Submitted:
13 February 2025
Posted:
16 February 2025
You are already at the latest version
Abstract
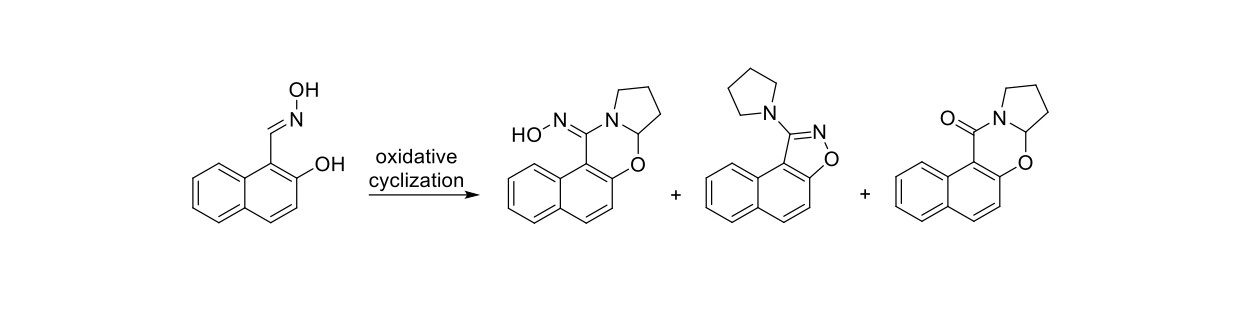
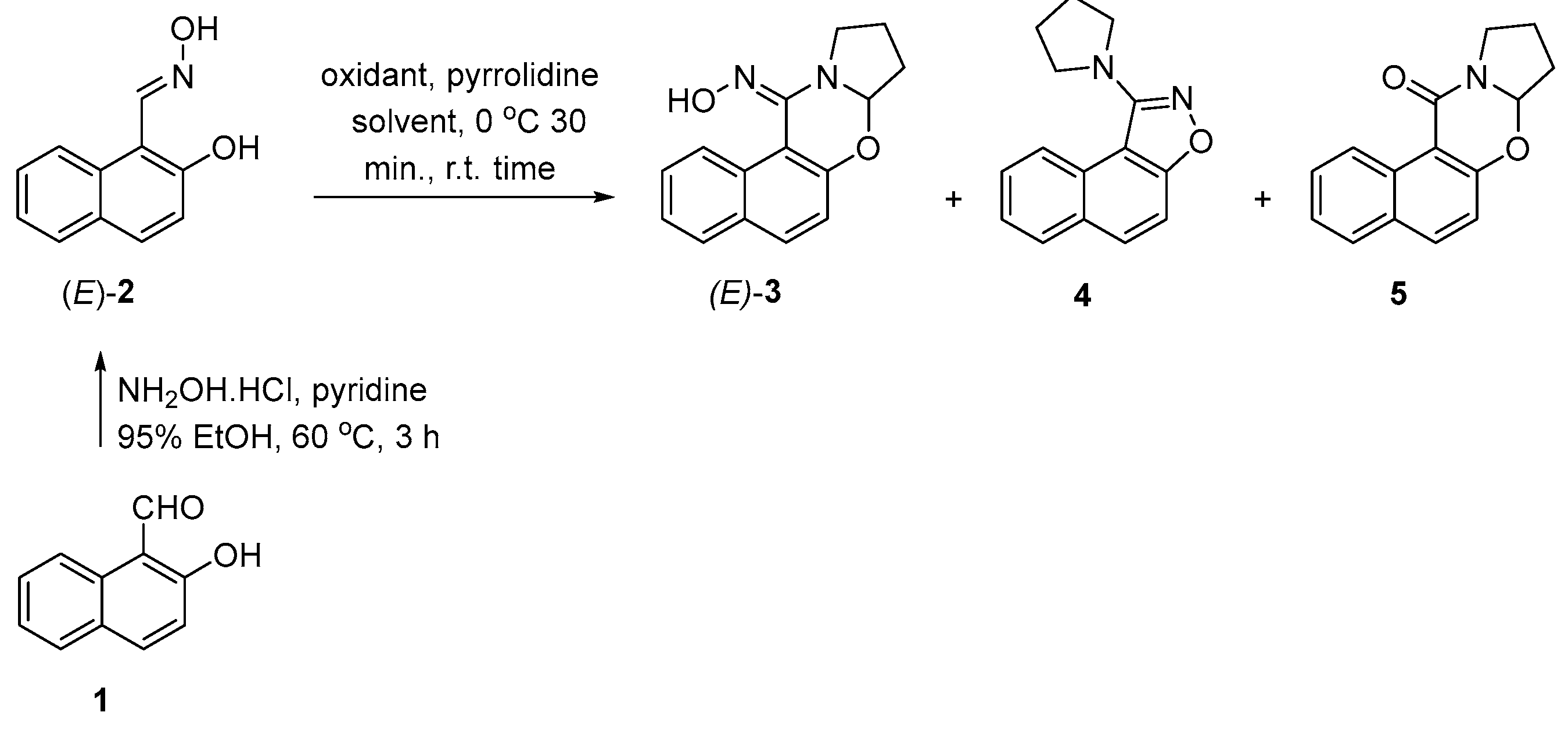
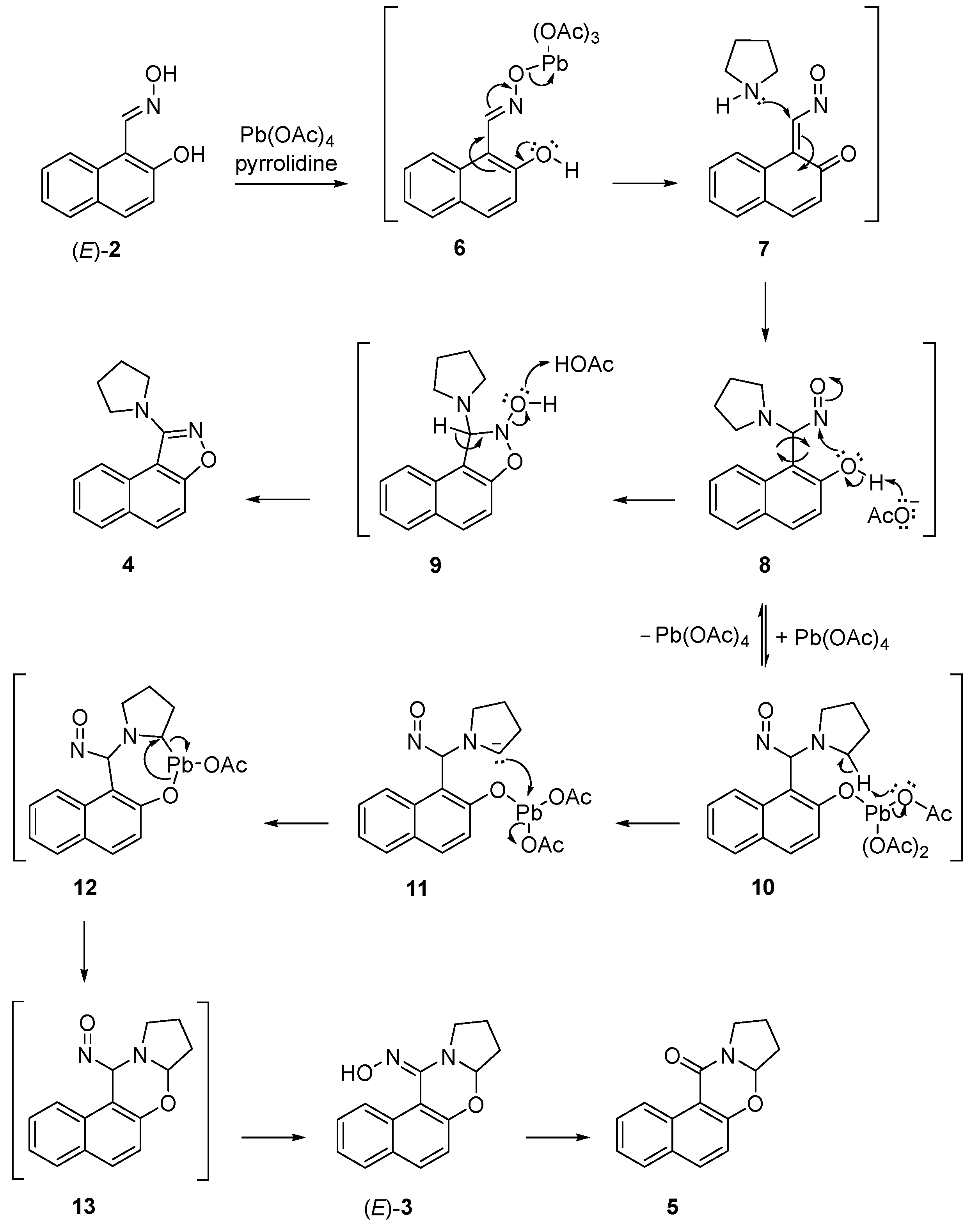
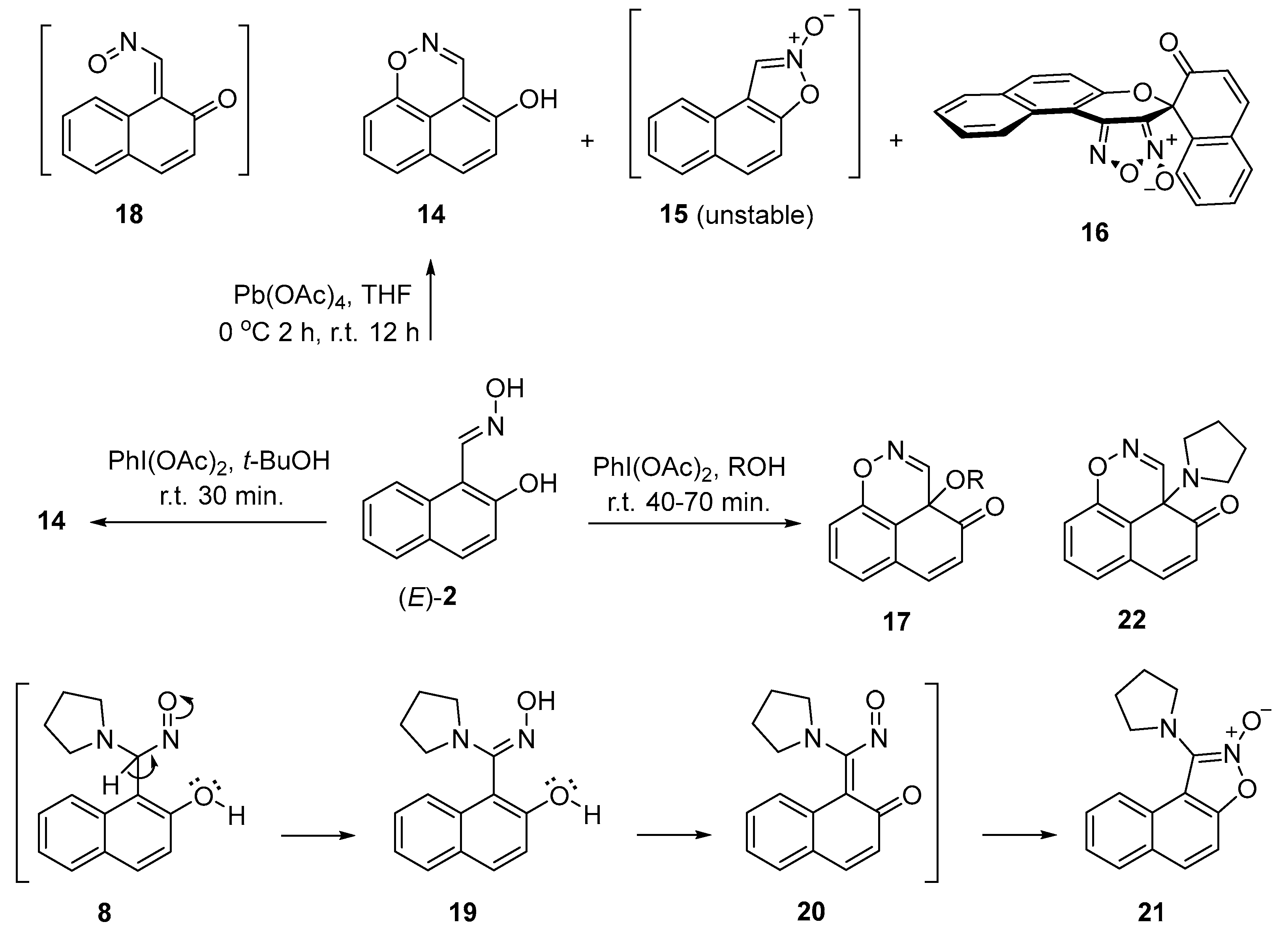
In this study, we examined the oxidation of (E)-2-hydroxy-1-naphthaldehyde oxime with lead tetraacetate in tetrahydrofuran that produced novel (E)-7a,8,9,10-tetrahydro-12H-naphtho[1,2-e]pyrrolo[2,1-b][1,3]oxazin-12-one oxime and 1-(pyrrolidin-1-yl)naphtho[1,2-d]isoxazole, and, known 7a,8,9,10-tetrahydro-12H-naphtho[1,2-e]pyrrolo-[2,1-b][1,3]oxazin-12-one, in 15, 18 and 10% yields, respectively. The oxime is readily hydrolysed to its corresponding ketone. Modifying the oxidants and reaction conditions did not improve the product yields. Based on previous studies in our laboratory, we proposed that the reactions proceed via the formation of an o-naphthoquinone nitrosomethide intermediate. 1H and 13C NMR, HRMS, IR, and UV-VIS spectra provided information that supported the structure of the products.
Keywords:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. (E)-7a,8,9,10-Tetrahydro-12H-naphtho[1,2-e]pyrrolo[2,1-b][1,3]oxazin-12-one oxime (3), 1-(pyrrolidin-1-yl)naphtho[1,2-d]isoxazole (4) and 7a,8,9,10-Tetrahydro-12H-naphtho[1,2-e]pyrrolo-[2,1-b][1,3]oxazin-12-one (5)
4.1.1. (E)-7a,8,9,10-Tetrahydro-12H-naphtho[1,2-e]pyrrolo[2,1-b][1,3]oxazin-12-one oxime (3)
4.1.2. 1-(Pyrrolidin-1-yl)naphtho[1,2-d]isoxazole (4).
4.1.3. 7a,8,9,10-Tetrahydro-12H-naphtho[1,2-e]pyrrolo[2,1-b][1,3]oxazin-12-one (5).
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| LTA | Lead tetraacetate |
| PIDA | Phenyliodine(III) diacetate |
| PIFA | Phenyliodine(III) bis(trifluoroacetate) |
| μ-Oxo-bridged PIFA | μ-Oxo-bis[phenyl(trifluoromethoxy)iodine(III)] |
References
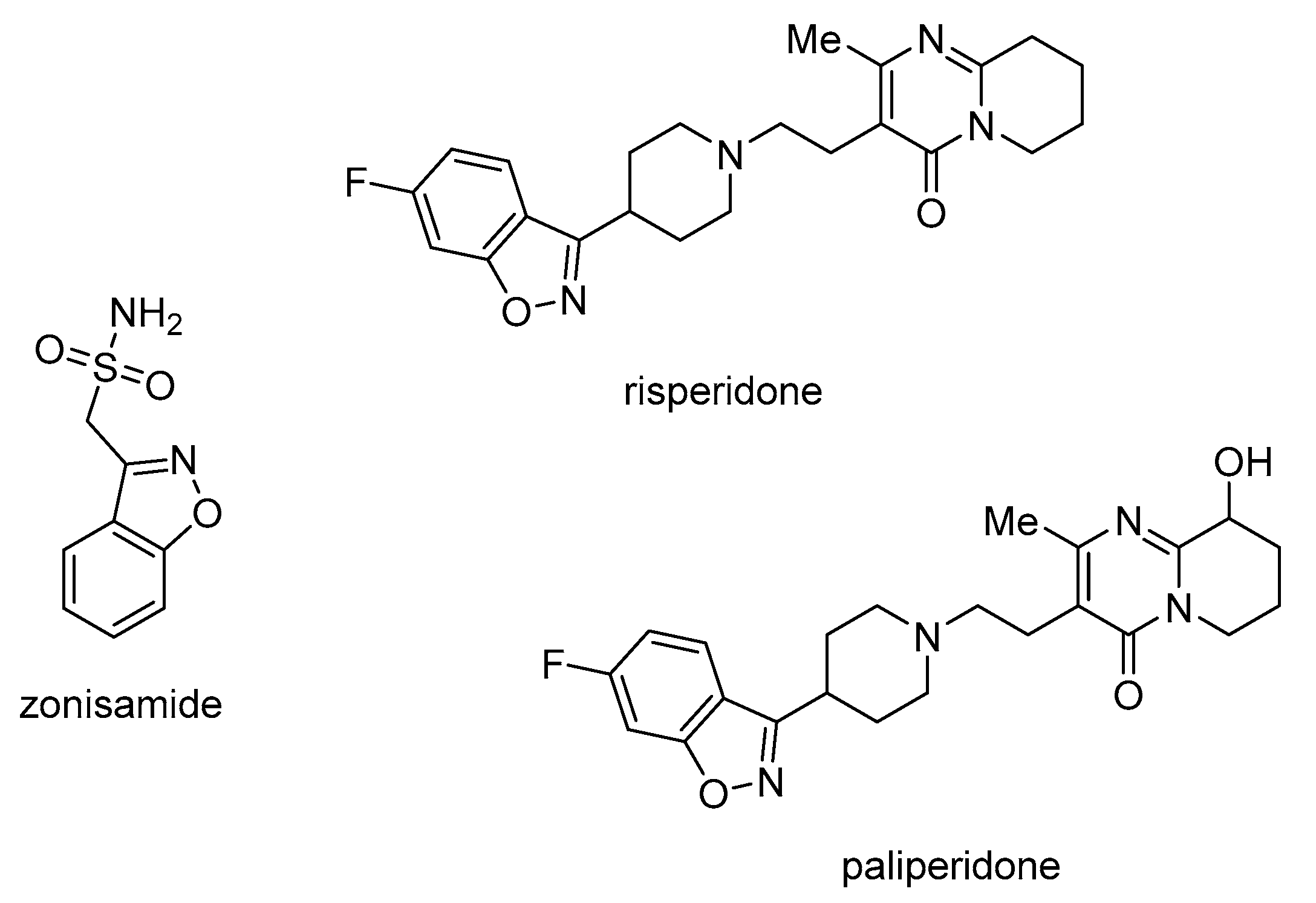
- Rakesh, K.P.; Shantharam, C.S.; Sridhara, M.B.; Manukumar, H.M.; Qin, H.L. Benzisoxazole: A Privileged Scaffold for Medicinal Chemistry. Med. Chem. Comm. 2017, 8, 2023–2039. [Google Scholar]
- Schulze-Bonhage, A. Zonisamide in the Treatment of Epilepsy Schulze-Bonhage Zonisamide. Expert. Opin. Pharmacother. 2010, 11, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Harrison, T.C.; Goa, K.L.; Beauclair, L. Long-Acting Risperidone. A Review of Its Use in Schizophrenia. CNS Drugs 2004, 18, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Solanki, P. V.; Uppelli, S.B.; Pandit, B.S.; Mathad, V.T. An Improved and Efficient Process for the Production of Highly Pure Paliperidone, a Psychotropic Agent, via DBU Catalyzed N-Alkylation. ACS Sustain. Chem. Eng. 2013, 1, 243–248. [Google Scholar] [CrossRef]
- Lukoyanov, A.A.; Aksenova, S.A.; Tabolin, A.A.; Sukhorukov, A.Yu. 3-Halo-5,6-Dihydro-4 H-1,2-Oxazine N -Oxides as Synthetic Equivalents of Unsaturated Nitrile Oxides in the [3 + 2]-Cycloaddition with Arynes: Synthesis of Substituted 3-Vinyl-1,2-Benzisoxazoles. Org. Biomol. Chem. 2024, 22, 3615–3621. [Google Scholar] [CrossRef]
- Shastri, R. Review on Synthesis of 3-Substituted 1,2-Benzisoxazole Derivatives. Chem. Sci. Trans 2016. [Google Scholar] [CrossRef]
- Giomi, D.; Cordero, F.M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier Science: Oxford, UK, 2008; Volume 4, pp. 365–485. [Google Scholar]
- Mahadevan, K.M.; Vaidya, V.P.; Vagdevi, H.M. Synthesis of Novel Naphtho[2,1-b]Furopyrimidine Derivatives. Indian. J. Chem. Sect. B 2003, 42, 1931–1936. [Google Scholar] [CrossRef]
- Dale, T.J.; Sather, A.C.; Rebek, J. Synthesis of Novel Aryl-1,2-Oxazoles from Ortho-Hydroxyaryloximes. Tetrahedron Lett. 2009, 50, 6173–6175. [Google Scholar] [CrossRef]
- Kitamura, M.; Sakata, R.; Tashiro, N.; Ikegami, A.; Okauchi, T. Synthesis of Diazonaphthoquinones from Naphthols by Diazo-Transfer Reaction. Bull. Chem. Soc. Jpn. 2015, 88, 824–833. [Google Scholar] [CrossRef]
- Kitamura, M.; Tashiro, N.; Sakata, R.; Okauchi, T. Synthesis of Diazonaphthoquinones from Naphthols by Diazo-Transfer Reaction with 2-Azido-1,3-Dimethylimidazolinium Chloride. Synlett 2010, 2010, 2503–2505. [Google Scholar] [CrossRef]
- Nimnual, P.; Tummatorn, J.; Thongsornkleeb, C.; Ruchirawat, S. Utility of Nitrogen Extrusion of Azido Complexes for the Synthesis of Nitriles, Benzoxazoles, and Benzisoxazoles. J Org Chem 2015, 80, 8657–8667. [Google Scholar] [CrossRef] [PubMed]
- Shelke, A.; Bhong, B.; Karade, N. Oxidation of 1-Amidoalkyl-2-Naphthols Using (Diacetoxyiodo)Benzene: Unusual Formation of 1-Arylnaphtho[1,2-d]Isoxazoles. Synthesis (Stuttg) 2014, 46, 752–756. [Google Scholar] [CrossRef]
- Chen, G.; Liu, H.; Li, S.; Tang, Y.; Lu, P.; Xu, K.; Zhang, Y. A Novel PPh 3 Mediated One-Pot Method for Synthesis of 3-Aryl or Alkyl 1,2-Benzisoxazoles. Org. Lett. 2017, 19, 1792–1795. [Google Scholar] [CrossRef]
- Van Eker, D.; Chauhan, J.; Murphy, W.A.; Conlon, I.L.; Fletcher, S. Chromatography-Free, Mitsunobu-Triggered Heterocyclizations of Salicylhydroxamic Acids to 3-Hydroxybenzisoxazoles. Tetrahedron Lett. 2016, 57, 5301–5303. [Google Scholar] [CrossRef]
- Cayley, A.; Gallagher, K.; Ménard-Moyon, C.; Schmidt, J.; Diorazio, L.; Taylor, R. Preparation of Novel Polycyclic Heterocycles Using a Tin(II) Chloride Dihydrate-Mediated Deacetalisation-Bicyclisation Sequence. Synthesis (Stuttg) 2008, 2008, 3846–3856. [Google Scholar] [CrossRef]
- Modak, A.; Dutta, U.; Kancherla, R.; Maity, S.; Bhadra, M.; Mobin, S.M.; Maiti, D. Predictably Selective (sp3)C-O Bond Formation through Copper Catalyzed Dehydrogenative Coupling: Facile Synthesis of Dihydro-Oxazinone Derivatives. Org. Lett. 2014, 16, 2602–2605. [Google Scholar] [CrossRef]
- Whiting, E.; Lanning, M.E.; Scheenstra, J.A.; Fletcher, S. Chromatography-Free Entry to Substituted Salicylonitriles: Mitsunobu-Triggered Domino Reactions of Salicylaldoximes. J. Org. Chem. 2015, 80, 1229–1234. [Google Scholar] [CrossRef]
- Supsana, P.; Tsoungas, P.G.; Varvounis, G. A Novel One-Pot Synthesis of Isomeric Naphtho[1,2-d]Isoxazole 2-Oxide and Naphtho[1,8-de][1,2]Oxazine Ring Systems. A Case of Simultaneous o- and Peri-Cyclisation in Naphthalene. Tetrahedron Lett. 2000, 41, 1845–1847. [Google Scholar] [CrossRef]
- Supsana, P.; Tsoungas, P.G.; Aubry, A.; Skoulika, S.; Varvounis, G. Oxidation of 1-Acyl-2-Naphthol Oximes: Peri - and o -Cyclisation and Spiro Cyclodimerisation of Naphthoquinone Nitrosomethide Intermediates. Tetrahedron 2001, 57, 3445–3453. [Google Scholar] [CrossRef]
- Dolka, C.; Hecke, K. Van; Meervelt, L. Van; Tsoungas, P.G.; Eycken, E.V. Van der; Varvounis, G. Novel Thermal and Microwave-Assisted Facile Route to Naphthalen-2(1H)-Ones via an Oxidative Alkoxylation-Ring-Opening Protocol. Org. Lett. 2009, 11, 2964–2967. [Google Scholar] [CrossRef]
- Kozielewicz, P.; Tzeli, D.; Tsoungas, P.G.; Zloh, M. Arene-Fused 1,2-Oxazole N-Oxides and Derivatives. The Impact of the N–O Dipole and Substitution on Their Aromatic Character and Reactivity Profile. Can It Be a Useful Structure in Synthesis? A Theoretical Insight. Struct. Chem. 2014, 25, 1837–1846. [Google Scholar] [CrossRef]
- Pal, S.; Khan, Md.N.; Karamthulla, S.; Choudhury, L.H. Synthesis of Pyranocoumarin Fused Spirooxindoles via Knoevenagel/Michael/Cyclization Sequence: A Regioselective Organocatalyzed Multicomponent Reaction. Tetrahedron Lett. 2015, 56, 359–364. [Google Scholar] [CrossRef]
- Tsoungas, G.P.; Cordopatis, P.; Gardikis, Y.; Potamitis, C.; Zervou, M. Xanthones in Heterocyclic Synthesis. An Efficient Route for the Synthesis of C-3 o-Hydroxyaryl Substituted 1,2-Benzisoxazoles and Their N-Oxides, Potential Scaffolds for Angiotensin(II) Antagonist Hybrid Peptides. Heterocycles 2011, 83, 1077. [Google Scholar] [CrossRef]
- Jadhav, V.K.; Deshmukh, A.P.; Wadagaonkar, P.P.; Salunkhe, M.M. Sodium Perborate: A Facile Synthesis of 1,2-Benzisoxazole 2-Oxides. Synth. Commun. 2000, 30, 1521–1527. [Google Scholar] [CrossRef]
- Hesse, M.; Meier, H.; Zeeh, B. Spectroscopic Methods in Organic Chemistry; Hesse, M. , Meier, H., Zeeh, B., Eds.; Georg Thieme Verlag: Stuttgart, 2008; ISBN 9783131060426. [Google Scholar]
- Bracken, C.; Baumann, M. Development of a Continuous Flow Photoisomerization Reaction Converting Isoxazoles into Diverse Oxazole Products. J. of Org. Chem. 2020, 85, 2607–2617. [Google Scholar] [CrossRef]
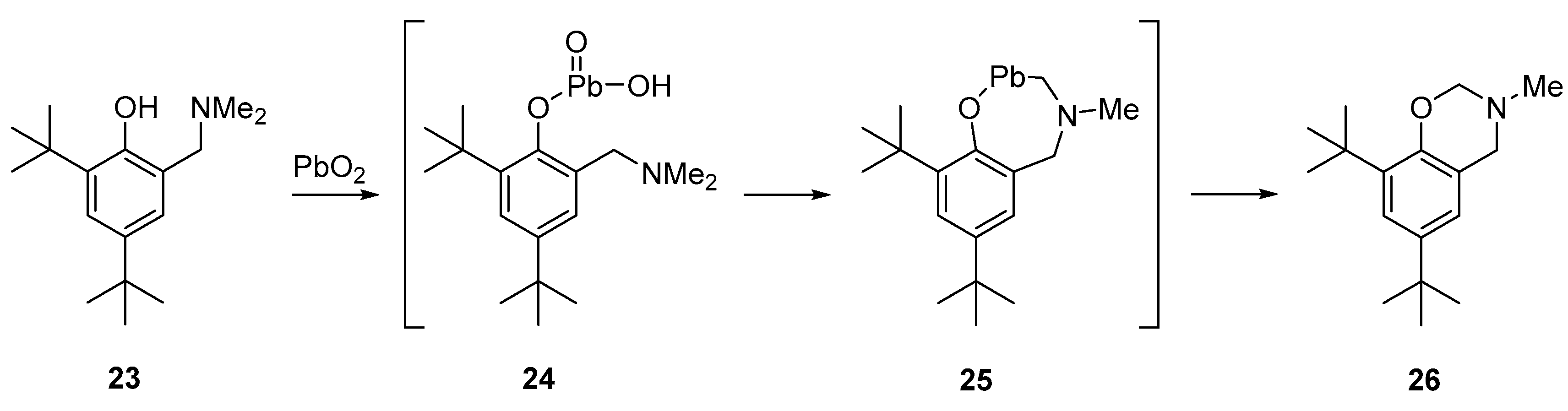
- Belostotskaya, I.S.; Vol’eva, V.B.; Komissarova, N.L.; Dekaprilevich, M.O.; Khrustalev, V.N.; Karmilov, A.Yu.; Ershov, V. V. Oxidation of 2-Dialkylaminomethyl-4,6-Di-Tert-Butylphenols. Russ. Chem. Bull. 1997, 46, 1272–1280. [Google Scholar] [CrossRef]
- Maegawa, T.; Oishi, R.; Maekawa, A.; Segi, K.; Hamamoto, H.; Nakamura, A.; Miki, Y. The Reaction of Ketoximes with Hypervalent Iodine Reagents: Beckmann Rearrangement and Hydrolysis to Ketones. Synthesis (Stuttg) 2022, 54, 4095–4103. [Google Scholar] [CrossRef]

| entry | Oxidant (equiv.) |
pyrrolidine (equiv.) | solvent | time (h) |
(E)-3 (yield%)2 | 4 (yield%)2 |
5 (yield%)2 |
|---|---|---|---|---|---|---|---|
| 1 | LTA (2) | (1.5) | THF | 12 | 15 | 18 | 10 |
| 2 | PIDA (2) | (1.5) | THF | 12 | 9 | 12 | 8 |
| 3 | PIFA (2) | (1.5) | THF | 12 | 6 | 8 | 7 |
| 4 | PIDA (2) | (1.5) | DCM | 12 | 10 | 14 | 7 |
| 5 | PIFA (2) | (1.5) | DCM | 12 | 6 | 9 | 4 |
| 6 | μ-oxo-bridged PIFA (1) | (1.5) | DCM | 12 | – | – | – |
| 7 | PIDA (2) | (1.5) | MeCN | 12 | 13 | 16 | 9 |
| 8 | PIDA (3) | (2.5) | THF | 8 | 7 | 14 | 10 |
| 9 | PIDA (3) | (2.5) | DCM | 8 | 8 | 15 | 11 |
| 10 | PIDA (3) | (2.5) | MeCN | 8 | 9 | 13 | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



