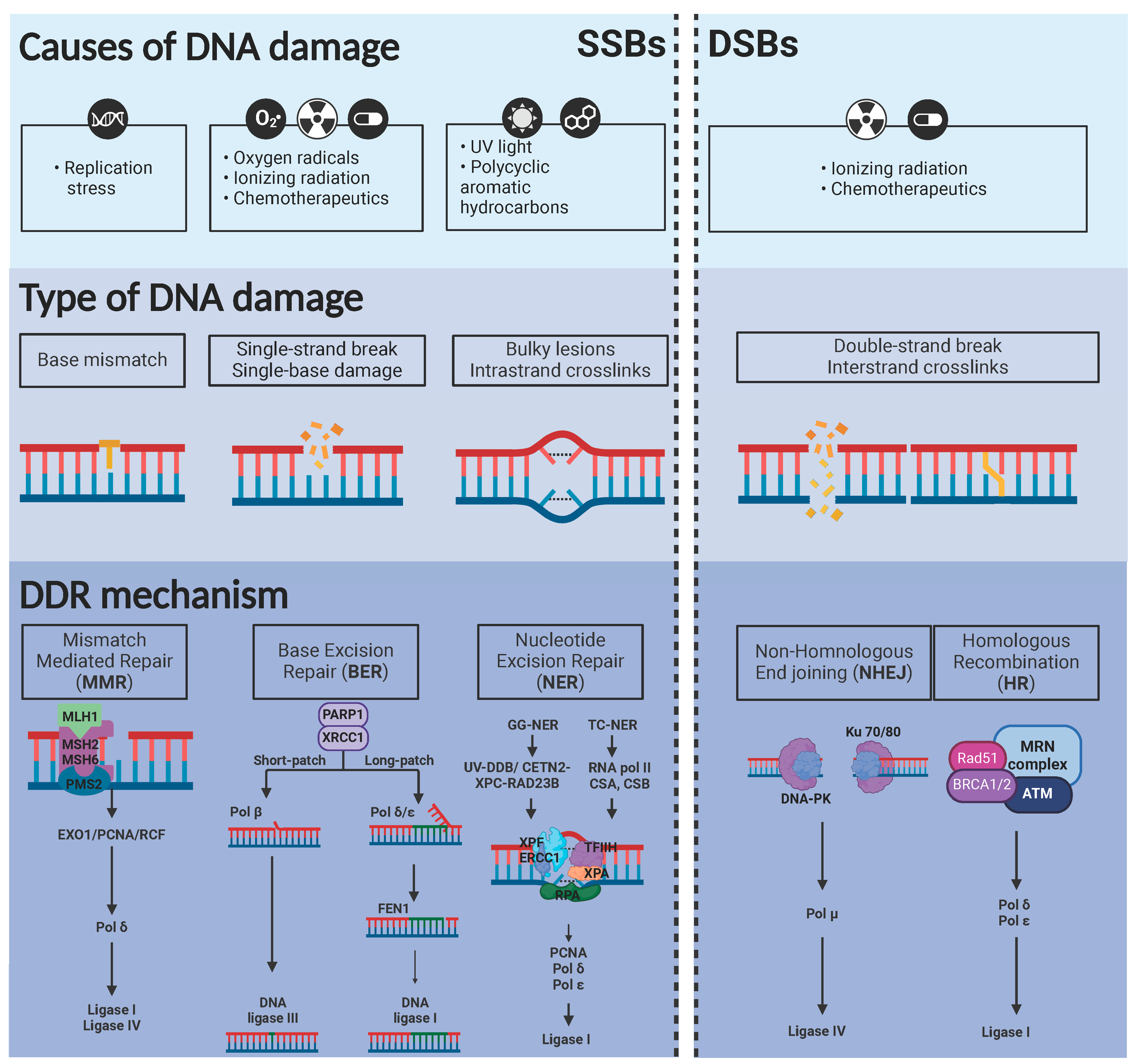
Figure 2.
Crosstalk between Chk1/ATR and Chk2/ATM. Replication stress and ionizing radiation can induce activation of Chk1/ATR and Chk2/ATM pathways, respectively, converging in cell cycle arrest. In addition, Chk1/ATR/Wee1 axis can recruit Rad51/BRCA1, triggering homologous recombination (HR).
Figure 2.
Crosstalk between Chk1/ATR and Chk2/ATM. Replication stress and ionizing radiation can induce activation of Chk1/ATR and Chk2/ATM pathways, respectively, converging in cell cycle arrest. In addition, Chk1/ATR/Wee1 axis can recruit Rad51/BRCA1, triggering homologous recombination (HR).
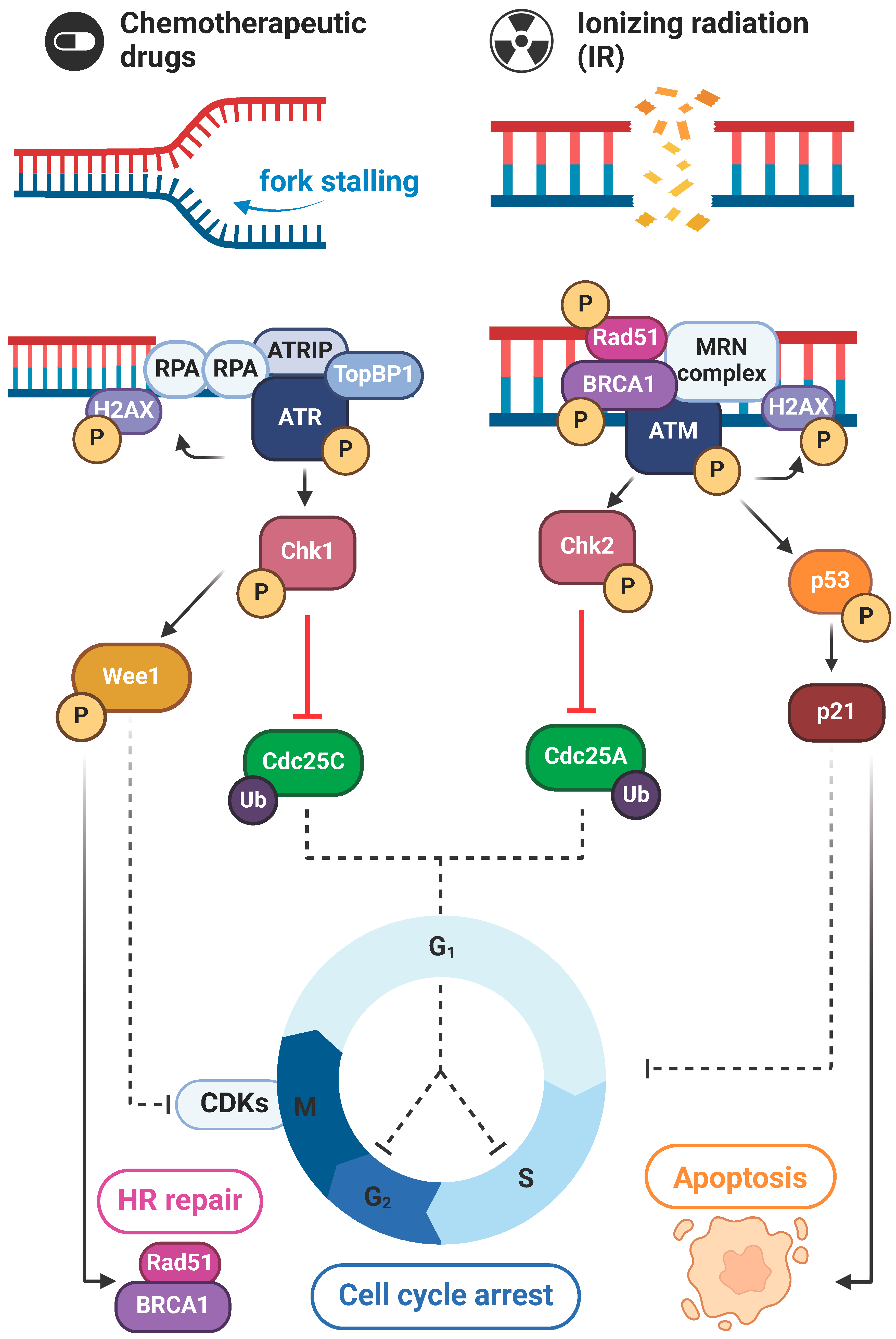
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



