Submitted:
27 March 2025
Posted:
28 March 2025
You are already at the latest version
Abstract
Systemic sclerosis (also called scleroderma, SSc) is a chronic autoimmune fibrotic disorder affecting the skin and internal organs to variable extent and categorized as limited cutaneous subset, when distal areas of skin are involved and diffuse cutaneous subset when more extensive proximal skin involvement is seen. Multiple pathogenic mechanisms have been demonstrated, including production of disease-specific autoantibodies, infiltration of involved tissues by immune cells, as well as environmental factors triggering the onset such as solvents and viruses. Although not strongly familial , susceptibility to SSc is associated with single nucleotide polymorphisms in immunoregulatory genes. In addition, several lines of evidence demonstrate abnormalities within the epithelial cell layer in SSc. Macroscopically the epidermis is pigmented, thickened and stiff and strongly promotes myofibroblasts in 3D co-culture. Moreover, multiple activating factors and pathways have been implicated in epithelium, including wound healing responses, induction of DAMPS and the release of pro-fibrotic growth factors and cytokines. Similar to SSc, data from studies of cutaneous wound healing indicate a major role for epidermal keratinocytes in promoting underlying fibroblast responses and coordinating resolution of the wound. Since the epithelium is strongly exposed to environmental factors and richly endowed with protective immune cells, we have proposed that disease initiating mechanisms in SSc involve dysregulation of this cell layer. Treatments designed to quiesce this cell layer or to block epithelial-fibroblast cross-talk could be of benefit in this severe and resistant disease.
Keywords:
Systemic Sclerosis
; Epidermis
; Fibrosis
; Fibroblast
; EMT
; ECM
1. Introduction
Systemic sclerosis (SSc) is a heterogenous chronic multi-systemic disorder, characterised by autoimmunity, vascular dysfunction and progressive fibrosis which spreads in a continuous fashion, affecting the skin and internal organs, most notably the lungs[1]. Although the aetiology of SSc is not fully elucidated, there exists an underlying interplay between immune cell activation (innate and adaptive), endothelial cell dysfunction and activation of fibroblasts, leading to persistence of myofibroblasts responsible for the skin and organ fibrosis[2]. In the resulting fibrotic microenvironment there is excess synthesis of type I and III collagen, which elevates stiffness and mechanical stress within the local extracellular matrix (ECM). Activation of myofibroblasts is essential to normal wound healing where it is co-ordinated and self-limiting, whereas in SSc there is persistence of the activated state of the myofibroblasts, due to the formation of the stiff and growth factor-enhanced micro-environment[3].
There has been significant research into the immune abnormalities, the role of endothelial dysfunction and fibroblast abnormalities within SSc, but the potential contribution of epithelial cells has received less attention. Within the fibrotic lesions, the pathogenic myofibroblasts are derived from multiple sources including; local tissue-resident fibroblasts, infiltrating monocyte derived cells (“fibrocytes”) [4] subcutaneous fat derived mesenchymal stem cells (MSCs) [5,6], perivascular stem cells (pericytes)[7], transdifferentiating tissue resident cells such as endothelial cells undergoing endothelial to mesenchymal transition (endoMT)[8], but also a role for epithelial cells undergoing partial epithelial to mesenchymal transition (EMT)[9]. Variation in the relative contribution of activated fibroblasts from different sources could explain heterogeneity of this disease and may have implications for treatment.
Moreover, there is now emerging evidence suggesting that dysregulation of the epithelial layer may contribute to the pro-fibrotic response of SSc [9,10,11,12,13,14]. Epithelial dysfunction is already implicated in other fibrotic disease such as idiopathic lung fibrosis [15,16] and renal fibrosis [17]. It is also well-established that in restoration of tissue homeostasis after wound healing, the cross-talk between epithelial cells and fibroblasts have an important role to play[18]. In this review we will aim to discuss the potential contribution of the dysregulated epithelial cell layer in the pathogenesis of SSc.
2. Altered Differentiation of the Epidermis in Scleroderma: Activated Wound Healing Phenotype
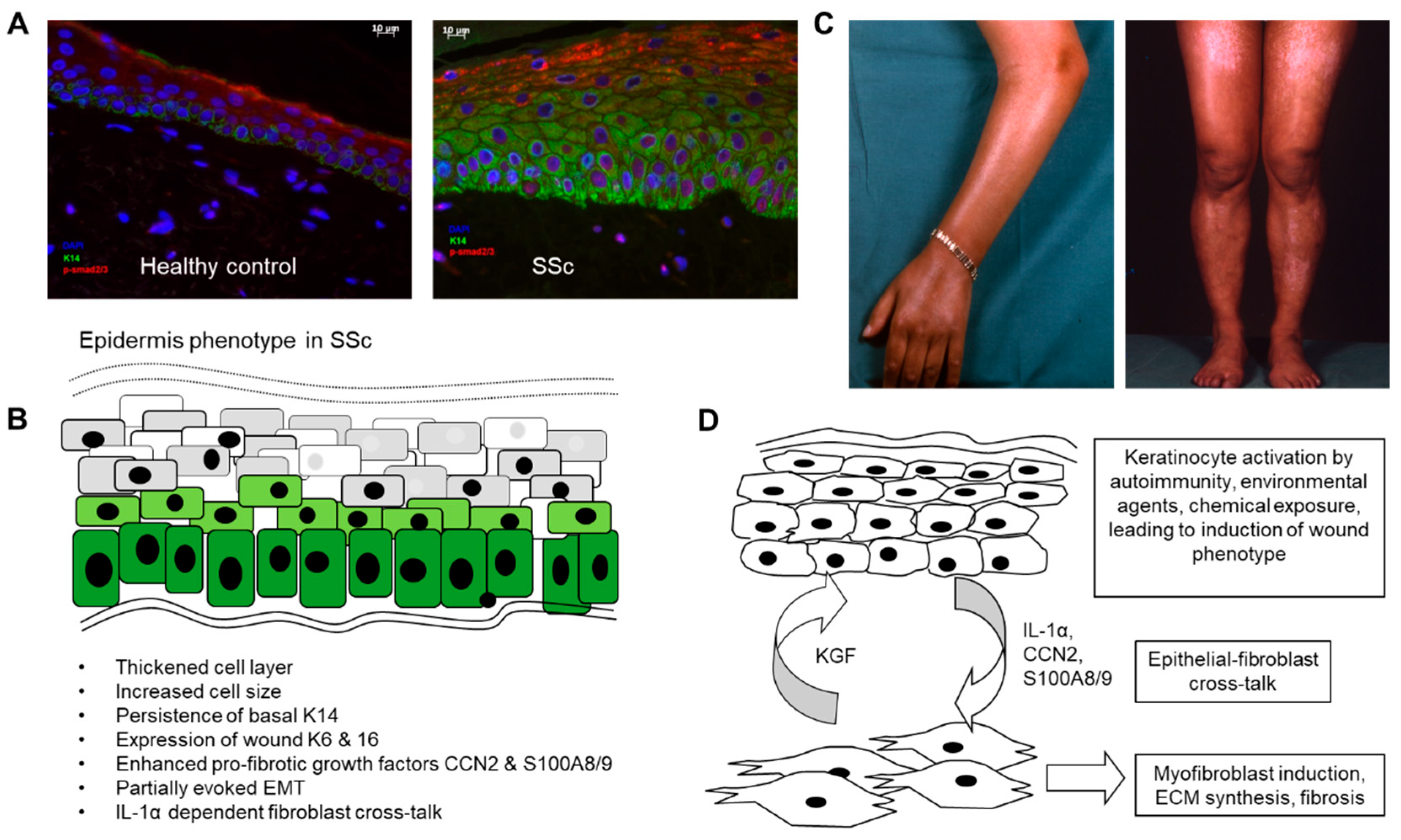
The epidermis is a highly specialised self-regenerating stratified squamous epithelium(reviewed in [19]). Keratinocytes are polar cells, in which function is dictated by micro-environment factors, including homo- and heterotypic cell-cell contact, cell-ECM adhesion, mechano-sensing and metabolic factors such as O2 level and pH[20]. In health the basal keratinocytes express cytokeratins 5 (K5) and 14 (K14), which are lost as the keratinocytes commit to differentiation and migrate upwards, switching to K1 and K10[21]. This process is markedly altered in the SSc epidermis where there is abnormal persistence of the basal K14 into the more superficial layers, combined with delayed expression of K10 [10,13]. Moreover, involucrin and loricrin, which have essential roles in keratinocyte differentiation involving the formation of the keratinocyte protein envelop, are altered in the SSc epidermis [12,14]. Keratinocyte maturation, size and epidermal thickness are consistently abnormal in biopsies from SSc patients [12] (Figure 1A).This pattern is recognised as a tissue repair/wound healing phenotype leading us and others to look for other changes characteristic of repair epithelial layers, such as thickening, and expression of cytokeratins usually restricted to wound environments [22].
The epidermis is involved in a co-ordinated wound-healing process that evolves over four stages; homeostasis, inflammation, proliferation, and remodelling[23]. Immediately after skin injury there is vascular constriction and fibrin clot formation. At this stage, disruption of the epidermal barrier causes release of pre-stored IL-1α by keratinocytes. Platelets degranulate into the damaged tissue, releasing PDGF, TFG-β and EGF. The formed clot provides a matrix for inflammatory cells infiltrating into the wound. IL-8 released downstream of Toll-like receptor (TLR) signalling and C3a act as chemokines for neutrophils, which are recruited into the wound and remove bacteria from the wound site. TFGβ released in the wound by the action of thrombin on LAP associated forms, promotes differentiation of infiltrating monocytes into macrophages. In the early inflammatory phase there is an M1 polarity, with macrophages releasing pro-inflammatory cytokines including IL-1β, 1L-6, followed by a switch to an M2 phenotype responsible for secretion of growth factors promoting fibroblast activation and neo-angiogenesis [24]. In the proliferative phase, or re-epithelialisation, keratinocytes become activated and they begin to express K6 and K16, which facilitate keratinocyte migration and enables the cells to withstand wound environments and cover wound defects[25]. Accordingly, in SSc, we have shown abnormal expression of K6 and K16 in epidermal keratinocytes[11] confirmed by other studies [13,14], consistent with an activated wound healing, pro-fibrotic phenotype occurring at the level of the epidermis.
Current data does not fully explain the altered keratinocytes phenotype in SSc. Profiling has demonstrated altered homeobox gene regulation suggesting fundamentally altered imprinting[14]. Pigmentary changes are also evident in SSc due to the enhanced delivery of pigmentary granules into the basal keratinocytes. Studies have attempted to explain and link these pigmentary changes to underlying pathogenic mechanisms in SSc [26]. The altered pigmentation is apparent in around 50 percent of SSccases, broadly categorised as being of two patterns: 1) vitiligo-like associated with perifollicular hyperpigmentation also termed “salt and pepper changes”, or 2) a more diffuse hyperpigmentation phenotype. The latter phenotype is typically seen in severe diffuse SSc patients with higher modified Rodnan skin scores, and also with a higher frequency of digital ulceration. Of relevance, absolute numbers of melanocytes are elevated in SSc patients of less than 5 years disease duration, and then fall in number with late stage SSc (27). CCN3 (also called NOV) a regulatory matricellular protein of reduced activity in fibrosis, is found to decrease in melanocytes in SSc as a potential contributory change [27]. The racial background is also important, with SSc associated pigmentary changes more prevalent in non-Caucasians [28]. It is plausable that elevated melanocyte content in SSc is induced by more chemotaxis of melanocytes or enhanced survival in the SSc epithelial layer. In our own work, we have highlighted increased stem cell factor (SCF, Kit-ligand) in SSc epidermis, which may recruit melanocytes since they are known to express the receptor c-Kit[29].
3. Release of Pro-Inflammatory and Pro-Fibrotic Factors by the SSc Epidermis
The essential role of the epidermis in innate and adaptive immunity is firmly established based on multiple publications and reviewed [30]). This cell layer represents a major innate immune barrier to invading pathogens and moreover is a site where highly specific adaptive immune responses can be initiated, as seen in the use of intra-epidermal vaccines[31]. Within the epidermal layers there are important immune cell populations including Langerhans cells, resident lymphocyte populations and keratinocytes themselves which may act as antigen presenting cells.
The functions of immune cells and endothelial cells can be influenced by exosomes released by keratinocytes as these cells are capable of secreting chemo-attractants, anti-microbials, cytokines as well as DAMPs. The functions of keratinocytes as first line of defence is mediated by the expression of receptors for DAMPS and PAMPS including cell surface and endosomal TLRS and intracellular pathways such as the NOD-like receptors, which confer responsiveness to viral nucleic acids, bacterial cell wall components and endotoxins. Once triggered via these pathways, keratinocytes release cytokines such as type-I interferons and chemo-attractants including IL-8. Furthermore, keratinocytes contain a reservoir of pre-formed IL-1α which they release upon activation, stimulating in an autocrine fashion ans signalling to adjacent dermal fibroblasts in a paracrine fashion to promote wound healing responses and KGF release by the fibroblasts [18,32]. The importance of keratinocyte derived IL-1α in epithelial-fibroblast cross talk has been demonstrated in several papers [11,14] and moreover in SSc, the feedback from dermal fibroblast synthesis of KGF has been confirmed as promoting the keratinocyte activation (Figure 1D).
Aberrant expression of both pro-inflammatory and pro-fibrotic factors has been demonstrated in and adjacent to the SSc epidermis (summarised in Figure 1B). Demonstrated by profiling of candidate secreted factors, SSc epidermis synthesises the matricellular protein CCN2 (also called CTGF) which is found deposited at the epidermal-dermal interface [12]. Over-expression of CCN2 is thought to be a hallmark of fibrotic processes, acting as a modifier of cell-matrix interactions [33]. Though those studies look at the fibroblast response, the work of Nikitorowicz-Buniak et al, showed more pronounced presence of CCN2 in the epidermal-dermal junction in those with early SSc, suggesting that epidermal production of CCN2 could contribute to the pro-fibrotic response in SSc [12]. TGFβ is a major pro-fibrotic growth factor and believed to have a role in SSc fibrosis[34]. However, when assayed, LAP-associated and total levels of TGFβ were not elevated in SSc epidermis [11], and furthermore the activated keratinocyte phenotype appears independent of this growth factor [35].
As mentioned above, IL-1α is considered a key initiator of keratinocyte activation during wound healing and of interest IL1-α polymorphisms are known to be linked to increased risk and severity of SSc[36]. Moreover, the receptor IL-1R is overexpressed by SSc fibroblasts and inhibition of IL-1R leads to decreased release of IL-6 and PDGF-A in those fibroblasts, consistent with a paracrine pathway between keratinocytes and dermal fibroblasts [37]. In tissue culture models SSc keratinocytes are able to stimulate fibroblasts through IL-1α release leading to IL-6 and IL-8 release by fibroblasts[14]. Despite these preclinical data, when studied in patients riloacapt, an IL-1 receptor biologic, failed to reduce skin score or drop IL-6 levels in SSc patients [38].
Another potential pathway involving epithelial-fibroblast cross talk is through S100A9 and its receptor TLR-4. S100A9 is calcium binding protein often present as a heterodimer with S100A8, released by activated myeloid cells as well as epithelial cells, promoting inflammatory cell recruitment and inducing cytokine release[39]. In fact in SSc overexpression of S100A9 has been confirmed in the epidermis, not seen in healthy controls. Moreover, when stimulated by S100A9 SSc fibroblasts oversecrete CCN2 , the matricellular pro-fibrotic protein[12]. With the proliferation of singe cell RNAseq studies in SSc overexpression of wound cytokeratins as well as high levels of S100A8 have been confirmed in SSc keratinocytes[13].
4. Cross-Talk and Transition Between Epithelial Cells and Mesenchymal Cells in SSc
Epithelial to mesenchymal transition (EMT) is a process of trans-differentiation in which epithelial cells lose polarity and switch to become a mesenchymal cell, occurring during embryogenesis (Type I EMT), during tissue regeneration and fibrosis (Type 2), and during cancer invasion (Type 3) [40]. Biomarkers exist to identify the different subtypes of EMT.
TGFβ, implicated in SSc fibrosis, is a known key driver of EMT: many studies confirming in tissue culture that this factor can induce mesenchymal markers in epithelial cells (44,45).
In fact in SSc tissues, epithelial cells do stain positive for FSP-1 and vimentin [9], and also demonstrate increased SNAI1, but not SNAI2,consistent with partially invoked EMT, without full commitment or invasion into the dermis. Additionally, data implicating SFRP4, a Wnt modulator, released by keratinocytes undergoing EMT [41,42]. It is possible therefore that cells undergoing EMT in the epidermis contribute some of the pro-fibrotic tendency in SSc [48] in a process involving TFGβ and Wnt signalling.
Further evidence for a pro-fibrotic cross talk, a study by McCoy et al was able to demonstrate increased expression of COL1A1 and α-SMA in healthy fibroblasts incubated with conditioned media from SSc keratinocytes[35].
As part of normal skin homeostasis, there is keratinocyte-fibroblast cross talk maintaining a fully differentiated, functional epidermis with normal lipid content and skin permeability. Early work demonstrated a double paracrine mechanism involved in the regulation of tissue repair, where fibroblasts secrete KGF, which stimulate keratinocytes to produce 1L-α and this in return stimulates fibroblast activation and production of KGF [43].
More recent work has shown that SSc fibroblasts show increased expression of KGF (likely through epigenetic alterations since it persists in tissue culture). It was demonstrated in SSc that fibroblast derived KGF induces oncostatin M (OSM) production by keratinocytes which in term leads to OSM mediated fibroblast activation through the phosphorylation of STAT3. This leads to increased collagen production and urokinase-type plasminogen activator (uPA), which enhances fibroblast migration[44]. This work further supports aberrancy in the epidermal-fibroblasts cross talk in SSc, since when normal keratinocytes are co-cultured with normal fibroblasts no increased STAT3 mediated activation of fibroblasts was seen.
Possible initiating biomechanisms leading to epidermis activation in SSc:
Whilst the evidence as presented, for abnormal activation of the keratinocyte layer in SSc is compelling, the initiating mechanisms remain to be determined. Possible explanations for would include diffusion of growth factors and cytokines from the underlying disease dermis, effects of the mechano-stimulation from the enhanced ECM stiffness, or else autoimmune or environmental factors. In a previous edition of “Cells”, we present data showing that SSc IgG autoantibodies bind and activate keratinocytes leading to IL-1α release. These mechanisms resemble the effects of anti-endothelial cell antibodies believed to have an initiating role in vascular damage in SSc [45].
In idiopathic lung fibrosis, outside the setting of SSc, epithelial-fibroblast interactions are now established as being crucial to the fibrotic process. Environmental exposures leading to injured alveolar epithelial cells, trigger myofibroblast activation and over-secretion of stiff ECM, creating a pro-fibrotic niche. It is possible that an “inside-out” type mechanism is occurring in some SSc patients through endothelial cell damage leading to paracrine fibrosis, whilst in others an “outside-in” mechanism resulting from epithelial cells damage and epithelial-fibroblast cross talk drives the disease process.
As alluded to already the epidermis is also richly endowed with immune cells including resident dendritic cells and transiting lymphocyte populations that could be initiating responses in this layer. Moreover, the effects of triggering injurious environmental factors such as industrial solvents might maximally induce innate responses in the epidermal cell layer [46].
Future directions
As the technology underlying biomedical research has progressed, ever more sophisticated approaches to understanding SSc as a disease process have been achievable. Latest profiling methodologies such as single cell, ATAC-seq and proteomics can be applied to the SSc epidermal cell layer in isolation, especially since it is readily available for biopsy or sampling. The high prevalence of gut and interstitial lung involvement could be explained though by the importance of their having a potentially activated or damaged epithelial cell layer.
Also, with the application of biologic therapies of known specific mode of action, tissues including the epithelial layer can be profiled before and after therapy to gain insights into the underlying molecular pathways involved as illustrated in [47].
Finally, targeted treatment could be preferentially delivered into the epithelial by topical application, microneedle delivery or by inhaled nebulised route. Improvement in the underlying deeper fibroblast rich tissues could implicate the epithelia as a driving influence.
References
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet (London, England) 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic sclerosis. Nature reviews Disease primers 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Shiwen, X.; Stratton, R.; Nikitorowicz-Buniak, J.; Ahmed-Abdi, B.; Ponticos, M.; Denton, C.; Abraham, D.; Takahashi, A.; Suki, B.; Layne, M.D.; et al. A Role of Myocardin Related Transcription Factor-A (MRTF-A) in Scleroderma Related Fibrosis. PLoS One 2015, 10, e0126015. [Google Scholar] [CrossRef] [PubMed]
- Grieb, G.; Bucala, R. Fibrocytes in Fibrotic Diseases and Wound Healing. Advances in wound care 2012, 1, 36–40. [Google Scholar] [CrossRef]
- Marangoni, R.G.; Korman, B.D.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis & rheumatology (Hoboken, N.J.) 2015, 67, 1062–1073. [Google Scholar] [CrossRef]
- Horsley, V. Adipocyte plasticity in tissue regeneration, repair, and disease. Curr Opin Genet Dev 2022, 76, 101968. [Google Scholar] [CrossRef]
- Rajkumar, V.S.; Sundberg, C.; Abraham, D.J.; Rubin, K.; Black, C.M. Activation of microvascular pericytes in autoimmune Raynaud's phenomenon and systemic sclerosis. Arthritis and rheumatism 1999, 42, 930–941. [Google Scholar] [CrossRef]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. The American journal of pathology 2015, 185, 1850–1858. [Google Scholar] [CrossRef]
- Nikitorowicz-Buniak, J.; Denton, C.P.; Abraham, D.; Stratton, R. Partially Evoked Epithelial-Mesenchymal Transition (EMT) Is Associated with Increased TGFbeta Signaling within Lesional Scleroderma Skin. PLoS One 2015, 10, e0134092. [Google Scholar] [CrossRef]
- Aden, N.; Shiwen, X.; Aden, D.; Black, C.; Nuttall, A.; Denton, C.P.; Leask, A.; Abraham, D.; Stratton, R. Proteomic analysis of scleroderma lesional skin reveals activated wound healing phenotype of epidermal cell layer. Rheumatology (Oxford, England) 2008, 47, 1754–1760. [Google Scholar] [CrossRef]
- Aden, N.; Nuttall, A.; Shiwen, X.; de Winter, P.; Leask, A.; Black, C.M.; Denton, C.P.; Abraham, D.J.; Stratton, R.J. Epithelial cells promote fibroblast activation via IL-1alpha in systemic sclerosis. The Journal of investigative dermatology 2010, 130, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Nikitorowicz-Buniak, J.; Shiwen, X.; Denton, C.P.; Abraham, D.; Stratton, R. Abnormally differentiating keratinocytes in the epidermis of systemic sclerosis patients show enhanced secretion of CCN2 and S100A9. The Journal of investigative dermatology 2014, 134, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, J.S.; Tabib, T.; Xiao, H.; Sadej, G.M.; Khanna, D.; Fuschiotti, P.; Lafyatis, R.A.; Das, J. Cell Type-Specific Biomarkers of Systemic Sclerosis Disease Severity Capture Cell-Intrinsic and Cell-Extrinsic Circuits. Arthritis & rheumatology (Hoboken, N.J.) 2023. [CrossRef]
- Russo, B.; Borowczyk, J.; Boehncke, W.H.; Truchetet, M.E.; Modarressi, A.; Brembilla, N.C.; Chizzolini, C. Dysfunctional Keratinocytes Increase Dermal Inflammation in Systemic Sclerosis: Results From Studies Using Tissue-Engineered Scleroderma Epidermis. Arthritis & rheumatology (Hoboken, N.J.) 2021, 73, 1311–1317. [Google Scholar] [CrossRef]
- Wolters, P.J.; Blackwell, T.S.; Eickelberg, O.; Loyd, J.E.; Kaminski, N.; Jenkins, G.; Maher, T.M.; Molina-Molina, M.; Noble, P.W.; Raghu, G.; et al. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? The Lancet. Respiratory medicine 2018, 6, 154–160. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Zeisberg, M.; Kalluri, R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med (Berl) 2004, 82, 175–181. [Google Scholar] [CrossRef]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-fibroblast interactions in wound healing. The Journal of investigative dermatology 2007, 127, 998–1008. [Google Scholar] [CrossRef]
- Watt, F.M. Mammalian skin cell biology: at the interface between laboratory and clinic. Science (New York, N.Y.) 2014, 346, 937–940. [Google Scholar] [CrossRef]
- Louis, B.; Tewary, M.; Bremer, A.W.; Philippeos, C.; Negri, V.A.; Zijl, S.; Gartner, Z.J.; Schaffer, D.V.; Watt, F.M. A reductionist approach to determine the effect of cell-cell contact on human epidermal stem cell differentiation. Acta Biomater 2022, 150, 265–276. [Google Scholar] [CrossRef]
- Fuchs, E.; Green, H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell 1980, 19, 1033–1042. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, M.; Zhang, L.J. Keratin 6, 16 and 17-Critical Barrier Alarmin Molecules in Skin Wounds and Psoriasis. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N Engl J Med 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R.J.; Lantis, J.; Kirsner, R.S.; Shah, V.; Molyneaux, M.; Carter, M.J. Macrophages: A review of their role in wound healing and their therapeutic use. Wound Repair Regen 2016, 24, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Osumi, M.; Ohnishi, T.; Ichikawa, E.; Takahashi, H. Changes in cytokeratin expression in epidermal keratinocytes during wound healing. Histochemistry and cell biology 1995, 103, 425–433. [Google Scholar] [CrossRef]
- Tabata, H.; Hara, N.; Otsuka, S.; Yamakage, A.; Yamazaki, S.; Koibuchi, N. Correlation between diffuse pigmentation and keratinocyte-derived endothelin-1 in systemic sclerosis. Int J Dermatol 2000, 39, 899–902. [Google Scholar] [CrossRef]
- Henrot, P.; Pain, C.; Taieb, A.; Truchetet, M.E.; Cario, M. Dysregulation of CCN3 (NOV) expression in the epidermis of systemic sclerosis patients with pigmentary changes. Pigment Cell Melanoma Res 2020, 33, 895–898. [Google Scholar] [CrossRef]
- Nusbaum, J.S.; Gordon, J.K.; Steen, V.D. African American race associated with body image dissatisfaction among patients with systemic sclerosis. Clinical and experimental rheumatology 2016, 34 Suppl 100, 70–73. [Google Scholar]
- Ahmed Abdi, B.; Lopez, H.; Karrar, S.; Renzoni, E.; Wells, A.; Tam, A.; Etomi, O.; Hsuan, J.J.; Martin, G.R.; Shiwen, X.; et al. Use of Patterned Collagen Coated Slides to Study Normal and Scleroderma Lung Fibroblast Migration. Scientific reports 2017, 7, 2628. [Google Scholar] [CrossRef]
- Piipponen, M.; Li, D.; Landen, N.X. The Immune Functions of Keratinocytes in Skin Wound Healing. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Kim, Y.C.; Quan, F.S.; Yoo, D.G.; Compans, R.W.; Kang, S.M.; Prausnitz, M.R. Improved influenza vaccination in the skin using vaccine coated microneedles. Vaccine 2009, 27, 6932–6938. [Google Scholar] [CrossRef]
- Werner, S.; Smola, H. Paracrine regulation of keratinocyte proliferation and differentiation. Trends Cell Biol 2001, 11, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Shi-wen, X.; Abraham, D.J.; Leask, A. CCN2 is required for bleomycin-induced skin fibrosis in mice. Arthritis and rheumatism 2011, 63, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Lafyatis, R. Transforming growth factor beta--at the centre of systemic sclerosis. Nature reviews. Rheumatology 2014, 10, 706–719. [Google Scholar] [CrossRef] [PubMed]
- McCoy, S.S.; Reed, T.J.; Berthier, C.C.; Tsou, P.S.; Liu, J.; Gudjonsson, J.E.; Khanna, D.; Kahlenberg, J.M. Scleroderma keratinocytes promote fibroblast activation independent of transforming growth factor beta. Rheumatology (Oxford, England) 2017, 56, 1970–1981. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Tochimoto, A.; Ichikawa, N.; Harigai, M.; Hara, M.; Kotake, S.; Kitamura, Y.; Kamatani, N. Association of IL1A gene polymorphisms with susceptibility to and severity of systemic sclerosis in the Japanese population. Arthritis and rheumatism 2003, 48, 186–192. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Harigai, M.; Suzuki, K.; Hara, M.; Kobayashi, K.; Ishizuka, T.; Matsuki, Y.; Tanaka, N.; Nakamura, H. Interleukin 1 receptor on fibroblasts from systemic sclerosis patients induces excessive functional responses to interleukin 1 beta. Biochem Biophys Res Commun 1993, 190, 154–161. [Google Scholar] [CrossRef]
- Mantero, J.C.; Kishore, N.; Ziemek, J.; Stifano, G.; Zammitti, C.; Khanna, D.; Gordon, J.K.; Spiera, R.; Zhang, Y.; Simms, R.W.; et al. Randomised, double-blind, placebo-controlled trial of IL1-trap, rilonacept, in systemic sclerosis. A phase I/II biomarker trial. Clinical and experimental rheumatology 2018, 36 Suppl 113, 146-149.
- Foell, D.; Wittkowski, H.; Vogl, T.; Roth, J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol 2007, 81, 28–37. [Google Scholar] [CrossRef]
- Kalluri, R. EMT: when epithelial cells decide to become mesenchymal-like cells. The Journal of clinical investigation 2009, 119, 1417–1419. [Google Scholar] [CrossRef]
- Bayle, J.; Fitch, J.; Jacobsen, K.; Kumar, R.; Lafyatis, R.; Lemaire, R. Increased expression of Wnt2 and SFRP4 in Tsk mouse skin: role of Wnt signaling in altered dermal fibrillin deposition and systemic sclerosis. The Journal of investigative dermatology 2008, 128, 871–881. [Google Scholar] [CrossRef]
- Tinazzi, I.; Mulipa, P.; Colato, C.; Abignano, G.; Ballarin, A.; Biasi, D.; Emery, P.; Ross, R.L.; Del Galdo, F. SFRP4 Expression Is Linked to Immune-Driven Fibrotic Conditions, Correlates with Skin and Lung Fibrosis in SSc and a Potential EMT Biomarker. J Clin Med 2021, 10. [Google Scholar] [CrossRef]
- Maas-Szabowski, N.; Shimotoyodome, A.; Fusenig, N.E. Keratinocyte growth regulation in fibroblast cocultures via a double paracrine mechanism. Journal of cell science 1999, 112 ( Pt 12), 1843–1853. [Google Scholar] [CrossRef]
- Canady, J.; Arndt, S.; Karrer, S.; Bosserhoff, A.K. Increased KGF expression promotes fibroblast activation in a double paracrine manner resulting in cutaneous fibrosis. The Journal of investigative dermatology 2013, 133, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.; Savage, C.O.; Isenberg, D.; Pearson, J.D. IgG anti-endothelial cell autoantibodies from patients with systemic lupus erythematosus or systemic vasculitis stimulate the release of two endothelial cell-derived mediators, which enhance adhesion molecule expression and leukocyte adhesion in an autocrine manner. Arthritis and rheumatism 1999, 42, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Muntyanu, A.; Milan, R.; Rahme, E.; Baron, M.; Netchiporouk, E.; Canadian Scleroderma Research, G. Organic solvent exposure and systemic sclerosis: A retrospective cohort study based on the Canadian Scleroderma Research Group registry. J Am Acad Dermatol 2023. [CrossRef] [PubMed]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet (London, England) 2016, 387, 2630–2640. [Google Scholar] [CrossRef]
Figure 1.
Role of the epidermis in systemic sclerosis (A) Altered differentiation and activation of the epidermal cell layer in SSc. SSc epidermis was found to be thickened and to have abnormal persisting expression of cytokeratin 14 into suprabasal layers (green stain). pSMAD2/3 indicating active TGFβ signalling was enhanced in the disease epidermis (red stain). (B) Summary of phenotypic changes described in various published articles and (C) pigmentary changes due to dysregulated melanosis in the epidermal layer. (D) Proposed mechanisms linking epidermal activation to the underlying fibrotic process in SSc.
Figure 1.
Role of the epidermis in systemic sclerosis (A) Altered differentiation and activation of the epidermal cell layer in SSc. SSc epidermis was found to be thickened and to have abnormal persisting expression of cytokeratin 14 into suprabasal layers (green stain). pSMAD2/3 indicating active TGFβ signalling was enhanced in the disease epidermis (red stain). (B) Summary of phenotypic changes described in various published articles and (C) pigmentary changes due to dysregulated melanosis in the epidermal layer. (D) Proposed mechanisms linking epidermal activation to the underlying fibrotic process in SSc.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



