
Submitted:
05 February 2025
Posted:
06 February 2025
You are already at the latest version
Abstract
Background/Objectives: Pulmonary arterial hypertension (PAH) is a progressive vascular disorder characterized by increased pulmonary vascular resistance, right ventricular dysfunction, and high mortality rates. Despite advancements in vasodilatory therapies, PAH remains a life-threatening condition with limited curative options. This review aims to explore emerging molecular mechanisms, novel therapeutic targets, and future research directions in PAH treatment, focusing on strategies to improve long-term patient outcomes. Methods: This systematic review synthesizes recent advancements in PAH pathophysiology and therapeutic development. A structured literature search was conducted in PubMed and ClinicalTrials.gov using keywords such as “Pulmonary Arterial Hypertension,” “vascular remodeling,” “metabolic dysfunction,” and “emerging therapies.” Studies published between 2015 and 2025 were included, with a focus on preclinical models, clinical trials, and translational research. Key areas of investigation include vascular remodeling, metabolic dysregulation, inflammation, and right ventricular dysfunction. The review also evaluates the potential of novel pharmacological agents, gene-based therapies, and AI-driven diagnostics for PAH management. Results: Recent studies highlight dysregulated BMPR2 signaling, epigenetic modifications, and inflammatory cytokine pathways as critical contributors to PAH progression. Emerging therapies such as JAK-STAT inhibitors, metabolic reprogramming agents, and mesenchymal stromal cell-derived extracellular vesicles (EVs) show promise in preclinical and early clinical trials. Additionally, AI-enhanced imaging and non-invasive biomarkers are improving PAH diagnostics. Future research directions emphasize precision medicine approaches and the development of RV-targeted therapies. Conclusions: PAH remains a complex and fatal disease requiring multifaceted therapeutic strategies beyond traditional vasodilation. Advances in molecular-targeted treatments, AI-driven diagnostics, and personalized medicine offer new hope for disease-modifying interventions. Future research must bridge translational gaps to bring novel therapies from bench to bedside, improving survival and quality of life in PAH patients.
Keywords:
1. Background
2. Introduction
2.1. Gaps in Current Research and Treatment
2.2. Objective of This Review
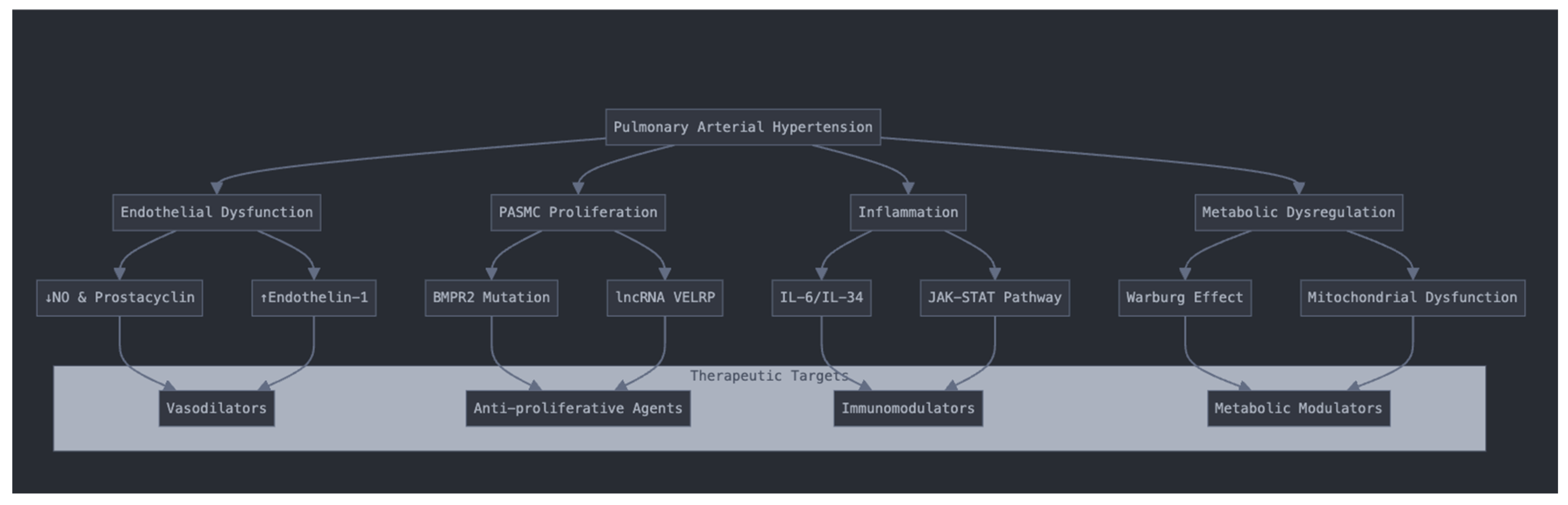
3. Pathophysiology and Molecular Mechanisms of PAH
3.1. Endothelial Dysfunction and Vascular Remodeling
3.2. Pulmonary Arterial Smooth Muscle Cell Proliferation and Resistance to Apoptosis
3.3. Inflammatory and Immune Dysregulation
3.4. Epigenetics and Genetic Modifications in PAH
3.5. Metabolic Dysregulation and Mitochondrial Dysfunction
4. Emerging Therapeutic Strategies for Pulmonary Arterial Hypertension (PAH)
4.1. Targeting Pulmonary Vascular Remodeling
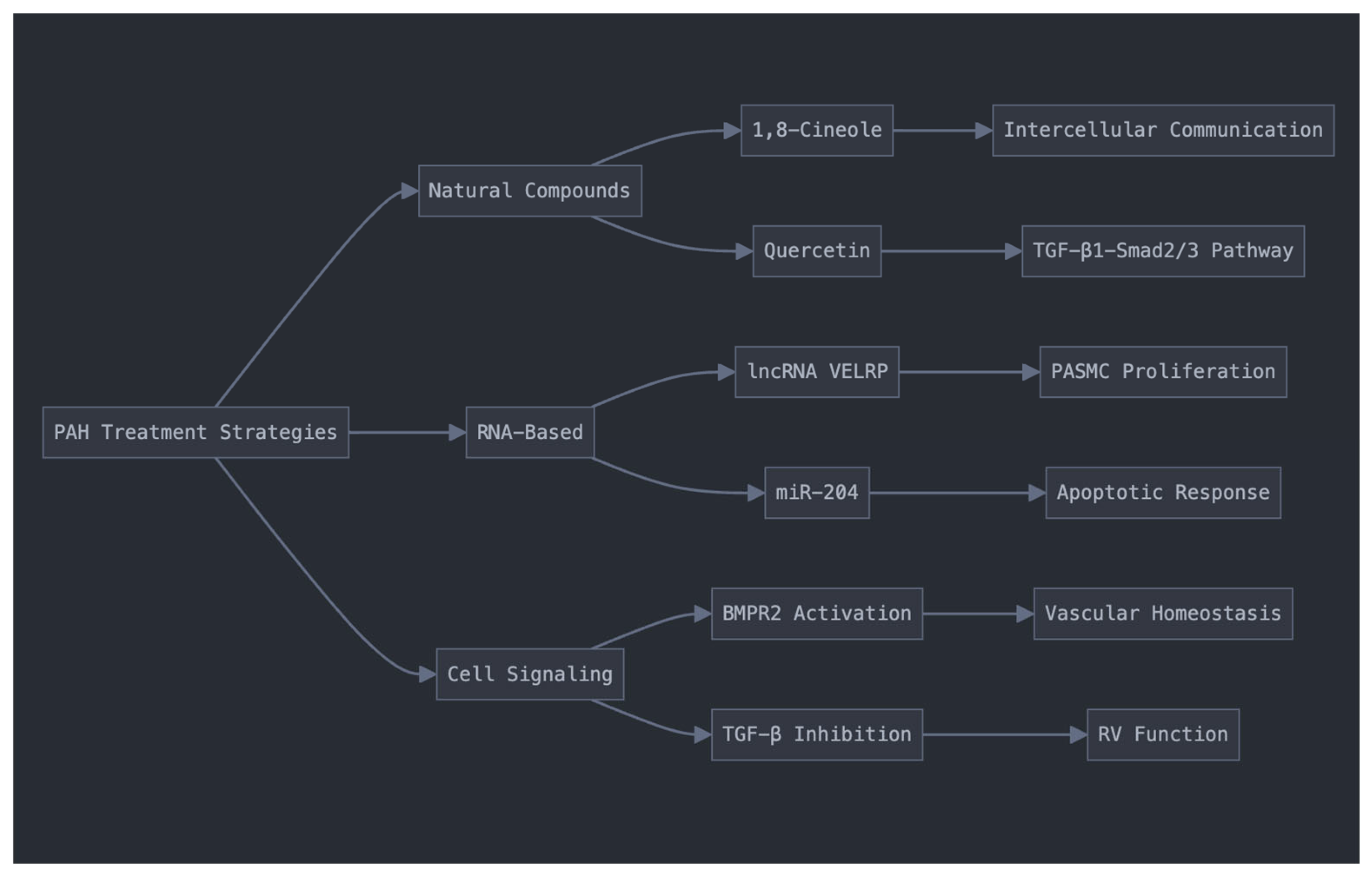
4.1.1. Natural Compounds
4.1.2. RNA-Based Interventions
4.1.3. Cell Signaling Pathway Modulation
4.2. Immunomodulatory and Anti-Inflammatory Therapies
4.3. Metabolic Modulation in PAH
4.4. Novel Pharmacological Interventions
4.5. Right Ventricular-Directed Therapies
5. Challenges in PAH Research
5.1. Heterogeneity of PAH Etiology
5.2. Lack of Early Diagnostic Biomarkers
5.3. Limited Translational Success
5.4. Right Ventricular Dysfunction Is Understudied
6. Future Research Directions
6.1. Precision Medicine Approaches
6.2. Novel Drug Discovery and Repurposing
6.2.1. Gene Therapy and RNA-Based Interventions
6.2.2. Stem Cell and Extracellular Vesicle (EV) Therapy
6.3. Advanced Imaging and Non-Invasive Diagnostics
6.4. Overcoming Barriers to Clinical Translation
7. Conclusions
7.1. Key Takeaways from This Review
7.1.1. Pathophysiology and Molecular Mechanisms
7.1.2. Emerging Therapies
7.1.3. Future Directions and Research Challenges
7.2. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PAH | Pulmonary Arterial Hypertension |
| RV | Right Ventricle |
| PVR | Pulmonary Vascular Resistance |
| PASMCs | Pulmonary Arterial Smooth Muscle Cells |
| BMPR2 | Bone Morphogenetic Protein Receptor Type 2 |
| ET-1 | Endothelin-1 |
| NO | Nitric Oxide |
| Jak-Stat | Janus-Kinase-Signal Transducer and Activator of Transcription |
| HDAC | Histone Deacetylase |
| TGF-β | Transforming Growth Factor-Beta |
| miRNA | MicroRNA |
| lncRNA | Long Non-Coding RNA |
| PDE5 | Phosphodiesterase Type 5 |
| cGMP | Cyclic Guanosine Monophosphate |
| PKG | Protein Kinase G |
| EVs | Extracellular Vesicles |
| MSC | Mesenchymal Stromal Cells |
| AI | Artificial Intelligence |
| scRNA-seq | Single-Cell RNA Sequencing |
| RHC | Right Heath Catheterization |
References
- C. Guignabert, “From basic scientific research to the development of new drugs for pulmonary arterial hypertension: insights from activin-targeting agents,” Breathe, vol. 21, no. 1, Jan. 2025. [CrossRef]
- Y. Zhang et al., “Endogenous hydrogen sulfide persulfidates endothelin type A receptor to inhibit pulmonary arterial smooth muscle cell proliferation,” Redox Biology, vol. 80, p. 103493, Mar. 2025. [CrossRef]
- X. Tian et al., “Loss of Type 2 Bone Morphogenetic Protein Receptor Activates NOD-Like Receptor Family Protein 3/Gasdermin E-Mediated Pyroptosis in Pulmonary Arterial Hypertension,” Journal of the American Heart Association, vol. 14, no. 3, p. e034726, Feb. 2025. [CrossRef]
- J. Hou et al., “Integrated Transcriptomic and Metabolomic Analysis of Rat PASMCs Reveals the Underlying Mechanism for Pulmonary Arterial Hypertension,” American Journal of Hypertension, p. hpaf015, Feb. 2025. [CrossRef]
- C. Wittig et al., “Shear stress unveils patient-specific transcriptional signatures in PAH: Towards personalized molecular diagnostics,” Theranostics, vol. 15, no. 5, pp. 1589–1605, Jan. 2025. [CrossRef]
- T. Fujiwara, S. Ishii, S. Minatsuki, M. Hatano, and N. Takeda, “Exploring Novel Therapeutics for Pulmonary Arterial Hypertension,” International Heart Journal, vol. 66, no. 1, pp. 3–12, 2025. [CrossRef]
- J. Tan et al., “Celastrol Ameliorates Hypoxia-Induced Pulmonary Hypertension by Regulation of the PDE5-cGMP-PKG Signaling Pathway,” Phytotherapy Research, vol. n/a, no. [CrossRef]
- M. C. van de Veerdonk, H. J. Bogaard, and N. F. Voelkel, “The right ventricle and pulmonary hypertension,” Heart Fail Rev, vol. 21, no. 3, pp. 259–271, May 2016. [CrossRef]
- J. Hudson and L. Farkas, “Epigenetic Regulation of Endothelial Dysfunction and Inflammation in Pulmonary Arterial Hypertension,” International Journal of Molecular Sciences, vol. 22, no. 22, Art. no. 22, Jan. 2021. [CrossRef]
- J. J. Ryan and S. L. Archer, “The Right Ventricle in Pulmonary Arterial Hypertension,” Circulation Research, vol. 115, no. 1, pp. 176–188, Jun. 2014. [CrossRef]
- C. Guignabert et al., “Pathology and pathobiology of pulmonary hypertension: current insights and future directions,” European Respiratory Journal, vol. 64, no. 4, Oct. 2024. [CrossRef]
- J. Hannemann and R. Böger, “Dysregulation of the Nitric Oxide/Dimethylarginine Pathway in Hypoxic Pulmonary Vasoconstriction—Molecular Mechanisms and Clinical Significance,” Front. Med., vol. 9, Feb. 2022. [CrossRef]
- Y.-H. Shen et al., “Panorama of artery endothelial cell dysfunction in pulmonary arterial hypertension,” Journal of Molecular and Cellular Cardiology, vol. 197, pp. 61–77, Dec. 2024. [CrossRef]
- W. Zhang et al., “An Overview of miRNAs Involved in PASMC Phenotypic Switching in Pulmonary Hypertension,” BioMed Research International, vol. 2021, no. 1, p. 5765029, 2021. [CrossRef]
- “Sex Dimorphism in Pulmonary Arterial Hypertension Associated With Autoimmune Diseases | Arteriosclerosis, Thrombosis, and Vascular Biology.” Accessed: Feb. 04, 2025. [Online]. Available: https://www.ahajournals.org/doi/10.1161/ATVBAHA.124.320886.
- C. Liu et al., “lncRNA VELRP Modulates Pulmonary Arterial Smooth Muscle Cell Proliferation and Promotes Vascular Remodeling in Pulmonary Hypertension,” Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 44, no. 12, pp. 2560–2576, Dec. 2024. [CrossRef]
- H. A. Bolayır et al., “Inflammatory and cardiac biomarkers in pulmonary arterial hypertension: The prognostic role of IL-34,” Heart & Lung, vol. 69, pp. 202–207, Jan. 2025. [CrossRef]
- D. Yerabolu et al., “Targeting Jak–Stat Signaling in Experimental Pulmonary Hypertension,” Am J Respir Cell Mol Biol, vol. 64, no. 1, pp. 100–114, Jan. 2021. [CrossRef]
- Roger, J. Milara, P. Montero, and J. Cortijo, “The Role of JAK/STAT Molecular Pathway in Vascular Remodeling Associated with Pulmonary Hypertension,” International Journal of Molecular Sciences, vol. 22, no. 9, Art. no. 9, Jan. 2021. [CrossRef]
- G. H. Kim, J. J. Ryan, G. Marsboom, and S. L. Archer, “Epigenetic Mechanisms of Pulmonary Hypertension,” Pulm Circ, vol. 1, no. 3, pp. 347–356, Jul. 2011. [CrossRef]
- Q. Yang, Z. Lu, R. Ramchandran, L. D. Longo, and J. U. Raj, “Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high-altitude long-term hypoxia: role of histone acetylation,” American Journal of Physiology-Lung Cellular and Molecular Physiology, vol. 303, no. 11, pp. L1001–L1010, Dec. 2012. [CrossRef]
- R.-J. Gao et al., “Quercetin regulates pulmonary vascular remodeling in pulmonary hypertension by downregulating TGF-β1-Smad2/3 pathway,” BMC Cardiovascular Disorders, vol. 24, no. 1, p. 535, Oct. 2024. [CrossRef]
- Boucherat et al., “HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension,” Sci Rep, vol. 7, no. 1, p. 4546, Jul. 2017. [CrossRef]
- W. Xu and S. C. Erzurum, “Endothelial Cell Energy Metabolism, Proliferation, and Apoptosis in Pulmonary Hypertension,” Compr Physiol, vol. 1, no. 1, pp. 357–372, Jan. 2011. [CrossRef]
- J. Ryan, A. Dasgupta, J. Huston, K.-H. Chen, and S. L. Archer, “Mitochondrial dynamics in pulmonary arterial hypertension,” J Mol Med, vol. 93, no. 3, pp. 229–242, Mar. 2015. [CrossRef]
- Y. Gong et al., “Inhibition of phosphodiesterase 5 reduces bone mass by suppression of canonical Wnt signaling,” Cell Death Dis, vol. 5, no. 11, pp. e1544–e1544, Nov. 2014. [CrossRef]
- N. Zhang et al., “[Quercetin improves pulmonary arterial hypertension in rats by regulating the HMGB1/RAGE/NF-κB pathway],” Nan Fang Yi Ke Da Xue Xue Bao, vol. 43, no. 9, pp. 1606–1612, Sep. 2023. [CrossRef]
- Q. Jin et al., “Long noncoding RNAs: emerging roles in pulmonary hypertension,” Heart Fail Rev, vol. 25, no. 5, pp. 795–815, Sep. 2020. [CrossRef]
- J. Liu, Y. Liu, F. Wang, and M. Liang, “miR-204: Molecular Regulation and Role in Cardiovascular and Renal Diseases,” Hypertension, vol. 78, no. 2, pp. 270–281, Aug. 2021. [CrossRef]
- B. J. Dunmore, R. J. Jones, M. R. Toshner, P. D. Upton, and N. W. Morrell, “Approaches to treat pulmonary arterial hypertension by targeting BMPR2: from cell membrane to nucleus,” Cardiovascular Research, vol. 117, no. 11, pp. 2309–2325, Oct. 2021. [CrossRef]
- L. Wang et al., “Dysregulated Smooth Muscle Cell BMPR2–ARRB2 Axis Causes Pulmonary Hypertension,” Circulation Research, vol. 132, no. 5, pp. 545–564, Mar. 2023. [CrossRef]
- P. Andre, S. R. Joshi, S. D. Briscoe, M. J. Alexander, G. Li, and R. Kumar, “Therapeutic Approaches for Treating Pulmonary Arterial Hypertension by Correcting Imbalanced TGF-β Superfamily Signaling,” Front. Med., vol. 8, Jan. 2022. [CrossRef]
- “Full article: Emerging biologics for the treatment of pulmonary arterial hypertension.” Accessed: Feb. 04, 2025. [Online]. Available: https://www.tandfonline.com/doi/full/10.1080/1061186X.2023.2199351.
- H. A. Bolayır et al., “Inflammatory and cardiac biomarkers in pulmonary arterial hypertension: The prognostic role of IL-34,” Heart & Lung, vol. 69, pp. 202–207, Jan. 2025. [CrossRef]
- X. Zhang et al., “Dysfunction in mitochondrial electron transport chain drives the pathogenesis of pulmonary arterial hypertension: insights from a multi-omics investigation,” Respiratory Research, vol. 26, no. 1, p. 29, Jan. 2025. [CrossRef]
- J. Tan et al., “Celastrol Ameliorates Hypoxia-Induced Pulmonary Hypertension by Regulation of the PDE5-cGMP-PKG Signaling Pathway,” Phytotherapy Research, vol. n/a, no. [CrossRef]
- E. Legchenko et al., “PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation,” Sci. Transl. Med., vol. 10, no. 438, p. eaao0303, Apr. 2018. [CrossRef]
- S. Goren, N. Kidwai, W. S. Aronow, and G. M. Lanier, “The Role of Intravenous Selexipag in Managing PAH and Bridging Gaps in Oral Treatment: A Narrative Review,” TCRM, vol. 21, pp. 55–60, Jan. 2025. [CrossRef]
- Y. Xiong, Y. Wang, T. Yang, Y. Luo, S. Xu, and L. Li, “Receptor Tyrosine Kinase: Still an Interesting Target to Inhibit the Proliferation of Vascular Smooth Muscle Cells,” Am J Cardiovasc Drugs, vol. 23, no. 5, pp. 497–518, Sep. 2023. [CrossRef]
- M. Kuntz, M. M. Leiva-Juarez, and S. Luthra, “Systematic Review of Randomized Controlled Trials of Endothelin Receptor Antagonists for Pulmonary Arterial Hypertension,” Lung, vol. 194, no. 5, pp. 723–732, Oct. 2016. [CrossRef]
- S. Appunni et al., “Molecular remodeling in comorbidities associated with heart failure: a current update,” Mol Biol Rep, vol. 51, no. 1, p. 1092, Oct. 2024. [CrossRef]
- “Right heart failure in pulmonary hypertension: Diagnosis and new perspectives on vascular and direct right ventricular treatment - Tello - 2021 - British Journal of Pharmacology - Wiley Online Library.” Accessed: Feb. 04, 2025. [Online]. Available: https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14866.
- F. T. Bekedam, M. J. Goumans, H. J. Bogaard, F. S. de Man, and A. Llucià-Valldeperas, “Molecular mechanisms and targets of right ventricular fibrosis in pulmonary hypertension,” Pharmacology & Therapeutics, vol. 244, p. 108389, Apr. 2023. [CrossRef]
- J. Dave, V. Jagana, R. Janostiak, and M. Bisserier, “Unraveling the epigenetic landscape of pulmonary arterial hypertension: implications for personalized medicine development,” J Transl Med, vol. 21, no. 1, p. 477, Jul. 2023. [CrossRef]
- S. Saha, S. Majumdar, and P. Bhattacharyya, “Pulmonary Hypertension,” in Pulmonomics: Omics Approaches for Understanding Pulmonary Diseases, S. Saha, S. Majumdar, and P. Bhattacharyya, Eds., Singapore: Springer Nature, 2023, pp. 201–239. [CrossRef]
- J. Weatherald et al., “The evolving landscape of pulmonary arterial hypertension clinical trials,” The Lancet, vol. 400, no. 10366, pp. 1884–1898, Nov. 2022. [CrossRef]
- Baroutidou et al., “Haemoptysis in Pulmonary Arterial Hypertension Associated with Congenital Heart Disease: Insights on Pathophysiology, Diagnosis and Management,” Journal of Clinical Medicine, vol. 11, no. 3, Art. no. 3, Jan. 2022. [CrossRef]
- R. Rafikov, V. de Jesus Perez, A. Dekan, T. V. Kudryashova, and O. Rafikova, “Deciphering the Complexities of Pulmonary Hypertension: The Emergent Role of Single-Cell Omics,” Am J Respir Cell Mol Biol, vol. 72, no. 1, pp. 32–40, Jan. 2025. [CrossRef]
- E. K. Reem, A. S. Antonella, B. Olivier, B. Sebastien, P. Steeve, and P. Francois, “Multiomics Integration for Identifying Treatment Targets, Drug Development, and Diagnostic Designs in PAH,” Advances in Pulmonary Hypertension, vol. 23, no. 2, pp. 33–42, Jan. 2025. [CrossRef]
- Cuthbertson, N. W. Morrell, and P. Caruso, “BMPR2 Mutation and Metabolic Reprogramming in Pulmonary Arterial Hypertension,” Circulation Research, vol. 132, no. 1, pp. 109–126, Jan. 2023. [CrossRef]
- Y. Hussain, J.-H. Cui, H. Khan, P. Makvandi, and W. Alam, “Biomacromolecule-mediated pulmonary delivery of siRNA and anti-sense oligos: challenges and possible solutions,” Expert Reviews in Molecular Medicine, vol. 23, p. e22, Jan. 2021. [CrossRef]
- H. Guo, Y. Su, and F. Deng, “Effects of Mesenchymal Stromal Cell-Derived Extracellular Vesicles in Lung Diseases: Current Status and Future Perspectives,” Stem Cell Rev and Rep, vol. 17, no. 2, pp. 440–458, Apr. 2021. [CrossRef]
- Q. Qu, Y. Pang, C. Zhang, L. Liu, and Y. Bi, “Exosomes derived from human umbilical cord mesenchymal stem cells inhibit vein graft intimal hyperplasia and accelerate reendothelialization by enhancing endothelial function,” Stem Cell Res Ther, vol. 11, no. 1, p. 133, Mar. 2020. [CrossRef]
- V. Anand, A. D. Weston, C. G. Scott, G. C. Kane, P. A. Pellikka, and R. E. Carter, “Machine Learning for Diagnosis of Pulmonary Hypertension by Echocardiography,” Mayo Clinic Proceedings, vol. 99, no. 2, pp. 260–270, Feb. 2024. [CrossRef]
- L. Deng et al., “MicroRNA-143 Activation Regulates Smooth Muscle and Endothelial Cell Crosstalk in Pulmonary Arterial Hypertension,” Circulation Research, vol. 117, no. 10, pp. 870–883, Oct. 2015. [CrossRef]
- N. L. Kazanskiy, S. N. Khonina, and M. A. Butt, “A review on flexible wearables – Recent developments in non-invasive continuous health monitoring,” Sensors and Actuators A: Physical, vol. 366, p. 114993, Feb. 2024. [CrossRef]
- J. Weatherald et al., “Clinical trial design, end-points, and emerging therapies in pulmonary arterial hypertension,” European Respiratory Journal, vol. 64, no. 4, Oct. 2024. [CrossRef]
- M. C. Fortin and J. Szilagyi, “In Vitro Toxicology: Next Generation Models and Methods to Improve Safety Evaluation,” in Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays, F. J. Hock and M. K. Pugsley, Eds., Cham: Springer International Publishing, 2024, pp. 2529–2557. [CrossRef]
- C. Napoli, G. Benincasa, and J. Loscalzo, “Epigenetic Inheritance Underlying Pulmonary Arterial Hypertension,” Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 39, no. 4, pp. 653–664, Apr. 2019. [CrossRef]
- S. Dhoble, V. Patravale, E. Weaver, D. A. Lamprou, and T. Patravale, “Comprehensive review on novel targets and emerging therapeutic modalities for pulmonary arterial Hypertension,” International Journal of Pharmaceutics, vol. 621, p. 121792, Jun. 2022. [CrossRef]
- J. M. Alves-Silva et al., “1,8-Cineole ameliorates right ventricle dysfunction associated with pulmonary arterial hypertension by restoring connexin43 and mitochondrial homeostasis,” Pharmacological Research, vol. 180, p. 106151, Jun. 2022. [CrossRef]
- J.-H. Xu, J.-P. Liang, C.-J. Zhu, and Y.-J. Lian, “Mesenchymal Stem Cell-Derived Extracellular Vesicles Therapy for Pulmonary Hypertension: A Comprehensive Review of Preclinical Studies,” Journal of Interventional Cardiology, vol. 2022, no. 1, p. 5451947, 2022. [CrossRef]
- C. J. Rhodes, A. J. Sweatt, and B. A. Maron, “Harnessing Big Data to Advance Treatment and Understanding of Pulmonary Hypertension,” Circulation Research, vol. 130, no. 9, pp. 1423–1444, Apr. 2022. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



