
Submitted:
18 March 2025
Posted:
19 March 2025
You are already at the latest version
Abstract
Parkinson’s disease encompasses a spectrum of motor and non-motor manifestations, with the latter often preceding motor symptoms and influencing disease trajectory. Sensory dysfunction, sleep-related disorders, autonomic impairment, neuropsychiatric symptoms, and cognitive decline reflect widespread neurodegeneration involving the brainstem, limbic system, and peripheral nervous system. Hyposmia and visual dysfunction are emerging as early biomarkers, while chronic pain and paraesthesias indicate central and peripheral nociceptive dysregulation. Sleep-related disorders, particularly REM sleep behaviour disorder and insomnia, frequently arise in the prodromal phase, highlighting disruptions in circadian regulation and neuroanatomical integrity. Neuropsychiatric symptoms—including depression, anxiety, apathy, and hallucinations—result from complex dysregulation of dopaminergic, serotonergic, and noradrenergic pathways, compounding disease burden and impairing quality of life. Autonomic dysfunction, affecting cardiovascular, gastrointestinal, thermoregulatory, and genitourinary systems, underscores the systemic nature of the disease. Despite growing recognition of these non-motor features, treatment strategies remain limited, constrained by a dopamine-centric therapeutic framework that does not fully address broader neurobiological dysfunction. The lack of reliable biomarkers hampers early diagnosis and precision medicine approaches, while the conventional dichotomy between motor and non-motor symptoms has hindered the development of integrative treatment paradigms. While previous reviews have examined individual non-motor manifestations in detail, this article provides a comprehensive synthesis, offering a clinically relevant perspective for medical students, primary care physicians, and non-specialists. Recognizing non-motor symptoms as core disease manifestations rather than secondary sequelae is essential to refine diagnostic criteria and develop targeted therapies. Parkinson’s disease is a complex, multisystem neurodegenerative disorder that necessitates a paradigm shift in research priorities and clinical management. A broader, integrative perspective is required to redefine how the disease is understood, taught, and treated, ensuring that motor and non-motor symptoms are addressed to improve patient outcomes.
Keywords:
Parkinson’s disease
; non-motor symptoms
; sensory dysfunction
; sleep disturbances
; neuropsychiatric disorders
; autonomic dysfunction
; REM sleep behaviour disorder
; pain
; cognitive impairment
; biomarkers
1. Introduction
Parkinson’s disease is no longer solely defined by its motor symptoms. While tremor, rigidity, and slowness of movement remain central to clinical diagnosis, non-motor symptoms often precede motor onset by many years, shaping disease progression, complicating management, and reducing quality of life [1,2,3,4,5,6]. Sensory dysfunction, autonomic failure, sleep-related disorders, neuropsychiatric symptoms, and cognitive decline are not incidental comorbidities but fundamental aspects of Parkinson’s disease [2,3,4,5,6]. These symptoms frequently emerge in the prodromal stage and, in many cases, contribute more significantly to disability than movement impairment itself. Despite their clinical significance, non-motor symptoms remain underrecognized, underdiagnosed, and unresponsive, primarily to dopamine-based therapies. The involvement of multiple neurotransmitter systems—including serotonin, norepinephrine, and acetylcholine pathways—demands a departure from the classical view of Parkinson’s disease as a disorder confined to dopamine depletion in the basal ganglia, requiring a broader understanding of its neurobiological underpinnings [1,2,3,4,5,6].
The understanding of Parkinson’s disease has evolved considerably over time. In 1912, Friedrich Lewy described extranigral proteinaceous inclusions, later termed Lewy bodies, providing the first evidence that Parkinson’s disease pathology extends beyond the substantia nigra [2,3,4,5,6]. This discovery challenged the notion of Parkinson’s disease as a purely motor disorder, laying the foundation for subsequent research into its widespread neurodegenerative processes. The identification of α-synuclein aggregates as a defining feature of the disease [7,8] led to Braak’s staging hypothesis [9], which proposed a progressive spread of pathology from the olfactory bulb and autonomic nuclei to the midbrain and cortical regions (Figure 1). This model provided a neuropathological framework to explain why symptoms such as impaired sense of smell, REM sleep behaviour disorder, and autonomic dysfunction often precede movement impairment by decades [2,3,4,5,6]. However, Braak’s hypothesis remains controversial. Not all individuals with Parkinson’s disease follow this sequential pattern, and the accumulation of α-synuclein does not always correlate with neurodegeneration or gliosis [9,10]. Additionally, alternative mechanisms—including neuroinflammation, mitochondrial dysfunction, and disruption of the gut-brain axis—challenge the notion of Parkinson’s disease as a single-pathway neurodegenerative disorder.
Non-motor symptoms in Parkinson’s disease are increasingly recognized as integral to its pathophysiology, influencing disease trajectory and quality of life [2,3,4,5,6]. While previous reviews have examined individual non-motor manifestations in detail, this article provides a comprehensive synthesis, offering a clinically relevant perspective for medical students, primary care physicians, and non-specialists (Figure 1 and Figure 2). Rather than an exhaustive account of each symptom, this review integrates current evidence on sensory dysfunction, autonomic impairment, sleep-related disorders, neuropsychiatric symptoms, and cognitive decline, highlighting their mechanistic overlap and therapeutic implications.
The expanding understanding of non-motor symptoms has necessitated a shift in research priorities and clinical management. These features are no longer considered secondary to the motor phenotype but central to refining diagnostic criteria, guiding targeted interventions, and reinterpreting Parkinson’s disease as a complex, multisystem neurodegenerative disorder. A deeper appreciation of these interconnections is essential to advancing precision medicine approaches and improving patient outcomes.
2. Autonomic Dysfunction in Parkinson's Disease
Autonomic dysfunction is a pervasive and debilitating feature of Parkinson's disease, affecting nearly every physiological system (see Table 1). More than 90% of patients develop autonomic symptoms, often preceding motor manifestations and progressively worsening as the disease advances [11]. Unlike motor symptoms primarily resulting from dopaminergic degeneration in the basal ganglia, autonomic dysfunction stems from widespread neurodegeneration across the central and peripheral nervous systems. The accumulation of α-synuclein in autonomic ganglia and peripheral nerves contributes to sympathetic and parasympathetic regulation dysfunction, impairing cardiovascular stability, gastrointestinal motility, urinary control, and thermoregulation [12]. Despite its profound impact on quality of life, autonomic dysfunction remains an underappreciated and underdiagnosed aspect of Parkinson's disease, often recognized only when symptoms become severe.
Cardiovascular dysregulation manifests as orthostatic hypotension, cardiac arrhythmias, and impaired vasomotor control, placing patients at increased risk of falls and cardiovascular complications [13,14]. Gastrointestinal dysfunction emerges early in the disease course, with symptoms such as sialorrhea, dysphagia, and constipation frequently appearing years before motor symptoms [13,14]. These symptoms reflect local neurodegeneration in the enteric nervous system and broader autonomic failure, particularly in parasympathetic pathways. Urinary dysfunction, including incontinence and urinary retention, further complicates the disease, often exacerbated by both autonomic failure and the side effects of dopaminergic therapies [13,14]. Cutaneous manifestations, such as seborrhoea, hyperhidrosis, and anhidrosis, highlight the widespread nature of autonomic impairment, as do thermoregulatory disturbances that result in heat intolerance or paradoxical hypothermia. Electrophysiological studies confirm the multi-organ involvement of autonomic dysfunction in Parkinson's disease, reinforcing the view that this disorder extends far beyond the motor system [15].
Despite its high prevalence and clinical significance, autonomic dysfunction in Parkinson's disease remains poorly understood and inadequately treated [13,14]. The traditional dopamine-centric approach to therapy fails to address the widespread neurochemical deficits underlying these symptoms. Current treatments offer only symptomatic relief, with no disease-modifying strategies available to mitigate the underlying autonomic degeneration. The continued fragmentation of research—dividing Parkinson's disease into motor and non-motor domains rather than considering it as a multisystem disorder—has hindered progress in developing targeted interventions. Future research must move beyond the basal ganglia and recognize that autonomic dysfunction is not a peripheral consequence of Parkinson's disease but a core component of its pathology. The failure to integrate autonomic dysfunction into routine diagnostic and therapeutic frameworks reflects a broader limitation in the clinical approach to Parkinson's disease, one that must be urgently addressed to improve patient outcomes (Figure 1 and Figure 2).
2.1. Sympathetic Autonomic Nervous System
The sympathetic autonomic nervous system undergoes profound dysfunction in Parkinson's disease, often manifesting before the onset of motor symptoms. Cardiovascular instability, characterized by orthostatic hypotension and impaired baroreflex function, emerges as a primary clinical consequence of sympathetic failure [16]. These abnormalities result from both preganglionic lesions affecting central autonomic regulation and postganglionic degeneration, leading to widespread sympathetic denervation [13]. The accumulation of α-synuclein and the formation of Lewy bodies within the sympathetic nervous system underscore the systemic nature of Parkinson's pathology, extending far beyond the basal ganglia and into peripheral autonomic circuits [17].
The presence of α-synuclein aggregates in the enteric nervous system, sympathetic ganglia, and postganglionic neurons of the heart, peri-adrenal tissues, and skin suggests a widespread autonomic pathology contributing to the disease's multisystem involvement [18]. Autonomic structures embryologically derived from the neural crest—including the adrenal gland, spinal ganglia, and sympathetic chain ganglia—also exhibit α-synuclein accumulation, indicating that neurodegeneration in Parkinson's disease is not confined to the central nervous system [19]. The degeneration of sympathetic nerve terminals results in norepinephrine deficiency, particularly in the heart and vasculature, which contributes to orthostatic hypotension and impairs compensatory vasoconstriction [20].
Pathological studies confirm that α-synuclein deposits selectively target sympathetic preganglionic neurons in the lateral horn of the thoracic spinal cord while sparing other nuclei, such as Clarke's dorsal nucleus. Similarly, the celiac ganglion, a key structure regulating postganglionic sympathetic output, exhibits significant α-synuclein accumulation, reinforcing the notion that Parkinson's disease involves both central and peripheral autonomic dysfunction [10]. Despite the clear pathological evidence linking sympathetic dysfunction to Parkinson's progression, therapeutic approaches remain largely symptomatic, focusing on managing orthostatic hypotension with volume expansion and vasopressors rather than targeting the underlying neurodegeneration.
The widespread involvement of the sympathetic nervous system in Parkinson's disease presents a crucial yet overlooked opportunity for early diagnosis and intervention. Biomarkers of sympathetic denervation, including cardiac imaging of norepinephrine uptake and skin biopsies detecting peripheral α-synuclein aggregates, could provide valuable insights into disease progression. However, the clinical translation of these findings remains slow, reflecting the broader failure to integrate autonomic dysfunction into Parkinson's disease management. Research efforts continue to focus overwhelmingly on motor symptoms despite clear evidence that autonomic failure significantly impacts morbidity and quality of life. Without a paradigm shift recognizing the full scope of Parkinson's neurodegeneration, patients will remain underserved, and therapeutic progress will remain stagnant.
2.2. Parasympathetic Autonomic Nervous System
Parasympathetic dysfunction represents a fundamental yet often overlooked aspect of Parkinson's disease, affecting multiple organ systems and contributing to a broad range of non-motor symptoms. Gastrointestinal dysmotility, delayed gastric emptying, and constipation frequently appear in the prodromal phase, preceding motor symptoms by years and serving as potential early indicators of disease onset [21]. As Parkinson's progresses, parasympathetic dysfunction becomes more pronounced, with patients experiencing urinary urgency, retention, and cardiovascular irregularities that reflect the systemic disruption of autonomic control. The loss of parasympathetic tone leads to chronotropic incompetence—characterized by an impaired ability to regulate heart rate—and increased resting heart rate variability, further underscoring the widespread neurodegenerative impact of Parkinson's disease [13].
At the core of parasympathetic dysfunction lies the degeneration of the dorsal motor nucleus of the vagus (DMV), one of the earliest sites of α-synuclein deposition in Parkinson's disease [9]. This structure plays a central role in autonomic regulation, coordinating parasympathetic output to the gastrointestinal tract, heart, and bladder. The presence of α-synuclein aggregates in the DMV, often detected before significant nigrostriatal degeneration, suggests that Parkinson's disease follows a pathological trajectory that extends well beyond the basal ganglia. This early parasympathetic involvement aligns with the broader Braak staging hypothesis, which proposes a caudo-rostral spread of α-synuclein pathology from the enteric and peripheral nervous systems to central structures. However, despite these findings, the clinical significance of DMV degeneration remains insufficiently explored, and its potential as a diagnostic or therapeutic target has yet to be fully realized.
The gastrointestinal system, heavily reliant on parasympathetic innervation, is particularly vulnerable to dysfunction in Parkinson's disease. The slowing of gastric motility leads to delayed gastric emptying, bloating, and nausea, while colonic dysmotility exacerbates constipation, a symptom that not only impairs quality of life but also contributes to systemic inflammation and possibly accelerates neurodegeneration. The enteric nervous system itself exhibits α-synuclein pathology, suggesting that gastrointestinal dysfunction is not merely a secondary consequence of autonomic failure but an integral component of Parkinson's disease pathophysiology. While the gut-brain axis has gained significant attention, research efforts have yet to produce effective interventions that translate into meaningful clinical benefits.
Cardiovascular consequences of parasympathetic dysfunction remain similarly underappreciated. The loss of vagal regulation of heart rate contributes to significant cardiac autonomic instability, increasing the risk of arrhythmias and sudden cardiac events [22]. Chronotropic incompetence, commonly observed in Parkinson's disease, reflects impaired vagal modulation and reduces patients' ability to adapt to physiological stress. While these abnormalities provide potential biomarkers for disease progression, they remain underutilized in clinical practice. The lack of targeted therapies to restore parasympathetic function further illustrates the limitations of current Parkinson's disease management, which remains focused on motor symptoms while neglecting autonomic dysfunction's substantial impact on morbidity and mortality.
Despite growing recognition of parasympathetic failure in Parkinson's disease, research remains fragmented, and treatment approaches remain primarily symptomatic [23]. Dopaminergic therapies provide little to no benefit for autonomic dysfunction, reinforcing the need to explore alternative neurochemical pathways, including cholinergic and serotonergic systems, which play critical roles in autonomic regulation. The continued reliance on symptomatic treatments, such as prokinetic agents for gastrointestinal symptoms or anticholinergics for urinary dysfunction, reflects a broader failure to address the underlying neurodegeneration driving these disturbances. Without a shift in research priorities, parasympathetic dysfunction will remain an overlooked yet debilitating component of Parkinson's disease, depriving patients of comprehensive care and limiting progress toward truly disease-modifying therapies.
2.3. Sialorrhea and Dysphagia
Sialorrhea and dysphagia are among the most disabling non-motor symptoms of Parkinson's disease, severely impairing quality of life and increasing the risk of serious complications (see Table 1). Sialorrhea, affecting between 37% and 84% of patients, arises early in the disease course and progressively worsens over time [24,25]. Contrary to common perception, excessive saliva production is not the underlying cause; impaired swallowing efficiency and inadequate saliva clearance due to orofacial bradykinesia lead to its accumulation [26]. The loss of voluntary and reflexive swallowing control results from a combination of brainstem and basal ganglia dysfunction, α-synuclein aggregation in key autonomic and motor control centres, and deficits in both dopaminergic and cholinergic neurotransmission, as supported by studies [25,27].
Dysphagia affects an estimated 40% to 87% of Parkinson's disease patients, appearing early in the disease course and becoming more pronounced with progression [28]. The gradual decline in swallowing efficiency is directly linked to bradykinesia, as slowed and uncoordinated muscle movements impair bolus transit through the oropharyngeal and oesophagal phases. This dysfunction dramatically increases the risk of malnutrition, dehydration, and aspiration pneumonia—one of the leading causes of mortality in Parkinson's disease [29]. The pathophysiological basis of dysphagia involves α-synuclein accumulation in the dorsal motor nucleus of the vagus and nucleus ambiguous, along with neurodegeneration in key dopaminergic and cholinergic pathways governing swallowing [30,31]. These structural and neurochemical disruptions lead to delayed swallow initiation, impaired pharyngeal clearance, and an increased risk of airway invasion.
Management of sialorrhea and dysphagia remains challenging, as conventional dopaminergic therapy offers inconsistent benefits and, in some cases, worsens symptoms by exacerbating oropharyngeal dyscoordination [32,33]. Treatment strategies rely on a combination of pharmacological and non-pharmacological approaches. Anticholinergic agents such as glycopyrrolate or botulinum toxin injections into the salivary glands have demonstrated efficacy in reducing sialorrhea, though they carry potential side effects, including xerostomia and dysphagia exacerbation. For dysphagia, rehabilitation techniques incorporating compensatory maneuvers, strengthening exercises, and modified diets are widely used to mitigate aspiration risk, though their long-term efficacy remains uncertain.
The systemic impact of these swallowing disorders underscores the urgent need for a comprehensive, multidisciplinary approach involving neurologists, speech therapists, physiotherapists, and nutritionists to optimize patient outcomes. Despite the clear clinical burden of sialorrhea and dysphagia, research into their mechanistic underpinnings remains inadequate, with no targeted neuroprotective interventions available to prevent or slow their progression. The persistence of these symptoms as major contributors to Parkinson's disease morbidity highlights a critical failure in current treatment paradigms, which continue to prioritize motor function over equally debilitating non-motor manifestations. Without a shift in focus toward the early detection and management of autonomic and motor dysfunction affecting swallowing, the preventable complications associated with sialorrhea and dysphagia will remain a leading cause of hospitalization and mortality in Parkinson's disease.
2.4. Constipation
Constipation is one of the most common and earliest non-motor symptoms of Parkinson's disease, affecting between 40% and 63% of patients [34]. Often appearing up to two decades before motor symptoms emerge, it is a prodromal marker and a potential risk factor for Parkinson's disease development (see Table 1) [35]. The presence of chronic constipation long before the clinical diagnosis suggests that Parkinson's pathology extends far beyond the brain from its earliest stages. The enteric nervous system, the dorsal motor nucleus of the vagus, and sacral parasympathetic nuclei all exhibit early α-synuclein aggregation, supporting the hypothesis that gastrointestinal dysfunction is not a secondary complication of neurodegeneration but an integral component of Parkinson's disease pathophysiology [36].
The disruption of autonomic regulation, particularly the loss of cholinergic and serotonergic control of gut motility, leads to a slowing of colonic transit and impaired coordination of peristalsis [34,36]. This dysfunction manifests as infrequent bowel movements, excessive straining, and dyssynergia defecation, which contribute to significant discomfort and reduced quality of life [37]. Beyond neuromuscular impairment, accumulating evidence suggests that gut microbiota alterations and intestinal barrier dysfunction exacerbate neuroinflammation and α-synuclein pathology, raising the question of whether gastrointestinal dysbiosis plays an active role in disease progression rather than being a passive consequence [38]. Small intestinal bacterial overgrowth and increased intestinal permeability further complicate the picture, potentially amplifying systemic inflammation and promoting neurodegenerative processes.
Despite its high prevalence and impact, constipation in Parkinson's disease remains inadequately managed, with current treatment strategies primarily focusing on symptomatic relief rather than addressing the underlying neuropathology [39]. Laxatives, stool softeners, and dietary fibre are commonly used, but their effectiveness is often limited, and their long-term impact on disease progression remains unexplored. Prokinetic agents targeting cholinergic dysfunction have shown promise, yet their role in Parkinson's disease treatment is not well established. Given the multifactorial nature of constipation in Parkinson's disease, a multidisciplinary approach is essential, integrating the expertise of neurologists, gastroenterologists, and nutritionists to optimize management and prevent severe complications such as megacolon and intestinal obstruction.
The persistence of constipation as a significant yet neglected feature of Parkinson's disease highlights a broader issue in the field: the failure to integrate autonomic dysfunction into routine disease management [39]. The gut-brain axis, despite its growing recognition, has yet to yield tangible therapeutic breakthroughs, and the mechanisms linking enteric neuropathy to central neurodegeneration remain poorly defined. Without a paradigm shift that prioritizes early intervention in autonomic dysfunction, Parkinson's disease management will continue to focus narrowly on motor symptoms while overlooking the significant burden of gastrointestinal pathology. Recognizing constipation not only as a quality-of-life issue but also as a potential driver of neurodegeneration could redefine treatment approaches and pave the way for novel disease-modifying strategies.
2.5. Neurogenic Sexual Dysfunction
Neurogenic sexual dysfunction is a highly prevalent yet frequently underreported non-motor symptom of Parkinson's disease, affecting approximately 68% of men and 53% of women [40]. Loss of libido is the most commonly reported issue across both sexes while men predominantly experience erectile dysfunction, and women frequently report vaginal dryness and difficulties achieving orgasm (see Table 1) [41]. Although age-related changes and comorbid depression contribute to the high prevalence of sexual dysfunction in Parkinson's disease, neurodegenerative mechanisms play a fundamental role in its development [41,42]. The interplay between disease-related neurochemical alterations and the side effects of dopaminergic treatments further complicates the clinical presentation, with some patients paradoxically exhibiting hypersexuality as a consequence of dopamine agonist therapy [43].
The neurobiological underpinnings of sexual dysfunction in Parkinson's disease extend beyond the well-established loss of dopaminergic neurons in the basal ganglia. The amygdala, a critical structure for regulating sexual impulse control, undergoes significant neurodegeneration, disrupting emotional and cognitive aspects of sexual behaviour. The mesocorticolimbic dopaminergic pathway, essential for motivation and reward processing, is also implicated, contributing to diminished sexual desire. The paraventricular nucleus and medial preoptic area of the hypothalamus, key regulators of sexual function, receive reduced dopaminergic input in Parkinson's disease, exacerbating erectile and arousal difficulties [44]. Sympathetic neurons within the spinal cord, responsible for modulating autonomic components of sexual function, also exhibit neurodegenerative changes, further impairing physiological responses. Experimental studies have demonstrated that dopaminergic dorsal putamen activity is essential for erection, as evidenced by primate models showing that its stimulation induces penile erection [45]. Additionally, deep brain stimulation of the thalamus and subthalamic nucleus has been reported to trigger spontaneous erections, reinforcing the involvement of these regions in sexual function [46].
Despite the clear pathophysiological basis of sexual dysfunction in Parkinson's disease, its clinical management remains inadequate. The loss of dopaminergic innervation in hypothalamic and spinal structures contributes to diminished sexual function, yet dopaminergic replacement therapy yields inconsistent results, sometimes improving symptoms while at other times exacerbating impulse control disorders, including compulsive sexual behaviour [43]. The involvement of dopamine D2 receptors in erectile function suggests that dopaminergic modulation plays a role in regulating cholinergic activity, though this mechanism remains insufficiently explored [43]. Current treatment approaches, including phosphodiesterase inhibitors for erectile dysfunction and hormone replacement therapy for female sexual dysfunction, offer symptomatic relief but fail to address the underlying neurodegeneration.
The reluctance of both patients and clinicians to openly discuss sexual dysfunction in Parkinson's disease results in significant underdiagnosis and undertreatment, further compounding its impact on quality of life [43]. Stigma, embarrassment, and a lack of standardized assessment tools contribute to the persistent research and clinical care gap. Given the profound psychosocial and relational consequences of sexual dysfunction, it should be recognized as a core non-motor symptom requiring targeted therapeutic interventions. The current approach remains fragmented, with limited consideration of the complex interplay between neurodegeneration, neurotransmitter dysregulation, and treatment side effects. Addressing sexual dysfunction in Parkinson's disease demands a multidisciplinary effort, integrating neurological, psychiatric, and urological expertise to develop more effective and individualized treatment strategies. Without a shift in focus, this distressing yet largely ignored aspect of Parkinson's disease will continue to be marginalized, depriving patients of comprehensive care and impairing their overall well-being.
2.6. Urinary Symptoms
Urinary dysfunction is a pervasive yet often overlooked non-motor symptom of Parkinson's disease, affecting approximately 61% of patients and profoundly impacting the quality of life [47,48]. In more severe cases, lower urinary tract symptoms manifest as urgency, frequency, nocturia, incontinence, and urinary retention (see Table 1). These disturbances frequently appear in the premotor phase, reinforcing that Parkinson's disease is a multisystem disorder extending beyond the basal ganglia long before motor symptoms emerge [49]. As the disease progresses, urinary dysfunction correlates with increased motor disability and more extensive dopaminergic denervation, yet the underlying mechanisms remain only partially understood.
The pathophysiology of urinary symptoms in Parkinson's disease involves complex interactions between the central and peripheral nervous systems. The basal ganglia, through its dopaminergic D1-GABAergic direct pathway, plays a crucial role in suppressing involuntary bladder contractions, ensuring appropriate timing of voiding. Dysfunction in this circuit leads to detrusor overactivity, one of the most common urinary disturbances in Parkinson's disease, resulting in urgency and incontinence [49]. However, beyond dopaminergic deficits, neurodegeneration extends to brain regions directly involved in bladder control. The pontine micturition centre, which coordinates voluntary voiding, exhibits α-synuclein pathology, as do preganglionic and postganglionic sympathetic neurons, sacral parasympathetic nuclei, and the pelvic plexus [48]. This widespread neurodegeneration disrupts the delicate balance between excitatory and inhibitory control of the bladder, leading to a spectrum of voiding dysfunctions that fluctuate with disease severity.
The presence of α-synuclein aggregates in the genitourinary tract further supports that Parkinson's disease involves peripheral autonomic structures not confined to the central nervous system [50,51]. These pathological changes suggest that bladder dysfunction is not simply a downstream consequence of motor impairment but a fundamental feature of Parkinson's disease progression. Despite this, urinary symptoms remain underdiagnosed and undertreated, often attributed to ageing rather than being recognized as a direct consequence of neurodegeneration.
Current treatment strategies for urinary dysfunction in Parkinson's disease remain primarily symptomatic [52]. Anticholinergic agents and β3-adrenergic receptor agonists provide some relief by reducing detrusor overactivity, yet cognitive side effects and inconsistent efficacy often limit their effectiveness. Dopaminergic therapies, which are the mainstay of Parkinson's disease treatment, offer little to no improvement in urinary symptoms, highlighting the need for alternative therapeutic approaches that target non-dopaminergic pathways [52]. Non-pharmacological interventions, including pelvic floor therapy and behavioural modifications, are frequently recommended, yet their long-term impact on disease progression remains uncertain.
The continued marginalization of urinary dysfunction in Parkinson's disease research reflects a broader failure to acknowledge the full complexity of the disease [53]. Although bladder symptoms significantly contribute to morbidity, they remain underrepresented in clinical trials, with few targeted treatments emerging from translational research. The lack of standardized assessment tools for urinary dysfunction further complicates diagnosis and management, leaving many patients inadequately treated. A more integrative approach that recognizes the neuroanatomical and neurochemical underpinnings of urinary dysfunction in Parkinson's disease is essential. Without a paradigm shift in both research and clinical practice, urinary symptoms will continue to be an underestimated yet debilitating burden for patients, limiting their independence and reducing overall quality of life.
2.7. Cardiac Alterations
Autonomic cardiac dysfunction is a common but often underrecognized feature of Parkinson's disease, emerging early in the disease course and progressively worsening as neurodegeneration advances [54]. The imbalance between increased sympathetic activity and decreased parasympathetic regulation leads to significant cardiovascular abnormalities, including heart rate variability, arrhythmias, prolonged P-R and Q-T intervals, and, in rare cases, cardiomyopathy [55,56]. These alterations not only contribute to increased morbidity but also elevate the risk of sudden cardiac events, underscoring the systemic nature of Parkinson's disease.
The pathophysiology of cardiac dysfunction in Parkinson's disease is driven by widespread autonomic degeneration, characterized by selective loss of sympathetic nerve terminals. Immunohistochemical analyses for tyrosine hydroxylase, a key enzyme in catecholamine synthesis, confirm marked sympathetic denervation of the heart. This loss has been further validated by imaging studies using (123I)-metaiodobenzylguanidine and (18F)-fluorodopa, which demonstrate reduced cardiac norepinephrine uptake, indicative of sympathetic failure [57]. Post-mortem studies reveal α-synuclein aggregates within the epicardial sympathetic axon terminals, with axonal degeneration progressing in a distal-to-proximal manner—first affecting the heart before extending to prevertebral and paravertebral sympathetic ganglia [58].
This pattern of degeneration suggests that cardiac autonomic dysfunction in Parkinson's disease is not merely a secondary consequence of motor impairment or medication side effects but rather a primary feature of disease pathology [23]. The involvement of the central and peripheral autonomic nervous systems reinforces the idea that Parkinson's disease extends far beyond the dopaminergic system, disrupting regulatory networks that control cardiovascular homeostasis. Despite this, cardiac dysfunction remains underdiagnosed in routine clinical assessments, largely due to the overshadowing focus on motor symptoms.
Current management strategies for Parkinson 's-related cardiac dysfunction are limited, with treatment primarily focusing on symptom control rather than targeting the underlying neurodegeneration [23]. Beta-blockers, pacemakers, and volume-expanding agents mitigate arrhythmias and orthostatic hypotension, yet these interventions fail to address the progressive loss of sympathetic innervation [59]. The absence of disease-modifying therapies underscores a critical gap in Parkinson's research—despite clear evidence linking α-synuclein pathology to cardiac dysfunction, no targeted approaches have emerged to preserve autonomic regulation of the heart.
The continued neglect of cardiac autonomic dysfunction in Parkinson's disease reflects a broader failure to integrate non-motor manifestations into disease management and research priorities [60]. The reliance on motor symptoms as the primary diagnostic and therapeutic target has led to an incomplete understanding of the disease, leaving cardiovascular complications inadequately addressed. Recognizing cardiac autonomic dysfunction as a core component of Parkinson's disease is essential for advancing both clinical care and translational research [13]. Without a shift in focus, patients will remain at heightened risk for cardiovascular morbidity and mortality, while therapeutic innovation will continue to lag behind the pressing needs of those affected by this complex and systemic disorder.
2.8. Orthostatic Hypotension
Orthostatic hypotension (OH) is a debilitating and often underdiagnosed cardiovascular manifestation of Parkinson's disease, affecting approximately 30% of patients. Defined by a drop of more than 20 mmHg in systolic blood pressure or 10 mmHg in diastolic pressure within three minutes of standing, OH significantly impairs daily function, increasing the risk of dizziness, fatigue, syncope, falls, and injury [61,62]. Unlike primary hypotensive disorders, neurogenic OH in Parkinson's disease arises from a fundamental breakdown in autonomic regulation, reflecting widespread sympathetic denervation and baroreflex failure [63].
The underlying pathophysiology of OH in Parkinson's disease stems from impaired norepinephrine release, leading to deficient vasoconstriction and a subsequent failure to maintain blood pressure upon standing [62,64]. Studies indicate that this dysfunction is primarily driven by sympathetic neural degeneration rather than baroreceptor failure, distinguishing Parkinson's-related OH from other forms of dysautonomia [65,66]. The depletion of norepinephrine at both central and peripheral levels compromises vascular tone, exacerbating cerebral autoregulation deficits that further contribute to orthostatic intolerance. The insular cortex, a critical autonomic control region, also shows significant Lewy body accumulation in Parkinson's disease, correlating with the severity of OH and the overall progression of autonomic dysfunction [63] .
Despite its high prevalence and profound impact on patient well-being, OH remains inadequately managed in Parkinson's disease [67]. Many cases go undiagnosed due to the subtle and fluctuating nature of symptoms, often misattributed to medication side effects or general frailty. The lack of routine autonomic testing in clinical practice further delays recognition, leaving patients vulnerable to repeated falls and associated complications. Even when diagnosed, treatment strategies are primarily symptomatic, relying on pharmacological agents such as fludrocortisone, midodrine, and droxidopa to increase blood pressure, while non-pharmacological interventions, including fluid loading, compression garments, and physical counter-manoeuvres, provide only partial relief. However, these approaches fail to address the underlying neurodegeneration, leaving OH as a persistent and often progressive burden.
The neglect of OH in Parkinson's disease research reflects a broader failure to prioritize autonomic dysfunction within the disease framework [67]. Despite clear evidence linking autonomic failure to disease progression and increased mortality risk, the field remains disproportionately focused on motor symptoms, overlooking the substantial impact of cardiovascular dysregulation. The absence of disease-modifying therapies targeting autonomic dysfunction underscores a critical gap in treatment development, leaving patients reliant on palliative measures rather than comprehensive solutions. A paradigm shift is urgently needed to integrate autonomic dysfunction, including OH, into both diagnostic criteria and therapeutic innovation, recognizing that Parkinson's disease extends far beyond its typical motor symptoms and requires a holistic approach to management.
2.9. Seborrhoea and Seborrheic Dermatitis
Seborrhoea and seborrheic dermatitis (SD) are among the earliest and most frequent non-motor manifestations of Parkinson’s disease, affecting between 52% and 59% of patients, with severity often increasing as the disease progresses [68]. Characterized by excessive sebum production, erythema, and flaking of the scalp, face, and upper trunk, these skin disorders highlight the systemic nature of Parkinson’s pathology. Despite their high prevalence, seborrheic symptoms remain largely unrecognized as part of the disease spectrum, often misattributed to age-related changes or primary dermatological conditions rather than an expression of underlying neurodegeneration [68].
Emerging evidence suggests a complex interplay between Parkinson’s disease pathology and seborrheic skin disorders. Malassezia yeast colonization, a key factor in SD, has been found in significantly higher concentrations on the scalp and skin of Parkinson’s patients than controls, suggesting an altered skin microbiome as a contributing factor [69]. However, microbial imbalance alone does not explain the strong association between SD and Parkinson’s disease. Pathophysiological studies reveal that α-synuclein aggregates accumulate in sebaceous glands, dermal nerve fibres, and autonomic skin structures, implicating cutaneous autonomic dysfunction as an early disease feature [70].
The disruption of sebaceous gland activity in Parkinson’s disease is closely linked to basal ganglia dysfunction [71]. Dopaminergic pathways regulate sebaceous gland function, and the neurodegeneration affecting these circuits likely contributes to excessive sebum production and altered lipid composition in Parkinson’s patients [71]. These findings reinforce the idea that Parkinson’s disease extends beyond the central nervous system, with autonomic skin involvement potentially serving as an early diagnostic marker. Since skin biopsies have successfully detected phosphorylated α-synuclein in peripheral nerves, there is growing interest in utilizing cutaneous biomarkers for early diagnosis, though this remains an underdeveloped area of research.
Despite the clear connection between neurodegeneration and seborrheic manifestations, current treatment strategies remain simplistic, focusing on antifungal and anti-inflammatory agents such as ketoconazole and topical corticosteroids [72]. While these approaches provide symptomatic relief, they fail to address the underlying autonomic dysfunction contributing to sebaceous gland dysregulation. The lack of targeted interventions highlights a broader issue in Parkinson’s disease research—non-motor symptoms, despite their prevalence and impact, continue to be treated as secondary concerns rather than integral aspects of disease pathology.
The failure to recognize seborrhoea and seborrheic dermatitis as part of Parkinson’s disease reflects a broader tendency to compartmentalize symptoms rather than acknowledge the disorder as a multisystem condition [68]. Dermatological manifestations offer potential early biomarkers and provide a window into the broader autonomic dysfunction in Parkinson’s disease. However, the persistent neglect of these symptoms in clinical practice and research underscores a lack of comprehensive disease management. A multidisciplinary approach involving neurologists and dermatologists is essential to improve diagnosis and treatment, yet without a shift in perspective, skin-related symptoms will remain an underutilized resource in Parkinson’s disease research and patient care.
2.10. Anhidrosis/Hyperhidrosis
Sweating abnormalities are a frequently overlooked yet debilitating non-motor symptom of Parkinson’s disease, manifesting as both excessive sweating (hyperhidrosis) and reduced sweating (anhidrosis). These disturbances are typically asymmetrical, with excessive sweating predominantly affecting the head, neck, and trunk, while the extremities exhibit decreased sweat production [73]. Unlike other autonomic dysfunctions that correlate with disease severity, sweating irregularities appear independent of overall motor progression, aligning with broader autonomic impairments such as sialorrhea and postprandial hypotension [74].
The pathophysiology of these sweating abnormalities remains incompletely understood, reflecting the complexity of autonomic dysfunction in Parkinson’s disease. Both central and peripheral mechanisms appear to be involved, significantly disrupting the sympathetic nervous system’s ability to regulate sudomotor function. Sympathetic vasoconstrictive and sweating control mechanisms are diminished at rest, while response latencies in the extremities are prolonged, suggesting target organ impairment rather than a simple reduction in central autonomic output [75]. Centrally, dysfunction of α-2 adrenergic receptors in the hypothalamus may contribute to hyperhidrosis, as evidenced by the fact that clonidine—a centrally acting α-2 adrenergic agonist—can improve excessive sweating, tremor and even some cognitive symptoms. However, this remains an area of ongoing investigation.
Peripheral autonomic dysfunction further complicates the picture. Early in Parkinson’s disease, preganglionic sympathetic fibres exhibit functional impairment, while postganglionic neurons become increasingly affected in later disease stages. The progressive degeneration of sympathetic postganglionic fibres ultimately reduces sweat gland innervation, impairing thermoregulation and vascular control [76]. Although some studies suggest that hyperhidrosis is more common in late-stage Parkinson’s disease, this remains a subject of debate [77]. Additionally, the reduced innervation of cutaneous blood vessels and sweat glands contributes to widespread autonomic dysregulation, exacerbating thermoregulatory and cardiovascular instability in affected patients.
Despite its clear impact on quality of life, managing sweating disturbances in Parkinson’s disease remains largely empirical. Hyperhidrosis is typically treated with systemic anticholinergic agents, botulinum toxin injections, or sympathetic denervation procedures, while anhidrosis and related thermoregulatory dysfunction lack effective interventions [78]. The failure to develop targeted therapies reflects a broader neglect of autonomic symptoms in Parkinson’s disease research. Current treatments focus on symptom suppression rather than addressing the underlying neurodegeneration driving sudomotor dysfunction [79].
The continued oversight of autonomic dysfunction in Parkinson’s disease highlights a critical gap in disease management [60]. Though often dismissed as minor inconveniences, sweating disturbances are precise indicators of widespread autonomic failure. Their presence in early disease stages suggests potential as biomarkers, yet they remain underutilized in clinical diagnostics. Without a fundamental shift in research priorities that recognizes the full extent of autonomic dysfunction, Parkinson’s disease treatment will remain disproportionately focused on motor symptoms, leaving critical non-motor manifestations inadequately addressed and contributing to the persistent gap between scientific understanding and patient care.
2.11. Thermoregulatory Alterations
Thermoregulatory dysfunction is a frequently overlooked yet clinically significant non-motor feature of Parkinson’s disease, manifesting as impaired heat dissipation, cold intolerance, and episodes of hypothermia [80]. Defined as a body temperature below 35°C, hypothermia in Parkinson’s disease can progress to moderate (<32°C) or severe (<28°C) states, posing a serious risk of cardiac arrhythmias and other systemic complications [81]. Unlike temperature fluctuations seen in healthy individuals, these alterations occur independently of ambient temperature, indicating a fundamental failure of autonomic regulation rather than a simple response to external conditions [80,81].
The pathophysiology of thermoregulatory impairment in Parkinson’s disease is rooted in autonomic dysfunction, particularly within the hypothalamus, a critical centre for thermoregulation. The hypothalamus balances heat production and dissipation by modulating sympathetic vasoconstriction, sudomotor activity, and metabolic rate. In Parkinson’s disease, neurodegeneration disrupts these regulatory mechanisms, leading to impaired sympathetic control over thermoregulatory responses [80]. Parasympathetic hyperactivity further exacerbates the dysfunction, particularly by amplifying cutaneous vasoconstriction deficits, which impair the body's ability to conserve or dissipate heat effectively [75].
Sympathetic neurograms in Parkinson’s disease patients consistently show reduced activity compared to healthy controls, with the severity of autonomic impairment correlating negatively with age and disease duration [82]. This progressive decline in sympathetic output compromises thermoregulatory homeostasis, rendering patients vulnerable to heat stress and cold intolerance [80]. Additionally, the loss of dopaminergic modulation of hypothalamic circuits likely alters the set points for temperature regulation, although this remains an area requiring further research.
Despite the potential severity of thermoregulatory disturbances, their clinical recognition remains inadequate, and management strategies are largely non-specific [80]. Current interventions focus on symptomatic control, including external temperature regulation, hydration, and behavioural modifications to avoid extreme temperatures. However, these approaches fail to address the underlying neurophysiological deficits driving autonomic failure in Parkinson’s disease. The absence of targeted treatments reflects a broader issue in Parkinson’s research, where non-motor symptoms remain secondary concerns despite their significant impact on morbidity and mortality [80].
The persistent neglect of thermoregulatory dysfunction in Parkinson’s disease underscores a critical flaw in disease management [80]. While movement-related symptoms dominate both clinical attention and therapeutic development, autonomic impairments such as thermoregulatory failure remain primarily unaddressed despite their role in increasing patient vulnerability to environmental stressors and systemic complications. Recognizing these disturbances as fundamental components of Parkinson’s pathology rather than peripheral concerns is essential for advancing comprehensive treatment strategies [83]. Without a paradigm shift that integrates autonomic dysfunction into both clinical care and research priorities, patients will continue to suffer from largely preventable complications that significantly diminish their quality of life.
3. Sensory Alterations in Parkinson’s Disease
Sensory dysfunction is a pervasive yet underappreciated aspect of Parkinson’s disease, affecting multiple sensory modalities and significantly impairing patients’ quality of life [73,74]. While Parkinson’s disease is primarily defined by its motor symptoms, accumulating evidence suggests that sensory deficits often precede motor dysfunction, highlighting their role as early indicators of disease onset [74]. These disturbances include olfactory dysfunction (hyposmia), gustatory impairment (ageusia), visual disturbances, chronic pain, and paraesthesias, each contributing to the complex and multifaceted nature of Parkinson’s pathology (see Table 2). Despite their high prevalence, sensory alterations remain insufficiently addressed in clinical practice and research, reinforcing the need for a more integrative approach to disease management (Figure 1 and Figure 2).
3.1. Hyposmia
Olfactory dysfunction is one of the most well-established non-motor symptoms of Parkinson’s disease, first recognized over three decades ago as a frequent yet overlooked aspect of the disease [75,76,77]. Hyposmia, characterized by a diminished sense of smell, affects between 75% and 90% of patients and often precedes the onset of motor symptoms by several years. This strong temporal association makes olfactory impairment one of the most reliable early biomarkers of Parkinson’s disease, with individuals experiencing hyposmia exhibiting a 3.84-fold increased risk of developing the disorder [78,79,80,81,82].
Hyposmia in Parkinson’s disease is not merely an isolated sensory impairment but rather a reflection of widespread neurodegeneration. The severity of olfactory dysfunction correlates with cognitive decline, depression, anxiety, and REM sleep behaviour disorder, suggesting a shared pathophysiological mechanism underlying these non-motor symptoms [83,84,85,86]. The neuropathological hallmark of Parkinson’s disease—Lewy pathology—originates in the olfactory bulb and spreads along neural pathways, leading to neuronal loss and reduced olfactory bulb volume. This process extends to limbic structures, including the amygdala and hippocampus, further implicating olfactory dysfunction as a harbinger of broader neurodegeneration [9,87,88].
Identifying α-synuclein aggregates in olfactory mucosal receptor neurons has raised the prospect of using nasal swabs as a potential diagnostic tool for Parkinson’s disease (see Table 2). Although preliminary studies support the feasibility of this approach, additional research is needed to refine its sensitivity and specificity before it can be implemented in routine clinical practice [89,90]. Beyond its association with Lewy pathology, olfactory dysfunction in Parkinson’s disease is also linked to disruptions in dopaminergic transmission. The nigrostriatal dopaminergic system, known for its role in motor function, also modulates sensory processing, including olfaction. Dopaminergic denervation correlates with the severity of hyposmia, with lower dopamine transporter activity in striatal regions corresponding to reduced olfactory performance [83,91]. Given that up to 80% of dopaminergic neurons are lost before the appearance of motor symptoms, olfactory dysfunction may provide a crucial window for early intervention [92].
Despite its strong potential as an early biomarker, hyposmia lacks disease specificity, occurring in other neurodegenerative disorders and even normal ageing. The absence of standardized olfactory assessments further complicates its clinical utility, underscoring the need for improved diagnostic tools to distinguish Parkinson’s-related olfactory dysfunction from other causes [93]. Although several pharmacological and non-pharmacological approaches have been proposed to mitigate hyposmia, their efficacy remains inconsistent, with no definitive treatment currently available. This gap in therapeutic strategies highlights the broader neglect of sensory dysfunction in Parkinson’s disease, reinforcing the need for targeted research aimed at restoring sensory processing and improving early disease detection.
The continued underestimation of hyposmia in Parkinson’s disease reflects a persistent failure to acknowledge the full spectrum of non-motor dysfunction [83]. Despite its substantial predictive value, olfactory impairment remains underutilized in clinical settings, and research efforts remain disproportionately focused on motor symptomatology. Integrating olfactory testing into routine neurological assessments could significantly enhance early diagnosis and allow for earlier therapeutic interventions. However, without a shift in research priorities that recognizes the importance of sensory dysfunction, Parkinson’s disease management will continue to fall short of addressing the full complexity of this neurodegenerative disorder.
3.2. Ageusia
Ageusia, the loss of taste perception, is a frequently overlooked sensory deficit in Parkinson’s disease, often dismissed as a secondary effect of olfactory dysfunction rather than a distinct neurological impairment (see Table 2). However, evidence suggests that ageusia results from independent neuroanatomical and molecular abnormalities, making it an essential but underrecognized component of the non-motor symptom spectrum. Prevalence estimates vary widely, ranging from 9% to 54%, depending on study methodology and disease stage [94,95,96,97]. Given its significant impact on appetite, nutrition, and overall quality of life, the failure to adequately assess and manage ageusia in Parkinson’s disease reflects a broader neglect of sensory dysfunction in clinical practice.
Although olfactory dysfunction strongly influences taste perception, growing research indicates that ageusia in Parkinson’s disease stems from direct neurodegeneration of gustatory pathways. Lewy body pathology progressively affects key regions involved in taste processing, including the orbitofrontal cortex, insular operculum, and related sensory integration centres, leading to diminished gustatory sensitivity [94,98]. This disruption is particularly pronounced in later disease stages, where structural and functional alterations in these regions become more evident.
The cholinergic system, crucial for sensory processing and cognitive function, is also implicated in Parkinson’s-related ageusia. Degeneration of basal forebrain cholinergic neurons reduces acetylcholine levels, impairing sensory integration and gustatory perception [99]. Additionally, cognitive decline associated with Parkinson’s disease exacerbates these sensory deficits, as cholinergic dysfunction affects key gustatory processing structures such as the orbitofrontal cortex, cingulate gyrus, amygdala, and hippocampus. This interplay between sensory and cognitive impairment suggests that taste dysfunction is not merely an isolated sensory symptom but an indicator of broader neurodegenerative processes [100].
At the molecular level, dysregulated expression of taste receptor genes further supports the hypothesis that Parkinson’s disease disrupts gustatory function at multiple levels. Genetic alterations, combined with Lewy body accumulation and widespread neurotransmitter dysfunction, contribute to the progressive impairment of taste perception [101]. However, despite clear mechanistic links, ageusia remains an underexplored aspect of Parkinson’s disease, with limited research dedicated to understanding its pathophysiology and therapeutic interventions.
The failure to acknowledge ageusia as a clinically significant symptom reflects a persistent gap in Parkinson’s disease management. While motor dysfunction remains the primary focus of both diagnosis and treatment, non-motor symptoms such as ageusia significantly impact patient well-being yet receive minimal clinical attention. Currently, no targeted therapies exist to address gustatory deficits in Parkinson’s disease, leaving patients to navigate its effects without effective intervention. This oversight underscores the urgent need for a paradigm shift in Parkinson’s research and treatment approaches—one that integrates sensory dysfunction as a fundamental aspect of disease progression rather than a peripheral concern. Without such a shift, Parkinson’s disease will continue to be managed through an outdated, motor-centric framework that fails to address the full complexity of its clinical manifestations.
3.3. Visual Disturbances
Visual dysfunction is one of the most prevalent yet often underappreciated non-motor symptoms of Parkinson’s disease, with more than 90% of patients experiencing at least one form of visual impairment (see Table 2). These disturbances worsen as the disease progresses, profoundly affecting daily activities such as reading, driving, and spatial navigation [102,103]. Patients frequently report blurred vision, excessive tearing, difficulty focusing, light sensitivity, and ocular fatigue, yet these symptoms are rarely considered primary targets for intervention. While early-stage Parkinson’s disease is often associated with subtle contrast sensitivity deficits, more pronounced visual disturbances, including perceptual distortions and hallucinations, emerge in later stages, reflecting broader cortical dysfunction [104,105].
The underlying pathophysiology of visual disturbances in Parkinson’s disease is complex, involving both central and retinal mechanisms. Cortical degeneration plays a significant role, as Lewy pathology and neurodegeneration within visual processing areas—such as the occipital and parietal lobes—contribute to impaired contrast sensitivity and visuospatial dysfunction. These deficits correlate with disease severity and cognitive impairment, suggesting that visual symptoms may be early indicators of neurodegenerative progression beyond the basal ganglia [104].
Retinal dysfunction is emerging as a crucial component of Parkinson’s disease pathophysiology, with increasing evidence supporting the retina as a window into neurodegeneration. Phosphorylated α-synuclein aggregates in the retina mirror Lewy pathology in the brain, with deposits observed in retinal ganglion cells even before the onset of motor symptoms [106,107]. Retinal dopaminergic amacrine cells, essential for contrast sensitivity and visual processing, undergo progressive degeneration, leading to abnormalities in colour discrimination and spatial perception that parallel the widespread dopaminergic dysfunction in Parkinson’s disease [107,108,109].
Advances in retinal imaging have opened new possibilities for non-invasive biomarkers in Parkinson’s disease. Optical coherence tomography studies consistently demonstrate retinal nerve fibre layer thinning, macular atrophy, and vascular changes in Parkinson’s patients. These alterations include reduced superficial capillary plexus density, foveal microvascular abnormalities, and decreased deep macular vascular density, all of which correlate with disease duration and severity [110,111,112]. While these findings suggest that retinal imaging could be an accessible biomarker for early diagnosis and disease progression monitoring, further standardization and validation are required before widespread clinical implementation.
Despite the profound impact of visual impairment on quality of life, current treatment strategies remain inadequate [113]. Pharmacological approaches, such as dopaminergic replacement therapy, provide inconsistent benefits, while non-pharmacological interventions, including contrast-enhancing visual aids and rehabilitative therapies, remain underutilized. The failure to prioritize visual dysfunction in Parkinson’s research underscores a broader issue: non-motor symptoms, despite their high prevalence and disabling effects, remain secondary considerations in clinical care.
The neglect of visual dysfunction reflects the persistent motor-centric bias in Parkinson’s disease management [113]. Although visual symptoms not only precede motor onset in some patients but also correlate with cognitive decline, they continue to be regarded as peripheral concerns rather than integral aspects of disease pathology. A more comprehensive approach incorporating retinal biomarkers, early visual assessments, and targeted interventions is needed. Without this paradigm shift, the field will continue to overlook key diagnostic opportunities and fail to provide meaningful symptom relief for the majority of Parkinson’s patients struggling with progressive visual impairment.
3.4. Pain
Pain is a highly prevalent yet frequently underestimated non-motor symptom of Parkinson’s disease, affecting between 20% and 98% of patients depending on study methodology and diagnostic criteria [114,115,116]. For many, pain is not merely a secondary complication but a defining feature of the disease, often preceding the onset of motor symptoms by several years (see Table 2). The nature of pain in Parkinson’s disease is heterogeneous, encompassing musculoskeletal pain linked to rigidity and postural abnormalities, dystonic pain arising from involuntary muscle contractions, and neuropathic or radicular pain indicative of peripheral nerve involvement [4,114]. Despite its impact on daily life, pain remains underdiagnosed mainly and poorly managed, highlighting the need for a more integrated approach to its assessment and treatment.
The pathophysiology of pain in Parkinson’s disease is multifaceted, involving both central and peripheral mechanisms. Lewy pathology extends beyond the basal ganglia to regions directly involved in pain processing, including the spinal cord, amygdala, periaqueductal grey, and nucleus accumbens. Neurodegeneration in these areas alters nociceptive modulation, increasing pain sensitivity and disrupting the endogenous analgesic system [117]. Additionally, Parkinson’s disease affects non-dopaminergic structures such as the subthalamic nucleus, anterior cingulate cortex, and insular cortex—regions implicated in pain perception and emotional processing—further amplifying the subjective experience of pain [118,119,120].
Dysregulation of pain-modulating neurotransmitters is another key contributor to heightened pain sensitivity in Parkinson’s disease. Beyond dopamine, disruptions in norepinephrine and serotonin transmission impair descending pain inhibition, intensifying nociceptive signalling and altering the brain’s ability to regulate pain perception [120,121]. Furthermore, neuroinflammation plays a critical role, as elevated levels of proinflammatory cytokines contribute to peripheral and central sensitization, exacerbating pain symptoms [122,123]. These overlapping mechanisms suggest that pain in Parkinson’s disease is not merely a consequence of motor dysfunction but a fundamental component of disease pathology, warranting targeted therapeutic strategies.
Despite its high prevalence, no definitive biomarkers have been identified to correlate pain severity with specific neuropathological changes in Parkinson’s disease [124]. While some patients experience pain long before motor symptoms emerge, the mechanisms underlying this early manifestation remain poorly understood. Identifying reliable biomarkers and delineating the neurochemical pathways involved in Parkinson’s-related pain is critical for developing precision-targeted therapies. Current treatment approaches remain largely symptomatic, relying on pharmacological and non-pharmacological interventions. However, conventional dopaminergic therapy provides inconsistent relief, underscoring the need for alternative strategies that address the non-dopaminergic contributions to pain processing.
The continued neglect of pain in Parkinson’s disease reflects a broader failure to prioritize non-motor symptoms in both research and clinical care [124]. Pain significantly impairs quality of life, yet it remains underreported and frequently misattributed to ageing or comorbid musculoskeletal conditions. The traditional focus on motor dysfunction has led to an incomplete understanding of Parkinson’s disease as a multisystem disorder, leaving critical gaps in pain management. A paradigm shift is needed—one that integrates pain as a core feature of Parkinson’s disease rather than an incidental complication. Patients will endure unnecessary suffering without such a shift, and treatment approaches will remain reactive rather than proactive.
3.5. Paraesthesias
Paraesthesias, characterized by abnormal sensations such as tingling, burning, or numbness, affect approximately 40% of Parkinson’s disease patients, often contributing to significant discomfort and disability [125]. Although these symptoms are frequently dismissed as secondary to ageing or peripheral neuropathy, mounting evidence suggests that they stem from both peripheral and central mechanisms directly linked to Parkinson’s pathophysiology [125]. Despite their high prevalence, paraesthesias remain poorly understood and inadequately addressed in routine clinical practice, underscoring a broader failure to integrate sensory dysfunction into Parkinson’s disease management (see Table 2).
The pathophysiological basis of paraesthesias in Parkinson’s disease involves α-synuclein accumulation within both sensory and autonomic neurons, contributing to progressive neurodegeneration. Skin biopsies have confirmed the presence of α-synuclein aggregates in peripheral nerve fibres, including cutaneous autonomic structures, reinforcing the role of peripheral neurodegeneration in sensory disturbances [126,127,128,129]. The involvement of Schwann cells, essential for nerve conduction and regeneration, further suggests that Parkinson’s disease disrupts neuronal function and the supportive glial environment required for peripheral sensory integrity [130].
As peripheral nerves degenerate, small-fibre neuropathy is a key contributor to paraesthesias. Loss of epidermal nerve fibres correlates with disease progression, with a marked reduction in epidermal nerve fibre density observed in Parkinson’s patients [131,132,133]. The autonomic nervous system is also implicated, as sympathetic and parasympathetic dysfunction alters cutaneous sensory transmission, leading to abnormal pain perception and temperature dysregulation. These findings highlight the need to consider Parkinson’s disease as a disorder with widespread neurodegeneration beyond the central nervous system, affecting sensory processing at multiple levels.
Despite growing recognition of sensory impairments in Parkinson’s disease, the clinical approach to paraesthesias remains inadequate [125]. The absence of standardized diagnostic tools means these symptoms are often overlooked or misattributed to unrelated conditions, delaying appropriate intervention. Moreover, treatment options remain limited, with current strategies relying primarily on symptomatic management rather than addressing the underlying disease mechanisms [125]. Pharmacological treatments such as neuropathic pain modulators provide inconsistent relief, while non-pharmacological approaches, including physical therapy and nerve stimulation, are underutilized.
The persistent neglect of paraesthesias in Parkinson’s disease reflects a broader issue within the field—non-motor symptoms continue to be relegated to secondary importance despite their significant impact on quality of life [125]. If Parkinson’s disease research remains overly focused on motor dysfunction, crucial aspects of sensory pathology will remain unexplored, leading to suboptimal patient care. A paradigm shift is needed, one that prioritizes comprehensive neurodegenerative models incorporating sensory impairment as an integral component of disease progression. Until then, patients will continue to experience these debilitating symptoms without effective solutions, trapped in a clinical framework that fails to acknowledge the full scope of their disease.
4. Sleep-Related Disorders in Parkinson’s Disease
Sleep-related disorders are among the most pervasive and debilitating non-motor manifestations of Parkinson’s disease, affecting between 60% and 98% of patients [134]. These disturbances significantly impair motor function, cognitive performance, and emotional stability, creating a vicious cycle that accelerates disease progression and erodes quality of life [134]. Despite their high prevalence, sleep dysfunction remains underrecognized and undertreated, often overshadowed by motor symptoms in clinical assessments. The spectrum of sleep disturbances in Parkinson’s disease is highly heterogeneous, encompassing nocturia, insomnia, excessive daytime sleepiness, restless legs syndrome, obstructive sleep apnoea, and REM sleep behaviour disorder (RBD, see Table 3). Many of these symptoms emerge in the prodromal phase, preceding motor signs by years or even decades, highlighting their potential as early disease biomarkers (Figure 1 and Figure 2) [134].
Among these disturbances, RBD is particularly significant. It is characterized by a loss of REM atonia, leading to dream enactment behaviours that often result in self-injury or harm to bed partners. RBD is not merely a symptom of Parkinson’s disease but a harbinger of neurodegeneration, strongly associated with synucleinopathies such as dementia with Lewy bodies and multiple system atrophy [134]. Longitudinal studies indicate that individuals with idiopathic RBD have an 80–90% likelihood of developing Parkinson’s disease or a related disorder within 10–15 years, making it one of the most reliable clinical markers of prodromal disease [134]. Its strong association with autonomic dysfunction, neuropsychiatric symptoms, and cognitive decline further underscores its role in Parkinson’s disease progression [134].
Insomnia, reported in up to 80% of Parkinson’s patients, represents another major challenge. Nocturnal akinesia, rigidity, and pain contribute to sleep fragmentation while underlying neurodegenerative processes disrupt sleep initiation and maintenance [134,135,136]. Chronic sleep deprivation exacerbates cognitive decline, emotional instability, and autonomic dysfunction, reinforcing a cycle that worsens disease burden [134]. Similarly, excessive daytime sleepiness, affecting up to 76% of patients, increases with disease progression and is further exacerbated by dopamine agonist therapy [134]. These medications, while beneficial for motor symptoms, contribute to unpredictable sleep attacks, posing significant safety risks, particularly for driving and daily activities [134]. Restless legs syndrome, another common sleep-related manifestation, disrupts sleep with involuntary leg movements and sensations of discomfort, while obstructive sleep apnoea not only fragments sleep but also contributes to oxidative stress, cognitive impairment, and accelerated neurodegeneration [134].
The pathophysiology of these sleep disturbances reflects widespread neurodegeneration beyond the basal ganglia. Loss of dopaminergic neurons in the substantia nigra and ventral tegmental area disrupts sleep-wake stability. At the same time, degeneration of the raphe nuclei and locus coeruleus—hallmarks of Braak stages 1 and 2—impairs serotonergic and noradrenergic tone, leading to early-onset sleep dysfunction [9,10,134]. The flip-flop switch model of sleep regulation posits that a balance between cholinergic neurons promoting REM sleep and noradrenergic/serotonergic neurons suppressing it is essential for stable sleep-wake transitions [134]. In Parkinson’s disease, this balance is disrupted, resulting in fragmented sleep architecture, increased nocturnal awakenings, and reduced REM sleep [134]. The emerging role of the glymphatic system, which facilitates the clearance of toxic protein aggregates during sleep, further implicates sleep dysfunction in disease progression. Glymphatic impairment may contribute to α-synuclein accumulation and accelerate neurodegeneration, positioning sleep disturbances not just as symptoms but as active drivers of Parkinson’s disease pathology [137].
Circadian rhythm dysregulation also plays a pivotal role in Parkinson’s-related sleep dysfunction. Dopaminergic degeneration and altered melatonin secretion disrupt sleep-wake cycles, metabolic homeostasis, and emotional regulation [134]. The retinohypothalamic and mesencephalic-pineal axis, essential for regulating circadian rhythms, is functionally compromised in Parkinson’s disease, with similar disruptions observed in primate models [134]. Other affected structures, including the lateral hypothalamus, subthalamic nucleus, and area postrema, contribute to the complex and multifactorial nature of sleep disturbances in Parkinson’s disease [134].
Despite the strong link between sleep dysfunction and disease progression, treatment strategies remain largely symptomatic and inconsistent. Given the heterogeneity of sleep-related symptoms, a one-size-fits-all approach is ineffective. Pharmacological interventions, including melatonin, dopamine agonists, and sedatives, provide variable benefits and often introduce additional side effects [134]. Non-pharmacological therapies, such as cognitive-behavioural therapy for insomnia, bright light therapy for circadian dysfunction, and continuous positive airway pressure for obstructive sleep apnoea, remain underutilized despite mounting evidence supporting their efficacy [134]. The failure to develop targeted treatments reflects a broader issue—the persistent marginalization of non-motor symptoms in Parkinson’s disease research and care.
The neglect of sleep disorders in Parkinson’s disease underscores a critical gap in both clinical and research priorities. These disturbances are not merely secondary symptoms but may serve as modifiable contributors to disease progression, exacerbating neurodegeneration through increased oxidative stress, impaired neuroplasticity, and disrupted protein clearance pathways [134]. Sleep dysfunction should be recognized as a core aspect of Parkinson’s disease, integrated into diagnostic frameworks, and prioritized in treatment strategies. Without a paradigm shift in clinical practice and research focus, patients will endure fragmented sleep, excessive fatigue, and circadian instability without practical solutions. The failure to address sleep disturbances reflects a broader inertia in Parkinson’s disease research, where non-motor symptoms remain underappreciated despite their profound impact on patients’ lives [134]. Advancing our understanding of sleep dysfunction, developing targeted therapies, and integrating sleep management into routine Parkinson’s disease care must become urgent priorities if we are to redefine the trajectory of this complex disorder.
5. Neuropsychiatric Manifestations in Parkinson’s Disease
Although Parkinson’s disease is primarily recognized for its motor symptoms, neuropsychiatric manifestations are equally disabling and often precede motor onset by years or even decades (see Table 4). Depression, apathy, anxiety, visual hallucinations, and phantosmia emerge at various stages of the disease, yet they remain underdiagnosed and undertreated, significantly diminishing quality of life and increasing the likelihood of institutionalization [138]. The traditional focus on motor dysfunction has relegated neuropsychiatric symptoms to a secondary role in clinical management despite their substantial impact on patient autonomy, caregiver burden, and disease progression [138]. Pathophysiologically, these symptoms stem from α-synuclein accumulation and the disruption of key neuronal circuits, particularly within the mesocorticolimbic, serotonergic, and noradrenergic systems, yet research efforts to integrate these mechanisms into comprehensive treatment strategies remain fragmented [139]. Recognizing neuropsychiatric symptoms as early biomarkers of Parkinson’s disease and developing targeted interventions to address them is crucial to improving diagnosis and therapeutic outcomes (Figure 1 and Figure 2) [139,140].
5.1. Depression
Depression is one of the most common neuropsychiatric symptoms of Parkinson’s disease, affecting between 35% and 45% of patients (see Table 4). It may present at any stage of the disease, frequently emerging before the onset of motor dysfunction and persisting throughout disease progression [141]. Unlike primary depressive disorders, Parkinson’s-related depression is often characterized by irritability, anhedonia, cognitive slowing, and profound apathy rather than overt sadness, complicating its recognition and differentiation from other non-motor symptoms. Sleep disturbances, fatigue, and psychomotor retardation further contribute to functional impairment, exacerbating the overall disease burden [141,142].
The underlying pathophysiology of Parkinson’s-related depression extends beyond dopamine depletion, implicating widespread neurodegeneration in key mood-regulating structures. Structural imaging studies reveal significant grey matter atrophy in the bilateral thalami and amygdalae, regions critical for emotional processing, while cortical gyrification deficits in the frontal and parietal lobes correlate with depressive severity [143]. Braak’s staging suggests that early degeneration of the mesolimbic dopaminergic system, as well as noradrenergic neurons in the locus coeruleus and serotonergic neurons in the raphe nucleus, underlies mood dysregulation in Parkinson’s disease [9,10]. However, serotonin transporter reductions do not consistently correlate with depression severity, raising questions about additional mechanisms contributing to these symptoms [144]. Interestingly, increased SNCA gene expression, which encodes α-synuclein, has been observed in patients with Parkinson’s-related depression, suggesting a molecular link between neurodegeneration and mood disorders [145].
Despite its high prevalence and significant impact, depression in Parkinson’s disease remains inadequately addressed in clinical practice. Conventional antidepressants, particularly selective serotonin reuptake inhibitors, offer inconsistent benefits, likely due to the unique pathophysiology of Parkinson’s-associated depression [146]. Dopaminergic treatments such as pramipexole have demonstrated antidepressant effects, yet their use remains limited by concerns over impulse control disorders [146]. Non-pharmacological approaches, including cognitive-behavioural therapy, transcranial magnetic stimulation, and structured exercise programs, show promise but rarely integrate into standard care.
The continued underrecognition of depression in Parkinson’s disease reflects a broader systemic failure to prioritize non-motor symptoms in both research and treatment paradigms. While motor symptoms define the disease clinically, neuropsychiatric symptoms often dictate the patient’s quality of life, influencing disease progression, caregiver distress, and healthcare utilization [146]. Without a shift in focus that integrates neuropsychiatric dysfunction into the core framework of Parkinson’s disease management, patients will remain underserved, enduring a silent burden that is both preventable and treatable. Future research must move beyond dopamine-centric models to explore novel therapeutic targets, leveraging advanced imaging and biomarker studies to refine diagnostic criteria and develop precision medicine approaches. Until then, Parkinson’s disease will continue to be managed as an incomplete disorder—one where movement is prioritized, but the mind is left behind.
5.2. Apathy
Apathy, a profound lack of motivation that is independent of mood, consciousness, or cognition, is one of the most disabling non-motor symptoms of Parkinson’s disease [147]. Affecting between 40% and 52% of patients, apathy often emerges early in the disease course, with or without coexisting depression or anxiety, yet remains significantly underrecognized and undertreated [148,149]. It manifests in three distinct domains—behavioural and cognitive inertia, emotional blunting, and diminished social interaction—each contributing to a progressive disengagement from daily activities and exacerbating functional decline (see Table 4). Apathy is diagnosed when these deficits persist for at least four weeks and impair at least two domains, making it a critical yet frequently overlooked determinant of disease burden [150,151].
The impact of apathy extends far beyond motivation deficits, profoundly influencing cognition, emotional well-being, and survival. Patients with apathy exhibit more significant cognitive impairment, more severe depressive symptoms, and an increased risk of mortality compared to those without these symptoms. While early-onset apathy in Parkinson’s disease is often associated with concurrent depression, its later emergence signals a heightened risk for cognitive decline and progression to dementia [152,153]. This evolution from a motivational deficit to a harbinger of cognitive deterioration highlights its potential role as a biomarker of disease progression.
Neuroimaging studies have revealed widespread dysfunction in the mesocorticolimbic network, integral to goal-directed behaviour and reward processing. Structural and functional imaging analyses show reduced connectivity within the prefrontal and orbitofrontal cortex, nucleus accumbens, and ventral tegmental area, reinforcing the notion that apathy in Parkinson’s disease results from a breakdown in dopaminergic, limbic, and motivational circuits [154,155,156]. At a molecular level, increased iron deposition and α-synuclein oligomers in cerebrospinal fluid suggest that oxidative stress and neuroinflammation may further exacerbate motivational deficits, although the precise mechanisms remain unclear [157]. The strong association between apathy and cognitive dysfunction raises the question of whether targeting motivational pathways early in the disease could delay or even prevent dementia onset [158].
Despite its prevalence and clinical relevance, apathy remains one of the least effectively managed non-motor symptoms of Parkinson’s disease [159]. Traditional dopaminergic therapies provide inconsistent benefits, as apathy is often resistant to levodopa, suggesting a dysfunction extending beyond the nigrostriatal system. While dopamine agonists such as pramipexole have shown some efficacy, their use is frequently limited by side effects, including impulse control disorders. Non-pharmacological approaches, including structured cognitive interventions, exercise programs, and targeted behavioural therapy, hold promise but remain vastly underutilized in routine clinical care.
Apathy represents a fundamental challenge in Parkinson’s disease research and management, underscoring the urgent need for more targeted therapeutic strategies. Its strong link to cognitive decline and neurodegeneration raises critical questions about whether it is merely a symptom of disease progression or an independent driver of neural dysfunction [159]. Early identification and treatment should be prioritized in clinical practice if apathy accelerates cognitive decline. However, the lack of standardized assessment tools and the persistent misconception that apathy is an inevitable part of Parkinson’s disease continue to hinder meaningful intervention. Future research must refine biomarkers to predict and track apathy and develop novel treatment paradigms to restore motivational drive. Until then, apathy will remain an insidious yet overlooked contributor to disability in Parkinson’s disease, silently eroding quality of life while eluding effective management.
5.3. Anxiety
Anxiety is a frequent and often debilitating non-motor symptom of Parkinson’s disease, characterized by excessive worry, an exaggerated perception of threat, and diminished coping mechanisms (see Table 4). Affecting approximately 31% of patients, anxiety in Parkinson’s disease is not merely a secondary reaction to diagnosis but an intrinsic feature of the disease, emerging as early as the prodromal phase and persisting throughout disease progression [160]. Unlike in primary anxiety disorders, Parkinson’s-related anxiety exhibits a unique profile, with generalized anxiety disorder, social phobia, and panic disorder being the most prevalent manifestations [160]. The presence of anxiety accelerates disease progression, exacerbates motor dysfunction, interferes with treatment response, and increases overall mortality risk [161]. Despite its profound impact on patients' quality of life, anxiety remains largely underdiagnosed and undertreated, often dismissed as a reactive component of living with a neurodegenerative disorder.
The emergence of anxiety in Parkinson’s disease is closely tied to structural and functional alterations in key neural circuits. Neuroimaging studies reveal significant atrophy in the left amygdala, fronto-cingulate cortex, and parietal regions and heightened connectivity within the fear circuit and salience network [162]. These alterations disrupt emotional regulation and threat perception, likely contributing to the heightened anxiety response observed in Parkinson’s disease. Furthermore, α-synuclein pathology extends beyond the dopaminergic system, with accumulating evidence linking α-synuclein deposits in the amygdala to anxiety symptoms. Elevated erythrocytic α-synuclein levels have been detected in Parkinson’s patients with anxiety, reinforcing its potential role in disease pathophysiology [163,164]. Beyond neurodegeneration, oxidative stress and systemic inflammation have also been implicated as key contributors to Parkinson’s-related anxiety, suggesting that peripheral immune dysfunction may play a previously underestimated role in neuropsychiatric symptoms [165].
Anxiety’s early onset, often predating motor symptoms, raises the intriguing possibility that it could serve as a biomarker for Parkinson’s disease. Advanced imaging techniques, serum biomarker analysis, and machine learning approaches have begun identifying specific structural and network changes associated with Parkinson’s-related anxiety, paving the way for earlier detection and intervention [161]. However, the field remains fragmented, with a lack of standardized diagnostic criteria to distinguish Parkinson’s-specific anxiety from primary anxiety disorders. This diagnostic ambiguity hinders targeted treatment development and leaves many patients inadequately managed.
Current treatment approaches for Parkinson’s-related anxiety remain suboptimal [166]. While selective serotonin reuptake inhibitors and benzodiazepines are frequently prescribed, their efficacy is inconsistent, and benzodiazepines, in particular, pose significant risks, including cognitive decline and increased fall risk. Dopaminergic medications, often thought to alleviate non-motor symptoms, may paradoxically worsen anxiety in some patients, complicating pharmacological management. Non-pharmacological interventions, including cognitive behavioural therapy and mindfulness-based strategies, have shown promise yet remain vastly underutilized in routine clinical care.
Anxiety in Parkinson’s disease is more than a comorbidity; it is an intrinsic feature of disease progression that profoundly impacts quality of life, clinical outcomes, and mortality. Its presence long before motor onset suggests that neuropsychiatric dysfunction may be a harbinger of broader neural degeneration rather than a downstream effect of Parkinson’s pathology [166]. Nevertheless, despite its prevalence and impact, anxiety remains an overlooked component of clinical care, often dismissed as an inevitable consequence of chronic illness. Future research must prioritize the identification of robust biomarkers to enable early detection, refine treatment strategies to account for the unique neurobiological underpinnings of Parkinson’s-related anxiety, and integrate behavioural therapies into comprehensive disease management. Until then, anxiety will remain a critical yet neglected factor driving disability and suffering in Parkinson’s disease.
5.4. Visual Hallucinations
Visual hallucinations represent one of the most striking and disruptive non-motor symptoms of Parkinson’s disease, characterized by false sensory perceptions that occur without external stimuli [167]. These hallucinations typically involve structured images, such as people or animals, distinct from visual illusions from actual stimuli misinterpretations (see Table 4). Their prevalence varies widely across populations and methodologies, with reported rates ranging from 27% to 50% [168,169]. Minor visual hallucinations—brief, non-distressing perceptual disturbances—often emerge early in Parkinson’s disease, affecting approximately 22% of patients, and are associated with cognitive decline and reduced quality of life [168,170]. As the disease progresses, hallucinations become more frequent and complex, eventually evolving into distressing visual experiences that signal advanced neurodegeneration.
The mechanisms underlying visual hallucinations in Parkinson’s disease remain incompletely understood, but multiple neurobiological factors contribute to their development. Dopaminergic therapy, particularly the use of dopamine agonists, is a well-established risk factor, yet hallucinations also occur in drug-naïve patients, indicating a primary neurodegenerative basis [171,172]. Cholinergic dysfunction plays a crucial role, with neuroimaging studies demonstrating reduced cholinergic transmission in the left ventral visual stream and superior temporal lobe, regions essential for visual perception and processing [173]. Additionally, impaired inhibitory neurotransmission further contributes to hallucinatory phenomena, as patients with Parkinson’s disease exhibit lower GABA levels in the primary visual cortex, disrupting sensory filtering and reality discrimination [174].
Network dysfunction extends beyond localized neurotransmitter deficits. Structural and functional imaging studies reveal altered connectivity between the lateral geniculate nucleus, visual cortex, prefrontal cortex, and medial thalamus, underscoring the role of higher-order cortical dysfunction in visual hallucinations [175,176]. The presence of Lewy body pathology in the amygdala is strongly associated with hallucinations, particularly in Parkinson’s disease dementia, while degeneration of the nucleus basalis of Meynert further exacerbates visual processing deficits [177,178]. These findings suggest that hallucinations are not simply a consequence of isolated neurotransmitter imbalances but reflect widespread network failure across multiple brain regions involved in sensory integration, attention, and cognitive control.
Visual hallucinations hold significant clinical relevance, as they are strongly linked to disease severity, cognitive decline, and progression to dementia. Their presence in early Parkinson’s disease may serve as an indicator of more aggressive disease trajectories, highlighting the need for early intervention [179]. However, despite their importance, current treatment strategies remain inadequate. Reducing dopaminergic therapy can alleviate symptoms but often exacerbates motor dysfunction, placing clinicians in a therapeutic dilemma. Cholinesterase inhibitors have shown some benefit, particularly in patients with concurrent cognitive impairment, yet their efficacy remains inconsistent. Non-pharmacological approaches, such as cognitive behavioural therapy and environmental modifications, are underexplored despite their potential to improve coping mechanisms and reduce hallucination severity.
The persistence of visual hallucinations as an unresolved challenge in Parkinson’s disease underscores the need for a paradigm shift in their management [177]. Rather than treating hallucinations reactively once they emerge, future research should prioritize identifying predisposing factors and implementing preventive strategies. Integrating advanced neuroimaging, electrophysiological markers, and machine learning approaches may better understand the neural networks involved, leading to targeted interventions. Until then, visual hallucinations will remain an ominous predictor of disease progression, reflecting the broader failure to address non-motor symptoms in Parkinson’s disease adequately.
5.5. Phantosmia
Phantosmia, the perception of odours without an external source, represents a lesser-known but intriguing non-motor symptom of Parkinson’s disease (see Table 4). Its reported prevalence varies widely, ranging from 0.5% to 18.2% of patients, reflecting inconsistencies in study methodologies and diagnostic criteria [180]. These olfactory hallucinations can be either pleasant, like the scent of flowers or fruit, or unpleasant, including odours resembling rotten eggs, smoke, or garbage. They occur more frequently in women and often co-occur with visual and auditory hallucinations, suggesting a broader dysfunction in sensory processing. Unlike other non-motor symptoms, phantosmias do not correlate with motor impairment, cognitive function, or even measured olfactory and gustatory deficits, making their pathophysiological basis particularly enigmatic [181,182]. Their presence at any disease stage, including the prodromal phase, raises the possibility that they could be an early biomarker, though current evidence remains inconclusive [183].
Despite the increasing recognition of phantosmias in Parkinson’s disease, their underlying mechanisms remain poorly understood. One prevailing hypothesis implicates a disruption in hierarchical predictive processing within the olfactory bulb and anterior olfactory nucleus, regions involved in sensory perception and integration [184]. However, unlike hyposmia, which has been linked to α-synuclein accumulation in olfactory pathways, no direct association between Lewy pathology and olfactory hallucinations has been established in Parkinson’s disease. This dissociation challenges conventional neurodegenerative models and suggests phantosmia may arise from dysfunctional sensory gating rather than direct neuronal loss.
The absence of specific diagnostic markers or reliable pathophysiological explanations has hindered the development of effective treatments. Current management strategies remain largely empirical, relying on general neuropsychiatric approaches rather than targeted interventions. Given that phantosmias are not exclusive to Parkinson’s disease and can occur in other neurological and psychiatric disorders, their diagnostic specificity remains questionable. Future research must clarify whether these hallucinations represent a distinct feature of Parkinson’s disease or an epiphenomenon of broader sensory network dysfunction. Until then, phantosmia remains an underexplored phenomenon, highlighting the limitations of current non-motor symptom research in Parkinson’s disease.
6. Cognitive Dysfunction in Parkinson’s Disease
Cognitive dysfunction is a pervasive non-motor symptom of Parkinson’s disease, with executive deficits emerging early and progressively worsening as the disease advances (see Table 5). In treatment-naïve patients, cognitive impairments are present in approximately 27% of cases, escalating to 93% in later stages, highlighting the relentless nature of neurodegeneration in Parkinson’s disease [146,185]. The severity of these deficits is modulated by disease duration, motor symptom burden, and medication status, with more pronounced impairments observed in individuals with advanced motor dysfunction or those undergoing dopaminergic withdrawal [146].
Patients frequently report difficulties in adaptive responses, working memory, visuospatial perception, task initiation, and a decline in processing speed and attentional control [185]. The earliest structural changes occur in the caudate nucleus, where dopaminergic depletion in the nigrostriatal pathway disrupts the basal ganglia-thalamo-cortical loop, particularly impairing the prefrontal cortex [186,187,188]. As the disease progresses, degeneration extends to the mesocorticolimbic system, further exacerbating cognitive dysfunction and limiting executive flexibility [189]. These deficits impair dual-task activities, such as walking while speaking, revealing the impact of cognitive dysfunction on daily motor control and functional independence [190].
Beyond the clinical impact on individual patients, cognitive impairment in Parkinson’s disease has far-reaching consequences, reducing autonomy, increasing social strain, and often leading to premature employment loss [186,191,192]. The high burden on caregivers and healthcare systems underscores the urgent need for early recognition and intervention strategies. However, cognitive decline in Parkinson’s disease is often misattributed to ageing or overlooked until significant deterioration occurs, delaying appropriate management (Figure 1 and Figure 2).
6.1. Inattention and Task-Switching Performance
Deficits in attentional control disrupt goal-directed behaviour, impairing the ability to switch between tasks and resolve conflicts [193]. Inattention, defined as an inability to sustain focus or engage with cognitively demanding tasks, is a common early symptom, affecting up to 20.86% of untreated Parkinson’s disease patients and sometimes preceding motor symptoms by two years [194,195,196]. The neural substrates of attention involve an intricate network connecting the frontal lobe to the parietal and temporo-occipital regions, with key roles played by the medial prefrontal cortex, anterior cingulate, and prelimbic cingulate areas. Dopaminergic signalling within the mesocortical pathway is fundamental to attentional control, and experimental dopaminergic inhibition in healthy individuals reproduces Parkinson’s disease-related attentional deficits [197,198].
Stroop task studies suggest that attentional deficits in Parkinson’s disease result from limited cognitive resources rather than a fundamental failure of executive control. Task complexity and cognitive load appear to exacerbate these difficulties, with Parkinson’s disease patients showing disproportionately slower responses and increased task-switching challenges [199,200]. Electrophysiological studies reveal altered gamma and α-band activity, with increased bilateral gamma oscillations in bottom-up attention and heightened left α-2 connectivity in top-down attention, suggesting an impaired ability to filter distractions [201,202,203]. Functional imaging highlights dysfunction within the dorsal frontoparietal network, further corroborating the role of disrupted connectivity in Parkinson’s disease -related inattention [204].
While dopamine depletion is central to attention deficits, emerging evidence suggests that lower-order attentional shifts depend on corticostriatal dopaminergic transmission, whereas higher-order shifts rely on parietofrontal circuits influenced by noradrenergic and serotonergic systems [205,206,207]. This distinction underscores the necessity of targeting multiple neurotransmitter systems for therapeutic intervention. However, despite growing recognition of attentional deficits in Parkinson’s disease, these impairments are frequently misdiagnosed as early cognitive decline or learning difficulties, delaying targeted intervention.
Current treatment strategies remain suboptimal. While dopaminergic therapies can partially alleviate attentional deficits, their effectiveness varies across patients, and their long-term impact on cognition is debated. Alternative approaches, including ADHD-related pharmacological treatments, cognitive training, and environmental modifications, have been proposed, yet their efficacy remains poorly established. The heterogeneity of cognitive dysfunction in Parkinson’s disease necessitates a personalized treatment paradigm, integrating pharmacological and behavioural strategies to optimize patient outcomes. However, without standardized cognitive assessments and targeted interventions, many patients will continue to experience a silent but debilitating decline in executive function, further complicating disease management.
6.2. Bradyphrenia
Bradyphrenia, characterized by slowed thought processes, impaired attention, and reduced cognitive spontaneity, represents a core cognitive alteration in Parkinson’s disease. Unlike dementia, bradyphrenia manifests as a distinct deficit in cognitive speed and executive flexibility, making it a sensitive yet nonspecific marker of cerebral pathology and frontosubcortical circuit dysfunction [186,208]. Its neurobiological basis has been linked to alterations in norepinephrine metabolism and deficits in white matter integrity, further implicating disruptions in cortico-subcortical connectivity [186,188].
The prevalence of bradyphrenia varies depending on age, baseline intelligence, and cardiovascular risk factors, with studies indicating that up to 25% of Parkinson’s disease patients without dementia exhibit attention-executive or visuospatial deficits [209]. While typically emerging alongside motor symptoms, its course fluctuates, sometimes progressing insidiously throughout the disease. Given its subtle nature, bradyphrenia often escapes clinical detection in early disease stages despite its significant impact on daily life and decision-making abilities.
Assessment involves a combination of cognitive processing speed, flexibility, attention, working memory, and global cognitive function tests, which help delineate bradyphrenia from other cognitive impairments in Parkinson’s disease [210,211,212]. Computational modelling studies suggest that while Parkinson’s disease patients retain cognitive information efficiently, they struggle with sensorimotor adaptation, leading to rigid thinking, difficulty switching strategies, and impaired motor learning [210]. This cognitive inflexibility, often described as the mental equivalent of bradykinesia, can severely impact problem-solving and adaptation to novel situations.
Despite its parallels to motor slowing, bradyphrenia appears to have distinct neurochemical underpinnings, as levodopa treatment—though beneficial for motor symptoms—has been linked to decreased cognitive efficiency in some patients, suggesting that dopamine replacement therapy does not uniformly mitigate cognitive slowing [208,213]. These findings raise questions about the role of non-dopaminergic pathways, including noradrenergic and cholinergic systems, in regulating cognitive speed and adaptability.
Attention plays a crucial role in learning and executive function, making it essential to differentiate bradyphrenia from confounding factors such as sensorimotor impairments, anxiety, arousal deficits, language barriers, educational background, apathy, and depression [209]—the failure to distinguish bradyphrenia from other cognitive deficits risks misdiagnosis, leading to ineffective interventions. Current treatment strategies remain inadequate, relying on pharmacological and non-pharmacological approaches to optimise cognitive performance. However, the lack of standardized therapeutic protocols and the absence of targeted interventions underscore the pressing need for further research to refine diagnostic criteria and treatment strategies.
6.3. Dementia
Parkinson’s disease dementia (PDD) represents a significant cognitive decline that interferes with daily functioning, affecting 20–40% of patients, with up to 80% developing it over time [214]. The risk is exceptionally high among elderly individuals, those with the akinetic-rigid Parkinson’s disease subtype, and patients with cardiovascular risk factors or visual hallucinations [214,215]. Additionally, mild cognitive impairment in early Parkinson’s disease, observed in 30–70% of newly diagnosed patients, serves as a predictive marker for dementia progression, with a shorter transition time to full-blown cognitive decline [214].
The underlying pathophysiology of PDD involves widespread dysfunction of cortico-striato-thalamo-cortical circuits, resembling the cognitive decline seen in dementia with Lewy bodies. Deficits predominantly affect executive, visuospatial, and attentional functions, with patients exhibiting disinhibition, apathy, hallucinations, and delusions, while memory impairments remain secondary [216,217]. Although α-synuclein pathology remains the primary hallmark of PDD, coexisting Alzheimer’s-related pathologies—such as tau, amyloid, and TDP-43 proteinopathies—contribute significantly to disease progression and severity [217,218]. Subcortical degeneration, involving dopaminergic, cholinergic, noradrenergic, and serotonergic systems, further exacerbates cognitive impairment. Additionally, synaptic alterations and neuronal loss in limbic cortical areas, including the amygdala, accelerate the neurodegenerative process [219,220].
Beyond proteinopathies and neurodegeneration, ageing-related lesions, neuroinflammation, mitochondrial dysfunction, oxidative stress, and metabolic disturbances have been implicated in the complex pathology of PDD [221,222]. The interplay between these factors suggests a multifactorial aetiology, making it challenging to develop effective disease-modifying interventions. Despite extensive research, no proven strategy currently exists to delay or prevent mild cognitive impairment progression to dementia in Parkinson’s disease [2].
Emerging pharmacological and non-pharmacological therapies have shown promise in improving cognitive and behavioural symptoms. However, their effects remain modest, and treatment approaches lack personalization. Given the heterogeneity of PDD, future research must prioritize identifying predictive biomarkers and refining therapeutic strategies to slow cognitive decline, enhance quality of life, and prolong patient independence in advanced disease stages. A shift toward precision medicine—integrating neuroimaging, cerebrospinal fluid markers, and machine learning models—could help tailor interventions and address the individual variability in PDD progression.
6.4. Impulse Control Disorders and Impulsive-Compulsive Behaviours
Impulse control disorders (ICDs) in Parkinson’s disease manifest as compulsive behaviours that disrupt social and occupational functioning, with consequences ranging from interpersonal conflicts to severe legal and financial repercussions [223,224]. These disorders affect approximately 20% of early Parkinson’s disease patients, with prevalence increasing over time, ranging between 6% and 45% depending on study populations and assessment methods [225]. Risk factors include male sex, a prior history of ICDs, substance abuse, younger Parkinson’s disease onset, and longer disease duration. Notably, early-onset Parkinson’s disease patients exhibit an exceptionally high susceptibility, with 58.3% displaying ICDs, often associated with dopamine agonist use [226].
ICDs encompass a spectrum of compulsive behaviours, including pathological gambling, hypersexuality, compulsive shopping, and binge eating, all of which significantly impair quality of life [227,228,229]. Their pathophysiology is rooted in dopaminergic dysregulation, which can be classified into four distinct syndromes: dopamine deficiency syndrome, characterized by maladaptive reward-seeking behaviour; dopamine dependency syndrome, associated with addictive patterns; dopamine dysregulation syndrome, linked to stereotyped compulsions; and broader impulsivity disorders, encompassing risky decision-making and poor inhibitory control [230,231].
While mesocortical and mesolimbic dopamine dysfunction is the primary driver of ICDs, dysregulation of the opiate and serotonin systems further contributes to the loss of inhibitory control [230]. Dopamine agonists enhance reward-based learning and impulsive decision-making, heightening risk preference and impairing working memory, thereby exacerbating compulsive tendencies [232,233,234].
The current approach to ICD management primarily involves modifying or discontinuing dopamine replacement therapy, particularly dopamine agonists, yet this strategy is complicated by the potential worsening of motor symptoms and withdrawal effects [235]. Given these limitations, non-pharmacological interventions, including cognitive-behavioural therapy and impulse control retraining, are being explored. Alternative pharmacological approaches, such as opioid antagonists, serotonin reuptake modulators, and novel neurostimulation techniques, offer emerging therapeutic potential but require further validation. As ICDs continue to pose a substantial challenge in Parkinson’s disease, future research must focus on identifying predictive biomarkers, refining risk stratification, and personalized treatment strategies to mitigate their impact without compromising motor symptom control.
7. Conclusions and Perspectives
Recognizing non-motor symptoms as central to Parkinson’s disease has significantly reshaped the understanding of its pathophysiology, yet clinical management remains frustratingly insufficient [3,5]. Despite growing evidence implicating multiple neurotransmitter systems and widespread neural dysfunction, therapeutic strategies remain largely anchored in a dopamine-centric model that fails to address the full complexity of the disease [5]. Patients experience a relentless progression of autonomic dysfunction, cognitive decline, sleep-related disorders, and neuropsychiatric symptoms that often precede motor impairments by decades, yet these manifestations continue to be overlooked, poorly diagnosed, and insufficiently treated (Figure 1 and Figure 2). The failure to integrate these disabling non-motor symptoms into standard care and research priorities exposes a critical gap in translational efforts and underscores the urgent need for a paradigm shift in scientific investigation and clinical practice [6].
Efforts to diagnose Parkinson’s disease in its earliest stages have increasingly focused on non-motor symptoms, but the search for reliable biomarkers remains elusive [2,3,4,5,6,24,114,116,214]. Sensory deficits, REM sleep behaviour disorder, and autonomic dysfunction are among the earliest clinical indicators, yet their predictive value and specificity remain uncertain. α-synuclein aggregation, long considered the pathological hallmark of Parkinson’s disease, is inconsistently associated with neuronal loss and symptom severity, raising fundamental questions about its role as a driver or byproduct of neurodegeneration. The widely accepted Braak staging hypothesis, which proposes a progressive cauda-rostral spread of pathology, has provided a compelling framework for disease progression. However, inconsistencies in its applicability to all patients and the failure to translate this model into effective therapeutic interventions highlight the limitations of viewing Parkinson’s disease as a singular pathological entity. The assumption that Parkinson’s disease follows a uniform trajectory has impeded the development of alternative hypotheses and hindered the pursuit of innovative diagnostic and therapeutic approaches [2,3,4,5,6,24,114,116,214].
Cognitive impairment remains one of the most devastating and least treatable aspects of Parkinson’s disease. While mild executive dysfunction often emerges early, a substantial proportion of patients progress to full-blown dementia, yet treatment options remain rudimentary. Cholinesterase inhibitors, repurposed from Alzheimer’s disease, offer only modest benefits, failing to address the intricate interplay between dopamine, acetylcholine, and other neurotransmitter systems involved in cognitive decline. The artificial distinction between Parkinson’s disease dementia and dementia with Lewy bodies has further complicated efforts to develop effective interventions, as both likely represent overlapping spectra of the same neurodegenerative process [2,3,4,5,6,24,114,116,214]. Additionally, the presence of Alzheimer’s-related tau and amyloid pathology in a subset of patients suggests a convergence of mechanisms that has been largely ignored in clinical trials. Cognitive dysfunction should no longer be regarded as an inevitable late-stage complication but as a core feature of Parkinson’s disease requiring earlier and more targeted interventions.
Sleep-related disorders and autonomic dysfunction, despite their profound impact on quality of life, remain some of the most neglected aspects of Parkinson’s disease research and treatment [134]. Orthostatic hypotension, excessive salivation, constipation, and urinary dysfunction affect nearly all patients at some stage, yet current management strategies are largely symptomatic, addressing secondary consequences rather than underlying mechanisms [5]. Autonomic dysfunction is one of the earliest detectable features of Parkinson’s disease, yet its role in disease progression remains poorly understood. Similarly, sleep-related disorders, including rapid eye movement, sleep behaviour disorder and excessive daytime sleepiness, often appear years before motor symptoms, presenting an opportunity for early intervention [5]. However, research on sleep dysfunction has been disproportionately focused on its association with neurodegeneration rather than its potential as a diagnostic and therapeutic target [134]. The continued relegation of these symptoms to secondary status in clinical care and research has significantly hindered progress in developing effective treatments.
Neuropsychiatric symptoms—including depression, anxiety, apathy, and psychosis—further complicate Parkinson’s disease and remain among the most challenging to manage. Although serotonin dysfunction is strongly implicated in mood disturbances, selective serotonin reuptake inhibitors yield inconsistent results, suggesting that current pharmacological approaches fail to target the specific neurochemical alterations underlying these symptoms [2,3,4,5,6,24,114,116,214]. Apathy, often misdiagnosed as depression, appears to stem from mesolimbic dopamine dysfunction, yet no approved treatments exist to address its distinct neurobiological basis. The frequent coexistence of neuropsychiatric symptoms with cognitive impairment suggests a shared pathology, yet these conditions continue to be treated in isolation, with little effort to develop integrative management strategies. The longstanding division between neurology and psychiatry in both research and clinical practice has contributed to the neglect of these symptoms, leaving patients in a therapeutic void.
The emerging focus on the gut-brain axis has introduced novel perspectives on Parkinson’s disease pathogenesis, yet robust clinical evidence remains lacking. The discovery of α-synuclein aggregates in the enteric nervous system aligns with Braak’s hypothesis, but whether gut dysbiosis actively contributes to neurodegeneration or reflects systemic disease progression remains uncertain [9,10]. The enthusiasm surrounding microbiome research has often outpaced available evidence, with many studies relying on small cohorts and correlative findings rather than establishing causality. While modifying gut microbiota to influence disease progression is intriguing, clinical trials have yet to provide compelling evidence that such interventions are effective. The widespread speculation that targeting the gut could alter disease trajectory remains, at this stage, an unproven hypothesis rather than a transformative breakthrough.
Despite these challenges, the most pressing issue remains the stagnation of therapeutic development. No neuroprotective therapy has succeeded in altering the course of Parkinson’s disease, largely because research continues to target the wrong biological pathways at the wrong stages [2,3,4,5,6,24,114,116,214]. The longstanding approach of treating Parkinson’s disease as a homogeneous disorder has undermined clinical trials, as different subtypes likely require distinct therapeutic strategies. Future treatments must move beyond dopamine replacement and embrace a precision medicine approach that identifies patient subgroups based on genetic, neurochemical, and biomarker profiles. Expanding the traditional neurocentric model to incorporate neuroinflammation, mitochondrial dysfunction, and peripheral immune activation is essential, as mounting evidence suggests that Parkinson’s disease extends far beyond the brain [2,3,4,5,6,24,114,116,214]. Advances in multi-omics technologies, artificial intelligence-driven analysis, and network-based approaches hold promise for uncovering novel therapeutic targets, yet their integration into mainstream Parkinson’s disease research has been slow.
The field now stands at a crossroads, forced to choose between incremental refinements of existing models and a radical rethinking of Parkinson’s disease pathophysiology. The continued adherence to outdated frameworks has resulted in repeated failures, underscoring the urgent need for disruption. Non-motor symptoms must no longer be considered secondary to movement impairments but recognized as defining features that drive disease burden and progression [2,3,4,5,6,24,114,116,214]. The persistence of a dopamine-dominated treatment paradigm is no longer justifiable in light of overwhelming evidence implicating broader neurochemical dysfunction. Only by challenging entrenched assumptions and adopting a systems biology perspective can Parkinson’s disease research move beyond its current limitations. The next breakthrough will not emerge from yet another dopamine-based therapy but from a fundamental shift in how this complex, multi-system disorder is conceptualized and treated.
This review did not seek to catalogue every non-motor symptom of Parkinson’s disease but rather to challenge the prevailing perception of the disorder among medical students, primary care clinicians, and non-specialists. Parkinson’s disease is not merely a movement disorder—it is a multisystem neurodegenerative disease that demands a fundamental shift in how it is taught, recognized, and managed. Persisting with a dopaminergic-centric model limits both diagnosis and treatment, delaying recognition of early, disabling non-motor symptoms and restricting therapeutic innovation. The future of Parkinson’s disease care requires a redefined research agenda, a transformation in medical education, and a clinical approach that prioritizes the disease’s full spectrum—beyond motor dysfunction—to improve outcomes and quality of life.
Author Contributions
Conceptualization, O.A.-C.; methodology, O.A.-C.; investigation, O.A.-C., L.P.-Z., K.D.-M., M.V.R., K.L.-A., A.S.-B., J.A.-G., I.P.-S., E.O.-R. and L.O.S.-R.; data curation, L.P.-Z. and O.A.-C.; writing—original draft preparation, O.A.-C., L.P.-Z., K.D.-M., M.V.R., K.L.-A., A.S.-B., J.A.-G., I.P.-S., E.O.-R. and L.O.S.-R.; writing—review and editing, L.P.-Z., K.D.-M., M.V.R., K.L.-A., A.S.-B., J.A.-G., I.P.-S., E.O.-R. and L.O.S.-R.; visualization, L.P.-Z., K.D.-M., M.V.R., K.L.-A., A.S.-B., J.A.-G., I.P.-S., E.O.-R. and L.O.S.-R.; supervision, O.A.-C.; project administration, O.A.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We extend our gratitude to Hospital General Dr. Manuel Gea González for its invaluable support and hospitality, which, in its own way, contributed to the completion of this work. Figures were created with https://www.BioRender.com. The manuscript's clarity and readability were enhanced with the assistance of the Hemingway Editor.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| α-syn | α-synuclein |
| CBT | Cognitive behavioural therapy |
| CPAP | Continuous positive airway pressure |
| CSF | Cerebrospinal fluid |
| CSP | Cortical silent period |
| DBS | Deep brain stimulation |
| DMV | Dorsal motor nucleus of the vagus |
| DRT | Dopamine replacement therapy |
| EDS | Excessive daytime sleepiness |
| EEG | Electroencephalogram |
| FOp | Frontal insular operculum |
| ICD | Impulse control disorder |
| MAO-B | Monoamine oxidase B |
| NBM | Nucleus basalis of Meynert |
| NPDs | Neuropsychiatric disorders |
| NSAID | Nonsteroidal anti-inflammatory drug |
| OB | Olfactory bulb |
| OFC | Orbitofrontal cortex |
| OH | Orthostatic hypotension |
| OSA | Obstructive sleep apnoea |
| PDD | Parkinson’s disease dementia |
| PFC | Prefrontal cortex |
| RBD | REM sleep behaviour disorder |
| REM | Rapid eye movement |
| RLS | Restless legs syndrome |
| SC | Spinal cord |
| SD | Seborrheic dermatitis |
| SN | Substantia nigra |
| SNRI | Serotonin/norepinephrine reuptake inhibitor |
| SSRI | Selective serotonin reuptake inhibitor |
| TCA | Tricyclic antidepressant |
| TMS | Transcranial magnetic stimulation |
| TNF-α | Tumour necrosis factor alpha |
| UPDRS | Unified Parkinson’s Disease Rating Scale |
| VTA | Ventral tegmental area |
References
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Ffytche, D.H.; Weintraub, D.; Ballard, C. Cognitive decline in Parkinson disease. Nat Rev Neurol 2017, 13, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.; National Institute for Clinical, E. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol 2006, 5, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, P.; Manuel Rojo-Abuin, J.; Rizos, A.; Rodriguez-Blazquez, C.; Trenkwalder, C.; Perkins, L.; Sauerbier, A.; Odin, P.; Antonini, A.; Chaudhuri, K.R.; et al. Distribution and impact on quality of life of the pain modalities assessed by the King's Parkinson's disease pain scale. NPJ Parkinsons Dis 2017, 3, 8. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat Rev Neurosci 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Todorova, A.; Jenner, P.; Ray Chaudhuri, K. Non-motor Parkinson's: integral to motor Parkinson's, yet often neglected. Pract Neurol 2014, 14, 310–322. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Kaufmann, H.; Goldstein, D.S. Autonomic dysfunction in Parkinson disease. Handb Clin Neurol 2013, 117, 259–278. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhou, X.; Zhou, X.; Xiang, Y.; Zhu, L.; Qin, L.; Wang, Y.; Pan, H.; Zhao, Y.; Sun, Q.; et al. Characteristics of Autonomic Dysfunction in Parkinson's Disease: A Large Chinese Multicenter Cohort Study. Front Aging Neurosci 2021, 13, 761044. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, G.; Liu, J. Autonomic dysfunction in Parkinson's disease: Implications for pathophysiology, diagnosis, and treatment. Neurobiol Dis 2020, 134, 104700. [Google Scholar] [CrossRef]
- Stewart, C.B.; Ledingham, D.; Foster, V.K.; Anderson, K.N.; Sathyanarayana, S.; Galley, D.; Pavese, N.; Pasquini, J. The longitudinal progression of autonomic dysfunction in Parkinson's disease: A 7-year study. Front Neurol 2023, 14, 1155669. [Google Scholar] [CrossRef]
- Jain, S. Multi-organ autonomic dysfunction in Parkinson disease. Parkinsonism Relat Disord 2011, 17, 77–83. [Google Scholar] [CrossRef]
- Park, J.W.; Okamoto, L.E.; Kim, S.H.; Lee, C.N.; Park, K.W.; Baek, S.H.; Sung, J.H.; Jeon, N.; Koh, S.B.; Gamboa, A.; et al. Sympathetic dysfunction as an early indicator of autonomic involvement in Parkinson's disease. Clin Auton Res 2024, 34, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.A.; Chelimsky, T.C. Pure autonomic failure. Handb Clin Neurol 2019, 161, 413–422. [Google Scholar] [CrossRef]
- Isonaka, R.; Sullivan, P.; Goldstein, D.S. Pathophysiological Significance of alpha-Synuclein in Sympathetic Nerves: In Vivo Observations. Neurology 2025, 104, e210215. [Google Scholar] [CrossRef]
- Wakabayashi, K. Where and how alpha-synuclein pathology spreads in Parkinson's disease. Neuropathology 2020, 40, 415–425. [Google Scholar] [CrossRef]
- Fumimura, Y.; Ikemura, M.; Saito, Y.; Sengoku, R.; Kanemaru, K.; Sawabe, M.; Arai, T.; Ito, G.; Iwatsubo, T.; Fukayama, M.; et al. Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J Neuropathol Exp Neurol 2007, 66, 354–362. [Google Scholar] [CrossRef]
- Goncalves, V.C.; Cuenca-Bermejo, L.; Fernandez-Villalba, E.; Martin-Balbuena, S.; da Silva Fernandes, M.J.; Scorza, C.A.; Herrero, M.T. Heart Matters: Cardiac Dysfunction and Other Autonomic Changes in Parkinson's Disease. Neuroscientist 2022, 28, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Nakamura, T.; Hirayama, M.; Ueda, M.; Katsuno, M.; Sobue, G. Cardiac parasympathetic dysfunction in the early phase of Parkinson's disease. J Neurol 2017, 264, 333–340. [Google Scholar] [CrossRef]
- Palma, J.A.; Kaufmann, H. Treatment of autonomic dysfunction in Parkinson disease and other synucleinopathies. Mov Disord 2018, 33, 372–390. [Google Scholar] [CrossRef]
- van Wamelen, D.J.; Leta, V.; Johnson, J.; Ocampo, C.L.; Podlewska, A.M.; Rukavina, K.; Rizos, A.; Martinez-Martin, P.; Chaudhuri, K.R. Drooling in Parkinson's Disease: Prevalence and Progression from the Non-motor International Longitudinal Study. Dysphagia 2020, 35, 955–961. [Google Scholar] [CrossRef]
- Polychronis, S.; Nasios, G.; Dardiotis, E.; Messinis, L.; Pagano, G. Pathophysiology and Symptomatology of Drooling in Parkinson's Disease. Healthcare (Basel) 2022, 10. [Google Scholar] [CrossRef]
- Isaacson, J.; Patel, S.; Torres-Yaghi, Y.; Pagan, F. Sialorrhea in Parkinson's Disease. Toxins (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Cheng, Y.Q.; Ge, N.N.; Zhu, H.H.; Sha, Z.T.; Jiang, T.; Zhang, Y.D.; Tian, Y.Y. Dihydroergotoxine mesylate for the treatment of sialorrhea in Parkinson's disease. Parkinsonism Relat Disord 2019, 58, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Suttrup, I.; Warnecke, T. Dysphagia in Parkinson's Disease. Dysphagia 2016, 31, 24–32. [Google Scholar] [CrossRef]
- Umemoto, G.; Furuya, H. Management of Dysphagia in Patients with Parkinson's Disease and Related Disorders. Intern Med 2020, 59, 7–14. [Google Scholar] [CrossRef]
- Labeit, B.; Berkovich, E.; Claus, I.; Roderigo, M.; Schwake, A.L.; Izgelov, D.; Mimrod, D.; Ahring, S.; Oelenberg, S.; Muhle, P.; et al. Dysphagia for medication in Parkinson's disease. NPJ Parkinsons Dis 2022, 8, 156. [Google Scholar] [CrossRef]
- Cosentino, G.; Avenali, M.; Schindler, A.; Pizzorni, N.; Montomoli, C.; Abbruzzese, G.; Antonini, A.; Barbiera, F.; Benazzo, M.; Benarroch, E.E.; et al. A multinational consensus on dysphagia in Parkinson's disease: screening, diagnosis and prognostic value. J Neurol 2022, 269, 1335–1352. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Liria, R.; Parra-Egeda, J.; Vega-Ramirez, F.A.; Aguilar-Parra, J.M.; Trigueros-Ramos, R.; Morales-Gazquez, M.J.; Rocamora-Perez, P. Treatment of Dysphagia in Parkinson's Disease: A Systematic Review. Int J Environ Res Public Health 2020, 17. [Google Scholar] [CrossRef]
- Yang, C.L.; Huang, J.P.; Tan, Y.C.; Wang, T.T.; Zhang, H.; Qu, Y. The effectiveness and safety of botulinum toxin injections for the treatment of sialorrhea with Parkinson's disease: a systematic review and meta-analysis. BMC Pharmacol Toxicol 2023, 24, 52. [Google Scholar] [CrossRef]
- Safarpour, D.; Stover, N.; Shprecher, D.R.; Hamedani, A.G.; Pfeiffer, R.F.; Parkman, H.P.; Quigley, E.M.; Cloud, L.J.; Other Non-motor Features Working Group of the Parkinson Study, G. Consensus practice recommendations for management of gastrointestinal dysfunction in Parkinson disease. Parkinsonism Relat Disord 2024, 124, 106982. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Boeve, B.F.; Mielke, M.M. When Do alpha-Synucleinopathies Start? An Epidemiological Timeline: A Review. JAMA Neurol 2018, 75, 503–509. [Google Scholar] [CrossRef]
- Stocchi, F.; Torti, M. Constipation in Parkinson's Disease. Int Rev Neurobiol 2017, 134, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.J.; Yu, S.Y.; Zuo, L.J.; Lian, T.H.; Hu, Y.; Wang, R.D.; Piao, Y.S.; Guo, P.; Liu, L.; Jin, Z.; et al. Parkinson disease with constipation: clinical features and relevant factors. Sci Rep 2018, 8, 567. [Google Scholar] [CrossRef]
- Frazzitta, G.; Ferrazzoli, D.; Folini, A.; Palamara, G.; Maestri, R. Severe Constipation in Parkinson's Disease and in Parkinsonisms: Prevalence and Affecting Factors. Front Neurol 2019, 10, 621. [Google Scholar] [CrossRef]
- Pedrosa Carrasco, A.J.; Timmermann, L.; Pedrosa, D.J. Management of constipation in patients with Parkinson's disease. NPJ Parkinsons Dis 2018, 4, 6. [Google Scholar] [CrossRef]
- Raciti, L.; De Cola, M.C.; Ortelli, P.; Corallo, F.; Lo Buono, V.; Morini, E.; Quattrini, F.; Filoni, S.; Calabro, R.S. Sexual Dysfunction in Parkinson Disease: A Multicenter Italian Cross-sectional Study on a Still Overlooked Problem. J Sex Med 2020, 17, 1914–1925. [Google Scholar] [CrossRef]
- Benigno, M.D.S.; Amaral Domingues, C.; Araujo Leite, M.A. Sexual Dysfunction in Parkinson's Disease: A Systematic Review of the Arizona Sexual Experience Scale Sexual Dysfunction in Parkinson Disease: A Systematic Review of the Arizona Sexual Experience Scale. J Geriatr Psychiatry Neurol 2023, 36, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Haktanir, D.; Yilmaz, S. Sexual Dysfunction and Related Factors in Patients With Parkinson's Disease. J Psychosoc Nurs Ment Health Serv 2023, 61, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Codling, D.; Shaw, P.; David, A.S. Hypersexuality in Parkinson's Disease: Systematic Review and Report of 7 New Cases. Mov Disord Clin Pract 2015, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Jost, W.H. Autonomic dysfunctions in idiopathic Parkinson's disease. J Neurol 2003, 250 Suppl 1, I28–30. [Google Scholar] [CrossRef]
- Robinson, B.W.; Mishkin, M. Penile erection evoked from forebrain structures in Macaca mulatta. Arch Neurol 1968, 19, 184–198. [Google Scholar] [CrossRef]
- Temel, Y.; Visser-Vandewalle, V.; Ackermans, L.; Beuls, E.A. Thalamus and penile erection. Int J Impot Res 2004, 16, 505–511. [Google Scholar] [CrossRef]
- Pavy-Le Traon, A.; Cotterill, N.; Amarenco, G.; Duerr, S.; Kaufmann, H.; Lahrmann, H.; Tison, F.; Wenning, G.K.; Goetz, C.G.; Poewe, W.; et al. Clinical Rating Scales for Urinary Symptoms in Parkinson Disease: Critique and Recommendations. Mov Disord Clin Pract 2018, 5, 479–491. [Google Scholar] [CrossRef]
- Li, F.F.; Cui, Y.S.; Yan, R.; Cao, S.S.; Feng, T. Prevalence of lower urinary tract symptoms, urinary incontinence and retention in Parkinson's disease: A systematic review and meta-analysis. Front Aging Neurosci 2022, 14, 977572. [Google Scholar] [CrossRef]
- Valentino, F.; Bartolotta, T.V.; Cosentino, G.; Mastrilli, S.; Arnao, V.; Aridon, P.; Scurria, S.; Pavone, A.; Pavone, C.; D'Amelio, M. Urological dysfunctions in patients with Parkinson's disease: clues from clinical and non-invasive urological assessment. BMC Neurol 2018, 18, 148. [Google Scholar] [CrossRef]
- Stefanis, L. alpha-Synuclein in Parkinson's disease. Cold Spring Harb Perspect Med 2012, 2, a009399. [Google Scholar] [CrossRef]
- Nam, D.; Lee, J.Y.; Lee, M.; Kim, J.; Seol, W.; Son, I.; Ho, D.H. Detection and Assessment of alpha-Synuclein Oligomers in the Urine of Parkinson's Disease Patients. J Parkinsons Dis 2020, 10, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Cui, X.; Yoshimura, N.; Mao, W.; Xu, E.; Wang, Q.; Ou, T. Assessment and Management of Urinary Dysfunction in 187 Patients with Parkinson's Disease. J Parkinsons Dis 2020, 10, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Yeo, L.; Singh, R.; Gundeti, M.; Barua, J.M.; Masood, J. Urinary tract dysfunction in Parkinson's disease: a review. Int Urol Nephrol 2012, 44, 415–424. [Google Scholar] [CrossRef]
- Li, S.T.; Dendi, R.; Holmes, C.; Goldstein, D.S. Progressive loss of cardiac sympathetic innervation in Parkinson's disease. Ann Neurol 2002, 52, 220–223. [Google Scholar] [CrossRef]
- Cuenca-Bermejo, L.; Almela, P.; Navarro-Zaragoza, J.; Fernandez Villalba, E.; Gonzalez-Cuello, A.M.; Laorden, M.L.; Herrero, M.T. Cardiac Changes in Parkinson's Disease: Lessons from Clinical and Experimental Evidence. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Scorza, F.A.; Fiorini, A.C.; Scorza, C.A.; Finsterer, J. Cardiac abnormalities in Parkinson's disease and Parkinsonism. J Clin Neurosci 2018, 53, 1–5. [Google Scholar] [CrossRef]
- Satoh, A.; Serita, T.; Seto, M.; Tomita, I.; Satoh, H.; Iwanaga, K.; Takashima, H.; Tsujihata, M. Loss of 123I-MIBG uptake by the heart in Parkinson's disease: assessment of cardiac sympathetic denervation and diagnostic value. J Nucl Med 1999, 40, 371–375. [Google Scholar] [PubMed]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson's disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef]
- Denfeld, Q.E.; Turrise, S.; MacLaughlin, E.J.; Chang, P.S.; Clair, W.K.; Lewis, E.F.; Forman, D.E.; Goodlin, S.J.; American Heart Association Cardiovascular Disease in Older Populations Committee of the Council on Clinical, C. ; Council on, C.; et al. Preventing and Managing Falls in Adults With Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circ Cardiovasc Qual Outcomes 2022, 15, e000108. [Google Scholar] [CrossRef]
- Pfeiffer, R.F. Autonomic Dysfunction in Parkinson's Disease. Neurotherapeutics 2020, 17, 1464–1479. [Google Scholar] [CrossRef]
- Lamotte, G.; Lenka, A. Orthostatic Hypotension in Parkinson Disease: What Is New? Neurol Clin Pract 2022, 12, e112–e115. [Google Scholar] [CrossRef] [PubMed]
- Cutsforth-Gregory, J.K.; Low, P.A. Neurogenic Orthostatic Hypotension in Parkinson Disease: A Primer. Neurol Ther 2019, 8, 307–324. [Google Scholar] [CrossRef]
- Fathy, Y.Y.; Jonker, A.J.; Oudejans, E.; de Jong, F.J.J.; van Dam, A.W.; Rozemuller, A.J.M.; van de Berg, W.D.J. Differential insular cortex subregional vulnerability to alpha-synuclein pathology in Parkinson's disease and dementia with Lewy bodies. Neuropathol Appl Neurobiol 2019, 45, 262–277. [Google Scholar] [CrossRef]
- Xing, Y.; Li, Q.; Xu, E.; Zeng, J.; Li, Q.; Mei, S.; Hua, Y. Impaired Cerebral Autoregulation in Parkinson's Disease: An Orthostatic Hypotension Analysis. Front Neurol 2022, 13, 811698. [Google Scholar] [CrossRef]
- Goldstein, D.S. Dysautonomia in Parkinson's disease: neurocardiological abnormalities. Lancet Neurol 2003, 2, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Ogaki, K.; Shiina, T.; Sakuramoto, H.; Nozawa, N.; Suzuki, K. Impact of autonomic symptoms on the clinical course of Parkinson's disease. Neurol Sci 2024, 45, 3799–3807. [Google Scholar] [CrossRef] [PubMed]
- Velseboer, D.C.; de Haan, R.J.; Wieling, W.; Goldstein, D.S.; de Bie, R.M. Prevalence of orthostatic hypotension in Parkinson's disease: a systematic review and meta-analysis. Parkinsonism Relat Disord 2011, 17, 724–729. [Google Scholar] [CrossRef]
- Tomic, S.; Kuric, I.; Kuric, T.G.; Popovic, Z.; Kragujevic, J.; Zubonja, T.M.; Rajkovaca, I.; Matosa, S. Seborrheic Dermatitis Is Related to Motor Symptoms in Parkinson's Disease. J Clin Neurol 2022, 18, 628–634. [Google Scholar] [CrossRef]
- Laurence, M.; Benito-Leon, J.; Calon, F. Malassezia and Parkinson's Disease. Front Neurol 2019, 10, 758. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson's Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef]
- Sinclair, E.; Trivedi, D.K.; Sarkar, D.; Walton-Doyle, C.; Milne, J.; Kunath, T.; Rijs, A.M.; de Bie, R.M.A.; Goodacre, R.; Silverdale, M.; et al. Metabolomics of sebum reveals lipid dysregulation in Parkinson's disease. Nat Commun 2021, 12, 1592. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.W.; Pope, S.M.; Jaboori, K.A. Diagnosis and treatment of seborrheic dermatitis. Am Fam Physician 2015, 91, 185–190. [Google Scholar]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson's disease. Lancet Neurol 2021, 20, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Nolano, M.; Provitera, V.; Estraneo, A.; Selim, M.M.; Caporaso, G.; Stancanelli, A.; Saltalamacchia, A.M.; Lanzillo, B.; Santoro, L. Sensory deficit in Parkinson's disease: evidence of a cutaneous denervation. Brain 2008, 131, 1903–1911. [Google Scholar] [CrossRef]
- Haehner, A.; Hummel, T.; Reichmann, H. Olfactory loss in Parkinson's disease. Parkinsons Dis 2011, 2011, 450939. [Google Scholar] [CrossRef]
- Ansari, K.A.; Johnson, A. Olfactory function in patients with Parkinson's disease. J Chronic Dis 1975, 28, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Doty, R.L. Olfactory dysfunction in Parkinson disease. Nat Rev Neurol 2012, 8, 329–339. [Google Scholar] [CrossRef]
- Haehner, A.; Boesveldt, S.; Berendse, H.W.; Mackay-Sim, A.; Fleischmann, J.; Silburn, P.A.; Johnston, A.N.; Mellick, G.D.; Herting, B.; Reichmann, H.; et al. Prevalence of smell loss in Parkinson's disease--a multicenter study. Parkinsonism Relat Disord 2009, 15, 490–494. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, E.J.; Zhang, W.; Lu, Y.; Liu, R.; Huang, X.; Ciesielski-Jones, A.J.; Justice, M.A.; Cousins, D.S.; Peddada, S. Meta-analyses on prevalence of selected Parkinson's nonmotor symptoms before and after diagnosis. Transl Neurodegener 2015, 4, 1. [Google Scholar] [CrossRef]
- Fullard, M.E.; Morley, J.F.; Duda, J.E. Olfactory Dysfunction as an Early Biomarker in Parkinson's Disease. Neurosci Bull 2017, 33, 515–525. [Google Scholar] [CrossRef]
- Sui, X.; Zhou, C.; Li, J.; Chen, L.; Yang, X.; Li, F. Hyposmia as a Predictive Marker of Parkinson's Disease: A Systematic Review and Meta-Analysis. Biomed Res Int 2019, 2019, 3753786. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Roos, D.S.; Twisk, J.W.R.; Raijmakers, P.; Doty, R.L.; Berendse, H.W. Hyposmia as a marker of (non-)motor disease severity in Parkinson's disease. J Neural Transm (Vienna) 2019, 126, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Zhao, Y.; He, Y.; Zhou, Y.; Yang, J.; Zhou, X.; Zhu, L.; Zhou, X.; Liu, Z.; Xu, Q.; et al. Olfactory Dysfunction Predicts Disease Progression in Parkinson's Disease: A Longitudinal Study. Front Neurosci 2020, 14, 569777. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.C.; Chang, M.H.; Yang, C.P.; Chen, Y.H.; Lin, C.H. The Association of Olfactory Dysfunction With Depression, Cognition, and Disease Severity in Parkinson's Disease. Front Neurol 2021, 12, 779712. [Google Scholar] [CrossRef]
- Fujita, H.; Shiina, T.; Sakuramoto, H.; Nozawa, N.; Ogaki, K.; Suzuki, K. Sleep and Autonomic Manifestations in Parkinson's Disease Complicated With Probable Rapid Eye Movement Sleep Behavior Disorder. Front Neurosci 2022, 16, 874349. [Google Scholar] [CrossRef]
- Sengoku, R.; Matsushima, S.; Bono, K.; Sakuta, K.; Yamazaki, M.; Miyagawa, S.; Komatsu, T.; Mitsumura, H.; Kono, Y.; Kamiyama, T.; et al. Olfactory function combined with morphology distinguishes Parkinson's disease. Parkinsonism Relat Disord 2015, 21, 771–777. [Google Scholar] [CrossRef]
- Park, S.; Lee, J.; Kim, J.Y.; Park, J.H.; Lee, J.E.; Park, S.I.; Yim, Y. Clinical significance of MRI-measured olfactory bulb height as an imaging biomarker of idiopathic Parkinson's disease. PLoS One 2024, 19, e0312728. [Google Scholar] [CrossRef]
- Stefani, A.; Iranzo, A.; Holzknecht, E.; Perra, D.; Bongianni, M.; Gaig, C.; Heim, B.; Serradell, M.; Sacchetto, L.; Garrido, A.; et al. Alpha-synuclein seeds in olfactory mucosa of patients with isolated REM sleep behaviour disorder. Brain 2021, 144, 1118–1126. [Google Scholar] [CrossRef]
- Srivastava, A.; Wang, Q.; Orru, C.D.; Fernandez, M.; Compta, Y.; Ghetti, B.; Zanusso, G.; Zou, W.Q.; Caughey, B.; Beauchemin, C.A.A. Enhanced quantitation of pathological alpha-synuclein in patient biospecimens by RT-QuIC seed amplification assays. PLoS Pathog 2024, 20, e1012554. [Google Scholar] [CrossRef]
- Yoo, H.S.; Lee, S.; Jeong, S.H.; Ye, B.S.; Sohn, Y.H.; Yun, M.; Lee, P.H. Clinical and Dopamine Depletion Patterns in Hyposmia- and Dysautonomia-Dominant Parkinson's Disease. J Parkinsons Dis 2021, 11, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 2001, 7, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhang, J.; Zhao, X.; Nie, W.; Xu, X.; Liu, M.; Zhang, X. Olfactory dysfunction and its related molecular mechanisms in Parkinson's disease. Neural Regen Res 2024, 19, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Deeb, J.; Fernando, M.; Noyce, A.; Visentin, E.; Findley, L.J.; Hawkes, C.H. Abnormality of taste and smell in Parkinson's disease. Parkinsonism Relat Disord 2009, 15, 232–237. [Google Scholar] [CrossRef]
- Kashihara, K.; Hanaoka, A.; Imamura, T. Frequency and characteristics of taste impairment in patients with Parkinson's disease: results of a clinical interview. Intern Med 2011, 50, 2311–2315. [Google Scholar] [CrossRef]
- Cossu, G.; Melis, M.; Sarchioto, M.; Melis, M.; Melis, M.; Morelli, M.; Tomassini Barbarossa, I. 6-n-propylthiouracil taste disruption and TAS2R38 nontasting form in Parkinson's disease. Mov Disord 2018, 33, 1331–1339. [Google Scholar] [CrossRef]
- Jagota, P.; Chotechuang, N.; Anan, C.; Kitjawijit, T.; Boonla, C.; Bhidayasiri, R. Umami and Other Taste Perceptions in Patients With Parkinson's Disease. J Mov Disord 2022, 15, 115–123. [Google Scholar] [CrossRef]
- Oppo, V.; Melis, M.; Melis, M.; Tomassini Barbarossa, I.; Cossu, G. "Smelling and Tasting" Parkinson's Disease: Using Senses to Improve the Knowledge of the Disease. Front Aging Neurosci 2020, 12, 43. [Google Scholar] [CrossRef]
- Crowley, S.J.; Kanel, P.; Roytman, S.; Bohnen, N.I.; Hampstead, B.M. Basal forebrain integrity, cholinergic innervation and cognition in idiopathic Parkinson's disease. Brain 2024, 147, 1799–1808. [Google Scholar] [CrossRef]
- Mantovani, E.; Zanini, A.; Cecchini, M.P.; Tamburin, S. The Association Between Neurocognitive Disorders and Gustatory Dysfunction: A Systematic Review and Meta-Analysis. Neuropsychol Rev 2024, 34, 192–213. [Google Scholar] [CrossRef]
- Garcia-Esparcia, P.; Schluter, A.; Carmona, M.; Moreno, J.; Ansoleaga, B.; Torrejon-Escribano, B.; Gustincich, S.; Pujol, A.; Ferrer, I. Functional genomics reveals dysregulation of cortical olfactory receptors in Parkinson disease: novel putative chemoreceptors in the human brain. J Neuropathol Exp Neurol 2013, 72, 524–539. [Google Scholar] [CrossRef] [PubMed]
- van der Lijn, I.; de Haan, G.A.; Huizinga, F.; van der Feen, F.E.; Rutgers, A.W.F.; Stellingwerf, C.; van Laar, T.; Heutink, J. Self-Reported Visual Complaints in People with Parkinson's Disease: A Systematic Review. J Parkinsons Dis 2022, 12, 785–806. [Google Scholar] [CrossRef] [PubMed]
- van der Lijn, I.; de Haan, G.A.; van der Feen, F.E.; Huizinga, F.; Stellingwerf, C.; van Laar, T.; Heutink, J. Prevalence and nature of self-reported visual complaints in people with Parkinson's disease-Outcome of the Screening Visual Complaints questionnaire. PLoS One 2023, 18, e0283122. [Google Scholar] [CrossRef]
- Brown, T.; Kanel, P.; Griggs, A.; Carli, G.; Vangel, R.; Albin, R.L.; Bohnen, N.I. Regional cerebral cholinergic vesicular transporter correlates of visual contrast sensitivity in Parkinson's disease: Implications for visual and cognitive function. Parkinsonism Relat Disord 2025, 131, 107229. [Google Scholar] [CrossRef]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov Disord 2008, 23, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Carew, J.; Serrano, G.; Adler, C.H.; Shill, H.A.; Sue, L.I.; Sabbagh, M.N.; Akiyama, H.; Cuenca, N.; Arizona Parkinson's Disease, C. Phosphorylated alpha-synuclein-immunoreactive retinal neuronal elements in Parkinson's disease subjects. Neurosci Lett 2014, 571, 34–38. [Google Scholar] [CrossRef]
- Ortuno-Lizaran, I.; Beach, T.G.; Serrano, G.E.; Walker, D.G.; Adler, C.H.; Cuenca, N. Phosphorylated alpha-synuclein in the retina is a biomarker of Parkinson's disease pathology severity. Mov Disord 2018, 33, 1315–1324. [Google Scholar] [CrossRef]
- Ortuno-Lizaran, I.; Sanchez-Saez, X.; Lax, P.; Serrano, G.E.; Beach, T.G.; Adler, C.H.; Cuenca, N. Dopaminergic Retinal Cell Loss and Visual Dysfunction in Parkinson Disease. Ann Neurol 2020, 88, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Li, J.; Zhou, L.; Liu, X.; Xiao, Z.; He, R.; Chu, H.; Tang, Y.; Liu, P.; Lu, X. Retinopathy in Parkinson's disease: A potential biomarker for early diagnosis and clinical assessment. Neuroscience 2025, 565, 202–210. [Google Scholar] [CrossRef]
- Eslami, F.; Ghiasian, M.; Mohamadrahimi, B.; Jiriaee, N.; Eslamighayour, A. Optical coherence tomography (OCT) findings in patients with Parkinson's disease presenting to Farshchian Hospital (Sina) in 2019 compared to the normal population. J Fr Ophtalmol 2025, 48, 104379. [Google Scholar] [CrossRef]
- Bilgin, S.; Uysal, H.A.; Bilgin, S.; Yaka, E.C.; Kusbeci, O.Y.; Sener, U. Assessment of Changes in Vascular Density in the Layers of the Eye in Patients With Parkinson's Disease. Ann Neurosci, 1177. [Google Scholar] [CrossRef]
- Erdem, M.; Soker, E.B.; Ozdogru, D.; Balal, M.; Ciloglu, E. Evaluation of retinal microvascular changes with OCT-A in Parkinson disease and essential tremor. Medicine (Baltimore) 2024, 103, e40752. [Google Scholar] [CrossRef] [PubMed]
- Hamedani, A.G.; Abraham, D.S.; Maguire, M.G.; Willis, A.W. Visual Impairment Is More Common in Parkinson's Disease and Is a Risk Factor for Poor Health Outcomes. Mov Disord 2020, 35, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Mylius, V.; Perez Lloret, S.; Cury, R.G.; Teixeira, M.J.; Barbosa, V.R.; Barbosa, E.R.; Moreira, L.I.; Listik, C.; Fernandes, A.M.; de Lacerda Veiga, D.; et al. The Parkinson disease pain classification system: results from an international mechanism-based classification approach. Pain 2021, 162, 1201–1210. [Google Scholar] [CrossRef]
- Nogueira, A.C.R.; Pereira, K.C.; Rodrigues, V.F.; Alves, D.P.A.; Marques, J.B.; Monteiro, E.R.; Jesus, I.R.T. Pain characterization in patients with Parkinson's disease. Pain Pract 2024, 24, 786–797. [Google Scholar] [CrossRef]
- Shalash, A.; Mohamed, S.R.; Badr, M.Y.; Elgamal, S.; Elaidy, S.A.; Elhamrawy, E.A.; Abdel-Tawab, H.; Elshebawy, H.; Abdelraheem, H.S.; Roushdy, T.; et al. Pain Characteristics of Parkinson's Disease Using Validated Arabic Versions of the King's Parkinson's Disease Pain Scale and Questionnaire: A Multicenter Egyptian Study. J Mov Disord 2024, 17, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, D.; Gambioli, F.; De Bartolo, M.I.; Mancinelli, R.; Biagioni, F.; Carotti, S.; Falato, E.; Leodori, G.; Puglisi-Allegra, S.; Vivacqua, G.; et al. Pain in Parkinson's disease: a neuroanatomy-based approach. Brain Commun 2024, 6, fcae210. [Google Scholar] [CrossRef]
- Tseng, M.T.; Lin, C.H. Pain in early-stage Parkinson's disease: Implications from clinical features to pathophysiology mechanisms. J Formos Med Assoc 2017, 116, 571–581. [Google Scholar] [CrossRef]
- Tai, Y.C.; Lin, C.H. An overview of pain in Parkinson's disease. Clin Park Relat Disord 2020, 2, 1–8. [Google Scholar] [CrossRef]
- Cattaneo, C.; Jost, W.H. Pain in Parkinson's Disease: Pathophysiology, Classification and Treatment. J Integr Neurosci 2023, 22, 132. [Google Scholar] [CrossRef]
- Gandolfi, M.; Geroin, C.; Antonini, A.; Smania, N.; Tinazzi, M. Understanding and Treating Pain Syndromes in Parkinson's Disease. Int Rev Neurobiol 2017, 134, 827–858. [Google Scholar] [CrossRef]
- Roversi, K.; Callai-Silva, N.; Roversi, K.; Griffith, M.; Boutopoulos, C.; Prediger, R.D.; Talbot, S. Neuro-Immunity and Gut Dysbiosis Drive Parkinson's Disease-Induced Pain. Front Immunol 2021, 12, 759679. [Google Scholar] [CrossRef]
- Li, J.; Wei, Y.; Zhou, J.; Zou, H.; Ma, L.; Liu, C.; Xiao, Z.; Liu, X.; Tan, X.; Yu, T.; et al. Activation of locus coeruleus-spinal cord noradrenergic neurons alleviates neuropathic pain in mice via reducing neuroinflammation from astrocytes and microglia in spinal dorsal horn. J Neuroinflammation 2022, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.; Gallop, K.; Correll, C.U.; Carvalho, A.F.; Veronese, N.; Wright, E.; Stubbs, B. Pain perception in Parkinson's disease: A systematic review and meta-analysis of experimental studies. Ageing Res Rev 2017, 35, 74–86. [Google Scholar] [CrossRef]
- Corra, M.F.; Vila-Cha, N.; Sardoeira, A.; Hansen, C.; Sousa, A.P.; Reis, I.; Sambayeta, F.; Damasio, J.; Calejo, M.; Schicketmueller, A.; et al. Peripheral neuropathy in Parkinson's disease: prevalence and functional impact on gait and balance. Brain 2023, 146, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, C.H.; Garcia, J.; Wang, N.; Shih, L.C.; Freeman, R. The diagnostic discrimination of cutaneous alpha-synuclein deposition in Parkinson disease. Neurology 2016, 87, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Doppler, K.; Jentschke, H.M.; Schulmeyer, L.; Vadasz, D.; Janzen, A.; Luster, M.; Hoffken, H.; Mayer, G.; Brumberg, J.; Booij, J.; et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson's disease. Acta Neuropathol 2017, 133, 535–545. [Google Scholar] [CrossRef]
- Leon-Bejarano, F.; Mendez, M.O.; Alba, A.; Rodriguez-Leyva, I.; Gonzalez, F.J.; Rodriguez-Aranda, M.D.C.; Guevara, E.; Guirado-Lopez, R.A.; Ramirez-Elias, M.G. Raman Spectroscopy Study of Skin Biopsies from Patients with Parkinson's Disease: Trends in Alpha-Synuclein Aggregation from the Amide I Region. Appl Spectrosc 2022, 76, 1317–1328. [Google Scholar] [CrossRef]
- Li, J.; Duan, S.; Yang, J.; Zheng, H.; Yuan, Y.; Tang, M.; Wang, Y.; Liu, Y.; Xia, Z.; Luo, H.; et al. Detection of skin alpha-synuclein using RT-QuIC as a diagnostic biomarker for Parkinson's disease in the Chinese population. Eur J Med Res 2024, 29, 114. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, L.; Sun, L.; Zhi, Y.; Ding, J.; Yuan, Y.S.; Shen, F.F.; Li, X.; Ji, P.; Wang, Z.; et al. Phosphorylated alpha-synuclein deposits in sural nerve deriving from Schwann cells: A biomarker for Parkinson's disease. Parkinsonism Relat Disord 2019, 60, 57–63. [Google Scholar] [CrossRef]
- Nolano, M.; Provitera, V.; Manganelli, F.; Iodice, R.; Stancanelli, A.; Caporaso, G.; Saltalamacchia, A.; Califano, F.; Lanzillo, B.; Picillo, M.; et al. Loss of cutaneous large and small fibers in naive and l-dopa-treated PD patients. Neurology 2017, 89, 776–784. [Google Scholar] [CrossRef]
- Vacchi, E.; Senese, C.; Chiaro, G.; Disanto, G.; Pinton, S.; Morandi, S.; Bertaina, I.; Bianco, G.; Staedler, C.; Galati, S.; et al. Alpha-synuclein oligomers and small nerve fiber pathology in skin are potential biomarkers of Parkinson's disease. NPJ Parkinsons Dis 2021, 7, 119. [Google Scholar] [CrossRef]
- Melli, G.; Vacchi, E.; Biemmi, V.; Galati, S.; Staedler, C.; Ambrosini, R.; Kaelin-Lang, A. Cervical skin denervation associates with alpha-synuclein aggregates in Parkinson disease. Ann Clin Transl Neurol 2018, 5, 1394–1407. [Google Scholar] [CrossRef]
- Arias-Carrion, O.; Ortega-Robles, E.; Ortuno-Sahagun, D.; Ramirez-Bermudez, J.; Hamid, A.; Shalash, A. Sleep-Related Disorders in Parkinson's Disease: Mechanisms, Diagnosis, and Therapeutic Approaches. CNS Neurol Disord Drug Targets 2025, 24, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Liu, H.; Hu, X.; Yu, Q.; Kuang, G.; Liu, L.; Zhang, S.; Wang, X.; Li, J.; Yu, D.; et al. Insomnia in Parkinson's Disease: Causes, Consequences, and Therapeutic Approaches. Mol Neurobiol 2025, 62, 2292–2313. [Google Scholar] [CrossRef]
- Stefani, A.; Hogl, B. Sleep in Parkinson's disease. Neuropsychopharmacology 2020, 45, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, F.; Liu, X.; Zu, J.; Zhang, W.; Zhou, S.; Zhu, J.; Zhang, T.; Cui, G.; Xu, C. Association between serum neurofilament light chain levels and sleep disorders in patients with Parkinson's disease. Neurosci Lett 2023, 812, 137394. [Google Scholar] [CrossRef] [PubMed]
- Burchill, E.; Watson, C.J.; Fanshawe, J.B.; Badenoch, J.B.; Rengasamy, E.; Ghanem, D.A.; Holle, C.; Conti, I.; Sadeq, M.A.; Saini, A.; et al. The impact of psychiatric comorbidity on Parkinson's disease outcomes: a systematic review and meta-analysis. Lancet Reg Health Eur 2024, 39, 100870. [Google Scholar] [CrossRef]
- Amstutz, D.; Sousa, M.; Maradan-Gachet, M.E.; Debove, I.; Lhommee, E.; Krack, P. Psychiatric and cognitive symptoms of Parkinson's disease: A life's tale. Rev Neurol (Paris), 1016. [Google Scholar] [CrossRef]
- Macias-Garcia, P.; Rashid-Lopez, R.; Cruz-Gomez, A.J.; Lozano-Soto, E.; Sanmartino, F.; Espinosa-Rosso, R.; Gonzalez-Rosa, J.J. Neuropsychiatric Symptoms in Clinically Defined Parkinson's Disease: An Updated Review of Literature. Behav Neurol 2022, 2022, 1213393. [Google Scholar] [CrossRef]
- Cong, S.; Xiang, C.; Zhang, S.; Zhang, T.; Wang, H.; Cong, S. Prevalence and clinical aspects of depression in Parkinson's disease: A systematic review and meta-analysis of 129 studies. Neurosci Biobehav Rev 2022, 141, 104749. [Google Scholar] [CrossRef]
- Qin, Y.; Li, J.; Quan, W.; Song, J.; Xu, J.; Chen, J. Risk of Parkinson's disease and depression severity in different populations: A two-sample Mendelian randomization analysis. Brain Behav 2024, 14, e3642. [Google Scholar] [CrossRef]
- Badenoch, J.B.; Paris, A.; Jacobs, B.M.; Noyce, A.J.; Marshall, C.R.; Waters, S. Neuroanatomical and prognostic associations of depression in Parkinson's disease. J Neurol Neurosurg Psychiatry 2024, 95, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, J.; Ceravolo, R.; Brooks, D.J.; Bonuccelli, U.; Pavese, N. Progressive loss of raphe nuclei serotonin transporter in early Parkinson's disease: A longitudinal (123)I-FP-CIT SPECT study. Parkinsonism Relat Disord 2020, 77, 170–175. [Google Scholar] [CrossRef]
- Brazdis, R.M.; von Zimmermann, C.; Lenz, B.; Kornhuber, J.; Muhle, C. Peripheral Upregulation of Parkinson's Disease-Associated Genes Encoding alpha-Synuclein, beta-Glucocerebrosidase, and Ceramide Glucosyltransferase in Major Depression. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Sandoval-Rincon, M.; Saenz-Farret, M.; Miguel-Puga, A.; Micheli, F.; Arias-Carrion, O. Rational pharmacological approaches for cognitive dysfunction and depression in Parkinson's disease. Front Neurol 2015, 6, 71. [Google Scholar] [CrossRef]
- den Brok, M.G.; van Dalen, J.W.; van Gool, W.A.; Moll van Charante, E.P.; de Bie, R.M.; Richard, E. Apathy in Parkinson's disease: A systematic review and meta-analysis. Mov Disord 2015, 30, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Song, E.; Zhu, M.; Appel-Cresswell, S.; McKeown, M.J. Apathy scores in Parkinson's disease relate to EEG components in an incentivized motor task. Brain Commun 2024, 6, fcae025. [Google Scholar] [CrossRef]
- Wee, N.; Kandiah, N.; Acharyya, S.; Chander, R.J.; Ng, A.; Au, W.L.; Tan, L.C. Baseline predictors of worsening apathy in Parkinson's disease: A prospective longitudinal study. Parkinsonism Relat Disord 2016, 23, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; MacAskill, M.; Pascoe, M.; Anderson, T.; Heron, C.L. Dimensions of apathy in Parkinson's disease. Brain Behav 2023, 13, e2862. [Google Scholar] [CrossRef]
- Robert, P.; Lanctot, K.L.; Aguera-Ortiz, L.; Aalten, P.; Bremond, F.; Defrancesco, M.; Hanon, C.; David, R.; Dubois, B.; Dujardin, K.; et al. Is it time to revise the diagnostic criteria for apathy in brain disorders? The 2018 international consensus group. Eur Psychiatry 2018, 54, 71–76. [Google Scholar] [CrossRef]
- Ou, R.; Lin, J.; Liu, K.; Jiang, Z.; Wei, Q.; Hou, Y.; Zhang, L.; Cao, B.; Zhao, B.; Song, W.; et al. Evolution of Apathy in Early Parkinson's Disease: A 4-Years Prospective Cohort Study. Front Aging Neurosci 2020, 12, 620762. [Google Scholar] [CrossRef]
- Le Heron, C.; Horne, K.L.; MacAskill, M.R.; Livingstone, L.; Melzer, T.R.; Myall, D.; Pitcher, T.; Dalrymple-Alford, J.; Anderson, T.; Harrison, S. Cross-Sectional and Longitudinal Association of Clinical and Neurocognitive Factors With Apathy in Patients With Parkinson Disease. Neurology 2024, 102, e209301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, H.; Cao, X.; Wang, L.; Gan, C.; Sun, H.; Shan, A.; Yuan, Y.; Zhang, K. Association between the functional connectivity of ventral tegmental area-prefrontal network and pure apathy in Parkinson's disease: a cross-sectional study. Quant Imaging Med Surg 2024, 14, 4735–4748. [Google Scholar] [CrossRef] [PubMed]
- Lucas-Jimenez, O.; Ojeda, N.; Pena, J.; Cabrera-Zubizarreta, A.; Diez-Cirarda, M.; Gomez-Esteban, J.C.; Gomez-Beldarrain, M.A.; Ibarretxe-Bilbao, N. Apathy and brain alterations in Parkinson's disease: a multimodal imaging study. Ann Clin Transl Neurol 2018, 5, 803–814. [Google Scholar] [CrossRef]
- Martinez-Horta, S.; Sampedro, F.; Pagonabarraga, J.; Fernandez-Bobadilla, R.; Marin-Lahoz, J.; Riba, J.; Kulisevsky, J. Non-demented Parkinson's disease patients with apathy show decreased grey matter volume in key executive and reward-related nodes. Brain Imaging Behav 2017, 11, 1334–1342. [Google Scholar] [CrossRef]
- Wang, F.; Yu, S.Y.; Zuo, L.J.; Cao, C.J.; Hu, Y.; Chen, Z.J.; Piao, Y.S.; Wang, Y.J.; Wang, X.M.; Chen, S.D.; et al. Excessive Iron and alpha-Synuclein Oligomer in Brain are Relevant to Pure Apathy in Parkinson Disease. J Geriatr Psychiatry Neurol 2016, 29, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.P.; McDonald, K.R.; Allsop, D.; Diggle, P.J.; Leroi, I. Apathy as a behavioural marker of cognitive impairment in Parkinson's disease: a longitudinal analysis. J Neurol 2020, 267, 214–227. [Google Scholar] [CrossRef]
- Mele, B.; Van, S.; Holroyd-Leduc, J.; Ismail, Z.; Pringsheim, T.; Goodarzi, Z. Diagnosis, treatment and management of apathy in Parkinson's disease: a scoping review. BMJ Open 2020, 10, e037632. [Google Scholar] [CrossRef]
- Broen, M.P.; Narayen, N.E.; Kuijf, M.L.; Dissanayaka, N.N.; Leentjens, A.F. Prevalence of anxiety in Parkinson's disease: A systematic review and meta-analysis. Mov Disord 2016, 31, 1125–1133. [Google Scholar] [CrossRef]
- Jia, M.; Yang, S.; Li, S.; Chen, S.; Wu, L.; Li, J.; Wang, H.; Wang, C.; Liu, Q.; Wu, K. Early identification of Parkinson's disease with anxiety based on combined clinical and MRI features. Front Aging Neurosci 2024, 16, 1414855. [Google Scholar] [CrossRef]
- Carey, G.; Lopes, R.; Viard, R.; Betrouni, N.; Kuchcinski, G.; Devignes, Q.; Defebvre, L.; Leentjens, A.F.G.; Dujardin, K. Anxiety in Parkinson's disease is associated with changes in the brain fear circuit. Parkinsonism Relat Disord 2020, 80, 89–97. [Google Scholar] [CrossRef]
- Torres, E.R.S.; Stanojlovic, M.; Zelikowsky, M.; Bonsberger, J.; Hean, S.; Mulligan, C.; Baldauf, L.; Fleming, S.; Masliah, E.; Chesselet, M.F.; et al. Alpha-synuclein pathology, microgliosis, and parvalbumin neuron loss in the amygdala associated with enhanced fear in the Thy1-aSyn model of Parkinson's disease. Neurobiol Dis 2021, 158, 105478. [Google Scholar] [CrossRef]
- Yu, Z.; Liu, G.; Li, Y.; Arkin, E.; Zheng, Y.; Feng, T. Erythrocytic alpha-Synuclein Species for Parkinson's Disease Diagnosis and the Correlations With Clinical Characteristics. Front Aging Neurosci 2022, 14, 827493. [Google Scholar] [CrossRef]
- Lian, T.; Zhang, W.; Li, D.; Guo, P.; He, M.; Zhang, Y.; Li, J.; Guan, H.; Zhang, W.; Luo, D.; et al. Parkinson's disease with anxiety: clinical characteristics and their correlation with oxidative stress, inflammation, and pathological proteins. BMC Geriatr 2024, 24, 433. [Google Scholar] [CrossRef]
- Shi, Y.; Dobkin, R.; Weintraub, D.; Cho, H.R.; Caspell-Garcia, C.; Bock, M.; Brown, E.; Aarsland, D.; Dahodwala, N. Association of Baseline Depression and Anxiety with Longitudinal Health Outcomes in Parkinson's Disease. Mov Disord Clin Pract 2024, 11, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Shek-Kwan Chang, R.; Cheung, C.; Pang, S.; Lau, K.K.; Suckling, J.; Rowe, J.B.; Yu, K.; Ka-Fung Mak, H.; Chua, S.E.; et al. The default mode network is disrupted in Parkinson's disease with visual hallucinations. Hum Brain Mapp 2014, 35, 5658–5666. [Google Scholar] [CrossRef]
- Clegg, B.J.; Duncan, G.W.; Khoo, T.K.; Barker, R.A.; Burn, D.J.; Yarnall, A.J.; Lawson, R.A. Categorising Visual Hallucinations in Early Parkinson's Disease. J Parkinsons Dis 2018, 8, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Fenelon, G.; Mahieux, F.; Huon, R.; Ziegler, M. Hallucinations in Parkinson's disease: prevalence, phenomenology and risk factors. Brain 2000, 123 ( Pt 4) Pt 4, 733–745. [Google Scholar] [CrossRef]
- Murphy, N.; Killen, A.; Gupta, R.K.; Graziadio, S.; Rochester, L.; Firbank, M.; Baker, M.R.; Allan, C.; Collerton, D.; Taylor, J.P.; et al. Exploring Bottom-Up Visual Processing and Visual Hallucinations in Parkinson's Disease With Dementia. Front Neurol 2020, 11, 579113. [Google Scholar] [CrossRef]
- Ignatavicius, A.; Matar, E.; Lewis, S.J.G. Visual hallucinations in Parkinson's disease: spotlight on central cholinergic dysfunction. Brain 2025, 148, 376–393. [Google Scholar] [CrossRef]
- Zhu, J.; Shen, B.; Lu, L.; Lan, W.; Pan, Y.; Zhang, L.; Dong, J.; Wang, M.; Zhang, L. Prevalence and risk factors for visual hallucinations in Chinese patients with Parkinson's disease. J Neurol Sci 2017, 372, 471–476. [Google Scholar] [CrossRef]
- d'Angremont, E.; van der Zee, S.; Slingerland, S.; Slomp, A.C.; de Vries, E.F.J.; van Laar, T.; Sommer, I.E. Cholinergic deficiency in Parkinson's disease patients with visual hallucinations. Brain 2024, 147, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Firbank, M.J.; Parikh, J.; Murphy, N.; Killen, A.; Allan, C.L.; Collerton, D.; Blamire, A.M.; Taylor, J.P. Reduced occipital GABA in Parkinson disease with visual hallucinations. Neurology 2018, 91, e675–e685. [Google Scholar] [CrossRef]
- Thomas, G.E.C.; Zeidman, P.; Sultana, T.; Zarkali, A.; Razi, A.; Weil, R.S. Changes in both top-down and bottom-up effective connectivity drive visual hallucinations in Parkinson's disease. Brain Commun 2023, 5, fcac329. [Google Scholar] [CrossRef]
- Zarkali, A.; McColgan, P.; Leyland, L.A.; Lees, A.J.; Weil, R.S. Longitudinal thalamic white and grey matter changes associated with visual hallucinations in Parkinson's disease. J Neurol Neurosurg Psychiatry 2022, 93, 169–179. [Google Scholar] [CrossRef]
- Kalaitzakis, M.E.; Christian, L.M.; Moran, L.B.; Graeber, M.B.; Pearce, R.K.; Gentleman, S.M. Dementia and visual hallucinations associated with limbic pathology in Parkinson's disease. Parkinsonism Relat Disord 2009, 15, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Ikeda, T.; Ishida, C.; Komai, K.; Yamada, M. Delusions and visual hallucinations in a patient with Parkinson's disease with dementia showing pronounced Lewy body pathology in the nucleus basalis of Meynert. Neuropathology 2019, 39, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Warren, J.D.; Lees, A.J. Using the presence of visual hallucinations to differentiate Parkinson's disease from atypical parkinsonism. J Neurol Neurosurg Psychiatry 2008, 79, 652–655. [Google Scholar] [CrossRef]
- Gillette, B.; Reid, J.A.; Shermetaro, C. Phantosmia. In StatPearls, Treasure Island (FL), 2025.
- Solla, P.; Masala, C.; Pinna, I.; Ercoli, T.; Loy, F.; Orofino, G.; Fadda, L.; Defazio, G. Frequency and Determinants of Olfactory Hallucinations in Parkinson's Disease Patients. Brain Sci 2021, 11. [Google Scholar] [CrossRef]
- Bannier, S.; Berdague, J.L.; Rieu, I.; de Chazeron, I.; Marques, A.; Derost, P.; Ulla, M.; Llorca, P.M.; Durif, F. Prevalence and phenomenology of olfactory hallucinations in Parkinson's disease. J Neurol Neurosurg Psychiatry 2012, 83, 1019–1021. [Google Scholar] [CrossRef]
- Landis, B.N.; Burkhard, P.R. Phantosmias and Parkinson disease. Arch Neurol 2008, 65, 1237–1239. [Google Scholar] [CrossRef]
- Ercoli, T.; Bagella, C.F.; Frau, C.; Ruiu, E.; Othmani, S.; Gusinu, G.; Masala, C.; Sechi, L.A.; Solla, P.; Defazio, G. Phantosmia in Parkinson's Disease: A Systematic Review of the Phenomenology of Olfactory Hallucinations. Neurol Int 2023, 16, 20–32. [Google Scholar] [CrossRef]
- Wallace, E.R.; Segerstrom, S.C.; van Horne, C.G.; Schmitt, F.A.; Koehl, L.M. Meta-Analysis of Cognition in Parkinson's Disease Mild Cognitive Impairment and Dementia Progression. Neuropsychol Rev 2022, 32, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Lanni, K.E.; Ross, J.M.; Higginson, C.I.; Dressler, E.M.; Sigvardt, K.A.; Zhang, L.; Malhado-Chang, N.; Disbrow, E.A. Perceived and performance-based executive dysfunction in Parkinson's disease. J Clin Exp Neuropsychol 2014, 36, 342–355. [Google Scholar] [CrossRef]
- Dirnberger, G.; Frith, C.D.; Jahanshahi, M. Executive dysfunction in Parkinson's disease is associated with altered pallidal-frontal processing. Neuroimage 2005, 25, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Stern, Y.; Mayeux, R.; Cote, L. Reaction time and vigilance in Parkinson's disease. Possible role of altered norepinephrine metabolism. Arch Neurol 1984, 41, 1086–1089. [Google Scholar] [CrossRef]
- Kehagia, A.A.; Barker, R.A.; Robbins, T.W. Cognitive impairment in Parkinson's disease: the dual syndrome hypothesis. Neurodegener Dis 2013, 11, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.D.; Ren, X.; Ellis, T.D.; Toraif, N.; Barthelemy, O.J.; Neargarder, S.; Cronin-Golomb, A. Dual tasking in Parkinson's disease: Cognitive consequences while walking. Neuropsychology 2017, 31, 613–623. [Google Scholar] [CrossRef]
- Proud, E.; Morris, M.E.; Bilney, B.; Miller, K.J.; Nijkrake, M.J.; Munneke, M.M.; McGinley, J.L. Effects of dual-task interference on dexterity performance in people with mild to moderately severe Parkinson's disease: An observational analysis. J Hand Ther 2025, 38, 97–102. [Google Scholar] [CrossRef]
- Kay, K.R.; Uc, E.Y. Real-life consequences of cognitive dysfunction in Parkinson's disease. Prog Brain Res 2022, 269, 113–136. [Google Scholar] [CrossRef]
- Focker, J.; Cole, D.; Beer, A.L.; Bavelier, D. Neural bases of enhanced attentional control: Lessons from action video game players. Brain Behav 2018, 8, e01019. [Google Scholar] [CrossRef]
- Arabaci, G.; Parris, B.A. Inattention and task switching performance: the role of predictability, working memory load and goal neglect. Psychol Res 2020, 84, 2090–2110. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ou, R.; Yuan, X.; Liu, H.; Hou, Y.; Wei, Q.; Song, W.; Cao, B.; Chen, Y.; Shang, H. Executive dysfunctions and behavioral changes in early drug-naive patients with Parkinson's disease. J Affect Disord 2019, 243, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Pont-Sunyer, C.; Hotter, A.; Gaig, C.; Seppi, K.; Compta, Y.; Katzenschlager, R.; Mas, N.; Hofeneder, D.; Brucke, T.; Bayes, A.; et al. The onset of nonmotor symptoms in Parkinson's disease (the ONSET PD study). Mov Disord 2015, 30, 229–237. [Google Scholar] [CrossRef]
- Melugin, P.R.; Nolan, S.O.; Kandov, E.; Ferrara, C.F.; Farahbakhsh, Z.Z.; Siciliano, C.A. Medial prefrontal dopamine dynamics reflect allocation of selective attention. bioRxiv, 1101. [Google Scholar] [CrossRef]
- Arias-Carrion, O.; Poppel, E. Dopamine, learning, and reward-seeking behavior. Acta Neurobiol Exp (Wars) 2007, 67, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Woodward, T.S.; Bub, D.N.; Hunter, M.A. Task switching deficits associated with Parkinson's disease reflect depleted attentional resources. Neuropsychologia 2002, 40, 1948–1955. [Google Scholar] [CrossRef]
- Gul, A.; Yousaf, J.; Ahmad, H. Frontal-subcortical defects correlate with task switching deficits in Parkinson;s disease. Neurosciences (Riyadh) 2017, 22, 224–227. [Google Scholar] [CrossRef]
- Bin Yoo, H.; Concha, E.O.; De Ridder, D.; Pickut, B.A.; Vanneste, S. The Functional Alterations in Top-Down Attention Streams of Parkinson's disease Measured by EEG. Sci Rep 2018, 8, 10609. [Google Scholar] [CrossRef]
- Katsuki, F.; Constantinidis, C. Bottom-up and top-down attention: different processes and overlapping neural systems. Neuroscientist 2014, 20, 509–521. [Google Scholar] [CrossRef]
- Tommasi, G.; Fiorio, M.; Yelnik, J.; Krack, P.; Sala, F.; Schmitt, E.; Fraix, V.; Bertolasi, L.; Le Bas, J.F.; Ricciardi, G.K.; et al. Disentangling the Role of Cortico-Basal Ganglia Loops in Top-Down and Bottom-Up Visual Attention: An Investigation of Attention Deficits in Parkinson Disease. J Cogn Neurosci 2015, 27, 1215–1237. [Google Scholar] [CrossRef]
- Bocquillon, P.; Bourriez, J.L.; Palmero-Soler, E.; Destee, A.; Defebvre, L.; Derambure, P.; Dujardin, K. Role of basal ganglia circuits in resisting interference by distracters: a swLORETA study. PLoS One 2012, 7, e34239. [Google Scholar] [CrossRef]
- Wulaer, B.; Kunisawa, K.; Tanabe, M.; Yanagawa, A.; Saito, K.; Mouri, A.; Nabeshima, T. Pharmacological blockade of dopamine D1- or D2-receptor in the prefrontal cortex induces attentional impairment in the object-based attention test through different neuronal circuits in mice. Mol Brain 2021, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Moscovitch, M.; Alain, C. A systematic review and meta-analysis of memory-guided attention: Frontal and parietal activation suggests involvement of fronto-parietal networks. Wiley Interdiscip Rev Cogn Sci 2021, 12, e1546. [Google Scholar] [CrossRef] [PubMed]
- Kehagia, A.A.; Barker, R.A.; Robbins, T.W. Revisiting the effects of Parkinson's disease and frontal lobe lesions on task switching: the role of rule reconfiguration. J Neuropsychol 2014, 8, 53–74. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Stern, Y.; Sano, M.; Cote, L.; Williams, J.B. Clinical and biochemical correlates of bradyphrenia in Parkinson's disease. Neurology 1987, 37, 1130–1134. [Google Scholar] [CrossRef]
- Peavy, G.M. Mild cognitive deficits in Parkinson disease: where there is bradykinesia, there is bradyphrenia. Neurology 2010, 75, 1038–1039. [Google Scholar] [CrossRef]
- Steinke, A.; Lange, F.; Seer, C.; Hendel, M.K.; Kopp, B. Computational Modeling for Neuropsychological Assessment of Bradyphrenia in Parkinson's Disease. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Letanneux, A.; Velay, J.L.; Viallet, F.; Pinto, S. Altered Inhibitory Mechanisms in Parkinson's Disease: Evidence From Lexical Decision and Simple Reaction Time Tasks. Front Hum Neurosci 2021, 15, 624026. [Google Scholar] [CrossRef]
- Bonelli, R.M.; Cummings, J.L. Frontal-subcortical dementias. Neurologist 2008, 14, 100–107. [Google Scholar] [CrossRef]
- Wang, W.; Baker, K.; Umamahesan, C.; Gilmour, S.; Charlett, A.; Taylor, D.; Young, A.H.; Dobbs, R.J.; Dobbs, S.M. Bradyphrenia and Tachyphrenia in Idiopathic Parkinsonism Appear, in Part, Iatrogenic: An Observational Study with Systematic Review Background. J Clin Med 2023, 12. [Google Scholar] [CrossRef]
- Gibson, L.L.; Weintraub, D.; Lemmen, R.; Perera, G.; Chaudhuri, K.R.; Svenningsson, P.; Aarsland, D. Risk of Dementia in Parkinson's Disease: A Systematic Review and Meta-Analysis. Mov Disord 2024, 39, 1697–1709. [Google Scholar] [CrossRef]
- Emre, M.; Aarsland, D.; Brown, R.; Burn, D.J.; Duyckaerts, C.; Mizuno, Y.; Broe, G.A.; Cummings, J.; Dickson, D.W.; Gauthier, S.; et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord 2007, 22, 1689–1707. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathobiology of Cognitive Impairment in Parkinson Disease: Challenges and Outlooks. Int J Mol Sci 2023, 25. [Google Scholar] [CrossRef]
- Astrom, D.O.; Simonsen, J.; Raket, L.L.; Sgarbi, S.; Hellsten, J.; Hagell, P.; Norlin, J.M.; Kellerborg, K.; Martinez-Martin, P.; Odin, P. High risk of developing dementia in Parkinson's disease: a Swedish registry-based study. Sci Rep 2022, 12, 16759. [Google Scholar] [CrossRef]
- Russell, A.; Drozdova, A.; Wang, W.; Thomas, M. The impact of dementia development concurrent with Parkinson's disease: a new perspective. CNS Neurol Disord Drug Targets 2014, 13, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Caballol, N.; Marti, M.J.; Tolosa, E. Cognitive dysfunction and dementia in Parkinson disease. Mov Disord 2007, 22 Suppl 17, S358–366. [Google Scholar] [CrossRef]
- Emre, M. Dementia associated with Parkinson's disease. Lancet Neurol 2003, 2, 229–237. [Google Scholar] [CrossRef]
- Gruszka, A.; Hampshire, A.; Barker, R.A.; Owen, A.M. Normal aging and Parkinson's disease are associated with the functional decline of distinct frontal-striatal circuits. Cortex 2017, 93, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Novikov, N.I.; Brazhnik, E.S.; Kitchigina, V.F. Pathological Correlates of Cognitive Decline in Parkinson's Disease: From Molecules to Neural Networks. Biochemistry (Mosc) 2023, 88, 1890–1904. [Google Scholar] [CrossRef]
- Schreiber, L.; Odlaug, B.L.; Grant, J.E. Impulse control disorders: updated review of clinical characteristics and pharmacological management. Front Psychiatry 2011, 2, 1. [Google Scholar] [CrossRef]
- Theis, H.; Prange, S.; Bischof, G.N.; Hoenig, M.C.; Tittgemeyer, M.; Timmermann, L.; Fink, G.R.; Drzezga, A.; Eggers, C.; van Eimeren, T. Impulsive-compulsive behaviour in early Parkinson's disease is determined by apathy and dopamine receptor D3 polymorphism. NPJ Parkinsons Dis 2023, 9, 154. [Google Scholar] [CrossRef]
- Jellinger, K.A. Behavioral disorders in Parkinson disease: current view. J Neural Transm (Vienna) 2025, 132, 169–201. [Google Scholar] [CrossRef] [PubMed]
- Turcano, P.; Jacobson, J.; Ghoniem, K.; Mullan, A.; Camerucci, E.; Stang, C.; Piat, C.; Bower, J.H.; Savica, R. Impulse control disorders and use of dopamine agonists in early onset Parkinson's disease. Front Neurol 2024, 15, 1404904. [Google Scholar] [CrossRef] [PubMed]
- Poletti, M.; Logi, C.; Lucetti, C.; Del Dotto, P.; Baldacci, F.; Vergallo, A.; Ulivi, M.; Del Sarto, S.; Rossi, G.; Ceravolo, R.; et al. A single-center, cross-sectional prevalence study of impulse control disorders in Parkinson disease: association with dopaminergic drugs. J Clin Psychopharmacol 2013, 33, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Joutsa, J.; Martikainen, K.; Vahlberg, T.; Voon, V.; Kaasinen, V. Impulse control disorders and depression in Finnish patients with Parkinson's disease. Parkinsonism Relat Disord 2012, 18, 155–160. [Google Scholar] [CrossRef]
- Rodriguez-Violante, M.; Gonzalez-Latapi, P.; Cervantes-Arriaga, A.; Camacho-Ordonez, A.; Weintraub, D. Impulse control and related disorders in Mexican Parkinson's disease patients. Parkinsonism Relat Disord 2014, 20, 907–910. [Google Scholar] [CrossRef]
- Weintraub, D.; Claassen, D.O. Impulse Control and Related Disorders in Parkinson's Disease. Int Rev Neurobiol 2017, 133, 679–717. [Google Scholar] [CrossRef]
- Wolters, E.; van der Werf, Y.D.; van den Heuvel, O.A. Parkinson's disease-related disorders in the impulsive-compulsive spectrum. J Neurol 2008, 255 Suppl 5, 48–56. [Google Scholar] [CrossRef]
- Voon, V.; Mehta, A.R.; Hallett, M. Impulse control disorders in Parkinson's disease: recent advances. Curr Opin Neurol 2011, 24, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Napier, T.C.; Corvol, J.C.; Grace, A.A.; Roitman, J.D.; Rowe, J.; Voon, V.; Strafella, A.P. Linking neuroscience with modern concepts of impulse control disorders in Parkinson's disease. Mov Disord 2015, 30, 141–149. [Google Scholar] [CrossRef]
- Wolfschlag, M.; Cedergren Weber, G.; Weintraub, D.; Odin, P.; Hakansson, A. Impulse control disorders in Parkinson's disease: a national Swedish registry study on high-risk treatments and vulnerable patient groups. J Neurol Neurosurg Psychiatry, 1136. [Google Scholar] [CrossRef]
- Weintraub, D.; Mamikonyan, E. Impulse Control Disorders in Parkinson's Disease. Am J Psychiatry 2019, 176, 5–11. [Google Scholar] [CrossRef]
Figure 1.
Temporal progression of non-motor symptoms in Parkinson’s disease in relation to Braak staging. This figure depicts the sequential onset and progression of non-motor symptoms in Parkinson’s disease, aligned with Braak’s neuropathological staging. The gradient colour intensities illustrate the increasing severity of each symptom over time. Non-motor symptoms are categorized into autonomic dysfunction, sensory disturbances, sleep-related disorders, neuropsychiatric manifestations, and cognitive impairments. These symptoms often precede motor dysfunction, reflecting the multisystem involvement of Parkinson’s disease and highlighting the need for earlier recognition and intervention. Understanding the temporal trajectory of non-motor symptoms can aid in refining diagnostic criteria and optimizing therapeutic strategies. Abbreviations: EDS, excessive daytime sleepiness; NPDs, neuropsychiatric disorders; OSA, obstructive sleep apnoea; RBD, REM sleep behaviour disorder; RLS, restless legs syndrome.
Figure 1.
Temporal progression of non-motor symptoms in Parkinson’s disease in relation to Braak staging. This figure depicts the sequential onset and progression of non-motor symptoms in Parkinson’s disease, aligned with Braak’s neuropathological staging. The gradient colour intensities illustrate the increasing severity of each symptom over time. Non-motor symptoms are categorized into autonomic dysfunction, sensory disturbances, sleep-related disorders, neuropsychiatric manifestations, and cognitive impairments. These symptoms often precede motor dysfunction, reflecting the multisystem involvement of Parkinson’s disease and highlighting the need for earlier recognition and intervention. Understanding the temporal trajectory of non-motor symptoms can aid in refining diagnostic criteria and optimizing therapeutic strategies. Abbreviations: EDS, excessive daytime sleepiness; NPDs, neuropsychiatric disorders; OSA, obstructive sleep apnoea; RBD, REM sleep behaviour disorder; RLS, restless legs syndrome.
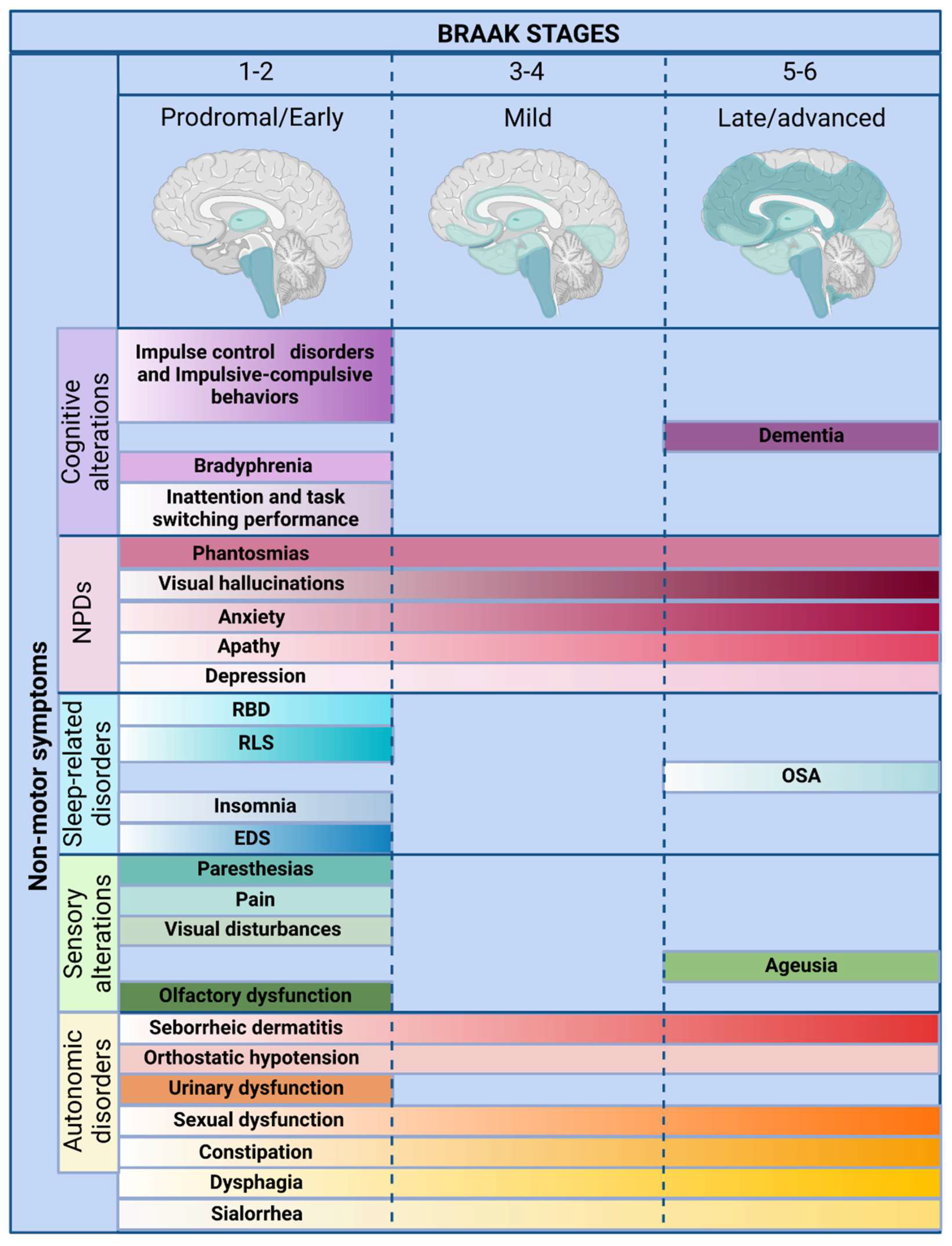
Figure 2.
Anatomical substrates and pathophysiological mechanisms of non-motor symptoms in Parkinson’s disease. This figure illustrates the anatomical structures implicated in Parkinson’s disease and their corresponding pathophysiological alterations underlying non-motor symptoms. Neurodegeneration, α-synuclein aggregation, and structural atrophy contribute to a broad spectrum of non-motor manifestations, including autonomic dysfunction, sensory disturbances, sleep-related disorders, neuropsychiatric symptoms, and cognitive impairment. The widespread involvement of cortical, subcortical, and brainstem regions underscores the multisystem nature of Parkinson’s disease, reinforcing the need for a more comprehensive diagnostic and therapeutic approach that extends beyond dopaminergic dysfunction. Abbreviations: α-syn, α-synuclein; EDS, excessive daytime sleepiness; FOp, frontal insular operculum; NBM, nucleus basalis of Meynert; OB, olfactory bulb; OFC, orbitofrontal cortex; OSA, obstructive sleep apnoea; PFC, prefrontal cortex; RBD, REM sleep behaviour disorder; RLS, restless legs syndrome; SC, spinal cord; SN, substantia nigra; VTA, ventral tegmental area.
Figure 2.
Anatomical substrates and pathophysiological mechanisms of non-motor symptoms in Parkinson’s disease. This figure illustrates the anatomical structures implicated in Parkinson’s disease and their corresponding pathophysiological alterations underlying non-motor symptoms. Neurodegeneration, α-synuclein aggregation, and structural atrophy contribute to a broad spectrum of non-motor manifestations, including autonomic dysfunction, sensory disturbances, sleep-related disorders, neuropsychiatric symptoms, and cognitive impairment. The widespread involvement of cortical, subcortical, and brainstem regions underscores the multisystem nature of Parkinson’s disease, reinforcing the need for a more comprehensive diagnostic and therapeutic approach that extends beyond dopaminergic dysfunction. Abbreviations: α-syn, α-synuclein; EDS, excessive daytime sleepiness; FOp, frontal insular operculum; NBM, nucleus basalis of Meynert; OB, olfactory bulb; OFC, orbitofrontal cortex; OSA, obstructive sleep apnoea; PFC, prefrontal cortex; RBD, REM sleep behaviour disorder; RLS, restless legs syndrome; SC, spinal cord; SN, substantia nigra; VTA, ventral tegmental area.
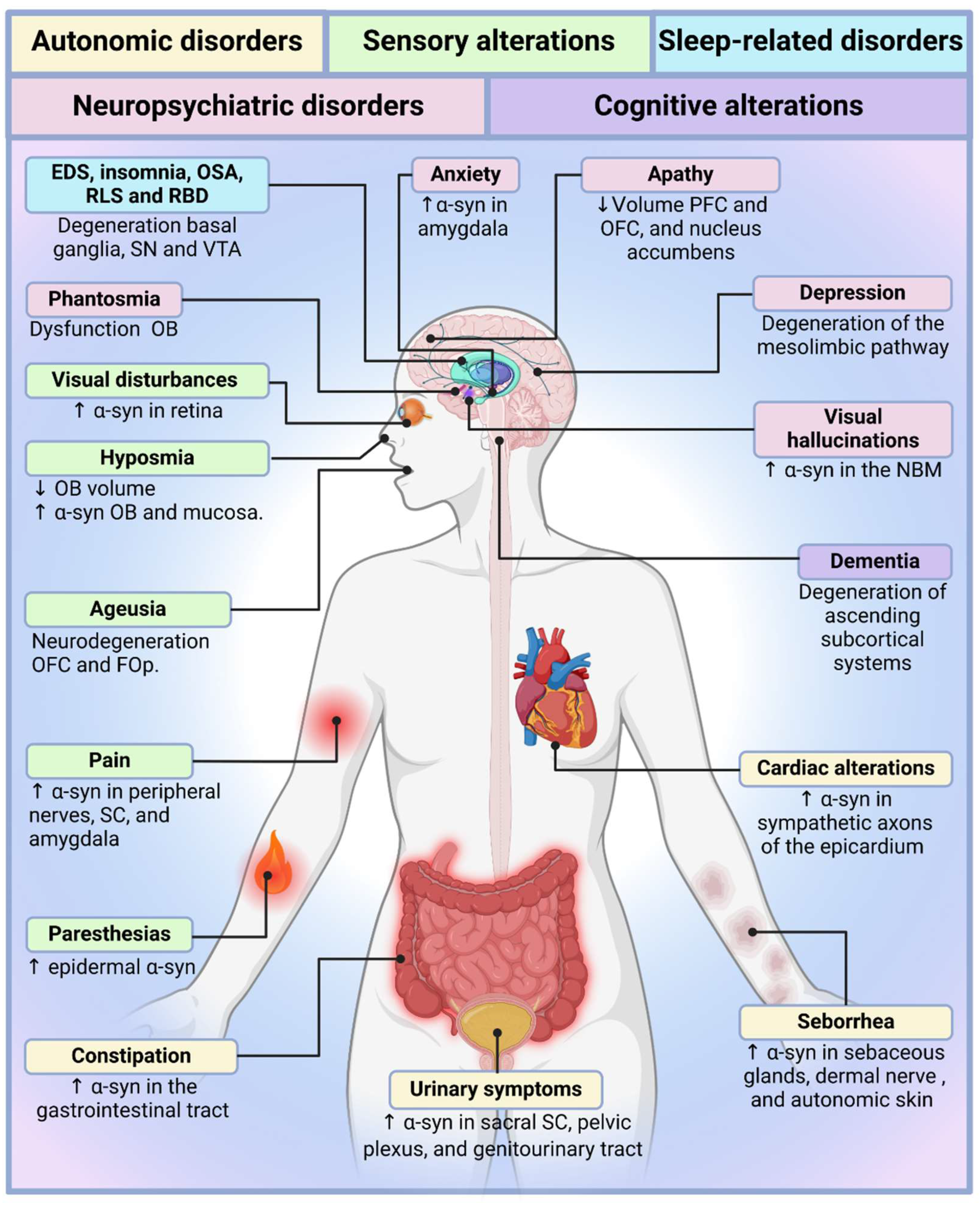

Table 1.
Autonomic Disorders.
| Symptom | Prevalence & Stage | Biomarkers | Pharmacological Treatment |
Non-pharmacological Treatment |
|---|---|---|---|---|
| Sialorrhea | ↑ 37-84%, early | ↑ α-synuclein (basal ganglia) | Botulinum toxin, glycopyrrolate | Speech therapy, postural adjustments |
| Dysphagia | ↑ 40-87%, early | ↑ α-synuclein (enteric nervous system) | Botulinum toxin, Levodopa | Swallowing therapy, neuromodulation |
| Constipation | ↑ 40-63%, Prodromal | ↑ α-synuclein (sacral nuclei) | Lubiprostone, Prokinetics | Dietary fibre |
| Sexual dysfunction | ↑ 65-90%, Early | No α-synuclein correlation | Sildenafil, Hormone therapy | Psychotherapy, Couples therapy |
| Urinary dysfunction | ↑ 25-61%, 5-6 years post-motor onset | ↑ α-synuclein (pelvic plexus) | Antimuscarinics, Beta-3 agonists | Bladder training, Pelvic floor exercises |
| Orthostatic hypotension | ↑ 30-50%, Early/Late | ↑ α-synuclein (autonomic nervous system) | Droxidopa, midodrine, fludrocortisone | ↑ Salt/fluids, compression stockings |
| Seborrheic dermatitis | ↑ 52-59%, early & progressive | ↑ α-synuclein (sebaceous glands, dermal nerves) | Ketoconazole, cannabidiol | Skincare, microbiome modulation |
↑ = Increased; ↓ = Decreased.
Table 2.
Sensory Disorders.
| Symptom | Prevalence & Stage | Biomarkers | Pharmacological Treatment |
Non-pharmacological Treatment |
|---|---|---|---|---|
| Olfactory dysfunction | ↑ 90%, prodromal | ↑ α-synuclein (olfactory bulb, mucosa), ↓ functional activity | Intranasal insulin, DBS | Olfactory training |
| Ageusia | ↑ 4-54%, mild-advanced | ↓ Taste receptor gene expression, neurodegeneration | No specific treatment | Dietary adjustments |
| Visual disturbances | ↑ 90%, prodromal | ↑ α-synuclein (retina), ↓ dopamine, retinal atrophy | Dopaminergic therapy, artificial tears | Prism glasses, vision therapy |
| Pain | ↑ 20-98%, prodromal | ↑ α-synuclein (spinal cord, nerves), ↓ dopamine, ↑ neuroinflammation | NSAIDs, anticonvulsants, opioids | Physical therapy, CBT, DBS |
| Paresthesias | ↑ 40%, prodromal | ↑ α-synuclein (epidermal nerves), ↓ nerve fiber density | Dopaminergic meds, anticonvulsants | Sensory retraining |
↑ = Increased; ↓ = Decreased; NSAID = Nonsteroidal anti-inflammatory drug; DBS = Deep Brain Stimulation; CBT = Cognitive Behavioural Therapy.
Table 3.
Sleep-related Disorders.
| Symptom | Prevalence & Stage | Biomarkers | Pharmacological Treatment |
Non-pharmacological Treatment |
|---|---|---|---|---|
| EDS | ↑ 21-76%, early | ↓ Hypocretin-1, ↑ Tau, ↑ α-synuclein | Modafinil, istradefylline, melatonin | Sleep hygiene, CBT, exercise |
| Insomnia | ↑ 60-80%, early | ↑ α-synuclein, associated with motor & cognitive symptoms | Melatonin, benzodiazepines | CBT-I, neuromodulation |
| OSA | ↑ 45-66%, advanced | ↑ Leptin, ghrelin, IL-6 | Clonazepam, melatonin | CPAP therapy, sleep hygiene |
| RLS | ↑ 20-40%, early | ↓ Dopamine & serotonin, ↑ iron in brain | Dopaminergic agents, gabapentin | Exercise, thermotherapy |
| RBD | ↑ 33-58%, preclinical | ↑ Cognitive impairment risk, α-synuclein | Clonazepam, melatonin | Sleep safety modifications |
↑ = Increased; ↓ = Decreased; EDS = Excessive Daytime Sleepiness; OSA = Obstructive Sleep Apnoea; RLS = Restless Legs Syndrome; RBD = REM Sleep Behaviour Disorder; CBT-I = Cognitive Behavioural Therapy for Insomnia; CPAP = Continuous Positive Airway Pressure.
Table 4.
Neuropsychiatric Disorders.
| Symptom | Prevalence & Stage | Biomarkers | Pharmacological Treatment |
Non-pharmacological Treatment |
|---|---|---|---|---|
| Depression | ↑ 35-45%, prodromal-late | ↓ Thalamus & amygdala function | SSRIs, SNRIs, TCAs | CBT, TMS, ECT |
| Apathy | ↑ 40-52%, early-late | ↓ Mesocortical activity, VTA dysfunction | Dopamine agonists, rivastigmine | DBS, TMS |
| Anxiety | ↑ 31%, prodromal-late | ↑ TNF-α, ↓ nitric oxide | SSRIs, benzodiazepines | CBT, meditation |
| Visual Hallucinations | ↑ 27-50%, early-advanced | ↓ Acetylcholine, cognitive decline | Rivastigmine, clozapine | CBT, music therapy |
| Phantosmias | ↑ 0.5-18.2%, early-late | Correlation with hallucinations | Antiseizure, antipsychotics | Surgical intervention |
↑ = Increased; ↓ = Decreased; VTA = Ventral Tegmental Area; TNF-α = Tumour Necrosis Factor Alpha; SSRI = Selective Serotonin Reuptake Inhibitor; SNRI = Serotonin/Norepinephrine Reuptake Inhibitor; TCA = Tricyclic Antidepressant; CBT = Cognitive Behavioural Therapy; TMS = Transcranial Magnetic Stimulation; ECT = Electroconvulsive Therapy; DBS = Deep Brain Stimulation.
Table 5.
Cognitive Disorders.
| Symptom | Prevalence & Stage | Biomarkers | Pharmacological Treatment |
Non-pharmacological Treatment |
|---|---|---|---|---|
| Inattention & Task-Switching | ↑ 20%, Preclinical | EEG alterations, gene mutations | Atomoxetine, methylphenidate | Cognitive training, physical activity |
| Bradyphrenia | ↑ 25%, Early | ↑ CSF metabolites, linked to constipation & bradykinesia | Levodopa, MAO-B inhibitors | Processing speed training |
| Dementia | ↑ 80%, Late | ↑ Cortical atrophy, ↑ α-synuclein, ↓ Amyloid-β | Donepezil, memantine, emerging therapies | Exercise, non-invasive brain stimulation |
| Impulse Control Disorders | ↑ 20%, Early | ↑ Dopamine tone, ↑ OFC metabolism | Adjust DRT, antipsychotics | CBT, DBS |
↑ = Increased; ↓ = Decreased; EEG = Electroencephalogram; CSF = Cerebrospinal Fluid; OFC = Orbitofrontal Cortex; MAO-B = Monoamine Oxidase B; DRT = Dopamine Replacement Therapy; CBT = Cognitive Behavioural Therapy; DBS = Deep Brain Stimulation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



