Submitted:
10 February 2025
Posted:
10 February 2025
You are already at the latest version
Abstract
Left ventricular assist devices (LVAD) have been widely adopted for the treatment of both pediatric and adult patients with end-stage heart failure. By unloading the failing ventricle, maintaining sufficient end-organ perfusion, and improving the heart's functional capacity, they are increasingly used as bridges to recovery, cardiac transplantation, or destination therapy, thus improving patients' overall survival rate and life quality. However, beyond their life-sustaining functions, these devices pumping total cardiac output may have a range of short and long-term harmful effects initiated by interactions of blood cells with artificial surfaces at the LVAD/Host interface or by non-physiological flow conditions generated by these devices. One of the major challenges in LVAD therapy is the lack of satisfactory sensitivity and specificity of the currently available biomarkers, making it difficult to differentiate the patients who will benefit from LVAD placement from those at high risk for adverse events and poor outcomes. To this end, myocardial miRNAs have emerged as promising targets for investigating heart failure-related molecular pathways. In addition, extracellular circulatory miRNAs whose concentrations change with pathological state and disease progression may also mediate intercellular signaling pathways that are important in the functional and structural changes associated with the development and progression of HF. Herein, we provide a comprehensive overview of the currently known cardiac miRNAs involved in LVAD-induced myocardial reverse remodeling, remission, and recovery, and circulatory cell-free and platelet-enriched miRNAs that may serve as potential non-invasive biomarkers for pre-LVAD decision-making or monitoring and predicting patient response to LVAD therapy.
Keywords:
Heart failure
; LVAD
; microRNA (miRNA)
; circulatory biomarkers
; mechanical circulatory support
1. Introduction
Heart failure (HF) is a life-threatening clinical syndrome characterized by a heterogeneous pathophysiology and multiple etiologies [1]. The major causes of HF are ischemic (ICM) or dilated cardiomyopathy (DCM), hypertensive heart disease, and valvular heart diseases (Figure 1) [1]. The global burden of HF is significant and a pressing issue of utmost urgency. It affects 1-3% of the general adult population, with over 64 million people estimated to suffer from HF worldwide [2,3,4]. Furthermore, the prevalence of HF is on the rise. This is driven by improved treatment outcomes, an aging population, and sedentary lifestyles, along with an increase in other HF risk factors such as hypertension, coronary artery disease, obesity, and diabetes [1,2,3,4]. Despite improved diagnostics and expanding knowledge, HF remains a leading cause of death worldwide, with 1-year mortality estimated at 15–30% and a 5-year survival as low as 25%, thus making this disease as “malignant” as many common types of cancer [1,2,3,4,5]. The economic burden created by HF is also significant. This underscores the urgent need for continued research and development of novel, patient-specific treatment strategies.
The central feature in HF progression is represented by adverse remodeling of the cardiac left ventricular (LV) wall in response to the increased mechanical and biochemical stress initiated by abnormal hemodynamic and local neurohormonal and cytokine factors [6,7,8]. These stress factors profoundly affect different components of the myocardium, altering their gene expression, metabolic processes, signaling pathways, extracellular matrix structure, and protein function [6,8,9]. At the very onset, the remodeling responses may be adaptive but become pathological and cause further myocardial damage when sustained over time. Consequently, the ventricular volume, thickness, geometry, electrical integrity, and biochemical composition of the myocardial wall, and the orientation, size, and shape of myocardial muscle fibers are all modified in states of chronic HF [6,9,10,11,12,13].
Clinically, that is manifested by inefficient myocardial contraction and progressive elevation in left ventricular end-diastolic (LVEDV) and end-systolic volume (LVESV), followed by a significant decrease in left ventricular ejection fraction (LVEF) (Figure 1) [1,7,11,14,15]. Together with the inability of the cardiac muscle to pump a sufficient amount of blood and oxygen throughout the body, including the heart itself, this is associated with significantly higher mortality in HF patients, frequent hospitalization, and substantially decreased quality of life [16,17].
Although the adverse remodeling was initially considered to be an irreversible process, clinical studies in the past few decades have shown that treatment modalities that counteract the underlying mechanical and biochemical stress factors may effectively attenuate or, to a certain extent, even reverse the functional and structural changes associated with the development and progression of HF [16,17]. This phenomenon, referred to as “reverse remodeling” has become a primary goal in treating HF [18,19,20,21,22,23]. It is associated with better clinical outcomes and improved prognosis, offering hope for the future of HF treatment. [18,19,20,21,22,23]. Idealy reverse remodeling should lead to myocardial recovery, i.e., the sustained normalization of myocardial structure and function at the cellular, molecular, and transcriptional levels accompanied by freedom from future HF events. However, the complete myocardial recovery accompanied by normalizing systolic functions is currently accomplished in rare cases [16,24]. Most of the so-called “optimally treated” HF patients who are receiving guideline-directed medical therapy (GDMT) are in a clinically stabilized remission state. In other words, they experience long-term disease stabilization when they are monitored on an outpatient basis, tolerate treatment well, and do not require hospitalization for decompensated HF. This may be accompanied by myocardial or cardiac remission, with or without improved LVEF correspondingly [24].
Existing pharmacological therapies that promote myocardial reverse remodeling include inhibitors of angiotensin-converting enzyme (ACEI), renin-angiotensin-aldosterone [RAS] system, beta-blockers (BB) or sodium-glucose cotransporter 2 inhibitors (SGLT2I) [16,17]. Reverse remodeling can also be achieved by myocardial revascularization (coronary artery bypass surgery), mitral valve repair and replacement, cardiac resynchronization therapy (CRT), or mechanical circulatory support (MCS) with left ventricular assist devices (LVAD) [16,17,19,25,26,27].
Among available therapeutic options, LVAD support of HF patients was associated with the most significant degree of ventricular remodeling at the structural, cellular, and molecular levels. Furthermore, in selected HF patients with favorable clinical factors such as younger age, shorter duration of HF, nonischemic cardiomyopathy (NICM), smaller left ventricular end-diastolic diameter (LVEDD), and normal or mildly impaired renal function, a sustained improvement in myocardial structure and function is noted, allowing the weaning from LVAD assisted device without the need for transplantation or subsequent reintroduction of LVAD support [28]. This means that the stage of disease and degree of structural damage, molecular alterations, and hemodynamic dysfunctions will not only determine the type and clinical significance of HF [28]. They can also influence the possibility and extent of the myocardial reverse remodeling, remission, and recovery once the appropriate therapeutic approach has been applied. In most cases, despite the improved cardiac function, the underlying molecular processes are not in their physiological state, and these patients are still in danger of disease progression [24]. Various transcriptional and proteomic profiling studies of paired apical myocardial tissue samples explanted from human transplant recipients before and after a period of mechanical circulatory support with LVAD assist devices and pharmacological (e.g., isoproterenol and angiotensin II infusion), surgical (e.g., thoracic aortic banding), and transgenic animal models of HF have identified a number of genes that differ between failing and nonfailing (NF) human hearts or differentiate responders and nonresponders to LVAD support [29,30,31]. They also led to the discovery of a subset of persistently dysregulated HF genes that did not normalize during reverse LV remodeling. The latter are grouped into multiple functionally distinct gene modules related to cellular metabolism, sarcomere, cytoskeletal, and extracellular matrix (ECM) components, or excitation-contraction coupling processes [32,33]. Coregulation of distinct sets of genes involved in tissue repair has also been observed [32,33].
New areas of investigation continue to develop with an emphasis on biomarkers and advanced imaging techniques that may be helpful for real-time evaluation of pathophysiologic changes and prognosis in individual HF patients [30,34,35,36,37,38]. Until now, various proteins and protein-coding mRNAs have been the most extensively studied despite accounting for only a minority of the human genome [30,34,35,36,37,38]. Thus, a growing number of damage, remodeling, and neurohormonal activation proteins implicated in the development of HF have been described, some of which are well-validated and established as prognostic biomarkers in clinical practice (Figure 2) [34,35,36,37,38]. However, an in-depth understanding of the underlying mechanisms of myocardial reverse remodeling, remission, and recovery in HF is still missing. The development of next-generation sequencing (NGS) techniques has enabled in-depth coverage of the transcriptome landscape [39]. This has allowed for a more comprehensive investigation of noncoding RNAs, which have become an attractive target for defining novel noninvasive biomarkers (Supplementary Table S1) and regulatory mechanisms in HF and other cardiovascular pathologies (Figure 3) [39,40,41]. Although unable to encode proteins, noncoding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), exhibit stable and highly organized spatial, temporal, and tissue-specific gene expression [39,40,41]. They are crucial components of stress response in the heart that fine-tune the genomic interactions with various environmental factors and, thus, significantly influence cardiac homeostasis and function [39,40,41].
Therefore, this review focuses on the biological effects of LVAD support of end-stage HF patients, emphasizing the promising role of miRNAs as biomarkers of HF and potential predictors of structural, functional, and clinical aspects of myocardial reverse remodeling, remission, and recovery elicited by LVAD-induced mechanical unloading. The expression profile of corresponding circulatory miRNAs and their potential as noninvasive biomarkers for monitoring and predicting patient response to LVAD therapy will also be examined.
2. Left Ventricular Assist Devices
Heart transplantation has been a historically mainstay of therapy for end-stage heart failure patients. It has been, however, significantly limited due to a severe shortage of donor heart supply, long waiting times, and strict recipient criteria applied to avoid poor outcomes. The shortage of organ donors and the increasing incidence of HF have prompted efforts toward developing alternative approaches, namely short and long-term mechanical circulatory support (MCS) devices [42,43]. In general, MCS may be distinguished by hemodynamic characteristics, site of blood draw and return, technique of insertion, and utilization of gas exchange units [42]. Two broad categories of durable MCS are available: ventricular assist devices (VAD), which may provide support to either the left (LVAD), right (RVAD), or both ventricles (BiVAD, biventricular assist devices) and total artificial hearts (TAHs) [42,43,44]. The FDA does not currently approve durable MCSs for RV support, and most long-term MCSs are continuous-flow devices designed for LV support. They are indicated for patients with end-stage HF refractory to conventional medical treatment.
Implementing LVADs in routine clinical practice has significantly advanced the treatment of end-stage HF [21,27,45,46,47,48]. By reducing the mechanical pressure and volume of the failing heart, maintaining sufficient blood supply throughout the body, and improving the heart’s functional capacity, they are used as bridges to cardiac transplantation (BTT) and recovery (BTR) or as destination therapy (DT) in patients who are not eligible for heart transplantation [21,27,45,46,47,48]. This has remarkably increased HF patients’ survival rates and life quality [49,50,51,52]. A concomitant technological advance and the development of smaller, durable, intracorporeal, and magnetically levitated devices paralleled with the improvement of patient selection and post-implant device management guidelines, have led to their widespread use in a steadily increasing number of heart patients and, at earlier points of disease progression [49,50,51,52,53,54,55,56]. In addition, these advancements have opened the door to minimally invasive surgical procedures for LVAD implantation, which have further improved patient outcomes [57,58]. However, beyond life-sustaining functions, these devices pumping total cardiac output may have short- and long-term harmful effects induced by mechanical blood flow patterns and the interactions occurring at the LVAD/Host interface. Most frequently, they are related to the incidence of device malfunction, thrombus, stroke, infection, gastrointestinal bleeding, multisystem organ failure, and right heart failure [51]. The hazard for most of these adverse events is the highest in the early postoperative period (≤ 90 days) and diminishes significantly after that [51]. Nevertheless, with current technological advances, survival rates are higher, adverse event rates are lower than in prior eras, and the life expectancy of LVAD-treated HF patients is increasing.
Approximately 3000 LVADs are implanted worldwide each year. According to the Society of Thoracic Surgeons (STS) and Interagency Registry for Mechanically Assisted Circulatory Support (Intermacs), 27,493 patients were implanted with continuous-flow LVAD devices in the last decade (2013-2022) [50]. In addition to remarkable contributions to the treatment of end-stage HF, the clinical experience with LVADs has been invaluable for identifying structural, cellular, and molecular aspects of myocardial reverse remodeling, remission, and recovery [26,27,30,35,53,59].
Initial studies of paired apical myocardial tissue samples explanted from human transplantation recipients before and after a period of mechanical circulatory support with LVAD assist devices showed for the first time that the structural aspects of cardiac remodeling, defined and quantified by shifts of the ventricular end-diastolic pressure-volume relationship (EDPVR), can be substantially reversed towards its normal values, even in the most advanced stages of HF [60,61]. Since then, intensive research has been carried out on the possibility of myocardial reverse remodeling, and LVAD implantation has been firmly established as a cornerstone in the treatment of advanced HF patients, providing a reliable and effective therapeutic option [26,27,30,35,53,59].
3. Noncoding RNAs
Besides mRNA acting as blueprints for the translation of proteins that account for ~1.5% of cellular transcripts, it is now clear that more than 75% of the human genome is transcribed, generating an extraordinary range of RNAs with no protein-coding capacity [42,43]. A complete catalog of these protein noncoding RNAs (ncRNAs) is yet to be available, and their characteristic sub-cellular localizations, biological functions, and interactions with other components of cellular gene regulatory networks still need to be better understood. According to GeneCards knowledgebase (Version 5.20; updated: May 14, 2024; https://www.genecards.org/), a searchable, integrative database that provides comprehensive information on all annotated and predicted human genes, and its GeneCaRNA Suite (https://www.genecards.org/genecarna) that integrates the resources from HGNC (The HUGO Gene Nomenclature Committee; http://www.genenames.org/), NCBI Gene (The National Center for Biotechnology Information Gene database; www.ncbi.nlm.nih.gov/gene), and Ensembl (ENSG; https://useast.ensembl.org/index.html) databases together with information from RNAcentral transcript source (The non-coding RNA sequence database; https://rnacentral.org/ ) there are currently 291,492 annotated genome sequences encoding protein non-coding RNAs (ncRNAs) [44,45,46,47,48,49]. This includes long-known, highly abundant, and functionally important ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs) involved in protein synthesis, as well as other constitutively expressed small “housekeeping” ncRNAs necessary for cell viability, such as small nuclear RNAs (snoRNAs), small nuclear RNAs (snRNAs), and telomere RNA (TRNA) involved in post-transcriptional RNA processing, mRNA splicing, and chromosome end synthesis correspondingly. The pervasive transcription of the human genome also gives rise to diverse arrays of regulatory ncRNAs. They can be classified based on the transcript length and structure, cellular location, mechanism of action, or association with DNA regulatory elements, annotated genome sequences, and DNA repeats [50,51,52,53,54]. Based on the length and structure of their transcript sequences, we differentiate short ncRNAs [e.g., micro RNAs (miRNA), small interfering RNA (siRNA), piwi-interacting RNA (piRNA), etc.] containing less than 200 nucleotides (nt) and long ncRNAs (lncRNAS; > 200 nt) with linear or circular (circRNA) transcript structures [50,51,52,53,54]. Derived as sense, antisense, or bidirectional transcripts from either mitochondrial DNA or intronic, exonic, and intergenic regions of the nuclear genome, regulatory ncRNAs are found in association with diverse membrane-bound (e.g., nucleus, mitochondria, endoplasmatic reticulum) or membrane-less subcellular compartments [e.g., stress granules (SG), Cajal bodies (CB), processing bodies (PB), and another ribonucleoprotein (RNP) granules] where they can be either biologically active or sequestered with their binding partners (e.g., DNA, RNA, proteins, lipid components) [50,51,52,53,54]. The tissue-specific spatiotemporal distribution of regulatory ncRNAs is intimately related to their functions in development, cell biology, and diseases, ranging from DNA replication and mRNA translation regulation to genome stability defense. This is accomplished through intricate ncRNA–RNA (e.g., lncRNA-miRNA-mRNA), ncRNA–DNA, and ncRNA–protein interactions and tightly regulated by dynamic changes in physiological conditions and/or exogenous environmental stress factors [51,52,55,56,57,58]. Regulatory ncNRAs have also been detected in a variety of biological bodily fluids (e.g., serum/plasma, blood, and urine) following their secretion through extracellular vesicles and, as such, can be used as potential noninvasive diagnostic and prognostic biomarkers or therapeutic tools in various pathological processes, including HF and other cardiovascular diseases [57,59,60].
The two most well-known classes of regulatory ncRNAs are miRNAs, which are involved in sequence-specific post-transcriptional regulation of mRNA stability and translation, and lncRNA molecules that acting as a signal, protein and RNA-binding decoys, molecular guides, or structural scaffolds, play a crucial role in a variety of mechanisms involved in epigenetic control of gene expression [51,52,55,56,57,58].
4. MicroRNAs
Endogenous miRNA transcripts of ~ 22 nucleotides in length are important regulatory factors involved in normal cellular homeostasis and pathological conditions, including myocardial disease processes. Genes coding for miRNAs may be found by themselves or in polycistron clusters in various coding and non-coding regions of the human genome [61,62]. About half of the known mammalian miRNAs are within the introns of protein-coding genes or the introns or exons of other non-coding RNAs, including lncRNAs [61,62]. In most cases (canonical miRNA pathway), they are transcribed by RNA polymerase II as primary miRs (pri-miRs) that are cleaved by RNase III Drosha and its co-factor Pasha (Partner of Drosha, or DGCR8 in vertebrates) into a stem-loop structure called precursor miRNAs (pre-miRNAs) [55]. These pre-miRNAs are then exported from the nucleus to the cytoplasm by Exportin 5 and cleaved by Dicer, another RNAse III family member, into small double-stranded RNAs (dsRNAs). Subsequently, either arm of this dsRNA, designated as -3p or -5p, in conjunction with a member of the Argonaute (AGO) protein family, gets incorporated into a miRNA-induced silencing complex (miRISC) that represses the targeted gene expression [55]. In that process, the mature and biologically active miRNA strand acts as a guide that directly interacts with complementary MRE (miRNA response elements) sites predominantly located within the 3′ untranslated region (3′-UTR) of target mRNAs leading to their translational repression (imperfect base pairing) or degradation (complete base pairing) [55]. Still, some miRNAs have been associated with increased expression of target genes through their interaction with the promoter sequences [63]. Several so-called non-canonical pathways, i.e., Drosha/DGCR8- and Dicer-independent pathways involved in the nuclear biogenesis of miRNAs, have also been described [64]. It has been estimated that human miRNAs may regulate as many as one-half of the human protein-coding genes and play fundamental roles in regulating different cellular functions [65]. Typically, a miRNA can regulate the expression of tens or hundreds of protein-coding genes, but a particular gene can also be precisely controlled by a number of different miRNAs, resulting in a complex and combinatorial mode of miRNA action in gene regulation [66]. The potential for generating multiple mature miRNAs, or isomiRs with tissue-dependent expression, resulting from alternative post-transcriptional processing of the same miRNA precursor, allows for additional functional redundancy [67,68]. Also, various RNA-binding proteins and other ncRNAs (e.g., lncRNAs) and epigenetic factors control the interactions of miRNAs with their targets [69]. Many miRNAs are ubiquitously expressed throughout the human body, while others are tissue-specific [70,71]. The distribution pattern of mature miRNA levels within a particular cell or tissue is tightly regulated at multiple levels, including transcriptional and post-transcriptional steps. Furthermore, their expression pattern is subjected to change during cellular differentiation or in response to environmental and pathophysiological stimuli and can, as such, serve as a signature of cell identity or biomarker of underlying disease processes. Currently, there are 1917 human microRNAs hairpin sequences annotated in mirBase repository version 22.1 (https://www.mirbase.org/browse/results/?organism=hsa), while the GeneCards knowledgebase (Version 5.20; updated: May 14, 2024; https://www.genecards.org/) enlists 6903 miRNA related human genes [72]. So far, up to 150 of human encoded miRNAs have an established role in the cardiovascular system, while 30-35 of them have been comprehensively analyzed and validated in experimental models in vivo, highlighting both their clinical significance in the myocardium and vasculature and their biomarker potential for patients diagnosis and treatment [73,74]. In recent years, numerous studies have supported the involvement of miRNAs in pathological mechanisms leading to the development and progression of HF [74,75,76,77,78,79]. Correspondingly, myocardial and circulatory miRNAs differentially expressed as the consequence of LVAD-induced mechanical unloading of a failing human heart have also been the center of intensive scrutiny.
5. Myocardial miRNA Signature in LVAD-Supported HF Patients
Research on the role of miRNAs as biomarkers and mediators of myocardial recovery in LVAD-supported patients has been limited, employing various methods like real-time PCR [80,81,82,83,84,85,86,87,88], microarray analysis [89,90,91], and high-throughput RNA sequencing [83,86,87,90,92,93,94]. The findings from these studies, summarized in Table 1, indicate that miRNAs may play a critical role in the heart remodeling process associated with LVAD-induced mechanical unloading.
Schipper et al. were the first to report significant changes in the expression of myocardial miRNAs accompanying LVAD-induced heart remodeling [80]. In their HF patient cohort, they found that preoperative expression levels of miR-1, miR-133a, and miR-133b were decreased in the myocardium from both the ischemic (ICM) and nonischemic dilated cardiomyopathy (NICM) patient group compared to the nonfailing myocardial tissue [80]. The decrease in miRNA expression levels was more pronounced in the ICM group. Interestingly, LVAD support did not stop further reduction in their expression levels in the myocardium of NICM patients [80]. At the same time, the expression of miR-1, miR-133a, and miR-133b was increased in the ICM group (Table 1) [80]. However, the observed restoration was only partial and did not reach the expression values of the control myocardium, either in LVAD patients supported for a short or a long (i.e., more than a year) period of time [80]. The role of miR-1 and miR-133a in heart remodeling is significant. Various in vitro and in vivo studies suggest that these miRNAs, by specifically regulating multiple targets and molecular pathways, have indispensable roles in repressing fetal gene programs in the postnatal heart, suppressing pathological cardiac remodeling and fibrosis, and regulating cardiomyocyte growth response, Ca2+ handling, apoptosis, conduction disturbances, and arrhythmogenesis [76,95,96]. Therefore, their expression patterns were frequently found to be dysregulated in the hearts of patients with hypertrophic, ischemic, or idiopathic cardiomyopathy, cardiac arrhythmias, and end-stage HF [76,95,96].
Ikeda et al. also detected reduced miR-1, miR-133a, and miR-133b expression in failing human myocardium [97]. However, these data were contrasted by some microarray-based studies showing opposite trends [89,98]. This variability in the transcriptome signature is encountered across studies of HF and can be explained by differences in methodology, the clinical characteristics of included subjects, inter-individual susceptibility to disease progression due to heterogeneous genetic or environmental etiologies, and the response to pharmacologic and nonpharmacologic therapies of a failing myocardium. Thus, improving the metrics to identify unique genomic signatures for HF of different etiologies and, more importantly, different prognoses may have a superior potential for better clinical diagnosis, patient monitoring, and treatment outcomes.
Matkovich et al. demonstrated that integrating miRNA profiling with myocardial mRNA transcriptome analysis significantly enhances the RNA signature’s ability to classify the clinical status of HF patients [109]. Their study identified eighty-one miRs expressed in human ICM and NICM cardiovascular tissue, with 28 stress-responsive miRNAs (Table 1) that can evoke cardiac hypertrophy, ventricular remodeling, and heart failure, including miR-1 and miR-133b, significantly upregulated in failing myocardium. A strong trend for upregulation was also observed for miR-21, miR-23a, miR-133a, and miR-638. Conversely, none of the miRNAs detected in their study met the same level of stringency for downregulation in HF [109]. Post-LVAD support, many of these upregulated miRNAs showed a trend for normalization, indicating the myocardium’s ability to recover after biomechanical unloading [89]. Eight miRNAs, i.e., let-7f, miR-27b, miR-30a-5p, miR-30c, miR-30d, miR-103, miR-130a, and miR-378, returned to baseline levels, showing a full normalization while the expression levels of let-7g, miR-1, miR-22, miR-24, miR-26b, miR-27a, miR-29b, miR-30b, miR-126, miR-143, miR-195, and miR-499 displayed a significant reduction in recovering, i.e., LVAD-supported myocardium [89]. The remaining miRNAs showed intermediate expression signatures that, although downregulated in the post-LVAD group, were not significantly different from the ones registered in the HF patients without LVAD support [89]. Parallel mRNA microarray analysis detected 155 (35%) upregulated and 289 (65%) downregulated mRNAs compared to NF controls. However, only 29 of them were normalized by at least 25% following LVAD support. Nevertheless, when compared with the miR signature, unsupervised hierarchical clustering of the overall mRNA expression pattern was better at distinguishing between NF and HF patient groups. However, it failed to differentiate between recovering and non-failing hearts. In contrast, the obtained miR signature was more sensitive to the mechanical unloading status of end-stage HF but failed to determine some LVAD-supported hearts from those in the NF group. Nonetheless, the combined RNA profiling method proved effective in classifying different HF types and functional states, albeit with limitations due to the use of patient cohorts with no paired pre- and post-LVAD samples.
Ramani et al. studied myocardial miRNA expression in HF patients with nonischemic etiology and found distinct expression patterns between recovery and LVAD-dependent groups [81]. Their study included a test and validation HF cohort, which differed based on demographic and clinical parameters. Specifically, the validation cohort had more male patients, greater LV diameters, and a longer disease duration before LVAD implantation. Also, the validation cohort was subjected to more extended LVAD support and had more patients supported with continuous (cf-LVAD) blood flow devices as opposed to more prevalent pulsatile blood flow (pf-LVAD) support in the test cohort. Half of the patients in the test and validation cohorts underwent LVAD removal (recovery group), while the other half remained LVAD-dependent [81]. Notably, histological cardiomyocyte size determination in the test cohort revealed that the recovery group had significantly smaller cardiomyocyte cross-sectional areas than the LVAD-dependent group. In the test cohort, initial PCR array analysis identified ten differentially expressed miRNAs (Table 1), with miR-103, miR-142-3p, miR-181b, and miR-376a upregulated in the recovery group, while miR-15b, miR-23a, miR-27a, miR-26a, miR-195, and miR-424 were downregulated [81]. Conversely, miR-1, miR-21, miR-133a, miR-133b, and miR-208, previously reported to play an essential role in HF-related pathways , showed a similar expression pattern in the recovery and LVAD-dependent groups. Subsequent qRT-PCR analysis of the test cohort further confirmed significantly downregulated expression of miR-15b, miR-23a, miR-26a, and miR-195 in the recovery group. However, only miR-23a and miR-195 showed the same expression signatures in the validation cohort. Additional comparisons between cohorts indicated that miRs 23a and 195 had similar signatures in recovery and non-failing groups. Finally, the analysis of a separate cohort of six paired pre- and post-LVAD myocardial tissue samples revealed no difference in the expression levels of miR-23a and miR-195 [81]. Also, excluding the upregulated expression of miR-21 and miR-208 in pre-LVAD heart tissue, no difference in the expression levels of any other miRNAs was detected. As suggested by the editorial comment of Mann and Burkhoff, these results indicate that the expression signatures of miR-23a and miR-195 observed in the recovery group might reflect the less severe nature of these patients at the time of LVAD implantation rather than be a marker of reverse remodeling or myocardial recovery induced by mechanical unloading and concomitant medical therapy [88]. Both miR-23a and miR-195 have previously been reported as pro-hypertrophic and pro-apoptotic miRNAs upregulated during pressure-induced cardiac hypertrophy [99,100]. MiR-195 also alters cardiac energy metabolism, fibroblast transdifferentiation, and cardiac fibrosis [101,102].
Comparative sequence-based coding and non-coding transcriptome profiling in pre- and post-LVAD myocardial tissue samples reported by Yang et al. revealed only modest changes in miRNA signatures [92]. They detected 160 (100 up- and 60 down-regulated) and 147 (106 up- and 41 down-regulated) differentially expressed miRNAs identified in the Ischemic and nonischemic patient groups, respectively. Notably, a small fraction of miRNAs (seven in ICM and eleven in NICM) exhibited significant post-LVAD improvement (Table 1), with only two of them in ICM (miR-365a-3p and miR-378a-3p) and five in NICM (miR-93-3p, miR-193b-5p, miR-425-5p, miR-548d-5p, and miR-760), normalizing to their near non-failing myocardium levels [92]. Their findings contrast Matkovich et al.’s results, which show more dramatic expression differences [89]. Paralleled mRNA transcriptome analysis of the same samples identified 2262 and 1929 differentially expressed mRNAs, respectively, with a small fraction (68 miRNAs in ICM and 45 miRNAs in NICM ) normalized during LVAD support. Despite these findings, the study encountered challenges distinguishing between advanced HF etiologies and differentiating pre- and post-LVAD samples using miRNA and mRNA profiles. Ultimately, while combined mRNA/miRNA profiles could differentiate non-failing from HF tissues, they were ineffective in segregating pre- and post-LVAD myocardial tissues from either ICM or NICM origins. This is also in sharp contrast with the study data of Matkovich et al., where combined miRNA and mRNA profiling provided distinctions between HF samples before and after LVAD support [89]. Contrarily, as clustering highly depends on the selected genes, later bioinformatic reanalysis of transcriptional data from this study, performed by Li et al., revealed distinct miRNA and mRNA expression signatures in both ICM and NICM samples compared with NF controls [94]. In their study, only 55 miRNAs, including miR-21-5p, miR-125b-1-3p, and miR-106b-5p, showed consistent upregulation in ICM and NICM. In contrast, 38 miRNAs, including miR-20a-5p, miR-17-5p, and let-7e-5p, were downregulated in both disease states. Similarly, 180 mRNA genes (135 up- and 45 down-regulated) showed the same differential expression pattern in ICM and NICM. Some of these genes are involved in miRNA-mRNA networks regulating the key HF disease pathways, such as Wnt signaling, calcium signaling, and cell cycle regulation [114]. The recent findings of Zhu et al. highlight the variations in the outcomes of these studies [103]. Based partly on sequencing data from Yang et al., their results indicate that complex lncRNA/circRNA-miRNA-mRNA networks regulate HF with different etiologies. These networks comprise pathways common to all HF types, suggesting a shared pathological mechanism for advanced HF. There are also distinct etiology type-specific regulatory networks as well as patient-specific regulatory pathways that manifest differently across individuals regardless of their underlying HF etiology [103]. Thus, besides the differences in platforms used for transcriptional profiling (i.e., microarray versus deep sequencing analysis), clinical characteristics of cardiomyopathy subtypes, and individual patient selection, applied bioinformatics tools can greatly influence the outcome of this and other profiling studies.
In addition to mRNA and miRNA sequencing, Yung et al. also conducted a comprehensive profiling of lncRNAs, thus revealing additional transcriptional complexity associated with HF and myocardial recovery following LVAD-induced mechanical unloading [92]. They detected 679 lncRNAs (569 up and 110 down-regulated) differentially expressed in ICM and 570 (438 up- and 132 down-regulated) in the NICM patient group. Among them, 55 lncRNAs in ICM (8.1%) and 56 in NICM (9.8%) were improved, while 26 lncRNAs in ICM (3.8%) and 30 in NICM (5.3%) were normalized with LVAD support [92]. Thus, a significantly higher proportion of differentially expressed lncRNAs in failing hearts were found to improve or normalized by LVAD support compared to the expression signature of miRs or mRNAs. Their clustering analyses showed that differences in lncRNA expression signatures, even with no stringent selection criteria applied, could effectively distinguish cardiomyopathies of different etiology and discern between myocardial samples obtained before and after LVAD treatment for either type of cardiomyopathy investigated [92]. Until now, this is the only report providing a comprehensive analysis of lncRNAs in failing human hearts before and after LVAD-induced mechanical unloading.
Similar to the results of Yang et al., comparative RNA-sequencing analysis of myocardial miRNAs in patients with end-stage HF before and at different postoperative time points after LVAD implantation reported by Akata et al. detected only marginal differences in miRNA expression between ICM and DCM hearts (Table 1) and no significant differences in miRNA cistron expression in the patient-matched myocardial samples taken before and after LVAD support [90]. Interestingly, the cardiac-specific mir-208a, mir-208b, and mir-499 were unaltered in either DCM HF or ICM HF compared to NF control. However, the study revealed that miRNA changes in HF tissue partially resemble those of the fetal myocardium. These results align with previous findings that reactivation of a fetal miRNA program may substantially contribute to gene expression alterations in failing human hearts. This insight could have significant implications for future research and clinical practice [98,104].
Muthiah et al. also detected no difference in the expression of myocardial miRNAs among LVAD-supported patient groups [93]. Their study explored the interplay between clinical, cellular, and transcriptional signatures of reverse myocardial remodeling in advanced HF patients. Following the LVAD support, no change in the extent of interstitial fibrosis was detected, probably due to the chronicity of HF (median 74 months). However, significant regression of cardiomyocyte hypertrophy (i.e., decreased cardiomyocyte cell size), improved LV and RV ejection fraction, decreased LV wall stress, and a considerable fall in circulatory NT-proBNP levels were observed. Nevertheless, despite these beneficial findings, there was an apparent disconnect between the molecular and functional phenotypes of myocardial recovery obtained with hemodynamic unloading since no significant fold-change in myocardial miRNA signatures was detected [93]. Notably, most of the miRNAs investigated in this study (Table 1) were previously associated with genomic control of cardiac hypertrophy. The lack of transcriptional adaptations following LVAD support was also confirmed by unsupervised hierarchical clustering and principle components analysis of miRNA expression signatures, which did not resolve etiology or implant/explant status in the primary or secondary cardiomyopathy patient groups. There was also no differential miR expression among patients separated according to their functional recovery status based on pre-defined criteria of LVEF improvement [93]. In light of these results, Eduardo Rame J’s comment suggests that in this study, and in general, the response to myocardial unloading may be predominantly mediated by long non-coding RNAs (lncRNAs) or metabolic interactions [105]. Other possibilities may be that the unsupported right ventricle continues to inform the heart’s overall cellular expression or that a miRNA phenotype of molecular reprogramming does take place with mechanical unloading but with limited durability [105]. The true answer may be somewhere in between and is currently unknown. As the available literature shows, this molecular functional phenotype dissociation also exists in other contexts of myocardial reverse remodeling and functional recovery following LVAD support that is not coupled to a reversal or normalization of the molecular phenotype is not an unusual finding.
Differential expression of miRNA signatures, although on a smaller scale, in LVAD-supported HF patients was also studied by Lok et al. [82]. They reported increased expression levels of miR-137 in cardiomyocytes and LV myocardial tissue stromal cells following LVAD support [82]. Also, the expression of miR-137 was inversely correlated with α-1-antichymotrypsin (ACT) mRNA levels in myocardial tissue. Subsequent analysis showed that miR-137 directly targets ACT, thus indicating their role in the pathophysiology of HF and reverse remodeling during mechanical support [82]. Similarly, Morley-Smith et al. reported enriched expression of miR-483-3p and miR-1202 in the pre-LVAD myocardium of end-stage HF patients with ICM and NICM etiology [85]. The LVAD support resulted in noticeable, although nonsignificant, up-regulation of miR-483–3p, while no change in myocardial miR-1202 expression was observed [85].
RNA sequencing study performed by Barsanti et al. also reported a significant change of miRNA signature in end-stage HF patients supported by LVADs, identifying 23 differentially expressed miRNAs [83]. However, their patient cohort involved only post-LVAD patients, while myocardial samples from HF patients undergoing heart transplants were used as controls. Nevertheless, hierarchical clustering showed that the obtained miRNA signatures were well distinguished between LVAD and control samples despite their wide intrinsic variability. Thirteen miRNAs (miR-23a-5p, miR-27a-5p, miR-142-3p, miR-142-5p, miR-144-5p, miR-146b-3p, miR-223-3p, miR-335-3p, miR-338-3p, miR-378g, miR-628-5p, miR-4461 and miR-5683) were found upregulated and ten downregulated (miR-29b-3p, miR-135a-5p, miR-216a-5p, miR-374b-5p, miR-376a-3p, miR-3195, miR-4284, miR-4485, miR-4532, and miR-4792) in the LVAD supported group [83]. The expression signatures of miR-29b-3p, miR-142-5p, miR-142-3p, miR-135a-5p, and miR-223-3p validated by qRT-PCR in patient subgroups correlated with sequencing data for all the tested miRNAs. A significant difference was confirmed for miR-135a-5p, miR-142-3p, and miR-223-3p, while the expression values of miR-142-5p and miR-29b-3p showed a clear, although nonsignificant, consistence with sequencing data [83]. Notably, miR-27a-5p, miR-142-3p, miR-142-5p, miR-223-3p, miR-338-3p, and miR-378g that were upregulated in the LVAD group were positively associated with cardiac index values assessed before LVAD implant and off-pump during the heart transplant [83]. Contrarily, miR-216a-5p, downregulated in the LVAD group, showed a negative association with the cardiac index values. As expected, the cardiac index values that measure improvement in heart function before and after LVAD support seemed to be significantly improved in the LVAD-supported group with respect to control patients. Furthermore, miR-29b-3p and miR-374b-5p showed a significant positive correlation with pulmonary vascular resistance, calculated as the ratio between pulmonary artery pressure and cardiac output values. The later measurements were available only for the patients in the control group. Nevertheless, both miR-29b-3p and miR-374b-5p were found to be downregulated in LVAD-supported compared to the control patient group [132]. In addition, in silico identification and pathway enrichment analysis of miRNAs correlated with cardiac index and pulmonary vascular resistance revealed a number of putative targets associated with hypertrophic and dilated cardiomyopathy, Wnt, PI3K-Akt, TGF-beta, and mTOR signaling pathway, regulation of actin cytoskeleton, ECM-receptor interaction, and cytokine-cytokine receptor interactions [83]. Interestingly, fourteen miRNAs analyzed in this study were previously identified by Akata et al. as differentially modulated in fetal myocardium compared to nonfailing postnatal hearts [90]. However, only four of them showed changes similar to those identified in LVAD-supported hearts, with miR-335-3p exhibiting upregulated and miR-29b, miR-135a, and miR-374b-5p downregulated expression in fetal myocardium compared to nonfailing postnatal hearts [90]. Furthermore, none of the miRNAs marked as improved or normalized by LVAD support in Yang et al. [92] was found to be significantly changed in this study [83]. On the contrary, miR-27a-5p, which was significantly upregulated in this study’s LVAD group, was also upregulated after LVAD implantation in Akat et al. [90]. In contrast, miR-216a was strongly upregulated both in tissue and plasma samples of advanced HF patients, as reported by Akat et al. [90], but was downregulated in this study, thus once again highlighting the discrepancies commonly encountered in corresponding transcriptome profiling studies [83].
Given their smaller, quieter design and superior clinical outcomes, continuous-flow LVAD devices (cf-LVAD) are now routinely used in clinical practice for short and long-term support in advanced HF. However, continuous-flow LVADs use a motor at fixed speeds, leading to constant blood ejection into the systemic circulation, while previously prevalent pulsatile-flow LVAD devices (pf-LVAD) mimic the natural rhythmic action of the heart. Some studies indicated that continuous-flow LVAD support devices may have a lower rate of left ventricular recovery associated with diminished pulsatility, increased pressure gradients on the aortic valve, and decreased compliance in smaller arterial vessels [106].
Lok et al. examined the differences in miRNA expression profiles affected by each type of LVAD device and their role in pathological cardiac remodeling [84]. They compared the miRNA expression patterns in pf-LVAD versus cf-LVAD-supported HF patients with NICM etiology [84]. The investigated miRNAs were selected based on the literature search and experimental microarray data from the test cohort of LVAD patients. Ultimately, 26 miRNAs were studied in the validation cohort using qRT-PCR methodology (Table 1). Of all the differentially expressed miRNAs in LVAD-supported myocardium, only 30% were common to both LVAD devices, with some showing the same expression pattern changes following mechanical unloading. The expression of miR-129* and miR-146a was significantly decreased in pre-LVAD-supported patients and increased during mechanical support. Conversely, miR-155, miR-221, and miR-222 were upregulated in pre-LVAD and decreased in the myocardium of the recovery hearts [84]. At the same time, miR-let-7i, miR-21, miR-378, and miR-378* changed significantly only during pf-LVAD support, with miR-let-7i and miR-21 showing decreased expression and miR-378 and amiR-378* increasing expression following device implantation. In cf-LVAD-supported hearts, let-7i and miR-21 showed the same trend as in pf-LVAD, but only miR-137 changed significantly, displaying elevated post-LVAD expression levels. However, comparing pre- and post-LVAD expression levels of these five miRNAs within individual patients showed no significant changes between pf- and cf-LVAD devices, thus indicating that observed myocardial changes or clinical outcomes specific to each device do not relate to differences in miRNA expression levels [84]. Previously, this group showed that the expression of miR-1, miR-133a, and miR-133b is partially restored during pf-LVAD support [80]. However, this was only observed in ICM patients and not in the NICM patient group. Similarly, results were reported in this study, showing no significant change in their expression in myocardial tissue of cf-LVAD-supported nonischemic patients with dilated cardiomyopathy [84].
Right ventricular failure (RVF) is a common complication of LVAD-supported HF patients, with early RVF (i.e., the first 30 days following LVAD placement) occurring in as high as 35 % of patients and late RVF (> 30 days post-LVAD implantation) that happens in 10 % of cases [107,108]. The RVF is associated with increased perioperative mortality, prolonged intensive care unit and hospital lengths of stay, higher rates of HF admission and hemocompatibility-related adverse events (HRAEs), and worse survival even after cardiac transplantation [107,108]. Several RVF risk scores have been developed, combining clinical, laboratory, and hemodynamic measurements [109,110,111]. Still, the pathological mechanisms of LVAD-related RVF’s pathophysiology remain poorly understood, and reliable biomarkers informing RV function have not been identified. Furthermore, RV adverse and reverse remodeling mechanisms differ in many ways from corresponding LV processes, so assumptions of function and molecular pathways based on studies of LV remodeling can be generalized and applied to the RV only to some extent [112,113]. Differences in protein-coding gene expressions between the RV and LV have been comprehensively studied in healthy and diseased hearts from animal models and human samples [113,114,115]. In contrast, data involving miRNA expression in the RV are still scarce, particularly in LVAD support settings [91,116,117].
Chamber-specific transcriptomic changes in end-stage HF patients subjected to heart transplantation were recently reported by Parikh et al. [91]. They used transmural LV and RV ventricular free wall (LVFW and RVFW) tissue samples explanted from the nonischemic hearts with dilated cardiomyopathy with and without LVAD support and nonfailing hearts for a microarray-based global mRNA and miRNA expression analysis. Histologically, both LV and RV of DCM hearts without LVAD support showed increased fibrosis and higher cardiomyocyte cross-sectional area compared to nonfailing myocardium, which was decreased following LVAD support. The cardiac enlargement was confirmed by significantly up-regulated mRNA levels of the brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC). However, the levels of these biomarkers were reduced following LVAD support [91]. The miRNA transcriptome analysis revealed 39 (Table 1) differentially expressed miRNAs (16 up-regulated and 23 down-regulated) in the LV tissue of DCM hearts without LVAD support compared to NF myocardium. In contrast, 19 miRNAs (Table 1) were differentially expressed (12 up-regulated and 7 down-regulated) in RV. Interestingly, six of these miRs, i.e., miR-10b, miR-95, miR-182, miR-373, miR-431, and miR-451, were detected in both LV and RV myocardium. They all showed the same mode of expression, with miR-431 showing downregulated, and miR-10b, miR-95, miR-182, miR-373, and miR-451 upregulated expression in both LV and RV of LVAD unsupported hearts compared to control NF myocardium [91]. Following LVAD support, twenty-one (54%) miRNAs in LV and five (26%) in RV showed a decrease in fold change compared to unsupported hearts (Table 1), while eight LV (21%) and seven RV miRNAs (37%) showed no change when compared to the NF control. However, only miR-4458 in the LV and miR-21*, miR-1972, and miR-4461 in the RV were normalized after LVAD implantation, thus indicating that LVAD-induced mechanical unloading has a more pronounced effect on miRNA normalization in the RV compared to the LV myocardium [91]. Both miR-4458 and miR-21* have been previously identified as negative modulators of cardiac hypertrophy. However, there are also reports of the pro-hypertrophic role of miR-21* in a model of angiotensin II-induced heart disease [91]. Paralled microarray analysis of mRNA transcripts revealed 922 differentially expressed mRNAs (392 up-regulated and 530 down-regulated ) in LVAD unsupported LV, whereas 858 mRNAs (238 up-regulated and 620 down-regulated) were differentially expressed in the RV compared to the NF myocardium. Among them, 567 differentially expressed mRNAs were commonly altered in both the LV and RV. A number of these differentially expressed mRNAs were found to be a target of eighteen miRNAs expressed in LV and thirteen in RV (Table 1), correspondingly. [91]. Following LVAD support, 617 mRNA transcripts in LV and 493 mRNAs in RV showed a decreased expression compared to NF myocardium. Also, 205 mRNAs (133 genes) in LV and 116 mRNAs (corresponding to 79 genes) in RV were significantly normalized. Enrichment analysis of these mRNA transcripts revealed their function in apoptosis, immunity-related processes, oxygen homeostasis, and regulation of the ECM composition. LVAD-induced mechanical unloading improved immune response in both ventricles, while oxygen homeostasis and ECM remodeling processes were normalized in LV [91]. This was accompanied by up-regulation of tissue inhibitors of matrix metalloproteinase (TIMP), thus leading to increased inhibition of matrix metalloproteinases (MMP) enzymes in the setting of an overall reduction of myocardial fibrosis [91]. Collectively, these results indicate a chamber-specific heart response to LVAD-induced mechanical unloading. However, no paired pre- and post-LVAD tissue samples were analyzed in this study, and inter-individual variability may still play a role in the observed alterations of post-LVAD miRNA and mRNA signatures.
LVAD therapy, as a bridge to heart transplant, has also become a part of standard care for pediatric end-stage HF patients unresponsive to medical management. The etiology and pathogenesis of HF among children are different from that of adult patients. ICM represents the primary cause of HF in adults. At the same time, congenital heart disease (CHD) and DCM are the most common reasons for HF and heart transplantation in the pediatric population [118]. The incidence and prevalence of pediatric HF is 0.87 to 7.4 per 100,000 people per year, and it is estimated that more than 11,000 children with HF are hospitalized each year [119]. The study of cardiac biomarkers in adult HF patients has exponentially increased over the last two decades. However, from a molecular point of view, pediatric HF disease represents a separate condition, and the data collected on adult HF patients cannot be easily translated into pediatric cases. Even so, there is a significant gap regarding biomarkers of HF in the pediatric population [120,121,122]. Also, little is known about the miRNA profiles in LVAD-supported pediatric HF patients.
Ragusa et al. were the first to report concomitant myocardial miRNA and mRNA expression profiles in paired pre- and post-LVAD samples collected from pediatric HF patients [86]. The next-generation sequencing (NGS) detected 437 miRs in each sample, but only six were differentially expressed (two up-regulated and four down-regulated) after LVAD support (Table 1). Following real-time PCR validation, only the expression of miR-19a-3p, miR-1246, and miR-199b-5p decreased significantly in post-LVAD samples, thus confirming the sequencing results. In silico assessment of their gene targets suggested that they are involved in cardiac protection (adiponectin system), neurohormonal cardiomyocyte regulation (angiotensin system, natriuretic peptide receptors, adrenergic system), and cardiac sarcomere function, i.e., the expression and regulation of cardiac troponins (cTns) [86]. Subsequent transcriptional analysis of troponin mRNAs in myocardial tissue revealed a significant increase in the expression of adult (cTnT3), fetal (cTnT4), and non-canonical (cTnT10/11/12 ) isoforms of cardiac Troponin T, adult cardiac troponin I (cTnI), and cardiac troponin C (cTnC) in pediatric HF patients following LVAD support. The fetal/slow skeletal troponin I (ssTnI) was also present, but no significant difference in its expression was detected between pre- and post-LVAD tissue [86]. In contrast, the analysis of troponin mRNAs in paired pre- and post-LVAD myocardium of adult HF patients resulted in a significant post-LVAD increase of all cTnT variants, as well as of cTnI and cTnC, while ssTnI mRNA was no longer expressed. When compared with pediatric HF cases, expression of cTnT1, cTnT3, cTnT4, cTnT 10/11/12, cTnI, and cTnC were significantly higher in adult HF cases, both in pre-LVAD and post-LVAD tissue. At the same time, no differences were found in ssTnI expression in pre-LVAD tissue, while post-LVAD ssTnI was still expressed only in pediatric HF cases [86]. Significantly, in pediatric HF patients, miR-19a-3p, miR-1246, and miR-199b-5p expression levels were negatively related to cTnT3, cTnT4, and cTnI [86]. In contrast, only miR-19a-3p and miR-1246h were negatively associated with expression levels of cTnC. Subsequent in vitro validation of miR targets in cardiac muscle cell line (HL-1) confirmed a down-regulatory effect only for miR-19a-3p on cTnC [86]. Therefore, it may be suggested that LVAD-induced changes in myocardial sarcomere organization through miR-mediated epigenetic regulation of cTn isoform expression may have a cardioprotective role in pediatric HF patients, potentially opening up new avenues for treating this condition. The same research group also reported the components of the adiponectin (ADPN) system, including ADPN and its receptors AdipoR1 and AdipoR2 and co-receptor T-cadherin (T-cad), as targets of miR-19a-3p, miR-199b-5p, and miR-1246 in pediatric HF patients [87]. They detected significantly higher ADPN plasma levels in pediatric HF patients than in healthy children, which were not modified after one month of LVAD support. However, no significant change in cardiac levels of ADPN between pre- and post-LVAD myocardial tissue was detected. Still, LVAD support increased the mRNA expression of ADPN receptors, AdipoR1, AdipoR2, and T-cad in post-LVAD myocardial tissue. Furthermore, myocardial AdipoR2 expression levels were inversely related to miR-19a-3p, miR-199b-5p, and miR-1246 expression, while miR-1246 was also negatively associated with T-cad. Contrarily, no relationship between AdipoR1 and any of the LVAD-modified miRNAs was observed. Subsequently, the in vitro validation of miR-mediated epigenetic regulation of the myocardial ADPN system in the cardiac muscle cell line (HL-1) confirmed the regulatory role of miR-1246 and miR-199b-5p on AdipoR2 and miR-199b-5p on T-cad [87]. The authors, therefore, hypothesize that LVAD-induced increase of the ADPN receptors in cardiac tissue from pediatric HF patients could counterbalance the impaired signaling and development of “ADPN resistance” observed among HF patients [87]. The study’s findings align with previous reports showing decreased expression of adiponectin AdipoR1 and AdipoR2 receptors in the myocardium of patients with advanced HF [123,124].
In summary, the pre- and post-LVAD myocardial miRNA signature exhibits significant variability across studies, mainly due to methodological differences, patient characteristics, type and time of LVAD support, and HF etiology. Some studies have reported differential expression of specific miRNAs associated with HF recovery and mechanical unloading linked with altered expression of their downstream targets, while others noted normalization or lack of significant change following LVAD support. Some miRNAs were found to be associated with cardiac index and pulmonary vascular resistance values or indicated in LVAD-induced changes in myocardial sarcomere organization and increased levels of AdipoR2 and T-Cad receptors, thus counterbalancing the so-called ADPN resistance. Others mirror their fetal gene expression signature or reflect the nature of HF in recovery and LVAD-dependent patient groups. Age (pediatric vs. adult HF cases) and chamber-specific myocardial miRNA responses to LVAD-induced mechanical unloading were also noted. Aside from the limited number of patients and controls included, a consistent finding across studies is that myocardial miRNA expression changes minimally with LVAD support, and no differences among device types (continuous-flow vs. pulsatile-flow LVADs) were detected. Accordingly, integrating miRNA profiling with mRNA data significantly enhanced the RNA signature’s ability to differentiate failing from non-failing hearts and classify the clinical status of HF patients.
The cellular diversity of cardiac tissue, with nine major cell types and more than twenty cell subtypes present within the human heart, and limited access to samples impede a more detailed characterization of molecular pathways and regulatory networks leading to HF. Recent advances in low-input RNA sequencing have allowed definitions of cellular transcriptomes at a single-cell resolution scale, thus offering new insights into the cellular foundation of cardiac homeostasis and disease processes [125,126]. Wang et al. demonstrated through single-cell RNA sequencing that patients receiving LVAD support exhibit differential transcriptional profiles linked to cardiac function improvement [127]. In one patient with enhanced function, their cell-specific gene expression profiles, particularly in endothelial cells, resembled those of non-failing control hearts, while in the second patient with no significant functional improvement, no such changes were observed across various cell types, including cardiomyocytes and fibroblasts [127]. It is reasonable to expect that when applied to ncRNA analysis, this approach will greatly facilitate the discovery of cell and HF type-specific miRNA profiles underlying myocardial reverse remodeling, remission, and recovery.
6. Biomarker Potential of Circulating miRNAs Associated with LVAD-Induced Mechanical Unloading
Many miRNAs released from cells upon apoptosis, necrosis, or secretion activity are found in the blood and other body fluids. They are surprisingly stable and bound to RNA-binding protein complexes and lipoprotein particles or loaded into extracellular vesicles, including apoptotic bodies, microvesicles, endosomes, and exosomes. Taken up by recipient cells, they may participate in paracrine and endocrine regulation of targeted genes [128]. Their abnormal expression is associated with various disease processes, making them promising novel noninvasive diagnostic markers and thus highlighting their potential in clinical applications.
Several studies have reported differential expression of circulatory miRs in LVAD-supported end-stage HF patients [84,85,90,129,130,131,132,133,134]. Their findings are summarized in Table 2.
Akat et al. conducted a comprehensive transcriptional analysis of small circulatory RNAs from healthy controls and patients with stable and advanced HF, revealing a striking up to 140-fold increase in heart and muscle-specific miRs in advanced HF compared to healthy individuals, alongside elevated cardiac troponin I (cTnI) levels indicating myocardial injury [90]. Notably, the changes in circulatory levels of heart and muscle-specific miRs nearly completely reversed three and six months following the initiation of LVAD support. Conversely, compared with healthy controls, circulating miRNAs in patients with stable HF showed less than fivefold differences, while heart and muscle-specific miRs and cTnI were only captured near the detection limit. In healthy subjects, only a few miRs, including mir-122 from the liver and miR-1-1 from the muscle, were among the top 85% of sequence reads. Conversely, the combined abundance of circulatory heart and muscle-specific miRs was less than 0.1%. However, it increased to over 1% in advanced HF patients with cardiac-specific miR-208b, miR-208a, and miR-499, and the muscle-specific miR-1-1 and miR-133b showing 143-, 78-, 28-, 18-, and 21-fold higher expression at LVAD implantation, respectively [90]. The relative abundance of circulatory heart-specific miR-208b, miR-208a, and miR-499 followed the ratio determined in heart tissue, and it was consistent with it being their only source. Interestingly, mir-208a, mir-208b, and mir-499 myocardial signatures were unaltered in either DCM or ICM patients compared to NF myocardium [90]. There was also a 25-fold increase in circulatory miR-216a levels in advanced HF patients compared to controls. Still, as confirmed by absolute amounts and individual–paired samples analysis, this rise in circulatory miR-216a was more likely linked to its release by endothelial cells, which express miR-216a at higher levels than the whole heart tissue [90]. Following LVAD implantation, miR-208b, miR-208a, miR-499, miR-1-1, and miR-133b declined as early as three months after LVAD support, nearly approaching their control values. However, at LVAD explantation, their levels rose again with alterations comparable in magnitude to those observed at the time of device implantation [90]. Conversely, the circulatory heart and muscle-specific miRs levels in stable HF patients were comparable to those in healthy controls. In this case, the highest difference was noted regarding the 5-fold increase in miR-375 and a 4-fold decrease in miR-203. In addition, miR-210, miR-1908, and miR-1180 showed similar changes in patients with advanced and stable HF, showing a two-fold [miR-210, miR-1908] and four-fold [mir-1180] higher expression compared to healthy controls [90]. Finally, higher levels of the heart-specific miR-208a, miR-208b, and miR-499 were positively correlated with the protein levels of cTnI, indicating myocardial injury. In contrast, no correlation was established between the circulatory levels of miR-208a, miR-208b, and miR-499 with those of B-type natriuretic peptide (BNP) [90].
No association of circulatory post-implant miRNA levels with plasma levels of NT-proBNP or any other protein biomarker examined was reported by Wang et al. [131]. They investigated the expression of twenty-three miRNAs (Table 2), previously shown to play a role in HF pathogenesis , in peripheral blood samples isolated from end-stage HF patients before and after cfLVAD-induced mechanical unloading. Subsequently, nineteen miRNAs that demonstrated adequate expression in more than 80% of samples were selected for further analysis. Except for miR-155, all plasma levels remained relatively unchanged following LVAD implantation. Among them, seven miRs (miR-21, miR-92a, miR-103, miR-159a, miR-195, miR-320, and miR-423) were downregulated compared to their pre-LVAD values. The most extensive reduction was observed for miR-103. Still, no statistical significance was demonstrated for any of them. In contrast, twelve miRNAs (miR-15b, miR-16, miR-24, miR-27a, miR-29a, miR-126, miR-133a, miR-146a, miR-146b, miR-155, miR-221, and miR-222) were upregulated with miR-126 and miR-155 showing the largest increase following LVAD implantation. However, only the elevation of miR-155 plasma levels was found to be statistically significant [131]. Notably, the observed miRNA signatures were not associated with improvements in the plasma levels of preselected HF biomarkers, i.e., NT-proBNP, galectin-3, sST2, GDF-15, and NGAL [131]. Also, pre-LVAD miRNA levels stratified by tertiles of baseline NT-proBNP showed no significant difference between patients in the highest NT-proBNP tertile (accompanied by more extended length of stay, higher degrees of renal insufficiency, poorer cardiac index values, and greater elevations of galectin-3, sST2, GDF-15, and NGAL), and those in the lowest tertile. Furthermore, when the changes in plasma miRs levels following LVAD implantation were analyzed according to prespecified changes [≤25% decrease (no improvement) vs. >25% decrease (improvement)] in the levels of galectin-3, NT-proBNP, and sST2, no marked difference was observed. Also, the levels of miR-155, mir-27a, miR-126, miR-133a, and miR-146b were upregulated in both cohorts of HF patients with and without post-implant improvement in galectin-3, NT-proBNP, and sST2 levels [117].
Contrarily, Morley-Smith, and colleagues detected early and sustained up-regulation of circulating miR-483-3p expression levels in advanced HF patients with ischemic and nonischemic disease etiology measured at baseline (pre-LVAD) and at different times following LVAD implantation that mirrored the reduction in N-terminal proBNP (NT-proBNP) plasma levels and improvement of LV function [85]. Also, the baseline levels of circulating miR-1202 measured in their patient cohort correlated well with change in NT-proBNP levels at three months of LVAD support, even stratifying the patient cohort into poor versus good responders to LVAD induced mechanical unloading with greater accuracy than either baseline NT-proBNP or patients pre-operative INTERMACS profile. Initially, PCR microarray analysis showed differential, predominantly upregulated expression of twelve circulatory miRNAs (Table 1) six months following LVAD implantation, but after real-time qPCR validation, only miR-483-3p and miR-1202 were confirmed for further screening in a larger cohort of LVAD patients. Further, both miRNAs were enriched in the myocardium, suggesting the heart as the possible source of their plasma fraction. Contrary to miR-483-3p, plasma levels of miR-1202 showed only small changes from baseline at all time points following LVAD implantation in the test cohort. Considering all plasma samples examined, there was a trend toward a correlation between plasm expression of miR-1202 and NT-proBNP levels. However, no such correlation between miR-483-3p and NT-proBNP was established. No direct correlation between plasma and myocardial levels of miR-483-3p and miR-1202 and between either miRNA and HF etiology, duration of LVAD support, or patient gender was established. Nevertheless, these results indicate circulatory levels of miR-483-3p and miR-1202 as potentially valuable noninvasive biomarkers for monitoring (miR-483-3p) and predicting (miR-1202) patient response to LVAD therapy that warrants further evaluation in a larger patient cohort for potential clinical use [85].
Qian et al. also assessed the diagnostic potential of circulatory miRNAs associated with HF and LVAD-induced mechanical unloading [130]. They performed a real-time qPCR-based analysis of preselected eighty-nine (Table 2) human cardiovascular-related miRs [130]. As a result, miR-30a-5p, miR-100, miR-499b-3p, miR-320a, and miR-433, previously upregulated in HF compared to control, showed significant downregulation following LVAD implantation. Conversely, the levels of miR-654-5p, previously downregulated in HF, were upregulated compared to their pre-LVAD values [130]. The rest of the miRNAs in the screening panel were not significantly associated with LVAD-induced mechanical unloading. Notably, all HF patients selected for this screening analysis of paired plasma samples obtained before and after LVAD implantation (i.e., discovery cohort) had at least a 30 % decrease in circulatory NT-proBNP levels following device implantation. The diagnostic potential of circulating miR-30a-5p and miR-654-5p as the most significantly changed miRNAs upon treatment was further tested in independent training and validation cohort of HF patients, and the best-suited two-circulating miRNA model (miR-30a-5p/miR-654-5p), with the highest sensitivity (98.75%) and the best specificity (95.0%), was chosen for potential HF diagnosis [130]. Notably, the two-circulating miRNA model achieved somewhat higher prognostic accuracy in diagnosing HF patients when compared with the traditional approach using NT-proBNP serum levels, showing a 1.25 % versus 2.5 % false negative rate achieved by the NT-proBNP approach [130]. Consistent with the results in the discovery phase, a significant elevation of miR-30a-5p and downregulation of miR-654-5p was confirmed in plasma samples of both training and validation HF cohorts compared with control. A similar expression pattern was observed regarding the patient’s pathologic characteristics, i.e., NYHA Functional Classification (I+II vs. III+IV) and cardiomyopathy subtypes (ICM vs. NICM vs. other) showing upregulation of miR-30a-5p and downregulation of miR-654-5p in each subgroup compared to control. Still, no difference in their expression between the NYHA classification subgroups or cardiomyopathy types was observed [130]. Additional comparison of circulatory miR-30a-5p and miR-654-5p levels with plasma NT-proBNP values showed a significant negative correlation of miR-654-5p and a positive correlation of miR-30a-5p with NT-proBNP plasma levels of HF patients enrolled in the discovery phase [130]. Also, more prominent changes in circulatory miR-30a-5p and miR-654-5p levels, especially regarding miR-654-5p, were observed in the HF patient subgroup with higher NT-proBNP reduction, thus further suggesting their regulation by LVAD-induced mechanical unloading. The analysis of the external dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52601) previously reported by Akat et al., further verified these results [90,130]. Specifically, circulatory miR-30a-5p levels declined while miR-654-5p increased in the plasma of end-stage HF patients following LVAD support, thus approaching its control levels, which was associated with improved myocardial function [130]. Furthermore, an overlapping set of targeted genes (eleven for miR-30a-5p and nine for miR-654-5p) was detected, thus suggesting the role of these miRNAs in the regulation of HF-related pathways such as apoptosis, p53 signaling, viral myocarditis, atherosclerosis, and cardiomyocyte adrenergic signaling. Altogether, these results indicate that circulatory miRs may have a high sensitivity and specificity for diagnosing HF across a variety of patient characteristics, as opposed to NT-pro-BNP levels, which may be altered by factors such as age, obesity, and renal function [135]. Also, as epigenetic regulators, miRNAs can be directly linked to various cellular mechanisms and HF-related molecular pathways, thus offering insight into disease pathophysiology and progression that is unavailable by NT-pro-BNP or other protein biomarkers.
Lok et al. reported differential expression of circulatory miR-21, miR-146a, miR-221, and miR-222 in blood samples of end-stage HF patients at different times of LVAD support, i.e., before and 1, 3, and 6 months after cf-LVAD implantation and prior to transplantation [84]. The time response in miR-21 expression levels was homogenous between all the patients. There was a two-fold upregulation in pre-LVAD samples and decreased expression following LVAD support but without normalization to its control values. In contrast, the miR-146a, miR-221, and miR-222 showed discrepant expression patterns between the patients. Levels of miR-146a were increased in pre-LVAD blood samples but revealed a fluctuating expression following device implantation with a tendency to decrease. A fluctuating expression pattern was also noted for circulatory miR-221 and miR-222, showing elevated expression after one month of LVAD support, followed by their down-regulation [84].
Alteration in circulatory miRNA levels may also be associated with the levels of blood cells in advanced HF. Adhikari et al. reported that miR-16, a member of miR-15/107 family (i.e., miR-15 -16, -195, -497, -103, -107, and miR-503) and an important regulator of cardiomyocyte proliferation, survival, and angiogenesis which is highly enriched in peripheral blood cells, targets vacuolar protein sorting 4a (VPS4a) [136]. However, in vitro experiments confirmed that miR-16 does not bind efficiently to VPS4a carrying the SNP, rs16958754. Furthermore, the expression levels of circulatory miR-16 were found decreased, while those of VPS4a were increased in the peripheral circulation of end-stage HF patients, thus lowering the number of platelets and reticulocytes [129]. Other tested miR-15/107 family members, i.e., miR-15a, miR-195, and miR-103, were also downregulated in the peripheral circulation of end-stage HF [129]. Also, LVAD support leads to a further reduction in circulatory miR-15a and miR-195 levels. Contrarily, the circulatory miR-16 levels were upregulated seven days following LVAD support, leading to decreased expression of VPS4a. Compared to blood, the expression of VPS4a, miR-16, miR-15a, miR-195, and miR-103 showed no change in the heart tissue from end-stage HF compared to non-failing controls [129]. Overall, these results suggest a potential role for the miR-16/VPS4a signaling pathway in regulating platelet and/or reticulocyte numbers in HF.
The arterial pulse is essential to cardiovascular homeostasis. Therefore, the potential negative impact of cf-LVAD devices on peripheral vascular function, leading to vessel wall damage and endothelial dysfunction, is also a matter of significant interest [136]. Dlouha et al. explored dynamic changes in plasma levels of flow-sensitive miRs involved in vascular remodeling (miR-21, miR-146a, miR-146b, and miR-155) and endothelial dysfunction (miR-10a, miR-10b, miR-126, miR-663a, miR-663b) in response to the long-term support by cf-LVAD devices [132]. Blood samples were collected at baseline from two control groups, i.e., NF subjects and stable HF patients on optimal therapy, and from cf-LVAD-supported end-stage HF patients before the LVAD implantation and at follow-up time points 3, 6, 9, and 12 months after implantation. Although the markers of cardiac damage, inflammation, and renal function steadily improved during the LVAD support, no significant association between miRNA levels and individual clinical events or hemodynamic parameters was observed. The baseline plasma levels of miR-21, miR-146b, miR-155, miR-663a, and miR-663b were significantly higher in LVAD patients than in nonfailing controls, probably reflecting the severity of HF accompanied by low flow status and endothelial dysfunction [118]. Subsequently, during the follow-up period after LVAD implantation, the plasma levels of miR-146b, miR-155, and miR-663b were upregulated, but no significant change in their expression was observed. Conversely, the plasma levels of cardiac enriched miR-21 showed a decreasing trend compared to healthy controls, possibly demonstrating the ventricular regenerative response to mechanical unloading (. The trend towards stable, lower plasma levels of miR-10a and miR-10b in the end-stage HF patients following LVAD support was also observed. However, the plasma levels of miR-126 and miR-146a were the only ones that changed significantly following LVAD support, showing increased expression compared to both control groups. A significant difference in miRNA expression was also detected between the end-stage HF patients with ischemic and nonishemic etiology, with ICM patients showing higher miR-155 plasma levels in the 3rd and miR-10b, miR-21, miR-146a, and miR-146b, at the 12th month following cf-LVAD implantation. Also, a positive association was observed between plasma levels of miR-155 and Belcaro score, i.e., semiquantitative classification of preclinical atherosclerosis. Conversely, circulatory miR-126 showed an inverse correlation with endothelial function, measured as the reactive hyperemia index value. These results indicate that long-term, pulse-diminished cf-LVAD support upregulates plasma levels of circulating flow-sensitive miRNAs, contributing to endothelial dysfunction and vascular remodeling.
The potential role of circulating miRs as biomarkers of HF was also investigated in pediatric patients [137]. So far, Ragusa et al. were the only ones to evaluate the effects of LVAD-induced mechanical unloading on circulatory miRNA levels in HF children [133]. Their initial NGS screening of pediatric HF patients before and one month after pf-LVAD implantation detected the expression of 340 circulatory miRs, with 169 of them present in all tested samples. Although hierarchical clustering couldn’t batch them in the pre- and post-LVAD group, thirteen circulatory miRs (Table 2) were expressed differently before and after LVAD implantation. Specifically, nine of these circulatory miRs were downregulated (miR-30a-5p, miR-127-3p, miR-409-3p, miR-432-5p, miR-483-3p, miR-483-5p, miR-485-3p, miR-3135b, and miR-4433b-3p) and four upregulated (miR-16-5p, miR-150-3p, miR-375, and miR-941) one month following LVAD implantation [133]. The validation and subsequent time course real-time qPCR analysis in a larger patient group confirmed the sequencing data for six miRNAs. Namely, miR-409-3p, miR-483-3p, and miR-485-3p were downregulated up to undetectable levels, while miR-432-5p showed a trend to decrease. At the same time, the circulatory levels of miR-150-3p and miR-375 increased after one month of LVAD implantation [133]. Later in silico analysis indicated that all six miRs were involved in the synergic regulation of several factors affecting platelet activation and blood coagulation cascade. However, in vitro studies confirmed the regulatory effect of only miR-409, causing the downregulation of thrombin (F2) and coagulation factor 7 (F7). Of note, all patients in their study had thrombotic events requiring LVAD changes during the first three months after device implantation. These results indicate a potential role for circulatory miRs, particularly 409-3p, as an early biomarker for monitoring hemostasis-related adverse events in pediatric LVAD-supported HF patients.
Bleedings and thromboembolic events represent one of the leading long-term complications among LVAD-supported patients. The underlying molecular mechanisms are still not fully known. We have already seen that patients with cf-LVAD support are characterized by increased endothelial dysfunction, especially in microvasculature. This, in turn, decreases the bioavailability of nitric oxide, creates favorable conditions for platelet and leukocyte activation and adhesion, and triggers an inflammatory response that finally leads to an increased thromboembolic risk. The balance between bleeding and thrombosis is vital in managing LVAD-supported patients. Patients do not typically fit a bleeding or prothrombotic profile, but they instead move along the bleeding/thrombotic spectrum with an increased risk of thromboembolic event following an episode of bleeding or an increased risk of bleeding episode as a result of a prior thrombotic event [138,139].
Platelets are the essential mediators of hemostasis. Their activation, adhesion, and aggregation upon tissue injury stimulate coagulation factors and other mediators that aid in hemostasis and wound healing [140]. Emerging evidence also supports the pivotal role of platelets in angiogenesis, inflammation, tissue repair, and regeneration [140,141]. Besides, these small anucleated cells inherit many mature proteins, mRNAs, and non-coding RNAs from their parent megakaryocytes while gradually forming in the bone marrow. They can also take up a part of their RNA cargo, including ncRNAs, from contacting cells or the circulating blood and process the inherited and ingested RNAs into fully functional miRs [140,141]. In addition, many platelet-derived ncRNAs may be stored in platelet secretory granules and secreted [i.e., in the form of platelet-derived microvesicles (PMVs) or other membrane encapsulated extracellular vesicles (EVs)] after platelet activation following endothelial injury, infection, and/or metabolic disorders, thus altering the gene expression in a nearby or distant recipient cells [140,141]. As such, they can be involved in a series of pathological processes, including dysregulation of endothelial homeostasis, smooth muscle cell migration, and proliferation, fibroblast proliferation, alteration of ECM, inflammatory cell infiltration, and cardiomyocyte apoptosis, leading, among others, to myocardial damage, and adverse cardiac remodeling [140,141]. Therefore, studying platelet-derived miRNAs might be beneficial in clinical settings, revealing a new set of therapeutic targets and more specific noninvasive diagnostic and prognostic biomarkers of HF and other cardiovascular diseases [142].
Lombardi et al. investigated the involvement of platelet-enriched miRs in hemocompatibility-related adverse events, i.e., surgical bleeding and thromboembolic complications, among LVAD-supported HF patients. [134]. They reported differential expression of several well-known platelet-enriched miRs (Table 2) whose levels were measured in platelet-rich (PRP) and platelet-poor (PPP) plasma fractions before the LVAD implantation and longitudinally at different time points of LVAD support. Six miRs were found differentially expressed between pre-LVAD and control plasma samples, with miR-126-5p and miR-374b-5p significantly downregulated in PRP and PPP fractions and miR-223-3p in PRP, respectively, while miR-20b-5p and miR-451a were found decreased in platelet-poor plasma samples. Conversely, the levels of miR-320a-3p were higher in preimplant PRP samples compared to the control blood tissue [134]. The expression levels of miR-223-3p and miR-320a-3p of the same pattern as in the PRP also reached borderline significance in the PPP fraction. Notably, the expression levels of all these miRNAs were higher in the PRP fraction of all the samples analyzed, thus supporting their platelet origin with the parallel existence of a minor circulating fraction potentially generated by thrombocyte activation and carried through circulation by lipoproteins of microvesicles. Interestingly, the pre-LVAD patient group exhibited significantly higher miR-20b-5p levels and lower expression of miR-25-3p in the ischemic versus nonischemic patient cohort [134]. Furthermore, a substantial elevation of miR-19b-3p in the chronic versus acute HF patients was noted. Also, a lower expression of miR-144-3p was detected among the patients with an Impella device installed before the beginning of LVAD support. Collectively, these data indicate the existence of different miRNA-related platelet response that depends on HF etiology, disease duration, and concomitant treatment modalities [134]. LVAD installment significantly altered miRNA expression levels. Specifically, during the first twelve months following the implantation, there was a progressive rise in the miR-25-3p and miR-451a levels (reaching significance in the 6th or 12th-month time point of LVAD support), possibly connected to a parallel increase in thrombocyte activation. However, no similar time pattern was observed for miR-320a-3p and miR-144-3p, known factors of platelet activity ). That, on the other hand, could be the sign of only partial or transient thrombocyte activation [134]. This apparent lack of a clear platelet miRNA expression pattern may represent constant thrombocyte adaptation to the evolving clinical picture and local or systemic tissue changes triggered by LVAD-induced mechanical unloading. Notably, the miR-126 expression levels in PRP plasma fraction approximately 12 months [480 (416–512) days] following LVAD implantation showed a good correlation with the levels of D-dimers and AT-III and the LVEF values as the left ventricular function metrics [134]. Interestingly, the HF patients suffering from post-implant bleeding had significantly higher pre-implant PRP levels of hemostasis-related miR-151a-3p and miR-454-3p compared to patients with no HRAE episodes. Moreover, altered expression of miR-151a-3 and miR-454-3p was noted in these patients earlier before the occurrence of bleeding, with miR-151a-3p showing more than five-fold higher and miR-454-3p more than eight hundred lower expressions in bleeders vs. non-bleeders at approximately six-month [210 (184–223) days] following LVAD implantation. Nonetheless, no difference in the expression of these miRNAs between the bleeders and the control patient group was noted [134]. In silico analysis and literature review revealed that miR-25, miR-144, miR-320, and miR-451a share molecular pathways, including that of peroxisome proliferator-activated receptor (PPAR), a known regulator of platelet activation and TNF-α and mammalian target of rapamycin (mTOR), which are involved in bleeding. Together, these data indicate a dynamic re-programming of platelet miRs during LVAD support, possibly revealing the existence of a platelet miRs signature with potential implications for clinical practice, i.e., to identify LVAD patients at high risk for postoperative bleeding and related HRAE events.
As can be seen, transcriptional profiling of circulatory and platelet miRs in pediatric and adult LVAD-supported HF patients, although not consistent in the data so far provided, may reveal potential clinically useful diagnostic and prognostic noninvasive biomarkers for monitoring and estimating patient response to LVAD therapy, the occurrence of myocardial injury, endothelial dysfunction, and vascular remodeling, or predicting of hemocompatibility-related adverse events associated with LVAD induced mechanical unloading. Nonetheless, as in the case of myocardial miRNA profiling, a relatively small number of patients were investigated, and the differences in methodologies applied, patient characteristics, time of LVAD support, and HF etiology still preclude gathering more reliable and consistent data clinically applicable for specific HF etiology, patient age, and concurrent treatment modalities.
7. Conclusions
Transcriptional profiling of cardiac, circulatory cell-free, and platelet-enriched miRNAs in pediatric and adult LVAD-supported HF patients shows promise in identifying cellular and molecular pathways underlying myocardial reverse remodeling, remission, and recovery and for developing clinically useful noninvasive biomarkers with high sensitivity and specificity for monitoring and predicting patient response to LVAD therapy. MiRNA-based diagnostic and prognostic tools will likely help improve patient stratification to adequately select the end-stage HF patients eligible for LVAD placement and those more suitable for alternative clinical interventions, thereby significantly impacting patient care.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
F.P and K.L.W – Design of the study; Writing original draft– review & editing, Conceptualization; G.I - Writing – review & editing, Conceptualization. All the authors have contributed to and approved the final version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 8-OHdG | 8-hydroxy-2’-deoxyguanosine |
| ACC/AHA | American College of Cardiology/American Heart Association; |
| ACE | angiotensin-converting enzyme |
| ADM | adrenomedullin |
| ADMA | asymmetric dimethyl arginine |
| ARBs | angiotensin receptor blockers |
| ARN | angiotensin receptor/ neprilysin inhibitors |
| BDNF | brain-derived neurotrophic factor |
| BNP | brain natriuretic peptide |
| BTR | bridge to recovery |
| BTT | bridge to transplantation |
| CA125 | cancer antigen 125 |
| cf-LVAD | continuous flow LVAD devices |
| CHD | congenital heart disease |
| CRP | C-reactive protein |
| CRT | cardiac resynchronization therapy |
| DCM | dilated cardiomyopathy |
| DT | destination therapy |
| ET-1 | endothelin 1 |
| FGF-21 | fibroblast growth factor 21 |
| GAL-3 | galectin-3 |
| GDF-15 | growth differentiation factor 15 |
| GSTP1 | glutathione transferase P1 |
| HCM | hypertrophic cardiomyopathy |
| HF | heart failure |
| H-FABP | heart-type fatty acid-binding protein |
| HFmrEF | heart failure with mildly reduced ejection fraction |
| HFpEF | heart failure with preserved ejection fraction |
| HFrEF | heart failure with reduced ejection fraction |
| hs-cTnT/CTnI | high-sensitivity cardiac troponins cTnT and cTnI |
| HTx | heart transplantation |
| HTx-ctrl | heart transplant control |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| ICM | ischemic cardiomyopathy |
| IL-1 | interleukin |
| IL-1 RA | interleukin-1 receptor antagonist |
| IL-15 | interleukin 15 |
| IL-18 | interleukin 18 |
| IL-33 | interleukin 33 |
| IL-6 | interleukin 6 |
| IL-8 | interleukin 8 |
| INTERMACS | interagency registry for mechanically assisted circulatory support |
| KIM-1 | kidney injury molecule-1 |
| lncRNA | long non-coding RNA |
| LV | left ventricle |
| LVAD | left ventricular assist device |
| LVEF | left ventricular ejection fraction |
| LVNC | LV non-compaction |
| MCS | mechanical circulatory support |
| MMP-1 | matrix metalloproteinase 1 |
| MMP-2 | matrix metalloproteinase 2 |
| MMP-9 | matrix metalloproteinase 9 |
| MPO | myeloperoxidase |
| MRA | mineralocorticoid receptor antagonist |
| MR-proADM | mid-regional pro-adrenomedullin |
| NF | non-failing control |
| NGAL | neutrophil gelatinase-associated lipocalin |
| NGS | next generation sequencing |
| NICM | nonischemic cardiomyopathy |
| NO | nitric oxide |
| NT-proBNP | N-terminal brain natriuretic pro-peptide |
| NTproCNP | N-Terminal pro C-Type Natriuretic Peptide |
| NYHA class | New York Heart Association functional classification for heart failure |
| Ox LDL | oxidized low-density lipoprotein |
| pf-LVAD | pulsatile flow LVAD devices |
| PICP | procollagen type I carboxyterminal peptide |
| PIIINP | pro-collagen type III amino terminal peptide |
| qPCR | quantitative real-time PCR |
| RMC | restrictive cardiomyopathy |
| RV | right ventricle |
| SE | standard error |
| SGLT2 | sodium-glucose linked cotransporter 2 |
| sST2 | soluble suppression of tumorigenesis-2 |
| TIMP-1 | tissue inhibitor of matrix metalloproteinase 1 |
| TNF-α | transforming growth factor alpha |
| VCAM-1 | vascular cell adhesion protein 1 |
| vWF | Von Willebrand factor |
| α1-AT | alpha-1 antitrypsin |
References
- Bozkurt, B., Coats, A. J. S., Tsutsui, H., Abdelhamid, C. M., Adamopoulos, S., Albert, N., Anker, S. D., Atherton, J., Böhm, M., Butler, J., Drazner, M. H., Michael Felker, G., Filippatos, G., Fiuzat, M., Fonarow, G. C., Gomez-Mesa, J. E., Heidenreich, P., Imamura, T., Jankowska, E. A., Januzzi, J., Khazanie, P., Kinugawa, K., Lam, C. S. P., Matsue, Y., Metra, M., Ohtani, T., Francesco Piepoli, M., Ponikowski, P., Rosano, G. M. C., Sakata, Y., Seferović, P., Starling, R. C., Teerlink, J. R., Vardeny, O., Yamamoto, K., Yancy, C., Zhang, J. & Zieroth, S. (2021). Universal definition and classification of heart failure: a report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur J Heart Fail. 23, 352-380. [CrossRef]
- Khan, M. S., Shahid, I., Bennis, A., Rakisheva, A., Metra, M. & Butler, J. (2024). Global epidemiology of heart failure. Nat Rev Cardiol. [CrossRef]
- Savarese, G., Becher, P. M., Lund, L. H., Seferovic, P., Rosano, G. M. C. & Coats, A. J. S. (2023). Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. 118, 3272-3287. [CrossRef]
- Shahim, B., Kapelios, C. J., Savarese, G. & Lund, L. H. (2023). Global Public Health Burden of Heart Failure: An Updated Review. Card Fail Rev. 9, e11. [CrossRef]
- Stewart, S., MacIntyre, K., Hole, D. J., Capewell, S. & McMurray, J. J. (2001). More ’malignant’ than cancer? Five-year survival following a first admission for heart failure. Eur J Heart Fail. 3, 315-322. [CrossRef]
- González, A., Richards, A. M., de Boer, R. A., Thum, T., Arfsten, H., Hülsmann, M., Falcao-Pires, I., Díez, J., Foo, R. S. Y., Chan, M. Y., Aimo, A., Anene-Nzelu, C. G., Abdelhamid, M., Adamopoulos, S., Anker, S. D., Belenkov, Y., Ben Gal, T., Cohen-Solal, A., Böhm, M., Chioncel, O., Delgado, V., Emdin, M., Jankowska, E. A., Gustafsson, F., Hill, L., Jaarsma, T., Januzzi, J. L., Jhund, P. S., Lopatin, Y., Lund, L. H., Metra, M., Milicic, D., Moura, B., Mueller, C., Mullens, W., Núñez, J., Piepoli, M. F., Rakisheva, A., Ristić, A. D., Rossignol, P., Savarese, G., Tocchetti, C. G., Van Linthout, S., Volterrani, M., Seferovic, P., Rosano, G., Coats, A. J. S. & Bayés-Genís, A. (2022). Cardiac remodelling - Part 1: From cells and tissues to circulating biomarkers. A review from the Study Group on Biomarkers of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 24, 927-943. [CrossRef]
- Aimo, A., Vergaro, G., González, A., Barison, A., Lupón, J., Delgado, V., Richards, A. M., de Boer, R. A., Thum, T., Arfsten, H., Hülsmann, M., Falcao-Pires, I., Díez, J., Foo, R. S. Y., Chan, M. Y. Y., Anene-Nzelu, C. G., Abdelhamid, M., Adamopoulos, S., Anker, S. D., Belenkov, Y., Ben Gal, T., Cohen-Solal, A., Böhm, M., Chioncel, O., Jankowska, E. A., Gustafsson, F., Hill, L., Jaarsma, T., Januzzi, J. L., Jhund, P., Lopatin, Y., Lund, L. H., Metra, M., Milicic, D., Moura, B., Mueller, C., Mullens, W., Núñez, J., Piepoli, M. F., Rakisheva, A., Ristić, A. D., Rossignol, P., Savarese, G., Tocchetti, C. G., van Linthout, S., Volterrani, M., Seferovic, P., Rosano, G., Coats, A. J. S., Emdin, M. & Bayes-Genis, A. (2022). Cardiac remodelling - Part 2: Clinical, imaging and laboratory findings. A review from the Study Group on Biomarkers of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 24, 944-958. [CrossRef]
- Cohn, J. N., Ferrari, R. & Sharpe, N. (2000). Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 35, 569-582. [CrossRef]
- Zeglinski, M. R., Moghadam, A. R., Ande, S. R., Sheikholeslami, K., Mokarram, P., Sepehri, Z., Rokni, H., Mohtaram, N. K., Poorebrahim, M., Masoom, A., Toback, M., Sareen, N., Saravanan, S., Jassal, D. S., Hashemi, M., Marzban, H., Schaafsma, D., Singal, P., Wigle, J. T., Czubryt, M. P., Akbari, M., Dixon, I. M. C., Ghavami, S., Gordon, J. W. & Dhingra, S. (2018). Myocardial Cell Signaling During the Transition to Heart Failure: Cellular Signaling and Therapeutic Approaches. Compr Physiol. 9, 75-125. [CrossRef]
- González, A., Schelbert, E. B., Díez, J. & Butler, J. (2018). Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J Am Coll Cardiol. 71, 1696-1706. [CrossRef]
- Triposkiadis, F., Giamouzis, G., Boudoulas, K. D., Karagiannis, G., Skoularigis, J., Boudoulas, H. & Parissis, J. (2018). Left ventricular geometry as a major determinant of left ventricular ejection fraction: physiological considerations and clinical implications. Eur J Heart Fail. 20, 436-444. [CrossRef]
- Husti, Z., Varró, A. & Baczkó, I. (2021). Arrhythmogenic Remodeling in the Failing Heart. Cells. 10. [CrossRef]
- Frangogiannis, N. G. (2019). The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ Res. 125, 117-146. [CrossRef]
- Bristow, M. R., Kao, D. P., Breathett, K. K., Altman, N. L., Gorcsan, J., 3rd, Gill, E. A., Lowes, B. D., Gilbert, E. M., Quaife, R. A. & Mann, D. L. (2017). Structural and Functional Phenotyping of the Failing Heart: Is the Left Ventricular Ejection Fraction Obsolete? JACC Heart Fail. 5, 772-781. [CrossRef]
- Vancheri, F., Longo, G. & Henein, M. Y. (2024). Left ventricular ejection fraction: clinical, pathophysiological, and technical limitations. Front Cardiovasc Med. 11, 1340708. [CrossRef]
- Heidenreich, P. A., Bozkurt, B., Aguilar, D., Allen, L. A., Byun, J. J., Colvin, M. M., Deswal, A., Drazner, M. H., Dunlay, S. M., Evers, L. R., Fang, J. C., Fedson, S. E., Fonarow, G. C., Hayek, S. S., Hernandez, A. F., Khazanie, P., Kittleson, M. M., Lee, C. S., Link, M. S., Milano, C. A., Nnacheta, L. C., Sandhu, A. T., Stevenson, L. W., Vardeny, O., Vest, A. R. & Yancy, C. W. (2022). 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 145, e895-e1032. [CrossRef]
- McDonagh, T. A., Metra, M., Adamo, M., Gardner, R. S., Baumbach, A., Böhm, M., Burri, H., Butler, J., Čelutkienė, J., Chioncel, O., Cleland, J. G. F., Coats, A. J. S., Crespo-Leiro, M. G., Farmakis, D., Gilard, M., Heymans, S., Hoes, A. W., Jaarsma, T., Jankowska, E. A., Lainscak, M., Lam, C. S. P., Lyon, A. R., McMurray, J. J. V., Mebazaa, A., Mindham, R., Muneretto, C., Francesco Piepoli, M., Price, S., Rosano, G. M. C., Ruschitzka, F. & Kathrine Skibelund, A. (2021). 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 42, 3599-3726. [CrossRef]
- Falcão-Pires, I., Ferreira, A. F., Trindade, F., Bertrand, L., Ciccarelli, M., Visco, V., Dawson, D., Hamdani, N., Van Laake, L. W., Lezoualc’h, F., Linke, W. A., Lunde, I. G., Rainer, P. P., Abdellatif, M., Van der Velden, J., Cosentino, N., Paldino, A., Pompilio, G., Zacchigna, S., Heymans, S., Thum, T. & Tocchetti, C. G. (2024). Mechanisms of myocardial reverse remodelling and its clinical significance: A scientific statement of the ESC Working Group on Myocardial Function. Eur J Heart Fail. [CrossRef]
- Koitabashi, N. & Kass, D. A. (2011). Reverse remodeling in heart failure--mechanisms and therapeutic opportunities. Nat Rev Cardiol. 9, 147-157. [CrossRef]
- Kim, G. H., Uriel, N. & Burkhoff, D. (2018). Reverse remodelling and myocardial recovery in heart failure. Nat Rev Cardiol. 15, 83-96. [CrossRef]
- Boulet, J. & Mehra, M. R. (2021). Left Ventricular Reverse Remodeling in Heart Failure: Remission to Recovery. Structural Heart. 5, 466-481. [CrossRef]
- Chudý, M. & Goncalvesová, E. (2022). Prediction of Left Ventricular Reverse Remodelling: A Mini Review on Clinical Aspects. Cardiology. 147, 521-528. [CrossRef]
- Hellawell, J. L. & Margulies, K. B. (2012). Myocardial reverse remodeling. Cardiovasc Ther. 30, 172-181. [CrossRef]
- Reis Filho, J. R., Cardoso, J. N., Cardoso, C. M. & Pereira-Barretto, A. C. (2015). Reverse Cardiac Remodeling: A Marker of Better Prognosis in Heart Failure. Arq Bras Cardiol. 104, 502-506. [CrossRef]
- Ojo, A., Tariq, S., Harikrishnan, P., Iwai, S. & Jacobson, J. T. (2017). Cardiac Resynchronization Therapy for Heart Failure. Interv Cardiol Clin. 6, 417-426. [CrossRef]
- Burkhoff, D., Topkara, V. K., Sayer, G. & Uriel, N. (2021). Reverse Remodeling With Left Ventricular Assist Devices. Circ Res. 128, 1594-1612. [CrossRef]
- Hamad, E. A., Byku, M., Larson, S. B. & Billia, F. (2023). LVAD therapy as a catalyst to heart failure remission and myocardial recovery. Clin Cardiol. 46, 1154-1162. [CrossRef]
- Pan, S., Aksut, B., Wever-Pinzon, O. E., Rao, S. D., Levin, A. P., Garan, A. R., Fried, J. A., Takeda, K., Hiroo, T., Yuzefpolskaya, M., Uriel, N., Jorde, U. P., Mancini, D. M., Naka, Y., Colombo, P. C. & Topkara, V. K. (2015). Incidence and predictors of myocardial recovery on long-term left ventricular assist device support: Results from the United Network for Organ Sharing database. J Heart Lung Transplant. 34, 1624-1629. [CrossRef]
- Ponzoni, M., Coles, J. G. & Maynes, J. T. (2023). Rodent Models of Dilated Cardiomyopathy and Heart Failure for Translational Investigations and Therapeutic Discovery. Int J Mol Sci. 24. [CrossRef]
- Galeone, A., Buccoliero, C., Barile, B., Nicchia, G. P., Onorati, F., Luciani, G. B. & Brunetti, G. (2023). Cellular and Molecular Mechanisms Activated by a Left Ventricular Assist Device. Int J Mol Sci. 25. [CrossRef]
- Drakos, S. G., Badolia, R., Makaju, A., Kyriakopoulos, C. P., Wever-Pinzon, O., Tracy, C. M., Bakhtina, A., Bia, R., Parnell, T., Taleb, I., Ramadurai, D. K. A., Navankasattusas, S., Dranow, E., Hanff, T. C., Tseliou, E., Shankar, T. S., Visker, J., Hamouche, R., Stauder, E. L., Caine, W. T., Alharethi, R., Selzman, C. H. & Franklin, S. (2023). Distinct Transcriptomic and Proteomic Profile Specifies Patients Who Have Heart Failure With Potential of Myocardial Recovery on Mechanical Unloading and Circulatory Support. Circulation. 147, 409-424. [CrossRef]
- Margulies, K. B., Matiwala, S., Cornejo, C., Olsen, H., Craven, W. A. & Bednarik, D. (2005). Mixed messages: transcription patterns in failing and recovering human myocardium. Circ Res. 96, 592-599. [CrossRef]
- Topkara, V. K., Chambers, K. T., Yang, K. C., Tzeng, H. P., Evans, S., Weinheimer, C., Kovacs, A., Robbins, J., Barger, P. & Mann, D. L. (2016). Functional significance of the discordance between transcriptional profile and left ventricular structure/function during reverse remodeling. JCI Insight. 1, e86038. [CrossRef]
- Yan, C. L. & Grazette, L. (2023). A review of biomarker and imaging monitoring to predict heart failure recovery. Front Cardiovasc Med. 10, 1150336. [CrossRef]
- Holzhauser, L., Kim, G., Sayer, G. & Uriel, N. (2018). The Effect of Left Ventricular Assist Device Therapy on Cardiac Biomarkers: Implications for the Identification of Myocardial Recovery. Curr Heart Fail Rep. 15, 250-259. [CrossRef]
- Motiwala, S. R. & Gaggin, H. K. (2016). Biomarkers to Predict Reverse Remodeling and Myocardial Recovery in Heart Failure. Curr Heart Fail Rep. 13, 207-218. [CrossRef]
- Castiglione, V., Aimo, A., Vergaro, G., Saccaro, L., Passino, C. & Emdin, M. (2022). Biomarkers for the diagnosis and management of heart failure. Heart Fail Rev. 27, 625-643. [CrossRef]
- Jaltotage, B., Dwivedi, G., Ooi, D. E. L. & Mahadavan, G. (2021). The Utility of Circulating and Imaging Biomarkers Alone and in Combination in Heart Failure. Curr Cardiol Rev. 17, e160721193557. [CrossRef]
- Robinson, E. L., Baker, A. H., Brittan, M., McCracken, I., Condorelli, G., Emanueli, C., Srivastava, P. K., Gaetano, C., Thum, T., Vanhaverbeke, M., Angione, C., Heymans, S., Devaux, Y., Pedrazzini, T. & Martelli, F. (2022). Dissecting the transcriptome in cardiovascular disease. Cardiovasc Res. 118, 1004-1019. [CrossRef]
- Dangwal, S., Schimmel, K., Foinquinos, A., Xiao, K. & Thum, T. (2017). Noncoding RNAs in Heart Failure. Handb Exp Pharmacol. 243, 423-445. [CrossRef]
- Singh, D. D., Kim, Y., Choi, S. A., Han, I. & Yadav, D. K. (2023). Clinical Significance of MicroRNAs, Long Non-Coding RNAs, and CircRNAs in Cardiovascular Diseases. Cells. 12. [CrossRef]
- Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., Oyama, R., Ravasi, T., Lenhard, B., Wells, C., Kodzius, R., Shimokawa, K., Bajic, V. B., Brenner, S. E., Batalov, S., Forrest, A. R., Zavolan, M., Davis, M. J., Wilming, L. G., Aidinis, V., Allen, J. E., Ambesi-Impiombato, A., Apweiler, R., Aturaliya, R. N., Bailey, T. L., Bansal, M., Baxter, L., Beisel, K. W., Bersano, T., Bono, H., Chalk, A. M., Chiu, K. P., Choudhary, V., Christoffels, A., Clutterbuck, D. R., Crowe, M. L., Dalla, E., Dalrymple, B. P., de Bono, B., Della Gatta, G., di Bernardo, D., Down, T., Engstrom, P., Fagiolini, M., Faulkner, G., Fletcher, C. F., Fukushima, T., Furuno, M., Futaki, S., Gariboldi, M., Georgii-Hemming, P., Gingeras, T. R., Gojobori, T., Green, R. E., Gustincich, S., Harbers, M., Hayashi, Y., Hensch, T. K., Hirokawa, N., Hill, D., Huminiecki, L., Iacono, M., Ikeo, K., Iwama, A., Ishikawa, T., Jakt, M., Kanapin, A., Katoh, M., Kawasawa, Y., Kelso, J., Kitamura, H., Kitano, H., Kollias, G., Krishnan, S. P., Kruger, A., Kummerfeld, S. K., Kurochkin, I. V., Lareau, L. F., Lazarevic, D., Lipovich, L., Liu, J., Liuni, S., McWilliam, S., Madan Babu, M., Madera, M., Marchionni, L., Matsuda, H., Matsuzawa, S., Miki, H., Mignone, F., Miyake, S., Morris, K., Mottagui-Tabar, S., Mulder, N., Nakano, N., Nakauchi, H., Ng, P., Nilsson, R., Nishiguchi, S., Nishikawa, S., Nori, F., Ohara, O., Okazaki, Y., Orlando, V., Pang, K. C., Pavan, W. J., Pavesi, G., Pesole, G., Petrovsky, N., Piazza, S., Reed, J., Reid, J. F., Ring, B. Z., Ringwald, M., Rost, B., Ruan, Y., Salzberg, S. L., Sandelin, A., Schneider, C., Schönbach, C., Sekiguchi, K., Semple, C. A., Seno, S., Sessa, L., Sheng, Y., Shibata, Y., Shimada, H., Shimada, K., Silva, D., Sinclair, B., Sperling, S., Stupka, E., Sugiura, K., Sultana, R., Takenaka, Y., Taki, K., Tammoja, K., Tan, S. L., Tang, S., Taylor, M. S., Tegner, J., Teichmann, S. A., Ueda, H. R., van Nimwegen, E., Verardo, R., Wei, C. L., Yagi, K., Yamanishi, H., Zabarovsky, E., Zhu, S., Zimmer, A., Hide, W., Bult, C., Grimmond, S. M., Teasdale, R. D., Liu, E. T., Brusic, V., Quackenbush, J., Wahlestedt, C., Mattick, J. S., Hume, D. A., Kai, C., Sasaki, D., Tomaru, Y., Fukuda, S., Kanamori-Katayama, M., Suzuki, M., Aoki, J., Arakawa, T., Iida, J., Imamura, K., Itoh, M., Kato, T., Kawaji, H., Kawagashira, N., Kawashima, T., Kojima, M., Kondo, S., Konno, H., Nakano, K., Ninomiya, N., Nishio, T., Okada, M., Plessy, C., Shibata, K., Shiraki, T., Suzuki, S., Tagami, M., Waki, K., Watahiki, A., Okamura-Oho, Y., Suzuki, H., Kawai, J. & Hayashizaki, Y. (2005). The transcriptional landscape of the mammalian genome. Science. 309, 1559-1563. [CrossRef]
- Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., Tanzer, A., Lagarde, J., Lin, W., Schlesinger, F., Xue, C., Marinov, G. K., Khatun, J., Williams, B. A., Zaleski, C., Rozowsky, J., Röder, M., Kokocinski, F., Abdelhamid, R. F., Alioto, T., Antoshechkin, I., Baer, M. T., Bar, N. S., Batut, P., Bell, K., Bell, I., Chakrabortty, S., Chen, X., Chrast, J., Curado, J., Derrien, T., Drenkow, J., Dumais, E., Dumais, J., Duttagupta, R., Falconnet, E., Fastuca, M., Fejes-Toth, K., Ferreira, P., Foissac, S., Fullwood, M. J., Gao, H., Gonzalez, D., Gordon, A., Gunawardena, H., Howald, C., Jha, S., Johnson, R., Kapranov, P., King, B., Kingswood, C., Luo, O. J., Park, E., Persaud, K., Preall, J. B., Ribeca, P., Risk, B., Robyr, D., Sammeth, M., Schaffer, L., See, L. H., Shahab, A., Skancke, J., Suzuki, A. M., Takahashi, H., Tilgner, H., Trout, D., Walters, N., Wang, H., Wrobel, J., Yu, Y., Ruan, X., Hayashizaki, Y., Harrow, J., Gerstein, M., Hubbard, T., Reymond, A., Antonarakis, S. E., Hannon, G., Giddings, M. C., Ruan, Y., Wold, B., Carninci, P., Guigó, R. & Gingeras, T. R. (2012). Landscape of transcription in human cells. Nature. 489, 101-108. [CrossRef]
- Stelzer, G., Rosen, N., Plaschkes, I., Zimmerman, S., Twik, M., Fishilevich, S., Stein, T. I., Nudel, R., Lieder, I., Mazor, Y., Kaplan, S., Dahary, D., Warshawsky, D., Guan-Golan, Y., Kohn, A., Rappaport, N., Safran, M. & Lancet, D. (2016). The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr Protoc Bioinformatics. 54, 1.30.31-31.30.33. [CrossRef]
- Yates, A. D., Achuthan, P., Akanni, W., Allen, J., Allen, J., Alvarez-Jarreta, J., Amode, M. R., Armean, I. M., Azov, A. G., Bennett, R., Bhai, J., Billis, K., Boddu, S., Marugán, J. C., Cummins, C., Davidson, C., Dodiya, K., Fatima, R., Gall, A., Giron, C. G., Gil, L., Grego, T., Haggerty, L., Haskell, E., Hourlier, T., Izuogu, O. G., Janacek, S. H., Juettemann, T., Kay, M., Lavidas, I., Le, T., Lemos, D., Martinez, J. G., Maurel, T., McDowall, M., McMahon, A., Mohanan, S., Moore, B., Nuhn, M., Oheh, D. N., Parker, A., Parton, A., Patricio, M., Sakthivel, M. P., Abdul Salam, A. I., Schmitt, B. M., Schuilenburg, H., Sheppard, D., Sycheva, M., Szuba, M., Taylor, K., Thormann, A., Threadgold, G., Vullo, A., Walts, B., Winterbottom, A., Zadissa, A., Chakiachvili, M., Flint, B., Frankish, A., Hunt, S. E., G, I. I., Kostadima, M., Langridge, N., Loveland, J. E., Martin, F. J., Morales, J., Mudge, J. M., Muffato, M., Perry, E., Ruffier, M., Trevanion, S. J., Cunningham, F., Howe, K. L., Zerbino, D. R. & Flicek, P. (2020). Ensembl 2020. Nucleic Acids Res. 48, D682-d688. [CrossRef]
- Braschi, B., Denny, P., Gray, K., Jones, T., Seal, R., Tweedie, S., Yates, B. & Bruford, E. (2019). Genenames.org: the HGNC and VGNC resources in 2019. Nucleic Acids Res. 47, D786-d792. [CrossRef]
- (2019). RNAcentral: a hub of information for non-coding RNA sequences. Nucleic Acids Res. 47, D221-d229. [CrossRef]
- Barshir, R., Fishilevich, S., Iny-Stein, T., Zelig, O., Mazor, Y., Guan-Golan, Y., Safran, M. & Lancet, D. (2021). GeneCaRNA: A Comprehensive Gene-centric Database of Human Non-coding RNAs in the GeneCards Suite. J Mol Biol. 433, 166913. [CrossRef]
- Brown, G. R., Hem, V., Katz, K. S., Ovetsky, M., Wallin, C., Ermolaeva, O., Tolstoy, I., Tatusova, T., Pruitt, K. D., Maglott, D. R. & Murphy, T. D. (2015). Gene: a gene-centered information resource at NCBI. Nucleic Acids Res. 43, D36-42. [CrossRef]
- Morris, K. V. & Mattick, J. S. (2014). The rise of regulatory RNA. Nat Rev Genet. 15, 423-437. [CrossRef]
- Hombach, S. & Kretz, M. (2016). Non-coding RNAs: Classification, Biology and Functioning. Adv Exp Med Biol. 937, 3-17. [CrossRef]
- Nemeth, K., Bayraktar, R., Ferracin, M. & Calin, G. A. (2024). Non-coding RNAs in disease: from mechanisms to therapeutics. Nat Rev Genet. 25, 211-232. [CrossRef]
- Ninomiya, K., Yamazaki, T. & Hirose, T. (2023). Satellite RNAs: emerging players in subnuclear architecture and gene regulation. Embo j. 42, e114331. [CrossRef]
- St Laurent, G., Wahlestedt, C. & Kapranov, P. (2015). The Landscape of long noncoding RNA classification. Trends Genet. 31, 239-251. [CrossRef]
- Shang, R., Lee, S., Senavirathne, G. & Lai, E. C. (2023). microRNAs in action: biogenesis, function and regulation. Nat Rev Genet. 24, 816-833. [CrossRef]
- Statello, L., Guo, C. J., Chen, L. L. & Huarte, M. (2021). Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 22, 96-118. [CrossRef]
- Loganathan, T. & Doss, C. G. (2023). Non-coding RNAs in human health and disease: potential function as biomarkers and therapeutic targets. Funct Integr Genomics. 23, 33. [CrossRef]
- Wu, R., Su, Y., Wu, H., Dai, Y., Zhao, M. & Lu, Q. (2016). Characters, functions and clinical perspectives of long non-coding RNAs. Mol Genet Genomics. 291, 1013-1033. [CrossRef]
- Poller, W., Dimmeler, S., Heymans, S., Zeller, T., Haas, J., Karakas, M., Leistner, D. M., Jakob, P., Nakagawa, S., Blankenberg, S., Engelhardt, S., Thum, T., Weber, C., Meder, B., Hajjar, R. & Landmesser, U. (2018). Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J. 39, 2704-2716. [CrossRef]
- Garcia-Padilla, C., Lozano-Velasco, E., Garcia-Lopez, V., Aranega, A., Franco, D., Garcia-Martinez, V. & Lopez-Sanchez, C. (2022). Comparative Analysis of Non-Coding RNA Transcriptomics in Heart Failure. Biomedicines. 10. [CrossRef]
- Rodriguez, A., Griffiths-Jones, S., Ashurst, J. L. & Bradley, A. (2004). Identification of mammalian microRNA host genes and transcription units. Genome Res. 14, 1902-1910. [CrossRef]
- Godnic, I., Zorc, M., Jevsinek Skok, D., Calin, G. A., Horvat, S., Dovc, P., Kovac, M. & Kunej, T. (2013). Genome-wide and species-wide in silico screening for intragenic MicroRNAs in human, mouse and chicken. PLoS One. 8, e65165. [CrossRef]
- Ramchandran, R. & Chaluvally-Raghavan, P. (2017). miRNA-Mediated RNA Activation in Mammalian Cells. Adv Exp Med Biol. 983, 81-89. [CrossRef]
- Stavast, C. J. & Erkeland, S. J. (2019). The Non-Canonical Aspects of MicroRNAs: Many Roads to Gene Regulation. Cells. 8. [CrossRef]
- Friedman, R. C., Farh, K. K., Burge, C. B. & Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92-105. [CrossRef]
- Gebert, L. F. R. & MacRae, I. J. (2019). Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 20, 21-37. [CrossRef]
- Londin, E., Loher, P., Telonis, A. G., Quann, K., Clark, P., Jing, Y., Hatzimichael, E., Kirino, Y., Honda, S., Lally, M., Ramratnam, B., Comstock, C. E., Knudsen, K. E., Gomella, L., Spaeth, G. L., Hark, L., Katz, L. J., Witkiewicz, A., Rostami, A., Jimenez, S. A., Hollingsworth, M. A., Yeh, J. J., Shaw, C. A., McKenzie, S. E., Bray, P., Nelson, P. T., Zupo, S., Van Roosbroeck, K., Keating, M. J., Calin, G. A., Yeo, C., Jimbo, M., Cozzitorto, J., Brody, J. R., Delgrosso, K., Mattick, J. S., Fortina, P. & Rigoutsos, I. (2015). Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc Natl Acad Sci U S A. 112, E1106-1115. [CrossRef]
- Tomasello, L., Distefano, R., Nigita, G. & Croce, C. M. (2021). The MicroRNA Family Gets Wider: The IsomiRs Classification and Role. Front Cell Dev Biol. 9, 668648. [CrossRef]
- Zealy, R. W., Wrenn, S. P., Davila, S., Min, K. W. & Yoon, J. H. (2017). microRNA-binding proteins: specificity and function. Wiley Interdiscip Rev RNA. 8. [CrossRef]
- Sood, P., Krek, A., Zavolan, M., Macino, G. & Rajewsky, N. (2006). Cell-type-specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci U S A. 103, 2746-2751. [CrossRef]
- Ludwig, N., Leidinger, P., Becker, K., Backes, C., Fehlmann, T., Pallasch, C., Rheinheimer, S., Meder, B., Stähler, C., Meese, E. & Keller, A. (2016). Distribution of miRNA expression across human tissues. Nucleic Acids Res. 44, 3865-3877. [CrossRef]
- Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. (2019). miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155-d162. [CrossRef]
- Laggerbauer, B. & Engelhardt, S. (2022). MicroRNAs as therapeutic targets in cardiovascular disease. J Clin Invest. 132. [CrossRef]
- Gargiulo, P., Marzano, F., Salvatore, M., Basile, C., Buonocore, D., Parlati, A. L. M., Nardi, E., Asile, G., Abbate, V., Colella, A., Chirico, A., Marciano, C., Paolillo, S. & Perrone-Filardi, P. (2023). MicroRNAs: diagnostic, prognostic and therapeutic role in heart failure-a review. ESC Heart Fail. 10, 753-761. [CrossRef]
- Shen, N. N., Wang, J. L. & Fu, Y. P. (2022). The microRNA Expression Profiling in Heart Failure: A Systematic Review and Meta-Analysis. Front Cardiovasc Med. 9, 856358. [CrossRef]
- Gholaminejad, A., Zare, N., Dana, N., Shafie, D., Mani, A. & Javanmard, S. H. (2021). A meta-analysis of microRNA expression profiling studies in heart failure. Heart Fail Rev. 26, 997-1021. [CrossRef]
- Parvan, R., Hosseinpour, M., Moradi, Y., Devaux, Y., Cataliotti, A. & da Silva, G. J. J. (2022). Diagnostic performance of microRNAs in the detection of heart failure with reduced or preserved ejection fraction: a systematic review and meta-analysis. Eur J Heart Fail. 24, 2212-2225. [CrossRef]
- Vilella-Figuerola, A., Gallinat, A., Escate, R., Mirabet, S., Padró, T. & Badimon, L. (2022). Systems Biology in Chronic Heart Failure-Identification of Potential miRNA Regulators. Int J Mol Sci. 23. [CrossRef]
- Figueiredo, R., Adão, R., Leite-Moreira, A. F., Mâncio, J. & Brás-Silva, C. (2022). Candidate microRNAs as prognostic biomarkers in heart failure: A systematic review. Rev Port Cardiol. 41, 865-885. [CrossRef]
- Schipper, M. E., van Kuik, J., de Jonge, N., Dullens, H. F. & de Weger, R. A. (2008). Changes in regulatory microRNA expression in myocardium of heart failure patients on left ventricular assist device support. J Heart Lung Transplant. 27, 1282-1285. [CrossRef]
- Ramani, R., Vela, D., Segura, A., McNamara, D., Lemster, B., Samarendra, V., Kormos, R., Toyoda, Y., Bermudez, C., Frazier, O. H., Moravec, C. S., Gorcsan, J., 3rd, Taegtmeyer, H. & McTiernan, C. F. (2011). A micro-ribonucleic acid signature associated with recovery from assist device support in 2 groups of patients with severe heart failure. J Am Coll Cardiol. 58, 2270-2278. [CrossRef]
- Lok, S. I., van Mil, A., Bovenschen, N., van der Weide, P., van Kuik, J., van Wichen, D., Peeters, T., Siera, E., Winkens, B., Sluijter, J. P., Doevendans, P. A., da Costa Martins, P. A., de Jonge, N. & de Weger, R. A. (2013). Post-transcriptional regulation of α-1-antichymotrypsin by microRNA-137 in chronic heart failure and mechanical support. Circ Heart Fail. 6, 853-861. [CrossRef]
- Barsanti, C., Trivella, M. G., D’Aurizio, R., El Baroudi, M., Baumgart, M., Groth, M., Caruso, R., Verde, A., Botta, L., Cozzi, L. & Pitto, L. (2015). Differential regulation of microRNAs in end-stage failing hearts is associated with left ventricular assist device unloading. Biomed Res Int. 2015, 592512. [CrossRef]
- Lok, S. I., de Jonge, N., van Kuik, J., van Geffen, A. J., Huibers, M. M., van der Weide, P., Siera, E., Winkens, B., Doevendans, P. A., de Weger, R. A. & da Costa Martins, P. A. (2015). MicroRNA Expression in Myocardial Tissue and Plasma of Patients with End-Stage Heart Failure during LVAD Support: Comparison of Continuous and Pulsatile Devices. PLoS One. 10, e0136404. [CrossRef]
- Morley-Smith, A. C., Mills, A., Jacobs, S., Meyns, B., Rega, F., Simon, A. R., Pepper, J. R., Lyon, A. R. & Thum, T. (2014). Circulating microRNAs for predicting and monitoring response to mechanical circulatory support from a left ventricular assist device. Eur J Heart Fail. 16, 871-879. [CrossRef]
- Ragusa, R., Di Molfetta, A., Del Turco, S., Cabiati, M., Del Ry, S., Basta, G., Mercatanti, A., Pitto, L., Amodeo, A., Trivella, M. G., Rizzo, M. & Caselli, C. (2021). Epigenetic Regulation of Cardiac Troponin Genes in Pediatric Patients with Heart Failure Supported by Ventricular Assist Device. Biomedicines. 9. [CrossRef]
- Ragusa, R., Di Molfetta, A., Mercatanti, A., Pitto, L., Amodeo, A., Trivella, M. G., Rizzo, M. & Caselli, C. (2024). Changes in adiponectin system after ventricular assist device in pediatric heart failure. JHLT Open. 3, None. [CrossRef]
- Mann, D. L. & Burkhoff, D. (2011). Myocardial expression levels of micro-ribonucleic acids in patients with left ventricular assist devices signature of myocardial recovery, signature of reverse remodeling, or signature with no name? J Am Coll Cardiol. 58, 2279-2281. [CrossRef]
- Matkovich, S. J., Van Booven, D. J., Youker, K. A., Torre-Amione, G., Diwan, A., Eschenbacher, W. H., Dorn, L. E., Watson, M. A., Margulies, K. B. & Dorn, G. W., 2nd (2009). Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation. 119, 1263-1271. [CrossRef]
- Akat, K. M., Moore-McGriff, D., Morozov, P., Brown, M., Gogakos, T., Correa Da Rosa, J., Mihailovic, A., Sauer, M., Ji, R., Ramarathnam, A., Totary-Jain, H., Williams, Z., Tuschl, T. & Schulze, P. C. (2014). Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci U S A. 111, 11151-11156. [CrossRef]
- Parikh, M., Shah, S., Basu, R., Famulski, K. S., Kim, D., Mullen, J. C., Halloran, P. F. & Oudit, G. Y. (2022). Transcriptomic Signatures of End-Stage Human Dilated Cardiomyopathy Hearts with and without Left Ventricular Assist Device Support. Int J Mol Sci. 23. [CrossRef]
- Yang, K. C., Yamada, K. A., Patel, A. Y., Topkara, V. K., George, I., Cheema, F. H., Ewald, G. A., Mann, D. L. & Nerbonne, J. M. (2014). Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 129, 1009-1021. [CrossRef]
- Muthiah, K., Humphreys, D. T., Robson, D., Dhital, K., Spratt, P., Jansz, P., Macdonald, P. S. & Hayward, C. S. (2017). Longitudinal structural, functional, and cellular myocardial alterations with chronic centrifugal continuous-flow left ventricular assist device support. J Heart Lung Transplant. 36, 722-731. [CrossRef]
- Li, X., Liu, C. Y., Li, Y. S., Xu, J., Li, D. G., Li, X. & Han, D. (2016). Deep RNA sequencing elucidates microRNA-regulated molecular pathways in ischemic cardiomyopathy and nonischemic cardiomyopathy. Genet Mol Res. 15. [CrossRef]
- Chistiakov, D. A., Orekhov, A. N. & Bobryshev, Y. V. (2016). Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J Mol Cell Cardiol. 94, 107-121. [CrossRef]
- Song, Z., Gao, R. & Yan, B. (2020). Potential roles of microRNA-1 and microRNA-133 in cardiovascular disease. Rev Cardiovasc Med. 21, 57-64. [CrossRef]
- Ikeda, S., Kong, S. W., Lu, J., Bisping, E., Zhang, H., Allen, P. D., Golub, T. R., Pieske, B. & Pu, W. T. (2007). Altered microRNA expression in human heart disease. Physiol Genomics. 31, 367-373. [CrossRef]
- Thum, T., Galuppo, P., Wolf, C., Fiedler, J., Kneitz, S., van Laake, L. W., Doevendans, P. A., Mummery, C. L., Borlak, J., Haverich, A., Gross, C., Engelhardt, S., Ertl, G. & Bauersachs, J. (2007). MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 116, 258-267. [CrossRef]
- van Rooij, E., Sutherland, L. B., Liu, N., Williams, A. H., McAnally, J., Gerard, R. D., Richardson, J. A. & Olson, E. N. (2006). A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 103, 18255-18260. [CrossRef]
- Sigutova, R., Evin, L., Stejskal, D., Ploticova, V. & Svagera, Z. (2022). Specific microRNAs and heart failure: time for the next step toward application? Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 166, 359-368. [CrossRef]
- Carvalho, A., Ji, Z., Zhang, R., Zuo, W., Qu, Y., Chen, X., Tao, Z., Ji, J., Yao, Y. & Ma, G. (2023). Inhibition of miR-195-3p protects against cardiac dysfunction and fibrosis after myocardial infarction. Int J Cardiol. 387, 131128. [CrossRef]
- Zhang, X., Ji, R., Liao, X., Castillero, E., Kennel, P. J., Brunjes, D. L., Franz, M., Möbius-Winkler, S., Drosatos, K., George, I., Chen, E. I., Colombo, P. C. & Schulze, P. C. (2018). MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation. 137, 2052-2067. [CrossRef]
- Zhu, M., Zhang, C., Zhang, Z., Liao, X., Ren, D., Li, R., Liu, S., He, X. & Dong, N. (2022). Changes in transcriptomic landscape in human end-stage heart failure with distinct etiology. iScience. 25, 103935. [CrossRef]
- Dirkx, E., da Costa Martins, P. A. & De Windt, L. J. (2013). Regulation of fetal gene expression in heart failure. Biochim Biophys Acta. 1832, 2414-2424. [CrossRef]
- Eduardo Rame, J. (2017). Hemodynamic unloading and the molecular-functional phenotype dissociation in myocardial recovery. J Heart Lung Transplant. 36, 715-717. [CrossRef]
- Cheng, A., Williamitis, C. A. & Slaughter, M. S. (2014). Comparison of continuous-flow and pulsatile-flow left ventricular assist devices: is there an advantage to pulsatility? Ann Cardiothorac Surg. 3, 573-581. [CrossRef]
- Lampert, B. C. & Teuteberg, J. J. (2015). Right ventricular failure after left ventricular assist devices. J Heart Lung Transplant. 34, 1123-1130. [CrossRef]
- Rodenas-Alesina, E., Brahmbhatt, D. H., Rao, V., Salvatori, M. & Billia, F. (2022). Prediction, prevention, and management of right ventricular failure after left ventricular assist device implantation: A comprehensive review. Front Cardiovasc Med. 9, 1040251. [CrossRef]
- Frankfurter, C., Molinero, M., Vishram-Nielsen, J. K. K., Foroutan, F., Mak, S., Rao, V., Billia, F., Orchanian-Cheff, A. & Alba, A. C. (2020). Predicting the Risk of Right Ventricular Failure in Patients Undergoing Left Ventricular Assist Device Implantation: A Systematic Review. Circ Heart Fail. 13, e006994. [CrossRef]
- Løgstrup, B. B., Nemec, P., Schoenrath, F., Gummert, J., Pya, Y., Potapov, E., Netuka, I., Ramjankhan, F., Parner, E. T., De By, T. & Eiskjaer, H. (2020). Heart failure etiology and risk of right heart failure in adult left ventricular assist device support: the European Registry for Patients with Mechanical Circulatory Support (EUROMACS). Scand Cardiovasc J. 54, 306-314. [CrossRef]
- Balcioglu, O., Ozgocmen, C., Ozsahin, D. U. & Yagdi, T. (2024). The Role of Artificial Intelligence and Machine Learning in the Prediction of Right Heart Failure after Left Ventricular Assist Device Implantation: A Comprehensive Review. Diagnostics (Basel). 14. [CrossRef]
- Lippmann, M. R. & Maron, B. A. (2022). The Right Ventricle: From Embryologic Development to RV Failure. Curr Heart Fail Rep. 19, 325-333. [CrossRef]
- Havlenova, T., Skaroupkova, P., Miklovic, M., Behounek, M., Chmel, M., Jarkovska, D., Sviglerova, J., Stengl, M., Kolar, M., Novotny, J., Benes, J., Cervenka, L., Petrak, J. & Melenovsky, V. (2021). Right versus left ventricular remodeling in heart failure due to chronic volume overload. Sci Rep. 11, 17136. [CrossRef]
- Frisk, C., Das, S., Eriksson, M. J., Walentinsson, A., Corbascio, M., Hage, C., Kumar, C., Ekström, M., Maret, E., Persson, H., Linde, C. & Persson, B. (2024). Cardiac biopsies reveal differences in transcriptomics between left and right ventricle in patients with or without diagnostic signs of heart failure. Sci Rep. 14, 5811. [CrossRef]
- Dewar, M. B., Ehsan, F., Izumi, A., Zhang, H., Zhou, Y. Q., Shah, H., Langburt, D., Suresh, H., Wang, T., Hacker, A., Hinz, B., Gillis, J., Husain, M. & Heximer, S. P. (2024). Defining Transcriptomic Heterogeneity between Left and Right Ventricle-Derived Cardiac Fibroblasts. Cells. 13. [CrossRef]
- Batkai, S., Bär, C. & Thum, T. (2017). MicroRNAs in right ventricular remodelling. Cardiovasc Res. 113, 1433-1440. [CrossRef]
- Toro, V., Jutras-Beaudoin, N., Boucherat, O., Bonnet, S., Provencher, S. & Potus, F. (2023). Right Ventricle and Epigenetics: A Systematic Review. Cells. 12. [CrossRef]
- Hsu, D. T. & Pearson, G. D. (2009). Heart failure in children: part I: history, etiology, and pathophysiology. Circ Heart Fail. 2, 63-70. [CrossRef]
- Shaddy, R. E., George, A. T., Jaecklin, T., Lochlainn, E. N., Thakur, L., Agrawal, R., Solar-Yohay, S., Chen, F., Rossano, J. W., Severin, T. & Burch, M. (2018). Systematic Literature Review on the Incidence and Prevalence of Heart Failure in Children and Adolescents. Pediatr Cardiol. 39, 415-436. [CrossRef]
- Senekovič Kojc, T. & Marčun Varda, N. (2022). Novel Biomarkers of Heart Failure in Pediatrics. Children (Basel). 9. [CrossRef]
- Amdani, S., Auerbach, S. R., Bansal, N., Chen, S., Conway, J., Silva, J. P. D., Deshpande, S. R., Hoover, J., Lin, K. Y., Miyamoto, S. D., Puri, K., Price, J., Spinner, J., White, R., Rossano, J. W., Bearl, D. W., Cousino, M. K., Catlin, P., Hidalgo, N. C., Godown, J., Kantor, P., Masarone, D., Peng, D. M., Rea, K. E., Schumacher, K., Shaddy, R., Shea, E., Tapia, H. V., Valikodath, N., Zafar, F. & Hsu, D. (2024). Research Gaps in Pediatric Heart Failure: Defining the Gaps and Then Closing Them Over the Next Decade. J Card Fail. 30, 64-77. [CrossRef]
- Ragusa, R., Di Molfetta, A., Amodeo, A., Trivella, M. G. & Caselli, C. (2020). Pathophysiology and molecular signalling in pediatric heart failure and VAD therapy. Clin Chim Acta. 510, 751-759. [CrossRef]
- Khan, R. S., Kato, T. S., Chokshi, A., Chew, M., Yu, S., Wu, C., Singh, P., Cheema, F. H., Takayama, H., Harris, C., Reyes-Soffer, G., Knöll, R., Milting, H., Naka, Y., Mancini, D. & Schulze, P. C. (2012). Adipose tissue inflammation and adiponectin resistance in patients with advanced heart failure: correction after ventricular assist device implantation. Circ Heart Fail. 5, 340-348. [CrossRef]
- Mado, H., Szczurek, W., Gąsior, M. & Szyguła-Jurkiewicz, B. (2021). Adiponectin in heart failure. Future Cardiol. 17, 757-764. [CrossRef]
- Litviňuková, M., Talavera-López, C., Maatz, H., Reichart, D., Worth, C. L., Lindberg, E. L., Kanda, M., Polanski, K., Heinig, M., Lee, M., Nadelmann, E. R., Roberts, K., Tuck, L., Fasouli, E. S., DeLaughter, D. M., McDonough, B., Wakimoto, H., Gorham, J. M., Samari, S., Mahbubani, K. T., Saeb-Parsy, K., Patone, G., Boyle, J. J., Zhang, H., Zhang, H., Viveiros, A., Oudit, G. Y., Bayraktar, O. A., Seidman, J. G., Seidman, C. E., Noseda, M., Hubner, N. & Teichmann, S. A. (2020). Cells of the adult human heart. Nature. 588, 466-472. [CrossRef]
- Tucker, N. R., Chaffin, M., Fleming, S. J., Hall, A. W., Parsons, V. A., Bedi, K. C., Jr., Akkad, A. D., Herndon, C. N., Arduini, A., Papangeli, I., Roselli, C., Aguet, F., Choi, S. H., Ardlie, K. G., Babadi, M., Margulies, K. B., Stegmann, C. M. & Ellinor, P. T. (2020). Transcriptional and Cellular Diversity of the Human Heart. Circulation. 142, 466-482. [CrossRef]
- Wang, L., Yu, P., Zhou, B., Song, J., Li, Z., Zhang, M., Guo, G., Wang, Y., Chen, X., Han, L. & Hu, S. (2020). Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol. 22, 108-119. [CrossRef]
- Mitchell, P. S., Parkin, R. K., Kroh, E. M., Fritz, B. R., Wyman, S. K., Pogosova-Agadjanyan, E. L., Peterson, A., Noteboom, J., O’Briant, K. C., Allen, A., Lin, D. W., Urban, N., Drescher, C. W., Knudsen, B. S., Stirewalt, D. L., Gentleman, R., Vessella, R. L., Nelson, P. S., Martin, D. B. & Tewari, M. (2008). Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 105, 10513-10518. [CrossRef]
- Adhikari, N., Guan, W., Capaldo, B., Mackey, A. J., Carlson, M., Ramakrishnan, S., Walek, D., Gupta, M., Mitchell, A., Eckman, P., John, R., Ashley, E., Barton, P. J. & Hall, J. L. (2014). Identification of a new target of miR-16, Vacuolar Protein Sorting 4a. PLoS One. 9, e101509. [CrossRef]
- Qian, L., Zhao, Q., Yu, P., Lü, J., Guo, Y., Gong, X., Ding, Y., Yu, S., Fan, L., Fan, H., Zhang, Y., Liu, Z., Sheng, H. & Yu, Z. (2022). Diagnostic potential of a circulating miRNA model associated with therapeutic effect in heart failure. J Transl Med. 20, 267. [CrossRef]
- Wang, T., O’Brien, E. C., Rogers, J. G., Jacoby, D. L., Chen, M. E., Testani, J. M., Bowles, D. E., Milano, C. A., Felker, G. M., Patel, C. B., Bonde, P. N. & Ahmad, T. (2017). Plasma Levels of MicroRNA-155 Are Upregulated with Long-Term Left Ventricular Assist Device Support. Asaio j. 63, 536-541. [CrossRef]
- Dlouha, D., Ivak, P., Netuka, I., Novakova, S., Konarik, M., Tucanova, Z., Lanska, V., Hlavacek, D., Wohlfahrt, P., Hubacek, J. A. & Pitha, J. (2021). The effect of long-term left ventricular assist device support on flow-sensitive plasma microRNA levels. Int J Cardiol. 339, 138-143. [CrossRef]
- Ragusa, R., Di Molfetta, A., D’Aurizio, R., Del Turco, S., Cabiati, M., Del Ry, S., Basta, G., Pitto, L., Amodeo, A., Trivella, M. G., Rizzo, M. & Caselli, C. (2020). Variations of circulating miRNA in paediatric patients with Heart Failure supported with Ventricular Assist Device: a pilot study. Sci Rep. 10, 5905. [CrossRef]
- Lombardi, M., Bonora, M., Baldetti, L., Pieri, M., Scandroglio, A. M., Landoni, G., Zangrillo, A., Foglieni, C. & Consolo, F. (2023). Left ventricular assist devices promote changes in the expression levels of platelet microRNAs. Front Cardiovasc Med. 10, 1178556. [CrossRef]
- Richards, A. M. (2018). N-Terminal B-type Natriuretic Peptide in Heart Failure. Heart Fail Clin. 14, 27-39. [CrossRef]
- Poredos, P., Jezovnik, M. K., Radovancevic, R. & Gregoric, I. D. (2021). Endothelial Function in Patients With Continuous-Flow Left Ventricular Assist Devices. Angiology. 72, 9-15. [CrossRef]
- Pasławska, M., Grodzka, A., Peczyńska, J., Sawicka, B. & Bossowski, A. T. (2024). Role of miRNA in Cardiovascular Diseases in Children-Systematic Review. Int J Mol Sci. 25. [CrossRef]
- Loyaga-Rendon, R. Y., Kazui, T. & Acharya, D. (2021). Antiplatelet and anticoagulation strategies for left ventricular assist devices. Ann Transl Med. 9, 521. [CrossRef]
- Leebeek, F. W. G. & Muslem, R. (2019). Bleeding in critical care associated with left ventricular assist devices: pathophysiology, symptoms, and management. Hematology Am Soc Hematol Educ Program. 2019, 88-96. [CrossRef]
- Leng, Q., Ding, J., Dai, M., Liu, L., Fang, Q., Wang, D. W., Wu, L. & Wang, Y. (2022). Insights Into Platelet-Derived MicroRNAs in Cardiovascular and Oncologic Diseases: Potential Predictor and Therapeutic Target. Front Cardiovasc Med. 9, 879351. [CrossRef]
- Gutmann, C., Joshi, A., Zampetaki, A. & Mayr, M. (2021). The Landscape of Coding and Noncoding RNAs in Platelets. Antioxid Redox Signal. 34, 1200-1216. [CrossRef]
- Kabłak-Ziembicka, A., Badacz, R., Okarski, M., Wawak, M., Przewłocki, T. & Podolec, J. (2023). Cardiac microRNAs: diagnostic and therapeutic potential. Arch Med Sci. 19, 1360-1381. [CrossRef]
Figure 1.
Schematic presentation of heart failure etiology, progression, classification, prevalence, and therapeutic approaches. ACC/AHA - American College of Cardiology/American Heart Association; ACE – angiotensin-converting enzyme; ARBs – angiotensin receptor blockers; ARN – angiotensin receptor/ neprilysin inhibitors; BTR - bridge to recovery; BTT – bridge to transplantation; CRT – cardiac resynchronization therapy; DT – destination therapy; HF – heart failure; HFmrEF – heart failure with mildly reduced ejection fraction; HFpEF – heart failure with preserved ejection fraction; HFrEF – heart failure with reduced ejection fraction; LVAD – left ventricle assist devices; MCS – mechanical circulatory support; MRA - mineralocorticoid receptor antagonist; SGLT2 – sodium-glucose linked cotransporter 2. Created in BioRender. Paic, F. (2025) https://BioRender.com/u96e763.
Figure 1.
Schematic presentation of heart failure etiology, progression, classification, prevalence, and therapeutic approaches. ACC/AHA - American College of Cardiology/American Heart Association; ACE – angiotensin-converting enzyme; ARBs – angiotensin receptor blockers; ARN – angiotensin receptor/ neprilysin inhibitors; BTR - bridge to recovery; BTT – bridge to transplantation; CRT – cardiac resynchronization therapy; DT – destination therapy; HF – heart failure; HFmrEF – heart failure with mildly reduced ejection fraction; HFpEF – heart failure with preserved ejection fraction; HFrEF – heart failure with reduced ejection fraction; LVAD – left ventricle assist devices; MCS – mechanical circulatory support; MRA - mineralocorticoid receptor antagonist; SGLT2 – sodium-glucose linked cotransporter 2. Created in BioRender. Paic, F. (2025) https://BioRender.com/u96e763.
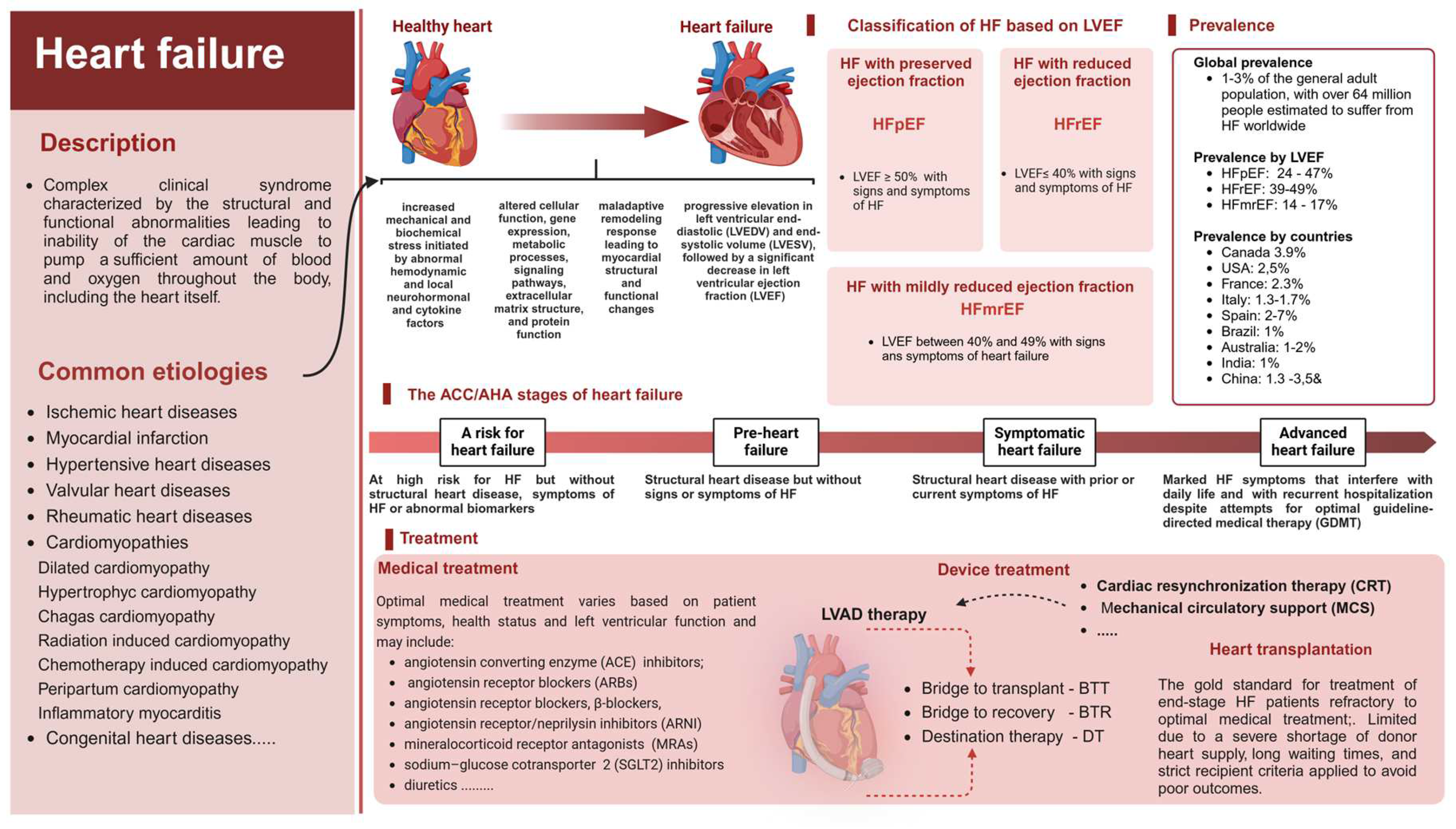
Figure 2.
Established and novel circulating biomarkers of heart failure. ADM – adrenomedullin; ADMA - asymmetric dimethyl arginine; α1-AT - alpha-1 antitrypsin; BDNF - brain-derived neurotrophic factor; BNP - brain natriuretic peptide; CA125 - cancer antigen 125; CRP - C-reactive protein; ET-1 – endothelin 1; FGF-21 - fibroblast growth factor 21; GAL-3 - galectin-3;GDF-15 - growth differentiation factor 15; GSTP1 - glutathione transferase P1; H-FABP - heart-type fatty acid-binding protein; hs-cTnT/CTnI -high-sensitivity cardiac troponins cTnT and cTnI; ICAM-1 - Intercellular Adhesion Molecule 1; IL-1 – interleukin; IL-6 – interleukin 6; IL-8 – interleukin 8; IL-15 – interleukin 15; IL-18 – interleukin 18; IL-33 – interleukin 33; IL-1 RA- interleukin-1 receptor antagonist; KIM-1 - kidney injury molecule-1; lncRNA – long non-coding RNA; MMP-1, matrix metalloproteinase 1; MMP-2 -matrix metalloproteinase 2; MMP-9 - matrix metalloproteinase 9; MPO – myeloperoxidase; MR-proADM - mid-regional pro-adrenomedullin; NGAL - neutrophil gelatinase-associated lipocalin; NO -nitric oxide; NT-proBNP - N-terminal brain natriuretic pro-peptide; NTproCNP - N-Terminal pro C-Type Natriuretic Peptide; Ox LDL - oxidized low-density lipoprotein; PICP - procollagen type I carboxyterminal peptide; PIIINP - pro-collagen type III aminoterminal peptide;sST2 soluble suppression of tumorigenesis-2; TIMP-1 - tissue inhibitor of matrix metalloproteinase 1; TNF-α – transforming growth factor alpha; vWF - Von Willebrand factor; VCAM-1 - vascular cell adhesion protein 1; 8-OHdG - 8-hydroxy-2’ -deoxyguanosine; Created in BioRender. Paic, F. (2025) https://BioRender.com/c26u158.
Figure 2.
Established and novel circulating biomarkers of heart failure. ADM – adrenomedullin; ADMA - asymmetric dimethyl arginine; α1-AT - alpha-1 antitrypsin; BDNF - brain-derived neurotrophic factor; BNP - brain natriuretic peptide; CA125 - cancer antigen 125; CRP - C-reactive protein; ET-1 – endothelin 1; FGF-21 - fibroblast growth factor 21; GAL-3 - galectin-3;GDF-15 - growth differentiation factor 15; GSTP1 - glutathione transferase P1; H-FABP - heart-type fatty acid-binding protein; hs-cTnT/CTnI -high-sensitivity cardiac troponins cTnT and cTnI; ICAM-1 - Intercellular Adhesion Molecule 1; IL-1 – interleukin; IL-6 – interleukin 6; IL-8 – interleukin 8; IL-15 – interleukin 15; IL-18 – interleukin 18; IL-33 – interleukin 33; IL-1 RA- interleukin-1 receptor antagonist; KIM-1 - kidney injury molecule-1; lncRNA – long non-coding RNA; MMP-1, matrix metalloproteinase 1; MMP-2 -matrix metalloproteinase 2; MMP-9 - matrix metalloproteinase 9; MPO – myeloperoxidase; MR-proADM - mid-regional pro-adrenomedullin; NGAL - neutrophil gelatinase-associated lipocalin; NO -nitric oxide; NT-proBNP - N-terminal brain natriuretic pro-peptide; NTproCNP - N-Terminal pro C-Type Natriuretic Peptide; Ox LDL - oxidized low-density lipoprotein; PICP - procollagen type I carboxyterminal peptide; PIIINP - pro-collagen type III aminoterminal peptide;sST2 soluble suppression of tumorigenesis-2; TIMP-1 - tissue inhibitor of matrix metalloproteinase 1; TNF-α – transforming growth factor alpha; vWF - Von Willebrand factor; VCAM-1 - vascular cell adhesion protein 1; 8-OHdG - 8-hydroxy-2’ -deoxyguanosine; Created in BioRender. Paic, F. (2025) https://BioRender.com/c26u158.
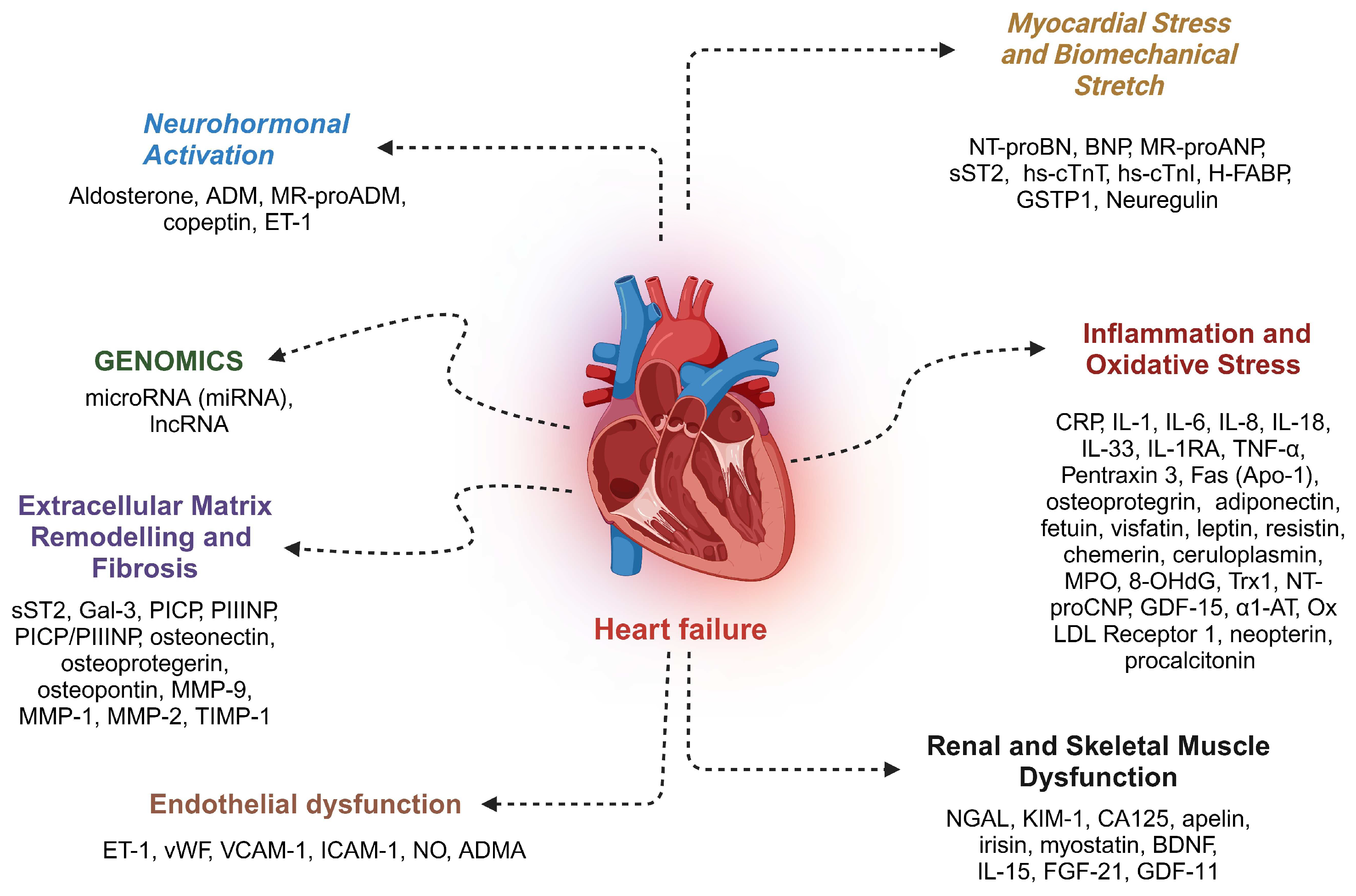
Figure 3.
miRNAs implicated in regulation of cardiac hypertrophy, apoptosis, fibrosis and cardiac arrythmias in heart diseases, i.e., dilated cardiomyophaty, diabetic cardiomyopathy, miocardyal infraction, atrial fibrillation and pressure overload induced heart failure (hypertensove heart disease and transverse aortic constriction). Created in BioRender. Paic, F. (2025) https://BioRender.com/t01x701.
Figure 3.
miRNAs implicated in regulation of cardiac hypertrophy, apoptosis, fibrosis and cardiac arrythmias in heart diseases, i.e., dilated cardiomyophaty, diabetic cardiomyopathy, miocardyal infraction, atrial fibrillation and pressure overload induced heart failure (hypertensove heart disease and transverse aortic constriction). Created in BioRender. Paic, F. (2025) https://BioRender.com/t01x701.
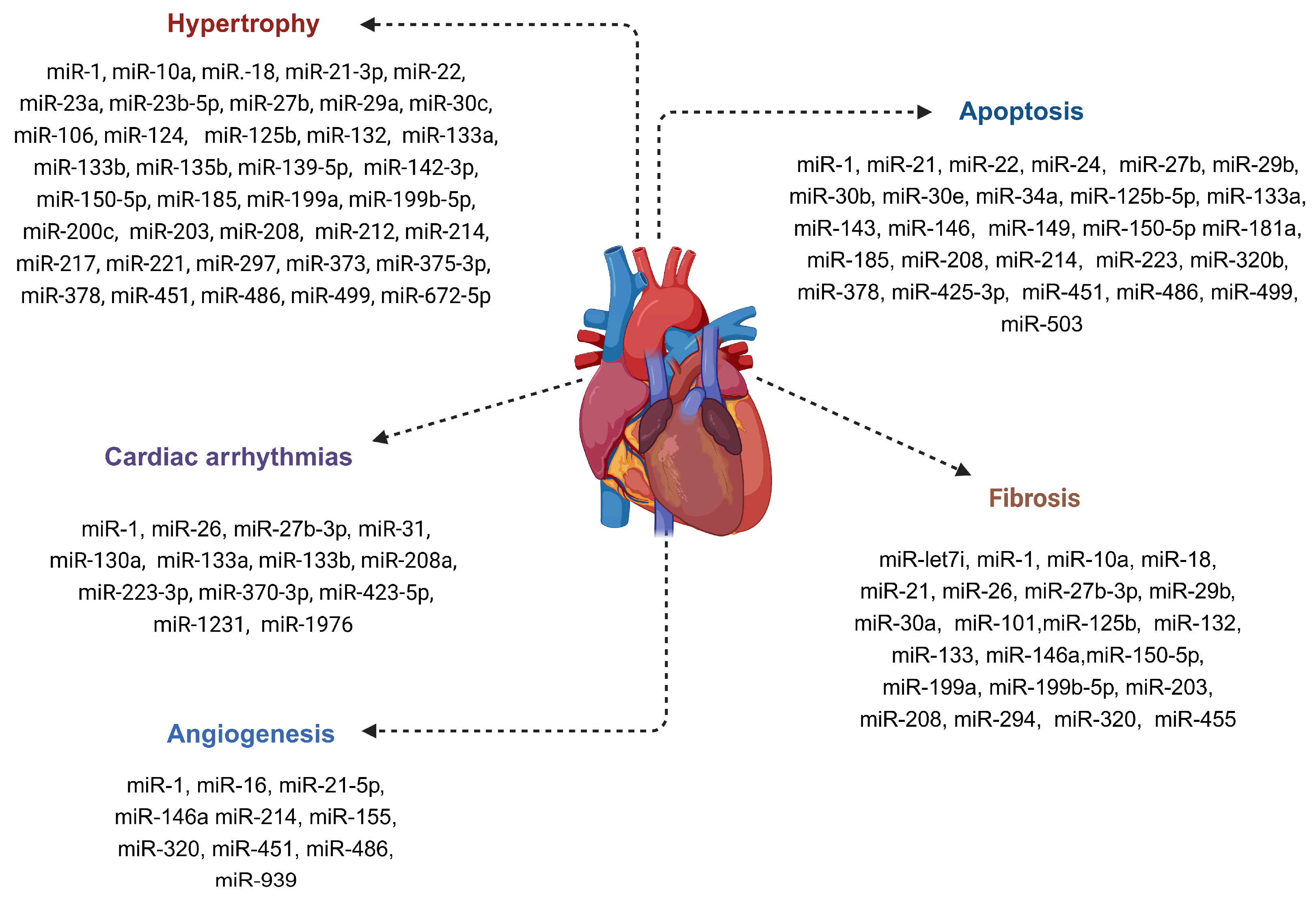

Table 1.
Summary of human miR expression studies in LVAD-supported LV myocardium.
| Study | Methodology | Baseline patient characteristics | HF etiology | LVAD device | Time of LVAD support and/or duration of HF in days | miRNAs of interest | Major findings |
|---|---|---|---|---|---|---|---|
| Schipper et al. [80] | qPCR, 4 miRs |
LVAD:17[ male 88%; age 40 ±13 years, NYHA class IV (%) 100] NF: 6 |
ICM – 8 (47%) NICM - 9 (53%) |
pf-LVAD (HeartMate, Thoratec, Pleasanton, CA) | LVAD support: 262±129 (range 57-557) HF duration - not reported |
miR-1, 133a, 133b and miR-208 | Upregulation and partial normalization of miR-1, mir-133a, and miR-133b in ICM; further decrease in DCM; similar changes of miR-208 - expression level too low for reliable statistical analysis |
| Ramani et al. [101] | PCR-based array; qPCR, 376 miRs |
Test cohort: recovered 7 [male 50 %; age 40±12 years; EF(% + SE) 18±5] dependent 7 (male 50 %, age 42±15 years; LVEF (%) 15±5) Validation cohort; recovered 7 [male 71 %, age 27±8 years; LVEF (%+SE) 18±5); dependent (male 71 %; age 33±9 years; EF (%+SE) 17±5] Paired pre- and post-LVAD samples: 6 [male 50 %; age 54±18 years; LVEF (%+SE) 18±5] NF: 7 [male 50 %; age 36±10 years; LVEF (%+SE) 65±10] |
NICM - 100 % | pf-LVAD (Thoratec, Pleasanton, CA) and rotary cf-LVAD support Test cohort: recovered -rotary 14 %; dependent – rotary – 13 % Validation cohort: recovered – rotary 71 %; dešendent – rotary 42 % |
LVAD support: Test cohort: recovered 53±31; dependent 61±29 ; Validation cohort: recovered 433±250; dependent 369±167; Paired pre- and post-LVAD samples: 144±67 HF duration: Test cohort:: recovered 62±49; dependent 75±58 Validation cohort: recovered 680±1117 ; dependent 771±802 Paired pre- and post-LVAD samples: 210±108 |
miR-1, 15b, 21, 23a, 26a, 27a, 103, 133a, 133b, 142-3p, 181b, 195, 208, 376a, and miR-424 | Downregulated expression of miR-23a and miR-195 in the LVAD-recovered versus LVAD-dependent myocardial tissue probably reflects the less serious nature of HF at the time of LVAD implantation; no notable change in miR expression between pre- and post-LVAD patient group |
| Lok et al. [82] | qPCR, 1 miR |
LVAD - 18 [male 78 %; age 43.0 (28.0–48.0) years; NYHA IV (%) 100]; 15 underwent HTx NF: 10 |
NICM - 100 % | cf-LVAD (Heart-Mate II, Thoratec, CA) | LVAD support (HTx patients): 282 (207–521) HF duration: 1675 (416–1954) |
miR-137↑ | α-1-antichymotrypsin (ACT) confirmed as a direct target for miR-137; miR-137 expression is inversely correlated with ACT mRNA levels in myocardial tissue |
| Barsanti et al. [83]. | RNA sequencing, qPCR, 23 miRs | HTx-LVAD: 8 [male 100 %; age 46 ± 4 years; LVEF (%+SE) 21 ± 2] HTx-ctrl: 9 [male 55.5 %; age 52 ± 3 years; LVEF (%+SE) 26 ± 2] All patients; NYHA class III-IV; LVEF < 35 % at HTX or LVAD implantation |
HTx-LVAD – ICM - 12.5 %, DICM - 87.5 % HTx-ctrl: NICM – 22 %, NICM – 78 % |
cf-LVAD: 7 [ 4 HeartMate II (Thoratec, Pleasanton, CA), 2 De Bakey (MicroMed Technology, Houston, TX), 1 INCOR (Berlin Heart AG, Germany)] pf-LVAD: 1 Best BEAT (NewCorTec, Pomezia, Italy) |
HTX-LVAD support: 357 ± 66 HF duration: not reported |
miR-23a-5p↑, 27a-5p↑, 29b-3p↓, 135a-5p↓, 142-3p↑, 142-5p↑, 144-5p↑, 146b-3p↑, 216a-5p↓, 223-3p↑, 335-3p↑, 338-3p↑, 374b-5p↓, 376a-3p↓, 378g↑, 628-5p↑, 3195↓, 4284↓, 4461↑, 4532↓, 4485↓, 4792↓ and miR-5683↑ | No paired LVAD samples; 13 upregulated and 10 downregulated miRs in the LVAD group with respect to the HTx-ctrl group; a positive correlation was found between some miRs and cardiac index (miR-27a-5p, miR-142-3p, miR-142-5p, miR-223-3p, miR-338-3p, and miR-378g) and pulmonary vascular resistance values (miR-29b-3p and miR-374b-5p) |
| Lok et al. [84]. | PCR-based array; qPCR, 26 miRs | Test cohort – pf-LVAD: 5 [male 100 %; age 38 ± 7 years; NYHA IV (%) 100]; cf-LVAD: 5 [males 80 %; age 49 ± 6 years; NYH class IV (%) 100] Validation cohort - pf-LVAD: 17 [male 82 %; age 45 ± 3 years; NYHA class IV (%) 100]; cf-LVAD : 17 [male 94 %, age 39 ± 3 years; NYHA IV (%) 100] |
Test cohort: NICM – 100 % Validation cohort: NICM 100 % |
Test cohort – pf-LVAD: HeartMate (X)VE 60 %; Thoratec 20 %; Novacor 20 %; cf-LVAD: HeartMate II 100% Validation cohort – pf-LVAD: HeartMate-(X)VE 70 %; Thoractec 18 %; Novacor 6 %; HeartMate-IP 6 %; cf-LVAD: HeartMate-II 100 % |
Test cohort – pf-LVAD support 204 (180–301); HF duration 261 (26–1023); cf-LVAD support 489 (238–897); HF duration 1747 (1531–3316)Validation cohort – pf-LVAD support 282 (197–512); HF duration 1754 (750–2489); cf-LVAD support 206 (190–317); HF duration 398 (49–1344) | let-7i, miR-1-1, 17*, 21, 22, 23a*, 23a, 25, 29b-1*, 92a, 129*↑, 133a, 133b, 136, 137, 142-5p, 146a↑, 155↓, 199a-5p, 199b-5p, 208a, 221↓, 222↓, 320d, 378, and miR-378* | Five miRNAs (miR-129*, 146a, 155, 221, and miR-222) displayed a similar expression pattern among cf- and pf-LVAD devices, whereas others only changed significantly during pf-LVAD (miR-let-7i, 21, 378, and miR-378*) or cf-LVAD support (miR-137); no significant pre- and post-LVAD changes within individual patients |
| Morley-Smith et al.[105]. | qPCR, 2 miRs | Paired pre- and post-LVAD samples - n = 10; no gender and age data available | ICM - 53 % NICM - 47 % |
cf-LVAD (Heart-Mate II, Thoratec, CA) | LVAD support: 200 (133–299) days | miR-483-3p and miR-1202 | Noticeable, although nonsignificant, up-regulation of miR-483–3p, no change in myocardial miR-1202 expression |
| Ragusa et al. [106] | NGS/RNA sequencing, qPCR, 463 miRs | Pediatric HF:13 [male 46 %; age 29 (5–123) months; LVEF (%) 19 (13.75–20.75)] Adult HF: 21 [male 66.7 %, age 60 (50-64) years, LVEF (%) 22.5 (19.5–25)] |
DCM -69.2 % LVNC – 15.4 % RCM – 15.4 % |
Pediatric HF: pf-LVAD n=3 (Thoratec, Berlin Heart Excor); cf-LVAD n=1 (Jarvik); BiVAD n=3 (BiVAD; Thoratec, Berlin Heart Excor) | Not reported | NGS: miR-19a-3p↓, 29b-1-5p↑, 199b-5p↓, 199a-5p↓, 338-3p↑, and miR-1246 ↓ qPCR: miR-1246↓, -19a-3p↓ and miR-199b-5p↓ |
Downregulated expression of miR-19a-3p, miR-199b-5p, and miR-1246 in post-LVAD tissue; down-regulatory effect of miR-19a-3p on cTnC expression; no data for miRNA expression in adult HF patients |
| Ragusa et al. [107] | NGS/RNA sequencing, qPCR, 463 miRs | Pediatric HF:13 [male 46 %; age 29 (5–123) months; LVEF (%) 19 (13.75–20.75)] Plasma samples: 9 pediatric HF patients and 107 healthy children |
DCM 70 % LVNC – 15 % RCM – 15 % |
Pediatric HF: pf-LVAD n=3 (Thoratec, Berlin Heart Excor); cf-LVAD n=1 (Jarvik); BiVAD n=3 (BiVAD; Thoratec, Berlin Heart Excor) | Not reported | NGS: miR-19a-3p↓, 29b-1-5p↑, 199b-5p↓, 199a-5p↓, 338-3p↑, and miR-1246 ↓ qPCR: miR-1246↓, -19a-3p↓ and miR-199b-5p↓ |
myocardial AdipoR2 expression levels were inversely related to miR-19a-3p, miR-199b-5p, and miR-1246 expression; miR-1246 was also negatively associated with T-cad; no relationship was observed among miRNAs and AdipoR1; in vitro validation confirmed regulatory role of miR-1246 and miR-199b-5p on AdipoR2 and miR-199b-5p on T-cad |
| Matkovich et al. [109]. | Microarray, 467 miRs |
LVAD: 10 [male 80 %; age 53 ± 14; ] end-stage HF: 17 [male 64 %; age 56 ± 6; LVEF (%+SE) 14 ± 6 ] NF: 11 [male 36 %; age 56 ± 6, LVEF (%+SE) 62 ± 5 ] |
LVAD: ICM – 40%, NICM – 60% End-stage HF: ICM - 41%, NICM - 59 % |
Not reported | LVAD support- average 51 days Hf duration: LVAD - 1.7 ± 1.0 months; end-stage HF - 68 ± 60 months |
let-7f, let-7g↓, let-7i, miR-1↓, 15a, 16, 21, 22↓, 23a, 24↓, 26a, 26b↓, 27a↓, 27b, 29a, 29b↓, 30b↓, 30a-5p, 30c, 30d, 103, 125b, 126↓, 130a, 133a, 133b, 143↓,195↓, 199a-3p, 378, 499↓, and miR-638 | Eight miRs showed full normalization, while twelve miRs showed significant decreases in expression levels between the failing and LVAD-supported myocardium, no paired LVAD samples; combined miRNA/mRNA signature sufficiently effective in classifying different HF types and functional states |
| Akat et al. [90] | NGS/RNA sequencing; > 500 miRs | NICM 21 [male 95 %; age57 (33–78); LVEF median (range) 15(10–30)] ICM: 13 [male 92 %; age 66 (51–78) LVEF median (range) 17.5(10–22) ] NF; 8 [male 63 %; age 48 (2–80) LVEF, median(range) 57/65(6 unknown) ] Fetal hearts:5 fetal (gestational age 19–24 weeks), LVAD:: 8 NICM, 7 ICM |
NICM- 62 % ICM – 38 % |
pf-LVAD: HeartMate I, Thoratec Corp., Pleasanton, California) cf-LVAD; HeartMate II, (Thoratec Corp., Pleasanton, California); HeartWare (HeartWare International, Framingham, MA, USA) |
Not reported | let-7-f, let-7g, let-7i, miR-1, 15a, 15b, 16, 17*, 19a, 21, 22. 23a*, 23a, 24, 25, 26a, 26b, 27a, 27a*↑, 27b, 29a, 29b, 29b-1*, 30b, 30c, 30d, 92a, 103, 125b, 126, 130a, 133a, 133b, 136-3p, 136-5p, 137, 142-3p, 142-5p, 143, 146a, 155, 181b, 195, 199a-5p, 199a-3p, 199b-5p, 204, 208a, 208b, 216a, 217, 221, 222, 223, 335-3p, 338-3p, 376a, 374b, 376a-3p, 378, 378*, 424, 483-3p, 499, 628 and miR-2114 | Marginal difference in miRs signature between ICM and NICM and no difference in miRNA cistron expression among paired myocardial samples before and after LVAD support; miRNA changes in HF tissues partially resembled that of fetal myocardium |
| Parikh et al. [111]. | Microarray, 58 miRs | LVAD: 8 [male 87.5%); age 57 (45–59) years; LVEF (5) 13 (10–20); LVEF<50% 50 %; NYHA II/IIV 87,5 % ] HF without LVAD: 8 [male 87.5 %; age 50 (43–54) years; LVEF (%) 18 (15–26); LVEF<50% 87.5%; NYHA II/IIV 87.5 %] NF: 6 [age 45 (40–51) years] |
LVAD: NICM – 100 % HF without LVAD: NICM – 100 % |
Not reported | LVAD support: 156 days (131–268 ) HF duration: LVAD - 48 (30–120); HF without LVAD - 39 (6–108) |
LV; miR-10b↓, 95, 103b, 135b, 182↓, 187, 208a, 218,223↓, 224, 299-5p↓, 329, 373, 374b↓, 431, 451↓, 495, 548x↓, 601↓, 628-5p↓, 940↓, 1226, 1226*↓, 1825↓, 3128, 3187-3p↓, 3201, 3910↓, 4269, 4270↓, 4458, 4521, 4539↓, 4687-3p↓, 4689↓, 4741↓, 4793-3p, ENSG00000202498↓ and ENSG00000202498_x↓ RV: miR-10b↓, 21*, 92a-1, 95, 124, 138, 181a-2↓, 182↓, 216a, 217, 373, 431, 451↓, 1247, 1972, 3065-3p, 4461, 4524, and HBII-52-32_x↓ |
miR-4458 in the LV and miR-21*, miR-1972, miR-4461 in the RV were significantly normalized following LVAD implantation. |
| Yang et al. [112] | NGS/RNA sequencing, 1007 miRs | LVAD: 16 [male 81 %, age 60 (54-65) years; LVEF < 35 %] NF: 8 [male 87.5 %; age 53.5 (52-58) years ] |
ICM - 50%, NICM - 50% | Not reported | 305±50 days (range 111 - 690) | miR-23b-5p↑, 93-3p↓, 130b-5p↓, 183-3p↓, 193b-5p↑, 301a-5p↓, 302a-3p↑, 363-3p↓, 365a-3p↑, 378a-3p↑, 378e↑, 378f↑, 425-5p↓, 429, 548d-5p↓, 665↑, 760, and miR-4484↑ | Only seven miRs [miR-365a-3p and miR-378a-3p in ICM and miR-93-3p, miR-193b-5p, miR-425-5p, miR-548d-5p, and miR-760 in NICM] normalized with LVAD support |
| Muthiah et al. [93] | RNA sequencing, 100 miRs | HF: 37 [male 86.5 %; age 49.6 ± 13.1 years; LVEF (%) 23.7±7.7; INTERMACS – I 32.4 %, II 62.1 %, III 5.4 % ] 12 paired pre- and post-LVAD samples were used for RNA sequencing |
NICM- 54 % ICM – 35 % HCM – 8 % CHD – 3 % |
cf-LVAD HeartWare (HeartWare International, Framingham, MA, USA); Five patients had biventricular support with a HeartWare centrifugal-flow LVAD placed in the left LV and RV. |
HF duration: 83 ±68.9 months Paired samples LVAD support: 307 ±132 days |
let-7f-1, miR-1-1, 1-2, 10b-5p, 15a, 15b, 16-1, 21, 23a, 23b, 24-1, 26a-1, 27b, 29a,29a-5p, 29b-1, 30a, 30c-1, 30d,30e-5p, 34a, 34b, 34c, 92a-1, 100, 101-1-5p, 103a-1, 125b-1, 129-1, 130a, 133a-1, 133a-2, 133b,133b-5p, 140-5p, 145-5p, 151a-5p, 155, 182, 192-5p, 195, 199b, 199a-1, 206, 210, 211, 212, 208a, 208b, 214, 214-5p, 221, 328, 378a, 378b, 378c, 378d-1, 378d-2, 378e, 378f, 378g, 378h, 378i, 378j, 423, 451b, 452-5p, 455-5p, 489, 526a-1, and miR-1307-5p | No significant changes in miRs expression were detected |
CHD – congenital heart disease; cf-LVAD – continuous flow LVAD devices; DCM – dilated cardiomyopathy; HCM – hypertrophic cardiomyopathy; HTx -heart transplantation; HTx-ctrl – heart transplant control, ICM – ischemic cardiomyopathy; INTERMACS - interagency registry for mechanically assisted circulatory support; LV – left ventricle; LVEF - left ventricular ejection fraction; LVAD – left ventricular assist device; LVNC- LV non-compaction; NF – non-failing control; NGS - next generation sequencing; NICM – nonischemic cardiomyopathy; NYHA class - New York Heart Association functional classification for heart failure; pf-LVAD – pulsatile flow LVAD devices; qPCR – quantitative real-time PCR; RMC - restrictive cardiomyopathy; RV – right ventricle; SE - standard error; Bold - upregulated in HF compared to NF, ↓ downregulated following LVAD support. ↑ upregulated following LVAD support; italics – normalized after LVAD support; underlined – miRNAs that regulate mRNA transcripts altered in HF myocardium without LVAD support.
Table 2.
Summary of circulatory miR expression studies in LVAD-supported HF patients.
| Study | Methodology | Baseline patient characteristics | HF etiology | LVAD device | Time of LVAD support and/or duration of HF in days | miRNAs of interest | Major findings |
|---|---|---|---|---|---|---|---|
| Akat et al. [110] | RNA sequencing | Plasma - Stable HF: 14 [male 79 %, age 63(49–71) years; LVEF,median(range) 22(10–43)] Advanced HF at LVAD implantation: 24 [male 92 %; age 66(33–78) years; LVEF, median(range) 18(10–24)] Advanced HF 3 (n=10) and 6 months after LVAD implantation (n =10), and at LVAD explantation (n=/) NF: 13 [male 69 %; age 60(32–70) years] Serum – Advanced HF: 7; LVAD explantation: 7; NF 4 |
ICM NICM |
pf-LVAD: HeartMate I, Thoratec Corp., Pleasanton, California) cf-LVAD; HeartMate II, (Thoratec Corp., Pleasanton, California); HeartWare (HeartWare International, Framingham, MA, USA) |
Measured at baseline and 3 and 6 months following LVAD implantation | miR-1-1, 22, 122 , 126, 133b, 203, 208a, 208b, 210, 216a, 375, 499, 1180, and miR-1908 | Up to 140-fold increase in heart and muscle-specific miRs in advanced HF compared to healthy individuals, alongside elevated cardiac troponin I (cTnI) levels indicating myocardial injury; the levels dropped as early as 3 months after the initiation of LVAD support, approaching normal levels, but rose again at LVAD explantation; higher levels of miR-208a, miR-208b, and miR-499 in advanced HF positively correlated with the protein levels of cTnI |
| Morley-Smith et al. [105] | PCR array; qPCR, 1113 miRs | Whole cohort: 19 [male 79 %, age 52 (30–61) years; LVEF (%), median (IQR) 15 (15–25) ] Good responders: 7 [male 71 %; age 40 (14–62) years; LVEF (%), median (IQR) 20 (10–30)] Poor responders: 6 [male 83 %; age 55 (36–60) years; LVEF (%), median (IQR) 15 (13–23)] |
Whole cohort: ICM 53 %; NICM 47 % Good responders: ICM 29 %; NICM 71 % Poor responders: ICM 50 %; NICM 50 % |
Cf-LVAD Thoratec HeartMate II | Measured at baseline and 3, 6, 9, and 12 months following LVAD implantation | PCR array - miR-33a↑, 1254↑, 219-1-3p↑, 483-3p↑, 548 l↓, 557↑, 938↑, 1202↑, 1250↑, 1275↑, 4266↑, and miR-4325↓ qPCR – miR-488-3p↑ and miR-1202 |
Plasma miR-483-3p levels exhibit significant up-regulation with LVAD support that mirrors the suppression of NT-proBNP levels; miR-1202 levels correlate with change in NT-proBNP at three months following LVAD support, thus stratifying the patients into poor vs. good responders; potentially valuable noninvasive biomarkers for monitoring (miR-483-3p) and predicting (miR-1202) patient response to LVAD therapy |
| Adhikari et al. [129] | qPCR, 4 miRs | end-stage HF: 10 [male 57 %; age 60 years; ] NF: 9 [male 57 %; age 45±5 ] Paired pre- and post-LVAD blood samples: 9 [males 89 %; age 60 years; LVEF < 25 % ] NF: 6 [males 60%; age 54±4 ] |
Blood – ICM- 55.5 % NICM -44.5 |
Not reported | Measured at baseline and 7 days following LVAD implantation | miR-15a↓, 16↑, 103, and miR- 195↓ | Downregulated expression of circulatory miR-16 in HF patients; miR-16 targets VPS4a; LVAD support increases the levels of miR 16 thus decreasing the levels of VPS4a |
| Lok et al. [104] | Microarray, qPCR, 4 miRs | Test cohort: 5 [male 40%; age 42 ± 6 ; NYHA class IV(%) 100 %] Validation cohort: 18 [male 78 %; age 45 ± 3; NYHA class IV (%) 100% ] |
NICM 100 %; | cf-LVAD HeartMate-II | Measured at baseline and 1, 3, and 6 months following LVAD implantation | miR-21↓, 146a, 221 and miR-222 | A two-fold upregulation of miR-21 in pre-LVAD samples and decreased expression following LVAD support but did not normalize, fluctuating expression pattern of miR-146a, miR-221, and miR-222 with a tendency to reduction following LVAD support |
| Qian et al. [137] | qPCR, 89 miRs | Discovery phase - In hospital/Out-of-hospital: 40 [age 64.40±11.88; LVEF (%) 34.05±5.77] Training phase – HF 30; NF 15 Validation cohort: HF 50; NF 25 |
ICM- 26 % NICM- 53 % Other – 21 |
External dataset reported by Akat et al. [110]. | External dataset reported by Akat et al. [110]. |
Discovery phase - let-7a, let-7b, let-7c, let-7e, let-7f, let-7g, let-7i, miR-10a, 15a, 15b, 16, 16, 17, 18a, 18b, 19a, 19b, 20b, 21, 23a, 24, 26, 27a, 27b, 29a, 29b, 30a-5p↓, 30d, 92, 93, 99b, 100↓, 103a, 106a, 106b, 122, 125b, 126, 129, 130a, 133a, 133b, 136, 140, 143, 145, 150, 151-5p, 155, 181b, 181a, 182, 191, 195, 199a, 199a-3p, 208a, 214, 221, 222, 302a, 302c, 302d, 302e, 320a↓, 320b, 324-5p, 342, 346, 369-5p, 329, 382, 423-3p, 423-5p, 433, 484, 486, 493, 495, 499a, 499b-5p, 499b-3p↓, 543, 622, 638, 654-5p↑, 665, 675, 762, and miR-885-5p Training and validation phase - miR-30a-5p and miR-654-5p |
Circulatory miR-30a-5p, miR-100, miR-499b, miR-320a, and miR-433 showed significant downregulation, while the levels of miR-654-5p were upregulated in HF patients compared to control; a significantly negative correlation of circulatory miR-654-5p and a positive correlation of miR-30a-5p with NT-proBNP plasma levels of HF patients; a novel 2-circulating miRNA (miR-30a-5p/miR-654-5p) model with diagnostic and prognostic potential |
| Wang et al. [138] | qPCR, 23 miRs | HF – 40 [male 72.5%; age 67 (51–74); LVEF (%) 20 (15-20); INTERMACS I/II 57.5% ] NF - 7 |
Not reported | Cf-LVAD - Heartmate II (62.5%), HeartWare | SMeasured at serial blood draws - median 96.5 (72–150) days post-LVAD implantation | miRNA-1, 10a, 15b, 16, 21, 24, 27a, 27b, 29a, 92a, 103, 126, 133a, 146a, 146b, 155↑, 159a, 195, 221, 222, 320, 423, and miR-872 | Upregulated expression of circulatory miR-155 following LVAD support |
| Dlouha et al. [139] | qPCR, 9 miRs | LVAD treatment- 33 [male 85 %; age 55.7 ± 11.6 ] NF 13 [male 61.5 %; age 50.1 ± 13.5 years; LVEF (%+SE) 18.9 ± 3.2] |
ICM: 48.5 % NICM – 51.5 % |
cfLVAD: axial HeartMate II – 42 % centrifugal HeartMate III 58 % |
Measured at baseline and 3, 6, 9, and 12 months after LVAD implantation | miR-10a, 10b, 21, 126↑, 146a↑, 146b, 155, 663a, and miR-663b | Iow pulsatile flow up-regulates plasma levels of circulating flow-sensitive miRNAs; increased plasma levels of miR-126 and miR-146a following cf-LVAD support; positive association between miR-155 and Belcaro score; an inverse correlation between miR-126 and endothelial function, measured as the reactive hyperemia index |
| Ragusa et al. [140] | NGS/RNA sequencing, qPCR, 340 miRs | NGS analysis – HF 5 [male 40 %; age 13.8±6.25 years; LVEF (%+SE) 16.6±1.7 ] LVAD treatment – 8 [male 37.5 %; age 25.25 ± 10.9 years; LVEF (%+SE) 21.7 ± 5.6] |
NGS HF - NICM 80 %; LVNC 20 % LVAD treatment – NICM 75 %, LVNC 25 % |
Berlin Heart Chamber | Measured at baseline and 4 hrs, and 1, 3, 7, 14, and 30 days after LVAD implantation LVAD support (179 ± 34.71 days) |
miR-16-5p, miR-30a-5p, 127-3p, miR-150-3p↑, miR-375↑, 409-3p↓, 432-5p, 483-3p, 483-5p↓, 485-3p↓, miR-941. 3135b, and miR-4433b-3p | miR-409-3p, miR-483-3p, and miR-485-3p were downregulated up to undetectable levels, while miR-432-5p showed a trend to decrease, while the circulatory levels of miR-150-3p and miR-375 increased after one month of LVAD implantation; in vitro data confirmed coagulation factors 7 (F7) and F2a as targets of hsa-miR-409-3p; a potential role for circulatory miRs, particularly 409-3p, as an early biomarker for monitoring hemostasis-related adverse events in pediatric LVAD-supported HF patients |
| Lombardi et al. [141] | qPCR, 12 miRs | HF-15 [male 100 %; age 64 (63–71) years; chronic HF 73 % ] Control – 5 (male 100 %; age 53 (47–61) years] |
ICM – 60 % NICM - 40 % |
cf-LVAD: Heartmate III (HM3; Abbott, United States) 53 %; HVAD (Medtronic Inc., United States) 47 % | measured before and 1, 6, and 12 months following LVAD implantation | miR-19b-3p, 20b-5p, 25-3p↑, 126-5p, 144-3p, 151a-3p, 223-3p, 320a, 374b-5p, 382-5p, 451a↑, 454-3p, 16-5pa and miR-103a-3pa |
significantly different pre-implant levels of platelet miR-126, miR-374b, miR-223, and miR-320a in HF patients compared to controls. The LVAD patients suffering from bleeding had significantly higher pre-implant levels of platelet miR-151a and miR-454. The same miRs were also differentially expressed in these patients following LVAD implantation early before the clinical manifestation of the bleeding events. The expression levels of platelet miR-25, miR-144, miR-320, and miR-451a changed significantly over the course of LVAD support, indicating partial or increasing platelet activation. |
ICM – ischemic cardiomyopathy; INTERMACS - interagency registry for mechanically assisted circulatory support; LV – left ventricle; LVEF - left ventricular ejection fraction; LVAD – left ventricular assist device; LVNC- LV non-compaction; NF – non-failing control; NGS - next generation sequencing; NICM – nonischemic cardiomyopathy; NYHA class - New York Heart Association functional classification for heart failure; pf-LVAD – pulsatile flow LVAD devices; qPCR – quantitative real-time PCR; RV – right ventricle; SE - standard error; Bold - upregulated in HF compared to NF, ↓ downregulated following LVAD support. ↑ upregulated following LVAD support; italics – normalized after LVAD support.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



