
Submitted:
02 April 2025
Posted:
02 April 2025
You are already at the latest version
Abstract
Phosphorus is one of the most abundant minerals in the body and plays a pivotal role in
numerous cellular processes, including maintenance of skeletal health, integrity of phospholipid
bilayers, cell signaling, and synthesis of nucleic acid and adenosine triphosphate (ATP). About
85% of total phosphate is deposited in bone as hydroxyapatite crystals, 14% is present in soft
tissues as various organic phosphates, and the remaining 1% is found in extracellular space
mainly as inorganic phosphate. The plasma inorganic phosphorus concentration is tightly
maintained between 2.5 - 4.5 mg/dL by intertwined interactions between fibroblast growth factor
23 (FGF-23), parathyroid hormone (PTH) and vitamin D that tightly regulate the precise balance
in the phosphorus trafficking across the gastrointestinal tract, kidneys and bones. Disruption of
the tight hemostatic control of phosphorus balance can lead to altered cellular and organ
functions that are associated with high morbidity and mortality.
In the past three decades, there has been a steady increase in the prevalence of kidney failure
(KF) population needing various kidney replacement therapies. The individuals with KF have
unacceptably high mortality, and well over half of deaths are related to cardiovascular disease.
Abnormal phosphorus metabolism is one of the major factors that is independently associated
with vascular calcification and cardiovascular mortality in KF. While overt hyperphosphatemia is observed late during the progression of chronic kidney disease (CKD), a series of adaptive processes involving FGF-23, PTH and vitamin D occur in early stages of CKD attempting to enhance phosphate excretion and maintain plasma phosphorus level in the normal range. However, as CKD worsens, the ability to adequately eliminate phosphorus diminishes, eventually resulting in hyperphosphatemia, which is almost ubiquitous in individuals with KF. Notably, these hormonal imbalances and the associated adverse consequences are driven by the underlying hyperphosphatemic state in KF. Hence, it appears logical to strictly control serum phosphorus and its associated hormonal imbalance. Conventional dialysis is inadequate in removing phosphorus and most patients require dietary restrictions and pharmacologic interventions to manage hyperphosphatemia. Despite high phosphate content, plant-based diets are more efficient in controlling hyperphosphatemia as compared with animal-based diets. However, diet control comes with many challenges with adherence and may place patients at risk for inadequate protein intake and malnutrition that in turn may increase the risk of morbidity and mortality. Phosphate binders help to reduce phosphorus level but come with a sizable pill burden, have high financial cost, associated with poor adherence and psychosocial issues. Additionally, long-term use of binders may increase the risk of calcium, lanthanum or iron overload or promote gastrointestinal side effects that exacerbate malnutrition and affect quality of life. Given aforesaid challenges with phosphorus binders, novel therapies targeting small intestinal phosphate absorption pathways have been investigated. Recently, tenapanor, an agent that blocks paracellular absorption of phosphate via inhibition of enteric sodium-hydrogen-exchanger-3 (NHE3) was approved for the treatment of hyperphosphatemia in KF. While various clinical tools are now available to manage hyperphosphatemia, there is lack of convincing clinical data to demonstrate improvement in outcomes in KF with lowering of phosphorus level to near normal range. Conceivably, deleterious effects associated with hyperphosphatemia could be attributable to disruptions in phosphorus sensing mechanisms and hormonal imbalance thereof. Further exploration of mechanisms that precisely control phosphorus sensing and regulation may facilitate development of strategies to diminish the deleterious effects of phosphorus load and improve overall outcomes in KF.
Keywords:
hyperphosphatemia
; phosphorus
; kidney failure
; chronic kidney disease
; dietary management
Introduction
Phosphorus is one of the most abundant minerals in the body and plays a pivotal role in numerous cellular processes, including maintenance of skeletal health, integrity of phospholipid bilayers, cell signaling, and synthesis of nucleic acids and adenosine triphosphate (ATP). The total body store of phosphorus in adult approximates 700-800 g comprising about 1% to 1.4% of fat-free mass [1]. About 85% of total phosphate is deposited in bone as hydroxyapatite crystals, 14% is present in soft tissues as various organic phosphates, and the remaining 1% is found in extracellular space as inorganic phosphate [2,3]. Of the phosphorus in the extracellular space, about 15-20% of the inorganic phosphorus is bound to plasma proteins, and the rest is complexed with sodium, magnesium, and calcium or circulates in blood as monohydrogen (HPO42−) or dihydrogen forms (H2PO4−). The plasma inorganic phosphate concentration (commonly referred to plasma phosphorus concentration) is narrowly maintained between 2.5 - 4.5 mg/dL by intertwined interactions between fibroblast growth factor 23 (FGF-23), parathyroid hormone (PTH) and vitamin D that tightly regulate the precise balance in phosphate trafficking across the gastrointestinal tract, kidneys and bones. Disruption of the tight hemostatic control of phosphorus balance can lead to altered cellular and organ functions that are associated with high morbidity and mortality [3].
In the past three decades, there has been a steady increase in the prevalent population of kidney failure (KF) needing kidney replacement therapies [4]. The individuals with KF have unacceptably high mortality (145.6 deaths per 1000 person-years reported in 2022), and well over half (55.9%) of deaths are related to cardiovascular disease [4]. Abnormal phosphate metabolism is one of the major factors that is independently associated with vascular calcification and cardiovascular mortality in KF [3,5,6,7]. While overt hyperphosphatemia is observed late during the progression of chronic kidney disease (CKD), a series of adaptive processes involving FGF-23, PTH and vitamin D occur in the early stages of CKD attempting to enhance phosphate excretion and maintain plasma phosphorus level in the normal range [8]. Recognizing the mechanism of phosphate homeostasis is paramount to comprehend the consequences of hyperphosphatemia and the rationale for its management in KF.
Phosphate Homeostasis
Phosphate Balance in Health:
In the steady state, the amount of net intestinal uptake of dietary phosphorus equals the amount excreted by kidneys to maintain normal phosphorus balance [9,10,11] (Figure 1). Kidney and small intestine are the major organs involved in absorption, excretion and reabsorption of phosphorus. Additionally, the balance between phosphorus inflow and outflow amid the extracellular fluid and bone and soft tissue is a contributory factor in the maintenance of normal serum phosphate concentration.
A typical western diet contains about 1500 mg of phosphorus. Additionally, about 300 mg of phosphorus is exuded into the intestine via pancreatic and intestinal secretions. Approximately 1200 mg of ingested dietary phosphate is absorbed in the proximal intestine giving a net daily phosphorus absorption of approximately 900 mg. Approximately 600 mg of phosphorus that is not absorbed in the intestine or released into the intestinal lumen eventually appears in the feces. Absorbed phosphorus enters the extracellular fluid pool and about 300 mg of it moves in and out mainly of bone and to a lesser extent of soft tissues maintaining a neutral balance. Approximately 900 mg of phosphorus, which is equivalent to the net amount absorbed from the intestine, is excreted in the urine (Figure 1) [12].
Intestinal Phosphorus Handling:
Dietary phosphorus absorption occurs in the proximal small intestine, mainly jejunum via two distinct pathways: the saturable transcellular pathway and the non-saturable paracellular pathway [11,13,14,15,16]. Transcellular pathway is the active intestinal phosphate transport involving sodium-dependent phosphate cotransporter IIb (NaPi-IIb, Npt2b), that is regulated by many factors including dietary phosphorus intake and 1,25 dihydroxy vitamin D [17,18]. As a typical modern diet is abundant in phosphorus, saturable transcellular pathway is quickly overwhelmed and does not remain the primary modality of intestinal phosphate absorption. Consequently, paracellular pathway, which lacks the saturation limit, becomes the major route of phosphorus absorption in humans [19,20,21]. The paracellular phosphate absorption, driven by the phosphate concentration gradient, occurs passively through tight junction complexes claudins and occludins [15,21,22,23]. The permeability of phosphate through the paracellular pathway is influenced by the sodium-hydrogen exchanger 3 (NHE3), which is highly expressed in the small intestine [23,24]. Indeed, inhibition of enterocyte NHE3 by tenapanor has demonstrated reduced intestinal phosphorus absorption resulting in reduction of serum phosphorus level in patients with KF [23,25,26].
Renal Phosphate Handling
Kidneys are the primary organs responsible for phosphorus clearance and can excrete up to 4,000 mg of phosphorus in a day. Following glomerular filtration, 80-85% of the filtered phosphate is reabsorbed in the proximal tubule (PT). Consequently, less than 15-20% of filtered phosphate is typically excreted in the urine in healthy individuals [27]. Renal phosphate absorption is facilitated by three sodium phosphorus cotransporters namely sodium-phosphate co-transporter type IIa (NaPi-IIa, Npt2a), IIc (NaPi-IIc, Npt2c) and sodium-potassium co-transporter type III (Pit-2, Ram-1), all situated in the apical brush border membrane (BBM) of the PT [28]. These channels are regulated predominantly by FGF-23 and PTH.
FGF-23 is a 32 kDa glycoprotein, belonging to the endocrine FGF family, mainly produced by osteoblasts and osteocytes [29,30]. Circulating FGF-23 requires the co-receptor αKlotho to substantially enhance the binding affinity to FGF receptor 1c (FGFR1c). Binding of FGF-23 to FGFR1c leads to downstream signaling pathways triggering internalization and degradation of NaPi-IIa and NaPi-IIc from the apical membrane, promoting increased urinary phosphate excretion. Furthermore, FGF-23 inhibits expression of 1α-hydroxylase (CYP27B1), the rate-limiting enzyme for vitamin D hormone (1,25(OH)2D3) synthesis. Both the phosphaturic and the 1,25(OH)2D3-lowering effect of FGF-23 protect against hyperphosphatemia by increasing urinary elimination of phosphate and reducing intestinal phosphate absorption respectively [29,30] (Figure 2). In addition to FGF-23, PTH is a key phosphaturic hormone. PTH binds to PTHR1 receptor in PT resulting in removal of NaPi-IIa and NaPi-IIc from the apical BBM via clathrin-coated pits [31,32]. However, unlike FGF-23, PTH increases transcriptional activation of 1α-hydroxylase (CYP27B1), thereby promoting 1α,25(OH)2D3) synthesis [30] (Figure 2).
Endocrine Regulation of Phosphate Homeostasis in Health and Kidney Disease
In normal individuals, a dietary phosphate load triggers release of FGF-23 from bones. A recent study showed that PT glycolysis acts as a phosphate sensor in the kidney. In renal PT cells, phosphorus and not glucose is the rate-limiting step for glycolysis. Consequently, following dietary phosphorus load, renal phosphorus filtration and consequent absorption in PT promotes glycolysis, thereby increasing production of glycerol-3-phosphate (G-3-P). Subsequently, via a yet unknown mechanism, G-3-P is secreted into the circulation where it stimulates FGF-23 production in bone. FGF-23, in turn, downregulates RT NaPi-IIa and NaPi-IIc, thereby reducing phosphate uptake, glycolytic flux, and thus, further G-3-P production and so completes the negative feedback loop [33]. Additionally, by reducing 1,25(OH)2D levels, FGF-23 indirectly reduces intestinal phosphate absorption by downregulation of NaPi-IIb. Moreover, reduction of 1,25(OH)2D increases PTH release that exerts its phosphaturic action by promoting recycling of NaPi-IIa and NaPi-IIc from apical BBM in PT [29,30,31].
In the early stages of CKD, rising FGF-23 level following dietary phosphate load is still able to maintain normal serum phosphorus level by enhancing fractional excretion of phosphorus (Figure 2). Moreover, FGF-23 mediated reduction 1,25(OH)2D levels leads to secondary hyperparathyroidism, and the rise in PTH further augments renal phosphate excretion. However, as CKD advances, overt hyperphosphatemia ensues, as aforementioned adaptive effects of FGF-23, PTH and 1,25(OH)2D to enhance phosphate excretion are not able to overcome the phosphorus retention from continued dietary phosphorus intake and progressive reduction in glomerular filtration rate (GFR). In addition, other factors such as continued phosphate reabsorption by PT, decreased phosphate uptake into bone due to low 1,25(OH)2D levels, and ongoing phosphorus release from bone by elevated PTH further exacerbate hyperphosphatemia, thus creating a vicious cycle (Figure 2).
Complications and Outcomes Associated with Hyperphosphatemia
Hyperphosphatemia is a common complication of advanced kidney disease [34]. While phosphate level remains in the normal range in early stages of CKD, disturbances in phosphate metabolism occur early, triggering adaptive mechanisms to increase fractional excretion of urinary phosphate and reduce intestinal phosphate absorption. As dietary intake overwhelms excretory capacity of kidneys with progressive CKD, hyperphosphatemia ensues [35]. Hyperphosphatemia in CKD and KF has been associated with high risk of cardiovascular disease (CVD), metabolic bone disease (MBD), and high cardiovascular (CV) and overall mortality [3,5,6,7,36,37,38]. Furthermore, hyperphosphatemia is associated with faster progression of CKD in non-dialysis CKD patients [39].
Hyperphosphatemia: Cardiovascular Risks and Mortality
Patients with KF have unacceptably high mortality, and CVD accounts for more than half of the deaths in these patients [4,40]. Furthermore, mortality from CVD in KF patients is about 20 times higher than that in the general population [41]. Numerous studies have shown a strong association between hyperphosphatemia and CVD in CKD. Mechanistically, hyperphosphatemia is linked to an increased risk for CVD through multiple physiologic mechanisms. First, high phosphate concentrations and calcium phosphate products may increase vascular and soft tissue calcification [42,43]. While to some extent there is passive precipitation of calcium-phosphate in soft tissues, high extracellular phosphate induces expression of osteoblastic genes in vascular smooth muscle cells (VSMC) causing transformation of VSMC into osteoblast-like cells fostering vascular calcification [44]. Moreover, hyperphosphatemia triggers remodeling of the extracellular matrix around VSMC promoting calcification in the medial layer of the vasculature [45,46,47]. In addition, phosphate exposure activates pro-inflammatory cellular signaling in VSMC thereby initiating oxidative stress causing DNA damage that can lead to more inflammation and vascular calcification [48,49].
Secondly, phosphate retention raises FGF-23 and PTH concentrations. Both FGF-23 and PTH have been independently associated with direct pathogenic CV effects [50,51]. Increased FGF-23 levels have been demonstrated to enhance pro-inflammatory cytokines, which further worsens vascular calcification. Clinical and experimental studies have observed FGF-23 to be an independent risk factor for hypertension, left ventricular hypertrophy, congestive heart failure and CV mortality [50,52,53,54,55]. Likewise, excess PTH is associated with proinflammatory effects, hypertension, impaired myocardial energy production, cardiac fibrosis, left ventricular hypertrophy, and heart failure [56,57,58,59,60,61].
Hyperphosphatemia and Risk of Mortality and Progression of Renal Disease:
Various epidemiological studies have observed strong association between high phosphorus levels and morbidity and mortality in CKD and KF patients [36]. A 2011 meta-analysis reported an 18% increased risk of death for every increase of 1 mg/dl in serum phosphate in CKD patients [38]. In another prospective cohort study, a serum phosphate level ≥3.5 mg/dL was associated with significantly increased mortality risk among CKD patients [62]. Furthermore, compared to CKD patients with normal phosphorus level, the risk of death increased linearly with each subsequent 0.5 mg/dL increase in serum phosphate level and nearly doubled in patients with a serum phosphate level ≥4.5 mg/dL [62]. Yet, another study observed a 1.62 folds increase in mortality risk in CKD patients for every 1 mg/dl increase in serum phosphate [63]. Even in individuals with preserved kidney function, higher serum phosphate is associated with high mortality. In a post-hoc analysis of the data from the Cholesterol And Recurrent Events (CARE) study, a graded independent relation between higher serum phosphate and the risk of death and cardiovascular events was observed [64]. Similarly, in a meta-analysis of 24 clinical trials in patients without CKD, serum phosphate was associated with higher mortality [65].
In addition to risk of mortality, higher serum phosphate concentrations are associated with a faster progression of CKD [66]. In experimental studies, phosphorus loading leads to kidney fibrosis in normal animals, and promotes progression of kidney disease in CKD animals [67,68]. Likewise, in individuals without kidney disease, the risk of KF was significantly higher in patients with highest quartile phosphate compared to those with the lowest quartile, suggesting that relatively high phosphate levels within the normal range can be a risk factor for development of CKD [69]. Among patients with CKD, those with higher serum phosphorus level had faster progression to KF than the persons with lower phosphate level [70]. Furthermore, a meta-analysis of 12 cohort studies observed a 1.36 risk of KF and 1.2 fold increase in mortality for every 1-mg/dL increase in serum phosphate level in CKD patients [71].
Hyperphosphatemia and Metabolic Bone Disease:
High phosphate is closely associated with metabolic bone disease (MBD), a widespread complication of CKD and KF. Even before development of overt hyperphosphatemia, phosphorus dietary phosphorus load in CKD leads to increase in FGF-23 and PTH, key factors driving development of CKD-MBD [72,73]. High PTH causes further phosphate release from bone, worsening overall phosphorus balance and stimulating additional FGF23 and PTH production, leading to development of high turnover bone disease, with high risk of musculoskeletal pain and bone fractures [72,73]. Indeed, observational data noted a significant increase in bone fracture risk in hemodialysis patients with a baseline serum phosphate greater than 6.1 mg/dL [73]. Likewise, data from the Dialysis Outcomes and Practice Patterns Study (DOPPS) showed higher frequency of femoral neck fracture in KF patients than in the general population [74]. Similarly, Fusaro, et al. observed a significant risk of fracture with elevated phosphate level in CKD patients [75]. Direct role of phosphate in the pathogenesis of MBD was demonstrated in experimental data, which observed that inorganic phosphate induces apoptosis of cultured osteoblast-like cells and inhibits RANK–RANKL signaling-mediated cell differentiation of cultured osteoclast- like cells [76,77].
Management of Hyperphosphatemia
Many studies have underscored the association of hyperphosphatemia with increased risk of CVD), MBD and mortality [3,5,6,7,36,37,38]. Furthermore, despite known association of hyperphosphatemia and poor outcomes, there has been a steady rise in mean serum phosphate concentrations amongst KF patients in the United States [36,78]. Hence, it appears logical to control serum phosphate and its associated hormonal perturbations in individuals with CKD and KF. The most recent Kidney Disease Improving Global Outcome (KDIGO) guidelines recommend lowering elevated phosphate levels toward the normal range [79]. Current management strategies include removal of phosphate by dialysis, reduction in dietary phosphate intake and reducing intestinal absorption of phosphate.
Removal of Phosphate by Dialysis:
As discussed in an earlier section, hyperphosphatemia and its metabolic consequences in KF are initiated by inability of kidneys to adequately excrete phosphate. Hence dialysis, as kidney replacement therapy, is utilized as a tool to remove phosphate from blood with a hope to mitigate hyperphosphatemia related adverse outcomes. However, both traditional in-center HD and peritoneal dialysis (PD) are grossly inadequate for phosphate removal (Table 1).
As most phosphorus is stored in bone or intracellular space, only 1% of total body phosphorus is available in extracellular space and accessible for removal by traditional HD. Consequently, in traditional three times a week dialysis, phosphate is mainly removed from extracellular fluid and with minimal removal from intracellular or tissue pools [80]. In an earlier study, DeSoi and Umans found a rapid early phosphate removal with nadir of serum phosphate around 2 hours followed by a rapid rebound to 88–100% of their pre-dialysis serum phosphate within 4 hours post-dialysis [81]. Similar results were observed in other studies that revealed a rapid decline in serum phosphate during early phase of HD, followed by plateauing of serum phosphate in the final 2-4 hours of the treatment [82,83,84,85,86]. In the ensuing post HD period, phosphate fluxes from intracellular compartments and bones back to extracellular compartment thereby equilibrating serum phosphate level [82,83,84,85,86]. All the aforesaid studies did not control for dietary measures or use of phosphorus binders. Interestingly, in a recent study comprising 13 HD subjects, who had been off phosphate binders for 10 days and consumed a standardized low phosphate (900 mg/day) diet for 3 weeks prior to the assessments, there was an average drop in serum phosphate of −2.85 mg/dL post-dialysis. Furthermore, ‘rebound’ or return to baseline levels occurred slowly during the 24 to 48 hours, suggesting that the low dietary intake prolonged the return of serum phosphate level to baseline in post dialysis period [87].
While standard HD is inadequate in effective phosphorus removal, other HD modalities may be more efficient in the management of hyperphosphatemia. For instance, compared to standard HD, more frequent short daily HD or nocturnal HD are shown to be more effective in serum phosphate management, likely due to rate of phosphate moving from different physiologic compartments and continued removal of phosphate from ultrafiltration [88]. Indeed, convective clearance is more effective in phosphate removal, as shown by Minutolo and colleagues in a single blind cross over study of 12 patients undergoing two different dialysis treatments. One dialytic treatment utilized both diffusive and convective fluid fluctuations using post-dilution reinfusions of bicarbonate at high rates of ultrafiltration in one of the three treatments per week. A standard bicarbonate hemodialysis treatment was performed as the other study treatment. They observed higher removal of inorganic phosphate in patients undergoing the combined diffusive and convective hemodialysis treatment compared to those undergoing standard HD alone [89].
Compared to HD, phosphate removal in peritoneal dialysis (PD) is more complex. Unlike HD, where the dialysis membranes have well defined and relatively uniform characteristics, the peritoneal membranes vary among different patients and in the same individual over dialysis vintage [90]. Hence, the transfer rate of solutes including phosphate vary among different individuals and depend upon peritoneal membrane permeability and dialysis prescription (number of exchanges, dwell time, dialysate composition and ultrafiltration) [91]. Moreover, compared to other small solutes of similar molecular weight, phosphate peritoneal clearance is lower [92] underscoring the important role of peritoneal solute transfer characteristics and PD prescriptions in influencing phosphate removal and, therefore, serum phosphate control. In a prospective observational study including 380 adult peritoneal dialysis patients, Courivaud C. et al. [91] observed that slower peritoneal transporter status was associated with reduced weekly peritoneal phosphate clearance and consequently higher serum phosphate levels.
Additionally, irrespective of peritoneal phosphate transfer rate, patients receiving continuous ambulatory peritoneal dialysis (CAPD) with long dwell time showed a greater peritoneal phosphate clearance compared to patients treated with automated peritoneal dialysis (APD) with shorter dwell time. Accordingly, peritoneal phosphate removal could be enhanced by increasing dialysate volume and dwell time in CAPD. In APD, phosphate clearance can be increased by increasing volume, number, and duration of dialysis cycles as well as by addition of longer daytime exchanges. Similarly, in an observational cross-sectional study, Debowska M. et al. [93] assessed phosphate clearance by CAPD, continuous cyclic peritoneal dialysis (CCPD), and APD. Patients treated with CAPD showed a greater total weekly phosphate removal compared to CCPD and APD as well as lower serum phosphate level compared to APD patients. In a similar fashion, a recent multicenter prospective cohort study conducted on 737 patients, transition from CAPD to APD was followed by an increase in serum phosphate whereas the opposite occurred after switching from APD to CAPD underscoring the importance of long dwell time in the management of serum phosphate levels [94] Therefore, notwithstanding overall inadequate phosphorus removal by PD, individualized PD prescriptions are important taking into account the peritoneal phosphate transfer rates, especially in slow transporters and particularly those with low residual renal function undergoing APD [91].
Dietary Management of Hyperphosphatemia:
Reduction in kidney function promotes hyperphosphatemia due to impaired phosphate excretion in the setting of ongoing dietary phosphate intake. Hyperphosphatemia is independently associated with dire consequences including bone and mineral disease, high risk of CV disease, and mortality [95,96,97]. Hence, it is imperative to address the dietary phosphorus intake to minimize these complications associated with hyperphosphatemia and increase a better outcome in patients with CKD and KF [98].
The typical daily phosphate intake in a Western diet is about 1500 mg, of which about 1200 mg is absorbed in the gastrointestinal tract depending upon the bioavailability of phosphate in food (Figure 1) [95]. Based upon the current guidelines, patients with KF are typically recommended a daily phosphorus intake of 900 mg/day [99]. Furthermore, the guidelines highly emphasize considering the source of phosphorus in the dietary recommendation for hyperphosphatemia [99]. Traditional approaches for dietary phosphorus restriction can compromise both nutritional adequacy and quality of life. The challenge lies in creating a diet that is both nutritionally adequate, palatable, cost effective and promotes adherence with minimal impact on quality of life (QOL). Hence, understanding the different dietary forms of phosphorus and their bioavailability is essential for dietary planning in KF patients.
Forms of Phosphorus in the Diet: Organic and Inorganic
Phosphorus in the diet exists primarily in two forms: organic and inorganic. Organic phosphorus is predominantly found in plant- and animal-based food sources and is bound to proteins, lipids, or other organic molecules. Phosphorus from animal sources that is mainly present as phosphoproteins and phospholipids, is more bioavailable than plant-derived phosphorus [96]. In plants, phosphorus is primarily stored as phytate (inositol hexaphosphate), which is poorly absorbed by humans due to the absence of the enzyme phytase, which is required for its breakdown [96,97]. In contrast, inorganic phosphorus is commonly found in food additives, preservatives, supplements, and pharmaceutical agents and exists as free phosphate ions (Table 2) [97].
The distinction between these forms is crucial because their bioavailability, or the percentage absorbed in the gastrointestinal tract, varies significantly. Inorganic phosphorus is more readily absorbed than organic phosphorus, which has important implications for phosphorus balance, particularly in individuals with CKD and KF [96]. Specifically, inorganic phosphorus has a bioavailability of 80-100%, while organic phosphorus is absorbed at rates of 30-60% from animal sources and 20-40% from plant sources (Table 2) [100].
Intervention
In clinical practice, dietary recalls and food diaries are typically utilized to estimate dietary phosphate intake in patients with CKD and KF. However, considerable amount of hidden phosphorus is present in preservatives, additives, and pharmaceutical preparations that is not often recognized by individuals and healthcare providers (Table 2) [101,102]. These hidden sources can contribute a considerable portion of daily phosphorus intake (up to 500-1000 mg), [102]. Moreover, inorganic phosphates are nearly 100% absorbed in the gastrointestinal tract, making them a major contributor to hyperphosphatemia.
The American food industry plays a major role in exacerbating phosphorus-related issues in CKD patients due to its heavy reliance on processed foods and additives. Unlike many other countries, the U.S. diet is dominated by ultra-processed foods, fast food, and convenience meals, all of which contain high levels of inorganic phosphorus from preservatives and additives such as sodium phosphate [103]. In contrast, many other countries emphasize whole, minimally processed foods, which contain primarily organic phosphorus from plant and animal sources, with considerably lower absorption rates. Moreover, one of the challenges in tracking phosphorus intake is the lack of transparency in food labeling. The FDA does not require phosphorus content to be listed on nutrition labels, making it nearly impossible for patients and dietitians to accurately track intake [104]. Even when food labels list the phosphorus content, they fail to account for the amount and bioavailability. As a result, tracking phosphorus intake becomes even more complicated, potentially leading to gross underestimation of actual intake.
Furthermore, Inorganic phosphate is added to medications and nutrition supplements and is another, generally unrecognized, source of phosphate exposure. In a Canadian hemodialysis population, 11% of prescribed medication contained a phosphate salt with a median phosphate burden of 111 mg per day [105]. With a median daily pill burden of 19, prescription medications can substantially contribute to the daily phosphate load in dialysis patients [106]. Moreover, phosphate is usually added to multivitamin supplements with estimations between 20 and 150 mg per supplement. The use of a multivitamin supplement will further contribute to a higher dietary phosphate load.
A diet that reduces phosphorus intake while maintaining nutritional balance is imperative (Table 1). The source of phosphorus, as well as its bioavailability, is important to consider when incorporating dietary changes. As discussed above, the bioavailability of phosphorus in plant-based foods is much lower than that in animal-based foods. Thus, the use of plant-based foods may help to reduce phosphorus levels more effectively while promoting nutritional adequacy [98]. In an experimental study with rats, it was shown that a plant-based diet significantly lowered phosphorus levels compared to an animal-based diet [107]. Likewise, in a study in human subjects with CKD stages 3-4, those individuals who followed a vegetarian diet exhibited lower serum phosphate levels, reduced serum FGF-23 levels, and less urinary phosphate excretion compared to those on a meat-based diet containing the same amount of phosphate [108]. Even a partial replacement of animal-based with plant-based significantly lowered serum phosphorus levels in patients with CKD [109]. Furthermore, data from both prospective and retrospective cohort studies indicates that people following a plant-based diet, including vegetarians, had significantly reduced serum phosphate levels in cases of kidney failure [110,111].
The critical role of plant-based diet in CKD and KF management has been underscored by recent international guidelines. The International Society of Renal Nutrition and Metabolism (ISRNM) recommends incorporation of plant-based protein sources into the dietary plans of CKD/KF patients while considering individual preferences and cultural dietary habits [112]. Likewise, the 2024 KDIGO Clinical Practice Guideline for the Evaluation and Management of CKD emphasizes a balanced, diverse diet, prioritizing plant-based foods while limiting animal proteins and processed foods [113]. Hence, a well-planned diet should minimize processed foods and additives and emphasize plant-based sources. This can not only help control phosphorus load but also addresses comorbidities such as diabetes, hypertension and metabolic acidosis, which are prevalent among CKD and KF patients [98,114,115]. Additionally, low-protein diets can slow kidney function and reduce the risk of kidney failure in non-dialysis CKD patients [116,117].
One of the concerns of strict dietary phosphate restriction is the risk of malnutrition. In contemporary practice, patients with KF are often discouraged from consuming plant-based proteins and instead focus on animal-based proteins. This recommendation is based on the misperception that plant-based protein sources have a high phosphorus content and can lead to hyperphosphatemia. In reality, the bioavailability of phosphorus is not appropriately acknowledged. While animal-based foods are dense in protein, the phosphorus content is highly bioavailable. Whereas plant-based foods have a high phosphorus content but low bioavailability. While both types of proteins contain phosphorus, a considerably smaller amount is absorbed from plant-based foods. The aforementioned studies have shown that plant-based proteins actually help improve phosphorus load [98,107,108,109,110,111]. Furthermore, a plant-based diet in patients with kidney failure is associated with improved nutritional status [118]. Moreover, a plant-based diet is richer in micronutrients such as vitamins and minerals, and it is also linked to better-controlled metabolic acidosis. In conclusion, consuming a plant-based diet is much more effective in controlling hyperphosphatemia than consuming an animal-based diet.
Dietary modifications for managing kidney disease can be both costly and challenging for patients to maintain [119]. These specialized diets often require careful planning and can be more expensive than standard dietary options, potentially leading to financial strain [119]. In contrast, processed foods are typically more affordable and convenient, aligning with the fast-paced nature of American lifestyles. However, it is important to recognize that while processed foods may offer short-term savings, they can contribute to long-term health issues, potentially increasing healthcare costs over time [120]. Plant-based diets, such as vegan or vegetarian options can be more cost-effective in the long-run. For instance, a recent modelling study comparing food prices from the international comparison program for 150 countries showed that healthy and sustainable dietary patterns were up to 25-29% lower in cost [121]. Variants of vegetarian and vegan dietary patterns were generally more affordable and vegan diets reduced food costs by up to one-third compared to meat and dairy products [122]. Moreover, a low-fat vegan diet has been associated with a 16% decrease in total food costs, offering potential economic benefits alongside health improvements [123]. However, it is crucial to note that the affordability of plant-based diets can vary based on geographic location, availability of fresh produce, and individual dietary needs [124]. In some cases, specialized plant-based products may be more expensive, offsetting potential savings [124]. In summary, while dietary modifications for health conditions can be costly and require substantial adherence, plant-based diets may offer a balance between health benefits and cost savings [125,126]. Furthermore, dietary counseling can focus on other simple food preparation techniques that can be utilized to lower dietary phosphate content. For instance, boiling reduces phosphate content due to the demineralization of food, as the minerals move from the food into the boiling water [127]. Reported phosphate reductions by boiling vary between 35% and 50% [128].
Medical nutrition therapy (MNT) comprising of dietary education and counseling by a registered dietitian (RD) are crucial in the management of hyperphosphatemia [129]. Indeed, the 2020 KDOQI Clinical Practice Guideline for Nutrition in CKD update recommends MNC as an important strategy to optimize nutritional status, and to minimize risks associated with comorbidities and metabolic derangements in patients with CKD and KF [130]. Likewise, KDIGO underscores the role of renal dietitians in formulating dietary modifications according to disease severity and comorbidities, addressing sodium, phosphorus, potassium, and protein intake [113]. Key components of effective dietary management include careful planning, regular assessment of nutritional status, and monitoring adherence. CKD patients often face comorbidities requiring specific dietary management, which can be overwhelming [131]. The dietitian’s role goes beyond advice, providing individualized, holistic counseling based on the patient’s health, preferences, and pre-existing conditions. Effective education, motivation, and alternative food options tailored to the patient’s preferences are essential for adherence [132]. It is important for patients to be active participants in the management of their dietary modifications and the dietary advice should be based upon a shared decision making between the patient and their care team.
In conclusion, dietary phosphate restriction in CKD patients requires a rational approach rather than an indiscriminate prescription of reduced dietary protein intake. The nature of dietary phosphate, including hidden sources, should be carefully examined and avoided (Table 1). Appropriate dietary counseling and educational programs involving self-care and shared decision making are vital. Encouraging patients to reduce meat consumption and to shift to a grain-based vegetarian diet may allow sufficient protein intake without adversely affecting serum phosphate. Furthermore, it must be stressed that dietary phosphate restriction is difficult to accept and usually insufficient to achieve adequate control of serum phosphate. Therefore, other strategies such as pharmacological measures, and phosphate removal by dialysis in patients with KF should be discussed when deemed necessary.
Reducing Intestinal Phosphate Absorption:
While dietary restriction and dialysis are able to reduce phosphorus level to certain extent, most individuals with KF need additional pharmaceutical interventions targeting small intestinal phosphate absorption for the management of hyperphosphatemia. The current pharmacological approaches include use of phosphate binders and intestinal phosphate transport inhibitors (Table 1).
Phosphate binders: More than 80% of patients with KF take one or combination of phosphate binders [133]. These agents bind with dietary phosphates to form insoluble complexes in the intestinal lumen and excreted in the feces [134,135,136,137,138]. Phosphate binders can be calcium or non-calcium based. Calcium-based binders in common use include calcium carbonate and calcium acetate. Non–calcium-based binders include now-defunct aluminum-based binders, resin-based binders (sevelamer), and metal based binders (lanthanum carbonate, sucroferric oxyhydroxide, and ferric citrate).
Aluminum is an avid phosphate binder and was the pillar of hyperphosphatemia management in the early years. However, its use has been largely abandoned after realization of systemic toxicity due to its accumulation in brain, bone and bone marrow causing encephalopathy, dementia, osteomalacia and anemia respectively [139,140,141]
Calcium-based Binders: After retraction of aluminum use, calcium- based binders became the predominant agents for hyperphosphatemia management in KF [142,143]. The avidity to bind phosphate per gram of calcium and capacity to lower serum phosphate is similar between calcium carbonate and calcium acetate [143,144,145]. While there was no evidence of a difference in the risk of hypercalcemia between the two agents, concern for risks of hypercalcemia and vascular calcification with the use of calcium-based binders remains a matter of concern. Whereas studies comparing outcomes of calcium-based binders with non-calcium binders, mainly sevelamer, have yielded mixed results, the overall findings from these studies seemed to show either a potential for benefit or an absence of harm associated with calcium-free phosphate-binding agents compared with calcium-based agents [145,146,147,148,149,150,151,152]. Based upon these studies, KDIGO-2017 guidelines recommend restricting the use of calcium-based binders in the management of hyperphosphatemia in KF [79]. Aside from hypercalcemia, the major adverse events associated with calcium-based agents are gastrointestinal symptoms, which are similar between calcium carbonate and calcium acetate, and less frequent than that associated with sevelamer [145,146,147].
Resin based Binders: Sevelamer-based binders are the original non–calcium-based phosphate binders and currently the most often used phosphate binder in clinical practice [153,154,155,156,157]. Sevelamer is a cationic polymeric ion exchange resin that binds to dietary phosphate without being degraded or absorbed, thereby suppressing intestinal phosphate absorption. [158]. It was first released as sevelamer hydrochloride but given concerns about metabolic acidosis due to the hydrochloride moiety, the formulation was later changed to sevelamer carbonate, which appears to have a similar effect on phosphate lowering [159,160]. While sevelamer is effective in reducing blood phosphorus levels, high pill burden, and gastrointestinal side effects may lead to non-adherence, and consequent poor control of serum phosphate level [161]. Over 25% of individuals experience adverse effects, including constipation, abdominal discomfort, nausea, and dyspepsia [162,163]. Rarely, sevelamer can cause lower gastrointestinal bleeding due to deposition of sevelamer crystals in colonic mucosa and ensuing mucosal damage [164,165].
In addition to chelating phosphate, sevelamer binds bile salts, resulting in a significant reduction in serum total cholesterol and low-density lipoprotein cholesterol, but may concurrently interfere with the absorption of the fat-soluble vitamins A, D, E, and K, and other nutrients [166,167,168].
- Metal-Based Phosphate Binders:
Lanthanum carbonate, a chewable, calcium free metal cation that has minimal intestinal absorption, was approved in 2004 for phosphate chelation. While its efficacy of phosphate binding is similar to those of other binders, one advantage of lanthanum carbonate is lower pill burden compared with those of previous phosphate binders [169,170,171]. In a multicentered randomized controlled trial in non-dialysis CKD patients, lanthanum was observed to be effective in reducing phosphate level [172]. Likewise, in another study randomizing HD patients to calcium carbonate and lanthanum groups, phosphate levels fell similarly between the two groups, while hypercalcemia was limited to calcium carbonate groups [173]. Key adverse effects of lanthanum reported in a systematic review included vomiting, diarrhea, intradialytic hypotension, cramps, myalgia, and abdominal pain [174].
Based on the experience with aluminum-based preparations, buildup of heavy metals in the body is a cause of concern. In fact, accumulation of lanthanum in the liver and many other organs has been reported in uremic rats [175]. While lanthanum is present in the lysosomes of hepatocytes, there are no reports of cells or tissue damage in the liver and no increase in the incidence of adverse events associated with any organ function including liver after up to 6 years of treatment in KF patients [176]. Moreover, Lanthanum also accumulates in the bones of chronic dialysis patients, with a 50- to 80-fold increase in bone content after 1 to 3 years of lanthanum carbonate therapy [176,177]. However, paired bone biopsies confirmed no accumulation of lanthanum in patients treated with lanthanum carbonate over a long period of time [178].
- Iron-based binders: Ferric Citrate and Sucroferric Oxyhydroxide
Ferric citrate is a new type of phosphate binder that binds phosphate in exchange to citrate to form insoluble ferric phosphate, which is excreted in the feces, thereby lowering the serum phosphate level. In a phase III RCT that compared ferric citrate with non-iron-containing phosphate binders (sevelamer or calcium-based), ferric citrate was noninferior to sevelamer or calcium-based binders in controlling serum phosphate [179]. Additionally, it increased serum ferritin, reduced the need for intravenous iron and erythropoietin- stimulating agents, and improved overall anemia management [179,180]. Furthermore, ferric citrate reduced FGF23 levels and lowered the PTH level, thereby exerting effect on secondary hyperparathyroidism [181]. One concern commonly raised regarding use of ferric citrate in KF patients is risk of aluminum toxicity, as citrate can potentially enhance the absorption of aluminum. However, a RCT comparing safety profile of ferric citrate with control group (sevelamer and calcium acetate) in KF patients on HD did not observe aluminum toxicity among recipients of ferric citrate [182].
Sucroferric oxyhydroxide is a chewable phosphate binder, comprising a mixture of polynuclear iron-oxyhydroxide, sucrose, and starch. After oral administration, the sucrose and starch are broken down, releasing polynuclear iron-oxyhydroxide that binds to phosphates in the intestinal lumen to form an insoluble compound. One advantage of the chewable sucroferric oxyhydroxide is its ability to disintegrate rapidly upon contact with water or saliva [183]. Sucroferric oxyhydroxide has high phosphate-binding capacity across the physiological gastrointestinal pH range [184]. Moreover, in the phase III RCT comparing sucroferric oxyhydroxide and sevelamer in PD and HD patients similar efficacy of serum phosphate reduction was observed with the two agents [185,186]. However, sucroferric oxyhydroxide reduced pill burden by 75% and consecutively improved treatment adherence [186]. Adverse effects are primarily gastrointestinal including diarrhea, nausea, constipation, and vomiting [186]. Similar results were observed in another study that randomized CKD patients to sucroferric oxyhydroxide and sevelamer. There was a significant and sustained 30% reduction in serum phosphate, and a significant 64% decrease in FGF-23 at 24 weeks with sucroferric oxyhydroxide. Furthermore, there was significant reduction in PTH level at week 24, but returned to nearly the baseline level at week 52 [187]. In the post hoc analysis and extension of the phase III trial [186], compared to sevelamer, there was a significant increase in transferrin saturation and hemoglobin level with sucroferric oxyhydroxide in the first 24 weeks from the baseline; ferritin level also trended up but did not achieve statistical significance [188]. Conversely, no significant changes in iron-related parameters were noted over the 6-month of observation in another study [189].
With availability of a wide variety of phosphate binders, the choice of binder for an individual patient has become difficult. The ideal phosphate binder should be inexpensive, have low pill burden, efficient in binding dietary phosphate and safe with minimal systemic absorption and few side effects. Unfortunately, none of the binders discussed above meet all these criteria. In a recent network meta-analysis examining 22 different strategies and 16 different phosphate lowering agents, sevelamer was noted to be the most common binder used clinically. [153] Moreover, all investigated drugs exhibited superior or comparable efficacy in phosphorus reduction compared to placebo [153]. The current KDIGO guidelines recommend restricting calcium based binders and considering non-calcium binders due to concern about calcium load and consequent vascular calcification with the former [79]. As there is no convincing outcome data comparing one non-calcium binder with another, the choice of non-calcium binders can be difficult and should consider the cost, side effects, pill burden and collateral benefits, if any. For instance, ferric citrate may assist with anemia management, sucroferric oxyhydroxide and lanthanum carbonate may provide a lower pill burden, and sevelamer may have low density lipoprotein-lowering and anti-inflammatory effects [167,169,170,171,179,180,186].
- Drugs inhibiting intestinal phosphate transport:
As discussed in earlier sections, intestinal phosphate absorption occurs via two distinct pathways: the 1,25 dihydroxy vitamin D -dependent saturable transcellular pathway through NaPi-IIb and the non-saturable paracellular pathway that is dependent upon NHE-3 [11,13,14,15,16]. Moreover, intestinal phosphate chelation may result in compensatory upregulation of NaPi-IIb [148]. Therefore, inhibition of phosphate transporter activities is a plausible alternative or complementary approach to reduce phosphate load in KF patients (Table 2) [190].
Nicotinamide (also known as niacinamide), is a form of vitamin B3, that reduces intestinal NaPi-IIb expression, and can potentially lower sodium-dependent intestinal phosphate absorption [191,192]. However, recent randomized trials have demonstrated limited efficacy of nicotinamide in reducing phosphate level, poor tolerance, and some safety concerns [193,194]. For instance, in a RCT, nicotinamide as an add-on therapy to phosphate binders in HD patients did lower phosphate level at 24 weeks but the effect was not maintained at 52 weeks [194]. Moreover, nicotinamide was associated with higher rates of side effects, including diarrhea, pruritus, and thrombocytopenia. Hence use of nicotinamide as a sole agent for the management of hyperphosphatemia is not recommended.
Tenapanor is an inhibitor of the NHE3 that reduces paracellular intestinal sodium and phosphate absorption [23,24,26,195]. In a phase-III randomized, double-blind placebo-controlled trial, 8-weeks treatment with tenapanor significantly reduced serum phosphate by a mean of 1.0-1.2 mg/dl in hyperphosphatemic patients on HD [26]. Furthermore, in another double-blind phase-III trial, 4-weeks treatment with tenapanor as an add on therapy to binders achieved a larger mean change in serum phosphate compared binders plus placebo in HD or PD patients who were hyperphosphatemic despite receiving phosphate binder therapy [158]. In both studies, adverse events were mainly restricted to stool softening and increased bowel movements resulting from increased stool sodium and water content due to inhibition of intestinal NHE-3 by tenapanor [26,195,196]. In October 2023 the US Food and Drug Administration approved tenapanor as an add-on therapy for the use in adult dialysis patients who have an inadequate response to phosphate binders or who are intolerant of any dose of phosphate binder therapy [197].
Controversies and Challenges in the Management of Hyperphosphatemia
The current paradigm in the management of hyperphosphatemia in KF focusses on reduction of serum phosphate level principally by dietary phosphate restriction and pharmaceutical measures to diminish intestinal phosphate absorption. Indeed KDIGO-2017 guidelines recommend lowering phosphate to the normal range in people with KF [79]. Consequently, rather than using hard endpoints, clinical trials for phosphate lowering agents use reduction in serum phosphate level as the primary end point [26,160,172,173,179,185,186,187,194,195,196]. Similarly, regulatory agencies approve these drugs based upon their capacity to reduce phosphate level and not on the outcome data [197]. Adherence to the guidelines-recommended phosphate target often requires colossal efforts that include substantial dietary restrictions and considerably increase pill burden at a considerable cost. Generally, individuals with KF take loads of prescription medications. Indeed, in a cross-sectional study the median daily pill burden of 19 was noted among KF patients [198]. Phosphate binders accounted for 49% of this pill burden [198]. Similarly, in a Japanese dialysis cohort phosphate binders comprised 33% of total pill burden [199] High pill burden and the need to take the binders with every meal and snack can have considerable psychosocial consequences and impact quality of life [198,200]. While pill burden can be reduced with appropriate choice of binders, add on therapy with new agents such as tenapanor as well as appropriate dietary measures, cost of novel agents may be prohibitive. Indeed, Phosphate-lowering therapies represent a sizable financial burden, with annual cost totaling $1500 per user and $658 millions aggregate for the prevalent dialysis population [4]. Furthermore, like pharmacologic measures, dietary phosphate restriction can be cumbersome. KF patients, in addition to watching phosphorus, need to adhere to myriads of dietary restrictions such as sodium, potassium, calories (as half of KF individuals have diabetes) and fluid intake. Moreover, strict phosphate control may limit necessary protein intake. Such arduous restrictions run the risk of malnutrition which is associated with lower survival in KF. This can be mitigated with dietary counselling, avoiding non-protein sources of phosphorus, using plant-based proteins that contain phosphate of low bioavailability, and diligent use of phosphate-lowering medications.
Given immense efforts and financial resources devoted to lower serum phosphate level, it would be logical to presume that there are high-powered studies demonstrating improvement in health outcomes in KF persons with tight control of hyperphosphatemia. However, such data is unfortunately lacking. The 2017-KDIGO recommendations to lower phosphate level are based upon low quality grade 2-C evidence [79]. Moreover, the appropriate target phosphate level for optimizing bone health and improving cardiovascular risk without causing unintended consequences remains vague. Two recent large RCTs have tried to define the specific phosphate targets. The recently halted HiLo trial defined tight control as <5.5 mg/dl, while the ongoing PHOSPHATE trial (NCT03573089) lowers this cutoff closer to 4.5 mg/dl and should provide some guidance in the ensuing years [201,202].
While managing serum phosphate level, it should be realized that development of overt hyperphosphatemia is a late event in CKD, preceded by a chain of adaptive events including elevated FGF-23, PTH and low 1,25 dihydroxy-vitamin D levels, which have been independently associated with high CV risks, BMD and mortality. Conceivably, deleterious effects associated with hyperphosphatemia could be attributable to disruptions in phosphorus sensing mechanisms and hormonal imbalance thereof, rather than with overt hyperphosphatemia. By the time hyperphosphatemia ensues, CV disease may already be far advanced, and reduction of hyperphosphatemia is inadequate in mitigating CV risks and mortality, akin to the ineffectiveness of traditional CV risk factors in reducing CV mortality in CKD [203]. Further exploration of mechanisms that precisely control phosphorus sensing and regulation may facilitate development of strategies to diminish the deleterious effects of phosphorus load early in the CKD course, before overt hyperphosphatemia is evident to improve overall outcomes in KF. Sans high quality evidence, tight phosphate control at the expense of quality of life may not be justified. Therefore, until then management of hyperphosphatemia should be based upon shared decision-making between the individuals, dieticians, physicians and other care provides with focus not only on clinical outcomes such as comorbidities and other clinical issues related to CKD/KF but also considering patient-oriented goals, such as patient preference, quality of life, nutritional status, pill burden, and financial constraints.
Conflicts of Interest
Both authors have nothing to disclose.
References
- Heaney RP. Phosphorus. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:447-58).
- Hu, M.C.; Moe, O.W. Phosphate and Cellular Senescence. Adv. Exp. Med. Biol. 2022, 1362, 55–72.
- Phosphorus. Facts Sheet For Health Professionals. https://ods.od.nih.gov/factsheets/Phosphorus-HealthProfessional/#en1, updated May 4, 2023. Accessed Mar. 24, 2025.
- The United States Renal Data System Annual Data Report 2024. www.usrds.org. Accessed March 24, 2025.
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic insights into vascular calcification in CKD. J. Am. Soc. Nephrol. 2013, 24, 179–189. [CrossRef]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17.
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Ren. Physiol. 2014, 307, F891–F900. [CrossRef]
- Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370-1378.
- Zhou W, Simic P, Zhou IY, Caravan P, Vela Parada X, Wen D, Washington OL, Shvedova M, Pierce KA, Clish CB, Mannstadt M, Kobayashi T, Wein MN, Jüppner H, Rhee EP. Kidney glycolysis serves as a mammalian phosphate sensor that maintains phosphate homeostasis. J Clin Invest.2023;133(8):e164610. [CrossRef]
- Sabbagh Y, Giral H, Caldas Y, Levi M, Schiavi SC. Intestinal phosphate transport. Adv Chronic Kidney Dis. 2011; 18(2): 85–90.
- Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004; 19(3): 429–35.
- Berndt TJ, Kumar R: Clinical Disturbances of Phosphate Homeostasis In Seldin and Giebisch’s The Kidney,Fifth Edition. [CrossRef]
- Walton J, Gray TK. Absorption of inorganic phosphate in the human small intestine. Clin Sci. 1979; 56(5): 407–12. [CrossRef]
- Danisi G, Straub RW. Unidirectional influx of phosphate across the mucosal membrane of rabbit small intestine. Pflugers Arch. 1980; 385(2): 117–22. [CrossRef]
- Davis GR, Zerwekh JE, Parker TF, Krejs GJ, Pak CY, Fordtran JS. Absorption of phosphate in the jejunum of patients with chronic renal failure before and after correction of vitamin D deficiency. Gastroenterology. 1983; 85(4): 908–16. [CrossRef]
- Marks J. Debnam ES, Unwin RJ. The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr Opin Nephrol Hypertens 22: 481–487, 2013. [CrossRef]
- Sabbagh Y, O’Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009; 20 (11): 2348–58.
- Marks J. Debnam ES, Unwin RJ Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol 299: F285–F296, 2010. [CrossRef]
- Larsson TE, Kameoka C, Nakajo I, Taniuchi Y, Yoshida S, Akizawa T, et al. NPT-IIb inhibition does not improve hyperphosphatemia in CKD. Kidney Int Rep. 2018; 3(1): 73–80. [CrossRef]
- Saurette M, Alexander RT. Intestinal phosphate absorption: the paracellular pathway predominates? Exp Biol Med. 2019; 244(8): 646–54.
- Knöpfel T, Himmerkus N, Günzel D, Bleich M, Hernando N, Wagner CA. Paracellular transport of phosphate along the intestine. Am J Physiol Gastrointest Liver Physiol. 2019; 317(2): G233–G41. [CrossRef]
- Lee DB, Walling MW, Corry DB. Phosphate transport across rat jejunum: influence of sodium, pH, and 1,25-dihydroxyvitamin D3. Am J Physiol. 1986; 251(1 Pt 1): G90–5. [CrossRef]
- King AJ, Siegel M, He Y, Nie B, Wang J, Koo-McCoy S, et al. Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Sci Transl Med. 2018; 10(456):eaam6474. [CrossRef]
- Dudeja PK, Rao DD, Syed I, Joshi V, Dahdal RY, Gardner C, et al. Intestinal distribution of human Na+/H+ exchanger isoforms NHE-1, NHE-2, and NHE-3 mRNA. Am J Physiol.1996; 271(3 Pt 1): G483–93. [CrossRef]
- Rosenbaum DP, Yan A, Jacobs JW. Pharmacodynamics, safety, and tolerability of the NHE3 inhibitor tenapanor: two trials in healthy volunteers. Clin Drug Investig. 2018; 38(4): 341–51. [CrossRef]
- Block GA, Rosenbaum DP, Yan A, Chertow GM. Efficacy and safety of tenapanor in patients with hyperphosphatemia receiving maintenance hemodialysis: a randomized phase 3 trial. J Am Soc Nephrol. 2019; 30(4): 641–52. [CrossRef]
- Tatsumi, S.; Miyagawa, A.; Kaneko, I.; Shiozaki, Y.; Segawa, H.; Miyamoto, K. Regulation of renal phosphate handling: Inter-organ communication in health and disease. J. Bone Min. Metab. 2016, 34, 1–10. [CrossRef]
- Lederer, E. Renal phosphate transporters. Curr. Opin. Nephrol. Hypertens. 2014, 23, 502–506.
- Erben RG. Physiological Actions of Fibroblast Growth Factor-23. Front Endocrinol 9: 267, 2018. [CrossRef]
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012 Jan;92(1):131–155. [CrossRef]
- Lee M, Partridge NC. Parathyroid hormone signaling in bone and kidney. Curr Opin Nephrol Hypertens. 2009 Jul;18(4):298–302. [CrossRef]
- Bargagli, M.; Arena, M.; Naticchia, A.; Gambaro, G.; Mazzaferro, S.; Fuster, D.; Ferraro, P.M. The Role of Diet in Bone and Mineral Metabolism and Secondary Hyperparathyroidism. Nutrients 2021, 13, 2328.
- Zhou W, Simic P, Zhou IY, Caravan P, Vela Parada X, Wen D, Washington OL, Shvedova M, Pierce KA, Clish CB, Mannstadt M, Kobayashi T, Wein MN, Jüppner H, Rhee EP. Kidney glycolysis serves as a mammalian phosphate sensor that maintains phosphate homeostasis. J Clin Invest.2023;133(8):e164610. [CrossRef]
- Ritz E, Gross M-L Hyperphosphatemia in renal failure. Blood Purif 2005, 23:6–9,.
- Chen W, Bushinsky D. Chronic kidney disease–mineral and bone disorder. In: Nissenson, A.R., Fine, R.N.B.T. Handbook of Dialysis Therapy, 5th edn. Elsevier, 2017, pp 685–697.e1.
- Block GA, Hulbert-Shearon TE, Levin NW, Port FK: Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis, 31: 607-617, 1998 10.1053/ajkd.1998.v31.pm9531176.
- Dhingra R, Sullivan LM, Fox CS, Wang TJ, D’Agostino RB, Sr., Gaziano JM, Vasan RS: Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med, 167: 879-885, 2007. 10.1001/archinte.167.9.879.
- Palmer SC, Hayen A, Macaskill P, Pellegrini F, Craig JC, Elder GJ, Strippoli GF (2011) Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA 305(11):1119–1127.
- Schwartz S, Trivedi BK, Kalantar-Zadeh K, Kovesdy CP. Association of disorders in mineral metabolism with progression of chronic kidney disease. Clin J Am Soc Nephrol. 2006;1: 825-831. [CrossRef]
- Tong J., Liu M., Li H., et al. Mortality and associated risk factors in dialysis patients with cardiovascular disease. Kidney Blood Press Res. 2016;41(4):479–487. [CrossRef]
- Cozzolino M., Mangano M., Stucchi A., Ciceri P., Conte F., Galassi A. Cardiovascular disease in dialysis patients. Nephrol Dial Transplant. 2018;33(suppl 3):iii28–iii34. [CrossRef]
- Ganesh SK, Stack AG, Levin NW, Hulbert-Shearon T, Port FK (2001) Association of elevated serum PO(4), Ca x PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol 12(10):2131–2138.
- Cozzolino M, Dusso AS, Slatopolsky E (2001) Role of calcium-phosphate product and bone-associated proteins on vascular calcification in renal failure. J Am Soc Nephrol 12(11):2511–2516.
- Zhang D., Bi X., Liu Y., et al. High phosphate-induced calcification of vascular smooth muscle cells is associated with the TLR4/NF-κb signaling pathway. Kidney Blood Press Res. 2017;42(6):1205–1215. [CrossRef]
- Amann K (2008) Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin J Am Soc Nephrol 3(6):1599–1605.
- El-Abbadi MM, Pai AS, Leaf EM et al. (2009) Phosphate feeding induces arterial medial calcification in uremic mice: role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int 75(12):1297–1307.
- Pai AS, Giachelli CM (2010) Matrix remodeling in vascular calcification associated with chronic kidney disease. J Am Soc Nephrol 21(10):1637–1640.
- Luong TTD, Schelski N, Boehme B, Makridakis M, Vlahou A, Lang F, Pieske B, Alesutan I, Voelkl J (2018) Fibulin-3 attenuates phosphate-induced vascular smooth muscle cell calcification by inhibition of oxidative stress. Cell Physiol Biochem 46:1305–1316. [CrossRef]
- Voelkl J, Lang F, Eckardt K et al. (2019) Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci 76(11):2077–2091. [CrossRef]
- Faul C., Amaral A.P., Oskouei B., et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–4408. [CrossRef]
- Wannamethee S.G., Welsh P., Papacosta O., Lennon L., Whincup P.H., Sattar N. Elevated parathyroid hormone, but not vitamin D deficiency, is associated with increased risk of heart failure in older men with and without cardiovascular disease. Circulation Heart Fail. 2014;7(5):732–739. [CrossRef]
- Vogt I, Haffner D, Leifheit-Nestler M. FGF23 and Phosphate–Cardiovascular Toxins in CKD. Toxins Basel. 2019;11(11):647. [CrossRef]
- Ix J.H., Katz R., Kestenbaum B.R., et al. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study) J Am Coll Cardiol. 2012;60(3):200–207. [CrossRef]
- Edmonston D, Grabner A, Wolf M: FGF23 and klotho at the intersection of kidney andcardiovascular disease. Nat Rev Cardiol, 21: 11-24, 2024 10.1038/s41569-023-00903-0.
- Zhong Z, Feng S, Fu D, et al. Serum fibroblast growth factor 23 concentration and the risk of mortality in patients undergoing peritoneal dialysis. Perit Dial Int 2024; 44:194.
- Cheng S.P., Liu C.L., Liu T.P., Hsu Y.C., Lee J.J. Association between parathyroid hormone levels and inflammatory markers among US adults. Mediators Inflamm. 2014;2014:709024. [CrossRef]
- Goldsmith D.J., Covic A.A., Venning M.C., Ackrill P. Blood pressure reduction after parathyroidectomy for secondary hyperparathyroidism: further evidence implicating calcium homeostasis in blood pressure regulation. Am J Kidney Dis. 1996;27(6):819–825. [CrossRef]
- Smogorzewski M., Perna A.F., Borum P.R., Massry S.G. Fatty acid oxidation in the myocardium: effects of parathyroid hormone and CRF. Kidney Int. 1988;34(6):797–803. [CrossRef]
- Rodríguez-Ayala E., Avila-Díaz M., Foyo-Niembro E., Amato D., Ramirez-San-Juan E., Paniagua R. Effect of parathyroidectomy on cardiac fibrosis and apoptosis: possible role of aldosterone. Nephron Physiol. 2006;103(3):112–118. [CrossRef]
- Saleh F.N., Schirmer H., Sundsfjord J., Jorde R. Parathyroid hormone and left ventricular hypertrophy. Eur Heart J. 2003;24(22):2054–2060. [CrossRef]
- Li Y., Chen C., Liu H.L., Qian G. Vitamin D, parathyroid hormone, and heart failure in a Chinese elderly population. Endocr Pract. 2015;21(1):30–40. [CrossRef]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, D.J.; Andress, D.L. Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 520–528.
- Voormolen, N.; Noordzij, M.; Grootendorst, D.C.; Beetz, I.; Sijpkens, Y.W.; van Manen, J.G.; Boeschoten, E.W.; Huisman, R.M.;Krediet, R.T.; Dekker, F.W.; et al. High plasma phosphate as a risk factor for decline in renal function and mortality in pre-dialysis patients. Nephrol. Dial. Transpl. 2007, 222, 909–916. [CrossRef]
- Tonelli M, Curhan G, Pfeffer M, Sacks F, Thadhani R, Melamed ML, Wiebe N, Muntner P (2009) Relation between alkaline phosphatase, serum phosphate, and all-cause or cardiovascular mortality. Circulation 120(18):1784–1792.
- Li JW, Xu C, Fan Y, Wang Y, Xiao YB (2014) Can serum levels of alkaline phosphatase and phosphate predict cardiovascular diseases and total mortality in individuals with preserved renal function? A systemic review and meta-analysis. PLoS One 9(7):e102276.
- Schwartz S, Trivedi BK, Kalantar-Zadeh K, Kovesdy CP. Association of disorders in mineral metabolism with progression of chronic kidney disease. Clin J Am Soc Nephrol. 2006;1(4):825-831. [CrossRef]
- Maique J, Flores B, Shi M et al. (2020) High phosphate induces and Klotho attenuates kidney epithelial senescence and fibrosis. Front Pharmacol 11:1273. [CrossRef]
- Shen ZJ, Hu J, Shiizaki K et al. (2016) Phosphateinduced renal fibrosis requires the prolyl isomerase Pin1. PLoS One 11:e01500 . [CrossRef]
- Sim, J.J.; Bhandari, S.K.; Smith, N.; Chung, J.; Liu, I.L.; Jacobsen, S.J.; Kalantar-Zadeh, K. Phosphorus and risk of renal failure in subjects with normal renal function. Am. J. Med. 2013, 126, 311–318.
- Zoccali, C.; Ruggenenti, P.; Perna, A.; Leonardis, D.; Tripepi, R.; Tripepi, G.; Mallamaci, F.; Remuzzi, G.; REIN Study Group. Phosphate may promote CKD progression and attenuate renoprotective effect of ACE inhibition. J. Am. Soc. Nephrol. 2011, 22, 1923–1930.
- Da, J.; Xie, X.; Wolf, M.; Disthabanchong, S.; Wang, J.; Zha, Y.; Lv, J.; Zhang, L.; Wang, H. Serum phosphorus and progression of CKD and mortality: A meta-analysis of cohort studies. Am. J. Kidney Dis. 2015, 66, 258–265.
- Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370-1378. [CrossRef]
- Barrera-Baena P, Rodríguez-García M, Rodríguez-Rubio E, et al. Serum phosphate is associated with increased risk of bone fragility fractures in haemodialysis patients. Nephrol Dial Transplant. 2023;39(4):618-626. [CrossRef]
- Tentori, F.; McCullough, K.; Kilpatrick, R.D.; Bradbury, B.D.; Robinson, B.M.; Kerr, P.G.; Pisoni, R.L. High rates of death and hospitalization follow bone fracture among hemodialysis patients. Kidney Int. 2014, 85, 166–173. [CrossRef]
- Fusaro, M.; Holden, R.; Lok, C.; Iervasi, G.; Plebani, M.; Aghi, A.; Gallieni, M.; Cozzolino, M. Phosphate and bone fracture risk in chronic kidney disease patients. Nephrol. Dial. Transpl. 2021, 36, 405–412.
- Meleti, Z.; Shapiro, I.M.; Adams, C.S. Inorganic phosphate induces apoptosis of osteoblast-like cells in culture. Bone 2000, 27, 359–366. [CrossRef]
- Mozar, A.; Haren, N.; Chasseraud, M.; Louvet, L.; Mazière, C.; Wattel, A.; Mentaverri, R.; Morlière, P.; Kamel, S.; Brazier, M.; et al. High extracellular inorganic phosphate concentration inhibits RANK-RANKL signaling in osteoclast-like cells. J. Cell. Physiol.2008, 215, 47–54.
- Guedes M, Bieber B, Dasgupta I, et al. Serum Phosphorus Level Rises in US Hemodialysis Patients Over the Past Decade: A DOPPS Special Report. Kidney Med. 2022;5(2):100584. [CrossRef]
- Kidney Disease: Improving Global Outcomes CKDMBDUWG: KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl (2011), 7: 1-59, 2017 10.1016/j.kisu.2017.04.001.
- Kuhlmann MK. Phosphate elimination in modalities of hemodialysis and peritoneal dialysis. Blood Purif. 2010;29(2):137–44. [CrossRef] [PubMed]
- DeSoi CA, Umans JG. Phosphate kinetics during high-flux hemodialysis. J Am Soc Nephrol. Nov 1993;4(5):1214–8. [PubMed]
- Sugisaki H, Onohara M, Kunitomo T. Dynamic behavior of plasma phosphate in chronic dialysis patients. Trans Am Soc Artif Intern Organs. 1982;28:302–7. [PubMed]
- Zucchelli P, Santoro A. Inorganic phosphate removal during different dialytic procedures. Int J Artif Organs. 1987;10(3):173–8. [PubMed]
- Haas T, Hillion D, Dongradi G. Phosphate kinetics in dialysis patients. Nephrol Dial Transplant. 1991;6(Suppl 2):108–13. [PubMed]
- Man NK, Chauveau P, Kuno T, Poignet JL, Yanai M. Phosphate removal during hemodialysis, hemodiafiltration, and hemofiltration. A reappraisal. ASAIO Trans. Jul-Sep 1991;37(3):M463–5. [PubMed]
- Daugirdas JT. Removal of Phosphorus by Hemodialysis. Semin Dial. Nov-Dec 2015;28(6):620–3. [CrossRef]
- Kooienga L. Phosphorus balance with daily dialysis. Semin Dial. Jul-Aug 2007;20(4):342–5. [CrossRef] [PubMed]
- Minutolo R, Bellizzi V, Cioffi M, et al. Postdialytic rebound of serum phosphorus: pathogenetic and clinical insights. J Am Soc Nephrol. Apr 2002;13(4):1046–54. [PubMed]
- Stremke ER, Trevino L, Simit Doshi S, Moorthi RN, Gallant KMH. Moe SM, Post-Dialysis Serum Phosphate Equilibrium in Hemodialysis Patients on a controlled diet and no binders Hemodial Int . 2022 April ; 26(2): 255–263. [CrossRef]
- Perl, J.; Bargman, J.M. Peritoneal dialysis: From bench to bedside and bedside to bench. Am. J. Physiol. Renal Physiol. 2016, 311, F999–F1004. [CrossRef]
- Courivaud, C.; Davenport, A. Phosphate Removal by Peritoneal Dialysis: The Effect of Transporter Status and Peritoneal Dialysis Prescription. Perit. Dial. Int. 2016, 36, 85–93. [CrossRef]
- Bammens, B.; Evenepoel, P.; Verbeke, K.; Vanrenterghem, Y. Removal of middle molecules and protein-bound solutes by peritoneal dialysis and relation with uremic symptoms. Kidney Int. 2003, 64, 2238–2243. [CrossRef]
- Debowska, M.; Gomez, R.; Pinto, J.; Waniewski, J.; Lindholm, B. Phosphate clearance in peritoneal dialysis. Sci. Rep. 2020, 10, 17504.
- Peruzzo, D.; Guedes, M.; Larkin, J.W.; Yokoyama, G.; Dos Santos, T.L.; Pecoits-Filho, R.; Ribeiro, S.C.; Ramos, A.; Barretti, P.; de Moraes, T.P.; et al. Peritoneal dialysis modality transition and impact on phosphate and potassium serum levels. PLoS ONE 2021, 16, e0257140. [CrossRef]
- Calvo MS, Park YK. Changing phosphorus content of the U.S. diet: potential for adverse effects on bone. J Nutr. 1996;126:1168S–1180S. [CrossRef]
- Kalantar-Zadeh, K., Gutekunst, L., Mehrotra, R., Kovesdy, C. P., Bross, R., Shinaberger, C. S., Noori, N., Hirschberg, R., Benner, D., Nissenson, A. R., & Kopple, J. D. (2010). Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clinical Journal of the American Society of Nephrology, 5(3), 519-530. [CrossRef]
- Calvo, M. S., & Uribarri, J. (2011). Dietary phosphorus intake and the risk for cardiovascular disease in the general population. Advances in Chronic Kidney Disease, 18(4), 266-272.
- Dang Z, He Y, Xie R, Chen P, Dong F, Plant-based Diet and Chronic Kidney Disease: A Systematic Review and Meta-analysis, Journal of Renal Nutrition (2025).
- Huml AM, Sullivan CM, Leon JB, et al. The adequacy of phosphorus binder prescriptions among American hemodialysis patients. Ren Fail. 2012;34:1258–1263. KDIGO-2017. [CrossRef]
- Brown-Tortorici, A. R., Narasaki, Y., You, A. S., Norris, K. C., Streja, E., Peralta, R. A., Guerrero, Y., Daza, A., Arora, R., Lo, R., Nakata, T., Nguyen, D. V., Kalantar-Zadeh, K., & Rhee, C. M. (2022). The interplay between dietary phosphorus, protein intake, and mortality in a prospective hemodialysis cohort. Nutrients, 14(15), 3070. [CrossRef]
- Fenton, T. R., et al. (2017). Phosphorus additives in food and their impact on human health. Current Opinion in Clinical Nutrition & Metabolic Care, 20(3), 192-196.
- Younes, M., Aquilina, G., Castle, L., Engel, K.-H., Fowler, P., Frutos Fernandez, M. J., Fürst, P., Gürtler, R., Husøy, T., Mennes, W., Moldeus, P., Oskarsson, A., Shah, R., Waalkens-Berendsen, I., Wölfle, D., Aggett, P., Cupisti, A., Fortes, C., Kuhnle, G., Lillegaard, I. T., Scotter, M., Giarola, A., Rincon, A., Tard, A., & Gundert-Remy, U. (2019). Re-evaluation of phosphoric acid–phosphates – di-, tri- and polyphosphates (E 338–341, E 343, E 450–452) as food additives and the safety of proposed extension of use. EFSA Panel on Food Additives and Flavourings (FAF). Adopted 4 June 2019; Published 12 June 2019. [CrossRef]
- Capra BT, Hudson S, Helder M, Laskaridou E, Johnson AL, Gilmore C, Marinik E, Hedrick VE, Savla J, David LA, Davy KP, Davy BM. Ultra-processed food intake, gut microbiome, and glucose homeostasis in mid-life adults: Background, design, and methods of a controlled feeding trial. Contemp Clin Trials. 2024 Feb;137:107427. Epub 2024 Jan 4. [CrossRef] [PubMed] [PubMed Central]
- FoodNavigator-USA. (2014, July 17). Until phosphorus gets on the USDA’s radar, labeling policy won’t change – NKF. FoodNavigator-USA. https://www.foodnavigator-usa.com/Article/2014/07/17/Until-phosphorus-gets-on-the-USDA-s-radar-labeling-policy-won-t-change-NKF/ (Accessed on March 23, 2025).
- Nelson SM, Sarabia SR, Christilaw E, et al. Phosphate-containing prescription medications contribute to the daily phosphate intake in a third of hemodialysis patients. J Ren Nutr. 2017;27:91–96.
- Sherman RA, Ravella S, Kapoian T. A dearth of data: the problem of phosphorus in prescription medications. Kidney Int. 2015;87:1097–1099. [CrossRef]
- Moe SM, Chen NX, Seifert MF, et al. A rat model of chronic kidney disease-mineral bone disorder. Kidney Int. 2009;75(2):176-184. [CrossRef]
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6(2):257-264.
- Azadbakht L, Esmaillzadeh A. Soy-protein consumption and kidney-related biomarkers among type 2 diabetics: a crossover, randomized clinical trial. J Ren Nutr. 2009;19(6):479-486.
- Wu T, Chang C, Hsu W, et al. Nutritional status of vegetarians on maintenance haemodialysis. Nephrology (Carlton). 2011;16(6):582-587. [CrossRef]
- Garcia-Torres R, Young L, Murray DP, Kheda M, Nahman NS Jr. Dietary protein source and phosphate levels in patients on hemodialysis. J Ren Nutr. 2020:1-7 . [CrossRef]
- Kistler BM, Moore LW, Benner D, et al. The International Society of Renal Nutrition and Metabolism commentary on the National Kidney Foundation and Academy of Nutrition and Dietetics KDOQI clinical practice guideline for nutrition in chronic kidney disease. J Ren Nutr 2021;31(2):116-120.e1. [CrossRef]
- Stevens PE, Ahmed SB, Carrero JJ, et al. KDIGO 2024 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. 2024;105(4):S117- S314. [CrossRef]
- Kalantar-Zadeh, K., & Kopple, J. D. (2013). Preventing and correcting metabolic acidosis in chronic kidney disease: Implications for bone and mineral disease. American Journal of Kidney Diseases, 61(3), 508-516. [CrossRef]
- Goraya, N., & Wesson, D. E. (2020). Whole-Food Low-Protein Plant-Based Nutrition to Prevent or Slow Progression of Chronic Kidney Disease. Journal of Renal Nutrition, 30(6), 480–488.
- KDOQI Clinical Practice Guideline for Nutrition in CKD: 2020 Update. (2020). American Journal of Kidney Diseases, 76(3), S1–S107.
- Zarantonello, D., & Brunori, G. (2023). Plant-Based Diets in Preventing and Mitigating Chronic Kidney Disease. Journal of Clinical Medicine, 12(19), 6137. [CrossRef]
- Joshi, S., Shah, S., & Kalantar-Zadeh, K. (2019). Adequacy of plant-based proteins in chronic kidney disease. Journal of Renal Nutrition, 29(2), 112-117. [CrossRef]
- Consultant360. (n.d.). Dietary modification in renal disease. Retrieved from https://www.consultant360.com/blog/consultant360/nephrology/dietary-modification-renal-disease (Accessed on March 23, 2025).
- Escoffier. (n.d.). The true costs of processed foods on health and the planet. Retrieved from https://www.escoffier.edu/blog/world-food-drink/true-costs-processed-foods-health-planet/#:~:text=So%20although%20processed%20food%20is,And%20agricultural%20byproducts%20are%20destroyed. (Accessed on March 23, 2025).
- Springmann, M., Clark, M. A., Rayner, M., Scarborough, P., & Webb, P. (2021). The global and regional costs of healthy and sustainable dietary patterns: A modelling study. The Lancet Planetary Health, 5(11), e797–e807. [CrossRef]
- Silva, A. R., Rodrigues, A. M., & Lima, M. L. (2021). Do plant-based consumers spend more on food? Frontiers in Nutrition, 8, 732129. [CrossRef]
- Kahleova, H., Levin, S., & Barnard, N. D. (2023). Vegan diet and food costs among adults with overweight: A secondary analysis of a randomized clinical trial. JAMA Network Open, 6(9), e2332106. [CrossRef]
- GFI. (2022). Reducing the price of alternative proteins. Retrieved from https://gfi.org/reducing-the-price-of-alternative-proteins/ (Accessed on March 23, 2025).
- Food & Wine. (2024, April 5). Why going vegetarian is a recipe for a cheaper grocery bill. Retrieved from https://www.foodandwine.com/vegetarian-cheapest-groceries-report-11685113 (Accessed on March 23, 2025).
- The Guardian. (2024, August 28). “Don’t be scared of beans”: how readers are handling US grocery inflation. Retrieved from https://www.theguardian.com/environment/article/2024/aug/28/inflation-groceries-tips (Accessed on March 23, 2025).
- Kimura M, Itokawa Y. Cooking losses of minerals in foods and its nutritional significance. J Nutr Sci Vitaminol (Tokyo). 1990;36 Suppl 1: S25–S32. discussion S33.
- Jones WL. Demineralization of a wide variety of foods for the renal patient. J Ren Nutr. 2001;11:90–96.
- Naber, T., & Purohit, S. (2021). Chronic Kidney Disease: Role of Diet for a Reduction in the Severity of the Disease. Nutrients, 13(9), 3277. [CrossRef]
- Ikizler TA, Burrowes JD, Byham-Gray LD ∙ Katrina L. Campbell KL, Carrero JJ, Chan W, Fouque D, Friedman AN, Ghaddar S, Goldstein-Fuchs DJ, Kaysen GA, Kopple JD. Teta D, Wang AY-M, Cuppari L. KDOQI Clinical Practice Guideline for Nutrition in CKD: 2020 Update. [CrossRef]
- Noce, A., Marrone, G., Wilson Jones, G., Di Lauro, M., Pietroboni Zaitseva, A., Ramadori, L., Celotto, R., Mitterhofer, A. P., & Di Daniele, N. (2021). Nutritional Approaches for the Management of Metabolic Acidosis in Chronic Kidney Disease. Nutrients, 13(12), 4299.
- Kalantar-Zadeh, K., Bellizzi, V., Piccoli, G. B., Shi, Y., Lim, S. K., Riaz, S., Urbina Arronte, R., Lau, W. P., & Fouque, D. (2023). Caring for Patients With Advanced Chronic Kidney Disease: Dietary Considerations. Nutrition in Clinical Practice, 38(1), 22–34.
- US-DOPPS Practice Monitor, May 2021; http://www.dopps.org/DPM.
- Fresenius Medical Care North America. PhosLo® gelcaps (calcium acetate): 667 mg [prescribing information]. Waltham, MA: Fresenius Medical Care North America; 2011.
- Fresenius Medical Care North America. VELPHORO ® (sucroferric oxyhydroxide) [prescribing information]. Waltham, MA: Fresenius Medical Care North America; 2013.
- Shire US Inc. FOSRENAL® (lanthanum carbonate) [prescribing information]. Lexington, MA: Shire US Inc.; 2016.
- Keryx Biopharmaceuticals Inc. AURYXIA® (ferric citrate) tablets [prescribing information]. Cambridge, MA: Keryx Biopharmaceuticals Inc.; 2017.
- Genzyme Corp. RENVELA® (sevelamer carbonate) [prescribing information]. Cambridge, MA: Genzyme Corp.; 2020.
- Salusky IB, Foley J, Nelson P, Goodman WG. Aluminum accumulation during treatment with aluminum hydroxide and dialysis in children and young adults with chronic renal disease. N Engl J Med1991;324:527-31.
- Elliott HL, Dryburgh F, Fell GS, Sabet S, Macdougall AI. Aluminium toxicity during regular haemodialysis. BMJ 1978;1:1101-3. [CrossRef]
- Parkinson IS, Ward MK, Feest TG, Fawcett RW, Kerr DN. Fracturing dialysis osteodystrophy and dialysis encephalopathy: an epidemiological survey. Lancet 1979;1:406-9. [CrossRef]
- Young EW, Albert JM, Satayathum S, et al. Predictors and consequences of altered mineral metabolism: the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2005;67:1179-87. [CrossRef]
- K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003;42: Suppl 3:S1-S201.
- Janssen MJ, van der Kuy A, ter Wee PM, van Boven WP. Aluminum hydroxide, calcium carbonate and calcium acetate in chronic intermittent hemodialysis patients. Clin Nephrol 1996;45:111-9.
- Navaneethan SD, Palmer SC, Craig JC, Elder GJ, Strippoli GF. Benefits and harms of phosphate binders in CKD: a systematic review of randomized controlled trials. Am J Kidney Dis 2009;54:619-37. [CrossRef]
- Suki WN, Zabaneh R, Cangiano JL, et al. Effects of sevelamer and calcium-based phosphate binders on mortality in hemodialysis patients. Kidney Int 2007;72: 1130-7. [CrossRef]
- Tonelli M, Wiebe N, Culleton B, et al. Systematic review of the clinical efficacy and safety of sevelamer in dialysis patients. Nephrol Dial Transplant 2007;22: 2856-66. [CrossRef]
- Block GA, Wheeler DC, Persky MS, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. 2012;23:1407–1415.
- Hill KM, Martin BR, Wastney ME, et al. Oral calcium carbonate affects calcium but not phosphorus balance in stage 3-4 chronic kidney disease. Kidney Int. 2013;83:959–966. [CrossRef]
- Spiegel DM, Brady K. Calcium balance in normal individuals and in patients with chronic kidney disease on low- and high-calcium diets. Kidney Int. 2012;81:1116–1122. [CrossRef]
- Di Iorio B, Bellasi A, Russo D. Mortality in kidney disease patients treated with phosphate binders: a randomized study. Clin J Am Soc Nephrol. 2012;7: 87–493.
- Di Iorio B, Molony D, Bell C, et al. Sevelamer versus calcium carbonate in incident hemodialysis patients: results of an open-label 24-monthandomized clinical trial. Am J Kidney Dis. 2013;62:771–778.
- Zheng C, Liu J, Wang T, Hu H, Chen Y. A network meta-analysis of therapies for hyperphosphatemia in CKD based on randomized trials. Scientific Reports | (2025) 15:2012. [CrossRef]
- Perry, C. M. & Plosker, G. L. Sevelamer carbonate: A review in hyperphosphataemia in adults with chronic kidney disease. Drugs 74, 771–792. (2014). [CrossRef]
- Raggi, P., Vukicevic, S., Moysés, R. M., Wesseling, K. & Spiegel, D. M. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin. J. Am. Soc. Nephrol. 5(Suppl 1), S31-40. (2010). [CrossRef]
- Vervloet, M. G. et al. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 13, 27–38. (2017). [CrossRef]
- Lenglet, A. et al. Does the administration of sevelamer or nicotinamide modify uremic toxins or endotoxemia in chronic hemodialysis patients?. Drugs 79, 855–862. (2019). [CrossRef]
- Wrong O, Harland C. Sevelamer. Nephrol Dial Transplant 2008;23:2101-2.
- Pai AB, Shepler BM. Comparison of sevelamer hydrochloride and sevelamer carbonate: risk of metabolic acidosis and clinical implications. Pharmacotherapy 2009;29:554-61. [CrossRef]
- Delmez J, Block G, Robertson J, et al. A randomized, double-blind, crossover design study of sevelamer hydrochloride and sevelamer carbonate in patients on hemodialysis. Clin Nephrol 2007;68:386-91. [CrossRef]
- Fissell, R.B.; Karaboyas, A.; Bieber, B.A.; Sen, A.; Li, Y.; Lopes, A.A.; Akiba, T.; Bommer, J.; Ethier, J.; Jadoul, M.; et al. Phosphate binder pill burden, patient-reported non-adherence, and mineral bone disorder markers: Findings from the DOPPS. Hemodial. Int. 2016, 20, 38–49.
- Goldsmith, D. R., Scott, L. J., Cvetković, R. S. & Plosker, G. L. Sevelamer hydrochloride: A review of its use for hyperphosphataemia in patients with end-stage renal disease on haemodialysis. Drugs 68, 85–104. (2008). [CrossRef]
- Pai, A. B. & Shepler, B. M. Comparison of sevelamer hydrochloride and sevelamer carbonate: Risk of metabolic acidosis and clinical implications. Pharmacotherapy 29, 554–561. (2009). [CrossRef]
- Swanson BJ, Limketkai BN, Liu TC, Nazari K, Park JY, Santiago WC, Torbenson MS, Voltaggio LY, Martha M, Arnold CA. Sevelamer Crystals in the Gastrointestinal Tract (GIT) A New Entity Associated With Mucosal Injury.; Am J Surg Pathol. 37 :1686-1693, 2013. [CrossRef]
- Elkalashy A, Rainwater RR, Ali U, Elbahnasawy E, Singh M, Karakala N, Gastrointestinal mucosal cell injury caused by sevelamer crystals- Case series and literature review, J Renal Nutrition (2025). [CrossRef]
- Braunlin W, Zhorov E, Guo A, et al. Bile acid binding to sevelamer HCl Kidney Int, 62 (2002), pp. 611-619.
- Burke SK, Dillon MA, Hemken DE, et al. Meta-analysis of the effect of sevelamer on phosphorus, calcium, PTH, and serum lipids in dialysis patients Adv Ren Replace Ther, 10 (2003), pp. 133-145.
- Susantitaphong P, Jaber BL. Potential interaction between sevelamer and fat-soluble vitamins: a hypothesis. Am J Kidney Dis, 59 (2012), pp. 165-167.
- Vemuri N, Michelis MF, Matalon A. Conversion to lanthanum carbonate monotherapy effectively controls serum phosphorus with a reduced tablet burden: a multicenter open-label study. BMC Nephrol. 2011;12:49. [CrossRef]
- Dellanna F, Reichel H, Seibt F. Efficacy and safety of lanthanum carbonate in German patients on dialysis. Clin Nephrol. 2012;78:382–390. [CrossRef]
- Wilson RJ, Keith MS, Preston P, Copley JB. The real-world dose-relativity of sevelamer hydrochloride and lanthanum carbonate monotherapy in patients with end-stage renal disease. Adv Ther. 2013;30:1100–1110. [CrossRef]
- Takahara Y, Matsuda Y, Takahashi S, et al. Ef cacy and safety of lanthanum carbonate in pre-dialysis CKD patients with hyperphosphatemia: a randomized trial. Clin Nephrol. 2014;82:181–190. [CrossRef]
- Toida T, Fukudome K, Fujimoto S, et al. Effect of lanthanum carbonate vs. calcium carbonate on serum calcium in hemodialysis patients: a crossover study. Clin Nephrol. 2012;78:216–223.
- Zhang C, Wen J, Li Z, Fan J. Efficacy and safety of lanthanum carbonate on chronic kidney disease mineral and bone disorder in dialysis patients: a systematic review. BMC Nephrol. 2013;14:226. [CrossRef]
- Lacour B, Lucas A, Auchere D, et al. Chronic renal failure is associated with increased tissue deposition of lanthanum after 28-day oral administration. Kidney Int. 2005;67:1062–1069.
- Hutchison AJ, Barnett ME, Krause R, et al. Lanthanum carbon- ate treatment, for up to 6 years, is not associated with adverse effects on the liver in patients with chronic kidney disease stage 5 receiving hemodialysis. Clin Nephrol. 2009;71:286–295.
- Spasovski GB, Sikole A, Gelev S, et al. Evolution of bone and plasma concentration of lanthanum in dialysis patients before, during 1 year of treatment with lanthanum carbonate and after 2 years of follow-up. Nephrol Dial Transplant. 2006;21:2217–2224.
- Hutchison, A.J.; Wilson, R.J.; Garafola, S.; Copley, J.B. Lanthanum carbonate: Safety data after 10 years. Nephrology 2016, 21, 987–994.
- Lewis JB, Sika M, Koury MJ, et al. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol. 2015;26:493–503.
- Umanath K, Jalal DI, Greco BA, et al. Ferric citrate reduces intravenous iron and erythropoiesis-stimulating agent use in ESRD. J Am Soc Nephrol. 2015;26:2578–258. [CrossRef]
- Yokoyama, K.; Hirakata, H.; Akiba, T.; Fukagawa, M.; Nakayama, M.; Sawada, K.; Kumagai, Y.; Block, G.A. Ferric citrate hydrate for the treatment of hyperphosphatemia in nondialysis-dependent CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 543–552. [CrossRef]
- Van Buren PN, Lewis JB, Dwyer JP, Greene T, Middleton J, Sika M, Umanath K, Abraham JD, Arfeen SS, Bowline IG, Chernin G, Fadem SZ, Goral S, Koury M, Sinsakul MV, Weiner DE; Collaborative Study Group. The Phosphate Binder Ferric Citrate and Mineral Metabolism and Inflammatory Markers in Maintenance Dialysis Patients: Results From Prespecified Analyses of a Randomized Clinical Trial. Am J Kidney Dis. 2015 Sep;66(3):479-88. Epub 2015 May 7. [CrossRef] [PubMed] [PubMed Central]
- Floege J. Phosphate binders in chronic kidney disease: a systematic review of recent data. J Nephrol. 2016;29:329–340. [CrossRef]
- Cozzolino M, Funk F, Rakov V, et al. Preclinical pharmacokinetics, pharmacodynamics and safety of sucroferric oxyhydroxide. Curr Drug Metab. 2014;15:953–965.
- Floege J, Covic AC, Ketteler M, et al. Long-term effects of the iron-based phosphate binder, sucroferric oxyhydroxide, in dialysis patients. Nephrol Dial Transplant. 2015;30:1037–1046. [CrossRef]
- Floege J, Covic AC, Ketteler M, et al. A phase III study of the efficacy and safety of a novel iron based phosphate binder in dialysis patients. Kidney Int. 2014;86:638–647.
- Ketteler, M.; Sprague, S.M.; Covic, A.C.; Rastogi, A.; Spinowitz, B.; Rakov, V.; Walpen, S.; Floege, J. Effects of sucroferric oxyhydroxide and sevelamer carbonate on chronic kidney disease-mineral bone disorder parameters in dialysis patients. Nephrol. Dial. Transpl. 2019, 34, 1163–1170. [CrossRef]
- Covic, A.C.; Floege, J.; Ketteler, M.; Sprague, S.M.; Lisk, L.; Rakov, V.; Rastogi, A. Iron-related parameters in dialysis patients treated with sucroferric oxyhydroxide. Nephrol. Dial. Transpl. 2017, 32, 1330–1338.
- Lioulios, G.; Stangou, M.; Sarafidis, P.A.; Tsouchnikas, I.; Minasidis, I.; Vainas, A.; Faitatzidou, D.; Sampani, E.; Papagianni, A. Chronic therapy with sucroferric oxyhydroxide does not affect iron and anemia markers in dialysis patients. Blood Purif. 2020, 49. [CrossRef]
- Schiavi SC, Tang W, Bracken C, et al. Npt2b deletion attenuates hyperphosphatemia associated with CKD. J Am Soc Nephrol. 2012;23:1691–1700. [CrossRef]
- Müller D, Mehling H, Otto B, et al. Niacin lowers serum phosphate and increases HDL cholesterol in dialysis patients. Clin J Am Soc Nephrol 2007; 2:1249.
- Cheng SC, Young DO, Huang Y, et al. A randomized, double-blind, placebo-controlled trial of niacinamide for reduction of phosphorus in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1131.
- Ix JH, Isakova T, Larive B, et al. Effects of Nicotinamide and Lanthanum Carbonate on Serum Phosphate and Fibroblast Growth Factor-23 in CKD: The COMBINE Trial. J Am Soc Nephrol 2019; 30:1096. [CrossRef]
- Ketteler M, Wiecek A, Rosenkranz AR, et al. Modified-release nicotinamide for the treatment of hyperphosphataemia in haemodialysis patients: 52-week efficacy and safety results of the phase 3 randomized controlled NOPHOS trial. Nephrol Dial Transplant 2023; 38:982.
- Gallant KMH, Sprague SM, Rosenbaum DP, Spiegel DM, Kozuka K, Edelstein S, Chertow GM. Tenapanor: A phosphate absorption inhibitor for the management of hyperphosphatemia in patients with kidney failure. J Renal Nutrition 35: 2025: pp 25-34. [CrossRef]
- Pergola PE, Rosenbaum DP, Yang Y, Chertow GM. A randomized trial of tenapanor and phosphate binders as a dual-mechanism treatment for hyperphosphatemia in patients on maintenance dialysis (AMPLIFY). J Am Soc Nephrol 32: 1465–1473, 2021. [CrossRef]
- https://ir.ardelyx.com/news-releases/news-release-details/fda-approves-xphozahr-tenapanor-first-class-phosphate-absorption#:~:text=FDA%20approval%20of%20XPHOZAH%20is,met%20their%20primary%20and%20key. Accessed March 24, 2025.
- Chiu YW, Teitelbaum I, Misra M, de Leon EM, Adzize T, Mehrotra R: Pill burden, adherence, hyperphosphatemia, and quality of life in maintenance dialysis patients. Clin J Am Soc Nephrol Jun 2009;4(6):1089-1096. [CrossRef]
- Nagano N, Ito K, Ono T, Ariyoshi Y, Masima S, Kobayashi H, Ando T, Tsutsui T, Ogawa T: Prescription characteristics of phosphate binders in a high pill burden for hemodialysis patients. Renal Replacement Therapy 2021/02/04 2021;7(1):5. [CrossRef]
- Forfang D, Edwards DP, Kalantar-Zadeh K: The Impact of Phosphorus Management Today on Quality of Life: Patient Perspectives. Kidney Med, 4: 100437, 2022. [CrossRef]
- Edmonston DL, Isakova T, Dember LM, Brunelli S, Young A, Brosch R, Beddhu Chakraborty H, Wolf M: Design and Rationale of HiLo: A Pragmatic, Randomized Trial of Phosphate Management for Patients on Maintenance Hemodialysis. Am J Kidney Dis, 2020 10.1053/j.ajkd.2020.10.008.
- Wald R, Walsh MW: In Search of the Optimal Target for Phosphate Control: Episode 1. J Am Soc Nephrol Mar 2021;32(3):526-528. [CrossRef]
- Greg LP, Hedayati SS. Management of Traditional Cardiovascular Risk Factors in CKD: What Are the Data? Am J Kidney Dis. 72(5): 728-744. 2018. [CrossRef]
Figure 1.
Normal phosphate homeostasis.
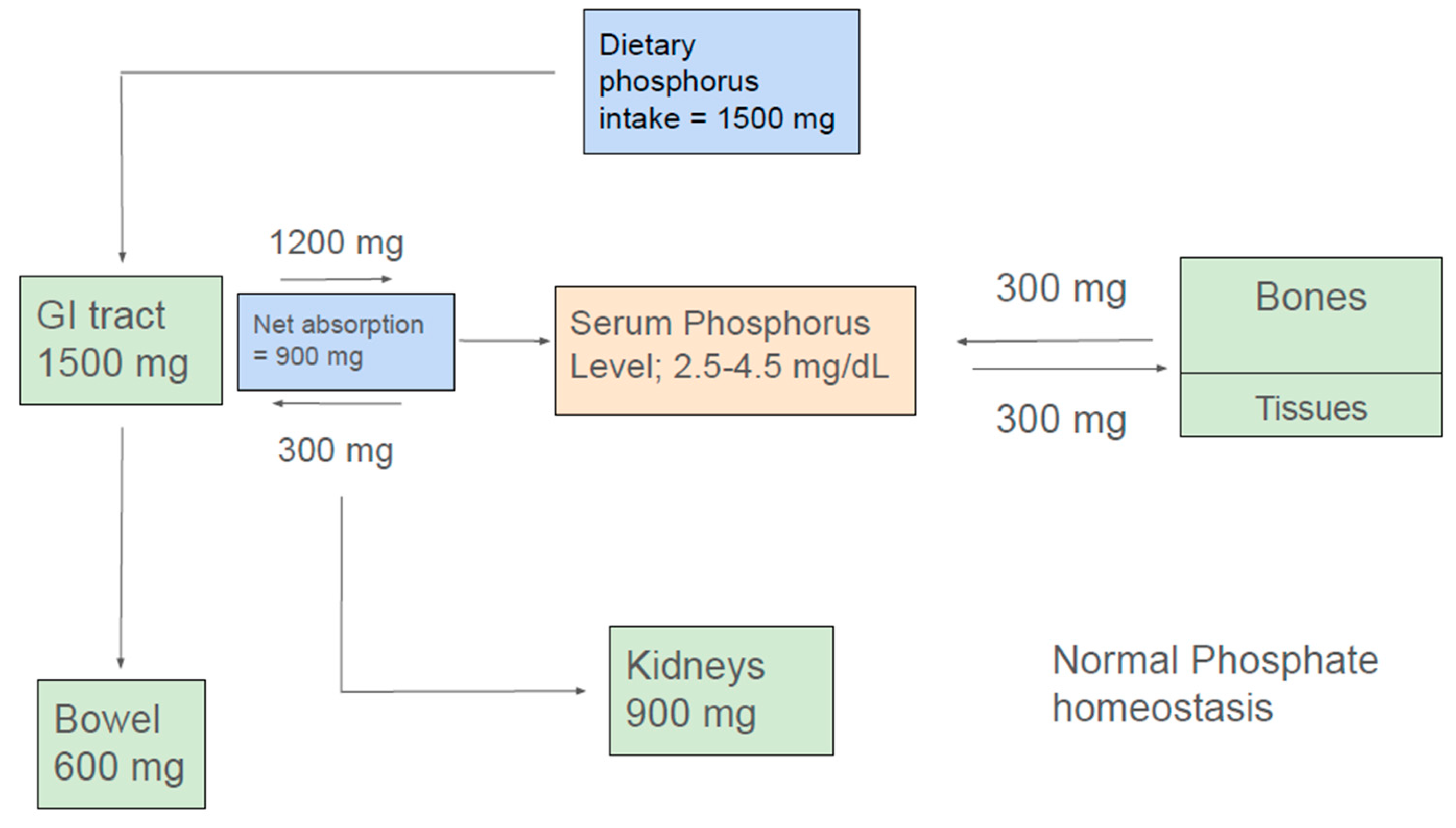
Figure 2.
Endocrine regulation of phosphate in kidney disease.
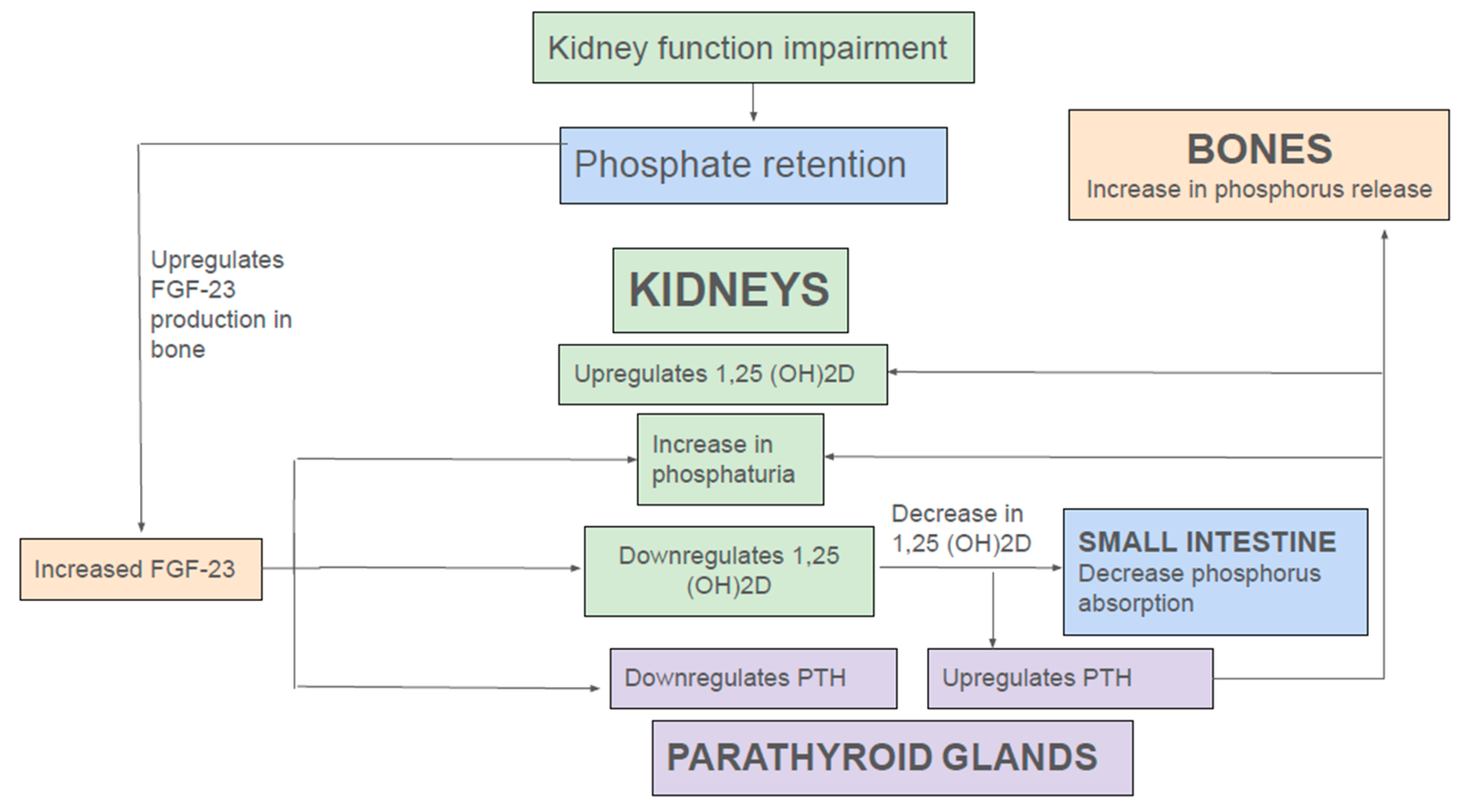

Table 1.
Current Strategies to Manage Hyperphosphatemia in chronic kidney disease and kidney failure.
Table 1.
Current Strategies to Manage Hyperphosphatemia in chronic kidney disease and kidney failure.
| Dietary interventions | Comments |
|---|---|
| Limit processed meats, processed cheese and dairy products | Contain highly bioavailable (80-100%) inorganic phosphates |
| Choose fresh meat or fish without added phosphates | Contain organic phosphates of intermediate bioavailability (40-60%) |
| Choose plant based food (Like legumes, soy products, nuts, whole grains) | Contain organic phosphate complexed with phytates. Has low bioavailability (20-40%) |
| Be aware of hidden phosphates in prescription/ over-the counter medicines and supplements | May contain considerable amount of highly bioavailable (80-100%) inorganic phosphates |
| Reduction of intestinal phosphate absorption | |
| Phosphate Binders | Comments |
| Calcium acetate: Daily dose 1334-2001 mg) | High pill burden. Risk of calcium load and vascular calcification. Cheap |
| Calcium carbonate: Daily dose 1250-3750 mg) | High pill burden. Risk of calcium load and vascular calcification. Cheap |
| Sevelamer hydrochloride and sevelamer carbonate: Daily dose 2400-9600 mg | High pill burden. Gastrointestinal side effects. More expensive than Ca-based binders |
| Lanthanum carbonate: Daily dose 1500 mg Sucroferric oxyhydroxide: Daily dose 7.5-15 g Ferric citrate: Daily dose 630-1260 mg |
Low pill burden. Chewable tablets or powder preparations. Gastrointestinal side effects. No evidence of hepatotoxicity Low pill burden. Chewable. Low iron absorption Primary Gastrointestinal side effects. Low pill burden. High iron absorption. May help with anemia management. Primary Gastrointestinal side effects. Risk of aluminum toxicity as citrate increases absorption of aluminum |
| Intestinal phosphate transport inhibitors | Comments |
| Nicotinamide Tenapanor: Dose 10-30 mg twice a day |
Blocks small intestinal active transport of phosphate via NaPi-IIb. Limited efficacy and poor tolerance due to side effects (Diarrhea, pruritus, and thrombocytopenia). Not recommended for hyperphosphatemia management Inhibits small intestinal paracellular transport of phosphate by blocking sodium/hydrogen exchanger 3. Used as an add-on therapy with phosphate binders. The major adverse effect is an increase in stool frequency and Diarrhea |
| Removal of phosphate by Dialysis | |
| Dialysis modality | Comments |
| In-center hemodialysis 3-times a week | Inadequate in removing daily phosphate load. Patients need additional measures (Diet and phosphate lowering agents) to manage hyperphosphatemia |
| Short-daily or nocturnal hemodialysis | Much better in controlling hyperphosphatemia |
| Peritoneal dialysis | Inadequate in removing daily phosphate load. Patients need additional measures (Diet and phosphate lowering agents) to manage hyperphosphatemia |
Table 2.
Dietary Sources of Phosphorus.
| Sources of Phosphate | Nature of Phosphate | Bioavailability | Examples |
|---|---|---|---|
| Plant-based Foods | Organic phosphates Complexed with phytates |
20-40% | Nuts and seeds Legumes Whole grain Leafy greens Soy products |
| Animal-based Foods | Organic Phosphates | 40-60% | Chicken/Poultry Red meat Fish/ sea food Milk/ dairy products |
| Preservatives/Additives | Inorganic Phosphates | 80-100% | Soft drinks Processed foods Canned foods |
| Medicines/ supplements | Inorganic Phosphates | 80-100% | OTC* multivitamins/supplements Prescription Medicines |
*OTC: Over-the-counter.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



